Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Generation and development of...
3. Different transfection vectors
4. Key considerations for...
5. Other factors of construction...
6. Concluding remarks and future...
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2024; 14(19):7424-7447. doi:10.7150/thno.101941 This issue Cite
Review
Revolutionizing cancer treatment: the emerging potential and potential challenges of in vivo self-processed CAR cell therapy
Ruijie Lv1#, Yanting Guo1#, Weici Liu2#, Guangjian Dong1, Xiangyin Liu1, Caihui Li1, Yi Ren3, Zipeng Zhang4, Shi-Yong Neo5, Wenjun Mao2 ![]() , Jing Wu1
, Jing Wu1 ![]()
1. Department of Pharmacy, The First Affiliated Hospital of Shandong First Medical University & Shandong Provincial Qianfoshan Hospital; Jinan 250014, China; School of Pharmacy, Shandong First Medical University & Shandong Academy of Medical Sciences, Jinan, Shandong 250117, China.
2. Department of Thoracic Surgery, The Affiliated Wuxi People's Hospital of Nanjing Medical University, Wuxi People's Hospital, Wuxi Medical Center, Nanjing Medical University, Wuxi, Jiangsu 214023, China.
3. Department of Clinical Pharmacy, School of Pharmacy, Shandong Second Medical University, Weifang, Shandong 261042, China.
4. Medical Science and Technology Innovation Center Shandong First Medical University & Shandong Academy of Medical Sciences Jinan 250117, China.
5. Singapore Immunology Network, Agency for Science, Technology and Research, Singapore 138648, Republic of Singapore.
#These authors contributed equally to this work.
Received 2024-8-5; Accepted 2024-10-23; Published 2024-10-28
Abstract

Chimeric antigen receptor (CAR) cell immunotherapies, including CAR-T, CAR-Macrophages, CAR-Natural Killer, CAR-γδ T, etc., have demonstrated significant advancements in the treatment of both hematologic malignancies and solid tumors. Despite the notable successes of traditional CAR cell manufacturing, its application remains constrained by the complicated production process and expensive costs. Consequently, efforts are focused on streamlining CAR cell production to enhance efficacy and accessibility. Among numerous proposed strategies, direct in vivo generation of CAR cells represents the most substantial technical challenge, yet holding great promise for achieving clinical efficacy. Herein, we outlined the current state-of-the-art in vivo CAR therapy, including CAR technology development, transfection vectors, and influence factors of construction of CAR in vivo. We also reviewed the types and characteristics of different delivery systems and summarized the advantages of in vivo CAR cell therapy, such as rapid preparation and cost-effectiveness. Finally, we discussed the limitations, including technical issues, challenges in target and signal design, and cell-related constraints. Meanwhile, strategies have correspondingly been proposed to advance the development of CAR cell therapy, in order to open the new horizons on cancer treatment.
Keywords: CAR, Immunotherapy, Construction, Delivery technology, Vector particles
1. Introduction
Immunotherapy has transformed therapeutic approaches, establishing itself as the fourth pillar of cancer treatment, alongside surgery, radiotherapy, and chemotherapy [1]. It has significantly improved the prognosis for metastatic cancer, offering long-term remissions and even potential cures for patients. In 2023, immunotherapy, including ICIs and CAR-T cell therapies, continued to improve cancer treatment outcomes. ICIs raised the five-year survival rate for advanced lung cancer to 23%, up from 5% [2]. CAR-T cell therapy showed a 58% survival rate in patients with large B-cell lymphoma, with 41% achieving long-term remission [3]. Immunotherapy focuses on various components of the immune system, employing strategies such as tumor-infiltrating lymphocytes (TIL), CAR T cells, CAR natural killer (NK) cells, and T cell receptors [4,5].
CAR cell therapy is an immunotherapy that utilizes genetically engineered immune cells to target and destroy both cancerous and certain non-cancerous cells. Over 700 clinical trials are currently evaluating the efficacy of CAR-T cell therapy in solid tumors, as listed on clinicaltrials.gov [6]. CAR-Macrophages (CAR-M) cell therapy leverages the innate properties of macrophages, equipping them with specific anti-tumor functions. Similarly, NK cells can recognize and eliminate cancer cells without prior activation [7,8], and this capability is further enhanced in CAR-NK cell therapy, which aims to improve therapeutic outcomes by more precisely targeting cancer cells. Notably, NK cells secrete a diverse array of cytokines, contributing to robust anti-tumor immunity [9]. While different CAR cell therapies share key features, such as the use of genetic engineering to target specific cancer antigens and initiate immune activation and cancer cell elimination, they vary in the type of immune cells used, the cancers they target, their mechanisms of action, persistence, and risk of graft-versus-host disease (GvHD). For instance, CAR-T cell therapy is primarily used for hematologic malignancies, providing long-lasting effects but posing a higher risk of GvHD. In contrast, CAR-NK cell therapy is effective against both hematologic and solid tumors, with a lower risk of adverse toxicity effects but still limited by poor persistency in vivo. CAR-M cell therapy excels homing into solid tumors and reshaping the immune microenvironment [10].
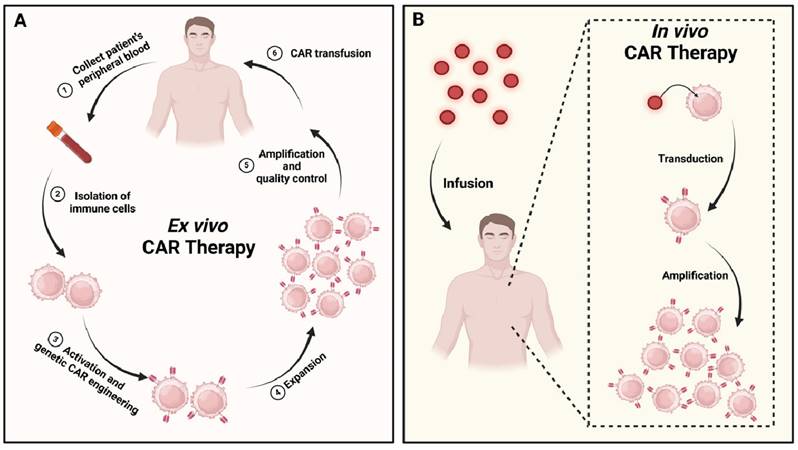
Despite the promising advances, CAR cell therapies face significant challenges due to the personalized cell engineering and manufacturing processes required. These processes are essential but extend the treatment timeline to several weeks or even months. The delays, combined with the high costs of cell engineering, production, and treatment monitoring, make CAR cell therapies prohibitively expensive for widespread use. An emerging solution to these challenges is the in vivo construction of CAR cells. This approach circumvents the time-consuming ex vivo engineering process [11], simplifies treatment by reducing the number of complex steps involved, and lowers overall costs, making it more economically feasible compared to ex vivo construction (Figure 1) [12]. While the development of CAR cell therapies presents a promising avenue for cancer treatment, innovative strategies are needed to overcome the existing technical and economic barriers. This review article provides an overview of the structure and evolution of CARs, various types of CAR cell therapies, key vector technologies used in CAR cell therapy delivery and factors influencing CAR construction.
Ex vivo vs. in vivo CAR cell therapies: a comparative. (A) The ex vivo approach begins with the isolation of immune cells from the patient's blood. These cells are then activated, expanded, and genetically modified in a controlled laboratory environment. Following stringent quality control measures, the engineered CAR cells are reinfused into the patient. (B) In contrast, the in vivo method involves directly infusing vector particles (represented as red dots) into the patient. These vectors interact with the patient's immune cells within the body, selectively transferring the genetic material necessary to encode the CAR.
2. Generation and development of CAR technology
CAR cell therapy marks a significant leap in cancer treatment, evolving from earlier TCR-based therapies, which were limited by Human Leukocyte Antigen HLA restrictions and tumor escape mechanisms [13,14]. To overcome these challenges, CARs were engineered by combining functional components, leading to advancements in CAR cell therapy [15]. Initially, CARs had a simple structure with an antigen-binding domain and a signal transduction domain. Over time, improvements such as adding costimulatory molecules and refining signal strength have greatly enhanced their efficacy [16]. Today, CAR cell therapy includes NK cells and macrophages, enabling personalized treatments for various cancers [17,18].
2.1. The structure of CAR
The modular design of CARs includes four key components: the antigen-binding domain, hinge, transmembrane domain, and intracellular signaling domain [19]. Each part plays a distinct role, allowing for optimal design flexibility. The antigen-binding domain, typically a single-chain variable fragment (scFv) made from the variable regions of monoclonal antibodies, is critical for recognizing and binding tumor antigens [20]. The (Gly4Ser)3 linker, commonly used to connect antibody fragments, ensures proper folding and antigen binding [21]. This precision enhances CARs' ability to target cancer cells, significantly improving therapeutic outcomes [22]. The hinge and transmembrane domains connect the extracellular and intracellular components of the CAR. The hinge provides flexibility to avoid steric hindrance, aiding antigen capture near the membrane [23]. Hinge domains often include sequences from CD8, CD28, IgG1, or IgG4 [24]. The transmembrane domain, commonly derived from proteins like CD3ζ, CD28, or CD8α, anchors the CAR, ensuring stability and function [25]. The intracellular signaling domain contains an activation domain, usually derived from CD3ζ, and costimulatory domains from molecules like CD28 or 4-1BB, which are critical for effective CAR activation and have received FDA approval [26,27]. The CAR structure used in CAR-T cell therapy mirrors those in CAR-NK and CAR-M cell therapies, all of which combine antigen recognition, transmembrane anchoring, and intracellular signaling to enable precise tumor targeting and elimination by genetically modified immune cells.
2.2. The development process of CAR cells
CAR cells are currently classified into five generations, each distinguished by its intracellular signaling structures (Figure 2) [28-31]. The first generation introduced a basic CAR configuration, featuring an extracellular antigen recognition domain from the variable region of a monoclonal antibody, and an intracellular signaling domain that typically utilizes the CD3ζ chain [32]. This design enables CARs to recognize tumor antigens and trigger an immune response through CD3ζ signaling [33,34], laying the foundation for genetically engineered immune cells to target and destroy tumor cells [35].
The development process of CAR cells from the first to the fifth generation: CAR cells are categorized into five generations, each defined by distinct intracellular signal transduction structures.
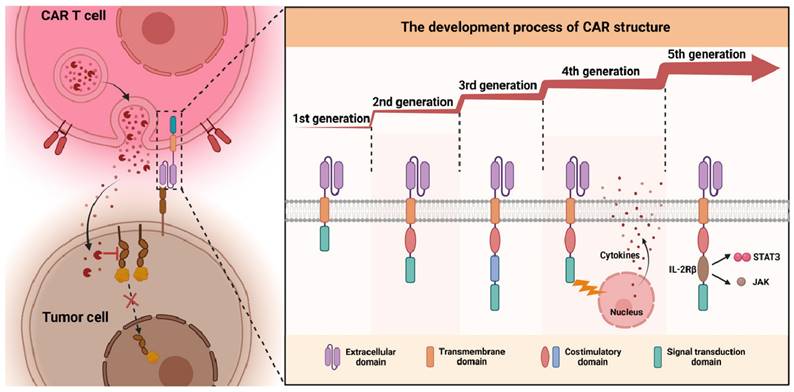
The second generation of CAR cells introduced costimulatory molecules like CD28 or 4-1BB alongside the CD3ζ chain, enhancing cell survival, proliferation, and tumor-killing ability [36]. Carl et al. developed a second-generation CAR-T targeting CD19 with a 4-1BB costimulatory domain, leading to CTL-019, the first FDA-approved CAR-T product [37]. The third generation incorporated additional costimulatory molecules, such as OX40 and ICOS, along with CD28 or 4-1BB, to boost CAR cell activation and persistence, but results have been mixed, with some studies showing no significant improvements over the second generation [38-41]. The fourth generation, called “combined antigen receptors” or TRUCKs (T cells Redirected for Universal Cytokine Killing), retains traditional CAR functions but adds the ability to secrete specific cytokines, such as IL-12. This helps modulate the tumor microenvironment, enhancing anti-tumor efficacy by recruiting and activating other immune cells like NK cells and macrophages [42]. The fifth generation, referred to as “Universal CAR,” features an optimized design with a tumor antigen recognition region, a costimulatory signaling region, and an enhanced signaling domain. This allows for better CAR cell activation and persistence, crucial for overcoming tumor microenvironment challenges and improving tumor cell eradication [43,44]. Each generation builds upon the last, optimizing CAR technology for improved efficacy and expanding treatment possibilities for cancer patients.
In summary, the progressive evolution of CAR structures has primarily focused on augmenting cell activity, persistence, and functionality while improving their performance within the challenging tumor microenvironment. Each generational innovation has aimed to resolve the limitations of its predecessor, gradually enhancing the clinical potential and applicability of CAR cell therapy.
2.3. Different CAR cell therapies
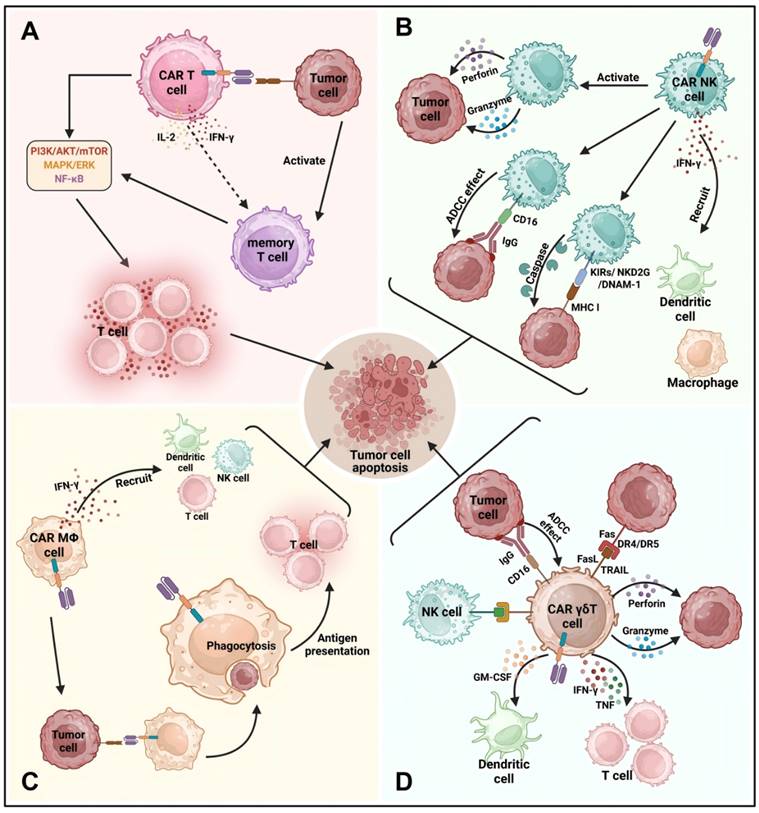
In this review, we briefly outline the mechanisms and application of CAR-T, CAR-NK, CAR-M, and CAR-γδ T cell therapies (Figure 3).
