Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2023; 13(15):5207-5222. doi:10.7150/thno.89033 This issue Cite
Research Paper
Upregulated FOXM1 stimulates chondrocyte senescence in Acot12-/-Nudt7-/- double knockout mice
Jinsoo Song1†, Ee Hyun Kim2†, Jun-Ho Yang1, Donghyeon Kim1, Akhmad Irhas Robby3, Se-ah Kim4, Sung Young Park3, Ji Hyun Ryu4, Eun-Jung Jin1 ![]()
1. Department of Biological Sciences, College of Health Sciences, Wonkwang University; Iksan, Chunbuk, 570-749, Korea.
2. Graduate School of Pharmaceutical Sciences, College of Pharmacy, Ewha Womans University; Seoul 03760, Korea.
3. Department of Chemical and Biological Engineering, Korea National University of Transportation; Chungju 27469, Korea.
4. Department of Carbon Convergence Engineering, Wonkwang University; Iksan, Chunbuk, 570-749, Korea.
†These authors contributed equally.
Received 2023-8-10; Accepted 2023-9-13; Published 2023-9-25
Abstract

Rationale: One of the hallmarks of osteoarthritis (OA), the most common degenerative joint disease, is increased numbers of senescent chondrocytes. Targeting senescent chondrocytes or signaling mechanisms leading to senescence could be a promising new therapeutic approach for OA treatment. However, understanding the key targets and links between chondrocyte senescence and OA remains unclear.
Methods: Senescent chondrocytes were identified from Nudt7-/-, Acot12-/-, double-knockout mice lacking Acot12 and Nudt7 (dKO) and applied to microarray. The presence of forkhead transcription factor M1 (FOXM1) was detected in aged, dKO, and destabilization of the medial meniscus (DMM) cartilages and articular chondrocytes, and the effect of FoxM1 overexpression and acetyl-CoA treatment on cartilage homeostasis was examined using immunohistochemistry, quantitative real-time PCR (qRT-PCR), cell apoptosis and proliferation assay, and safranin O staining. Delivery of Rho@PAA-MnO2 (MnO2 nanosheet) or heparin-ACBP/COS-GA-siFoxM1 (ACBP-siFoxM1) nanoparticles into DMM cartilage was performed.
Results: Here, we propose the specific capture of acetyl-CoA with the delivery of (FoxM1 siRNA (siFoxM1) to prevent cartilage degradation by inhibiting the axis of chondrocyte senescence. dKO stimulate chondrocyte senescence via the upregulation of FoxM1 and contribute to severe cartilage breakdown. We found that the accumulation of acetyl-CoA in the dKO mice may be responsible for the upregulation of FoxM1 during OA pathogenesis. Moreover, scavenging reactive oxygen species (ROS) induced by chondrocyte senescence via the implantation of MnO2 nanosheets or delivery of siFoxM1 functionalized with acetyl-CoA binding protein (ACBP) to capture acetyl-CoA using an injectable bioactive nanoparticle (siFoxM1-ACBP-NP) significantly suppressed DMM-induced cartilage destruction.
Conclusion: We found that the loss of Acot12 and Nudt7 stimulates chondrocyte senescence via the upregulation of FoxM1 and accumulation of acetyl-CoA, and the application of siFoxM1-ACBP-NP is a potential therapeutic strategy for OA treatment.
Keywords: osteoarthritis, Acot12-/-Nudt7-/-, FoxM1, Rho@PAA-MnO2, heparin-ACBP/COS-GA-siFoxM1
Introduction
Osteoarthritis (OA), the most common progressive chronic joint disease, is characterized by structural modification primarily to articular cartilage and subchondral bone, but also to synovia, ligaments, and muscles, indicating that OA pathogenesis is a complex process that involves and affects the cartilage and entire joint tissues [1]. The physiological and pathological changes in the joint tissue during the occurrence and development of OA are mediated by the interplay of various factors involved in maintaining cartilage homeostasis, including proinflammatory cytokines such as interleukin (IL)-1β, tumor necrosis factor (TNF)-α, IL-6, and anti-inflammatory cytokines such as IL-4 and IL-10; growth factors such as transforming growth factor-β and fibroblast growth factor-21; and adipokines [2]. An imbalance among these factors within the cartilage impairs the stable turnover of the cartilage matrix and results in cartilage degeneration and destruction. Traditionally, the management of OA has been constrained to relieve the joint pain using the non-steroidal anti-inflammatory drugs (NSAIDs) or analgesics rather than targeting the components of structural disease progression [3]. However, since their side-effects often restrict their use, there is an urgent need for new biological targets for intervention and treatments designed to prevent or reverse the symptoms of OA. Given correlation between chondrocyte senescence and OA [4,5], targeting senescent chondrocytes or responsible signaling mechanism could be a novel approach for OA therapy. The impact of senescence on OA is complex and just beginning to be understood. Senescent cartilage cells express high levels of senescence biomarkers such as p53 and the cyclin-dependent kinase inhibitors p21 and p16INK4a [6]. Recent study demonstrated rejuvenating effects of exosomes derived from umbilical cord-derived mesenchymal stem cells on senescent OA chondrocytes through modulating p53 signaling pathway [7]. Age-related reactive oxygen species (ROS) production of chondrocytes plays a role in cartilage degradation as well as chondrocyte cell death [8]. ROS accumulate dysfunctional mitochondria, induce DNA damage, and lead to premature senescence, degradation of matrix and loss of bone mass in OA [9].
