Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2024; 14(11):4536-4553. doi:10.7150/thno.95796 This issue Cite
Research Paper
ISG15 accelerates acute kidney injury and the subsequent AKI-to-CKD transition by promoting TGFβR1 ISGylation
Na Cui1#, Chengyu Liu2#, Xiang Tang1, Liangliang Song1, Zixuan Xiao3, Chen Wang1, Yancai Wu1, Yihao Zhou4, Chentai Peng1, Yuxia Liu4, Ling Zheng4, Xinran Liu1, Kun Huang1, Hong Chen1 ![]()
1. Tongji School of Pharmacy, Huazhong University of Science and Technology, Wuhan, China, 430030.
2. Department of Transfusion Medicine, Wuhan Hospital of Traditional Chinese and Western Medicine, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China, 430000.
3. ISA Wenhua Wuhan High School, Fenglin Road, Junshan New Town, Wuhan Economics & Technological Development Zone, Wuhan, Hubei, China, 430119.
4. Hubei Key Laboratory of Cell Homeostasis, College of Life Sciences, Wuhan University, Wuhan, China, 430072.
#Equal contribution.
Received 2024-2-29; Accepted 2024-7-17; Published 2024-7-22
Abstract

Rationale: Acute kidney injury (AKI) has substantial rates of mortality and morbidity, coupled with an absence of efficacious treatment options. AKI commonly transits into chronic kidney disease (CKD) and ultimately culminates in end-stage renal failure. The interferon-stimulated gene 15 (ISG15) level was upregulated in the kidneys of mice injured by ischemia-reperfusion injury (IRI), cisplatin, or unilateral ureteral obstruction (UUO), however, its role in AKI development and subsequent AKI-to-CKD transition remains unknown.
Methods: Isg15 knockout (Isg15 KO) mice challenged with bilateral or unilateral IRI, cisplatin, or UUO were used to investigate its role in AKI. We established cellular models with overexpression or knockout of ISG15 and subjected them to hypoxia-reoxygenation, cisplatin, or transforming growth factor- β1 (TGF-β1) stimulation. Renal RNA-seq data obtained from AKI models sourced from public databases and our studies, were utilized to examine the expression profiles of ISG15 and its associated genes. Additionally, published single cell RNA-seq data from human kidney allograft biopsies and mouse IRI model were analyzed to investigate the expression patterns of ISG15 and the type I TGF-β receptor (TGFβR1). Western blotting, qPCR, co-immunoprecipitation, and immunohistochemical staining assays were performed to validate our findings.
Results: Alleviated pathological injury and renal function were observed in Isg15 KO mice with IRI-, cisplatin-, or UUO-induced AKI and the following AKI-to-CKD transition. In hypoxia-reoxygenation, cisplatin or TGF-β1 treated HK-2 cells, knockout ISG15 reduced stimulus-induced cell fibrosis, while overexpression of ISG15 with modification capacity exacerbated cell fibrosis. Immunoprecipitation assays demonstrated that ISG15 promoted ISGylation of TGFβR1, and inhibited its ubiquitination. Moreover, knockout of TGFβR1 blocked ISG15's fibrosis-exacerbating effect in HK-2 cells, while overexpression of TGFβR1 abolished the renal protective effect of ISG15 knockout during IRI-induced kidney injury.
Conclusions: ISG15 plays an important role in the development of AKI and subsequent AKI-to-CKD transition by promoting TGFβR1 ISGylation.
Keywords: ISG15, TGFβR1, acute kidney injury, fibrosis, AKI-to-CKD transition
Introduction
Over 10 million individuals worldwide receive new diagnoses of acute kidney injury (AKI), a potentially fatal syndrome characterized by a rapid decline in kidney function, resulting in approximately 1.7 million deaths every year [1, 2]. Ischemia-reperfusion injury (IRI), unilateral urethral obstruction (UUO), platinum-based chemotherapeutic agents (cisplatin), contrast agents and other factors are important triggers for AKI [3-8]. AKI may develop into irreversible renal fibrosis, which can trigger serious chronic kidney diseases (CKD) [9, 10].
Fibrosis entails the gradual accumulation of extracellular matrix constituents in the kidney, prolonged or extensive fibrosis in AKI contributes to CKD and end-stage renal failure [11, 12]. Transforming growth factor-β1 (TGF-β1) is recognized as the most crucial pro-fibrotic factor and the primary signaling pathway driving renal fibrosis [8, 13]. TGF-β1 binds and phosphorylates type II TGF-β receptor (TGFβR2), which subsequently phosphorylates the type I TGF-β receptor (TGFβR1) [8, 13]. Activation of TGFβR1 then phosphorylates Smad2/3, facilitating their activation and translocation into the nucleus where they bind to relevant co-factors, thereby regulating the transcription of fibrotic genes such as α-smooth muscle actin (α-SMA) and fibronectin 1 (Fn1), thus promoting renal fibrosis [14, 15]. On the other hand, activation of TGFβR1 may also trigger Smad-independent signaling, inducing the expression of fibrotic genes [8, 14, 16]. Understanding the role of TGFβR1 in this process is essential for developing therapies mitigating fibrosis and preserving renal function in the AKI and the following transition to CKD.
Interferon-stimulated gene 15 (ISG15), an ubiquitin-like protein, initially presents as a 17 kDa precursor protein which undergoes rapid protease-mediated cleavage to yield a mature 15 kDa form (ISG15GG) exposing a C-terminal LRLRGG motif [17]. Structurally, ISG15 comprises two β-grasp domains akin to ubiquitin, positioned at its N- and C-terminal, linked by a short hinge region [18]. Notably, both the hinge region and the C-terminal LRLRGG residues are indispensable for ISG15 modification [19]. ISG15 engages in covalent conjugation with target proteins via this LRLRGG motif through a three-step process termed ISGylation [20]. Initially, ISG15 is activated by the E1 enzyme UBA7, followed by conjugation facilitated by the E2 enzyme UBCH8, and ultimately ligation mediated by three identified E3 enzymes: HERC5/6, TRIM25, and HHARI. ISGylation is intricately involved in the regulation of various biological processes [19, 21]. Concurrently, free ISG15 has been implicated in diverse signaling pathways, including immune regulation [22]. Despite its involvement in numerous critical cellular processes, the precise role of ISG15 in AKI and subsequent AKI-to-CKD transition remains elusive.
In this study, Isg15 knockout (Isg15 KO) mice were challenged with IRI, cisplatin, or UUO injury. Injured Isg15 KO mice showed attenuated pathologies and fibrosis, with improved renal function. Additionally, knockout of ISG15 suppressed the cell fibrosis induced by hypoxia-reoxygenation (HR), cisplatin and TGF-β1, while overexpression of ISG15 with conjugation capacity (ISG15GG) has the opposite effect. The Venn analysis of RNA-seq data from three types of AKI animal models, along with in silico analysis of protein interactions, suggests a correlation between ISG15 and TGFβR1. Further exogenous and endogenous co-immunoprecipitation (co-IP) assays demonstrated that ISG15 covalently modified TGFβR1 and suppressed its ubiquitination, thereby enhancing the protein stability of TGFβR1. Notably, TGFβR1 overexpression aggregated renal fibrosis in vivo and in vitro. Moreover, overexpression of TGFβR1 blocked the renal protective effect of ISG15 knockout in IRI-treated mice. Consistently, overexpression of ISG15 failed to promote fibrosis in TGFβR1 knockout HK-2 cells. Together, our results reveal a novel role of ISG15 in regulating renal fibrosis and suggest a potential therapeutic approach against AKI and following AKI-to-CKD transition.
Materials and Methods
Animals
Breeding pairs of Isg15+/- mice in C57BL/6 background were generated by Cyagen Biosciences (Suzhou, Jiangsu, China). Age-matched male Isg15 KO mice and wildtype (WT) littermates were used. Genotyping was performed (primers listed in Table S1). Expected Mendelian ratio of mice with different genotypes was found in this study. Male C57BL/6 mice (10-week-old, 25 ± 3 g) were obtained from the Hubei Center for Disease Control and Prevention. Mice were randomly assigned, and housed in a specific-pathogen-free, temperature-controlled (22 ± 1℃) animal facility with a 12-hr light/dark cycle, with free access to water/food. Animals were handled according to the Guidelines of the China Animal Welfare Legislation, as approved by the Committee on Ethics in the Care and Use of Laboratory Animals, College of Life Sciences, Wuhan University (WDSKY0201705-2, Wuhan, China).
AKI models and treatments
Ischemic AKI was induced using both bilateral IRI (BIRI) and unilateral renal IRI (UIRI) models. For the BIRI model, mouse was anesthetized and underwent midline abdominal incisions, both kidneys were clamped to block blood flow for 22 minutes. After ischemia, clamps were released to start reperfusion. For the UIRI model was used as we previously described [23, 24], left renal pedicle of mouse was bluntly clamped for 30 minutes, reperfusion was achieved by removing the clamp. Sham operated mice (Sham) were used as respective controls. Mice were sacrificed at day 1, 2 or 3 after BIRI; or at day 1, 3, 7 and 21 after UIRI. Death of some animals were observed at Day 3 of BIRI, but not during the UIRI study. For cisplatin-induced AKI, a single dose of 30 mg/kg body-weight cisplatin (T1564, Targetmol, Shanghai, China) was injected intraperitoneally and the mice were euthanized at Day 3 after the injection [25]. For the UUO model, the left ureter of the mouse was ligated at proximal and distal points and then cut between the ligated points [26]. Mice were sacrificed 7 days later and kidneys were collected.
