Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Tumor heterogeneity is an...
Immune checkpoint inhibitors are...
The prostate cancer...
Potentiating immune checkpoint...
Enhancing the effectiveness of...
Discussion and future...
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2024; 14(18):6913-6934. doi:10.7150/thno.100555 This issue Cite
Review
Immunological facets of prostate cancer and the potential of immune checkpoint inhibition in disease management
Stian Bakke Hansen1, Bilal Unal1,2, Omer Faruk Kuzu1,2, Fahri Saatcioglu1,2 ![]()
1. Department of Biosciences, University of Oslo, Oslo, Norway.
2. Institute for Cancer Genetics and Informatics, Oslo University Hospital, Oslo, Norway.
Received 2024-7-5; Accepted 2024-8-27; Published 2024-10-21
Abstract

Prostate cancer (PCa) is the most common non-cutaneous cancer in men and a major cause of cancer-related deaths. Whereas localized PCa can be cured by surgery and radiotherapy, metastatic disease can be treated, but is not curable. Inhibition of androgen signaling remains the main therapeutic intervention for treatment of metastatic PCa, in addition to chemotherapy, radionuclide therapy and emerging targeted therapies. Although initial responses are favorable, resistance to these therapies invariably arise with development of castration resistant PCa (CRPC) and lethal phenotypes. Recent findings have implicated the crosstalk between PCa cells and the tumor microenvironment (TME) as a key factor for disease progression and metastasis, and the immune system is becoming an increasingly attractive target for therapy. Given the striking success of immune checkpoint inhibitors (ICIs) in various cancer types, preclinical and clinical studies have begun to explore their potential in PCa. It has become clear that the PCa TME is largely immunosuppressive, and ICI therapy does not have efficacy for PCa. Intense effort is therefore being made in the field to understand the mechanisms of suppression and to turn the immunosuppressive TME into an immune active one that would enable ICI efficacy. Herein we examine this recent body of knowledge and how the mutational landscape of PCa integrates with an immunosuppressive TME to circumvent ICI-mediated T-cell activity and tumor killing. We then review the emerging potential success of combinatorial ICI approaches, utility of careful patient selection, and potential novel strategies to improve the efficacy of ICI for PCa therapy.
Keywords: Cold tumor, Combination therapy, Immune checkpoint inhibitor, Immunotherapy, Prostate cancer, Tumor microenvironment.
Introduction
Prostate cancer (PCa) is the most common non-cutaneous cancer in men and a leading cause of cancer-related deaths in developed countries [1]. More than 1.4 million new cases and ~375,000 deaths worldwide were ascribed to PCa in 2020, with an expected increase of 17% and 19%, respectively, by 2025 [2]. Most patients are diagnosed at an early stage for which the mainstay treatment is surgery and radiotherapy, but the risk of potential overtreatment, along with the inherent dangers of treatment-related disease progression, has made active surveillance one common option from a quality of life perspective [3, 4]. Despite successful treatment in most patients, PCa recurs in 20-40% of cases within 10 years after the treatment [4, 5]. When recurrence occurs, it may be amenable to salvage radiotherapy and androgen-deprivation therapy (ADT), which can be achieved through chemical or surgical castration. However, a significant proportion of patients will eventually relapse into castration-resistant PCa (CRPC) to which any further treatment is rather palliative. Disease management at this point involves novel hormonal therapy (NHT) with antiandrogens or the androgen synthesis inhibitor abiraterone, concurrent with or followed by chemotherapy. Bone metastatic disease is eligible for systemic Radium-233 treatment, and mCRPC patients that present with specific biomarkers may be assigned to receive the novel 177Lu-PSMA-617 radioligand therapy or poly (ADP-ribose) polymerase (PARP)-inhibitors [4, 6]. Resistance and emergence of advanced disease invariably ensues, such as neuroendocrine PCa and double negative PCa, and new strategies to overcome this are urgently needed.
Immunotherapy is a treatment modality that aims to manage disease by targeting and activating the immune system. In cancer, this involves priming the patient's lymphocytes for destroying the growing tumor rather than attacking features within the cancer cells that are prone to mutation and diversion via bypass pathways. Immune checkpoint inhibitors (ICIs) offer one such strategy and are based on the observation that tumor-infiltrating T cells are often dysfunctional characterized by low activity and expression of co-inhibitory surface receptors [7]. These checkpoints serve to maintain self-tolerance in normal physiology, but are subverted in favor of the cancer cell by a complex interplay of chronic inflammation and the cytokine milieu within the tumor microenvironment (TME). Checkpoints include cytotoxic T-lymphocyte associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1), which have been studied extensively. Novel targets are emerging, such as the T-cell immunoglobulin and mucin domain 3 (TIM-3), lymphocyte activation gene-3 (LAG-3), and V-domain Ig suppressor of T cell activation (VISTA), that are being investigated for translational applications. The ICI approach aims to release the brakes on the immune system by blocking such co-inhibitory molecules, and a number of monoclonal antibodies are now in clinical use since the first approval of an anti-CTLA-4 agent for advanced melanoma in 2011 [8]. The resounding success of these checkpoint blockers in melanoma and some other cancers sparked hope for their benefits in PCa [9-11]; unfortunately, this expectation has yet to be fulfilled.
The main reason appears to be that PCa has characteristics of a “cold” tumor type with low T-cell infiltrate and intensive immunosuppressive mechanisms. Success of immunotherapy for other cancer types has therefore been difficult to realize for PCa; at present, a dendritic cell-based vaccine (Sipuleucel-T, or Provenge) is the only immune-based treatment specifically approved for PCa [12]. Two PD-1 blocking agents have received tissue-agnostic approval based on DNA repair status or mutational burden since 2017 [13], yet their performance in PCa is at best questionable, and attempts at targeting other checkpoint molecules have similarly failed in performing under recent data from clinical trials despite showing promise in early stages. A greater understanding of the immunological features of PCa in disease progression is therefore necessary to reactivate the immune compartment in the TME and thereby enhance the clinical benefit of ICIs. Herein we discuss previous setbacks in ICI monotherapies for PCa and how the mutational landscape of PCa integrates with an immunosuppressive TME to circumvent ICI-mediated T-cell activity and tumor killing. We then focus on the emerging benefit of combinatorial approaches, utility of biomarker-informed patient selection, and potential novel strategies to improve the efficacy of ICIs in PCa.
Tumor heterogeneity is an important hurdle for prostate cancer treatment
Despite many breakthroughs that have emerged in cancer research over the years, the development of treatment resistance and relapse into an aggressive form with poor prognosis remains a major challenge in the clinic. This can mainly be attributed to the heterogeneity of tumors and the complexities of the TME that serve to fuel growth and evolution [14]. In essence, the presence of substantial differences at the cellular and molecular levels between patients suffering from a specific subtype of cancer (inter-patient heterogeneity) has been an important driving force for the design of personalized treatment options, the idea of precision medicine. However, the idea of screening a patient's genotype and matching it with a tailored treatment option falters when the heterogeneity extends within a single patient, especially later in the disease course. For example, the striking heterogeneity between PCa foci gives rise to a clear variability in classification, risk stratification, predicted recurrence, and estimated androgen receptor (AR) pathway activity for each patient [15]. These findings emphasize the problem of making a clinical decision based on a single lesion that may not be the origin for the lethal stages of PCa.
The immune landscape in cancer is a complex spectrum with many aspects concerning the quantity and characteristics of immune infiltrates; some simplification can be made by assigning positions on each extreme of the spectrum. Neoplasms may be classified by their immunological phenotype into either “hot” or “cold” tumors, wherein the former is recognized by high mutational burden, extensive T-cell infiltration, and markers of both T-cell mediated killing and exhaustion [16]. Due to the nature of repeated T-cell stimulation, such tumors are more likely to respond to ICIs. Cold tumors, on the other hand, are characterized by little or no T-cell infiltration, where the cells are restricted to the margins of the tumor (immune-excluded) or missing altogether (immune-desert). Lack of responsive lymphocytes would portend inefficacy of checkpoint blockers, which in fact is what is observed in the clinic. Melanomas, non-small cell lung cancer (NSCLC), and liver cancer, are considered immunologically hot tumors, whereas cancers such as PCa, breast cancer, and pancreatic cancer are recognized as immunologically cold [17-19]. Distinction between these immunophenotypes is highly context dependent; all cancer types can present on both sides of the spectrum and thus understanding the mechanisms involved in establishing a cold TME becomes more important than assigning a cancer to any category. This basic understanding is becoming paramount to crack the code that could dramatically improve the success of ICIs for cancer in general, as well as for PCa.
Immune checkpoint inhibitors are largely ineffective in PCa
In line with the immunophenotype of PCa, ICI therapy has not yielded any significant response in patients with localized or advanced forms of the disease, because there is little release in the TME. Despite showing favorable prostate-specific antigen (PSA) response rates and increases in progression-free survival (PFS) compared to placebo control, the anti-CTLA-4 treatment ipilimumab failed to improve overall survival (OS) in chemotherapy-naïve as well as in patients previously treated with docetaxel and radiotherapy in two separate Phase III studies [20, 21]. Severe treatment-related complications were observed in a number of patients that could not justify the benefits of the treatment. Long-term follow-up for the post-chemotherapy study did however find an increase of survivors that becomes pronounced from three years onward [22]. In addition, a small Phase II study found an expansion of T cells of effector memory and Th1-like subtypes in patients treated with ipilimumab plus ADT [23], indicating that the ICI evokes a response in a subset of patients the basis of which is not entirely understood. Anti-PD-1 therapy with nivolumab showed tolerable safety profiles in a Phase I study published in 2010 and another from 2012, yet their results demonstrate little activity in PCa [24, 25].
Pembrolizumab is another PD-1 targeting antibody that has found a firm standing in the clinic since its approval for advanced melanoma in 2014 [8]. Its efficacy, coupled with good safety profiles in other cancer types raised hopes for its successful use in PCa, but it has so far failed to meet expectations. Efforts to identify predictive biomarkers are still ongoing. Intuitively, the expression of programmed death-ligand 1 (PD-L1) in tumor tissue would present a rational marker for stratification of cancer patients for response to anti-PD-1 therapy. With this in mind, a large study published in 2018 observed a significant increase in PD-L1 immunoreactivity in metastatic CRPC (mCRPC) compared to primary PCa [26], where expression is rare, and the Phase II KEYNOTE-199 study took this rationale into practice for mCRPC patients [27]. Patients with a treatment history involving docetaxel were enrolled in PD-L1 positive or negative cohorts for administration of 200 mg pembrolizumab every 3 weeks until specified endpoints were reached. Objective response rates (ORR) were equivalent in the two cohorts, with a 5% response in the PD-L1 positive cohort, only 2% higher than the PD-L1 negative group. With a durable response or stable disease in 10% of the participants, it is clear that a small number of mCRPC patients respond favorably to single treatment, but the characteristics of this subpopulation can clearly not be defined by PD-L1 status alone.
In June 2020, the FDA approved the use of pembrolizumab as monotherapy for treatment of advanced solid tumors with a high-mutational burden (TMB), defined by a score of 10 mutations per megabase or higher, that have progressed on prior treatment and for which no effective alternatives are available [28]. Mutational burden is often predictive of responses to ICIs, because mutations produce neoantigens that may be targeted by effectors of the immune system and thereby recruit lymphocytes to the tumor. The FDA-approval was considered in the wake of the advancing Phase II KEYNOTE-158 trial (ClinicalTrials.gov: NCT02628067) and a retrospective large-scale whole-exome sequencing analysis, whose 175 mutations/exome criterion is judged equivalent to the established 10 mut/Mb setpoint [28, 29]. Consistent with KEYNOTE-158 [29], patients with a high mutational burden demonstrated greater responses to the treatment than their counterparts [28].
This is also the case for PCa-patients; however, yet again the overall response rate is very low, peaking at 9% versus 6% in the low-TMB cohort, consistent with a study in ipilimumab-treated patients which suggested that TMB is not a singular decisive factor for ICI responses in PCa [30]. The retrospective analysis also suffered from an underrepresentation of PCa cases, with a modest 11 pembrolizumab-treated tissue samples eligible for whole-exome sequencing. A study followed up on this and suggests that pembrolizumab may still be a viable option instead of chemotherapy in mCRPC when the mutational burden is high [31], but additional markers need to be evaluated to maximize treatment benefits against the cost. Note that mismatch repair deficiencies have been suggested reasonable predictors across tumor types [32], and the responses within mismatch repair deficient cohorts appear to be beneficial in PCa [33, 34] although the studies in question are limited by sample size.
The prostate cancer immunophenotype is characterized by low mutational burden and active suppression
The mutational landscape of PCa
The mutational landscape of PCa is multidimensional but rather sparse. Overall, patients present with a low mutation frequency, covering less than 1 mutation per megabase (mut/Mb) in primary disease and up to an average of 4 mut/Mb for advanced metastatic disease, albeit with significant variation between patients [35, 36]. Hypermutated phenotypes with higher frequencies do associate with tumors that have mismatch repair deficiencies (commonly dMMR), most often due to alterations in MSH2, MSH6 and MLH1 genes, yet these make up only a small subset of cancer patients [37]. PCa is therefore significantly different from the archetypical hot cancers such as melanoma and lung cancer that present with a median frequency close to 10 mut/Mb and much higher maximal representation [38]. Instead of a high mutational burden, structural lesions at the chromosomal level are widely recognized in PCa with fusion events being most common and often causing overexpression of oncogenes in the E26 transformation-specific (ETS) transcription factor family downstream of androgen-regulated promoters [35, 39]. The TMPRSS2:ERG fusion alone coincides with around 50% of all PCa cases and makes up 90% of ETS-related fusion events, but other androgen-sensitive fusion partners such as NDRG1 and SLC45A3 are also frequently observed [40].
Other large-scale chromosomal events in PCa involve amplification of regions 8q and Xq and more commonly deletions in regions 8p, 10q, 13q, and 17p; these regions encompass genes that are strongly linked to PCa progression, including the AR gene, the MYC oncogene, and tumor suppressors PTEN and TP53 [35, 41]. Coincidental amplification or deletion involving two or more of these has been linked with aggressive cancer traits in patients and preclinical models [42-45]. The Cancer Genome Atlas (TCGA) Research Network published a paper in 2015 that delineated the molecular taxonomy of primary PCa that resulted in the classification of primary disease into seven main clusters, which are characterized by gene fusions or single gene mutations [36]. According to this classification, ETS-related fusion events and overexpression (ERG, ETV1, ETV4, FLI1) constitute 4 of the 7 clusters, the final 3 defined by coding mutations or copy-number variations in the Speckle-type POZ protein (SPOP), Forkhead box protein A1 (FOXA1), and Isocitrate dehydrogenase 1 (IDH1) loci. The dependency on AR signaling in PCa pathology is accentuated by the fact that the proteins encoded by these genes have been associated with AR protein activity, either by physical protein-protein interactions or by being AR target genes [46-49].
Genetic abnormalities give rise to antigens that can be broadly classified as tumor-associated or tumor-specific antigens (TAA and TSA, respectively), depending on their expression pattern in healthy tissues. Several detailed reviews have been published in recent years that incorporate advances in cancer vaccine strategies with a discussion of antigen selection for maximizing benefit in the clinic [50, 51]. TAAs are autologous but characterized by abnormal expression in cancer versus benign tissue: overexpressed antigens, cell of origin lineage-specific differentiation antigens, and cancer-testis (CT) antigens. The latter group covers a number of potential targets such as melanoma-associated antigen 1 (MAGE-A1), New York esophageal squamous cell carcinoma 1 (NY-ESO-1), and Kita-Kyushu lung cancer antigen 1 (KK-LC-1), that are normally restricted to germline tissue but overexpressed in cancer [51, 52]. PCa is no exception, and CT antigens of the MAGE-A and CSAG protein subfamilies are particularly abundant in advanced PCa [53].