Senescent cells showed a high metabolic activity, despite the decline in their proliferative potential. Chondrocytes depend mainly on anaerobic metabolism to develop, maintain, and repair the cartilage matrix. Anaerobic glycolysis is stimulated in OA chondrocytes to generate sufficient ATP for maintaining homeostasis in the cartilage matrix [10,11]. Pyruvate kinase M2, a critical enzyme involved in the glycolytic step that converts phosphoenolpyruvate to pyruvate with the generation of ATP, is upregulated in OA chondrocytes compared to healthy chondrocytes [12]. Furthermore, in OA tissues, the tricarboxylic acid cycle is also upregulated, most likely increasing the production of ATP required to repair cartilage damage [13-15]. The fatty acid β-oxidation pathway, a catabolic process in which fatty acid molecules are broken down to provide energy, is another metabolic pathway that is affected during OA pathogenesis [16,17]. Mitochondrial β-oxidation of fatty acids plays an essential role in the development and progression of OA [18,19]. Dysregulation of oxidized fatty acid derivatives impairs the homeostasis of the cartilage matrix and reduces cell viability, as observed in osteoarthritic cartilage [16,20]. Exposure of human chondrocytes to linoleic acid stimulates the secretion of IL-6 and the production of prostaglandin E2, resulting in biological responses that are closely related to the severity of OA [21]. Palmitic acid induces chondrocyte apoptosis, which is associated with the accumulation of lipid droplets [22,23]. Accumulated fatty acids in OA cartilage suggest that mitochondrial dysfunction may be closely related to the pathogenesis of OA, as mitochondria use lipids as the main fuel to produce chemical energy that controls many cellular processes; i.e. accumulated fatty acids may affect the biological pathways involved in cartilage degradation, including oxidative stress, inflammation, and chondrocyte apoptosis through mitochondrial dysfunction. Recently, we demonstrated a functional interaction between peroxisomes and mitochondria in the regulation of lipid metabolism [24]. Furthermore, impaired peroxisomal function in the regulation of fatty acid β-oxidation results in an imbalance in cartilage matrix homeostasis, which is closely associated with the pathogenesis of OA. Peroxisomes are small ubiquitous membrane-bound organelles that play many vital roles for various metabolic pathways, including the β-oxidation of very long chain fatty acids (VLCFA) and LCFA, synthesis of ether lipid, and bile acid metabolism. Among various peroxisomal enzymes, our laboratory recently suggested that nudix hydrolase 7 (NUDT7), which hydrolyzes the diphosphate of coenzyme A (CoA), plays an important role in maintaining cartilage homeostasis [25]. Deficiency of Nudt7 impairs the peroxisomal β-oxidation and accumulates the acetyl-CoA in cartilage leading to the inflammation and apoptotic cell death via activation of phosphoglycerate mutase 1, one of key enzymes in aerobic glycolysis. Moreover, our laboratory showed that in Acyl-CoA thioesterase 12 knockout (Acot12-/-) cartilage, accumulation of acetyl-CoA stimulates de novo lipogenesis and consequently contributes to the degradation of the cartilage matrix [26]. These data suggest the importance of peroxisomal β-oxidation of fatty acids, particularly acetyl-CoA, in maintaining cartilage homeostasis during OA pathogenesis. However, the mechanism by which peroxisomal β-oxidation of fatty acids drives metabolic dysfunction in OA cartilage remains unclear. Furthermore, the underlying regulatory and signaling mechanisms remain largely unknown. In the current study, we generated double-knockout mice lacking Acot12 and Nudt7 (Acot12-/- Nudt7-/-, dKO) to evaluate the regulatory role and signaling mechanism of peroxisomal β-oxidation of fatty acids in the pathogenesis of OA. Our results revealed that dKO mice spontaneously developed severe degradation of the cartilage matrix due to an increased level of cellular senescence via upregulation of forkhead transcription factor M1 (FOXM1) via accumulation of acetyl-CoA. Recently, targeted biomaterials specific to cartilage to synovium have been applied to improve the therapeutic efficiency for OA treatment. Functional biomaterials such as hydrogels, non-hydrogel polymers, and inorganic nanomaterials are widely used for OA delivery, chondrocyte encapsulation, and cartilage engineering [27-29]. Most commonly used hydrogels in OA treatment are polysaccharides (e.g. chitosan, hyaluronic acid, heparin) and proteins (e.g. collagens, gelatin). The advantage of using these biomaterial-based hydrogels is that they overcome the limitation of poor drug retention in knee joints. This increases the therapeutic effectiveness by allowing the drug to stay in the joint or cartilage longer and be released slowly. In the current study, we generated the delivery system targeting senescent chondrocytes using biomaterials such as chitosan and heparin using siFoxM1 and acetyl-CoA binding protein (ACBP) and evaluated the therapeutic effect.
