Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2024; 14(5):1815-1828. doi:10.7150/thno.89701 This issue Cite
Research Paper
Precision peptide theranostics: developing N- to C-terminus optimized theranostics targeting cholecystokinin-2 receptor
Marwa N. Rahimi1,2, Alicia Corlett3, Jessica Van Zuylekom4, Marc Antoine Sani5, Benjamin Blyth4, Philip Thompson6, Peter D. Roselt1,2, Mohammad B. Haskali1,2 ![]()
1. Department of Radiopharmaceutical Sciences, Cancer Imaging, The Peter MacCallum Cancer Centre, Victoria 3000, Australia.
2. Sir Peter MacCallum Department of Oncology, The University of Melbourne, Victoria 3010, Australia.
3. Cancer Imaging, Peter MacCallum Cancer Centre, Melbourne, Victoria 3000, Australia.
4. Models of Cancer Translational Research Centre, The Peter MacCallum Cancer Centre, Victoria 3000, Australia.
5. The Bio21 Institute, School of Chemistry, The University of Melbourne, Melbourne, Victoria, 3010 Australia.
6. Medicinal Chemistry, Monash Institute of Pharmaceutical Sciences, Faculty of Pharmacy and Pharmaceutical Sciences, Monash University (Parkville Campus), Parkville, Victoria 3052, Australia.
Received 2023-8-31; Accepted 2024-1-10; Published 2024-2-24
Abstract

Peptides are ideal for theranostic development as they afford rapid target accumulation, fast clearance from background tissue, and exhibit good tissue penetration. Previously, we developed a novel series of peptides that presented discreet folding propensity leading to an optimal candidate [68Ga]Ga-DOTA-GA1 ([D-Glu]6-Ala-Tyr-NMeGly-Trp-NMeNle-Asp-Nal-NH2) with 50 pM binding affinity against cholecystokinin-2 receptors (CCK2R). However, we were confronted with challenges of unfavorably high renal uptake.
Methods: A structure activity relationship study was undertaken of the lead theranostic candidate. Prudent structural modifications were made to the peptide scaffold to evaluate the contributions of specific N-terminal residues to the overall biological activity. Optimal candidates were then evaluated in nude mice bearing transfected A431-CCK2 tumors, and their biodistribution was quantitated ex vivo.
Results: We identified and confirmed that D-Glu3 to D-Ala3 substitution produced 2 optimal candidates, [68Ga]Ga-DOTA-GA12 and [68Ga]Ga-DOTA-GA13. These radiopeptides presented with high target/background ratios, enhanced tumor retention, excellent metabolic stability in plasma and mice organ homogenates, and a 4-fold reduction in renal uptake, significantly outperforming their non-alanine counterparts.
Conclusions: Our study identified novel radiopharmaceutical candidates that target the CCK2R. Their high tumor uptake and reduced renal accumulation warrant clinical translation.
Introduction
Over the last 60 years, advances in molecular and cell biology techniques have transformed the foundations of our modern medical scientific understanding. This has allowed researchers and clinicians to work concertedly to unravel the cellular pathways that drive cancer and develop new techniques for non-invasive, high-resolution, in vivo imaging technologies [1-3]. Targeted molecular imaging is an indispensable research and clinical tool that provides diagnostic, prognostic, and predictive information in oncology as well as cardiology, neurology, and infectious and inflammatory disorders [4]. Unlike anatomical imaging techniques such as computed tomography (CT) and magnetic resonance imaging (MRI), molecular imaging techniques such as positron emission tomography (PET) examine the cellular abnormalities that are the basis of malignancies, quantifying target expression using imaging probes [5, 6]. The evolution of PET and single photon emission computed tomography (SPECT) imaging methods has afforded new opportunities to produce exceptional streamlined diagnostic and therapeutic radiopharmaceuticals using the same targeting vector [4, 7]. By combining therapeutics and diagnostics using the same vector but different radionuclides, we can produce theranostic radiopharmaceuticals [8].
In recent years, work in theranostic radiopharmaceuticals has seen unprecedented acceleration with various radioisotope-labeled ligands being introduced for the diagnosis and therapy of metastatic neuroendocrine tumors, prostate cancer and many other malignancies [9, 10]. These successes have inspired the development of many other peptide-based radiopharmaceuticals that deliver systemic treatment designed to eliminate both residual primary lesions and metastatic cancer cell deposits [11, 12].
The cholecystokinin-2 receptor (CCK2R) is a highly promising target in nuclear medicine and has been the focus of radiopharmaceutical development over the past two decades [13]. CCK2R is overexpressed in multiple tumor states, including medullary thyroid carcinomas [14], small cell lung cancer [14], somatostatin-2 negative neuroendocrine tumors [15], stromal ovarian cancer [16, 17] and gastrointestinal stromal tumors [18, 19]. Additionally, targeting CCK2R has shown promising results for gastroenteropancreatic and bronchopulmonary neuroendocrine tumors, especially insulinomas and vipomas, as well as bronchial and ileal carcinoids [13, 18]. The absence of CCK2R expression in normal, healthy surrounding tissues and overexpression in the tumor environment provides favorable opportunities for high-contrast diagnostic imaging and radionuclide therapy of these tumors, making CCK2R an excellent target for the development of radiolabeled peptides for diagnosis and therapy in nuclear medicine.
Gastrin (G17), the natural ligand of CCK2R, and minigastrin (MG) (Figure 1), the truncated variant of the endogenous molecule, have undergone several stages of development by various groups to identify critical binding features, including truncating the lengthy peptide and optimizing several other characteristics to make it suitable for use as a theranostic radiopharmaceutical in nuclear medicine. The pentameric glutamic acid chain located on the N-terminus of the MG peptide contributes to the increased biological potency and higher binding affinity by promoting the peptide to fold in a hairpin structure, thereby stabilising the peptide in a biologically active conformation [20-22]. The C-terminus of the MG peptide houses the critical tetrapeptide sequence [Trp-Met-Asp-Phe-NH2], responsible for the recognition of the peptide by the CCK2R target [23]. Many modifications have been explored in these regions, seeking to address issues presented with the overall metabolic stability and biodistribution of the peptide. These endeavors have produced the clinically translated minigastrin analogs CP04 (PP-F11) and PP-F11N [24, 25]. Substitution of the penta-L-glutamic acid chain to its D-glutamic acid (D-Glu) and the addition of a sixth D-Glu residue in both CP04 and PP-F11N resulted in enhanced metabolic stability to the parent MG peptide, a known technique to safeguard peptides against enzymatic attack in the cellular environment.
Chemical structures (above) and amino acid sequences (below) of CCK2R binding peptides. Minigastrin (MG), the lead molecule, and structural modifications made to produce high affinity binding Gastramide (GA) peptides.
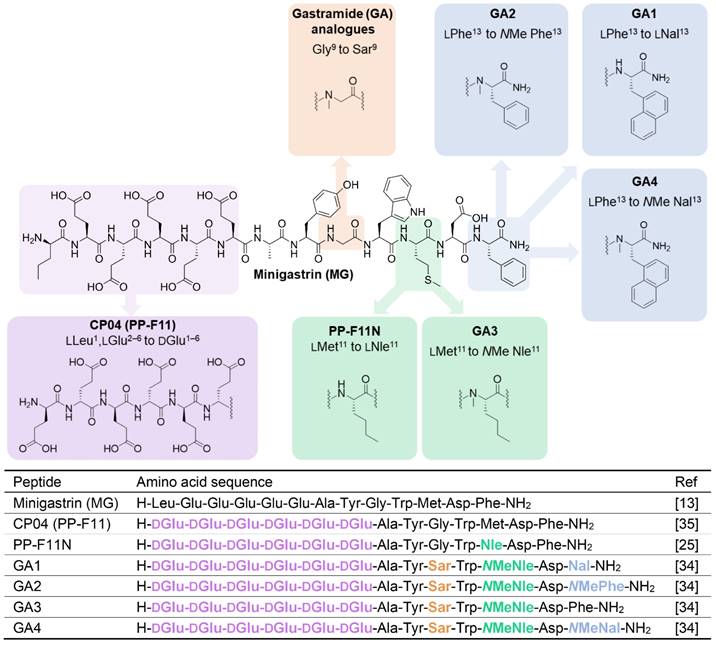
Renal reabsorption of radiolabeled peptides is a clinical problem, often restricting the maximum therapeutic dose of the theranostic, with the kidneys being the dose-limiting organ in peptide receptor radionuclide therapy (PRRT) [26, 27]. There have been several strategies explored by groups to address this key issue of renal accumulation of CCK2R targeting peptides. It is established that high kidney retention of the MG analogs is related to the presence of N-terminal ionic glutamic acid residues [28]. Therefore, a popular approach to suppress unfavorable kidney uptake has been the truncation of the implicated pentameric glutamic acid chain in the CP04 sequence to produce MG11 [21, 29-32].
