Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2024; 14(11):4218-4239. doi:10.7150/thno.95749 This issue Cite
Research Paper
Aryl hydrocarbon receptor confers protection against macrophage pyroptosis and intestinal inflammation through regulating polyamine biosynthesis
Yajing Gao1,2#, Kwei-Yan Liu1,2,3#, Wenfeng Xiao1,2#, Xueru Xie1,2, Qiuyan Liang1,2, Zikun Tu1,2, Lan Yang1,2, Hongmiao Yu1,2, Haiyan Guo1,2, Saihua Huang1,2, Xiao Han1,2, Jinrong Fu1,2, Yufeng Zhou1,2 ![]()
1. Department of Critical Care Medicine, Children's Hospital of Fudan University, National Children's Medical Center, and the Shanghai Key Laboratory of Medical Epigenetics, International Co-laboratory of Medical Epigenetics and Metabolism, Ministry of Science and Technology, Institutes of Biomedical Sciences, Fudan University, Shanghai 200032, China.
2. National Health Commission (NHC) Key Laboratory of Neonatal Diseases, Fudan University, Shanghai, China.
3. National Institute of Environmental Health Sciences, National Health Research Institutes, Taiwan.
# These authors contributed equally to this work.
Received 2024-2-27; Accepted 2024-6-27; Published 2024-7-8
Abstract

Rationale: The aryl hydrocarbon receptor (AhR) functions in the regulation of intestinal inflammation, but knowledge of the underlying mechanisms in innate immune cells is limited. Here, we investigated the role of AhR in modulating the functions of macrophages in inflammatory bowel disease pathogenesis.
Methods: The cellular composition of intestinal lamina propria CD45+ leukocytes in a dextran sulfate sodium (DSS)-induced mouse colitis model was determined by single-cell RNA sequencing. Macrophage pyroptosis was quantified by analysis of lactate dehydrogenase release, propidium iodide staining, enzyme-linked immunosorbent assay, western blot, and flow cytometry. Differentially expressed genes were confirmed by RNA-seq, RT-qPCR, luciferase assay, chromatin immunoprecipitation, and immunofluorescence staining.
Results: AhR deficiency mediated dynamic remodeling of the cellular composition of intestinal lamina propria (LP) CD45+ immune cells in a colitis model, with a significant increase in monocyte-macrophage lineage. Mice with AhR deficiency in myeloid cells developed more severe dextran sulfate sodium induced colitis, with concomitant increased macrophage pyroptosis. Dietary supplementation with an AhR pre-ligand, indole-3-carbinol, conferred protection against colitis while protection failed in mice lacking AhR in myeloid cells. Mechanistically, AhR signaling inhibited macrophage pyroptosis by promoting ornithine decarboxylase 1 (Odc1) transcription, to enhance polyamine biosynthesis. The increased polyamine, particularly spermine, inhibited NLRP3 inflammasome assembly and subsequent pyroptosis by suppressing K+ efflux. AHR expression was positively correlated with ODC1 in intestinal mucosal biopsies from patients with ulcerative colitis.
Conclusions: These findings suggest a functional role for the AhR/ODC1/polyamine axis in maintaining intestinal homeostasis, providing potential targets for treatment of inflammatory bowel disease.
Keywords: Aryl hydrocarbon receptor, macrophage, pyroptosis, ODC1, spermine
Introduction
Aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor that responds to a diverse range of exogenous compounds, such as environmental pollutants, as well as endogenous signals derived from dietary components, microbiota, and cellular metabolites [1]. In the absence of ligands, AhR forms a stable complex with Hsp90, XAP2, and p23 in the cytoplasm. Upon ligand binding, AhR translocates from the cytoplasm to the nucleus where it dimerizes with aryl hydrocarbon receptor nuclear translocator, to enhance binding to promoter regions of downstream genes at xenobiotic response elements (XREs) [2]. Recent studies have identified cell-intrinsic roles for AhR in the development, differentiation, survival, and maintenance of several immune cell types, highlighting the pivotal function of this molecule in regulating physiological homeostasis and immune responses [3-5]; however, the detailed molecular mechanisms through which AhR modulates inflammatory responses remain poorly understood.
Inflammatory bowel disease (IBD), comprised of Crohn's disease (CD) and ulcerative colitis (UC), is an idiopathic and relapsing inflammatory disease of the gastrointestinal tract [6]. There is accumulating evidence that environmental, dietary, microbial, and immunological cues regulate intestinal inflammation in mouse models by targeting AhR in epithelial cells [7, 8], innate lymphoid cells (ILCs) [9, 10], intraepithelial lymphocytes [11], and T cells [12-14]; however, little is known about the exact role of AhR in regulating intestinal macrophage functions in IBD pathogenesis. To maintain intestinal homeostasis, the gut is equipped with one of the largest populations of macrophages in the body [15]. Once pathogens break through the epithelial barrier and invade the intestinal mucosa, they can be recognized by macrophages via pattern recognition receptors, which induce a series of inflammatory responses [16]. Overactivated macrophages participate in IBD pathogenesis by controlling the initiation and amplification of local inflammation [17]. In response to lipopolysaccharide (LPS), macrophages derived from AhR-null mice exhibited hyper-activated expression of the pro-inflammatory cytokines (e.g., IL-1β, IL-6) [18, 19], whereas production of the anti-inflammatory cytokine, IL-10, was decreased [20]. Altered expression of cytokines by macrophages may underlie AhR-mediated regulation of inflammatory responses; however, the mechanisms through which AhR modulates macrophage activation and cytokine release remain to be fully elucidated.
Pyroptosis is a lytic form of programmed cell death, characterized by pore formation in the cell membrane, cell swelling, and ultimate release of large amounts of cell contents, including pro-inflammatory cytokines [21]. The main pyroptosis signaling pathway is mediated by inflammasome dependent caspase-1 activation, resulting in a process involving IL-1β, IL-18, and gasdermin D (GSDMD) maturation [22, 23]. Dysregulation of pyroptosis is often associated with excessive inflammatory responses, that lead to various inflammatory diseases, including IBD [24, 25]. Genome-wide association study analysis has pointed to several IBD susceptibility genes encoding proteins associated with pyroptosis, including IL-1 antagonist receptor (IL-1-RN), IL-1β, IL-18 receptor accessory protein (IL-18RAP), and IL-18 receptor 1 (IL-18R1) [26, 27]. Further, large numbers of pro-inflammatory macrophages are reported to infiltrate the gut mucosa of patients with UC and CD [28, 29], while increased levels of pro-inflammatory cytokines, including IL-1β and IL-18, are detected in active IBD and correlate with inflammation severity [2, 30-32].
In the current study, we discovered that, in a colitis mouse model, AhR deficiency mediated dynamic remodeling of the cellular composition of intestinal lamina propria (LP) CD45+ immune cells, with a significant increase in the monocyte-macrophage lineage. Significantly, we show that AhR signaling protects against macrophage pyroptosis and intestinal inflammation via the AhR-ODC1-polyamine axis, both in vitro and in vivo.
Results
AhR deficiency exacerbated dextran sulfate sodium (DSS)-induced colitis and LPS-induced inflammation
To investigate the potential role of AhR in regulating inflammatory responses in vivo, age- and sex-matched AhR-null (AhR-/-) mice and co-housed wild type (AhR+/+) littermates were orally administered 2% DSS to induce acute colitis, as described previously (Figure 1A) [33]. Relative to controls administered with water, DSS-treated mice exhibited typical symptoms of colitis, including diarrhea, hematochezia, and body weight loss. Relative to DSS-treated littermate controls, DSS-treated AhR-/- mice showed more severe colitis, characterized by greater body weight loss, higher disease activity index (DAI) score, and shorter colon length (Figure 1B-E). Histopathological analysis demonstrated that treatment of AhR-/- mice with DSS led to more severe transmural inflammation, with disruption of the mucosal epithelium, focal areas of extensive ulceration, and loss of goblet cells (Figure 1F-G).
AhR deficiency exacerbated DSS-induced colitis and LPS-induced inflammation. (A) Schematic representation of the DSS-induced colitis model. AhR-null (AhR-/-) mice and co-housed wide type (AhR+/+) littermates were orally administered 2% (w/v) DSS in drinking water for 7 days, followed by regular drinking water for the next 2 days. Mice were sacrificed on day 9. (B) Body weight changes were monitored daily and are presented as percentage of initial body weight (n = 6 per group). (C) DAI scores were determined daily for each AhR-/- and AhR+/+ mouse, based on stool consistency, fecal bleeding, and body weight loss (n = 6 per group). (D) Representative morphology images of colon specimens from each group. (E) Colon lengths were measured and recorded on day 9 (n = 6 per group). (F) Representative histopathological images of H&E and PAS-stained colon sections. Scale bar, 200 μm. (G) Semiquantitative histopathology scoring (n = 6 per group). (H) Survival plots for AhR+/+ (n = 10) and AhR-/- mice (n = 10) intraperitoneally injected with LPS (20 mg/kg). (I) IL-1β and (J) IL-6 concentrations in serum from AhR+/+ and AhR-/- mice 16 h after LPS (10 mg/kg) challenge; n = 8 per group. Statistical analysis of the data was performed using one-way ANOVA with Welch's correct (E) or not (G), two-way ANOVA (B, C, I, J) followed by either Tukey's, Sidak's or Dunnett's T3 multiple comparisons test, and Log-rank (Mantel-Cox) test (H). Data are presented as the mean ± SEM. ns, not significant; *P < 0.05, **P < 0.01, *** P < 0.001, and **** P < 0.0001. AhR, aryl hydrocarbon receptor; DSS: dextran sulfate sodium salt; DAI: disease activity index; H&E, hematoxylin and eosin; PAS, periodic acid-Schiff.
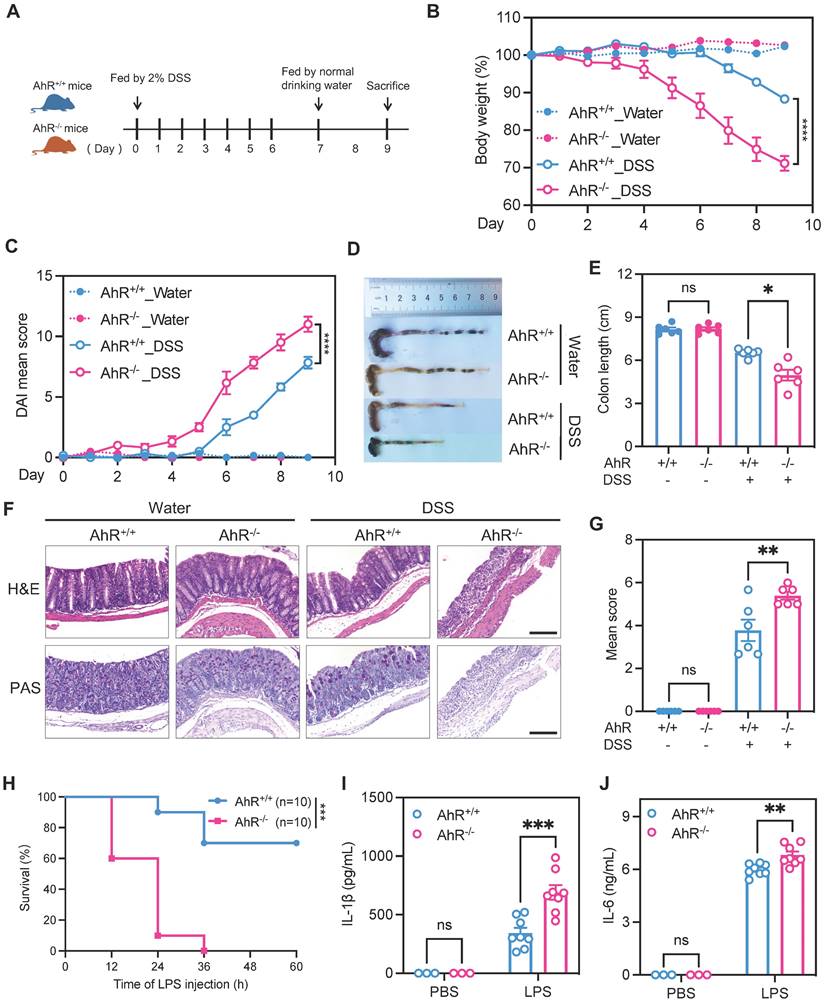
An LPS-induced sepsis model, generated by intraperitoneal injection of AhR+/+ and AhR-/- mice with LPS (20 mg/kg), was used to confirm the function of AhR in acute inflammation in vivo. After 36 h, AhR+/+ mice showed 70% survival, whereas all AhR-/- mice had died (Figure 1H). The exacerbated inflammatory responses in LPS-injected AhR-/- mice were further emphasized by marked increases in plasma concentrations of IL-1β and IL-6 (Figure 1I-J).
Collectively, these results indicate that AhR deficiency exacerbated DSS-induced colitis and LPS-induced inflammation, suggesting a protective role for AhR in controlling inflammatory responses and immune homeostasis.
