Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2024; 14(5):2246-2264. doi:10.7150/thno.93192 This issue Cite
Research Paper
Myocardin reverses insulin resistance and ameliorates cardiomyopathy by increasing IRS-1 expression in a murine model of lipodystrophy caused by adipose deficiency of vacuolar H+-ATPase V0d1 subunit
Wenlin Yuan1, Hui Lin2,3, Yuan Sun2,5, Lihuan Liu1, Meijuan Yan1, Yujuan Song1, Xiaofan Zhang1, Xiangling Lu1, Yipei Xu1, Qiyue He1, Kunfu Ouyang6, Chenglin Zhang1, Yong Pan1, Yu Huang7, Ying Li1 ![]() , Xifeng Lu1,2,3,4
, Xifeng Lu1,2,3,4 ![]() , Jie Liu1
, Jie Liu1 ![]()
1. Department of pathophysiology, Shenzhen University Medical School, Shenzhen University, Shenzhen, China.
2. Clinical Research Center, The First Affiliated Hospital of Shantou University Medical College, Shantou, China.
3. Division of Pharmacology and Vascular Medicine, Department of Internal Medicine, Erasmus Medical Center, Rotterdam, the Netherlands.
4. Department of Pharmacology, Shantou University Medical College, Shantou, China.
5. Department of Pharmacology, College of Pharmacy, Shenzhen Technology University, Shenzhen, China.
6. Department of Cardiovascular Surgery, Peking University Shenzhen Hospital, Shenzhen, China.
7. Department of Biomedical Sciences, City University of Hong Kong, Hong Kong, China.
Received 2023-12-13; Accepted 2024-3-2; Published 2024-3-11
Abstract

Aim: Adipose tissue (AT) dysfunction that occurs in both obesity and lipodystrophy is associated with the development of cardiomyopathy. However, it is unclear how dysfunctional AT induces cardiomyopathy due to limited animal models available. We have identified vacuolar H+-ATPase subunit Vod1, encoded by Atp6v0d1, as a master regulator of adipogenesis, and adipose-specific deletion of Atp6v0d1 (Atp6v0d1AKO) in mice caused generalized lipodystrophy and spontaneous cardiomyopathy. Using this unique animal model, we explore the mechanism(s) underlying lipodystrophy-related cardiomyopathy.
Methods and Results: Atp6v0d1AKO mice developed cardiac hypertrophy at 12 weeks, and progressed to heart failure at 28 weeks. The Atp6v0d1AKO mouse hearts exhibited excessive lipid accumulation and altered lipid and glucose metabolism, which are typical for obesity- and diabetes-related cardiomyopathy. The Atp6v0d1AKO mice developed cardiac insulin resistance evidenced by decreased IRS-1/2 expression in hearts. Meanwhile, the expression of forkhead box O1 (FoxO1), a transcription factor which plays critical roles in regulating cardiac lipid and glucose metabolism, was increased. RNA-seq data and molecular biological assays demonstrated reduced expression of myocardin, a transcription coactivator, in Atp6v0d1AKO mouse hearts. RNA interference (RNAi), luciferase reporter and ChIP-qPCR assays revealed the critical role of myocardin in regulating IRS-1 transcription through the CArG-like element in IRS-1 promoter. Reducing IRS-1 expression with RNAi increased FoxO1 expression, while increasing IRS-1 expression reversed myocardin downregulation-induced FoxO1 upregulation in cardiomyocytes. In vivo, restoring myocardin expression specifically in Atp6v0d1AKO cardiomyocytes increased IRS-1, but decreased FoxO1 expression. As a result, the abnormal expressions of metabolic genes in Atp6v0d1AKO hearts were reversed, and cardiac dysfunctions were ameliorated. Myocardin expression was also reduced in high fat diet-induced diabetic cardiomyopathy and palmitic acid-treated cardiomyocytes. Moreover, increasing systemic insulin resistance with rosiglitazone restored cardiac myocardin expression and improved cardiac functions in Atp6v0d1AKO mice.
Conclusion: Atp6v0d1AKO mice are a novel animal model for studying lipodystrophy- or metabolic dysfunction-related cardiomyopathy. Moreover, myocardin serves as a key regulator of cardiac insulin sensitivity and metabolic homeostasis, highlighting myocardin as a potential therapeutic target for treating lipodystrophy- and diabetes-related cardiomyopathy.
Introduction
Cardiovascular diseases (CVDs), including coronary heart disease and congestive heart failure (HF), are the leading cause of death worldwide. Obesity and diabetes are major risk factors for CVDs. About 2.6 billion people globally are overweight or obese, and 537 million adults were living with diabetes in 2021. In addition to inducing atherosclerosis, metabolic disorders can directly cause cardiac hypertrophy, fibrosis and cardiac diastolic and systolic dysfunctions, resulting in cardiomyopathy. However, the mechanisms underlying the pathogenesis of cardiomyopathy remain largely unclear.
Adipose tissue is an active metabolic organ that maintains the balance between lipid storage and energy utilization. Maladaptation of adipose tissue induces metabolic disorders, including insulin resistance, dyslipidemia, hepatic steatosis, and type 2 diabetes, which cause damage to multiple organs and contribute to the development of a variety of diseases [1-3]. The heart has high energy demands, and its function is tightly associated with the health of adipose tissue [4, 5]. Adipose tissue dysfunction, which occurs in both obesity and lipodystrophy, is recognized to have detrimental effects on the heart. Clinical investigations demonstrated that the incidence and prevalence of heart failure (HF) are high in obese patients, due to the direct adverse effects of obesity on left ventricular (LV) structure and function, and the increased risk of coronary heart disease. The pathological changes characterized by LV hypertrophy, cardiac fibrosis, diastolic and systolic dysfunction, and metabolic disorders originating from obesity alone are also known as obesity cardiomyopathy [6, 7]. Interestingly, lipodystrophy manifested by generalized or partial loss of adipose tissue, can also cause cardiomyopathy, regardless of whether the etiology of lipodystrophy is congenital or acquired [8-10]. Just like obesity cardiomyopathy, myocardial hypertrophy is the most prominent pathological change of lipodystrophy cardiomyopathy. Dilated cardiomyopathy and severe biventricular HF have also been described in patients with congenital lipodystrophy [8, 10].
Adipogenesis plays a central role in determining AT mass and functions. Recent studies revealed vacuolar H+-ATPase (V-ATPase) subunits ATP6AP2 and ATP6V0A1 regulates adipogenesis and adipose functions [11, 12]. V-ATPase is highly conserved and expressed in virtually all eukaryotes as a multi-subunit complex, composed of a transmembrane Vo domain (subunits a, c, c', c'', d, e) that transports H+ across membrane, and a cytosolic V1 domain (subunits A-H) responsible for ATP hydrolysis. V-ATPase regulates the pH of intracellular vesicles, such as endosomes, lysosomes and Golgi apparatus [13-16], playing critical roles in receptor-mediated signaling, vesicle trafficking, receptor recycling, protein degradation and ion homeostasis [17-19]. Beyond pumping protons across membranes, V-ATPase also acts as a central hub for monitoring and responding to changes in cellular nutrient levels and energy status by modulating the activity of mammalian target of rapamycin complex 1 and AMP activated kinase [20, 21]. It is not yet clear whether other V-ATPase subunits play a role in regulating adipogenesis and AT functions. In the current study, we screened the expression of V-ATPase subunit during adipogenesis and the consequences of inhibiting V-ATPase subunits expression on adipogenesis using 3T3-L1 model. In this approach, we found that the expression of Vod1, which is encoded by Atp6v0d1, was increased as preadipocyte differentiates, while inhibiting its expression blocked differentiation of 3T3-L1 cells into mature adipocytes.
In the present study, we first identified Vod1 subunit as a master regulator of adipogenesis in 3T3-L1, and adipose-specific deletion of Atp6v0d1 (Atp6v0d1AKO) in mice resulted in a complete loss of adipose tissues in adulthood. The Atp6v0d1AKO mice spontaneously developed cardiac hypertrophy as early as at 12 weeks and progressed to HF at 28 weeks, resembling generalized lipodystrophy cardiomyopathy in patients. Using this animal model, we investigated the mechanisms underlying the pathogenesis of cardiomyopathy in Atp6v0d1AKO mice. Here, we revealed a previously unrecognized role of myocardin in regulating cardiac insulin sensitivity and glucose and lipid metabolism through increasing the expression of insulin signaling/docking molecule insulin receptor substrate (IRS)-1. Myocardin was downregulated in the heart of Atp6v0d1AKO mice, and restoring myocardin expression had a therapeutic effect against the development of cardiac hypertrophy and HF in Atp6v0d1AKO mice.
Methods
Animal care
All animals were procured from the Animal Center of Guangdong Province, China, and were kept in specific pathogen-free conditions with unrestricted access to food and water. All animal procedures were sanctioned by the Institutional Care and Ethical Committee of Shenzhen University, China, and were performed in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health publication No. 85-23, revised 1996) and the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes.
Generation of the Atp6v0d1 Knockout (Atp6v0d1AKO) mice
Atp6v0d1 floxed mice were generated using CRISPR/Cas9 system and provided by the Model Animal Research Center of Nanjing University (Nanjing, China). To genotype the floxed Atp6v0d1 allele, forward primer (5'-GGCTACTGTGTGAGGAATGGAC-3') and reverse primer (5'-GGTTGGACTGCAGC-AAAGG-3') were used. Atp6v0d1 floxed mice were crossed with Adipo-Cre mice (Jackson Laboratory) to generate adipose-specific Atp6v0d1 knockout mice (Adipo-Cre+/0 Atp6v0d1flox/flox), denoted as Atp6v0d1AKO. Mice were housed with a 12-h light/12-h dark cycle and fed ad libitum. Blood samples were collected via submandibular bleeding after 6 h of fasting, and blood glucose levels were determined using a glucometer (Roche). To measure glucose tolerance and insulin tolerance, mice were fasted for 16 h and injected intraperitoneally with 1 g/kg body weight glucose or 0.75 U/kg body weight insulin. Blood samples were collected via the tail vein at 0, 15, 30, 60, 90, and 120 min after injection.
Rosiglitazone and virus administration
Rosiglitazone (5 mg/kg/day) or the vehicle (0.5% sodium carboxymethyl cellulose) was administered to male control or Atp6v0d1AKO mice (aged 16 weeks) by intragastric gavage every other day. At 16 weeks and 26 weeks of age, control or Atp6v0d1AKO mice were administered AAV9-cTnt-myocardin or AAV9 vectors at a dose of 2.5×1011 viral genomes (vg)/mice via tail vein injection of at 16 weeks and 26 weeks of age. At 36 weeks of age, all mice were euthanized with an overdose of pentobarbital (150 mg/kg).
Construction of shRNA lentiviral vector and lentivirus infection
To achieve knockdown of Atp6v0d1 expression in 3T3-L1 cells, oligonucleotides against Atp6v0d1 (5'-GCGTTCAATAGCTGAACTTGT-3') were synthesized and inserted into the shRNA expressing lentiviral vector PLVx-U6-ccdB-EF1a-copGFP by homologous recombination. Scramble shRNA (5'-CCTAAGGTTAAGTCGCCCTCT-3') that does not target any known coding genes was cloned into the lentiviral vector. To produce lentivirus, shRNA expressing lentiviral shRNA expressing vectors were co-transfected with the packaging plasmids (pPSPAX2 & pCMV-VSVG at 2:1) into HEK293T cells, and lentiviral particles were collected and titered as previously described [22]. To transduce 3T3-L1 cells, collected lentivirus were added into culture medium at multiplicity of infection (MOI) of 2, together with 5 mg/mL polybrene.