Roques and coworkers were early explorers of the CCK2R landscape. They conducted a comprehensive investigation of promising CCK2R ligands, primarily on the CCK8 and CCK4 scaffold [33]. They substituted N-methylated norleucine (NMe-Nle) for methionine (Met) and introduced bulky aromatic naphthylalanine (Nal) to the scaffold, which vastly improved affinity, selectivity, and bioavailability of peptides to CCK2R [33]. These modifications were incorporated in MG11 to produce the high-affinity ligand MGS5 [32]. Its truncated length due to deletion of the N-terminal glutamic acid residues and N-methylation of the C-terminus led to reduced kidney accumulation by a factor greater than 25, resulting in significant improvement of the tumor-to-kidney ratio [29].
Recent work by our group dramatically improved upon the biological and pharmacological properties of CP04 by employing the modifications explored by Roques and coworkers concurrently and independently of MGS5 developments. By incorporating conformational constraints such as N-methylation of several residues and enhancing the amphiphilic character of the peptides, we developed a novel class of optimized peptides which we denoted as Gastramide (GA). GA peptide analogs afforded low pM affinity against CCK2R (Figure 1) and confirmed the important contributions of secondary peptide conformations in enhancing the biological affinity of peptides to their target [34]. This investigation resulted in the first generation of optimized candidates GA1, GA3, and GA4 (Figure 1) [34]. Despite these astounding improvements to the overall stability and affinity of the peptides, reducing their uptake and retention in the kidneys would further increase their suitability for clinical theranostic applications [26].
In the present work, our structural investigation of the GA scaffold resumes with a comprehensive evaluation of the established GA1. Previously shown to have low picomolar affinity (IC50 50 pM for CCK2R), GA1 is an ideal candidate for conducting a structure activity relationship (SAR) study to determine the importance of individual N-terminal glutamic acid (Glu) residues in contributing to target affinity. We aim to improve pharmacokinetic properties and reduce renal uptake of the peptide by identifying nonessential residues and substituting them with more favorable alternatives, to produce optimal theranostic candidates.
Methods, Results, and Discussion
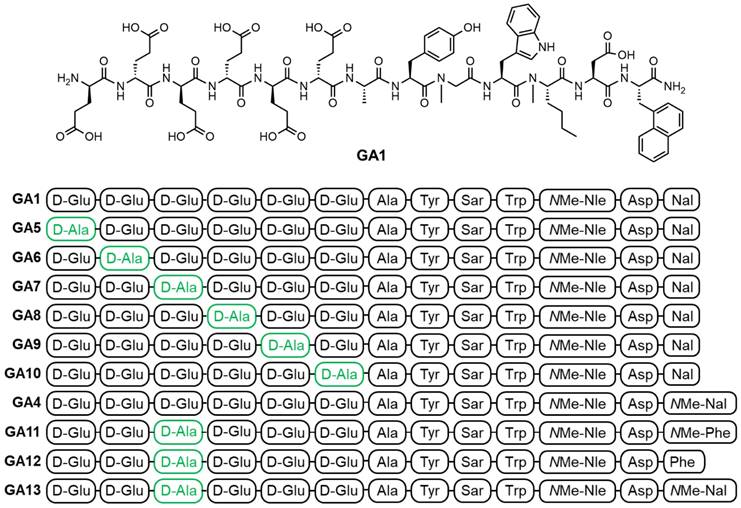
Peptide synthesis, DOTA coupling and natGa/68Ga-labeling: We prepared a D-Ala scan library (GA5-GA10) based on GA1 by systematically substituting each one of the N-terminal D-Glu residues (Figure 2). This enabled the evaluation of the synergistic effect of each D-Glu on the peptides' overall potency. Peptides tolerating D-Ala substitution were then modified at the C-terminal residue (GA4, GA11-13) to prepare metabolically stable and potent theranostic candidates (Figure 2).
The chemical structure of linear peptides GA1 (top) and peptide sequences of alanine analogs GA1, GA4-GA13 (bottom). Residues highlighted in green indicate positions of D-Ala substitution.
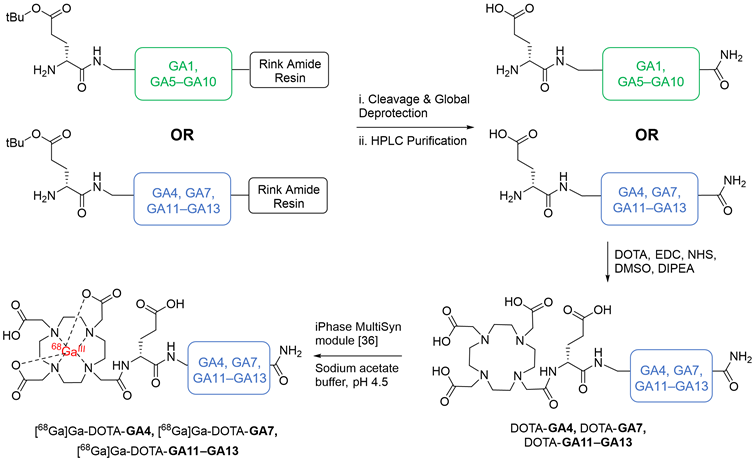
The established minigastrin analogue CP04 (PP-F11) (Figure 1), its derivative GA1, D-Ala substituted GA5-GA10, and other CCK2R scaffolds GA4, GA11-13 were synthesized following the method described in our supplementary data and afforded moderate yields using standard DIC/Oxyma based Fmoc solid phase peptide synthesis conditions (Figure 3) [34]. The highly acidic N-terminus of the peptide led to frequent solubility issues in aqueous compositions. All peptides synthesized were dissolved in an acetonitrile:water mixture and purified immediately after trituration with ethyl ether to minimize undesirable aggregation. All linear peptides were synthesized, isolated, and lyophilized to afford an amorphous white powder with a >20 % yield and >94% purity (Table 1).
The chemical synthesis of linear peptides GA1, alanine analogs GA5-GA10 (in green), and GA4, GA7, GA11-13 (in blue). Peptides GA4, GA7, GA11-13 were carried forward for DOTA conjugation and subsequent radiolabeling, yielding DOTA-GA4, DOTA-GA7, DOTA-GA11-GA13 and their 68Ga labeled peptides.
Analytical data for all linear peptides generated in this study.
| Peptide | Exact Mass (Calc.) | % Yielda | Mass obtained (mg)b | ESI-MS m/zc | HPLC Purity (%)d |
|---|---|---|---|---|---|
| GA1 | 1721.71 | 35.00 | 43.21 | 830.90 | >95% |
| GA5 | 1663.70 | 20.68 | 22.60 | 831.80 | 94% |
| GA6 | 1663.70 | 21.68 | 27.24 | 830.90 | >95% |
| GA7 | 1663.70 | 37.56 | 47.62 | 831.15 | 94% |
| GA8 | 1663.70 | 36.64 | 44.82 | 830.90 | >95% |
| GA9 | 1663.70 | 32.92 | 42.10 | 830.90 | >95% |
| GA10 | 1663.70 | 22.32 | 27.03 | 867.15 | >95% |
| GA4 | 1735.73 | 25.20 | 31.39 | 831.15 | >95% |
| GA11 | 1627.70 | 32.04 | 41.34 | 806.05 | >95% |
| GA12 | 1613.69 | 32.36 | 39.87 | 838.15 | >95% |
| GA13 | 1677.72 | 31.76 | 41.11 | 830.90 | >95% |
a % Yield reported represents % pure peptide obtained following assembly and subsequent HPLC purification. b Mass Obtained (mg) represents the mass of pure peptide obtained after HPLC purification. c ESI-MS base peak corresponds to [M-2H]2+. d Analytical conditions: Kinetex C18 XB 5µm 4.6x150mm, flow rate 1.5mL/min, gradient 15-90% MeCN in water containing 0.1% formic acid over 10 min with column heating at 40°C.
The synthesis and purification of GA5, GA6 and GA10 encountered specific challenges. Substitution of the alanine at positions 1, 2 and 6 on the parent GA1 scaffold, reducing the acidic nature at the N-terminus of the peptide, led to increased product aggregation and lower overall yield post-purification (Table 1). In comparison, the substitution of alanine at the remaining positions did not face similar aggregation issues.