Intrarenal adenovirus delivery
TGFβR1 overexpression adenovirus and control adenovirus (Genechem, Shanghai, China) were delivered into the mouse kidney by renal intraparenchymal injection, followed by IRI modeling experiments. Initially, the mice were temporarily anesthetized, and their abdominal cavity was opened to expose the unilateral kidneys. Then, 150 μL of TGFβR1 or control adenovirus (6 × 1010 pfu/mL) was aspirated with a 31G needle. Four to six sites were selected from each kidney the slow injection of the virus into the renal cortex, as previously described [27].
Assessment of renal function
Renal function was evaluated by measuring serum creatinine (CREA) and blood urea nitrogen (BUN) levels using a creatinine reagent kit and a BUN reagent kit [28], respectively (both from Jiancheng Bio., Nanjing, China).
Histological and immunohistochemical studies
Paraffin-embedded sections were deparaffinized and rehydrated as previously reported [29, 30]. For pathological evaluation, H&E stained renal sections were assessed and evaluated as previously described [29, 30]. For evaluation of fibrosis, a Masson staining kit (Solarbio, Wuhan, China) was used. For immunohistochemical staining, primary antibodies for α-SMA, Fn1 (information listed in Table S2) were applied overnight at 4 °C. Sections were then incubated with respective biotinylated secondary antibody, followed by incubation in ABC-peroxidase solution (Vector laboratories, Burlingame, CA), and visualized using 3,3′-diaminobenzidine (DAB, CWbiotech, Beijing, China). Quantitative analysis of positively stained cells was performed as previously reported [29, 30]. For immunofluorescent staining, tissue sections and cryosections were incubated overnight with respective primary antibodies (information listed in Table S2). After washing, sections were incubated with respective Alexa Fluor secondary antibody (Thermo Fisher, Waltham, MA). Sections were covered with DAPI and anti-fading medium. Images were taken by a confocal microscope (Nikon AX2, Japan).
Cell culture
Human renal tubular cell line HK-2 (GDC0152, from CCTCC, China Center for Type Culture Collection, Wuhan, China) was cultured in DMEM/F12 media (Hyclone, Palo Alto, CA) plus 10% FBS. HEK293T cells (CL-0005, Procell Biotech) were cultured in DMEM media (Hyclone) containing 10% FBS. HK-2 and HEK293T cells were analyzed with authenticated STR locus and tested for mycoplasma contamination, by CCTCC or Procell Biotech. In vitro HR experiments were performed as previously described with slight modifications [31]. At 80% confluence, HK-2 cells were placed in a 37 ℃ incubator under 1% O2 with the regular media replaced by the glucose- and FBS-free media to mimic hypoxia. After 24-hr culturing under the hypoxia condition, HK-2 cells were returned to the regular culture condition for another hour to simulate reperfusion. For cisplatin treatments, HK-2 cells were treated with 5 μg/mL cisplatin for 24 hrs. For TGF-β1 (Peprotech, 100-21-10) treatment, HK-2 cells were treated with 10 ng/mL TGF-β1 for 24 hrs.
Plasmids and transfection
Plasmids encoding human ISG15GG (myc-ISG15GG), a mature form capable of ISGylation, and ISG15AA (myc-ISG15AA), which lacks ISGylation activity and represents free ISG15, were generously provided by Dr. Jin-Hyun Ahn (University of Erlangen-Nuremberg). The CRISPR-Cas9-based protocols for genome engineering were used as described [32]. CRISPR-Cas9 vectors containing gRNA targeting ISG15 (5'-ATCTGCCTTACCATGGCTG-3'), TGFβR1 (5'-CACCGCATACAAACGGCCTATCTCG-3'), were used to knockout ISG15, TGFβR1, respectively. Expression plasmids for HA-tagged E1, E2, E3 and Flag-tagged TGFβR1 were constructed (information listed in Table S1). Most cells were transfected with indicated plasmids for six hours, then treated with/without HR, cisplatin, TGF-β1 before collection.
Quantitative real-time PCR (qPCR)
qPCR was performed as we previously described [33]. Total RNA was isolated from the kidneys or cultured cells using RNAiso Plus (TaKaRa Biotech, Dalian, China). Total RNA was reverse transcribed into cDNA using an M-MLV first strand synthesis system (Invitrogen, Grand Island, NY). The abundance of specific gene transcripts was assessed by qPCR. The mRNA levels of specific genes were normalized to β-actin or Rn18s. Primers used are listed in Table S3.
Western blots
Freshly collected kidney or cultured cells were sonicated in ice-cold RIPA buffer (Beyotime), and protein concentrations were quantitated as we previously described [34, 35]. 20-80 μg protein from each sample was separated by SDS-PAGE. The proteins were transferred onto PVDF membranes for immune detection. Antibodies used are shown in Table S2.
Co-Immunoprecipitation (Co-IP)
Cells were lysed with pre-lysis buffer (25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 5% glycerol), cell lysate was sonicated and centrifuged at 12000 g for 10 min. Then cell lysates were incubated with the indicated antibodies (Table S2) or respective IgGs with Dynabeads Protein G (Thermo Scientific, Waltham, MA) overnight at 4 °C. After washing (pre-lysis buffer with additional 50 mM NaCl), the beads were boiled in loading buffer and subjected to immunoblotting [36, 37].
RNA sequencing (RNA-seq) and analysis
Total renal RNA was isolated from IRI-treated mice for RNA-seq, and the data has been uploaded to the NCBI Gene Expression Omnibus database (GSE183455) [33]. And published RNA-seq data deposited in NCBI GEO database (GSE216376 and GSE207587) were retrieved, and analyzed via GEO integrated analyzing tool (GEO2R) or R3.5.2 for the heatmap. Log2 fold change of differentially expressed genes and ISG15 were visualized.
Protein-Protein Interaction (PPI) Modeling of ISG15 and TGFβR1
ISG15- TGFβR1 interaction was modeled using Alphafold 2. Query sequence and multiple sequence alignment (MSA) from MMseqs2 served as input without templates [38]. MSA generation and AlphaFold 2 predictions were performed using ColabFold (https://colab.research.google.com/ssssub/sokrypton/ColabFold). Modeling results were analyzed using PDBePISA (www.ebi.ac.uk/msd-srv/prot_int/pistart.html). The binding strength of ISG15 and TGFβR1, TGFβR2, were analyzed by ΔiG (solvation energies) and root-mean-square deviation (RMSD).
Single cell RNA-seq (scRNA-seq) data analysis
The scRNA-seq data of human kidney allograft biopsy and mice IRI were obtained respectively from GSE109564 and GSE139107, which contain the gene expression matrix data and cell cluster annotations. Then, we performed data normalization and scaling according to standard pre-processing workflow of the Seurat (version V5, https://satijalab.org/seurat/).
Statistics
Data were analyzed with GraphPad Prism (version 8). Statistical analysis was performed using two-tailed Student's t test for two experimental groups, and one-way ANOVA for multiple experimental groups without adjustment. Data are reported as the mean values with error bars showing the standard deviation (SD). For cell culture experiments, at least three independent experiments were performed with similar results. For animal study, the n number of biological independent animals in different groups are provided in figure legends. A p value of less than 0.05 was considered statistically significant.
Results
ISG15 is upregulated during renal IRI-induced kidney injury
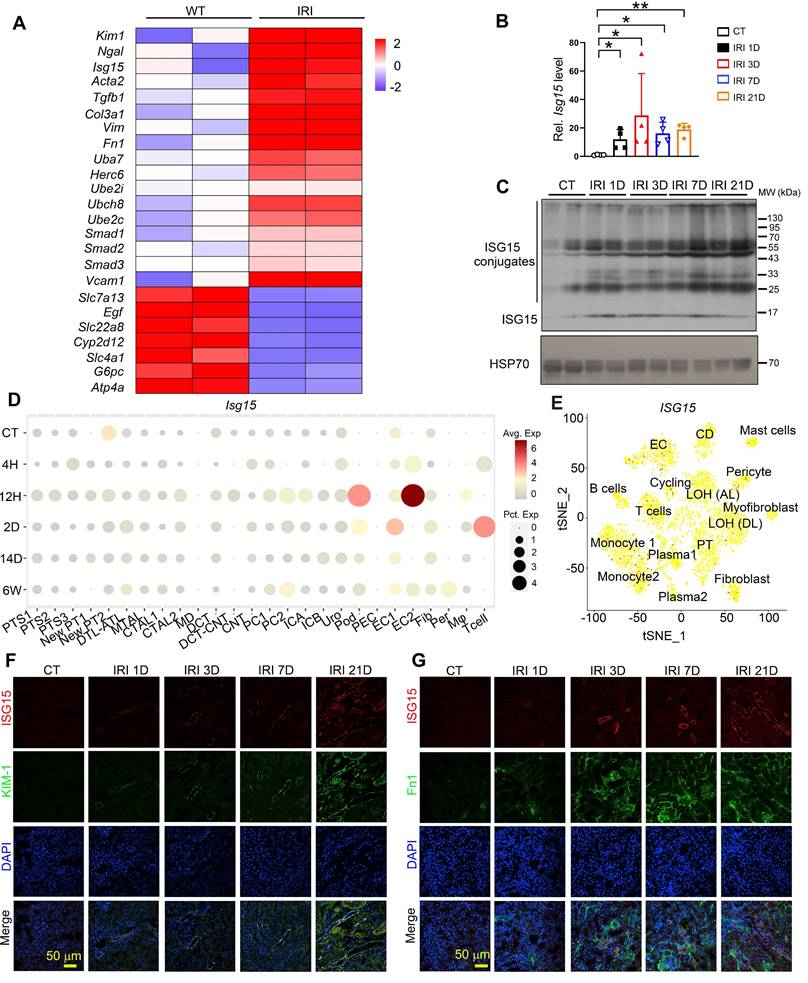
To investigate the role of ISG15 in IRI-induced kidney injury, we analyzed our previous RNA-seq data of IRI model, and found that Isg15 and its E1/E2/E3 ligases levels were increased in IRI injured kidneys (Figure 1A). Further validation through qPCR and Western blots confirmed that the gene and protein levels of ISG15 increased at different time points following IRI (Figure 1B-C). Moreover, to explore the cellular sources of ISG15 in IRI injured kidney, we analyzed published scRNA-seq data of a human kidney allograft biopsy and a mouse IRI model (Figure 1D-E). ISG15 was ubiquitously expressed in a variety of cells. However, with prolonged injury duration, sustained high expression of ISG15 is observed in the proximal tubules and fibroblasts (Figure 1D-E). Co-staining of ISG15 with KIM1 or Fn1 consistently demonstrated weak ISG15 staining in tubular cells and fibroblast under non-injury conditions; after IRI, a significantly increased ISG15 level was observed in renal tubules and fibroblast of IRI mice (Figure 1F-G).
