Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. The immune response
3. Ultrasound and inflammation -...
4. Ultrasound and inflammation -...
5. Summary
Abbreviations
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2024; 14(8):3150-3177. doi:10.7150/thno.96270 This issue Cite
Review
Ultrasound and neuroinflammation: immune modulation via the heat shock response
Graham M. Seasons1,2, ![]() , Carly Pellow1,3, Hedwich F. Kuipers1,2,4, G. Bruce Pike1,2,3
, Carly Pellow1,3, Hedwich F. Kuipers1,2,4, G. Bruce Pike1,2,3
1. Hotchkiss Brain Institute, University of Calgary, Alberta, T2N 4N1, Canada.
2. Department of Clinical Neurosciences, Cumming School of Medicine, University of Calgary, Alberta, T2N 1N4, Canada.
3. Department of Radiology, Cumming School of Medicine, University of Calgary, Alberta, T2N 1N4, Canada.
4. Department of Cell Biology & Anatomy, Hotchkiss Brain Institute and Snyder Institute for Chronic Diseases, University of Calgary, Alberta, T2N 1N4, Canada.
Received 2024-3-14; Accepted 2024-5-9; Published 2024-5-19
Abstract

Current pharmacological therapeutic approaches targeting chronic inflammation exhibit transient efficacy, often with adverse effects, limiting their widespread use - especially in the context of neuroinflammation. Effective interventions require the consideration of homeostatic function, pathway dysregulation, and pleiotropic effects when evaluating therapeutic targets. Signalling molecules have multiple functions dependent on the immune context, and this complexity results in therapeutics targeting a single signalling molecule often failing in clinical translation. Additionally, the administration of non-physiologic levels of neurotrophic or anti-inflammatory factors can alter endogenous signalling, resulting in unanticipated effects. Exacerbating these challenges, the central nervous system (CNS) is isolated by the blood brain barrier (BBB), restricting the infiltration of many pharmaceutical compounds into the brain tissue. Consequently, there has been marked interest in therapeutic techniques capable of modulating the immune response in a pleiotropic manner; ultrasound remains on this frontier. While ultrasound has been used therapeutically in peripheral tissues - accelerating healing in wounds, bone fractures, and reducing inflammation - it is only recently that it has been applied to the CNS. The transcranial application of low intensity pulsed ultrasound (LIPUS) has successfully mitigated neuroinflammation in vivo, in models of neurodegenerative disease across a broad spectrum of ultrasound parameters. To date, the underlying biological effects and signalling pathways modulated by ultrasound are poorly understood, with a diverse array of reported molecules implicated. The distributed nature of the beneficial response to LIPUS implies the involvement of an, as yet, undetermined upstream signalling pathway, homologous to the protective effect of febrile range hyperthermia in chronic inflammation. As such, we review the heat shock response (HSR), a protective signalling pathway activated by thermal and mechanical stress, as the possible upstream regulator of the anti-inflammatory effects of ultrasound.
1. Introduction
Inflammation is a natural process that is part of the immune response to stress, pathogens, and general physiological insults. While acute inflammation can be beneficial [1], it can have a deleterious impact if dysregulated [2]. Across biological systems, inflammation is a defining and contributing factor of chronic disease, making it a common therapeutic target. To date, very few therapeutics have resulted in successful treatment outcomes for chronic inflammation, or are poorly tolerated and subject to patient comorbidities [3]. Additionally, in the case of the central nervous system (CNS), the parenchymal tissue is separated from the vascular system through a complex of endothelial cells connected via tight junctions, maintained by tight junction proteins, called the blood brain barrier (BBB). This barrier prevents pathogens, immune cells, and many small molecules from infiltrating the brain. However, while this is protective in homeostatic conditions, it also prevents therapeutics from accessing the parenchyma, and targeting diseases and disorders of the neurological system. Consequently, treatments that are capable of reaching the brain are of great interest; this has led to increased interest in ultrasound as a method to temporarily disrupt the BBB for the targeted delivery of therapeutics [4]. In addition to this, ultrasound is being considered as a means of minimally invasive neurosurgery [5], tumour ablation [6], as well as directly leveraging biological effects on the immune system [7]. However, the modulation of the immune system that is induced by ultrasound remains poorly characterized, with no coherent hypothesis linking the upregulation of various disparate anti-inflammatory signalling pathways. A common, highly phylogenetically conserved signalling pathway is the heat shock response (HSR) [8], polysemous in its activation in response to both thermal and mechanical stress, which regulates a broad spectrum of protective signalling pathways essential for cell survival. The HSR is characterized by the upregulation in transcription of HSR-related genes, and changes in HSR-related protein affinity, the most well-known of which are heat shock proteins (HSPs). HSPs are chaperone proteins, responsible for refolding, sequestering, and degrading dysregulated proteins, thereby mitigating toxic conformations and subsequent apoptosis. While one component of a much broader signalling response, HSPs are often treated as a synecdoche for the HSR, used to identify heat shocked cells and demonstrated to have a potent anti-apoptotic effect [9], both in conjunction with, and apart from, the broader signalling response. Traditionally implicated in the protective effect of febrile hyperthermia, recent research has demonstrated a homologous overlap in immune modulation of the HSR beyond with the mechanical effects of ultrasound, possibly constituting a common upstream immune regulator. As a rapidly growing and promising field, it is important to further map and comprehend the cellular signalling pathways driving the beneficial response to ultrasound, as to date, many of the mechanisms remain unclear.
1.1 Magnetic resonance guided focused ultrasound
Transcranial high intensity magnetic resonance guided focused ultrasound (MRgFUS) is a minimally invasive surgical technique [5] that can focus mechanical energy deep in the brain, inducing an increase in temperature beyond a lesional threshold at a target location, while sparing the surrounding tissue. MRgFUS procedures are monitored with MR thermometry, which leverages the relationship between temperature and the proton resonance frequency shift [10] to monitor shifts in temperature through changes in phase maps, facilitating real-time thermal imaging. While current applications have focused on essential tremor (ET) [5] and tumour ablation [6] to induce cell death, ultrasound-induced bioeffects extend to the immune system. High intensity focused ultrasound (HIFU) has been suggested to increase tumour cell antigen presentation for targeting by the adaptive immune system, as well as activating endogenous danger signalling pathways [11]. Studies on the immune effect of HIFU have largely remained concentrated on cancer and stimulating a T cell response; however, it has also been proposed that HIFU can modulate radiation-induced inflammation and reduce concomitant side effects [12] - a translational result which would be of interest in the treatment of a broad spectrum of inflammatory diseases. Despite this, HIFU remains limited to ablative therapy, with the primary purpose of inducing cell death, making it unsuitable for use in inflamed, but otherwise healthy tissue. The practical translation of ablative MRgFUS procedures to the modulation of inflammation requires the selection of parameters that induce hyperthermia below the physiological limits of tissue damage, analogous to fever, the body's own protective mechanism of hyperthermia. Consequently, while postulated for HIFU, ultrasound-mediated mild hyperthermia instead constitutes a promising and actionable method of ameliorating inflammation across pathologies.
1.2 MRgFUS mediated resolution of radiation induced edema
Interestingly, a recent case study detailed a persistent stereotactic radiosurgery (SRS)-related edema, which was resolved after undergoing an MRgFUS thalamotomy [13]. In 2015, a 62-year-old man with chronic demyelinating polyneuropathy underwent a SRS thalamotomy to treat tremor, which was transiently successful, regressing to approximately pre-treatment baseline levels of tremor approximately three years following treatment. It is at this time that they were enrolled in a study on MRgFUS thalamotomy. Pre-MRgFUS T2w imaging revealed a large hyperintense region of edema surrounding and extending beyond the SRS target, which was absent from pre-SRS imaging three years prior. SRS is another minimally invasive neurosurgical technique used in the treatment of ET [14], characterized by the focusing of high-dose ionizing radiation to create a targeted brain lesion; as SRS cannot be monitored intra-operatively, lesions can be accompanied by unforeseen radiosurgery related edema, manifesting as a hyperintense zone around the lesion site on T2-weighted (T2w) magnetic resonance (MR) images [13,15,16]. Radiosurgery-related edema has been associated with the development of clinical side effects [15], and although no adverse effects were observed in the case study, the T2w hyperintensity exhibited a similar morphology to that associated with inflammation in prior reports [16,17]. One-day post-MRgFUS the hyperintensity had both an increased volume and signal intensity on T2w images. However, three months post-MRgFUS the hyperintensity was completely resolved, resembling that of a typical MRgFUS thalamotomy patient [13]. Patient medication remained stable leading up to and following the thalamotomy.
While edema cannot be explicitly connected to localized inflammation, radiation is known to induce an inflammatory response in the brain [18], and the resulting T2w hyperintensity has been correlated with long-term macrophage infiltration and inflammation [16,17]. Additionally, corticosteroids have been used to treat SRS-related edema [15], indicating a potential inflammatory component to the corresponding imaging finding. As such, the evidence presented in the case study supports the potential of HIFU as a modulator of radiation-induced inflammation [12]. Regardless, the biological mechanisms behind the finding remain unclear, and bear further investigation in vivo. The following review attempts to cast light on the potential underlying mechanisms behind the described imaging finding, represented in Figure 1, focusing on the ameliorating potential of both the mechanical and thermal effects of ultrasound in chronic inflammation, confluently linked through the HSR.
Depiction of a chronic neuroinflammatory lesion located deep in the brain characterized by cellular disruption, and the proposed ability of ultrasound mediated hyperthermia as a method to access and resolve neuroinflammation leading to restored homeostasis. Highlighted cells (microglia, astrocytes, and endothelial cells) are proposed to be stimulated by ultrasound mediated hyperthermia to induce the resolution of neuroinflammation.
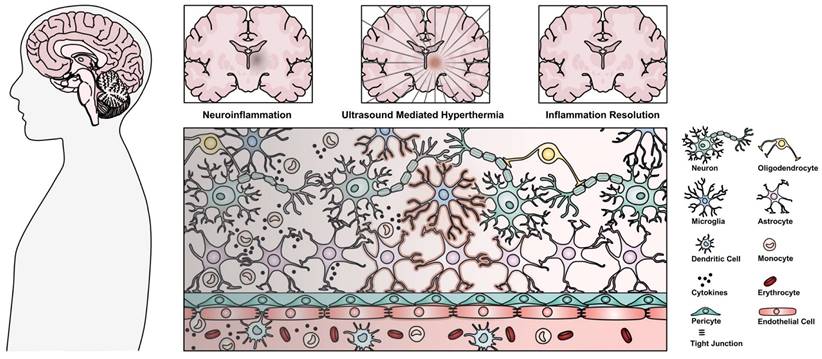
2. The immune response
The immune system is composed of two distinct, but interrelated responses: innate and adaptive, with the innate system present ubiquitously, whereas the adaptive system is bifurcated and present primarily in specialized locations. While the innate immune system non-specifically reacts to and attempts to eliminate any foreign agent, based on molecular patterns, the adaptive immune system is instructed, by the innate immune system, to recognize specific antigens (peptides capable of activating an immune receptor, and initiating a subsequent immune response). For a considerable time, the CNS was considered a site of immune privilege, absent of any native immune cells, and separated from the peripheral immune system due to the impermeability of the BBB [19]. The concept of the CNS as an immune privileged site has since changed, more accurately classified as immune restricted, where the direct stimulation of the adaptive response by CNS resident immune cells is very limited [19]. Peripheral immune cells stem from a common hematopoietic lineage that differentiates into myeloid and lymphoid cell lineages, which compose the innate, and adaptive immune responses respectively. The CNS lacks direct counterparts of the immune cell subtypes found in the periphery. However, (some of) their roles are observed by glial cells, a subclass of neural support cells, which include oligodendrocytes, astrocytes, and microglia. Microglia and astrocytes primarily constitute the immunocompetent cells of the CNS and can display an array of immune functions, although oligodendrocytes are not bystanders [20]. Microglia are considered the resident macrophages of the CNS, stemming from hematopoietic lineage and infiltrating the CNS during development [21], capable of self-renewal, producing and responding to cytokines, phagocytosis, and antigen presentation [22]. At rest they are in an immune surveillant state, whereas they are dynamically capable of changing phenotype, both functional and morphological, in response to stimuli. Classically, two microglial phenotypes or activation states have been characterized: M1 and M2, corresponding to an inflammatory and a reparative state, respectively. However, this is an overly simplistic view [22], as microglia can co-express both pro- and anti-inflammatory markers used for characterization. Moreover, depending on the stimulatory ligand in vitro, the morphological phenotype and behaviour of microglia is altered [23], exhibiting varied phagocytosis, migration, and transcriptomic profiles between colloquially described M1 states, indicating a more complex functional heterogeneity than a simple paradigm of binary activation. Alternately, astrocytes are derived from neuroepithelial stem cells, and in a homeostatic environment are involved in maintaining proper BBB integrity, monitoring the cellular environment, and supporting other CNS cells [24]. Similar to microglia, astrocytes are also involved in cytokine production and detection, as well as phagocytosis, albeit on a reduced scale [19]. Astrocytes are also colloquially accepted to adopt both pro- and anti-inflammatory states in response to stimuli - termed reactive astrogliosis, using A1 and A2 to denote each respectively [25]. As with microglia, the phenotypic activation states of astrocyte are likely more plastic than this proposed dichotomy, corresponding to heterogeneous populations of activation states [26].
Lymphocytes, of which T cells are an important subset, are the dominant actors in executing the adaptive immune response. The adaptive response is targeted to a specific antigen that is presented in the context of the major histocompatibility complex (MHC) by antigen presenting cells (APCs). The innate and adaptive immune systems act in a synergistic fashion, where innate immune cells act as APCs to activate and instruct adaptive immune cells. Exogenous antigens are processed following phagocytosis of the foreign agent by APCs and displayed through MHC II for recognition by cluster of differentiation four positive (CD4+) T cells [27]. Proper activation of T cells requires efficient coordination of cell-to-cell interactions between APCs and T cells. Within the peripheral immune system, the dominant APCs are dendritic cells (DCs), monocytes, and macrophages. After phagocytosing antigens at sites of infiltration of foreign agents, APCs traffic via the lymphatic system to secondary lymphoid tissues, which allow for the localized interaction of APCs and a dense population of lymphocytes, which subsequently facilitates the egress of stimulated, antigen-specific, T cells into circulation, enabling the recognition and destruction of pathogenic cells, and instruction of the local immune response [27]. In order to assure proper recognition of presented non-self antigens, and prevent excessive recognition of self antigens, T cells are selected in a 2-step process during development. In the thymic cortex, they are first tested against a specialized section of the cortical epithelium for (self-)MHC recognition, and subsequently for autoreactivity of self-tissue proteins in the thymic medulla [28]; cells that fail either stage undergo apoptosis, ensuring proper self/non-self differentiation. In parallel with the recognition of exogenous antigens by CD4+ T cells, endogenous, as well as intracellular foreign antigens are displayed by MHC I for detection by CD8+ T cells, a mechanism which is not restricted to professional APCs - this generally occurs in response to a viral infection [27].
While neither microglia nor astrocytes can directly initiate an adaptive immune response, there is cross-communication between the CNS and the peripheral immune system and both cell types are thought to be able to re-activate peripherally initiated T cell responses. Both astrocytes and microglia are capable of presenting antigens, as they are able to express both MHC I and II [29,30]. However, they are not considered professional APCs in the classical sense, because they lack high expression of costimulatory molecules. In addition, they can produce various cytokines that affect the local inflammatory milieu, supporting or inhibiting T cell responses. There are several additional mechanisms by which peripheral and CNS inflammatory responses can affect each other. Within the CNS, glial and neuronal cells release extracellular vesicles (EVs) which can communicate cellular state, and transfer cytokines, proteins, and microRNAs (miRNAs) between cells. The contents of these EVs correspond to the source cell, and its reactive or degenerative state [31]. EVs have gained interest in recent years as a therapeutic target, as they are implicated in a number of neurodegenerative diseases and processes [31], and are capable of crossing the BBB [32], leading to proposals of using EVs as diagnostic markers [33]. In crossing the BBB, EVs can also transfer inflammation from the CNS to the periphery; following an intracerebral injection of interleukin-1β (IL-1β) in a mouse model, astrocyte-derived EVs were found circulating in the blood stream and inducing an inflammatory response in the liver, as well as leading to leukocyte infiltration in the brain [32]. As such, while the CNS immune cells do not directly initiate an adaptive immune response via antigen presentation, they can induce leukocyte infiltration through the dispersion of pro-inflammatory EVs and subsequently act as APCs in a localized reactivation of an adaptive immune response. To aid this process, while usually leukocyte trafficking across the BBB is limited, its integrity becomes compromised during inflammation or injury, allowing infiltration of various peripheral immune cell types [19,34]. While implicated in facilitating the activation of the adaptive immune system from the CNS, the permeability of the BBB to EVs also affords opportunity for novel therapeutics, with proposals of using EVs for drug delivery to deep brain structures [33].
2.1 Peripheral inflammation translocation to CNS
Peripheral inflammation can also transfer to the CNS, independent of immune cell infiltration. In the innate immune system there are a number of receptors that recognize damage or pathogen associated molecular patterns (DAMP/PAMPs), inducing an inherent, immediate inflammatory response [27]. While many different types of immune receptors recognize DAMP/PAMPs, classified as pattern recognition receptors (PRR), the toll like receptors (TLRs) remain one of the most well studied and characterized families, and are capable of responding to a wide variety of both exogenous and endogenous stimulatory molecules [35]. This genetically engrained response allows for the rapid mobilization of immune cells in an attempt to eliminate and control the deleterious stimulus; this casts a broad net, responding to a type of immune challenge through the release of cytokines and chemokines [27]. The innate immune response is not pathogen specific, with different TLRs instead recognizing classes of DAMPs/PAMPs - these TLRs can be segregated into two groups: cell membrane, and intracellular receptors. Cell membrane receptors (TLR1, TLR2, TLR4, TLR5, TLR6, TLR10) respond to bacteria, proteins, lipids, and lipoproteins, while the intracellular compartmental receptors (TLR3, TLR7, TLR8, TLR9) respond to nucleic acids (DNA, RNA, miRNA) [36]; the TLR response is highly conserved across species and cell types, although the specific receptors expressed are not [36], with more TLRs expressed in mice than humans.