The mechanisms of CAR-T, CAR-NK, CAR-M, and CAR-γδ T cell therapies. (A) When CAR-T cells bind to target antigens, they activate intracellular signaling pathways like PI3K/AKT/mTOR, MAPK/ERK, and NF-κB, leading to T cell proliferation, cytokine production, and cytotoxic activity through perforin and granzyme release, inducing cancer cell apoptosis. CAR-T cells also secrete cytokines such as IFN-γ and IL-2, boosting the antitumor response, with some differentiating into memory T cells for long-term immune surveillance and reduced cancer recurrence. (B) Similarly, activated CAR-NK cells produce perforin and granzyme, promoting cancer cell death through caspase-mediated apoptosis. CAR-NK cells express receptors such as KIRs, NKG2D, and DNAM-1, and mediate antibody-dependent cellular cytotoxicity (ADCC) via CD16, playing a crucial role in targeting HER2 and EGFR in solid tumors. Their efficient ADCC is linked to better outcomes in various cancers. (C) CAR-M cell therapy uses engineered macrophages to enhance phagocytosis and antigen presentation, adapting to tumor environments while promoting pro-inflammatory signaling and suppressing tumor-promoting polarization. CAR-M cells secrete IFN-γ, recruiting and activating immune cells, and upregulate MHC-I and MHC-II, improving T cell activation and immune infiltration into tumors. (D) CAR-γδ T cells exhibit antitumor effects via TCR, NKRs, and CD16, triggering ADCC and directly killing tumor cells by releasing TRAIL, FasL, perforin, and granzyme. They enhance cytotoxic T cell and NK cell functions through IFN-γ, TNF, and CD137 signaling, and produce GM-CSF to regulate dendritic cell infiltration, further augmenting the antitumor immune response.
2.3.1 CAR-T cell therapy
CAR technology, initially developed for T cells, has revolutionized immunotherapy by enhancing its synergy with other immune cells [45]. Over time, CAR cell therapy expanded to various immune cells, offering new medical possibilities. The process involves creating a recombinant plasmid that merges an antibody fragment targeting tumor antigens with an immune receptor tyrosine activation motif (ITAM). This plasmid is transfected into patient T cells, enabling them to target tumor cells [46]. CAR-T cell therapy has been highly effective in treating cancers like leukemia, lymphoma, and glioma [47,48]. The treatment involves extracting, modifying, and reinfusing T cells, which then target cancer cells. Once the CAR-T cells bind to the tumor antigen, they activate pathways such as PI3K/AKT/mTOR, leading to T cell proliferation, cytokine release, and cytotoxic activity. Cytokines like IFN-γ and IL-15 enhance the immune response, and memory T cells provide long-term protection, reducing cancer recurrence risks [49-51]. Although no CAR-T cell products have been approved for the treatment of solid tumors, numerous clinical trials are underway exploring their application in this area. The following targets are currently being investigated (Table 1).
Solid tumor targets of CAR-T
| Target | Cancer type | Clinical trials NCT number |
|---|---|---|
| EpCAM | Malignant neoplasm of nasopharynx, colon, esophageal, pancreatic, prostate, gastric, liver, and breast cancer | NCT03013712, NCT02915445, NCT02729493, NCT02725125, NCT05028933, NCT04151186 |
| PD-L1 | Mesothelioma, colorectal, lung, and liver cancer | NCT03330834, NCT05089266, NCT04489862, NCT03672305, NCT03060343 |
| HER2 | Metastatic malignant neoplasm in the brain, GBM, ependymoma, glioma, sarcoma, and CNS tumor Gastric, breast, ovarian, bladder, head and neck, lung, esophageal, colorectal, pancreatic, and salivary gland cancer | NCT04903080, NCT04650451, NCT03696030, NCT03500991, NCT04995003, NCT04903080, NCT04511871, NCT03740256, NCT02442297, NCT03198052, NCT01109095, NCT00902044, NCT03423992, NCT03267173, NCT05681650 |
| EGFR | Glioma, lung, liver, GC, and other EGFR positive advanced solid tumor | NCT05060796, NCT04153799, NCT03618381, NCT03182816, NCT04976218, NCT02331693, NCT03198052, NCT02873390, NCT02862028 |
| B7-H3 | GBM, sarcoma, melanoma, liver, lung, breast, pancreatic, lung, ovarian, and adrenocortical cancer | NCT05366179, NCT05241392, NCT04483778, NCT04385173, NCT04077866, NCT04897321, NCT04670068, NCT03198052, NCT05341492, NCT05143151, NCT04691713, NCT04864821, NCT05515185, NCT05323201, NCT05366179, NCT05241392, NCT04483778, NCT04385173, NCT04077866, NCT04897321, NCT04670068, NCT03198052, NCT05341492, NCT05143151, NCT04691713, NCT04864821, NCT05515185, NCT05323201 |
| MSLN | Malignant pleural mesothelioma, pleural mesothelioma, glioma, lung, liver, ovarian, pancreatic, breast, cervical, colorectal, GC, and other MSLN-positive advanced solid tumors | NCT03356808, NCT03916679, NCT03799913, NCT03545815, NCT03323944, NCT03182803, NCT03054298, NCT03030001, NCT02930993, NCT05057715, NCT04577326, NCT03198052, NCT03814447, NCT03638193, NCT03615313, NCT02792114, NCT02706782, NCT02465983, NCT02159716, NCT01897415, NCT01583686, NCT03497819, NCT05373147, NCT04562298, NCT04503980, NCT04203459, NCT05166070, NCT05089266, NCT03267173, NCT04489862, NCT02959151, NCT03356795, NCT05623488, NCT05693844, NCT03941626 |
| MUC1 | Sarcoma, glioma, cervical, lung, esophageal, colorectal, gastric, liver, pancreatic, breast, and OC | NCT03706326, NCT03525782, NCT03179007, NCT02617134, NCT02587689, NCT04025216, NCT04020575, NCT03356808, NCT03356795, NCT03356782, NCT03267173, NCT03198052 |
| ROR1 | Breast and lung cancer | NCT05274451, NCT05748938, NCT05638828 |
| GD2 | Glioma, NB, sarcoma, embryonal tumor, melanoma, and cervical cancer | NCT05298995, NCT03721068, NCT03373097, NCT04099797, NCT02992210, NCT02107963, NCT01953900, NCT01822652, NCT00085930, NCT03356795, NCT03423992, NCT03356782, NCT03356808, NCT05437315, NCT05620342 |
CNS: Central nervous system; GBM: Glioblastoma; GC: Gastric cancer; NB: Neuroblastoma; OC: Ovarian cancer;
As of now, six CAR-T cell therapies have received FDA approval, three of which target B-cell maturation antigen (BCMA), while the others target CD19 (Table 2). Despite significant breakthroughs in tumor treatment, CAR-T cell therapy faces several challenges, such as the high cost and complex manufacturing process ahead of widespread clinical application, severe adverse reactions in company with CAR-T cell therapy, and insufficient response and even resistance in certain applications.
Current FDA approvals of CAR-T cell therapies
| Number | Name | Trade Name | Target | Approval date | Indication | Approved Countries | Clinical benefit | Generation |
|---|---|---|---|---|---|---|---|---|
| 1 | Axicabtagene ciloleucel | Yescarta | CD19 | 2017.10 | Large B-cell lymphoma [88,91,92] | USA | CR:54% | Second |
| 2021.6 | China | / | ||||||
| 2 | Brexucabtagene autoleucel | Tecartus | CD19 | 2020.7 | Mantle cell lymphoma [93] | USA | CR:67% | Second |
| 3 | Idecabtagene vicleucel | Abecma | BCMA | 2021.3 | Multiple myeloma [94] | USA | CR:25% | Second |
| 4 | Tisagenlecleucel | Kymriah | CD19 | 2017.8 | Acute Lymphoblastic Leukemia [95] | USA | CR:>90% | Second |
| 5 | Lisocabtagene maraleucel | Breyanzi | CD19 | 2021.2 | Large B-cell Lymphoma [96] | USA | CR:54% | Second |
| 6 | Ciltacabtagene autoleucel | Carvykti | BCMA | 2022.2 | Multiple myeloma [97] | USA | CR:78% | Third |
2.3.2 CAR-NK cell therapy
NK cells are crucial to the innate immune system, able to eliminate tumor cells without prior sensitization [52]. Both autologous and allogeneic NK cell infusions show promise in treating relapsed/refractory acute myeloid leukemia (AML) [53]. CAR-NK cells combine CAR's precision with NK cells' innate tumor-killing ability, improving targeting of AML [54]. They offer a favorable safety profile with lower risks of cytokine release syndrome (CRS) and neurotoxicity. NK cells also express less PD-1, reducing immunosuppression in the tumor environment [55,56]. CAR-NK cells have lower immunogenicity and reduce the risk of graft-versus-host disease (GVHD), while maintaining durable antitumor effects.
NK cells naturally kill tumor cells using cytotoxic molecules like perforin and granzyme, without needing antigen recognition like T cells [57,58]. They use receptors like NKG2D and DNAM-1 to trigger cancer cell death, and higher expression of these receptors improves cancer outcomes [59,60]. NK cells also help immune responses by releasing IFN-γ and mediate antibody-dependent cellular cytotoxicity (ADCC) through CD16 receptors, aiding in therapies against HER2 and EGFR in solid tumors [61,62].
CAR-NK cells enhance NK cell activity by targeting specific cancer antigens, using CAR constructs with NK-specific domains like NKG2D and DAP-10 to increase cytotoxicity and cytokine production [63]. Despite the challenges posed by the tumor microenvironment, CAR-NK therapy shows promise in clinical trials [64]. There have been over 20 reported clinical trials involving CAR-NK cells, three of which have been fully completed (Table 3). Combining CAR-NK cell therapy with other treatments could improve outcomes by overcoming immune suppression in tumors [65]. In tumor immunotherapy, particularly for solid tumors, both preclinical and clinical research on CAR-NK cells urgently require the development of more efficient infection methods and safer non-viral transfection technologies to overcome the inhibitory tumor microenvironment and continuously accumulate clinical experience. By improving infection techniques, CAR-NK cells can be more efficiently engineered to express CARs, enhancing their ability to target and destroy tumor cells. Efficient infection methods ensure a higher transduction rate, equipping more NK cells with the CARs needed to recognize and kill cancer cells, even within the suppressive tumor environment. Furthermore, non-viral transfection methods, such as electroporation or nanoparticle delivery systems, offer a safer alternative by reducing risks associated with viral vectors, such as insertional mutagenesis and immune responses. This approach enables repeated dosing or modification of NK cells without the drawbacks of viral methods, promoting their persistence and functionality in the tumor microenvironment [66].
Current completed clinical trials for CAR-NK cell therapies
| NCT Number | Study Title | Conditions | Interventions | Phases | Study Start Date | Completion Date |
|---|---|---|---|---|---|---|
| NCT03056339 | Umbilical & cord blood (CB) derived CAR-engineered NK cells for B lymphoid malignancies | B-lymphoid malignancies| acute lymphocytic leukemia| chronic lymphocytic leukemia| non-hodgkin lymphoma | Drug: fludarabine| drug: cyclophosphamide| drug: mesna | biological: IC9/CAR.19/IL15-transduced CB-NK cells|drug: AP1903 | phase1|phase2 | 2017.6 | 2023.6 |
| NCT04538599 | RD13-01 for patients with r/r CD7+ T/NK cell hematologic malignancies | Hematologic malignancies | Drug: RD13-01 cell infusion | phase1 | 2020.9 | 2021.11 |
| NCT05563545 | Anti-CD19 CAR-engineered NK cells in the treatment of relapsed/refractory acute lymphoblastic leukemia | Acute lymphoblastic leukemia | Biological: CAR-NK-CD19 cells | phase1 | 2022.7 | 2022.11 |
2.3.3 CAR-M cell therapy
CAR-M cell therapy employs genetically engineered macrophages to enhance their phagocytic capacity and improve antigen presentation to tumor cells. This approach not only boosts antigen presentation, thereby increasing T cell cytotoxicity and facilitating tumor cell engulfment, but it also adapts to environmental signals, potentially modifying its phenotype. Compared to CAR-T cell therapy, CAR-M therapy presents advantages such as reduced off-target toxicity and shorter treatment durations. Several CAR-M cell therapy candidates are currently in different phases of preclinical and clinical research and development (Table 4). CAR recognition of tumor antigens initiates macrophage-mediated antibody-dependent cellular cytotoxicity (ADCC) [67]. This process involves antibodies against carcinoembryonic antigen (CEA) on macrophages binding to the Fc region of CEA antibodies on cancer cells, thereby inducing antibody-dependent cellular cytotoxicity (ADCC) to eliminate CEA-expressing tumor cells. Upon binding to specific tumor antigens, CAR receptors activate intracellular signaling pathways that promote phagocytosis, directly killing cancer cells and facilitating rapid antigen presentation to activate T cell-mediated immunity [68]. CAR-mediated signaling prevents macrophage polarization towards a tumorigenic phenotype while activating pro-inflammatory pathways. CAR-M cells secrete pro-inflammatory cytokines, such as interferon-gamma (IFN-γ), which recruit and activate other immune cells to further target cancer cells. As CAR-M cells eliminate more cancer cells, they stimulate adaptive immune responses, providing effective and durable antitumor immunity. CAR-M activation typically upregulates MHC-I and MHC-II expression, enhancing the presentation of tumor-associated antigens and promoting T cell activation. Furthermore, CAR-M cells enhance the infiltration of CD4+ and CD8+ T cells, NK cells, and dendritic cells within tumors, augmenting immune-mediated cytotoxicity against solid tumors [69].
CAR-M cell therapies based clinical trials
| NCT Number | Product name | Manufacturer | Target | Indication | Phases | Study Start Date | Completion Date |
|---|---|---|---|---|---|---|---|
| NCT04660929 | CT-0508+ Pembrolizumab | Carisma Therapeutics | HER2 | HER2 overexpressing solid tumors | Phase1 | 2021/2/2 | 2024/12/31 |
| NCT03608618 | MCY-M11 | MaxCyte | Mesothelin | Relapsed/refractory ovarian cancer and peritoneal mesothelioma | Phase1 | 2018/8/27 | 2021/8/24 |
| NCT05138458 | MT-101 | Myeloid Therapeutics | CD5 | Refractory or relapsed peripheral T-cell lymphomas | Phase1 Phase2 | 2021/12/15 | 2025/10/1 |
| NCT06562647 | SY001 | Cell Origin Biotech (Hangzhou) Co., Ltd. | Mesothelin | Overexpressing solid tumors | NA | 2023/4/12 | 2025/4/1 |
| / | CT-1119 | Carisma Therapeutics | HER2 | Mesothelin-positive solid tumors | preclinical | / | / |
| / | CAR-iMAC | Cell Origin | EGFRvIII, GPC3 | Hepatocellular carcinoma | preclinical | / | / |
Compared to CAR-T and CAR-NK, CAR-M cell therapies presents unique advantages as a novel cell-based immunotherapy [10]. These advantages include the ability to establish a pro-inflammatory environment within the tumor and to reverse the immunosuppressive tumor microenvironment. In preclinical animal studies, CAR-M has demonstrated effective anti-tumor capabilities. However, the efficacy and safety of CAR-M cell therapy still require validation in clinical settings.
2.3.4 CAR-γδ T cell therapy
In cell therapy, CAR-T cell therapy primarily targets αβ T cells, making up about 95% of the T cell population [70]. In contrast, γδ T cells, which play a complex role in tumor immunology, exhibit significant antitumor activity, especially in humans [71,72]. They activate through γδ T cell receptors (TCRs), natural killer receptors (NKRs), and CD16, enabling antibody-dependent cellular cytotoxicity (ADCC) against tumor cells [73]. γδ T cells can directly eliminate tumors via TCR and NKR engagement and secrete cytotoxic granules containing perforin and granzymes.
Moreover, γδ T cells enhance antitumor responses by secreting IFN-γ and TNF, improving αβ T cell function and MHC I expression on tumor cells. They also stimulate NK cells and produce GM-CSF to regulate dendritic cell infiltration [74]. However, γδ T cells can have protumor effects, particularly through IL-17, which promotes tumor growth and angiogenesis in certain contexts [75]. IL-17+ γδ T cells have been observed in advanced cancer stages, contributing to immune evasion [76,77].