ISG15 is up-regulated in the kidney of mice after renal IR injury. (A) RNA-seq data of ISG15 and its related ligase genes, and fibrotic genes in the kidneys of IRI-treated mice. (B) qPCR results of Isg15 of non-injured (CT) or IRI-treated mice at 1, 3, 7, or 21 days, n = 4 per group, *P < 0.05; **P < 0.01. (C) Western blot of ISG15 at indicated times after IRI, n = 2 per group. (D) single cell RNA-seq data of ISG15 in the kidneys of IRI mice. CT, control; 4H, 4 hours; 12H, 12 hours; 2D, 2 days; 14D, 14 days; 6W, 6 weeks. (E) tSNE plot of ISG15 expressed cell clusters from single cell RNA-seq of a human kidney allograft biopsy. CD, collecting duct; EC, endothelial cell; LOH (AL), loop of Henle, ascending limb; LOH (DL), loop of Henle, distal limb; PT, proximal tubule. (F) Representative images of ISG15 (red), KIM-1 (green) and DAPI (blue) staining of the kidney. (G) Representative images of ISG15 (red), Fn1 (green) and DAPI (blue) staining of the kidney. CT, non-injured mice; IRI 1D/3D/7D/21D, mice at IRI 1/3/7/21 days, n = 4 per group. Scale bar = 50 μm.
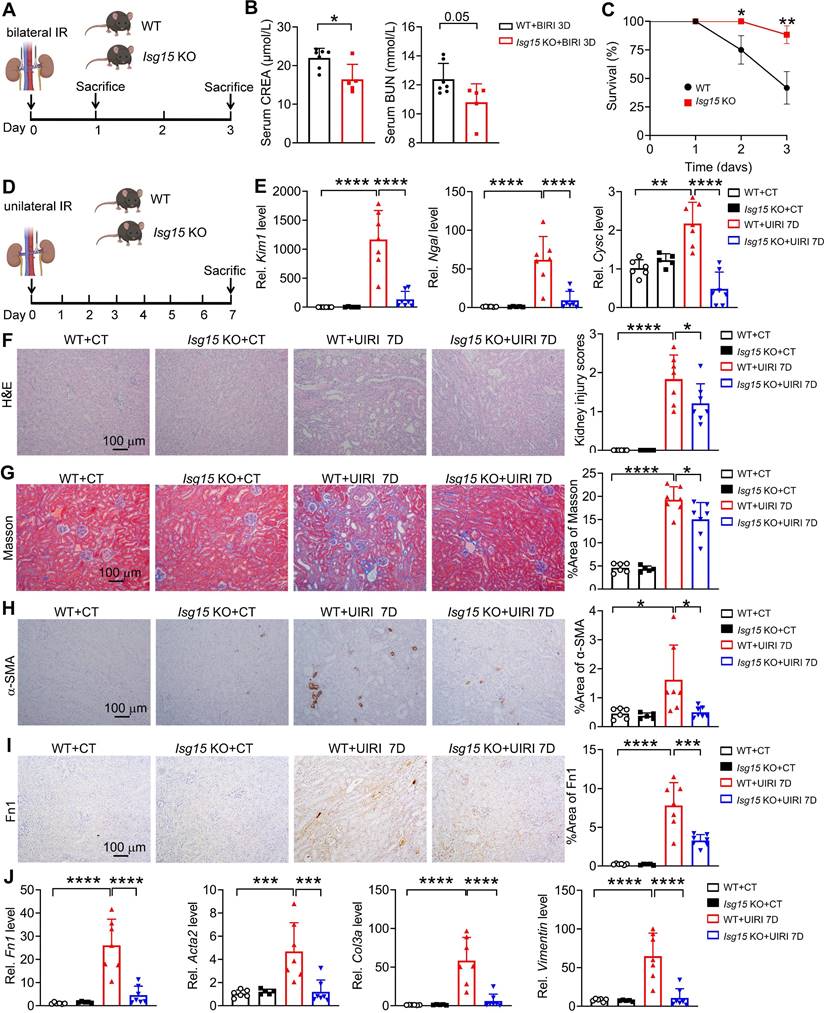
Knockout of ISG15 attenuates renal IRI induced kidney injury
To study the role of ISG15 in renal IRI, Isg15 knockout (Isg15 KO; Isg15-/-) mice and their age-matched wildtype (WT; Isg15+/+) littermates were studied (Figure S1A-B). The deletion efficiency of ISG15 in the kidney was examined, ISG15 gene and protein levels were barely detectable in the kidneys of Isg15 KO mice (Figure S1C-D).
In order to gain a detailed understanding of the specific role of ISG15 in AKI and following AKI-to-CKD transition, we established multiple kidney injury models and collected samples at different time points. At first, knockout of ISG15 did not affect renal function in mice subjected to bilateral IRI (BIRI) at day 1 (Figure S2); however, at day 3 post-BIRI, ISG15 knockout significantly improved renal function and reduced mortality caused by BIRI (Figure 2A-C). Due to the high mortality rate observed in mice subjected to BIRI, we established unilateral IRI (UIRI) models for 7 days (Figure 2D). ISG15 knockout significantly reduced kidney damage in mice at 7 days post-UIRI (Figure 2E-F). Fibrosis markers, such as collagen deposition (Masson staining), α-smooth muscle actin (α-SMA, gene Acta2) and Fibronectin (Fn1) were analyzed, while Isg15 KO markedly decreased the collagen deposition, and levels of α-SMA and Fn1 after renal UIRI (Figure 2G-I). Additionally, the transcriptional levels of ISGylation related E1/E2/E3 ligases (UBA7, UBCH8, HERC6) were also increased in the kidneys of UIRI-injured mice (Figure S3A). And, the mRNA levels of fibrotic genes (Fn1, Acta2, Col3a, Vimentin) were markedly elevated after renal UIRI (Figure 2J); Isg15 KO significantly inhibited the transcriptional levels of Fn1, Acta2, Col3a, Vimentin (Figure 2J).
Knockout of ISG15 suppresses IRI-induced kidney injury. (A) Experimental design chart of bilateral IRI (BIRI). (B) Serum creatinine (left) and BUN (right) levels for indicated experimental groups at 3 days after BIRI 3D. WT+BIRI 3D, n = 7; Isg15 KO+BIRI 3D, n = 5. (C) Survival rate of WT and Isg15 KO mice were subjected to BIRI. WT+BIRI, n = 12; Isg15 KO+BIRI, n = 14. (D) Experimental design chart of unilateral IRI (UIRI). (E) qPCR results of Kim1, Ngal, Cysc for WT and Isg15 KO mice at non-injured conditions (CT) or at 7 days after UIRI. (F) Representative H&E images (left) with injury scores (right) of the kidney of WT and Isg15 KO mice at non-injured conditions (CT) or at 7 days after UIRI. Scale bar = 100 μm. (G-I) Representative images and quantitative results of Masson staining (G), immunostaining for α-SMA (H), Fn1 (I) of the kidney of WT and Isg15 KO mice at non-injured conditions (CT) or at 7 days after UIRI. Scale bar = 100 μm. (J) qPCR results of Fn1, Acta2, Col3a and Vimentin in the kidney of WT and Isg15 KO mice at non-injured conditions (CT) or at 7 days after UIRI. WT+CT, n = 6; Isg15 KO+CT, n = 5; WT+UIRI 7D, n = 7; Isg15 KO+UIRI 7D, n = 7. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
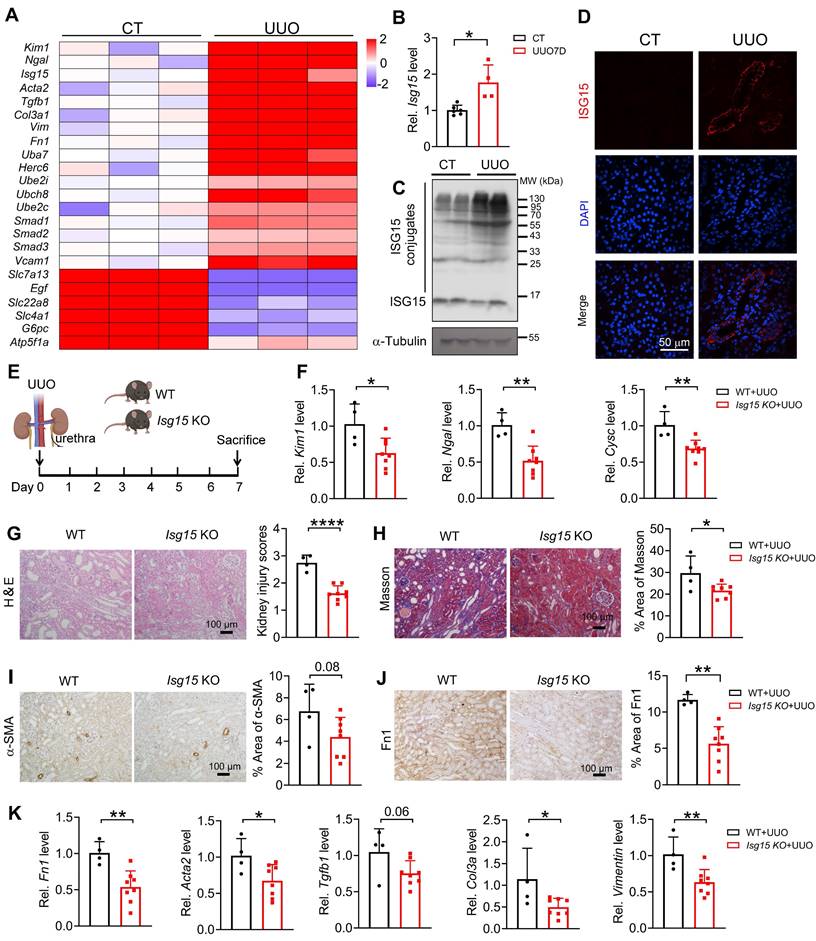
Knockout of ISG15 improves UUO- or cisplatin- induced kidney injury
To investigate whether ISG15 also responds in other renal injury models, UUO- or cisplatin-induced kidney injury model was applied. Similar to IRI model, we analyzed published RNA-seq data of UUO injured kidney, Isg15 and its related E1/E2/E3 ligases levels were significantly increased after UUO injury (Figure 3A). Consistent with the RNA-seq data, ISG15 transcriptional and protein levels, as well as its related E1/E2/E3 ligases levels were upregulated in the tubular of UUO injured kidneys (Figure 3B-D and Figure S3B). Isg15 KO consistently decreased the mRNA levels of the kidney injury markers Kim1, Ngal, Cysc, and inhibited renal pathological injury in the UUO injured mice (Figure 3E-G). Masson, α-SMA and Fn1 staining showed reduced fibrosis in Isg15 KO mice compared to WT mice after UUO injury (Figure 3H-J). Meanwhile, the mRNA levels of fibrotic factors Fn1, Acta2, Col3a, Vimentin were decreased in UUO-injured Isg15 KO mice (Figure 3K).