Upon activation by an appropriate ligand, an adaptor protein is recruited to the cytoplasmic Toll/IL-1 receptor (TIR) domain of the TLR [37], which controls the subsequent intracellular signalling pathway activated. TLRs 1, 2, 4, and 6 all use the myeloid differentiation primary response 88 (MyD88) adaptor like (MAL) adaptor protein, which recruits MyD88 and subsequently activates a signalling cascade through the interleukin-1 receptor-associated kinases (IRAKs), and tumour necrosis factor receptor-associated factor 6 (TRAF6); this activates downstream c-Jun N-terminal kinase (JNK) and nuclear factor kappa B (NF-κB) inflammatory pathways [35]. NF-κB is bound in the cytoplasm by inhibitor of NF-κB (IκB), which is degraded through phosphorylation by the IκB kinase (IKK) complex, resulting in the nuclear translocation of NF-κB, and the subsequently induced transcription of tumour necrosis factor alpha (TNFα), IL-1β, and IL-6 [35]. All TLRs which use either MAL/MyD88 or MyD88 adaptor proteins (TLR1, TLR2, TLR4-10) utilize this signalling pathway. In addition, intracellular TLRs 7-9 use a direct signalling pathway through TRAF6, which leads to the nuclear translocation of interferon regulatory factor 7 (IRF7) [35], and the induction of interferon alpha (IFNα) [38]. While MyD88 is involved in most TLR signalling, the TIR domain-containing adaptor-inducing interferon beta (TRIF) is used by both TLR4, and TLR3, and activation of this pathway results in NF-κB nuclear translocation and IFNβ production via IRF3 [35].
Peripheral TLR activation can result in neuroinflammation. In a murine model of sepsis, induced by an intraperitoneal injection of 5 mg/kg of lipopolysaccharide (LPS, a TLR4 ligand) [34,39,40], persistent neuroinflammation was observed 10 months post-injection, while peripheral inflammation was resolved within 7 days [39]. While LPS is used as a model of sepsis due to its ability to induce severe, widespread peripheral inflammation, it can also act as a model of more general peripheral infection, especially when investigating neuroinflammation. The persistent neuroinflammation was characterized by an elevated level of TNFα in the brain parenchyma, with protein levels observed at 24 hours post-injection sustained at 10 months. In a TNFα receptor (TNFR1/2) knockout mouse, peripheral LPS injection did not result in elevated TNFα levels in the brain [39], indicating the TNF signalling pathway is responsible for translocation of inflammation into the CNS, and is widely induced upon activation of any TLR. As such, it acts as a model of chronic sterile CNS inflammation.
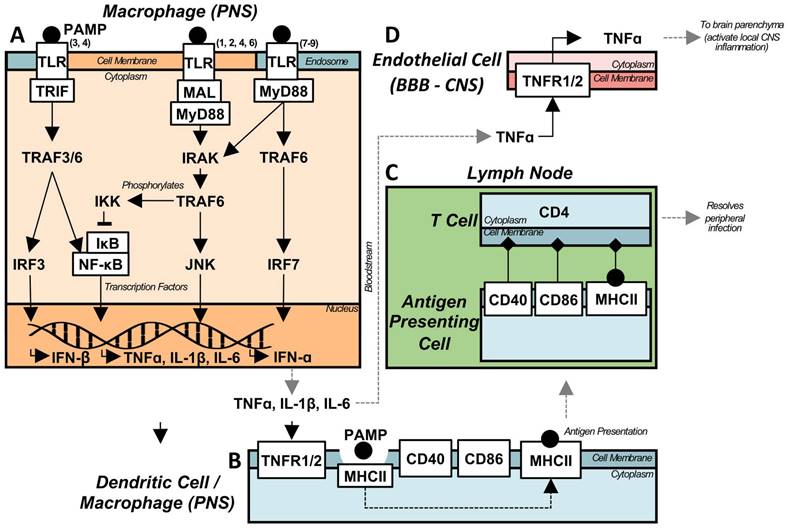
While the LPS model was first described to induce holistic brain inflammation, with early timepoints following stimulation resulting in inflammation of the substantia nigra (SN), hippocampus, and cortex [39], further research has demonstrated preferential, localized, chronic inflammation. TNFα messenger RNA (mRNA) levels are significantly elevated in the hippocampus and midbrain 1 month post-LPS compared to control, while this is not observed in the cortex or striatum [40]. It has been proposed that the regional effects induced by peripheral inflammation are mediated by microglia numbers [39], as well as anti-inflammatory signalling pathway component (i.e. CD200/CD200R) concentrations [40]. Despite the relatively simple transfer of peripherally induced LPS inflammation to the CNS via a single cytokine (Figure 2), the etiology of the induced chronic inflammatory state is less clear.
Acute peripheral infection can translocate to the CNS and cause chronic neuroinflammation. (A) Depiction of TLR signalling for both the intracellular, endosomal TLRs (3, 7-9) and the cell surface receptors (1, 2, 4-10). During an infection, PAMPs are recognized by TLRs, causing cytoplasmic signalling adaptors to associate with their respective, activated, TLR. The majority of TLRs utilize MyD88, either directly (TLRs 5, 7-10), or via an additional adaptor protein MAL (TLRs 1, 2, 4, 6). However, TLRs 3 and 4 utilize a MyD88-indepenent pathway via TRIF. The majority of TLR and MyD88 signalling is facilitated by IRAK, which subsequently activates TRAF6 leading to downstream NF-kB nuclear translocation, through IKK phosphorylation of IkB, and JNK pathway activation leading to pro-inflammatory cytokine transcription. Alternately, the endosomal TLRs are capable of producing type I interferons through the activation of IRF7. The MyD88-independent pathway activates TRAF3/6 through TRIF, leading to both NF-kB and IRF3 activation, again leading to pro-inflammatory cytokine and type I interferon transcription. (B) Following the peripheral activation of immune cells by DAMPs, and the subsequent production of inflammatory cytokines, APCs respond to the pro-inflammatory environment, and become activated, expressing co-stimulatory molecules necessary for activating the adaptive immune response. PAMPs are phagocytosed, and processed for display by MHC II, after which the activated APC traffics to lymphatic tissue. (C) The APC binds to the respective T cell receptors, activating the pathogen specific, adaptive immune response. Activated lymphocytes then traffic to infected tissue and target infected cells to assist in the resolution of peripheral infection. (D) Pro-inflammatory cytokines produced from peripheral infection, both circulating and generated by circulating monocytes, are capable of activating immune receptors on the endothelial cells of the BBB. Specifically, TNFa activates its receptors, leading to the production of TNFa within the parenchyma, activating pro-inflammatory cytokine production and positive feedback loops, eventually leading to chronic, sterile neuroinflammation.
2.2 Heterogeneity of PRR activation
Traditionally, research into neurodegenerative disease has focused on the mechanisms behind the initiation of an inflammatory, or neurodegenerative state, and the corresponding ligands or molecules involved in this initiation. This can be restrictive in developing a broad understanding of the progression of inflammation, as the paradigm of a specific PRR activation inducing a characterized result does not always account for the downstream inflammatory cascade in vivo, or the larger environmental context. Ethanol and amyloid beta (Aβ) are both characterized as TLR4 [41], and TLR2/4 [35] ligands respectively, and both receptors are implicated in a wide spectrum of neurodegenerative diseases [42]. However, they do not solely induce TLR2/4 stimulation. Recently, transgenic Aβ overexpression mouse models displayed inflammation, and demonstrated increased microglial reactivity surrounding nucleic acid positive Aβ clusters, and concomitant increase in type-I IFN production and IRF7 transcription, indicating likely intracellular compartmental TLR signalling. Blocking downstream IFN signalling by inhibiting type-I IFN receptors (IFNAR) improved pathology, indicating the necessity of considering all components contributing to the perpetuation of an inflammatory cascade [43].
In a model of chronic ethanol-induced neurotoxicity, ethanol induced the release of microglial EVs containing the miRNA let-7b and high mobility group box protein 1 (HMGB1), which was found to stimulate TLR7 and induce IFNα, TNFα, and IL-1β production in a rat hippocampal-entorhinal slice culture; blockade of TLR7 reduced the activation of the MyD88 signalling pathway [44]. While TLR4 blockade prior to ethanol administration will eliminate the inflammatory response [41], during chronic ethanol exposure endogenous TLR ligands are released, expanding the inflammatory purview beyond a single TLR. In Alzheimer's disease (AD) patients a similar endogenous signalling pathway is induced, characterized by increased levels of let-7b in the cerebral spinal fluid (CSF) [45], although it should be noted that the pro-inflammatory effect of let-7b and miRNAs in general is dependent on environmental, or EV context [44,46]. While there has been a focus on let-7b as an endogenous TLR7 ligand, many other miRNAs have the ability to activate TLR7, including miRNA-21 [47,48], which is upregulated by HMGB1 [47] and overexpressed in inflammation [48]. Consequently, the confluent downstream activation of TLRs by induced endogenous ligands should be considered when investigating the in vivo effect of an exogenous ligand.
Furthermore, TLRs act in a synergistic fashion. TLR4 stimulation has been shown to lead to the endosomal localization of TLR7, resulting in increased expression [49] and co-stimulation of receptors that can amplify the inflammatory response [50]. As the presumed method of translocation of peripheral inflammation into the CNS, it is important to understand the TNFα signalling cascade in inflammation. Through the TNFR receptors, TNFα induces a positive, pro-inflammatory feedback loop, where increased TNFα levels increase the transcription of the cytokine. Additionally, TNFα has been shown to induce reactive oxygen species (ROS), HMGB1 [51] (a TLR4 ligand), type-I IFN in an IFNAR-mediated positive feedback loop [52], and an upregulation in miRNA release. Additionally, downstream effects of TNFα signalling include the activation and priming of different PRR responses, such as that mediated by the nucleotide-binding oligomerization domain (NOD)-like receptor family pyrin domain containing 3 (NLRP3) inflammasome, a cytosolic complex which potently induces IL-1β, and caspase-1 [53]. The diversity in pathways upregulated in inflammation necessitates the consideration of both the local, cellular context, and that of the larger environment.
2.3 Sex differences
Mirroring the heterogeneity in the signalling pathways that are activated in inflammation, the immune response varies with sex [54], age [55], and the circadian rhythm [56]. In early postnatal development, female-derived (FD) murine microglia exhibit increased triggering receptor expressed on myeloid-2 (TREM2) expression, associated with increased phagocytosis, as well as a less reactive phenotype when compared to male-derived (MD) cells [57]. In adult mice, FD microglia display a neuroprotective or reparative phenotype [57], which is sustained following transplantation into male mice, resulting in reduced lesion volume in a model of ischemic stroke [58]. However, in response to DAMP/PAMPs the response of FD immune cells is more robust [54,57,59], which can lead to improved pathogen clearance [54,59] and an increased susceptibility to chronic inflammation and autoimmunity, especially in aged populations [54,55]. Immune cell phenotype is modulated by the expression of sex hormones in a dose dependent fashion, where high levels of endogenous estrogen contribute to a reparative phenotype in microglia, astrocytes, monocytes, and T cells which is lost with age [54,57,60], potentially contributing to the increased female bias of many autoimmune and neurodegenerative diseases.
In a mouse model of AD, peripheral immune cell infiltration was greater in 12-month-old female mice compared to their male counterparts [61]. Endothelial nitric oxide synthase (eNOS) is an important mediator of vascular health and cerebral blood flow (CBF) [62], and its impairment has been associated with increased BBB permeability and leukocyte infiltration into the CNS [63], as well as concomitantly increased inflammation [64]. In young animals, FD endothelial cells express more eNOS [65], which exhibits a symbiotic relationship with estrogen (E2; 17β-estradiol) [66] via activation of estrogen receptor α (ERα) [67], leading to a decreased risk for cardiovascular disease [68] - conversely, a reduction of estrogen with age results in decreased eNOS and increased risk for cardiovascular disease [68], as well as an exacerbated inflammatory response [54,69]. While the relationship between low estrogen levels and increased inflammation has been characterized in humans [54], mouse estrogen is largely constant over the lifespan [70], with age instead correlating with a decrease in ERα expression in bone marrow derived macrophages [69]. As eNOS is directly modulated by E2 through ERα [67], decreased estrogen signalling in both middle aged mice and humans, leads to a loss of eNOS-mediated neuroprotection with age, and potentially exacerbating inflammation. Contextualizing these results, a broad array of endogenous factors potentially contributing to sex differences in the immune system exist, in addition to changes with age: increased type-I IFN production occurs in the ageing brain, reducing the production of brain derived neurotrophic factor (BDNF), a neurotrophin with anti-inflammatory properties [71] downstream of eNOS [72], from the choroid plexus [73].
Aside from the influence of sex hormones, there are chromosomally mediated sex differences in immune responses. Due to the presence of two X chromosomes, there are multiple copies of the same gene present in the genome of female immune cells. Typically, these doubly present genes are subject to X chromosome inactivation, a process by which one of the X chromosomes is inactivated, leading to comparable mRNA and protein expression levels as those in male cells [59]. However, this process is incomplete, and distally located genes can escape inactivation. In the immune system this is of consequence, as TLR7 is distally located on the X chromosome, and can escape inactivation [59]. This partially explains the increased production of IFNα in response to viral infections [59], and the increased female bias of autoimmune interferonopathies such as systemic lupus erythematosus [54,74]. Needless to say, chronic inflammation is pathologically complex, with an intricate web of contributing endogenous and exogenous factors - this complexity necessitates the development of therapeutic methods targeting a broad spectrum of pathways.
3. Ultrasound and inflammation - mechanical mechanisms of resolution
HIFU ablation of tissue involves the confluence of multiple biophysical mechanisms including hyperthermia, and mechanical stimulation, which each have immunomodulatory potential. Much of this research has concentrated on cancer model systems [11], where the desired outcome is the destruction or disruption of pathological tissue, complicating the direct translation of the signalling pathways involved into other etiological contexts where ablation or lesioning is not a desired outcome. Consequently, the bulk of immunological studies concentrating on the effect of ultrasound on neurodegenerative diseases and inflammatory conditions have used LIPUS. LIPUS is a heterogeneous term, encompassing a wide variety of ultrasound parameters, a number of which have been elucidated in Table 1. Typical parameters reported are: spatial peak temporal average intensity (ISPTA) of 10 to 528 mW/cm2, centre frequency (fC) of 1 to 1.875 MHz, pulse repetition frequency (fR) of 1 Hz to 6 kHz, pulse burst length of 17 μs to 0.2 s, and individual treatment duration ranging from 5 to 20 minutes in vivo, and up to several hours in vitro. Comparatively, HIFU is intended for ablation, with typical parameters of: 0.5 to 8 MHz, acoustic power ranging from 100-10,000 W/cm2, with short continuous wave sonication times of 10-25s, and focal temperature increases in excess of 50-60 oC [5,6]. While high intensity therapeutic ultrasound has been previously defined at an intensity of ≥ 3W/cm2 [75] for therapeutic application in an in vitro model of inflammation [76] (Table 1), studies at this intensity remain sparse. Essentially, LIPUS uses parameters at or below those used in clinical diagnostic ultrasound [75] to apply a mechanical force to local cells in the targeted area, including resident neurons, glia, and infiltrating immune cells, thereby activating pathways which modulate the inflammatory milieu and immune response, while HIFU uses intensities orders of magnitude higher for the purpose of thermal ablation. Investigations into LIPUS as a treatment for chronic inflammation have demonstrated promising results, in both the CNS and a diverse array of peripheral tissues. To date, the mechanisms behind the mitigating effect of LIPUS on inflammation are poorly understood, as studies are conducted in a wide variety of tissues with heterogeneous ultrasound parameters and disease states. Developing a holistic understanding thus requires the integration of information from numerous disparate studies (see Table 1). Recent in vivo studies have investigated the efficacy of LIPUS in neurodegenerative disease and models of cerebral trauma, with beneficial results consistently observed. While there is a consistent finding of upregulated BDNF [64,77-80] corresponding to decreased inflammation with LIPUS application in inflammatory models, the underlying mechanisms are unclear.
Summary of experiments investigating the biophysical impacts of therapeutic ultrasound, where all effects are compared to a non-US group (i.e. LIPUS compared to SHAM, LPS+LIPUS compared to SHAM+LIPUS). For a more extensive compilation of the effect of LIPUS across cell types, see the review by Xu et al. [7].
