Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2025; 15(7):2814-2833. doi:10.7150/thno.106018 This issue Cite
Research Paper
FBXW7/GSK3β-mediated proline-rich 11 degradation promotes oxidative DNA damage and inhibits tumor progression in renal cell carcinoma
Siming Chen1,2#, Kangping Xiong1#, Jianmin Liu1#, Shijie Yao3#, Mingxing Li1, Jingtian Yu1, Gang Wang2,4, Sheng Tu1, Wan Jin4,5, Jiageng Shi1, Yu Xiao4, Yi Zhang5,6, Kaiyu Qian2 ![]() , Lingao Ju4†
, Lingao Ju4† ![]() , Xinghuan Wang1,7,8
, Xinghuan Wang1,7,8 ![]()
1. Department of Urology, Zhongnan Hospital of Wuhan University, Wuhan, China.
2. Hubei Key Laboratory of Urological Diseases, Zhongnan Hospital of Wuhan University, Wuhan, China.
3. Department of Gynecological Oncology, Zhongnan Hospital of Wuhan University, Wuhan, China.
4. Department of Biological Repositories, Human Genetic Resources Preservation Center of Hubei Province, Zhongnan Hospital of Wuhan University, Wuhan, China.
5. Euler Technology, ZGC Life Sciences Park, Beijing, China.
6. Center for Quantitative Biology, School of Life Sciences, Peking University, Beijing, China.
7. Wuhan Research Center for Infectious Diseases and Cancer, Chinese Academy of Medical Sciences, Wuhan, China.
8. Medical Research Institute, Frontier Science Center for Immunology and Metabolism, Taikang Center for Life and Medical Sciences, Wuhan University, Wuhan, China.
#These authors contributed equally to this work.
†Lead contact.
Received 2024-10-30; Accepted 2025-1-24; Published 2025-2-3
Abstract

Rationale: Renal cell carcinoma (RCC) is a highly malignant and common urological tumor. In our previous study, we reported the upregulation of PRR11 in RCC, emphasizing its important role in cell cycle regulation and apoptosis. In this follow-up study, we aim to further investigate the carcinogenic mechanism of PRR11.
Methods: Immunoprecipitation-mass spectrometry (IP-MS), ubiquitination assays, and in vitro phosphorylation assays were used to investigate the phosphorylation and ubiquitination-mediated degradation of PRR11 by FBXW7 and GSK3β. RNA-seq analysis of PRR11 knockdown RCC cells and cellular functional assays, including flow cytometry and comet assays, were performed to explore downstream signaling pathways and regulatory functions. Mouse subcutaneous tumor, tail vein lung metastasis, and popliteal lymph node metastasis models were established to validate PRR11's role in vivo.
Results: Our results reveal that GSK3β recognizes and phosphorylates the CDC4 phosphodegron (CPD) consensus motif of PRR11, enabling FBXW7 to bind to PRR11 and catalyze its K48-linked ubiquitination and degradation. Moreover, PRR11 activates AKT signaling, which inhibits GSK3β activity. This inhibition prevents the phosphorylation of CPD motifs on PRR11, thereby obstructing FBXW7-mediated ubiquitination and degradation. The interaction between PRR11 and AKT creates a positive feedback loop that increases the level of both proteins, which ultimately accelerates RCC progression by inhibiting oxidative DNA damage.
Conclusion: The FBXW7/GSK3β-PRR11-AKT axis plays a pivotal role in the development of RCC by regulating oxidative DNA damage. Targeting PRR11 may be a potential therapeutic strategy for RCC.
Keywords: Renal cell carcinoma, PRR11, FBXW7, GSK3β, AKT signaling, oxidative DNA damage.
Introduction
Renal cell carcinoma (RCC) accounts for 4.01% of all adult malignancies and 28.48% of urinary malignancies, making it the third most prevalent cancer of the urinary system after bladder and prostate cancer [1, 2]. A considerable number of patients present with advanced disease, with up to 17% already harboring distant metastases at the time of diagnosis [2]. Moreover, the prognosis for patients with advanced RCC is especially poor, with an average 5-year survival rate of approximately 8% [3]. Oxidative stress is a hallmark of tumor cells and is pervasive throughout tumorigenesis and progression [4]. Several studies have demonstrated the critical role that oxidative DNA damage plays in the development of RCC [5, 6]. However, the specific mechanism underlying oxidative DNA damage in RCC remains largely elusive, emphasizing the importance of investigating RCC mechanisms for the development of potential diagnostic and therapeutic approaches.
In our previous study, we identified proline-rich 11 (PRR11) as a poor prognostic marker for RCC, demonstrating its role in promoting RCC cell proliferation and metastasis. Additionally, PRR11 can affect cell cycle progression by inducing E2F1 protein degradation via interaction with E2F1 [7]. Here, we further explored its molecular mechanisms in RCC. PRR11, located on chromosome 17q22-23, encodes a protein with an N-terminal structural domain containing a proline-rich region and a C-terminal structural domain with numerous motifs [8, 9]. Notably, the C-terminal domain of PRR11 harbors abundant ubiquitination and phosphorylation sites crucial for PRR11 protein function [9]. Subsequent research has consistently confirmed the oncogenic role of PRR11 in various tumors, attracting because of its significance as an oncogene [10]. In cutaneous squamous cell carcinoma, PRR11 promotes tumor progression by activating the EGFR signaling pathway [11]. In non-small cell lung cancer, PRR11 regulates cytoskeleton-nucleoskeleton assembly and chromatin remodeling by recruiting ARP2/3 complex and can promote tumor metastasis by facilitating filamentous pseudopod formation via ARP2/3 complex [12, 13]. High PRR11 expression is also strongly associated with poor prognosis in other urinary system malignancies, including prostate and bladder cancer [14, 15]. However, the carcinogenic mechanism of high levels of PRR11 remains to be explored.
An essential member of the F-box family, F-box and WD repeat domain containing 7 (FBXW7), serves as the target protein recognition element of the SKP1-Cullin1-F-box (SCF) E3 ubiquitin ligase complex [16]. FBXW7 has been shown to degrade many key regulators of cellular function by targeting them at cell cycle checkpoints, tumor cell proliferation, DNA damage repair, genomic instability, regulation of cell survival, and tumor microenvironment regulation [17-19]. For the most part, FBXW7 binds to a substrate only when the substrate with the conserved CDC4 phosphodegron (CPD) motif is phosphorylated by one or more kinases. T/S-P-X-X-S/T is the typical sequence of CPD, in which a particular kinase phosphorylates serine or threonine residues before FBXW7 recognizes them [20]. Currently, numerous investigations have verified that FBXW7 mediates the ubiquitination of numerous important tumor proteins, including NOTCH1, c-MYC, c-JUN, and CCNE1 [21]. Therefore, FBXW7 is currently recognized as a classical tumor suppressor protein. In addition, FBXW7 was found to be widely mutated in human malignant tumors, with a total mutation rate of nearly 6% [22]. In RCC, FBXW7 overexpression induces cell apoptosis and inhibits tumor proliferation and metastasis [23]. However, many important FBXW7 targets involved in tumor development remain to be characterized.
In this study, we report that FBXW7 mediates the ubiquitination and degradation of PRR11 through the GSK3β-mediated phosphorylation of PRR11 at Thr287, Ser291, Thr326, and Thr330. Furthermore, we demonstrated the important role of the AKT-GSK3β axis in preventing FBXW7-mediated PRR11 degradation. Importantly, we revealed that PRR11 affects the oxidative DNA damage process in tumors and activates the AKT pathway. This regulatory network forms a positive feedback loop that accelerates RCC development.
Results
GSK3β promotes PRR11 phosphorylation and ubiquitin-dependent degradation
Previously, we demonstrated that PRR11 is an independent and adverse prognostic factor for RCC that promotes RCC tumorigenesis [7]. Given the potential importance of PRR11 in RCC, we aimed to identify our PRR11-binding partners of interest. HA-PRR11 was transfected into 293T cells, followed by IP-MS analysis. We identified GSK3β as a potential interacting factor for PRR11, and in addition, previously reported PRR11 interacting proteins such as ARPC1A and ARPC1B were also identified, confirming the validity of our screen for identifying PRR11 interacting proteins [13] (Figure 1A, Figure S1A and Dataset S1). Co-immunoprecipitation (co-IP) assays confirmed the interaction between exogenous HA-PRR11 and Flag-GSK3β (Figure S1B), which was further validated in RCC cells for endogenous PRR11 and GSK3β interactions (Figure 1B and Figure S1C-D). Importantly, GST pull-down assay showed direct binding of recombinant GST-PRR11 and recombinant His-GSK3β in vitro (Figure 1C). To explore the precise binding region of PRR11 and GSK3β more deeply, we constructed fragmented HA-PRR11 for co-IP assays and revealed that the amino acids 251-360 of PRR11 could interact with GSK3β (Figure 1D). Interestingly, this region contains numerous ubiquitination and phosphorylation sites, including two CPD motifs. Considering the reported susceptibility of PRR11 to proteasomal degradation [9] and the binding of GSK3β to CPD motif-containing regions, we hypothesized that GSK3β might mediate PRR11 degradation via phosphorylation. Subsequent investigations revealed that GSK3β overexpression attenuated the steady-state expression of PRR11, an effect effectively blocked by proteasome inhibitor MG132, λ-protein phosphatase (λ-PPase), and the GSK3β inhibitors CHIR-99021 and LiCl (Figure 1E). Moreover, knockdown or overexpression of GSK3β in ACHN and Caki-1 cells increased or decreased PRR11 protein levels, respectively, without affecting PRR11 mRNA levels (Figure S1E-G). We then found that CHIR-99021 treatment reduced the degradation rate of endogenous PRR11 in ACHN cells (Figure S1H). Notably, GSK3β overexpression significantly promoted the degradation rate of exogenous HA-PRR11 protein, whereas the catalytic-deficient mutant of GSK3β (GSK3β-S9D and GSK3β-Y216A) failed to accelerate PRR11 degradation (Figure 1F and Figure S1I). Additionally, in vivo ubiquitination assays showed that GSK3β overexpression promoted PRR11 ubiquitination in a dose-dependent manner, whereas the catalytic-deficient mutant of GSK3β significantly inhibited PRR11 ubiquitination (Figure 1G and Figure S1J).
GSK3β interacts with PRR11 to regulate PRR11 stability. (A) HA-PRR11 plasmid was overexpressed in 293T cells, and the results were analyzed by co-IP with HA antibody, followed by silver staining (top) and mass spectrometry (bottom). (B) Western blot analysis of PRR11 in ACHN cells after IP analysis with GSK3β antibody. (C) Western blot analysis of His-GSK3β, GST-PRR11 after GST pull-down assays. (D) Diagram of the truncation of PRR11 (top). Full-length or truncated mutants of HA-PRR11 and Flag-GSK3β plasmid were transfected into 293T cells, and Western blot analysis of Flag-GSK3β was performed after IP analysis with HA antibody (bottom). PR: Proline-rich domain; P: CDC4 Phosphodegron (CPD) motif; D-box: D-box motif; KEN box: KEN box motif. (E) MG132 (10 μM), λ-PPase (8 U/μL), or GSK3β inhibitors CHIR-99021 (10 μM) and LiCl (20 mM) were added to 293T cells transfected with HA-PRR111 and Flag-GSK3β, after which Western blot analysis of HA-PRR11 was performed. (F) After exposing 293T cells transfected with HA-PRR11, Flag-GSK3β-WT, or Flag-GSK3β-Mut (GSK3β-S9D and GSK3β-Y216A) to CHX (50 μg/mL) for the indicated durations, Western blot analysis was conducted for HA-PRR11 (left). Quantification of PRR11 half-life (right, n = 3 biologically independent experiments). (G) 293T cells transfected with HA-PRR11, Myc-Ub, Vector, Flag-GSK3β-WT, or Flag-GSK3β-Mut were incubated with MG132 (10 μM) for 6 h, and Western blot analysis of Myc-Ub was performed after IP analysis with HA antibody. (H) GSK3β phosphorylation recognition sequence was compared with the conserved PRR11 sequence (left). Design the corresponding PRR11 dephosphorylation mimic mutants (PRR11-1A: T287A/S291A, PRR11-2A: T326A/T330A, PRR11-1A/2A: T287A/S291A/T326A/T330A), PRR11 phosphorylation mimic mutants (PRR11-1D: T287D/S291D, PRR11-2D: T326D/T330D, PRR11-1D/2D: T287D/S291D/T326D/T330D) and PRR11 phosphorylation motif deletion mutants (PRR11-1Δ: I286_S291del, PRR11-2Δ: L325_T330del, PRR11-1Δ/2Δ: I286_S291del/L325_T330del) according to the conserved sequence recognized by PRR11 (right). (I) 293T cells were transfected with the indicated plasmids and Western blot analysis of HA-PRR11 was performed. (J) HA-PRR11-WT was phosphorylated in vitro with active GSK3β and ATP-γ-S, and immunoblotted after alkylation with PNBM. (K) HA-PRR11-WT and HA-PRR11-1A/2A were phosphorylated in vitro with active GSK3β and ATP-γ-S, and immunoblotted after alkylation with PNBM. (L) 293T cells transfected with Myc-Ub, HA-PRR11-WT, or HA-PRR11-1D/2D were treated with DMSO or CHIR-990211 (10 μM) and incubated with MG132 (10 μM) for 6 h before cell collection, and then Western blot analysis of Myc-Ub was performed after IP analysis with HA antibody. (M) 293T cells transfected with Myc-Ub, Flag-GSK3β, HA-PRR11-WT, or HA-PRR11-1A/2A were incubated with MG132 (10 μM) for 6 h, and Western blot analysis of Myc-Ub was performed after IP analysis with HA antibody. Protein levels were quantitatively detected with ImageJ software, and linear regression was used to analyze the protein half-life (F). Data are presented as mean values ± SD.