Single-cell RNA sequencing (scRNA-seq) analysis of LP CD45+ cells revealed an increase in the pro-inflammatory monocyte-macrophage lineage in AhR-/- mice during colitis
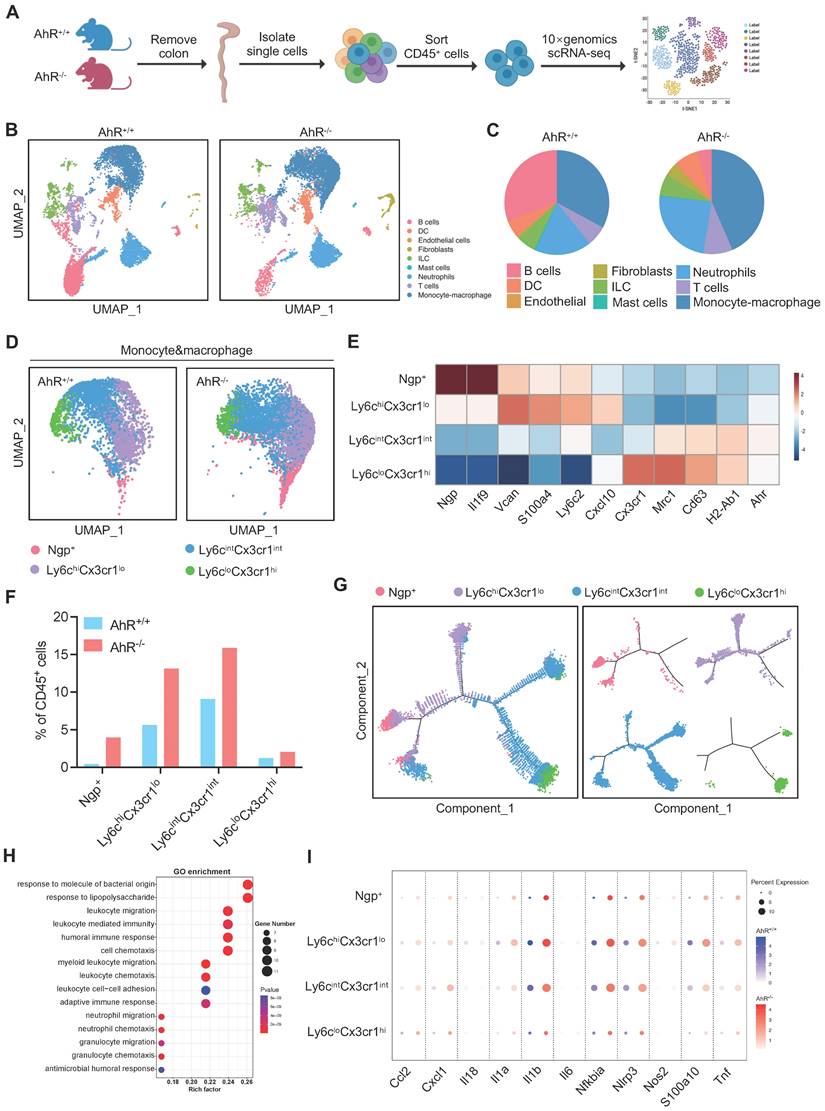
Next, we sought to identify potential effector cells contributing to the exacerbated inflammatory responses in AhR-/- mice. Recent studies highlight the essential role of AhR in various immune cell types during IBD. Hence, a comprehensive and unbiased approach with single-cell resolution to investigate AhR functions in immune cells is essential for understanding the pathogenesis of IBD. Therefore, we conducted scRNA-seq to analyze intestinal LP CD45+ leukocytes from the colonic tissue of AhR+/+ and AhR-/- mice with colitis (Figure 2A). We purified 37613 high-quality LP CD45+ leukocytes, including 16787 from AhR-/- mice and 20844 from AhR+/+ littermates. The transcriptomic diversity of the resulting data was projected onto two dimensions by uniform manifold approximation and projection (UMAP). Unsupervised clustering analysis identified nine distinct cell types, which were annotated based on known lineage markers, including B cells, dendritic cells (DCs), ILCs, mast cells, neutrophils, T cells, and monocyte-macrophage lineage (Figure 2B, Figure S1A-C). The proportion of B cells relative to CD45+ immune cells was lower in AhR-/- mice than in AhR+/+ mice, while those of DCs, ILCs, and T cells were comparable (Figure 2C). Myeloid cells, including monocyte-macrophage lineage and neutrophils, were predominate in the colonic LP. Notably, 38.5% of LP CD45+ cells were intestinal monocyte-macrophage lineage in AhR-/- mice, compared with only 22.1% in AhR+/+ controls (Figure 2C). scRNA-seq transcription profiles showed that, relative to monocyte-macrophage lineage, neutrophils exhibited minimal AhR expression (Figure S1D). Since the monocyte-macrophage lineage are the most abundant cells in the LP, and infiltrating macrophages are known to produce pro-inflammatory cytokines resulting in local uncontrolled inflammation during IBD, we next analyzed the heterogeneity of the intestinal monocyte-macrophage pool. UMAP clustering was applied to divide the monocyte-macrophage lineage into four distinct clusters, based on their respective transcriptional signatures (Figure 2D-E). Among these clusters, Ngp+ monocytes exhibited high expression of Ngp and Il1f9, and were identified as newly-recruited monocytes; Ly6chiCx3cr1lo monocytes exhibited high expression of Vcan, Ly6c2, S100a4, and Cxcl10, similar to hemopoietic system-derived classical inflammatory monocytes; Ly6cintCx3cr1int macrophages exhibited high expression of major histocompatibility complex class II (MHC-II) genes (e.g., H2-Aa, H2-Ab1, H2-DMb1, and H2-Eb1), and were identified as inflammation-elicited plastic macrophages; and Ly6cloCx3cr1hi macrophages expressed high levels of Cx3cr1, Cd163, Mrc1, and Tgfbr2, similar to tissue resident macrophages (Figure 2E, Figure S2A-B). Moreover, the proportions of Ngp+ monocytes, Ly6chiCx3cr1lo monocytes, and Ly6cintCx3cr1int macrophages were significantly higher in the LP of AhR-/- mice than those in AhR+/+ controls, whereas the proportion of tissue resident macrophages (Ly6cloCx3cr1hi) was comparable in both mouse groups (Figure 2F). Further, a pseudotime-organized sequence of “differentiation/activation” events showed that recruited monocytes (Ngp+ and Ly6chiCx3cr1lo monocytes) differentiate into tissue resident macrophages (Ly6cloCx3cr1hi). During intestinal inflammation, this differentiation process is disrupted, leading to accumulation of Ly6cintCx3cr1int macrophages, which produce inflammatory mediators (Figure 2G). Notably, gene ontology enrichment analysis demonstrated that enriched genes were highly associated with the response to the LPS pathway in monocyte-macrophage lineage of AhR-/- mice (Figure 2H). Moreover, we observed that the expression levels of inflammatory cytokines (e.g., Il1β, Il1α, Il18, Il6, Tnf), chemokines (e.g., Cxcl1), and antibacterial peptides (e.g., S100a10) were significantly higher in Ngp+, Ly6chiCx3cr1lo and Ly6cintCx3cr1int monocyte-macrophage subsets from AhR-/- mice than those in AhR+/+ mice (Figure 2I). These results suggest that AhR deletion leads to an increased accumulation of monocyte-macrophage lineage in the gut microenvironment, concomitant with enhanced inflammatory responses.
Distinct transcriptional signatures of LP CD45+ immune cells from AhR-/- mice and AhR+/+ littermates determined by high-throughput single cell RNA sequencing (scRNA-seq) analysis. (A) Experimental schematic of scRNA-seq. Briefly, 8-week-old AhR+/+ and AhR-/- mice were administered 2% DSS in drinking water for 7 days to induce colitis, and intestinal LP CD45+ immune cells isolated for scRNA-seq analysis on day 8 using CD45+ immunological microbeads. (B) UMAP and graph visualization of LP CD45+ cells (16787 AhR-/- and 20844 AhR+/+ cells) defined nine clusters. (C) Pie charts of cell-type fractions for LP CD45+ cells from AhR-/- mice and AhR+/+ mice, colored according to cell type. (D) UMAP and graph visualization of the four monocyte-macrophage lineage cell subsets detected in the two mouse strains. (E) Heatmap showing normalized expression of representative DEGs in each monocyte-macrophage lineage subset. (F) Proportions of AhR-/- and AhR+/+ monocyte-macrophage lineages within each cluster relative to CD45+ immune cells. (G) Pseudotime trajectory analysis of the four monocyte-macrophage lineage cell subsets. (H) Gene ontology terms for the most enriched pathways in intestinal monocyte-macrophage lineages from AhR-/- and AhR+/+ mice. (I) Dot plots showing representative DEGs between AhR-/- and AhR+/+ intestinal monocyte-macrophage lineage cell subsets. AhR, aryl hydrocarbon receptor; DEGs, differentially expressed genes; LP, lamina propria; scRNA-seq, single cell RNA sequence; UMAP, uniform manifold approximation and projection.
AhR deficiency in macrophages exacerbated DSS-induced colitis
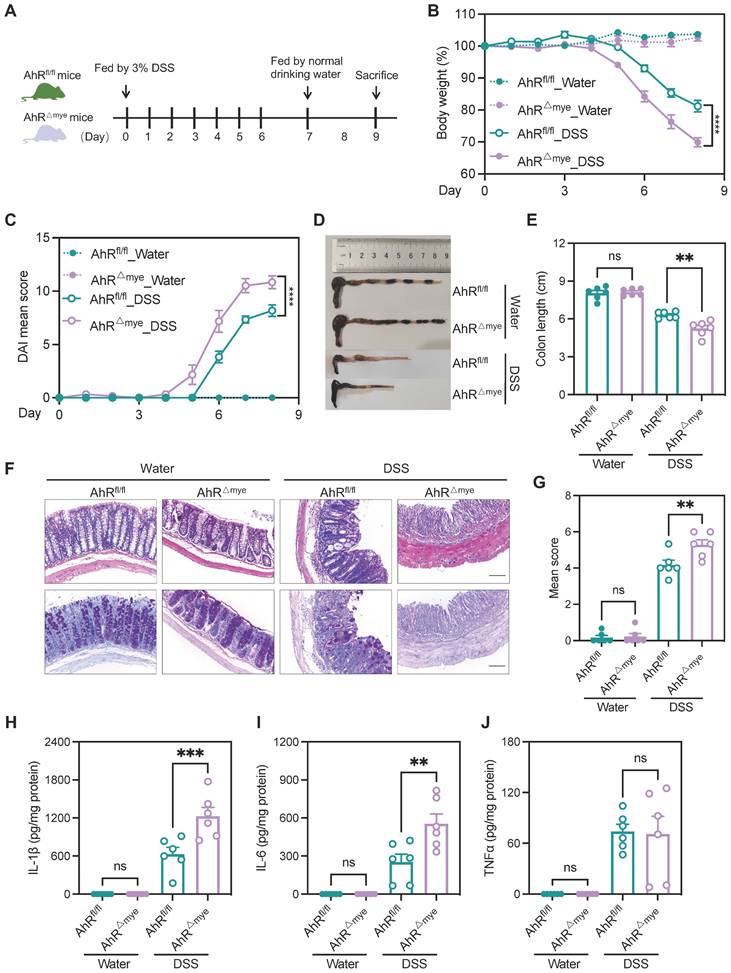
To investigate AhR functions in modulating the role of macrophages in IBD pathogenesis, we generated mice with myeloid cell-specific knock-out of AhR (AhRfl/fl;LysM-Cre+, hereafter AhR△Mye mice) by crossing lysozyme-cre mice with AhRfl/fl mice (Figure S3A-B); co-housed littermates AhRfl/fl mice were used as controls. The efficiency of AhR knockout in macrophages was confirmed by western blot analysis of bone marrow derived macrophages (BMDMs) (Figure S3C).
These cells failed to induce expression of the AhR target gene, Cyp1a1, on stimulation with the AhR ligands, 6-Formylindolo[3,2-b] carbazole (FICZ) or indeno[1,2,3-cd]pyrene (IP) (Figure S3D), confirming efficient and specific AhR deletion in macrophages. Additionally, no apparent abnormalities of external morphology were noted in AhR△Mye mice relative to AhRfl/fl mice. Gut microbiota balance is critical for intestinal homeostasis regulation; however, 16s rDNA analysis revealed that microbial community diversity and phylum composition were comparable between 8-week-old AhR△Mye mice and their AhRfl/fl littermates (Figure S4A-C). Next, age- and sex-matched AhR△Mye mice and co-housed AhRfl/fl littermates were orally administered 3% DSS to induce acute colitis (Figure 3A). Relative to controls receiving water, DSS-treated mice exhibited typical symptoms of colitis, with diarrhea, hematochezia, and body weight loss. Further, DSS-treated AhR△Mye mice showed significantly greater body weight loss, and had higher DAI scores and shorter colons than DSS-treated AhRfl/fl littermates (Figure 3B-E). Histopathological analysis showed that treatment of AhR△Mye mice with DSS led to more severe transmural inflammation, with disruption of the mucosal epithelium, focal areas of extensive ulceration, and more extensive goblet cell loss (Figure 3F-G). Moreover, levels of pro-inflammatory cytokines (e.g., IL-1β and IL-6) were higher in colonic tissue from DSS-treated AhR△Mye mice than those in AhRfl/fl littermates, while TNFα levels did not differ significantly between these two groups (Figure 3H-J).
