Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Experimental Section
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2024; 14(1):116-132. doi:10.7150/thno.88061 This issue Cite
Research Paper
Reprogramming Exosomes to Escape from Immune Surveillance for Mitochondrial Protection in Hepatic Ischemia-Reperfusion Injury
Shanshan Liu1,8†, Xinyu Xiao1†, La Zhang2†, Jianwei Wang1†, Wei Zhao1, Haichuan Liu2, Rui Liao2, Zhi Li3, Mengxia Xu3, Jiao Guo1, Baoyong Zhou2 ![]() , Chengyou Du2
, Chengyou Du2 ![]() , Qiling Peng1,4
, Qiling Peng1,4 ![]() , Ning Jiang5,6,7
, Ning Jiang5,6,7 ![]()
1. School of Basic Medical Science, Chongqing Medical University, Chongqing 400016, P. R. China.
2. Department of Hepatobiliary Surgery, the First Affiliated Hospital of Chongqing Medical University, Chongqing 400016, P. R. China.
3. Traditional Chinese Medicine Hospital of Bijie City, Guizhou province, 551700, People's Republic of China.
4. Bijie Municipal Health Bureau, Guizhou province, 551700, People's Republic of China.
5. Department of Pathology, Chongqing Medical University, Chongqing 400016, P. R. China.
6. Molecular Medicine Diagnostic and Testing Center, Chongqing Medical University, Chongqing 400016, P. R. China.
7. Department of Pathology, the First Affiliated Hospital of Chongqing Medical University, Chongqing 400016, P. R. China.
8. Department of Plastic and Maxillofacial Surgery, The Second Affiliated Hospital of Chongqing Medical University, Chongqing 400016, P. R. China.
† These authors contributed equally to this work.
Received 2023-7-13; Accepted 2023-10-6; Published 2024-1-1
Abstract

Background: Therapeutic interventions such as synthetic drugs and microRNA (miR) modulators have created opportunities for mitigating hepatic ischemia/reperfusion injury (HIRI) by alleviating mitochondrial dysfunction. However, delivering multi-therapeutic ingredients with low toxicity to hepatocytes still lags behind its development.
Methods: In this study, we endowed exosomes with delivery function to concentrate on hepatocytes for multidimensionally halting mitochondria dysfunction during HIRI. Concretely, exosomes were reprogrammed with a transmembrane protein CD47, which acted as a “camouflage cloak” to mimic the “don't eat me” mechanism to escape from immune surveillance. Besides, HuR was engineered bridging to the membrane by fusing with CD47 and located in the cytoplasm for miR loading.
Results: This strategy successfully delivered dual payloads to hepatocytes and efficiently protected mitochondria by inhibiting the opening of mitochondrial permeability transition pore (mPTP) and upregulating mitochondrial transcription factor A (TFAM), respectively.
Conclusions: The reprogramming of exosomes with CD47 and HuR for targeted delivery of CsA and miR inhibitors represents a promising therapeutic strategy for addressing HIRI. This approach shows potential for safe and effective clinical applications in the treatment of HIRI.
Keywords: Exosomes, Hepatic ischemia/reperfusion injury, Macrophages, CD47, Mitochondria, Cyclosporin A
Introduction
Hepatic ischemia/reperfusion injury (HIRI), an inevitable pathological insult during liver surgery such as liver resection and transplantation, presents one of the major causes of perioperative morbidity and mortality [1]. HIRI is a two-phase event, in which cell damage initially results from oxygen deficiency by insufficient blood flow and is exacerbated upon restoration of oxygen supply [2]. As a characteristic of the ischemic phase, the damage of mitochondria which are particularly sensitive to hypoxia, embodies in oxidative phosphorylation decline, ATP depletion, and metabolite accumulation [3]. Once blood supply is restored, excessive metabolites, especially reactive oxygen species (ROS), intensify mitochondrial dysfunction, as manifested by the depolarization of membrane potential, opening of mitochondrial permeability transition pore (mPTP), and damage of mitochondrial DNA (mtDNA) [3]. Accompanied by mitochondrial damage aggravating, pro-apoptotic factors such as cytochrome C (CytC) are released, ultimately leading to hepatocyte death and triggering a cascade of HIRI events. In general, mitochondria are critical instigators and later effectors of HIRI, making the degree of mitochondrial dysfunction directly proportional to the severity of HIRI. Accordingly, the critical importance of mitochondria in HIRI highlights mitochondria-targeted approaches as potential therapeutic avenues for future treatments.
Accumulated evidence has revealed the crucial role of mitochondria as therapeutic targets in ameliorating HIRI [4,5]. Strategies for mitigating mitochondrial damage generally concentrate on modulating mitochondrial morphology, reversing mitochondrial respiratory chain dysfunction, especially suppressing the opening of mPTP and maintaining mitochondrial homeostasis [6]. Previous studies have confirmed the therapeutic efficacy of cyclosporin A (CsA), a polypeptide, which indirectly interferes with mPTP opening by competitively inhibiting cyclophilin D (CypD), a key regulator of mPTP [6,7]. Besides, microRNA (miR)-based therapy has demonstrated potential in maintaining mitochondrial homeostasis by regulating the expression of mitochondrial regulator proteins, such as mitochondrial transcription factor A (TFAM) [8,9]. TFAM plays a crucial role in sustaining mtDNA stability, and an increase of its expression has been shown to relieve HIRI significantly [10]. Given the complexity of mitochondrial damage, carrier systems to co-deliver therapeutic ingredients are potential approaches to effectively halt the progress of HIRI. However, the inevitable side effects of conventional carrier systems are required to be addressed, which arise from insufficient targeting precision, low solubility and stability, and poor biocompatibility [11]. Hence, developing an innovative delivery system for CsA and miR-modulators, which inhibit CypD and upregulate TFAM, respectively, to alleviate HIRI is paramount.
Recently, nanoparticle (NP)-based therapeutics have depicted prospective approaches in alleviating HIRI, such as Ceria NPs [12] and platinum NPs [13]. However, as foreign particles, NPs still exist some issues in toxicity, efficacy, and stability [14]. Gratifyingly, as one of the small extracellular vesicles with remarkable biocompatibility, stability, and favorable safety profile, exosomes (Exos) have emerged as promising drug delivery vehicles compared with traditional ones [15]. Owing to their unique “tropism” capability, Exos could target and engage with cells from their parental tissue selectively, thereby enhancing the precision of therapeutic delivery [16]. Nevertheless, macrophages still craft a hurdle to the effectiveness of Exos in delivering to hepatocytes [17]. Therefore, the ideal delivery vehicle should possess the capability to evade immune surveillance of macrophages, particularly Kupffer cells.
To accommodate the demands of escaping from immune surveillance and co-loading CsA and miR-modulators with fewer side effects, inspired by the ability of CD47 expressed on tumor cells emitting a “don't eat me” signal to macrophages and human antigen R (HuR) interacting with AU-rich elements (AREs) to bind target RNA [18,19], we established a nimbly reprogrammed Exos wearing the CD47 “camouflage cloak” and grafting the “AREs-magnetism” HuR. In detail, reprogrammed Exos overexpressed CD47-HuR and efficiently delivered CsA and miR inhibitor (miRi) targeting TFAM to hepatocytes for assuaging HIRI, referred to as CsA/miRi@ExosCD47-HuR, as shown in Scheme 1. In summary, our study introduces an innovative method of utilizing reprogrammed Exos as efficient drug carriers for both pharmaceutical ingredients and RNA molecules, which seeds hope for HIRI therapy against mitochondrial dysfunction.
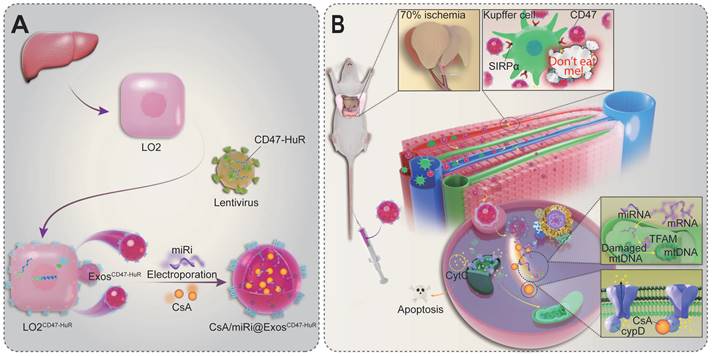
Scheme of reprogrammed exosomes for alleviating hepatic ischemia/reperfusion injury via mitochondria protection. (A) The human liver cells (LO2) were functionalized with CD47-HuR fusion protein (LO2CD47-HuR). The Cyclosporin A (CsA) and mitochondrial transcription factor A (TFAM) targeting microRNA inhibitor (miRi) were loaded into LO2CD47-HuR-derived exosomes by electroporation to prepare CsA/miRi@ExosCD47-HuR. (B) The mechanism of CsA/miRi@ExosCD47-HuR to alleviate HIRI by inhibiting the opening of mitochondrial permeability transition pore (mPTP) and upregulating TFAM.