| Model | Experiment | LIPUS Parameters | Biological effect |
|---|---|---|---|
| in vitro [76] macrophages: bone marrow derived HUS | measured gene expression 3h post-US LPS: 100ng/mL measured inflammatory response to LPS 3h post-US, 1.5h with LPS (4.5h from start of US) | fc: 1 MHz fR: 100 Hz ISPTA: 3 W/cm2 Duration: 5 min Duty cycle: 20% Burst length: N/A Transducer: plane | US: - apoptosis, ↑Irg1 (mRNA), ↑itaconate (mRNA), ↑Nrf2 (mRNA) US+LPS: ↓IL-1b (mRNA), ↓IL-6 (mRNA), ↓Tnfα (mRNA) Verified cell media did not exceed 37 oC |
| in vitro [81] microglia: BV-2 astrocytes: CTX TNA2 LIPUS | LPS: 1 μg/mL LIPUS 12h post-LPS Evaluation at 4, 8, 12, 24 h post-LIPUS | fc: 1 MHz fR: 100 Hz ISPTA: 10, 20, 30 mW/cm2 Duration: 15 min (3x5/5 on/off) Duty cycle: 50% Burst length: 5 ms Transducer: plane | Microglia LIPUS: ↑BDNF, ↑GDNF, ↑VEGF, ↑p-CREB/CREB, ↑p-Akt/Akt, ↑p-TrkB/TrkB LPS+LIPUS: ↑BDNF, ↑GDNF, ↓VEGF, ↓TNFa, ↓IL-1b, ↓IL-6, ↑p-CREB/CREB, ↑p-Akt/Akt, ↑p-TrkB/TrkB, ↓TLR4, ↓MyD88, ↓p-JNK/JNK, ↓p-p65/p65, ↓iNOS TrkB/Akt/CREB/BDNF pathway required for BDNF production Akt inhibits JNK Astrocytes LIPUS: ↑BDNF, ↑GDNF, ↑VEGF, |
| in vitro [84] astrocytes: CTX TNA2 LIPUS | Evaluation at 4, 8, 12, 24 h post-LIPUS | fc: 1 MHz fR: 10 Hz ISPTA: 110 mW/cm2 Duration: 15 min (3x5/5 on/off) Duty cycle: 50% Burst length: 50 ms Transducer: plane | LIPUS: ↑BDNF, ↑p-TrkB/TrkB, ↑p-Akt/Akt, ↑Ca2+ influx, ↓p-CREB/CREB, ↑p-NF-kB p65/NF-kB p65, ↑p-p65/p65 |
| in vitro [76] preosteoblasts: MC3T3-E1 LIPUS | mRNA measurement 0, 6, 24, 48h post-LIPUS | fc: 1.5 MHz fR: 1 kHz ISATA: 30 mW/cm2 Duration: 20 min Duty cycle: 25% Burst length: 0.2 s Transducer: N/A | LIPUS: ↑CD200 (mRNA) at 24h, ↑bone morphology genes |
| in vitro [190] osteoblasts: MC3T3-E1 LIPUS | LPS: 1 μg/mL LIPUS/sham simultaneous with LPS | fc: 1.5 MHz fR: 100 kHz ISATA: 30 mW/cm2 Duration: 2 h Duty cycle: N/A Burst length: 0.2 ms Transducer: N/A | LPS: ↑CCL2 (mRNA), ↑CXCL1 (mRNA), ↑ CXCL10 (mRNA), ↑RANKL (mRNA), ↑NF-kB (mRNA) LPS+LIPUS: - CCL2 (mRNA), ↓CXCL1 (mRNA), ↓CXCL10 (mRNA), ↓RANKL (mRNA), ↓NF-kB (mRNA), ↓ERK/Akt/IKK/IRF3, ↓TLR4/MyD88 association |
| in vitro [114] endothelial: ECV 304 | Applied laminar fluid shear stress to endothelial cell line | Laminar fluid shear stress: 15 dynes/cm2 (1.6 Pa) Duration: 5, 15, 30, 60 mins | Shear Stress: prolonged shear stress led to increased association between eNOS and HSP90, but did not lead to an increase in HSP90 |
| in vivo [77] Female Sprague-Dawley rats LIPUS | 6-OHDA: 12μg 2μL of 6-OHDA injected into right striatum 6-OHDA model of Parkinson's Disease Cellular evaluation at 8 weeks post-injection | fc: 1 MHz fR: 1 Hz ISPTA: 528 mW/cm2 Duration: 15 min (3x5/5 on/off) Duty cycle: 5% Burst length: 50 ms Transducer: focused, single hemisphere | 6-OHDA+LIPUS: ↓GFAP+ astrocytes, ↓Iba1+ microglia, ↓IL-1b, ↓p-NF-kB, ↑GDNF, - BDNF (insignificant increase), ↑BBB stability (ZO-1, claudin-5) LIPUS started 2 weeks following 6-OHDA injection, LIPUS administered 5 days/week for next 6 weeks Observed no tissue damage with LIPUS regimen |
| in vivo [97] Beagles (2-8 years old) LIPUS | Wound healing in mucoperiosteal flap surgery | fc: 1.5 MHz fR: 1 kHz I: 30 mW/cm2 Duration: 20 min/day Duty cycle: N/A Burst length: 0.2 ms Transducer: 13 mm diameter | LIPUS: ↑HSP70 expression in gingival tissues (absent in control), ↑cementum and mandibular bone regeneration LIPUS continued for period of 4 weeks following surgery |
| in vivo [78] Female C57BL/6 mice (7 weeks old) LIPUS | LIPUS in model of repetitive restraint stress (RRS): 22 days LIPUS in cuprizone (0.2% in chow) model of demyelination: 35 days Analysis 24 hours following last LIPUS treatment | fc: 1.5 MHz fR: 1 kHz ISPTA: 25 mW/cm2 Duration: 20 min/day Duty cycle: 20% Burst length: N/A Transducer: plane | LIPUS: ↑BDNF (hippocampus) RRS+LIPUS: ↓anhedonia, ↑BDNF (hippocampus) CUPRIZONE+LIPUS: ↑MBP (myelin basic protein, cortex), ↑NG2 (neural/glial antigen 2, cortex) NG2 expressed in oligodendrocyte progenitors, and microglia |
| in vivo [79] Male C57BL/6 mice (8 weeks old) LIPUS | LPS: 250 μg/kg LPS administration from days 5-11 LIPUS treatment from days 7-13 Biological analysis at 4 (GFAP, Iba1, BDNF, CREB) and 48 h post-LIPUS | fc: 1 MHz fR: 1 Hz ISPTA: 528 mW/cm2 Duration: 15 min (3x5/5 on/off) Duty cycle: 5% Burst length: N/A Transducer: focal, (D: 38 mm, ROC:63.5 mm; half-max pressure amplitude of focal zone - D: 3 mm, L: 26 mm) | LPS+LIPUS: ↑spatial learning and memory (Morris water maze, novel object recognition), ↓Aβ (hippocampus, but not cortex), ↓cleaved caspase-3, ↓GFAP, - Iba1 (non-significant decrease), ↓TNFa, ↓IL-1b, ↓IL-6, ↓p-NF-kB, ↓TLR4, ↑BDNF, ↑p-CREB LIPUS: ↑BDNF, ↑p-CREB, ↑p-NF-kB (cortex), ↑TLR4 Increase in TLR4 and p-NF-kB in LIPUS group did not correspond to a significant increase in TNFa, IL-1b, or IL-6 |
| in vivo [64] Vascular Dementia Male C57BL/6 mice (10-12 weeks old), eNOS-/- (C57BL/6 background) Alzheimer's Disease Male 5XFAD transgenic mice (14-16 weeks old, C57BL/6 background) LIPUS | LIPUS applied every other day following surgery (i.e. 1, 3, 5; BCAS or sham for vascular dementia) LIPUS applied on days: 1, 3, 5, 28, 30, 32, 56, 58, 60, 84, 86 for AD model, testing on day 86 | fc: 1.875 MHz fR: 6 kHz ISPTA: 90 mW/cm2 Duration: 20 min (x3: right, midline, left)/day Duty cycle: 5% Burst length: 17μs Transducer: focal, (focus: 9 cm, beam width: 3.6-4 mm) | VaD+LIPUS - Molecular (day 3/7): ↓WM lesions, ↑angiogenesis and OPC genes (mRNA), ↑eNOS (both inactivated and activated; correlated with increased NGF, BDNF, VEGF), ↑GFAP, ↑Olig2+ VaD+LIPUS - Behavioural (day 28): ↑cognitive function (Y-maze, passive avoidance, novel object recognition, not all significant) VaD+LIPUS - CBF: ↑CBF with LIPUS over time (start at day 2, continues to day 28) VaD+LIPUS+eNOS-/- (with respect to VaD): - behavioural, - CBF, - WM lesions, - NGF, - BDNF, - VEGF 5XFAD+LIPUS: ↑HSP90, ↑CBF, ↑immune and angiogenesis genes (mRNA), ↑cognitive function (Y-maze), ↓Iba1+, ↑eNOS, ↑BDNF, ↓Aβ Note: HSP90 not measured in VaD group, only 5XFAD |
| in vivo [80] Male C57BL/6 mice (8 weeks old) LIPUS | Cortical Impact Injury (CCI) - model of traumatic brain injury (TBI) LIPUS applied 5 mins post-CCI, daily thereafter Analysis at day 1, 4, and 148 | fc: 1 MHz fR: N/A ISPTA: 528 mW/cm2 Duration: 5 min/day Duty cycle: N/A Burst length: N/A Transducer: focal, (focal diameter: 3 mm, focal width: 26 mm, diameter: 38 mm, ROC: 63.5 mm) | TBI+LIPUS (day 1): -MMP-9, ↓neutrophils, ↓Iba1+, -p-Foxo1, ↓neurological deficits TBI+LIPUS (day 4): ↓MMP-9, ↓neutrophils, ↓Iba1+, ↑p-Foxo1, ↓neurological deficits TBI+LIPUS (day 148): insignificant ↑BDNF+ neurons, ↑neurons, ↓T2w edema volume, ↓neurological deficits |
3.1 Brain derived neurotrophic factor
Several mechanisms have been proposed to underly LIPUS mediated upregulation of BDNF, through in vitro studies. The application of LIPUS to BV-2 microglial cells led to the upregulation of BDNF, along with phosphorylated cyclic adenosine monophosphate response element-binding protein (p-CREB), phosphorylated protein kinase B (p-Akt), and phosphorylated tyrosine protein kinase B (p-TrkB), both in LPS and control conditions; with LPS administration, LIPUS additionally downregulated pro-inflammatory cytokines, TLR4, MyD88, and downstream kinase and NF-κB transcription [81]. TrkB and BDNF engage in a positive feedback loop, with BDNF acting as a ligand for TrkB, subsequently activating the downstream signalling factor Akt, and CREB, upregulation of which is associated with increased BDNF production - both Akt and CREB were required for BDNF expression in response to LIPUS [81]. Akt is a typically anti-inflammatory signalling molecule, the activation of which has been demonstrated to reduce inflammation in an LPS model of sepsis [82]. Within the intracellular context of LPS-stimulated microglia, Akt is proposed to increase CREB expression, in addition to directly inhibiting the phosphorylation of JNK, and the nuclear translocation of NF-κB [83]. A similar upregulation of BDNF in cultured astrocytes was observed [81] in response to LIPUS, although the mechanism of action is likely different [84]. LIPUS stimulated BDNF production by naïve astrocytes is thought to be induced by increased Ca2+ influx, and calcium kinase signalling pathways. Interestingly, while this pathway again relies on Akt, CREB is downregulated in response to LIPUS in astrocytes, indicating the existence of a distinct pathway from their microglial counterpart. This difference is emphasized by the increased nuclear translocation of NF-κB, a pro-inflammatory transcription factor, inhibition of which led to the elimination of LIPUS-mediated BDNF production [84]. Furthermore, a similar Ca2+ dependent mechanism is thought to be responsible for the confluent increase of BDNF with neuronal activity [85], potentially indicating parallel signalling pathways and co-interaction of neurons and astrocytes in BDNF production, beyond the BDNF/TrkB positive feedback loop, as a neuromodulatory process. Increased BDNF has been demonstrated in response to LIPUS in a model of cuprizone-induced demyelination, and chronic restraint stress, as well as accelerated remyelination, a process attributed to neuromodulation [78]. In considering LIPUS mediated neuromodulation in vivo, changes in local BDNF and neuronal activity cannot be divorced from the influence of surrounding glial cells.
It is important to note that while studying the astrocytic origin of BDNF production, an inflammatory challenge prior to LIPUS stimulation was not used, as described in the study by Liu et al. mentioned previously [84]. In vivo LIPUS treatment of mice resulted in increases in BDNF, CREB, NF-κB, and TLR4, with no significant increase in pro-inflammatory cytokines. However, after an LPS challenge, LIPUS treatment resulted in decreased TLR4 and NF-κB activation, while maintaining the upregulation of BDNF and CREB [79]. This could indicate that the induction of separate signalling pathways depends on the context of the glial phenotype, cellular environment, and the local cell populations, or that microglial BDNF production through the TrkB/Akt/CREB pathway is dominant in the overall effect of LIPUS. Regardless of the cell of origin, or mechanism of production, LIPUS treatment both in vitro and in vivo modulates the endogenous release of neurotrophic factors associated with the reduction of inflammation, constituting one potential branch of a therapeutic pathway, while avoiding potential unwanted side effects [86].
3.2 Blood brain barrier
Coincident with the release of neurotrophic factors, LIPUS has been observed to reduce peripheral immune cell infiltration, and increase the stability of the BBB [77,80]. Peripheral immune cell infiltration and BBB disruption are implicated in the etiology and exacerbation of neurodegenerative diseases [87,88], aging [88], and in the progression of vascular dementia (VaD) and AD, as pro-inflammatory cytokines reduce the expression of tight junction proteins integral to maintaining BBB function [77]. In a model of traumatic brain injury (TBI), LIPUS was applied immediately, and daily thereafter, which resulted in a decrease in reactive microglia, infiltrating neutrophils, metalloproteinase 9 (MMP-9) expression, and an increase in forkhead box protein O1 (FOXO1) expression [80]. MMP-9 is associated with increased cleavage of tight junctions and disruption of endothelial cells, and its upregulation is associated with the development of edema [89]; alternately, FOXO1 is a downstream factor of the Akt pathway [90], and is associated with reduced apoptosis of endothelial cells and preventing tight junction loss, corresponding to reduced BBB permeability [91]. In combination, the effects of LIPUS led to decreased long term neurological deficits and edema. However, this study applied LIPUS longitudinally, starting in an acute stage from the induction of injury, an immediacy in treatment that is not available in chronic inflammatory diseases. The 6-hydroxodopamine (6-OHDA) model of Parkinson's disease (PD) exhibits BBB disruption, and has been used in the investigation of LIPUS on BBB stability in the chronic stage [77]. Starting LIPUS treatments two weeks following intracerebral 6-OHDA injection, and continuing for the next six weeks, resulted in a reduction of glial cell reactivity and pro-inflammatory cytokine expression, while the expression of tight junction proteins zonula occludens-1 (ZO-1) and claudin-5 were increased [77]. Interestingly, BDNF was not significantly increased in this model, potentially indicating the dominant therapeutic factor was tight junction protein related; it should be noted that glial cell line-derived neurotrophic factor (GDNF) was significantly upregulated following LIPUS in this model, which is neuroprotective and linked to the upregulation of claudin-5 [92].
Endothelial cells are integral to maintaining proper vascular and BBB function, as well as secreting factors to maintain homeostasis, such as eNOS. Recently, eNOS has been found to be essential to the beneficial effect of LIPUS in a model of VaD, reducing white matter lesions, increasing neurotrophic factors, improving cognitive function and CBF; these benefits were abolished in an eNOS knockout mouse [64]. eNOS is bound to caveolin-1 (Cav-1) in the caveolae of endothelial cells, and is released upon mechanical stimulation [93] through the aid of a heat shock protein 90 (HSP90) and Akt dependent process [94]. Furthermore, caveolae and Cav-1 is increased in response to mechanical or thermal stimulation [93], potentially corresponding to a concomitant increase in eNOS. While an eNOS knockout could not be generated for the 5xFAD transgenic AD mouse model, there was similar correlation between the LIPUS-induced upregulation of eNOS, and the production of BDNF, phagocytosis of Aβ, improved cognitive performance and CBF in this model, which was also accompanied by an increase in HSP90 [64].
Pharmaceutically, sildenafil, an inhibitor of phosphodiesterase-5 used commonly to treat erectile dysfunction, has many of the same neuroprotective benefits as those observed with LIPUS, and acts as a parallel from which to deduce potential underlying pathways. Mechanistically, in a model of experimental autoimmune encephalomyelitis (EAE), sildenafil functioned by decreasing inducible NOS (iNOS), a pro-inflammatory species, while increasing the expression of eNOS [72,95]. Sildenafil increases the expression of adenosine monophosphate (AMP)-activated protein kinase (AMPK), which signals in a positive feedback loop with eNOS, where AMPK activates eNOS, and eNOS produced NO again increases AMPK activation [72]. Furthermore, AMPK decreases mammalian target of rapamycin (mTOR) expression, resulting in the upregulation of CREB and BDNF [72]. In addition to the generation of neurotrophic factors, the eNOS/AMPK pathway is associated with increased autophagy activity [72], a process dysregulated in aging, neurodegenerative diseases, and inflammation [96]; however, as autophagy was measured using absolute autophagy related protein (microtubule-associated protein 1A/1B-light chain 3, beclin-1, autophagy related 5) levels, instead of autophagic flux [96], a definitive statement on the influence of this pathway on autophagy cannot be made. The eNOS/AMPK axis is a potent anti-inflammatory pathway, though, associated with inhibiting NF-κB nuclear translocation, through the upregulation of IκBa, neurotrophin release, and likely stimulation of autophagy [72,95]. Consequently, LIPUS, likely stimulates endogenous anti-inflammatory pathways through similar signalling pathways.
3.3 CD200/CD200R1 signalling
In peripheral tissues, LIPUS has been used to accelerate both wound and fracture healing [7,97]. Investigating the effect of LIPUS on bone fracture healing in pre-osteoblasts, both the upregulation of bone morphology genes and CD200 mRNA was observed [76]. While implicated as a possible modulator of bone mass in fracture healing, the CD200/CD200R axis has been proposed as a therapeutic treatment for neuroinflammation, indicating increased CD200 as a possible LIPUS induced anti-inflammatory pathway. CD200 is an inhibitory transmembrane glycoprotein from the immunoglobulin domain super family, ubiquitously expressed across immune cells; signalling through its receptor, CD200R1, leads to the suppression of the inflammatory response [98]. Intracellularly, CD200/CD200R1 signalling is propagated through a docking protein 2 (Dok2) and Ras GTPase activating protein (RasGAP)-mediated pathway [98], which leads to the suppression of the phosphorylation of Akt, and extracellular signal-regulated kinase 1 and 2 (ERK1/2), which is induced in inflammation and associated with pro-inflammatory cytokine production [99]. Due to its anti-inflammatory function, and dysregulation in inflammation [40,100-103], targeting CD200 has been proposed as a potential treatment for neurodegenerative diseases. Through the administration of either blocking or agonistic antibodies, the CD200/CD200R1 pathway has been observed to promote inflammation resolution [34,100,103]; however, the determination of CD200 function was predicated on intervention prior to the induction of, or during an acute inflammatory state. As such, immune cells, especially microglia, were primed with no suppressive mechanism in the case of inhibitory antibody administration, or their inflammatory function was suppressed over that of the homeostatic state, with CD200R1 agonist administration. A recent study discovered that the inflammatory state of the immune cells suppressed by CD200/CD200R1 signalling is critical to this pathway's function when activated in these cells - peripheral blood mononuclear cells (PBMCs) pre-treated with IFNα exhibited altered CD200/CD200R1 signalling compared to untreated PBMCs, either losing the suppressive effect on PBMC activation, or potentiating the inflammatory response [99]. Reactive, pro-inflammatory microglia require intracellular caspase-3 activation to mount an immune response, and its blockade prevents microglial activation in response to inflammatory stimuli [105]. As mentioned above, immunosuppressive CD200/CD200R1 signalling requires both Dok2 and RasGAP to inhibit both Akt and ERK1/2 activation, and exert its anti-inflammatory effect; with caspase-3 activation, RasGAP is cleaved, which prevents binding to Dok2 resulting in the differential suppression of the Akt pathway, while ERK1/2 and downstream mTOR activation are unaffected [99]. Consequently, CD200/CD200R1 loses its homeostatic function in inflammation, a notion often overlooked when investigating the pathway prior to an inflammatory stimulus.