Another synthetic challenge common to all peptides discussed in this work was the deletion of glutamic acid residues, which led to the formation of side products with deletion sequences. During on-resin solid phase peptide synthesis, amino acid couplings were confirmed via small-scale cleavage and deprotection, and the resulting product mass was confirmed using mass spectrometry. All linear peptides generated incomplete coupling of D-Glu residues at positions 3 and 4, with deletion products being observed during initial HPLC purification. Amongst the structures discussed in this work, deletion product peptides constituted 5-30% of the final isolated peptide, and their structural and chemical similarity to the full-length peptide led to difficulties separating the products during HPLC purification. The challenge of glutamic acid deletions at positions 3 and 4 was addressed by increasing the coupling times of these residues under microwave conditions and performing double couplings of the glutamic acid residues. These specific modifications eliminated the deletion products altogether, leading to the yields reported in Table 1.
Competition binding studies
Once all N-terminal free peptides were in hand, CP04, the parent GA1, and its D-Ala substituted analogs GA5-GA10 were evaluated for their ability to disrupt the binding between CCK2R and [177Lu]Lu-DOTA-CP04 in vitro, using the epidermoid carcinoma cell line A431 transfected to stably express CCK2R (A431-CCK2R) [35, 36]. [177Lu]Lu-DOTA-CP04 was used as the competitive radioligand to determine the CCK2R binding affinity of the linear peptides as it has an established high binding affinity to the receptor of interest and thus serves as an ideal positive control in these assays [34].
The apparent binding affinities (IC50) (Figure 4) demonstrate the impact of D-Ala substitution on each of the glutamic acid residues on the critical hexameric peptide sequence. Linear CP04 without gallium coordinated DOTA at the N-terminus showed a greater than 3-fold decrease in affinity for the CCK2R target, from 0.76 nM to 2.33 nM. This loss of affinity very clearly demonstrates the importance of the radiometal-DOTA complex in improving the affinity of the peptide to the receptor of interest. Similarly, GA1 had previously been assessed in its natGa-DOTA-GA1 form with an affinity of 0.05 nM for CCK2R but observed a 5-fold reduction in its affinity to the receptor without the gallium-coordinated DOTA at the N-terminus [34]. Despite this, GA1 with a free N-terminus established an important baseline for the subsequent peptides, with an affinity of 0.32 nM.
(A) Binding curves for IC50 determination of CP04, GA1 and D-Ala scan peptides GA5-GA10 against [177Lu]Lu-DOTA-CP04 in A431-CCK2R cells. Curves are representative of 2-4 biological replicates performed in three technical replicates (n=3). (B) Binding affinities (IC50) for the human CCK2R, expressed as an average across 2-4 biological replicates performed in three technical replicates.
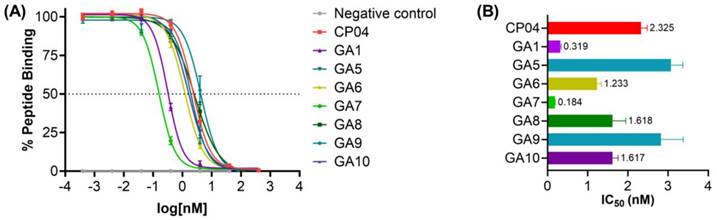
GA5, with D-Ala substituted at terminal position 1, saw a significant loss of binding affinity to CCK2R (Figure 4). This clearly demonstrates the important role of the N-terminal D-Glu residue in improving the binding affinity of the peptide sequence to the target, as observed by the greater than 10-fold loss of activity. Similarly, GA9 observed an equivalent loss of activity for the receptor. Evaluating the biological activity of substitutions made at positions 2, 4, and 6 in peptides GA6, GA8, and GA10, respectively, found that the removal of the D-Glu moiety was also poorly tolerated. The resulting activity across these peptides exhibited an approximately 5-fold loss in binding affinity to the target receptor across all three peptides (Figure 4).
GA7, with the substitution of D-Ala at position 3, yielded the most active ligand of the library of peptides evaluated. With an IC50 of 0.18 nM, GA7 displayed a greater than 10-fold improvement upon the activity of the established CP04. It also exceeded the already outstanding affinity of its predecessor GA1 by almost 2-fold. Position 3 therefore, is not only highly tolerable of modifications, but the substitution of a neutral hydrophobic moiety such as D-Ala greatly improves the binding between the ligand and receptor. Considering the 3-dimensional β-hairpin structure of this scaffold, it is possible that the highly polar and bulky D-Glu at this position repels favorable intramolecular interactions and may also negatively influence the binding in CCK2R. Furthermore, alanine residues are helix-stabilising in peptides and may infer conformational stability when substituted in position 3 of GA7 [37].
The D-Ala substitutions at positions 1 and 5 may suggest at the vital nature of the D-Glu residues in the formation and stability of a secondary structure; the loss of these critical residues results in a loss of activity. Previous work in this scaffold has shown that the linear peptide preferentially folds into a β-hairpin structure through backbone and residue intramolecular interactions and that this secondary structure is critical for its exquisite activity [34]. Furthermore, Moroder et al., and Peggion et al., demonstrated that the N-terminus of CCK and Gastrin peptides adopt α-helical orientation, which induces ordered conformations of the C-terminus involving a β-turn [38-41]. Interestingly, Peggion and coworkers also demonstrated that Glu-residues at positions 1 and 2 of des-Trp1,Nle2-MG form hydrogen bonds to Glu residues 5 and 6, to form the anticipated α-helical structure of N-terminus [40]. Recently, cryo-electron microscopy confirmed that the 17 amino acid long hormone gastrin-17 indeed forms a β-hairpin structure upon binding to CCK2R with the five C-terminal residues inserting deeply into the binding pocket of CCK2R [42]. Furthermore, the N-terminal section of gastrin-17 also orientates into a shallow binding cavity of CCK2R [42].
The C-terminal residue of gastrin-17 based peptides is prone to metabolic degradation with N-methylation of this position, leading to a metabolically stable analogue named GA4 (Figure 2). This peptide presented advantageous biostability resulting in the highest tumor uptake in biodistribution mouse studies when evaluated alongside other potential ligands [34]. However, GA4 also had the highest renal uptake of compounds tested [34]. This is in large part due to the highly acidic hexameric terminal peptide chain, known to promote kidney uptake [29, 43]. GA4 serves as an important lead scaffold to investigate the impact of D-Ala substitution, reducing the overall acidic nature of the peptide and screening the impact of this structural change on the peptide binding affinity of the peptide to CCK2R target. GA7, GA11-GA13 (Figure 2) were designed to implement the favorable D-Ala substitution at position 3 alongside changes to the critical C-terminal residue. By systematically incorporating bulky aromatic naphthylalanines and N-methyl amino acids, we intended to enhance the peptide's structural conformity, increase affinity, and improve metabolic stability.
DOTA coupled peptides DOTA-GA4, DOTA-GA7, DOTA-GA11-GA13 were then coordinated to natural gallium (natGa), producing natGa-DOTA-GA4, natGa-DOTA-GA7, natGa-DOTA-GA11-GA13, respectively. Previous work from our laboratory had demonstrated that a DOTA conjugation in solution was a far more effective strategy to produce peptides in good yields compared to the traditional on-resin coupling of DOTA-tris (tert-butyl ester) [34]. As such, GA4, GA7, GA11-GA13 were conjugated to DOTA in solution (Figure 3) to obtain DOTA-peptides DOTA-GA4, DOTA-GA7, DOTA-GA11-GA13 at yields of 64-86% from the respective linear peptides (Table S2).
Natural gallium was then coordinated to peptides DOTA-GA4, DOTA-GA7, DOTA-GA11-GA13 in a sodium ascorbate-buffered solution (pH 4.5) to generate the non-radioactive reference peptides natGa-DOTA-GA4, natGa-DOTA-GA7, natGa-DOTA-GA11-GA13. natGa-DOTA-peptides were then purified using preparative HPLC to remove salts and afford high-purity products for ligand binding assays (Table S3).
Modification of the N-terminus of all CP04-based peptides with natGa-DOTA had previously been determined to improve binding affinity of the peptide to CCK2R [34]. Peptides GA1, GA3, and GA4 have previously been investigated and shown to have a sub-nanomolar affinity to the CCK2R target [34]. To determine the impact of D-Ala substitution at position 3, natGa-DOTA peptides were assayed against [177Lu]Lu-DOTA-CP04 and shown to tolerate the substitution extremely well, with less than 2-fold loss of binding affinity across all peptides tested with varying substitutions at C-terminus (Figure 5). Affinity assays demonstrated conclusively that substitution of D-Ala for D-Glu at position 3 on this scaffold has little to no negative impact on the affinity of the peptides to the target receptor while minimizing overall negative ionic charge.