Results
Exos reprogrammed with the CD47-HuR fusion protein and the identification of miR modulators
Inspired by the “camouflage cloak” function of CD47 [18] to evade immune surveillance and the capacity of HuR to bind AREs [19], a functionalized fusion protein was successfully developed using advanced genetic engineering techniques (Figure 1A). Specifically, the target fusion gene CD47-HuR was integrated into LO2 cells via lentiviral infection, leading to overexpression of the CD47-HuR fusion protein in LO2 cells, which were referred to as LO2CD47-HuR cells (Figure S1A and B). Exos secreted from LO2 or LO2CD47-HuR cells were extracted by ultracentrifugation, with ExosCD47-HuR inheriting overexpression of the CD47-HuR fusion protein (Figure S1C), signifying the successful integration of CD47-HuR onto Exos. To fully harness the potential of HuR, the TargetScan database [20] was searched for miRs with a high content of AREs that could regulate TFAM expression in Homo sapiens (human, hsa) or Mus musculus (mouse, mmu). Subsequently, hsa-miR-590-3p and mmu-miR-7057-3p were identified, as shown in Figure S2A and E. In addition, western blot and quantitative real-time polymerase chain reaction (qRT-PCR) were performed to validate whether the miR modulators possessed this functionality in LO2 and NCTC-1469 cells (Figure S2B-D and F-H). As expected, our results demonstrated that the miR inhibitor (miRi) could significantly upregulate TFAM expression. Thus, the reprogrammed ExosCD47-HuR were successfully fabricated, and miR modulators that could increase TFAM expression were obtained.
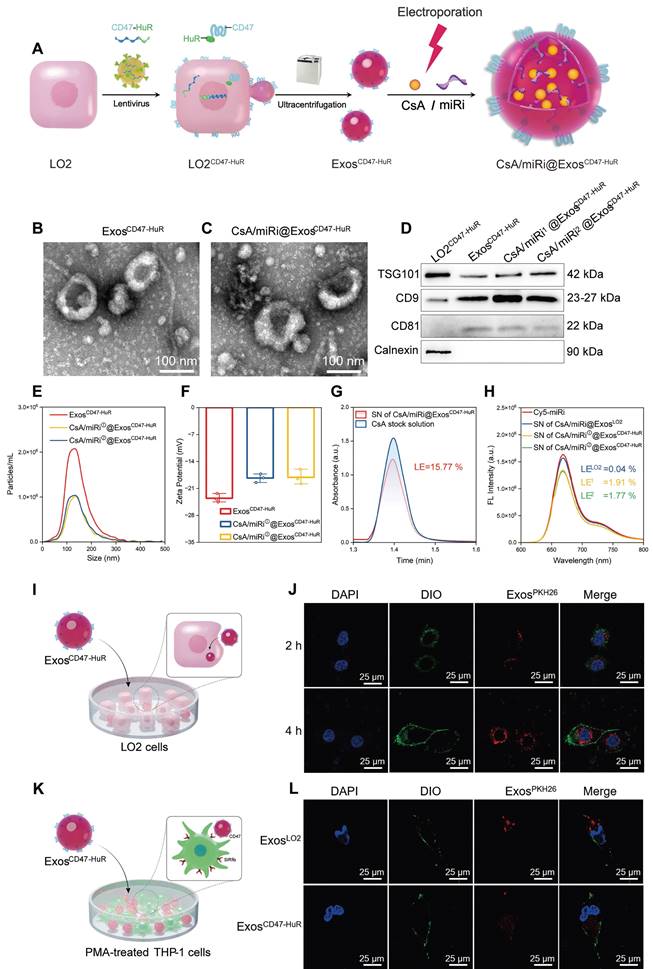
Preparation and molecular characterization of reprogrammed Exos. (A) Schematic illustration of CsA/miRi@ExosCD47-HuR preparation. Transmission electron microscope (TEM) images of ExosCD47-HuR isolated from lentivirus-infected LO2 (LO2CD47-HuR) cells (B) and CsA/miRi@ExosCD47-HuR which were loaded with CsA and miRi simultaneously through electroporation (C). Scale bars: 100 nm. (D) Exosomal biomarkers were assessed using western blot analysis, proteins of LO2CD47-HuR cells (lane 1), ExosCD47-HuR (lane 2), CsA/miRi①@ExosCD47-HuR (lane 3), and CsA/miRi②@ExosCD47-HuR (lane 4). TSG101, CD9, and CD81 were Exo-specific biomarkers that enriched in Exos, while the endoplasmic reticulum marker, Calnexin, was devoid. (E) Particle size (nm) and concentration (particles/mL) of Exos were measured by nanoparticle tracking analysis (NTA). (F) Zeta potential (ξ) histogram of ExosCD47-HuR, CsA/miRi①@ExosCD47-HuR, CsA/miRi②@ExosCD47-HuR, respectively. (G) The absorbance of CsA stock solution and supernatant (SN) of CsA/miRi@ExosCD47-HuR after electroporation and ultracentrifugation using high-performance liquid chromatography with ultraviolet detection (HPLC-UV) at a wavelength of 210 nm. Loading efficiency (LE) of CsA was quantified by HPLC-UV. The LE was calculated by the formula of (WCsA-Wfree CsA)/WExos × 100%, where WCsA was the total weight of CsA, Wfree CsA was the weight of unbounded CsA, and WExos was the weight of Exos. (H) Fluorescent spectrum analyses of aqueous solution of Cy5-labeled miRi, SN of CsA/miRi@ExosLO2, SN of CsA/miRi①@ExosCD47-HuR, and SN of CsA/miRi②@ExosCD47-HuR after electroporation and ultracentrifugation, with excitation at 650 nm and emission at 670 nm. The LE was calculated by: (WmiRi-Wfree miRi)/WExos × 100%, where WmiRi was the weight of total miRi, Wfree miRi was the weight of unbounded miRi, and WExos was the weight of Exos. (I) Schematic illustration of ExosCD47-HuR being internalized by LO2 cells. (J) Confocal images display the in vitro uptake of ExosCD47-HuR by LO2 cells within 4 h. Nuclei were stained with 4', 6-diamidino-2-phenylindole (DAPI, blue), Exos were labeled with PKH-26 (red), and cell membranes were stained with 3,3'-dioctadecyloxacarbocyanine perchlorate (DIO, green). Scale bar: 25 µm. (K) Schematic illustration of ExosCD47-HuR escaping phagocytosis from THP-1-M cells. (L) Exos functionalized with CD47-HuR have a reduced likelihood of being internalized by THP-1-M cells. Nuclei were stained with DAPI (blue), Exos were labeled with PKH-26 (red), and cell membranes were stained with DIO (green). THP-1-M cells were differentiated with 100 ng/mL Phorbol-12-myristate-13-acetate (PMA) for 48 h to differentiate macrophages in vitro. Scale bar: 25 µm. miRi① represents hsa-miR-590-3p inhibitor, miRi② represents mmu-miR-7057-3p inhibitor.
Characterization and antiphagocytic function of CsA/miRi@ExosCD47-HuR in vitro
After extracting ExosCD47-HuR from donor cells (LO2CD47-HuR), CsA and miRi were concurrently incorporated into Exos via electroporation (Figure 1A). Subsequently, transmission electron microscopy (TEM) revealed the characteristic round-shaped morphology of ExosCD47-HuR (Figure 1B and C), while western blot showed the expression of Exo-specific markers (TSG101, CD9, CD81) [21] and an endoplasmic reticulum marker (Calnexin) [22] (Figure 1D). Nanoparticle tracking analysis (NTA) of ExosCD47-HuR revealed an average size of 142 nm at a concentration of approximately 2.0 × 106 particles/mL (Figure 1E), and the zeta potential (ξ) was negative (- 23.53 mV) (Figure 1F). Notably, electroporation did not affect their morphology (Figure 1C), protein expression (Figure 1D), average size (Figure 1E), or zeta potential (ξ) (Figure 1F). Furthermore, the loading efficiency (LE) of CsA in ExosCD47-HuR was determined to be 15.77% by high-performance liquid chromatography with ultraviolet detection (HPLC-UV) (Figure 1G), while the LE of miRi altered from 0.04% in ExosLO2 to approximately 2.0% in ExosCD47-HuR, as measured by a fluorescence spectrophotometer (Figure 1H), indicating that the overexpression of CD47-HuR increased the LE of miRi into Exos. Having fabricated reprogrammed Exos, in vitro experiments were conducted to assess their internalization. Confocal laser scanning microscopy (CLSM) confirmed the cellular uptake of Exos (labeled with the fluorescent dye PKH26) after incubation with LO2 cells or phorbol-12-myristate-13-acetate (PMA) treated THP-1 cells (THP-1-M). ExosCD47-HuR were effectively internalized by LO2 cells rather than THP-1-M cells after 4 h of incubation (Figure 1I-L, Fig S3), and the uptake efficiency of ExosLO2 without CD47-HuR overexpression was higher in THP-1-M cells, suggesting that ExosCD47-HuR could evade immune surveillance. The in vitro release assay of CsA and miRi from ExosCD47-HuR indicated that 50% of CsA was released within the initial 12 h, while 80% of it was released within 24 h, whereas 50% of miRi was released within the first 24 h, with 80% of miRi being released within 36 h (Figure S4). Under these circumstances, CsA/miRi@ExosCD47-HuR with high loading of dual cargos and high distribution in target cells could be used for further in vitro and in vivo investigations to verify therapeutic benefits.