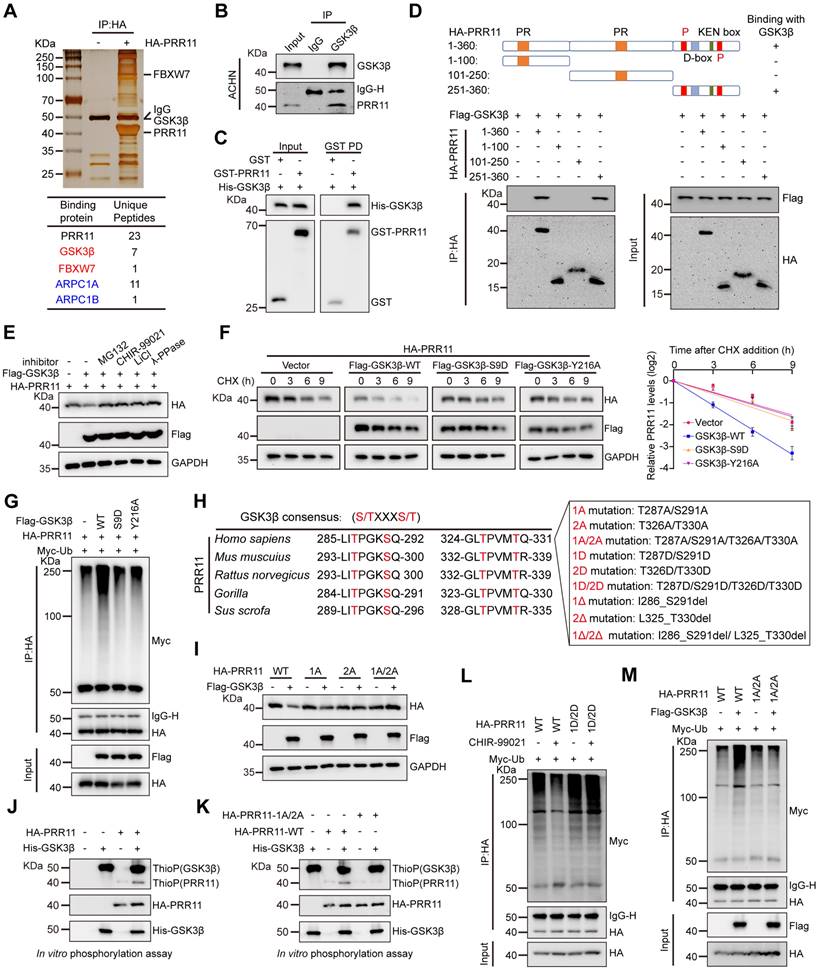
GSK3β typically binds to a substrate-specific motif (S/T-X-X-X-S/T) to catalyze phosphorylation [24]. Therefore, we conducted a search and identified a GSK3β-specific phosphorylation motif in the PRR11 protein sequence that is conserved in mammals. Therefore, we constructed PRR11 dephosphorylation mimic mutants (PRR11-1A: T287A/S291A, PRR11-2A: T326A/T330A, PRR11-1A/2A: T287A/S291A/T326A/T330A), PRR11 phosphorylation mimic mutants (PRR11-1D: T287D/S291D, PRR11-2D: T326D/T330D, PRR11-1D/2D: T287D/S291D/T326D/T330D) and PRR11 CPD motif deletion mutants (PRR11-1Δ: I286_S291del, PRR11-2Δ: L325_T330del, PRR11-1Δ/2Δ: I286_S291del/L325_T330del) (Figure 1H). Notably, while GSK3β overexpression attenuated the protein levels of PRR11-WT, it did not affect the protein levels of dephosphorylation mimic mutants (Figure 1I). To investigate whether GSK3β could directly phosphorylate PRR11, we performed an in vitro phosphorylation assay using active GSK3β and showed that PRR11 could be directly phosphorylated by GSK3β (Figure 1J). In addition, mutating T287/S291/T326/T330 of PRR11 to alanine largely reduced the phosphorylation of PRR11 by GSK3β, indicating that T287/S291/T326/T330 of PRR11 are indeed phosphorylation sites for GSK3β (Figure 1K). Similarly, the GSK3β inhibitor CHIR-99021 inhibited the ubiquitination of PRR11-WT but not PRR11-1D/2D (Figure 1L). In addition, PRR11-WT underwent GSK3β-mediated ubiquitination, whereas PRR11-1A/2A mutants resisted the effects of GSK3β (Figure 1M). Collectively, these results suggest that GSK3β mediates the ubiquitination and degradation of PRR11 by phosphorylating the T287/S291/T326/T330 sites of PRR11.
FBXW7 destabilizes PRR11 via ubiquitination
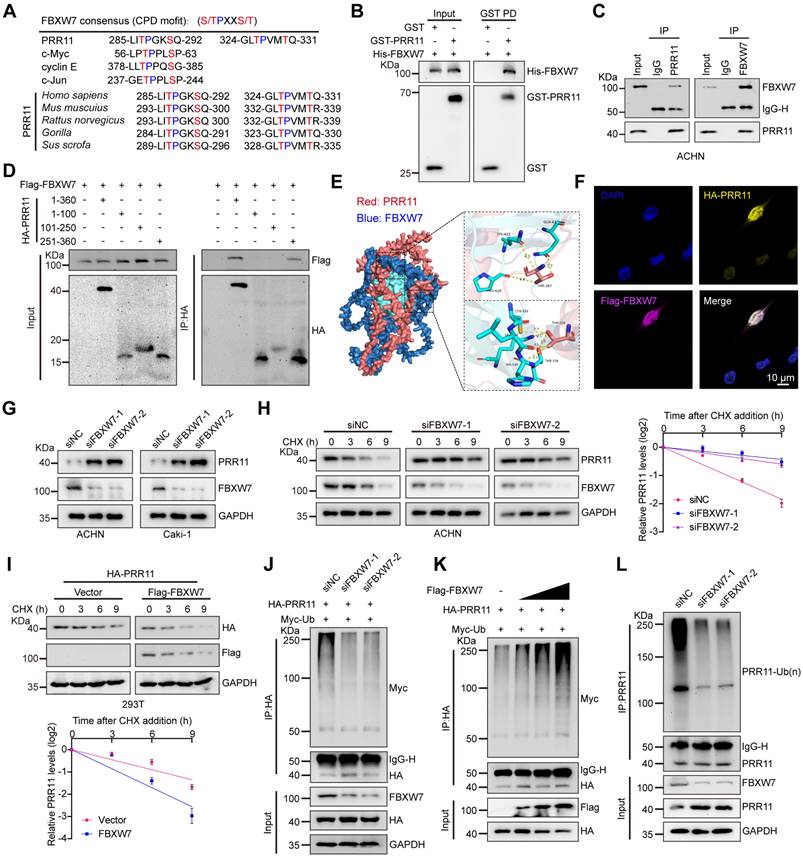
Given the role of FBXW7 in degrading various key proteins, such as c-MYC, CCNE1, and c-JUN, through recognition of their CPD (T/S-P-X-X-S/T) motifs and since the phosphorylation site of GSK3β on PRR11 coincides with the FBXW7-binding motif (Figure 2A), we explored FBXW7 as a potential interacting protein of PRR11 through IP-MS analysis (Figure 1A and Figure S2A). Therefore, we identified FBXW7 as a potential E3 ligase involved in GSK3β-mediated PRR11 degradation. Indeed, we demonstrated a direct binding relationship between recombinant GST-PRR11 and recombinant His-FBXW7 by GST pull-down assay (Figure 2B). Next, we confirmed the interaction of FBXW7 with PRR11 using both endogenous and exogenous co-IP assays (Figure 2C and Figure S2B-C). Additionally, similar to GSK3β, FBXW7 binds to PRR11 via its amino acid domain 251-360 (Figure 2D). Protein molecular docking simulation experiments also showed that the C-terminal structural domain of PRR11 mainly interacts with the WD40 structural domain of FBXW7. Fine-structure interaction data indicated that Thr287 and Thr326 of PRR11 interact tightly with the WD40 structural domain of FBXW7 through multiple hydrogen bonds (Figure 2E). Table S1 showed the detailed information of the molecular docking interaction site. Immunofluorescence analysis revealed that FBXW7 and PRR11 colocalized in the nucleus (Figure 2F). Silencing FBXW7 in ACHN and Caki-1 cells resulted in elevated PRR11 protein expression, whereas overexpression of FBXW7 led to decreased PRR11 protein levels (Figure 2G and Figure S2D), with no effect on PRR11 mRNA levels (Figure S2E). Furthermore, PRR11 protein levels were reduced by FBXW7 in a dose-dependent manner (Figure S2F), and proteasome inhibitor MG132 prevented FBXW7 from degrading the PRR11 protein (Figure S2G). Consistently, silencing FBXW7 in RCC cells slowed the PRR11 half-life (Figure 2H and Figure S2H), whereas overexpressing FBXW7 in 293T cells accelerated the PRR11 half-life (Figure 2I). Moreover, silencing FBXW7 attenuated PRR11 ubiquitination in 293T cells (Figure 2J), whereas overexpression of FBXW7 promoted PRR11 ubiquitination in a dose-dependent manner (Figure 2K). Similarly, FBXW7 silencing in ACHN cells reduced endogenous PRR11 ubiquitination (Figure 2L). These findings collectively indicate that FBXW7 targets PRR11 for ubiquitination and subsequent degradation.
FBXW7 promotes ubiquitination and degradation of PRR11. (A) Comparison of conserved PRR11 sequences with known CPD motifs recognized by FBXW7. (B) Western blot analysis of His-FBXW7, GST-PRR11 after GST pull-down assays. (C) Western blot analysis of PRR11 and FBXW7 in ACHN cells after IP analysis with PRR11 (left) and FBXW7 (right) antibodies. (D) Full-length or truncated mutants of HA-PRR11 and Flag-FBXW7 plasmid were transfected into 293T cells, and Western blot analysis of Flag-FBXW7 was performed after IP analysis with HA antibody. (E) Protein docking model of FBXW7 (blue) and PRR11 (red). Hydrogen bonds are shown in yellow, and the WD40 domain of FBXW7 is shown in cyan. (F) Subcellular localization of overexpression of HA-PRR11 (yellow) and Flag-FBXW7 (magenta) in ACHN cells was detected by immunofluorescence. (G) Western blot analysis of PRR11 expression in RCC cells after FBXW7 knockdown. (H) ACHN cells transfected with siFBXW7-1 or siFBXW7-2 were treated with CHX (50 μg/mL) for the indicated durations, after which Western blot analysis of PRR11 expression was performed (left). PRR11 half-life quantification (right, n = 3 biological independent experiments). (I) 293T cells transfected with Flag-FBXW7 and HA-PRR11 were treated with CHX (50 μg/mL) for indicated durations, after which Western blot analysis was performed for HA-PRR11 (top). Quantification of PRR11 half-life (bottom, n = 3 biologically independent experiments). (J) 293T cells transfected with siFBXW7, HA-PRR11, and Myc-Ub were incubated with MG132 (10 μM) for 6 h, and Western blot analysis of Myc-Ub was performed after IP analysis with HA antibody. (K) 293T cells transfected with Flag-FBXW7 (0.2 μg, 1.0 μg, or 4.0 μg), HA-PRR11, and Myc-Ub were incubated with MG132 (10 μM) for 6 h, and Western blot analysis of Myc-Ub was performed after IP analysis with HA antibody. (L) FBXW7 knockdown ACHN cells were incubated with MG132 (10 μM) for 6 h, IP analysis using PRR11 antibody and Western blot analysis to detect endogenous ubiquitination. Protein levels were quantitatively detected with ImageJ software, and Linear Regression for analyzing protein half-life (H-I). Data are presented as mean values ± SD.