Engineered with CARs, γδ T cells can specifically target tumor antigens, kill tumor cells, and recruit other immune cells, making them promising for solid tumors due to their ability to penetrate the tumor microenvironment [78]. Unlike the more common αβ T cells, γδ T cells, which constitute about 0.5%-5% of T cells, possess both innate and adaptive immune features, allowing for rapid recognition of non-MHC-restricted antigens [79,80]. CAR-γδ T cell therapies show significant potential, although clinical trials are limited. For example, the phase I trial ADI-001 (NCT04735471) targeting CD20 for B-cell non-Hodgkin lymphoma has shown promising efficacy and safety [81]. Compared to CAR-T cell therapy, CAR-γδ T cell immunotherapy offers advantages such as MHC-independent recognition, applicability to various tumor types, and reduced risk of cytokine release syndrome, highlighting their potential as a therapeutic approach [82].
2.3.5 Other CAR cell therapies
In addition to CAR-T, CAR-NK, CAR-M, and CAR-γδ T cell therapies, several other CAR cell therapies are emerging. Chimeric Antigen Receptor Natural Killer T (CAR-NKT) cells involve genetically engineering NKT cells to express chimeric antibodies, allowing for targeted destruction of tumor cells while preserving their innate antitumor properties [7]. This dual functionality enhances overall immune responses against tumors, offering long-term protection and improved persistence through interleukin 15 (IL-15) inclusion [64,83]. Similarly, CAR-DC cell therapy utilizes the antigen-presenting capabilities of dendritic cells (DCs), modifying them with CARs to directly target cancer cells. This enhances T cell responses through effective antigen presentation, cytokine secretion, and recruitment of additional immune cells [84]. In contrast, CAR-Treg cell therapy focuses on regulatory T cells (Tregs) engineered to recognize specific antigens, suppressing immune activation through mechanisms like CTLA-4 engagement with CD80 and the release of inhibitory cytokines [85]. This approach is useful for preventing immune rejection and maintaining immune homeostasis. Lastly, Chimeric Autoantibody Receptor T (CAAR-T) cells are designed to selectively target autoreactive B cells by recognizing autoantibody proteins on their surface [86]. This method effectively eliminates memory B cells and plasma cells that produce pathogenic autoantibodies without causing widespread immune suppression [87]. CAAR-T therapy shows promise in treating autoimmune disorders like Pemphigus Vulgaris, with ongoing clinical trials assessing optimal dosing and efficacy [88].
Research on CAR-T cell therapies for solid tumors is still limited compared to their success in hematologic malignancies. Challenges include a lack of tumor-specific antigens, low CAR-T cell trafficking efficiency, and an immunosuppressive microenvironment. In contrast, CAR-NK and CAR-M cells offer advantages in treating solid tumors due to their strong antitumor properties and reduced side effects like cytokine release syndrome (CRS). However, issues such as "on-target, off-tumor" toxicity and antigen escape remain. CAR-M cell therapy combines innate and adaptive immune responses, enhancing tumor regression through interactions with T and NK cells. Combining CAR-M with CAR-NK or CAR-T cells could improve efficacy against various tumor antigens. Advanced technologies like artificial intelligence (AI) and radiomics are being utilized to optimize CAR cell therapies, with AI predicting new cancer-associated antigens and improving CAR-T cell manufacturing, while radiomics provides insights into the tumor microenvironment. Additionally, CAR cell therapies are being investigated for autoimmune diseases. For instance, CAR-T cells targeting CD19 have shown promise in lupus nephritis, and CAR-Tregs have been effective in multiple sclerosis and type 1 diabetes by suppressing immune attacks while maintaining tolerance [89,90]. These developments highlight CAR cell therapy's potential for both cancer and autoimmune diseases.
In summary, the combined application of CAR-NK cells and CAR-M cells brings new hope for the treatment of solid tumors. Future research should focus on optimizing their roles within the tumor microenvironment to overcome existing challenges and achieve more effective treatments. CAR cell therapy shows great promise, and with continuous technological advancements and deeper clinical trials, it is poised to bring new hope and potential cures to more cancer patients in the future.
3. Different transfection vectors
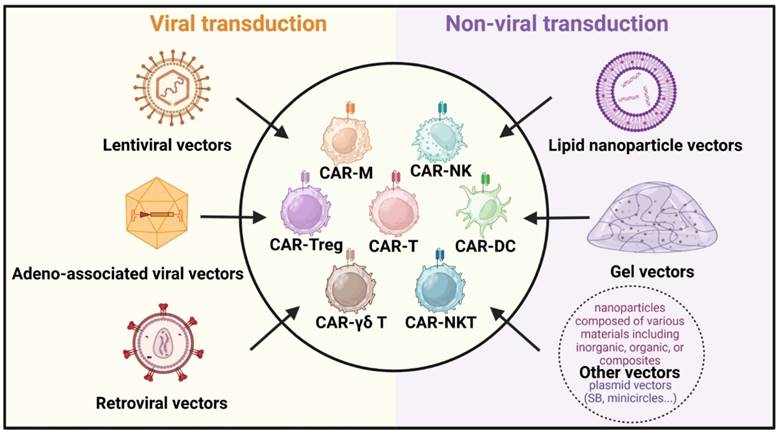
In vivo construction of CAR cells has become a pivotal area of interest in CAR cell therapy due to its rapid preparation time and cost-effectiveness. It enhances the survival rate and functional efficacy of CAR cells within the body, offering a promising future for immunotherapy. Further studies on various delivery platforms, each with its own unique set of benefits and challenges, could influence therapeutic effectiveness. Beyond traditional viral transduction methods like lentiviral, retroviral, and adeno-associated viral vectors, non-viral delivery strategies such as lipid nanoparticles and gels have emerged (Figure 4) [98]. These innovative approaches are designed to improve the safety and efficacy of CAR cell therapy, opening new avenues for treating various tumors.
Delivery strategies for in vivo CAR construction. The primary vectors for viral transduction (left) include lentiviral vectors, adeno-associated viral vectors, and retroviral vectors. Non-viral transduction (right) vectors consist of lipid nanoparticle vectors, gel vectors, and other nanoparticle vectors like plasmid vectors (Sleeping Beauty, minicircles, etc.).
3.1. Lentiviral vectors and Retroviral vectors
Lentiviral (LV) transduction is widely used in conventional CAR cell engineering for the efficient delivery of CAR transgenes [99]. Derived from the human immunodeficiency virus (HIV), these retroviruses have been engineered to remove virulence genes and incorporate exogenous target genes as classical LV vectors. LVs are typically pseudotyped with the glycoprotein from vesicular stomatitis virus (VSV-G), resulting in a diameter of 120-150 nm. VSV-G mediates cellular entry through the low-density lipoprotein receptor (LDLR) and its family members, which are expressed on many cell types [100]. This class of pseudotyped viruses can integrate foreign genes into the host genome, ensuring stable expression and the capability to infect both dividing and non-dividing cells [101]. Once inside the cell, the LV genome is reverse transcribed to DNA in the cytoplasm, forming a pre-integration complex that enters the nucleus, where the DNA integrates into the cellular genome (Figure 5A). LV vectors serve as a pivotal gene delivery method in in vivo CAR construction, introducing CAR-encoding genes into target cells to enable CAR expression. The primary advantage of LV vectors is their ability to achieve stable gene integration into the host genome, thus ensuring sustained gene expression [102]. However, challenges such as potential pathogenicity and the risk of random genome integration, which could lead to gene mutations, remain. Of particular concern is the fact that several cases of leukemia have been reported by the FDA following the use of LV in CAR-T cell therapy. Consequently, the FDA mandates that patients be informed of these associated risks. Despite these risks, LV vectors are a promising option for gene and cell therapy [103]. In 2018, 54% of CAR-T cell generation clinical studies in the US utilized LV vectors as carriers [104]. The two market-leading CAR-T cell therapies, Kymriah and Yescarta, which target CD19, also employ LV as their vector system. Additionally, over 100 ongoing clinical trials utilizing LV, predominantly focusing on immunology, hematological diseases, and cancer, underline its broad potential applications [105]. Pioneering work by Christian J. Buchholz et al. demonstrated the induction of CAR-T cells in situ in immunodeficient mice using second-generation anti-CD19-CAR gene-encapsulated LV, exhibiting anti-tumor activity [106]. Samuel K. Lai et al. developed a bispecific conjugate directing LV vectors to T cells for specific in vivo engineering of CAR-T cells, showcasing that LV-modified in vivo CAR-T cells possessed antineoplastic activity [107]. Kathryn R. Michels and her team developed an in vivo CAR T-cell engineering platform, VivoVec, based on a lentiviral vector. Their candidate drug, UB-VV100, employs surface-engineered lentiviral particles that can bind, activate, and transduce T cells, enabling these cells to express an anti-CD19 CAR and a rapamycin-activated cytokine receptor (RACR) system. Preclinical studies demonstrated that UB-VV100 successfully activated T cells, achieved CAR T-cell transduction, and effectively eliminated tumor cells. Additionally, in vivo studies on humanized mice and canine models showed that the biodistribution of UB-VV100 was largely confined to immune cells and did not induce significant tissue pathology. This research provides a novel in vivo approach to engineering CAR T cells, potentially simplifying the therapeutic process [108]. Future research will likely focus on enhancing the safety and specificity of LV vectors, minimizing potential side effects, and optimizing vector design to meet diverse treatment needs. The continued utilization of LV in genetic engineering, particularly for the in vivo construction of chimeric antigen receptors, opens new avenues for gene and cellular therapies with profound clinical implications. As technology advances and research progresses, the application of LV as an in vivo construction vector is expected to expand, offering greater potential in the field of gene therapy.
The Vector entry modes into the cell. (A) LVs contain one or more viral glycoproteins and two copies of a single-stranded RNA (ssRNA) genome encapsulated within a nucleocapsid. Once inside, the transferred gene undergoes reverse transcription, is transported into the nucleus, and integrates into the host genome. (B) After internalization, AAVs are enclosed in endocytosed vesicles. The acidification of these vesicles triggers viral escape into the cytoplasm, where the viruses utilize the cellular microtubule transport system to approach the nucleus. AAVs then interact with the nuclear pore complex to gain entry into the nucleus. Once inside, the single-stranded DNA undergoes conformational changes to form double-stranded DNA, which may subsequently integrate into the host genome. (C) Retroviruses bind to host cell surface receptors via their glycoproteins. Subsequent membrane fusion introduces the viral capsids into the cells. In the nucleus, the capsids uncoat, and the viral RNA is reverse transcribed and integrated into the host genome. (D) In synthetic vectors, CAR-encoding nucleic acids are complexed with NPs, LNPs, or gel-based carriers. After escaping the endosome, mRNA payloads are available for translation, while packaged DNA may reach the nucleus for potential integration into host chromatin.
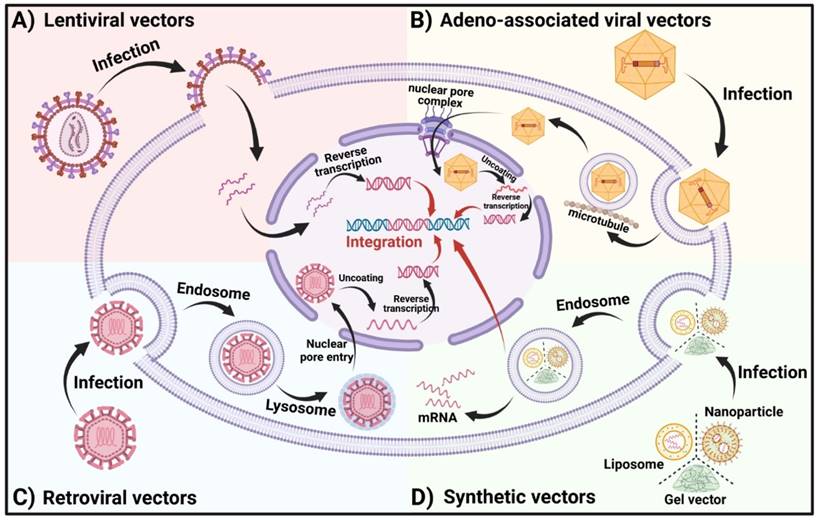
Retroviruses (RVs) are a class of viruses characterized by single-stranded RNA genomes and their ability to reverse-transcribe RNA into DNA, mediated by the enzyme reverse transcriptase [109]. Upon contact with a host cell, viral glycoproteins bind to cell surface receptors, facilitating membrane fusion and the entry of the viral capsid into the cell. Inside the host cell, reverse transcriptase synthesizes cDNA from the viral RNA, which is then transported into the nucleus and integrated into the host's chromosomal DNA with the assistance of integrase (Figure 5C). RVs are extensively utilized as gene delivery vectors in medicine due to their robust transduction capabilities across various cell types and relatively lower risk of integration compared to other vectors. A significant application of RVs includes the in vivo construction of chimeric antigen receptors (CARs). Despite their many advantages, RVs have some drawbacks, such as the potential for insertional mutagenesis, which could lead to genomic instability [110]. Nevertheless, RVs have demonstrated considerable success in clinical settings; for instance, RV-based CAR-T therapies have been notably effective in treating leukemia and lymphoma [111,112]. Furthermore, researchers from North Carolina State University and North Carolina University have developed an implant called MASTER, which employs modified RVs to transfer the CAR gene into T cells directly within the host, reprogramming them into CAR-T cells. This technique can generate and deploy CAR-T cells in vivo within just one day, significantly streamlining the production process [113].
Lentiviral vectors and retroviral vectors both belong to the retrovirus family and are capable of integrating exogenous genes into the host cell genome, enabling long-term stable expression. The primary differences between the two lie in their host range, gene integration sites, safety, and transduction efficiency. Lentiviral vectors can infect both dividing and non-dividing cells, with more random gene insertion and higher safety, making them widely applicable across various cell types. In contrast, retroviral vectors can only infect dividing cells, and their gene insertion tends to occur near promoter or enhancer regions, posing a higher risk of insertional mutagenesis, thus limiting their application.
As gene therapy technology continues to advance, RVs are expected to play an increasingly vital role as in vivo gene delivery mechanisms. They hold significant promise as key tools in biomedicine, paving new paths for treating genetic disorders, cancer, and various other diseases.