Antiapoptosis effects of CsA/miRi@ExosCD47-HuR by protecting mitochondria in vitro
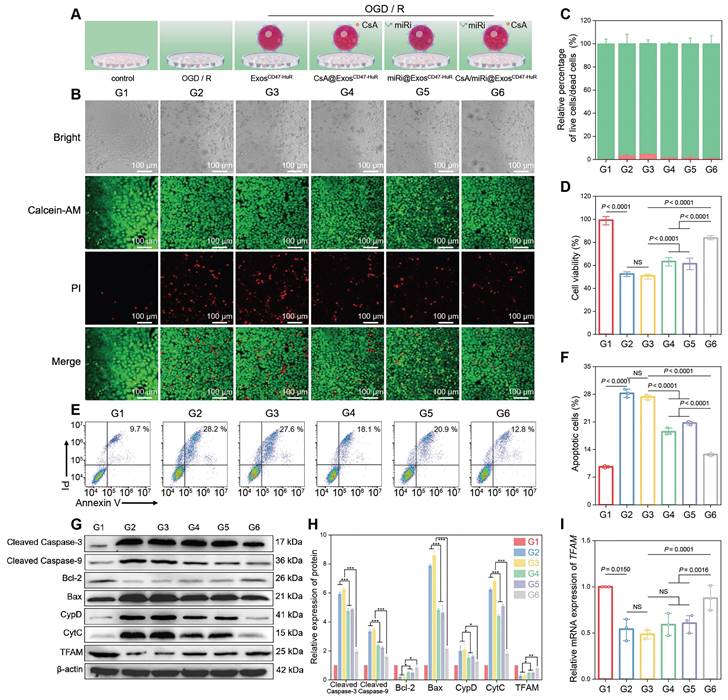
Having established well-crafted Exos loaded with CsA and miRi, further studies were conducted to investigate whether CsA/miRi@ExosCD47-HuR could alleviate the damage induced by oxygen-glucose deprivation/ reoxygenation (OGD/R) in vitro. First, OGD for 6 h and R for 6 h was selected to maximize the antiapoptosis effects, because the expression of apoptosis-related proteins (cleaved caspase 3, cleaved caspase 9, Bcl-2, Bax, CypD, and CytC) [23] and TFAM reached a plateau in LO2 cells (Figure S5). Furthermore, CsA/miRi@ExosCD47-HuR and other treatments were incubated with LO2 cells for 24 h before OGD/R, as shown in Figure 2A. Afterward, CLSM was used to analyze LO2 cells stained with calcein AM and propidium iodide (PI) and effectively visualize live and dead cells (Figure 2B and C). Furthermore, the cell counting kit-8 assay revealed that less than 51.5% of cells survived following OGD/R, whereas over 83.7% of cells remained viable after pretreatment with CsA/miRi@ExosCD47-HuR (Figure 2D, Figure S6D). More importantly, we observed a significant decrease in cell apoptosis after treatment with CsA/miRi@ExosCD47-HuR, indicating that these factors significantly maintained cell viability (Figure 2E and F, Figure S6A and C). Furthermore, the expression of apoptosis-related proteins was evaluated by western blot and revealed that CsA/miRi@ExosCD47-HuR could robustly alleviate apoptosis, while CsA@ExosCD47-HuR and miRi@ExosCD47-HuR exerted less pronounced effects (Figure 2G and H). Overall, the antiapoptotic effects of CsA/miRi@ExosCD47-HuR on the OGD/R model confirmed that among the different treatments, CsA/miRi@ExosCD47-HuR provided optimal protection of LO2 cells.
Therapeutic effects and anti-apoptosis mechanism of drug-loaded ExosCD47-HuR in vitro. (A) Schematic illustration of group design. Groups were designed as: normal LO2 cells (G1); LO2 cells after OGD/R (G2); LO2 cells pretreated with ExosCD47-HuR before OGD/R (G3); LO2 cells pretreated with CsA@ExosCD47-HuR before OGD/R (G4); LO2 cells pretreated with miRi@ExosCD47-HuR before OGD/R (G5); LO2 cells pretreated with CsA/miRi@ExosCD47-HuR before OGD/R (G6). miRi refers to hsa-miR-590-3p inhibitor. (B) Confocal laser scanning microscope (CLSM) images of LO2 cells co-stained with calcein AM/PI after pretreatments and OGD/R. Green: live cells; Red: dead cells. Scale bar: 100 μm. (C) Relative percentage of live and dead LO2 cells across different treatment groups, calculated by ImageJ. (D) Cell viability of LO2 cells after OGD/R declined to 50%, and recovered to 80% after pretreatment with CsA/miRi@ExosCD47-HuR. (E) LO2 cell apoptosis was analyzed using Annexin V/PI staining by flow cytometry, with quantitative analysis shown in (F). (G) Western blot of apoptosis-related protein and TFAM expression in LO2 cells. (H) Quantification of apoptosis-related protein and TFAM expression. (I) qRT-PCR analysis of the mRNA level of TFAM in LO2 cells after being treated with different formulations. Data are expressed as mean ± S.D. (n = 3), ***P < 0.001, **P < 0.01, *P < 0.05, NS means no difference (non-repeated ANOVA followed by Tukey's test).
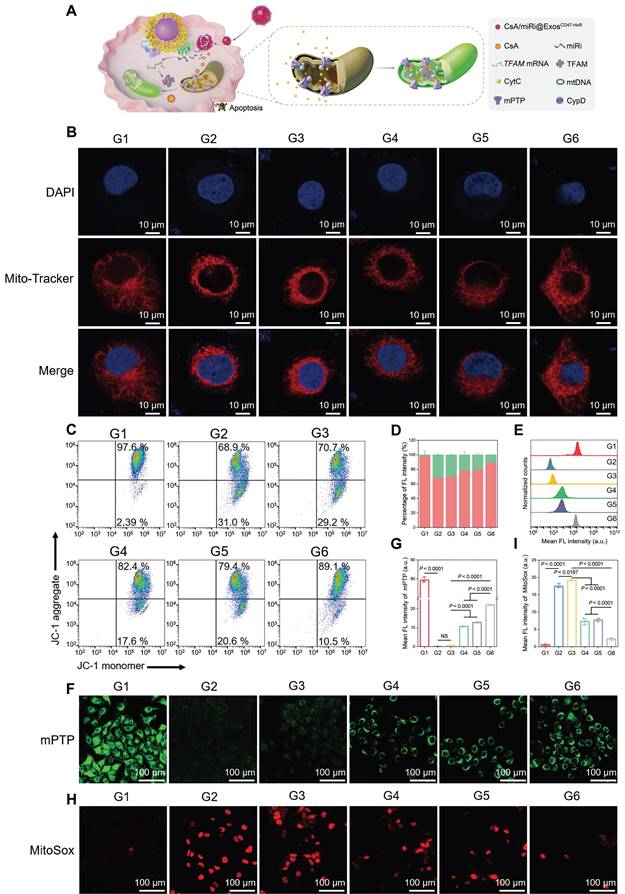
As a classic pathway of programmed cell death, apoptosis can be triggered by the activation of the mitochondrial pathway [24]. Concomitantly, TFAM, a key regulator of mitochondrial gene expression [25], was upregulated at the protein and mRNA levels after treatment with CsA/miRi@ExosCD47-HuR, as determined by western blot and qRT-PCR (Figure 2G-I). Therefore, we examined whether CsA/miRi@ExosCD47-HuR exerted antiapoptotic effects by protecting mitochondria, as shown in Figure 3A. First, Mito Tracker (red) staining was used to monitor mitochondrial morphology in LO2 cells that were pretreated with CsA/miRi@ExosCD47-HuR and other Exos after OGD/R (Figure 3B, Figure S6B). Consequently, CsA/miRi@ExosCD47-HuR partially restored the filamentous network of mitochondria in LO2 cells, suggesting its potential to ameliorate mitochondrial damage by directly maintaining mitochondrial morphology. Subsequently, mitochondrial functional assays were performed to examine mitochondrial membrane potential (Δψm), mPTP opening, and mitochondrial ROS levels. Specifically, the change in Δψm was monitored by JC-1 staining, and aggregates represent healthy polarized mitochondria (red), whereas monomers (green) indicate unhealthy depolarized mitochondria [26]. Flow cytometry and CLSM showed that OGD/R exhibited decreased JC-1 aggregates and increased JC-1 monomers, while CsA/miRi@ExosCD47-HuR effectively restored Δψm (Figure 3C and D, Figure S7A and B). Concurrently, the extent of mPTP opening was assessed via a fluorescence assay kit, which revealed that CsA@ExosCD47-HuR, miRi@ExosCD47-HuR, and CsA/miRi@ExosCD47-HuR could inhibit mPTP opening, as indicated by flow cytometry and CLSM (Figure 3E-G, Figure S7C). Moreover, representative CLSM images of MitoSox (red) which is a mitochondrial-specific ROS indicator, showed the same outcomes, as shown in Figure 3H and I. Accordingly, a significant amount of ROS (DCFH-DA probe) was found in the cytoplasm of LO2 cells after OGD/R, while incubating LO2 cells with CsA/miRi@ExosCD47-HuR significantly reduced the fluorescence intensity, indicating the potent scavenging of ROS, as shown in Figure S7D and E. In summary, our results demonstrated a significant reduction in apoptosis, particularly through the alleviation of mitochondrial dysfunction after treatment with CsA/miRi@ExosCD47-HuR in the OGD/R model, suggesting that the use of CsA/miRi@ExosCD47-HuR might be a therapeutic strategy for mitigating OGD/R-induced cell damage.
Protective effects against mitochondrial dysfunction of drug-loaded ExosCD47-HuR in vitro. (A) Schematic illustration of CsA/miRi@ExosCD47-HuR protecting mitochondria. (B) Representative CLSM images showing mitochondrial morphology stained with MitoTracker (red) and DAPI (blue). Scale bar: 10 μm. (C) Mitochondrial membrane potential (Δψm) detected by flow cytometry after stained with JC-1 dye. (D) The percentages of JC-1 aggregates (mitochondrial membrane potential intact, red) and JC-1 monomers (mitochondrial membrane potential lost, green) indicate the extent of mitochondrial-dependent apoptosis. The extent of mPTP opening was evaluated by calcein AM (green) staining and then detected by flow cytometry (E) and CLSM (F). Scale bar: 100 μm. (G) Quantification of the extent of mPTP opening from CLSM. (H) Representative confocal images of reactive oxygen species generated from mitochondria stained with MitoSox (red). Scale bar: 100 μm. (I) Quantitative analysis of MitoSox fluorescence intensity. Groups were designed as: normal LO2 cells (G1); LO2 cells after OGD/R (G2); LO2 cells pretreated with ExosCD47-HuR before OGD/R (G3); LO2 cells pretreated with CsA@ExosCD47-HuR before OGD/R (G4); LO2 cells pretreated with miRi@ExosCD47-HuR before OGD/R (G5); LO2 cells pretreated with CsA/miRi@ExosCD47-HuR before OGD/R (G6). miRi refers to hsa-miR-590-3p inhibitor. Data are expressed as mean ± S.D. (n = 3), NS means no difference (non-repeated ANOVA followed by Tukey's test).