AhR deficiency in macrophages exacerbated DSS-induced colitis. (A) Schematic representation of DSS-induced colitis model. AhR△Mye mice and co-housed AhRfl/fl littermates were administered 3% (w/v) DSS in drinking water for 7 days, followed by regular drinking water for 1 day. Mice were sacrificed on day 8. (B) Body weight changes were monitored daily and are presented as a percentage of initial body weight (n = 6 per group). (C) DAI was scored daily for each AhR△Mye and AhRfl/fl mouse based on stool consistency, fecal bleeding, and body weight loss (n = 6 per group). (D) Representative morphology images of colon specimens from each group. (E) Colon lengths were measured and recorded on day 8 (n = 6 per group). (F) Representative histopathological images of H&E and PAS-stained colon sections. Scale bar, 200 μm. (G) Semiquantitative scoring of histopathology. (H-J) Expression levels of IL-1β, IL-6, and TNFα in colon tissue quantified by ELISA; n = 6 per group. Statistical analysis of the data was performed using one-way ANOVA (E, G, H, I, J), and two-way ANOVA (B, C) followed by Tukey's multiple comparisons test. Data are presented as the mean ± SEM. ns, not significant; **P < 0.01, *** P < 0.001, and **** P < 0.0001. AhR, aryl hydrocarbon receptor; DSS: dextran sulfate sodium salt; DAI: disease activity index; H&E, hematoxylin and eosin; PAS, periodic acid-Schiff.
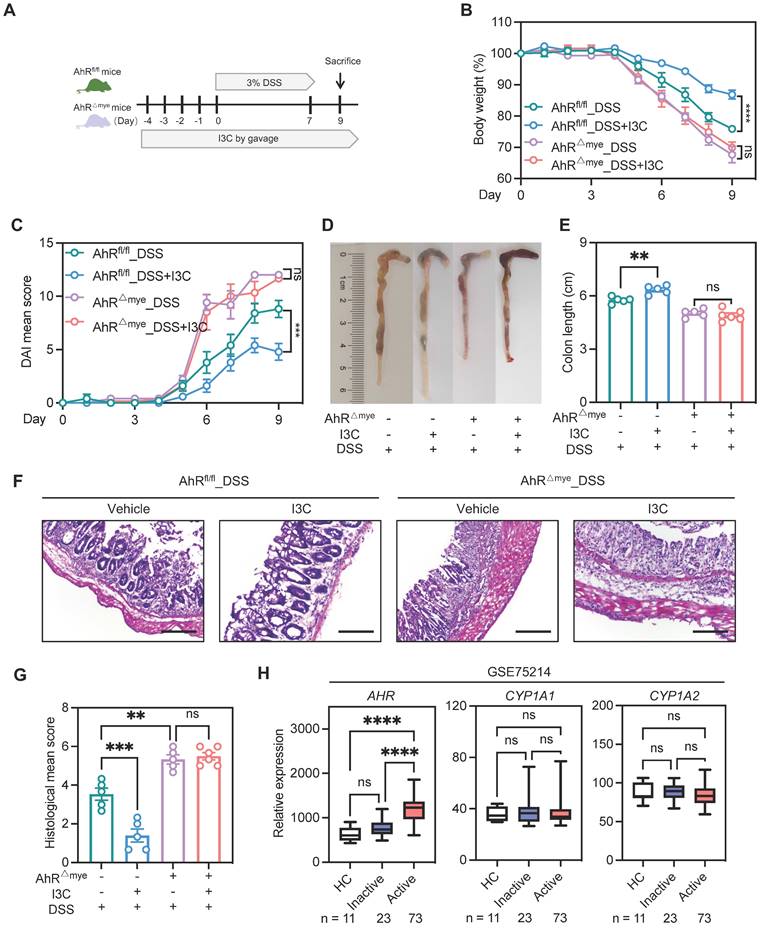
Pharmacologic activation of AhR is reported to rescue the intestinal inflammatory phenotype [34]. Therefore, to further confirm the contribution of macrophage AhR in colitis, we treated AhR△Mye mice and AhRfl/fl mice with indole-3-carbinol (I3C), a plant-derived AhR ligand precursor, by daily oral gavage beginning from 4 days before DSS treatment until the end of the experiments (Figure S5A). Mice treated with I3C showed AhR activation, evidenced by the induction of the AhR target gene, Cyp1a1, in colonic tissue (Figure S5B). Administration of I3C alone had no obvious impact on mouse body weight, colon length, or colonic structure (Figure S5C-F). Next, mice were treated with I3C and DSS (Figure 4A); AhRfl/fl mice treated with I3C effectively ameliorated the colitis disease symptoms, with reduced body weight loss, lower DAI score, and extended colon length compared with the DSS group (Figure 4B-E). The beneficial effects mediated by I3C were also confirmed by histopathology scores (Figure 4F-G). In contrast, AhR△Mye mice treated with I3C did not exhibit colonic inflammation rescue, suggesting that the effect of I3C is partially mediated through targeting AhR in macrophages (Figure 4B-G). Further, we analyzed transcriptome profiles from independent cohorts of patients with UC (GSE75214) and found that AHR mRNA levels were elevated in active UC mucosa relative to those in healthy controls, while no significant difference was detected in the AhR target gene, CYP1A1 and CYP1A2 (Figure 4H). Collectively, these data suggest that macrophage-specific AhR activation is required to confer protection from DSS-induced colonic damage and colitis.
Pharmacologic activation of AhR alleviated inflammation in the DSS-induced colitis model, dependent on macrophages. (A) Schematic illustration of DSS-induced colitis. Age- and sex-matched AhR△Mye and AhRfl/fl mice were administered 3% DSS in drinking water for 7 days followed by normal drinking water for 2 days. Mice in I3C treatment groups received daily gavage of I3C for 14 days. (B) Body weight changes were monitored daily and are depicted as a percentage of initial body weight. (C) DAI was scored daily for each mouse, based on stool consistency, fecal bleeding, and body weight loss. (D) Representative images showing the morphology of colons from each group. (E) Colon lengths were measured and recorded on day 9. (F) Representative histopathological images of H&E-stained colon sections. Scale bar, 200 μm. (G) Semiquantitative scoring of histopathology. n = 5-6 per group. (H) Database (GSE75214) analysis of AHR, CYP1A1, and CYP1A2 mRNA in intestinal mucosal biopsies from patients with UC and healthy controls. Statistical analysis of the data was performed using one-way ANOVA (E, G, H CYP1A2) and two-way ANOVA (B, C) followed by Tukey's multiple comparisons test, or Welch ANOVA tests followed by Dunnett's T3 multiple comparisons test (H AHR), or Kruskal-Wallis test followd by Dunn's multiple comparisons test (H CYP1A1). Data are shown as the mean ± SEM. ns, not significant; **P < 0.01, ***P < 0.001, and ****P < 0.0001. AhR, aryl hydrocarbon receptor; DSS: dextran sulfate sodium salt; DAI: disease activity index; H&E, hematoxylin and eosin; HC, healthy control; I3C, indole-3-carbinol; UC, ulcerative colitis.
AhR deficiency enhanced IL-1β secretion by promoting macrophage pyroptosis
We detected increased numbers of F4/80-labeled macrophages in the DSS group, suggesting increased macrophages that resided in and were recruited to the colon (Figure 5A). Infiltrating macrophages are known to produce pro-inflammatory cytokines, resulting in local uncontrolled inflammation during IBD [29]. Therefore, we examined whether AhR deficiency disturbed cytokine expression. The results showed the increased presence of F4/80+IL-1β+ macrophages infiltrating in DSS-treated AhR△Mye mice relative to AhRfl/fl mice (Figure 5A), consistent with increased IL-1β expression in colon tissues (Figure 3H). IL-1β is a secreted effector protein produced by activation of the NLRP3 inflammasome, which is dominantly released by macrophages undergoing pyroptosis [22]. As AhR deficiency significantly increased IL-1β levels, we hypothesized that AhR regulates macrophage pyroptosis and cytokine production in the context of NLRP3 activation. Thus, BMDMs from WT or AhR-/- mice were primed with LPS before treatment with the commonly used activator, nigericin (Nig) or ATP (Figure 5B). Pyroptosis, as measured by lactate dehydrogenase (LDH) release, was significantly increased in primary AhR-/- BMDMs relative to WT BMDMs (Figure 5C, Figure S6A). In addition, more AhR-/- BMDMs were positive for staining with propidium iodide (PI; a dead cell dye indicator), indicating more cell death among AhR-/- BMDMs (Figure 5D). NLRP3 inflammasome mediated pyroptosis also leads to caspase-1 activation and GSDMD cleavage, accompanied by secretion of mature IL-1β. As expected, AhR deficiency promoted caspase-1 activation (caspase1-p20), GSDMD cleavage (GSDMD-NT), and mature IL-1β secretion (IL-1β p17) in LPS-primed macrophages treated with Nig or ATP (Figure 5E-G, Figure S6B).
AhR deficiency enhanced IL-1β secretion by promoting macrophage pyroptosis. (A) Immunofluorescence labeling of IL-1β (red), F4/80+ (green) macrophages, and DAPI (blue) in colon sections from control or DSS-treated AhR△Mye and AhRfl/fl mice. Scale bar, 50 μm. (B-I) BMDMs isolated from WT or AhR knockout (AhR-/-) mice were stimulated with LPS (200 ng/mL) for 4 h, followed by treatment with Nig for 45 min. (B) Schematic of experimental design. (C) Cytotoxicity was assessed by LDH release. (D) Cell morphology assessed by PI staining. Scale bar, 50 μm. (E) IL-1β levels in cell culture supernatants determined by ELISA. (F) Cell lysates and supernatants were immunoblotted to detect full-length and cleaved caspase-1 and GSDMD, and mature IL-1β (IL-1β p17). (G) Quantitative protein analysis for F. (H) Representative immunofluorescence images of ASC speck formation (red). Nuclei were stained with DAPI (blue). Scale bar, 20 μm. (I) Bar chart showing percentages of BMDMs containing a visible ASC speck. (J-K) AhR-/- BMDMs were pretreated with VX-765 (50 μM) or vehicle control for 30 min, then stimulated with LPS for 4 h followed by Nig for 45 min. (J) Proteins from cell lysates and supernatants were immunoblotted for full-length and cleaved caspase-1 and GSDMD, and mature IL-1β (IL-1β p17). (K) Cytotoxicity was assessed by LDH release. (L) Immunoblot analysis of full-length and cleaved GSDMD and caspase-1 in colon tissues from control or DSS-treated AhR△Mye and AhRfl/fl mice. n = 3-6 per group. Statistical analysis of the data was performed using one-way ANOVA (K) and two-way ANOVA (A, C, D, E, G, I, J, L) followed by either Tukey's or Sidak's multiple comparison tests. Data are shown as the mean ± SEM from three independent experiments. ns, not significant; *P < 0.05, **P < 0.01, and ****P< 0.0001. AhR, aryl hydrocarbon receptor; ASC, apoptosis associated speck like protein containing a CARD; BMDM, bone barrow derived macrophage; LDH, lactate dehydrogenase; Nig, nigericin; PI, propidium iodide; WT, wild type.
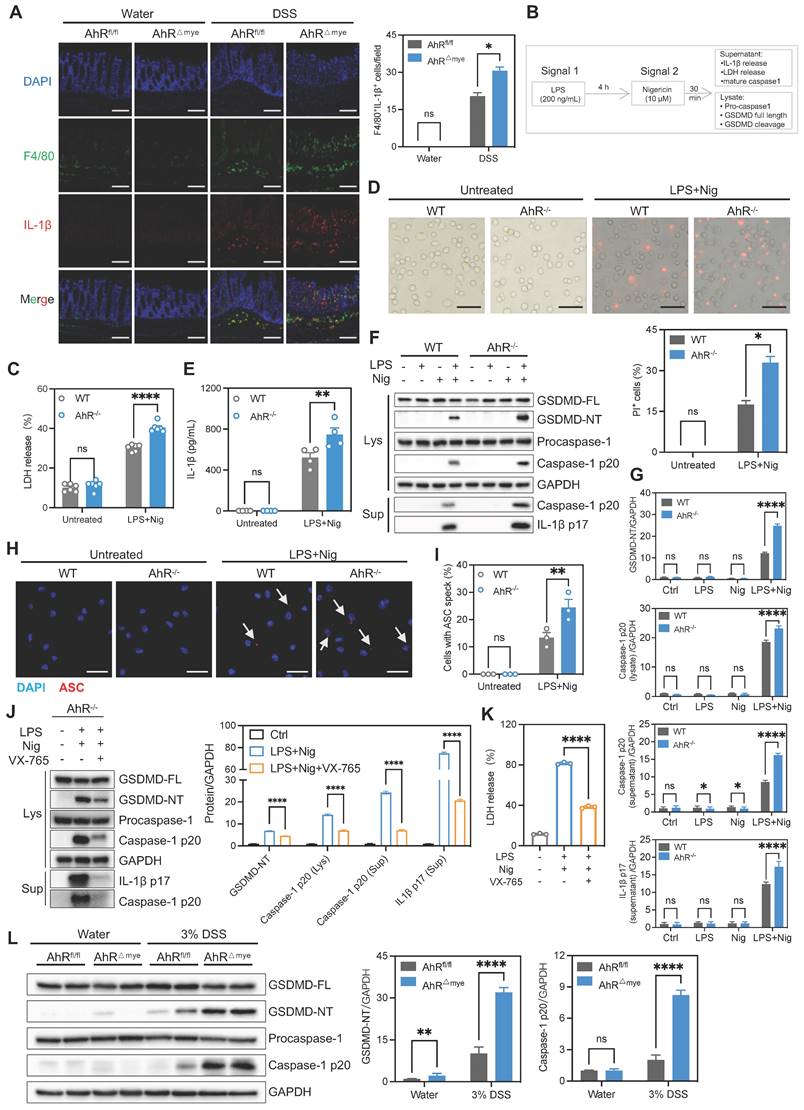
To validate our findings indicating that AhR is involved in NLRP3 inflammasome activation, we assessed speck formation by apoptosis associated speck like protein containing a CARD (ASC), as an indicator of inflammasome assembly. We found that LPS plus Nig-induced ASC speck formation was enhanced in AhR-deficient cells (Figure 5H-I). Total ASC protein levels were not altered (Figure S6C). Moreover, treatment with the caspase-1 inhibitor, VX-765, markedly restored LDH release and IL-1β production in AhR-deficient cells, confirming that cell death induction was indeed caspase-1-dependent (Figure 5J-K). Next, we evaluated in vivo pyroptosis activation in both control and DSS-treated mice. Immunoblotting analysis showed substantially increased caspase-1 p20 and GSDMD-NT in colonic tissues from colitis mice relative to water control mice. Further, the up-regulation of caspase-1 p20 and GSDMD-NT was more aggravated in DSS-treated AhR△Mye than in AhRfl/fl mice (Figure 5L). Additionally, mRNA expression levels of pyroptosis-associated genes, including NLRP3, Caspase-1, GSDMD, and IL-1β were elevated in active UC mucosa compared with healthy controls (GSE75214) (Figure S6D). Together with our previous results, these findings suggest that AhR deficiency in macrophages promoted macrophage pyroptosis and enhanced IL-1β secretion and may be associated with IBD severity.
