Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2024; 14(19):7554-7568. doi:10.7150/thno.99565 This issue Cite
Research Paper
Rc3h1 negatively regulates osteoclastogenesis by limiting energy metabolism
Liuyuan Chen1#, Yuangang Su1,2#, Chaofeng Wang1,2, Qian Huang1, Weiwei Chen1,2, Na Hai1,2, Jikang Wang1,2, Haoyu Lian1,2, Jinmin Zhao1,2, Jiake Xu1,3,4 ![]() , Qian Liu1
, Qian Liu1 ![]()
1. Guangxi Key Laboratory of Regenerative Medicine, Orthopaedic Department, The First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi 530021, China.
2. Collaborative Innovation Centre of Regenerative Medicine and Medical BioResource Development and Application Co-constructed by the Province and Ministry, Life Sciences Institute, Guangxi Medical University, Nanning, Guangxi 530021, China.
3. Faculty of Pharmaceutical Sciences, Shenzhen University of Advanced Technology, and Chinese Academy of Sciences, Shenzhen, China.
4. School of Biomedical Sciences, The University of Western Australia, Perth, Western Australia, Australia.
# Liuyuan Chen and Yuangang Su contributed equally to this study.
Received 2024-6-13; Accepted 2024-10-22; Published 2024-11-4
Abstract

Rationale: Osteoclasts are giant bone-resorbing cells that need vigorous mitochondrial respiration to support their activation. Rc3h1, an RNA-binding protein, precisely governs the homeostasis of mRNA. However, the precise role of Rc3h1 in regulating iron metabolism and mitochondrial respiration in osteoclasts is not yet understood.
Methods: We generated Rc3h1-deficient mice in osteoclast precursors and mature osteoclasts. The bone mass and osteoclast activity in bone tissues were evaluated. Moreover, we assessed the differentiation, bone resorption, iron content, and mitochondrial function of osteoclasts in vitro. In the end, the target gene of Rc3h1 and its role in mediating the effect of Rc3h1 on mitochondrial respiration in osteoclasts were further investigated.
Results: Mice lacking Rc3h1 exhibit low bone mass. In addition, Rc3h1 deletion in osteoclasts significantly promotes osteoclast activation. Mechanistically, Rc3h1 post-transcriptionally represses the expression of transferrin receptor 1 (Tfr1), restricting iron absorption and mitochondrial respiration in osteoclasts. Inhibition of Tfr1 in Rc3h1-deficient osteoclasts diminishes excessive osteoclast formation and mitochondrial respiration.
Conclusion: These findings suggest that Rc3h1 has a negative effect on osteoclast activation via limiting iron resorption and mitochondrial respiration. Finally, targeting the Rc3h1/Tfr1 axis might represent a potential therapeutic approach for bone-loss diseases.
Keywords: Rc3h1, osteoclast, Tfr1, mitochondria, osteoporosis
Introduction
Bone is a metabolically dynamic organ maintained by continuous bone renewal, completed by osteoclasts (OCs)-mediated old bone resorption and subsequent new bone formation by osteoblasts [1]. Bone resorption exceeds bone formation in bone remodeling, caused by over-activated osteoclasts, resulting in bone mass loss. With the stimulation of two master regulators, macrophage colony-stimulating factor (M-CSF) and receptor activators of nuclear factor kappa-Β ligand (RANKL), bone marrow-derived macrophages (BMMs) differentiate into tartrate-resistant acid phosphatase (TRAP)-positive mononuclear cells and fuse to multinucleated osteoclasts, meanwhile migrating to the bone surface to resorb old bone [2]. This process needs constant energy supply and metabolic reprogramming to meet osteoclast formation and bone resorption [3,4]. Mitochondrial oxidative phosphorylation (OXPHOS) is the main source of energy supply in osteoclast differentiation [5]. OXPHOS is positively associated with osteoclast differentiation, and targeting pathways modulating OXPHOPS might serve as an attractive target to preserve bone mass [6-8].
Rc3h1, also known as Roquin1, is an RNA-binding protein that works directly by binding with certain stem-loop structural elements in the 3' untranslated regions (3'-UTR) of mRNA [9,10]. Rc3h1 is widely recognized as a regulator of T cell fates by suppressing the expression of key genes that play a critical role in Th1, Th17, and Tfh activation at the post-transcriptional level [11-13]. Rc3h1 in macrophages specifically identifies the stem-loop motif in the 3'-UTR of TNF-α mRNA, inhibiting the synthesis of TNF-α [10]. Rc3h1 could be seen as a 'gatekeeper' that limits immoderate immune responses and inflammatory processes. Also, Rc3h1 serves as a regulator for maintaining the homeostasis of micro-RNA levels [14]. Additionally, Rc3h1 functions as an E3 ligase and involves the ubiquitinated degradation of the α1 subunit of adenosine monophosphate-activated protein kinase (AMPK) through its RING domain [15]. Rc3h1 was reported to be a major regulator for cellular iron homeostasis by limiting the expression of transferrin receptor 1 (Tfr1), encoded by TFRC [16]. The absence of Tfr1 in osteoclasts reduces mitochondrial biogenesis and OXPHOS by lowering Tfr1-mediated iron absorption [17]. Furthermore, higher iron uptake promotes mitochondrial biogenesis and function in osteoclasts or adipocytes [18-20]. Nevertheless, the role of Rc3h1 in regulating iron metabolism and mitochondrial respiration in osteoclasts remains unclear.
This work involved the deletion of Rc3h1 in both osteoclast precursors and mature osteoclasts to investigate the effect of Rc3h1 on bone mass and osteoclast formation. It was found that the absence of Rc3h1 led to significant bone loss, accompanied by an increased number of osteoclasts in bone tissues. Furthermore, Rc3h1 post-transcriptionally inhibits the expression of Tfr1, subsequently contributing to attenuated iron absorption and OXPHOS. Taken together, our findings reveal Rc3h1 as a novel regulator in controlling iron metabolism and mitochondrial respiration in osteoclasts.
Results
Deletion of Rc3h1 in osteoclast precursors leads to low bone mass
We evaluated the expression of Rc3h1 in the bone tissue of the femoral head of postmenopausal osteoporosis patients using the dataset GSE230665 [21]. The level of Rc3h1 expression was elevated in postmenopausal osteoporosis patients (PMOPs) compared to healthy postmenopausal women (Figure S1A-B). In addition, the western blot analysis revealed an upregulation of Rc3h1 during in vitro osteoclast formation induced by M-CSF and RANKL, as shown in Figure 1A. These results suggested that Rc3h1 potentially plays an important role in osteoporosis.
Low bone mass was exhibited in Rc3h1LysM mice. (A) Protein level of Rc3h1 during osteoclast formation induced by M-CSF and RANKL from C57BL/6J mice. (B) The protein expression of Rc3h1 in Rc3h1flox and Rc3h1LysM osteoclasts. (C-D) Representative micro-CT images and quantitative analysis of proximal tibia trabecular and cortical bone in 12-week-old male Rc3h1flox (n = 8) and Rc3h1LysM mice (n = 11). (E-F) Representative micro-CT images and quantitative analysis of proximal tibia trabecular and cortical bone in 12-week-old female Rc3h1flox (n = 7) and Rc3h1LysM (n = 7) mice. (G) TRAP staining and analysis of proximal tibia from Rc3h1flox (n = 5) and Rc3h1LysM (n = 5) mice. Scale bar= 200 μm. (H-I) The ELISA of CTX-1 and PINP from Rc3h1flox (n = 8) and Rc3h1LysM (n = 11) serum. (J) Double calcein label of proximal tibia bone in Rc3h1flox (n = 5) and Rc3h1LysM (n = 5) mice. Scale bar= 50 μm. *P < 0.05, **P < 0.01, ***P < 0.001.