(A) Binding curves for IC50 determination of peptides natGa-DOTA-GA4, natGa-DOTA-GA7 and natGa-DOTA-GA11-GA13 against [177Lu]Lu-DOTA-CP04 in A431-CCK2R cells. Curves are representative of 2-4 biological replicates performed in three technical replicates (n=3). (B) Binding affinities (IC50) for the human CCK2R, expressed as an average across 2-4 biological replicates performed in three technical replicates

Circular Dichroism (CD) Spectroscopy
N-terminal free peptides GA1, GA5-GA10, and natural gallium co-ordinated peptides natGa-DOTA-GA4, natGa-DOTA-GA7, natGa-DOTA-GA11-GA13 were analyzed by CD spectroscopy to investigate the impact of chemical structure changes on conformational preferences of the peptides (Figure 6). GA1, GA5-GA10 presented CD spectra of fully folded β-hairpin peptides with two maxima at ca. 200 and 230 nm and a minimum at ca. 220, both in water and DPC micelles (Figure 6A) [34, 44]. However, the minimum at ca. 220 nm and the maximum at 230 nm were more pronounced in DPC compared to water for the GA1 peptide and its analogs (Figure 6A).
CD Spectroscopy of (A) N-terminal free linear peptides GA1, GA5-GA10 and (B) natural gallium co-ordinated natGa-DOTA-GA4, natGa-DOTA-GA7, natGa-DOTA-GA11-GA13 in water and DPC micelles.
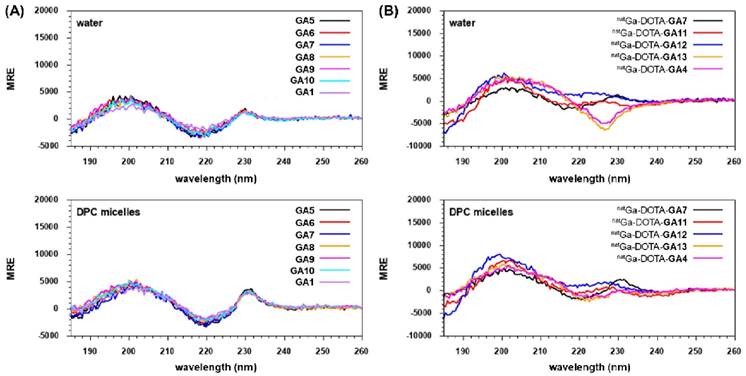
Interestingly, natGa-DOTA-peptides demonstrated more pronounced differences between structural conformations during CD spectroscopy. natGa-DOTA-GA4 and its D-Ala substituted variant natGa-DOTA-GA13 exhibited one broad maximum at ca. 200 and a minimum at ca. 227 nm in water (Figure 6B). All other GA1, D-Ala modified analogs, and natGa chelated peptides demonstrated the expected two maxima at ca. 200 and 230 nm and a minimum at ca. 220. However, the intensities of their respective maxima and minima varied in water. In DPC micelles, natGa-DOTA-GA7 presented the most intense maxima at ca. 230 nm and the most intense minima at ca. 220 nm in comparison to other peptides (Figure 6B).
All natGa-DOTA-peptides also revealed the fingerprint of fully folded β-hairpin peptides with two maxima at ca. 200 and 230 nm and a minimum at ca. 220 in DPC micelles (Figure 6B). It has been suggested that peptides based on the gastrin hormone interact with the cell membrane in a way that induces the preferred conformation, thus facilitating recognition by CCK2R [35, 45]. This is further supported by our CD experiments, where all evaluated peptides adopted intensified and distinct structural conformations in DPC micelles.
LogD7.4 and Serum Protein Binding Assays
Lipophilicity, expressed as the octanol-water partition coefficient, constitutes an important property in drug action, influencing both pharmacokinetic and pharmacodynamics processes. The distribution coefficient (LogD7.4) of [68Ga]Ga-DOTA-peptides was determined by measuring radioactivity distribution in the aqueous phase (phosphate buffer pH7.4) and the organic phase (n-octanol).
Peptides in this series were radiolabeled according to the protocol outlined in supplementary data. All radiolabeled peptides displayed high hydrophilicity with low LogD7.4 values ranging from -4.1 to -2.5 (Table 2). A low LogD7.4 is an important parameter required in the development of peptide-based theranostics with a suitable biodistribution profile [46]. Substitution of the D-Glu in [68Ga]Ga-DOTA-GA4 by D-Ala in analogs [68Ga]Ga-DOTA-GA7 and [68Ga]Ga-DOTA-GA13 did not influence lipophilicity a great deal as demonstrated by their similar LogD7.4 values: -2.97 - -2.59 (Table 2). The reduced hydrophilicity of these peptides is likely due to the hydrophobicity afforded by the bulky naphthylalanine (Nal) residue shared by all three peptides. The contribution of the Nal residue towards the overall lipophilicity of the corresponding peptides can be further demonstrated by comparison to peptides [68Ga]Ga-DOTA-GA11 and [68Ga]Ga-DOTA-GA12 with N-methylated phenylalanine (13NMe-Phe) and phenylalanine (13Phe) respectively as their terminal residue. Both these peptides presented with decreased LogD7.4 values in comparison to peptides [68Ga]Ga-DOTA-GA4, [68Ga]Ga-DOTA-GA7, and [68Ga]Ga-DOTA-GA13 (Table 2).
LogD7.4 and protein binding (%) of [68Ga]Ga-DOTA-GA4, [68Ga]Ga-DOTA-GA7, [68Ga]Ga-DOTA-GA11-GA13. LogD7.4 values (n=5) obtained after incubation in PBS/n-octanol mix. Serum protein binding results after peptides were incubated with pooled human serum for 1 h at 37oC.
| Peptide | LogD7.4 | Plasma Protein Binding (%) |
|---|---|---|
| [68Ga]Ga-DOTA-GA4 | -2.97 ± 0.13 | 82.43 ± 0.38 |
| [68Ga]Ga-DOTA-GA7 | -2.88 ± 0.09 | 78.69 ± 2.11 |
| [68Ga]Ga-DOTA-GA11 | -3.56 ± 0.01 | 59.75 ± 1.04 |
| [68Ga]Ga-DOTA-GA12 | -4.06 ± 0.14 | 63.62 ± 0.45 |
| [68Ga]Ga-DOTA-GA13 | -2.59 ± 0.02 | 83.16 ± 0.13 |
Serum or plasma protein binding is recognized as an important factor in theranostic development as their interactions influence the pharmacokinetics and pharmacodynamics of viable candidates. Correlative relationships between lipophilicity and serum protein binding of radiotracers have previously been established, showing strong linear trends where protein binding in serum increases with increasing lipophilicity [47, 48]. The [68Ga]Ga-DOTA-peptides examined in this series demonstrated a moderate to high degree of plasma protein binding (59-84%), with higher plasma protein binding corresponding to more lipophilic (higher LogD7.4) peptides (Table 2).
In Vitro Metabolic Assays
Metabolic stability is an important determinant of the efficacy of peptide radiopharmaceuticals and their progression as promising theranostic candidates. In vitro, metabolic degradation through hepatic and renal enzymes can be assessed by utilizing liver and kidney tissue homogenates. Radiolabeled peptides [68Ga]Ga-DOTA-GA4, and D-Ala substituted variants [68Ga]Ga-DOTA-GA7, [68Ga]Ga-DOTA-GA11-GA13 were incubated in liver and kidney mouse homogenates, in human serum at 37°C, and in HEPES buffer (pH 7.4) as a negative control. Following a time course experiment, the metabolites were analyzed using HPLC to quantify the % breakdown of the respective parent peptide.
All peptides tested remained intact when incubated with human serum and HEPES buffer at 37°C over the 90 min time course of the experiment (Figure 7A). [68Ga]Ga-DOTA-GA7 and [68Ga]Ga-DOTA-GA12 both underwent significant metabolism in liver and kidney homogenates over the course of the experiment (Figure 7B, 7C). [68Ga]Ga-DOTA-GA12 was particularly prone to degradation, with approximately 35% peptide intact when incubated with liver microsomes (T1/2 64.3 min) and only 20% peptide intact in kidney homogenates (T1/2 46.3) after 90 min. [68Ga]Ga-DOTA-GA7 demonstrated a more stable metabolic profile, with approximately 80% of peptide intact when incubated with liver microsomes (T1/2 352.15 min) and 70% of the parent peptide intact in kidney homogenates at 90 min (T1/2 225.83 min). The remaining peptides [68Ga]Ga-DOTA-GA4, [68Ga]Ga-DOTA-GA11, and [68Ga]Ga-DOTA-GA13 were extremely stable in liver and kidney homogenates, with no observable metabolites detected at the 90 min timepoint (Figure 7).