3.2. Adeno-associated viral vectors
Adeno-associated viruses (AAVs) are small, non-enveloped viruses with a single-stranded DNA genome, which have emerged as one of the preferred vectors in gene therapy due to their efficient transduction capabilities, favorable safety profile, and wide host cell range [114]. Following cellular entry through internalization, AAVs are encased within endocytosed vesicles. The acidification of these vesicles facilitates viral escape into the cytoplasm. The viruses then utilize the cellular microtubule transport system to move close to the nucleus, where they interact with the nuclear pore complex to enter the nucleus [115,116]. Once inside, the single-stranded DNA undergoes structural changes to become double-stranded DNA, which may integrate into the host genome (Figure 5B). The AAV genome comprises genes that code for specific antigen receptors, and these genes are engineered to facilitate the expression of the receptors upon the infection of host cells. This allows the modified cells to target and bind to specific antigens, offering novel strategies and approaches for cancer immunotherapy, viral therapy, and other disease treatments. A key advantage of AAV vectors is the existence of multiple serotypes, which exhibit different tissue tropisms, allowing for more precise targeting in gene therapy. For example, AAV9 has shown strong tropism for muscle and nervous tissues, making it a promising candidate for treating neuromuscular diseases, while AAV8 is often used for targeting the liver [117]. This diversity enables the development of AAV-based therapies that can be tailored to specific tissue types or disease contexts. Strecker and colleagues developed a tumor-specific delivery system using an adeno-associated virus (AAV vector, HER2-AAVaPD-1, to deliver an anti-PD-1 immunoadhesin (aPD-1) directly to tumor cells. Additionally, they engineered HER2-targeted CAR-NK cells, specifically NK-92/5.28.z cells. When used in combination, these therapies significantly extended survival and controlled tumor growth in mouse models, without causing noticeable immune-related side effects. This dual approach offers a promising new strategy for the immunotherapy of glioblastoma [118]. The utility of AAV was first demonstrated by R.J. Samulski et al., who cloned the AAV genome into expression plasmids, laying the foundation for its application in gene therapy. When these plasmids were transfected into mammalian cell lines, they generated a substantial quantity of infectious viruses [119]. Notably, Luxturna, an AAV2-based vector targeting the retinoid isomerase RPE65 gene associated with Leber congenital amaurosis and progressive blindness, became the second AAV-based therapy to gain commercial approval in 2017 [120]. Similarly, Zolgensma, an AAV-based treatment approved for spinal muscular atrophy (SMA), further demonstrates the clinical efficacy of AAV vectors [121]. AAVs exhibit a lower risk of toxicity compared to other viral vectors. Despite the expanding use of AAV vectors in clinical applications, challenges remain in their large-scale production. High titers are required for effective in vivo delivery, and the production process can be labor-intensive and costly. Advances in manufacturing techniques, such as the development of optimized production platforms using suspension cell cultures and transient transfection methods, are being explored to address these limitations and make AAV therapies more accessible [122]. In March 2021, Wu et al. reported that AAVs encoding third-generation CAR genes can efficiently reprogram immune effector cells to produce CAR-T cells in vivo, illustrating that AAVs can facilitate direct CAR-T cell formation within the body [123]. In the context of CAR-T cell therapy, AAV vectors provide a novel platform for delivering CAR constructs directly into T cells in vivo, bypassing the need for ex vivo manipulation. This in vivo reprogramming strategy reduces the complexity and costs associated with traditional CAR-T cell production, potentially making CAR-T cell therapies more widely accessible. Nevertheless, like other gene delivery vectors, AAVs possess distinct advantages and limitations that require thorough evaluation [124]. However, despite their low immunogenicity relative to other viral vectors, AAVs can still elicit immune responses, especially in patients with pre-existing immunity to the vector capsid proteins. Such immune responses can reduce the transduction efficiency and limit the efficacy of subsequent administrations, which is a significant challenge in clinical settings, particularly for therapies requiring long-term or repeated dosing [125]. Strategies to mitigate these immune responses, such as the use of immunosuppressive drugs or modified AAV capsids, are being actively explored.
3.3. Lipid nanoparticle vectors
Liposomes have been recognized as a powerful medical tool for over five decades [126]. Composed primarily of lipid molecules such as phospholipids, cholesterol, and surfactants, lipid nanoparticles (LNPs) form the core structure of liposomes [127]. Phospholipids are the main components of LNPs, forming a bilayer membrane that encloses an internal aqueous phase. Cholesterol acts as a stabilizing and supportive element within the lipid bilayer, while surfactants on the LNP surface enhance stability and biocompatibility. These surfactants, which can be non-ionic, anionic, or cationic, are utilized to modulate the stability and charge of LNPs. The capability of LNPs to encapsulate drugs and deliver them precisely to targeted sites in the body highlights their immense potential for treating a wide range of diseases [128].
In recent years, LNPs have been employed to reprogram CAR cells in vivo. Margaret M. Billingsley and colleagues developed an in vivo CAR T cell engineering platform that employs ionizable LNPs conjugated with antibodies (Ab-LNPs) to target pan-T cell markers, facilitating efficient T cell transfection. This innovative approach enables the generation of functional CAR T cells within the body [129]. Jonathan A. Epstein et al. developed a method to generate transient anti-cardiac fibrosis CAR T cells in vivo using targeted LNP delivery of modified mRNA [130]. Jiang et al. designed a specialized LNP (CAR&Siglec-GΔITIMs LNP) that co-delivers CAR mRNA and truncated sialic-binding immunoglobulin-like lectin-G mRNA (Siglec-GΔITIMs mRNA). This LNP selectively adsorbs plasma proteins following intravenous injection and specifically edits liver macrophages to enhance phagocytosis and initiate immune responses within tumors, thereby effectively halting the progression of hepatocellular carcinoma [131]. As a pivotal nanocarrier system, LNPs offer numerous advantages, including a simple structure, versatile compositions, and notable benefits [132-135]. The efficiency of drug loading and targeted release is influenced by the adjustment of lipid composition and structure, which enhances drug bioavailability and efficacy. LNPs also exhibit excellent biocompatibility and biodegradability, rarely causing immune or toxic reactions. Despite the widespread use of LNPs in drug delivery, especially in gene therapy and vaccine development, their immunogenicity remains a concern. LNP formulations, particularly those used for mRNA vaccines, have been shown to trigger inflammatory responses. These immune responses can vary based on the composition of the lipids, with some ionizable lipids being more immunogenic than others [136]. Modifying lipid structures or incorporating additional excipients to reduce immunogenicity is an ongoing area of research aimed at improving the clinical safety profile of LNP-based therapies [137]. Targeting can be improved by modifying surfactants or lipid molecules, reducing drug toxicity to normal tissues, and enhancing therapeutic effectiveness. The size, shape, and surface properties of LNPs can be precisely tailored to meet various drug requirements through adjustments in formulation and processing conditions. Recent advancements in LNP technology have focused on the development of new lipid materials to enhance stability and functionality. For instance, the incorporation of ionizable lipids in LNPs has dramatically improved the efficiency of nucleic acid delivery by facilitating endosomal escape. Additionally, functionalizing the LNP surface with targeting ligands, such as peptides or antibodies, enables precise targeting of specific cell types, further enhancing therapeutic potential. PEGylation (the attachment of polyethylene glycol chains) is another widely used strategy to extend the circulation time of LNPs in vivo by reducing their recognition by the immune system [138]. Furthermore, the preparation method for LNPs is straightforward and cost-effective, lay a foundation for large-scale production and clinical applications. The success of LNPs in clinical applications is best exemplified by their use in mRNA vaccines for COVID-19. Both Pfizer-BioNTech and Moderna's vaccines utilize LNPs to deliver mRNA encoding the spike protein of the SARS-CoV-2 virus, enabling the body's immune system to recognize and combat the virus. These vaccines have demonstrated the safety and efficacy of LNPs as drug carriers at a global scale, marking a significant milestone in the field of nanomedicine. This success has further fueled research into applying LNPs for a range of other therapies, including cancer treatments and genetic disorders [139].
However, LNPs face several challenges, including potential instability in the biological environment, which can lead to uneven drug release or reduced efficacy. Some LNPs may also induce cytotoxicity or immune responses, which can compromise clinical safety. Additionally, the drug loading capacity of LNPs is currently limited, potentially hindering the efficient delivery of certain drugs. Future advancements are anticipated to address these issues by enhancing stability, reducing toxicity, increasing drug loading capacity, and developing more functional nanoparticles. Improving LNPs to create a safer and more efficient drug delivery platform will drive further innovations and progress in clinical therapies.
3.4. Gel vectors
As water-based materials with a three-dimensional network structure, gels are highly recognized in the medical field for their exceptional biocompatibility and versatility as carriers for bioactive substances, particularly in immunotherapy [140,141]. This biomaterial-based hydrogel is designed to encapsulate and deliver viral particles, enhancing their retention at the target site and enabling controlled release. The system aims to optimize the local delivery of viral vectors, ensuring efficient transfection and improving therapeutic outcomes. The utilization of gels in the in vivo construction of CAR-T cells, a pivotal strategy in immunotherapy, has gained substantial attention. Gels provide a scaffold that not only supports cellular structures but also prolongs the release duration of therapeutic agents [142]. Hydrogels possess unique mechanical properties, including high water content and the ability to mimic the extracellular matrix, making them ideal for the controlled release of drugs and immune-modulating agents [143]. Their viscoelastic nature enables them to conform to irregularly shaped surgical cavities, ensuring precise delivery of therapeutic agents in solid tumors, such as gliomas. Furthermore, the pore size of hydrogels can be fine-tuned to regulate the diffusion of bioactive molecules, offering an additional level of control in the therapeutic process [144]. This capability is forging new paths in immunotherapy research and application, potentially revolutionizing cancer treatment. By constructing an injectable nanoparticle-hydrogel superstructure (NP-hydrogel superstructure), this study aims to in situ induce the generation of glioma stem cell (GSC)-specific CAR macrophages/microglia (MΦs) in the tumor resection cavity, thereby preventing the recurrence of glioblastoma (GBM) [145].
Wu et al. developed an innovative injectable supramolecular hydrogel system loaded with CAR plasmids to continuously modify CAR-T cells at solid tumor sites [146]. Additionally, other researchers introduced a gene nanocarrier-injectable hydrogel superstructure, effectively employed in the immunotherapy of malignant glioma [145]. In post-surgery glioma models, the hydrogel served as a 'filler' to deliver macrophage-targeted editing nanocarriers and CD47 antibodies into the tumor cavity. This approach aims to edit local macrophages to enhance their phagocytic activity against GSCs while blocking the tumor's “don't eat me” signals. This dual strategy activates the adaptive immune system to clear residual GSCs post-surgery and establish immune memory, thereby preventing glioma recurrence. As a delivery vector, gels offer numerous benefits: they act as three-dimensional scaffolds for drug loading, regulate cell microenvironments by releasing growth factors to enhance CAR activity and anti-tumor effects, and maintain excellent biocompatibility, supporting cell survival and function while minimizing immune rejection [147-150]. Exploiting the unique properties of gels can significantly advance the construction of CAR cells for further immunotherapy. Despite their numerous benefits, the use of hydrogels in clinical settings presents several challenges. For instance, the rate of hydrogel degradation can vary significantly depending on the material composition, potentially leading to either premature release of therapeutic agents or insufficient breakdown to facilitate immune clearance [151]. Additionally, achieving consistent mechanical strength and ensuring that hydrogels maintain their structural integrity under physiological conditions remains a key area of research [152]. Overcoming these challenges will be critical in advancing the clinical application of hydrogels for CAR-based immunotherapy.
3.5. Other vectors
In addition to LVs, AAVs, RVs, LNPs, and gel-based vectors, a variety of other vectors are employed for in vivo CAR construction. Nanoparticles (NPs), defined by their nanoscale dimensions, can be synthesized from inorganic, organic, or composite materials. Their extensive specific surface area, coupled with distinctive physical and chemical properties, endows NPs with remarkable versatility in drug delivery and biological imaging. Furthermore, their degradability and cell-targeting capabilities render them particularly suitable for in vivo CAR construction. For instance, Matthias T. Stephan et al. developed biodegradable NPs capable of programming immune cells to recognize and destroy cancer cells in vivo. NPs-programmed T cells have demonstrated rapid clearance of cancer cells in leukemia mouse models, significantly improving their prognosis [153]. The NPs are engineered to deliver a CAR gene, which specifically targets T cells through molecular markers. This interaction promotes the integration of the CAR gene into the nuclear genome of the T cells, subsequently enabling the cells to express CARs. This innovation allows CAR-T cells to function as a precise and readily deployable “drug” for cancer therapy [154]. The advancement of suitable vectors for in vivo CAR construction plays an increasingly pivotal role in medical progress and patient well-being.
Plasmid vectors such as Sleeping Beauty (SB) and minicircles are emerging as valuable tools for non-viral gene delivery in CAR cell therapy, offering a safer and more cost-effective alternative to viral vectors like lentiviruses or retroviruses. The SB transposon system facilitates stable integration of CAR constructs into the genome with a reduced risk of insertional mutagenesis, making it a promising option for clinical applications. Similarly, minicircles, which are smaller plasmids lacking bacterial sequences, lower immunogenicity and enhance gene expression efficiency. These advantages make plasmid vectors particularly attractive for in vivo CAR generation, as they support more efficient and durable CAR cell production while mitigating safety concerns and reducing overall costs.
Inamdar et al. utilized a porous collagen scaffold-based implantable device to recruit host T cells in situ within tumors. By employing strategies such as CAR incorporation, this device facilitates the reprogramming and amplification of T cells for the treatment of solid tumors. Experimental results demonstrate that the device effectively enhances T cell tumor recognition capabilities, leading to tumor regression, thereby providing a novel approach for CAR-T cell therapy [155]. “Drydux” is an innovative biomaterial scaffold designed for the rapid and efficient in situ generation of tumor-specific CAR-T cells. This scaffold enhances the in vivo retention, functionality, and durability of the cells, enabling sustained tumor remission across various animal tumor models. It holds the potential to revolutionize CAR-T cell therapy for solid tumors [156].
4. Key considerations for constructing CAR in vivo
CARs within the complex environment of the human body presents significant challenges, including navigating intricate blood circulation systems and interacting with diverse cell types. The success of CAR cell therapies depends on the meticulous selection of appropriate cell types and vectors, as well as the employment of effective construction methods. Factors influencing the in vivo construction of CARs include the choice of cell types, vector selection, and specific methodologies. Each of these elements is critical in determining the ultimate efficacy of CAR cell therapies in treating various diseases.
4.1. Cell types
Selecting suitable cell types for in vivo modification is crucial for the success of CAR cell therapies. A thorough understanding of the impact of various cell types on CAR construction necessitates considering factors such as cell abundance, distribution, phagocytic capacity, and modification potential. Accurate analysis can aid in selecting the most appropriate cell type to achieve the desired therapeutic effect. For instance, macrophages play a critical role in tumor invasion, metastasis, immunosuppression, and angiogenesis [157]. Saar Gill et al. modified macrophages with HER2-targeted CARs, achieving a remarkable tumor-killing effect in mouse models [158]. Specifically, HER2-CAR-M was found to convert immunosuppressive M2 macrophages into pro-inflammatory M1 macrophages, thereby enhancing the cytotoxic effects of T cells in the tumor microenvironment. Another notable example involves Kupffer cells (KCs), which constitute 80-90% of macrophages and are primary responders to foreign particles in the sinusoids of liver. Jiang et al. developed a novel strategy that transforms KCs into CAR-KCs for HCC treatment. LNPs were used to specifically target liver macrophages to deliver mRNA encoding CAR and a modified form of CD24-Siglec-G (Siglec-GΔITIMs) lacking ITIMs. This innovative approach enhanced the phagocytic activity of liver macrophages in HCC mouse models, significantly reducing tumor burden and improving survival rates. The appropriate choice of cell types facilitates CAR cell therapies, making them an effective and adaptable strategy for HCC treatment [128].
4.2. Construction methods
Construction methods, including gene editing technologies and transfection techniques, are also fundamental for the successful in vivo construction of CARs [159]. Gene editing employs precise tools to modify, edit, or regulate an organism genome, facilitating the insertion, deletion, or alteration of specific genes. This approach effectively changes an organism's genetic traits [160]. Prominent gene editing tools, such as CRISPR/Cas9, TALEN, and ZFN, enable targeted DNA modifications at specific genomic locations. These tools hold significant potential for treating genetic diseases, advancing biological research, and enhancing bioengineering efforts [161]. Conversely, transfection technology involves the transfer of external DNA or RNA into target cells [162]. Techniques such as viral, chemical, and electrical transfection are employed to introduce the CAR gene into cells, facilitating the creation of CAR cells.
Further enhancing the application of these gene editing tools, Jennifer R. Hamilton et al. demonstrated how these technologies can be tailored for precision targeting. By using antibody fragments displayed on membrane-derived particles encapsulating CRISPR-Cas9 proteins and guide RNAs, they successfully directed genome editing tools to specific cells [163]. This antibody-targeted Cas9 delivery vector preferentially enables genome editing in homologous target cells within a mixed cell population in vivo. Additionally, Cas9-encapsulated delivery vectors (Cas9-EDVs) effectively generated genome-edited CAR-T cells in humanized mice. This approach underscores a programmable delivery modality with broad therapeutic potential, utilizing retroviral virus-like particle (VLP) assembly for transient delivery of Cas9 ribonucleoproteins (RNPs). More importantly, Cas9-EDVs achieved targeted genome editing in CAR-T cells without off-target effects on hepatocytes. These findings highlight the potential of EDVs as a programmable platform for delivering molecular cargo specifically to desired cell types for complex genome engineering in vivo. Understanding the variables that influence the in vivo construction of CARs is essential for optimizing the effectiveness of CAR cell therapy.