Since the testes constitute an immune privileged site and because germline cells express low to no MHC class I molecules on their surface [51], CT antigens evade interaction with the immune system until their ectopic expression in cancer cells leads to their recognition as foreign. With minimal peripheral tolerance mechanisms at play, such molecules have the potential to elicit strong immune responses despite being self-antigens and have proven strong candidates for cancer vaccines [51]. A major challenge is the elevated risk of inducing autoimmunity, and careful vaccine design involving these antigens is paramount to favor benefit above risk. Recently, the epigenetic reader protein Tudor domain containing 1 (TDRD1) was recognized as central to the biogenesis of small nuclear ribonucleoproteins (snRNPs) in PCa [54]. The germline protein is ectopically expressed in more than half of clinical cases, and its ablation disrupts the cellular snRNP machinery as well as suppressing proliferation in vitro, while also increasing antiandrogen sensitivity. These observations serve as reminders that CT antigens and TAAs in general have inherent value as actionable targets aside from their potential as active vaccine components, adding another layer to their clinical relevance.
TSAs are by definition highly immunogenic because they are recognized as foreign. Such antigens may be derived from viral oncogenes or tumor-specific mutations that generate neoantigens, which are also promising candidates for the active component in therapeutic cancer vaccines [50]. Independent studies have observed increased survival and potentially favorable responses to ICIs in patients with higher estimated neoantigen load and T-cell activation signature in different cancer types [55-57]. Furthermore, recent publications suggest that neoantigen-specific B-cells and CD4+ helper T cells, in particular T follicular helper cell subsets, can strongly promote antitumor immunity by enhancing effector cell function, sparking the possibility for synergy between neoantigen vaccines and ICI treatment [58, 59]. Because its mutational landscape favors a low neoantigen burden, however, the typical PCa presents with a panel of antigens that by themselves may not be optimal inducers of the immune response; furthermore, without a population of high-quality neoantigens on display for effector cells, the tumor can more easily escape immune recognition and destruction. As such, the immunogenicity of mutated gene products, or rather lack thereof, would discourage mass B- and T-cell infiltration and render the PCa tumor immunologically cold.
Characteristics of an immunosuppressive microenvironment
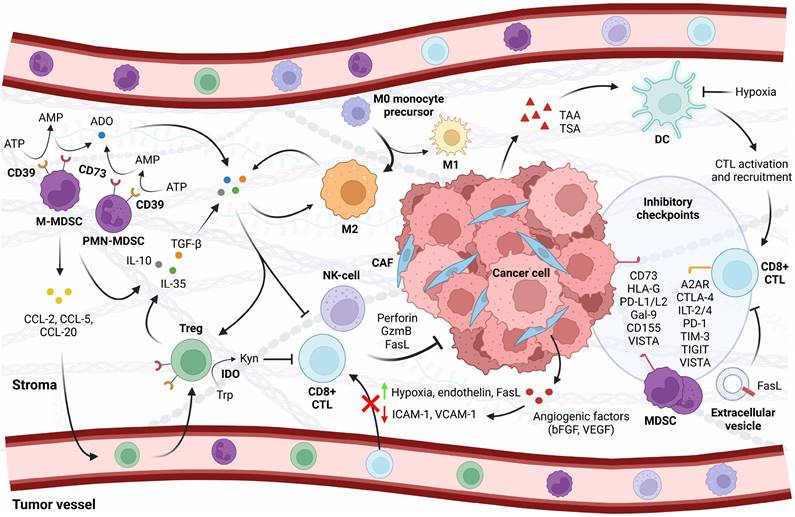
In addition to mutational burden, the immunophenotype of a cancer is subject to active immune suppression through physical cell-to-cell contact and chemical modulation. Indeed, earlier studies in murine models demonstrated that primary tumors quickly establish an immunosuppressive environment, gradually impairing the immune system in its ability to act upon insults over time (e.g. [60]). Such restraints may occur at any point during recruitment and trafficking up to effector cell action, resulting in a population of inactive cells or a scarcity of immune cells altogether. The immune landscape in PCa is generally sparse in tumor-reactive effectors, although it shows significant heterogeneity in infiltration that may yet show prognostic value [61, 62]. The immune cell composition of PCa encompasses a number of cell types including CD8+ cytotoxic T-lymphocytes (CTLs) and B-cells, natural killer (NK) cells, and neutrophils, as well as M1-polarized macrophages that exert pro-inflammatory activities [62, 63]. Importantly, however, PCa tumors and peripheral blood in patients are enriched with cellular subtypes with immunosuppressive gene signatures: anti-inflammatory M2-polarized macrophages, regulatory T cells (Tregs), and myeloid-derived suppressor cells (MDSC) of monocytic or granulocytic origin [64, 65]. These immunosuppressive cell types function by mobilizing immune checkpoints and by secreting immunosuppressive cytokines such as interleukin (IL)-10, IL-35, and transforming growth factor ꞵ (TGF-ꞵ) that inhibit effector cell maturation or function (Figure 1) [66-68].
Characteristics of the PCa TME. Antigen recognition by dendritic cells leads to the activation and recruitment of CD8+ cytotoxic T lymphocytes (CTLs) to the tumor. A modified cytokine milieu discourages CTL recruitment and favors the infiltration of CD4+ regulatory T cells (Tregs), pro-tumorigenic M2-polarized macrophages, and myeloid-derived suppressor cells (MDSCs) that cooperate in maintaining an immunosuppressive environment. Exposure to angiogenic factors from cancer cells and surrounding fibroblasts depletes adhesion molecules in the endothelial lining that are required for CTLs to extravasate; furthermore, cells that successfully infiltrate the stroma are subject to inhibition by hypoxia, local metabolite depletion, and pro-tumorigenic cytokines. Upregulation of Fas ligand in the vasculature and on tumor-derived exosomes initiates apoptosis in CTLs without affecting cells with stronger apoptotic defenses. As a final barrier to antitumor immunity, cancer cells and the immunosuppressive cell types engage inhibitory checkpoints that are currently attractive targets in the clinic. Also shown are natural killer (NK) cells, which are similar to CTLs in their mode of action and subject to similar constraints. A2AR = adenosine A2A receptor, Ado = adenosine, AMP = adenosine monophosphate, ATP = adenosine trisphosphate, bFGF = basic fibroblast growth factor, CAF = cancer-associated fibroblast, CCL-2/-5/-20 = chemokine ligand-2/-5/-20, CD73/-155 = cluster of differentiation 73/-155, CTL = cytotoxic T lymphocyte, CTLA-4 = cytotoxic T-lymphocyte-associated protein 4, DC = dendritic cell, FasL = Fas ligand, Gal-9 = galectin 9, GzmB = granzyme B, HLA-G = human leukocyte antigen G, ICAM-1 = intercellular adhesion molecule 1, IDO = indoleamine 2,3-dioxygenase, IL-10/-35 = interleukin-10/-35, ILT-2/-4 = inhibitory receptors Ig-like transcript 2/-4, Kyn = kynurenine, MDSC = myeloid-derived suppressor cell (M = monocytic, PMN = polymorphonuclear), NK-cell = natural killer cell, PD-1 = programmed death protein 1, PD-L1/-2 = programmed death-ligand 1/-2, TAA = tumor-associated antigen, TGF-β = transforming growth factor β, TIGIT = T-cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine-based inhibitory motif domains, TIM-3 = T cell immunoglobulin and mucin domain-containing protein 3, Treg = regulatory T cell, Trp = tryptophan, TSA = tumor-specific antigen, VCAM-1 = vascular cell adhesion molecule 1, VEGF = vascular endothelial growth factor, VISTA = V-domain Ig suppressor of T cell activation.
A recent single-cell RNA-Seq study on primary PCa identified a deficit in T-cell cytotoxicity score compared with a group of hot tumors, along with an increase in T-cell exhaustion markers relative to the healthy prostate [63]. This coincided with an increased Treg activity score that correlated with a monocytic MDSC-like signature and reciprocal expression of chemokine receptor CCR6 with its cognate ligand CCL20, suggesting that they cooperate to maintain a suppressive TME. Although this particular study did not explicitly evaluate of CD8+ T cell density, another study found that the median density for PCa patients (51 cells/mm2) is dramatically lower than reported in its melanoma counterpart (approx. 2500 cells/mm2 in anti-PD-1 responders) [69-71].
In contrast, a paper published in 2020 addressed the CD8+ infiltration in 84 different tumor types and detected a much smaller difference in median density between PCa and melanoma, yet the range was greater for melanoma which peaked at a maximum of about four times higher than PCa with its 499 cells/mm2 [72]. Despite the limitation of small sample sizes, such data demonstrate that the cold characteristics of PCa are manifested in a functional loss together with quantitative deficiency of CTLs; the prognostic value of intraprostatic CD8+ T cells is therefore actively being investigated. This is a rather complex task, however, as some studies associate high numbers of CTLs with increased time to biochemical recurrence and overall survival after surgery [70, 73], while others find a shorter time to cancer progression with high cell density [74, 75], suggesting a context dependency the basis of which is currently unclear.
In one of these studies, the negative prognosis for CD8+ cell density was dependent on high expression in the adjacent epithelium of CD73, an immune checkpoint that acts upon the adenosinergic pathway to suppress antitumor responses (Figure 1) [75]. Coincident with low CD73, the association between CD8+ density and time to recurrence was therefore not significant. These findings suggested that CD73 may mechanistically aid the conversion of CTLs to non-conventional immunosuppressive CD8+CD25+ subtypes, which have previously been described in PCa [75, 76]. Similar to CD4+ Tregs, these subpopulations are capable of suppressing effector T-cell function by contact-dependent and -independent mechanisms; however, the mechanisms are not completely elucidated and current evidence challenges the relevance of IL-10 and TGF-β that are well characterized products of conventional Tregs [76, 77]. It is therefore important to recognize that the CD8+ T-cell compartment is more dynamic when evaluating its clinical implications. Current data are more consistent with regards to the prognostic value of Tregs [78] and MDSCs [79], whose overrepresentation is associated with worse prognosis for PCa patients.
Activated T cells upregulate receptors including CXCR3 and CCR5 to recognize chemokine ligands in the TME, which then aid their functional maturation and navigation into the site of insult [80]. Elimination of these cytokines by proteolytic cleavage [81], transcriptional repression [82], or epigenetic silencing [83, 84] is a means by which tumors can impair infiltration; by precise fine-tuning of the cytokine milieu the PCa cells tip the scales in favor of immunosuppressive rather than antitumor effector cells [85-87]. Ectopic expression of components in the Polycomb repressive complex 1 (PRC1) promoted self-renewal and metastasis in double-negative mCRPC (DNPC), while concurrently recruiting Tregs, tumor associated macrophages (TAMs), and MDSCs, mainly by production of the cytokine CCL2 [88]. Inhibition of PRC1 suppressed recruitment of immunoinhibitory cell types and dramatically increased the efficacy of double checkpoint immunotherapy (anti-CTLA-4 + anti-PD-1) in murine models for DNPC, indicated by the significant decline in tumor burden as well as emergence of CTLs and CD4+ effectors [88]. These findings suggest that PRC1 could serve as a rational target for PCa, and also underscores the potential for combination therapies to improve clinical outcomes with ICIs in PCa, as discussed below.
Infiltration of immune cells into the TME can be hindered at the endothelial level by modifying expression of adhesion proteins such as intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1), which are necessary for leukocyte trafficking and extravasation [89]. Consistently, prolonged exposure to angiogenic factors such as vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF) effectively reduced the inflammatory upregulation of these molecules in the endothelial lining, thereby preventing T-cell adhesion and transendothelial migration (Figure 1) [90]; other studies suggested that angiogenic factors cause clustering defects with similar outcome [91]. This effect has been denoted tumor endothelial cell anergy due to the emerging insensitivity to inflammatory cytokines. Anti-angiogenic treatment counters this suppression and enhances infiltration [92]. The angiogenic signature is suggested to be prognostic in PCa and suggests potential targets of combinational benefit [63, 93].
Furthermore, the endothelial compartment appears to have a profound selectivity between infiltrating immune cells. The common lymphatic endothelial and vascular endothelial receptor (CLEVER-1) is upregulated in hepatocellular carcinoma with a preference for Tregs more than other T-cell subsets [94]. Endothelin B receptor overexpression has been associated with lower CTL recruitment but larger Treg populations in gliomas [95], while a receptor antagonist increases overall T-cell infiltration in preclinical models of ovarian cancer [96]. There is sparse knowledge on the immunological impact of vascular endothelin receptors in PCa, but endothelin ligands are increased in patients and found to aid in tumor growth and metastasis [97], suggesting that a similar mechanism may be involved. Interestingly, tumor-derived cytokines have been found to upregulate Fas ligand in the surrounding vasculature of tumors including PCa, selectively engaging the extrinsic pathway of apoptosis in CD8+ T cells but not in Tregs, which have stronger anti-apoptotic barriers (Figure 1) [98]. Studies on PCa cell lines have also reported the release of Fas ligand in soluble form [99] or as part of tumor-derived exosomes that induce cell death in CD8+ lymphocytes [100].
Immune cells that successfully extravasate are rendered inactive by a hostile microenvironment. The tumor vasculature is abnormal with an endothelial lining that is loosely connected, highly irregular, and under pressure that causes individual vessels to collapse [101]. The result is an oxygen deficit accompanied by acidosis which inhibits functional CTL maturation [102]. Activation of hypoxia-inducible factor 1 alpha (HIF-1ɑ) engages a transcriptional program that impairs dendritic cell activation and CTL function [103, 104], and leads to recruitment and activation of Tregs and MDSCs as well as polarization of immunosuppressive M2 macrophages in a number of cancer types [105-107]. Such hypoxic zones are prevalent in clinical and preclinical PCa and behave as immune-privileged sites that may be targeted for therapeutic benefit [108]. Administration of the hypoxia-activated prodrug evofosfamide leads to a collapse of these areas in the TRAMP-C2 PCa mouse model and improves T-cell infiltration at the expense of immunosuppressive cell types [108]. Hypoxia reduction sensitizes the tumors to CTLA-4/PD-1 checkpoint blockade as demonstrated by a robust increase in CD8+ T-cell effector function and tumor rejection.
Protumorigenic cytokine profile and hypoxia stimulate immune checkpoints including CTLA-4, TIM-3, lymphocyte activation gene-3, and VISTA on T cells or other components of the TME [109, 110], which may directly inhibit CTL function [109, 111, 112] or stimulate suppressor cell activity (Figure 1) [110]. PD-1 has been identified on the surface of PCa-infiltrating CD8+ T cells [113], its ligands PD-L1 and PD-L2 are frequently overexpressed in the prostate TME [114, 115]; in addition, the compensatory upregulation of VISTA in PCa that is treated with ipilimumab suggests an emergent role of this checkpoint that warrants further investigation [116].
Although current immunotherapies are mainly directed at tumor-specific T cells, an increasing body of evidence points towards innate NK cells as important contributors to tumor immunity, and means of harnessing their potential is a point of interest [117]. NK cell infiltration and activity has been associated with PCa stages and prognosis [118, 119], emphasizing their relevance. Similar to their adaptive counterparts, NK cells are subject to immune checkpoint activity; consistently, Pasero et al. (2016) observed a shift in surface profile of activating receptors (e.g. NKp46 and NKG2D) that were underrepresented and inhibitory receptors (e.g. ILT2 and NKG2A) were overexpressed in tumor-infiltrating NK cells of PCa patients [119]. This coincided with an immature phenotype and impaired NK cell function. This was supported by another study where an altered phenotype in the circulating fraction of NK cells in patients was observed, including NKG2D downregulation and upregulation of exhaustion markers TIM-3 and PD-1, again suggestive of an impaired NK cell state [120]. Taken together with reports that PCa cells actively employ mechanisms to evade recognition and NK cell-mediated killing [121, 122], these observations suggest that remobilizing the NK cell compartment to attack the evasive tumor may be a fruitful therapeutic approach.