Results
Loss of Acot12 and Nudt7 involves OA pathogenesis
We previously reported that two peroxisomal enzymes, Acot12 and Nudt7 are important for maintaining cartilage homeostasis [25,26]. Moreover, the accumulation of acetyl-CoA due to impaired β-oxidation of LCFA and VLCFA in Acot12-/- - or Nudt7-/- cartilage could be a key player in the regulation of cartilage homeostasis through the modulation of catabolic and inflammatory responses (Figure S1A). We generated Acot12-/-Nudt7-/- mice (Figure S1B). Unlike our previous reports demonstrating no difference in the skeletal development of Acot12-/- [26] or Nudt7-/- mice [25], skeletal development in dKO mice, that is, the length of the tibia and femur of postnatal mice and endochondral ossification, was significantly altered (Figure 1A). Moreover, micromass cultures of dKO mouse limb bud mesenchymal cells showed significantly increased cellular condensation and chondrogenic differentiation, as assessed by Peanut agglutinin (PNA)- and Alcian blue staining, respectively, compared to Acot12+/+Nudt7+/+ (wild-type, WT) mesenchymal cells (Figure 1B). Severe cartilage degradation was observed in 12- and 20-month-old dKO mice compared to WT mice (Figure 1C). Tibialis anterior (TA) muscle mass, analyzed by measuring cross-sectional area, also significantly decreased with dysregulation of peroxisomal and mitochondrial gene profiles in 12- and 20-month-old dKO mice (Figure S2). These data suggest that Acot12-/- Nudt7-/- dKO exacerbates the degeneration of the musculoskeletal system, such as cartilage and muscle, possibly by functional interaction with peroxisomes and mitochondria.
Musculoskeletal and cartilage homeostasis in dKO mice. (A) Alizarin red/Alcian blue staining of WT and dKO newborn mice (n = 4 per group). (B) PNA and Alcian blue staining of micromass cultures of WT and dKO limb mesenchymal cells. (C) Safranin O staining of cartilage of 6-, 12-, and 20-month-old WT and dKO mice. The degree of cartilage degradation is quantified according to the OARSI grade. Scale bar = 200 μm. (D) Safranin O staining of DMM cartilage of WT and dKO mice. Cartilage thickness was measured (n = 5 per group). Scale bar = 200 μm. (E) Immunohistochemistry of C1,2C in WT and dKO DMM cartilage at eight weeks post-surgery (n = 4 per group). (F-H) Expression level of catabolic and anabolic genes in maintaining cartilage homeostasis with WT and dKO DMM cartilage (n = 3 per group). (I) Safranin O staining and immunohistochemistry of C1,2C in WT, or dKO cartilage with or without restoration of lenti-HA-Nudt7, -Acot12, or both. cartilage thickness was measured. Scale bar = 50 μm. *P < 0.05; **P < 0.01; *** P < 0.001; **** P < 0.0001.