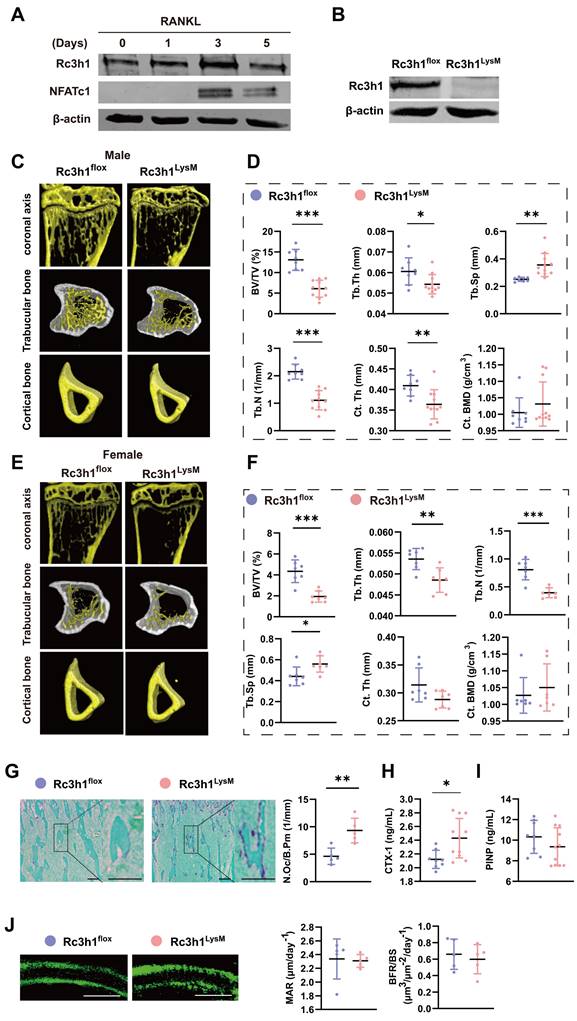
Our team has previously discovered that mice with a point mutation of Rc3h1 develop significant bone loss and increased osteoclasts in bone tissue [22]. To elucidate the role of Rc3h1 in osteoclasts and bone diseases, we obtained Rc3h1 conditional knockout mice in osteoclast precursors by crossing Rc3h1-flox mice, where the exon 3 of the Rc3h1 gene was flanked with two loxP sites, with myeloid-specific LysM-Cre mice. Genotyping and WB analysis were used to identify wild-type (Rc3h1flox/flox; LysM-Cre- referred to as Rc3h1flox) and knockout mice (Rc3h1flox/flox; LysM-Cre+ referred to as Rc3h1LysM), and to validate the knockout efficiency of Rc3h1 (Figure S1C and Figure 1B). Generally, Rc3h1LysM mice did not show marked differences in body size and weight compared to WT littermates (data not shown). Micro-CT analysis of the proximal tibia of 12-week-old WT and Rc3h1LysM mice revealed that deletion of Rc3h1 resulted in significant trabecular bone loss, reaching an approximately 2-fold reduction in BV/TV in both male and female mice compared to WT mice (Figure 1C-D and Figure 1E-F). In addition, the number of trabeculae (Tb.N) and the thickness of trabeculae (Tb. Th) were also significantly attenuated, while the trabecular separation (Tb. Sp) was enlarged in Rc3h1LysM mice. Only male mice exhibited reduced cortical bone thickness (Ct. Th). The cortical bone mineral density (Ct. BMD) in Rc3h1LysM mice was not noticeably altered in both genders.
Bone histological analysis revealed that the proximal tibial bone surface of Rc3h1LysM was stained for more TRAP activity (Figure 1G), a marker of osteoclasts. Serum CTX-1 in Rc3h1LysM mice was also increased (Figure 1H). The elevated osteoclast activity might affect the differentiation of osteoblasts [23, 24]. However, the serum PINP levels, mineral deposition rate (MAR), and bone formation rate (BFR/BS) in Rc3h1LysM mice were similar to those in the WT group (Figure 1I and 1J). These results showed that the loss of bone mass in Rc3h1-deficient mice may be attributable to enhanced osteoclast activity rather than osteoblast activity.
Loss of Rc3h1 in differentiated osteoclasts results in decreased bone mass
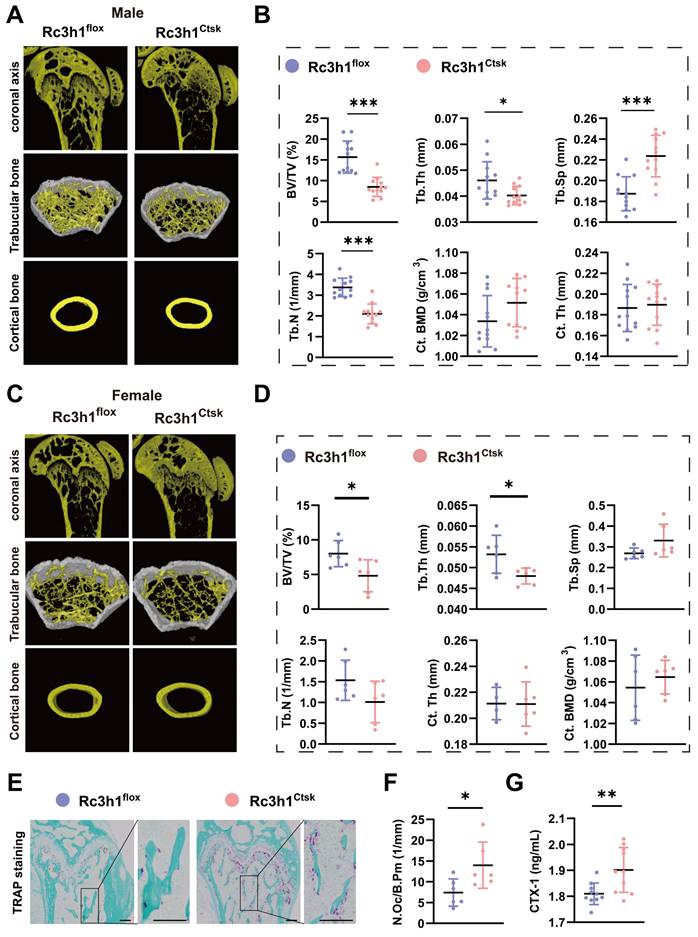
To further explore the role of Rc3h1 specifically in differentiated osteoclasts, the Rc3h1flox/flox; Ctsk-Cre- (Rc3h1flox) and Rc3h1flox/flox; Ctsk-Cre+ (Rc3h1Ctsk) mice were generated. The distal femurs were harvested from 12-week-old Rc3h1flox and Rc3h1Ctsk mice, and the bone tissues were subsequently scanned by Micro-CT.
As shown in Figure 2A-D, the BV/TV and Tb. Th were decreased in male and female Rc3h1Ctsk mice. The Tb. N was declined, and the Tb. Sp was increased only in male Rc3h1Ctsk mice. The Ct. Th and Ct.BMD were not altered in both genders of Rc3h1Ctsk mice compared to their WT littermates. Consistent with the Micro-CT findings, the TRAP staining of the distal femur from Rc3h1Ctsk demonstrated more osteoclast activity (Figure 2E-F). The serum CTX-1 level in Rc3h1LysM mice also was increased (Figure 2G).
Deletion of Rc3h1 in differentiated osteoclasts leads to bone loss. (A-B) Representative 3D micro-CT reconstruction images and quantitative analysis of distal femur trabecular and cortical bone in 12-week-old male Rc3h1flox (n = 12) and Rc3h1Ctsk (n = 12) mice. (C-D) Micro-CT scanning and analysis of distal femur trabecular and cortical bone in 12-week-old female Rc3h1flox (n = 6) and Rc3h1Ctsk (n = 6) mice. (E-F) TRAP staining was used to quantitatively analyze the number of osteoclasts in distal femur bone tissue in 12-week-old male Rc3h1flox (n = 6) and Rc3h1Ctsk (n = 6) mice. (G) The serum CTX-1 was measured by ELISA. Scale bar= 200 μm (n = 10). *P < 0.05, **P < 0.01, ***P < 0.001.