Knockout of ISG15 suppresses UUO-induced kidney injury. (A) RNA-seq data of ISG15 and its related ligase genes, and fibrotic genes in the kidneys of UUO injured mice. (B-C) qPCR (B) and WB (C) results of ISG15 of non-injured (CT) or UUO injured mice. (D) Representative images of ISG15 (red), and DAPI (blue) staining of the kidney of CT or UUO injured mice. CT, n = 6; UUO, n = 5. (E) Experimental design chart of UUO injury. (F) qPCR results of Kim1, Cysc, Ngal in the kidneys of WT and Isg15 KO mice after UUO injury. (G) Representative H&E images (left) with injury scores (right) of the kidney of WT and Isg15 KO mice after UUO injury. Scale bar = 100 μm. (H-J) Representative images and quantitative results of Masson staining (H), immunostaining for α-SMA (I), Fn1 (J) of the kidney of WT and Isg15 KO mice after UUO injury. Scale bar = 100 μm. (K) qPCR results of Fn1, Tgfb1, Acta2, Col3a and Vimentin in the kidney of WT and Isg15 KO mice after UUO injury. WT+UUO, n = 4; Isg15 KO+UUO, n = 8. *P < 0.05; **P < 0.01.
Meantime, in published RNA-seq data of cisplatin-injured kidney, Isg15 and its related E1/E2/E3 ligases, TGF-β1 signaling gene levels were increased at day 3 post cisplatin injection (Figure S4A). Consistently, ISG15 transcriptional and protein levels were upregulated in the tubular of cisplatin-injured kidneys (Figure S4B-D). Knockout of Isg15 decreased serum creatinine and BUN levels, improved tubular injury, reduced fibrosis in cisplatin-injured mice (Figure S4E-L).
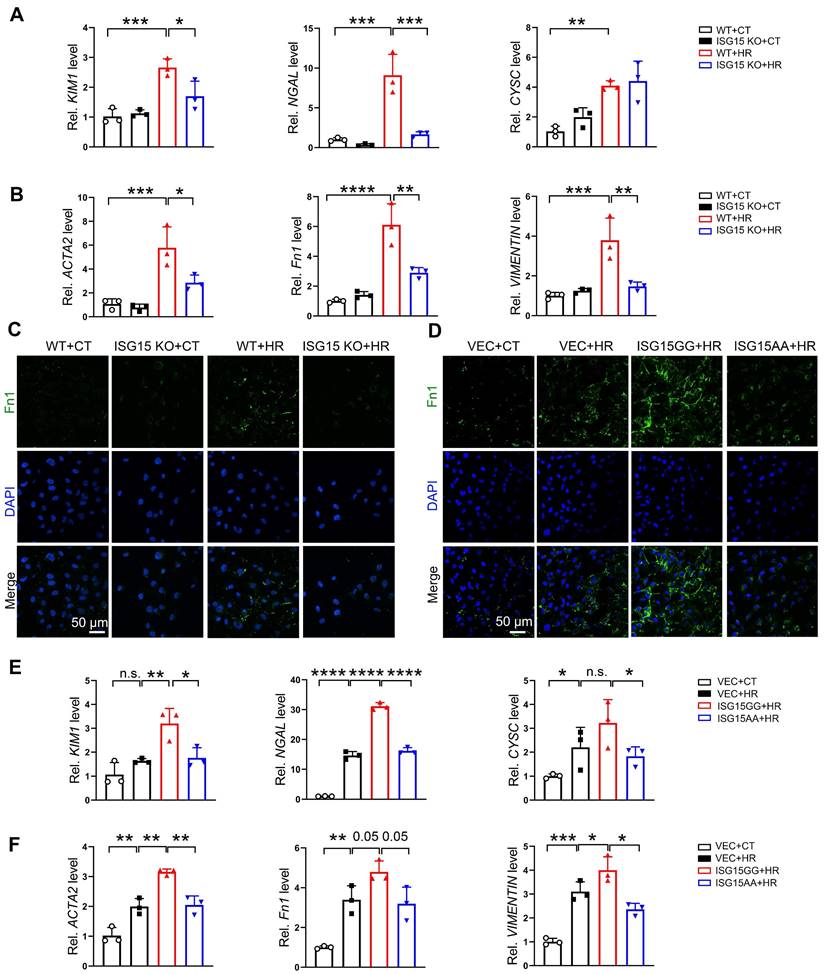
ISG15 exacerbates HR induced renal fibrosis in cultured renal tubular cells
To clarify the role of ISG15 in the AKI and AKI-to-CKD transition, we investigated the effects of ISG15 on HR-injured cells in vitro. ISG15 knockout or overexpressed HK-2 cells were generated (Figure S5A-B). The mRNA levels of the kidney injury and fibrotic factors, as well as Fn1 protein level, were up-regulated in HR treated cells, while knockout of ISG15 suppressed the KIM1, NGAL, ACTA2, Fn1, VIMENTIN, and Fn1 protein level in HR-treated HK-2 cells (Figure 4A-C). Notably, in HR-treated HK-2 cells, overexpression of ISG15GG (mature form with ISGylation capability) aggravated HR induced cell fibrosis, while overexpression of ISG15AA (lacking ISGylation ability and represents free ISG15) showed no obvious effect (Figure 4D-F).
ISG15 aggravates HR induced fibrosis in cultured HK-2 cells. (A-B) qPCR results of KIM1, NGAL, CYSC, ACTA2, Fn1 and VIMENTIN in ISG15 knockout HK-2 cells with or without HR treatment. (C) Representative Fn1 images in ISG15 knockout HK-2 cells with or without HR treatment. (D) Representative Fn1 images in ISG15 overexpressed HK-2 cells with HR treatment. Scale bar = 50 μm. (E-F) qPCR results of KIM1, NGAL, CYSC, ACTA2, Fn1 and VIMENTIN in ISG15 overexpressed HK-2 cells with HR treatment. The experiments were repeated three times, and at least three biological replicates per group were used. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; n.s., not significant.
ISG15 aggravates TGF-β1 or cisplatin induced renal fibrosis in renal tubular epithelial cells
The effects of ISG15 on renal cell fibrosis was further examined in HK-2 cells treated with TGF-β1, a classic fibrosis inducer. TGF-β1 induced up-regulation of KIM1, NGAL, ACTA2, Fn1, VIMENTIN mRNA levels, as well as Fn1 protein level, which was inhibited by ISG15 knockout (Figure 5A-C). As expected, overexpression of ISG15GG aggravated TGF-β1 induced renal fibrosis, while overexpression of ISG15AA showed no obvious effect on fibrosis (Figure 5D-F). Similarly, ISG15 knockout inhibited cisplatin-induced cell damage, whereas ISG15GG overexpression exacerbates cisplatin-induced cell damage (Figure S6).
ISG15 aggravates TGF-β1 induced fibrosis in cultured HK-2 cells. (A-B) qPCR results of KIM1, NGAL, CYSC, ACTA2, Fn1 and VIMENTIN in ISG15 knockout HK-2 cells with or without TGF-β1 treatment. (C) Representative Fn1 images in ISG15 knockout HK-2 cells with or without TGF-β1 treatment. (D) Representative Fn1 images in ISG15 overexpressed HK-2 cells with TGF-β1 treatment. Scale bar = 50 μm. (E-F) qPCR results of KIM1, NGAL, CYSC, ACTA2, Fn1 and VIMENTIN in ISG15 overexpressed HK-2 cells with TGF-β1 treatment. The experiments were repeated three times, and at least three biological replicates per group were used. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; n.s., not significant.