Potentiating immune checkpoint inhibitors
Combining multiple checkpoint inhibitors and other immune-targeting agents
With the fluctuating success of ICI monotherapies, efforts have been ongoing to assess the clinical effects of combinatorial treatment. Following its approval for BRAF V600 wild-type advanced melanoma in 2015, the combination of nivolumab and ipilimumab has expanded its range to other cancer types including hepatocellular carcinoma, advanced renal cell carcinoma, and NSCLC for patient subgroups [8]. On several occasions nivolumab plus ipilimumab has been superior to either monotherapy, and it is suggested that combination treatment may achieve synergy due to differential mechanism of action for CTLA-4 and PD-1 blockade that integrate for a unique outcome [123, 124]. Taking into account the compensatory upregulation of PD-L1 that has been demonstrated in PCa patients treated with ipilimumab [116], the ongoing CheckMate 650 trial is evaluating whether the dual targeting of PD-1 and CTLA-4 may improve the clinical performance in patients with mCRPC (ClinicalTrials.gov: NCT02985957) [125]. Initially, 90 patients were evenly divided into chemotherapy naïve and post-chemotherapy cohorts and treated with 1 mg/kg nivolumab and 3 mg/kg ipilimumab every three weeks for up to four doses, followed by 480 mg nivolumab every four weeks until cancer progression or threshold toxicity. Preliminary data from the trial inspired a carefully optimistic point of view, with a small number of patients achieving complete responses and objective response rates of 25% and 10% for pre-chemotherapy and post-chemotherapy patients, respectively [125].
Although caution must be exercised when comparing studies of different design, the survival benefits for the pre-chemotherapy population was higher than for either treatment alone [125]. The median 15.2-month OS is notably better than the 11.2 months for ipilimumab alone [21]; with its objective response rate, it also improved on the outcome of nivolumab monotherapy [25]. Because of high discontinuation rates during combination dosing or monotherapy maintenance, however, the current Phase II study was expanded to cover dose and schedule modifications. 259 post-chemotherapy patients were randomly assigned 2:2:1:2 to four cohorts, with cohort D1 and D2 receiving a modified combination treatment [126]. Patients in D1 received 3 mg/kg nivolumab plus 1 mg/kg ipilimumab every three weeks up to four doses, whereas D2 received 1 mg/kg nivolumab every three weeks up to eight doses and 3 mg/kg ipilimumab every six weeks up to four doses. Both were followed up with 480 mg nivolumab every four weeks.
So far, 15% and 26% of patients have discontinued due to toxicity in the two cohorts, which represents a slight improvement over withdrawal in the first report (36% in the post-chemotherapy cohort) [125, 126]. Preliminary analyses report 9% and 15% objective response rate for cohorts D1 and D2, along with a median OS of 15.9 and 13.5 months, respectively. Interestingly, the chemotherapy arm (D4) was associated with lower discontinuation rates and greater PSA responses than each of the combination cohorts [126]. 24% of patients with baseline measurable disease achieved a >50% PSA decline in cohort D4, compared to 14% and 18% in cohorts D1 and D2. Given a 14.8 month median OS, this raises a question as to when the trade-off between efficacy and toxicity makes the checkpoint combination more beneficial.
The upregulation of VISTA that emerged in PCa patients treated with ipilimumab raises a question of how it fits in with the timeline in cancer development [116]: does it emerge as a backup checkpoint when other checkpoints fail to protect the tumor, or does it have other functions that directly fuel cancer growth and metastasis? Could VISTA be a positive modulator of antitumor immunity that aims to counter a suppressive environment as suggested in other cancers [127]? These questions are still open, but a recent study found a significant increase in VISTA-expression on circulating CD8+ T cells in PCa patients undergoing stereotactic body radiation therapy (SBRT) [128], further supporting a role for the checkpoint in PCa that warrants further investigation. With this background, a VISTA-targeting antibody, KVA12123, alone or in combination with pembrolizumab, has entered the recruitment stage for a Phase I/II study in patients with refractory or relapsed advanced solid tumors (VISTA-101, NCT05708950). Other immune checkpoints are gaining more interest as potential targets in recent years, including the adenosinergic CD73-axis [75, 129] and the non-classical Human leukocyte antigen (HLA)-G [119, 130, 131], both of which are being investigated in clinical trials for PCa (see Table 1).
Ongoing or recently completed clinical trials with ICI combinations in PCa. Target group is specific for mCRPC unless otherwise specified.
| Clinical trial ID | Common name | Combination1 | Phase | Status | Enrollment2/study type | Start | Est. study completion |
|---|---|---|---|---|---|---|---|
| NCT02985957 | CheckMate 650 | Ipilimumab (CTLA-4) + Nivolumab (PD-1) | 2 | Active, not recruiting | 351 Randomized, Open Label | March, 2017 | January, 2025 |
| NCT05708950* | VISTA-101 | KVA12123 (VISTA) + Pembrolizumab (PD-1) | 1/2 | Recruiting | 314 Randomized, Open Label | March, 2023 | December, 2024 |
| NCT02740985* | REFMAL 435 | AZD4635 (A2AR) + Durvalumab (PD-L1) | 1 | Completed | 313 (45) Non-randomized, Open Label | June, 2016 | March, 2023 |
| NCT04089553 | - | AZD4635 (A2AR) + Durvalumab (PD-L1); AZD4635 + Oleclumab (CD73) | 2 | Completed | 59 Randomized, Open Label | August, 2019 | April, 2023 |
| NCT02788773 | - | Durvalumab (PD-L1) + Tremelimumab (CTLA-4) | 2 | Active, not recruiting | 52 (39) Randomized, Open Label | August, 2016 | June, 2024 |
| NCT02465060* | NCI MATCH Molecular Analysis for Therapy Choice | Nivolumab (PD-1) + Relatlimab (LAG-3) | 2 | Active, not recruiting | 6452 Non-randomized, Open Label Genetic screening based (main purpose) | August 2015 | December, 2025 |
| NCT03333616** | - | Ipilimumab (CTLA-4) + Nivolumab (PD-1) | 2 | Active, not recruiting | 100 (5) Single Group, Open Label | December, 2017 | May, 2025 |
| NCT03651271* | AMADEUS | Ipilimumab (CTLA-4) + Nivolumab (PD-1) | 2 | Completed | 100 Non-randomized, Open Label Stratified by CD8+ density | October, 2018 | June, 2023 |
| NCT04717154 | INSPIRE | Ipilimumab (CTLA-4) + Nivolumab (PD-1) | 2 | Active, not recruiting | 69 Single Group, Open Label Patients with immunogenic signature by sequencing | January, 2021 | February, 2026 |
| NCT03061539 | NEPTUNES | Ipilimumab (CTLA-4) + Nivolumab (PD-1) | 2 | Active, not recruiting | 380 Non-randomized, Open Label Patients with immunogenic signature by IHC and sequencing | February, 2018 | June, 2027 |
| NCT03454451* | - | CPI-006 (CD73) + Ciforadenant (A2AR); CPI-006 (CD73) + Pembrolizumab (PD-1) | 1/1b | Completed | 117 Randomized, Open Label | April 2018 | February, 2023 |
| NCT04485013* PCa is currently reserved as keyword for the study, but not yet assigned to treatment arms. | - | Pembrolizumab (PD-1) + TTX-080 (HLA-G) | 1a/1b | Active, not recruiting | 240 Non-randomized, Open Label | July, 2020 | June, 2024 |
| NCT02861573 Arms G and H: mCRPC and treatment-emergent neuroendocrine mCRPC. | KEYNOTE 365 | Pembrolizumab (PD-1) + Vibostolimab (TIGIT) | 1b/2 | Recruiting | 1200 Non-randomized, Open Label | November, 2016 | October, 2027 |
A2AR = adenosine A2A receptor, CD73 = cluster of differentiation 73, CTLA-4 = cytotoxic T-lymphocyte-associated protein 4, HLA-G = human leukocyte antigen-G, LAG-3 = lymphocyte activation gene-3, PD-1 = programmed cell death protein 1, PD-L1 = programmed death-ligand 1, TIGIT = T cell immunoreceptor with immunoglobulin and ITIM domain, VISTA = V-domain Ig suppressor of T cell activation.
1Combination may be one of several treatment arms in a larger study. Parentheses indicate the targeted checkpoint.
2Parentheses indicate the number of PCa patients included in a treatment arm for a multicancer study, or the number of patients receiving the specified treatment in a PCa-specific study with multiple arms, where this information is available.
*Advanced solid malignancies and metastatic cancers (including but not exclusive to mCRPC).
**Rare genitourinary tumors (includes rare and aggressive PCa subtypes).
Advances in technology allow for more precise targeting of anticancer agents for greater therapeutic benefit; particularly the development of bispecific antibodies (bsAbs) is bringing the field of immunotherapy a step forward [132]. Unlike the aforementioned antibodies which are designed to bind a specific epitope, bsAbs are engineered with binding sites that have different specificities and thereby coordinate separate functions including blocking [133], drug delivery [134], and T-cell engagement to TAAs [135]. T-cell engaging bsAbs targeting prostate-specific membrane antigen (PSMA) [136], delta-like protein 3 (DLL3) [137], and HER2 [138] are showing promise in preclinical and early clinical trials for PCa, as is a trispecific PSMA-targeting T-cell engager that is derived from the half-life extended TriTAC platform [139]. Even bsAbs targeting costimulatory molecules are demonstrating clinical activity concurrent with checkpoint blockers [140].
Data on double checkpoint targeting bsAbs in PCa are currently sparse, but constructs that simultaneously interfere with the PD-1 and LAG-3 or CTLA-4 axes have demonstrated activity in preclinical models [141]; consistently, a PD-1/CTLA-4 bsAb performed well in a Phase I trial that included mCRPC patients [142]. 13% of evaluable patients achieved objective responses, including two CRPC patients with confirmed PSA responses. This bsAb called vudalimab (XmAb®20717) has now advanced into two Phase II studies that encompass mCRPC, one of which evaluates the antibody alone or in combination with chemotherapy or olaparib in 5 molecular subtypes (NCT05005728) [143, 144]. Similarly another PD-1/CTLA-4 bsAb derivative is in a Phase I study for mCRPC [145]. Once the mechanistic role of VISTA in PCa is more fully understood, dual targeting with VISTA/CTLA-4 or VISTA/PD-1 bsAb may also prove beneficial in the clinic.
Because the immunosuppressive TME is more than the sum of its checkpoints, an alternative strategy would be to target both checkpoints and cytokines that protect the tumor, such as TGF-ꞵ, with a single agent [146]. Bintrafusp alfa (M7824) is a bifunctional protein that fuses the extracellular domains of the TGF-ꞵ receptor II to the C-terminal end of a humanized anti-PD-L1 heavy chain; it has demonstrated potent activity in preclinical models along with manageable safety profiles in heavily pretreated cancer patients [147, 148]. Independent studies have shown that M7824 depletes soluble TGF-ꞵ and increases T-cell trafficking into tumor sites along with antigen-specific CD8+ T-cell mediated cytolytic activity, but it also enforces changes in the microenvironment in favor of the immune system to potentially increase the efficacy of cancer vaccines [148, 149]. With this rationale, a Phase II study is currently evaluating a triple-attack in patients with biochemically recurrent PCa (NCT03315871). The triple treatment comprises the viral vector based PROSTVAC-V/F regimen [150] in combination with a CV301 prime-boost system of viruses expressing TAAs carcinoembryonic antigen (CEA) and mucin-1 (MUC-1) plus TRICOM [triad of costimulatory molecules (B7.1, ICAM-1, and lymphocyte function-associated antigen 3 (LFA-3)], and bintrafusp alfa. The combination of checkpoint blockers and antitumor vaccination holds significant potential deserving of further research (see Table 2) [59, 151].
Ongoing or recently completed Phase II clinical trials with vaccination in combination with checkpoint inhibition or bifunctional checkpoint targeting agents that have previously been described. Target group is specific for mCRPC unless otherwise specified.
| Clinical trial ID | Common name | Combination1 | Phase | Status | Enrollment/ study type | Start | Est. study completion |
|---|---|---|---|---|---|---|---|
| NCT03315871 BCR PCa | - | PROSTVAC-V/F (viral prime-boost vaccine presenting PSA and TRICOM) + M7824 (PD-L1, TGF-ꞵ trap) + CV301 (viral prime-boost vaccine presenting CEA, MUC-1, and TRICOM) | 2 | Active, not recruiting | 40 Non-randomized, Open Label | March, 2018 | January, 2025 |
| NCT03493945* | QuEST1 | BN-Brachyury (viral prime-boost vaccine presenting brachyury transcription factor and TRICOM) + M7824 (PD-L1, TGF-ꞵ trap); BN-Brachyury + M7824 + N-803 (IL-15/IL-15R alpha superagonist complex); BN-Brachyury + M7824 + N-803 + Epacadostat (IDO1 inh.) | 1/2 | Active, not recruiting | 53 Randomized, Open Label | May, 2018 | December, 2024 |
| NCT03600350 Non-metastatic BCR PCa | - | pTVG-HP (plasmid DNA vaccine encoding PAP) + Nivolumab (PD-1) + GM-CSF (APC growth factor) | 2 | Active, not recruiting | 19 Single Group, Open Label | September, 2018 | December, 2027 |
| NCT02933255 mCRPC and localized advanced PCa | - | PROSTVAC-V/F (viral prime-boost vaccine presenting PSA and TRICOM) + Nivolumab (PD-1) | 1/2 | Completed | 24 Non-randomized, Open Label | April, 2018 | December, 2023 |
| NCT04989946 Newly diagnosed, high risk PCa | - | Degarelix (GnRH antagonist) + pTVG-AR (plasmid DNA vaccine encoding AR ligand-binding domain) + Nivolumab (PD-1) | 1/2 | Recruiting | 60 Randomized, Open Label | December, 2021 | December, 2028 |
| NCT02499835 | - | pTVG-HP (plasmid DNA vaccine encoding PAP) + Pembrolizumab (PD-1) | 1/2 | Completed | 66 Randomized, Open Label | July, 2015 | July, 2023 |
| NCT04090528 | - | pTVG-HP (plasmid DNA vaccine encoding PAP) + Pembrolizumab (PD-1); pTVG-HP + pTVG-AR (plasmid DNA vaccine encoding AR ligand-binding domain) + Pembrolizumab | 2 | Active, not recruiting | 60 Randomized, Open Label | October, 2019 | October, 2026 |
AR = androgen receptor, BCR PCa = Biochemically recurrent prostate cancer, GnRH = gonadotropin-releasing hormone, HOXB13 = homeobox B13, IDO1 = indoleamine 2,3-dioxygenase 1, IL-15/IL-15R = interleukin-15/IL-15 receptor, KLK2/3 = kallikrein related peptidase 2/3, M7824 = bintrafusp alfa (anti-PD-L1 + TGF-ꞵ receptor II ligand binding domain), MMAE = monomethyl auristatin E, NK3 homeobox 1, PAP = prostatic acid phosphatase, RNA-LPX = RNA lipoplex, TRICOM = triad of costimulatory molecules (B7.1, intercellular adhesion molecule 1 (ICAM-1), and lymphocyte function-associated antigen 3 (LFA-3)), PD-1 = programmed cell death protein 1, PD-L1 = programmed death-ligand 1.
1Combination may be one of several treatment arms in a larger study. Parentheses indicate the targeted checkpoint or the properties of the treatment.
*Advanced solid malignancies and metastatic cancers (including but not exclusive to mCRPC).