Several further studies looked into the function of CD200/CD200R1 signalling in the context of neuroinflammation, in models of EAE [30], chronic neuropathic pain [106], and aging [102]. Subcutaneous injection of 100 μg of CD200Fc, a recombinant CD200 protein, was conducted every other day following disease onset in EAE, to the conclusion of the study, or from day 20-30 to target the chronic phase [30]. Administration of CD200Fc at disease onset significantly reduced EAE severity, microglial activation, and prevented macrophage infiltration, as measured by the microglial/macrophage marker CD11b - T cells were largely unaffected. Additionally, pro-inflammatory cytokine levels in CNS and splenic tissue were reduced with CD200Fc administration, while in vitro experiments demonstrated a dose dependent effect, with functional benefits observed at lower concentrations in microglia, and higher concentrations [30] in astrocytes. While T cell behaviour was not affected, microglia and dendritic cells isolated from CD200Fc treated mice, in the chronic stage of EAE, demonstrated impaired antigen presenting ability through both reduced MHCII expression, and an inability to stimulate naïve T cells in an in vitro assay. The beneficial effect of CD200Fc administration in EAE potentially acts through the reduced activation of T cells upon CNS infiltration [30], which is supported by a reduction in IL-2 production; however, CD200Fc did not alter the expression of markers of activated CD11b+ cells, such as CD86 and CD40, indicating that, in chronic inflammation, CD200Fc affects the adaptive immune response without altering CD11b+ cell reactivity. Flow cytometric analysis at the chronic stage of EAE revealed that CD200Fc administration induced CD11b+ cell apoptosis, based on Annexin V staining, while sparing oligodendrocytes and astrocytes. While caspase-3 activation was also increased in CD11b+ cells with CD200Fc administration [30] this was not used as a marker for apoptosis, as in microglia caspase-3 is necessary for, and associated with, a pro-inflammatory, reactive phenotype [105]. As observed in the EAE model, CD200Fc administration in chronic inflammation is associated with the depletion of the local reactive microglial population, and inhibition of antigen presentation in acute stages of inflammation, contributing to reduced disease severity. Classically, the CD200/CD200R1 interaction is associated with the inhibition of the ERK1/2 and Akt signalling pathways, suppressing a pro-inflammatory phenotype. However, in chronic inflammation, CD200/CD200R1 signalling can become rewired through the cleavage of RasGAP by caspase-3 [99], leading to selective suppression of Akt, propagation of ERK1/2, and concomitantly increased caspase-3 activation. Low caspase-3 activity is associated with RasGAP cleavage at position 455, generating an apoptotic C-terminal fragment, and an anti-apoptotic N-terminal fragment; high caspase-3 activity results in the further cleavage of the N-terminal fragment, resulting in C-terminal-induced apoptosis [107], constituting a potential pathway behind the observed CD200Fc induced apoptosis of reactive CD11b+ cells. With CD200Fc treatment, caspase-3 activation was increased in CD11b+ cells with CD200Fc treatment, while this was not observed in oligodendrocytes or astrocytes, indicating a potentially rewired signalling pathway in microglia. While apoptosis of reactive microglia is beneficial in reducing the inflammatory response, it should be noted that microglia can promote remyelination as well [22], implying an inherent limitation of CD200Fc treatment in eliminating reactive microglia, rather than altering cell phenotype [108].
Similarly, in a chronic constriction injury model of neuropathic pain, CD200Fc administration resulted in the reduced expression of pro-inflammatory cytokine mRNA, as well as reduced CD11b protein in the spinal cord [106], although it is unclear whether this observation is derived from CD11b+ cell apoptosis, or decreased cellular infiltration. Curiously, both CD200 and CD200R1 expression is upregulated in the neuropathic pain model, and restored to homeostatic levels following the intrathecal administration of 10 μg of CD200Fc [106], potentially indicating the inability of basal levels of CD200/CD200R1 signalling to ameliorate inflammation. In naïve aged (20-22 month old) male mice, intrahippocampal injection of 10 μg of CD200Fc induced a reduction in MHC II expression on CD11b+ cells in the hippocampus, suggesting reduced capability of antigen presentation, but did not affect CD11b or CD68 expression, markers of a pro-inflammatory phenotype [102]. However, CD200Fc treatment reduced IFNγ-inducible protein 10 (IP-10) and monocyte chemoattractant protein-1 (MCP-1) mRNA expression, chemokines associated with monocyte and lymphocyte recruitment, in hippocampal tissue. As T cell infiltration into the CNS occurs in aged mice, the reduction in inflammation is likely derived from impaired antigen presentation, and T cell reactivation upon CNS infiltration; while lymphocyte infiltration was not investigated directly, suppressed IP-10 production following CD200Fc treatment may further indicate decreased T cell infiltration [102]. CD200Fc administration during the course of inflammation differentially affects activated and quiescent immune cells, inducing apoptosis in reactive CD11b+ cells, and reducing their antigen presentation capacity, while suppressing immune activation in quiescent cells - as such CD200Fc may preferentially affect T cell driven inflammation. Furthermore, CD200R1 agonists may act in a dose dependent fashion, as in vitro treatment with 3 μg/ml of OX108, a CD200R1 agonistic antibody, to a cell culture of reactive PBMCs resulted in a loss of immunosuppressive CD200/CD200R1 signalling, instead propagating inflammatory signalling compared to controls, whereas higher doses of CD200Fc were associated with reduced pro-inflammatory cytokine production in microglia (10 μg/ml) and astrocytes (100 μg/ml) respectively, accompanied by decreased disease severity [30,102,106]. This suggests that the effect of CD200R1 agonists on downstream signalling pathways may be dependent on the strength of receptor activation. The overexpression of CD200, and treatment with CD200R1 agonists, results in increased production of GDNF [104] and the mitigation of inflammation [30,102,106], which may belie the function of endogenous CD200/CD200R1 signalling in activated immune cells [99]. CD200 also plays a role in sex differences in the immune response, with its loss accentuating the difference between male and female TLR7 mediated IFNα production [109], and the induction of reactive microgliosis in the absence of an immunological challenge [34]. This is mirrored by an accompanying, regionally specific increase in basal neuroinflammation in aged female mice, as well as increased caspase-3 activity following stroke [110], compared to aged male mice [111,112] - consequently, loss of homeostatic CD200/CD200R1 function may exacerbate sex-differences in inflammation, and propagate DAMP mediated pro-inflammatory feedback loops in females, via TLR7.
4. Ultrasound and inflammation - hyperthermic mechanisms of resolution
In the context of wound healing, LIPUS has been beneficially associated with the induction of HSP70 in gingival tissue following mucoperiosteal flap surgery, as well as increased bone regeneration [97]. The HSR is a phylogenetically conserved, stress-induced protective mechanism activated in response to hyperthermia, oxidative stress, and shear stress, among others [8]. Currently, the heat shock response is not often considered in the mechanistic mitigation of chronic inflammation observed following LIPUS treatment, due to the expected insignificant increases in the temperature of target tissue [7]. However, LIPUS has been observed to induce the upregulation of HSP90 [64] and HSP70 [97], both of which were correlated with beneficial downstream effects, including increased CBF and reduced inflammation. Murine physiology dictates that an increase in CBF will concomitantly increase brain temperature, due to the reduced surface area to volume ratio of the brain, retaining, as opposed to diffusing, heat [113]; however, the LIPUS mediated CBF increase is unlikely to induce the HSR alone. Applying laminar fluid stress of 15 dynes/cm2 (1.6 Pa) to an in vitro culture of endothelial cells resulted in an increased association between HSP90, and eNOS, without increasing the expression of HSP90 [114]. The peak rarefactional pressure in low intensity ultrasound is on the order of mega-pascals [115], necessitating the consideration of both altered intracellular HSP behaviour and affinity, as well as increased expression following LIPUS treatment [64,97].
As a commonly activated protective pathway, across mechanical or thermal stimulation, the endogenous activation of the HSR requires further investigation as a treatment for chronic inflammation. While LIPUS is the dominant ultrasound modality under investigation when considering treating inflammatory diseases, ultrasound-mediated hyperthermia constitutes an underexplored treatment modality. Hyperthermia is a state where tissue is elevated above resting biophysical levels; presently, the most common use of therapeutic ultrasound, to focally increase tissue temperature, is in the context of HIFU, where temperatures can exceed 60 oC [11,13] in thermal ablation, and reach acceptable peak values of 40-45 oC [11] in mechanical ablation. The febrile range constitutes a physiologically tolerable increase in body temperature, often in response to infection in the form of a fever, to stimulate a series of heat sensitive signalling pathways - at the ultrasound focus in HIFU, these temperatures far exceed the febrile range, reaching lesional potential for use in functional neurosurgery [5] or tumour ablation [6]. However, these targets may overlap with comorbid inflammation [13], as inflammation is a risk factor for tumour development, and tumours themselves can generate a local, persistent inflammatory response associated with growth [116]; margin effects of ultrasound-mediated hyperthermia, in the febrile range, may affect surrounding inflammatory cells. In the direct application of hyperthermia to pathological inflammation, tissue health must be minimally affected, requiring an alternate treatment modality to HIFU. Ultrasound-mediated mild hyperthermia uses the same principles of high intensity transcranial MRgFUS, while instead limiting temperature increases to physiologically tolerable levels in the febrile range, thereby enabling functional application in neurodegenerative or inflammatory disease. The intersection of febrile range temperatures in inflamed tissue, by deliberate targeting, or edge, effects, necessitates the comprehension of the mechanisms and impact of hyperthermia on chronic inflammation. Additionally, ultrasound-mediated hyperthermia is compatible with real time imaging using MR thermometry, allowing for precise, localized targeting and treatment monitoring in the febrile range.
4.1 Low intensity pulsed ultrasound as a regulator of the heat shock response
Fever is a highly conserved physiological mechanism associated with an enhanced response to infection, acting as an adjuvant to the inflammatory response in acute temporal proximity to an immune challenge [8,117] inducing increased pro-inflammatory cytokine release [118]; however, extreme fever-related inflammation is deleterious. Within the febrile response, hyperthermia is often delayed from the activation of the innate immune system, and it has been proposed that this enables the endogenous regulation of the immune response, acting as a shutdown mechanism to prevent unregulated inflammation and tissue damage [118]. Accompanying the febrile response is an increase in core temperature of 1-4 oC, which activates the HSR. Heat shock factor 1 (HSF1) is a transcriptional factor which propagates and initiates downstream HSR signalling [8], and is bound to HSP90 in the cytoplasm, maintaining a quiescent state [119]. Upon stimulation with hyperthermic temperatures, HSP90 and HSF1 dissociate [120], and HSF1 trimerizes and translocates to the nucleus, facilitating the transcription of HSR-related genes [119] and HSPs, notably HSP90, HSP70, HSP60, and HSP27 [8]. HSF1 activation occurs over a dynamic thermal range, coincident with temperature and durational increases, resulting in the proliferation of HSP production [8]; temperatures observed in HIFU, well above the febrile range, denature HSPs, thereby mitigating their efficacy and functional propagation [11]. Intracellular HSPs execute a chaperone function, binding perturbed proteins for refolding, degradation, or sequestration from the surrounding intracellular environment, protecting the cell from the toxicity induced by dysregulated proteins [8]. Currently, there is a wide array of heterogeneous evidence as to the impact of exogenous, or extracellular HSPs, with conflicting reports of pro- or anti-inflammatory functions, although this is likely a function of cell activation state [121] and release mechanism [122].
As hyperthermia has been found to be detrimental in disease etiology and inflammation in models of acute CNS insult, such as mild traumatic brain injury [123], it has largely been underexplored as a therapeutic strategy for chronic inflammation, despite promising results in peripheral tissues [124]. Aside from the upregulation of HSPs, HSF1 directly modulates the release of pro-inflammatory cytokines [53,121,125,126] and promotes neurotrophic factor release [127]. Elevated temperatures have been associated with the reduction of pro-inflammatory cytokines during fever [8], indicating a mechanism of endogenous regulation of inflammation instigated by the HSR. Heat shock related gene expression, or transcription, is mediated by the heat shock element (HSE), a transcriptional regulatory element in the promoter of genes modulated in a stress response [126]. Notably, IL-1β [128], TNFα [129], and BDNF [127] all express HSE sites, enabling modulation of their transcription by HSF1. For IL-1β and TNFα, HSF1 acts as an inhibitory factor, preventing cytokine transcription by binding to the HSE [128,129], thereby inhibiting the inflammatory response; this has been show to result in a marked reduction in the levels of pro-inflammatory cytokines produced by human monocytes in response to LPS stimulation, and can be induced both by hyperthermia and exogenous HSF1 treatment. While acting as an inhibitor for IL-1β and TNFα, HSF1 interaction with the BDNF HSE binding site resulted in the upregulation of BDNF in the hippocampus, and a neuronal cell culture [127]. When detailing the upregulation of BDNF in microglia in response to LIPUS, the mechanism of action was not investigated outside of the BDNF/TrkB positive feedback loop [81]. Consequently, the in vivo mechanism of BDNF upregulation with ultrasound treatment may be mediated by HSF1 and the HSR.
Downstream of HSF1, HSPs perform several neuroprotective functions which may aid in the resolution of chronic inflammation. HSP70 is a protein of particular interest due to its specific upregulation through HSF1 and its implications in numerous neurological diseases. Thought of as neuroprotective when acting intracellularly, the extracellular role of HSP70 is contentious, with evidence of both protective [126] and inflammatory functions [122]. HSP70 is capable of acting as an endogenous ligand for TLR4 [126] and TLR7 [130], through direct, or EV, release; however, the induced signalling pathways are not identical to those mediated by PAMPs. Upon HSP70-mediated TLR7 activation, a cell-membrane bound, rapid, transcription-independent signalling pathway is induced, which upregulates phagocytic activity via phosphatidylinositol-3-kinase (PI3K), an upstream Akt signalling molecule [82], and p38 mitogen-activated protein kinase (MAPK), a generally pro-inflammatory although context-dependent molecule [131] [130]. As such, while HSP70-TLR7 interaction increased the relative expression of TNFα compared to homeostasis in unstimulated macrophage cells [130], it is unclear whether a pro- or anti-inflammatory effect is induced, or how the signalling pathways will change in chronic inflammation, although increased phagocytosis is thought to be beneficial in neurodegenerative diseases [96,104].
Interpreting the consequences of HSP70 interactions with TLR7 necessitates additional context for the role of PRRs, as the signalling response bifurcates according to the activating ligand. The PI3K-Akt pathway is an essential component of both eNOS release and phosphorylation [132], which acts as a potent anti-inflammatory signal. In whole brain LIPUS treatment of a vascular dementia model, the blockade of eNOS was sufficient to abrogate the anti-inflammatory effect of LIPUS in mice [64]. Phosphorylated Akt is essential for eNOS activation, and acts as a downstream target for both the inducible and constitutive forms of HSP70; blockade of constitutive HSP70 with small interfering RNA resulted in a decrease in PI3K production in human umbilical vein endothelial cells in response to vascular endothelial growth factor (VEGF), while the knockdown of the inducible form reduced Akt phosphorylation independent of PI3K activity [132]. In both cases, the activation of eNOS was eliminated. Traditionally, the connection between HSPs and eNOS has canonically been linked to HSP90 [114], which associates with phosphorylated Akt, thereby preventing its inactivation, and allowing downstream eNOS activation. However, the requirement of HSP70 expression for eNOS activation in both in vitro models of VEGF administration, and in vivo mouse models of hind limb ischemia [132], potentially indicates a larger integration with the HSR. Cellular stress, and the HSR, has been shown to alter protein function at homeostasis [114], independent of transcriptional regulation. HSP70 affinity and release may be altered during periods of cellular stress, resulting in increased interactions between constitutive HSP70 and TLR7, and the subsequent activation of PI3K [130]. Similarly, inducible HSP70 may be necessary as a co-chaperone for HSP90 [133] in maintaining Akt phosphorylation during cellular stress. However, while the HSR and HSPs are necessary for the induction and facilitation of many anti-inflammatory signalling pathways, the functional ability to induce gene transcription in response to stress varies with age.
Aging and chronic inflammation are associated with cellular senescence, and the subsequent impaired ability to induce protective signalling pathways, such as the HSR, in response to stress signals. A key protein downregulated with aging and associated with cellular senescence, is the deacetylase sirtuin-1 (Sirt1) [134]; the role of Sirt1 as a transcriptional regulator is of interest in the context of chronic inflammation. Cellular senescence is characterized by an inability to respond to the environment and signalling molecules, limiting the ability to transcriptionally respond to external stimuli, and, in chronic inflammation, commonly resulting in a persistent pro-inflammatory phenotype [135]. In homeostasis, abundant Sirt1 acts as a transcriptional regulator, interacting with HSF1 and increasing its binding stability to the HSE of target genes by preventing its acetylation, allowing for rapid and sufficient induction of the HSR - in aging, reduced levels of Sirt1 concomitantly increase HSF1 acetylation, impairing the HSR, the subsequent induction of HSPs, phenotypic plasticity, and stress tolerance as a result [136]. While aging reduces HSP production regardless of sex [137], females are preferentially affected [138]. Estrogen signalling is reduced with age in females, and directly corresponds to eNOS production [66]; Sirt1 and phosphorylated eNOS act in a positive feedback loop, with eNOS activating Sirt1, and vice-versa [139], potentially explaining the preferential HSP decline in females as a function of estrogen signalling. Contextualizing Sirt1 as a component of the HSR necessitates the consideration of potential non-canonical HSP70 interactions, with TLR7, and in forming a stabilizing complex with HSP90 and phosphorylated Akt. Consequently, while previous studies have identified eNOS as the main regulator of the anti-inflammatory effect of ultrasound [64], a wider context emphasizes an overarching signalling pathway stemming from the HSR acting in a protective, and self-propagating fashion. While eNOS may be necessary, in a model of vascular dementia, the constituent signalling components (HSP70, PI3K, Akt, Sirt1) are also essential; the resultant effect on HSF-1 transcription, and upregulation of HSP70 indicates a closed loop starting with the foundational, canonical components of the HSR, which may be able to reduce cellular senescence, and ameliorate sex differences in chronic inflammation, and aging.
4.2 Heat shock response
A recurring theme in the immune system is the context dependent outcome of signalling pathways or stimuli; this carries over to the interaction between HSP70 and TLR4. Treatment of unstimulated macrophages with EV-HSP70 or exogenous HSP70 resulted in an increase in the release of pro-inflammatory cytokines, and NF-κB translocation, and acted as an adjuvant in the inflammatory response to a mycobacterial infection [122]. In this context, HSP70 contributes to a more aggressive inflammatory response of infected cells, improving pathogen clearance, and likely functions as a DAMP, activating unstimulated cells for phagocytosis. However, other experiments demonstrated HSP70 administration following LPS monocyte stimulation significantly inhibited TNFα production, while HSP70 pre-treatment before LPS stimulation prevented any increase in TNFα expression [126]. Consequently, while there are conflicting reports, HSP70 seems to inhibit or reduce inflammation. Mechanistically, HSP70 binds to TLR4 and induces the downstream activation of HSF1, which intracellularly inhibits pro-inflammatory cytokine release, thereby regulating excessive inflammation [126]; this is reflected in vivo, as HSP70 induction in sepsis, via hyperthermia, is associated with reduced mortality [140].