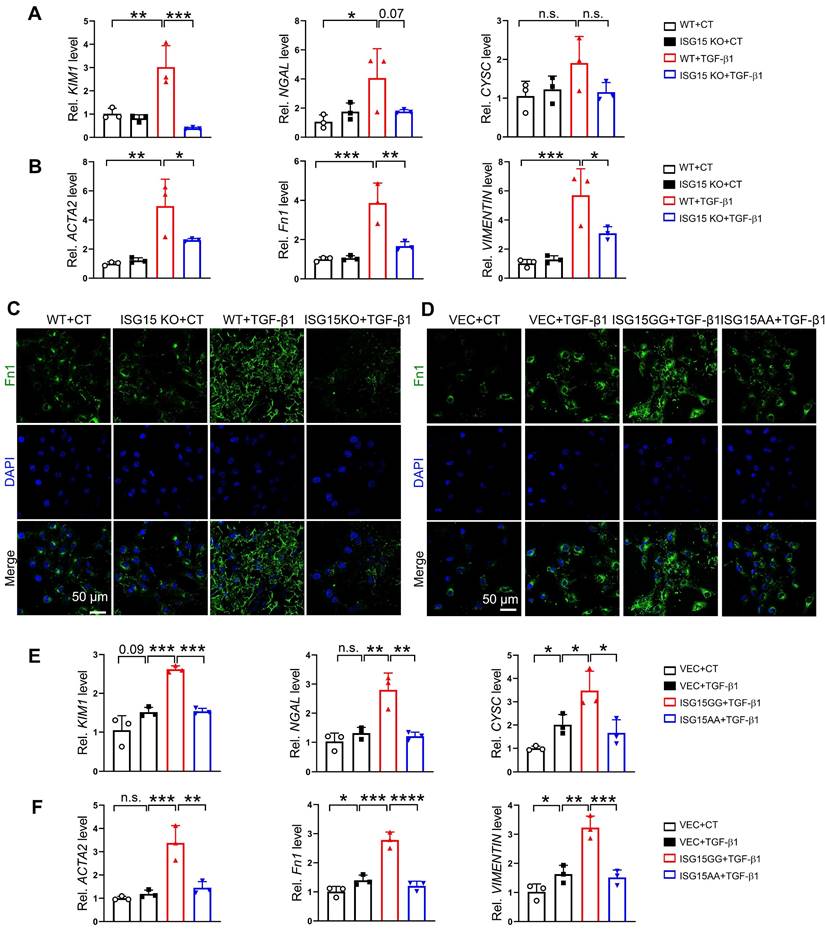
ISG15 ISGylates TGFβR1 in renal tubular epithelial cells
To uncover the downstream effectors of ISG15, we analyzed the RNA-seq data of IRI-, cisplatin-, or UUO models to extract upregulated genes. Subsequently, we identified a crucial receptor in the fibrosis pathway, TGFβR1 (Figure 6A), which may suggest a potential interaction between ISG15 and TGFβR1. Tgfbr1 transcriptional levels were upregulated in IRI-, cisplatin-, or UUO-injured kidneys (Figure 6B-D). Moreover, scRNA-seq data revealed that TGFβR1 was ubiquitously expressed in a variety of cells (Figure 6E), which was similar to ISG15 distribution. Leveraging AlphaFold2 predictions, we found evidence of a possible interaction between ISG15 and TGFβR1 (ΔiG = -7.3 kcal/mol, with ΔiG < -2 kcal/mol indicating interaction, Figure 6F and Table S4). We further confirmed the ISG15-TGFβR1 interaction by co-IP assays. Exogenous co-IP experiments demonstrated that ISG15 covalently conjugated TGFβR1 (Figure 6G-H). Additionally, overexpression of USP18, a de-ISGylation enzyme, decreased ISGylation level of TGFβR1 (Figure S7).
ISG15 binds and ISGylates TGFβR1 receptor. (A) Venn diagram analysis of three RNA-seq data. (B-D) qPCR results of Tgfbr1 in the kidneys after IRI (B), cisplatin (C), UUO (D) injury at indicated time, n = 4-6 per group. *P < 0.05; n.s., not significant. (E) tSNE plot of TGFβR1 expressed cell clusters from scRNA-seq of a human kidney allograft biopsy. CD, collecting duct; EC, endothelial cell; LOH (AL), loop of Henle, ascending limb; LOH (DL), loop of Henle, distal limb; PT, proximal tubule. (F) Binding model of ISG15 and TGFβR1. (G-H) Co-IP results of ISG15 and TGFβR1 in HEK293T cells transfected with indicated plasmids. (I) Effects of MG132 on ISG15 knockout-mediated proteolysis of TGFβR1. The cells were treated with MG132 (10 μM) for 6 h before immunoblots. (J) Effects of ISG15 knockout on the protein levels of TGFβR1 under the treatment of a ribosome inhibitor CHX (100 μM) for 6 h before immunoblots. (K) ISG15 overexpression reduced ubiquitination of endogenous TGFβR1 in HEK293T cells treated with MG132. HSP70 serves as the loading control. (L) ISG15 knockout enhanced ubiquitination of endogenous TGFβR1 in HK-2 cells treated with MG132. HSP70 serves as the loading control. The experiments were repeated three times.
Notably, decreased TGFβR1 and p-Smad2 levels were observed in the Isg15 KO mice injured with IRI, cisplatin, or UUO injury (Figure S8). Treatments with proteasome inhibitor (MG132) and protein synthesis inhibitor (cycloheximide, CHX) were performed to examine whether ISG15 affect the stability of TGFβR1. MG132 completely blocked ISG15 knockout-mediated TGFβR1 degradation, whereas CHX showed no effect (Figure 6I-J). To address whether ISG15 regulates TGFβR1 stability by ubiquitination, ubiquitination assays were performed. Overexpression of ISG15 enhanced TGFβR1 ISGylation, and inhibited ubiquitination of TGFβR1 (Figure 6K), while ISG15 knockout showed the opposite effects (Figure 6L).
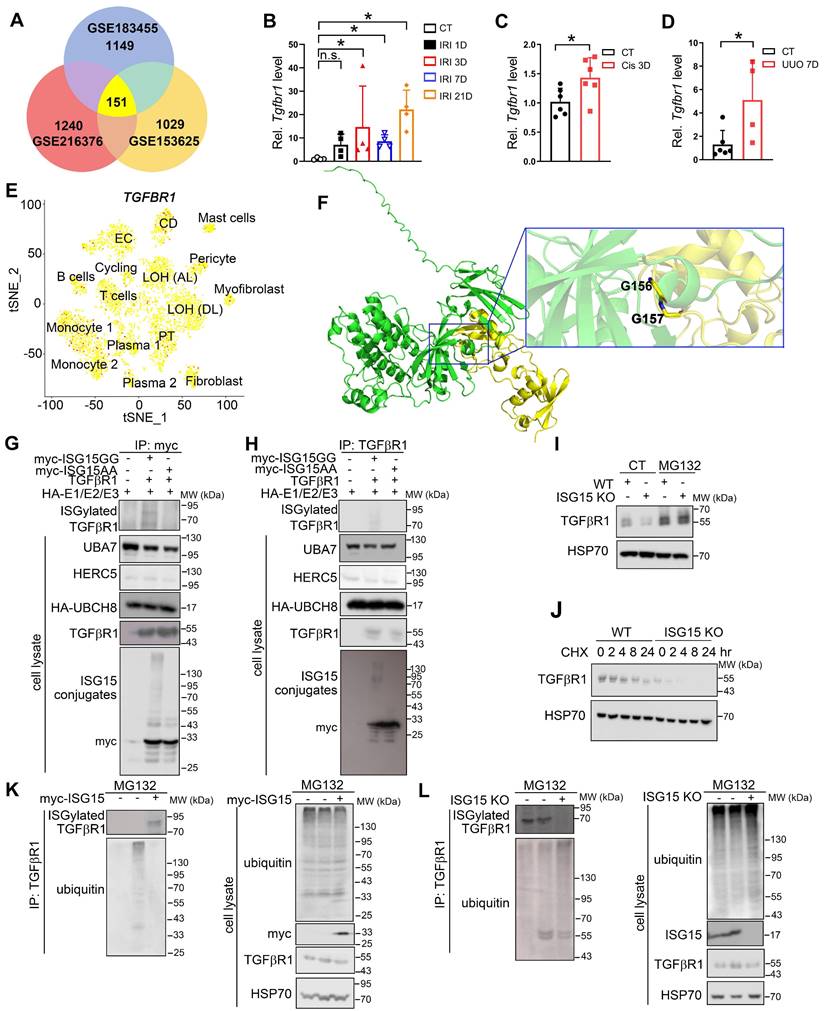
TGFβR1 aggregates AKI and subsequent AKI-to-CKD transition in vitro and in vivo
To verify the effect of TGFβR1 in AKI and subsequent AKI-to-CKD transition, we constructed a TGFβR1 overexpression animal model by renal intraparenchymal injection of TGFβR1 overexpressing adenovirus (Figure S9). Overexpression of TGFβR1 exacerbated renal pathological injury, and increased mRNA levels of Kim1, Ngal, Cysc, Acta2, Col3a, Vimentin in the kidneys of mice subjected to UIRI (Figure 7A-C). Additionally, overexpression of TGFβR1 aggravated the renal fibrosis in the kidney of mice after UIRI 7D (Figure 7D-F). In vitro, overexpression of TGFβR1 led to increased transcriptional levels of KIM1, NAGL, CYSC, ACTA2, Fn1, and VIMENTIN, along with elevated Fn1 protein level in TGF-β1 treated HK-2 cells (Figure 7G-H). Additionally, knockout of TGFβR1 reduced renal cell injury and fibrosis in TGF-β1 treated HK-2 cells (Figure 7I-J).