FBXW7-mediated destabilization of PRR11 via its CPD motif
By further investigating whether the CPD motif of PRR11 is associated with FBXW7 binding and ubiquitination, we found that PRR11 dephosphorylation mimic mutants and CPD motif deletion mutants reduced the interaction of FBXW7 with PRR11 (Figure 3A and Figure S3A), whereas PRR11 phosphorylation mimic mutants enhanced this interaction (Figure S3B). Importantly, PRR11 dephosphorylation mimic mutants and CPD motif deletion mutants presented a prolonged protein half-life and inhibited ubiquitination (Figure 3B-C and Figure S3C, E), whereas phosphorylation mimic mutants presented a shorter half-life and enhanced ubiquitination (Figure S3D, F).
FBXW7 degrades PRR11 via the CPD motif of PRR11. (A) Flag-FBXW7, Vector, HA-PRR11-WT, or PRR11 dephosphorylation mimic mutants (HA-PRR11-1A, HA-PRR11-2A, and HA-PRR11-1A/2A) plasmid were transfected into 293T cells. After IP analysis with HA antibody, Western blot analysis of Flag-FBXW7 was performed. (B) 293T cells transfected with Flag-FBXW7, HA-PRR11-WT, or PRR11 dephosphorylation mimic mutants were treated with CHX (50 μg/mL) for indicated times, and then Western blot analysis was performed for HA-PRR11 (left). Quantification of PRR11 half-life (right, n = 3 biologically independent experiments). (C) 293T cells transfected with Flag-FBXW7, Myc-Ub, HA-PRR11-WT, or PRR11 dephosphorylation mimic mutants were incubated with MG132 (10 μM) for 6 h, and Western blot analysis of Myc-Ub was performed after IP analysis with HA antibody. (D) 293T cells transfected with HA-PRR11, Vector, Flag-FBXW7, Myc-Ub-WT, or Myc-Ub-Mut (K6O, K11O, K27O, K29O, K33O, K48O, or K63O) were incubated with MG132 (10 μM) for 6 h, and Western blot analysis of Myc-Ub was performed after IP analysis with HA antibody. K6O, K11O, K27O, K29O, K33O, K48O, and K63O appearing represent ubiquitin mutants (including K6-only, K11-only, K27-only, K29-only, K33-only, K48-only, and K63-only) that retain only a single lysine residue, respectively. (E) 293T cells transfected with HA-PRR11, Myc-Ub-Mut (K48O or K48R), Vector, or Flag-FBXW7 were incubated with MG132 (10 μM) for 6 h, and Western blot analysis of Myc-Ub was performed after IP analysis with HA antibody. K48O and K48R appearing represent ubiquitin mutant retaining only the K48 lysine residue and ubiquitin mutant in which only lysine residue 48 is mutated to arginine, respectively. (F) Representative images of immunohistochemical staining of PRR11 and FBXW7 proteins in HKidE180Su02 cohort in RCC adjacent tissue and each AJCC stage (The seventh edition of AJCC: stage I, II, III, IV) (left). 149 RCC samples were quantified and the correlation between PRR11 and FBXW7 protein levels was analyzed based on Pearson test (right). (G) Statistical plots of the expression levels of PRR11 and FBXW7 in HKidE180Su02 cohort in different AJCC stages. (H-I) The RCC samples were divided into high expression and low expression groups based on median PRR11 and FBXW7 protein levels, respectively, and survival analyses were performed by the log-rank test of Kaplan-Meier analysis. PRR11 average optical density (AOD) = integral optical density (IOD)/area. Protein levels were quantitatively detected with ImageJ software, and linear regression was used to analyze the protein half-life (B). The p-values were calculated with two-tailed Student's t-test (G). Data are presented as mean values ± SD.
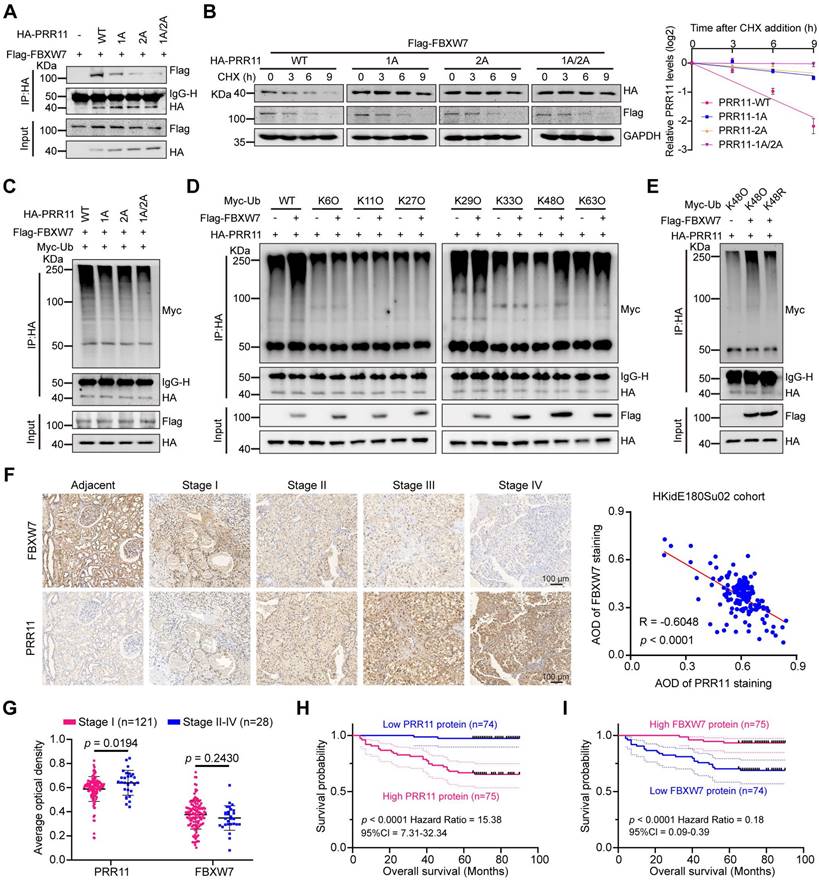
FBXW7 is frequently mutated in human cancers, with R505 and R479 identified as common mutation hotspots [16]. To investigate their effects, we constructed FBXW7-R505C and FBXW7-R479Q mutant plasmids. Additionally, an FBXW7 catalytic-deficient mutant (FBXW7-ΔF-box) was constructed to examine the role of FBXW7 catalytic activity in regulating PRR11. Compared to FBXW7-WT, both FBXW7-R505C and FBXW7-R479Q mutants exhibited significantly reduced binding to PRR11 and inhibited the degradation of PRR11. Although FBXW7-ΔF-box retained its binding ability to PRR11, it significantly weakened the degradation of PRR11 (Figure S4A-B). Furthermore, all FBXW7 mutants reduced the ubiquitination of PRR11 and extended its protein half-life (Figure S4C-D). To determine the type of ubiquitin chain in which PRR11 is ubiquitinated by FBXW7, we generated a series of mutant ubiquitin plasmids. The results showed that FBXW7 efficiently catalyzed the K48-linked ubiquitin chain on PRR11 (Figure 3D), whereas the K48R ubiquitin mutant blocked FBXW7-induced ubiquitination of PRR11 (Figure 3E). These findings collectively suggest that phosphorylation of the PRR11 CPD motif is essential for FBXW7-mediated K48 ubiquitination of PRR11.
To further characterize the relationship between PRR11 and FBXW7, we quantified PRR11 and FBXW7 protein expression by IHC in 149 clinical RCC samples from the HKidE180Su02 cohort and found that there was a significant negative correlation between PRR11 and FBXW7 protein levels (Figure 3F). In addition, PRR11 expression was significantly positively correlated with tumor stage and pathological grade, and although FBXW7 expression did not significantly differ with tumor stage or tumor size, it exhibited a negative correlation trend (Figure 3G and Table S2-S3). In terms of prognosis, PRR11 expression was strongly correlated with the poor prognosis of RCC patients, and in contrast to PRR11, high expression of FBXW7 significantly improved the overall survival of RCC patients (Figure 3H-I). These findings indicate that PRR11 expression is negatively correlated with FBXW7 expression in RCC and that higher PRR11 expression is associated with advanced tumor stage, pathological grade, and poor prognosis, whereas elevated FBXW7 expression improves overall survival in RCC patients.
FBXW7 promotes PRR11 ubiquitination in a GSK3β-mediated phosphorylation-dependent manner
Subsequently, we continued to explore the synergistic regulation of PRR11 by GSK3β and FBXW7. By GST pull-down assay, we found that there is no competition for binding of GSK3β and FBXW7 to PRR11 interactions (Figure S5A). Interestingly, the interaction between FBXW7 and PRR11 was attenuated by the addition of λ-PPase to cellular protein extracts (Figure S5B). More importantly, both CHIR-99021 treatment and GSK3β knockdown weakened the interactions between FBXW7 and PRR11 (Figure 4A and Figure S5C). Conversely, overexpression of GSK3β enhanced the FBXW7-PRR11 interaction (Figure S5D). As expected, in FBXW7-downregulated cells, GSK3β overexpression rescued the effect of FBXW7 silencing on the PRR11 protein level (Figure 4B). Consistently, CHIR-99021 treatment significantly prevented the decrease in PRR11 abundance induced by FBXW7 overexpression (Figure S5E). Furthermore, knockdown of GSK3β or treatment with CHIR-99021 abolished FBXW7-mediated PRR11 ubiquitination (Figure 4C and Figure S5F), whereas GSK3β overexpression enhanced FBXW7-mediated PRR11 ubiquitination (Figure S5G).
FBXW7 promotes ubiquitination and degradation of PRR11 via GSK3β-mediated phosphorylation. (A) HA-PRR11, Flag-FBXW7, or Flag-FBXW7+siGSK3β were transfected into 293T cells. After IP analysis with Flag antibody, Western blot analysis was performed on HA-PRR11. (B) 293T cells transfected with siFBXW7-1, Flag-GSK3β, or siFBXW7-1+Flag-GSK3β were incubated with CHX (50 μg/mL) for indicated times, and then Western blot analysis was performed for PRR11 (left). Quantification of PRR11 half-life (right, n = 3 biologically independent experiments). (C) 293T cells transfected with HA-PRR11, Myc-Ub, Flag-FBXW7, or Flag-FBXW7+siGSK3β were treated with MG132 (10 μM) for 6 h, and Western blot analysis of Myc-Ub was performed after IP analysis with HA antibody. (D) 293T cells were transfected with Myc-FBXW7, Flag-GSK3β, HA-PRR11-WT, or PRR11 dephosphorylation mimic mutants (HA-PRR11-1A, HA-PRR11-2A, and HA-PRR11-1A/2A), and Western blot analysis of PRR11 was performed. (E) 293T cells transfected with Myc-Ub, Flag-FBXW7, Flag-GSK3β, HA-PRR11-WT, or PRR11 dephosphorylation mimic mutants were treated with MG132 (10 μM) for 6 h, and Western blot analysis of Myc-Ub was performed after IP analysis with HA antibody. (F) Schematic illustration of FBXW7-GSK3β can only promote phosphorylated PRR11 degradation. Protein levels were quantitatively detected with ImageJ software, and linear regression was used to analyze the protein half-life (B).