Staining of lipid droplets in differentiated 3T3-L1 cells
3T3-L1 preadipocytes were obtained from ZenBio (North Carolina, USA) and cultured in preadipocyte medium (ZenBio) at 37 °C under 5% CO2 in a humidified incubator. To induce differentiation into mature adipocytes, 3T3-L1 cells were cultured to 100% confluence (Day 0). Two days later (Day 2), the culture medium was replaced with differentiation medium (ZenBio) and cells were cultured for a further three days. Subsequently, the cells were cultured in adipocyte medium (ZenBio), which was refreshed daily until Day 11 when full differentiation into mature adipocytes was achieved. Lipid droplets were stained with oil red O (ORO) and LipidSpotTM 610 as previously described [23]. In brief, for ORO staining, differentiated 3T3-L1 cells were fixed with 4% PFA, rinsed twice with 60% isopropanol and then stained with ORO (Sigma, 0.6% in 60% isopropanol) for 30 min at room temperature. Stained samples were then rinsed twice with 60% isopropanol and then with PBS. For fluorescence staining, cells were fixed with 4% paraformaldehyde (PFA) and then permeabilized with 0.1% Triton X-100 for 10 min. The cells were incubated first with LipidSpotTM 610 (1:1,000, Biotium) for 10 min followed by DAPI solution (Solarbio) for 5 min at room temperature, avoiding light exposure. Images were captured with the Cytation 5 Imaging Multimode Reader (Biotek).
Hepatic lipid extraction and measurement
Hepatic lipids were extracted using Folch's method [24]. Briefly, liver samples (50 mg) were homogenized with 625 μL chloroform/methanol mixture (2:1). After adding 125 μL chloroform, the homogenate was mixed and centrifuged at 14,000 ×g for 10 min at 4 ℃. The supernatant was transferred into a new Eppendorf (EP) tube and 250 μL methanol was added before the mixture was mixed again and centrifuged at 2,400 ×g for 5 min. Extracted lipids in the chloroform phase were collected and transferred into a new EP tube. The extracted lipids were then dried under nitrogen gas and resuspended in 100 μL PBS containing 1% Triton X-100 (Sigma-Aldrich). The levels of cholesterol, triglycerides, and non-esterified fatty acids (NEFAs) were measured using commercial kits (BioSino).
Measurement of ALT and AST
Blood samples were pre-cleared by centrifugation at 3,000 ×g for 5 min at 4 ℃. Plasma alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activities were measured using commercial kits (BioSino) following the manufacturer's instructions.
H&E staining and ORO staining of tissue sections
H&E staining was performed on 4 μm sections prepared from tissues fixed with 4% PFA and embedded in paraffin. Neutral lipids were detected in ORO staining of 7 μm sections prepared from tissues embedded in optimal cutting temperature (OCT) compound. After fixation with 4% PFA, sections were stained with 0.3% ORO following standard procedures. Images were captured with the Cytation 5 Imaging Multimode Reader (Biotek).
Isolation and culture of rat neonatal cardiomyocytes
Neonatal rat ventricular myocytes (NRVMs) were isolated and cultured according to previously described methods [25]. In brief, Sprague-Dawley rats (aged 1-2 days) were humanely euthanized by isoflurane inhalation followed by cervical dislocation. The ventricles were then removed and digested in PBS solution containing 0.75 mg/ml collagenase (type II, Worthington, USA). After pre-plating the cells for 2 h, the supernatant containing purified cardiomyocytes was collected and cultured in DMEM supplemented with 10% fetal bovine serum (FBS, MIKX), 1% penicillin-streptomycin, and bromodeoxyuridine (1:100, to inhibit fibroblast growth) for an additional 48 h before further treatment.
Isolation of adult mouse cardiomyocytes
Control and Atp6v0d1AKO mice were anesthetized with pentobarbital sodium (50 mg/kg i.p.), and their hearts were quickly removed. The hearts were then subjected to retrograde aortic perfusion with Ca2+-free buffer [150 mM NaCl, 5.4 mM KCl, 1.2 mM MgSO4, 10 mM glucose, 5 mM HEPES, 10 mM 2,3-butanedione monoxime (BDM; Sigma), and 5 mM taurine (Sigma)] at a constant rate of 5-6 mL/min and a temperature of 37 °C for 2 min. Collagenase type II (1 mg/mL; Worthington) was then added to the perfusion solution to initiate enzymatic digestion. After 15 min, the ventricles were quickly removed, cut into several pieces, and further digested by gentle agitation for 3 min at 37 ℃ in the same enzyme solution. This process released single myocytes, which were collected in the supernatant and then pelleted by low-speed centrifugation.
Echocardiography
Echocardiography was performed on non-anesthetized mice using a Vevo 2100 system (Visual Sonics, Toronto, Ontario, Canada), as previously described [26]. For two-dimensional (2-D) guided M-mode echocardiography, the heart image was captured in the parasternal short-axis view. The following parameters were measured from this view: percentage of left ventricular (LV) fractional shortening (FS), LV ejection fraction (EF), LV internal dimensions at both diastole and systole (LVIDd and LVIDs, respectively), and LV posterior wall dimensions at both diastole and systole (LVPWd and LVPWs, respectively). LV fractional shortening (FS, %) was calculated as [(LVIDd - LVIDs)/LVIDd] × 100, and LV ejection fraction (EF, %) was calculated as [(LVIDd2 - LVIDs2)/ LVIDd2] × 100.
Western blot analysis
Samples of mouse tissue or cells were homogenized in RIPA buffer (Solarbio, Beijing, China) supplemented with protease inhibitor cocktail (Cat #HY-K0010, MCE, Monmouth Junction, NJ, USA) and Phosphatase Inhibitor Cocktail II & III (Cat #HY-K0022& HY-K0023, MCE). Lysates were cleared by centrifugation at 14,000 ×g for 10 min at 4 ℃. The total protein concentration in the supernatant was determined by BCA assay (Pierce). Equal amounts of protein (20-50 μg in total) were separated by SDS-PAGE and then transferred to PVDF membranes (Merck Millipore, Bedford, MA, USA). The blots were then probed with the antibodies listed in Supplementary Table 1, detected by Western Chemiluminescent HRP Substrate (Merck Millipore, Bedford, MA) and visualized using a UVP ChemStudio PLUS imaging system (CA, USA). Band intensities were quantified using ImageJ (NIH, USA).
Immunofluorescence
Control and Atp6v0d1AKO hearts were fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned at 5 μm intervals. For antigen retrieval, the sections were dehydrated in a graded series of ethanol solutions and then microwaved in citrate buffer (0.4 g/L citric acid and 3 g/L sodium citrate) for 3 min. Permeabilization and blocking were performed using 5% BSA (IgG free) and 0.2% Triton X-100 in PBS at room temperature for 30 min. The sections were then incubated overnight at 4 ℃ with primary antibodies. The samples were further incubated with corresponding secondary antibodies and visualized by adding 4′,6-Diamidine-2′-phenylindole dihydrochloride (DAPI, Cat #10236276001, Sigma-Aldrich, Burlington, MA, USA). Confocal images were captured using a Zeiss LSM880 microscope (Carl Zeiss, Germany).
Real-time quantitative PCR
Total RNA from heart tissue and cell samples was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the standard protocol. Genomic DNA contamination was removed from total RNA (500 ng-1 µg) using gDNA wiper mix (Vazyme, Nanjing, China), followed by reverse transcription using HiScript III qRT Supermix (Vazyme, Nanjing, China) according to the manufacturer's instructions. Quantitative real-time PCR (qPCR) was performed with ChamQ Universal SYBR qPCR master mix and gene-specific primers on a QuantStudio 3 real-time PCR System (Applied Biosystems, Waltham, MA, USA). Data were normalized against the GAPDH gene and the relative expression level of each gene was calculated using the 2-∆∆Ct method. Primers used in the study are listed in Supplementary Table 2.
Oroboros O2K measurement
The oxygen concentration and oxygen flux of intact cells were measured using the Oxygraph-2k (O2k, OROBOROS Instruments, Innsbruck, Austria) according to the manufacturer's instructions [27]. Experiments were conducted using isolated adult cardiomyocytes in culture medium (106 cells/mL) and performed in two O2k chambers maintained at 37 ℃. The oxygen consumption rate (OCR) was measured under basal conditions and in response to oligomycin (oligo), FCCP, and rotenone plus antimycin A (Rot/AM). Basal OCR was measured before oligomycin injection and maximum OCR was measured after FCCP injection and the non-mitochondrial respiration (measurement after rotenone and antimycin A injection) was subtracted.
Small interfering RNA-mediated gene knockdown
Small interfering RNA (siRNA) and scrambled siRNA were chemically synthesized by Ribobio (Guangzhou, China). The designed siRNA sense sequences were as follows: si-Myocd, 5′-CCTGGTTAATATGCACATT-3′; si-Irs1, 5′-GAGAAGAAGTGGC-GGCACA-3′. Cells were transfected with of siRNA (30 pmol/L) using Lipofectamine™ RNAiMAX Transfection Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions and cultured for 48 h. The medium was then changed and the culture was continued for a further 24 h before collection for total RNA.
Reporter plasmids and luciferase assays
The IRS1 promoter, cloned from rat heart chromatin, was inserted between the NheI and HindIII restriction enzyme sites into the Promega pGL4.10 promoterless luciferase vector, resulting in the construction of the pGL4.10-IRS1 vector. For dual luciferase reporter assay, HEK293T cells were transiently transfected with Lipofectamine 3000 reagent (Thermo Fisher Scientific) according to the manufacturer's instructions. Briefly, cells were seeded into 24-well plates, and cultured to 80% confluence before transfection with either 500 ng pGL4.10 promoterless control or pGL4.10-IRS1 and 500 ng SRF-expressing plasmid or pcDNA3.1. Cells were co-transfected with Renilla plasmid as an internal control. After transfection, cells were cultured for an additional 24 h and lysed for the measurement of luciferase and Renilla activity using the TransDetect® Double-Luciferase Reporter Assay Kit (TransGen) according to the manufacturer's instructions.
Chromatin immunoprecipitation
For chromatin immunoprecipitation (ChIP) assays, The Magna ChIP A/G Chromatin Immunoprecipitation Kit (Millipore Sigma) was utilized according to the manufacturer's instructions. Briefly, NRVM were fixed with 1% formaldehyde for 10 min at room temperature and then quenched with glycine for 5 min at room temperature. After sonication, equal amounts of cell lysate were immunoprecipitated with either anti-SRF antibody or IgG (#2729, Cell Signaling Technology, Boston, MA) at 4 ℃ overnight. The DNA fragments were eluted from the protein/DNA complexes by routine washing steps and then analyzed by quantitative PCR using the primers listed in the Supplementary Table 2. The IRS1 promoter region flanking binding sites served as target regions, and the distal regions located approximately 1 kb and 2 kb from the transcription start site served as control regions.