Rc3h1 deficiency facilitates the formation of osteoclasts
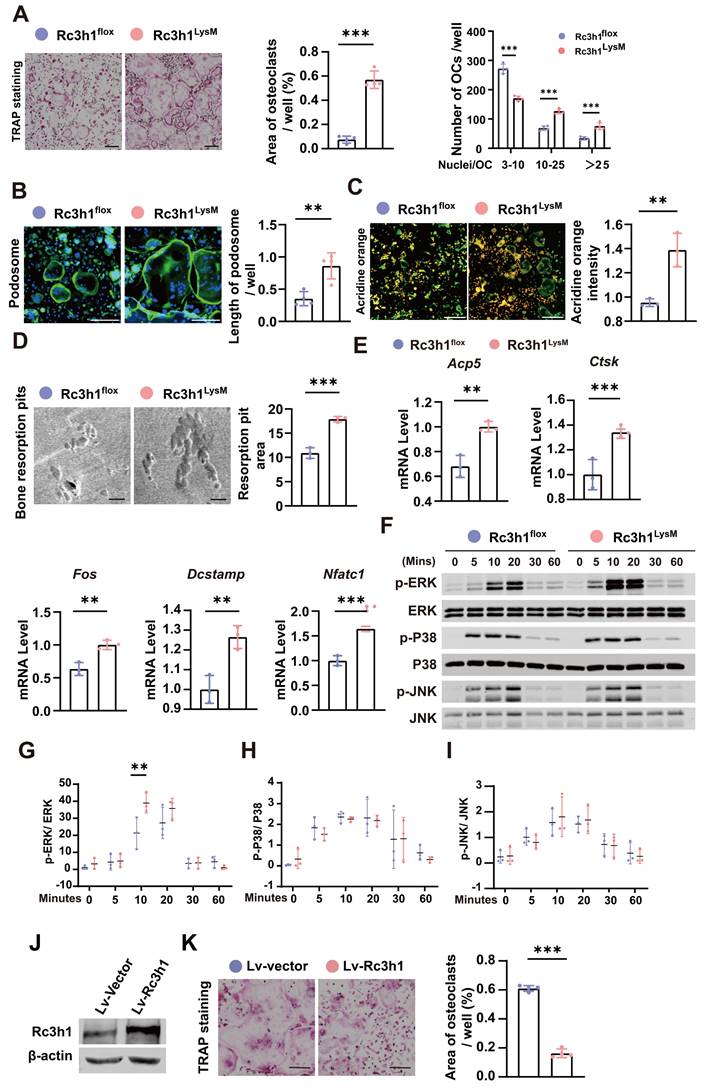
BMMs were isolated from the tibias and femurs of Rc3h1flox and Rc3h1LysM mice and cultured with M-CSF and RANKL to generate mature osteoclasts. As shown in Figure 3A-B, Rc3h1LysM osteoclasts demonstrated a more robust capability to spread and form larger morphologies of mature osteoclasts and F-actin rich podosome-belts. There was an increase in the activation of NFATc1 and its translocation from the cytoplasm to the nucleus (Figure S2A-B). Rc3h1 deletion enhanced the capacity of osteoclasts to acidify the extracellular circumstances and degrade the bone matrix, as shown by higher acid content and more bone resorptive pits (Figure 3C and D). The expression of genes, including Acp5, Ctsk, Fos, Dc-stamp, and Nfatc1, required for osteoclast differentiation, was also increased in Rc3h1LysM osteoclasts compared to controls (Figure 3E). The protein levels of CTSK, NFATc1, ACP5 and c-FOS were also raised in Rc3h1-deficient osteoclasts (Figure S2C-D). Furthermore, deficiency of Rc3h1 overly activated the RANKL-induced ERK signaling pathway (Figure 3F-I). We simultaneously overexpressed Rc3h1 in BMMs and induced osteoclasts in M-CSF and RANKL until mature osteoclasts formed in controls. The expression of Rc3h1 was evaluated in control and Rc3h1-overexpressed osteoclasts (Figure 3J). We observed a decrease in osteoclastogenesis in Rc3h1-overexpressed osteoclasts compared to the control group, which had already formed mature and large osteoclasts (Figure 3K). These results indicate that loss of Rc3h1 fuels osteoclast formation and function in an intrinsic manner.
Loss of Rc3h1 accelerates osteoclast formation. (A) In vitro TRAP staining was performed on osteoclasts from Rc3h1flox and Rc3h1LysM mice. Scale bar= 200 μm. (B) Podosome staining on Rc3h1flox and Rc3h1LysM osteoclasts. Scale bar= 200 μm. (C) Representative images of acidification in osteoclasts from Rc3h1flox and Rc3h1LysM osteoclasts. Scale bar= 300 μm. (D) Representative images of scanning electron microscopy on bone resorption pits from Rc3h1flox and Rc3h1LysM osteoclasts. Scale bar= 100 μm. (E) Expression levels of osteoclast-related genes on BMMs with the induction of M-CSF and RANKL for 3 days assessed using qPCR. (F-I) Proteins and phosphorylation states of ERK, P38, and JNK upon the stimulation of RANKL. (J-K) The WB of Rc3h1 and TRAP staining of osteoclasts stimulated with RANKL and M-CSF for 6 days from BMMs transfected with Lv-vector or Lv-Rc3h1. *P < 0.05, **P < 0.01, ***P < 0.001.
Rc3h1 targets the mRNA of Tfr1 and restrains Tfr1-mediated iron resorption in osteoclasts
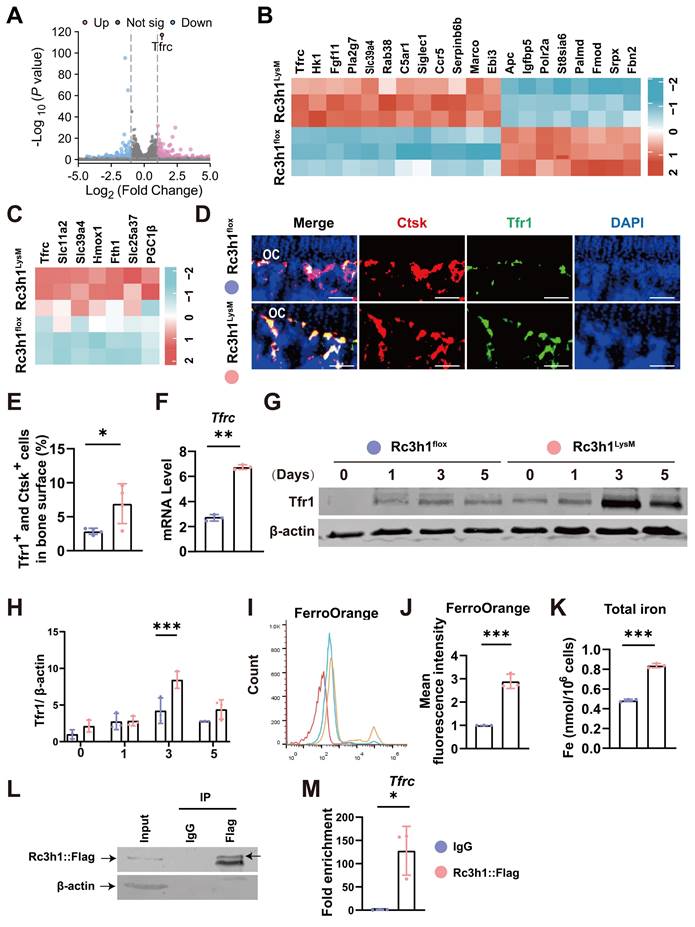
As an RNA-binding protein, Rc3h1 promotes the mRNA of target genes decay by directly binding the 3′-untranslated region of mRNA [10,25]. Rc3h1 targets the mRNA of Icos and Ox40 in follicular helper T cells, cell cycle-promoting genes in breast cancer cell lines, and Tfr1 in HAP1, HUVEC, L-M, and MEF cell lines [16,26,27], suggesting Rc3h1 controls the abundance of mRNA in a cell-specific manner. Subsequently, we utilized RNA-seq to identify possible targets of Rc3h1 in osteoclasts from Rc3h1LysM and WT mice. As shown in Figure 4A-B, Tfrc, which encodes the Tfr1 protein, is the most significantly upregulated gene. A previous study has found that Rc3h1 is a major mediator of Tfr1-mediated iron metabolism, and the mRNA is one of the targets of Rc3h1 in HAP1, HUVEC, L-M, and MEF cell lines [16]. Tfr1 is widely recognized as the primary carrier of iron and plays an essential role in regulating cellular iron homeostasis [28,29]. Thus, we focused on the iron metabolism-related genes and found these genes were upregulated (Figure 4C). Next, the expression of Tfr1 on osteoclasts from bone tissues was higher in Rc3h1LysM than in WT littermates (Figure 4D-E). Consistently, we verified the increased mRNA level of Tfr1 (Figure 4F) and the protein level of Tfr1 (Figure 4G-H) in Rc3h1LysM osteoclasts. Meanwhile, the Fe2+ and total iron content in Rc3h1LysM osteoclasts were significantly elevated (Figure 4I-K). Eventually, we confirmed the interaction of Rc3h1 with the mRNA of Tfr1 using an RNA immunoprecipitation (RIP) assay (Figure 4L and 4M). In conclusion, Rc3h1 negatively controls cellular iron content through limiting Tfr1-mediated iron resorption in osteoclasts.