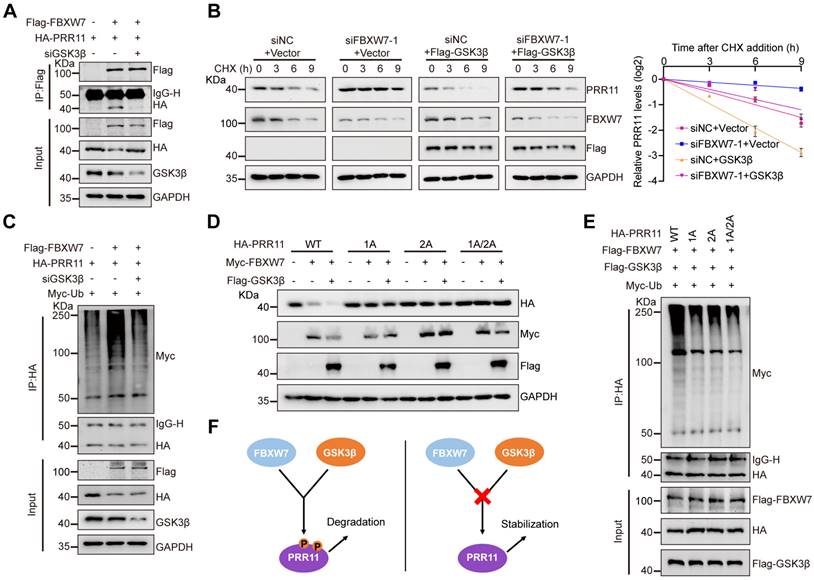
Notably, PRR11 dephosphorylation mimic mutants abrogated the synergistic degradation of PRR11 protein levels by FBXW7 and GSK3β (Figure 4D). In addition, these mutants attenuated the degree of PRR11 ubiquitination mediated by the co-expression of FBXW7 and GSK3β compared to PRR11-WT (Figure 4E). Similarly, the GSK3β/FBXW7-mediated ubiquitination of PRR11 at K48 was inhibited by these mutants (Figure S5H). This further demonstrated the necessity of PRR11 phosphorylation for FBXW7-mediated degradation (Figure 4F). In summary, our results suggest that FBXW7 collaborates with GSK3β to promote PRR11 ubiquitination and degradation.
PRR11 silencing induced oxidative DNA damage in RCC cells
Numerous studies have shown that PRR11 is involved in cell cycle arrest and apoptotic processes [7, 8, 25]. Notably, stress-induced DNA damage can trigger both apoptosis and cell cycle arrest [26]. Through Pearson correlation analysis of TCGA-KIRC data, we identified the top 200 genes closely related to PRR11 (Dataset S2). Subsequent functional annotation analyses (GO and KEGG) revealed enrichment of PRR11 in processes related to homologous recombination, DNA replication, DNA double-strand breaks, DNA damage and cell cycle (Figure S6A-B, Table S4-S5 and Dataset S3). RNA-seq assays of PRR11-silenced ACHN cells corroborated these findings and, as indicated by GSEA, revealed associations between PRR11 and DNA replication/damage/repair, oxidative phosphorylation, and cell cycle (Figure 5A and Table S6 and Dataset S4).
PRR11 knockdown promotes oxidative DNA damage in RCC. (A) RNA-seq analysis was performed on PRR11 knockdown ACHN cells, followed by GSEA for all genes. GSEA was performed using the R package “clusterProfiler”. Statistical significance was determined by two-tailed Fisher's exact test, and the p-value was corrected by Benjamini & Hochberg's method. The top 12 gene sets were selected according to p.adjust and the normalized enrichment score (NES) is displayed. (B) γH2AX staining after PRR11 silencing in ACHN and Caki-1 cells was analyzed by immunofluorescence (left), and the number of γH2AX foci was quantified (right, n = 4 biologically independent experiments). (C) Representative images of alkaline comet assays after PRR11 silencing in ACHN and Caki-1 cells (left). ACHN and Caki-1 cells per group (n = 150) were quantified using CASP software (right, n = 3 biologically independent experiments). (D-E) ROS levels in ACHN cells after PRR11 knockdown or overexpression were detected using flow cytometry (left), followed by statistical analysis (right, n = 3 biologically independent experiments). (F) Flow cytometry detection of MMP levels in ACHN cells after PRR11 silencing (left), followed by statistical analysis (right, n = 3 biologically independent experiments). (G) Western blot analysis of DNA damage markers and oxidative stress markers after PRR11 silencing or PRR11 overexpression in ACHN cells. (H) γH2AX staining after PRR11 silencing or/and ROS scavenger NAC (5 mM) treatment in ACHN and Caki-1 cells was analyzed by immunofluorescence (top), and the number of γH2AX foci was quantified (bottom, n = 4 biologically independent experiments). (I) Representative images of alkaline comet assays after PRR11 silencing or/and ROS scavenger NAC (5 mM) treatment in ACHN and Caki-1 cells (top). ACHN and Caki-1 cells per group (n = 150) were quantified using CASP software (bottom, n = 3 biologically independent experiments). (J) Western blot analysis of DNA damage markers after PRR11 silencing or/and ROS scavenger NAC (5 mM) treatment in ACHN and Caki-1 cells. The p-values were calculated with one-way ANOVA with Dunnett's multiple comparisons test (B-D, F), one-way ANOVA with Tukey's multiple comparisons test (H-I) and two-tailed Student's t-test (E). Data are presented as mean values ± SD.
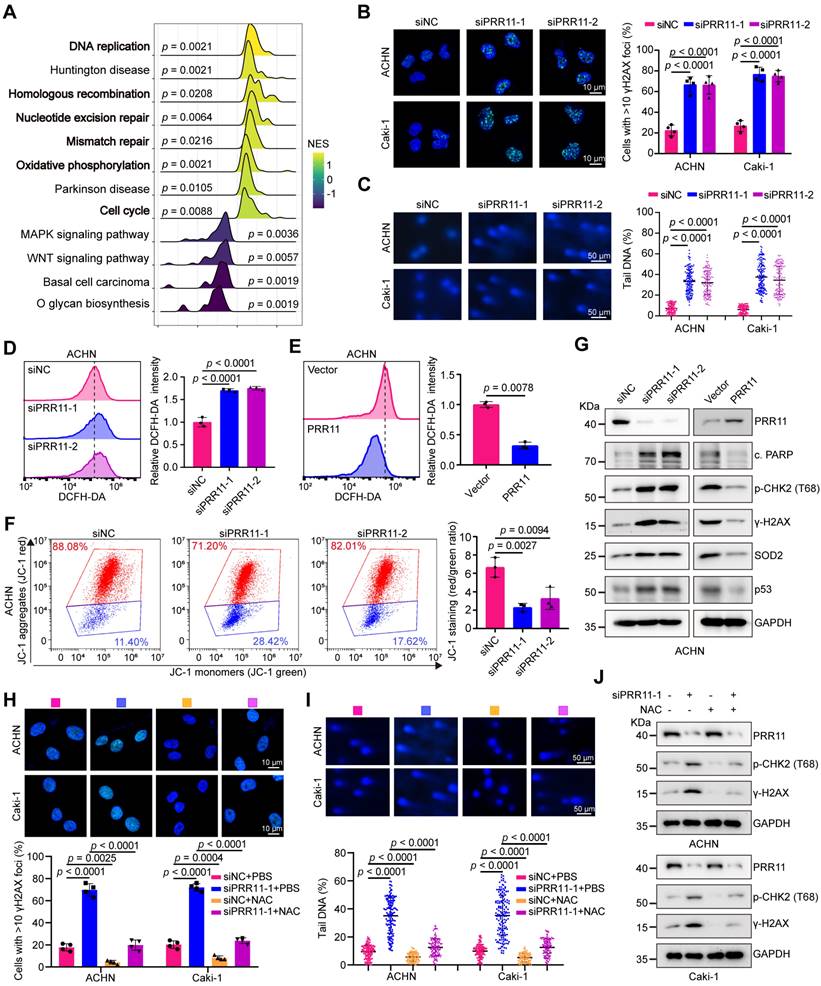
To further investigate the role of PRR11 in RCC, we established and validated knockdown and overexpression RCC cell lines (Figure S6C-D). Consistent with these findings, the knockdown of PRR11 increased the number of foci with the DNA damage marker γH2AX, whereas the overexpression of PRR11 reduced the number of foci with this marker (Figure 5B and Figure S6E). Direct observation of DNA damage via the alkaline comet assay confirmed that PRR11 silencing increased the comet tail DNA content, whereas PRR11 overexpression decreased the comet tail moment (Figure 5C and Figure S6F). Given the pivotal role of ROS in mitochondria-mediated apoptosis and DNA damage [27], we investigated the impact of PRR11 on ROS levels in RCC cells. Flow cytometry analysis revealed that PRR11 knockdown increased ROS levels, whereas PRR11 overexpression inhibited ROS production (Figure 5D-E and Figure S7A-B). Moreover, assessment of the mitochondrial membrane potential (MMP) using JC-1 revealed fewer JC-1 aggregates in PRR11 knockdown RCC cells and more JC-1 aggregates in PRR11 overexpressing RCC cells, suggesting a reduced MMP upon PRR11 silencing (Figure 5F and Figure S7C-E). Western blot analysis further supported these findings, showing increased levels of cleaved PARP, pT68-CHK2, γ-H2AX, SOD2 and p53 in PRR11-silenced RCC cells, whereas PRR11 overexpression had the opposite effect (Figure 5G and Figure S7F).
Excessive accumulation of ROS often triggers DNA damage in cells [28]. To investigate whether PRR11 knockdown-induced DNA damage is caused by elevated ROS, we treated RCC cells with the ROS scavenger NAC. NAC treatment significantly inhibited ROS accumulation caused by PRR11 knockdown (Figure S8A-B). Importantly, the addition of NAC counteracted the PRR11 knockdown-induced decrease in MMP and increase in DNA damage (Figure 5H-J and Figure S8C-D). Thus, these data collectively suggest that PRR11 knockdown promotes DNA damage through ROS accumulation in RCC cells.
PRR11 regulates the AKT pathway and activated AKT stabilizes PRR11 protein
To explore the downstream signals of PRR11 in RCC, we further analyzed the RNA-seq data of ACHN cells after PRR11 knockdown. KEGG pathway analysis results revealed that PRR11 was enriched in AKT signaling pathway (Figure 6A, Table S7 and Dataset S5). As expected, Western blot analysis suggested that PRR11 silencing upregulated PTEN expression and reduced the levels of pT308-AKT and pS473-AKT, as well as those of the downstream effectors of the AKT pathway, pS2448-mTOR and pS9-GSK3β (Figure 6B). Conversely, overexpression of PRR11 had the opposite effect (Figure S9A).
FBXW7/GSK3β-PRR11 axis activates the AKT pathway and AKT activation inhibits PRR11 degradation. (A) KEGG pathway enrichment analysis was performed on DEGs in the RNA-seq expression matrix of PRR11 silencing ACHN cells. KEGG analysis was performed using the R package "clusterProfiler". The p-value was calculated by two-tailed Fisher's exact test, and p < 0.05 was used as a screening criterion. (B) Protein levels of each important factor of the AKT signaling pathway were analyzed by Western blot analysis after PRR11 knockdown in RCC cells. (C) Western blot analysis of AKT phosphorylation levels after transfection of ACHN and Caki-1 cells with siGSK3β or/and siPRR11. (D) AKT activity was measured in ACHN cells transfected with siFBXW7 (top)/siGSK3β (bottom) or/and siPRR11 (n = 3 biologically independent experiments). (E) Changes in PRR11 protein levels were analyzed by Western blot analysis after transfection of AKT-CA or AKT-DN in RCC cells. (F) AKT-CA or AKT-DN plasmid was transfected into ACHN cells. After IP analysis with PRR11 antibody, Western blot analysis was performed on GSK3β and FBXW7. (G) Western blot analysis of PRR11 after transfection of ACHN cells with Flag-GSK3β (top)/Flag-FBXW7 (bottom) or/and HA-AKT-CA. (H) 293T cells transfected with the Flag-GSK3β or/and HA-AKT-CA were treated with CHX (50 μg/mL) for the indicated durations, after which Western blot analysis was performed for PRR11 (left). Quantification of the PRR11 half-life (right, n = 3 biologically independent experiments). (I) 293T cells transfected with the Flag-FBXW7 or/and HA-AKT-CA were treated with CHX (50 μg/mL) for the indicated durations, after which Western blot analysis was performed for PRR11 (left). Quantification of the PRR11 half-life (right, n = 3 biologically independent experiments). (J) 293T cells transfected with HA-PRR11, Myc-Ub, Flag-GSK3β, or/and GFP-AKT-CA were incubated with MG132 (10 μM) for 6 h, and Western blot analysis of Myc-Ub was performed after IP analysis with HA antibody. (K) 293T cells transfected with HA-PRR11, Myc-Ub, Flag-FBXW7, or/and GFP-AKT-CA were incubated with MG132 (10 μM) for 6 h, and Western blot analysis of Myc-Ub was performed after IP analysis with HA antibody. Protein levels were quantitatively detected with ImageJ software, and linear regression was used to analyze the protein half-life (H-I). The p-values were calculated with one-way ANOVA with Tukey's multiple comparisons test (D). Data are presented as mean values ± SD.