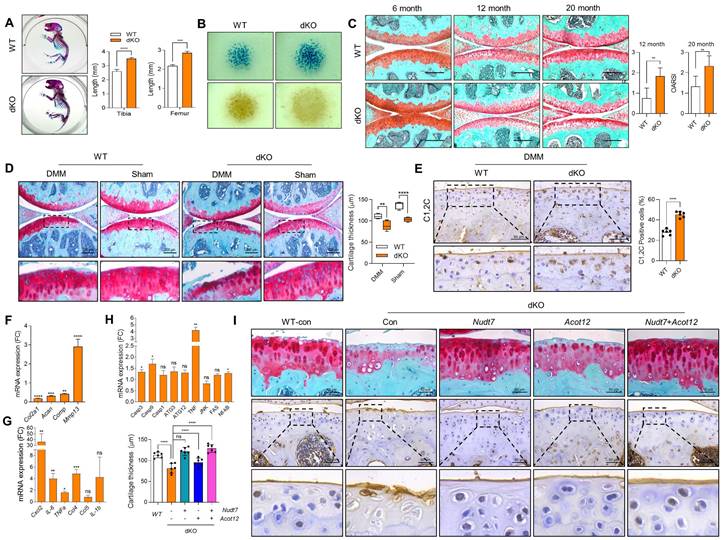
To investigate the effect of dKO on the pathogenesis of OA, destabilization of the medial meniscus (DMM) was performed to induce OA pathological conditions. Severe cartilage degradation, as assessed by safranin O staining, was observed in DMM-induced dKO cartilage compared to control cartilage (Figure 1D). A dramatic reduction in cartilage thickness was also observed in the DMM-induced dKO cartilage. The intensity and number of Collagen C1,2C positive cells in DMM-induced dKO cartilage were significantly increased compared to that in WT cartilage (Figure 1E). Moreover, the transcription levels of cartilage matrix genes such as Col2a1, Acan, and Comp decreased (Figure 1F), whereas the expression levels of a major cartilage degrading enzyme, Mmp13, inflammatory chemokines and cytokines such as Cxcl2, IL-6, TNFα, and Ccl4, and apoptotic genes such as Casp3 and Casp9 significantly increased in DMM-induced dKO cartilage (Figure 1G-H). To determine whether restoration of either Acot12 or Nudt7 in dKO cartilage could prevent cartilage degradation, Nudt7- (Lenti-HA-Nudt7) or Acot12-encoding lentiviruses (Lenti-HA-Acot12) were introduced into the knee joint cavity of dKO mice (Figure 1I). Intra-articular injection of Lenti-HA-Acot12 or -Nudt7 significantly reduced cartilage degradation, and a dramatic reduction in cartilage degradation was induced by the co-injection of Lenti-HA-Acot12 and -Nudt7. As observed in Nudt7-/- and Acot12-/- immature murine articular chondrocytes (iMACs), cell proliferation significantly decreased (Figure S3 and Figure S4A), and increased apoptosis (Figure S4B) and dysregulation of mitochondrial dynamics (Figure S4C) were observed in the dKO iMACs. However, in dKO iMACs, we did not observe significant lipid accumulation compared to that in WT iMACs, as seen in Nudt7-/- or Acot12-/- iMACs (Figure S4D-E). The expression levels of genes involved in lipid metabolism, including Fasn, Scd1, and Elovs, were also decreased in dKO iMACs, whereas they were increased in either Nudt7-/- or Acot12-/- iMACs (Figure S4F).
Loss of Acot12 and Nudt7 significantly stimulates chondrocyte senescence via FoxM1 upregulation
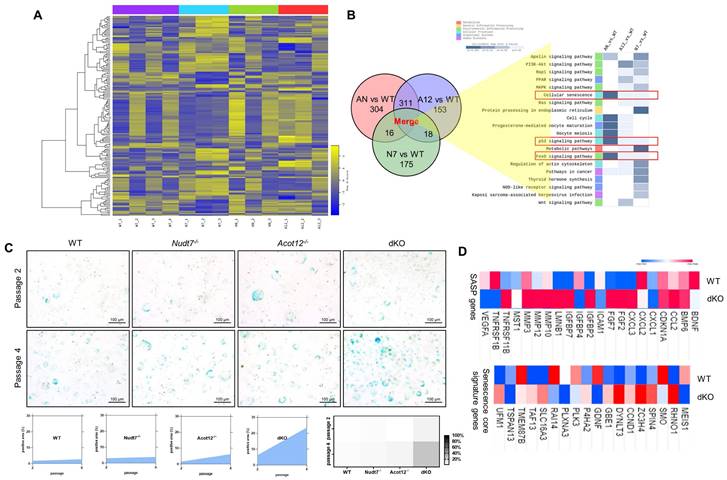
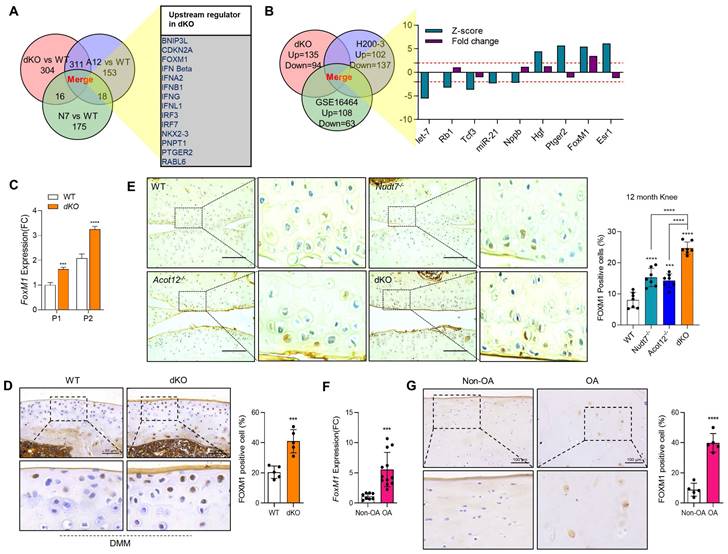
To identify the factors responsible for cartilage degradation modulated by dKO, we performed microarray analysis using iMACs isolated from WT, Nudt7-/--, Acot12-/--, and dKO iMACs mice, extracted common genes whose expression was significantly altered in Nudt7-/--, Acot12-/--, and dKO iMACs compared to WT iMACs, and analyzed the enriched signaling pathways (Figure 2A-B). Cellular senescence, the p53 signaling pathway, and the FoxO signaling pathway were suggested as common enriched signaling pathways, with the highest significance in dKO iMACs. In vitro analysis of passaging (P2, P4) iMACs showed an increased number of senescence-associated β-galactosidase (SA-β-gal)-positive cells in Nudt7-/--, Acot12-/--, and dKO iMACs compared to WT iMACs (Figure 2C). The most dramatic increase in the number of senescent cells was observed in dKO iMACs. Moreover, the transcription levels of senescence-associated secretory phenotype (SASP) and senescence core signature genes were also significantly upregulated in dKO iMACs