RNA-sequencing and bioinformatics
Total RNA was isolated from heart tissues of 36-week-old control and Atp6v0d1AKO mice. The quality and integrity of total RNA were evaluated using the Agilent Technologies 2100 Bioanalyzer. RNA-Seq libraries were sequenced on the Illumina NovaSeq 6000. The level of mRNA expression was normalized using FPKM (fragments per kilobase of transcript per million mapped reads). Clean reads were mapped to the reference gene using featureCounts (v1.5.0-p3), and to the reference genome using Hisat2 (v2.0.5). Differential expression analysis was performed using the DESeq2 R package (1.20.0). The resulting P-values were adjusted using the Benjamini and Hochberg approach to control for the false discovery rate. The threshold for significantly differential expression was set as Padj < 0.05 and |log2(foldchange)| > 0.5. Gene Ontology (GO) enrichment analysis of the differentially expressed genes was performed using the clusterProfiler R package (3.8.1), with enrichment declared for corrected P < 0.05. Reactome, an open-source, curated and peer-reviewed pathway database (https://reactome.org/) [28], was used to perform pathways enrichment on significant gene sets.
Statistical analysis
All results are analyzed using GraphPad Prism software and presented as the means ± SEM. Unpaired t-test and one-way ANOVA were used for two-group or multiple-group comparisons. The details of statistical analysis for figures and Supplementary Figures are performed as indicated in the figure legends.
Results
Atp6v0d1 regulates adipogenesis and its loss leads to lipodystrophy
We found an increase in Atp6v0d1 expression during differentiation of 3T3-L1 preadipocytes into mature adipocytes (Figure S1A and S1E), in parallel with elevated expression of Pparg, Cebpa, and Cebpb, which are markers of adipogenesis (Figure S1B-S1D). Silencing Atp6v0d1 expression led to downregulation of PPARγ, CEBPα and FASN, along with the reduced lipid droplet accumulation, an indicator of mature adipocytes, in 3T3-L1 adipocytes (Figure S1F and S1G), suggesting ATP6V0D1 as a master regulator of adipogenesis. Moreover, analysis of adipose tissues from diet-induced-obesity mice revealed a marked increase in ATP6V0D1 abundance (Figure S1H and S1I), suggesting Vod1 subunit is involved in maintaining adipose homeostasis.
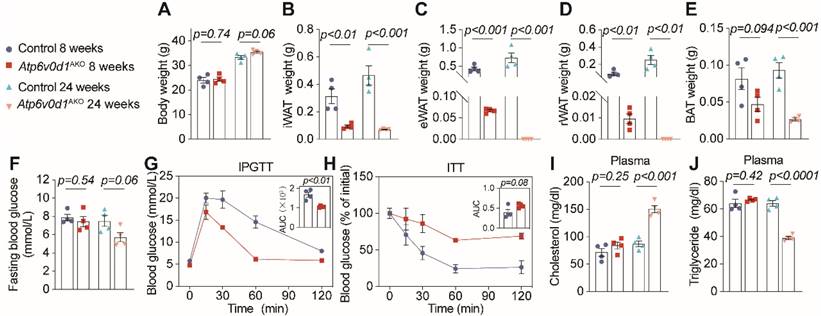
We further generated adipocyte-specific Atp6v0d1 knockout mice, denoted as Atp6v0d1AKO, by mating Atp6v0d1 floxed mice with Adiponectin-Cre mice (Figure S2A-S2C). Despite unaltered body weight (Figure 1A and Figure S2D), Atp6v0d1AKO mice exhibited markedly diminished adipose depots compared to their littermate controls at both 8 and 24 weeks of age (Figure 1B-1E and Figure S2E-S2F). Notably, at 24 weeks of age, Atp6v0d1AKO mice even had no visible epididymal and retroperitoneal white adipose depots, accompanied by reduction in plasma leptin and adiponectin levels (Figure S2G and S2H). These data suggested that ATP6V0D1 deficiency in adipocytes led to a progressive loss of adipose depots, likely due to the disruption of adipogenesis. Consistent with this, hepatic steatosis emerged at 8 weeks of age in Atp6v0d1AKO mice (Figure S2I-S2L), progressing to liver injury as measured by AST to ALT levels at 24 weeks (Figure S2M). Furthermore, 24-week-old Atp6v0d1AKO mice displayed significant disturbances of glucose and lipid metabolism, manifested as increased glucose tolerance but reduced insulin sensitivity (Figure 1F-H), increased plasma cholesterol (Figure 1I), and reduced triglycerides levels (Figure 1J). The plasma cholesterol remained high, and high NEFA levels and systemic insulin resistance were also observed in Atp6v0d1AKO mice aged 36 weeks (Figure S3A-S3E).
Atp6v0d1AKO mice exhibit lipodystrophy and insulin resistance. A, Body weight. B, Weights of inguinal WAT (iWAT). C, Epididymal white adipose tissue (eWAT). D, Retroperitoneal WAT (rWAT). E, Brown adipose tissue (BAT). F, Fasting blood glucose. G, Intraperitoneal glucose tolerance test (IPGTT) comparisons using area under curve (AUC). H, Insulin tolerance test comparisons using AUC. I, Plasma cholesterol. J, Plasma triglyceride. Data represent the mean ± SEM (n = 4) (two-tailed unpaired Student's t-test).
Adipocyte ATP6V0D1 deficiency leads to cardiac hypertrophy and contractile dysfunction
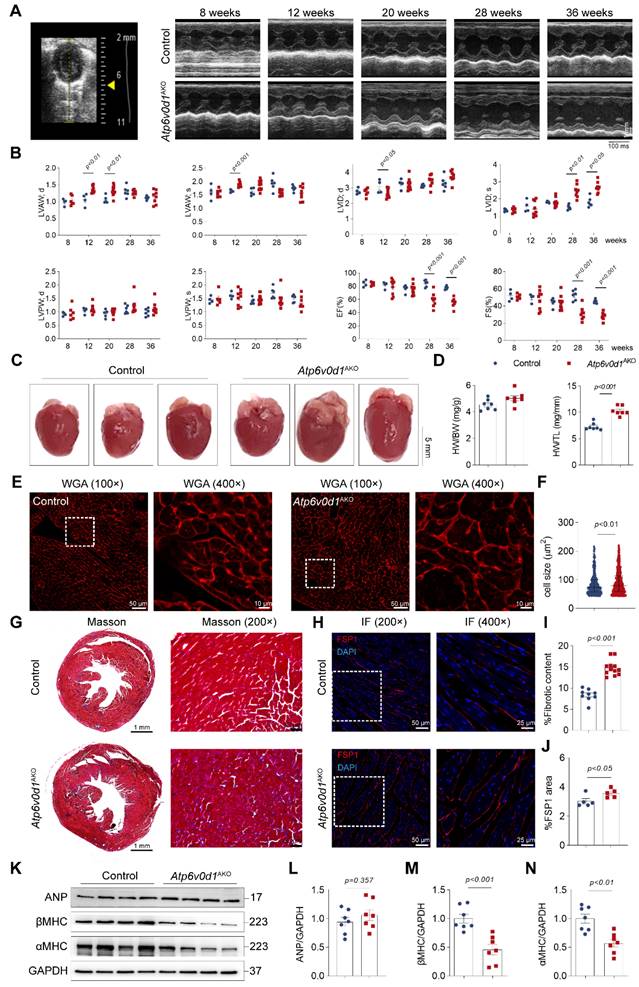
We then monitored the cardiac structure and function in Atp6v0d1AKO mice and control mice (Atp6v0d1flox/flox littermates) with echocardiography. At 8 weeks, the thickness of the anterior and posterior walls of the left ventricle (LVAW and LVPW, respectively) at systole (s) and diastole (d), the internal LV diameters at systole (LVIDs) and diastole (LVIDd), and the LV ejection fraction (EF%) and fractional shortening (FS%) were comparable in control and Atp6v0d1AKO mice (Figure 2A-2B and Figure S4A). At 12 weeks, the LVAWs and LVAWd in Atp6v0d1AKO mice were significantly larger than those in control (Figure 2B). In contrast, the LVIDd in Atp6v0d1AKO mice was smaller than that in their control littermates (Figure 2B). The EF% and FS% were comparable in these two groups of mice until 24 weeks (Figure 2B and Figure S4B-S4C), suggesting that the Atp6v0d1AKO mice developed cardiac hypertrophy with preserved contractile function. At 28 weeks, apparent cardiac contractile dysfunction was observed in Atp6v0d1AKO mice as evidenced by significantly larger LVIDs, and remarkedly reduced EF% and FS% (59.4% and 31.2%, respectively, in Atp6v0d1AKO mice versus 84.3% and 52.6%, respectively, in control mice), indicating the development of HF (Figure 2A-2B). There was only a minor and insignificant decrease in EF% and FS% in Atp6v0d1AKO mice aged 36 weeks compared with those in Atp6v0d1AKO mice aged 28 weeks (Figure 2A-2B), suggesting the progress of HF is slow. The hearts of Atp6v0d1AKO mice aged 36 weeks were visibly larger than those of their litter controls (Figure 2C). However, there was no difference in the HW to body weight (BW) ratio (HW/BW) between Atp6v0d1AKO mice and their littermate controls (Figure 2D); however, the significantly greater weight of the liver in Atp6v0d1AKO mice indicated that HW/BW ratio may not be suitable to reflect cardiac hypertrophy (Figure S3F). Therefore, we analyzed the ratio of HW to tibia length (HW/TL), and found that HW/TL in Atp6v0d1AKO mice was much larger than that of the littermate controls (Figure 2D). Histological images also showed that the heart size in Atp6v0d1AKO mice was larger than that in control mice (Figure 2C). WGA staining of the heart slices showed a significant increase in the size of cardiomyocytes in Atp6v0d1AKO mice compared to control mice (Figure 2E and 2F). These results further confirmed cardiac hypertrophy in Atp6v0d1AKO mice. Moreover, Masson Trichrome staining revealed that fibrosis occurred in Atp6v0d1AKO hearts (Figure 2G and 2I). Positive areas for immunostaining of FSP, the biomarker for fibroblasts, were also increased in Atp6v0d1AKO hearts (Figure 2H and 2J), suggesting enhanced proliferation of cardiac fibroblasts. Intriguingly, despite the apparent cardiac dysfunction in Atp6v0d1AKO mice at 36 weeks, there was no difference in the myocardial protein abundance of ANP, a fetal gene that is activated in HF, compared with control mice, while α- and β-MHC proteins were both significantly decreased (Figure 2K-2N). Furthermore, the mRNA levels of Nppa and Nppb, which encode ANP and BNP, respectively, were consistent with those of the corresponding proteins, while the mRNA levels of Myh6 and Myh7, which encode α-MHC and β-MHC, respectively, were significantly decreased in Atp6v0d1AKO hearts (Figure S4D). These data indicated that Atp6v0d1AKO mice develop cardiac hypertrophy and HF with relatively low ANP expression.
Atp6v0d1AKO mice exhibit cardiac hypertrophy and fibrosis. A, Representative M-mode tracings from the echocardiography of control and Atp6v0d1AKO mice aged 8, 12, 20, 28 and 36 weeks. B, Echocardiographic parameters recorded in (A) (n = 4-8). LVAWs, thickness of left ventricular anterior wall at systole; LVAWd, thickness of left ventricular anterior wall at diastole; LVPWs, thickness of left ventricular posterior wall at systole; LVPWd, thickness of left ventricular posterior wall at diastole; LVIDs, systolic left ventricular internal dimension; LVIDd, diastolic left ventricular internal dimension; EF left ventricular ejection fraction; FS, fractional shortening. C, Representative images of hearts from control and Atp6v0d1AKO mice aged 36 weeks. D, HW/BW and HW/TL between control and Atp6v0d1AKO mice. HW, heart weight; BW, body weight; TL, tibial length. E, Representative images of WGA stain from control and Atp6v0d1AKO mice aged 36 weeks. F, Quantification of the cell size for each group. G, Representative transverse sections of control and Atp6v0d1AKO hearts with Masson trichrome staining. H, Immuno-fluorescence imaging of FSP1. Control and Atp6v0d1AKO heart sections showing red signal for anti-FSP1 antibody. Cell nuclei are stained with DAPI (blue). I, Quantification of fibrotic content in control and Atp6v0d1AKO heart sections. J, % Fluorescent area for FSP1 area in control and Atp6v0d1AKO heart sections. K-N, Representative immunoblots and quantification of α-MHC, β-MHC and ANP protein levels in control and Atp6v0d1AKO heart tissues. Data are normalized to GAPDH and analyzed by two-tailed unpaired Student's t-test. Data represent the mean ± SEM (n = 7).