Metabolic stability of peptides [68Ga]Ga-DOTA-GA4, and D-Ala variants [68Ga]Ga-DOTA-GA7, [68Ga]Ga-DOTA-GA11-GA13 incubated in (A) human serum, (B) kidney homogenates, and (C) liver homogenates. Samples were assayed at various time points over 90 min, and the intact peptides were assessed using liquid chromatography. The results are mean values (n=2). Half-life (t1/2) is displayed where appropriate in minutes.
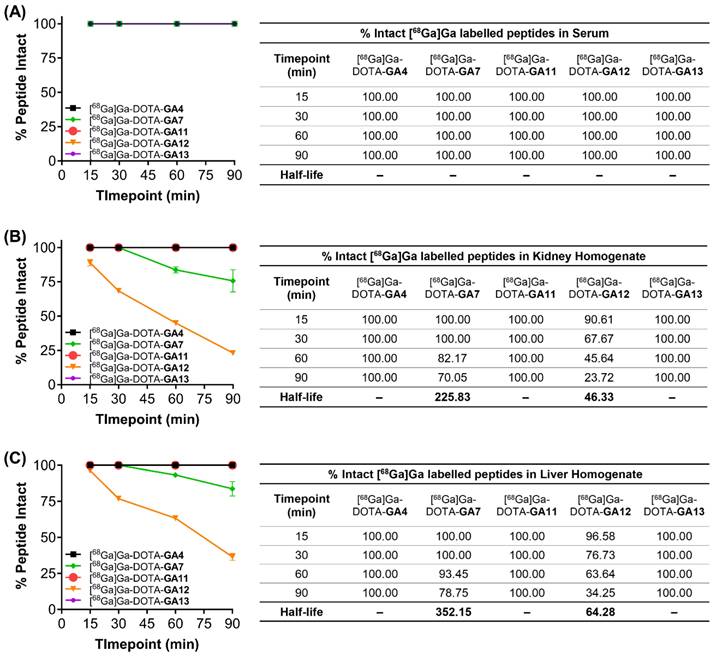
These studies demonstrate the metabolically labile nature of the C-terminal phenylalanine residue on [68Ga]Ga-DOTA-GA12. Enzymatic cleavage at the Phe residue can be prevented by substituting the natural Phe moiety for the non-natural N-methylated Phe, as demonstrated in [68Ga]Ga-DOTA-GA11. The incorporation of N-methylated residues is a known strategy to enhance protease resistance [49-51]. This is demonstrated yet again in [68Ga]Ga-DOTA-GA7 and its N-methylated naphthylalanine variant at position 13, [68Ga]Ga-DOTA-GA13. Although not as prone to metabolic degradation as [68Ga]Ga-DOTA-GA12, N-methylation of the C-terminal naphthylalanine residue in [68Ga]Ga-DOTA-GA13 prevents enzymatic degradation of the peptide, leaving 100% of the parent peptide intact.
Small Animal PET Imaging and Biodistribution
In order to assess these promising ligands as potential theranostics, the library of [68Ga]Ga-DOTA-peptides was evaluated using static small animal PET imaging and quantifying biodistribution studies. This study was performed to investigate tumor uptake as well as the accumulation of radioligands in other organs. Mice bearing A431-CCK2R tumors were imaged at 1- and 2 hours post tracer injection, and tracer biodistribution was evaluated at the 1 hour and 2.5 hour time points to quantify tumor uptake and retention.
The work presented employed the A431-CCK2R/NSG mouse model; we have observed that this model consistently yields reduced tumor uptake of radioligands compared to the AR42J/Balbc model. This was demonstrated using the clinically relevant CP04 and the lead GA peptide in this series, GA4. When tested in AR42J xenografts, CP04 demonstrated only moderate tumor uptake at 5.5 ± 0.8% ID/g, and in the A431-CCK2R/NSG mouse model, CP04 had a significantly lower tumor uptake of 2.5 ± 0.3% ID/g (Table 3). The marked difference between tumor uptake in the two models was more noticeably illustrated with GA4. Our previous work evaluated [68Ga]Ga-DOTA-GA4 (13b as published in Corlett et al.) in AR42J tumor xenografts with a tumor uptake of 31.1 ± 5.3% ID/g [34]. In this study, the same radioligand was assessed at a similar specific activity and time post-injection in the A431-CCK2R/NSG mouse model and showed a tumor uptake of only 8.77 ± 1.21% ID/g (Table 3).
Comparison of tumor uptake of [68Ga]Ga labeled CP04 and GA4 in AR42J and A431-CCK2R xenograft models. All data tumor uptake values are 1 hour post-injection. GA4 exhibited reduced uptake in A431-CCK2R xenografts when treated at the same dose as in AR42J xenografts. 1Data shown for [177Lu]Lu-CP04.
| AR42J | A431-CCK2R | |||
|---|---|---|---|---|
| CP04 (% ID/g) | GA4 (% ID/g) | CP04 (% ID/g)1 | GA4 (% ID/g) | |
| Tumor | 5.5 ± 0.8 [34] | 31.1 ± 5.3 [34] | 2.5 ± 0.3 [62] | 8.8 ± 1.2 |
The A431-CCK2R model reportedly has variable uptake of the same ligand in different studies. A study by Laverman and coworkers showed that 111In-radiolabeled CP04 (reported molar activity: 11 MBq/nmol) had a tumor uptake of 9.7 ± 1.8% ID/g in an A431-CCK2R mouse model, while Roosenburg and coworkers investigated the same radioligand (reported molar activity: 30 MBq/nmol) and demonstrated a tumor uptake of 1.89 ± 0.74% ID/g in the same tumor model [43, 52]. Despite the molar activity being lower for the study performed by Laverman and co-workers, the tumor uptake was higher than that observed by Roosenburg and co-workers. This comparison clearly demonstrates the variability in the A431-CCK2R model which cannot be explained only in terms of molar activity changes.
Differences in radioligand tumor uptake can also be attributed to different mouse models used in each investigation. Preclinical mouse models critically underpin early stages of the progression of a theranostic candidate. Differences in mouse size, physiology, and variable homology between models can produce inconsistent therapeutic outcomes. This is highlighted particularly in a comparative analysis of biodistribution of PP-F11 in studies undertaken by Laverman and Roosenburg, who used athymic BALB/c and SCID/Bg mice respectively [43, 52].
Nevertheless, Roosenburg and co-workers reported that 68Ga-radiolabeled CP04 had tumor uptake of 5.21 ± 2.19% ID/g in their A431-CCK2R mouse model at 1 hr post injection, which is analogous to our results [52]. Collectively, the experimental and reported data confirm that the moderate uptake observed for GA4 in this work could be due to variability in the model. A head-to-head comparison between CP04 and GA4 performed by Haskali and co-workers clearly confirms that GA4 is far superior to CP04 in tumor uptake (up to 6-fold increase in tumor uptake for the 68Ga-labeled counterparts). Thus, we have decided to benchmark all ongoing development presented herein against the optimal GA4 scaffold. In the future, an independently performed head-to-head comparison of GA4 and the reported developments in this manuscript should be done against MGS5 to evaluate their relative performance.
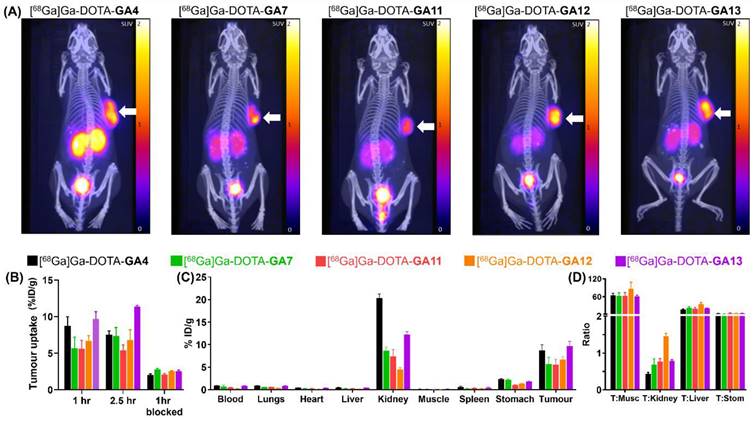
The maximum standardised uptake value (SUVmax) for the reference standard [68Ga]Ga-DOTA-GA4 was 1.41, at 1 hour post-injection (Supplementary Data, pg. S51). The D-Ala substituted peptides [68Ga]Ga-DOTA-GA7, [68Ga]Ga-DOTA-GA11-GA13 displayed a similar range of SUVmax (1.12-1.86) at the same time point (Supplementary Data, pg. S52-S55), highlighting the potency of all these ligands (Figure 7A). In particular, [68Ga]Ga-DOTA-GA13, which had SUVmax of 1.86, had a considerably higher tumor uptake compared to the reference compound [68Ga]Ga-DOTA-GA4.