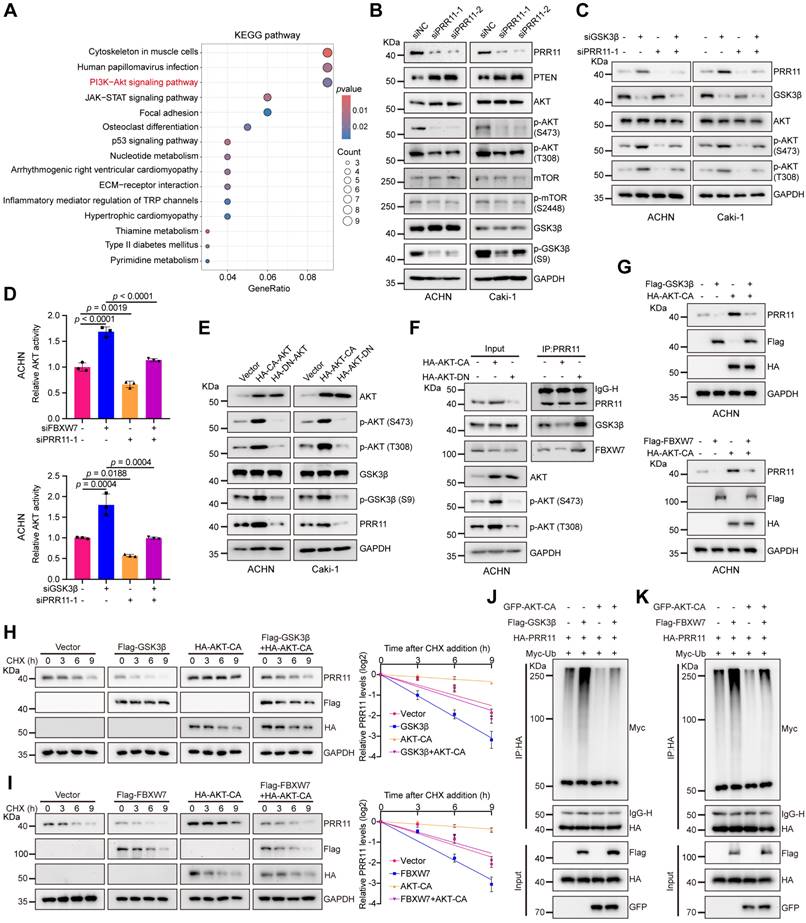
Deletion of FBXW7 synergizes with activated AKT signaling to promote cholangiocarcinogenesis. Interestingly, FBXW7 decreased the p-AKT level in some cholangiocarcinoma cell lines, whereas the dominant-negative form of FBXW7 (FBXW7-ΔF-box) significantly upregulated the p-AKT level [29]. It has also been shown that in pancreatic cancer FBXW7 could interfere with AKT signaling activity by targeting SIK2 degradation [30]. In addition, GSK3β-mediated phosphorylation of Rictor significantly inhibited AKT signaling was also demonstrated [31]. Based on the above studies and our conclusion that FBXW7/GSK3β regulates PRR11 protein stability, we explored whether FBXW7/GSK3β has an effect on PRR11-mediated AKT activity in RCC. We found that knockdown of FBXW7 or GSK3β enhanced the phosphorylation levels of AKT, an effect that could be reversed by downregulation of PRR11 (Figure 6C and Figure S9B). Importantly, quantification of AKT activity using the AKT kinase activity kit also demonstrated that downregulation of FBXW7 or GSK3β significantly enhanced AKT activity, an effect that was counteracted by PRR11 knockdown (Figure 6D and Figure S9C).
Notably, AKT is known to phosphorylate GSK3β, resulting in its inactivation [32]. Combined with previous findings showing that GSK3β promotes PRR11 degradation, we hypothesized that an AKT-GSK3β-PRR11 feedback loop is formed. To test whether AKT activation or inhibition affects the protein stability of PRR11, we transfected constitutively active AKT (AKT-CA) and dominant-negative AKT (AKT-DN) plasmids into RCC cells. The activation of AKT indeed increased the PRR11 protein level, whereas AKT inactivation attenuated the PRR11 protein level (Figure 6E). Importantly, an endogenous co-IP assay in RCC cells revealed that AKT activation attenuated PRR11 interactions with FBXW7 and GSK3β, whereas AKT inactivation strengthened these interactions (Figure 6F and Figure S9D). Then to explored whether AKT activation affects FBXW7/GSK3β-mediated ubiquitination and degradation of PRR11. We first demonstrated that activation of AKT prevented FBXW7/GSK3β-mediated PRR11 degradation by using AKT-CA in ACHN cells (Figure 6G). The in-depth mechanism of AKT on PRR11 via FBXW7/GSK3β was then continued to be explored in 293T cells. By CHX and ubiquitination assays we further found that AKT-CA inhibited the ubiquitination and degradation of PRR11 by FBXW7/GSK3β (Figure 6H-K and Figure S9E). In summary, PRR11 participates in AKT pathway activation, and activated AKT stabilizes the PRR11 protein by inactivating GSK3β, thus forming a positive feedback loop.
PRR11-AKT axis regulates oxidative DNA damage and promotes RCC
Subsequently, we further investigated the biological role of the PRR11-AKT axis in the tumor behavior of RCC. The addition of the AKT agonist SC79 to ACHN and Caki-1 cells rescued the increase in ROS levels in PRR11 knockdown cells (Figure 7A and Figure S10A). Similarly, the AKT inhibitor MK2206 counteracted the decrease in ROS levels caused by PRR11 overexpression (Figure 7B and Figure S10B). Additionally, knockdown of PRR11 increased the number of nuclear γH2AX foci, which was reversed by SC79 treatment (Figure 7C), while MK2206 treatment had the opposite effect (Figure S10C). Western blot analysis further verified that SC79 treatment rescued the upregulation of DNA oxidative damage markers (pT68-CHK2, γ-H2AX, and SOD2) and the decrease in key factors of AKT signaling pathway (pS473-AKT, pS2448-mTOR, and pS9-GSK3β) caused by PRR11 silencing (Figure 7D). Similarly, MK2206 treatment also counteracted the effect of PRR11 overexpression on the corresponding protein (Figure S10D). This observation suggested that PRR11 regulates oxidative DNA damage in RCC by activating the AKT pathway.
PRR11-AKT axis regulates oxidative DNA damage and affects proliferation and migration in RCC. (A-B) Flow cytometry detection of ROS levels from the indicated groups with PRR11 silencing/overexpression and AKT agonist SC79 (20 μM)/AKT inhibitor MK2206 (10 μM) treatment in Caki-1 cells (left), followed by statistical analysis (right, n = 3 biologically independent experiments). (C) Immunofluorescence analysis of the number of γH2AX foci in RCC cells treated with or without PRR11 silencing and/or SC79 treatment (left), followed by statistical analysis (right, n = 4 biological independent experiments). (D) Western blot analysis of oxidative DNA damage and AKT pathway associated protein levels in RCC cells treated with or without PRR11 silencing and/or SC79 treatment. (E) MTT assay of the proliferative capacity of ACHN cells treated with or without PRR11 silencing and/or AKT agonist SC79 treatment (n = 6 biologically independent experiments). (F) Colony formation assay of the proliferative capacity of ACHN cells treated with or without PRR11 silencing and/or AKT agonist SC79 treatment (left), followed by statistical analysis (right, n = 3 biologically independent experiments). (G) Transwell assay showing the migration capacity of ACHN cells treated with or without PRR11 silencing and/or AKT agonist SC79 treatment (left), followed by statistical analysis (right, n = 3 biologically independent experiments). The p-values were calculated with one-way ANOVA with Tukey's multiple comparisons test (A-C, E-G). Data are presented as mean values ± SD.
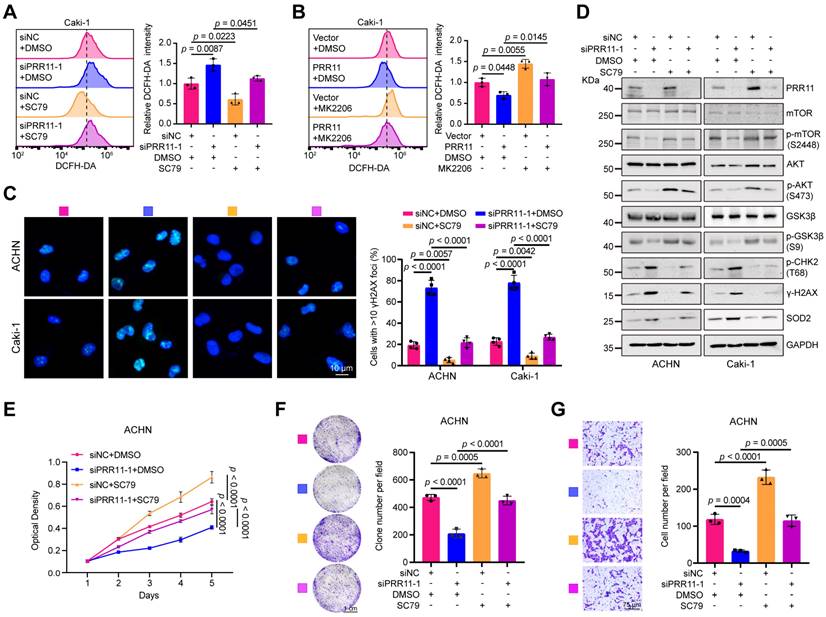
To investigate whether the PRR11-AKT axis affects RCC proliferation and metastasis, we conducted colony formation and MTT assays. Silencing and overexpression of PRR11 attenuated and enhanced the proliferative capacity of ACHN and Caki-1 cells, respectively, and these effects were reversed by SC79 and MK2206 (Figure 7E-F and Figure S11A-F). In addition, SC79 treatment counteracted the decrease in migratory capacity induced by PRR11 silencing, whereas MK2206 treatment inhibited the enhancing effect of PRR11 on migratory capacity (Figure 7G and Figure S11G-I).
To further investigate the impact of the PRR11-AKT axis on RCC tumor behavior in vivo, we established shNC and shPRR11 stable ACHN cells and verified the knockdown efficiency (Figure S12A). A subcutaneous tumor-bearing model was used to study its effect on tumor proliferation, and a tail vein lung metastasis model and popliteal lymph node metastasis model were used to study its effect on tumor hematogenous and lymphatic metastasis (Figure 8A). In subcutaneous tumor-bearing experiments, treatment with the SC79 reversed the PRR11 knockdown-induced reductions in tumor volume and weight (Figure 8B-C and Figure S12B), further supporting the establishment of the PRR11-AKT axis. 18F-FDG PET/CT is a commonly used tumor test in the clinic and plays an important role in the diagnosis and treatment of a variety of malignant tumors [33]. PET/CT imaging showed that tumor activity was significantly reduced in the PRR11 knockdown group, and SC79 treatment reversed the effect of PRR11 on tumors (Figure 8D). IHC demonstrated that PRR11 silencing reduced the abundance of pS473-AKT and Ki67 while increasing γ-H2AX levels, and these effects were counteracted by SC79 treatment (Figure 8E and Figure S12C). In addition, we established a tail vein lung metastasis model and found that PRR11 silencing significantly suppressed the fluorescence intensity and number of metastatic tumor nodules, and this suppression was significantly counteracted by SC79 treatment (Figure 8F-G and Figure S12D). Consistently, the popliteal lymph node metastasis model generated via MRI scanning and dissection also showed that popliteal lymph nodes were smaller in the shPRR11 group than in the shNC group, and this effect was counteracted by SC79 treatment (Figure 8H-I and Figure S12E). H&E staining clearly revealed lymph node tumor infiltrating lesions (Figure S12F). These findings suggest that PRR11 promotes the proliferation and migration of RCC cells by activating the AKT pathway. In conclusion, PRR11 affects oxidative DNA damage by activating the AKT pathway, thus promoting the proliferation and migration of RCC cells.