In vivo distribution and antiphagocytic capabilities of ExosCD47-HuR
The cytoprotective effects of CsA/miRi@ExosCD47-HuR against apoptosis through the attenuation of mitochondrial dysfunction was validated in vivo. First, the drug delivery capability of reprogrammed Exos to hepatocytes was evaluated by injecting 1,1'-dioctadecyl-3,3,3',3'-tetramethylindotricarbocyanine iodide (DIR)-labeled ExosLO2 or ExosCD47-HuR, and the maximum fluorescence intensity was observed at 24 h postinjection (Figure 4A). Consistently, quantitative analysis showed that the amount of ExosCD47-HuR in the liver was approximately 1.54-fold higher than that of ExosLO2 at 24 h postinjection (Figure 4B). Furthermore, the fluorescence intensity in the liver persisted even at 72 h postinjection. In contrast, there was a lower fluorescence intensity and earlier disappearance of ExosLO2 (Figure 4A), suggesting effective evasion of phagocytosis by ExosCD47-HuR. In addition, the ex vivo images showed a high level of fluorescence in liver tissue, particularly in the group that was injected with ExosCD47-HuR (Figure 4C, right). To further examine this effect, mice were injected with 1,1'-dioctadecyl-3,3,3',3'-tetramethylindodicarbocyanine,4-chlorobenzenesulfonate salt (DID)-labeled Exos, liver tissues were collected at 24 h postinjection and macrophages were labeled by the macrophage marker F4/80 [27]. Consistent with the in vitro results, CLSM showed reduced uptake of ExosCD47-HuR (red) by macrophages (green) (Figure 4D). Overall, ExosCD47-HuR were more capable of phagocytic escape than ExosLO2, highlighting the superior ability of ExosCD47-HuR with an extended half-life in liver tissue to deliver drugs precisely.
In vivo distribution and phagocytosis evasion of ExosCD47-HuR. (A) In vivo fluorescence images show the accumulation of 1,1'-dioctadecyl-3,3,3',3'-tetramethylindotricarbocyanine iodide (DIR)-labeled ExosLO2 and ExosCD47-HuR in the liver of BALB/c mice. (B) Quantification of fluorescence intensity in the liver at various time points, with the ExosCD47-HuR exhibiting a superior accumulation than ExosLO2. Data are mean ± S.D. (n = 3), ***P < 0.001, **P < 0.01, *P < 0.05 (Student's t-test). (C) Tissue distribution analyses show that ExosCD47-HuR is preferentially targeted to the liver. (D) Representative CLSM images show that ExosCD47-HuR can optimize their phagocytosis evasion ability. Macrophages were labeled green with F4/80, Exos were labeled red with 1'-Dioctadecyl-3,3,3',3'-tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate salt (DID), and nuclei were labeled blue with DAPI. (E-G) Protein and mRNA expression of TFAM at different reperfusion time points: 3 h, 6 h, 12 h, and 24 h, with the most significant decrease at 6 h. (H, I) Levels of aspartate transaminase (AST) and alanine transaminase (ALT) peaked at 6 h after reperfusion. (J) A step-wise overview of CsA/miRi@ExosCD47-HuR injection, ischemia/reperfusion procedure, and follow-up sample collection for BALB/c mice. Data are expressed as mean ± S.D. (n = 3), (non-repeated ANOVA followed by Tukey's test).
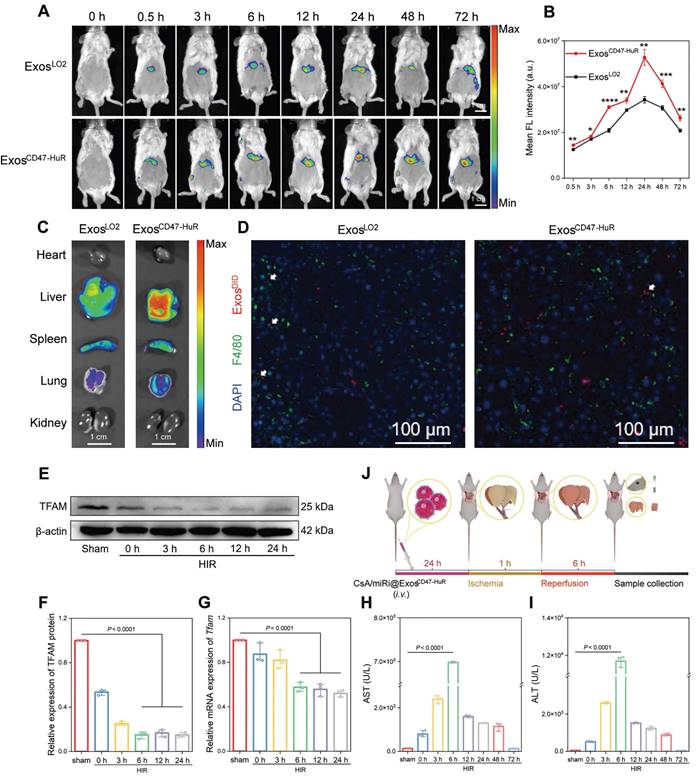
Therapeutic efficacy and biosafety of CsA/miRi@ExosCD47-HuR in the HIRI model
ExosCD47-HuR can evade phagocytosis and persist for a long time in the livers of mice, suggesting that ExosCD47-HuR can systematically deliver CsA and miRi to hepatocytes, which might exert protective effects on mitochondria in vivo. First, a model of 70% warm hepatic ischemia for 60 min was established according to a previous method [28], followed by reperfusion for 3, 6, 12, 24, 48, and 72 h to determine the optimal reperfusion time. Then, liver tissue and blood samples were prepared for protein or RNA extraction and biochemical analysis, and the results showed that TFAM protein and mRNA levels decreased with increasing reperfusion times (Figure 4E-G). Moreover, liver enzymes, such as aspartate aminotransferase (AST) and alanine aminotransferase (ALT), peaked at 6 h postreperfusion, suggesting maximal liver damage at this time point (Figure 4H and I). As a result, BALB/c mice were subjected to partial warm hepatic ischemia for 60 min followed by a 6 h of reperfusion in the HIRI model.
Subsequently, the therapeutic potential of CsA/miRi@ExosCD47-HuR was verified by injecting different groups of Exos into BALB/c mice 24 h before HIRI (Figure 4J). As expected, CsA/miRi@ExosCD47-HuR exhibited significantly superior therapeutic efficacy compared with those in the other groups (Figure 5). Hematoxylin & Eosin (H&E) staining and the Suzuki score were used to visualize the morphology of the liver and quantify disordered organization, including congestion, cytoplasmic parenchymal vacuolization, and necrosis, in HIRI mice, as shown in Figure 5A and B. In addition, AST and ALT analyses showed that CsA/miRi@ExosCD47-HuR could protect liver function (Figure 5C and D). Overall, treatment with drug-loaded ExosCD47-HuR ameliorated the damage to ischemic liver regions with superior efficacy.
Therapeutic efficacy of CsA/miRi@ExosCD47-HuR in vivo. (A) Hematoxylin & Eosin (H&E) staining of residual liver ischemia tissue with drug-loaded ExosCD47-HuR restored liver injury. (B) Quantification of liver damage calculated by Suzuki score shows significant therapy efficiency. (C, D) Levels of AST and ALT with administration of different Exos (CsA@ExosCD47-HuR, miRi@ExosCD47-HuR, or CsA/miRi@ExosCD47-HuR) decreased significantly. (E, F) Liver cell apoptosis by terminal deoxynucleotidyl transferase (TdT) dUTP nick end labeling (TUNEL) staining. (G, H) Apoptosis-related proteins and TFAM expression detected by western blot. (I) mRNA level of Tfam by qRT-PCR from liver tissue. Groups were designed as: sham (G1); BALB/c mice with HIRI (G2); BALB/c mice pretreated with ExosCD47-HuR before HIRI (G3); BALB/c mice pretreated with CsA@ExosCD47-HuR before HIRI (G4); BALB/c mice pretreated with miRi@ExosCD47-HuR before HIRI (G5); BALB/c mice pretreated with CsA/miRi@ExosCD47-HuR before HIRI (G6). miRi refers to mmu-miR-7057-3p inhibitor. Data are expressed as mean ± S.D. (n = 3), ***P < 0.001, **P < 0.01, *P < 0.05, NS means no difference (non-repeated ANOVA followed by Tukey's test).