While often proposed as the main actors in the HSR, studies investigating hyperthermia after ablation of HSF1, and concomitantly HSP70, demonstrated the downregulation of pro-inflammatory cytokines [141], which was absent from non-heat shocked cells with constitutively active overexpression of HSF1 [142]. The implication is that the beneficial reduction in inflammation due to heat shock is independent of both HSF1 and induced HSP70, contrary to commonly proposed mechanisms of resolution; however, this conclusion might be too simplistic. In models of inflammation and infection, the HSR is activated as a protective mechanism, resulting in the upregulation of HSF1, and subsequent HSP70 transcription [126], the loss of which increases mortality in models of sepsis [140]. Exposure to heat shock, or exogenous HSP70 treatment, leads to HSF1 upregulation - potentially sensitized through a hypoxia inducible factor 1α (HIF1α)-mediated interaction [143] - as well as a rapid recruitment of constitutive heat shock element binding factor (CHBF) to the transcriptional complex [144], which is absent following sole stimulation with LPS [126]. Both HSF1 and CHBF interact with the HSE on the TNFα promoter, thereby inhibiting its transcription. The similar anti-inflammatory function of CHBF provides a potential mechanism for the apparent HSF1-independent downregulation of pro-inflammatory cytokines. Despite this, the protective influence of HSF1 on the HSR is still apparent, but functional benefits may only be discernible under physiologic stressors, as the affinity and cellular behaviour of HSPs can be altered by shear stress, and VEGF [114]. This bears emphasis, as prior research has described a HSP70-mediated cessation of NF-κB inhibition, through the stabilization of IKK [145], preventing its insolubilization during hyperthermia [146] - while this would be detrimental in chronic inflammation, the model described returned IKK activity to baseline levels in response to heat shock [145], but not DAMP or PAMP stimulation. NF-κB plays an important role in cell homeostasis independent of inflammatory function as well [147], and HSP70 stabilizes this pathway in response to heat shock, while accomplishing an inverse function in inflammation [148].
Further investigation of the CHBF has revealed identical immunological function to the Ku autoantigen [149], a signalling molecule integral to the repair of double strand DNA (dsDNA) breaks, composed of Ku70/80 subunits. The presence of Ku70/80 in heat shock related pathways has generally been considered a suppressor of the HSR, with overexpression of the Ku70 subunit directly inhibiting HSP70 induction [150], although this could be a non-physiological response to supra-endogenous levels of a specific subunit [150], as Ku70 ablation also inhibits HSP70 expression [151]; while seemingly contradictory, Ku70/80 fills a complex regulatory function, facilitating HIF1α- [144] and HSR- [152] induced gene transcription. Gene transcription is regulated by RNA polymerase II (Pol II), which transcribes mRNA precursors from the DNA template - in stimulus induced gene transcription, Pol II is paused after binding, primed to facilitate rapid transcription [153]. An activating stimulus, such as hyperthermia or hypoxia, facilitates the agglomeration of a signalling complex capable of releasing the transcriptional brake on Pol II, resulting in the rapid transcription of downstream targets [144,151-153]. Ku70/80 are key components in promoting stimulus-induced gene transcription, and are specifically required for HSP70 protein expression [151,152]; however, Ku70/80 overexpression to supra-physiological levels can result in transcriptional suppression through outcompeting other transcription factors for binding to DNA [154]. Regardless, the reparative function of the Ku autoantigen in dsDNA breaks, a source of inflammation induced by ionizing radiation [18], implies a protective function. As a competitor of HSF1 for HSE binding [126], as well as a facilitator of HSP70 transcription [151,152], the effective increase in CHBF levels following heat shock may result in the preferential targeting of pro-inflammatory cytokines in inflammation, as well as facilitating the HSF1-mediated induction of HSP70, resulting in the dual upregulation of HSF1 and CHBF in response to exogenous HSP70 stimulation [126], while acting as an inducible HSP70 suppressor in homeostatic conditions. Contextually, the HSR is composed of multiple HSE binding factors capable of inhibiting pro-inflammatory cytokine transcription, and is not solely dependent on HSF1; furthermore, there appears to be a distinct difference between thermal activation of the HSR, accompanied by extracellular HSP70 release, and pathogen-induced stress, through the activation of different transcription factors [155]. Several functional changes likely occur during the non-pathogen induced activation of the HSR, with variations in the concentration, downstream effect, and release of HSPs, as well as HSE binding factors, constituting an anti-inflammatory response. As such, the HSR and HSPs can holistically be described as homeostatic regulators.
Intracellularly, HSP70 mitigates inflammation downstream of HSF1. The inflammasome is an inflammatory intracellular signalling complex implicated in a number of neurodegenerative diseases [156]; upon activation of PRRs by PAMP/DAMPs, NLRP3 transcription is increased [53] - a key component of the well characterized NLRP3 inflammasome. Further endogenous signalling results in the agglomeration of NLRP3, the apoptosis-associated speck-like protein containing a caspase recruitment domain adaptor protein, and pro-caspase-1, resulting in IL-1β activation and the induction of NLRP3 inflammasome activation in surrounding cells [53]. Whole body heat shock of 42 oC for one hour induced HSP70 overexpression in a mouse model, which was found to bind to NLRP3, inhibiting inflammasome formation and IL-1β production, independent of HSF1 [53]; of note, the hyperthermic parameters that were used upregulated HSP70, but no other HSPs investigated. In cardiac tissue HSP70 is predominantly induced at higher temperatures [157] while HSP90 has a lower threshold of activation [158], indicating HSP expression is both tissue and temperature specific - this holds within CNS cells [159]. A downstream signalling molecule induced by inflammasome activation [160], as well as TNFα signalling [51], is the endogenous TLR4 ligand HMGB1, which demonstrated an inverse relationship in expression to HSP70 in acute pancreatitis, with high HMGB1 and low HSP70 expression being associated with mortality [161]; HMGB1 release is abrogated following hyperthermia [125], potentially acting through HSP70/HSF1-mediated inflammasome and TNFα inhibition. Hyperthermia has further been shown to induce nuclear factor erythroid 2-related factor 2 (NRF2) [162], downstream of immune-responsive gene 1 (IRG1) and itaconate [163], a factor associated with reduced caspase-3 activation and neuroinflammation in a model of spinal cord injury [163]. NRF2 is a transcription factor capable of cross talk with HSF1 [164], and IRG1 and itaconate are both potent inhibitors of IL-1β and NLRP3 inflammasome activation [163]. While hyperthermia has to date only been associated with increased NRF2 in this pathway, high intensity ultrasound (HUS) has been demonstrated to induce IRG1 and itaconate production as well [165], potentially indicating a common signalling pathway responsible for NRF2 upregulation.
NRF2, aside from its association with the IRG1 and itaconate pathway, is capable of accelerating and changing the dynamics of the potent anti-inflammatory feedback regulator TNFα-induced protein 3 (TNFAIP3/A20). TNFAIP3 is a negative feedback regulator of NF-κB signalling, which contributes to the dynamic, oscillatory expression pattern of NF-κB, inhibiting IKK activation and downstream NF-κB nuclear translocation [166]. While often characterized in binary terms, NF-κB activation is modulated by a complex series of signalling pathways, and underlying self-regulated inhibitory molecules, which induce a damped, periodic behaviour in NF-κB nuclear translocation at both the single cell [167,168], and population level [169]. Conceptually, the dynamics of NF-κB transcription enables the encoding of gene expression in a context-dependent fashion, facilitating pleiotropic downstream effects. In the context of the HSR, TNFAIP3 has demonstrated temperature sensitive expression in response to a TNFα inflammatory challenge in SK-N-AS neuroblastoma cells, with an earlier upregulation in cells incubated at 40 oC compared to 37 oC [168]. Increased TNFAIP3 expression was correlated with an early upregulation of a number of NF-κB related genes, associated with both the immune system and the inflammatory response - a difference which was absent at later timepoints [168], indicating a time and state dependent alteration in inflammatory signalling. The acceleration in TNFa-induced TNFAIP3 expression in response to hyperthermia could be mediated by the upregulation of NRF2 downstream of the HSR. The TNFAIP3 promoter contains an antioxidant response element (ARE); NRF2 has been shown to bind to TNFAIP3's ARE, and subsequently upregulate its expression in bone marrow derived macrophages [170]. As an essential regulator of inflammation, perturbations in TNFAIP3 are associated with a number of autoimmune diseases [166], and its absence leads to mortality [171]. Consequently, the upregulation of TNFAIP3 has a potentially beneficial effect in treating inflammation and is downstream of pathways induced by the HSR. While attempts have been made to model the cross-talk between the HSR and NF-κB signalling pathways [167], largely predicated on the presumed inhibitory functions of HSFs and HSPs, it remains to be determined whether the assumptions and simplifications made hold true in chronic inflammation. Regardless, it is evident that the dynamics of signalling pathways are more complex than generally considered and are functionally capable of different downstream effects in response to the same stimulus; consequently, this may offer a partial explanation for the ability of the HSR to act as both an inflammation-promoting, and resolving mechanism depending on the cellular context.
Emphasizing the time and context dependent effect of hyperthermia, is the exacerbation of neural injury when hyperthermia is applied immediately following mild traumatic brain injury (TBI) in a murine model [123] - LIPUS, in contrast, has been demonstrated to reduce edema and improve therapeutic outcomes in both an acute and longitudinal timeframe [80]. Recently, EVs have received increased interest due to their implication as both a mechanism of disease and resolution, and may provide insight into the mechanistic immunological differences between LIPUS and hyperthermia - EV release is increased by both LIPUS [172] and hyperthermia [173], while EV contents are differentially modulated. FUS hyperthermia treatment of in vitro glioma cells induced a proteomic shift in EV contents, inducing a beneficial, pro-inflammatory, anti-cancer profile [173], similar to the immune adjuvant effect of acutely applied hyperthermia in a model of peripheral LPS infection [117]; this was accompanied by a decrease in major vault protein, which has been associated with sorting miRNAs for EV loading [174]. Alternately, bone marrow derived dendritic cells exposed to LIPUS exhibited an anti-inflammatory shift in EV miRNA contents [175], and subsequently reduced inflammation. Consequently, the context dependent effect of the HSR may manifest as changes in EV loading machinery, in addition to changing the dynamics and timing of certain immune signalling pathways. While there have been conflicting results on the pro- or anti-inflammatory effect of HSP70, whether exogenous or from EVs, these may be localized to the method of isolation, timing, and cellular origin or context when administered, as EV contents will impact downstream signalling. Regardless, the anti-inflammatory effect, and potential of HSPs is well established.
Beyond inhibiting NLRP3 inflammasome formation, HSP70 has a more fundamental effect on immune cell reactivity. Microglia require caspase-3 signalling for pro-inflammatory cytokine release [105], while caspase-9/caspase-3 activation induces apoptosis in neuronal cells [176]. Caspase-9, activated via the apoptotic protease-activating factor 1 (Apaf-1), is an endogenous activator of caspase-3, cleaving the pro-caspase form and subsequently inducing apoptosis [177]. Overexpression of HSP70 does not affect pro-caspase-9 transcription, but instead prevents its cytosolic activation, likely through interactions with Apaf-1, thereby inhibiting both caspase-9 and downstream capsase-3 signalling [9]. Apaf-1-mediated caspase activation is a constituent of the intrinsic apoptosis pathway, while caspase-mediated microglial reactivity is dependent on the extrinsic pathway, which is characterized by upstream activation of caspase-8 leading to pro-caspase-3 cleavage [105]; in a model of ischemic stroke, transgenic HSP70-overexpressing mice were found to have decreased caspase-8 activation [178], indicating the ability of HSP70 to inhibit both intrinsic and extrinsic pathways of apoptosis. Notably, the apoptotic pathways exhibit sex-driven differences in activation, with female mice exhibiting significantly more caspase-8 and capsase-3 activity following ischemic stroke, and reduced neurological deficits after administration of a pan-caspase inhibitor [110]; this effect was not observed in male mice. Additionally, in female rats, the ability to upregulate HSP70, and HSP27 in the brain following heat shock is reduced compared to their male counterparts [138], indicating a potentially reduced ability to abrogate caspase-3 mediated inflammation - this is consequential when considering that caspase-3 is implicated in CD200/CD200R1 dysregulation [99], a pathway which at homeostasis mitigates female biased sex-differences [109] in the immune response. Regardless, HSP70 is implicated in preventing neuronal apoptosis [9], and microglial activation, while potentially acting as a rectifier of immune-based sex differences.
Downstream of caspase signalling, hyperthermia-induced HSP70 in astrocytes exposed to oxygen-glucose deprivation interacts with JNK and p38, inhibiting their phosphorylation, subsequent nuclear translocation, and downstream inflammatory cytokine production - further, HSP70 prevents IκB phosphorylation, resulting in the continued sequestration of NF-κB in the cytoplasm [148]. Transcriptional changes confluent with hyperthermia included a reduction in MMP-9 mRNA expression [148], demonstrating a potential pathway leading to increased BBB stability, although hyperthermia is associated with increased acute BBB permeability [179]. The combination of both aging and inflammation can result in lymphocyte infiltration in the CNS, and is associated with detrimental outcomes; interestingly, HSPs can act as antigens for T cells, and modulate their response [180]. As HSPs are phylogenetically conserved, from bacteria to mammals, there is a self-reactivity of T cells to endogenous HSPs, likely mediated through cross-recognition of T cells sensitized to bacterial HSPs [180]. In disease, HSP-reactive T cells exhibit an immunoregulatory phenotype, and contribute to inflammation resolution [180]. Stressed cells induce HSPs, which can be processed and presented as antigens via MHC I/II on APCs [180], which act as a signal of cellular distress. Induced in the later stages of inflammation, or infection, self-HSP-reactive T cells act in an ameliorating fashion, recognizing cells stressed by excessive inflammation, and correspondingly secreting anti-inflammatory cytokines to promote resolution [181]. Hyperthermia could act as an inflammatory shutdown mechanism, through the adaptive immune response, by the induction of the HSR, and subsequently this regulatory T cell response.
In disease, HSP70 and HSP90 are two of the more studied HSPs, although there remains interest across other HSP family members, potentially displaying anti-inflammatory function. Of note, HSP27 is a small intracellular HSP which has been shown to improve cognitive function in mouse models of AD, without altering caspase-3 activation [182], and is thought to inhibit JNK phosphorylation [183] - regardless of the mechanism, HSP27 is thought to be protective. Similarly, HSP60 has been shown to induce regulatory T cells, and mitigate chronic inflammatory disease pathology [184,185], and can further act as a ligand for the TREM2 receptor [185], which induces a phagocytic, anti-inflammatory phenotype in myeloid cells. Induction of HSP release is a constitutive process, and is endogenously activated in the febrile range; ultrasound-mediated hyperthermia has been demonstrated to induce HSP transcription at lower temperatures than solely temperature-based methods [186]. For example, a similar adjuvant effect was observed with electric stimulation (ES)-mediated hyperthermia, compared to ES alone, in a model of chronic prostatitis [187].
Likewise, additional downstream targets of the HSR are implicated in its anti-inflammatory effects. Hyperthermia-mediated resolution of inflammation in a model of chronic prostatitis was not only accompanied by the upregulation of HSP70, but also an increase in suppressor of cytokine signalling 3 (SOCS3) expression [187]. The SOCS protein family are potent inhibitors of pro-inflammatory cytokine feedback signalling, especially following TLR activation [188]. Both SOCS1 and SOCS3 are upregulated downstream of the HSR, with HSP70 inducing SOCS1 expression through TLR4 activation [189], targeting TRAF6, MAPK, and the p65 subunit of NF-κB [188], which are implicated in a number of pro-inflammatory pathways. Additionally, the HSR-related upregulation of SOCS1 may explain the previously reported reduction in TLR4 expression [79], and subunit association [190] following ultrasound, through the direct degradation of the MAL adaptor protein [191], essential for MyD88 dependent TLR2/4 signalling. In LIPUS, this mechanism has previously been attributed to mechanical perturbation of the cell membrane, although this has not been verified [190]. Preventing degradation of IκB, and subsequent NF-κB nuclear translocation, has also been attributed to phosphorylated SOCS3 activity, which shields the phosphorylation sites of IκB from IKK [192]. Consequently, both SOCS1 and SOCS3 act as potent inhibitors of pro-inflammatory cytokine signalling in response to stress and are upregulated as part of the HSR downstream of HSP70. Colloquially, the HSR is often simplified and attributed to the upregulation of HSF1 and its target genes in response to stress, especially HSP70; however, it is clear that the HSR indirectly upregulates a number of signalling pathways (Figure 3), initiating a protective cascade, while modulating intracellular protein function depending on the stressor, and phenotypic state.
Comparison of the observed anti-inflammatory effects of LIPUS, with proposed HSR induced anti-inflammatory signalling pathways as an explanatory mechanism. (A) Experimentally observed changes in key signalling pathways proposed as the mechanisms behind LIPUS mediated resolution of inflammation. (B) Signalling pathways upregulated during the HSR, and mild hyperthermia, capable of inhibiting inflammasome formation, and downregulating caspase-3 activation (left). Under inflammatory conditions, CD200/CD200R signalling can become dysregulated, with caspase-3 cleaving RasGAP, leading to the differential inhibition of pAkt, while losing the ability to inhibit ERK activation. Microglial activation is dependent on the caspase-8/caspase-3 pathway, allowing HSP70 to both inhibit activation, and restore homeostatic CD200/CD200R signalling (middle). HSP70 also inhibits the neuronal apoptosis pathway (right). (C) In addition to the proposed LIPUS mediated inhibition of JNK phosphorylation, and NF-kB activation, through pAkt, HSP27 and HSP70 inhibit pJNK, while HSP70 additionally prevents p38 MAPK phosphorylation, and IKK mediated degradation of IkB. (D) HSP70 has been described to bind to TLR4, upregulating SOCS1/3 which directly inhibits MAL and subsequent MyD88 TLR4 signalling complex formation, and pro-inflammatory cytokine production. Additionally, HSP70 and TLR4 interactions are associated with increased HSF1, and CHBF transcription, both of which are capable of binding to the heat shock elements of target genes, downregulating pro-inflammatory cytokines, while upregulating neurotrophic factors. Crosstalk between stress response pathways, such as the hypoxia and heat stress responses, generates a compensatory, symbiotic cascade of anti-inflammatory signalling pathways, with CHBF, HIF1a, and HSF1 facilitating mutual gene expression. TLR7 and HSP70 have been shown to interact, and activate the PI3k/pAkt pathway, which is implicated in stabilizing HSF1 and upregulating the HSR. Through extracellular vesicles, and direct protein secretion, adjacent cells can communicate and influence the cellular environment, and activation states. In studies of LIPUS, eNOS was characterized as an essential component for an anti-inflammatory effect, and was correlated with the upregulation of BDNF, while a mechanism was not proposed. Pharmaceutical upregulation of eNOS is predicated on a complex of phosphorylated Akt, and HSP90, with potential co-chaperone behaviour of HSP70, eventually leading to NO release. NO stimulates the AMPK/mTOR/CREB/BDNF pathway, potentially explaining the observed relationship between eNOS and BDNF release. Simultaneously, NO upregulates Sirt1, a transcriptional regulator necessary for sufficient induction of the HSR.