Combining checkpoint inhibitors with standard of care treatment and targeted therapy
Conventional chemotherapeutics have been found to promote an immunomodulatory effect that exceeds direct tumor cell cytotoxicity [152]. This includes the induction of immunogenic cell death within tumors [152] and direct activation of effector cells [153]. Docetaxel, a standard line of treatment for CRPC, was found to promote the differentiation of antitumorigenic M1 macrophages in vitro and enhance CD8+ T-cell effector function in murine models for colon cancer [154]; however, CRPC seems to adapt to docetaxel-induced damage by activating pro-tumorigenic M2 macrophages [155]. This is a potential mechanism for chemotherapy resistance in late-stage PCa that constitutes a significant hurdle. Paradoxically, docetaxel-based therapy has proven clinically favorable in remodeling the TME, with paired pre- and post-therapy tumor samples showing statistically significant increases in CD8+ T-cell infiltration for patients with locally advanced PCa [153]. Taken together, the immunological effects of such treatments have very important implications that have previously been alluded to: Effects of immunotherapy may be improved with established treatment strategies that concurrently remodel the tumor immune landscape. This has been suggested by a study that combined sipuleucel-T with ipilimumab in mCRPC [156] and could be a new paradigm for clinical trials that investigate the combinatorial potential of ICIs, such as the Phase II CheckMate 9KD Trial that investigates the efficacy of nivolumab in combination with either docetaxel [157], the PARP-inhibitor rucaparib [158], or enzalutamide in mCRPC patients (NCT03338790).
Studies have shown that the success of ADT in early metastatic disease is associated with a remodeling of the immune infiltrate that could render the cancer susceptible to ICIs [159], but the window of opportunity to progression and therapy resistance could be extremely narrow. This raises the question as to how checkpoint blockade fits within the treatment timeline for PCa patients, whether early or treatment-naïve patients would achieve more durable responses than late-stage CRPC patients, and trials are ongoing on different ends of the spectrum. As an example, the Phase II PEAPOD_FOS trial is assessing the efficacy of PD-1 blockade in combination with cabazitaxel and carboplatin for patients with aggressive variant mCRPC (NCT05563558), while another Phase II trial is evaluating the combination of PD-1 blockade with ADT and docetaxel in newly metastatic hormone-sensitive PCa (mHSPC, NCT03951831). It is possible that studies would benefit from directly comparing responses at different portions of the treatment spectrum, but patients with mCRPC that have progressed on prior treatments with limited options are more likely to enroll in these studies than patients that have yet to undergo treatment. The PROSTRATEGY trial is assessing whether a double ICI approach could provide significant survival benefit with simultaneous chemotherapy and ADT when introduced to patients with mHSPC (NCT03879122). Other ongoing trials with dual checkpoint inhibition and standard of care treatment modalities are presented in Table 3.
Ongoing or recently completed Phase II and Phase III clinical trials with dual checkpoint inhibition in combination with targeted therapy or standard of care treatment in PCa. Target group is specific for mCRPC unless otherwise specified.
| Clinical trial ID | Common name | Combination1 | Phase | Status | Enrollment/study type | Start | Est. study completion |
|---|---|---|---|---|---|---|---|
| NCT05169684 | - | BMS-986218 (CTLA-4) + Nivolumab (PD-1) + Docetaxel (microtubule inh.) | 2 | Completed | 10 Randomized, Open Label | February, 2022 | December, 2023 |
| NCT03866382** | ICONIC | Cabozantinib (tyrosine kinase inh.) + Ipilimumab (CTLA-4) + Nivolumab (PD-1) | 2 | Recruiting | 314 Single Group, Open Label | April, 2019 | February, 2025 |
| NCT05150236 | EVOLUTION (ANZUP2001) | 177Lu-PSMA-617 (radioligand) + Ipilimumab (CTLA-4) + Nivolumab (PD-1) | 2 | Active, not recruiting | 93 Randomized, Open Label | April, 2022 | December, 2024 |
| NCT05655715 | CheckPRO | SBRT + Ipilimumab (CTLA-4) + Nivolumab (PD-1) | 2 | Recruiting | 90 Randomized, Open Label | November, 2019 | January, 2025 |
| NCT04709276 Neuroendocrine or aggressive variant PCa | CHAMP | Cabazitaxel (microtubule inh.) + Carboplatin (alkylating) + Ipilimumab (CTLA-4) + Nivolumab (PD-1) | 2 | Active, not recruiting | 43 Single Group, Open Label | June, 2021 | June, 2027 |
| NCT03879122 Metastatic hormone-sensitive PCa | PROSTRATEGY | ADT + Docetaxel (microtubule inh.) + Ipilimumab (CTLA-4) + Nivolumab (PD-1) | 2/3 | Active, not recruiting | 135 Randomized controlled, Open Label | February, 2019 | December, 2024 |
| NCT04169841* | GUIDE2REPAIR | Durvalumab (PD-L1) + Olaparib (PARP inh.) Tremelimumab (CTLA-4) | 2 | Active, not recruiting | 270 Single Group, Open Label Molecular screening for mutations in homologous repair genes | February, 2020 | August, 2027 |
| NCT03518606* | MOVIE | Durvalumab (PD-L1) + Tremelimumab (CTLA-4) + Vinorelbine (microtubule inh.) | 1/2 | Active, not recruiting | 126 Non-randomized, Open Label | June, 2018 | December, 2024 |
177Lu-PSMA-617 = Lutetium radionuclide conjugated with a prostate specific membrane antigen (PSMA) targeting ligand, ADT = androgen deprivation therapy, CTLA-4 = cytotoxic T-lymphocyte-associated protein 4, PARP = poly ADP-ribose polymerase, PD-1 = programmed cell death protein 1, PD-L1 = programmed death-ligand 1, SBRT = stereotactic body radiation therapy.
1Combination may be one of several treatment arms in a larger study. Parentheses indicate the targeted checkpoint or the properties of the treatment.
*Advanced solid malignancies and metastatic cancers (including but not exclusive to mCRPC).
**Rare genitourinary tumors (includes rare and aggressive PCa subtypes).
Radiotherapy is a mainstay treatment for PCa patients with progressive localized disease and as salvage treatment for biochemically recurrence after radical prostatectomy [4]. Due to the mutagenic and cytotoxic nature of targeted radiation, it is capable of mobilizing immune responses against the primary tumor and even tumors away from the irradiation site, an effect that may potentially improve ICI responses in the clinic [160, 161]. Efforts are therefore ongoing to investigate the efficacy of radiotherapy in combination with PD-1/PD-L1 blockers, e.g. as first-line treatment with atezolizumab (Tecentriq) and ADT (NCT04262154), and as salvage treatment with pembrolizumab (Pembro-SRT, NCT04931979). Preliminary results from a study combining nivolumab with brachytherapy and external beam radiotherapy (EBRT) in high-grade PCa are encouraging, and have therefore advanced into a Phase II trial (NCT03543189) [162]. In addition to targeted radiation, systemic radiotherapy using radioactive isotopes has been used in the clinic for advanced PCa, with the FDA approvals of Radium-233 for bone metastatic CRPC and the radioligand 177Lu-PSMA-617 for PSMA-positive mCRPC [6, 163]. Combination treatments with these systemic radiotherapies and ICIs are currently being explored, and alternative radiation sources like an Actinium-225 construct targeting the novel PCa marker CD46 may emerge as promising candidates in the future [164].
Notably, the observation that Radium-233 alters the expression pattern of PD-1 in infiltrating CD8+ T-cell subsets makes it a candidate for combination therapy [165]. However a Phase II study combining Radium-233 with pembrolizumab failed to increase immune infiltration into bone metastases and did not affect the secondary outcomes of OS and PFS [166]. Another trial tested a combination of Radium-233 with atezolizumab and found increased toxicity compared to each treatment alone without any survival benefit for mCRPC patients [167]. Other trials are ongoing, including a randomized Phase I/II study that combines the PD-L1 blocker avelumab with Radium-233 and M3814, an inhibitor of double-stranded break repair enzymes, with the hypothesis that this may enhance direct tumor killing and immune mobilization (NCT04071236). 177Lu-PSMA-617 was approved more recently and is therefore not as extensively evaluated in combination treatments, but it has shown a favorable safety profile and indications of activity in combination with pembrolizumab [168]; it is currently being tested with dual checkpoint inhibition in the Phase II EVOLUTION trial (see Table 3). In vivo studies and a patient case suggest increased efficacy of the radioligand when preceded by EBRT, which could add another layer of control with checkpoint blockers in management of PCa [169].
Along with the traditional strategies introduced above, targeted therapies are becoming increasingly attractive in PCa treatment. Given the central role of AR signaling in the growth and maintenance of PCa, AR itself is emerging as a promising target for ICI combination therapies [170]. Two Phase III studies assessed the combination of pembrolizumab with enzalutamide vs placebo and enzalutamide in mCRPC (KEYNOTE-641) and mHSPC (KEYNOTE-991), both of which were recently discontinued due to lack of efficacy [171, 172]. A similar trial assessed PD-L1 blockade with atezolizumab and enzalutamide, which also failed to achieve primary endpoints, but exploratory analyses associated better responses with CD8+ infiltration, TMB, PD-L1 expression, and phosphatase and tensin homolog (PTEN) loss, all of which have been alluded to previously [173]. Rather than discouraging further studies into the combination of NHT and ICIs, these results should encourage efforts into understanding mechanisms of treatment resistance in greater detail and biomarkers that can prospectively predict treatment responses. Such is the rationale for ongoing biomarker-selected studies like the Phase II GUNS study, where patients with hypermutated phenotypes, microsatellite instability, Lynch syndrome or CDK12 alterations are assigned to a cohort receiving ADT with NHT and PD-L1 blockade (NCT04812366).
The only PCa approved targeted treatment, aside from conventional AR-targeting agents, involves the PARP-inhibitors olaparib and rucaparib. They were approved by the FDA for use in DNA damage repair deficient mCRPC that has progressed on NHT, and rucaparib has docetaxel treatment as a final prerequisite [8]. The Phase Ib/II KEYNOTE-365 trial had a treatment arm with pembrolizumab and olaparib in chemotherapy-experienced mCRPC patients, and observed an acceptable safety profile with indications of clinical activity in molecularly unselected disease with an 8.5% objective response and 14-month median OS [174]. Olaparib is also showing promise in concert with the PD-L1 blocker durvalumab (Imfinzi) especially in DNA damage repair deficient mCRPC [175], in line with the synthetic lethal activity of PARP-inhibitors in the clinic.
Rucaparib demonstrated tolerable safety and encouraging activity in combination with PD-1 blockade in the CheckMate 9KD trial, where objective responses were 10.3% and 15.4% for post-chemotherapy and naïve cohorts, respectively, and OS was 13.9 and 20.2 months [158]. Responses were greater in patients with homologous repair deficiencies, especially in BRCA1/2-mutated subpopulations where 33.3% objective responses were observed in each cohort. When the combination advanced to the KEYLYNK-10 Phase III trial against enzalutamide and abiraterone, however, primary endpoints were not met and the trial was stopped [176]. Despite this setback and limitations in study design that disregard ICI monotherapy, the results presented outside of the trial favor further investigation into the PARP-inhibitor/ICI combination regimen, especially where patients are expected to harbor mutations that increase neoantigen load (see NCT04336943).
Although other targeted agents have not performed well as monotherapies, a number of candidates are being tested for potential benefit when combined with checkpoint blockers in PCa. Targets range from members of dysregulated pathways such as AKT (NCT03673787), receptor tyrosine kinases (RTKs) (NCT04848337), and cyclin-dependent kinases (CDKs) (NCT04751929), to epigenetic readers that support the function of transcriptional master regulators which in turn fuel oncogenic networks (NCT04471974) [177]. The RTK inhibitor cabozantinib demonstrated manageable safety and minor clinical activity when combined with atezolizumab in the mCRPC cohort of the ongoing Phase Ib COSMIC-021 trial, with a 32% ORR and a confirmed PSA response in 50% of evaluable patients [178]. This outcome laid the foundation for the Phase III CONTACT-02 trial, where mCRPC patients failing a single NHT were randomized to receive cabozantinib and atezolizumab or a second cycle of NHT (NCT04446117).
Preliminary results from the trial have shown a significant increase in median PFS with 6.3 vs 4.2 months and a median OS of 16.7 vs 14.6 months in the control group, but the study has sparked debate due to the modest values and questionable study design [179, 180]. The primary concern is the inclusion of a control regimen that delivers a second NHT, which is not considered the best standard of care treatment for the study group when taxane chemotherapy exists as a more viable treatment option [180]. With a median follow-up of 12 months, the reported OS is immature and unfit for making justified claims about the benefits of treatment. The study may yet show promise, but the final report will need to present more robust data for it to have significant implications.
Enhancing the effectiveness of checkpoint inhibition by novel combination strategies in pre-clinical models of PCa
As reviewed above, since clinical trials for ICI monotherapy in PCa have largely been unsuccessful so far, current emphasis is on combination therapies with agents that are already in the clinic. As we wait for the decisive outcome of these trials, it is important to explore additional combinatorial approaches based on the information that has accumulated on the biology of PCa over the years, which has to be supported by robust findings in preclinical models. This has in fact been ongoing for some time and below is an overview of such preclinical studies that may find translational applications in the future (Table 4).
ICI combination treatment findings in PCa pre-clinical models.
| Combination | Pre-clinical PCa model | Reference |
|---|---|---|
| Cabozantinib + CTLA-4 + PD-1 or BEZ235 + CTLA-4 + PD-1 | PB-Cre+; PtenL/L p53L/L Smad4L/L mTmGL/+ LSL-LUCL/+ (CPPSML) transgenic model | [181] |
| Uric acid + CTLA-4 + PD-1 | PB-Cre+; PtenL/L p53L/L Smad4L/L (Pten/p53/Smad4-deficient) transgenic model | [182] |
| YY001 + PD-1 | RM-1 cells subcutaneous and orthotopic syngeneic model MSK-PCa2 patient derived organoid tumor injected to humanized CD34+ mouse model | [183] |
| BAY1082439 + PD-1 | Pb-Cre+PtenL/ L(Pten-null) transgenic model | [184] |
| Degarelix + copanlisib + PD-1 | Pb-Cre; PTENL/L Trp53L/L (PTEN/p53-deficient) transgenic model | [185] |
| MKC8866 + PD-1 | Myc-CaP WT or PTEN CRISPR knock-out cells or RM-1 cells subcutaneous syngeneic model | [186] |
| CP1 + PD-1 | Myc-CaP WT or PTEN CRISPR knock-out cells orthotopic syngeneic model | [187] |
| Enzalutamide + anti-B7-H3 + PDL-1 or Enzalutamide + anti-B7-H3 + CTLA-4 | DX1 cells subcutaneous syngeneic model | [188] |
| Radiotherapy + anti-CD40 + CTLA-4 | TRAMP-C1 and DVL3 cells subcutaneous syngeneic model | [189] |
| Radium-223 + Degarelix + CTLA-4 + PD-1 | Myc-CaP cells injected in the femur syngeneic model | [190] |
| Bicalutamide + docetaxel + PD-1 | RM-1 cells subcutaneous syngeneic model | [153] |
| Irreversible electroporation + CTLA-4 + PD-1 | TRAMP-C2 cells subcutaneous syngeneic model | [192] |
| Cryoablation + CTLA-4 | TRAMP-C2 cells subcutaneous syngeneic model | [193] |
| Cryoablation + CTLA-4 | Myc-CaP subcutaneous syngeneic model | [194] |
| anti-CD73 + CTLA-4 or anti-CD73 + PD-1 | RM-1 cells subcutaneous syngeneic model | [195] |
| anti-RANKL + PD-1 + CTLA-4 | TRAMP-C1 cells subcutaneous syngeneic model | [196] |
| A485 + PD-L1 | TRAMP-C2-Ras cells subcutaneous syngeneic model | [197] |
| BAY1895344 + PD-L1 | RM-1-BM cells subcutaneous syngeneic model | [198] |
| JQ1 + CTLA-4 | Myc-CaP subcutaneous syngeneic model | [199] |
| EPZ6438 + PD-1 | B6-HiMYC transgenic model | [200] |
CTLA-4 = cytotoxic T-lymphocyte-associated protein 4, PD-1 = programmed cell death protein 1, PD-L1 = programmed death-ligand 1, Cabozantinib: multi-kinase inhibitor, BEZ235: PI3K)/mTOR dual inhibitor, YY001: the prostaglandin E2 receptor EP4 antagonist, BAY1082439: PI3Kα/β/δ inhibitor, Degarelix: androgen deprivation therapy agent, copanlisib: PI3K inhibitor, anti-RANKL: inhibitor of receptor activator of nuclear factor kappa beta, A485: a small molecule inhibitor of p300/CBP, BAY1895344: Ataxia telangiectasia protein kinase (ATR) inhibitor, JQ1: BET inhibitor, EPZ6438: Enhancer of zeste homolog 2 (EZH2) inhibitor.