compared to those in WT iMACs (Figure 2D). Gene profile analysis suggested that cell cycle: G2M DNA damage checkpoint regulation, ATM signaling, and cell cycle control of chromosomal replication were the top canonical pathways in dKO iMACs (Figure S5). According to the microarray analysis of common genes whose expression was significantly altered in Nudt7-/--, Acot12-/--, and dKO iMACs compared to WT iMACs, Bnip3l, Cdkn2a, Ifn, Irf3, Irf7, FoxM1, Nkk2.3, Pnpt1, Ptger2, and Rabl6 were suggested as possible upstream regulators (Figure 3A). Furthermore, we extracted the common genes whose expressions were significantly altered in the microarray of dKO iMACs (up = 135, down = 94), RNA sequencing of OA chondrocytes isolated from OA patients (up = 102, down = 137), and GSE16464 [3D culture of normal vs. OA chondrocytes (up = 108, down = 63)], and identified Rb1, Tcf3, Nppb, Hgf, Ptger2, FoxM1, and Esr1 (Figure 3B and Figure S5). FoxM1 was commonly altered in Nudt7-/-, Acot12-/-, and dKO iMACs as well as in OA chondrocytes. The expression levels of FoxM1 increased significantly as passaging progressed, with higher expression levels in dKO iMACs than in WT iMACs (Figure 3C). We also confirmed the upregulated expression level of FOXM1 in DMM-induced dKO cartilage compared to that in DMM-induced WT cartilage (Figure 3D). Compared to Nudt7-/-or Acot12-/- cartilage, as well as to WT cartilage, a significantly increased number of FOXM1-positive cells was observed in dKO cartilage (Figure 3E), and the introduction of either lenti-HA-Acot12 or -Nudt7 into dKO cartilage reduced these increased FOXM1-positive cells (Figure S6). The expression level of FoxM1 was also significantly upregulated in patients with OA (Figure 3F). Immunohistochemical analysis showed that the number of FOXM1-positive cells in the OA area of OA cartilage was increased by up to three-fold compared to that in the non-OA area of OA cartilage (Figure 3G).
Chondrocyte senescence in dKO cartilage. (A) Heatmap analysis of microarray data from WT, Nudt7-/-, Acot12-/-, and dKO iMACs. (B) Common pathways analysis of microarray data. (C) SA-β-gal staining of passage 2 and passage 4 iMACs isolated from WT, Nudt7-/-, Acot12-/-, and dKO embryos. (D) Transcriptional level of SASP and senescence score signature genes in WT and dKO cartilage.
FoxM1 in aged, dKO, and DMM cartilages. (A) Ven diagram of significantly altered genes from microarray data of Nudt7-/-, Acot12-/-, and dKO iMACs compared to WT iMACs. (B) Ven diagram of significantly altered genes from microarray data of dKO iMACs compared to those from human OA patent sequencing data. (C) Transcriptional level of FoxM1 in passaged (P1, P2) WT and dKO iMACs (n = 3 per group). (D) Immunohistochemistry of FOXM1 in DMM cartilage of WT and dKO mice (n = 5). FOXM1-positive cell numbers are indicated by dot graph. (E) Immunohistochemistry of FOXM1 in 12-month-old WT, Nudt7-/-, Acot12-/-, and dKO cartilage (n = 4 per group). FOXM1-positive cell number were indicated by bar-dot graph. (F) Transcriptional level of FoxM1 in normal and OA patient chondrocytes (n = 8 in normal, n = 12 in OA). (G) Immunohistochemistry of FOXM1 in normal and OA patient cartilage (n = 5 per group). FOXM1-positive cell numbers were indicated by dot graph. *** P < 0.001; **** P < 0.0001.