At 1 hour post-injection, a tumor uptake of 8.77% ID/g was observed for [68Ga]Ga-DOTA-GA4. Its D-Ala substituted variant [68Ga]Ga-DOTA-GA13 demonstrated a similarly high uptake of 9.68% ID/g (Table 4). There was a slight increase in tumor uptake at the 2.5 hour mark for [68Ga]Ga-DOTA-GA13, rising to 11.38% ID/g in contrast to [68Ga]Ga-DOTA-GA4, for which a small reduction to 7.54% ID/g was observed at the same time point. The remaining peptides [68Ga]Ga-DOTA-GA7, [68Ga]Ga-DOTA-GA11-GA12 demonstrated good tumor uptake at both 1 hour (5.60-6.70% ID/g) and 2.5 hours (5.42-7.36% ID/g) post-injection (Table 4).
Uptake of radiolabeled peptides in tumors of mice at 1 hour, and 2.5 hours post-injection. Blockade of radiolabeled peptides reported at 1 hour post-injection. Mice treated with CCK2R antagonist YM022 at dose of 3mg/Kg dose for blockade experiments.
| Uptake in A431-CCK2R Tumors (% ID/g) | ||||
|---|---|---|---|---|
| Peptide | 1 hr | 2.5 hr | 1 hr Blocked | Blockade (1hr) |
| [68Ga]Ga-DOTA-GA4 | 8.77 ± 1.21 | 7.54 ± 0.50 | 2.05 ± 0.14 | 77% |
| [68Ga]Ga-DOTA-GA7 | 5.71 ± 1.50 | 7.36 ± 1.16 | 2.76 ± 0.14 | 52% |
| [68Ga]Ga-DOTA-GA11 | 5.60 ± 1.16 | 5.42 ± 0.70 | 2.07 ± 0.15 | 63% |
| [68Ga]Ga-DOTA-GA12 | 6.70 ± 0.69 | 6.81 ± 1.40 | 2.53 ± 0.11 | 62% |
| [68Ga]Ga-DOTA-GA13 | 9.68 ± 1.02 | 11.38 ± 0.17 | 2.53 ± 0.17 | 74% |
The specific uptake of [68Ga]Ga-DOTA-peptides was then demonstrated in blocking experiments by co-injection of the CCK2R antagonist YM022 (Figure 7B). Co-administration of YM022 reduced tumor uptake to 2.05-2.76% ID/g, indicating a >50% blockade at the 1 hour mark for all radiopeptides, with [68Ga]Ga-DOTA-GA4 and analogue [68Ga]Ga-DOTA-GA13 showing the most substantial reduction in uptake (Table 4). All radioligands tested presented low uptake in the stomach, an organ known to have high physiological CCK2R expression (Figure 7C) [53-56]. Furthermore, all peptides investigated presented good renal clearance with low background uptake, leading to high tumor-to-background imaging ratios of 11.43 ± 0.90 for [68Ga]Ga-DOTA-GA4 and 17.27 ± 1.84, 14.37 ± 2.23, 24.93 ± 4.57, and 19.33 ± 1.49 for D-Ala substituted peptides [68Ga]Ga-DOTA-GA7 and [68Ga]Ga-DOTA-GA11-GA13 respectively (Supplementary Data, pg. S51-S55). No substantial uptake of [68Ga]Ga-DOTA-peptides was observed in any other organ/healthy tissue, including blood, lungs, heart, muscle, spleen and liver, which eloquently presents the suitability of these radioligands for clinical translation (Table 5).
The biodistribution of radiolabeled standard [68Ga]Ga-DOTA-GA4, and analogs [68Ga]Ga-DOTA-GA7, [68Ga]Ga-DOTA-GA11-GA13 in key organs of mice 1 hour post-injection (n=3).
| Uptake in Organs (% ID/g) | |||||
|---|---|---|---|---|---|
| Organ | [68Ga]Ga-DOTA-GA4 | [68Ga]Ga-DOTA-GA7 | [68Ga]Ga-DOTA-GA11 | [68Ga]Ga-DOTA-GA12 | [68Ga]Ga-DOTA-GA13 |
| Blood | 0.92 ± 0.05 | 0.68 ± 0.31 | 0.46 ± 0.08 | 0.29 ± 0.01 | 0.83 ± 0.07 |
| Lungs | 0.92 ±0.04 | 0.57 ± 0.03 | 0.59 ± 0.02 | 0.34 ± 0.04 | 0.85 ± 0.11 |
| Heart | 0.45 ± 0.03 | 0.30 ± 0.02 | 0.27 ± 0.08 | 0.14 ± 0.01 | 0.40 ± 0.03 |
| Liver | 0.51 ± 0.01 | 0.30 ± 0.01 | 0.29 ± 0.01 | 0.19 ± 0.02 | 0.44 ± 0.03 |
| Kidneys | 20.34 ± 0.89 | 8.67 ± 0.80 | 7.38 ± 1.54 | 4.52 ± 0.38 | 12.26 ± 0.63 |
| Muscle | 0.14 ± 0.01 | 0.12 ± 0.02 | 0.09 ± 0.02 | 0.09 ± 0.02 | 0.16 ± 0.03 |
| Spleen | 0.62 ± 0.14 | 0.24 ± 0.02 | 0.34 ± 0.08 | 0.25 ± 0.04 | 0.43 ± 0.13 |
| Stomach | 2.37 ± 0.12 | 2.22 ± 0.14 | 1.04 ± 0.06 | 1.38 ± 0.08 | 1.81 ± 0.12 |
| Tumor | 8.77 ± 1.21 | 5.71 ± 1.50 | 5.60 ± 1.16 | 6.70 ± 0.69 | 9.68 ± 1.02 |
A major consideration in this investigation was to evaluate the impact of alanine substitution in the peptide scaffold on renal uptake of the radiolabeled peptides. High kidney uptake of radioligands is a significant issue, as it can lead to nephrotoxicity in patients. Renal uptake is primarily mediated by the resorption of the tracer or its metabolites through receptor or transporter-mediated processes [57-59]. Various approaches have been employed to decrease kidney uptake and offer renal protection. These include the use of radioprotectors, utilizing albumin-binding moieties, and competitive inhibition via co-administration of positively charged amino acids such as L-lysine and L-arginine [60, 61]. However, we believe that the most efficient and immediate method of reducing renal uptake is decreasing the radioligand's positive charge by fine-tuning the hexameric N-terminal sequence.
Biodistribution analysis of the series of radioligands confirmed that whilst [68Ga]Ga-DOTA-GA4 showed a kidney uptake of 20.34% ID/g, this was reduced by 2-2.5 fold across the range of alanine substituted [68Ga]Ga-DOTA-GA7, and [68Ga]Ga-DOTA-GA11-GA13 peptides to 8.67, 7.38, 4.52, 12.26% ID/g respectively (Table 5). These results confirmed that substituting alanine for glutamic acid in peptides significantly reduces renal uptake of peptides by lowering their overall ionic nature. In particular, [68Ga]Ga-DOTA-GA12 demonstrated the highest tumor-to-kidney ratio (Figure 7D) with the lowest renal uptake (4.52% ID/g) observed in comparison to all other candidates evaluated, making it a truly promising candidate for clinical translation.
The N-methylated [68Ga]Ga-DOTA-GA13 remained fully intact against enzymatic degradation compared to its non-methylated pair [68Ga]Ga-DOTA-GA7. This modification also improved tumor uptake of the radiopeptide in mice (Table 4) but resulted in an increase in renal uptake (Table 5). The peptides [68Ga]Ga-DOTA-GA11 and [68Ga]Ga-DOTA-GA12 share N-methylated Phe and free Phe terminal residues, respectively. Unlike the 1-Nal substituted peptides, N-methylation of [68Ga]Ga-DOTA-GA11 gave reduced uptake in tumor and increased uptake in the kidneys compared to its non-methylated [68Ga]Ga-DOTA-GA12. This highlights the need to strike a careful balance between metabolic stability, renal uptake and tumor retention.