AhR suppressed IL-1β production by promoting Odc1 transcription
We next explored how AhR regulates the inflammasome to suppress IL-1β production. As IL-1β production is regulated transcriptionally and post-transcriptionally, we first examined the effect of AhR on IL-1β expression; however, IL-1β mRNA or protein levels were not differed significantly between WT and AhR-/- BMDMs (Figure S6E-F). Since AhR deficiency promoted mature IL-1β production, we speculated that the increase in IL-1β production by AhR-deficient cells was due to enhanced secretion, rather than increased synthesis, of IL-1β. We found that AhR did not interact directly with NLRP3 inflammasome components (Figure S6G). Since AhR is a ligand-dependent transcription factor, we next performed RNA-seq of WT and AhR-/- BMDMs, to comprehensively investigate AhR-dependent changes in gene expression associated with IL-1β secretion. Among the genes differentially expressed in AhR-/- macrophages, we noted markedly reduced expression of ornithine decarboxylase 1 (Odc1) which is reported to negatively regulate M1 macrophage activation and IL-1β production [35] (Figure 6A, Figure S7A). Reduced Odc1 expression was confirmed by real-time PCR and western blot analysis in WT and AhR-/- BMDMs (Figure 6B, Figure S7B), as well as in colon tissues in the context of DSS-induced colitis (Figure S7C-E). In addition, we found that ODC1 mRNA levels were elevated in active UC mucosa relative to those in healthy controls, and that AHR expression levels were positively correlated with those of ODC1 (GSE75214) (Figure 6C-D). As the rate-limiting enzyme in polyamine biosynthesis, ODC1 converts L-ornithine to putrescine, which is subsequentially converted to spermidine and spermine by spermidine and spermine synthases, respectively (Figure 6E). Importantly, AhR depletion resulted in decreased levels of the three major mammalian polyamines, putrescine, spermidine, and spermine, in BMDMs (Figure 6F). Furthermore, we assessed the polyamine levels in colon tissues from DSS-treated AhR-/- and AhR+/+ mice. The results showed that, upon DSS treatment, the levels of putrescine, spermidine and spermine were reduced in the colon of AhR-/- mice compared to AhR+/+ mice (Figure S7F-H).
AhR suppressed IL-1β production by promoting Odc1 transcription. (A) Heatmap illustrating differentially expressed genes between WT and AhR-/- BMDMs. (B) Odc1 expression levels in WT and AhR-/- BMDMs determined by RT-qPCR. (C) Database (GSE75214) analysis of ODC1 mRNA expression in intestinal mucosal biopsies from patients with UC and healthy controls. (D) Spearman correlation (two-tailed) analysis between AHR and ODC1 expression levels in intestinal mucosal biopsies (GSE75214). (E) Schematic illustration of polyamine biosynthesis. (F) Intracellular polyamine levels measured by liquid chromatography-mass spectrometry. (G) AhR-/- BMDMs were transfected with empty vector control or pcDNA3.1-ODC1 (500 ng) and IL-1β release measured by ELISA. (H) Proteins from cell lysates and supernatants were immunoblotted for full-length and cleaved caspase-1 and GSDMD, and mature IL-1β (IL-1β p17). (I) Cytotoxicity was assessed by LDH release. (J and K) WT BMDMs were pretreated with DMSO or AhR ligands (IP or FICZ) for 24 h, and mRNA expression levels of Odc1 and the AhR target gene, Cyp1a1, measured by RT-qPCR. (L) Schematic diagram of AhR binding sites in the Odc1 promoter region. (M) Luciferase reporter assay in HEK293T cells co-transfected with Renilla, pGL3-basic-ODC1 promoter (-2000 to +200) reporter, pEGFP-C3 empty vector, or pEGFP-C3-AhR. Values are presented as means, normalized to transfection efficiency by dividing firefly luciferase activity by Renilla luciferase activity. (N) ChIP analyses of the binding efficiency of AhR to the Odc1 promoter region in RAW264.7 cells. n = 3-6 per group. Statistical analysis of the data was performed using two-tailed unpaired t test (M), one-way ANOVA (J, K) and two-way ANOVA (B, F, G, I, N) followed by either Tukey's or Sidak's multiple comparison tests, or Kruskal-Wallis test (C) followed by Dunn's multiple comparisons test. Data are shown as the mean ± SEM from three independent experiments. ns, not significant; *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. AhR, aryl hydrocarbon receptor; BMDM, bone barrow derived macrophage; ChIP, chromatin immunoprecipitation; ELISA, enzyme-linked immunosorbent assay; FICZ, 6-Formylindolo[3,2-b]carbazole; HC, healthy control; IP, indeno[1,2,3-cd]pyrene; LDH, lactate dehydrogenase; Nig, nigericin; ODC1, ornithine decarboxylase 1; PI, propidium iodide; RT-qPCR, quantitative reverse transcription polymerase chain reaction; UC, ulcerative colitis; WT, wild type; XRE, xenobiotic response element.
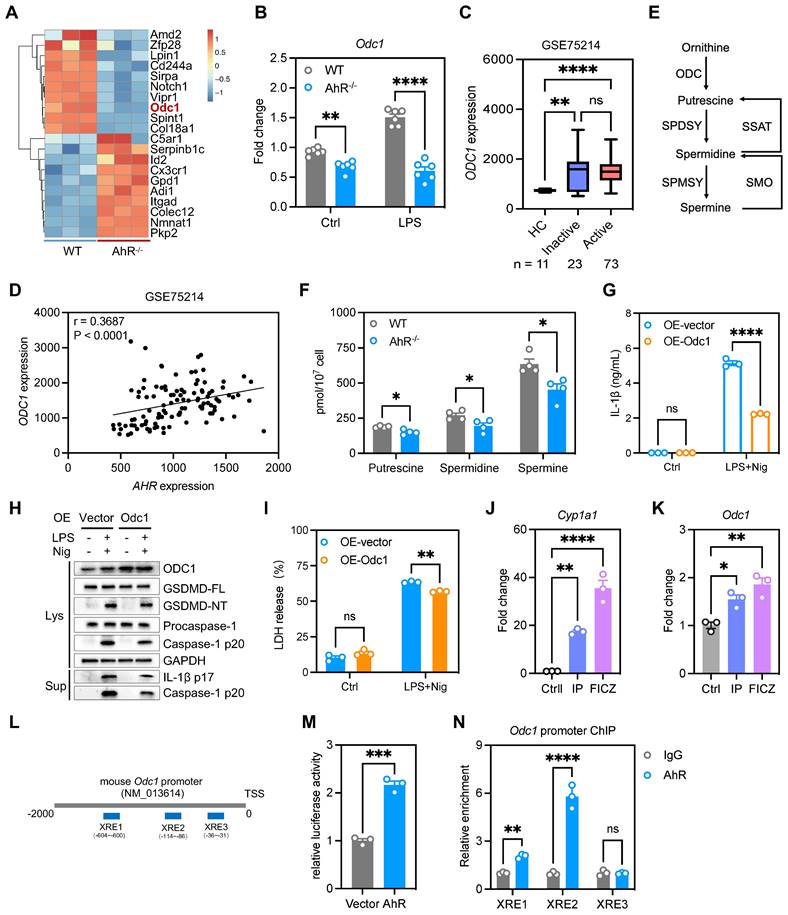
To investigate whether the increased IL-1β secretion in AhR-/- BMDMs was due to their reduced Odc1 expression, we next over-expressed Odc1 in AhR-/- BMDMs. Odc1 overexpression significantly inhibited LPS plus Nig-induced secretion of IL-1β compared with empty vector controls (Figure 6G), accompanied by decreased caspase-1 p20 and GSDMD-NT levels, suggesting that decreased Odc1 expression, at least in part, contributed to the increased IL-1β secretion in AhR-null BMDMs on NLRP3 inflammasome activation (Figure 6H, Figure S7I). Moreover, Odc1 overexpression inhibited AhR-/- BMDMs pyroptosis, as evidenced by reduced LDH release (Figure 6I). In contrast, Odc1 depletion promoted caspase-1 p20 activation and GSDMD cleavage, along with increasing IL-1β secretion and LDH release (Figure S8A-D). We also detected the pyroptosis levels in macrophages with ODC1 knockdown, followed by AhR agonist (FICZ) administration in the presence of LPS plus Nig treatment. The results showed that ODC1 knockdown enhanced macrophage pyroptosis, as evidenced by increased activation of pyroptosis-related proteins and LDH release. AhR agonist administration inhibited macrophage pyroptosis, whereas the inhibitory effect was abrogated with ODC1 knockdown (Figure S8E-F). Furthermore, treatment with endogenous (FICZ) and exogenous (IP) AhR ligands induced mRNA expression of Odc1, and that of a specific AhR target, Cyp1a1, which served as a positive control (Figure 6J-K).
AhR is reported to control gene transcription activity by direct binding to XRE in promoter regions in its target genes [36]; therefore, we next determined whether AhR bound to the Odc1 promoter and modulated its transcription activity. Three putative XREs were identified in the mouse Odc1 promoter: XRE-1 (nt -604 to -600), XRE-2 (nt -114 to -86), and XRE-3 (nt -35 to -31) (Figure 6L). To assess the function of these XREs in AhR-induced Odc1 expression, Odc1 promoter regions containing XREs (nt -2000 to +200) were sub-cloned into the pGL3-basic luciferase reporter plasmid. HEK293T cells transfected, in parallel, with the Odc1 promoter reporter construct were co-transfected with either AhR expression vector or empty vector as a control. AhR expression significantly increased luciferase reporter activity of Odc1 promoter (Figure 6M). Next, we performed chromatin immunoprecipitation (ChIP) assays using a commercially available antibody against AhR and primers flanking the putative AhR binding sites. Significant DNA enrichment of XRE1 and XRE2, but not XRE3, was detected in AhR versus IgG-precipitated material (Figure 6N). These data indicate that AhR controls Odc1 transcription and that decreased Odc1 expression is at least one of the causes for the increased IL-1β secretion detected in AhR-null BMDMs.
Spermine prevented macrophage pyroptosis and IL-1β production
ODC1 is the first and rate-limiting enzyme in polyamine biosynthesis, with spermine the final product of polyamine metabolism [37]. It is unclear whether polyamines regulate macrophage pyroptosis. We hypothesized that addition of exogenous spermine to cell cultures would reverse the enhanced pyroptosis and IL-1β production observed. To test this hypothesis, we treated BMDMs with difluoromethylornithine (DFMO), an irreversible and specific ODC inhibitor, for 3 days and then added exogenous spermine to the culture. The addition of spermine indeed reversed LDH release in DFMO-treated cells (Figure S9A), implying that the effects of ODC1 in inflammation may depend on polyamine biosynthesis pathways. Moreover, exogenous spermine inhibited GSDMD cleavage and abrogated caspase-1 p20 and IL-1β secretion in macrophages undergoing pyroptosis (Figures 7A-B). In addition, exogenous spermine inhibited pyroptosis-induced LDH release (Figure 7C) and reduced the proportion of PI-positive cells (Figure 7D, Figure S9B).
Spermine prevented macrophage pyroptosis and IL-1β production. Cells were pretreated with spermine (50 μM) together with LPS (200 ng/mL) for 4 h, then stimulated with nigericin (10 mM) for 30-45 min. (A) IL-1β secretion in supernatants determined by ELISA. (B) Proteins in cell lysates and supernatants were immunoblotted to detect full-length and cleaved caspase-1, GSDMD, and IL-1β. (C) Cytotoxicity was detected by LDH release assay. (D) Cell morphology determined by PI staining. Scale bar, 50 μm. (E) Relative intracellular K+ concentration detected by K+ ion fluorescence probe; data are shown as MFI. Statistical analysis of the data was performed using one-way ANOVA (A, C, E) and two-way ANOVA (B) followed by Tukey's multiple comparison tests. Data are shown as the mean ± SEM from three independent experiments. **P < 0.01, and ****P < 0.0001. ELISA, enzyme-linked immunosorbent assay; LDH, lactate dehydrogenase; MFI, mean fluorescence intensity; PI, propidium iodide; Spm, spermine.