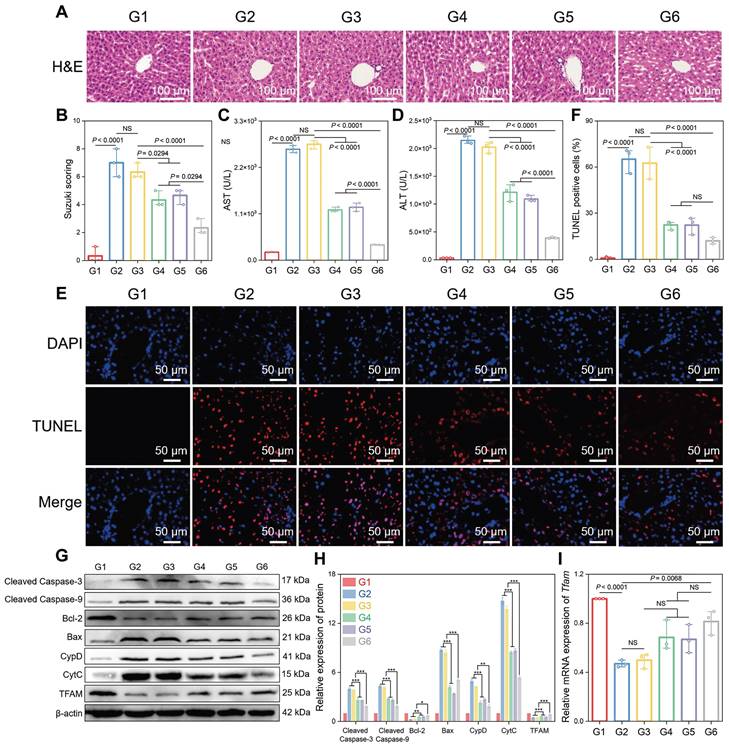
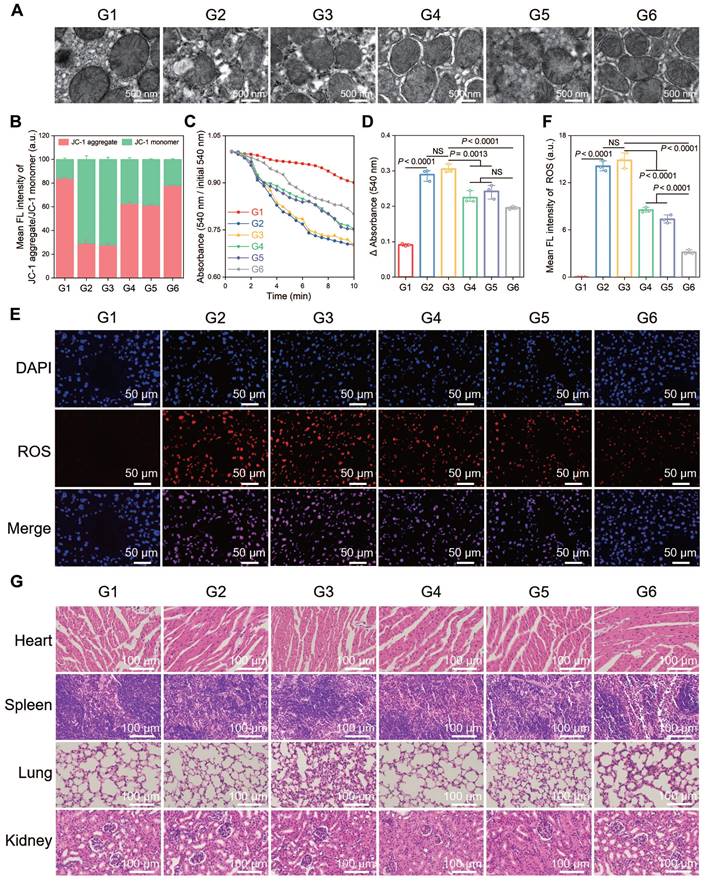
A terminal deoxynucleotidyl transferase (TdT) dUTP nick end labeling (TUNEL) assay was performed to observe cell apoptosis, and a significant increase in the number of TUNEL-positive cells was observed after HIRI, which was notably decreased in the CsA/miRi@ExosCD47-HuR group (Figure 5E and F). Western blot analysis of apoptosis-related proteins presented the same results (Figure 5G and H). Additionally, western blot and qRT-PCR analysis of residual liver tissue showed the upregulation in TFAM protein and mRNA levels after treatment with CsA/miRi@ExosCD47-HuR, revealing that CsA/miRi@ExosCD47-HuR possessed anti-apoptotic potential which might depend on the upregulation of TFAM (Figure 5G-I). Moreover, the impact of CsA/miRi@ExosCD47-HuR on mitigating mitochondrial damage was examined. First, electron microscopic showed that HIRI damaged mitochondrial morphology in the liver, which could be ameliorated by CsA/miRi@ExosCD47-HuR (Figure 6A). To further examine the alterations in mitochondrial function, Δψm and mPTP opening in mouse hepatocytes were assessed using a JC-1 assay and a colorimetric approach, respectively (Figure 6B-D), and ROS levels were evaluated by the DCFH-DA assay (Figure 6E and F). There was a significant decrease in Δψm, the extent of mPTP opening, and an increase in ROS levels in the HIRI groups compared to the sham group. In contrast, treatment with drug-loaded ExosCD47-HuR increased Δψm, inhibited mPTP opening, and reduced ROS production, suggesting their capacity to effectively protect mitochondrial function during HIRI in vivo. Finally, major organs (heart, spleen, lung, and kidneys) in the sham and other treatment groups were harvested to evaluate the potential side effects of Exos with or without cargos by H&E staining (Figure 6G). Mice treated with CsA/miRi@ExosCD47-HuR exhibited no discernible organ impairment or apparent inflammation or necrosis, indicating that CsA/miRi@ExosCD47-HuR possessed good biosafety for the treatment of HIRI. Taken together, our findings demonstrated that CsA/miRi@ExosCD47-HuR could evade immune surveillance by the “camouflage cloak” of CD47. Once endocytosed by hepatocytes, Exos could release the encapsulated cargos, which protected mitochondria and alleviated liver damage caused by HIRI.
Protective effects against mitochondrial dysfunction and biosafety of CsA/miRi@ExosCD47-HuR in vivo. (A) Morphological analysis of mouse liver mitochondria by electron microscopy. Scale bar: 500 nm. (B) Evaluation of Δψm by JC-1 dye in purified liver mitochondria. (C) Detection of mPTP opening with an absorbance assay at 540 nm, with measurements repeated every 30 sec for 20 times. (D) Changes in absorbance values were recorded within 10 min to indirectly evaluate the extent of mPTP opening. (E) ROS level detected by DCFH-DA probe in liver ischemia tissues. Scale bar: 50 μm. (F) Quantitative analysis of DCFH-DA fluorescence intensity. (G) H&E staining of major organs (heart, spleen, lung, and kidneys, respectively) to analyze the biosafety of Exos. Scale bar: 100 μm. Groups were designed as: sham (G1); BALB/c mice with HIRI (G2); BALB/c mice pretreated with ExosCD47-HuR before HIRI (G3); BALB/c mice pretreated with CsA@ExosCD47-HuR before HIRI (G4); BALB/c mice pretreated with miRi@ExosCD47-HuR before HIRI (G5); BALB/c mice pretreated with CsA/miRi@ExosCD47-HuR before HIRI (G6). miRi refers to mmu-miR-7057-3p inhibitor. Data are expressed as mean ± S.D. (n = 3), ***P < 0.001, **P < 0.01, *P < 0.05 NS means no difference (non-repeated ANOVA followed by Tukey's test).
Discussion
We successfully reprogrammed a potent and adaptable drug delivery system that could bolster the loading capability of dual therapeutic ingredients and evade immune surveillance to achieve targeted transport, which stabilized mitochondrial function and ultimately prevented the progression of HIRI. After engineering Exos to be “invisible” to macrophages, we demonstrated the feasibility of CsA/miRi@ExosCD47-HuR to protect mitochondrial function in LO2 cell models of OGD/R and mouse models of partial (70%) warm HIRI. This study is the first attempt to reprogram Exos for the precise dispatch of polypeptides and RNA and offers therapeutic prospects in the clinical treatment of HIRI.
As a crucial cause of liver damage during surgical procedures [2], HIRI is closely associated with the prognosis of patients undergoing hepatic resection, liver transplantation, or trauma procedures. Multiple strategies have been explored to lessen the harmful consequences of HIRI, such as ischemic preconditioning, which involves subjecting an organ to a brief period of ischemia before a more sustained ischemia/reperfusion period [29]. Ischemic preconditioning has been demonstrated to provide benefits and alleviate HIRI in rodent models and patients undergoing liver transplantation or major resection of the liver [30]. However, the absence of a standardized protocol for the optimal duration and number of ischemic preconditioning cycles has hindered the widespread promotion of this technique [31]. In addition, hypothermic machine perfusion, which is another preconditioning method, is limited by the requirement for specialized equipment and expertise [32]. Various pharmacological interventions, such as antioxidants, adenosine agonists, pentoxifylline, protease inhibitors, and prostaglandins, have been studied to counteract the detrimental impact of HIRI [33]. Although the aforementioned preventive measures can effectively reduce liver damage, challenges still exist in determining specific interventions and minimizing side effects [33]. While reviewing most methods, the primary priority was to preserve ATP and reduce oxidative stress, particularly by scavenging ROS. As energy factories and major sources of ROS production in cells, mitochondria are targets for ameliorating liver dysfunction. In the initial stage of HIRI, mitochondrial dysfunction is characterized by the opening of mPTP and subsequent CytC release, which leads to consequent hepatocyte death [34]. In addition, a decrease in TFAM, a nuclear-encoded protein that facilitates the maintenance of mtDNA integrity and prevents mutation accumulation, is a characteristic of the early phase of HIRI and further aggravates respiratory chain dysfunction, leading to mitochondrial deficiencies [10, 35, 36]. Under these circumstances, suppressing mPTP opening and upregulating TFAM expression in dysfunctional mitochondria is crucial in the initial stage to prevent subsequent irreparable damage. As a polypeptide that competitively inhibits CypD, CsA can suppress the opening of mPTP. On the other hand, as a noteworthy therapeutic molecule [37, 38], miRi can upregulate TFAM, which was demonstrated in our study. However, systemic administration of CsA inevitably causes widespread toxicity due to its poor solubility and nonselective distribution in vivo [39]. Recent delivery systems for miRi, such as viral vectors, poly(lactide-co-glycolide) particles, and neutral lipid emulsions, have limitations about loading capacity, targeting efficiency to cells, and toxicity [40]. In general, searching for a golden curecourier to safely and effectively deliver CsA and miRi to hepatocytes has considerable potential for the treatment of HIRI [41].