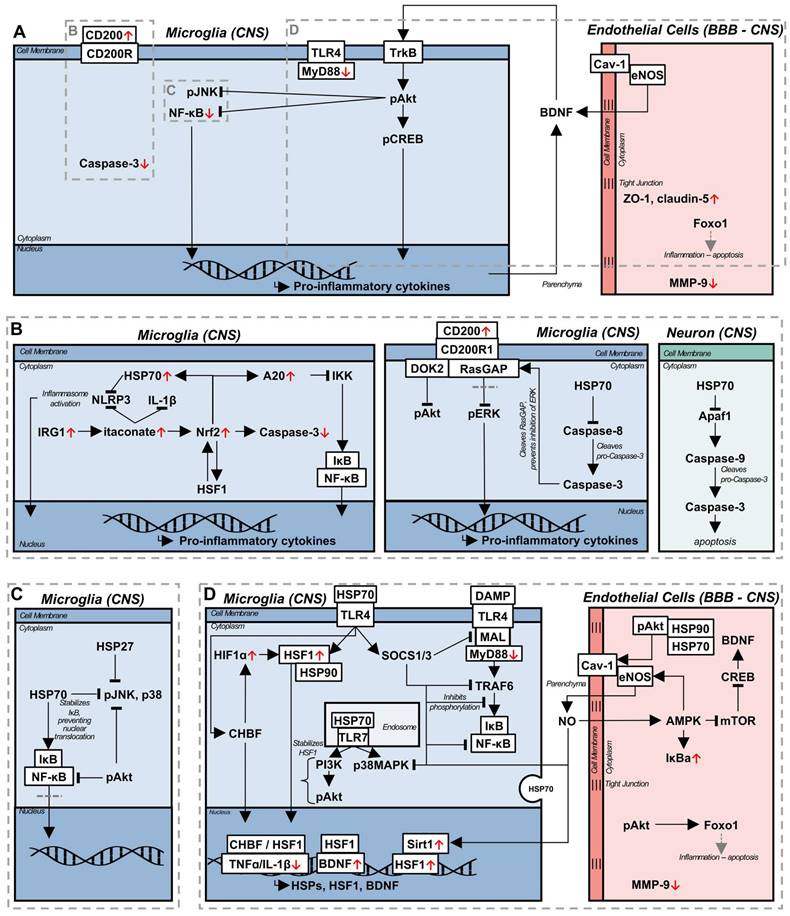
5. Summary
Investigation into ultrasound as a treatment for chronic neuroinflammation has raised interest in recent years, after demonstrated successes in accelerating healing in wounds, bone fractures, and reducing inflammation in peripheral tissues [7]. Promising results peripherally have been followed by the application of LIPUS in CNS models of chronic inflammation and neurodegenerative disease with similarly beneficial results, albeit using heterogeneous ultrasound parameters. The success of LIPUS has been attributed to the upregulation of neurotrophic factors, such as BDNF and GDNF, the upregulation of inhibitory signalling pathways (CD200/CD200R1), increased BBB stability, increased eNOS activation, and the inhibition of pro-inflammatory cytokine release. However, the broad spectrum of described ameliorating pathways established with heterogeneous experimental parameters indicates a potential common upstream pathway mediating these changes.
The effect of hyperthermia has been largely disregarded in the translation of thermal therapy from peripheral application to the CNS. While mild hyperthermia has an agonistic effect on the immune response in acute inflammation, it appears to act as a regulatory factor in chronic inflammation, mitigating the release of pro-inflammatory cytokines and promoting resolution. The HSR constitutes the upstream mechanism of this effect, inducing a protective, anti-inflammatory environment in response to stress, upregulating HSF1, and HSPs. Evaluation of common signalling pathways between hyperthermia and LIPUS demonstrate considerable overlap, with the HSR implicated in the upregulation of neurotrophic factors, BBB stability, inhibition of pro-inflammatory cytokine release, and inflammasome disruption. Furthermore, in addition to the upregulation of eNOS production, the HSR has the capacity to rescue inhibitory CD200/CD200R1 signalling, integral in mediating sex differences in inflammation, from a chronic inflammation induced change of function to potentiating the inflammatory response. With this contextual information, the observation of HSP upregulation in models of both wound healing [97] and Alzheimer's disease [64] following LIPUS treatment indicates a potentially confluent integration of the HSR in LIPUS-mediated inflammation resolution - as HSP family and function are mediated by the amount of cellular stress induced, seeming discrepancies in downstream outcomes are easily reconciled.
As HSPs are implicated in immune regulation, the prolonged nature of chronic inflammation potentially indicates a dysregulated HSR, and an upregulation of HSPs incapable of ameliorating the inflammatory response. In sterile inflammation, HSPs may act as immunoregulatory factors via both the innate immune system, through disruption of pro-inflammatory cytokine feedback loops and inflammasome activation, and the adaptive immune response, through the induction of regulatory T cells - if the upregulation of the HSR is impaired or disrupted, inflammation may become chronic. Endogenous stimulation of the HSR thereby constitutes a promising inlet to a cascade of beneficial downstream therapeutic pathways in chronic inflammation, while avoiding potential undesirable side effects of pharmacologically inducing or inhibiting individual signalling molecules.
In describing the potential of the HSR as a common regulator of the beneficial immunological response to both LIPUS and hyperthermia, the mechanisms behind the contextual effect of hyperthermia have not been explored. While there is considerable homology between LIPUS and hyperthermia, only hyperthermia has been recorded as capable of agonizing the immune response in acute inflammation, or infection - to date, there is a paucity of data attempting to explain this phenomenon. While changes in signalling dynamics have been reported in response to hyperthermia, and require further investigation, the immunological context is also influenced by secreted EVs. As a proposed biomarker of disease, and a potential therapeutic target, EV contents give a snapshot of the cellular environment - interestingly, their contents and loading complexes are influenced by both LIPUS, and hyperthermia. While acknowledging the gaps in the literature, hyperthermia and LIPUS may differentially affect EV loading complexes, and EV contents as a result; this may explain an aspect of the temporal plasticity of the immune response to hyperthermia and should constitute a future avenue of investigation.
Similarly, tissue oxygenation levels constitute another area of future research, as oxygenation can be affected by inflammation [193], hyperthermia [194], and LIPUS [64]. Contextually, hypoxia can form a positive feedback loop with inflammation, through the production of reactive oxygen species and DAMPs from chronic, sustained damage [193] - cellular survival is dependent on the mutualistic activation of both the hypoxia, and heat shock, response pathways. HIF1α is the primary regulator of the hypoxia response [195], responsible for altering cellular metabolism, oxygen consumption, and is essential in upregulating HSF1 and downstream HSPs during both hypoxia [195], and hyperthermia [143]. HIF1α is abundant at homeostasis, but normoxic conditions leads to its degradation, which can be abrogated by the induction of HSP90 [196], potentially explaining its upregulation following LIPUS [197], and hyperthermia. Holistically, the HSR and the hypoxia response are linked in a bidirectional modulatory relationship, demonstrated in their activity following heat acclimation in humans [198], and co-interactions. Additionally, hyperthermia and LIPUS are potentially capable of increasing tissue oxygenation, whether through transient, local changes [194], or long term eNOS mediated increase in CBF [64]; however, with respect to inflammation, it is unlikely that transient fluctuations in tissue oxygenation exert an immunological effect [193]. Regardless, while outside the scope of this review, the bioeffects of hyperthermia and ultrasound - and the cross-regulation of stress response pathways - in the context of oxygenation remain of interest in oncology and require further study in inflammation.
Ultrasound-mediated hyperthermia has been demonstrated to induce greater HSP transcription at lower temperatures than hyperthermia alone, indicating an amplifying effect of the costimulatory thermal and mechanical stress. This is essential in the context of aging, where the HSR, and downstream HSP transcription is compromised [137] - ultrasound is capable of initiating the HSR, and potentially improving transcriptional efficacy lost in aging through the upregulation of the metabolic protein Sirt1 [136,199], although the specific mechanisms are unclear. Additionally, ultrasound-mediated febrile range hyperthermia is largely understudied in chronic inflammation, especially with regard to the CNS, despite the inherent benefits associated with the technique. By using both focused ultrasound, and MR thermometry, ultrasound-mediated hyperthermia can be targeted both functionally, and anatomically, while allowing for intra-treatment monitoring. This is important, as some inflammatory and neurodegenerative diseases are localized, requiring the use of spatial as well as functional landmarks for targeting; MRgFUS neurosurgical ablation has resulted in the amelioration of a potentially inflammatory abnormality in the human brain, by an as yet unclear mechanism, necessitating further investigation. Combining both ultrasound and febrile range hyperthermia allows for the induction of the HSR, as well as a concomitant broad spectrum of HSPs and anti-inflammatory pathways, constituting a promising potential therapeutic modality.
Abbreviations
CNS: central nervous system; BBB: blood brain barrier; HSPs: heat shock proteins; LIPUS: low intensity pulsed ultrasound; HSR: heat shock response; MRgFUS: magnetic resonance guided focused ultrasound; ET: Essential tremor; HIFU: high intensity focused ultrasound; SRS: stereotactic radiosurgery; T2w: T2-weighted; MR: magnetic resonance; MHC: major histocompatibility complex; APC: antigen presenting cell; CD: cluster of differentiation; DC: dendritic cell; EV: extracellular vesicle; miRNA: microRNA; IL: interleukin; DAMP: damage associated molecular pattern; PAMP: pathogen associated molecular pattern; PRR: pattern recognition receptor; TLR: toll like receptor; DNA: deoxyribonucleic acid; RNA: ribonucleic acid; TIR: toll/interleukin-1 receptor; MyD88: myeloid differentiation primary response 88; MAL: MyD88 adaptor like; IRAK: interleukin-1 receptor-associated kinase; TRAF: tumour necrosis factor receptor-associated factor; JNK: c-Jun N-terminal kinase; NF-κB: nuclear factor kappa B; IκB: inhibitor of NF-κB; IKK: IκB kinase; TNF: tumour necrosis factor; IRF: interferon regulatory factor; IFN: interferon; TRIF: TIR domain containing adaptor-inducing interferon beta; LPS: lipopolysaccharide; SN: substantia nigra; TNFR: TNFα receptor; mRNA: messenger RNA; CD200R: CD200 receptor; Aβ: amyloid beta; IFNAR: type I IFN receptor; HMGB1: high mobility group box protein 1; AD: Alzheimer's disease; CSF: cerebral spinal fluid; ROS: reactive oxygen species; NOD: nucleotide-binding oligomerization domain; NLRP3: NOD-like receptor family pyrin domain containing 3; FD: female derived; TREM2: triggering receptor expressed on myeloid-2; MD: male derived; eNOS: endothelial nitric oxide synthase; CBF: cerebral blood flow; E2: 17β-estradiol; ERα: estrogen receptor α; BDNF: brain derived neurotrophic factor; ISPTA: spatial peak temporal average intensity; fR: pulse repetition frequency; CREB: cyclic adenosine monophosphate response element-binding protein; Akt: protein kinase B; TrkB: tyrosine protein kinase B; TBI: traumatic brain injury; MMP-9: metalloproteinase 9; FOXO1: forkhead box protein O1; 6-OHDA: 6-hydroxydopamine; PD: Parkinson's disease; GDNF: glial cell line-derived neurotrophic factor; VaD: vascular dementia; Cav-1: caveolin-1; HSP: heat shock protein; EAE: experimental autoimmune encephalomyelitis; iNOS: inducible nitric oxide synthase; AMP: adenosine monophosphate; AMPK: AMP activated protein kinase; mTOR: mammalian target of rapamycin; ERK: extracellular signal-regulated kinase; PBMC: peripheral blood mononuclear cell; Dok2: docking protein 2; RasGAP: Ras GTPase activating protein; IP-10: IFNγ-inducible protein 10; MCP-1: monocyte chemoattractant protein-1; HSF1: heat shock factor 1; HSE: heat shock element; PI3K: phosphatidylinositol-3-kinase; MAPK: mitogen-activated protein kinase; Sirt1: sirtuin-1; HIF1α: hypoxia inducible factor 1α; CHBF: constitutive heat shock element binding factor; dsDNA: double strand DNA; Pol II: RNA polymerase II; NRF2: nuclear factor erythroid 2-related factor 2; IRG1: immune responsive gene 1; HUS: high intensity ultrasound; TNFAIP3/A20: TNFα-induced protein 3; ARE: antioxidant response element; Apaf-1: apoptotic protease-activating factor 1; ES: electric stimulation; SOCS: suppressor of cytokine signalling 3.
Competing Interests
The authors have declared that no competing interest exists.
References
1. McMahon D, Bendayan R, Hynynen K. Acute effects of focused ultrasound-induced increases in blood-brain barrier permeability on rat microvascular transcriptome. Sci Rep. 2017;7:45657
2. Tovar-y-Romo LB, Penagos-Puig A, Ramírez-Jarquín JO. Endogenous recovery after brain damage: molecular mechanisms that balance neuronal life/death fate. J Neurochem. 2016;136:13-27
3. McInnes IB, Gravallese EM. Immune-mediated inflammatory disease therapeutics: past, present and future. Nat Rev Immunol. 2021;21:680-6
4. Song K-H, Harvey BK, Borden MA. State-of-the-art of microbubble-assisted blood-brain barrier disruption. Theranostics. 2018;8:4393-408
5. Lipsman N, Schwartz ML, Huang Y, Lee L, Sankar T, Chapman M. et al. MR-guided focused ultrasound thalamotomy for essential tremor: a proof-of-concept study. Lancet Neurol. 2013;12:462-8
6. Zhou Y-F. High intensity focused ultrasound in clinical tumor ablation. World J Clin Oncol. 2011;2:8-27
7. Xu M, Wang L, Wu S, Dong Y, Chen X, Wang S. et al. Review on experimental study and clinical application of low-intensity pulsed ultrasound in inflammation. Quant Imaging Med Surg. 2021;11:443-62
8. Singh IS, Hasday JD. Fever, hyperthermia and the heat shock response. Int J Hyperthermia. 2013;29:423-35
9. Sabirzhanov B, Stoica BA, Hanscom M, Piao C-S, Faden AI. Over-expression of HSP70 attenuates caspase-dependent and caspase independent pathways and inhibits neuronal apoptosis. J Neurochem. 2012;123:542-54
10. Ishihara Y, Calderon A, Watanabe H, Okamoto K, Suzuki Y, Kuroda K. et al. A precise and fast temperature mapping using water proton chemical shift. Magn Reson Med. 1995;34:814-23
11. Hu Z, Yang XY, Liu Y, Morse MA, Lyerly HK, Clay TM. et al. Release of endogenous danger signals from HIFU-treated tumor cells and their stimulatory effects on APCs. Biochem Biophys Res Commun. 2005;335:124-31
12. Cirincione R, Di Maggio FM, Forte GI, Minafra L, Bravatà V, Castiglia L. et al. High-intensity focused ultrasound- and radiation therapy-induced immuno-modulation: comparison and potential opportunities. Ultrasound Med Biol. 2017;43:398-411
13. Mazerolle EL, Seasons GM, Warwaruk-Rogers R, Romo P, Nordal R, Sevick RJ. et al. Focused ultrasound resolves persistent radiosurgery related change in a patient with tremor. Radiol Case Rep. 2019;14:1233-6
14. Ohye C, Higuchi Y, Shibazaki T, Hashimoto T, Koyama T, Hirai T. et al. Gamma knife thalamotomy for parkinson disease and essential tremor: a prospective multicenter study. Neurosurgery. 2012;70:526-36
15. Elaimy AL, Demakas JJ, Arthurs BJ, Cooke BS, Fairbanks RK, Lamoreaux WT. et al. Gamma knife radiosurgery for essential tremor: a case report and review of the literature. World J Surg Oncol. 2010;8:20
16. Steiner L, Forster D, Leksell L, Meyerson BA, Boëthius J. Gammathalamotomy in intractable pain. Acta Neurochir (Wien). 1980;52:173-84
17. Alomari A, Rauch PJ, Orsaria M, Minja FJ, Chiang VL, Vortmeyer AO. Radiologic and histologic consequences of radiosurgery for brain tumors. J Neurooncol. 2014;117:33-42
18. Sharma NK, Sharma R, Mathur D, Sharad S, Minhas G, Bhatia K. et al. Role of ionizing radiation in neurodegenerative diseases. Front Aging Neurosci [Internet]. 2018 [cited 31 January 2022]; 10. Available at: https://www.frontiersin.org/article/10.3389/fnagi. 2018 00134
19. Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest. 2012;122:1164-71
20. Peferoen L, Kipp M, Valk P, Noort JM, Amor S. Oligodendrocyte-microglia cross-talk in the central nervous system. Immunology. 2014;141:302-13
21. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S. et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841-5
22. Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. 2018;18:225-42
23. He Y, Taylor N, Yao X, Bhattacharya A. Mouse primary microglia respond differently to LPS and poly(I:C) in vitro. Sci Rep. 2021;11:10447
24. Jessen KR. Glial cells. Int J Biochem Cell Biol. 2004;36:1861-7
25. Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG. et al. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32:6391-410
26. Escartin C, Galea E, Lakatos A, O'Callaghan JP, Petzold GC, Serrano-Pozo A. et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci. 2021;24:312-25
27. Chaplin DD. Overview of the immune response. J Allergy Clin Immunol. 2010;125:S3-23
28. Nitta T, Murata S, Ueno T, Tanaka K, Takahama Y. Thymic microenvironments for T-cell repertoire formation. Adv Immunol. 2008;99:59-94
29. Rostami J, Fotaki G, Sirois J, Mzezewa R, Bergström J, Essand M. et al. Astrocytes have the capacity to act as antigen-presenting cells in the Parkinson's disease brain. J Neuroinflammation. 2020;17:119
30. Liu Y, Bando Y, Vargas-Lowy D, Elyaman W, Khoury SJ, Huang T. et al. CD200R1 agonist attenuates mechanisms of chronic disease in a murine model of multiple sclerosis. J Neurosci. 2010;30:2025-38
31. Upadhya R, Zingg W, Shetty S, Shetty AK. Astrocyte-derived extracellular vesicles: neuroreparative properties and role in the pathogenesis of neurodegenerative disorders. J Control Release Off J Control Release Soc. 2020;323:225-39
32. Dickens AM, Tovar-y-Romo LB, Yoo S-W, Trout AL, Bae M, Kanmogne M. et al. Astrocyte-shed extracellular vesicles regulate the peripheral leukocyte response to inflammatory brain lesions. Sci Signal. 2017;10:eaai7696
33. Zhao S, Sheng S, Wang Y, Ding L, Xu X, Xia X. et al. Astrocyte-derived extracellular vesicles: A double-edged sword in central nervous system disorders. Neurosci Biobehav Rev. 2021;125:148-59
34. Xie X, Luo X, Liu N, Li X, Lou F, Zheng Y. et al. Monocytes, microglia, and CD200-CD200R1 signaling are essential in the transmission of inflammation from the periphery to the central nervous system. J Neurochem. 2017;141:222-35
35. Frederiksen HR, Haukedal H, Freude K. Cell type specific expression of toll-like receptors in human brains and implications in Alzheimer's disease. BioMed Res Int. 2019;2019:7420189
36. Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1-14
37. Takeda K, Akira S. TLR signaling pathways. Semin Immunol. 2004;16:3-9
38. Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529-31
39. Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong J-S. et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453-62
40. Wang L, Gong X, Liu Y, Du T, Zhang Z, Zhang T. et al. CD200 maintains the region-specific phenotype of microglia in the midbrain and its role in Parkinson's disease. Glia. 2020;68:1874-90
41. Alfonso-Loeches S, Pascual-Lucas M, Blanco AM, Sanchez-Vera I, Guerri C. Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. J Neurosci. 2010;30:8285-95
42. Fiebich BL, Batista CRA, Saliba SW, Yousif NM, de Oliveira ACP. Role of microglia TLRs in neurodegeneration. Front Cell Neurosci [Internet]. 2018 [cited 15 June 2022]; 12. Available at: https://www.frontiersin.org/article/10.3389/fncel. 2018 00329
43. Roy ER, Wang B, Wan Y, Chiu G, Cole A, Yin Z. et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J Clin Invest. 130: 1912-30.