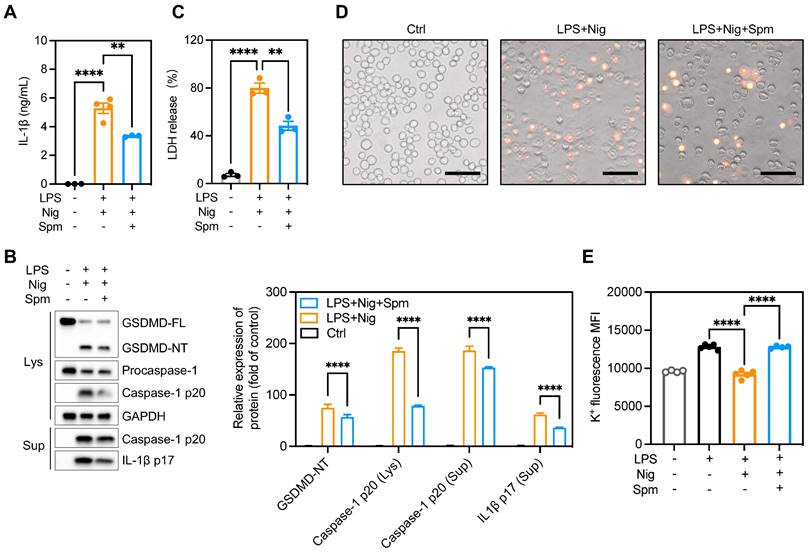
The main pathways related to NLRP3 inflammasome activation include: K+ efflux, mitochondria dysfunction, reactive oxygen species (ROS) production and lysosomal rupture [38]. Previous studies have demonstrated that polyamines such as spermidine and spermine block K+ efflux from cells by voltage-dependent binding to inwardly rectifying K+ (Kir) channels and displacing K+ ions from the pore [39, 40]. To investigate the role of spermine on K+ efflux, we analyzed relative intracellular K+ concentration during NLRP3 inflammasome activation using a K+ ion fluorescence probe. Spermine treatment significantly restrained K+ efflux in macrophages undergoing pyroptosis (Figure 7E). Additionally, we assessed speck formation by ASC, which serves as an indicator of inflammasome assembly. We observed that ASC speck formation induced by LPS plus Nig was inhibited upon spermine treatment (Figure S9C-D). These data suggest that spermine inhibits K+ efflux associated with NLRP3 inflammasome activation in macrophages.
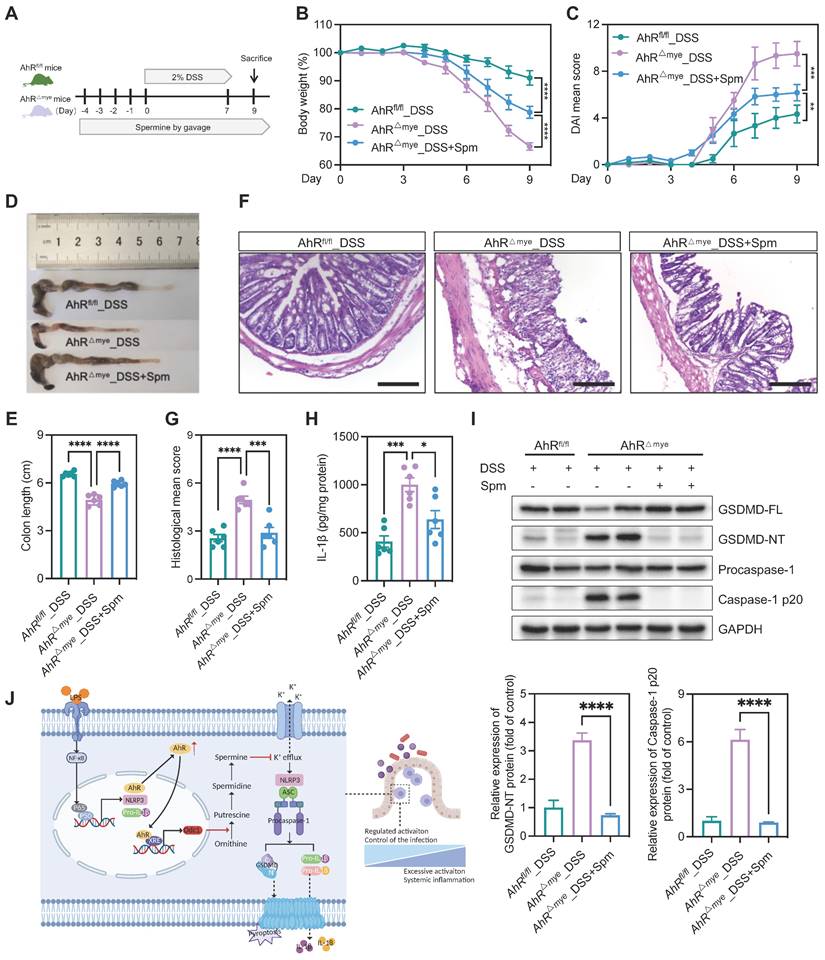
We then tested the in vivo effect of spermine supplementation in the DSS-induced colitis model. Mice were treated with spermine by daily oral gavage, beginning from 4 days before DSS treatment until the end of the experiments (Figure 8A). Administration of spermine alone had no significant impact on body weight, colon length, or colonic structure (Figure S10A-E). We also assessed the effect of spermine supplementation in this model and found that spermine levels were increased in the colon of mice following spermine oral gavage compared to control vehicle mice (Figure S10F). AhRfl/fl and AhR△Mye mice began to lose body weight on day 4 after starting DSS treatment and AhR△Mye mice showed more body weight loss than their AhRfl/fl littermates (Figure 8B). Remarkably, DSS-induced body weight loss, DAI score, and colon shorting were rescued in AhR△Mye mice treated with spermine (Figure 8B-E). Further, immune cell infiltration and epithelial damage were significantly aggravated in AhR△Mye mice compared with AhRfl/fl mice, and these features were rescued in AhR△Mye mice receiving spermine (Figures 8F-G). DSS-induced pyroptosis was significantly increased in AhR△Mye mice compared with AhRfl/fl mice, and treatment with spermine significantly reduced the expression of caspase-1 p20 and GSDMD-NT, as well as IL-1β production in colon tissue from AhR△Mye mice (Figure 8H-I). Thus, spermine treatment significantly inhibited macrophage pyroptosis and ameliorated DSS-induced colitis in AhR△Mye mice.
Spermine confers protection against intestinal inflammation. (A) Schematic illustration of DSS-induced colitis. Age- and sex-matched AhR△Mye and AhRfl/fl mice were administered 2% DSS in drinking water for 7 days, followed by normal water for 2 days. Mice in spermine treatment groups received daily gavage of Spm for 14 days. (B) Body weight changes were monitored daily and are presented as the percentage of initial body weight. (C) DAI was scored daily for each AhR△Mye and AhRfl/fl mouse based on stool consistency, fecal bleeding, and body weight loss. (D) Representative morphology images of colon specimens from each group. (E) Colon lengths were measured and recorded on day 9. (F) Representative histopathological images of H&E-stained colon sections. Scale bar, 200 μm. (G) Semiquantitative scoring of histopathology. (H) IL-1β expression in colon tissues determined by ELISA. (I) Immunoblot analysis of full-length and cleaved GSDMD and caspase-1 in colon tissue from DSS-treated AhR△Mye and AhRfl/fl mice. n = 6 per group. (J) Proposed working model of AhR confers protection against macrophage pyroptosis and intestinal inflammation. Statistical analysis of the data was performed using one-way ANOVA (E, G, H, I) and two-way ANOVA (B, C) followed by Tukey's multiple comparison tests. Data are shown as the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. AhR, aryl hydrocarbon receptor; DSS, dextran sulfate sodium; DAI, disease activity index; ELISA, enzyme-linked immunosorbent assay; HE, hematoxylin and eosin; Spm, spermine.
Discussion
In the present study, we identified a critical role for AhR in regulating macrophage pyroptosis. Using macrophage-specific AhR-deficient mice, we found that AhR△mye mice developed severe colitis on DSS treatment, characterized by a significant increase in macrophage pyroptosis and subsequent IL-1β release in colon tissues, compared with AhRfl/fl littermates. Mechanistic studies showed that AhR inhibited macrophage pyroptosis, in part via promoting Odc1 transcription, to enhance polyamine biosynthesis. The increased polyamine levels, particularly spermine, inhibited NLRP3 inflammasome assembly and subsequent pyroptosis by suppressing K+ efflux. Therefore, our findings reveal a potentially novel mechanism whereby the AhR-ODC1-polyamine-pyroptosis axis confers protection against experimental colitis and maintains intestinal immune homeostasis (Figure 8J).
AhR modulates intestinal homeostasis by targeting the development, differentiation, function, and maintenance of several key mucosal immune cell types in the gut [11, 12, 14, 41, 42]. While the molecular basis underlying these processes remains unclear, previous studies have shown that administration of the AhR agonist, FICZ, ameliorated colitis severity by downregulating pro-inflammatory cytokines and upregulating IL-22 production in DSS-, 2,6,4-trinitrobenzene sulfonic acid (TNBS)-, and T-cell transfer-induced colitis [34]. In contrast, treatment of mice with an AhR antagonist reduced IL-22 production and increased the severity of inflammation in a TNBS-induced mouse colitis model [34]. DSS-induced colitis severity was also attenuated by the toxic AhR ligand, 2,3,7,8-tetrachlorodi-benzo-p-dioxin (TCDD), via promoting regulatory T cell (Treg) differentiation and reducing Th17 cell induction through epigenetic regulation [14]. It is established that AhR functions are cell type- [43, 44], ligand- [45] and context- [46] dependent. Since AHR gene expression is increased, while CYP1A1 and CYP1A2 expression showed no difference, in inflamed tissue from patients with UC compared with healthy controls, it is tempting to speculate that AhR upregulation with inadequate activation may be a secondary protective feedback response. Therefore, it is necessary to improve AhR activation to fine-tune the imbalance between pro- and anti- inflammatory factors in patients with IBD. Using genetically modified mouse strains with cell type-specific AhR deletion, we found that mice with AhR deficient in macrophages developed more severe DSS-induced colonic damage. Moreover, dietary supplementation with an AhR pro-ligand, I3C, showed a protective effect in AhRfl/fl, but not AhR△mye colitis model mice, highlighting the critical role of macrophage AhR in regulating intestinal inflammation. In particular, AhR-mediated spermine production was shown to confer protection against macrophage pyroptosis and intestinal inflammation, adding a potential new dimension to understanding of AhR-regulated intestinal immune responses.
A constant balance between tissue-resident macrophages and recruited circulating monocytes is critical for maintaining homeostasis in a healthy gut and ensuring protective immunity during infection and autoimmunity [15]. ScRNA-seq analysis revealed that the process of circulating monocytes differentiation into tissue resident macrophages is disrupted during colitis, leading to an increased proportion of Ly6cintCx3xr1int macrophages in the LP, which are the main source of IL-1β and IL-6 [47]. In addition, the proportion of macrophage subsets and the expression of pro-inflammatory cytokines, including IL-1β and IL-6, were significantly elevated in AhR△mye mice compared with those in AhRfl/fl littermates. AhR can negatively regulate LPS-induced inflammatory responses through its interaction with the NF-κB pathway via Stat1, to inhibit IL-6 expression [18] and, via plasminogen activator inhibitor-2 (Pai-2), to inhibit IL-1β secretion [19]. AhR activation by TCDD negatively regulates NLRP3 inflammasome activity by inhibiting NLRP3 transcription [48]. We found that AhR-/- macrophages secreted much larger amounts of IL-1β on NLRP3 inflammasome activation, likely due to enhanced IL-1β processing, rather than synthesis. Since IL-1β is the secreted effector protein of pyroptosis, we found that AhR functions as a suppressor of macrophage pyroptosis via targeting polyamine biosynthesis. Pyroptosis-associated proteins were upregulated in colonic tissues from AhR△mye mice compared with those in AhRfl/fl littermates, which may have contributed to the development of intestinal mucosal inflammation in AhR△mye mice. Over-secretion of IL-1β by AhR-deficient macrophages suggests that AhR may function as a suppressor of macrophage pyroptosis, and even as a physiological immune suppressor. There is an apparent discrepancy between our observation of increased macrophage numbers in the colon of AhR△Mye mice following DSS challenge and the protective role of AhR against macrophage pyroptosis. The overall inflammatory environment created by DSS challenge and pyroptosis-induced cytokine release can attract immune cells, including macrophages, to the inflammation site. This amplification of the immune response can result in an increase in macrophage numbers, even if some macrophages undergo pyroptosis. Compensatory mechanisms may also enhance macrophage recruitment, proliferation, or survival to counteract the loss due to pyroptosis. Therefore, we also believe that AhR deficiency may affect the migration or recruitment of macrophages, possibly due to direct or indirect induction by macrophage pyroptosis.