TGFβR1 exacerbates AKI and the following AKI-to-CKD transition in vivo and in vitro. (A) Experimental design chart of TGFβR1 overexpression by renal intraparenchymal injection in UIRI-treated mice. (B) Representative H&E images (left) with injury scores (right) of the kidney of indicated groups. Scale bar = 100 μm. (C) qPCR results of Kim1, Ngal, Cysc, Acta2, Fn1, Col3a and Vimentin in the kidney of indicated groups. (D-F) Representative images and quantitative results of Masson staining (D), immunostaining for α-SMA (E), Fn1 (F) of the kidney of indicated groups. Scale bar = 100 μm. CT+Ad-ctrl, n = 5; UIRI+Ad-ctrl, n = 4; UIRI+Ad-Tgfbr1, n = 4. (G) qPCR results of KIM1, NGAL, CYSC, ACTA2, Fn1 and VIMENTIN in TGFβR1 knockout HK-2 cells with or without TGF-β1 treatment. (H) Representative Fn1 images in TGFβR1 overexpressed HK-2 cells with or without TGF-β1 treatment. Scale bar = 50 μm. (I) Representative Fn1 images in TGFβR1 overexpressed HK-2 cells with or without TGF-β1 treatment. Scale bar = 50 μm. (J) qPCR results of KIM1, NGAL, CYSC, ACTA2, Fn1 and VIMENTIN in TGFβR1 knockout HK-2 cells with or without TGF-β1 treatment. The experiments were repeated three times, and at least three biological replicates per group were used. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; n.s., not significant.

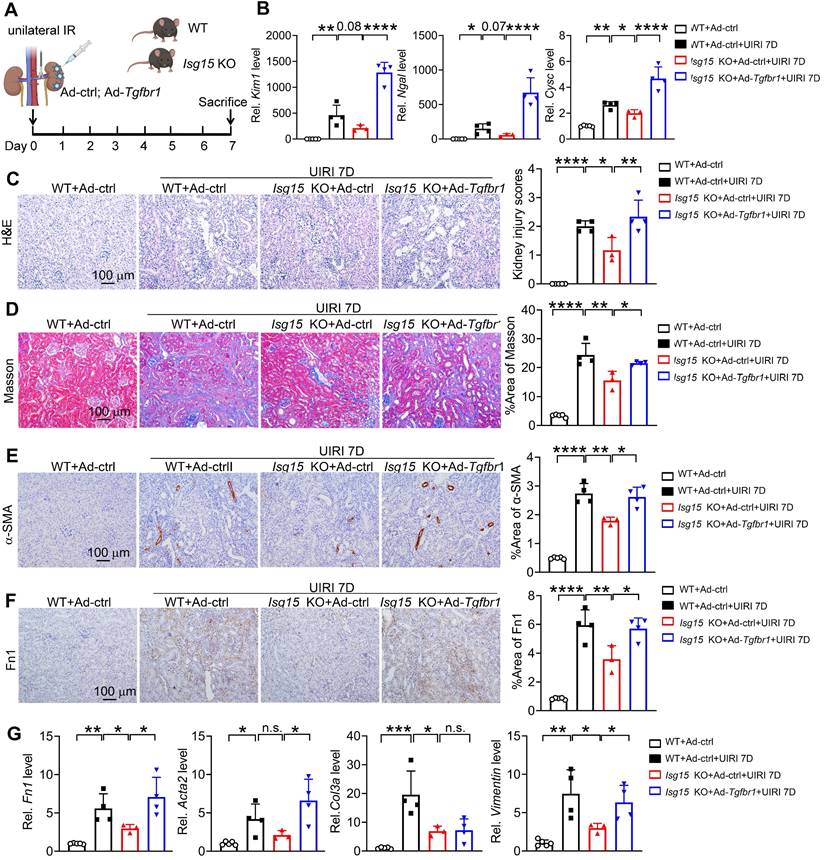
ISG15 accelerates AKI and subsequent AKI-to-CKD transition by ISGylating TGFβR1
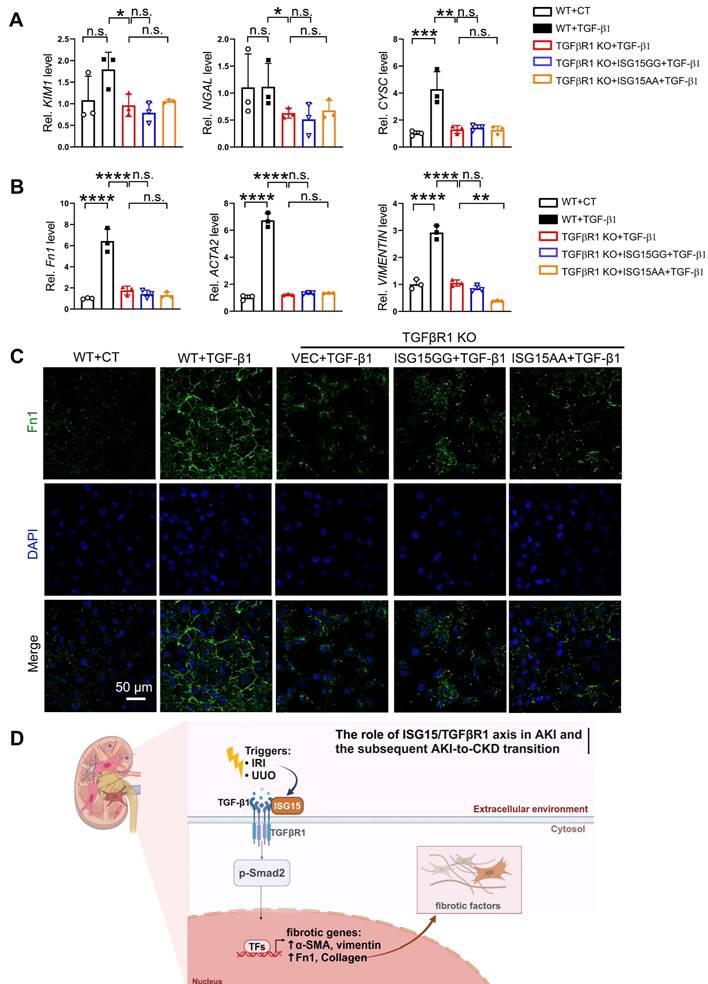
To confirm whether TGFβR1 is involved in the role of ISG15 in AKI and its transition to CKD, we overexpressed TGFβR1 in the kidneys of Isg15 KO mice. Compared with wildtype mice treated with UIRI, Isg15 KO mice showed reduced levels of the kidney injury markers Kim1, Ngal, Cysc along with less renal pathological injury, which was abolished by TGFβR1 overexpression (Figure 8A-C). Similarly, the renal fibrotic-suppressive effect of Isg15 KO was counteracted by overexpression of TGFβR1 (Figure 8D-G). In vitro, TGF-β1 stimulation led to an increase in the transcriptional levels of CYSC, ACTA2, Fn1, and VIMENTIN, along with an elevation in Fn1 protein level, which was suppressed by knockout of TGFβR1 (Figure 9A-C). Conversely, overexpression of ISG15 failed to increase the cell fibrosis in TGFβR1 knockout cells (Figure 9A-C). Similar findings were observed in cisplatin stimulated TGFβR1 KO cells transferred with ISG15 (Figure S10). Additionally, compared to TGFβR1 overexpression alone, co-overexpression of ISG15 and TGFβR1 upregulated the transcriptional levels of fibrotic factors in HK-2 cell treated by TGF-β1 (Figure S11). Collectively, these results suggest that TGFβR1 knockout effectively inhibits the fibrosis-promoting effect of ISG15.
Renal overexpression of TGFβR1 abolishes the renal protective effect of ISG15 knockout. (A) Experimental design chart of TGFβR1 overexpression by renal intraparenchymal injection in Isg15 KO mice subjected to UIRI. (B) qPCR results of Kim1, Ngal, Cysc in the kidney of indicated groups. (C) Representative H&E images (left) with injury scores (right) of the kidney of different groups. Scale bar = 100 μm. (D-F) Representative images and quantitative results of Masson staining (D), immunostaining for α-SMA (E), Fn1 (F) of the kidney of indicated groups. Scale bar = 100 μm. (G) qPCR results of Fn1, Acta2, Col3a and Vimentin in the kidney of different groups. WT+Ad-ctrl, n = 5; WT+UIRI+Ad-ctrl, n = 4; Isg15 KO+UIRI+Ad-ctrl, n = 3; Isg15 KO+UIRI+Ad-Tgfbr1, n = 4. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; n.s., not significant.
ISG15 aggravates renal injury by ISGylating TGFβR1. (A) qPCR results of KIM1, NGAL, CYSC in TGFβR1 knockout HK-2 cells with ISG15 overexpression under TGF-β1 treatment. (B) qPCR results of ACTA2, Fn1 and VIMENTIN in TGFβR1 knockout HK-2 cells with ISG15 overexpression under TGF-β1 treatment. (C) Representative Fn1 images in TGFβR1 knockout HK-2 cells with ISG15 overexpression under TGF-β1 treatment. Scale bar = 50 μm. (D) A schematic model of the ISG15 role involved in AKI and the following AKI-to-CKD transition. The experiments were repeated three times, and at least three biological replicates per group were used. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; n.s., not significant.