Tfr1 is a candidate target of Rc3h1 in osteoclasts. (A-B) Volcano plot and heatmap of differentially expressed genes in Rc3h1flox and Rc3h1LysM osteoclasts induced by M-CSF and RANKL for 3 days (n = 3). (C) Heatmap of iron metabolism-related genes in Rc3h1flox and Rc3h1LysM osteoclasts. (D-E) Immunofluorescent staining and quantitative analysis of Tfr1 on proximal tibia bone slides from Rc3h1flox and Rc3h1LysM mice (n = 4). Scale bar= 250 μm. (F) mRNA level of Tfr1 using qPCR in WT and Rc3h1-deficient osteoclasts. (G-H) Protein level of Tfr1 during the formation of osteoclasts in WT and Rc3h1-deficient osteoclasts. (I-J) Results of mean fluorescent intensity of FerroOrange from WT and Rc3h1-deficient osteoclasts using flow cytometry to detect cellular Fe2+content. (K) Total iron from lysed otseoclasts measured by absorbance at 593 nm. (L-M) Western bolts were presented to confirm the Rc3h1-IP efficiency, and qPCR was used to quantify the indicated mRNA in the IP and IgG groups. RIP was performed on RAW264.7 cells transfected with the Rc3h1-Puro-Flag vector after RANKL stimulation for 3 days (n= 3). *P < 0.05, **P < 0.01, ***P < 0.001.
Rc3h1 negatively modulates mitochondrial respiration
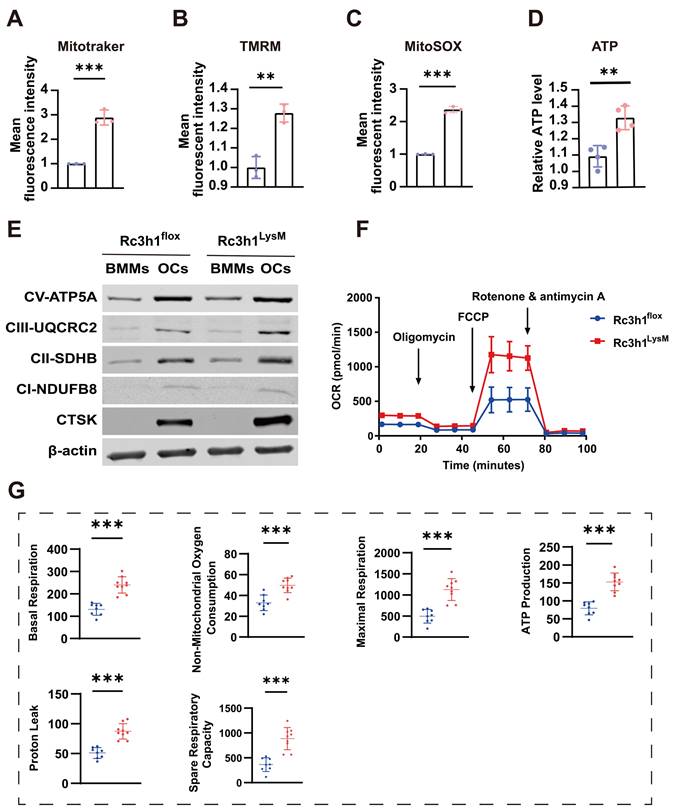
Tfr1-mediated iron absorption is crucial for mitochondrial OXPHOS [17, 19]. The differentiation of osteoclast relies heavily on the energy obtained from OXPHOS [30], and modulation of mitochondrial OXPHOS also determines the fate of osteoclast [7,18]. In Rc3h1-deficient osteoclasts, we observed a significant enhancement in Tfr1-mediated iron absorption. Firstly, we measured mitochondrial mass, membrane potential, and ROS levels in Rc3h1LysM and Rc3h1flox osteoclasts using flow cytometry. In Rc3h1LysM osteoclasts, the mean fluorescent intensity of mitotraker, tetramethylrhodamine (TMRM), and mitoSOX was significantly increased (Figure 5A-C). The absence of Rc3h1 resulted in a higher production of ATP (Figure 5D). We then performed WB analyses to examine the expression of the OXPHOS complexes. A modest increase in Rc3h1LysM osteoclasts was observed for complexes II and III (Figure 5E). Finally, the extracellular oxygen consumption rate (OCR) of Rc3h1-deficient osteoclasts and controls was assessed to measure mitochondrial respiration. Similarly, the Rc3h1LysM osteoclasts displayed a higher level of extracellular oxygen consumption rate compared to Rc3h1flox (Figure 5F-G). Our results indicated that Rc3h1 constraints mitochondrial respiration and ATP production in osteoclasts.
Loss of Rc3h1 in osteoclasts results in enhanced mitochondrial respiration. (A-C) Flow cytometry analysis of osteoclasts' mitochondrial mass, ROS, and membrane potential stained with mitogreen, mitoSOX, and TMRM, respectively. (D) Relative cellular ATP content in WT and Rc3h1-deficient osteoclasts. (E) Western blot evaluation of mitochondrial respiration complexes C-I to C-III, C-V and CTSK in Rc3h1flox and Rc3h1LysM osteoclasts. (F-G) Extracellular oxygen consumption rate (OCR) analysis and statistical analysis of Rc3h1flox and Rc3h1LysM osteoclasts using seahorse assay. All the osteoclasts were derived from Rc3h1flox and Rc3h1LysM BMMs after M-CSF and RANKL induction for 3 days. *P < 0.05, **P < 0.01, ***P < 0.001.
Tfr1 mediates the effects of Rc3h1 on mitochondrial respiration in osteoclasts
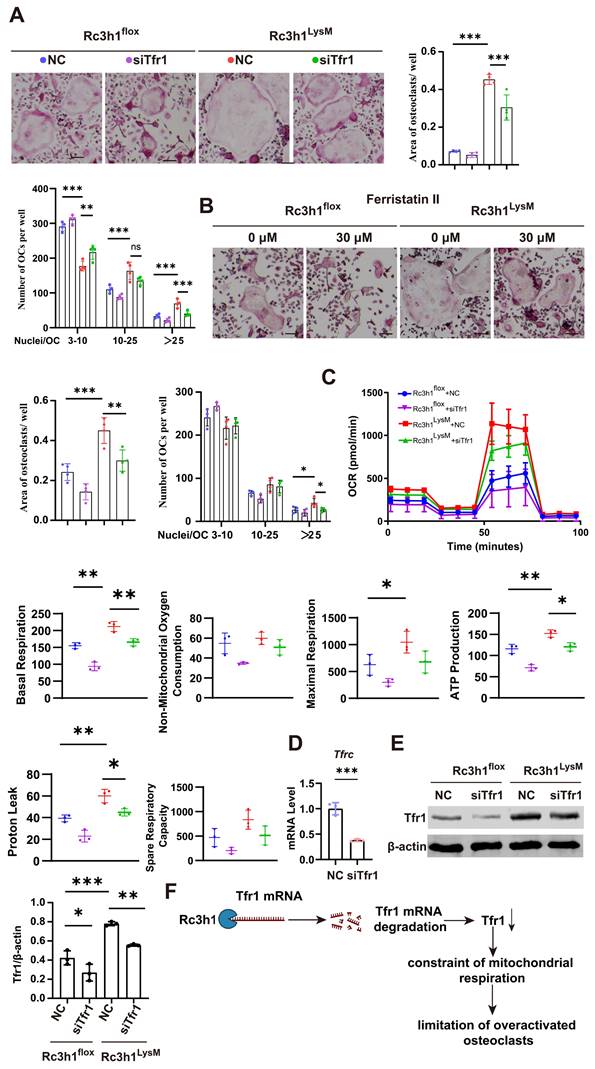
We investigated whether the elevated Tfr1 in Rc3h1-deficient osteoclasts mediates the enhanced mitochondrial respiration. To this end, the expression of Tfr1 was inhibited using either ferristatin II, a small-molecule inhibitor of Tfr1, or siRNA targeting Tfr1. Ferristatin II functions via facilitating the degradation of Tfr1 [31]. We observed that inhibition of Tfr1 by siRNA effectively mitigated the over-activated osteoclastogenesis induced by RANKL and M-CSF in Rc3h1LysM BMMs, as depicted in Figure 6A. Similarly, the osteoclastogenesis of Rc3h1LysM BMMs was reduced when treated with ferristatin II (Figure 6B). Simultaneously, the OCR in Rc3h1LysM osteoclasts was decreased when Tfr1 was inhibited by siRNA (Figure 6C). Additionally, the mRNA level of Tfr1 after knockdown by siTfr1 was evaluated through qPCR (Figure 6D). The protein level of Tfr1 in Rc3h1flox and Rc3h1LysM osteoclasts treated with siTfr1 also was measured by WB (Figure 6E).