Pyroptosis has been proposed to play an important role in the pathogenesis of colitis, although the results remain controversial. Some studies showed that NLRP3-/- deficiency resulted in decreased intestinal inflammation [16, 49], while other investigations have suggested that abnormal activation of the NLRP3 inflammasome leads to release of pro-inflammatory cytokines, such as IL-1β and IL-18, which disrupt the intestinal barrier [50, 51]. We found that the NLRP3 inflammasome was overactivated in mice in the DSS-treated group, and that spermine treatment suppressed expression levels of IL-1β and the pyroptosis executive proteins, caspase-1 p20 and GSDMD-NT. It is likely that pyroptosis activation in IBD occurs in the epithelium and plays a protective role, as it would be expected to maintain homeostasis through regulation of commensal microbiota and eradication of harmful bacterial; however, once the epithelial barrier is disrupted, gut microbiota will infiltrate into the LP and recruit immune cells; in this case, pyroptosis would be predicted to provoke a cytokine storm, which would damage the mucosa. Interestingly, a recent study reported that full-length GSDMD in epithelial cells plays a non-pyroptotic role in promoting the release of IL-1β-containing small extracellular vesicles [52]. Rana et al. also reported that GSDMB facilitates epithelial restitution and repair, dependent on PDGF-A-mediated FAK phosphorylation, but not pyroptosis [53]. The role of pyroptosis in IBD is emerging as the focus of increasing attention, and a clear understanding of the mechanism underlying pyroptosis will guide the development of future effective therapeutics for IBD.
Odc1 mRNA expression levels were markedly reduced in AhR-/- BMDMs, similar to a previous finding in normal human fibroblast (WI-38) cell line [54]. Additionally, we noticed that AHR is not induced in inactive disease, whereas ODC1 is. This suggests that ODC1 might also be AHR-independently regulated. Here are several alternative mechanisms to consider. First, microbiota-derived signals, such as LPS, can stimulate ODC activity through toll-like receptor signaling pathways independently of AhR activation [55]. Second, metabolic changes associated with inflammation, such as increased glycolysis or polyamine demand, can stimulate ODC1 expression to support cellular functions [56]. Third, inflammatory cytokines, such as TNF-α and IL-1β, are often elevated in inflammatory conditions and can activate downstream signaling pathways, including NF-κB, which in turn can enhance ODC1 transcription [57]. ODC1 is the first and rate-limiting enzyme in polyamine metabolism. Polyamines, including putrescine, spermidine, and spermine, are abundant within the gastrointestinal tract and are essential for cell viability, proliferation, function, and differentiation [58]. Natural polyamines are reported to have potential as an adjunctive treatment for colitis in mouse models. Nishiguchi et al. developed oral anti-inflammatory polyamine-based nanomedicines that exhibited great potential as biocompatible ROS scavenger drugs that suppressed inflammatory responses in a UC colitis mouse model [59]. Microbial-derived putrescine increased the abundance of anti-inflammatory macrophages in the colon, and ameliorated DSS-induced colitis [60]. Exogenous supplementation with putrescine or ornithine enhanced ILC3 production of IL-22, and protected mice against Citrobacter rodentium infection [61]. Spermidine supplementation reversed the DSS-induced colitis phenotype associated with increased expression of α-defensins and a shift in microbiome [62], M1/M2 macrophage polarization [63], and enhanced hypusination of intestinal epithelial cells [64]. Here we show that spermine supplementation can inhibit macrophage pyroptosis in vitro and ameliorate colon damage in vivo, which might be related to potassium channel regulation, suggesting its potential utility against IBD.
In summary, our study highlights a potentially novel regulatory pathway, involving the AhR-ODC1-polyamine-pyroptosis axis, in IBD. The abundance of natural AhR ligands and polyamines found in the environment provide a positive outlook for future therapeutic agents targeting AhR or polyamines in macrophages, as immunomodulators through their inhibitory effects on macrophage pyroptosis.
Methods
Mice
All mice used in this study were on a C57BL/6 background. AhR-null mice (AhR KO, B6.129-Ahrtm1Bra/J) and AhRfl/fl (B6.129(FVB)-Ahrtm3.1Bra/J) mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Lyz2-Cre mice [B6.129P2-Lyz2tm1(cre)Ifo/J] were obtained from Shanghai Model Organisms (Shanghai, China). AhRfl/fl mice bearing two loxP sites flanking the second exon of the AhR gene were cross-bred with Lyz2-Cre mice to specifically knockout AhR in the myeloid cell lineage, including macrophages (termed AhR△Mye mice). Mouse genotyping primers are listed in Supplementary Table S1. C57BL/6 mice were obtained from the SLAC Laboratory Animal Co. (Shanghai, China). All animal experiments complied with relevant laws and institutional guidelines, as overseen by the Committee on Animal Research and Ethics of Fudan University (Shanghai, China). All animals were housed, bred, and maintained under specific pathogen-free conditions.
DSS-induced mouse colitis model
A DSS-induced mouse colitis model was established as described previously, with slight modifications [33]. Briefly, age- and sex-matched AhR+/+, AhR-/-, AhRfl/fl, and AhR△Mye mice were administered DSS (molecular weight, 36-50 kDa; MP Biomedicals, Santa Ana, CA, USA) in drinking water for 7 days, followed by regular drinking water for 2 days. Our study examined male and female animals, and similar findings are reported for both sexes. Control mice were provided with normal drinking water. For pharmacological treatment, I3C (Sigma, Milpitas, CA, USA) or spermine (Sigma, Milpitas, CA, USA) were dissolved in DMSO and further diluted with ddH2O to generate working concentration solutions. Animals were treated with or without I3C (1 mg per mouse in 100 μL ddH2O) or spermine (50 mg/kg body weight in 100 μL ddH2O) by daily oral gavage, beginning from 4 days before DSS treatment until the end of the experiments. Mice in the control group received an equal volume gavage of vehicle. Mice were sacrificed in an automated CO2 delivery system, and their colons collected immediately for colon length measurement, protein extraction, and histology analysis.
Determination of disease activity index (DAI)
DAI was scored daily for each mouse, based on body weight loss, stool consistency, and gross bleeding. Scores were evaluated, as follows: body weight loss, depicted as the percentage of initial body weight: 0 (no loss), 1 (1% to 5% loss), 2 (6% to 10% loss), 3 (11% to 15% loss), 4 (> 15% loss); stool consistency: 0 (normal), 2 (pasty and semi-formed stools, did not adhere to the anus), 4 (liquid stools that did adhere to the anus); bleeding: 0 (no blood), 1 (positive hemoccult), 4 (gross bleeding).
Histologic analysis
Colon tissues were fixed in 4% neutral-buffered formalin. Paraffin-embedded colon sections (5 μm) were stained with hematoxylin and eosin according to manufacturer's protocols (Wuhan Servicebio Technology, Wuhan, China). Histological score was determined by combining scores for inflammatory cell infiltration (0 to 3) and tissue damage (0 to 3). The presence of inflammatory cells in the intestinal LP were evaluated as follows: 0, occasional inflammatory cells in the LP; 1, increased numbers of inflammatory cells in the LP; 2, confluence of inflammatory cells extending into the submucosa; 3, transmural extension of the infiltrate. Tissue damage was scored as follows: 0, no mucosal damage; 1, lymphoepithelial lesions; 2, surface mucosal erosion or focal ulceration; 3, extensive mucosal damage and extension into deeper structures of the bowel wall.
LPS-induced inflammation
Mice were injected intraperitoneally with 200 μL LPS (20 mg/kg, Escherichia coli 055: B5, Sigma, Milpitas, CA, USA) in sterile PBS. To determine survival rate, mice were constantly monitored every 6 h for a total of 60 h. In a separate experiment, mice were sacrificed 16 h after LPS (10 mg/kg) injection. Serum samples were collected for enzyme-linked immunosorbent assay (ELISA) of IL-1β and IL-6.
Cell culture, stimulation, and transfection
Mouse primary BMDMs were generated as previously described [65]. Briefly, bone marrow was extracted from femurs and tibias. Then, after lysis of erythrocytes, plated in complete medium consisting of Dulbecco's modified Eagle's medium (DMEM; Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum (FBS; Gibco, Carlsbad, CA, USA) and 10 ng/mL macrophage colony-stimulating factor-1 (M-CSF; R&D System, Minneapolis, MN, USA). Adherent macrophages were obtained after 7 days of culture. No differences in differentiation were observed between WT and AhR-/- BMDMs. RAW264.7 cells were purchased from the Cell Bank, Shanghai Institute for Biological Science, Chinese Academy of Science, and cultured in DMEM medium supplemented with 10% FBS. All cells were maintained in humidified cell incubators in 5% CO2 at 37 °C.
Transient transfection of siRNAs (GenePharma, Shanghai, China) was performed using Lipofectamine RNAiMAX reagent (Invitrogen, Carlsbad, CA, USA) and transfection of plasmids with Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's instructions. siRNA sequences are listed in Supplementary Table S2.
To activate the canonical NLRP3 inflammasome, BMDMs were primed with 200 ng/mL LPS (Sigma, Milpitas, CA, USA) for 4 h, followed by 10 μM Nig (Merck, Darmstadt, Germany) or 5 mM ATP (Sigma, Milpitas, CA, USA) for 30-60 min. For pharmacological treatment, VX-765 (50 μM; Merck, Darmstadt, Germany) was added 30 min prior to LPS treatment. Spermine (50 μM; Sigma, Milpitas, CA, USA) was added together with LPS for 4 h prior to Nig stimulation.
Microscopy imaging of cell death
To examine cell death morphology, cells were treated as indicated, then propidium iodide (PI; BD, Franklin Lake, NJ, USA) added to cell cultures at a final concentration of 1 μg/mL to monitor cell membrane integrity. Cells were washed with PBS and static field images of pyroptotic cells captured using a microscope (Leica, Weztlar, Germany).
Cytotoxicity assays
BMDMs were seeded at 3 × 104 cells per well in 96-well plates. After stimulation, cell supernatants were collected and centrifuged at 500 × g for 5 min to remove cellular debris. Cell death was determined by LDH release assay using a CytoTox 96 Non-Radioactive Cytotoxicity Assay kit (Promega, Madison, WI, USA), according to the manufacturer's instructions. Absorbance was read at 492 nm using a plate reader. Data were normalized relative to positive lysis wells (set as 100% lysis) treated with 0.1% Triton X-100.
Enzyme-linked immunosorbent assay (ELISA)
Mouse IL-1β, IL-6, and TNFα protein levels were measured using DuoSet ELISA kits (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's instructions. Absorbance was read at 450 nm using a plate reader.
Plasmid construction
pEGFP-C3-AhR was generated by inserting AhR cDNA fragments, generated from mouse cDNA by PCR amplification, into the HindIII and SacII restriction sites in the multiple cloning site of pEGFP-C3. A 2.0-kb fragment upstream of the Odc1 transcription start site was generated by PCR using mouse genomic DNA as template and cloned into the pGL3-Basic Vector using a ClonExpression® II One Step Cloning Kit (Vazyme, Nanjing, China), according to manufacturer's instructions. pcDNA3.1-flag-ODC1 was generated by cloning Odc1 cDNA fragments into the pcDNA3.1-flag vector using a ClonExpression® II One Step Cloning Kit (Vazyme, Nanjing, China), according to the manufacturer's instructions. Sense and anti-sense primers used are listed in Supplementary Table S3.
Luciferase reporter assay
Luciferase activity was measured using the Dual-luciferase Reporter Assay system (Promega, Madison, WI, USA), according to the manufacturer's instructions. Briefly, HEK293T cells (2 × 104 cells/well) were plated in 24-well plates, and co-transfected with a mixture of the luciferase reporter plasmid, pRT-TK-Renilla, and the indicated expression plasmids using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Post-transfection (48 h), luciferase activities were measured using a Dual luciferase Reporter Assay System. Data were normalized to transfection efficiency by dividing firefly luciferase activity by Renilla luciferase activity.
Visualization of ASC speck formation
BMDMs were seeded onto glass coverslips in 6-well plates. Cells were incubated with LPS (200 ng/mL) for 4 h followed by Nig (10 μM) for 45 min, then washed with PBS, fixed in 4% paraformaldehyde, and permeabilized in 0.3% Triton X-100. After three washes, cells were blocked with 5% BSA in PBS. Anti-ASC antibody (Adipogen, San Diego, CA, USA) was added at 5 μg/mL and incubated at 4 ℃ overnight. After washing, cells were incubated with alexa fluor 647-conjugated secondary antibody (1:500; Beyotime, Shanghai, China) for 1 h at room temperature. Finally, cells were stained with DAPI (Beyotime, Shanghai, China) and sealed for visual analyses. Images were captured by using a laser confocal microscope (Leica, Weztlar, Germany).
Immunofluorescence staining and confocal analysis
Mouse Colon tissues were fixed in 4% neutral-buffered formalin. Paraffin-embedded colon sections (5 μm) were deparaffinized, rehydrated, and fixed. After antigen retrieval, tissues were blocked with 3% BSA, then incubated with anti-mouse F4/80 (Servicebio, Wuhan, China) and anti-mouse IL-1β (Servicebio, Wuhan, China) at 4 °C overnight. After washing, tissue specimens were incubated with alexa fluor 488- and alexa fluor 647-conjugated secondary antibodies (Servicebio, Wuhan, China) for 1 h at room temperature. Nuclei were stained with DAPI. Images were captured using a laser confocal microscope (Leica, Weztlar, Germany).
Quantitative reverse transcription PCR (RT-qPCR)
Total RNA was extracted from cells using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA). A PrimeScript II 1st Strand cDNA Synthesis Kit (Takara, Japan) was used to synthesize first-strand cDNA. RT-qPCR was performed using SYBR® Premix Ex Taq™ II (Takara, Japan) on a Roche 480 Real Time PCR System. mRNA levels of target genes were normalized to those of β-actin. Differences in expression levels (fold-change) were calculated using the 2-ΔΔCT method. PCR primers used are listed in Supplementary Table S4.