Discussion
ISG15 is the earliest identified ubiquitin-like protein induced by interferons, acting both in a free form and as a protein modifier. ISG15 and the members of the enzymatic cascade that mediate ISG15 conjugation (ISGylation) are strongly induced by type I interferons [17, 20]. ISG15 also responds to viral and bacterial infections, lipopolysaccharide, retinoic acid and certain genotoxic stressors in addition to type I interferons [17, 39]. Despite its similarities to ubiquitin, ISG15's biological function is still poorly understood; however, ISG15 appears to play important roles in various biological and cellular functions, including innate immunity, anti-viral/bacterial infections, protein turnover, and tumorigenesis [22]. Moreover, a recent study suggests a novel DRD4-ISG15-NOX4 axis in the progression of AKI [40]. The results showed that dopamine D4 receptor (DRD4) was downregulated in AKI mice, which downregulated ISG15 expression, resulting in decreased ISGylation of NADPH oxidase 4 (NOX4) that competitively inhibited its ubiquitination and caused degradation of NOX4 [40].
In this study, we established various animal models of kidney injury and collected samples at multiple time points. The results showed that in the early stages of kidney injury, AKI, knockout of ISG15 ameliorated renal function and pathological change (Figure 2 and Figure S4). Moreover, in the later stages of kidney injury, AKI-to-CKD transition, ISG15 knockout also mitigated renal damage induced by IRI, and UUO (Figure 2-3). Additionally, in vitro studies showed that overexpression of ISG15 with modification capability (ISG15GG) promoted cell fibrosis and damage induced by HR, cisplatin or TGFβ1, while overexpression of the free form of ISG15 (ISG15AA) had no effect on cell fibrosis induced by these stimuli (Figure 4-5, Figure S6). These findings collectively underscore the predominant contribution of ISG15 to the initiation and progression of AKI.
IRI, cisplatin, and UUO treatments may induce kidney injury in mice through distinct pathways and with different progression rates. IRI induces kidney damage more rapidly compared to cisplatin- or UUO-induced kidney injury [41-43]. Although it is challenging to explain the effects of ISG15 in these kidney injuries using a single mechanism, our data nevertheless indicate that ISG15 modulates renal fibrosis, a common pathway influencing kidney injury in these models. Future efforts to elucidate the distinct and potentially overlapping pathways through which ISG15 operates in kidney injury initiated by different injuries are desired.
To date, hundreds of putative targets of ISGylation have been identified. However, the functional consequence of ISGylation is still poorly understood. Unlike ubiquitylation, ISGylation does not appear to directly target proteins for proteasome-mediated degradation [44]. It has been found that ISG15 can compete with ubiquitin for ubiquitin binding sites, thereby indirectly regulating protein degradation [45]. In addition, ISG15-ubiquitin mixed chains have been observed and may negatively regulate the turnover of ubiquitylated proteins [17]. However, for ISGylated proteins studied, only a small fraction of the total protein is modified by ISG15, making it a challenge to understand how much does ISGylation contribute to the overall function of a protein. In our study, in silico results suggested interaction between ISG15 and TGFβR1 (Figure 6F and Table S4), whereas no obvious interaction was predicted between ISG15 and another classic fibrosis receptor TGFβR2 (Table S4). Moreover, co-IP assays confirmed that ISG15 ISGylated TGFβR1, which may prevent TGFβR1 degradation by the ubiquitin-proteasome system (Figure 6).
Renal fibrosis is the principal pathological change and common pathway in the progression of AKI to CKD, ultimately leading to end-stage renal disease and renal failure [46]. It is the result of maladaptive repair following renal injury and manifests as a progressive process. TGF-β1 is recognized as the most crucial profibrotic factor and the primary signaling pathway driving renal fibrosis [47-49]. TGF-β1 is synthesized and secreted by a wide range of cell types throughout the body. Upon secretion, TGF-β1 activates the TGF-β receptor complex, initiating signaling cascades through both canonical (Smad-dependent) and non-canonical (Smad-independent) pathways [46]. The canonical pathway plays central regulatory roles in renal fibrosis [46]. In the canonical pathway, upon TGF-β1 activation, TGFβR2 recruits and phosphorylates the serine/threonine kinase domain of TGFβR1 within the cytoplasmic region, leading to the activation of TGFβR1 by binding and phosphorylating the GS domain [47, 48]. Subsequently, phosphorylated TGFβR1 activates the fibrotic signaling pathway, leading to the nuclear translocation of Smad2/3 and the regulation of fibrotic-related genes such as α-SMA, fibronectin, and collagen, promoting fibrosis [47, 48]. Increasing evidences show that TGFβR1 modification provides a new mechanism for the functional regulation of TGF-β responses. By regulating Smad activation and downstream transcription responses, TGFβR1 sumoylation elaborates the TGF-β responses that drives cancer progression [50]. Moreover, ubiquitin-specific protease 2a (USP2a) removes K33-linked ubiquitin chains from TGFβR1 and promotes SMAD2 recruitment [51]. Deletion or pharmacologic inhibition of USP2a impairs TGFβ-induced EMT and metastasis [51]. TGFβR1 is ubiquitinated by multiple E3s, including SMURF1/2, WWP1 and NEDD4-2, which results in the degradation of TGFβR1 and attenuation of TGF-β signaling [52-54]. In our study, we found that ISG15 promoted ISGylation level of TGFβR1 (Figure 6). And, knockout of ISG15 led to decreased TGFβR1 and its downstream p-Smad2 levels, reduces transcription levels of fibrotic genes such as α-SMA, Fn1, and Vimentin, ultimately inhibited renal fibrosis (Figure 2-3 and Figure S4, S8). Additionally, renal overexpression of TGFβR1 abolished the protective effect of ISG15 deficiency during kidney injury (Figures 8-9). Taken together, regulation or inhibition of TGFβR1 modification may be a key to treating AKI and the following AKI-to-CKD transition.
In summary, we reported that increased ISG15 and TGFβR1 expression in the models of IRI-, cisplatin- or UUO-induced kidney injury, and improved kidney function was found in Isg15 KO mice. We identified a novel modification of the key fibrosis receptor TGFβR1, namely ISGylation, which leads to sustained activation of the TGFβR1 signaling pathway, exacerbating renal fibrosis. Our study also provides a proof-of-concept into AKI/AKI-to-CKD treatment through targeting the modification of TGFβR1 (Figure 9D).
Abbreviations
AKI: acute kidney injury; IRI: ischemia-reperfusion injury; BIRI: bilateral ischemia-reperfusion injury; UIRI: unilateral ischemia-reperfusion injury; ISG15: Interferon-stimulated gene 15; UUO: unilateral ureteral obstruction; CKD: chronic kidney disease; TGF-β1: transforming growth factor-β1; TGFβR2: type II TGF-β receptor; TGFβR1: type I TGF-β receptor; α-SMA: α-smooth muscle actin; Fn1: fibronectin 1; CREA: creatinine; BUN: blood urea nitrogen; qPCR: quantitative real-time polymerase chain reaction; WT: wildtype; co-IP: co-Immunoprecipitation; ESRD: end-stage renal disease.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work is supported by the Natural Science Foundation of China (32371171 to H.C.; 82273838 to K.H.), the National Key R&D Program of China (2022YFA0806100 and 2023YFC2507900 to K.H.), and the Natural Science Foundation of Hubei Province (2021CFA004 to K.H.; 2022CFB247 to H.C.; 2021CFB250 to C.Y.L.). The graphic abstract was generated using biorender.com. And thanks for the technical support by the Huazhong University of Science & Technology Analytical & Testing center, Medical sub-center.