Similar to what has been observed in humans, combining anti-CTLA-4 and anti-PD-1 ICIs has resulted in only modest efficacy in mouse PCa models [181]. However, when immune checkpoint blockade was combined with MDSC-targeted therapy using multikinase inhibitors, such as cabozantinib and phosphoinositide 3-kinase (PI3K)/mTOR dual inhibitor BEZ235, there were robust synergistic tumor burden decreases in both primary and metastatic CRPC in murine models [181]. In another study involving MDSCs, lymphocyte-specific protein tyrosine kinase (LCK), a key protein for T cell activation, was shown to be nitrated and rendered inactive by reactive nitrogen species (RNS) generated by MDSCs in ICI resistant PCa tumors [182]. In a mouse model of CRPC, where Pten, p53, and Smad4 are specifically deleted in the prostate, CRPC exhibited resistance to anti-PD-1 and CTLA-4 agents. However, the effectiveness of ICI infusion was significantly enhanced when combined with uric acid which acts as an RNS neutralizing agent [182]. Taken together, these findings suggest that combining immune checkpoint blockade with MDSC-targeted therapies may be a viable treatment option for mCRPC [181, 182].
More recently, Peng et al. introduced a novel therapeutic strategy that targets the prostaglandin E2 receptor EP4 (PTGER4) that is found in various immune cells [183]. This strategy involves using a recently identified EP4 antagonist called YY001, effectively reversing the immunosuppressive characteristics of MDSCs while simultaneously boosting the infiltration and activity of CD8+ T cells. Combination of YY001 with anti-PD-1 therapy proved to be highly effective in inhibiting tumor progression. This combination led to long-term survival and the development of enduring immunologic memory. These findings suggest a transformation of the TME from an immunologically cold state, where immune response is limited, to an immunologically hot state, where the immune system actively targets and eliminates cancer cells [183].
Pten-deficient PCa mouse models typically exhibit resistance to anti-PD-1 immunotherapy. However, recent research suggests that intermittent administration of the PI3Kα/β/δ inhibitor BAY1082439, as opposed to continuous daily dosing, can effectively mitigate development of resistance [184]. This treatment regimen fosters increased infiltration of CD8+ T cells into the TME and augments the antitumor immune response, effectively transforming previously non-responsive cold tumors into "T-cell-inflamed" tumors. These findings represent a promising approach to enhance the efficacy of immunotherapy for Pten-null PCa [184]. Consistent with these findings, the combination of ADT degarelix and PI3K inhibitor copanlisib showed a partial antitumor response in a murine PCa model with Pten/p53 deficiency [185]. This response was achieved by increasing the frequency of activated TAMs in the TME. However, the addition of anti-PD-1 to copanlisib did not lead to a higher overall response rate. Nevertheless, when mice were treated with degarelix + copanlisib + anti-PD-1 combination therapy, there was a 60% increase in ORR within 28 days compared to untreated controls [185].
Implicating another central signaling pathway in the ICI response, we recently discovered that genetic deletion or small molecule inhibition of one of the canonical unfolded protein response pathways, IRE1α-XBP1s, increased response to PD-1 therapy in syngeneic mouse models [186]. CRISPR/Cas9-mediated deletion of IRE1α or treatment with its small molecule inhibitor MKC8866 (ORIN1001), which is currently in clinical trials, reprogrammed the TME, reversed immunosuppression, increased NK and CD8+ T-cell infiltration, augmented interferon responses, and enhanced the efficacy of anti-PD-1 therapy in various PCa syngeneic mouse models [186]. Furthermore, in the same study, we discovered a novel TAM gene signature that is associated with poor PCa survival that is significantly decreased by the combination of MKC8866 and anti-PD-1 therapy. These findings suggest that IRE1α inhibition could potentiate the effectiveness of anti-PD-1 immunotherapy in PCa. Further work is required to evaluate the translational potential of these findings.
In another study, anti-PD-1 immunotherapy combined with patient-derived prostate-specific microbe CP1 injection, there was notable improvement in survival rates and a reduction in tumor size in orthotopic models of MYC- and PTEN-mutant PCa syngeneic models [187]. CP1 injection enhanced the immunogenic cell death of cancer cells, boosted T cell cytotoxicity, and promoted the infiltration of activated CD8+ T cells as well as other cell types such as NK cells, M1 macrophages, and mature dendritic cells into the tumor. Consistently, durable antitumor effects and extended survival were achieved and the potential for a cure was observed in a syngeneic PCa model when anti-B7-H3 inhibitor was combined with enzalutamide and the blockade of PD-L1 or CTLA-4 [188].
In tumors with limited T-cell infiltration and poor response to radiation therapy, combining an agonistic anti-CD40 monoclonal antibody (mAb) led to a reprogramming of the TME [189]. This reprogramming involved increased IFN-γ signaling, activation of Th-1 pathways, and higher infiltration of CD8+ T cells into the TME. As a result, the combination therapy showed better tumor control compared to using radiation and anti-PD-1 alone Moreover, this regimen increased the presence of Tregs and engaged the CTLA-4 axis within the TME. When anti-CTLA-4 antibody was administered alongside radiation therapy and anti-CD40 mAb therapy, it overcame Treg-mediated immune suppression, resulting in a higher ratio of cytotoxic T cells, tumor rejection, and the development of long-term immunity [189]. Consistently, combining Radium-223 and degarelix with ICIs targeting PD-1 and CTLA-4 demonstrated superior efficacy compared to using each treatment alone in the Myc-CaP bone-tumor bearing mouse model [190].
The effectiveness of checkpoint therapy was also augmented by combining chemotherapy drugs with ICIs. For example, docetaxel treatment activated the cGAS/STING pathway in PCa, leading to the induction of IFN signaling and subsequent infiltration of lymphocytes into the tumor [153]. In a mouse model, a chemohormonal therapy based on docetaxel facilitated the intratumoral infiltration of T cells and sensitized the mouse tumors to anti-PD-1 blockade. To evaluate the clinical significance of these findings, a retrospective analysis was conducted on 30 metastatic CRPC patients. The results showed that combining docetaxel with anti-PD-1 antibody tislelizumab led to improved PSA progression-free survival for patients with a ≥25% PSA reduction compared to using tislelizumab alone [153].
Endogenous tumor-specific tissue-resident memory T (TRM) cells have emerged as a focal point in cancer immunotherapy research [191]. In a murine model of PCa, a novel dual therapy approach combining primary tumor destruction using irreversible electroporation, followed by anti-CTLA-4 treatment not only confirmed the establishment of TRM cells but also demonstrated their pivotal role in conferring protection against subsequent tumor challenges. Building upon this success, a triple-therapy strategy that included anti-PD-1 antibodies showed remarkable efficacy in cases that had initially shown resistance to treatment [192]. In another study, in the TRAMP-C2 model, the combination therapy of cryoablation and CTLA-4 blockade displayed remarkable synergy, leading to the rejection of a second tumor challenge [193]. Tumors exhibited increased infiltration of CD8+ T cells at the challenge sites and the combination therapy group showed a higher ratio of effector T cells to Treg cells. Furthermore, an independent study demonstrated that the combination of cryoablation, degarelix and CTLA-4 blockade resulted in a synergistic effect, leading to a notable delay in the growth of distant tumors and a reduction in the mortality rate [194].
In addition to these findings, combination of anti-CD73 antibodies with anti-PD-1 or anti-CTLA-4 treatment resulted in a substantial enhancement of antitumor activity in the RM-1 syngeneic mouse PCa model [195]. In another study, the addition of anti-RANKL (receptor activator of nuclear factor kappa beta) to the combination therapy of anti-PD-1 and anti-CTLA-4 led to enhanced anti-tumor responses, regardless of the ability of anti-CTLA-4 isotype to engage activating Fc receptors [196]. Both concurrent and delayed RANKL blockade proved to be highly effective. An early assessment during treatment showed that this triple combination therapy, when compared to the dual combination of anti-PD-1 and anti-CTLA-4, further increased the proportion of tumor-infiltrating CD4+ and CD8+ T cells capable of producing both IFN-γ and TNFα [196].
In a number of different studies, small molecule inhibitors were combined with ICIs in PCa pre-clinical models. For instance, the efficacy of PD-L1 blockade in the TRAMP-C2 model was significantly improved by using A485, a small molecule inhibitor that targets p300/CBP transcriptional coregulators [197]. A485 effectively blocked both the intrinsic and IFN-γ-induced PD-L1 expression. As a result, the combination of the inhibitor with PD-L1 blockade had a significantly enhanced efficacy in this model [197]. In another study, combination therapy involving ATR inhibitor (ATRi) BAY1895344 and anti-PD-L1 demonstrated greater inhibition and survival of tumor bearing mice compared to using either of the individual agents alone in RM-1-BM mouse PCa model [198]. The combined administration of ATRi and anti-PD-L1 therapy led to strong activation of the innate immune system and a synergistic therapeutic response that was T-cell dependent.
In another study, targeting BET bromodomains with the small molecule inhibitor JQ1 led to a reduction in PD-L1 expression and inhibited tumor progression in PCa models [199]. This effect was associated with an increase in MHC class I expression and immunogenicity of the tumor cells. Moreover, in the Myc-CaP syngeneic PCa model, combining JQ1 treatment with anti-CTLA-4 immunotherapy resulted in an additive effect, leading to an increased CD8/Treg ratio, which is beneficial for antitumor immune responses [199].
Morel et al. used the small molecule inhibitor EPZ6438 that targets enhancer of zeste homolog 2 (EZH2) of the Polycomb repressive complex 2 (PRC2) to activate a stress response involving double-stranded RNA-STING-Interferon-Stimulated Genes (ISG) pathway [200]. This activation leads to the upregulation of genes related to antigen presentation, Th1 chemokine signaling, and interferon response, including PD-L1. As a result of EZH2 inhibition, there was a significant increase in the infiltration of activated CD8+ T cells and M1 TAMs within the tumor [200]. This reversal of resistance to PD-1 checkpoint inhibition demonstrated the potential of EZH2 inhibition as a promising therapeutic strategy to enhance effectiveness of ICI therapy in PCa.
Discussion and future perspectives
ICIs have revolutionized the treatment landscape in some cancer types. However, they have so far failed to show efficacy in Phase III trials on PCa. This has been linked to the cold immunophenotype of the prostate TME, characterized by a quantitative deficiency in cytotoxic T cells and a plethora of immunosuppressive mechanisms that curb the antitumor response. A low mutational burden and scarcity of neoantigen formation that is necessary to mobilize the adaptive immune system limit antigen recognition on PCa cells. This in turn, does not allow “releasing the brakes on the immune system”, which is the goal of checkpoint blockers. Can this be reversed in some way in the case of PCa?
The current preclinical and clinical research findings suggest that understanding the immunophenotype of PCa mechanistically may be critical to improve the efficacy of ICIs for PCa. Whereas some studies have suggested that blockade of different checkpoints produces unique effects in the immune compartment [123, 124], dual checkpoint inhibition using monospecific antibodies or bispecific constructs has demonstrated clinical activity [125, 126, 133, 141, 142]. The compensatory upregulation of other checkpoints under monotherapy further supports this strategy [116], and the emergence of less well studied checkpoints to date suggests that new targets may be available when other options fail [119, 129, 131]. A significant challenge is that our knowledge on each checkpoint from a functional point of view is yet very limited, and some present with contradictory functions in the tumor landscape [127]. An important task for the future is to identify as to which targets are most actionable and which combination therapies would prove most effective. As a related but different strategy, therapeutic benefit may be achieved by blocking checkpoints and simultaneously depleting immunosuppressive cytokines from the TME [149], but evidence suggests that one can alternatively aim to circumvent immunosuppressive stimuli by supercharging costimulatory markers with TAA-CTL bridging bispecifics and complement with checkpoint targeting antibodies [140].
Given the low mutational burden of PCa, checkpoint inhibition may still not achieve its full potential until the immune system is primed to recognize the tumor as harmful, which involves a complex interplay between innate and adaptive immune compartments. Anticancer vaccines are the most straightforward option, but few vaccines have proven effective by themselves, and ideal antigen selection is difficult. Some studies have demonstrated a potential for ICIs and vaccines to synergize [148, 151], but the clinical benefit of this approach in PCa is not known. Current standard of care treatments have shown immunomodulatory and proinflammatory activities which has suggested that they could be combined with ICIs in PCa therapy [153, 159, 161, 170]. However, this raises an important issue regarding the timing of checkpoint combinations in a patient's treatment history; would a patient benefit more from combination regimens in the early phases of PCa or further down the course of the disease?
Perhaps the main issue in the ICI efficacy for PCa relates to the subject of patient selection in clinical trials. It is clear from previous trials that a select subset of patients respond more favorably to checkpoint mono- and combination therapies, but the full characteristics of these patient subsets are not known [22, 23, 30, 125, 173]. Although biomarkers such as PD-1 expression, CD8+ infiltration, TMB, and mismatch repair deficiency are repeatedly associated with better outcomes in response to different ICIs, attempts to stratify patients based on these characteristics have had limited success. Emerging biomarkers - CD73, VISTA, NK-cell markers and more - provide alternative routes to making informed decisions in future trials. Furthermore, a nested study design that takes multiple biomarkers into account may be required to optimize their utility in predicting treatment responses for PCa patients. The observation that blockade of different checkpoints enforces different changes in the immune landscape [123] suggests that biomarkers should not only inform a binary decision on ICI eligibility, but which checkpoints need to be co-targeted and how they can synergize with targeted therapies that are already showing promise in such combination regimens (e.g. [175, 178, 199]). This again requires deeper knowledge into the mechanism(s) underlying each checkpoint in the PCa TME and the concurrent impact of actionable targets in cancer cells.
Given that MKC8866 is currently in clinical trials (NCT03950570) for advanced cancer patients, our findings indicate that new clinical trials could be developed for PCa, incorporating anti-PD-1 immunotherapy with IRE1α inhibition. Beyond therapeutic potential, the TAM gene signature we identified demonstrates significant prognostic value in prostate cancer patients and is diminished by the combination of MKC8866 and anti-PD-1 therapy. This suggests that the TAM gene signature could be useful in predicting disease progression and tailoring treatment strategies. For instance, it may assist in stratifying prostate cancer patients for anti-PD-1 immunotherapy. Further research is needed to explore these possibilities.
In summary, current evidence suggests that ICIs hold an untapped potential in PCa; however, the cold immunophenotype poses a significant challenge to harnessing it. Chemotherapeutics and targeted therapies have demonstrated the potential to modify this phenotype for clinical benefit. Careful biomarker-based patient selection with informed combination regimens is therefore emerging as a means to optimizing ICI activity in the clinic. More knowledge is needed as to which biomarkers are suitable guides and actionable target nodes, but the progress thus far suggests that the future may present new possibilities to transform the cold and barren PCa TME to one that responds to checkpoint inhibition and other immunotherapies.
Abbreviations
ADT: Androgen deprivation therapy; AR: Androgen receptor; bsAb: Bispecific antibody; CRPC: Castration resistant prostate cancer; CTL: Cytotoxic T-lymphocyte; CTLA-4: Cytotoxic T-lymphocyte associated protein 4; ETS: E26 transformation-specific family; HLA: Human leukocyte antigen; ICAM-1: Intercellular adhesion molecule 1; ICI: Immune checkpoint inhibitor; LAG-3: Lymphocyte activation gene-3; MDSC: Myeloid-derived suppressor cell; mCRPC: Metastatic castration resistant prostate cancer; ORR: Objective response rate; OS: Overall survival; PARP: Poly (ADP-ribose) polymerase; PCa: Prostate cancer; PD-1: Programmed cell death protein 1; PD-L1/L2: Programmed death-ligand 1/2; PFS: Progression-free survival; PI3K: Phosphoinositide 3-kinase; PRC1/-2: Polycomb repressive complex 1/-2; PSA: Prostate-specific antigen; PSMA: Prostate-specific membrane antigen; TAA: Tumor-associated antigen; TAM: Tumor associated macrophage; TGF-β: Transforming growth factor β; TIM-3: T-cell immunoglobulin and mucin domain 3; TMB: Tumor mutational burden; TME: Tumor microenvironment; Treg: Regulatory T cell; TSA: Tumor-specific antigen; VISTA: V-domain Ig suppressor of T cell activation.