Protein preparation and immunoblotting
Cell and tissue protein lysates were extracted using radio immunoprecipitation assay lysis buffer (Thermo Fisher Scientific, Waltham, MA, USA) containing protease and phosphatase inhibitors (Thermo Fisher Scientific, Waltham, MA, USA). Proteins in cell culture supernatants were precipitated using methanol-chloroform, as described previously [66]. Protein concentrations were determined using BCA Reagent (Takara, Japan). Protein aliquots (30 μg) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis, transferred to PVDF membranes, and probed with antibodies directed against β-tubulin (Abcam, Cambridge, UK), GSDMD (L60) (Cell Signaling Technology, Boston, MA, USA), Caspase-1 p20 (Casper-1) (Adipogen, San Diego, CA, USA), IL-1β (Cell Signaling Technology, Boston, MA, USA), NLRP3 (Cryo-2) (Adipogen, San Diego, CA, USA), ASC (AL177) (Adipogen, San Diego, CA, USA), AhR (Enzo Lifesciences, Farmington, NYC, USA), ODC1 (Proteintech, Wuhan, China), or GAPDH (Proteintech, Wuhan, China). Bands were visualized using chemiluminescent HRP substrate (Thermo Fisher Scientific, Waltham, MA, USA) and a Molecular Imager® ChemiDocTM XRS+ Imaging System (Bio-Rad, Hercules, CA, USA).
Chromatin immunoprecipitation (ChIP)
ChIP assays were performed using a Simple ChIP® Plus Enzymatic Chromatin IP Kit (Cell Signaling Technology, Boston, MA, USA), according to the manufacturer's instructions. Briefly, cells were fixed with 1.5% formaldehyde for 20 min and quenched with glycine for 5 min. Chromatin was fragmented by micrococcal nuclease digestion and sonication and immunoprecipitated with an anti-AhR (Enzo Lifesciences, Farmington, NYC, USA) or a control IgG antibody, overnight at 4 °C. Antibody-chromatin complexes were then pulled down using ChIP-Grade Protein A/G Magnetic Beads and eluted with Elution buffer. After incubation at 65 °C for crosslink reversal, samples were digested with RNase A and proteinase K. Immunoprecipitated DNA was collected using DNA clean-up columns. DNA was quantified by qPCR using SYBR® Premix Ex Taq™ II (Takara, Japan), according to the manufacturer's instructions. The PCR primers used are listed in Supplementary Table S5.
RNA sequencing and data analysis
Total RNA was extracted from WT and AhR-/- BMDMs for RNA-seq analysis. Qualified RNA samples were used for library construction, and then sequenced (PE150) on an Illumina Novaseq™ 6000 (LC-Bio Technology CO., Ltd., Hangzhou, China), to generate 2 × 150 bp long reads, following the vendor's recommended protocol. After removing adaptors and low-quality bases from raw data, high-quality sequences (clean data) were mapped to the Mus musculus GRCm38 reference genome. Differentially expressed genes were selected as those with false discovery rate < 0.5, fold-change ≥ 1.5 or ≤ 0.5, and p value < 0.05, using the R package, edgeR. RNA-seq data have been deposited in the SRA database under accession number PRJNA952314.
ScRNA-seq library preparation and analysis
AhR-null and WT mice were administered 2% DSS in drinking water for 7 days, followed by normal drinking water to induced acute colitis. On day 8, colon tissues from three mice of the same group were randomly mixed as one sample, then, colons were digested and single cell suspensions obtained using 70-μm filters (BD, Franklin Lake, NJ, USA). After incubation with red blood cell lysis buffer, CD45+ immune cells were isolated by positive selection with anti-mouse CD45 magnetic beads (Miltenyi, Germany), according to the manufacturer's instructions. Finally, cells were resuspended in complete RPMI-1640 medium, counted for viability, and then immediately subjected to library preparation. ScRNA-Seq libraries were prepared using 10x Single Cell Genomics technology (10x Genomics, San Diego, CA, USA), according to the manufacturer's instructions. ScRNA-seq fastq was applied, with default parameters, to filter adaptor sequences and remove low-quality reads. Graph-based clustering was performed for cell clustering according to their gene expression profiles. Cells were visualized using a 2-dimensional UMAP algorithm with the RunUMAP function in Seurat. The FindAllMarkers function in Seurat was applied to identify marker genes for each cluster. Sequencing and bioinformatics analysis were conducted by OE Biotech Co., Ltd. (Shanghai, China). ScRNA-seq data have been deposited in the GEO database under accession number GSE242968.
16S rDNA sequencing
Microbial DNA was extracted from fecal samples from AhR△mye mice and AhRfl/fl littermates using a DNeasy PowerSoil kit (Qiagen, Hilden, Germany), following the manufacturer's instructions. DNA concentration and integrity were verified by NanoDrop 2000 analysis and agarose gel electrophoresis, respectively. Genomic DNA was used as template for PCR amplification with barcoded primers and Tks Gflex DNA Polymerase (Takara, Japan). After amplification and purification, PCR products were quantified using a Qubit dsDNA assay kit (Life Technologies, Carlsbad, CA, USA). Sequencing was performed on an Illumina NovaSeq 6000 (Illumina lnc., San Diego, CA; OE Biotech Company, Shanghai, China). 16S rDNA-seq data have been deposited in the SRA database under accession number PRJNA1002140.
Intracellular polyamine analysis
For harvest, BMDMs (1 × 107) were rinsed with 500 μL pre-cooled methanol:water (2:1, v/v) and subjected to three freeze-thaw cycles before sonication in an ice bath for 15 mins. Following centrifugation, supernatants were evaporated to dryness and re-dissolved in 50 μL of acetonitrile-water (1:4, v/v). Next, aliquots (10 μL) were vortex-mixed with 10 μL of NEM solution (20 mM) in phosphate buffer (0.1 M, pH 7.0) containing ascorbic acid (10 mM) and EDTA (10 mM) for 1 min. tBBT solution (10 μL, 0.23 M) was added, followed by 87 μL borate buffer (0.2 M, pH 8.8) containing TCEP (20 mM) and ascorbic acid (5 mM). After vortex-mixing, 33 μL 5-AIQC solution was added and samples incubated at 55 °C for 10 min. Mixtures were cooled and 2 μL formic acid added, followed by centrifugation at 13,000 ×g for 10 min at 4 °C, and supernatants filtered through 0.22 µm filters before UPLC-MS/MS analysis. Metabolite analysis was carried out using an Agilent 1290 UPLC coupled to an Agilent 6470 triple quadrupole mass spectrometer equipped with an electrospray ionization source (Agilent Technologies, Palo Alto, CA, USA). Chromatographic separation of polyamines was performed using a UPLC column (Agilent ZORBAX RRHD Eclipse XDB C18 column, 2.1 × 100 mm, 1.8 μm particles) with a solvent gradient of buffer A (water) and buffer B (methanol containing 0.1% (v/v) formic acid); flow rate, 0.5 mL/min. An optimized gradient elution scheme was employed as follows: 1% B (0-2 min), 1%-3.8% B (2-4 min), 3.8%-14% B (4-7.3 min), 14%-22% B (7.3-10.7 min), 22%-24% B (10.7-14.7 min), 24%-30% B (14.7-16 min), 30%-60% B (16-16.3 min), 60%-70% B (16.3-17.3 min), 70%-95% B (17.3-17.31 min), and 95% B (17.31-20 min). Electrospray ionization was performed in positive ion mode. Multiple reaction monitoring was applied for quantification of screening fragment ions. Peak determination and peak area integration were performed using MassHunter Workstation software (Agilent, Version B.08.00). Standard curves were constructed by least-squares linear regression analysis, using the peak area ratio of derivatized individual standards against the nominal concentration of the calibrator. Quantification was performed identically for all samples. Detection of polyamines was conducted by Shanghai Metabolome Institute (SMI)-Wuhan.
Intracellular potassium (K+) analysis
BMDMs were seeded in 48-well plates at 5 × 104 cells per well. After stimulation, cells were probed with IPG-4 AM (Ion Biosciences, San Marcos, TX, USA), a yellow-green fluorescent, intracellular K+ indicator, in a cell culture incubator at 37 °C for 90 min, according to the manufacturer's instructions. After staining, cells were washed and analyzed by flow cytometry (BD, Franklin Lake, NJ, USA). Mean fluorescence intensity was calculated using FlowJo software (BD, Version 10.8.1).
Statistical Analyses
All statistical analyses were performed using GraphPad Prism 9.0 software. Before comparisons between groups, data were tested for Gaussian distribution and homogeneity variance. For data in Gaussian distribution and with homogeneity variance, parametric test was used to analyze, such as independent t test, one or two-way ANOVA, etc. For data in non-Gaussian distribution, non-parametric test was used to analyze, such as the Mann-Whitney test, Kruskal-Wallis test, Spearman correlation, etc. For data in Gaussian distribution and without homogeneity variance, Welch's correction was used. P < 0.05 was considered statistically significant and data are presented as mean ± SEM.
Ethics Committee Approval
All animal experiments complied with all relevant ethical regulations for animal testing and research and were in accordance with protocols approved by the Committee on Animal Research and Ethics of Fudan University.
Data and materials availability
The raw data from the scRNA-seq, RNA-seq and 16s rDNA-seq have been deposited in the GEO or SRA database under accession numbers GSE242968, PRJNA952314, and PRJNA1002140, respectively. All data needed to evaluate the conclusions in the paper are present in the paper and the Supplementary Materials. Additional data are available from authors upon request.
Abbreviations
AhR: aryl hydrocarbon receptor; ARNT: aryl hydrocarbon receptor nuclear translocator; ASC: apoptosis associated speck like protein containing a CARD; ATP: adenosine Triphosphate; BMDMs: bone marrow derived macrophages; CD: Crohn's disease; ChIP: chromatin immunoprecipitation; DAI: disease activity index; DSS: dextran sulfate sodium; DFMO: difluoromethylornithine; ELISA: enzyme-linked immunosorbent assay; FICZ: 6-Formylindolo[3,2-b] carbazole; GSDMD: gasdermin D; IBD: inflammatory bowel disease; IL-1-RN: IL-1 antagonist receptor; IL-18RAP: IL18 receptor accessory protein; IL-18R1: IL18 receptor 1; IP: indeno[1,2,3-cd]pyrene; I3C: indole-3-carbinol; ILC: innate lymphoid cells; IEL: intraepithelial lymphocytes; LPS: lipopolysaccharide; LC-MS: liquid chromatograph mass spectrometer; LP: intestinal lamina propria; LDH: lactate dehydrogenase; M-CSF: macrophage colony-stimulating factor-1; Nig: nigericin; NLRP3: NOD-like receptor protein 3; Odc1: ornithine decarboxylase 1; PI: propidium iodide; PRRs: pattern recognition receptors; ROS: reactive oxygen species; scRNA-seq: single-cell RNA sequencing; Spd: spermidine; Spm: spermine; TNBS: 2,6,4-trinitrobenzene sulfonic acid; TCDD: 2,3,7,8-tetrachlorodibenzo-p-dioxin; UC: ulcerative colitis; UMAP: uniform manifold approximation and projection; WT: wild type; XREs: xenobiotic response elements.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by grants from the National Key R&D Program of China (2021YFC2701800, 2021YFC2701802 to YZ), National Natural Science Foundation of China (82241038, 81974248 to YZ, 81900751 to XH, 82100033 to SH), Program for Outstanding Medical Academic Leader (2019LJ19 to YZ), Shanghai Committee of Science and Technology (22QA1401500, 20ZR1408300 to XH, 21140902400 to YZ, 23ZR1407600, 21ZR1410000 to FJ). The International Joint Laboratory Program of National Children's Medical Center (EK1125180109 to YZ).