PRR11-AKT axis promotes RCC proliferation and metastasis in vivo. (A) A schematic diagram on the construction of in vivo models of tumor proliferation and metastasis and drug therapy. (B) Gross diagram of xenograft model from mice injected ACHN cells transfected with shNC or shPRR11 with or without SC79 treatment. (C) Tumor volume statistic for mice injected subcutaneously with ACHN cells transfected with shNC or shPRR11 and treated with or without SC79 (n = 8 per group). (D) 18F-FDG PET/CT scans were used to assess tumor growth and malignancy in each group of mice. Each group of images is represented from left to right as follows: CT image, PET image, PET/CT fusion image. (E) Representative images of Ki-67 and γ-H2AX IHC staining of tumor tissue from each group of mice in the xenograft model (top), and their quantitative statistical plots (bottom, n = 4 per group). (F) GFP fluorescence intensity between groups (control vs. shPRR11 vs. SC79 vs. shPRR11+SC79) in the model of tail vein injection lung metastasis established using ACHN cells (left). Average radiation efficiency in lung is evaluated by ImageJ software, and statistics are performed (right, n = 3 per group). (G) Representative images of H&E staining of mice lung tissues from each group. (H) Gross diagram of a popliteal lymph node metastasis model of mice injected with ACHN cells transfected with shNC or shPRR11 with or without SC79 treatment (left). Popliteal lymph node volume statistics for mice injected with ACHN cells transfected with shNC or shPRR11 and treated with or without SC79 (right, n = 5 per group). (I) MRI T2 axial scanning imaging of popliteal lymph nodes in mice. (J) Mechanistic diagram of this study. FBXW7-GSK3β mediates PRR11 degradation, thereby regulating the AKT pathway. In turn, AKT-GSK3β is involved in regulating PRR11 degradation, forming a positive feedback loop that regulates oxidative DNA damage and accelerates RCC progression. The p-values were calculated with one-way ANOVA with Tukey's multiple comparisons test (C, E-F, H). Data are presented as mean values ± SD.
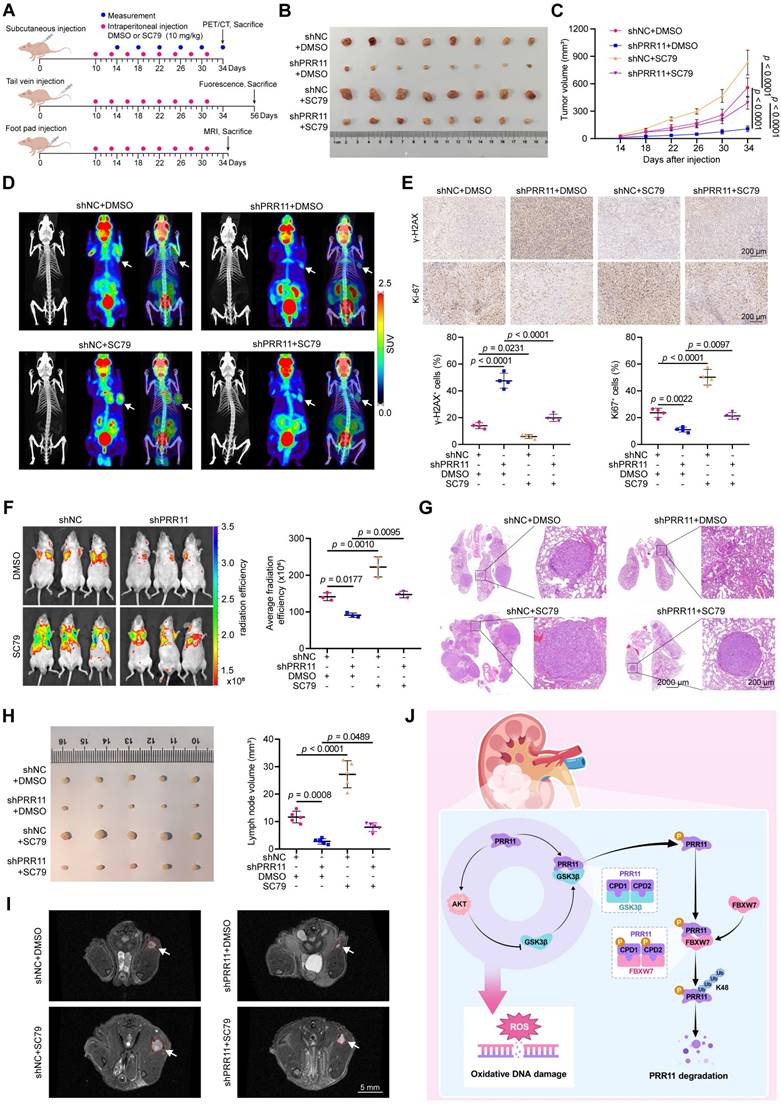
In summary, we found that PRR11-mediated activation of the AKT pathway affects oxidative DNA damage and accelerates RCC progression via a molecular mechanism in which FBXW7-GSK3β mediates PRR11 degradation. On the other hand, PRR11 activates the AKT signaling pathway, which in turn inhibits GSK3β activity, thereby preventing PRR11 degradation, forming a positive feedback loop and accelerating RCC progression (Figure 8J).
Discussion
Because RCC cells may exhibit sensitivity to defects in homologous recombination genes, there has been increasing interest in studies related to RCC-associated DNA damage [34]. DNA damage and subsequent genomic instability can lead to gene mutations and chromosomal damage, thereby promoting tumor development [35, 36]. On the other hand, massive and extensive DNA damage can cause fatal damage to tumor cells by inducing apoptosis or necrosis, cellular senescence, and cell cycle arrest [37, 38]. Importantly, oxidative DNA damage caused by ROS is the main cause of DNA damage [39].
In earlier studies, we found that PRR11 silencing caused cell apoptosis and cell cycle arrest in RCC [7]. Interestingly, oxidative DNA damage is also closely related to apoptosis and cell cycle progression [26]. Here, for the first time, we elucidated the important role of PRR11 silencing in causing oxidative DNA damage and thus inhibiting RCC development. Notably, PRR11 is a rapidly degradable protein with multiple ubiquitination and phosphorylation sites and is potentially degraded via the ubiquitin proteasome [9]. Based on these findings, we provide compelling evidence that the E3 ligase FBXW7 promotes the phosphorylation-dependent ubiquitination and degradation of PRR11 via a mechanism mediated by GSK3β.
AKT is a major regulator of cell survival that modulates biological functions by phosphorylating a wide range of kinases, enzymes, and transcription factors and is therefore frequently activated as a cancer driver [45, 46]. Notably, many RCC tumor inhibitors, including various cellular proteins, inhibit cell growth/migration inhibition and apoptosis by inhibiting the AKT signaling pathway [47-49]. GSK3β is regulated by AKT as its substrate, and AKT can inhibit GSK3β activity by phosphorylating the Ser9 site of GSK3β and thus inhibiting GSK3β activity [50]. Based on RNA-seq analysis and western blot experiments, we found that PRR11 amplification overactivated the AKT pathway through down-regulation of PTEN, which in turn affected the expression of signals downstream of the pathway, including the inactivation of GSK3β. Interestingly, GSK3β is involved in FBXW7-mediated PRR11 degradation. Importantly, we further demonstrated that inactivation or activation of the AKT pathway via GSK3β markedly affected the strength of the protein interactions between PRR11 and GSK3β and FBXW7. Based on these findings, we revealed the formation of a positive feedback loop between AKT-GSK3β-PRR11.This mutual activation between PRR11 and AKT is essential for maintaining mutual PRR11 overexpression, which enhances the procarcinogenic effect of PRR11-AKT on RCC.
The inactivation of AKT could impair DNA repair mechanisms, including non-homologous end joining (NHEJ) and homologous recombination (HR), leading to delayed DNA damage repair and ultimately resulting in genomic instability and the accumulation of mutations [51, 52]. Notably, AKT knockout mice show impaired DNA damage-dependent induction of p21 and increased tissue apoptosis, similar to the DNA-PK deficiency phenotype [53]. In addition, AKT inactivation could inhibit the expression of multiple antioxidant enzymes and regulate glutathione metabolism by down-regulating the KEAP1-NRF2 pathway, thereby promoting intracellular ROS accumulation [54-56]. Importantly, ROS accumulation could induce DNA damage and affect DNA damage response (DDR), which includes DNA strand breaks, base oxidation, and other forms of DNA structural damage [28]. Numerous studies have shown that AKT is an important regulator of genome stability and can modulate oxidative DNA damage [57]. For example, AKT regulates the response of p53 to oxidative stress to promote cell proliferation and tumorigenesis [58]. In addition, AKT can mediate the activation of protective responses to oxidative DNA damage [59]. We modeled AKT activation and inactivation by adding the AKT agonist SC79 and the AKT inhibitor MK2206 and found that AKT activation significantly attenuated the increase in oxidative DNA damage mediated by PRR11 silencing. Conversely, inactivation of AKT counteracted the PRR11 overexpression-mediated decrease in oxidative DNA damage. Consistent with these findings, we demonstrated that the activation of AKT significantly counteracted the reduction in the proliferation and migration of RCC cells caused by PRR11 silencing. Our experiments suggested that the PRR11-AKT feedback loop influenced the proliferation and migration of RCC cells by regulating oxidative DNA damage.
However, this study also has some limitations. We demonstrated that the PRR11-AKT positive feedback loop can regulate ROS accumulation and thus affect oxidative DNA damage. However, the exact mechanism of how the PRR11-AKT loop regulates ROS remains to be explored. Furthermore, since the AKT pathway genetically targets more pathway components than any other growth factor signaling pathway [60], it is reasonable to suspect that oxidative DNA damage is only part of its effect on RCC development. Last, we identified and demonstrated by RNA-seq that PRR11 activates the AKT signaling pathway by down-regulating PTEN in RCC, but the specific molecular biological mechanisms still need to be further elucidated in future.
In conclusion, our study revealed that FBXW7-GSK3β mediates PRR11 degradation, thereby regulating the AKT pathway. In turn, AKT-GSK3β is involved in regulating PRR11 degradation, forming a positive feedback loop that regulates oxidative DNA damage and accelerates RCC progression.
Methods
Cell culture and transfection
The RCC cell lines (ACHN and Caki-1) and HEK293T cells used in this study were obtained and authenticated by the Chinese Academy of Sciences Cell Bank (Shanghai, China). HEK293T cells were cultured in DMEM, ACHN cells were cultured in MEM and Caki-1 cells were cultured in McCoy's 5A. All cell lines were maintained in medium supplemented with 10% FBS and confirmed to be free of mycoplasma contamination. Lipofectamine 3000 (L3000015, Invitrogen) was used for transfection in this study.
Antibodies and reagents
Commercial sources provided the reagents, which were employed in accordance with instructions. Table S8 contains an inventory of the antibodies used in this study.
siRNAs and plasmids
siPRR11 (#1: 5'-ACGCAGGCCUUAAGGAGAATT-3', #2: 5'-GGCCUUAAGGAGAAAGUUUTT-3'), siFBXW7 (#1: 5'-GCAUAUGAUUUUAUGGUAATT-3', #2: 5'-UGAUACAUCAAUCCGUGUUUG-3') and siGSK3β (5'-ACACGAAAGUGAUUGGAAATT-3') were purchased from GenePharma (Suzhou, China). PRR11 cDNA was inserted into pcDNA3.1-2×Flag and pcDNA3.1-HA vectors. Flag-FBXW7 was generously gifted by Prof. Qing Guoliang from Wuhan University. Flag-GSK3β was purchased from Miaolingbio (Wuhan, China). Myc-FBXW7, Flag-FBXW7 (R479Q), Flag-FBXW7 (R505C) and Flag-FBXW7 (ΔF-box) were constructed based on Flag-FBXW7-WT by standard subcloning. Flag-GSK3β (S9D) and Flag-GSK3β (Y216A) were constructed based on Flag-GSK3β-WT by standard subcloning. HA-PRR11-1A (T287A/S291A), HA-PRR11-2A (T326A/T330A), HA-PRR11-1A/2A (T287A/S291A/T326A/T330A), HA-PRR11-1D (T287D/S291D), HA-PRR11-2D (T326D/T330D), HA-PRR11-1D/2D (T287D/S291D/T326D/T330D), HA-PRR11-1Δ (I286_S291del), HA-PRR11-2Δ (L325_T330del), HA-PRR11-1Δ/2Δ (I286_S291del/L325_T330del), HA-PRR11 (amino acids 1-100), HA-PRR11 (amino acids 101-250) and HA-PRR11 (amino acids 251-360) were constructed based on HA-PRR11-WT.
Quantitative reverse transcription PCR (qRT-PCR)
The ReverTra Ace qPCR RT Kit (FSQ-101, TOYOBO) was used to synthesize cDNA after RNA was extracted using the HiPure Total RNA Mini Kit (R4111-03, Magen). Next, cDNA was analyzed using iQ SYBR Green Supermix (1725125, Bio-Rad). The sequences of primers used are listed in Table S9.
Immunofluorescence
After being fixed for 30 min with paraformaldehyde, the RCC cells were cultured for 40 min at 20°C in a buffer consisting of 2% BSA and 0.3% Triton X-100. After the cells were stained with the corresponding primary antibody overnight at 4°C, the cells were treated with DAPI and the fluorescent secondary antibody. Imaging was performed via confocal laser microscopy (Nikon, Japan), and the images were processed using ImageJ software (version 1.52). For the γH2AX foci formation experiments, at least 100 cells per group were evaluated.