Tfr1 mediates the effects of Rc3h1 on osteoclastogenesis and mitochondrial respiration. (A-B) Representative TRAP staining images and quantitative analysis of osteoclasts induced by M-CSF and RANKL 5 days with the presence of siTfr1 or Ferristatin II. Scale bar= 200 μm. (C) OCR analysis of Rc3h1flox and Rc3h1LysM osteoclasts using seahorse assay with or without the presence of siTfr1. (D) The mRNA expression of Tfr1 in osteoclasts treated with negative control or siTfr1 using qPCR. (E) The protein expression of Tfr1 in osteoclasts treated with negative control or siTfr1. (F) The diagram of the mechanism of Rc3h1 regulating mitochondrial respiration in osteoclasts (created in BioRender.com). *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
Over-activated osteoclasts lead to excessive bone resorption, resulting in a disruption of the bone remodeling balance, which consequently causes osteoporosis [32]. Limiting the hyper-activation of osteoclasts has been one of the mainstream strategies for developing therapeutic agents for osteoporosis [33]. Rc3h1 has been widely reported to limit the overwhelming activation of T cells [34], proliferation of breast cancer cells [27], and excessive production of inflammatory factors secreted by macrophages [10]. It is reminiscent that Rc3h1 is likely capable of inhibiting the overt formation of osteoclasts. Indeed, a point mutation (M119R) in the Rc3h1 gene is associated with loss of bone mass in Sanroque mice [22]. Initially, we found upregulated Rc3h1 expression in osteoclasts under the pathologic condition of osteoporosis. Osteoclast-specific deletion of Rc3h1 causes bone loss. In vitro osteoclast differentiation showed that deletion of Rc3h1 in osteoclasts led to hyperactivation of osteoclasts. Mechanistically, Rc3h1 could target Tfr1 mRNA and promote its degradation, reducing cellular iron uptake and preventing excessive amplification of mitochondrial respiration, consequently repressing osteoclast hyperactivation.
The most exciting finding in our study might be the low bone mass phenotype in Rc3h1LysM mice. This phenotype is mainly caused by excessive bone resorption by osteoclasts, leading to imbalanced bone remodeling. Likewise, deletion of Rc3h1 accelerates osteoclast formation during RANKL induction in vitro, explaining the low bone mass phenotype in mice. Several critical coupling factors secreted by osteoclasts promote new bone formation by osteoblasts [35]. This coupling mechanism appears to not work in Rc3h1-deficient mice, where excessive bone resorption does not significantly affect new bone deposition. Another interesting phenomenon is that after the deletion of Rc3h1, major bone loss occurs in the trabecular bone region, with no significant change in cortical bone density. Trabecular and cortical bone exhibit distinct structural properties, metabolic activity, and rates of bone turnover [36,37]. Given the larger surface area available for bone remodeling in trabecular bone, the trabecular bone is the leading site where bone loss occurs during the initial phases of osteoporosis [38]. This may account for the notable reduction of bone mass in trabecular bone rather than cortical bone in Rc3h1-deficient mice.
Trabecular bone mass was reduced in both male and female Rc3h1LysM and Rc3h1Ctsk mice. Nevertheless, this phenotype appears to be more prominent in male mice in comparison to female mice, particularly when Rc3h1 is deleted in mature osteoclasts. It is wildly accepted that the drop in the estrogen level upon menopause could result in bone loss and overactivated osetoclastogenesis. In osteoclasts, estrogen suppresses the expression of Tfr1, as well as mitochondrial function and energy metabolism [17, 39].
Rc3h1 has been characterized as an RNA-binding protein that functions as a transcriptional repressor by facilitating the degradation of the mRNA of target genes [10,40]. As previously mentioned, Rc3h1 harbors a broad pool of target genes and selects its targets in a context- and cell-dependent manner [25,26,27]. Recently, Corral, VM, et al. reported that Rc3h1 regulates cellular iron metabolism by targeting and destabilizing the mRNA of Tfr1 in four different cell lines (HAP1, HUVEC, L-M, and MEF) [16]. Using RNA-seq, Tfr1 was the most significantly differential gene, and genes related to iron metabolism were also significantly altered, enlightening us to select Tfr1 as a candidate target of Rc3h1 in osteoclasts. Subsequent experiments revealed a markedly higher expression of Tfr1 and an elevated iron content in Rc3h1-deficient osteoclasts. Meanwhile, the RIP assay verified the capacity of Rc3h1 binding with the mRNA of Tfr1, substantiating that Tfr1 is one of the targets of Rc3h1 in osteoclasts.
Tfr1-mediated iron uptake is the predominant approach for the cellular acquisition of iron [41]. Tfr1 and iron are strongly associated with mitochondrial function in osteoclasts and other cells, mainly contributing to the facilitation of mitochondrial iron-sulfur cluster synthesis, heme synthesis, and the efficiency of electron transport [18,20,42]. Given the significantly increased Tfr1 and iron in Rc3h1-deleted osteoclasts, it is plausible to infer that mitochondrial metabolism might be strengthened. We found that mitochondrial mass, membrane potential, ROS, and extracellular oxygen consumption are elevated in Rc3h1-deficient osteoclasts. There are discrepancies between the results of the protein levels of mitochondrial complexes and other indicators of mitochondrial metabolism revealed by flow cytometry. Bhaba K. Das1 reported that the knockout of Tfr1 in osteoclasts decreased mitochondrial complexes II and III without affecting I, IV, and V [17]. One possible explanation is that the differential changes of subunits of ETC might be attributed to the varying distribution of iron-sulfur clusters or heme in the subunits of the electron transport chain [43]. An additional critical phenotype is that Rc3h1 deficiency leads to hyperactivation of osteoclasts. NFATc1, a major transcription factor that determines osteoclast differentiation, is phosphorylated in the cytoplasm, and then translocates to the nucleus to initiate transcriptional expression of osteoclast-related genes [44]. Increased NFATc1 translocation into the nucleus in Rc3h1-deleted osteoclasts indicates enhanced osteoclast formation. Whether the alteration of mitochondrial function affects the number of osteoclasts is controversial. PGC1β is a major regulator of mitochondrial biogenesis and function in osteoclast [7]. Zhang reported that loss of PGC1β in osteoclast precursor cells results in reduced mitochondrial biogenesis and respiration, leading to reduced osteoclast bone resorption without affecting osteoclast differentiation [45]. Another piece of evidence shows that deletion of PGC1β in osteoclasts inhibits osteoclast differentiation and mitochondrial function [18]. We also found that loss of Rc3h1 exhibited a larger cytoskeleton. It should be noted that counting the number of osteoclasts (TRAP-positive and nuclei ≥3) might underestimate the actual capacity of Rc3h1LysM osteoclast differentiation, especially when compared to control osteoclasts. Thus, vigorous mitochondrial respiration is most likely responsible for osteoclast hyperactivation in Rc3h1LysM osteoclasts. We found that the ERK signaling pathway was significantly enhanced in Rc3h1-deficient osteoclasts. Intracellular ROS originates mainly from the electron transport chain of mitochondria [46]. Our findings suggest that loss of Rc3h1 promotes Tfr1-mediated iron uptake in osteoclasts and the subsequent mitochondrial respiration. The enhanced mitochondrial OXPHOS contributes more intracellular ROS generation. The activation of the ERK signaling pathway mediated by ROS has been well studied [47]. A plausible explanation is that the increased activity of the ERK signaling pathway might be attributed to the augmented production of mitochondrial ROS.