Excessive lipid accumulation and reprogramed energy expenditure occur in Atp6v0d1AKO hearts
Oil red O (ORO) staining of heart cryosections showed excessive lipid deposition in Atp6v0d1AKO hearts (Figure 3A). In accordance with this finding, the mRNA, total protein levels and cell-surface expression of cluster of differentiation 36 (CD36), the major transporter of cardiac FA uptake, were significantly increased in Atp6v0d1AKO hearts compared with the controls (Figure 3B-3F), suggesting CD36 upregulation likely contributes to lipid infiltration in Atp6v0d1AKO hearts. We further mapped the oxidative respiratory status and capacity by measuring oxygen consumption rate (OCR) and fuel dependency using isolated cardiomyocytes from control and Atp6v0d1AKO mice. Basal OCR levels of isolated Atp6v0d1AKO cardiomyocytes were comparable to those of their littermate controls (Figure 3G and Figure S5A). However, FCCP (Trifluoromethoxy carbonylcyanide phenylhydrazone)-induced maximal respiration was reduced in Atp6v0d1AKO cardiomyocytes (Figure 3G), despite the comparable abundance of the mitochondrial respiratory complexes between hearts of Atp6v0d1AKO and control mice (Figure S5B). Following application of etomoxir, an inhibitor for carnitine palmitoyl-transferase (CPT1) that is responsible for importing long chain fatty acid into mitochondria for oxidation, the reduction in FCCP-induced maximal respiration was greater in Atp6v0d1AKO cardiomyocytes than that in controls (Figure 3H). The results suggested that the amount of FAO-supplied energy is increased in Atp6v0d1AKO cardiomyocytes, despite a reduction in maximal respiratory capacity.
Excessive lipid is accumulated and the energy expenditure is reprogramed in Atp6v0d1AKO hearts. A, Representative images of oil red O staining of sections of control and Atp6v0d1AKO hearts. B, qPCR quantification of CD36 mRNA levels in control and Atp6v0d1AKO heart tissues. C-D, Representative immunoblots and quantification of CD36 protein levels in control and Atp6v0d1AKO heart tissues. E-F, Representative immunoblots and quantification of cell-surface CD36 in heart lysates from both control and Atp6v0d1AKO heart tissues. G, Quantification of basal and maximal OCR levels of isolated cardiomyocytes from control and Atp6v0d1AKO hearts. H, Quantification of Eto-sensitive OCR levels of isolated cardiomyocytes from control and Atp6v0d1AKO hearts. OCR was measured using the Oroboros O2k with the addition of etomoxir (Eto) at the baseline. I, Heatmap analysis of fatty acid oxidation (FAO)-involved genes from the RNA-sequencing dataset of control and Atp6v0d1AKO hearts. J, qPCR quantification of the mRNA levels of FAO-related genes in control and Atp6v0d1AKO hearts. K, Heatmap analysis of glycolysis and glucose oxidation-related genes from the RNA-sequencing dataset of control and Atp6v0d1AKO hearts. L, qPCR quantification of the mRNA levels of glycolysis and glucose oxidation-related genes in control and Atp6v0d1AKO hearts. M, Representative immunoblots of GLUT1, GLUT4, PDK4, PDH, p-PDH protein levels in control and Atp6v0d1AKO heart tissues. N-Q, Quantification of specified densitometric ratios. Data represent the mean ± SEM (n = 4-7) (two-tailed unpaired Student's t-test).
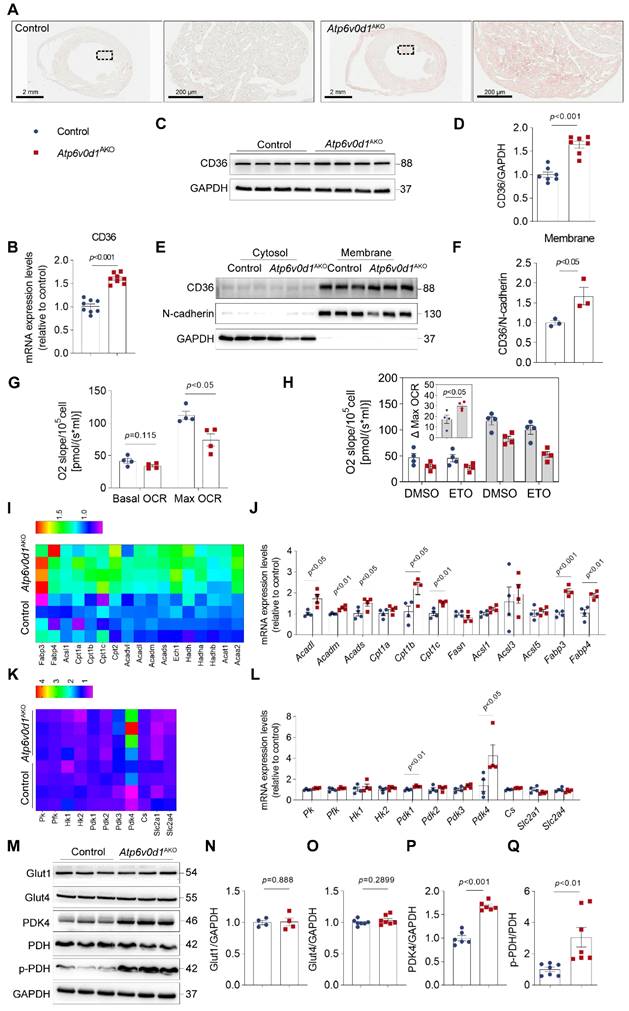
To understand the molecular mechanisms underlying the abnormal lipid metabolism and the development of hypertrophic cardiomyopathy, we examined the RNA expression profiles obtained from unbiased RNA sequencing (RNA-seq). Using |Log2FC|>0.5 and Padjusted<0.05 as cutoff values, we identified 527 upregulated genes and 262 downregulated genes (Figure S6A). GO and Reactome enrichment analysis indicates that lipid metabolism pathway and fatty acid metabolism pathway were among the top 15 most affected pathways in Atp6v0d1AKO hearts (Figure S6B and S6C). In accordance with the FAO shift, transcriptomic profiles revealed that FAO-related genes, including Acadl, Acadm, Acads, Cpt1b, Cpt1c, Acsl1, Fabp3 and Fabp4, were significantly upregulated in Atp6v0d1AKO hearts compared with control hearts (Figure 3I), which were confirmed by quantitative PCR (Figure 3J).
Lipid and glucose metabolism are competitive processed in terms of heart energy expenditure as described by the glucose fatty-acid cycle (Randle cycle) [29]. The increase in the utilization of FA as an energy source suggested a reciprocal repression of glucose uptake and oxidation in Atp6v0d1AKO hearts. We found that there were no differences in the mRNA and protein levels of the cardiac glucose transporters Glut1 and Glut4 (encoded by slc2a1 and slc2a4, respectively) between Atp6v0d1AKO and control hearts (Figure 3L-3O). However, RNA-seq analysis, qPCR and Western blot assays identified a critical gene in glucose oxidation cascade, pyruvate dehydrogenase kinase 4 (PDK4) that was remarkably increased in Atp6v0d1AKO compared with control hearts (Figure 3K-3M and 3P). PDK4 negatively regulates glucose oxidation by phosphorylation of pyruvate dehydrogenase (PDH), a rate-limiting enzyme that catalyzes oxidative decarboxylation of pyruvic acid to acetyl-CoA. Accordingly, we found the protein levels of phosphorylated PDH (p-PDH) rather than the total PDH were increased in Atp6v0d1AKO hearts (Figure 3M and 3Q). The results indicated that PDK4 downregulation may lead to decreased glucose oxidation by phosphorylating PDH in Atp6v0d1AKO mouse hearts.
Atp6v0d1AKO hearts exhibit insulin resistance
To gain a better understanding of the molecular aspects of the changes in glucose and lipid metabolism, we examined the insulin signal activity, and the protein abundance of Forkhead box O1 (FoxO1), PPARs (PPARα, PPARδ and PPARγ), and AMPK, which play critical roles in regulating cardiac lipid and glucose metabolism [30-32], in the hearts of control and Atp6v0d1AKO mice. The expression of insulin signaling/docking molecule insulin receptor substrate (IRS)-1 and IRS-2 were significantly decreased in Atp6v0d1AKO heart compared to that in control mice (Figure 4A-4C), suggesting Atp6v0d1AKO hearts developed insulin resistance. Insulin resistance has been shown to suppress PI3K-Akt signaling pathway [33]. However, we found PI3K activity indicated by the phosphorylation levels of PI3K (p-PI3K) and Akt-T308 was not changed (Figure 4D-4G), while the protein level of Akt phosphorylated at Ser473 (p-Akt-S473) and the p-Akt-S473/Akt ratio was remarkably increased in Atp6v0d1AKO hearts compared with the control (Figure 4D and 4E). In addition to insulin signal, PI3K-Akt activity can be regulated by multiple other signaling pathways. We thus examined PI3K-Akt activity in control and Atp6v0d1AKO hearts in response to insulin stimulation. The results showed that the protein level of phosphorylated AKT-S473 was significantly decreased, confirming insulin resistance in Atp6v0d1AKO hearts (Figure 4H-4I).
Atp6v0d1AKO hearts exhibit insulin resistance. A-C, Representative immunoblots and quantification of IRS1 and IRS2 protein levels. Data are normalized to GAPDH. Data represent the mean ± SEM (n = 7) (two-tailed unpaired Student's t-test). D, Total protein extracts from control and Atp6v0d1AKO hearts immunoblotted with the indicated antibodies, representative immunoblots of three independent experiments are shown. E-G, Statistical analysis of specified densitometric ratios. Data represent the mean ± SEM (n = 7) (two-tailed unpaired Student's t-test). H-I, Immunoblot analysis and quantification of AKT phosphorylation at Ser473 phosphorylation in heart tissues from control and Atp6v0d1AKO mice at 15 min after injection of insulin at 1 U/kg. Data represent the mean ± SEM (n = 3) (two-tailed unpaired Student's t-test). J, Total protein extracts from control and Atp6v0d1AKO hearts immunoblotted with the indicated antibodies, representative immunoblots of four independent experiments is shown. K-P, Statistical analysis of specified densitometric ratios. Data represent the mean ± SEM (n = 4 - 7) (two-tailed unpaired Student's t-test).