As intercellular communicators, Exos exhibit natural drug delivery properties [42]. However, the presence of macrophages which are responsible for engulfing foreign particles, especially Kupffer cells in the liver, continues to be a major challenge in the delivery of Exos to hepatocytes [43]. Thus, strategies to generate Exos that are “invisible” to immune surveillance, especially in the liver are urgently needed. Based on the mechanisms by which tumor cells evade macrophage phagocytosis as a result of their high expression of CD47 [44], we engineered Exos to transmit a “don't eat me” signal to achieve targeted transport to hepatocytes. As expected, our study demonstrated that the targeted delivery of ExosCD47-HuR by evading macrophage phagocytosis was superior to that of ExosLO2 both in vitro and in vivo. After achieving targeted transport, the major challenge was to enhance the loading of therapeutic agents in ExosCD47-HuR. About the loading of CsA, ExosCD47-HuR (LE = 15.77%) had a higher loading capability than the lipoprotein nanocarrier (LE = 7.30%) [45]. To load exogenous miRi into Exos, common methods include electroporation, sonication, and the use of Lipofectamine, but their loading capability is comparatively low [46]. To “seat more sites” for miRi in Exos, we borrowed the function of HuR, which can increase the efficiency of encapsulating miRi by interacting with the AREs of the target RNA [47]. Thus, we fused HuR with the C-terminus of CD47 to construct the novel carrier ExosCD47-HuR, which had a high loading capability for miRi compared with ExosLO2 (1.91% vs 0.04%), as validated in our study. After being loaded with dual cargos, there was an increase in the internalization of CsA/miRi@ExosCD47-HuR by hepatocytes, and CsA and miRi inside ExosCD47-HuR were released into the cytoplasm [48]. As anticipated, CsA/miRi@ExosCD47-HuR could significantly protect mitochondrial function by restricting mPTP opening and upregulating of TFAM, which collaboratively halted the progression of hepatocyte death.
In conclusion, we have established a novel curecourier to deliver CsA and miRi to hepatocytes to alleviate HIRI. By evading immune surveillance, our reprogrammed Exos can maintain mitochondrial homeostasis during oxygen supply/demand imbalance. By focusing on the protection of mitochondria, this versatile agent can prevent hepatocyte death. Therefore, our results provide a novel combination strategy of drug and transcriptional manipulation to treat HIRI in the clinic. In light of this, the tactic of reprogrammed curecourier to precisely deliver cargo lays the initial foundation for the treatment of mitochondrial damage-related diseases.
Experimental Section
Materials
LO2 cells (human liver cells) were derived in our laboratory. NCTC-1469 (mouse liver cells) and human THP-1 monocytes were purchased from Procell Life Science & Technology Company (Hubei, China). Dulbecco's modified Eagle's medium (DMEM), Roswell Park Memorial Institute-1640 (RPMI 1640), and Opti-MEM were supplied by Gibco Life Technologies (Grand Island, USA). Fetal bovine serum (FBS) and LipofectamineTM 2000 were purchased from VivaCell (Shanghai, China) and Invitrogen (Carlsbad CA, USA), respectively. Horse serums were purchased from Procell (Wuhan, China). 0.25% Trypsin-EDTA solution, cell counting kit-8 (CCK-8, C0005, TargetMol, USA), penicillin-streptomycin, phenylmethanesulfonyl fluoride (PMSF), Cell Lysis Buffer for Radio Immunoprecipitation Assay (RIPA) and 5,5',6,6'-tetrachloro-1,1',3,3'-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) were obtained from Beyotime Institute of Biotechnology (Shanghai, China). Exo-specific lysis buffer was purchased from Umibio Science & Technology Company (Shanghai, China). Cyclosporine A (CsA) was purchased from MCE company (New Jersey, USA). Phorbol-12-myristate-13-acetate (PMA) was supplied by Sigma-Aldrich (St Louis, USA). All other chemicals used in this work were of analytical grade without further purification and supplied by Chuandong Chemical Co., Ltd (Chongqing, China) unless specified. Deionized (DI) water (Millipore, USA, 18.2 MΩ) was used for all experiments.
Cell culture
LO2 cells transfected with lentivirus containing CD47-HuR fusion gene was designated as LO2CD47-HuR cells. Both LO2 and LO2CD47-HuR cells were cultured in DMEM supplemented with 10% Fetal bovine serum (FBS) and 1% penicillin-streptomycin. NCTC-1469 cells were cultured in DMEM supplemented with 10% horse serum and 1% penicillin-streptomycin. THP-1 cells were grown in RPMI-1640 medium containing 10% FBS and 0.05 mM 2-mercaptoethanol. All cells were cultured at 37 °C and 5% CO2 atmosphere.
Isolation and purification of exosomes (Exos)
After cells were cultured in serum-free cell culture medium for 48 h, a modified protocol to purify Exos was carried out as previously reported [49]. Initially, cell medium containing Exos was collected by centrifugation at 300 g for 10 min and 2,000 g for 20 min to remove cells and then followed by centrifugation at 10,000 g for 30 min to eliminate dead cells and cell debris. Afterward, the resulting clear supernatant was centrifuged for 70 min at 100,000 g using a Sorvall WX 100 + (Thermo Fisher Scientific, Japan) to isolate Exos. After removing the supernatant, Exos pellets were resuspended in 1 mL PBS and mixed with an additional 25 mL cold PBS. Then, Exos pellets were subjected to ultracentrifugation at 100,000 g again for 70 min to remove residual media components and filter the supernatant through a 0.22 μm filter. The purified Exos were finally resuspended in 100 μL PBS and immediately stored at - 80 ℃. Exos derived from LO2 cells were utilized in all in vivo and in vitro experiments.
Loading CsA and microRNA inhibitor (miRi) into Exos
Initially, 100 μg Exos, 100 μg miRi, and 100 μg CsA were mixed in 400 μL cold electroporation buffer (21% Opti-MEM reduced serum medium, 1.15 mM potassium phosphate, and 25 mM potassium chloride). The mixture mentioned above was transferred into an ice-cold cuvette (0.4 mm) and then subjected to electroporation using a Gene Pulser Xcell (BioRad, Hercules, CA) at 400 mV and 125 μF capacitance (pulse/10 ms). To remove unbound cargo, the mixture was washed twice with cold PBS via ultracentrifugation at 110,000 g for 70 min at 4 ℃ and subsequently resuspended in PBS. Exos loaded with CsA and miRi were called CsA/miRi@ExosCD47-HuR.
Characterization of Exos
The size, morphology, and structure of Exos were investigated by a transmission electron microscope (TEM, JEM-1200EX, Japan). Besides, solutions of Exos were diluted to an appropriate concentration (~106 particles/mL). Size distribution and zeta potential (ξ) of Exos were analyzed using a nanoparticle tracking analysis (NTA) instrument (ZetaView PMX110, Particle Metrix, Germany).
Loading efficiency (LE) of CsA and miRi
High-performance liquid chromatography (HPLC) was applied to confirm the LE of CsA. A solution containing 100 μg CsA was prepared in 8 mL solvent, which was used as the initial concentration for loading. The peak area of CsA was measured by an HPLC instrument (Waters e2695, USA). In detail, the test samples were dissolved in a 1:1 acetonitrile-water mixture and then injected into an HPLC system separately alongside control solutions. The chromatographic conditions included a Waters 2489 UV/Vis Detector and a SunFire C18 Sum (4.6 × 150 mm Column). Analysis was conducted at a wavelength of 210 nm with the column temperature set to 35 °C. The concentration of CsA (C62H111N11O12) in the solutions was determined by calculating the ratio of peak areas between the control and test samples. The LE was calculated by the formula of (WCsA-Wfree CsA)/WExos × 100%, where WCsA was the total weight of CsA, Wfree CsA was the weight of unbounded CsA, and WExos was the weight of Exos. The LE of miRi was assessed using a spectrophotometer. The miRi used in this study was labeled with Cy5, with excitation/emission wavelengths of 650 nm/670 nm, respectively. After electroporation, Exos were incubated at 37 °C for 1 h and ultracentrifuged (110,000 g, 4 °C, 70 min) to remove unbound CsA and miRi. An equal amount of Cy5-miRi dissolved in 2 mL DEPC water was referred to as a positive control. The LE was calculated by: (WmiRi-Wfree miRi)/WExos × 100%, where WmiRi was the weight of total miRi, Wfree miRi was the weight of unbounded miRi, and WExos was the weight of Exos.
Cell uptake of Exos
Exos were labeled with PKH26 dye (red). An appropriate amount of Exos was taken to determine their protein concentrations using a BCA assay. The “PKH26 linker (for red fluorescent cell labeling)” storage solution was diluted 10 times with “Diluent C” to prepare a dye working solution with a concentration of 100 μM. The operation was carried out in the dark and the working solution was prepared according to the experimental usage. 100 μg Exos was added to 50 μL working solution. After adding the dye working solution, the centrifuge tube was tightly capped and mixed for 1 min using a vortex mixer, followed by a 10 min incubation. Next, 10 mL PBS was added to the Exo-dye complex and mixed well. The Exos were extracted again using ultracentrifuge to remove excess dye. The precipitate, which was the stained Exos, was resuspended in 200 μL PBS. Cells were cultured in a 24-well plate and allowed to adhere for a minimum of 12 h. Then, PKH26 labeled Exos were added to the cells at a concentration of 50 μg/mL. After incubation for 2 or 4 h at 37 °C, the cells were washed with PBS buffer. To visualize the cellular uptake of Exos, cell membranes were stained with 3,3'-dioctadecyloxacarbocyanine perchlorate (DIO, green) and nuclei were stained with 4',6-diamidino-2-phenylindole (DAPI, blue). Confocal laser scanning microscopy (CLSM, Leica TCS SP8, Mannheim, Germany) was used for imaging.