44. Coleman LG, Zou J, Crews FT. Microglial-derived miRNA let-7 and HMGB1 contribute to ethanol-induced neurotoxicity via TLR7. J Neuroinflammation. 2017;14:22
45. Derkow K, Rössling R, Schipke C, Krüger C, Bauer J, Fähling M. et al. Distinct expression of the neurotoxic microRNA family let-7 in the cerebrospinal fluid of patients with Alzheimer's disease. PLOS ONE. 2018;13:e0200602
46. Ge X, Han Z, Chen F, Wang H, Zhang B, Jiang R. et al. miR-21 alleviates secondary blood-brain barrier damage after traumatic brain injury in rats. Brain Res. 2015;1603:150-7
47. Li X, Lin Z, Wang L, Liu Q, Cao Z, Huang Z. et al. RNA-Seq analyses of the role of miR-21 in acute pancreatitis. Cell Physiol Biochem. 2018;51:2198-211
48. Bayraktar R, Bertilaccio MTS, Calin GA. The interaction between two worlds: microRNAs and Toll-like receptors. Front Immunol [Internet]. 2019 [cited 15 June 2022]; 10. Available at: https://www.frontiersin.org/article/10.3389/fimmu. 2019 01053
49. Petes C, Odoardi N, Gee K. The toll for trafficking: Toll-like receptor 7 delivery to the endosome. Front Immunol [Internet]. 2017 [cited 28 June 2022]; 8. Available at: https://www.frontiersin.org/article/10.3389/fimmu. 2017 01075
50. Lavieri R, Piccioli P, Carta S, Delfino L, Castellani P, Rubartelli A. TLR costimulation causes oxidative stress with unbalance of proinflammatory and anti-inflammatory cytokine production. J Immunol. 2014;192:5373-81
51. Yang WS, Han NJ, Kim JJ, Lee MJ, Park S-K. TNF-α activates high-mobility group box 1 - Toll-like receptor 4 signaling pathway in human aortic endothelial cells. Cell Physiol Biochem. 2016;38:2139-51
52. Yarilina A, Park-Min K-H, Antoniv T, Hu X, Ivashkiv LB. TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nat Immunol. 2008;9:378-87
53. Martine P, Chevriaux A, Derangère V, Apetoh L, Garrido C, Ghiringhelli F. et al. HSP70 is a negative regulator of NLRP3 inflammasome activation. Cell Death Dis. 2019;10:256
54. Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol. 2016;16:626-38
55. Murtaj V, Belloli S, Di Grigoli G, Pannese M, Ballarini E, Rodriguez-Menendez V. et al. Age and sex influence the neuro-inflammatory response to a peripheral acute LPS challenge. Front Aging Neurosci. 2019;11:299
56. Wang X-L, Li L. Circadian clock regulates inflammation and the development of neurodegeneration. Front Cell Infect Microbiol [Internet]. 2021 [cited 14 September 2022]; 11. Available at: https://www.frontiersin.org/articles/10.3389/fcimb. 2021 696554
57. Lynch MA. Exploring sex-related differences in microglia may be a game-changer in precision medicine. Front Aging Neurosci [Internet]. 2022 [cited 26 July 2022]; 14. Available at: https://www.frontiersin.org/articles/10.3389/fnagi. 2022 868448
58. Villa A, Gelosa P, Castiglioni L, Cimino M, Rizzi N, Pepe G. et al. Sex-specific features of microglia from adult mice. Cell Rep. 2018;23:3501-11
59. Spiering AE, de Vries TJ. Why females do better: The X chromosomal TLR7 gene-dose effect in COVID-19. Front Immunol [Internet]. 2021 [cited 26 July 2022]; 12. Available at: https://www.frontiersin.org/articles/10.3389/fimmu. 2021 756262
60. Spence RD, Hamby ME, Umeda E, Itoh N, Du S, Wisdom AJ. et al. Neuroprotection mediated through estrogen receptor-α in astrocytes. Proc Natl Acad Sci U S A. 2011;108:8867-72
61. Unger MS, Schernthaner P, Marschallinger J, Mrowetz H, Aigner L. Microglia prevent peripheral immune cell invasion and promote an anti-inflammatory environment in the brain of APP-PS1 transgenic mice. J Neuroinflammation. 2018;15:274
62. Faraci FM, Heistad DD. Regulation of the cerebral circulation: role of endothelium and potassium channels. Physiol Rev. 1998;78:53-97
63. Koedel U, Paul R, Winkler F, Kastenbauer S, Huang PL, Pfister HW. Lack of endothelial nitric oxide synthase aggravates murine pneumococcal meningitis. J Neuropathol Exp Neurol. 2001;60:1041-50
64. Eguchi K, Shindo T, Ito K, Ogata T, Kurosawa R, Kagaya Y. et al. Whole-brain low-intensity pulsed ultrasound therapy markedly improves cognitive dysfunctions in mouse models of dementia - Crucial roles of endothelial nitric oxide synthase. Brain Stimul Basic Transl Clin Res Neuromodulation. 2018;11:959-73
65. Cattaneo MG, Vanetti C, Decimo I, Di Chio M, Martano G, Garrone G. et al. Sex-specific eNOS activity and function in human endothelial cells. Sci Rep. 2017;7:9612
66. McNeill AM, Zhang C, Stanczyk FZ, Duckles SP, Krause DN. Estrogen increases endothelial nitric oxide synthase via estrogen receptors in rat cerebral blood vessels. Stroke. 2002;33:1685-91
67. Geary GG, McNeill AM, Ospina JA, Krause DN, Korach KS, Duckles SP. Selected contribution: cerebrovascular nos and cyclooxygenase are unaffected by estrogen in mice lacking estrogen receptor-alpha. J Appl Physiol Bethesda Md 1985. 2001;91:2391-9 discussion 2389-2390
68. Stanhewicz AE, Wenner MM, Stachenfeld NS. Sex differences in endothelial function important to vascular health and overall cardiovascular disease risk across the lifespan. Am J Physiol-Heart Circ Physiol. 2018;315:H1569-88
69. Bowling MR, Xing D, Kapadia A, Chen Y-F, Szalai AJ, Oparil S. et al. Estrogen effects on vascular inflammation are age dependent. Arterioscler Thromb Vasc Biol. 2014;34:1477-85
70. Nelson JF, Felicio LS, Osterburg HH, Finch CE. Altered profiles of estradiol and progesterone associated with prolonged estrous cycles and persistent vaginal cornification in aging C578L/6J mice. Biol Reprod. 1981;24:784-94
71. Xu D, Lian D, Wu J, Liu Y, Zhu M, Sun J. et al. Brain-derived neurotrophic factor reduces inflammation and hippocampal apoptosis in experimental Streptococcus pneumoniae meningitis. J Neuroinflammation. 2017;14:156
72. Duarte-Silva E, Meiry da Rocha Araújo S, Oliveira WH, Lós DB, Bonfanti AP, Peron G. et al. Sildenafil alleviates murine experimental autoimmune encephalomyelitis by triggering autophagy in the spinal cord. Front Immunol. 2021;12:671511
73. Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A. et al. Aging. Aging-induced type I interferon signaling at the choroid plexus negatively affects brain function. Science. 2014;346:89-93
74. Pacheco GV, Cruz DC, González Herrera LJ, Pérez Mendoza GJ, Adrián Amaro GI, Nakazawa Ueji YE. et al. Copy number variation of TLR-7 gene and its association with the development of systemic lupus erythematosus in female patients from Yucatan Mexico. Genet Epigenetics. 2014;6:31-6
75. Xin Z, Lin G, Lei H, Lue TF, Guo Y. Clinical applications of low-intensity pulsed ultrasound and its potential role in urology. Transl Androl Urol. 2016;5:255-66
76. Tabuchi Y, Sugahara Y, Ikegame M, Suzuki N, Kitamura K, Kondo T. Genes responsive to low-intensity pulsed ultrasound in MC3T3-E1 preosteoblast cells. Int J Mol Sci. 2013;14:22721-40
77. Song W-S, Sung C-Y, Ke C-H, Yang F-Y. Anti-inflammatory and neuroprotective effects of transcranial ultrasound stimulation on Parkinson's disease. Ultrasound Med Biol. 2022;48:265-74
78. Guo H, Baker G, Hartle K, Fujiwara E, Wang J, Zhang Y. et al. Exploratory study on neurochemical effects of low-intensity pulsed ultrasound in brains of mice. Med Biol Eng Comput. 2021;59:1099-110
79. Chen T-T, Lan T-H, Yang F-Y. Low-intensity pulsed ultrasound attenuates lps-induced neuroinflammation and memory impairment by modulation of TLR4/NF-κB signaling and CREB/BDNF expression. Cereb Cortex. 2019;29:1430-8
80. Chen S-F, Su W-S, Wu C-H, Lan T-H, Yang F-Y. Transcranial ultrasound stimulation improves long-term functional outcomes and protects against brain damage in traumatic brain injury. Mol Neurobiol. 2018;55:7079-89
81. Chang J-W, Wu M-T, Song W-S, Yang F-Y. Ultrasound stimulation suppresses LPS-induced proinflammatory responses by regulating NF-κB and CREB activation in microglial cells. Cereb Cortex N Y N 1991. 2020;30:4597-606
82. Schabbauer G, Tencati M, Pedersen B, Pawlinski R, Mackman N. PI3K-Akt pathway suppresses coagulation and inflammation in endotoxemic mice. Arterioscler Thromb Vasc Biol. 2004;24:1963-9
83. Guha M, Mackman N. The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem. 2002;277:32124-32
84. Liu S-H, Lai Y-L, Chen B-L, Yang F-Y. Ultrasound enhances the expression of brain-derived neurotrophic factor in astrocyte through activation of TrkB-Akt and calcium-CaMK signaling pathways. Cereb Cortex. 2017;27:3152-60
85. Bading H, Ginty DD, Greenberg ME. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260:181-6
86. Koyama R, Ikegaya Y. To BDNF or not to BDNF: that is the epileptic hippocampus. Neurosci Rev J Bringing Neurobiol Neurol Psychiatry. 2005;11:282-7
87. Kortekaas R, Leenders KL, van Oostrom JCH, Vaalburg W, Bart J, Willemsen ATM. et al. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann Neurol. 2005;57:176-9
88. Hussain B, Fang C, Chang J. Blood-brain barrier breakdown: an emerging biomarker of cognitive impairment in normal aging and dementia. Front Neurosci [Internet]. 2021 [cited 24 October 2022]; 15. Available at: https://www.frontiersin.org/articles/10.3389/fnins. 2021 688090
89. Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA. et al. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci Off J Soc Neurosci. 2001;21:7724-32
90. Zhang X, Tang N, Hadden TJ, Rishi AK. Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta. 2011;1813:1978-86
91. Zhang T, Tian C, Wu J, Zhang Y, Wang J, Kong Q. et al. MicroRNA-182 exacerbates blood-brain barrier (BBB) disruption by downregulating the mTOR/FOXO1 pathway in cerebral ischemia. FASEB J. 2020;34:13762-75
92. Ibáñez CF, Andressoo J-O. Biology of GDNF and its receptors — relevance for disorders of the central nervous system. Neurobiol Dis. 2017;97:80-9
93. Kruglikov IL, Scherer PE. Caveolin-1 as a target in prevention and treatment of hypertrophic scarring. Npj Regen Med. 2019;4:1-7
94. Balligand J-L. Heat shock protein 90 in endothelial nitric oxide synthase signaling. Circ Res. 2002;90:838-41
95. Nunes AKS, Rapôso C, Rocha SWS, Barbosa KP de S, de Almeida Luna RL, da Cruz-Höfling MA. et al. Involvement of AMPK, IKβα-NFκB and eNOS in the sildenafil anti-inflammatory mechanism in a demyelination model. Brain Res. 2015;1627:119-33
96. Plaza-Zabala A, Sierra-Torre V, Sierra A. Autophagy and microglia: novel partners in neurodegeneration and aging. Int J Mol Sci. 2017;18:598
97. Ikai H, Tamura T, Watanabe T, Itou M, Sugaya A, Iwabuchi S. et al. Low-intensity pulsed ultrasound accelerates periodontal wound healing after flap surgery. J Periodontal Res. 2008;43:212-6
98. Kotwica-Mojzych K, Jodłowska-Jędrych B, Mojzych M. CD200:CD200R interactions and their importance in immunoregulation. Int J Mol Sci. 2021;22:1602
99. van der Vlist M, Ramos MIP, van den Hoogen LL, Hiddingh S, Timmerman LM, de Hond TAP. et al. Signaling by the inhibitory receptor CD200R is rewired by type I interferon. Sci Signal. 2021;14:eabb4324
100. Rabaneda-Lombarte N, Serratosa J, Bové J, Vila M, Saura J, Solà C. The CD200R1 microglial inhibitory receptor as a therapeutic target in the MPTP model of Parkinson's disease. J Neuroinflammation. 2021;18:88
101. Rabaneda-Lombarte N, Vidal-Taboada JM, Valente T, Ezquerra M, Fernández-Santiago R, Martí MJ. et al. Altered expression of the immunoregulatory ligand-receptor pair CD200-CD200R1 in the brain of Parkinson's disease patients. Npj Park Dis. 2022;8:1-15
102. Cox FF, Carney D, Miller A-M, Lynch MA. CD200 fusion protein decreases microglial activation in the hippocampus of aged rats. Brain Behav Immun. 2012;26:789-96
103. Zhang S, Wang X-J, Tian L-P, Pan J, Lu G-Q, Zhang Y-J. et al. CD200-CD200R dysfunction exacerbates microglial activation and dopaminergic neurodegeneration in a rat model of Parkinson's disease. J Neuroinflammation. 2011;8:154
104. Varnum MM, Kiyota T, Ingraham KL, Ikezu S, Ikezu T. The anti-inflammatory glycoprotein, CD200, restores neurogenesis and enhances amyloid phagocytosis in a mouse model of Alzheimer's disease. Neurobiol Aging. 2015;36:2995-3007
105. Burguillos MA, Deierborg T, Kavanagh E, Persson A, Hajji N, Garcia-Quintanilla A. et al. Caspase signalling controls microglia activation and neurotoxicity. Nature. 2011;472:319-24
106. Hernangómez M, Klusáková I, Joukal M, Hradilová-Svíženská I, Guaza C, Dubový P. CD200R1 agonist attenuates glial activation, inflammatory reactions, and hypersensitivity immediately after its intrathecal application in a rat neuropathic pain model. J Neuroinflammation. 2016;13:43
107. Yang J-Y, Widmann C. The RasGAP N-terminal fragment generated by caspase cleavage protects cells in a Ras/PI3K/Akt-dependent manner that does not rely on NFκB activation*. J Biol Chem. 2002;277:14641-6
108. Zhang L, Zhang J, You Z. Switching of the microglial activation phenotype is a possible treatment for depression disorder. Front Cell Neurosci [Internet]. 2018 [cited 2 August 2022]; 12. Available at: https://www.frontiersin.org/articles/10.3389/fncel. 2018 00306
109. Karnam G, Rygiel TP, Raaben M, Grinwis GCM, Coenjaerts FE, Ressing ME. et al. CD200 receptor controls sex-specific TLR7 responses to viral infection. PLoS Pathog. 2012;8:e1002710
110. Liu F, Li Z, Li J, Siegel C, Yuan R, McCullough LD. Sex differences in caspase activation after experimental stroke. Stroke J Cereb Circ. 2009;40:1842-8
111. Mangold CA, Wronowski B, Du M, Masser DR, Hadad N, Bixler GV. et al. Sexually divergent induction of microglial-associated neuroinflammation with hippocampal aging. J Neuroinflammation. 2017;14:141
112. Mangold CA, Masser DR, Stanford DR, Bixler GV, Pisupati A, Giles CB. et al. CNS-wide sexually dimorphic induction of the major histocompatibility complex 1 pathway with aging. J Gerontol A Biol Sci Med Sci. 2017;72:16-29
113. Wang H, Wang B, Normoyle KP, Jackson K, Spitler K, Sharrock MF. et al. Brain temperature and its fundamental properties: a review for clinical neuroscientists. Front Neurosci [Internet]. 2014 [cited 20 February 2019]; 8. Available at: https://www.frontiersin.org/articles/10.3389/fnins. 2014 00307/full
114. García-Cardeña G, Fan R, Shah V, Sorrentino R, Cirino G, Papapetropoulos A. et al. Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature. 1998;392:821-4
115. Leinenga G, Koh WK, Götz J. Scanning ultrasound in the absence of blood-brain barrier opening is not sufficient to clear β-amyloid plaques in the APP23 mouse model of Alzheimer's disease. Brain Res Bull. 2019;153:8-14
116. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860-7
117. Ostberg JR, Taylor SL, Baumann H, Repasky EA. Regulatory effects of fever-range whole-body hyperthermia on the LPS-induced acute inflammatory response. J Leukoc Biol. 2000;68:815-20
118. Evans SS, Repasky EA, Fisher DT. Fever and the thermal regulation of immunity: the immune system feels the heat. Nat Rev Immunol. 2015;15:335-49
119. Leach MD, Farrer RA, Tan K, Miao Z, Walker LA, Cuomo CA. et al. Hsf1 and Hsp90 orchestrate temperature-dependent global transcriptional remodelling and chromatin architecture in Candida albicans. Nat Commun. 2016;7:11704
120. Leach MD, Budge S, Walker L, Munro C, Cowen LE, Brown AJP. Hsp90 orchestrates transcriptional regulation by Hsf1 and cell wall remodelling by MAPK signalling during thermal adaptation in a pathogenic yeast. PLoS Pathog. 2012;8:e1003069
121. Lee C-T, Repasky EA. Opposing roles for heat and heat shock proteins in macrophage functions during inflammation: a function of cell activation state? Front Immunol. 2012;3:140
122. Anand PK, Anand E, Bleck CKE, Anes E, Griffiths G. Exosomal Hsp70 induces a pro-inflammatory response to foreign particles including mycobacteria. PLoS ONE. 2010;5:e10136
123. Truettner JS, Bramlett HM, Dietrich WD. Hyperthermia and mild traumatic brain injury: effects on inflammation and the cerebral vasculature. J Neurotrauma. 2018;35:940-52
124. Lee C-T, Kokolus KM, Leigh ND, Capitano M, Hylander BL, Repasky EA. Defining immunological impact and therapeutic benefit of mild heating in a murine model of arthritis. PLOS ONE. 2015;10:e0120327
125. Hagiwara S, Iwasaka H, Matsumoto S, Noguchi T. Changes in cell culture temperature alter release of inflammatory mediators in murine macrophagic RAW264.7 cells. Inflamm Res. 2007;56:297-303
126. Ferat-Osorio E, Sánchez-Anaya A, Gutiérrez-Mendoza M, Boscó-Gárate I, Wong-Baeza I, Pastelin-Palacios R. et al. Heat shock protein 70 down-regulates the production of toll-like receptor-induced pro-inflammatory cytokines by a heat shock factor-1/constitutive heat shock element-binding factor-dependent mechanism. J Inflamm. 2014;11:19
127. Franks H, Wang R, Li M, Wang B, Wildmann A, Ortyl T. et al. Heat shock factor HSF1 regulates BDNF gene promoters upon acute stress in the hippocampus, together with pCREB. J Neurochem. 2023Apr;165(2):131-148
128. Cahill CM, Waterman WR, Xie Y, Auron PE, Calderwood SK. Transcriptional repression of the prointerleukin 1β gene by heat shock factor 1*. J Biol Chem. 1996;271:24874-9
129. Singh IS, He J-R, Calderwood S, Hasday JD. A high affinity HSF-1 binding site in the 5'-untranslated region of the murine tumor necrosis factor-alpha gene is a transcriptional repressor. J Biol Chem. 2002;277:4981-8
130. Wang R, Town T, Gokarn V, Flavell RA, Chandawarkar RY. HSP70 enhances macrophage phagocytosis by interaction with lipid raft-associated TLR-7 and upregulating p38 MAPK and PI3K pathways. J Surg Res. 2006;136:58-69
131. Canovas B, Nebreda AR. Diversity and versatility of p38 kinase signalling in health and disease. Nat Rev Mol Cell Biol. 2021;22:346-66
132. Shiota M, Kusakabe H, Izumi Y, Hikita Y, Nakao T, Funae Y. et al. Heat shock cognate protein 70 is essential for Akt signaling in endothelial function. Arterioscler Thromb Vasc Biol. 2010;30:491-7
133. Chatterjee M, Andrulis M, Stühmer T, Müller E, Hofmann C, Steinbrunn T. et al. The PI3K/Akt signaling pathway regulates the expression of Hsp70, which critically contributes to Hsp90-chaperone function and tumor cell survival in multiple myeloma. Haematologica. 2013;98:1132-41
134. Rahman S, Islam R. Mammalian Sirt1: insights on its biological functions. Cell Commun Signal CCS. 2011;9:11
135. Saavedra D, Añé-Kourí AL, Barzilai N, Caruso C, Cho K-H, Fontana L. et al. Aging and chronic inflammation: highlights from a multidisciplinary workshop. Immun Ageing. 2023;20:25
136. Anckar J, Sistonen L. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem. 2011;80:1089-115
137. Blake MJ, Fargnoli J, Gershon D, Holbrook NJ. Concomitant decline in heat-induced hyperthermia and HSP70 mRNA expression in aged rats. Am J Physiol. 1991;260:R663-667
138. Rioux D. Sex differences in the induced expression of Hsp70 and Hsp27 in the brain and heart of rats. 2013 [cited 10 November 2022]; Available at: https://DalSpace.library.dal.ca//handle/10222/16023.