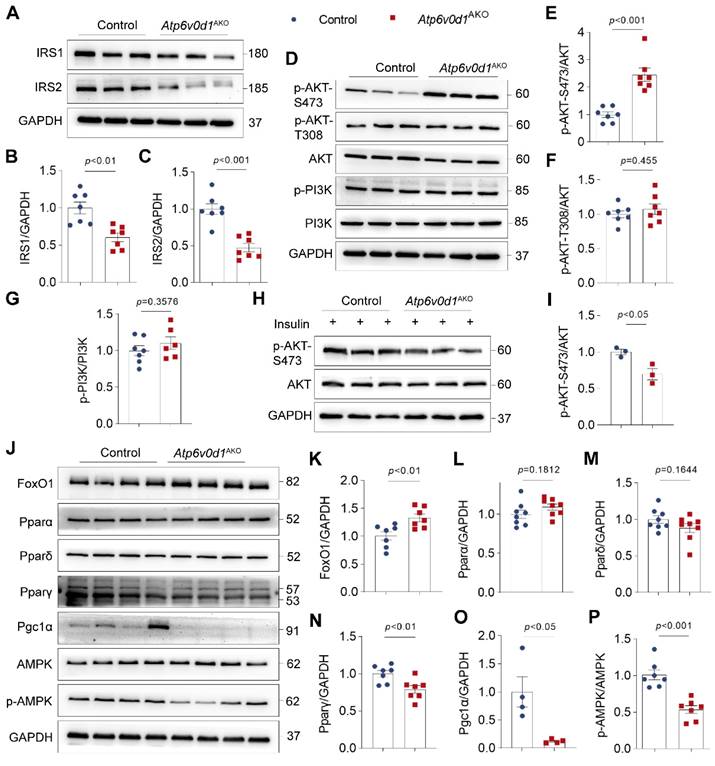
The protein abundance of FoxO1 was significantly increased (Figure 4J and 4K). There were no significant differences in the protein levels of PPARα and PPARδ, which are highly expressed in heart and play critical role in regulating lipid metabolism, between Atp6v0d1AKO and control hearts (Figure 4L and 4M), but the protein abundances of PPAR-γ and PGC-1, the PPARγ coactivator-1, which are important in lipogenesis and lipid synthesis in white adipose tissue, were significantly decreased in Atp6v0d1AKO relative to control hearts (Figure 4N and 4O). Moreover, the phosphorylated AMPK (p-AMPK) were significantly decreased, while the total AMPK expression was unchanged in Atp6v0d1AKO relative to control hearts (Figure 4P).
Increasing myocardin expression reverses cardiac insulin resistance and metabolic disorder, improving cardiac functions in Atp6v0d1AKO mice
Through further analysis of the RNA-seq data, we found that myocardin expression was remarkably reduced in Atp6v0d1AKO hearts. Quantitative PCR and Western blot analyses confirmed the remarkably decreased expression of myocardin in Atp6v0d1AKO hearts (Figure 5A-5C). Myocardin is a transcription cofactor that plays an essential role in regulating cardiomyocyte survival and maintaining cardiac functions by interacting with serum response factor (SRF) to control the transcription of multiple genes containing cis element CArG boxes [34]. To explore the possible role of myocardin downregulation in the pathogenesis of cardiomyopathy in Atp6v0d1AKO, we restored cardiac myocardin expression by tail-vein injection of AAV9 virus that expresses myocardin under the control of the cTNT promoter (AAV9-cTnt-Myocd) to Atp6v0d1AKO mice aged 16 weeks (Figure 5D). Subsequently, we detected 1.9- and 1.85-fold increases in myocardin protein levels in the hearts of control and Atp6v0d1AKO mice aged 36 weeks, respectively (Figure 5F-5G).
Increased myocardin expression ameliorates cardiac dysfunction in Atp6v0d1AKO mice. A, qPCR quantification of the mRNA levels of myocardin in control and Atp6v0d1AKO hearts. Data represent the mean ± SEM (n = 4) (two-tailed unpaired Student's t-test). B-C, Representative immunoblots and quantification of myocardin protein levels in control and Atp6v0d1AKO heart tissues. Data represent the mean ± SEM (n = 6) (two-tailed unpaired Student's t-test). D, Schematic illustration of AAV-mediated gene therapy, delivered via intravenous injection to control and Atp6v0d1AKO mice. E, qPCR quantification of the mRNA levels of myocardin. Data represent the mean ± SEM (n = 4 - 6) (one-way ANOVA). F, AAV9-mediated expression of myocardin in control and Atp6v0d1AKO hearts. G, Quantification of myocardin protein levels. Data represent the mean ± SEM (n = 4 - 6) (one-way ANOVA). H-I, Echo measurement of left ejection fraction (EF%) and fractional shortening (FS%) of control and Atp6v0d1AKO mice after tail vein injection of AAV9-cTnt-myocardin or AAV9-vectors. Data represent the mean ± SEM (n = 4 - 6) (one-way ANOVA). J, Representative images of hearts from control and Atp6v0d1AKO mice after tail vein injection of AAV9-cTnt-myocardin or AAV9-vectors. K-L, Heart weight (HW) to tibia ratio and lung weight (LW) to tibia ratio of control and Atp6v0d1AKO mice conducted with tail vein injection of AAV9-cTnt-myocardin or AAV9-vectors. Data represent the mean ± SEM (n = 4 - 6) (one-way ANOVA). M-N, Representative immunoblots and quantification of myocardin protein levels. Data represent the mean ± SEM (n = 4 - 6) (one-way ANOVA). O-P, Representative immunoblots and quantification of myocardin protein levels in normal chow diet (NCD) and high fat diet (HFD) mice. Data represent the mean ± SEM (n = 8) (two-tailed unpaired Student's t-test).
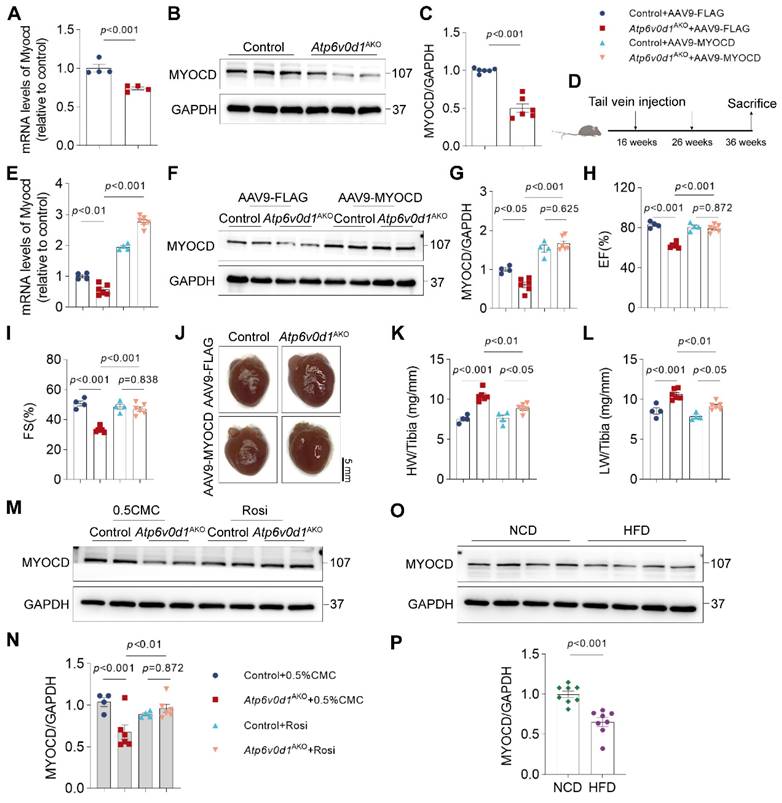
Although previous studies demonstrated that myocardin overexpression in cultured neonatal cardiomyocytes induced cardiomyocyte hypertrophy [35], we found that increasing myocardin expression level to this extent in adult mouse hearts had no significant effect on cardiac function and heart size, with comparable EF%, FS% and HW/TL in the control and myocardin-overexpressed (control + myocd) mice (Figure 5H-5L). However, restoring myocardin expression in the hearts of Atp6v0d1AKO mice (Atp6v0d1AKO + myocd) increased EF% and FS% to levels that were comparable to those in their littermate controls (Figure 5H-5I and Figure S8A), suggesting that the cardiac contractile functions of Atp6v0d1AKO mice were completely restored. Cardiac hypertrophy, as measured by HW/TL, was also attenuated by increasing myocardin expression (Figure 5J-5L). In addition, restoring myocardin expression in the hearts of Atp6v0d1AKO mice also reduced cardiomyocyte size and cardiac fibrosis (Figure S8B-S8D). These results collectively indicated that myocardin downregulation is critical for the onset and development of cardiomyopathy in Atp6v0d1AKO mice. Interestingly, we found that treatment with Rosiglitazone, a systemic insulin sensitizer which improved cardiac metabolism and function in Atp6v0d1AKO mice (Figure S7), completely restored myocardin expression in Atp6v0d1AKO hearts (Figure 5M-5N), suggesting a causal link between systemic insulin resistance and myocardin downregulation. In hearts of high fat diet-induced type 2 diabetes mice, we also found the expression of myocardin, as well as IRS1, was decreased (Figure 5O-5P and Figure S8E-S8F), providing further evidence for this hypothesis.
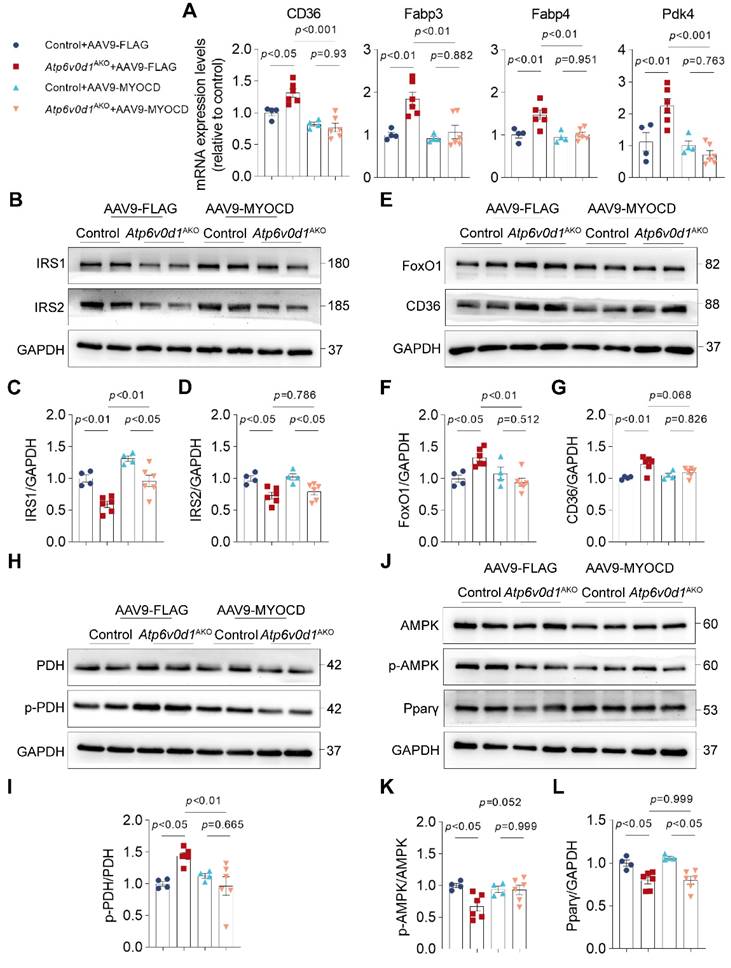
Given the critical role of disturbed cardiac metabolism in the development of cardiomyopathy, we decided to investigate the contribution of myocardin downregulation to metabolic disorders in Atp6v0d1AKO mouse hearts. Indeed, we found that restoring myocardin expression normalized the expression of genes involved in metabolic reprograming in Atp6v0d1AKO hearts, where the mRNA levels of Pdk4, CD36, Fabp3 and Fabp4 were significantly decreased (Figure 6A). Given the critical role of insulin signaling in regulating cardiac metabolism, we explored how myocardin regulates IRS expression in Atp6v0d1AKO mouse hearts. The results showed that myocardin upregulation increased the expression of IRS-1 (Figure 6B-6D). However, the expression of IRS-2 was not restored by myocardin upregulation (Figure 6B-6D), and serum insulin levels suggested that myocardin upregulation may not alter systemic insulin resistance (Figure S8G). In addition, the protein levels of FoxO1, CD36, p-PDH and p-AMPK were restored in Atp6v0d1AKO hearts (Figure 6E-6L). However, myocardin upregulation had no effect on PPARγ expression (Figure 6J and 6L). Previous studies demonstrated that PPARγ deficiency induced cardiac hypertrophy without interfering with cardiac contractile function [36]. Although myocardin upregulation completely restored cardiac contractile function in Atp6v0d1AKO mice (Figure 5H and 5I), the cardiac hypertrophy was ameliorated but not completely corrected, which may be attributed to the effect of PPARγ that was not counteracted by myocardin upregulation.