In vitro cargo release assay
The quantities of CsA and miRi were calculated using a standard curve. For the CsA release kinetics, CsA/miRi@ExosCD47-HuR (50 μgs of total protein) were suspended in 0.1 mL of PBS, loaded into Slide-A-Lyzer MINl dialysis units (molecular mass cutoff 10 kDa, Pierce), and dialyzed against 1 mL of PBS at 37 °C with continuous agitation at 250 rpm. The supernatant was harvested at 0, 2, 4, 8, 12, 24, 36, and 72 h. CsA content in samples was quantified using UV-visible spectroscopy at a wavelength of 210 nm, while the miRi content was determined using a spectrophotometer with excitation and emission wavelengths set at 650 nm and 670 nm, respectively.
OGD/R model
To establish the oxygen-glucose deprivation/reoxygenation (OGD/R) model, cellular hypoxia was ensured by exposure to the air condition of 1% O2, 5% CO2, and 94% N2. First, the hypoxia was induced for 2, 4, 6, 8, and 10 h, respectively, and the fixed reoxygenation time was set for 24 h. Then, the hypoxia time was fixed at 6 h, and reoxygenation was conducted for 3, 6, 12, and 24 h, respectively. Ultimately, the optimal time points for OGD/R were ascertained by the expression of TFAM and apoptosis-related protein.
Evaluation of the capability of escaping from the immune surveillance in vitro
THP-1 cells were seeded in the 24-well plates and incubated with 100 ng/mL of PMA for 48 h before the phagocytosis assay. ExosLO2 and ExosCD47-HuR labeled by PKH26 dye were co-cultured with differentiated THP-1 cells for 2 h and 4 h, respectively. Subsequently, the cells were stained with DIO and DAPI and observed using CLSM.
In vivo biodistribution
The biodistribution of Exos was investigated in vivo in this study. Exos were labeled with 1,1'-dioctadecyl-3,3,3',3'-tetramethylindotricarbocyanine iodide (DIR) dye (AAT Bioquest, USA) at a final concentration of 1 μM, with 200 μg of Exos in a volume of 200 μL, and were administered intravenously via the tail vein. Fluorescence signals were captured using a fluorescent imaging system (NightOWL II LB983, Berthold, Bad Wildbad, Germany) with excitation and emission wavelengths of 750/780 nm at 0.5 h, 3 h, 6 h, 12 h, 24 h, 48 h, and 72 h post-injection. Major organs, including the liver, lung, spleen, heart, and kidney, were harvested 24 h post-injection when the mice were humanely euthanized and were imaged using the same equipment. The mean fluorescence intensities were analyzed using Indigo software.
Establishment of BALB/c mouse hepatic ischemia/reperfusion injury (HIRI) model
Male BALB/c mice aged 6~8 weeks were procured from the Animal Center of Chongqing Medical University. The animal experiments were performed by the Guide for the Care and Use of Laboratory Animals and approved by the Animal Ethics Committee at Chongqing Medical University (Chongqing, China). Surgical induction of HIRI or sham surgery was conducted as previously reported [50]. Briefly, mice were anesthetized via intraperitoneal injection of sodium pentobarbital (60 mg/kg) and placed supine on a temperature-controlled surgery table set at 37 °C to maintain the mice's body temperature at 33 °C. A midline laparotomy was performed, and an atraumatic clip was employed to block clamp blood supply to the left lateral and median lobes of the liver. Following 60 min of partial hepatic ischemia, the clip was removed to initiate hepatic reperfusion. Sham control underwent identical procedures without vascular occlusion. The mice were kept hydrated with warm saline throughout the procedure. The mice were euthanized at several fixed reperfusion time points (3 h, 6 h, 12 h, 24 h, 36 h, 48 h, 72 h) after reperfusion to collect blood and the ischemic liver tissue for further analysis. Each animal experiment consisted of three replicates within each group of animals.
TUNEL
Saline or different kinds of drug-loaded ExosCD47-HuR were intravenously injected to mice via the tail vein 24 h before HIRI. After HIRI, the mice were euthanized and their liver tissues were carefully collected. The tissue sections were subjected to a dewaxing process using a series of solutions including xylene Ⅰ, xyleneⅡ, ethanol Ⅰ, ethanol Ⅱ, 85% ethanol, 75% ethanol, and distilled water wash. To ensure proper fixation, a histological pen was used to circle the tissue, and 100 μL 1×proteinase K working solution was added for 20 min at 37 °C. Following fixation, the slides were washed 3 times with PBS for 5 min each. Subsequently, 100 μL TDT equilibration buffer was added to each sample and incubated at 37 °C for 30 min. After washing with PBS, 50 μL labeling working solution was added to each sample and incubated at 37 °C for 1 h in a humid chamber. The sections were then stained with DAPI for 10 min at room temperature and washed again with PBS. The sections were slightly dried and sealed with an anti-fluorescence quenching sealing agent. Finally, the slides were examined under a fluorescence microscope and images were acquired.
Mitochondrial morphology by TEM
Liver tissues were carefully dissected into 0.5~1.0mm3 blocks using a precise cutting tool to minimize tissue distortion. The tissue samples were rapidly immersed in 3% precooled glutaraldehyde fixative within a time frame of no more than 1 min, to optimize the preservation of the tissue's native cellular structure. Firstly, it was washed 4 times with PBS for 15 min each to remove any extraneous material. Then, it was fixed with 1% osmium tetroxide for 2 h to preserve its ultrastructure. Afterward, it was washed again with PBS 4 times for 15 min each to remove any excess fixative. The sample was then dehydrated with a series of acetone gradients (50%, 70%, 90%, 100%) for 15 min each at 4 °C to remove water and prepare it for embedding. It was infiltrated with embedding medium and cured in a 60 °C oven, with the curing process consisting of 12 h at 35 °C, 12 h at 45 °C, and 2 days at 60 °C to ensure the sample was firmly embedded in the medium. The sample was sliced into ultra-thin sections of 60~80 nm, stained with lead citrate and uranyl acetate, washed with distilled water, and air-dried. Finally, the ultrastructure of the sample was observed under TEM, and images were captured.
Statistical analysis
OriginPro 2023 (version 10.0.0.154) was used to draw histograms and line plots. Data are shown as mean ± S.D. of triplicates unless otherwise indicated. Statistical analysis was performed using a two-tailed Student's t-test or one-way ANOVA with post hoc tests, as appropriate. No animals were excluded from the analysis. P value less than 0.05 was designated statistically significant. Fluorescence Images were quantitatively analyzed using ImageJ (version 1.52). Statistical analysis was performed with GraphPad Prism software (version 9.0.0).
Author contributions
Shanshan Liu: Conceptualization, Methodology, Investigation, Data Curation, Writing - Original draft preparation. Xinyu Xiao: Conceptualization, Methodology, Writing - Original draft preparation. La Zhang: Conceptualization, Writing - Original draft preparation. Jianwei Wang: Validation, Investigation. Wei Zhao: Methodology. Haichuan Liu: Methodology. Mengxia Xu: Visualization. Rui Liao: Visualization. Jiao Guo: Methodology. Zhi Li: Supervision. Baoyong Zhou: Methodology, Supervision. Chengyou Du: Writing - Review & Editing, Supervision. Qiling Peng: Funding acquisition, Writing - Review & Editing, Supervision. Ning Jiang: Funding acquisition, Writing - Review & Editing, Supervision.
Data availability
The data supporting the findings of this study are available within the article and its Supplementary Information. All other data are available from the corresponding author upon request. Source data are provided in this paper.
Supplementary Material
Supplementary methods, figures and tables.
Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (Grant No. 81972023, Grant No. 82203310), the Natural Science Foundation of Chongqing City (Grant No. CSTC2020jcyj-msxmX0144, CSTC2021jcyj-msxm0172, and CSTB2022NSCQ-MSX0054), the Science and Technology Research Program of Chongqing Education Commission of China (Grant No. KJQN201900425 and KJQN202000401), Creative Research Group of CQ University (Grant No. CXQT21017), and Program for Youth Innovation in Future Medicine from Chongqing Medical University.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zhai Y, Petrowsky H, Hong JC, Busuttil RW, Kupiec-Weglinski JW. Ischaemia-reperfusion injury in liver transplantation-from bench to bedside. Nat Rev Gastroenterol Hepatol. 2013;10:79-89
2. Peralta C, Jiménez-Castro MB, Gracia-Sancho J. Hepatic ischemia and reperfusion injury: effects on the liver sinusoidal milieu. J Hepatol. 2013;59:1094-106
3. Cannistrà M, Ruggiero M, Zullo A, Gallelli G, Serafini S, Maria M. et al. Hepatic ischemia reperfusion injury: A systematic review of literature and the role of current drugs and biomarkers. Int J Surg. 2016;33(Suppl 1):S57-70
4. Fu H, Xu H, Chen H, Li Y, Li W, Zhu Q. et al. Inhibition of glycogen synthase kinase 3 ameliorates liver ischemia/reperfusion injury via an energy-dependent mitochondrial mechanism. J Hepatol. 2014;61:816-24
5. Jeon BR, Lee SM. S-adenosylmethionine protects post-ischemic mitochondrial injury in rat liver. J Hepatol. 2001;34:395-401
6. Panel M, Ruiz I, Brillet R, Lafdil F, Teixeira-Clerc F, Nguyen CT. et al. Small-Molecule Inhibitors of Cyclophilins Block Opening of the Mitochondrial Permeability Transition Pore and Protect Mice From Hepatic Ischemia/Reperfusion Injury. Gastroenterology. 2019;157:1368-82
7. Hausenloy DJ, Yellon DM. Targeting Myocardial Reperfusion Injury-The Search Continues. N Engl J Med. 2015;373:1073-5
8. Song R, Dasgupta C, Mulder C, Zhang L. MicroRNA-210 Controls Mitochondrial Metabolism and Protects Heart Function in Myocardial Infarction. Circulation. 2022;145:1140-53
9. Zhao M, Liu S, Wang C, Wang Y, Wan M, Liu F. et al. Mesenchymal Stem Cell-Derived Extracellular Vesicles Attenuate Mitochondrial Damage and Inflammation by Stabilizing Mitochondrial DNA. ACS Nano. 2021;15:1519-38
10. Bi J, Zhang J, Ren Y, Du Z, Li Q, Wang Y. et al. Irisin alleviates liver ischemia-reperfusion injury by inhibiting excessive mitochondrial fission, promoting mitochondrial biogenesis and decreasing oxidative stress. Redox Biol. 2019;20:296-306
11. Guan Y, Yao W, Yi K, Zheng C, Lv S, Tao Y. et al. Nanotheranostics for the Management of Hepatic Ischemia-Reperfusion Injury. Small. 2021;17:e2007727
12. Ni D, Wei H, Chen W, Bao Q, Rosenkrans ZT, Barnhart TE. et al. Ceria Nanoparticles Meet Hepatic Ischemia-Reperfusion Injury: The Perfect Imperfection. Adv Mater. 2019;31:e1902956
13. Mu J, Li C, Shi Y, Liu G, Zou J, Zhang DY. et al. Protective effect of platinum nano-antioxidant and nitric oxide against hepatic ischemia-reperfusion injury. Nat Commun. 2022;13:2513
14. Majumder J, Taratula O, Minko T. Nanocarrier-based systems for targeted and site specific therapeutic delivery. Adv Drug Deliv Rev. 2019;144:57-77
15. Andaloussi S EL, Mäger I, Breakefield XO, Wood MJ. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov. 2013;12:347-57
16. Park EJ, Prajuabjinda O, Soe ZY, Darkwah S, Appiah MG, Kawamoto E. et al. Exosomal regulation of lymphocyte homing to the gut. Blood Adv. 2019;3:1-11
17. Parada N, Romero-Trujillo A, Georges N, Alcayaga-Miranda F. Camouflage strategies for therapeutic exosomes evasion from phagocytosis. J Adv Res. 2021;31:61-74
18. Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS. et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U S A. 2012;109:6662-7
19. Schultz CW, Preet R, Dhir T, Dixon DA, Brody JR. Understanding and targeting the disease-related RNA binding protein human antigen R (HuR). Wiley Interdiscip Rev RNA. 2020;11:e1581
20. Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015 4
21. Hu Y, Zhang Y, Ni CY, Chen CY, Rao SS, Yin H. et al. Human umbilical cord mesenchymal stromal cells-derived extracellular vesicles exert potent bone protective effects by CLEC11A-mediated regulation of bone metabolism. Theranostics. 2020;10:2293-308
22. Thura M, Al-Aidaroos AQ, Gupta A, Chee CE, Lee SC, Hui KM. et al. PRL3-zumab as an immunotherapy to inhibit tumors expressing PRL3 oncoprotein. Nat Commun. 2019;10:2484
23. Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. 2020;21:85-100
24. Boada-Romero E, Martinez J, Heckmann BL, Green DR. The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol. 2020;21:398-414
25. Chung KW, Dhillon P, Huang S, Sheng X, Shrestha R, Qiu C. et al. Mitochondrial Damage and Activation of the STING Pathway Lead to Renal Inflammation and Fibrosis. Cell Metab. 2019;30:784-99.e5
26. Woo JA, Liu T, Trotter C, Fang CC, De Narvaez E, LePochat P. et al. Loss of function CHCHD10 mutations in cytoplasmic TDP-43 accumulation and synaptic integrity. Nat Commun. 2017;8:15558
27. Shang M, Cappellesso F, Amorim R, Serneels J, Virga F, Eelen G. et al. Macrophage-derived glutamine boosts satellite cells and muscle regeneration. Nature. 2020;587:626-31
28. Qin JJ, Mao W, Wang X, Sun P, Cheng D, Tian S. et al. Caspase recruitment domain 6 protects against hepatic ischemia/reperfusion injury by suppressing ASK1. J Hepatol. 2018;69:1110-22
29. Teoh N, Dela Pena A, Farrell G. Hepatic ischemic preconditioning in mice is associated with activation of NF-kappaB, p38 kinase, and cell cycle entry. Hepatology. 2002;36:94-102
30. Banga NR, Homer-Vanniasinkam S, Graham A, Al-Mukhtar A, White SA, Prasad KR. Ischaemic preconditioning in transplantation and major resection of the liver. Br J Surg. 2005;92:528-38
31. Gurusamy KS, Kumar Y, Sharma D, Davidson BR. Ischaemic preconditioning for liver transplantation. Cochrane Database Syst Rev. 2008;2008:Cd006315
32. van Rijn R, Schurink IJ, de Vries Y, van den Berg AP, Cortes Cerisuelo M, Darwish Murad S. et al. Hypothermic Machine Perfusion in Liver Transplantation - A Randomized Trial. N Engl J Med. 2021;384:1391-401
33. Liu H, Man K. New Insights in Mechanisms and Therapeutics for Short- and Long-Term Impacts of Hepatic Ischemia Reperfusion Injury Post Liver Transplantation. Int J Mol Sci. 2021 22
34. Zhou B, Kreuzer J, Kumsta C, Wu L, Kamer KJ, Cedillo L. et al. Mitochondrial Permeability Uncouples Elevated Autophagy and Lifespan Extension. Cell. 2019;177:299-314.e16
35. Zhang W, Bi S, Li P, Liu J, Zhou C, Wang X. et al. In situ Observation of mtDNA Damage during Hepatic Ischemia-Reperfusion. Anal Chem. 2021;93:5782-8
36. Zhao M, Wang Y, Li L, Liu S, Wang C, Yuan Y. et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics. 2021;11:1845-63
37. Wang X, He Y, Mackowiak B, Gao B. MicroRNAs as regulators, biomarkers and therapeutic targets in liver diseases. Gut. 2021;70:784-95
38. Szabo G, Bala S. MicroRNAs in liver disease. Nat Rev Gastroenterol Hepatol. 2013;10:542-52
39. Zhang CX, Cheng Y, Liu DZ, Liu M, Cui H, Zhang BL. et al. Mitochondria-targeted cyclosporin A delivery system to treat myocardial ischemia reperfusion injury of rats. J Nanobiotechnology. 2019;17:18
40. Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16:203-22
41. Ledford H. Drug giants turn their backs on RNA interference. Nature. 2010;468:487
42. Ha D, Yang N, Nadithe V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: current perspectives and future challenges. Acta Pharm Sin B. 2016;6:287-96
43. Li L, He D, Guo Q, Zhang Z, Ru D, Wang L. et al. Exosome-liposome hybrid nanoparticle codelivery of TP and miR497 conspicuously overcomes chemoresistant ovarian cancer. J Nanobiotechnology. 2022;20:50
44. Barclay AN, Van den Berg TK. The interaction between signal regulatory protein alpha (SIRPα) and CD47: structure, function, and therapeutic target. Annu Rev Immunol. 2014;32:25-50
45. Chen L, Song Q, Chen Y, Meng S, Zheng M, Huang J. et al. Tailored Reconstituted Lipoprotein for Site-Specific and Mitochondria-Targeted Cyclosporine A Delivery to Treat Traumatic Brain Injury. ACS Nano. 2020;14:6636-48
46. O'Brien K, Breyne K, Ughetto S, Laurent LC, Breakefield XO. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat Rev Mol Cell Biol. 2020;21:585-606
47. Li Z, Zhou X, Wei M, Gao X, Zhao L, Shi R. et al. In vitro and in vivo RNA Inhibition by CD9-HuR Functionalized Exosomes Encapsulated with miRNA or CRISPR/dCas9. Nano Lett. 2019;19:19-28
48. Turturici G, Tinnirello R, Sconzo G, Geraci F. Extracellular membrane vesicles as a mechanism of cell-to-cell communication: advantages and disadvantages. Am J Physiol Cell Physiol. 2014;306:C621-33
49. El-Andaloussi S, Lee Y, Lakhal-Littleton S, Li J, Seow Y, Gardiner C. et al. Exosome-mediated delivery of siRNA in vitro and in vivo. Nat Protoc. 2012;7:2112-26
50. Abe Y, Hines IN, Zibari G, Pavlick K, Gray L, Kitagawa Y. et al. Mouse model of liver ischemia and reperfusion injury: method for studying reactive oxygen and nitrogen metabolites in vivo. Free Radic Biol Med. 2009;46:1-7
Author contact
![]() Corresponding authors: E-mail: sdlytczhzhcom (Baoyong Zhou); duchengyoucom (Chengyou Du); pqlpzyedu.cn (Qiling Peng); jiangningedu.cn (Ning Jiang)
Corresponding authors: E-mail: sdlytczhzhcom (Baoyong Zhou); duchengyoucom (Chengyou Du); pqlpzyedu.cn (Qiling Peng); jiangningedu.cn (Ning Jiang)