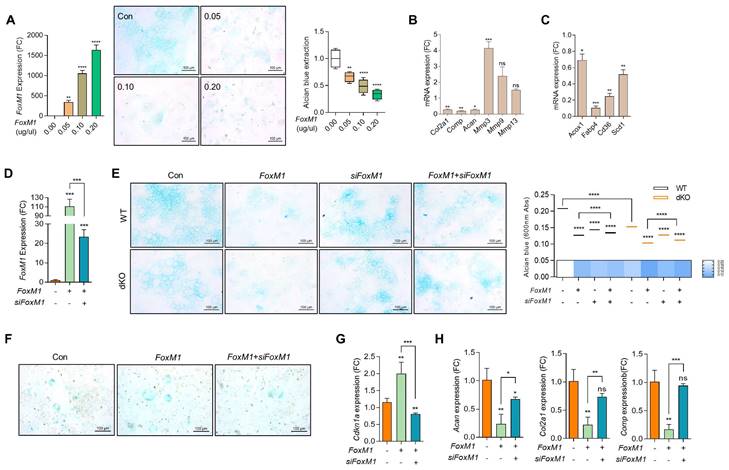
To investigate the role and function of FOXM1 in OA pathogenesis, the expression level of FOXM1 was modulated exogenously using FoxM1-encoding pcDNA construct (FoxM1; Figure S7). As observed in Nudt7-/--, Acot12-/--, and dKO iMACs, the expression levels of mitochondrial genes such as Cox2 and Cox4, peroxisomal genes such as Pmp70 and Cat, and lysosomal genes such as Lamp1 and Lamp2 were significantly suppressed by the introduction of FoxM1 into WT iMACs (Figure S8). The number of Alcian blue-positive cells (Figure 4A) and the expression levels of cartilage matrix genes such as Col2a1, Comp, and Acan were significantly decreased, whereas the expression levels of genes involved in cartilage degradation such as Mmp-3, -9, and -13 were significantly increased with the introduction of FoxM1 into WT iMACs (Figure 4B). Interestingly, as shown in dKO cartilage in Figure S4F, the expression levels of genes involved in lipid metabolism such as Fabp4, Cd36, and Scd1 were significantly decreased in FoxM1-introduced WT iMACs (Figure 4C). To determine whether small interfering RNA specific to Foxm1 (siFoxm1) could overcome this inhibitory action on cartilage homeostasis, three different siFoxM1 were introduced into dKO iMACs and their efficiency was examined (Figure S9, Figure 4D). In this study, a significant recovery in the number of Alcian blue-positive cells was observed by the introduction of siFoxM1 into FoxM1-introduced WT or dKO iMACs (Figure 4E left panel). Extraction and photometric quantification of Alcian blue staining (Figure 4E right upper panel) and Image J analysis of the Alcian blue-stained area (Figure 4E right lower panel) were also increased by the introduction of siFoxM1 into dKO iMACs. Consistent with the top canonical pathway observed in the dKO iMACs (Figure S5), the number of SA-β-gal-positive cells (Figure 4F) and expression level of Cdkn1a (Figure 4G) was increased by the introduction of FoxM1 into dKO iMACs. Decreased levels of cartilage matrix genes such as Col2a, Comp, and Acan by the introduction of FoxM1 were restored by the co-introduction of FoxM1-specific siRNA (Figure 4H).
FoxM1 stimulates chondrocyte senescence. (A) RT-PCR with dose-dependent introduction of pcDNA-FoxM1 (left panel), Alcian blue staining (middle panel), Quantification of Alcian blue staining (right panel) (n = 4 per group). FoxM1 were introduced into iMACs. (B) Transcriptional levels of cartilage matrix and cartilage degrading genes. (C) Transcriptional levels of genes in lipid metabolism in FoxM1-overexpressed iMACs (n = 3 per group). (D) Transcriptional level of FoxM1 (n = 3 per group). (E) Alcian blue staining in WT and dKO iMACs transfected with FoxM1, siFoxM1, or both (n = 4 per group). Quantification of Alcian blue staining extracted with 6M Guanidine-HCl was measured at a 650 nm absorbance. (F) SA-β-gal staining (n = 3 per group). G) Expression level of Cdkn1a (n = 4 per group). (H) Expression level of cartilage matrix genes (n = 3 per group).
Next, we exposed WT iMACs into IL-1β, one of the key cytokines involved in OA pathogenesis, to make an OA environment. Exposure of IL-1β into iMACs showed typical OA characteristics, that is, significantly decreased expression levels of cartilage matrix genes such as Col2a1 and Acan (Figure 5A) and increased chondrocyte apoptosis (Figure 5B). These alterations were exacerbated by co-treatment of FoxM1 in the presence of IL-1β. In vivo intra-articular injection of lenti-HA-FoxM1 into the cartilage of DMM-induced WT mice stimulated cartilage degradation with increased numbers of FOXM1, C1,2C, and MMP13-positive cells (Figure 5C). In contrast, intra-articular injection of siFoxM1 into the cartilage of DMM-induced dKO mice significantly suppressed cartilage degradation, with decreased numbers of FOXM1, C1,2C, and MMP13-positive cells.