Increased myocardin expression ameliorates metabolic reprogramming in Atp6v0d1AKO mice. A, qPCR quantification of the mRNA levels of FAO-related genes from hearts of control and Atp6v0d1AKO hearts after tail vein injection of AAV9-cTnt-myocardin or AAV9-vectors. B-D, Representative immunoblots and quantification of IRS1 and IRS2 protein levels from heart tissues of control and Atp6v0d1AKO mice after tail vein injection of AAV9-cTnt-myocardin or AAV9-vectors. Representative immunoblots of two independent experiments are shown. E, Representative immunoblots of FoxO1, and CD36 protein levels from heart tissues of control and Atp6v0d1AKO mice after tail vein injection of AAV9-cTnt-myocardin or AAV9-vectors. Representative immunoblots of two independent experiments are shown. F-G, Quantification of specified densitometric ratios. H, Representative immunoblots of p-PDH and PDH protein levels from heart tissues of control and Atp6v0d1AKO mice after tail vein injection of AAV9-cTnt-myocardin or AAV9-vectors. Representative immunoblots of two independent experiments are shown. I, Quantification of pPDH/PDH ratio. J, Representative immunoblots of p-AMPK, AMPK and Pparp-γ protein levels from heart tissues of control and Atp6v0d1AKO mice after tail vein injection of AAV9-cTnt-myocardin or AAV9-vectors. Representative immunoblots of two independent experiments are shown. K-L, Quantification of specified densitometric ratios. Data represent the mean ± SEM (n = 4 - 6) (one-way ANOVA).
Myocardin is critical for transcriptional activation of IRS-1
Myocardin regulates the transcription of genes containing the CArG element. We conducted a thorough analysis of the IRS1 promoter utilizing the JASPAR database (http://jaspar.genereg.net/). The DNA analysis uncovered the existence of a CArG-like element within the promoter regions of IRS-1 in mice, rats, and humans (Figure 7A). This discovery implies an unexplored role of myocardin in regulating the insulin signal within the heart. To test this hypothesis, we cloned the promoter region of IRS-1 containing a CArG-like element into a luciferase reporter plasmid, and co-transfected HEK293T cells with a myocardin overexpression plasmid (Myocd-OE) alone or together with a SRF overexpression plasmid (SFR-OE). Cotransfection of the myocardin plasmid moderately increased the luciferase activities compared with that associated with the empty plasmid, whereas cotransfection with myocardin and SRF plasmids dramatically increased the luciferase activities (Figure 7B). Both deletion and mutation of the CArG-like element in the promoter region of IRS-1strongly suppressed the luciferase activities in HEK293T cells (Figure 7C-7E), confirming that the CArG-like element was a key cis-element for IRS1 transcription. Chromatin immunoprecipitation (ChIP)-qPCR assays further confirmed that SRF bound specifically to the CArG-like element in the IRS-1 promoter in neonatal rat ventricular myocytes (NRVMs) (Figure 7F). Moreover, we knocked down myocardin expression with siRNA targeting myocardin (si-Myocd) to investigate its ability to decrease IRS-1 expression in cultured NRVMs. As anticipated, myocardin knockdown decreased IRS-1 expression (Figure 7G-7I). Consistent with the notion that insulin resistance inhibited Akt activity, Akt phosphorylation was decreased in myocardin knockdown cells (Figure 7H and 7I). Collectively, these results indicated that myocardin directly regulates IRS-1 expression.
Myocardin is critical for transcriptional activation of IRS-1. A, The IRS1 promoter contains the conserved CArG-like element. B, Firefly luciferase activities detected in HEK293T cells transfected with promoterless pGL4.10 or vectors expressing IRS1 promoter in the presence of SRF or pcDNA3.0 and using Renillin as a normalization control. Results are depicted as fold of change in activity compared to pGL4.10 alone. Data represent the mean ± SEM (n = 3) (one-way ANOVA). C, The promoter activities of rat IRS1 promoter truncations were determined via luciferase assay. D, Mutation of the conserved CArG-like element in IRS1 promoter. E, Firefly luciferase activities of wild type and mutated IRS1 promoter. Data represent the mean ± SEM (n = 3) (two-tailed unpaired Student's t-test). F, IgG and SRF ChIP-PCR of rat neonatal cardiomyocytes. Enrichment of the IRS1 promoter following pull-down is normalized to input. Data represent the mean ± SEM (n = 3) (one-way ANOVA). G, qPCR quantification of the mRNA levels of myocardin, IRS1, IRS2 and FoxO1 in rat neonatal cardiomyocytes infected with myocardin siRNA or scrambled siRNA. Data represent the mean ± SEM (n = 6) (two-tailed unpaired Student's t-test). H, Representative immunoblots of myocardin, IRS1, IRS2 and FoxO1 protein levels in rat neonatal cardiomyocytes infected with myocardin siRNA (si-Myocd) or scrambled siRNA (si-Scramble) as a control. I, Quantification analysis of indicated protein levels. Data represent the mean ± SEM (n = 4 - 8) (two-tailed unpaired Student's t-test). J, Representative immunoblots of IRS1 and FoxO1 protein levels in rat neonatal cardiomyocytes infected with IRS1 siRNA (si-Irs1) or scrambled siRNA (si-Scramble) as a control. K, Quantification analysis of IRS1 and FoxO1 protein levels. Data represent the mean ± SEM (n = 9) (two-tailed unpaired Student's t-test). L, Representative immunoblots of IRS1 and FoxO1 protein levels in rat neonatal cardiomyocytes infected with myocardin siRNA (si-Mycod) or scrambled siRNA (si-Scr) and Ad-IRS1 or Ad-GFP. M, Quantification analysis of FoxO1 protein levels. Data represent the mean ± SEM (n = 4) (one-way ANOVA). N, Schematic illustration of the working model for the reduced expression of myocardin in Atp6v0d1AKO hearts resulting in the decline in IRS-1 expression and the development of lipodystrophy cardiomyopathy. We discovered that myocardin enhances the interaction between SRF and the CArG element, resulting in the transcriptional activation of IRS-1. Atp6v0d1AKO hearts have reduced myocardin expression, leading to decreased IRS-1 expression and upregulation of the downstream factor FoxO1. The translocation of FoxO1 to the nucleus exacerbates fatty acid uptake and oxidation through the upregulation of CD36 and FAO-associated genes, while suppressing glucose oxidation by upregulation of PDK4, and subsequently phosphorylation of PDH. Treatment with Rosiglitazone or upregulated myocardin expression increased IRS-1 expression and restored the balance of genes involved in lipid and glucose metabolism. Hence, Atp6v0d1 deletion in hearts establishes a novel murine model of lipodystrophy cardiomyopathy, offering a new approach to understanding the pathogenesis of clinical adipose-related cardiomyopathy.
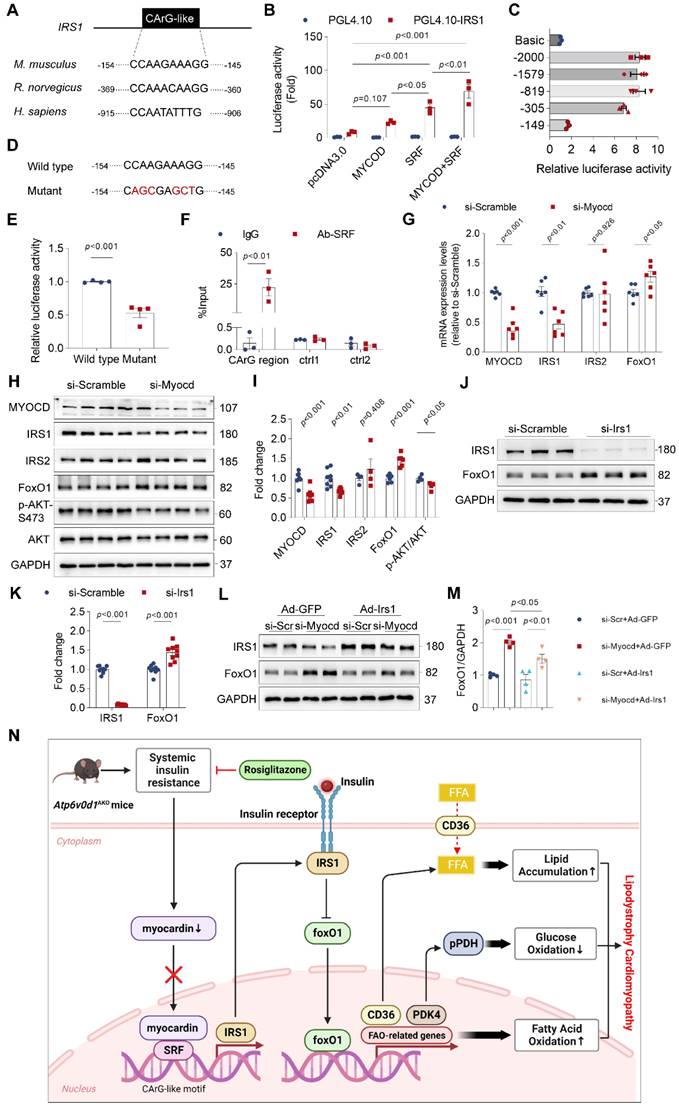
FoxO1 is a critical regulator of lipid and glucose metabolism. In myocardin knockdown cardiomyocytes, we also observed an increase in protein abundance of FoxO1 (Figure 7H and 7I). The results are consistent with our findings in vivo that increasing myocardin expression in hearts of Atp6v0d1AKO mice decreased FoxO1 expression (Figure 6E and 6F). Since myocardin promotes gene transcription, the changes in FoxO1 expression upon manipulation of myocardin are not the direct effect of myocardin. Previous studies suggested that the activation of insulin signal inhibits cardiac FoxO1 expression/activity [37]. To test the causal relationship between decrease in IRS-1 and increase in FoxO1 expression following myocardin knockdown, we examined the direct effect of IRS-1 knockdown on FoxO1 expression, and increasing IRS-1 on myocardin downregulation-induced FoxO1 increase. The data showed that FoxO1 protein abundance was increased following IRS-1 knockdown (Figure 7J and 7K). Adenovirus-mediated overexpression of IRS-1 in cardiomyocytes significantly suppressed the increased expression of FoxO1 induced by myocardin-knockdown (Figure 7L and 7M). The data collectively indicate IRS-1 mediates FoxO1 increase under the condition of myocardin downregulation.