Immunohistochemistry
Tissue microarrays for immunohistochemistry (HKidE180Su02, n = 149) were purchased from Shanghai Outdo Biotech. Formalin fixation, paraffin embedding, section dewaxing, and hydration were applied to the samples in that order. Subsequent procedures included DAB incubation, primary and secondary antibody incubation, and serum blocking.
Alkaline comet assay
Preheated 0.6% normal melting point agarose was dropped on the frosted surface of the same preheated slide, which was covered with a coverslip and solidified at low temperatures. The cell suspension at a concentration of 3 × 106 cells/ml was mixed with 0.6% low melting point agarose. The mixture was dropped on the first layer of agarose and covered with a coverslip to cool and solidify at low temperature. After removing the coverslips, the cells were lysed in lysis solution (2.5 mM NaCl, 100 mM EDTA, 10 mM Tris, pH = 10) at 4°C for 8 h. Slides containing cells were soaked in electrophoresis buffer (1 mM EDTA, 300 mM NaOH, pH > 13) for 30 min to allow DNA to unwind, followed by electrophoresis in electrophoresis buffer at 300 mA/30 V for 30 min. The slides were washed and neutralized, dehydrated with anhydrous ethanol, and finally stained with DAPI. Images were taken using a fluorescence microscope, and cellular comet tail DNA was quantified using CASP software (version 1.2.3b1). For each case, at least 150 cells were analyzed.
Flow cytometry
To detect reactive oxygen species (ROS), the collected RCC cells were stained with 10 μM DCFH-DA (D6883, Sigma) in PBS for 20 min, protected from light, and then washed with PBS. Flow cytometry (Beckman Cytoflex, USA) was used to measure ROS levels, and FlowJo software (version 10.8.1) was used to analyze the data.
To detect the MMP, collected RCC cells were stained with JC-1 reagent (C2006, Beyotime), and then samples were measured using flow cytometry.
Western blot analysis
Cells were lysed at low temperature in a mixture of Roche phosphatase inhibitor, protease inhibitor and RIPA buffer (P0013B, Beyotime) for 40 min, followed by high-speed centrifugation to collect the supernatant. SDS-PAGE gel was used to separate the denatured proteins, which were then electrotransferred to a PVDF membrane. The expression of the corresponding proteins was detected following sequential incubation with primary and secondary antibodies.
Co-immunoprecipitation (co-IP)
The BeaverBeadsTM Protein A/G Immunoprecipitation Kit (22202-100, Beaver) was used for co-IP detection. In brief, cells were treated with MG132 (10 μM, S2619, Selleck) for 6 h prior to collection, followed by preparation of cell lysates with cell lysate buffer containing proteasome inhibitors and Roche phosphatase inhibitors, and then incubated overnight with the target antibody at low temperature. After addition of Protein A/G magnetic beads, the antigen-antibody mixture was mixed at low temperature for 2 h. IP buffer was used to wash the protein-antibody-magnetic bead complex several times. Finally, the immune complex was eluted with 1× SDS buffer and analyzed by Western blotting.
Cycloheximide (CHX)-chase assay
To monitor the half-life and degradation efficiency of the target proteins, 50 μg/mL CHX (HY-12320, MCE) was added to the cell culture medium and mixed before cells were harvested at various intervals for Western blot analysis. The target protein expression levels were quantitatively analyzed using ImageJ software.
Ubiquitination assay
For PRR11 ubiquitination assay, cells were transfected with the indicated plasmids. 6 h before the cells were harvested, 10 μM MG132 was added to the cell culture media. Roche phosphatase inhibitor and proteasome inhibitor were added to RIPA buffer before cells were lysed. The next steps were consistent with those of the co-IP analysis.
In vitro phosphorylation assay
Following the methods of Moscat J et al. [61, 62], 48 h after the transfection of 293T cells with HA-labeled human PRR11 WT or PRR11 4A (T287A, S291A, T326A, T330A), the cells were lysed with RIPA buffer and HA-PRR11 was immunoprecipitated via magnetic beads containing the HA antibody. The immunoprecipitates were incubated with human active GSK3β (G09-10H, SignalChem) for 1 h at 30°C in kinase assay buffer (25 mM Tris-HCl, 25 mM KCl, 5 mM MgCl2, 1 mM DTT) containing 500 μM ATP-γ-S (ab138911, Abcam). The reaction was terminated with 0.1 mM EDTA and incubated with 0.5 mM p-nitrobenzyl mesylate (PNBM, ab138910, Abcam) at 30°C for 60 min. Finally, immunoblotting was performed using anti-thiophosphate antibody (ab92570, Abcam).
Immunoprecipitation-mass spectrometry (IP-MS) analysis
To identify the binding partners of PRR11, HA-Vector and HA-PRR11 plasmids were transfected into 293T cells for 48 h, and the proteins were then analyzed by co-IP. The samples were separated by SDS-PAGE and stained with a Fast Silver Stain Kit (P0017S, Beyotime). SpecAlly Life Technology Co., Ltd. (Wuhan, China) carried out IP-MS. A tims TOF Pro liquid mass spectrometry system (Bruker, USA) was used for mass spectrometry analysis. MaxQuant (version 2.2.0.0) was used to analyze the raw mass spectral data. The software's integrated Andromeda database search method was used. The database searched was the Human Protein Sequence Database obtained from UniProt.
RNA-seq and bioinformatics analysis
DNase I extraction of RNA was followed by DNA digestion. A NanoDropTM OneC Spectrophotometer (Thermo Fisher Scientific, USA) was used to for RNA quality control. 2 μg of RNA was used for stranded RNA sequencing library preparation. PCR products were enriched, quantified, and sequenced on a DNBSEQ-T7 sequencer (MGI Tech, China). Raw data were filtered, low-quality reads were discarded, and reads contaminated with adaptor sequences were trimmed. Clean data were mapped to the human genome (assembly GRCh38) using STAR software (version 2.5.3a), and reads were counted using featureCounts (Subread-1.5.1; Bioconductor).
Based on the TCGA-KIRC dataset, the top 200 genes associated with PRR11 were screened by Pearson correlation analysis. These 200 genes were subsequently analyzed by Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG). For the RNA-seq expression matrix, differential expression genes (DEGs) were identified using the R package “edgeR” after setting the screening threshold at p < 0.05. Subsequently, gene set enrichment analysis (GSEA) was performed for all genes in the expression matrix and KEGG analysis was performed for DEGs. The R package "clusterProfiler" was used to carry out GO, GSEA, KEGG. As a reference, the annotated gene set c2.cp.kegg.v2023.1.Hs.symbols.gmt was chosen. p < 0.05 was considered to indicate statistical significance.
Protein molecular docking
FBXW7 (UniProt ID: Q969H0) and PRR11 (UniProt ID: Q96HE9) were used as docking protein models. Protein-protein molecular docking was performed using HDOCK server (http://hdock.phys.hust.edu.cn/) [63]. Protein pre-processing (deletion of water molecules and redundant ligands, addition of hydrogen atoms) was performed and visualization was performed via PyMol 2.4 software. Docking Score, Confidence Score and Ligand RMSD were used as the evaluation criteria for docking and the model with the highest score was selected as the best docking model.
Proliferation assays
For MTT assay, RCC cells were seeded in 96-well plates at 3000 cells/well. 4 h before the assay, 20 μL MTT (5 mg/mL, HY-15924, MCE) solution was added to each well, the medium was discarded, the cells were dissolved in 150 μL of DMSO and shaken well, and the microplate reader (SpectraMax M2, USA) was used to measure the absorbance.
For colony formation assay, RCC cells were planted in 6-well plates with 1000 cells/well, fixed with formaldehyde after 2 weeks, and stained with crystal violet.
Migration assays
For transwell assay, 4 × 104 RCC cells were placed in upper chamber (Corning, USA) with serum-free medium, and migration was induced in the bottom chamber with serum-containing medium. The cells were finally fixed and stained.
AKT kinase activity assay
Briefly, proteins were first quantified using the BCA protein assay kit (P0011, Beyotime). AKT kinase activity was then assayed using the AKT kinase activity assay kit (ADI-EKS-400A, Enzo). AKT kinase assay was performed on 10 μg of proteins from each set of samples according to the manufacturer's instructions, and the absorbance of the reactants at 450 nm was measured by a microplate reader.
GST pull-down assay
Recombinant GST and His-FBXW7 proteins were purchased from CUSABIO Co., Ltd (Wuhan, China). Human PRR11 was subcloned into the pGEX-6p-1 construct. Recombinant GST tagged PRR11 was purified from Escherichia coli BL21. GST-PRR11 fusion protein was incubated with His-FBXW7 and His-GSK3β fusion proteins for 3 h at 4°C, respectively. The mixtures were incubated with glutathione-Sepharose beads for 2 h at 4°C for GST pull-down assay. GST pull-down products were eluted with 1× SDS buffer and denatured at 100°C for 8 min, then used for Western blotting.
Animal studies
The Laboratory Animal Welfare and Ethics Committee of Zhongnan Hospital of Wuhan University provided consent for all animal experimental procedures (approval number: ZN2022263). Four-week-old male BALB/c nude mice purchased from GemPharmatech Co., Ltd. (Nanjing, China) were acclimatized and fed for one week. Lentivirus from GenePharma (Suzhou, China) was used to transfect ACHN cells, and puromycin (540222, Sigma) was used to screen the stable PRR11 silencing cells (shPRR11) or negative control cells (shNC). The mice were randomly divided into four groups: shNC+DMSO, shPRR11+DMSO, shNC+SC79, and shPRR11+SC79.
For subcutaneous tumor-bearing experiments, 5 × 106 shNC or shPRR11 ACHN cells/100 μL of PBS were subcutaneously injected into nude mice. After 10 days of tumor growth, DMSO or SC79 (10 mg/kg, HY-18749, MCE) was intraperitoneally injected every 3 days for 8 times. Every 4 days following injection, the tumor size was recorded, and the tumor volume was calculated using the formula V = 1/2 × L × S2, where L represents the long diameter and S represents the short diameter. For positron emission tomography computed tomography (PET/CT) imaging, mice were injected with 18F-FDG (150 μCi/per mouse) in the tail vein, anesthetized with 2% isoflurane 1 h later, and then subjected to a 10-minute static PET scan using an Inliview-3000B system (Novelmedical, China). Data were reconstructed using a 3D ordered subsets expectation maximization algorithm.
For the lung metastasis model, ACHN cells were injected via the mice tail vein with 1 × 106 cells/100 μL of PBS. DMSO or SC79 was injected intraperitoneally as previously described. The fluorescence intensity of lung metastases in nude mice was measured by IVIS Spectrum Optical In vivo Imaging System (PerkinElmer, USA) after 8 weeks of feeding. The radiation efficiency was calculated as (p/sec/cm2/sr)/(μW/cm2).
For the popliteal lymph node metastasis model, ACHN cells (1 × 106 cells/50 μL of PBS) were injected into the right foot pads of the mice. DMSO or SC79 was added as previously described. Mouse popliteal lymph nodes were imaged using a 7.0T BioSpec 70/20 (Bruker, USA) small animal magnetic resonance imager (MRI). Subsequently, mice were sacrificed, and popliteal lymph nodes were dissected, measured, and fixed.
Statistical analysis
At least three independent replications of each experiment were conducted. The data used for the statistical analysis were analyzed with GraphPad Prism 8. The two-tailed Student's t-test was used to compare the two groups, and one-way ANOVA was used to evaluate multiple group differences, Tukey test was used to compare all column pairs, and Dunnett test was used to compare all columns with control column. p < 0.05 was considered to indicate statistical significance.
Supplementary Material
Supplementary figures and tables, dataset descriptions.
Supplementary datasets.
Acknowledgements
The excellent technical assistance of Ms. Yayun Fang, Ms. Mengxue Yu and Ms. Danni Shan is gratefully acknowledged. We thank Dr. Yuruo Chen for exceptional assistance in editing the diagram. This study was supported by the Fundamental Research Funds for the Central Universities (2042022dx0003) and the Research Fund of Zhongnan Hospital of Wuhan University (SWYBK01-03, RLYC2024001001, YYXKNLJS2024001). The funders played no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Data availability statement
The RNA-seq data generated in this study have been deposited in the GEO database (https://www.ncbi.nlm.nih.gov/geo/) with the access code GSE263951. The mass spectrometry proteomics data generated in this study are provided in Dataset S1. Correlation analysis data based on the TCGA-KIRC dataset were analyzed and obtained from the LinkedOmics online website (https://linkedomics.org/admin.php). The remaining data are available within the Article or Supplementary Information.