139. Potente M, Dimmeler S. NO Targets SIRT1. Arterioscler Thromb Vasc Biol. 2008;28:1577-9
140. Hotchkiss R, Nunnally I, Lindquist S, Taulien J, Perdrizet G, Karl I. Hyperthermia protects mice against the lethal effects of endotoxin. Am J Physiol. 1993;265:R1447-1457
141. Malhotra V, Eaves-Pyles T, Odoms K, Quaid G, Shanley TP, Wong HR. Heat shock inhibits activation of NF-κB in the absence of heat shock factor-1. Biochem Biophys Res Commun. 2002;291:453-7
142. Janus P, Pakuła-Cis M, Kalinowska-Herok M, Kashchak N, Szołtysek K, Pigłowski W. et al. NF-κB signaling pathway is inhibited by heat shock independently of active transcription factor HSF1 and increased levels of inducible heat shock proteins. Genes Cells. 2011;16:1168-75
143. Agarwal S, Ganesh S. Perinuclear mitochondrial clustering, increased ROS levels, and HIF1 are required for the activation of HSF1 by heat stress. J Cell Sci. 2020;133:jcs245589
144. Yang Y, Lu H, Chen C, Lyu Y, Cole RN, Semenza GL. HIF-1 Interacts with TRIM28 and DNA-PK to release paused RNA polymerase II and activate target gene transcription in response to hypoxia. Nat Commun. 2022;13:316
145. Lee K-H, Lee C-T, Kim YW, Han SK, Shim Y-S, Yoo C-G. Heat shock protein 70 negatively regulates the heat-shock-induced suppression of the IκB/NF-κB cascade by facilitating IκB kinase renaturation and blocking its further denaturation. Exp Cell Res. 2005;307:276-84
146. Lee K-H, Hwang Y-H, Lee C-T, Kim YW, Han SK, Shim Y-S. et al. The heat-shock-induced suppression of the IκB/NF-κB cascade is due to inactivation of upstream regulators of IκBα through insolubilization. Exp Cell Res. 2004;299:49-56
147. Wullaert A, Bonnet MC, Pasparakis M. NF-κB in the regulation of epithelial homeostasis and inflammation. Cell Res. 2011;21:146-58
148. Kim JY, Yenari MA, Lee JE. Regulation of inflammatory transcription factors by heat shock protein 70 in primary cultured astrocytes exposed to oxygen-glucose deprivation. Neuroscience. 2015;286:272-80
149. Kim D, Ouyang H, Yang S-H, Nussenzweig A, Burgman P, Li GC. A constitutive heat shock element-binding factor is immunologically identical to the Ku autoantigen (∗). J Biol Chem. 1995;270:15277-84
150. Yang S-H, Nussenzweig A, Yang W, Kim D, Li GC. Cloning and characterization of rat Ku70: involvement of Ku autoantigen in the heat-shock response. Radiat Res. 1996;146:603-11
151. Hazra J, Mukherjee P, Ali A, Poddar S, Pal M. Engagement of components of DNA-break repair complex and NFκB in Hsp70A1A transcription upregulation by heat shock. PLOS ONE. 2017;12:e0168165
152. Bunch H, Lawney BP, Lin Y-F, Asaithamby A, Murshid A, Wang YE. et al. Transcriptional elongation requires DNA break-induced signalling. Nat Commun. 2015;6:10191
153. Bunch H, Zheng X, Burkholder A, Dillon ST, Motola S, Birrane G. et al. TRIM28 regulates RNA polymerase II promoter proximal pausing and pause release. Nat Struct Mol Biol. 2014;21:876-83
154. Ono M, Tucker PW, Capra JD. Ku is a general inhibitor of DNA-protein complex formation and transcription. Mol Immunol. 1996;33:787-96
155. Sasi BK, Sonawane PJ, Gupta V, Sahu BS, Mahapatra NR. Coordinated transcriptional regulation of Hspa1a gene by multiple transcription factors: crucial roles for HSF-1, NF-Y, NF-κB, and CREB. J Mol Biol. 2014;426:116-35
156. Guo H, Callaway JB, Ting JP-Y. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677-87
157. Heads RJ, Latchman DS, Yellon DM. Stable high level expression of a transfected human HSP70 gene protects a heart-derived muscle cell line against thermal stress. J Mol Cell Cardiol. 1994;26:695-9
158. Ilangovan G, Osinbowale S, Bratasz A, Bonar M, Cardounel AJ, Zweier JL. et al. Heat shock regulates the respiration of cardiac H9c2 cells through upregulation of nitric oxide synthase. Am J Physiol-Cell Physiol. 2004;287:C1472-81
159. Foster JA, Brown IR. Differential induction of heat shock mRNA in oligodendrocytes, microglia, and astrocytes following hyperthermia. Mol Brain Res. 1997;45:207-18
160. Lamkanfi M, Sarkar A, Walle LV, Vitari AC, Amer AO, Wewers MD. et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol Baltim Md 1950. 2010;185:4385-92
161. Arriaga-Pizano L, Boscó-Gárate I, Martínez-Ordaz JL, Wong-Baeza I, Gutiérrez-Mendoza M, Sánchez-Fernandez P. et al. High serum levels of high-mobility group box 1 (HMGB1) and low levels of heat shock protein 70 (Hsp70) are associated with poor prognosis in patients with acute pancreatitis. Arch Med Res. 2018;49:504-11
162. Bozaykut P, Ozer NK, Karademir B. Nrf2 silencing to inhibit proteolytic defense induced by hyperthermia in HT22 cells. Redox Biol. 2016;8:323-32
163. Ni L, Xiao J, Zhang D, Shao Z, Huang C, Wang S. et al. Immune-responsive gene 1/itaconate activates nuclear factor erythroid 2-related factor 2 in microglia to protect against spinal cord injury in mice. Cell Death Dis. 2022;13:1-16
164. Dayalan Naidu S, Kostov RV, Dinkova-Kostova AT. Transcription factors Hsf1 and Nrf2 engage in crosstalk for cytoprotection. Trends Pharmacol Sci. 2015;36:6-14
165. Yamaguchi A, Maeshige N, Ma X, Uemura M, Noguchi H, Matsuda M. et al. Pulsed-ultrasound irradiation induces the production of itaconate and attenuates inflammatory responses in macrophages. J Inflamm Res. 2022;15:2387-95
166. Vereecke L, Beyaert R, van Loo G. Genetic relationships between A20/TNFAIP3, chronic inflammation and autoimmune disease. Biochem Soc Trans. 2011;39:1086-91
167. Kardyńska M, Paszek A, Śmieja J, Spiller D, Widłak W, White MRH. et al. Quantitative analysis reveals crosstalk mechanisms of heat shock-induced attenuation of NF-κB signaling at the single cell level. PLOS Comput Biol. 2018;14:e1006130
168. Harper CV, Woodcock DJ, Lam C, Garcia-Albornoz M, Adamson A, Ashall L. et al. Temperature regulates NF-κB dynamics and function through timing of A20 transcription. Proc Natl Acad Sci. 2018;115:E5243-9
169. Sheppard PW, Sun X, Emery JF, Giffard RG, Khammash M. Quantitative characterization and analysis of the dynamic NF-κB response in microglia. BMC Bioinformatics. 2011;12:276
170. Potteti HR, Venkareddy LK, Noone PM, Ankireddy A, Tamatam CR, Mehta D. et al. Nrf2 regulates anti-inflammatory A20 deubiquitinase induction by LPS in macrophages in contextual manner. Antioxidants. 2021;10:847
171. Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP. et al. Failure to regulate TNF-induced NF-κB and cell death responses in A20-deficient mice. Science. 2000;289:2350-4
172. Zeng Q, Hong S, Wang X, Cheng Y, Sun J, Xia W. Regulation of exosomes secretion by low-intensity pulsed ultrasound in lung cancer cells. Exp Cell Res. 2019;383:111448
173. Sheybani ND, Batts AJ, Mathew AS, Thim EA, Price RJ. Focused ultrasound hyperthermia augments release of glioma-derived extracellular vesicles with differential immunomodulatory capacity. Theranostics. 2020;10:7436
174. Teng Y, Ren Y, Hu X, Mu J, Samykutty A, Zhuang X. et al. MVP-mediated exosomal sorting of miR-193a promotes colon cancer progression. Nat Commun. 2017;8:14448
175. Li X, Li X, Lin J, Sun X, Ding Q. Exosomes derived from low-intensity pulsed ultrasound-treated dendritic cells suppress tumor necrosis factor-induced endothelial inflammation. J Ultrasound Med. 2019;38:2081-91
176. Cagnol S, Chambard J-C. ERK and cell death: mechanisms of ERK-induced cell death - apoptosis, autophagy and senescence. FEBS J. 2010;277:2-21
177. Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES. et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479-89
178. Matsumori Y, Northington FJ, Hong SM, Kayama T, Sheldon RA, Vexler ZS. et al. Reduction of caspase-8 and -9 cleavage is associated with increased c-FLIP and increased binding of Apaf-1 and Hsp70 after neonatal hypoxic/ischemic injury in mice overexpressing Hsp70. Stroke. 2006;37:507-12
179. Kiyatkin EA, Sharma HS. Permeability of the blood-brain barrier depends on brain temperature. Neuroscience. 2009;161:926-39
180. van Eden W, van der Zee R, Prakken B. Heat-shock proteins induce T-cell regulation of chronic inflammation. Nat Rev Immunol. 2005;5:318-30
181. Kimura Y, Yamada K, Sakai T, Mishima K, Nishimura H, Matsumoto Y. et al. The regulatory role of heat shock protein 70-reactive CD4+ T cells during rat listeriosis. Int Immunol. 1998;10:117-30
182. Tóth ME, Szegedi V, Varga E, Juhász G, Horváth J, Borbély E. et al. Overexpression of Hsp27 ameliorates symptoms of Alzheimer's disease in APP/PS1 mice. Cell Stress Chaperones. 2013;18:759-71
183. Stetler RA, Cao G, Gao Y, Zhang F, Wang S, Weng Z. et al. Hsp27 protects against ischemic brain injury via attenuation of a novel stress-response cascade upstream of mitochondrial cell death signaling. J Neurosci. 2008;28:13038-55
184. De Graeff-Meeder ER, van der Zee R, Rijkers GT, Schuurman HJ, Kuis W, Bijlsma JW. et al. Recognition of human 60 kD heat shock protein by mononuclear cells from patients with juvenile chronic arthritis. Lancet Lond Engl. 1991;337:1368-72
185. Stefano L, Racchetti G, Bianco F, Passini N, Gupta RS, Bordignon PP. et al. The surface-exposed chaperone, Hsp60, is an agonist of the microglial TREM2 receptor. J Neurochem. 2009;110:284-94
186. Hundt W, O'Connell-Rodwell CE, Bednarski MD, Steinbach S, Guccione S. In vitro effect of focused ultrasound or thermal stress on HSP70 expression and cell viability in three tumor cell lines. Acad Radiol. 2007;14:859-70
187. Zhu GQ, Jeon SH, Lee KW, Tian WJ, Cho HJ, Ha U-S. et al. Electric stimulation hyperthermia relieves inflammation via the suppressor of cytokine signaling 3-Toll like receptor 4 pathway in a prostatitis rat model. World J Mens Health. 2020;38:359-69
188. Huang S, Liu K, Cheng A, Wang M, Cui M, Huang J. et al. SOCS proteins participate in the regulation of innate immune response caused by viruses. Front Immunol [Internet]. 2020 [cited 7 November 2022]; 11. Available at: https://www.frontiersin.org/articles/10.3389/fimmu. 2020 558341
189. Mun H-S, Aosai F, Norose K, Piao L-X, Fang H, Akira S. et al. Toll-like receptor 4 mediates tolerance in macrophages stimulated with Toxoplasma gondii-derived heat shock protein 70. Infect Immun. 2005;73:4634-42
190. Nakao J, Fujii Y, Kusuyama J, Bandow K, Kakimoto K, Ohnishi T. et al. Low-intensity pulsed ultrasound (LIPUS) inhibits LPS-induced inflammatory responses of osteoblasts through TLR4-MyD88 dissociation. Bone. 2014;58:17-25
191. Mansell A, Smith R, Doyle SL, Gray P, Fenner JE, Crack PJ. et al. Suppressor of cytokine signaling 1 negatively regulates Toll-like receptor signaling by mediating Mal degradation. Nat Immunol. 2006;7:148-55
192. Nair S, Pandey AD, Mukhopadhyay S. The PPE18 protein of Mycobacterium tuberculosis inhibits NF-κB/rel-mediated proinflammatory cytokine production by upregulating and phosphorylating suppressor of cytokine signaling 3 protein. J Immunol Baltim Md 1950. 2011;186:5413-24
193. Kiers D, Gerretsen J, Janssen E, John A, Groeneveld R, van der Hoeven JG. et al. Short-term hyperoxia does not exert immunologic effects during experimental murine and human endotoxemia. Sci Rep. 2015;5:17441
194. Ohmoto Y, Fujisawa H, Ishikawa T, Koizumi H, Matsuda T, Ito H. Sequential changes in cerebral blood flow, early neuropathological consequences and blood-brain barrier disruption following radiofrequency-induced localized hyperthermia in the rat. Int J Hyperth Off J Eur Soc Hyperthermic Oncol North Am Hyperth Group. 1996;12:321-34
195. Baird NA, Turnbull DW, Johnson EA. Induction of the heat shock pathway during hypoxia requires regulation of heat shock factor by hypoxia-inducible factor-1*. J Biol Chem. 2006;281:38675-81
196. Katschinski DM, Le L, Heinrich D, Wagner KF, Hofer T, Schindler SG. et al. Heat induction of the unphosphorylated form of hypoxia-inducible factor-1α is dependent on heat shock protein-90 activity*. J Biol Chem. 2002;277:9262-7
197. Yang T, Liang C, Chen L, Li J, Geng W. Low-intensity pulsed ultrasound alleviates hypoxia-induced chondrocyte damage in temporomandibular disorders by modulating the hypoxia-inducible factor pathway. Front Pharmacol [Internet]. 2020 [cited 10 May 2023]; 11. Available at: https://www.frontiersin.org/articles/10.3389/fphar. 2020 00689
198. Kuennen M, Gillum T, Dokladny K, Bedrick E, Schneider S, Moseley P. Thermotolerance and heat acclimation may share a common mechanism in humans. Am J Physiol-Regul Integr Comp Physiol. 2011;301:R524-33
199. Shindo T, Ito K, Ogata T, Hatanaka K, Kurosawa R, Eguchi K. et al. Low-intensity pulsed ultrasound enhances angiogenesis and ameliorates left ventricular dysfunction in a mouse model of acute myocardial infarction. Arterioscler Thromb Vasc Biol. 2016;36:1220-9
Author contact
![]() Corresponding author: Graham M. Seasons graham.seasons1ca.
Corresponding author: Graham M. Seasons graham.seasons1ca.