Discussion
In the current study, we identified Atp6v0d1 as a crucial regulator of adipogenesis, and showed that loss of adipose Atp6v0d1 in mice resulted in a gradual loss of adipose tissues accompanied by systemic insulin resistance, hypercholesterolemia and hepatosteatosis, which are key features of generalized lipodystrophy [38, 39]. However, the link between Vod1 and lipodystrophy in human has yet to be confirmed. Considering that deletion of any of the V-ATPase subunits all cause embryonic lethality, genetic mutations that severely affect Vod1 functions may not be discovered. In addition, we found that Vod1 expression was increased in the AT of HFD-fed mice (Figure S1H and S1I). Collectively, our data suggest Vod1 subunit plays an important role in regulating adipogenesis and AT functions. Here, we found that Atp6v0d1AKO mice display a similar developmental trajectory of cardiomyopathy to that observed in Bscl2-/- mice, the most widely used animal model for studying lipodystrophy-related cardiomyopathy recapitulating human type 2 Berardinelli-Seip congenital lipodystrophy (BSCL2) disease, which progresses from compensated cardiac hypertrophy to heart failure [40]. Yet, loss of BSCL2 specifically in the myocardium also leads to the onset of cardiomyopathy [41], adding the complexity of this model as whether dysfunctional AT or altered cardiac functions as the direct cause for cardiomyopathy. Moreover, the cardiomyopathy in Atp6v0d1AKO mice can be rescued by increasing systemic insulin sensitivity with Rosiglitazone (Figure S6), similar to that observed in Bscl2-/- mice [42], demonstrating the prominent feature of lipodystrophy cardiomyopathy that had been observed in the well-established Bscl2-/- lipodystrophy mice.
However, Atp6v0d1AKO mice exhibit some distinct pathological changes in hearts compared with those in Bscl2-/- mice. First, lipid deposition in the heart, a pathological change that is widely observed in patients with lipodystrophy cardiomyopathy, is evident in Atp6v0d1AKO mice, whereas Bscl2-/- cardiomyopathy is characterized by reduced lipid accumulation [40]. Second, the heart energy expenditure in Atp6v0d1AKO mice is shifted preferentially to fatty acid oxidation. This, together with lipid accumulation implicates the occurrence of “lipotoxic” cardiomyopathy in Atp6v0d1AKO mice, which is in contrast to Bscl2-/- cardiomyopathy mainly caused by hyperglycemia and glucotoxicity [43]. Third, the cardiac hypertrophy in Atp6v0d1AKO cardiomyopathy is not accompanied by activation of the fetal genes, Nppa and Myh7, even when the contractile function is impaired. There is evidence that lower levels of ANP are associated with increased muscle mass in overweight and obese patients compared with those with normal weight [44]. Most obese patients with HF have comparatively low levels of natriuretic peptides [45], although the underlying mechanisms are unknown. From this perspective, Atp6v0d1AKO cardiomyopathy demonstrated the distinctive characteristics of obese cardiomyopathy in humans. Previous studies revealed that myocardin strongly transactivates the promoter for ANP [46]. Myocardin expression was decreased in Atp6v0d1AKO cardiomyopathy, and mimicking myocardin downregulation in cardiomyocytes decreased ANP expression (Figure S9). Our findings suggest that decreased myocardin expression may explain the distinct HF phenotype in obesity cardiomyopathy. Taken together, these findings highlight the potential of the murine Atp6v0d1AKO lipodystrophy model as a new approach to gain insights into the development and underlying mechanisms of adipose tissue-associated cardiomyopathy.
Insulin signaling in the heart plays a central role in regulating cardiac metabolism, maintaining normal cardiac functions and survival of cardiomyocytes [47]. Regulation of IRS has major roles in the control of cardiac metabolic homeostasis and function [47]. Loss of IRS-1 and IRS-2 in the heart following chronic insulin stimulation contributes to insulin resistance, abnormal lipid and glucose metabolism, and heart failure [48]. Phosphorylation of IRS protein by p38α MAP kinase, JNK, mTOR, or protein kinase C (PKC) stimulates its degradation [49]. However, transcriptional regulation of IRS, especially in myocardial tissues, remains to be elucidated. Here, we uncovered a previously unrecognized role of myocardin in regulating IRS-1 transcription. Through luciferase reporter and ChIP-qPCR assays, we revealed that myocardin facilitates the interaction of SRF with the conserved CArG element of the IRS-1 promoter region to promote transcriptional activation of IRS-1. Moreover, knockdown of myocardin expression decreased IRS-1 transcription. In the hearts of Atp6v0d1AKO mice, IRS-1 and myocardin expression levels were concomitantly decreased, while restored myocardin expression increased IRS-1 protein abundance. These results highlighted the critical role of myocardin downregulation in promoting insulin resistance in Atp6v0d1AKO cardiomyopathy.
In the hearts of Atp6v0d1AKO mice, the expression levels of genes associated with FA uptake (CD36) and FA oxidation (Acadl, Acadm, Acads, Cpt1b, Cpt1c, Acsl1, Fabp3 and Fabp4) were upregulated, resulting in a preferential shift of heart energy expenditure to FAO. Furthermore, PDK4 expression was increased and PDH was activated, suggesting that glucose oxidation is suppressed. Interestingly, myocardin upregulation largely restored the expression of the genes controlling FA uptake and oxidation, and glucose metabolism, suggesting a critical role for myocardin in causing lipid and glucose metabolism disorder in Atp6v0d1AKO cardiomyopathy. Accumulating evidence indicates that FoxO1 limits glucose oxidation by upregulating PDK4, and increases FA uptake and oxidation through upregulation of CD36 and FAO-associated genes [37].
FoxO1 expression was increased in Atp6v0d1AKO heart tissue, and myocardin upregulation decreased FoxO1 expression to normal levels. Moreover, knockdown of myocardin expression in cardiomyocytes increased FoxO1 expression. Collectively, these data indicate that FoxO1 mediates myocardin downregulation-induced lipid and glucose metabolism disorders.
FoxO1 is a downstream target of insulin, whereby insulin inhibits cardiac FoxO1 expression/activity through the IRS-1/Akt signaling in order to prevent gluconeogenesis. Previous studies showed that FoxO1 expression/activity was increased in HFD-induced diabetic cardiomyopathy [50]. We found FoxO1 upregulation in heart tissues of Atp6v0d1AKO mice. However, a causal link between insulin resistance and increased FoxO1 expression following myocardin downregulation in Atp6v0d1AKO mice remains to be established. Our data showed that knockdown of IRS-1 decreased FoxO1 expression. Moreover, increasing IRS-1 expression reversed myocardin downregulation-induced FoxO1 upregulation in cardiomyocytes. Therefore, the myocardin-mediated increase in FoxO1 expression is a result of myocardin-induced insulin resistance. Taken together, we propose that the myocardin inhibition-insulin resistance-increased FoxO1 expression signaling axis plays a central role in promoting lipid and glucose metabolism disorder and the development of cardiomyopathy in Atp6v0d1AKO mice (Figure 7N).
In this study, we found that Rosiglitazone treatment restored myocardin expression in Atp6v0d1AKO mice, suggesting that myocardin downregulation is a result of the systemic insulin resistance that occurs in diabetes, obesity and lipodystrophy. Indeed, we observed decreased myocardin expression in HFD-induced type 2 diabetic cardiomyopathy in mice. Even though myocardin downregulation may not be present in all types of metabolic cardiomyopathy, insulin resistance is prominent in the heart in these conditions [51]. Thus, upregulating myocardin is implicated as a potential therapeutic strategy for metabolic cardiomyopathies. The therapeutic effect of myocardin on treatment of diabetic and obesity cardiomyocytes, as well as the mechanism underlying systemic insulin resistance and reduction of myocardin in heart warrants future study.
Interestingly, V-ATPase in cardiomyocytes has also been shown to participate in regulating cardiac biological process and metabolism. For example, the study by Liu and colleagues demonstrated that lipid oversupply impairs V-ATPase function in heart by promoting CD36-mediated lipid uptake, which feeds forward to enhanced CD36 translocation, leading to insulin resistance and contractile dysfunction [52]. Activation of V-ATPase in the heart can ameliorate lipid-induced cardiomyopathy [53]. More recently, a study by Li, et al. showed that V-ATPase subunit, ATP6AP2 has a critical role in regulating autophagic flux in heart [54]. Reducing ATP6AP2 expression in cardiomyocytes impairs autophagic flux and subsequently activates NLRPs, resulting in cardiac dysfunction. At present, it remains unknown whether and how Vod1 subunit expressed in heart participate in regulation of cardiac function under physiological and pathological conditions. In a recent study, deletion of Vod2, an isoform of the Vod1 subunits in the heart indeed increased surface CD36 abundance in cardiomyocytes and increased cardiac fibrosis [53]. Given the critical role of Vod1 subunit in adipogenesis, and deleting Atp6v0d1 in adipocytes results in the pathogenesis of cardiomyopathy, it's intriguing to explore the effect of deleting Atp6v0d1 in cardiomyocytes on cardiac function.
In summary, by deleting Atp6v0d1 in adipocytes, we established a murine generalized lipodystrophy model that reproduces the major pathological features observed in patients with lipodystrophy and obesity cardiomyopathy. This model paves a new avenue for gaining insights into the pathogenesis of adipose dysfunction-related cardiomyopathy. Furthermore, we identified myocardin downregulation as a key mechanism of cardiac insulin resistance mediated by decreasing IRS-1 expression, highlighting the therapeutic potential of upregulating myocardin activity for lipodystrophy cardiomyopathy and insulin resistance-related HF.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We appreciate Dr Jessica Tamanini (Shenzhen University Health Science Center, China and ETediting, United Kingdom) for language editing.
Funding
This research was supported by the National Natural Science Foundation of China (Grant No. 32271151, 81970250, 82170881, 81800383), the Natural Science Foundation of Guangdong Province (Grant No. 2022A1515012310), Basic Research Foundation of Shenzhen (JCYJ20210324094006018, JCYJ20210324094808022 and JCYJ20190808170401660). Shenzhen Science and Technology Program (Grant No. RCBS20210609103118012). Shenzhen Key Laboratory of Metabolism and Cardiovascular Homeostasis Grant (ZDSYS20190902092903237).