Author contributions
This work was carried out in collaboration with all authors. H.C., K.H., C.Y.L., and N.C. conceptualized and designed the study. C.Y.L., N.C., X.T., L.L.S., Z.X.X., C.W., and Y.C.W. performed the experiments and analyzed the data; C.T.P., M.C.J., L.Z., K.H. assisted in the interpretation of the results. H.C., K.H., L.Z., C.Y.L., and N.C. wrote and edited the paper. H.C. and K.H. supervised studies. All authors have read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Guo C, Dong G, Liang X, Dong Z. Epigenetic regulation in AKI and kidney repair: mechanisms and therapeutic implications. Nat Rev Nephrol. 2019;15:220-39
2. Zarbock A, Forni LG, Ostermann M, Ronco C, Bagshaw SM, Mehta RL. et al. Designing acute kidney injury clinical trials. Nat Rev Nephrol. 2024;20:137-46
3. Vijayan A. Tackling AKI: prevention, timing of dialysis and follow-up. Nat Rev Nephrol. 2021;17:87-8
4. Pistolesi V, Regolisti G, Morabito S, Gandolfini I, Corrado S, Piotti G. et al. Contrast medium induced acute kidney injury: a narrative review. J Nephrol. 2018;31:797-812
5. De Clercq L, Ailliet T, Schaubroeck H, Hoste EAJ. Acute and Chronic Cardiovascular Consequences of Acute Kidney Injury: A Systematic Review and Meta-Analysis. Cardiorenal Med. 2023;13:26-33
6. Chalikias G, Tziakas D. Cardiovascular Consequences of Acute Kidney Injury. N Engl J Med. 2020;383:1094
7. Legrand M, Clark AT, Neyra JA, Ostermann M. Acute kidney injury in patients with burns. Nat Rev Nephrol. 2024;20:188-200
8. Richardson L, Wilcockson SG, Guglielmi L, Hill CS. Context-dependent TGFbeta family signalling in cell fate regulation. Nat Rev Mol Cell Biol. 2023;24:876-94
9. Kaushal GP, Shah SV. Challenges and advances in the treatment of AKI. J Am Soc Nephrol. 2014;25:877-83
10. Arai S, Kitada K, Yamazaki T, Takai R, Zhang X, Tsugawa Y. et al. Apoptosis inhibitor of macrophage protein enhances intraluminal debris clearance and ameliorates acute kidney injury in mice. Nat Med. 2016;22:183-93
11. Kurzhagen JT, Dellepiane S, Cantaluppi V, Rabb H. AKI: an increasingly recognized risk factor for CKD development and progression. J Nephrol. 2020;33:1171-87
12. Huang R, Fu P, Ma L. Kidney fibrosis: from mechanisms to therapeutic medicines. Signal Transduct Target Ther. 2023;8:129
13. Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov. 2012;11:790-811
14. Nastase MV, Zeng-Brouwers J, Wygrecka M, Schaefer L. Targeting renal fibrosis: Mechanisms and drug delivery systems. Adv Drug Deliv Rev. 2018;129:295-307
15. Budi EH, Duan D, Derynck R. Transforming Growth Factor-beta Receptors and Smads: Regulatory Complexity and Functional Versatility. Trends Cell Biol. 2017;27:658-72
16. Travis MA, Sheppard D. TGF-beta activation and function in immunity. Annu Rev Immunol. 2014;32:51-82
17. Perng YC, Lenschow DJ. ISG15 in antiviral immunity and beyond. Nat Rev Microbiol. 2018;16:423-39
18. Serrano I. Structural insights into the specific recognition and transfer of ISG15. Nat Struct Mol Biol. 2024;31:214
19. Malakhova OA, Zhang DE. ISG15 inhibits Nedd4 ubiquitin E3 activity and enhances the innate antiviral response. J Biol Chem. 2008;283:8783-7
20. Wallace I, Baek K, Prabu JR, Vollrath R, von Gronau S, Schulman BA. et al. Insights into the ISG15 transfer cascade by the UBE1L activating enzyme. Nat Commun. 2023;14:7970
21. Okumura F, Zou W, Zhang DE. ISG15 modification of the eIF4E cognate 4EHP enhances cap structure-binding activity of 4EHP. Genes Dev. 2007;21:255-60
22. Hermann M, Bogunovic D. ISG15: In Sickness and in Health. Trends Immunol. 2017;38:79-93
23. Xiong M, Chen H, Fan Y, Jin M, Yang D, Chen Y. et al. Tubular Elabela-APJ axis attenuates ischemia-reperfusion induced acute kidney injury and the following AKI-CKD transition by protecting renal microcirculation. Theranostics. 2023;13:3387-401
24. Chen H, Wang L, Wang W, Cheng C, Zhang Y, Zhou Y. et al. ELABELA and an ELABELA Fragment Protect against AKI. J Am Soc Nephrol. 2017;28:2694-707
25. Yang C, Xu H, Yang D, Xie Y, Xiong M, Fan Y. et al. A renal YY1-KIM1-DR5 axis regulates the progression of acute kidney injury. Nat Commun. 2023;14:4261
26. Fan Y, Yuan Y, Xiong M, Jin M, Zhang D, Yang D. et al. Tet1 deficiency exacerbates oxidative stress in acute kidney injury by regulating superoxide dismutase. Theranostics. 2023;13:5348-64
27. Xiong W, Xiong Z, Song A, Lei C, Ye C, Zhang C. Relieving lipid accumulation through UCP1 suppresses the progression of acute kidney injury by promoting the AMPK/ULK1/autophagy pathway. Theranostics. 2021;11:4637-54
28. Wang J, Xiong M, Fan Y, Liu C, Wang Q, Yang D. et al. Mecp2 protects kidney from ischemia-reperfusion injury through transcriptional repressing IL-6/STAT3 signaling. Theranostics. 2022;12:3896-910
29. Chen H, Li J, Jiao L, Petersen RB, Li J, Peng A. et al. Apelin inhibits the development of diabetic nephropathy by regulating histone acetylation in Akita mouse. J Physiol. 2014;592:505-21
30. Liu S, Sun Y, Jiang M, Li Y, Tian Y, Xue W. et al. Glyceraldehyde-3-phosphate dehydrogenase promotes liver tumorigenesis by modulating phosphoglycerate dehydrogenase. Hepatology. 2017;66:631-45
31. Chen H, Wan D, Wang L, Peng A, Xiao H, Petersen RB. et al. Apelin protects against acute renal injury by inhibiting TGF-beta1. Biochim Biophys Acta. 2015;1852:1278-87
32. Yang C, Zhang Y, Zeng X, Chen H, Chen Y, Yang D. et al. Kidney injury molecule-1 is a potential receptor for SARS-CoV-2. J Mol Cell Biol. 2021;13:185-96
33. Yang D, Fan Y, Xiong M, Chen Y, Zhou Y, Liu X. et al. Loss of renal tubular G9a benefits acute kidney injury by lowering focal lipid accumulation via CES1. EMBO Rep. 2023;24:e56128
34. Wang C, Xiong M, Yang C, Yang D, Zheng J, Fan Y. et al. PEGylated and Acylated Elabela Analogues Show Enhanced Receptor Binding, Prolonged Stability, and Remedy of Acute Kidney Injury. J Med Chem. 2020;63:16028-42
35. Sun Y, Wang Q, Zhang Y, Geng M, Wei Y, Liu Y. et al. Multigenerational maternal obesity increases the incidence of HCC in offspring via miR-27a-3p. J Hepatol. 2020;73:603-15
36. Chen H, Liu C, Wang Q, Xiong M, Zeng X, Yang D. et al. Renal UTX-PHGDH-serine axis regulates metabolic disorders in the kidney and liver. Nat Commun. 2022;13:3835
37. Chen Y, Shi J, Wang X, Zhou L, Wang Q, Xie Y. et al. An antioxidant feedforward cycle coordinated by linker histone variant H1.2 and NRF2 that drives nonsmall cell lung cancer progression. Proc Natl Acad Sci U S A. 2023;120:e2306288120
38. Tsaban T, Varga JK, Avraham O, Ben-Aharon Z, Khramushin A, Schueler-Furman O. Harnessing protein folding neural networks for peptide-protein docking. Nat Commun. 2022;13:176
39. Yuan W, Krug RM. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. EMBO J. 2001;20:362-71
40. Gao Y, Lu X, Zhang G, Liu C, Sun S, Mao W. et al. DRD4 alleviates acute kidney injury by suppressing ISG15/NOX4 axis-associated oxidative stress. Redox Biol. 2024;70:103078
41. Fu Y, Tang C, Cai J, Chen G, Zhang D, Dong Z. Rodent models of AKI-CKD transition. Am J Physiol Renal Physiol. 2018;315:F1098-F106
42. Chen Z, Li Y, Yuan Y, Lai K, Ye K, Lin Y. et al. Single-cell sequencing reveals homogeneity and heterogeneity of the cytopathological mechanisms in different etiology-induced AKI. Cell Death Dis. 2023;14:318
43. Kellum JA, Romagnani P, Ashuntantang G, Ronco C, Zarbock A, Anders HJ. Acute kidney injury. Nat Rev Dis Primers. 2021;7:52
44. Liu M, Li XL, Hassel BA. Proteasomes modulate conjugation to the ubiquitin-like protein, ISG15. J Biol Chem. 2003;278:1594-602
45. Desai SD, Haas AL, Wood LM, Tsai YC, Pestka S, Rubin EH. et al. Elevated expression of ISG15 in tumor cells interferes with the ubiquitin/26S proteasome pathway. Cancer Res. 2006;66:921-8
46. Li L, Fu H, Liu Y. The fibrogenic niche in kidney fibrosis: components and mechanisms. Nat Rev Nephrol. 2022;18:545-57
47. Kayhan M, Vouillamoz J, Rodriguez DG, Bugarski M, Mitamura Y, Gschwend J. et al. Intrinsic TGF-beta signaling attenuates proximal tubule mitochondrial injury and inflammation in chronic kidney disease. Nat Commun. 2023;14:3236
48. Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12:325-38
49. Yang X, Okamura DM, Lu X, Chen Y, Moorhead J, Varghese Z. et al. CD36 in chronic kidney disease: novel insights and therapeutic opportunities. Nat Rev Nephrol. 2017;13:769-81
50. Kang JS, Saunier EF, Akhurst RJ, Derynck R. The type I TGF-beta receptor is covalently modified and regulated by sumoylation. Nat Cell Biol. 2008;10:654-64
51. Zhao Y, Wang X, Wang Q, Deng Y, Li K, Zhang M. et al. USP2a Supports Metastasis by Tuning TGF-beta Signaling. Cell Rep. 2018;22:2442-54
52. Eichhorn PJ, Rodon L, Gonzalez-Junca A, Dirac A, Gili M, Martinez-Saez E. et al. USP15 stabilizes TGF-beta receptor I and promotes oncogenesis through the activation of TGF-beta signaling in glioblastoma. Nat Med. 2012;18:429-35
53. Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH. et al. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell. 2000;6:1365-75
54. Komuro A, Imamura T, Saitoh M, Yoshida Y, Yamori T, Miyazono K. et al. Negative regulation of transforming growth factor-beta (TGF-beta) signaling by WW domain-containing protein 1 (WWP1). Oncogene. 2004;23:6914-23
Author contact
![]() Corresponding author: Hong Chen, Ph.D. Tongji School of Pharmacy, Huazhong University of Science and TechnologyWuhan, China, 430030; hongchen2017edu.cn.
Corresponding author: Hong Chen, Ph.D. Tongji School of Pharmacy, Huazhong University of Science and TechnologyWuhan, China, 430030; hongchen2017edu.cn.