Acknowledgements
All graphics were created with BioRender.com.
We apologize that many significant primary research and review papers that have contributed to this field could not be included due to the space limitations.
Funding
This work was funded by grants from the Norwegian Research Council, Norwegian Cancer Society, and Norway South East Health Authority to FS.
Consent for publication
The Authors confirm: (1) that the work described has not been published before; (2) that it is not under consideration for publication elsewhere; (3) that its publication has been approved by all coauthors, (4) that its publication has been approved (tacitly or explicitly) by the responsible authorities at the institution where the work is carried out.
Author Contributions
All authors were involved in the planning, writing, and proofing of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ferlay J, Colombet M, Soerjomataram I, Parkin DM, Pineros M, Znaor A. et al. Cancer statistics for the year 2020: An overview. Int J Cancer. 2021;149:778-89
2. Ferlay J, Laversanne M, Ervik M, Lam F, Colombet M, Mery L. et al. Global Cancer Observatory: Cancer Tomorrow. Lyon, France: International Agency for Research on Cancer. 2020
3. Hamdy FC, Donovan JL, Lane JA, Mason M, Metcalfe C, Holding P. et al. 10-Year Outcomes after Monitoring, Surgery, or Radiotherapy for Localized Prostate Cancer. N Engl J Med. 2016;375:1415-24
4. Rebello RJ, Oing C, Knudsen KE, Loeb S, Johnson DC, Reiter RE. et al. Prostate cancer. Nat Rev Dis Primers. 2021;7:9
5. Suarez JF, Zamora V, Garin O, Gutierrez C, Pont A, Pardo Y. et al. Mortality and biochemical recurrence after surgery, brachytherapy, or external radiotherapy for localized prostate cancer: a 10-year follow-up cohort study. Sci Rep. 2022;12:12589
6. Fallah J, Agrawal S, Gittleman H, Fiero MH, Subramaniam S, John C. et al. FDA Approval Summary: Lutetium Lu 177 Vipivotide Tetraxetan for Patients with Metastatic Castration-Resistant Prostate Cancer. Clin Cancer Res. 2023;29:1651-7
7. Singer M, Wang C, Cong L, Marjanovic ND, Kowalczyk MS, Zhang H. et al. A Distinct Gene Module for Dysfunction Uncoupled from Activation in Tumor-Infiltrating T Cells. Cell. 2016;166:1500-11 e9
8. U.S. Food and Drug Administration: Center for Drug Evaluation and Research. Oncology (Cancer) / Hematologic Malignancies Approval Notifications. 2023
9. Motzer RJ, Tannir NM, McDermott DF, Aren Frontera O, Melichar B, Choueiri TK. et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med. 2018;378:1277-90
10. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-23
11. Antonia SJ, Borghaei H, Ramalingam SS, Horn L, De Castro Carpeno J, Pluzanski A. et al. Four-year survival with nivolumab in patients with previously treated advanced non-small-cell lung cancer: a pooled analysis. Lancet Oncol. 2019;20:1395-408
12. Cheever MA, Higano CS. PROVENGE (Sipuleucel-T) in prostate cancer: the first FDA-approved therapeutic cancer vaccine. Clin Cancer Res. 2011;17:3520-6
13. Iannantuono GM, Torino F, Rosenfeld R, Guerriero S, Carlucci M, Sganga S. et al. The Role of Histology-Agnostic Drugs in the Treatment of Metastatic Castration-Resistant Prostate Cancer. Int J Mol Sci. 2022;23:8535
14. Jamal-Hanjani M, Quezada SA, Larkin J, Swanton C. Translational implications of tumor heterogeneity. Clin Cancer Res. 2015;21:1258-66
15. Wei L, Wang J, Lampert E, Schlanger S, DePriest AD, Hu Q. et al. Intratumoral and Intertumoral Genomic Heterogeneity of Multifocal Localized Prostate Cancer Impacts Molecular Classifications and Genomic Prognosticators. Eur Urol. 2017;71:183-92
16. Liu YT, Sun ZJ. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics. 2021;11:5365-86
17. Maleki Vareki S. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J Immunother Cancer. 2018;6:157
18. Ye L, Li Y, Tang H, Liu W, Chen Y, Dai T. et al. CD8+CXCR5+T cells infiltrating hepatocellular carcinomas are activated and predictive of a better prognosis. Aging (Albany NY). 2019;11:8879-91
19. Vonderheide RH, Domchek SM, Clark AS. Immunotherapy for Breast Cancer: What Are We Missing? Clin Cancer Res. 2017;23:2640-6
20. Beer TM, Kwon ED, Drake CG, Fizazi K, Logothetis C, Gravis G. et al. Randomized, Double-Blind, Phase III Trial of Ipilimumab Versus Placebo in Asymptomatic or Minimally Symptomatic Patients With Metastatic Chemotherapy-Naive Castration-Resistant Prostate Cancer. J Clin Oncol. 2017;35:40-7
21. Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, van den Eertwegh AJ. et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15:700-12
22. Fizazi K, Drake CG, Beer TM, Kwon ED, Scher HI, Gerritsen WR. et al. Final Analysis of the Ipilimumab Versus Placebo Following Radiotherapy Phase III Trial in Postdocetaxel Metastatic Castration-resistant Prostate Cancer Identifies an Excess of Long-term Survivors. Eur Urol. 2020;78:822-30
23. Graff JN, Stein MN, Surana R, Al Rabadi L, Liu E, Fong L. et al. Phase II Study of Ipilimumab in Men With Metastatic Prostate Cancer With an Incomplete Response to Androgen Deprivation Therapy. Front Oncol. 2020;10:1381
24. Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH. et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28:3167-75
25. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF. et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443-54
26. Haffner MC, Guner G, Taheri D, Netto GJ, Palsgrove DN, Zheng Q. et al. Comprehensive Evaluation of Programmed Death-Ligand 1 Expression in Primary and Metastatic Prostate Cancer. Am J Pathol. 2018;188:1478-85
27. Antonarakis ES, Piulats JM, Gross-Goupil M, Goh J, Ojamaa K, Hoimes CJ. et al. Pembrolizumab for Treatment-Refractory Metastatic Castration-Resistant Prostate Cancer: Multicohort, Open-Label Phase II KEYNOTE-199 Study. J Clin Oncol. 2020;38:395-405
28. Marcus L, Fashoyin-Aje LA, Donoghue M, Yuan M, Rodriguez L, Gallagher PS. et al. FDA Approval Summary: Pembrolizumab for the Treatment of Tumor Mutational Burden-High Solid Tumors. Clin Cancer Res. 2021;27:4685-9
29. Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K. et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020;21:1353-65
30. Subudhi SK, Vence L, Zhao H, Blando J, Yadav SS, Xiong Q. et al. Neoantigen responses, immune correlates, and favorable outcomes after ipilimumab treatment of patients with prostate cancer. Sci Transl Med. 2020;12:eaaz3577
31. Graf RP, Fisher V, Weberpals J, Gjoerup O, Tierno MB, Huang RSP. et al. Comparative Effectiveness of Immune Checkpoint Inhibitors vs Chemotherapy by Tumor Mutational Burden in Metastatic Castration-Resistant Prostate Cancer. JAMA Netw Open. 2022;5:e225394
32. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK. et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409-13
33. Abida W, Cheng ML, Armenia J, Middha S, Autio KA, Vargas HA. et al. Analysis of the Prevalence of Microsatellite Instability in Prostate Cancer and Response to Immune Checkpoint Blockade. JAMA Oncol. 2019;5:471-8
34. Graham LS, Montgomery B, Cheng HH, Yu EY, Nelson PS, Pritchard C. et al. Mismatch repair deficiency in metastatic prostate cancer: Response to PD-1 blockade and standard therapies. PLoS One. 2020;15:e0233260
35. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM. et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215-28
36. Cancer Genome Atlas Research N. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015;163:1011-25
37. Ritch E, Fu SYF, Herberts C, Wang G, Warner EW, Schonlau E. et al. Identification of Hypermutation and Defective Mismatch Repair in ctDNA from Metastatic Prostate Cancer. Clin Cancer Res. 2020;26:1114-25
38. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214-8
39. Tomlins SA, Laxman B, Varambally S, Cao X, Yu J, Helgeson BE. et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia. 2008;10:177-88
40. Pflueger D, Rickman DS, Sboner A, Perner S, LaFargue CJ, Svensson MA. et al. N-myc downstream regulated gene 1 (NDRG1) is fused to ERG in prostate cancer. Neoplasia. 2009;11:804-11
41. van Dessel LF, van Riet J, Smits M, Zhu Y, Hamberg P, van der Heijden MS. et al. The genomic landscape of metastatic castration-resistant prostate cancers reveals multiple distinct genotypes with potential clinical impact. Nat Commun. 2019;10:5251
42. Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M. et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725-30
43. Couto SS, Cao M, Duarte PC, Banach-Petrosky W, Wang S, Romanienko P. et al. Simultaneous haploinsufficiency of Pten and Trp53 tumor suppressor genes accelerates tumorigenesis in a mouse model of prostate cancer. Differentiation. 2009;77:103-11
44. Hubbard GK, Mutton LN, Khalili M, McMullin RP, Hicks JL, Bianchi-Frias D. et al. Combined MYC Activation and Pten Loss Are Sufficient to Create Genomic Instability and Lethal Metastatic Prostate Cancer. Cancer Res. 2016;76:283-92
45. Tan HL, Sood A, Rahimi HA, Wang W, Gupta N, Hicks J. et al. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin Cancer Res. 2014;20:890-903
46. Lin HK, Hu YC, Lee DK, Chang C. Regulation of androgen receptor signaling by PTEN (phosphatase and tensin homolog deleted on chromosome 10) tumor suppressor through distinct mechanisms in prostate cancer cells. Mol Endocrinol. 2004;18:2409-23
47. Gonthier K, Poluri RTK, Weidmann C, Tadros M, Audet-Walsh E. Reprogramming of Isocitrate Dehydrogenases Expression and Activity by the Androgen Receptor in Prostate Cancer. Mol Cancer Res. 2019;17:1699-709
48. Gerhardt J, Montani M, Wild P, Beer M, Huber F, Hermanns T. et al. FOXA1 promotes tumor progression in prostate cancer and represents a novel hallmark of castration-resistant prostate cancer. Am J Pathol. 2012;180:848-61
49. An J, Wang C, Deng Y, Yu L, Huang H. Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants. Cell Rep. 2014;6:657-69
50. Hollingsworth RE, Jansen K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines. 2019;4:7
51. Zhao Y, Baldin AV, Isayev O, Werner J, Zamyatnin AA Jr, Bazhin AV. Cancer Vaccines: Antigen Selection Strategy. Vaccines (Basel). 2021;9:85
52. Fukuyama T, Hanagiri T, Takenoyama M, Ichiki Y, Mizukami M, So T. et al. Identification of a new cancer/germline gene, KK-LC-1, encoding an antigen recognized by autologous CTL induced on human lung adenocarcinoma. Cancer Res. 2006;66:4922-8
53. Suyama T, Shiraishi T, Zeng Y, Yu W, Parekh N, Vessella RL. et al. Expression of cancer/testis antigens in prostate cancer is associated with disease progression. Prostate. 2010;70:1778-87
54. Kim H, Barua A, Huang L, Zhou T, Bolaji M, Zachariah S. et al. The cancer testis antigen TDRD1 regulates prostate cancer proliferation by associating with the snRNP biogenesis machinery. Oncogene. 2023;42:1821-31
55. McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK. et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463-9
56. Li W, Amei A, Bui F, Norouzifar S, Lu L, Wang Z. Impact of Neoantigen Expression and T-Cell Activation on Breast Cancer Survival. Cancers (Basel). 2021;13:2879
57. Balachandran VP, Luksza M, Zhao JN, Makarov V, Moral JA, Remark R. et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. 2017;551:512-6
58. Cui C, Wang J, Fagerberg E, Chen PM, Connolly KA, Damo M. et al. Neoantigen-driven B cell and CD4 T follicular helper cell collaboration promotes anti-tumor CD8 T cell responses. Cell. 2021;184:6101-18 e13
59. Liu L, Chen J, Zhang H, Ye J, Moore C, Lu C. et al. Concurrent delivery of immune checkpoint blockade modulates T cell dynamics to enhance neoantigen vaccine-generated antitumor immunity. Nat Cancer. 2022;3:437-52
60. Ciavarra RP, Holterman DA, Brown RR, Mangiotti P, Yousefieh N, Wright GL Jr. et al. Prostate tumor microenvironment alters immune cells and prevents long-term survival in an orthotopic mouse model following flt3-ligand/CD40-ligand immunotherapy. J Immunother. 2004;27:13-26
61. Danaher P, Warren S, Lu R, Samayoa J, Sullivan A, Pekker I. et al. Pan-cancer adaptive immune resistance as defined by the Tumor Inflammation Signature (TIS): results from The Cancer Genome Atlas (TCGA). J Immunother Cancer. 2018;6:63
62. Wu Z, Chen H, Luo W, Zhang H, Li G, Zeng F. et al. The Landscape of Immune Cells Infiltrating in Prostate Cancer. Front Oncol. 2020;10:517637
63. Hirz T, Mei S, Sarkar H, Kfoury Y, Wu S, Verhoeven BM. et al. Dissecting the immune suppressive human prostate tumor microenvironment via integrated single-cell and spatial transcriptomic analyses. Nat Commun. 2023;14:663
64. Chi N, Tan Z, Ma K, Bao L, Yun Z. Increased circulating myeloid-derived suppressor cells correlate with cancer stages, interleukin-8 and -6 in prostate cancer. Int J Clin Exp Med. 2014;7:3181-92
65. JiaWei Z, ChunXia D, CunDong L, Yang L, JianKun Y, HaiFeng D. et al. M2 subtype tumor associated macrophages (M2-TAMs) infiltration predicts poor response rate of immune checkpoint inhibitors treatment for prostate cancer. Ann Med. 2021;53:730-40
66. Sawant DV, Yano H, Chikina M, Zhang Q, Liao M, Liu C. et al. Adaptive plasticity of IL-10(+) and IL-35(+) T(reg) cells cooperatively promotes tumor T cell exhaustion. Nat Immunol. 2019;20:724-35
67. Ju M, Fan J, Zou Y, Yu M, Jiang L, Wei Q. et al. Computational Recognition of a Regulatory T-cell-specific Signature With Potential Implications in Prognosis, Immunotherapy, and Therapeutic Resistance of Prostate Cancer. Front Immunol. 2022;13:807840
68. Hellsten R, Lilljebjorn L, Johansson M, Leandersson K, Bjartell A. The STAT3 inhibitor galiellalactone inhibits the generation of MDSC-like monocytes by prostate cancer cells and decreases immunosuppressive and tumorigenic factors. Prostate. 2019;79:1611-21
69. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L. et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568-71