5. Other factors of construction of CAR
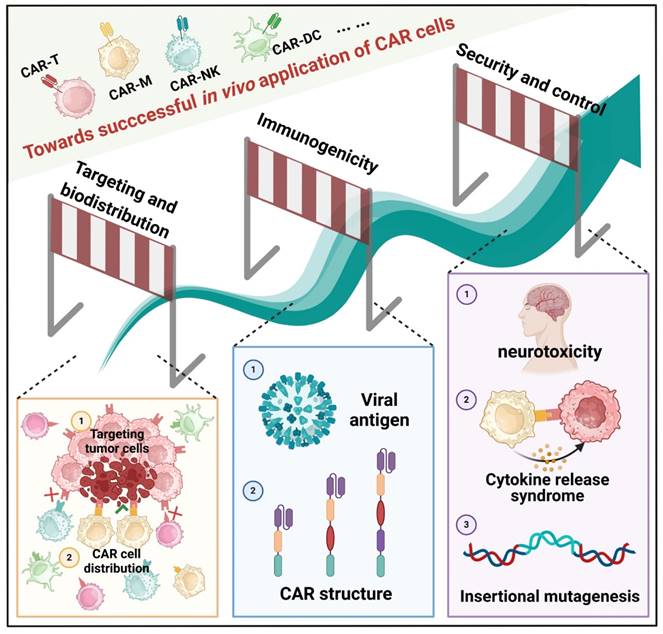
The in vivo application of CAR cells is a complex and challenging field that necessitates consideration of multiple critical factors. In the following sections, we will delve into issues related to the targeting and biodistribution of CAR cells, immunogenicity, and safety and control during in vivo application (Figure 6).
Crucial factors to consider for the application of CAR cells. 1) Targeting and biodistribution are crucial for optimizing therapeutic efficacy and advancing novel technologies. 2) CAR immunogenicity is a significant concern due to its diverse origins and multifaceted impact on treatment outcomes. 3) Safety and control-related challenges are of particular concern in in vivo CAR technology, including cytokine release syndrome (CRS), neurotoxicity, and potential insertional mutations during gene delivery. These may impair normal cellular function and induce tumorigenesis.
5.1. Targeting and biodistribution
CAR technology holds a significant role in tumor immunotherapy, with efficacy and safety critically hinging on precise targeting and effective biodistribution. The ability of CARs to target tumor cells fundamentally depends on their recognition of tumor-associated antigens [164]. These antigens are typically categorized into two types: tumor-specific antigens, such as neoantigens arising from gene mutations, and tumor-associated antigens, such as CD19 and BCMA, which are predominantly overexpressed in tumor cells compared to normal cells [165]. Optimizing CAR design by refining the antigen-binding region to increase affinity and specificity for tumor antigens can markedly enhance specificity [166]. Additionally, employing bispecific or multispecific CAR structures to recognize multiple tumor antigens simultaneously mitigates the risk of off-target effects due to the heterogeneous expression of single antigens [167].
Once the target antigen is identified, the biodistribution of CAR cells within the body becomes crucial to treatment outcomes. Distribution is influenced by several factors, including the expression of cell-surface molecules, the tumor microenvironment, and immune system regulation [168,169]. For instance, certain adhesion molecules enhance the interaction of CAR cells with vascular endothelial cells, facilitating their migration out of blood vessels and into tumor tissues. Additionally, chemokine receptors on CAR cells enable them to migrate towards chemokines secreted within the tumor microenvironment, significantly affecting CAR cell biodistribution [170]. The tumor microenvironment is typically immunosuppressive, enriched with inhibitory cytokines, immunosuppressive cells, and extracellular matrix components, which can impede CAR cell infiltration and survival. For example, PD-L1 molecules highly expressed in tumor tissues can bind to PD-1 receptors on CAR cells, inhibiting their activation and proliferation [171]. Moreover, the harsh conditions of low oxygen, limited nutrients, and acidic pH within tumor tissues can negatively impact the survival and function of CAR cells. To address these challenges, researchers have developed hypoxia-resistant CAR designs and metabolic reprogramming strategies [172].
The immune system plays a critical role in regulating the biodistribution of CAR cells [173]. In vivo, CAR cells are frequently identified as foreign entities, subjected to immune surveillance and elimination. Additionally, the immune system can indirectly influence CAR cell distribution and function through cytokine secretion and immune cell activation. For instance, regulatory T cells can suppress CAR cell activation and proliferation by secreting inhibitory cytokines, while macrophages and dendritic cells can promote CAR cell clearance through antigen presentation. To enhance the biodistribution and therapeutic efficacy of CAR cells, various strategies have been implemented. Researchers have genetically engineered CAR cells to express molecules such as chemokine receptors and anti-apoptotic proteins, thereby improving their infiltration and survival [174]. Additionally, combining CAR cell therapy with immunomodulators such as PD-1/PD-L1 inhibitors and CTLA-4 inhibitors can enhance the tumor microenvironment and boost CAR cell functionality [175,176]. Innovative drug delivery systems, including nanotechnology and hydrogels, have been employed to improve CAR cell aggregation and retention at tumor sites. Investigating CAR cell targeting and biodistribution is crucial for optimizing existing therapies and developing safer, more effective next-generation CAR technologies. Future efforts aim to achieve more precise CAR targeting and improved biodistribution, ultimately enhancing survival outcomes for cancer patients.
5.2. Immunogenicity
When the CAR is introduced into the body, it is recognized as a foreign substance, triggering an immune response, also known as immunogenicity. CAR immunogenicity remains a significant concern, as it can affect both the efficacy and safety of the treatment [177]. In the context of CAR cells, immunogenicity arises from several sources. Firstly, the structure of the CAR itself may be recognized as foreign by the immune system. CARs often contain antibody fragments from different species or synthetic peptides, which are not native to the human body and are likely to trigger an immune response [178]. Secondly, viral vectors or other gene delivery systems may introduce new antigens, which can also provoke an immune response [179]. Additionally, the large-scale expansion and persistence of CAR cells in the body may attract immune system attention [180]. The impact of CAR immunogenicity is multifaceted. On one hand, the immune response could reduce the survival and functionality of CAR cells, thereby diminishing the treatment's effectiveness. For instance, antibodies produced in the patient might bind to the CAR, preventing it from interacting with tumor antigens and impairing its tumor-killing capability. On the other hand, a strong immune response can cause serious adverse effects, such as cytokine release syndrome and neurotoxicity, posing significant health risks to the patient.
Scientists are exploring various strategies to mitigate CAR immunogenicity. In CAR design, using humanized antibody fragments or optimized peptide sequences can help reduce the presence of foreign components [181]. Selecting more appropriate gene delivery systems, such as low-immunogenic viral or non-viral vectors, can minimize the risk of immune responses [182]. Additionally, modulating the patient's immune system with immunosuppressive drugs can partially inhibit the immune attack on CAR cells. With an enhanced understanding of CAR immunogenicity, more effective strategies will be developed to address this issue.
5.3. Management of adverse toxicities
The safety and control issues arising from CAR in vivo technology have garnered increasing attention [183]. While immune responses are essential for CAR cell therapy to exert potent anti-tumor effects, they can also lead to serious adverse reactions, threatening patients' health and even their lives.
CRS is a common and serious complication of CAR cell therapy [184]. When CAR cells interact with tumor cells, they can rapidly activate immune cells, leading to the release of a large number of cytokines such as interleukin-6 (IL-6) and interleukin-1 (IL-1) [185]. Symptoms of CRS range from mild fever, fatigue, and myalgia to severe hypotension, respiratory failure, multiorgan dysfunction, and even life-threatening conditions. The severity of CRS is typically graded based on clinical symptoms and laboratory parameters. Neurotoxicity is another significant adverse reaction associated with CAR cell therapy [186]. Although the exact mechanism is not fully understood, neurotoxicity may involve the direct effects of cytokines, immune cell infiltration into the nervous system, and endothelial cell dysfunction. Symptoms of neurotoxicity are diverse and can include headache, delirium, confusion, seizures, aphasia, and ataxia. In severe cases, neurotoxicity can lead to permanent neurological damage.
Another critical safety issue is the risk of insertional mutagenesis during gene delivery, which can compromise normal cellular function and potentially induce tumorigenesis [187]. CAR genes are generally integrated into the cellular genome via viral vectors or alternative gene transfer methods. This stochastic insertion can disrupt adjacent genes, leading to aberrant function or gene inactivation [188,189]. Furthermore, CAR genes can be inserted into regulatory regions of the T cell genome, such as promoters, enhancers, or insulators, thereby influencing gene expression and regulation. In rare instances, CAR genes may integrate into oncogenes or tumor suppressor genes, resulting in dysfunction and heightening the risk of tumor development or progression. To mitigate the risks associated with insertional mutagenesis, scientists have implemented various strategies, including the optimization of gene transfer techniques, such as enhancing promoters and enhancers, and refining CAR structures [190]. They have also carefully selected appropriate vectors tailored to specific objectives, including lentiviral vectors, adeno-associated viral vectors, γ-retroviral vectors, mRNA vectors, and plasmid vectors [191]. The selection of insertion sites has been meticulously conducted, favoring “safe harbor” sites such as the AAVS1 site, CCR5 site, and Rosa26 site [192]. Moreover, rigorous quality control and testing of CAR-modified cells are crucial to ensuring their safety and efficacy.
In CAR cell therapy, despite its remarkable antitumor potential, functional exhaustion remains one of the primary obstacles limiting its clinical efficacy [193]. To address this issue, researchers have proposed various strategies to enhance CAR cell persistence and antitumor activity. Optimizing CAR structure is a key aspect of these efforts, with modifications to the intracellular signaling domains and the incorporation of costimulatory molecules such as CD28 or 4-1BB significantly improving CAR cell functional resilience [194]. Gene-editing technologies, including CRISPR/Cas9, have been widely employed to knock out exhaustion-related genes such as PD-1, LAG-3, and TIM-3 [195], or to enhance CAR cell metabolic activity to reduce exhaustion in hostile tumor microenvironments [196]. Additionally, the combined use of immune checkpoint inhibitors, such as PD-1/PD-L1 monoclonal antibodies, can block inhibitory signals and further prolong CAR cell efficacy [197]. To address antigen escape, multi-targeted or bispecific CAR designs have emerged as key solutions, with attempts also being made to create dynamically regulated CAR systems that adjust expression in response to environmental cues [198]. In improving the tumor microenvironment, pharmacological interventions and gene modification techniques have been employed to inhibit the function of immunosuppressive cells [199]. Moreover, combination therapies, such as CAR cells with tumor vaccines, radiotherapy, or chemotherapy, have been utilized to further enhance efficacy and delay exhaustion [200]. These multifaceted approaches provide a strong foundation for improving the therapeutic efficacy of CAR cells and driving advancements in this field.
6. Concluding remarks and future perspectives
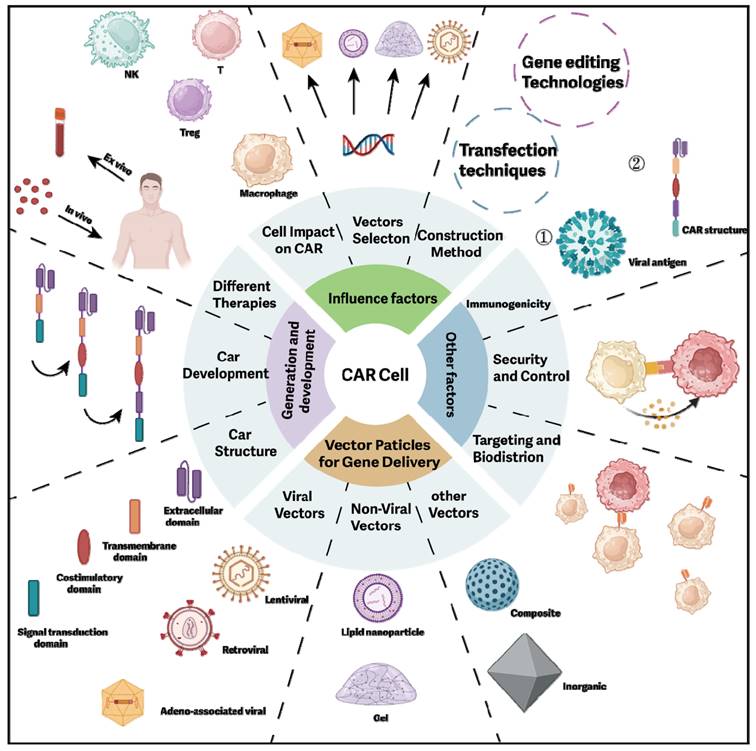
This article provides a comprehensive review of the CAR structure, its evolution, various forms, and delivery methods for CAR cells. Additionally, it examines the critical factors influencing CAR development, offering valuable insights for future research (Figure 7).
The comprehensive conclusion for CAR cell construction in vivo. This comprehensive summary covers the critical factors in constructing CAR cells, including gene editing, transfection techniques, vector selection, cell types, and CAR structure. It also illustrates the related elements of CAR cell construction and their interrelationships.
Selecting an appropriate vector for constructing CARs in vivo is paramount. A suitable vector facilitates the efficient transfer of the CAR gene into patients' immune cells, thereby enhancing the expression efficiency and stability of CAR cells. Continuous advancements in vector technology offer promising opportunities for the further development of CAR cell therapy. Beyond CAR-T cell therapy, CAR-NK, CAR-M, and other CAR cells represent emerging avenues for cancer treatment, demonstrating significant anti-tumor potential and undergoing extensive research and clinical trials.
However, traditional CAR preparation is complex and costly, limiting its widespread application. Several strategies are being developed to address these challenges: (1) Develop more efficient and cost-effective preparation technologies to streamline the process and reduce time and resource consumption. (2) Optimize cell collection methods to minimize individual differences, ensuring consistent quality and quantity of collected cells. (3) Improve the precision and efficiency of gene modification by exploring better conditions for cell expansion and genetic modification to enhance CAR cell functionality. (4) Transform the tumor microenvironment to promote CAR cell homing by understanding and modifying the tumor microenvironment. (5) Enhance gene delivery technology to improve in vivo delivery efficiency and specificity, and develop highly specific vectors and delivery systems. (6) Advance research on CAR construction factors by identifying more tumor-specific targets, better understanding signaling pathways, and designing optimal combinations.
To enhance the efficacy and safety of CAR cell therapy, researchers are exploring combination strategies with other treatment modalities. Taking CAR-T cell therapy as an example, the integration of immune checkpoint inhibitors (ICIs) such as PD-1 and PD-L1 monoclonal antibodies with CAR-T cells has been extensively studied. These inhibitors restore the ability of T cells to attack cancer cells, thereby improving the persistence and effectiveness of engineered cells within the tumor microenvironment. Clinical trials have demonstrated that in certain malignancies, such as glioblastoma and neuroblastoma, the combination of CAR-T cells with ICIs shows promising safety and efficacy, though results vary depending on the type of tumor. Moreover, cancer vaccines and oncolytic virus (OV) therapies are emerging as novel combination strategies that enhance CAR-T cell activation, proliferation, and persistence, thereby boosting their antitumor activity. Preclinical studies and early clinical trials indicate that cancer vaccines and oncolytic viruses can significantly improve CAR-T cell efficacy, particularly in solid tumors. Finally, monoclonal antibodies targeting cytokines, such as tocilizumab and anakinra, have shown efficacy in managing CAR-T cell-associated toxicities. These antibodies reduce the incidence of CRS and immune effector cell-associated neurotoxicity syndrome (ICANS) by inhibiting pro-inflammatory cytokines, thereby enhancing the safety of CAR-T cell therapy. Combining CAR-T cells with other treatment modalities, through multi-targeted and multi-mechanistic synergistic actions, holds the potential to overcome current efficacy and safety challenges, providing patients with more effective and durable treatment options.