In this study, we evaluate the essential role of Rc3h1 in osteoclast differentiation and function. The present study elucidates a post-transcriptional mechanism by which Rc3h1 regulates iron metabolism and mitochondrial respiration in osteoclasts. Our research suggests that Rc3h1 may be a therapeutic target for bone loss diseases.
Materials and Methods
Mice
The Rc3h1-flox (Rc3h1flox); LysM-Cre and Rc3h1-flox (Rc3h1flox); Ctsk-Cre mice were purchased from GemPharmatech Co., Ltd. (Nanjing, China). Briefly, we crossed Rc3h1flox mice with LysM-Cre or Ctsk-Cre mice to obtain the myeloid-specific and differentiated-osteoclast Rc3h1 knockout mice. C57BL/6J mice were obtained from the animal center of Guangxi Medical University. All the mice were maintained at the animal center of Guangxi Medical University with a 12-hour light cycle, free access to water, and a standard laboratory diet. All the animal studies were approved and supervised by the committee's Animal Care and Welfare Committee of Guangxi Medical University (Experimental Ethics Approval Number: 202005177).
Micro-computed tomography (Micro-CT) analysis
After removing the soft tissue of the tibia and femur from 12-week-old mice, the proximal tibia and distal femur were preserved overnight in 4% paraformaldehyde and washed three times with PBS before a micro-CT scan (SkyScan 1176, Bruker). The bone volume per tissue volume (BV/TV), trabecular number (Tb. N), trabecular space (Tb. Sp), trabecular thickness (Tb. Th), cortical bone mineral density (Ct. BMD), and cortical thickness (Ct. Th) were analyzed using the CTAn software (SkyScan). The 3D reconstruction of the tibia and femur was performed using CTvox software.
Bone histomorphometry
The tibias were decalcified for 14 days and embedded in paraffin for sectioning with a 5 μm thickness, and TRAP staining was performed. Images were acquired by a KFPRO scanner and visualized by K-Viewer software (KONFOONG BIOTECH INTERNATIONAL CO., LTD., Ningbo, China). For Tfr1 detection, double fluorescent staining for Ctsk and Tfr1 was performed, and the preparation for cryosection of the tibia bone was completed with reference to previous literature [48]. The fresh bone tissues were dissected, and the bone tissue was fixed in 4% paraformaldehyde for 24 hours immediately. Next, the bone tissues were decalcified in EDTA for 48 hours, and the EDTA was replaced with a cryoprotective solution (20% sucrose and 2% polyvinylpyrrolidone) 24 hours before embedding in the embedded solution (8% gelatin, 20% sucrose, and 2% PVP). After embedding, the bone tissues were preserved at -80 °C before section. The 50 μm thickness for the bone section was obtained. Following the permeabilization, the primary antibodies for Ctsk (sc-48353, Santa Cruz) and Tfr1 (sc-65882, Santa Cruz), followed by second antibodies, were used for the staining. The fluorescent images were acquired by the confocal microscope (Leica STELLARIS, Heidelberg, Germany). ImageJ software was used to analyze the bone histomorphometry.
BMMs isolation and osteoclast culture
BMMs isolation was carried out with reference to the other research [7]. Briefly, bone marrow cells were extracted from the tibia and femur, then lysed by the Red Blood Cell Lysis Buffer (C3702, Beyotime, China), and subsequently cultured in α-minimum essential medium plus 10% fetal bovine serum, 1% penicillin-streptomycin, and macrophage colony-stimulating factor. After 48 hours of culture, nonadherent cells were removed, and the complete media was refreshed. For osteoclast differentiation, BMMs were seeded into 96-well plates and induced by M-CSF and RANKL, and the medium containing M-CSF and RANKL was changed every other day to generate mature osteoclasts for 4-6 days. TRAP staining was conducted with a TRAP kit (Sigma-Aldrich). Cells positive for TRAP staining and ≥3 nuclei were counted as osteoclasts.
Extracellular acidification assay and bone resorption assay
For detecting acid production, BMMs stimulated with RANKL for 3 days were stained with acridine orange. For bone resorption, BMMs were seeded on the bovine cortical bone and cultured with RNAKL for 9 days. The bovine cortical bones were harvested and scanned with a scanning electron microscope as reported [49].
Lentiviral transfection
Rc3h1-overexpressing lentiviruses were purchased from Sangon Biotech (Shanghai) Co., Ltd. BMMs or RAW264.7 cells were infected with lentiviruses at a multiplicity of infection (MOI) of 50 or 150, respectively, with the help of 4 μg/mL polybrene. 48 hours later, the BMMs or RAW264.7 cells were screened with puromycin (4 or 10 μg/mL for BMMs and RAW264.7 cells respectively).
Total RNA isolation and qPCR
BMMs were seeded in 6-well plates and induced with RANKL for 3-5 days. According to the manufacturer's instructions, TRIzol reagent was used to extract the total RNA, and the total RNA concentration was measured with NanoDrop One/OneC (ThermoFisher). Reverse RNA transcription to cDNA was done with a Reverse Transcription Kit (k16225, Thermo Fisher) using a 20 μL reaction system. A quantitative polymerase chain reaction was performed on a PCR instrument (Roche, Basel, Switzerland). Gene expression was calculated using the 2-ΔΔCt method. The expression of the gene was normalized to the average expression level of Actb.
Western blots
BMMs stimulated with RANKL for 3-5 days were lysed using RIPA buffer, which included a protease inhibitor, PMSF, and a phosphatase inhibitor. The lysates were then heated at 100 °C for 10 minutes and mixed with loading buffer. Proteins were loaded to 8%-15% SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. Then, the membranes containing proteins were blocked with 5% fat-free milk for 1 hour, followed by incubation with the primary antibody overnight at 4 ◦C. After incubation, the membranes were washed with 1× tris-buffered saline plus 0.05% Tween for 5 minutes three times and then incubated with the second antibody for 1 hour. Images were obtained using the Image Quant LAS-4000 system (GE Healthcare, Chicago, Illinois, USA). The Image J software was used to quantify the protein level. The primary antibodies are as follows: anti-Rc3h1 (#70195, Abcam), anti-CTSK (#sc-48353, Santa Cruz, USA), anti-NFATc1 (#sc-7294, Santa Cruz, USA), anti-Tfr1 (#sc-65882, Santa Cruz, USA), anti-ERK1/2 (#4695, cell signaling, CHINA), anti-p-ERK1/2 (#4370, cell signaling, CHINA), anti-p38 (#8690, cell signaling, CHINA), anti-p-p38 (#4511, cell signaling, CHINA), anti-JNK (#9252, cell signaling, CHINA), anti-p-JNK (#4668, cell signaling, CHINA), anti-CI-NDUFB8 (#ab110242, Abcam, UK), anti-CII-SDHB (#ab14714, Abcam, UK), anti-CIII-UQCRC2 (#ab14745, Abcam, UK), and anti-V-ATP5A (#ab14748, Abcam, UK).
RNA-seq
BMMs were seeded into 6-well plates and stimulated with RANKL for 3 days. Total RNA was harvested using the TRIzol reagent. The RNA sequencing was completed using the Illumina HiseqTM platform by Sangon Biotech Co., Ltd. (Shanghai, China). The raw reads in FSTAQ format were passed through FastQC and subsequently mapped to the Mus musculus genome using HISAT2 software [50]. The gene expression level was estimated based on TPM (transcript per million) with StringTie software, and differential genes were defined as Q values (Benjamini-Hochberg-adjusted P values) ≤ 0.05.