Statement of animal rights
The study was approved by the Experimental Animal Welfare Ethics Committee, Zhongnan Hospital of Wuhan University (approval number: ZN2022263).
Author contributions
S.C., K.Q., L.J., and X.W. conceived and designed the study, S.C., K.X., S.Y., and J.L. performed the analysis procedures, S.C., K.X., S.Y., M.L., J.Y., G.W., S.T., J.S., K.Q., and J.L. analyzed the results, S.C., W.J., Y.X., Y.Z. and L.J. contributed analysis tools, S.C., Y.X., K.Q., L.J., and X.W. contributed to the writing of the manuscript. All authors reviewed the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73:17-48
2. Capitanio U, Montorsi F. Renal cancer. Lancet. 2016;387:894-906
3. Song XD, Tian YN, Li H, Liu B, Zhang AL, Hong Y. Research progress on advanced renal cell carcinoma. J Int Med Res. 2020;48:300060520924265
4. Hayes JD, Dinkova-Kostova AT, Tew KD. Oxidative Stress in Cancer. Cancer Cell. 2020;38:167-197
5. Kim MJ, Lee JS, Park SE, Yi HJ, Jeong IG, Kang JS. et al. Combination treatment of renal cell carcinoma with belinostat and 5-fluorouracil: a role for oxidative stress induced DNA damage and HSP90 regulated thymidine synthase. J Urol. 2015;193:1660-8
6. Jin J, Chen F, He W, Zhao L, Lin B, Zheng D. et al. YAP-Activated SATB2 Is a Coactivator of NRF2 That Amplifies Antioxidative Capacity and Promotes Tumor Progression in Renal Cell Carcinoma. Cancer Res. 2023;83:786-803
7. Chen S, He Z, Peng T, Zhou F, Wang G, Qian K. et al. PRR11 promotes ccRCC tumorigenesis by regulating E2F1 stability. JCI Insight. 2021;6:e145172
8. Ji Y, Xie M, Lan H, Zhang Y, Long Y, Weng H. et al. PRR11 is a novel gene implicated in cell cycle progression and lung cancer. Int J Biochem Cell Biol. 2013;45:645-56
9. Larance M, Ahmad Y, Kirkwood KJ, Ly T, Lamond AI. Global subcellular characterization of protein degradation using quantitative proteomics. Mol Cell Proteomics. 2013;12:638-50
10. Han W, Chen L. PRR11 in Malignancies: Biological Activities and Targeted Therapies. Biomolecules. 2022;12:1800
11. Wang S, Zhang X, Lei H, Song L, Huang Y, Kang T. et al. Proline-rich 11 (PRR11) promotes the progression of cutaneous squamous cell carcinoma by activating the EGFR signaling pathway. Mol Carcinog. 2023;62:613-627
12. Zhang L, Zhang Y, Lei Y, Wei Z, Li Y, Wang Y. et al. Proline-rich 11 (PRR11) drives F-actin assembly by recruiting the actin-related protein 2/3 complex in human non-small cell lung carcinoma. J Biol Chem. 2020;295:5335-5349
13. Wei Z, Wang R, Yin X, Zhang L, Lei Y, Zhang Y. et al. PRR11 induces filopodia formation and promotes cell motility via recruiting ARP2/3 complex in non-small cell lung cancer cells. Genes Dis. 2022;9:230-244
14. Ni W, Yi L, Dong X, Cao M, Zheng J, Wei Q. et al. PRR11 is a prognostic biomarker and correlates with immune infiltrates in bladder urothelial carcinoma. Sci Rep. 2023;13:2051
15. Cai C, He H, Duan X, Wu W, Mai Z, Zhang T. et al. miR-195 inhibits cell proliferation and angiogenesis in human prostate cancer by downregulating PRR11 expression. Oncol Rep. 2018;39:1658-1670
16. Yeh CH, Bellon M, Nicot C. FBXW7: a critical tumor suppressor of human cancers. Mol Cancer. 2018;17:115
17. Yumimoto K, Nakayama KI. Recent insight into the role of FBXW7 as a tumor suppressor. Semin Cancer Biol. 2020;67:1-15
18. Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS. et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature. 2011;471:104-9
19. Balamurugan K, Sharan S, Klarmann KD, Zhang Y, Coppola V, Summers GH. et al. FBXW7α attenuates inflammatory signalling by downregulating C/EBPδ and its target gene Tlr4. Nat Commun. 2013;4:1662
20. Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8:83-93
21. Fan J, Bellon M, Ju M, Zhao L, Wei M, Fu L. et al. Clinical significance of FBXW7 loss of function in human cancers. Mol Cancer. 2022;21:87
22. Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A. et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007;67:9006-12
23. Fu Y, Lin Y, Yang Z, Yang G, Li G, Liu Y. et al. FBXW7 overexpression suppresses renal cancer cell proliferation and induces apoptosis. Med Oncol. 2015;32:215
24. Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther. 2015;148:114-31
25. Han W, Chen L. Predictive significance of PRR11 in prognosis and immune infiltration of glioma patients. Mol Carcinog. 2023;62:975-990
26. Hafner A, Bulyk ML, Jambhekar A, Lahav G. The multiple mechanisms that regulate p53 activity and cell fate. Nat Rev Mol Cell Biol. 2019;20:199-210
27. Di Meo S, Reed TT, Venditti P, Victor VM. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid Med Cell Longev. 2016;2016:1245049
28. Srinivas US, Tan BWQ, Vellayappan BA, Jeyasekharan AD. ROS and the DNA damage response in cancer. Redox Biol. 2019;25:101084
29. Wang J, Wang H, Peters M, Ding N, Ribback S, Utpatel K. et al. Loss of Fbxw7 synergizes with activated Akt signaling to promote c-Myc dependent cholangiocarcinogenesis. J Hepatol. 2019;71:742-752
30. Zhang MX, Wang H, Sun GP. Tumor-suppressor Fbxw7 targets SIK2 for degradation to interfere with TORC2-AKT signaling in pancreatic cancer. Cell Biol Int. 2020;44:1900-1910
31. Chen CH, Shaikenov T, Peterson TR, Aimbetov R, Bissenbaev AK, Lee SW. et al. ER stress inhibits mTORC2 and Akt signaling through GSK-3β-mediated phosphorylation of rictor. Sci Signal. 2011;4:ra10
32. Nagini S, Sophia J, Mishra R. Glycogen synthase kinases: Moonlighting proteins with theranostic potential in cancer. Semin Cancer Biol. 2019;56:25-36
33. Fletcher JW, Djulbegovic B, Soares HP, Siegel BA, Lowe VJ, Lyman GH. et al. Recommendations on the use of 18F-FDG PET in oncology. J Nucl Med. 2008;49:480-508
34. Lai Y, Li Z, Lu Z, Zheng H, Chen C, Liu C. et al. Roles of DNA damage repair and precise targeted therapy in renal cancer (Review). Oncol Rep. 2022;48:213
35. Andor N, Maley CC, Ji HP. Genomic Instability in Cancer: Teetering on the Limit of Tolerance. Cancer Res. 2017;77:2179-2185
36. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
37. Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8:193-204
38. Roos WP, Thomas AD, Kaina B. DNA damage and the balance between survival and death in cancer biology. Nat Rev Cancer. 2016;16:20-33
39. Halliwell B, Adhikary A, Dingfelder M, Dizdaroglu M. Hydroxyl radical is a significant player in oxidative DNA damage in vivo. Chem Soc Rev. 2021;50:8355-8360
40. Davis RJ, Welcker M, Clurman BE. Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer Cell. 2014;26:455-64
41. King B, Trimarchi T, Reavie L, Xu L, Mullenders J, Ntziachristos P. et al. The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell. 2013;153:1552-66
42. Mao JH, Kim IJ, Wu D, Climent J, Kang HC, DelRosario R. et al. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science. 2008;321:1499-502
43. Close V, Close W, Kugler SJ, Reichenzeller M, Yosifov DY, Bloehdorn J. et al. FBXW7 mutations reduce binding of NOTCH1, leading to cleaved NOTCH1 accumulation and target gene activation in CLL. Blood. 2019;133:830-839
44. Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J. et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017;45:D777-D783
45. Revathidevi S, Munirajan AK. Akt in cancer: Mediator and more. Semin Cancer Biol. 2019;59:80-91
46. Song M, Bode AM, Dong Z, Lee MH. AKT as a Therapeutic Target for Cancer. Cancer Res. 2019;79:1019-1031
47. Chen S, Yu M, Ju L, Wang G, Qian K, Xiao Y. et al. The immune-related biomarker TEK inhibits the development of clear cell renal cell carcinoma (ccRCC) by regulating AKT phosphorylation. Cancer Cell Int. 2021;21:119
48. Peng XS, Yang JP, Qiang YY, Sun R, Cao Y, Zheng LS. et al. PTPN3 Inhibits the Growth and Metastasis of Clear Cell Renal Cell Carcinoma via Inhibition of PI3K/AKT Signaling. Mol Cancer Res. 2020;18:903-912
49. Guo H, German P, Bai S, Barnes S, Guo W, Qi X. et al. The PI3K/AKT Pathway and Renal Cell Carcinoma. J Genet Genomics. 2015;42:343-53
50. Liu R, Chen Y, Liu G, Li C, Song Y, Cao Z. et al. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020;11:797
51. Liu Q, Turner KM, Alfred Yung WK, Chen K, Zhang W. Role of AKT signaling in DNA repair and clinical response to cancer therapy. Neuro Oncol. 2014;16:1313-23
52. Shin S, Walker KA, Yoon SO. The PIKK-AKT connection in the DNA damage response. Sci Signal. 2022;15:eabm6211
53. Bozulic L, Surucu B, Hynx D, Hemmings BA. PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol Cell. 2008;30:203-13
54. Koundouros N, Poulogiannis G. Phosphoinositide 3-Kinase/Akt Signaling and Redox Metabolism in Cancer. Front Oncol. 2018;8:160
55. Kansanen E, Kuosmanen SM, Leinonen H, Levonen AL. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013;1:45-9
56. Shiau JP, Chuang YT, Tang JY, Yang KH, Chang FR, Hou MF. et al. The Impact of Oxidative Stress and AKT Pathway on Cancer Cell Functions and Its Application to Natural Products. Antioxidants (Basel). 2022;11:1845
57. Karimian A, Mir SM, Parsian H, Refieyan S, Mirza-Aghazadeh-Attari M, Yousefi B. et al. Crosstalk between Phosphoinositide 3-kinase/Akt signaling pathway with DNA damage response and oxidative stress in cancer. J Cell Biochem. 2019;120:10248-10272
58. Chibaya L, Karim B, Zhang H, Jones SN. Mdm2 phosphorylation by Akt regulates the p53 response to oxidative stress to promote cell proliferation and tumorigenesis. Proc Natl Acad Sci U S A. 2021;118:e2003193118
59. Castejón-Griñán M, Herraiz C, Olivares C, Jiménez-Cervantes C, García-Borrón JC. cAMP-independent non-pigmentary actions of variant melanocortin 1 receptor: AKT-mediated activation of protective responses to oxidative DNA damage. Oncogene. 2018;37:3631-3646
60. Glaviano A, Foo ASC, Lam HY, Yap KCH, Jacot W, Jones RH. et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol Cancer. 2023;22:138
61. Muta Y, Linares JF, Martinez-Ordoñez A, Duran A, Cid-Diaz T, Kinoshita H. et al. Enhanced SREBP2-driven cholesterol biosynthesis by PKCλ/ι deficiency in intestinal epithelial cells promotes aggressive serrated tumorigenesis. Nat Commun. 2023;14:8075
62. Linares JF, Zhang X, Martinez-Ordoñez A, Duran A, Kinoshita H, Kasashima H. et al. PKCλ/ι inhibition activates an ULK2-mediated interferon response to repress tumorigenesis. Mol Cell. 2021;81:4509-4526.e10
63. Yan Y, Tao H, He J, Huang SY. The HDOCK server for integrated protein-protein docking. Nat Protoc. 2020;15:1829-1852
Author contact
![]() Corresponding authors: Dr. Lingao Ju, email: julingao1990edu.cn; Dr. Kaiyu Qian, email: qky1009edu.cn; or Dr. Xinghuan Wang, email: wangxinghuanedu.cn.
Corresponding authors: Dr. Lingao Ju, email: julingao1990edu.cn; Dr. Kaiyu Qian, email: qky1009edu.cn; or Dr. Xinghuan Wang, email: wangxinghuanedu.cn.