Author contributions
Y.G., K.L., W.X., and Y.Z. conceived and designed the project. Y.G., K.L., W.X., J.F. and X.X. performed experiments and/or analyzed data. Y.G., K.L., W.X. performed single cell RNA sequencing analysis and RNA-seq analysis. Q.L., Z.T., L.Y., H.Y., and H.G. provided technical assistance. S.H. and X.H. contributed critical reagents and protocols. Y.G., K.L., and Y.Z. wrote the manuscript. Y.Z. planned, designed, supervised, and coordinated the overall research efforts.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sládeková L, Mani S, Dvořák Z. Ligands and agonists of the aryl hydrocarbon receptor AhR: Facts and myths. Biochemical pharmacology. 2023;213:115626
2. Ligumsky M, Simon PL, Karmeli F, Rachmilewitz D. Role of interleukin 1 in inflammatory bowel disease-enhanced production during active disease. Gut. 1990;31:686-9
3. Neavin DR, Liu D, Ray B, Weinshilboum RM. The Role of the Aryl Hydrocarbon Receptor (AHR) in Immune and Inflammatory Diseases. Int J Mol Sci. 2018;19:3851
4. Shinde R, McGaha TL. The Aryl Hydrocarbon Receptor: Connecting Immunity to the Microenvironment. Trends in immunology. 2018;39:1005-20
5. Gutiérrez-Vázquez C, Quintana FJ. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity. 2018;48:19-33
6. Kaplan GG. The global burden of IBD: from 2015 to 2025. Nat Rev Gastroenterol Hepatol. 2015;12:720-7
7. Metidji A, Omenetti S, Crotta S, Li Y, Nye E, Ross E. et al. The Environmental Sensor AHR Protects from Inflammatory Damage by Maintaining Intestinal Stem Cell Homeostasis and Barrier Integrity. Immunity. 2018;49:353-62.e5
8. Yu M, Wang Q, Ma Y, Li L, Yu K, Zhang Z. et al. Aryl Hydrocarbon Receptor Activation Modulates Intestinal Epithelial Barrier Function by Maintaining Tight Junction Integrity. Int J Biol Sci. 2018;14:69-77
9. Li S, Bostick JW, Ye J, Qiu J, Zhang B, Urban JF Jr. et al. Aryl Hydrocarbon Receptor Signaling Cell Intrinsically Inhibits Intestinal Group 2 Innate Lymphoid Cell Function. Immunity. 2018;49:915-28.e5
10. Yang W, Yu T, Huang X, Bilotta AJ, Xu L, Lu Y. et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nature communications. 2020;11:4457
11. Panda SK, Peng V, Sudan R, Ulezko Antonova A, Di Luccia B, Ohara TE. et al. Repression of the aryl-hydrocarbon receptor prevents oxidative stress and ferroptosis of intestinal intraepithelial lymphocytes. Immunity. 2023;56:797-812.e4
12. Apetoh L, Quintana FJ, Pot C, Joller N, Xiao S, Kumar D. et al. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nature immunology. 2010;11:854-61
13. Liu C, Li Y, Chen Y, Huang S, Wang X, Luo S. et al. Baicalein Restores the Balance of Th17/Treg Cells via Aryl Hydrocarbon Receptor to Attenuate Colitis. Mediators Inflamm. 2020;2020:5918587
14. Singh NP, Singh UP, Singh B, Price RL, Nagarkatti M, Nagarkatti PS. Activation of aryl hydrocarbon receptor (AhR) leads to reciprocal epigenetic regulation of FoxP3 and IL-17 expression and amelioration of experimental colitis. PloS one. 2011;6:e23522
15. Hegarty LM, Jones GR, Bain CC. Macrophages in intestinal homeostasis and inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2023;20:538-553
16. Bauer C, Duewell P, Mayer C, Lehr HA, Fitzgerald KA, Dauer M. et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut. 2010;59:1192-9
17. Moreira Lopes TC, Mosser DM, Gonçalves R. Macrophage polarization in intestinal inflammation and gut homeostasis. Inflamm Res. 2020;69:1163-72
18. Kimura A, Naka T, Nakahama T, Chinen I, Masuda K, Nohara K. et al. Aryl hydrocarbon receptor in combination with Stat1 regulates LPS-induced inflammatory responses. The Journal of experimental medicine. 2009;206:2027-35
19. Sekine H, Mimura J, Oshima M, Okawa H, Kanno J, Igarashi K. et al. Hypersensitivity of aryl hydrocarbon receptor-deficient mice to lipopolysaccharide-induced septic shock. Mol Cell Biol. 2009;29:6391-400
20. Zhu J, Luo L, Tian L, Yin S, Ma X, Cheng S. et al. Aryl Hydrocarbon Receptor Promotes IL-10 Expression in Inflammatory Macrophages Through Src-STAT3 Signaling Pathway. Front Immunol. 2018;9:2033
21. Shi J, Gao W, Shao F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends in biochemical sciences. 2017;42:245-54
22. Shao F, Fitzgerald KA. Molecular mechanisms and functions of pyroptosis. J Mol Biol. 2022;434:167461
23. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H. et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660-5
24. Chen X, Liu G, Yuan Y, Wu G, Wang S, Yuan L. NEK7 interacts with NLRP3 to modulate the pyroptosis in inflammatory bowel disease via NF-κB signaling. Cell death & disease. 2019;10:906
25. Yuan YY, Xie KX, Wang SL, Yuan LW. Inflammatory caspase-related pyroptosis: mechanism, regulation and therapeutic potential for inflammatory bowel disease. Gastroenterol Rep (Oxf). 2018;6:167-76
26. Hedl M, Zheng S, Abraham C. The IL18RAP region disease polymorphism decreases IL-18RAP/IL-18R1/IL-1R1 expression and signaling through innate receptor-initiated pathways. Journal of immunology (Baltimore, Md: 1950). 2014;192:5924-32
27. Yamamoto-Furusho JK, Santiago-Hernández JJ, Pérez-Hernández N, Ramírez-Fuentes S, Fragoso JM, Vargas-Alarcón G. Interleukin 1 β (IL-1B) and IL-1 antagonist receptor (IL-1RN) gene polymorphisms are associated with the genetic susceptibility and steroid dependence in patients with ulcerative colitis. J Clin Gastroenterol. 2011;45:531-5
28. Huang B, Chen Z, Geng L, Wang J, Liang H, Cao Y. et al. Mucosal Profiling of Pediatric-Onset Colitis and IBD Reveals Common Pathogenics and Therapeutic Pathways. Cell. 2019;179:1160-76.e24
29. Mitsialis V, Wall S, Liu P, Ordovas-Montanes J, Parmet T, Vukovic M. et al. Single-Cell Analyses of Colon and Blood Reveal Distinct Immune Cell Signatures of Ulcerative Colitis and Crohn's Disease. Gastroenterology. 2020;159:591-608.e10
30. Aschenbrenner D, Quaranta M, Banerjee S, Ilott N, Jansen J, Steere B. et al. Deconvolution of monocyte responses in inflammatory bowel disease reveals an IL-1 cytokine network that regulates IL-23 in genetic and acquired IL-10 resistance. Gut. 2021;70:1023-36
31. Friedrich M, Pohin M, Jackson MA, Korsunsky I, Bullers SJ, Rue-Albrecht K. et al. IL-1-driven stromal-neutrophil interactions define a subset of patients with inflammatory bowel disease that does not respond to therapies. Nature medicine. 2021;27:1970-81
32. Pizarro TT, Michie MH, Bentz M, Woraratanadharm J, Smith MF Jr, Foley E. et al. IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn's disease: expression and localization in intestinal mucosal cells. Journal of immunology (Baltimore, Md: 1950). 1999;162:6829-35
33. Han X, Huang S, Xue P, Fu J, Liu L, Zhang C. et al. LncRNA PTPRE-AS1 modulates M2 macrophage activation and inflammatory diseases by epigenetic promotion of PTPRE. Sci Adv. 2019;5:eaax9230
34. Monteleone I, Rizzo A, Sarra M, Sica G, Sileri P, Biancone L. et al. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology. 2011;141:237-48 48.e1
35. Hardbower DM, Asim M, Luis PB, Singh K, Barry DP, Yang C. et al. Ornithine decarboxylase regulates M1 macrophage activation and mucosal inflammation via histone modifications. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:E751-e60
36. Bock KW. Aryl hydrocarbon receptor (AHR)-mediated inflammation and resolution: Non-genomic and genomic signaling. Biochemical pharmacology. 2020;182:114220
37. Casero RA Jr, Murray Stewart T, Pegg AE. Polyamine metabolism and cancer: treatments, challenges and opportunities. Nat Rev Cancer. 2018;18:681-95
38. Xu J, Núñez G. The NLRP3 inflammasome: activation and regulation. Trends in biochemical sciences. 2023;48:331-44
39. Lee SJ, Nichols CG. Seeing spermine blocking of K+ ion movement through inward rectifier Kir2.2 channels. The Journal of general physiology. 2023;155:e202213144
40. Ficker E, Taglialatela M, Wible BA, Henley CM, Brown AM. Spermine and spermidine as gating molecules for inward rectifier K+ channels. Science (New York, NY). 1994;266:1068-72
41. Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M. et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nature immunology. 2011;13:144-51
42. Stockinger B, Shah K, Wincent E. AHR in the intestinal microenvironment: safeguarding barrier function. Nat Rev Gastroenterol Hepatol. 2021;18:559-70
43. Sun L, Fu J, Lin SH, Sun JL, Xia L, Lin CH. et al. Particulate matter of 2.5 μm or less in diameter disturbs the balance of T(H)17/regulatory T cells by targeting glutamate oxaloacetate transaminase 1 and hypoxia-inducible factor 1α in an asthma model. The Journal of allergy and clinical immunology. 2020;145:402-14
44. Liu KY, Gao Y, Xiao W, Fu J, Huang S, Han X. et al. Multidimensional Analysis of Lung Lymph Nodes in a Mouse Model of Allergic Lung Inflammation following PM2.5 and Indeno[1,2,3-cd]pyrene Exposure. Environmental health perspectives. 2023;131:37014
45. Houser CL, Lawrence BP. The Aryl Hydrocarbon Receptor Modulates T Follicular Helper Cell Responses to Influenza Virus Infection in Mice. Journal of immunology (Baltimore, Md: 1950). 2022;208:2319-30
46. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E. et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453:65-71
47. He J, Song Y, Li G, Xiao P, Liu Y, Xue Y. et al. Fbxw7 increases CCL2/7 in CX3CR1hi macrophages to promote intestinal inflammation. The Journal of clinical investigation. 2019;129:3877-93
48. Huai W, Zhao R, Song H, Zhao J, Zhang L, Zhang L. et al. Aryl hydrocarbon receptor negatively regulates NLRP3 inflammasome activity by inhibiting NLRP3 transcription. Nature communications. 2014;5:4738
49. Bauer C, Duewell P, Lehr HA, Endres S, Schnurr M. Protective and aggravating effects of Nlrp3 inflammasome activation in IBD models: influence of genetic and environmental factors. Dig Dis. 2012;30(Suppl 1):82-90
50. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010;32:379-91
51. Hirota SA, Ng J, Lueng A, Khajah M, Parhar K, Li Y. et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm Bowel Dis. 2011;17:1359-72
52. Bulek K, Zhao J, Liao Y, Rana N, Corridoni D, Antanaviciute A. et al. Epithelial-derived gasdermin D mediates nonlytic IL-1β release during experimental colitis. The Journal of clinical investigation. 2020;130:4218-34
53. Rana N, Privitera G, Kondolf HC, Bulek K, Lechuga S, De Salvo C. et al. GSDMB is increased in IBD and regulates epithelial restitution/repair independent of pyroptosis. Cell. 2022;185:283-98.e17
54. Bianchi-Smiraglia A, Bagati A, Fink EE, Affronti HC, Lipchick BC, Moparthy S. et al. Inhibition of the aryl hydrocarbon receptor/polyamine biosynthesis axis suppresses multiple myeloma. The Journal of clinical investigation. 2018;128:4682-96
55. Jiang F, Gao Y, Dong C, Xiong S. ODC1 inhibits the inflammatory response and ROS-induced apoptosis in macrophages. Biochemical and biophysical research communications. 2018;504:734-41
56. Shantz LM, Pegg AE. Translational regulation of ornithine decarboxylase and other enzymes of the polyamine pathway. Int J Biochem Cell Biol. 1999;31:107-22
57. Chung DH, Evers BM, Townsend CM Jr, Herndon DN, Ko TC, Uchida T. et al. Cytokine regulation of gut ornithine decarboxylase gene expression and enzyme activity. Surgery. 1992;112:364-9
58. Monelli E, Villacampa P, Zabala-Letona A, Martinez-Romero A, Llena J, Beiroa D. et al. Angiocrine polyamine production regulates adiposity. Nat Metab. 2022;4:327-43
59. Nishiguchi A, Taguchi T. Inflammation-targeting polyamine nanomedicines for the treatment of ulcerative colitis. J Mater Chem B. 2023;11:4005-13
60. Nakamura A, Kurihara S, Takahashi D, Ohashi W, Nakamura Y, Kimura S. et al. Symbiotic polyamine metabolism regulates epithelial proliferation and macrophage differentiation in the colon. Nature communications. 2021;12:2105
61. Peng V, Cao S, Trsan T, Bando JK, Avila-Pacheco J, Cleveland JL. et al. Ornithine decarboxylase supports ILC3 responses in infectious and autoimmune colitis through positive regulation of IL-22 transcription. Proceedings of the National Academy of Sciences of the United States of America. 2022;119:e2214900119
62. Gobert AP, Latour YL, Asim M, Barry DP, Allaman MM, Finley JL. et al. Protective Role of Spermidine in Colitis and Colon Carcinogenesis. Gastroenterology. 2022;162:813-27.e8
63. Ma L, Ni L, Yang T, Mao P, Huang X, Luo Y. et al. Preventive and Therapeutic Spermidine Treatment Attenuates Acute Colitis in Mice. J Agric Food Chem. 2021;69:1864-76
64. Gobert AP, Smith TM, Latour YL, Asim M, Barry DP, Allaman MM. et al. Hypusination Maintains Intestinal Homeostasis and Prevents Colitis and Carcinogenesis by Enhancing Aldehyde Detoxification. Gastroenterology. 2023;165:656-669
65. Zhou Y, Do DC, Ishmael FT, Squadrito ML, Tang HM, Tang HL. et al. Mannose receptor modulates macrophage polarization and allergic inflammation through miR-511-3p. The Journal of allergy and clinical immunology. 2018;141:350-64.e8
66. Wessel D, Flügge UI. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem. 1984;138:141-3
Author contact
![]() Corresponding author: Yufeng Zhou, MD, PhD, 399 Wanyuan Rd, Minhang, Shanghai 201102, China. Email: yfzhou1edu.cn. Tel.: 86-21-64932907.
Corresponding author: Yufeng Zhou, MD, PhD, 399 Wanyuan Rd, Minhang, Shanghai 201102, China. Email: yfzhou1edu.cn. Tel.: 86-21-64932907.