RNA-immunoprecipitation (RIP)
Approximate 1×107 RAW264.7 cells transfected with an Rc3h1-Flag-puromycin-expressing plasmid were stimulated with RANKL for 3 days. The RIP was performed according to the EZ-Magna RIP kit protocol (17-701, Merck, Sigma-Aldrich). Briefly, the cells were lysed by RIP lysis buffer and stored at -80 °C for less than 3 months for further use. Firstly, the magnetic beads were incubated with anti-Flag (390002, Zen-Bioscience, China) or IgG (17-701, Merck, Sigma-Aldrich) antibodies at room temperature for 30 minutes. Then, the lysates were incubated with magnetic beads at 4 °C overnight, and the magnetic beads were washed six times. Rc3h1-bound RNA purification was accomplished by the proteinase K buffer, followed by RNA extraction. Finally, the fold enrichment of RNA was analyzed by qPCR using the manufacturer's reference instructions.
Cellular iron detection
The Fe2+ detection was performed in flow cytometry as described below. To measure the total iron content, BMMs were stimulated with RANKL for 3 days. The cells were lysed, and total iron was calculated using the Cell Total Iron Colorimetric Assay Kit (E-BC-K880-M, Elabscience, China) according to the manufacturer's instructions.
Flow cytometry
For mitochondrial content, membrane potential, mitochondrial ROS detection, and Fe2+ detection, BMMs were seeded into 6-well plates and stimulated with RANKL for 3 days, then stained with the MitoTraker probes (M7514, ThermoFisher), TMRM (I34361, ThermoFisher), MitoSOX (M36008, ThermoFisher), or FerroOrange (F374, Dojindo, Japan) for 30 minutes at 37 °C according to the manufacturer's instructions. The cells were rinsed with PBS and lifted to be analyzed using Flow 6 (USA).
Extracellular Oxygen Consumption Assay
Dynamic analysis of mitochondrial function was based on measurements of extracellular oxygen consumption rate in osteoclasts. BMMs were seeded into XFe24 plates and stimulated with RANKL for 3 days. 1.5 μM oligomycin, 2 μM FCCP, 0.5 μM rotenone/ antimycin A were sequentially injected into wells followed by detection of OCR using the Seahorse XFe24 Extracellular Flux Analyzer.
ATP measurement
1×106 BMMs were seeded in 6-well plates and cultured with RANKL and M-CSF for 3 days. The relative ATP content was measured with an ATP detection kit (G7571, Promega) according to the manufacturer's instructions.
Inhibitor and siRNA of Tfr1
The inhibitor of Tfr1, Ferristatin II, was purchased from Sigma (C1144-5G). The siRNA for Tfr1 was designed and synthesized by Sangon Biotech Co., Ltd. (Shanghai, China). The siRNA, negative control, was transfected into BMMs with LipofectamineTM 3000 (L3000015, thermofisher). The sequence of siRNA is as follows: sense (5'-3'): CGUAUUAUGAAAGUGGAGUAUTT, antisense (5'-3'): AUACUCCACUUUCAUAAUACGTT.
Statistical analysis
The statistical analysis was conducted using GraphPad Prism software (version 8.0.2, San Diego, CA). The figures and legends provided information about the sample size for each genotype in each in vivo experiment. All in vitro experiments were performed with at least three independent replicates. The unpaired, two-tailed Student's t-test was used for statistical comparisons between the two groups. Multiple comparisons were assessed using an ordinary one-way ANOVA with Dunnett correction. All experimental data are expressed as the mean ± SD. A p-value < 0.05 is regarded as statistically significant.
Abbreviations
OCs: Osteoclasts
BMMs: Bone marrow-derived macrophages
M-CSF: Macrophage colony-stimulating factor
RANKL: Receptor activators of nuclear factor kappa-Β ligand
TRAP: Tartrate-resistant acid phosphatase
OXPHOS: Oxidative phosphorylation
OCR: Oxygen consumption rate
Tfr1: Transferrin receptor 1
RIP: RNA-immunoprecipitation
ATP: Adenosine triphosphate
ROS: Reactive oxygen species
PMOP: postmenopausal osteoporosis patients
Supplementary Material
Supplementary figures and table.
Acknowledgements
We thank all participants for their financial and technical support for this study, and we are particularly grateful for the technical support and instructions given by Kai Chen from the School of Molecular Sciences, The University of Western Australia. The graphical abstract was created in BioRender.com.
Funding
This research was supported by the Guangxi Science and Technology Base and Talent Special Project (Grant No. GuikeAD19254003), the National Natural Science Foundation of China (Grant No. 81960405, 82350710800, and 82374470), the Innovation Project of Guangxi Graduate Education (Grant No. YCSW2023225).
Author contributions
Liuyuan Chen: Writing original draft, Visualization, Investigation, Software. Yuangang Su: Methodology, Investigation, Software. Chaofeng Wang, Qian Huang: Methodology, Investigation. Weiwei Chen, Na Hai, Jikang Wang, and Haoyu Lian: Investigation. Jinmin Zhao: Project administration, Funding acquisition. Jiake Xu: Conceptualization, Writing - review & editing. Qian Liu: Supervision, Conceptualization, Writing - review, Funding acquisition.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wang L, You X, Zhang L, Zhang C, Zou W. Mechanical regulation of bone remodeling. Bone Res. 2022;10:16
2. Jacome-Galarza CE, Percin GI, Muller JT, Mass E, Lazarov T, Eitler J. et al. Developmental origin, functional maintenance and genetic rescue of osteoclasts. Nature. 2019;568:541-5
3. Lemma S, Sboarina M, Porporato PE, Zini N, Sonveaux P, Di Pompo G. et al. Energy metabolism in osteoclast formation and activity. Int J Biochem Cell Biol. 2016;79:168-80
4. Park-Min KH. Metabolic reprogramming in osteoclasts. Semin Immunopathol. 2019;41:565-72
5. Bae S, Lee MJ, Mun SH, Giannopoulou EG, Yong-Gonzalez V, Cross JR. et al. MYC-dependent oxidative metabolism regulates osteoclastogenesis via nuclear receptor ERRα. J Clin Invest. 2017;127:2555-68
6. Zeng R, Faccio R, Novack DV. Alternative NF-κB regulates RANKL-induced osteoclast differentiation and mitochondrial biogenesis via independent mechanisms. J Bone Miner Res. 2015;30:2287-99
7. Liu H, Zhai L, Liu Y, Lu D, Vander Ark A, Yang T. et al. The histone demethylase KDM5C controls female bone mass by promoting energy metabolism in osteoclasts. Sci Adv. 2023;9:eadg0731
8. Ling W, Krager K, Richardson KK, Warren AD, Ponte F, Aykin-Burns N. et al. Mitochondrial Sirt3 contributes to the bone loss caused by aging or estrogen deficiency. JCI Insight. 2021;6:e146728
9. Athanasopoulos V, Ramiscal RR, Vinuesa CG. ROQUIN signalling pathways in innate and adaptive immunity. Eur J Immunol. 2016;46:1082-90
10. Leppek K, Schott J, Reitter S, Poetz F, Hammond MC, Stoecklin G. Roquin promotes constitutive mRNA decay via a conserved class of stem-loop recognition motifs. Cell. 2013;153:869-81
11. Yu D, Tan AH, Hu X, Athanasopoulos V, Simpson N, Silva DG. et al. Roquin represses autoimmunity by limiting inducible T-cell co-stimulator messenger RNA. Nature. 2007;450:299-303
12. Essig K, Hu D, Guimaraes JC, Alterauge D, Edelmann S, Raj T. et al. Roquin suppresses the PI3K-mTOR signaling pathway to inhibit T helper cell differentiation and conversion of Treg to Tfr cells. Immunity. 2017;47:1067-82 e12