Moving forward, researchers aim to develop multi-target CAR cell therapies, where CAR cells capable of recognizing multiple tumor antigens, such as bi-specific or multi-specific CAR constructs, could enhance therapeutic efficacy and reduce tumor immune evasion. Developing safer and more efficient vectors, such as viral vectors with higher specificity and lower immunogenicity, or novel non-viral vectors, is crucial for improving the efficiency and accuracy of gene delivery. For example, a delivery platform based on protein-lipid vehicles (PLVs) integrates the advantages of both viral and non-viral vectors for the safe and effective in vivo delivery of DNA and RNA. By incorporating the fusion-associated small transmembrane protein (FAST) derived from orthoreovirus into the lipid formulation, this platform achieves broad gene delivery capabilities while demonstrating low immunogenicity and favorable tolerability. Additionally, further research on personalized treatments is essential. For example, tailoring specific CAR cell therapies based on the patient's tumor characteristics, immune status, and genetic background can enhance the specificity and effectiveness of the treatment. Exploring combination therapies of CAR cell therapy with immune checkpoint inhibitors, chemotherapy, radiotherapy, and targeted therapies could also improve the overall efficacy of cancer treatment. Moreover, future research should focus on the immunogenicity of CAR cells to develop more effective strategies to reduce immunogenicity and enhance the safety and efficacy of the treatment. Ideally, the painstaking efforts made within the immune-oncology space will further advance CAR cell therapy for better cancer treatment outcomes and patient survival rates.
With ongoing advancements in gene editing and transfection technologies, in vivo CAR construction is poised to become a key method of personalized treatment, leading to revolutionary advancements in cancer and disease treatment. Future research should focus on exploring diverse methods of in vivo CAR construction to enhance treatment efficacy, minimize side effects, and extend benefits to a larger patient population. Furthermore, through continuous technological innovation and multidisciplinary collaboration, it is anticipated that the current challenges can be overcome, bringing more hope for a cure to cancer patients. With an in-depth understanding of tumor biology and the immune system, CAR cell therapy is expected to achieve breakthroughs in a broader range of cancer types, paving the way for the development of precision oncology.
Abbreviations
AAV: adeno-associated virus; Ab-LNPs: lipid nanoparticles conjugated with antibodies; ADCC: antibody-dependent cellular cytotoxicity; AI: artificial intelligence; AML: acute myeloid leukemia; aPD-1: anti-PD-1 immunoadhesin; BCMA: B-cell maturation antigen; B7-H3: B7 homolog 3 protein; CAAR-T: Chimeric Autoantibody Receptor T; CAR: Chimeric antigen receptor; CAR-NKT: Chimeric Antigen Receptor Natural Killer T; CAR-T: Chimeric Antigen Receptor T; Cas9-EDVs: Cas9-encapsulated delivery vectors; CB: cord blood; CEA: carcinoembryonic antigen; CNS: Central nervous system; CRS: cytokine release syndrome; CSCs: cancer stem cells; DC: dendritic cell; ECM: extracellular matrix; EGFR: epidermal growth factor receptor; EpCAM: Epithelial cell adhesion molecule; FasL: Fas ligand; FAST: fusion-associated small transmembrane protein; GBM: glioblastoma; GC: Gastric cancer; GD2: disialoganglioside; GM-CSF: granulocyte-macrophage colony-stimulating factor; GSC: glioma stem cell; GvHD: graft-versus-host disease; HCC: hepatocellular carcinoma; HER2: human epidermal growth factor receptor 2; HIV: human immunodeficiency virus; HLA: Human Leukocyte Antigen; hPSC: human pluripotent stem cell; IBD: inflammatory bowel disease; ICANS: immune effector cell-associated neurotoxicity syndrome; ICIs: immune checkpoint inhibitors; IFN-γ: interferon-gamma; IgG: immunoglobulin G; IL-1: interleukin-1; IL-6: interleukin-6; IL-15: interleukin 15; iPSCs: induced pluripotent stem cells; ITAM: immune receptor tyrosine activation motif; KCs: Kupffer cells; KIRs: killer immunoglobulin-like receptors; LDLR: low-density lipoprotein receptor; LNPs: lipid nanoparticles; LV: Lentiviral; MICA/B: MHC class I chain-related protein A/B; MMPs: matrix metalloproteinases; MSLN: Mesothelin; MUC1: Mucin 1; MΦs: macrophages/microglia; NB: Neuroblastoma; NK: natural killer; NKRs: natural killer receptors; NP-hydrogel superstructure: nanoparticle-hydrogel superstructure; NPs: Nanoparticles; OC: Ovarian cancer; OV: oncolytic virus; PD-L1: Programmed cell death 1 ligand 1; PLVs: protein-lipid vehicles; PMN-MDSCs: polymorphonuclear myeloid-derived suppressor cells; RACR: rapamycin-activated cytokine receptor; RNPs: ribonucleoproteins; ROR1: receptor tyrosine kinase-like orphan receptor 1; RVs: Retroviruses; SB: Sleeping Beauty; scFv: single-chain variable fragment; SMA: spinal muscular atrophy; ssDNA: single-stranded DNA; ssRNA: single-stranded RNA; TCRs: T cell receptors; TIL: tumor-infiltrating lymphocytes; TME: tumor microenvironment; TRAIL: TNF-related apoptosis-inducing ligand; Tregs: regulatory T cells; TRUCK: T cells Redirected for Universal Cytokine Killing; VH: variable heavy; VL: variable light; VLP: virus-like particle; VSV-G: glycoprotein from vesicular stomatitis virus.
Acknowledgements
Illustrations used in the manuscript were created using BioRender.com.
Funding
This work was supported by Taishan Scholar Program of Shandong Province (tsqn202211333), Shandong Natural Science Foundation (ZR2022MH001), National Natural Science Foundation of China (No. 82473802), Shandong First Medical University Culture Foundation (202201-04) and Shandong Provincial Qianfoshan Hospital Culture Foundation (QYPY-RC2022NSFC1003).
Author contributions
Jing Wu and Wenjun Mao conceived and supervised this project. Ruijie Lv, Yanting Guo and Weici Liu wrote the original draft and designed the figures. Guangjian Dong, Xiangyin Liu, Caihui Li, Yi Ren and Zipeng Zhang revised the manuscript. Shiyong Neo edited the English and revised the title. Jing Wu got financial support. All authors reviewed and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sukari A, Nagasaka M. Immune checkpoint inhibitors: For how long do we need to release the brakes to achieve the optimum acceleration of immune-mediated anti-tumor response? Oral Oncol. 2020;101:104435
2. Spotlight on Immunotherapy: Pushing the Frontier of Cancer Medicine. August, 2023. https://cancerprogressreport.aacr.org/
3. Long-Term Data for Kite's Yescarta® CAR T-Cell Therapy Presented at ASH 2023 Demonstrate High Rate of Durable Response in Patients With High-Risk Large B-Cell Lymphoma. December 11, 2023. https://investors.gilead.com/
4. Wu L, Wei Q, Brzostek J, Gascoigne NRJ. Signaling from T cell receptors (TCRs) and chimeric antigen receptors (CARs) on T cells. Cell Mol Immunol. 2020;17:600-12
5. Wu J, Chen Z, Wickström SL, Gao J, He X, Jing X. et al. Interleukin-33 is a Novel Immunosuppressor that Protects Cancer Cells from TIL Killing by a Macrophage-Mediated Shedding Mechanism. Adv Sci (Weinh). 2021;8:e2101029
6. Drougkas K, Karampinos K, Karavolias I, Koumprentziotis IA, Ploumaki I, Triantafyllou E. et al. Comprehensive clinical evaluation of CAR-T cell immunotherapy for solid tumors: a path moving forward or a dead end? J Cancer Res Clin Oncol. 2023;149:2709-34
7. Zhang Y, Zhou W, Yang J, Yang J, Wang W. Chimeric antigen receptor engineered natural killer cells for cancer therapy. Exp Hematol Oncol. 2023;12:70
8. Simon B, Wiesinger M, März J, Wistuba-Hamprecht K, Weide B, Schuler-Thurner B. et al. The Generation of CAR-Transfected Natural Killer T Cells for the Immunotherapy of Melanoma. IJMS. 2018;19:2365
9. Nagaleekar VK, Sabio G, Aktan I, Chant A, Howe IW, Thornton TM. et al. Translational control of NKT cell cytokine production by p38 MAPK. J Immunol. 2011;186:4140-6
10. Pan K, Farrukh H, Chittepu VCSR, Xu H, Pan C-X, Zhu Z. CAR race to cancer immunotherapy: from CAR T, CAR NK to CAR macrophage therapy. J Exp Clin Cancer Res. 2022;41:119
11. Karsten H, Matrisch L, Cichutek S, Fiedler W, Alsdorf W, Block A. Broadening the horizon: potential applications of CAR-T cells beyond current indications. Front Immunol. 2023;14:1285406
12. Michels A, Ho N, Buchholz CJ. Precision medicine: In vivo CAR therapy as a showcase for receptor-targeted vector platforms. Mol Ther. 2022;30:2401-15
13. Sun Y, Li F, Sonnemann H, Jackson KR, Talukder AH, Katailiha AS. et al. Evolution of CD8+ T Cell Receptor (TCR) Engineered Therapies for the Treatment of Cancer. Cells. 2021;10:2379
14. Zhang B, Ren Z, Zhao J, Zhu Y, Huang B, Xiao C. et al. Global analysis of HLA-A2 restricted MAGE-A3 tumor antigen epitopes and corresponding TCRs in non-small cell lung cancer. Theranostics. 2023;13:4449-68
15. Chimeric antigen receptors. Nat Biotechnol. 2022;40:654
16. Dabas P, Danda A. Revolutionizing cancer treatment: a comprehensive review of CAR-T cell therapy. Med Oncol. 2023;40:275
17. Cortés-Selva D, Dasgupta B, Singh S, Grewal IS. Innate and Innate-Like Cells: The Future of Chimeric Antigen Receptor (CAR) Cell Therapy. Trends Pharmacol Sci. 2021;42:45-59
18. Neo SY, Xu S, Chong J, Lam K-P, Wu J. Harnessing novel strategies and cell types to overcome immune tolerance during adoptive cell therapy in cancer. J Immunother Cancer. 2023;11:e006434
19. Miao L, Zhang J, Huang B, Zhang Z, Wang S, Tang F. et al. Special Chimeric Antigen Receptor (CAR) Modifications of T Cells: A Review. Front Oncol. 2022;12:832765
20. Bailey SR, Maus MV. Gene editing for immune cell therapies. Nat Biotechnol. 2019;37:1425-34
21. Dwivedi A, Karulkar A, Ghosh S, Rafiq A, Purwar R. Lymphocytes in Cellular Therapy: Functional Regulation of CAR T Cells. Front Immunol. 2018;9:3180
22. Zhang G, Wang L, Cui H, Wang X, Zhang G, Ma J. et al. Anti-melanoma activity of T cells redirected with a TCR-like chimeric antigen receptor. Sci Rep. 2014;4:3571
23. James SE, Greenberg PD, Jensen MC, Lin Y, Wang J, Till BG. et al. Antigen sensitivity of CD22-specific chimeric TCR is modulated by target epitope distance from the cell membrane. J Immunol. 2008;180:7028-38
24. Harush O, Asherie N, Kfir-Erenfeld S, Adler G, Barliya T, Assayag M. et al. Preclinical evaluation and structural optimization of anti-BCMA CAR to target multiple myeloma. haematol. 2022;107:2395-407
25. Bridgeman JS, Hawkins RE, Bagley S, Blaylock M, Holland M, Gilham DE. The optimal antigen response of chimeric antigen receptors harboring the CD3zeta transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J Immunol. 2010;184:6938-49
26. Velasco Cárdenas RM-H, Brandl SM, Meléndez AV, Schlaak AE, Buschky A, Peters T. et al. Harnessing CD3 diversity to optimize CAR T cells. Nat Immunol. 2023;24:2135-49
27. Lin X, Lee S, Sharma P, George B, Scott J. Summary of US Food and Drug Administration Chimeric Antigen Receptor (CAR) T-Cell Biologics License Application Approvals From a Statistical Perspective. J Clin Oncol. 2022;40:3501-9
28. Mohanty R, Chowdhury CR, Arega S, Sen P, Ganguly P, Ganguly N. CAR T cell therapy: A new era for cancer treatment (Review). Oncol Rep. 2019;42:2183-95
29. Brandt LJB, Barnkob MB, Michaels YS, Heiselberg J, Barington T. Emerging Approaches for Regulation and Control of CAR T Cells: A Mini Review. Front Immunol. 2020;11:326
30. Holzinger A, Barden M, Abken H. The growing world of CAR T cell trials: a systematic review. Cancer Immunol Immunother. 2016;65:1433-50
31. Sun D, Shi X, Li S, Wang X, Yang X, Wan M. CAR-T cell therapy: A breakthrough in traditional cancer treatment strategies (Review). Mol Med Rep. 2024;29:47
32. Asmamaw Dejenie T, Tiruneh G/Medhin M, Dessie Terefe G, Tadele Admasu F, Wale Tesega W, Chekol Abebe E. Current updates on generations, approvals, and clinical trials of CAR T-cell therapy. Hum Vaccin Immunother. 2022;18:2114254
33. Wu W, Zhou Q, Masubuchi T, Shi X, Li H, Xu X. et al. Multiple Signaling Roles of CD3ε and Its Application in CAR-T Cell Therapy. Cell. 2020;182:855-871.e23
34. Yeku OO, Brentjens RJ. Armored CAR T-cells: utilizing cytokines and pro-inflammatory ligands to enhance CAR T-cell anti-tumour efficacy. Biochem Soc Trans. 2016;44:412-8
35. Daamen AR, Lipsky PE. Potential and pitfalls of repurposing the CAR-T cell regimen for the treatment of autoimmune disease. Ann Rheum Dis. 2024;83:696-9
36. Park TS, Rosenberg SA, Morgan RA. Treating cancer with genetically engineered T cells. Trends Biotechnol. 2011;29:550-7
37. Philipson BI, O'Connor RS, May MJ, June CH, Albelda SM, Milone MC. 4-1BB costimulation promotes CAR T cell survival through noncanonical NF-κB signaling. Sci Signal. 2020;13:eaay8248
38. Hurton LV, Singh H, Najjar AM, Switzer KC, Mi T, Maiti S. et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc Natl Acad Sci U S A. 2016;113:E7788-97
39. Jayaraman J, Mellody MP, Hou AJ, Desai RP, Fung AW, Pham AHT. et al. CAR-T design: Elements and their synergistic function. EBioMedicine. 2020;58:102931
40. Enblad G, Karlsson H, Gammelgård G, Wenthe J, Lövgren T, Amini RM. et al. A Phase I/IIa Trial Using CD19-Targeted Third-Generation CAR T Cells for Lymphoma and Leukemia. Clin Cancer Res. 2018;24:6185-94
41. Ramello MC, Benzaïd I, Kuenzi BM, Lienlaf-Moreno M, Kandell WM, Santiago DN. et al. An immunoproteomic approach to characterize the CAR interactome and signalosome. Sci Signal. 2019;12:eaap9777