70. Yang Y, Attwood K, Bshara W, Mohler JL, Guru K, Xu B. et al. High intratumoral CD8(+) T-cell infiltration is associated with improved survival in prostate cancer patients undergoing radical prostatectomy. Prostate. 2021;81:20-8
71. Kane N, Romero T, Diaz-Perez S, Rettig MB, Steinberg ML, Kishan AU. et al. Significant changes in macrophage and CD8 T cell densities in primary prostate tumors 2 weeks after SBRT. Prostate Cancer Prostatic Dis. 2023;26:207-9
72. Blessin NC, Spriestersbach P, Li W, Mandelkow T, Dum D, Simon R. et al. Prevalence of CD8(+) cytotoxic lymphocytes in human neoplasms. Cell Oncol (Dordr). 2020;43:421-30
73. Yanai Y, Kosaka T, Mikami S, Hongo H, Yasumizu Y, Takeda T. et al. CD8-positive T cells and CD204-positive M2-like macrophages predict postoperative prognosis of very high-risk prostate cancer. Sci Rep. 2021;11:22495
74. Ness N, Andersen S, Valkov A, Nordby Y, Donnem T, Al-Saad S. et al. Infiltration of CD8+ lymphocytes is an independent prognostic factor of biochemical failure-free survival in prostate cancer. Prostate. 2014;74:1452-61
75. Leclerc BG, Charlebois R, Chouinard G, Allard B, Pommey S, Saad F. et al. CD73 Expression Is an Independent Prognostic Factor in Prostate Cancer. Clin Cancer Res. 2016;22:158-66
76. Kiniwa Y, Miyahara Y, Wang HY, Peng W, Peng G, Wheeler TM. et al. CD8+ Foxp3+ regulatory T cells mediate immunosuppression in prostate cancer. Clin Cancer Res. 2007;13:6947-58
77. Olson BM, Jankowska-Gan E, Becker JT, Vignali DA, Burlingham WJ, McNeel DG. Human prostate tumor antigen-specific CD8+ regulatory T cells are inhibited by CTLA-4 or IL-35 blockade. J Immunol. 2012;189:5590-601
78. Karpisheh V, Mousavi SM, Naghavi Sheykholeslami P, Fathi M, Mohammadpour Saray M, Aghebati-Maleki L. et al. The role of regulatory T cells in the pathogenesis and treatment of prostate cancer. Life Sci. 2021;284:119132
79. Bronte G, Conteduca V, Landriscina M, Procopio AD. Circulating myeloid-derived suppressor cells and survival in prostate cancer patients: systematic review and meta-analysis. Prostate Cancer Prostatic Dis. 2023;26:41-6
80. Ozga AJ, Chow MT, Luster AD. Chemokines and the immune response to cancer. Immunity. 2021;54:859-74
81. Proost P, Mortier A, Loos T, Vandercappellen J, Gouwy M, Ronsse I. et al. Proteolytic processing of CXCL11 by CD13/aminopeptidase N impairs CXCR3 and CXCR7 binding and signaling and reduces lymphocyte and endothelial cell migration. Blood. 2007;110:37-44
82. William M, Leroux LP, Chaparro V, Graber TE, Alain T, Jaramillo M. Translational repression of Ccl5 and Cxcl10 by 4E-BP1 and 4E-BP2 restrains the ability of mouse macrophages to induce migration of activated T cells. Eur J Immunol. 2019;49:1200-12
83. Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W. et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527:249-53
84. Nagarsheth N, Peng D, Kryczek I, Wu K, Li W, Zhao E. et al. PRC2 Epigenetically Silences Th1-Type Chemokines to Suppress Effector T-Cell Trafficking in Colon Cancer. Cancer Res. 2016;76:275-82
85. Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D. et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med. 2011;208:1949-62
86. Muthuswamy R, Corman JM, Dahl K, Chatta GS, Kalinski P. Functional reprogramming of human prostate cancer to promote local attraction of effector CD8(+) T cells. Prostate. 2016;76:1095-105
87. Rani A, Dasgupta P, Murphy JJ. Prostate Cancer: The Role of Inflammation and Chemokines. Am J Pathol. 2019;189:2119-37
88. Su W, Han HH, Wang Y, Zhang B, Zhou B, Cheng Y. et al. The Polycomb Repressor Complex 1 Drives Double-Negative Prostate Cancer Metastasis by Coordinating Stemness and Immune Suppression. Cancer Cell. 2019;36:139-55 e10
89. Duru G, van Egmond M, Heemskerk N. A Window of Opportunity: Targeting Cancer Endothelium to Enhance Immunotherapy. Front Immunol. 2020;11:584723
90. Griffioen AW, Damen CA, Blijham GH, Groenewegen G. Tumor angiogenesis is accompanied by a decreased inflammatory response of tumor-associated endothelium. Blood. 1996;88:667-73
91. Bouzin C, Brouet A, De Vriese J, Dewever J, Feron O. Effects of vascular endothelial growth factor on the lymphocyte-endothelium interactions: identification of caveolin-1 and nitric oxide as control points of endothelial cell anergy. J Immunol. 2007;178:1505-11
92. Dirkx AE, oude Egbrink MG, Castermans K, van der Schaft DW, Thijssen VL, Dings RP. et al. Anti-angiogenesis therapy can overcome endothelial cell anergy and promote leukocyte-endothelium interactions and infiltration in tumors. FASEB J. 2006;20:621-30
93. West AF, O'Donnell M, Charlton RG, Neal DE, Leung HY. Correlation of vascular endothelial growth factor expression with fibroblast growth factor-8 expression and clinico-pathologic parameters in human prostate cancer. Br J Cancer. 2001;85:576-83
94. Shetty S, Weston CJ, Oo YH, Westerlund N, Stamataki Z, Youster J. et al. Common lymphatic endothelial and vascular endothelial receptor-1 mediates the transmigration of regulatory T cells across human hepatic sinusoidal endothelium. J Immunol. 2011;186:4147-55
95. Nakashima S, Sugita Y, Miyoshi H, Arakawa F, Muta H, Ishibashi Y. et al. Endothelin B receptor expression in malignant gliomas: the perivascular immune escape mechanism of gliomas. J Neurooncol. 2016;127:23-32
96. Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K. et al. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med. 2008;14:28-36
97. Nelson JB, Udan MS, Guruli G, Pflug BR. Endothelin-1 inhibits apoptosis in prostate cancer. Neoplasia. 2005;7:631-7
98. Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS. et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. 2014;20:607-15
99. Liu QY, Rubin MA, Omene C, Lederman S, Stein CA. Fas ligand is constitutively secreted by prostate cancer cells in vitro. Clin Cancer Res. 1998;4:1803-11
100. Abusamra AJ, Zhong Z, Zheng X, Li M, Ichim TE, Chin JL. et al. Tumor exosomes expressing Fas ligand mediate CD8+ T-cell apoptosis. Blood Cells Mol Dis. 2005;35:169-73
101. Stylianopoulos T, Jain RK. Combining two strategies to improve perfusion and drug delivery in solid tumors. Proc Natl Acad Sci U S A. 2013;110:18632-7
102. Nakagawa Y, Negishi Y, Shimizu M, Takahashi M, Ichikawa M, Takahashi H. Effects of extracellular pH and hypoxia on the function and development of antigen-specific cytotoxic T lymphocytes. Immunol Lett. 2015;167:72-86
103. Noman MZ, Janji B, Kaminska B, Van Moer K, Pierson S, Przanowski P. et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res. 2011;71:5976-86
104. Tran CW, Gold MJ, Garcia-Batres C, Tai K, Elford AR, Himmel ME. et al. Hypoxia-inducible factor 1 alpha limits dendritic cell stimulation of CD8 T cell immunity. PLoS One. 2020;15:e0244366
105. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP. et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. 2011;475:226-30
106. Chiu DK, Xu IM, Lai RK, Tse AP, Wei LL, Koh HY. et al. Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology. 2016;64:797-813
107. Park JE, Dutta B, Tse SW, Gupta N, Tan CF, Low JK. et al. Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microRNA-mediated metabolic shift. Oncogene. 2019;38:5158-73
108. Jayaprakash P, Ai M, Liu A, Budhani P, Bartkowiak T, Sheng J. et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J Clin Invest. 2018;128:5137-49
109. Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL. et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med. 2015;212:139-48
110. Deng J, Li J, Sarde A, Lines JL, Lee YC, Qian DC. et al. Hypoxia-Induced VISTA Promotes the Suppressive Function of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Cancer Immunol Res. 2019;7:1079-90
111. Mulati K, Hamanishi J, Matsumura N, Chamoto K, Mise N, Abiko K. et al. VISTA expressed in tumour cells regulates T cell function. Br J Cancer. 2019;120:115-27
112. Pagliano O, Morrison RM, Chauvin JM, Banerjee H, Davar D, Ding Q. et al. Tim-3 mediates T cell trogocytosis to limit antitumor immunity. J Clin Invest. 2022;132:e152864
113. Sfanos KS, Bruno TC, Meeker AK, De Marzo AM, Isaacs WB, Drake CG. Human prostate-infiltrating CD8+ T lymphocytes are oligoclonal and PD-1+. Prostate. 2009;69:1694-703
114. Gevensleben H, Dietrich D, Golletz C, Steiner S, Jung M, Thiesler T. et al. The Immune Checkpoint Regulator PD-L1 Is Highly Expressed in Aggressive Primary Prostate Cancer. Clin Cancer Res. 2016;22:1969-77
115. Zhao SG, Lehrer J, Chang SL, Das R, Erho N, Liu Y. et al. The Immune Landscape of Prostate Cancer and Nomination of PD-L2 as a Potential Therapeutic Target. J Natl Cancer Inst. 2019;111:301-10
116. Gao J, Ward JF, Pettaway CA, Shi LZ, Subudhi SK, Vence LM. et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat Med. 2017;23:551-5
117. Wolf NK, Kissiov DU, Raulet DH. Roles of natural killer cells in immunity to cancer, and applications to immunotherapy. Nat Rev Immunol. 2023;23:90-105
118. Tarle M, Kraljic I, Kastelan M. Comparison between NK cell activity and prostate cancer stage and grade in untreated patients: correlation with tumor markers and hormonal serotest data. Urol Res. 1993;21:17-21
119. Pasero C, Gravis G, Guerin M, Granjeaud S, Thomassin-Piana J, Rocchi P. et al. Inherent and Tumor-Driven Immune Tolerance in the Prostate Microenvironment Impairs Natural Killer Cell Antitumor Activity. Cancer Res. 2016;76:2153-65
120. Gallazzi M, Baci D, Mortara L, Bosi A, Buono G, Naselli A. et al. Prostate Cancer Peripheral Blood NK Cells Show Enhanced CD9, CD49a, CXCR4, CXCL8, MMP-9 Production and Secrete Monocyte-Recruiting and Polarizing Factors. Front Immunol. 2020;11:586126
121. Liu G, Lu S, Wang X, Page ST, Higano CS, Plymate SR. et al. Perturbation of NK cell peripheral homeostasis accelerates prostate carcinoma metastasis. J Clin Invest. 2013;123:4410-22
122. Mathew SO, Chaudhary P, Powers SB, Vishwanatha JK, Mathew PA. Overexpression of LLT1 (OCIL, CLEC2D) on prostate cancer cells inhibits NK cell-mediated killing through LLT1-NKRP1A (CD161) interaction. Oncotarget. 2016;7:68650-61
123. Das R, Verma R, Sznol M, Boddupalli CS, Gettinger SN, Kluger H. et al. Combination therapy with anti-CTLA-4 and anti-PD-1 leads to distinct immunologic changes in vivo. J Immunol. 2015;194:950-9
124. Wei SC, Anang NAS, Sharma R, Andrews MC, Reuben A, Levine JH. et al. Combination anti-CTLA-4 plus anti-PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc Natl Acad Sci U S A. 2019;116:22699-709
125. Sharma P, Pachynski RK, Narayan V, Flechon A, Gravis G, Galsky MD. et al. Nivolumab Plus Ipilimumab for Metastatic Castration-Resistant Prostate Cancer: Preliminary Analysis of Patients in the CheckMate 650 Trial. Cancer Cell. 2020;38:489-99 e3
126. Sharma P, Krainer M, Saad F, Castellano D, Bedke J, Kwiatkowski M. et al. Nivolumab plus ipilimumab for the treatment of post-chemotherapy metastatic castration-resistant prostate cancer (mCRPC): Additional results from the randomized phase 2 CheckMate 650 trial. J Clin Oncol. 2023;41:22
127. Huang X, Zhang X, Li E, Zhang G, Wang X, Tang T. et al. VISTA: an immune regulatory protein checking tumor and immune cells in cancer immunotherapy. J Hematol Oncol. 2020;13:83
128. Palermo B, Bottero M, Panetta M, Faiella A, Sperduti I, Masi S. et al. Stereotactic Ablative Radiation Therapy in 3 Fractions Induces a Favorable Systemic Immune Cell Profiling in Prostate Cancer Patients. Oncoimmunology. 2023;12:2174721
129. Tang K, Zhang J, Cao H, Xiao G, Wang Z, Zhang X. et al. Identification of CD73 as a Novel Biomarker Encompassing the Tumor Microenvironment, Prognosis, and Therapeutic Responses in Various Cancers. Cancers (Basel). 2022;14:5663
130. Dumont C, Jacquier A, Verine J, Noel F, Goujon A, Wu CL. et al. CD8(+)PD-1(-)ILT2(+) T Cells Are an Intratumoral Cytotoxic Population Selectively Inhibited by the Immune-Checkpoint HLA-G. Cancer Immunol Res. 2019;7:1619-32
131. Vittrant B, Bergeron A, Molina OE, Leclercq M, Legare XP, Hovington H. et al. Immune-focused multi-omics analysis of prostate cancer: leukocyte Ig-Like receptors are associated with disease progression. Oncoimmunology. 2020;9:1851950
132. Suurs FV, Lub-de Hooge MN, de Vries EGE, de Groot DJA. A review of bispecific antibodies and antibody constructs in oncology and clinical challenges. Pharmacol Ther. 2019;201:103-19
133. Sung E, Ko M, Won JY, Jo Y, Park E, Kim H. et al. LAG-3xPD-L1 bispecific antibody potentiates antitumor responses of T cells through dendritic cell activation. Mol Ther. 2022;30:2800-16
134. Green DJ, O'Steen S, Lin Y, Comstock ML, Kenoyer AL, Hamlin DK. et al. CD38-bispecific antibody pretargeted radioimmunotherapy for multiple myeloma and other B-cell malignancies. Blood. 2018;131:611-20
135. Lee SC, Ma JSY, Kim MS, Laborda E, Choi SH, Hampton EN. et al. A PSMA-targeted bispecific antibody for prostate cancer driven by a small-molecule targeting ligand. Sci Adv. 2021;7:eabi8193
136. Hummel HD, Kufer P, Grullich C, Seggewiss-Bernhardt R, Deschler-Baier B, Chatterjee M. et al. Pasotuxizumab, a BiTE((R)) immune therapy for castration-resistant prostate cancer: Phase I, dose-escalation study findings. Immunotherapy. 2021;13:125-41
137. Chou J, Egusa EA, Wang S, Badura ML, Lee F, Bidkar AP. et al. Immunotherapeutic Targeting and PET Imaging of DLL3 in Small-Cell Neuroendocrine Prostate Cancer. Cancer Res. 2023;83:301-15
138. Vaishampayan UN, Thakur A, Chen W, Deol A, Patel M, Dobson K. et al. Phase II Trial of Pembrolizumab and Anti-CD3 x Anti-HER2 Bispecific Antibody-Armed Activated T Cells in Metastatic Castration-Resistant Prostate Cancer. Clin Cancer Res. 2023;29:122-33
139. De Bono JS, Fong L, Beer TM, Gao X, Geynisman DN, Burris III HA. et al. Results of an ongoing phase 1/2a dose escalation study of HPN424, a tri-specific half-life extended PSMA-targeting T-cell engager, in patients with metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. 2021;39:5013