13. Raj T, Negraschus A, Heissmeyer V. Roquin-dependent gene regulation in immune-mediated diseases and future therapies. Int Immunol. 2023;35:159-70
14. Srivastava M, Duan G, Kershaw NJ, Athanasopoulos V, Yeo JH, Ose T. et al. Roquin binds microRNA-146a and Argonaute2 to regulate microRNA homeostasis. Nat Commun. 2015;6:6253
15. Ramiscal RR, Parish IA, Lee-Young RS, Babon JJ, Blagih J, Pratama A. et al. Attenuation of AMPK signaling by ROQUIN promotes T follicular helper cell formation. Elife. 2015;4:e08698
16. Corral VM, Schultz ER, Eisenstein RS, Connell GJ. Roquin is a major mediator of iron-regulated changes to transferrin receptor-1 mRNA stability. iScience. 2021;24:102360
17. Das BK, Wang L, Fujiwara T, Zhou J, Aykin-Burns N, Krager KJ. et al. Transferrin receptor 1-mediated iron uptake regulates bone mass in mice via osteoclast mitochondria and cytoskeleton. Elife. 2022;11:e73539
18. Ishii KA, Fumoto T, Iwai K, Takeshita S, Ito M, Shimohata N. et al. Coordination of PGC-1beta and iron uptake in mitochondrial biogenesis and osteoclast activation. Nat Med. 2009;15:259-66
19. Senyilmaz D, Virtue S, Xu X, Tan CY, Griffin JL, Miller AK. et al. Regulation of mitochondrial morphology and function by stearoylation of TFR1. Nature. 2015;525:124-8
20. Li J, Pan X, Pan G, Song Z, He Y, Zhang S. et al. Transferrin receptor 1 regulates thermogenic capacity and cell fate in brown/beige adipocytes. Adv Sci (Weinh). 2020;7:1903366
21. Xie L, Feng E, Li S, Chai H, Chen J, Li L. et al. Comparisons of gene expression between peripheral blood mononuclear cells and bone tissue in osteoporosis. Medicine (Baltimore). 2023;102:e33829
22. Lim BS, Landao E, Chim SM, Tickner J, Pavlos N, Xu JK. Roquin is a novel regulator of bone homeostasis, in: Annu. Meet. Am. Soc. Bone Miner. Res. Houston, TX, 2014. J Bone Miner Res. 2014;29:S1
23. Weivoda MM, Ruan M, Pederson L, Hachfeld C, Davey RA, Zajac JD. et al. Osteoclast TGF-β receptor signaling induces Wnt1 secretion and couples bone resorption to bone formation. J Bone Miner Res. 2016;31:76-85
24. Li X, Wang L, Huang B, Gu Y, Luo Y, Zhi X. et al. Targeting actin-bundling protein L-plastin as an anabolic therapy for bone loss. Sci Adv. 2020;6:eabb7135
25. Essig K, Kronbeck N, Guimaraes JC, Lohs C, Schlundt A, Hoffmann A. et al. Roquin targets mRNAs in a 3'-UTR-specific manner by different modes of regulation. Nat Commun. 2018;9:3810
26. Vogel KU, Edelmann SL, Jeltsch KM, Bertossi A, Heger K, Heinz GA. et al. Roquin paralogs 1 and 2 redundantly repress the Icos and Ox40 costimulator mRNAs and control follicular helper T cell differentiation. Immunity. 2013;38:655-68
27. Lu W, Zhou M, Wang B, Liu X, Li B. Roquin1 inhibits the proliferation of breast cancer cells by inducing G1/S cell cycle arrest via selectively destabilizing the mRNAs of cell cycle-promoting genes. J Exp Clin Cancer Res. 2020;39:255
28. Vogt AS, Arsiwala T, Mohsen M, Vogel M, Manolova V, Bachmann MF. On Iron Metabolism and Its Regulation. Int J Mol Sci. 2021;22:4591
29. Kawabata H. Transferrin and transferrin receptors update. Free Radic Biol Med. 2019;133:46-54
30. Li B, Lee WC, Song C, Ye L, Abel ED, Long F. Both aerobic glycolysis and mitochondrial respiration are required for osteoclast differentiation. FASEB J. 2020;34:11058-67
31. Byrne SL, Buckett PD, Kim J, Luo F, Sanford J, Chen J. et al. Ferristatin II promotes degradation of transferrin receptor-1 in vitro and in vivo. PLoS One. 2013;8:e70199
32. Li H, Xiao Z, Quarles LD, Li W. Osteoporosis: mechanism, molecular target and current status on drug development. Curr Med Chem. 2021;28:1489-507
33. Bone HG, Wagman RB, Brandi ML, Brown JP, Chapurlat R, Cummings SR. et al. 10 years of denosumab treatment in postmenopausal women with osteoporosis: results from the phase 3 randomised FREEDOM trial and open-label extension. Lancet Diabetes Endocrinol. 2017;5:513-23
34. Akef A, Muljo SA. How T cells go rogue in the absence of Roquins. Noncoding RNA Investig. 2018;2:20
35. Sims NA, Martin TJ. Osteoclasts provide coupling signals to osteoblast lineage cells through multiple mechanisms. Annu Rev Physiol. 2020;82:507-29
36. Szabó ME, Zekonyte J, Katsamenis OL, Taylor M, Thurner PJ. Similar damage initiation but different failure behavior in trabecular and cortical bone tissue. J Mech Behav Biomed Mater. 2011;4:1787-96
37. Li J, Bao Q, Chen S, Liu H, Feng J, Qin H. et al. Different bone remodeling levels of trabecular and cortical bone in response to changes in Wnt/β-catenin signaling in mice. J Orthop Res. 2017;35:812-9
38. Osterhoff G, Morgan EF, Shefelbine SJ, Karim L, McNamara LM, Augat P. Bone mechanical properties and changes with osteoporosis. Injury. 2016;47(Suppl 2):S11-20
39. Kim HN, Ponte F, Nookaew I, Ucer Ozgurel S, Marques-Carvalho A, Iyer S. et al. Estrogens decrease osteoclast number by attenuating mitochondria oxidative phosphorylation and ATP production in early osteoclast precursors. Sci Rep. 2020;10:11933
40. Glasmacher E, Hoefig KP, Vogel KU, Rath N, Du L, Wolf C. et al. Roquin binds inducible costimulator mRNA and effectors of mRNA decay to induce microRNA-independent post-transcriptional repression. Nat Immunol. 2010;11:725-33
41. Cheng Y, Zak O, Alsen P, Harrison SC, Walz T. Structure of the human transferrin receptor-transferrin complex. Cell. 2004;116:565-76
42. Li H, Liu Y, Shang L, Cai J, Wu J, Zhang W. et al. Iron regulatory protein 2 modulates the switch from aerobic glycolysis to oxidative phosphorylation in mouse embryonic fibroblasts. Proc Natl Acad Sci U S A. 2019;116:9871-6
43. Atamna H, Walter PB, Ames BN. The role of heme and iron-sulfur clusters in mitochondrial biogenesis, maintenance, and decay with age. Arch Biochem Biophys. 2002;397:345-53
44. Kim JH, Kim N. Regulation of NFATc1 in osteoclast differentiation. J Bone Metab. 2014;21:233-41
45. Zhang Y, Rohatgi N, Veis DJ, Schilling J, Teitelbaum SL, Zou W. PGC1β organizes the osteoclast cytoskeleton by mitochondrial biogenesis and activation. J Bone Miner Res. 2018;33:1114-25
46. Shadel GS, Horvath TL. Mitochondrial ROS signaling in organismal homeostasis. Cell. 2015;163:560-9
47. Chen K, Qiu P, Yuan Y, Zheng L, He J, Wang C. et al. Pseurotin A inhibits osteoclastogenesis and prevents ovariectomized-induced bone Loss by suppressing reactive oxygen species. Theranostics. 2019;9:1634-50
48. Kusumbe AP, Ramasamy SK, Starsichova A, Adams RH. Sample preparation for high-resolution 3D confocal imaging of mouse skeletal tissue. Nat Protoc. 2015;10:1904-14
49. Wang Q, Chen D, Jin H, Ye Z, Wang C, Chen K. et al. Hymenialdisine: a marine natural product that acts on both osteoblasts and osteoclasts and prevents estrogen-dependent bone loss in mice. J Bone Miner Res. 2020;35:1582-96
50. Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357-60
Author contact
![]() Corresponding authors: Qian Liu, Email: liuqianedu.cn; Jiake Xu, Email: jiake.xuac.cn.
Corresponding authors: Qian Liu, Email: liuqianedu.cn; Jiake Xu, Email: jiake.xuac.cn.