42. Li N, Geng S, Dong ZZ, Jin Y, Ying H, Li HW. et al. A new era of cancer immunotherapy: combining revolutionary technologies for enhanced CAR-M therapy. Mol Cancer. 2024;23:117
43. Zheng Z, Li S, Liu M, Chen C, Zhang L, Zhou D. Fine-Tuning through Generations: Advances in Structure and Production of CAR-T Therapy. Cancers (Basel). 2023;15:3476
44. Yuti P, Sawasdee N, Natungnuy K, Rujirachaivej P, Luangwattananun P, Sujjitjoon J. et al. Enhanced antitumor efficacy, proliferative capacity, and alleviation of T cell exhaustion by fifth-generation chimeric antigen receptor T cells targeting B cell maturation antigen in multiple myeloma. Biomed Pharmacother. 2023;168:115691
45. Harrer DC, Dörrie J, Schaft N. CARs and Drugs: Pharmacological Ways of Boosting CAR-T-Cell Therapy. Int J Mol Sci. 2023;24:2342
46. Ramos CA, Dotti G. Chimeric antigen receptor (CAR)-engineered lymphocytes for cancer therapy. Expert Opin Biol Ther. 2011;11:855-73
47. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021;11:69
48. Akahori Y, Wang L, Yoneyama M, Seo N, Okumura S, Miyahara Y. et al. Antitumor activity of CAR-T cells targeting the intracellular oncoprotein WT1 can be enhanced by vaccination. Blood. 2018;132:1134-45
49. Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. 2015;125:4017-23
50. Boulch M, Cazaux M, Cuffel A, Guerin MV, Garcia Z, Alonso R. et al. Tumor-intrinsic sensitivity to the pro-apoptotic effects of IFN-γ is a major determinant of CD4+ CAR T-cell antitumor activity. Nat Cancer. 2023;4:968-83
51. Alizadeh D, Wong RA, Yang X, Wang D, Pecoraro JR, Kuo CF. et al. IL15 Enhances CAR-T Cell Antitumor Activity by Reducing mTORC1 Activity and Preserving Their Stem Cell Memory Phenotype. Cancer Immunol Res. 2019;7:759-72
52. Rapp M, Wiedemann GM, Sun JC. Memory responses of innate lymphocytes and parallels with T cells. Semin Immunopathol. 2018;40:343-55
53. Kulkarni U, Arunachalam AK, Palani HK, Nair RR, Balasundaram N, Venkatraman A. et al. Haploidentical Natural Killer Cell Therapy as an Adjunct to Stem Cell Transplantation for Treatment of Refractory Acute Myeloid Leukemia. Cell Transplant. 2023;32:9636897231198178
54. Du Z, Ng YY, Zha S, Wang S. piggyBac system to co-express NKG2D CAR and IL-15 to augment the in vivo persistence and anti-AML activity of human peripheral blood NK cells. Mol Ther Methods Clin Dev. 2021;23:582-96
55. Mansouri V, Yazdanpanah N, Rezaei N. The immunologic aspects of cytokine release syndrome and graft versus host disease following CAR T cell therapy. Int Rev Immunol. 2022;41:649-68
56. Liu MY, Klement JD, Langan CJ, van Riggelen J, Liu K. Expression regulation and function of PD-1 and PD-L1 in T lymphoma cells. Cell Immunol. 2021;366:104397
57. Chu J, Gao F, Yan M, Zhao S, Yan Z, Shi B. et al. Natural killer cells: a promising immunotherapy for cancer. J Transl Med. 2022;20:240
58. Arias J, Yu J, Varshney M, Inzunza J, Nalvarte I. Hematopoietic Stem Cell- and Induced Pluripotent Stem Cell-Derived CAR-NK Cells as Reliable Cell-Based Therapy Solutions. Stem Cells Translational Medicine. 2021;10:987-95
59. Watzl C, Long EO. Signal Transduction During Activation and Inhibition of Natural Killer Cells. Curr Protoc Immunol. 2010 Aug; Chapter 11:Unit 11.9B
60. Zingoni A, Vulpis E, Loconte L, Santoni A. NKG2D Ligand Shedding in Response to Stress: Role of ADAM10. Front Immunol. 2020;11:447
61. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9:503-10
62. Lo Nigro C, Macagno M, Sangiolo D, Bertolaccini L, Aglietta M, Merlano MC. NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: biological evidence and clinical perspectives. Ann Transl Med. 2019;7:105-105
63. Xu Y, Liu Q, Zhong M, Wang Z, Chen Z, Zhang Y. et al. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. J Hematol Oncol. 2019;12:49
64. Ramezani F, Panahi Meymandi AR, Akbari B, Tamtaji OR, Mirzaei H, Brown CE. et al. Outsmarting trogocytosis to boost CAR NK/T cell therapy. Mol Cancer. 2023;22:183
65. Lin X, Sun Y, Dong X, Liu Z, Sugimura R, Xie G. IPSC-derived CAR-NK cells for cancer immunotherapy. Biomed Pharmacother. 2023;165:115123
66. Brunello L. Avoiding CAR-NK cell fratricide. Nat Cancer. 2022;3:1445
67. Zhang L, Tian L, Dai X, Yu H, Wang J, Lei A. et al. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J Hematol Oncol. 2020;13:153
68. Su S, Lei A, Wang X, Lu H, Wang S, Yang Y. et al. Induced CAR-Macrophages as a Novel Therapeutic Cell Type for Cancer Immune Cell Therapies. Cells. 2022;11:1652
69. Qian B-Z, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39-51
70. Cruz MS, Diamond A, Russell A, Jameson JM. Human αβ and γδ T Cells in Skin Immunity and Disease. Front Immunol. 2018;9:1304
71. Sebestyen Z, Prinz I, Déchanet-Merville J, Silva-Santos B, Kuball J. Translating gammadelta (γδ) T cells and their receptors into cancer cell therapies. Nat Rev Drug Discov. 2020;19:169-84
72. Silva-Santos B, Mensurado S, Coffelt SB. γδ T cells: pleiotropic immune effectors with therapeutic potential in cancer. Nat Rev Cancer. 2019;19:392-404
73. Couzi L, Pitard V, Sicard X, Garrigue I, Hawchar O, Merville P. et al. Antibody-dependent anti-cytomegalovirus activity of human γδ T cells expressing CD16 (FcγRIIIa). Blood. 2012Feb9;119(6):1418-27
74. Fisher JPH, Yan M, Heuijerjans J, Carter L, Abolhassani A, Frosch J. et al. Neuroblastoma killing properties of Vδ2 and Vδ2-negative γδT cells following expansion by artificial antigen-presenting cells. Clin Cancer Res. 2014;20:5720-32
75. Rei M, Pennington DJ, Silva-Santos B. The Emerging Protumor Role of γδ T Lymphocytes: Implications for Cancer Immunotherapy. Cancer Research. 2015;75:798-802
76. Wu P, Wu D, Ni C, Ye J, Chen W, Hu G. et al. γδT17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity. 2014;40:785-800
77. Lo Presti E, Toia F, Oieni S, Buccheri S, Turdo A, Mangiapane LR. et al. Squamous Cell Tumors Recruit γδ T Cells Producing either IL17 or IFNγ Depending on the Tumor Stage. Cancer Immunology Research. 2017;5:397-407
78. Mirzaei HR, Mirzaei H, Lee SY, Hadjati J, Till BG. Prospects for chimeric antigen receptor (CAR) γδ T cells: A potential game changer for adoptive T cell cancer immunotherapy. Cancer Lett. 2016;380:413-23
79. Li C, Zhou F, Wang J, Chang Q, Du M, Luo W. et al. Novel CD19-specific γ/δ TCR-T cells in relapsed or refractory diffuse large B-cell lymphoma. J Hematol Oncol. 2023;16:5
80. Carter JA, Preall JB, Grigaityte K, Goldfless SJ, Jeffery E, Briggs AW. et al. Single T Cell Sequencing Demonstrates the Functional Role of αβ TCR Pairing in Cell Lineage and Antigen Specificity. Front Immunol. 2019Jul31;10:1516
81. Nishimoto KP, Barca T, Azameera A, Makkouk A, Romero JM, Bai L. et al. Allogeneic CD20-targeted γδ T cells exhibit innate and adaptive antitumor activities in preclinical B-cell lymphoma models. Clin Transl Immunology. 2022Feb2;11(2):e1373
82. Van Acker HH, Campillo-Davo D, Roex G, Versteven M, Smits EL, Van Tendeloo VF. The role of the common gamma-chain family cytokines in γδ T cell-based anti-cancer immunotherapy. Cytokine Growth Factor Rev. 2018;41:54-64
83. Souza-Fonseca-Guimaraes F. New horizons for natural killer cell research in cancer, infection and inflammation. Clin Transl Immunology. 2021;10:e1275
84. Amberger DC, Schmetzer HM. Dendritic Cells of Leukemic Origin: Specialized Antigen-Presenting Cells as Potential Treatment Tools for Patients with Myeloid Leukemia. Transfus Med Hemother. 2020;47:432-43
85. Zhang S, Wu M, Wang F. Immune regulation by CD8+ Treg cells: novel possibilities for anticancer immunotherapy. Cell Mol Immunol. 2018;15:805-7
86. Lee J, Lundgren DK, Mao X, Manfredo-Vieira S, Nunez-Cruz S, Williams EF. et al. Antigen-specific B cell depletion for precision therapy of mucosal pemphigus vulgaris. J Clin Invest. 2020Dec1;130(12):6317-6324
87. Crunkhorn S. CAAR T cells to treat encephalitis. Nat Rev Drug Discov. 2024;23:22
88. Schmitt T, Hudemann C, Moztarzadeh S, Hertl M, Tikkanen R, Waschke J. Dsg3 epitope-specific signalling in pemphigus. Front Immunol. 2023Apr18;14:1163066
89. Konig MF, Paul S. More on CD19-CAR T Cells in Systemic Lupus Erythematosus. N Engl J Med. 2021;385:e67
90. Arjomandnejad M, Kopec AL, Keeler AM. CAR-T Regulatory (CAR-Treg) Cells: Engineering and Applications. Biomedicines. 2022;10:287
91. Badar T, Johnson BD, Hamadani M. Delayed neurotoxicity after axicabtagene ciloleucel therapy in relapsed refractory diffuse large B-cell lymphoma. Bone Marrow Transplant. 2021;56:683-5
92. Bouchkouj N, Kasamon YL, de Claro RA, George B, Lin X, Lee S. et al. FDA Approval Summary: Axicabtagene Ciloleucel for Relapsed or Refractory Large B-cell Lymphoma. Clin Cancer Res. 2019;25:1702-8
93. Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT. et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N Engl J Med. 2020;382:1331-42
94. Munshi NC, Anderson LD, Shah N, Madduri D, Berdeja J, Lonial S. et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N Engl J Med. 2021;384:705-16
95. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J. et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396:839-52
96. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H. et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med. 2018;378:439-48
97. Saez-Ibañez AR, Upadhaya S, Partridge T, Shah M, Correa D, Campbell J. Landscape of cancer cell therapies: trends and real-world data. Nat Rev Drug Discov. 2022;21:631-2
98. Kim S, Choi B, Kim Y, Shim G. Immune-Modulating Lipid Nanomaterials for the Delivery of Biopharmaceuticals. Pharmaceutics. 2023;15:1760
99. Hong M, Clubb JD, Chen YY. Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell. 2020;38:473-88
100. Krotova K, Nenavath GN, Packiriswamy N, Vandergaast R, Moy M, Ziegler C. et al. In Vivo Generation of Functional CD19 CAR-T Cells Using a Serum-Stable CD3-Targeted Lentiviral Vector. Blood. 2023;142:767
101. Borsotti C, Borroni E, Follenzi A. Lentiviral vector interactions with the host cell. Curr Opin Virol. 2016;21:102-8
102. Luis A. The Old and the New: Prospects for Non-Integrating Lentiviral Vector Technology. Viruses. 2020;12:1103
103. Milone MC, O'Doherty U. Clinical use of lentiviral vectors. Leukemia. 2018;32:1529-41
104. Vormittag P, Gunn R, Ghorashian S, Veraitch FS. A guide to manufacturing CAR T cell therapies. Curr Opin Biotechnol. 2018;53:164-81
105. Kohn LA, Kohn DB. Gene Therapies for Primary Immune Deficiencies. Front Immunol. 2021;12:648951
106. Frank AM, Braun AH, Scheib L, Agarwal S, Schneider IC, Fusil F. et al. Combining T-cell-specific activation and in vivo gene delivery through CD3-targeted lentiviral vectors. Blood Adv. 2020;4:5702-15
107. Huckaby JT, Landoni E, Jacobs TM, Savoldo B, Dotti G, Lai SK. Bispecific binder redirected lentiviral vector enables in vivo engineering of CAR-T cells. J Immunother Cancer. 2021;9:e002737
108. Michels KR, Sheih A, Hernandez SA, Brandes AH, Parrilla D, Irwin B. et al. Preclinical proof of concept for VivoVec, a lentiviral-based platform for in vivo CAR T-cell engineering. J Immunother Cancer. 2023Mar;11(3):e006292
109. Mo F, Mamonkin M. Generation of Chimeric Antigen Receptor T Cells Using Gammaretroviral Vectors. Methods Mol Biol. 2020;2086:119-30
110. Vargas JE, Chicaybam L, Stein RT, Tanuri A, Delgado-Cañedo A, Bonamino MH. Retroviral vectors and transposons for stable gene therapy: advances, current challenges and perspectives. J Transl Med. 2016;14:288
111. Schubert M-L, Schmitt A, Sellner L, Neuber B, Kunz J, Wuchter P. et al. Treatment of patients with relapsed or refractory CD19+ lymphoid disease with T lymphocytes transduced by RV-SFG.CD19.CD28.4-1BBzeta retroviral vector: a unicentre phase I/II clinical trial protocol. BMJ Open. 2019;9:e026644
112. Harris E, Elmer JJ. Optimization of electroporation and other non-viral gene delivery strategies for T cells. Biotechnology Progress. 2021;37:e3066
113. Agarwalla P, Ogunnaike EA, Ahn S, Froehlich KA, Jansson A, Ligler FS. et al. Bioinstructive implantable scaffolds for rapid in vivo manufacture and release of CAR-T cells. Nat Biotechnol. 2022;40:1250-8
114. Grossen P, Skaripa Koukelli I, van Haasteren J, H E Machado A, Dürr C. The ice age - A review on formulation of Adeno-associated virus therapeutics. Eur J Pharm Biopharm. 2023;190:1-23
115. Notarte KI, Catahay JA, Macasaet R, Liu J, Velasco JV, Peligro PJ. et al. Infusion reactions to adeno-associated virus (AAV)-based gene therapy: Mechanisms, diagnostics, treatment and review of the literature. J Med Virol. 2023;95:e29305
116. Liu Y, Kim YJ, Ji M, Fang J, Siriwon N, Zhang LI. et al. Enhancing gene delivery of adeno-associated viruses by cell-permeable peptides. Molecular Therapy - Methods & Clinical Development. 2014;1:12
117. Daya S, Berns KI. Gene Therapy Using Adeno-Associated Virus Vectors. Clin Microbiol Rev. 2008;21:583-93
118. Strecker MI, Wlotzka K, Strassheimer F, Roller B, Ludmirski G, König S. et al. AAV-mediated gene transfer of a checkpoint inhibitor in combination with HER2-targeted CAR-NK cells as experimental therapy for glioblastoma. Oncoimmunology. 2022;11:2127508
119. Samulski RJ, Berns KI, Tan M, Muzyczka N. Cloning of adeno-associated virus into pBR322: rescue of intact virus from the recombinant plasmid in human cells. Proc Natl Acad Sci U S A. 1982;79:2077-81
120. Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A. et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390:849-60
121. Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW. et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med. 2017;377:1713-22