FoxM1 overexpression exacerbates cartilage degradation. (A) Transcriptional level of Col2a1 and Acan with introduction of FoxM1 in the absence or presence of IL-β (n = 3 per group). (B) Apoptotic cell death with introduction of FoxM1 in the absence or presence of IL-β (n = 3 per group). (C) Safranin-O staining and immunohistochemistry of FOXM1, MMP13, C1,2C, and Neopeptide in DMM cartilage of WT and dKO mice infected with FoxM1- or siFoxM1-lentivirus (n = 6-9 per group). Positive cell numbers are indicated by bar-dot graph. *P < 0.05; **P < 0.01; *** P < 0.001.
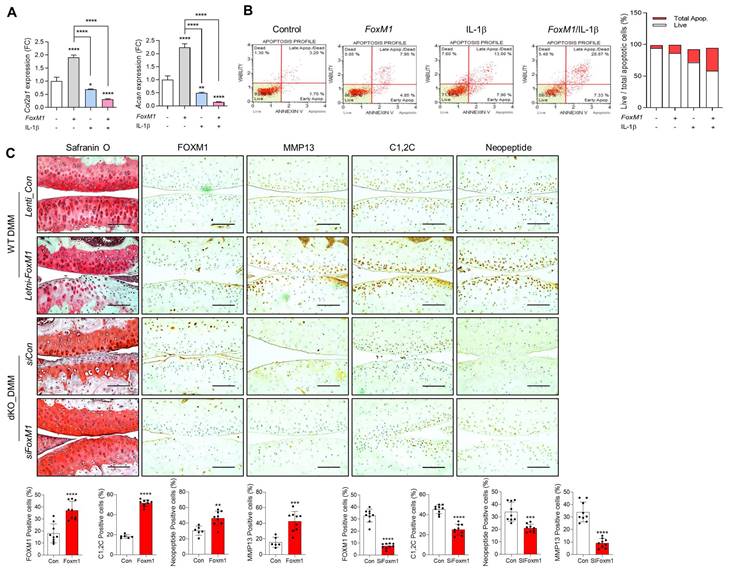
To identify the metabolic molecules underlying chondrocyte senescence and apoptosis induced by dKO mice during OA pathogenesis, we searched the human metabolome database and identified acetyl-CoA as a common metabolite (Figure 6A). To determine whether the elevated level of acetyl-CoA was responsible for FOXM1-induced cellular senescence, iMACs were treated with sodium acetate to accumulate acetyl-CoA (Figure S10). Exposure of iMACs to sodium acetate resulted in typical OA characteristics, that is, decreased Col2a1 expression and increased Mmp13 and Adamts5 expression. Exposure of iMACs to sodium acetate increased the expression levels of senescence signature genes concomitantly with an increase in FoxM1 levels. In addition, exposure of iMACs to acetyl-CoA also significantly increased the expression of senescence signature genes (Figure 6B), IL-1β-induced ROS (Figure 6C), FoxM1 (Figure 6D), and cartilage-degrading genes such as Adamts4 and Mmp13 (Figure 6E), and decreased the expression levels of cartilage matrix genes such as Col2a1, Comp, and Acan (Figure 6F). Intra-articular injection of acetyl-CoA into the DMM cartilage increased the number of FOXM1- (Figure 6G) and TUNEL- (Figure 6H) positive cells with severe cartilage degradation (Figure 6I).
Acetyl-CoA induces FoxM1 and increases chondrocyte senescence. (A) Metabolite analysis from human metabolome database. 50 μM Acetyl-CoA were exposed into iMACs. (B) Senescence genes signature, (C) Senescence-induced ROS level (n = 4), (D) Expression level of FoxM1, (E) Expression level of cartilage degrading genes, (F) Expression level of cartilage matrix genes (n = 3). (G) Immunohistochemistry of FOXM1 in DMM cartilage with or without introduction of Acetyl-CoA (n = 6-12 per group). FOXM1-postive cell numbers are indicated by the bar dot graph. (H) Tunel assay in DMM cartilage with or without introduction of Acetyl-CoA (n = 9-12). (I) Safranin-O staining of DMM cartilage with or without introduction of Acetyl-CoA (n = 7). The degree of cartilage degradation is quantified according to OARSI grade. Scale bar = 200 μm. *P < 0.05; **P < 0.01; *** P < 0.001, **** P < 0.0001.