The imaging and biodistribution characteristics of radiolabeled standard [68Ga]Ga-DOTA-GA4, and analogs [68Ga]Ga-DOTA-GA7, [68Ga]Ga-DOTA-GA11-GA13. (A) Imaging of mice at 1 hour post radiolabeled peptide injection, showing the CCK2R positive tumor on the right flank. The highest tumor uptake was observed for [68Ga]Ga-DOTA-GA4 and the alanine-substituted variant [68Ga]Ga-DOTA-GA13. (B) The tumor uptake study of [68Ga]Ga-DOTA-peptides at 1 hour, 2.5 hours, and 1 hour (blocked). (C) The biodistribution study of [68Ga]Ga-DOTA-peptides in different organs at 1 hour post-injection.(D) Tumor:Organ ratios of [68Ga]Ga-DOTA-peptides, notably showing reduced renal uptake and maintained high tumor uptake for [68Ga]Ga-DOTA-GA12. (T= tumor, Musc = muscle, Stom = stomach)
Conclusion
Our work presents detailed and careful optimisation of peptide-based ligands for managing CCK2R-positive tumors. Through a series of meticulous optimisations to the C-terminus of CP04, we previously developed peptides with high binding affinity against CCK2R and high metabolic stability. This study presents the first exploration of how systemic replacement of each glutamic acid residue in the N-terminus with D-Ala affects the binding affinity to CCK2R. We identified that D-Ala substitution of the third D-Glu residue in the gastramide (GA) scaffold led to candidates with improved affinity, excellent metabolic stability and reduced overall ionic charge. In particular, peptides [68Ga]Ga-DOTA-GA12 and [68Ga]Ga-DOTA-GA13 demonstrated high tumor uptake with a significant reduction in renal uptake in comparison to the previous generation of peptides, making them truly exciting candidates that warrant clinical translation. This study marks a significant milestone in our understanding of the structure activity relationship of CP04 based peptides for theranostic application against CCK2R.
Supplementary Material
Supplementary materials and methods, results, images.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Weissleder R. Molecular imaging in cancer. Science. 2006;312:1168-71
2. Weissleder R, Mahmood U. Molecular imaging. Radiology. 2001;219:316-33
3. Bruemmer KJ, Crossley SW, Chang CJ. Activity-based sensing: a synthetic methods approach for selective molecular imaging and beyond. Angew Chem Int Ed. 2020;59:13734-62
4. Weber WA, Czernin J, Anderson CJ, Badawi RD, Barthel H, Bengel F. et al. The future of nuclear medicine, molecular imaging, and theranostics. J Nucl Med. 2020;61:263S-72S
5. Sun X, Li Y, Liu T, Li Z, Zhang X, Chen X. Peptide-based imaging agents for cancer detection. Adv Drug Deliv Rev. 2017;110:38-51
6. Joshi BP, Wang TD. Exogenous molecular probes for targeted imaging in cancer: focus on multi-modal imaging. Cancers. 2010;2:1251-87
7. Polyak A, Ross TL. Nanoparticles for SPECT and PET imaging: towards personalized medicine and theranostics. Curr Med Chem. 2018;25:4328-53
8. Okamoto S, Shiga T, Tamaki N. Clinical perspectives of theranostics. Molecules. 2021;26:2232
9. Bodei L, Kwekkeboom DJ, Kidd M, Modlin IM, Krenning EP. Radiolabeled somatostatin analogue therapy of gastroenteropancreatic cancer. Semin Nucl Med: Elsevier. 2016 p. 225-38
10. Park S, Parihar AS, Bodei L, Hope TA, Mallak N, Millo C. et al. Somatostatin receptor imaging and theranostics: current practice and future prospects. J Nucl Med. 2021;62:1323-9
11. Zaknun JJ, Bodei L, Mueller-Brand J, Pavel M, Baum RP, Hörsch D. et al. The joint IAEA, EANM, and SNMMI practical guidance on peptide receptor radionuclide therapy (PRRNT) in neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2013;40:800-16
12. Ahmadi Bidakhvidi N, Goffin K, Dekervel J, Baete K, Nackaerts K, Clement P. et al. Peptide receptor radionuclide therapy targeting the somatostatin receptor: basic principles, clinical applications and optimization strategies. Cancers. 2021;14:129
13. von Guggenberg E, Kolenc P, Rottenburger C, Mikołajczak R, Hubalewska-Dydejczyk A. Update on preclinical development and clinical translation of cholecystokinin-2 receptor targeting radiopharmaceuticals. Cancers. 2021;13:5776
14. Reubi JC. Targeting CCK receptors in human cancers. Curr Top Med Chem. 2007;7:1239-42
15. Gotthardt M, Béhé MP, Grass J, Bauhofer A, Rinke A, Schipper ML. et al. Added value of gastrin receptor scintigraphy in comparison to somatostatin receptor scintigraphy in patients with carcinoids and other neuroendocrine tumours. Endocr Relat Cancer. 2006;13:1203-11
16. Reubi JC, Schaer J-C, Waser B. Cholecystokinin (CCK)-A and CCK-B/gastrin receptors in human tumors. Cancer Res. 1997;57:1377-86
17. Béhé M, Behr TM. Cholecystokinin-B (CCK-B)/gastrin receptor targeting peptides for staging and therapy of medullary thyroid cancer and other CCK-B receptor expressing malignancies. J Pept Sci. 2002;66:399-418
18. Reubi JC, Waser B. Concomitant expression of several peptide receptors in neuroendocrine tumours: molecular basis for in vivo multireceptor tumour targeting. Eur J Nucl Med Mol Imaging. 2003;30:781-93
19. Reubi JC, Maecke HR. Approaches to multireceptor targeting: hybrid radioligands, radioligand cocktails, and sequential radioligand applications. J Nucl Med. 2017;58:10S-6S
20. Copps J, Murphy RF, Lovas S. The production and role of gastrin-17 and gastrin-17-gly in gastrointestinal cancers. Protein Pept Lett. 2009;16:1504-18
21. Klingler M, Hnn AA, Guggenberg EV. Cholecystokinin-2 receptor targeting with radiolabeled peptides: Current status and future directions. Curr Med Chem. 2020;27:7112-32
22. Kolenc-Peitl P, Mansi R, Tamma M, Gmeiner-Stopar T, Sollner-Dolenc M, Waser B. et al. Highly improved metabolic stability and pharmacokinetics of indium-111-DOTA-gastrin conjugates for targeting of the gastrin receptor. J Med Chem. 2011;54:2602-9
23. Breeman WA, Fröberg A, de Blois E, Van Gameren A, Melis M, de Jong M. et al. Optimised labeling, preclinical and initial clinical aspects of CCK-2 receptor-targeting with 3 radiolabeled peptides. Nucl Med Biol. 2008;35:839-49
24. Kaloudi A, Nock BA, Lymperis E, Krenning EP, de Jong M, Maina T. Improving the in vivo profile of minigastrin radiotracers: a comparative study involving the neutral endopeptidase inhibitor phosphoramidon. Cancer Biother Radiopharm. 2016;31:20-8
25. Sauter AW, Mansi R, Hassiepen U, Muller L, Panigada T, Wiehr S. et al. Targeting of the cholecystokinin-2 receptor with the minigastrin analog 177Lu-DOTA-PP-F11N: does the use of protease inhibitors further improve in vivo distribution? J Nucl Med. 2019;60:393-9
26. Gotthardt M, van Eerd-Vismale J, Oyen WJ, De Jong M, Zhang H, Rolleman E. et al. Indication for different mechanisms of kidney uptake of radiolabeled peptides. J Nucl Med. 2007;48:596-601
27. Breeman WA, de Jong M, Kwekkeboom DJ, Valkema R, Bakker WH, Kooij PP. et al. Somatostatin receptor-mediated imaging and therapy: basic science, current knowledge, limitations and future perspectives. Eur J Nucl Med. 2001;28:1421-9
28. Béhé M, Kluge G, Becker W, Gotthardt M, Behr TM. Use of polyglutamic acids to reduce uptake of radiometal-labeled minigastrin in the kidneys. J Nucl Med. 2005;46:1012-5