122. Grieger JC, Samulski RJ. Adeno-associated virus vectorology, manufacturing, and clinical applications. Methods Enzymol. 2012;507:229-54
123. Nawaz W, Huang B, Xu S, Li Y, Zhu L, Yiqiao H. et al. AAV-mediated in vivo CAR gene therapy for targeting human T-cell leukemia. Blood Cancer J. 2021;11:119
124. Qu Y, Liu Y, Noor AF, Tran J, Li R. Characteristics and advantages of adeno-associated virus vector-mediated gene therapy for neurodegenerative diseases. Neural Regen Res. 2019;14:931-8
125. Mingozzi F, High KA. Immune responses to AAV in clinical trials. Curr Gene Ther. 2011;11:321-30
126. Mendonça MCP, Kont A, Kowalski PS, O'Driscoll CM. Design of lipid-based nanoparticles for delivery of therapeutic nucleic acids. Drug Discov Today. 2023;28:103505
127. Eygeris Y, Gupta M, Kim J, Sahay G. Chemistry of Lipid Nanoparticles for RNA Delivery. Acc Chem Res. 2022;55:2-12
128. Zhang Z, Yao S, Hu Y, Zhao X, Lee RJ. Application of lipid-based nanoparticles in cancer immunotherapy. Front Immunol. 2022;13:967505
129. Billingsley MM, Gong N, Mukalel AJ, Thatte AS, El-Mayta R, Patel SK. et al. In Vivo mRNA CAR T Cell Engineering via Targeted Ionizable Lipid Nanoparticles with Extrahepatic Tropism. Small. 2024;20:e2304378
130. Rurik JG, Tombácz I, Yadegari A, Méndez Fernández PO, Shewale SV, Li L. et al. CAR T cells produced in vivo to treat cardiac injury. Science. 2022;375:91-6
131. Yang Z, Liu Y, Zhao K, Jing W, Gao L, Dong X. et al. Dual mRNA co-delivery for in situ generation of phagocytosis-enhanced CAR macrophages augments hepatocellular carcinoma immunotherapy. J Control Release. 2023;360:718-33
132. Guevara ML, Persano F, Persano S. Advances in Lipid Nanoparticles for mRNA-Based Cancer Immunotherapy. Front Chem. 2020;8:589959
133. Pinto IS, Cordeiro RA, Faneca H. Polymer- and lipid-based gene delivery technology for CAR T cell therapy. J Control Release. 2023;353:196-215
134. Ye Z, Chen J, Zhao X, Li Y, Harmon J, Huang C. et al. In Vitro Engineering Chimeric Antigen Receptor Macrophages and T Cells by Lipid Nanoparticle-Mediated mRNA Delivery. ACS Biomater Sci Eng. 2022;8:722-33
135. In Vivo CAR Monocytes Show Efficacy in Mice. Cancer Discov. 2024Jan12;14(1):10
136. Pardi N, Hogan MJ, Porter FW, Weissman D. mRNA vaccines — a new era in vaccinology. Nat Rev Drug Discov. 2018;17:261-79
137. Zong Y, Lin Y, Wei T, Cheng Q. Lipid Nanoparticle (LNP) Enables mRNA Delivery for Cancer Therapy. Adv Mater. 2023Dec;35(51):e2303261
138. Kulkarni JA, Darjuan MM, Mercer JE, Chen S, van der Meel R, Thewalt JL. et al. On the Formation and Morphology of Lipid Nanoparticles Containing Ionizable Cationic Lipids and siRNA. ACS Nano. 2018;12:4787-95
139. Schoenmaker L, Witzigmann D, Kulkarni JA, Verbeke R, Kersten G, Jiskoot W. et al. mRNA-lipid nanoparticle COVID-19 vaccines: Structure and stability. International Journal of Pharmaceutics. 2021;601:120586
140. Mikhail AS, Morhard R, Mauda-Havakuk M, Kassin M, Arrichiello A, Wood BJ. Hydrogel drug delivery systems for minimally invasive local immunotherapy of cancer. Adv Drug Deliv Rev. 2023;202:115083
141. Liu C, Liao Y, Liu L, Xie L, Liu J, Zhang Y. et al. Application of injectable hydrogels in cancer immunotherapy. Front Bioeng Biotechnol. 2023Feb3;11:1121887
142. Zuo Y-H, Zhao X-P, Fan X-X. Nanotechnology-based chimeric antigen receptor T-cell therapy in treating solid tumor. Pharmacol Res. 2022;184:106454
143. Hennink WE, van Nostrum CF. Novel crosslinking methods to design hydrogels. Advanced Drug Delivery Reviews. 2002;54:13-36
144. Li J, Mooney DJ. Designing hydrogels for controlled drug delivery. Nat Rev Mater. 2016;1:16071
145. Chen C, Jing W, Chen Y, Wang G, Abdalla M, Gao L. et al. Intracavity generation of glioma stem cell-specific CAR macrophages primes locoregional immunity for postoperative glioblastoma therapy. Sci Transl Med. 2022;14:eabn1128
146. Zhu C, Ke L, Ao X, Chen Y, Cheng H, Xin H. et al. Injectable Supramolecular Hydrogels for In Situ Programming of Car-T Cells toward Solid Tumor Immunotherapy. Adv Mater. 2024;36:e2310078
147. Peng Y, Liang S, Meng Q-F, Liu D, Ma K, Zhou M. et al. Engineered Bio-Based Hydrogels for Cancer Immunotherapy. Adv Mater. 2024;36:e2313188
148. Cui R, Wu Q, Wang J, Zheng X, Ou R, Xu Y. et al. Hydrogel-By-Design: Smart Delivery System for Cancer Immunotherapy. Front Bioeng Biotechnol. 2021;9:723490
149. Zheng H, Li M, Wu L, Liu W, Liu Y, Gao J. et al. Progress in the application of hydrogels in immunotherapy of gastrointestinal tumors. Drug Deliv. 2023Dec;30(1):2161670
150. Seo HS. Short Review on Advances in Hydrogel-Based Drug Delivery Strategies for Cancer Immunotherapy. Tissue Eng Regen Med. 2022;19:263-280
151. Drury JL, Mooney DJ. Hydrogels for tissue engineering: scaffold design variables and applications. Biomaterials. 2003;24:4337-51
152. Van Vlierberghe S, Dubruel P, Schacht E. Biopolymer-Based Hydrogels As Scaffolds for Tissue Engineering Applications: A Review. Biomacromolecules. 2011;12:1387-408
153. Smith TT, Stephan SB, Moffett HF, McKnight LE, Ji W, Reiman D. et al. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nature Nanotech. 2017;12:813-20
154. Singh A. Materials modulate immunity and gut microbiome. Nat Mater. 2020;19:3-4
155. Inamdar VV, Hao S, Stephan SB, Stephan MT. Biomaterial-based scaffolds for direct in situ programming of tumor-infiltrating T lymphocytes. J Control Release. 2024;370:310-7
156. Pandit S, Agarwalla P, Song F, Jansson A, Dotti G, Brudno Y. Implantable CAR T cell factories enhance solid tumor treatment. Biomaterials. 2024;308:122580
157. Liu X, Li Y, Lyu R, Guo Y, Yin X, Liu J. et al. Type 1 diabetes mellitus, hyperlipidemia, and inflammatory bowel disease: a Mendelian randomization study. Acta Materia Medica. 2024;3:20-30
158. Pierini S, Gabbasov R, Gabitova L, Ohtani Y, Shestova O, Gill S. et al. Abstract 63: Chimeric antigen receptor macrophages (CAR-M) induce anti-tumor immunity and synergize with T cell checkpoint inhibitors in pre-clinical solid tumor models. Cancer Res 1 July. 2021;81(13_Supplement):63
159. Mirzaei HR, Pourghadamyari H, Rahmati M, Mohammadi A, Nahand JS, Rezaei A. et al. Gene-knocked out chimeric antigen receptor (CAR) T cells: Tuning up for the next generation cancer immunotherapy. Cancer Lett. 2018;423:95-104
160. Minter F. Two steps are better than one: improving gene editing to treat cancer. FEBS Open Bio. 2022;12:9-11
161. Yoon DE, Lee H, Kim K. Recent Research Trends in Stem Cells Using CRISPR/Cas-Based Genome Editing Methods. Int J Stem Cells. 2024;17:1-14
162. Alkharobi H. Exploring Various Transfection Approaches and Their Applications in Studying the Regenerative Potential of Dental Pulp Stem Cells. Curr Issues Mol Biol. 2023;45:10026-40
163. Hamilton JR, Chen E, Perez BS, Sandoval Espinoza CR, Kang MH, Trinidad M. et al. In vivo human T cell engineering with enveloped delivery vehicles. Nat Biotechnol. 2024
164. Long J, Wang Y, Jiang X, Ge J, Chen M, Zheng B. et al. Nanomaterials Boost CAR-T Therapy for Solid Tumors. Adv Healthc Mater. 2024;13:e2304615
165. Haubner S, Mansilla-Soto J, Nataraj S, Kogel F, Chang Q, de Stanchina E. et al. Cooperative CAR targeting to selectively eliminate AML and minimize escape. Cancer Cell. 2023Nov13;41(11):1871-1891.e6
166. Rhoden JJ, Dyas GL, Wroblewski VJ. A Modeling and Experimental Investigation of the Effects of Antigen Density, Binding Affinity, and Antigen Expression Ratio on Bispecific Antibody Binding to Cell Surface Targets. J Biol Chem. 2016;291:11337-47
167. Spiegel JY, Patel S, Muffly L, Hossain NM, Oak J, Baird JH. et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: a phase 1 trial. Nat Med. 2021;27:1419-31
168. Song EZ, Milone MC. Pharmacology of Chimeric Antigen Receptor-Modified T Cells. Annu Rev Pharmacol Toxicol. 2021;61:805-29
169. Jureczek J, Bergmann R, Berndt N, Koristka S, Kegler A, Puentes-Cala E. et al. An oligo-His-tag of a targeting module does not influence its biodistribution and the retargeting capabilities of UniCAR T cells. Sci Rep. 2019;9:10547
170. Chen P-H, Lipschitz M, Weirather JL, Jacobson C, Armand P, Wright K. et al. Activation of CAR and non-CAR T cells within the tumor microenvironment following CAR T cell therapy. JCI Insight. 2020;5:e134612 134612
171. Andreu-Saumell I, Rodriguez-Garcia A, Mühlgrabner V, Gimenez-Alejandre M, Marzal B, Castellsagué J. et al. CAR affinity modulates the sensitivity of CAR-T cells to PD-1/PD-L1-mediated inhibition. Nat Commun. 2024;15:3552
172. Van Der Vreken A, Vanderkerken K, De Bruyne E, De Veirman K, Breckpot K, Menu E. Fueling CARs: metabolic strategies to enhance CAR T-cell therapy. Exp Hematol Oncol. 2024;13:66
173. Wang X, Wu Z, Qiu W, Chen P, Xu X, Han W. Programming CAR T cells to enhance anti-tumor efficacy through remodeling of the immune system. Front Med. 2020;14:726-45
174. Ghahri-Saremi N, Akbari B, Soltantoyeh T, Hadjati J, Ghassemi S, Mirzaei HR. Genetic Modification of Cytokine Signaling to Enhance Efficacy of CAR T Cell Therapy in Solid Tumors. Front Immunol. 2021;12:738456
175. Liu D. CAR-T 'the living drugs', immune checkpoint inhibitors, and precision medicine: a new era of cancer therapy. J Hematol Oncol. 2019;12:113
176. Zhang R, Deng Q, Jiang Y-Y, Zhu H-B, Wang J, Zhao M-F. Effect and changes in PD-1 expression of CD19 CAR-T cells from T cells highly expressing PD-1 combined with reduced-dose PD-1 inhibitor. Oncol Rep. 2019;41:3455-63
177. Minute L, Teijeira A, Sanchez-Paulete AR, Ochoa MC, Alvarez M, Otano I. et al. Cellular cytotoxicity is a form of immunogenic cell death. J Immunother Cancer. 2020;8:e000325
178. Heard A, Chang J, Warrington JM, Singh N. Advances in CAR design. Best Pract Res Clin Haematol. 2021;34:101304
179. Monjezi R, Miskey C, Gogishvili T, Schleef M, Schmeer M, Einsele H. et al. Enhanced CAR T-cell engineering using non-viral Sleeping Beauty transposition from minicircle vectors. Leukemia. 2017;31:186-94
180. Petersen CT, Hassan M, Morris AB, Jeffery J, Lee K, Jagirdar N. et al. Improving T-cell expansion and function for adoptive T-cell therapy using ex vivo treatment with PI3Kδ inhibitors and VIP antagonists. Blood Adv. 2018;2:210-23
181. Nakamura T, Kobayashi E, Hamana H, Hayakawa Y, Muraguchi A, Hayashi A. et al. Evaluation of chimeric antigen receptor of humanized rabbit-derived T cell receptor-like antibody. Cancer Sci. 2022;113:3321-9
182. Wang F, Huang Y, Li J, Zhou W, Wang W. Targeted gene delivery systems for T-cell engineering. Cell Oncol (Dordr). 2024;47:1537-1560
183. Sicard A, Levings MK, Scott DW. Engineering therapeutic T cells to suppress alloimmune responses using TCRs, CARs, or BARs. American Journal of Transplantation. 2018;18:1305-11
184. Chen H, Wang F, Zhang P, Zhang Y, Chen Y, Fan X. et al. Management of cytokine release syndrome related to CAR-T cell therapy. Front Med. 2019;13:610-7
185. Zhang A, Sun Y, Du J, Dong Y, Pang H, Ma L. et al. Reducing Hinge Flexibility of CAR-T Cells Prolongs Survival In Vivo With Low Cytokines Release. Front Immunol. 2021;12:724211
186. Ahmed O. CAR-T-cell neurotoxicity: hope is on the horizon. Blood. 2019;133:2114-6
187. Shao L, Shi R, Zhao Y, Liu H, Lu A, Ma J. et al. Genome-wide profiling of retroviral DNA integration and its effect on clinical pre-infusion CAR T-cell products. J Transl Med. 2022;20:514
188. Hamada M, Nishio N, Okuno Y, Suzuki S, Kawashima N, Muramatsu H. et al. Integration Mapping of piggyBac-Mediated CD19 Chimeric Antigen Receptor T Cells Analyzed by Novel Tagmentation-Assisted PCR. EBioMedicine. 2018;34:18-26
189. Sumrada RA, Cooper TG. Point mutation generates constitutive expression of an inducible eukaryotic gene. Proc Natl Acad Sci U S A. 1985;82:643-7
190. Requejo Cier CJ, Valentini N, Lamarche C. Unlocking the potential of Tregs: innovations in CAR technology. Front Mol Biosci. 2023;10:1267762
191. Qin D-Y, Huang Y, Li D, Wang Y-S, Wang W, Wei Y-Q. Paralleled comparison of vectors for the generation of CAR-T cells. Anti-Cancer Drugs. 2016;27:711-22
192. Dabiri H, Safarzadeh Kozani P, Habibi Anbouhi M, Mirzaee Godarzee M, Haddadi MH, Basiri M. et al. Site-specific transgene integration in chimeric antigen receptor (CAR) T cell therapies. Biomark Res. 2023;11:67
193. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486-99
194. Lim WA, June CH. The Principles of Engineering Immune Cells to Treat Cancer. Cell. 2017;168:724-40
195. Ren J, Zhang X, Liu X, Fang C, Jiang S, June CH. et al. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget. 2017;8:17002-11
196. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z. et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest. 2013;123:4479-88
197. Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y. et al. The Immunoreceptor TIGIT Regulates Antitumor and Antiviral CD8 + T Cell Effector Function. Cancer Cell. 2014;26:923-37
198. Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D. et al. Intratumoral Tcf1+PD-1+CD8+ T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity. 2019;50:195-211.e10
199. Zhao L, Cao YJ. Engineered T Cell Therapy for Cancer in the Clinic. Front Immunol. 2019;10:2250
200. Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ. et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med. 2015;21:81-5
Author contact
![]() Corresponding authors: wujingedu.cn (Jing Wu), maowenjun1edu.cn (Wenjun Mao).
Corresponding authors: wujingedu.cn (Jing Wu), maowenjun1edu.cn (Wenjun Mao).