Author contributions
J. Liu and W. Yuan designed the research; W. Yuan, H. Lin, Y. Sun, L. Liu, Y. Song, M. Yan, X. Zhang, X. Lu, Y. Xu and Q. He performed the experiments; W. Yuan, Y. Li, C. Zhang and Y. Pan analyzed the data; Y. Huang and K. Ouyang contributed to the interpretation of the data; J. Liu, X. Lu and Y. Li interpreted data and wrote the manuscript. All authors read and approved the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Koenen M, Hill MA, Cohen P, Sowers JR. Obesity, Adipose Tissue and Vascular Dysfunction. Circ Res. 2021;128:951-68
2. Vekic J, Zeljkovic A, Stefanovic A, Jelic-Ivanovic Z, Spasojevic-Kalimanovska V. Obesity and dyslipidemia. Metabolism. 2019;92:71-81
3. Polyzos SA, Kountouras J, Mantzoros CS. Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism. 2019;92:82-97
4. Azevedo PS, Minicucci MF, Santos PP, Paiva SA, Zornoff LA. Energy metabolism in cardiac remodeling and heart failure. Cardiol Rev. 2013;21:135-40
5. Boengler K, Kosiol M, Mayr M, Schulz R, Rohrbach S. Mitochondria and ageing: role in heart, skeletal muscle and adipose tissue. J Cachexia Sarcopenia Muscle. 2017;8:349-69
6. Enomoto M, Ishizu T, Seo Y, Yamamoto M, Suzuki H, Shimano H. et al. Subendocardial Systolic Dysfunction in Asymptomatic Normotensive Diabetic Patients. Circ J. 2015;79:1749-55
7. Ren J, Wu NN, Wang S, Sowers JR, Zhang Y. Obesity cardiomyopathy: evidence, mechanisms, and therapeutic implications. Physiol Rev. 2021;101:1745-807
8. Romano MMD, Chacon PAI, Ramalho FNZ, Foss MC, Schmidt A. Cardiac Alterations in Patients with Familial Lipodystrophy. Arq Bras Cardiol. 2020;114:305-12
9. Sanon VP, Handelsman Y, Pham SV, Chilton R. Cardiac Manifestations of Congenital Generalized Lipodystrophy. Clin Diabetes. 2016;34:181-6
10. Scatteia A, Pagano C, Pascale C, Guarini P, Marotta G, Perrone-Filardi P. et al. Asymmetric hypertrophic cardiomyopathy in generalized lipodystrophy. Int J Cardiol. 2016;202:724-5
11. Wu CH, Mohammadmoradi S, Thompson J, Su W, Gong M, Nguyen G. et al. Adipocyte (Pro)Renin-Receptor Deficiency Induces Lipodystrophy, Liver Steatosis and Increases Blood Pressure in Male Mice. Hypertension. 2016;68:213-9
12. Zapata RC, Carretero M, Reis FCG, Chaudry BS, Ofrecio J, Zhang D. et al. Adipocytes control food intake and weight regain via Vacuolar-type H(+) ATPase. Nat Commun. 2022;13:5092
13. Nishi T, Forgac M. The vacuolar (H+)-ATPases-nature's most versatile proton pumps. Nat Rev Mol Cell Biol. 2002;3:94-103
14. Battelli MG, Bolognesi A, Polito L. Pathophysiology of circulating xanthine oxidoreductase: new emerging roles for a multi-tasking enzyme. Biochim Biophys Acta. 2014;1842:1502-17
15. Marshansky V, Futai M. The V-type H+-ATPase in vesicular trafficking: targeting, regulation and function. Curr Opin Cell Biol. 2008;20:415-26
16. Wang S, Schianchi F, Neumann D, Wong LY, Sun A, van Nieuwenhoven FA. et al. Specific amino acid supplementation rescues the heart from lipid overload-induced insulin resistance and contractile dysfunction by targeting the endosomal mTOR-v-ATPase axis. Mol Metab. 2021;53:101293
17. Kane PM. The long physiological reach of the yeast vacuolar H+-ATPase. J Bioenerg Biomembr. 2007;39:415-21
18. Vasanthakumar T, Rubinstein JL. Structure and Roles of V-type ATPases. Trends Biochem Sci. 2020;45:295-307
19. Wang S, Han Y, Nabben M, Neumann D, Luiken J, Glatz JFC. Endosomal v-ATPase as a Sensor Determining Myocardial Substrate Preference. Metabolites. 2022 12
20. Zhang CS, Jiang B, Li M, Zhu M, Peng Y, Zhang YL. et al. The lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab. 2014;20:526-40
21. Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011;334:678-83
22. Sacks D, Baxter B, Campbell BCV, Carpenter JS, Cognard C, Dippel D. et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int J Stroke. 2018;13:612-32
23. Farmer BC, Kluemper J, Johnson LA. Apolipoprotein E4 Alters Astrocyte Fatty Acid Metabolism and Lipid Droplet Formation. Cells. 2019 8
24. Schoonejans JM, Blackmore HL, Ashmore TJ, Pantaleão LC, Pellegrini Pisani L, Dearden L. et al. Sex-specific effects of maternal metformin intervention during glucose-intolerant obese pregnancy on body composition and metabolic health in aged mouse offspring. Diabetologia. 2022;65:2132-45
25. Yang L, Li RC, Xiang B, Li YC, Wang LP, Guo YB. et al. Transcriptional regulation of intermolecular Ca(2+) signaling in hibernating ground squirrel cardiomyocytes: The myocardin-junctophilin axis. Proc Natl Acad Sci U S A. 2021 118
26. Suhail H, Peng H, Xu J, Sabbah HN, Matrougui K, Liao TD. et al. Knockout of ACE-N facilitates improved cardiac function after myocardial infarction. J Mol Cell Cardiol Plus. 2023 3
27. Torres-Quesada O, Doerrier C, Strich S, Gnaiger E, Stefan E. Physiological Cell Culture Media Tune Mitochondrial Bioenergetics and Drug Sensitivity in Cancer Cell Models. Cancers (Basel). 2022 14
28. Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A. et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020;48:D498-d503
29. Heather LC, Clarke K. Metabolism, hypoxia and the diabetic heart. J Mol Cell Cardiol. 2011;50:598-605
30. Yan D, Cai Y, Luo J, Liu J, Li X, Ying F. et al. FOXO1 contributes to diabetic cardiomyopathy via inducing imbalanced oxidative metabolism in type 1 diabetes. J Cell Mol Med. 2020;24:7850-61
31. Montaigne D, Butruille L, Staels B. PPAR control of metabolism and cardiovascular functions. Nat Rev Cardiol. 2021;18:809-23
32. Sun Y, Zhou S, Guo H, Zhang J, Ma T, Zheng Y. et al. Protective effects of sulforaphane on type 2 diabetes-induced cardiomyopathy via AMPK-mediated activation of lipid metabolic pathways and NRF2 function. Metabolism. 2020;102:154002
33. Hill MA, Yang Y, Zhang L, Sun Z, Jia G, Parrish AR. et al. Insulin resistance, cardiovascular stiffening and cardiovascular disease. Metabolism. 2021;119:154766
34. Huang J, Min Lu M, Cheng L, Yuan LJ, Zhu X, Stout AL. et al. Myocardin is required for cardiomyocyte survival and maintenance of heart function. Proc Natl Acad Sci U S A. 2009;106:18734-9
35. Xing W, Zhang TC, Cao D, Wang Z, Antos CL, Li S. et al. Myocardin induces cardiomyocyte hypertrophy. Circ Res. 2006;98:1089-97
36. Duan SZ, Ivashchenko CY, Russell MW, Milstone DS, Mortensen RM. Cardiomyocyte-specific knockout and agonist of peroxisome proliferator-activated receptor-gamma both induce cardiac hypertrophy in mice. Circ Res. 2005;97:372-9
37. Chistiakov DA, Orekhov AN, Bobryshev YV. The impact of FOXO-1 to cardiac pathology in diabetes mellitus and diabetes-related metabolic abnormalities. Int J Cardiol. 2017;245:236-44
38. Nguyen ML, Sachdev V, Burklow TR, Li W, Startzell M, Auh S. et al. Leptin Attenuates Cardiac Hypertrophy in Patients With Generalized Lipodystrophy. J Clin Endocrinol Metab. 2021;106:e4327-e39
39. Brown RJ, Oral EA, Cochran E, Araújo-Vilar D, Savage DB, Long A. et al. Long-term effectiveness and safety of metreleptin in the treatment of patients with generalized lipodystrophy. Endocrine. 2018;60:479-89
40. Zhou H, Lei X, Yan Y, Lydic T, Li J, Weintraub NL. et al. Targeting ATGL to rescue BSCL2 lipodystrophy and its associated cardiomyopathy. JCI Insight. 2019 5
41. Zhou H, Li J, Su H, Li J, Lydic TA, Young ME. et al. BSCL2/Seipin deficiency in hearts causes cardiac energy deficit and dysfunction via inducing excessive lipid catabolism. Clin Transl Med. 2022;12:e736
42. Chang H, Di T, Wang Y, Zeng X, Li G, Wan Q. et al. Seipin deletion in mice enhances phosphorylation and aggregation of tau protein through reduced neuronal PPARγ and insulin resistance. Neurobiol Dis. 2019;127:350-61
43. Lugat A, Joubert M, Cariou B, Prieur X. [At the heart of diabetic cardiomyopathy: Bscl2 knockout mice to investigate glucotoxicity]. Med Sci (Paris). 2018;34:563-70
44. Obokata M, Reddy YNV, Pislaru SV, Melenovsky V, Borlaug BA. Evidence Supporting the Existence of a Distinct Obese Phenotype of Heart Failure With Preserved Ejection Fraction. Circulation. 2017;136:6-19
45. Mehra MR, Uber PA, Park MH, Scott RL, Ventura HO, Harris BC. et al. Obesity and suppressed B-type natriuretic peptide levels in heart failure. J Am Coll Cardiol. 2004;43:1590-5
46. Liu L, Zhao Q, Kong M, Mao L, Yang Y, Xu Y. Myocardin-related transcription factor A regulates integrin beta 2 transcription to promote macrophage infiltration and cardiac hypertrophy in mice. Cardiovasc Res. 2022;118:844-58
47. Guo S. Insulin signaling, resistance, and the metabolic syndrome: insights from mouse models into disease mechanisms. J Endocrinol. 2014;220:T1-t23
48. Qi Y, Xu Z, Zhu Q, Thomas C, Kumar R, Feng H. et al. Myocardial loss of IRS1 and IRS2 causes heart failure and is controlled by p38α MAPK during insulin resistance. Diabetes. 2013;62:3887-900
49. Copps KD, White MF. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia. 2012;55:2565-82
50. Zhang M, Sui W, Xing Y, Cheng J, Cheng C, Xue F. et al. Angiotensin IV attenuates diabetic cardiomyopathy via suppressing FoxO1-induced excessive autophagy, apoptosis and fibrosis. Theranostics. 2021;11:8624-39
51. Nishida K, Otsu K. Inflammation and metabolic cardiomyopathy. Cardiovasc Res. 2017;113:389-98
52. Liu Y, Steinbusch LKM, Nabben M, Kapsokalyvas D, van Zandvoort M, Schönleitner P. et al. Palmitate-Induced Vacuolar-Type H(+)-ATPase Inhibition Feeds Forward Into Insulin Resistance and Contractile Dysfunction. Diabetes. 2017;66:1521-34
53. Wang S, Han Y, Liu R, Hou M, Neumann D, Zhang J. et al. Glycolysis-Mediated Activation of v-ATPase by Nicotinamide Mononucleotide Ameliorates Lipid-Induced Cardiomyopathy by Repressing the CD36-TLR4 Axis. Circ Res. 2024;134:505-25
54. Li L, Cui YJ, Liu Y, Li HX, Su YD, Li SN. et al. ATP6AP2 knockdown in cardiomyocyte deteriorates heart function via compromising autophagic flux and NLRP3 inflammasome activation. Cell Death Discov. 2022;8:161
Author contact
![]() Corresponding authors: Jie Liu, M.D., Ph.D., Tel: +86-755-22673500, Email: liujedu.cn; Xifeng Lu, Ph.D., Email: xifengluedu.cn; Ying Li, Ph.D., Email: liying826edu.cn.
Corresponding authors: Jie Liu, M.D., Ph.D., Tel: +86-755-22673500, Email: liujedu.cn; Xifeng Lu, Ph.D., Email: xifengluedu.cn; Ying Li, Ph.D., Email: liying826edu.cn.