140. Stein MN, Zhang J, Kelly WK, Wise DR, Tsao K, Carneiro BA. et al. Preliminary results from a phase 1/2 study of co-stimulatory bispecific PSMAxCD28 antibody REGN5678 in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. 2023;41:154
141. Shi N, Zhou Y, Liu Y, Zhang R, Jiang X, Ren C. et al. PD-1/LAG-3 bispecific antibody potentiates T cell activation and increases antitumor efficacy. Front Immunol. 2022;13:1047610
142. Shum E, Reilley M, Najjar Y, Daud A, Thompson J, Baranda J. et al. 523 Preliminary clinical experience with XmAb20717, a PD-1 x CTLA-4 bispecific antibody, in patients with advanced solid tumors. J Immunother Cancer. 2021;9:A1054
143. Hou J, Goodman O, Berz D, Yao L, Soni N. 733 A phase 2 study of vudalimab (XmAb®20717), an anti-PD-1/CTLA-4 bispecific antibody, in patients with selected gynecological malignancies and high-risk metastatic castration-resistant prostate-cancer. J Immunother Cancer. 2022;10:A1603
144. Stein MN, Dorff TB, Goodman OB, Thomas RA, Silverman MH, Guo M. et al. A phase 2, multicenter, parallel-group, open-label study of vudalimab (XmAb20717), a PD-1 x CTLA-4 bispecific antibody, alone or in combination with chemotherapy or targeted therapy in patients with molecularly defined subtypes of metastatic castration-resistant prostate cancer. J Clin Oncol. 2022;40:TPS5097
145. Luke JJ, Sharma M, Chandana SR, Lugowska IA, Szczylik C, Zolnierek J. et al. Lorigerlimab, a bispecific PD-1×CTLA-4 DART molecule in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC): A phase 1 expansion (exp) cohort. J Clin Oncol. 2023;41:155
146. Polanczyk MJ, Walker E, Haley D, Guerrouahen BS, Akporiaye ET. Blockade of TGF-beta signaling to enhance the antitumor response is accompanied by dysregulation of the functional activity of CD4(+)CD25(+)Foxp3(+) and CD4(+)CD25(-)Foxp3(+) T cells. J Transl Med. 2019;17:219
147. Strauss J, Heery CR, Schlom J, Madan RA, Cao L, Kang Z. et al. Phase I Trial of M7824 (MSB0011359C), a Bifunctional Fusion Protein Targeting PD-L1 and TGFbeta, in Advanced Solid Tumors. Clin Cancer Res. 2018;24:1287-95
148. Knudson KM, Hicks KC, Luo X, Chen JQ, Schlom J, Gameiro SR. M7824, a novel bifunctional anti-PD-L1/TGFbeta Trap fusion protein, promotes anti-tumor efficacy as monotherapy and in combination with vaccine. Oncoimmunology. 2018;7:e1426519
149. Lan Y, Yeung TL, Huang H, Wegener AA, Saha S, Toister-Achituv M. et al. Colocalized targeting of TGF-beta and PD-L1 by bintrafusp alfa elicits distinct antitumor responses. J Immunother Cancer. 2022;10:e004122
150. Kantoff PW, Schuetz TJ, Blumenstein BA, Glode LM, Bilhartz DL, Wyand M. et al. Overall survival analysis of a phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J Clin Oncol. 2010;28:1099-105
151. Zahm CD, Moseman JE, Delmastro LE, D GM. PD-1 and LAG-3 blockade improve anti-tumor vaccine efficacy. Oncoimmunology. 2021;10:1912892
152. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell. 2015;28:690-714
153. Ma Z, Zhang W, Dong B, Xin Z, Ji Y, Su R. et al. Docetaxel remodels prostate cancer immune microenvironment and enhances checkpoint inhibitor-based immunotherapy. Theranostics. 2022;12:4965-79
154. Garnett CT, Schlom J, Hodge JW. Combination of docetaxel and recombinant vaccine enhances T-cell responses and antitumor activity: effects of docetaxel on immune enhancement. Clin Cancer Res. 2008;14:3536-44
155. Guan W, Hu J, Yang L, Tan P, Tang Z, West BL. et al. Inhibition of TAMs improves the response to docetaxel in castration-resistant prostate cancer. Endocr Relat Cancer. 2019;26:131-40
156. Sinha M, Zhang L, Subudhi S, Chen B, Marquez J, Liu EV. et al. Pre-existing immune status associated with response to combination of sipuleucel-T and ipilimumab in patients with metastatic castration-resistant prostate cancer. J Immunother Cancer. 2021;9:e002254
157. Fizazi K, Gonzalez Mella P, Castellano D, Minatta JN, Rezazadeh Kalebasty A, Shaffer D. et al. Nivolumab plus docetaxel in patients with chemotherapy-naive metastatic castration-resistant prostate cancer: results from the phase II CheckMate 9KD trial. Eur J Cancer. 2022;160:61-71
158. Fizazi K, Retz M, Petrylak DP, Goh JC, Perez-Gracia J, Lacombe L. et al. Nivolumab plus rucaparib for metastatic castration-resistant prostate cancer: results from the phase 2 CheckMate 9KD trial. J Immunother Cancer. 2022;10:e004761
159. Long X, Hou H, Wang X, Liu S, Diao T, Lai S. et al. Immune signature driven by ADT-induced immune microenvironment remodeling in prostate cancer is correlated with recurrence-free survival and immune infiltration. Cell Death Dis. 2020;11:779
160. Slovin SF, Higano CS, Hamid O, Tejwani S, Harzstark A, Alumkal JJ. et al. Ipilimumab alone or in combination with radiotherapy in metastatic castration-resistant prostate cancer: results from an open-label, multicenter phase I/II study. Ann Oncol. 2013;24:1813-21
161. Dudzinski SO, Cameron BD, Wang J, Rathmell JC, Giorgio TD, Kirschner AN. Combination immunotherapy and radiotherapy causes an abscopal treatment response in a mouse model of castration resistant prostate cancer. J Immunother Cancer. 2019;7:218
162. Yuan Z, Fernandez D, Dhillon J, Abraham-Miranda J, Awasthi S, Kim Y. et al. Proof-of-principle Phase I results of combining nivolumab with brachytherapy and external beam radiation therapy for Grade Group 5 prostate cancer: safety, feasibility, and exploratory analysis. Prostate Cancer Prostatic Dis. 2021;24:140-9
163. Gillessen S, Attard G, Beer TM, Beltran H, Bjartell A, Bossi A. et al. Management of Patients with Advanced Prostate Cancer: Report of the Advanced Prostate Cancer Consensus Conference 2019. Eur Urol. 2020;77:508-47
164. Bobba KN, Bidkar AP, Wadhwa A, Meher N, Drona S, Sorlin AM. et al. Development of CD46 targeted alpha theranostics in prostate cancer using (134)Ce/(225)Ac-Macropa-PEG(4)-YS5. Theranostics. 2024;14:1344-60
165. Kim JW, Shin MS, Kang Y, Kang I, Petrylak DP. Immune Analysis of Radium-223 in Patients With Metastatic Prostate Cancer. Clin Genitourin Cancer. 2018;16:e469-e76
166. Choudhury AD, Kwak L, Cheung A, Tripathi A, Pace AF, Van Allen EM. et al. Randomized phase II study evaluating the addition of pembrolizumab to radium-223 in metastatic castration-resistant prostate cancer. J Clin Oncol. 2021;39:98
167. Fong L, Morris MJ, Sartor O, Higano CS, Pagliaro L, Alva A. et al. A Phase Ib Study of Atezolizumab with Radium-223 Dichloride in Men with Metastatic Castration-Resistant Prostate Cancer. Clin Cancer Res. 2021;27:4746-56
168. Aggarwal R, Trihy L, Romero EH, Sam SL, Rastogi M, De Kouchkovsky I. et al. 1379P A phase Ib study of a single priming dose of 177Lu-PSMA-617 coupled with pembrolizumab in metastatic castration resistant prostate cancer (mCRPC). Ann Oncol. 2022;33:S1173-3
169. Arbuznikova D, Klotsotyra A, Uhlmann L, Domogalla LC, Steinacker N, Mix M. et al. Exploring the role of combined external beam radiotherapy and targeted radioligand therapy with [(177)Lu]Lu-PSMA-617 for prostate cancer - from bench to bedside. Theranostics. 2024;14:2560-72
170. Guan X, Polesso F, Wang C, Sehrawat A, Hawkins RM, Murray SE. et al. Androgen receptor activity in T cells limits checkpoint blockade efficacy. Nature. 2022;606:791-6
171. Merck. Merck Provides Update on Phase 3 Trials KEYNOTE-641 and KEYNOTE-789. Merck.com/news: Merck & Co, Inc. 2023
172. Merck. Merck Announces KEYNOTE-991 Trial Evaluating KEYTRUDA® (pembrolizumab) Plus Enzalutamide and Androgen Deprivation Therapy in Patients With Metastatic Hormone-Sensitive Prostate Cancer to Stop for Futility. Merck.com/news: Merck & Co, Inc.
173. Powles T, Yuen KC, Gillessen S, Kadel EE 3rd, Rathkopf D, Matsubara N. et al. Atezolizumab with enzalutamide versus enzalutamide alone in metastatic castration-resistant prostate cancer: a randomized phase 3 trial. Nat Med. 2022;28:144-53
174. Yu EY, Piulats JM, Gravis G, Fong PCC, Todenhofer T, Laguerre B. et al. Pembrolizumab plus Olaparib in Patients with Metastatic Castration-resistant Prostate Cancer: Long-term Results from the Phase 1b/2 KEYNOTE-365 Cohort A Study. Eur Urol. 2023;83:15-26
175. Karzai F, VanderWeele D, Madan RA, Owens H, Cordes LM, Hankin A. et al. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J Immunother Cancer. 2018;6:141
176. Antonarakis ES, Park SH, Goh JC, Shin SJ, Lee JL, Mehra N. et al. Pembrolizumab Plus Olaparib for Patients With Previously Treated and Biomarker-Unselected Metastatic Castration-Resistant Prostate Cancer: The Randomized, Open-Label, Phase III KEYLYNK-010 Trial. J Clin Oncol. 2023;41:3839-50
177. Coleman DJ, Gao L, King CJ, Schwartzman J, Urrutia J, Sehrawat A. et al. BET bromodomain inhibition blocks the function of a critical AR-independent master regulator network in lethal prostate cancer. Oncogene. 2019;38:5658-69
178. Agarwal N, Azad A, Carles J, Chowdhury S, McGregor B, Merseburger AS. et al. A phase III, randomized, open-label study (CONTACT-02) of cabozantinib plus atezolizumab versus second novel hormone therapy in patients with metastatic castration-resistant prostate cancer. Future Oncol. 2022;18:1185-98
179. Agarwal N, Azad A, Carles J, Matsubara N, oudard S, Saad F. et al. CONTACT-02: Phase 3 study of cabozantinib (C) plus atezolizumab (A) vs second novel hormonal therapy (NHT) in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. 2024;42:18
180. Plieth J. ASCO-GU - picking apart Exelixis's Contact-02 win. Oncologypipeline.com: ApexOnco. 2024
181. Lu X, Horner JW, Paul E, Shang X, Troncoso P, Deng P. et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature. 2017;543:728-32
182. Feng S, Cheng X, Zhang L, Lu X, Chaudhary S, Teng R. et al. Myeloid-derived suppressor cells inhibit T cell activation through nitrating LCK in mouse cancers. Proc Natl Acad Sci U S A. 2018;115:10094-9
183. Peng S, Hu P, Xiao YT, Lu W, Guo D, Hu S. et al. Single-Cell Analysis Reveals EP4 as a Target for Restoring T-Cell Infiltration and Sensitizing Prostate Cancer to Immunotherapy. Clin Cancer Res. 2022;28:552-67
184. Qi Z, Xu Z, Zhang L, Zou Y, Li J, Yan W. et al. Overcoming resistance to immune checkpoint therapy in PTEN-null prostate cancer by intermittent anti-PI3Kalpha/beta/delta treatment. Nat Commun. 2022;13:182
185. Chaudagar K, Hieromnimon HM, Khurana R, Labadie B, Hirz T, Mei S. et al. Reversal of Lactate and PD-1-mediated Macrophage Immunosuppression Controls Growth of PTEN/p53-deficient Prostate Cancer. Clin Cancer Res. 2023;29:1952-68
186. Unal B, Kuzu OF, Jin Y, Osorio D, Kildal W, Pradhan M. et al. Targeting IRE1α reprograms the tumor microenvironment and enhances anti-tumor immunity in prostate cancer. Nat commun. 2024;15:8895
187. Anker JF, Naseem AF, Mok H, Schaeffer AJ, Abdulkadir SA, Thumbikat P. Multi-faceted immunomodulatory and tissue-tropic clinical bacterial isolate potentiates prostate cancer immunotherapy. Nat Commun. 2018;9:1591
188. Shi W, Wang Y, Zhao Y, Kim JJ, Li H, Meng C. et al. Immune checkpoint B7-H3 is a therapeutic vulnerability in prostate cancer harboring PTEN and TP53 deficiencies. Sci Transl Med. 2023;15:eadf6724
189. Mukherjee D, Romano E, Walshaw R, Zeef LAH, Banyard A, Kitcatt SJ. et al. Reprogramming the immunosuppressive tumor microenvironment results in successful clearance of tumors resistant to radiation therapy and anti-PD-1/PD-L1. Oncoimmunology. 2023;12:2223094
190. Vardaki I, Corn P, Gentile E, Song JH, Madan N, Hoang A. et al. Radium-223 Treatment Increases Immune Checkpoint Expression in Extracellular Vesicles from the Metastatic Prostate Cancer Bone Microenvironment. Clin Cancer Res. 2021;27:3253-64
191. Yenyuwadee S, Sanchez-Trincado Lopez JL, Shah R, Rosato PC, Boussiotis VA. The evolving role of tissue-resident memory T cells in infections and cancer. Sci Adv. 2022;8:eabo5871
192. Burbach BJ, O'Flanagan SD, Shao Q, Young KM, Slaughter JR, Rollins MR. et al. Irreversible electroporation augments checkpoint immunotherapy in prostate cancer and promotes tumor antigen-specific tissue-resident memory CD8+ T cells. Nat Commun. 2021;12:3862
193. Waitz R, Solomon SB, Petre EN, Trumble AE, Fasso M, Norton L. et al. Potent induction of tumor immunity by combining tumor cryoablation with anti-CTLA-4 therapy. Cancer Res. 2012;72:430-9
194. Benzon B, Glavaris SA, Simons BW, Hughes RM, Ghabili K, Mullane P. et al. Combining immune check-point blockade and cryoablation in an immunocompetent hormone sensitive murine model of prostate cancer. Prostate Cancer Prostatic Dis. 2018;21:126-36
195. Allard B, Pommey S, Smyth MJ, Stagg J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin Cancer Res. 2013;19:5626-35
196. Ahern E, Harjunpaa H, O'Donnell JS, Allen S, Dougall WC, Teng MWL. et al. RANKL blockade improves efficacy of PD1-PD-L1 blockade or dual PD1-PD-L1 and CTLA4 blockade in mouse models of cancer. Oncoimmunology. 2018;7:e1431088
197. Liu J, He D, Cheng L, Huang C, Zhang Y, Rao X. et al. p300/CBP inhibition enhances the efficacy of programmed death-ligand 1 blockade treatment in prostate cancer. Oncogene. 2020;39:3939-51
198. Tang Z, Pilie PG, Geng C, Manyam GC, Yang G, Park S. et al. ATR Inhibition Induces CDK1-SPOP Signaling and Enhances Anti-PD-L1 Cytotoxicity in Prostate Cancer. Clin Cancer Res. 2021;27:4898-909
199. Mao W, Ghasemzadeh A, Freeman ZT, Obradovic A, Chaimowitz MG, Nirschl TR. et al. Immunogenicity of prostate cancer is augmented by BET bromodomain inhibition. J Immunother Cancer. 2019;7:277
200. Morel KL, Sheahan AV, Burkhart DL, Baca SC, Boufaied N, Liu Y. et al. EZH2 inhibition activates a dsRNA-STING-interferon stress axis that potentiates response to PD-1 checkpoint blockade in prostate cancer. Nat Cancer. 2021;2:444-56
Author contact
![]() Corresponding author: Fahri Saatcioglu, Postboks 1066 Blindern, 0316 Oslo, Tel: +47 22854569; Fax: +47 22857207, Email: fahrisno.
Corresponding author: Fahri Saatcioglu, Postboks 1066 Blindern, 0316 Oslo, Tel: +47 22854569; Fax: +47 22857207, Email: fahrisno.