29. Good S, Walter MA, Waser B, Wang X, Müller-Brand J, Béhé MP. et al. Macrocyclic chelator-coupled gastrin-based radiopharmaceuticals for targeting of gastrin receptor-expressing tumours. Eur J Nucl Med Mol Imaging. 2008;35:1868-77
30. Klingler M, Decristoforo C, Rangger C, Summer D, Foster J, Sosabowski JK. et al. Site-specific stabilization of minigastrin analogs against enzymatic degradation for enhanced cholecystokinin-2 receptor targeting. Theranostics. 2018;8:2896
31. Klingler M, Rangger C, Summer D, Kaeopookum P, Decristoforo C, von Guggenberg E. Cholecystokinin-2 receptor targeting with novel c-terminally stabilized HYNIC-Minigastrin analogs radiolabeled with technetium-99m. Pharmaceuticals. 2019;12:13
32. Klingler M, Summer D, Rangger C, Haubner R, Foster J, Sosabowski J. et al. DOTA-MGS5, a new cholecystokinin-2 receptor-targeting peptide analog with an optimized targeting profile for theranostic use. J Nucl Med. 2019;60:1010-6
33. Corringer P, Weng J, Ducos B, Durieux C, Boudeau P, Bohme A. et al. CCK-B agonist or antagonist activities of structurally hindered and peptidase-resistant Boc-CCK4 derivatives. J Med Chem. 1993;36:166-72
34. Corlett A, Sani M-A, Van Zuylekom J, Ang C-S, von Guggenberg E, Cullinane C. et al. A New Turn in Peptide-Based Imaging Agents: Foldamers Afford Improved Theranostics Targeting Cholecystokinin-2 Receptor-Positive Cancer. J Med Chem. 2021;64:4841-56
35. Lipiński PF, Garnuszek P, Maurin M, Stoll R, Metzler-Nolte N, Wodyński A. et al. Structural studies on radiopharmaceutical DOTA-minigastrin analogue (CP04) complexes and their interaction with CCK2 receptor. EJNMMI research. 2018;8:33
36. Maurin M, Garnuszek P, Baran P, Pawlak D, Mikołajczak R. The radiometal makes a difference. Synthesis and preliminary characterisation of DOTA-minigastrin analogue complexes with Ga, Lu and Y. Nuc Med Rev. 2015;18:51-5
37. Rohl CA, Fiori W, Baldwin RL. Alanine is helix-stabilizing in both template-nucleated and standard peptide helices. Proc Natl Acad Sci. 1999;96:3682-7
38. Moroder L, Romano R, Weyher E, Svoboda M, Christophe J. Circular dichroism study on fully bioactive CCK-peptides of increasing chain length. Z fur Naturforsch. 1993;48:1419-30
39. Peggion E, Jaeger E, Knof S, Moroder L, Wuensch E. Conformational aspects of gastrin-related peptides: A circular dichroism study. Biopolymers. 1981;20:633-52
40. Mammi S, Mammi NJ, Peggion E. Conformational studies of human Des-Trp1, Nle12 minigastrin in water-trifluoroethanol mixtures by proton NMR and circular dichroism. Biochem. 1988;27:1374-9
41. Mammi S, Peggion E. Conformational studies of human [15-2-aminohexanoic acid] little gastrin in sodium dodecyl sulfate micelles by proton NMR. Biochem. 1990;29:5265-9
42. Zhang X, He C, Wang M, Zhou Q, Yang D, Zhu Y. et al. Structures of the human cholecystokinin receptors in complex with agonists and antagonists. bioRxiv. 2021:2021.05. 19.444887.
43. Laverman P, Joosten L, Eek A, Roosenburg S, Peitl PK, Maina T. et al. Comparative biodistribution of 12 111 In-labelled gastrin/CCK2 receptor-targeting peptides. Eur J Nucl Med Mol Imaging. 2011;38:1410-6
44. Pastor MT, López de la Paz M, Lacroix E, Serrano L, Pérez-Payá E. Combinatorial approaches: A new tool to search for highly structured β-hairpin peptides. Proc Natl Acad Sci. 2002;99:614-9
45. Magnan R, Escrieut C, Gigoux V, De K, Clerc P, Niu F. et al. Distinct CCK-2 receptor conformations associated with β-arrestin-2 recruitment or phospholipase-C activation revealed by a biased antagonist. J Am Chem Soc. 2013;135:2560-73
46. Smith DA, Di L, Kerns EH. The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery. Nat Rev Drug Discov. 2010;9:929-39
47. Zoghbi SS, Anderson KB, Jenko KJ, Luckenbaugh DA, Innis RB, Pike VW. On quantitative relationships between drug-like compound lipophilicity and plasma free fraction in monkey and human. J Pharm Sci. 2012;101:1028-39
48. Lázníček M, Lázníčková A. The effect of lipophilicity on the protein binding and blood cell uptake of some acidic drugs. J Pharm Biomed Anal. 1995;13:823-8
49. Chatterjee J, Rechenmacher F, Kessler H. N-methylation of peptides and proteins: an important element for modulating biological functions. Angew Chem Int Ed. 2013;52:254-69
50. Sharma A, Kumar A, Abdel Monaim SA, Jad YE, El-Faham A, de la Torre BG. et al. N-methylation in amino acids and peptides: Scope and limitations. Biopolymers. 2018;109:e23110
51. Luisa Di Gioia M, Leggio A, Malagrinò F, Romio E, Siciliano C, Liguori A. N-methylated α-amino acids and peptides: synthesis and biological activity. Mini Rev Med Chem. 2016;16:683-90
52. Roosenburg S, Laverman P, Joosten L, Cooper M, Kolenc-Peitl P, Foster J. et al. PET and SPECT imaging of a radiolabeled minigastrin analogue conjugated with DOTA, NOTA, and NODAGA and labeled with 64Cu, 68Ga, and 111In. Mol Pharmaceutics. 2014;11:3930-7
53. Boyce M, Lloyd KA, Pritchard DM. Potential clinical indications for a CCK2 receptor antagonist. Curr Opin Pharmacol. 2016;31:68-75
54. Aly A, Shulkes A, Baldwin GS. Gastrins, cholecystokinins and gastrointestinal cancer. Biochim Biophys Acta Rev Cancer. 2004;1704:1-10
55. Lindström E, Björkqvist M, Håkanson R. Pharmacological analysis of CCK2 receptor antagonists using isolated rat stomach ECL cells. Br J Pharmacol. 1999;127:530-6
56. Zeng Q, Ou L, Wang W, Guo D-Y. Gastrin, cholecystokinin, signaling, and biological activities in cellular processes. Front Endocrinol. 2020;11:112
57. Melis M, Krenning EP, Bernard BF, Barone R, Visser TJ, de Jong M. Localisation and mechanism of renal retention of radiolabelled somatostatin analogues. Eur J Nucl Med Mol Imaging. 2005;32:1136-43
58. Ogawa K, Ishizaki A, Takai K, Kitamura Y, Makino A, Kozaka T. et al. Evaluation of Ga-DOTA-(D-Asp) n as bone imaging agents: D-aspartic acid peptides as carriers to bone. Sci Rep. 2017;7:13971
59. Kwon D, Zhang Z, Zeisler J, Kuo H-T, Lin K-S, Benard F. Reducing the Kidney Uptake of High Contrast CXCR4 PET Imaging Agents via Linker Modifications. Pharmaceutics. 2022;14:1502
60. Hennrich U, Kopka K. Lutathera®: the first FDA-and EMA-approved radiopharmaceutical for peptide receptor radionuclide therapy. Pharmaceuticals. 2019;12:114
61. Bergsma H, Konijnenberg MW, van der Zwan WA, Kam BL, Teunissen JJ, Kooij PP. et al. Nephrotoxicity after PRRT with 177 Lu-DOTA-octreotate. Eur J Nucl Med Mol Imaging. 2016;43:1802-11
62. Corlett A, Pinson J-A, Rahimi MN, Zuylekom JV, Cullinane C, Blyth B. et al. Development of Highly Potent Clinical Candidates for Theranostic Applications against Cholecystokinin-2 Receptor Positive Cancers. J Med Chem. 2023
Author contact
![]() Corresponding author: Department of Radiopharmaceutical Sciences, Cancer Imaging, The Peter MacCallum Cancer Centre, Melbourne, Victoria, Australia. Phone: (+61 3) 8559 6913, email: mo.haskaliorg.
Corresponding author: Department of Radiopharmaceutical Sciences, Cancer Imaging, The Peter MacCallum Cancer Centre, Melbourne, Victoria, Australia. Phone: (+61 3) 8559 6913, email: mo.haskaliorg.