In vivo targeting of senescent chondrocytes using heparin-ACBP/COS-GA-siFoxM1 nanoparticles reduces cartilage degradation
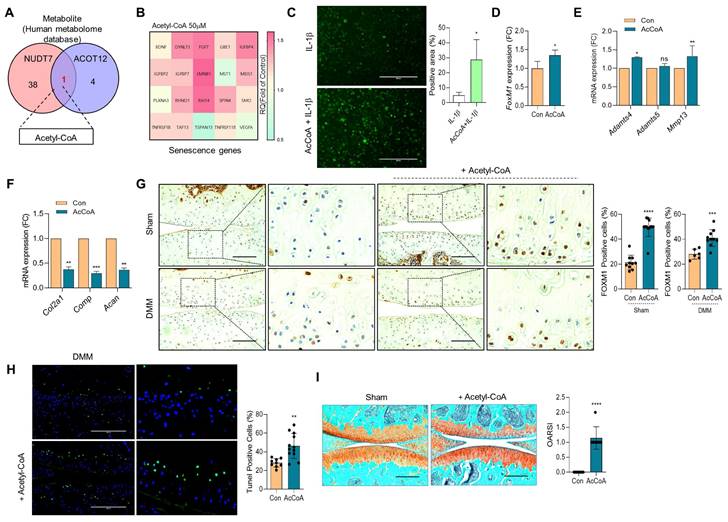
To investigate whether cartilage degradation could be inhibited by removing factors that induce chondrocyte senescence in dKO mice, we used a senescence-induced ROS-responsive off-on fluorescence-loaded polyacrylic acid (PAA)-MnO2 system (Figure 7A). Fluorescence is quenched by the aggregation of Rhodamine (Rho) and MnO2 while it is recovered via the cleavage of MnO2 to Mn2+ and the release of Rho 5G in the presence of senescence-induced ROS. Rho@PAA-MnO2 system was validated by decrease in particle size (Figure 7B) and increase in photoluminescence (PL) intensity after 0.5% or 1% H2O2 treatment (Figure 7C). Fluorescence recovery with H2O2 was confirmed by confocal microscopy at 488 nm (Figure S11). In vitro analysis of passaged iMACs showed an increase in the intensity of senescence-associated signals as cellular passaging progressed (P4~P9) (Figure S12). Compared with the passaged iMACs of WT mice, the passaged iMACs of Nudt7-/-, Acot12-/-, and dKO mice showed a strong senescence signal. In particular, the strongest senescence signal was detected in passaged iMACs isolated from dKO cartilage. The senescence signal significantly increased in the cartilage of DMM-induced Nudt7-/- and dKO mice, with the strongest signal in the DMM-induced dKO cartilage (Figure 7D). Moreover, we confirmed an increase in senescence-associated signals as the OA pathogenesis progressed in vivo. The senescence-associated signal increased with degradation of the cartilage matrix over time after DMM surgery (Figure S13). For heparin-ACBP/COS-GA-siFoxM1 (ACBP-siFoxM1) nanoparticles, gallic acid-conjugated chitosan oligosaccharide (COS-GA) was synthesized using standard carbodiimide chemistry. COS was conjugated with GA through the formation of amide linkages between the amine groups of COS and the carboxylic acid groups of GA (Figure 7E). The conjugation of GA to the COS backbone was confirmed by both UV-Vis and UV-Vis spectroscopies. The gallol protons of COS-GA were found in 6.9 to 7.1 ppm in 1H NMR spectra (Figure 7F). In addition, an absorbance peak at 261 nm caused by the gallol groups of COS-GA was observed (Figure 7G). As previously reported, the λmax of the absorbance of GA was significantly affected by pH and solvents [30]. Thus, COS-GA solutions and standard solutions with different concentrations were prepared in PBS (pH 7.4). The degree of GA substitution in the COS backbone was 14.4% measured by UV-Vis spectroscopic studies. The preparation procedure for COS-GA/siFoxM1 complexes is illustrated in Figure 7H. The electrostatic interactions between positively charged COS and negatively charged siFoxM1 were followed by gallol-mediated chemical crosslinking of the COS-GA backbones.
Cellular senescence detection by Rho-PAA-MnO2 sensor. (A) Scheme of a ROS-responsive fluorescence off-on system with Rho-PAA-MnO2 sensor. (B) Particle size of Rho@PAA-MnO2 (0.5% and 1%) after treatment with 1mM H2O2 for 30 min obtained by dynamic light scattering spectrometry (DLS). (C) Photoluminescence spectra of Rho@PAA-MnO2 (0.5% and 1%) after treatment with 1mM H2O2. (D) IVIS images of six-month-old WT, Nudt7-/-, Acot12-/-, and dKO mice implanted with senescence sensor into cartilage. (E-I) Preparation and characterizations of COS-GA/siFoxM1 and COS-GA/Hep/ACBP complexes. (E) Synthesis and chemical structures of COS-GA. (F) 1H NMR and (G) UV-Vis spectra of COS-GA. (H) Preparation of COS-GA/siFoxM1 complexes. SEM (first) and AFM (second and third) images of COS-GA/siFoxM1 complexes. (I) Preparation of COS-GA/Hep/ACBP complexes. SEM (first) and AFM (second and third) images of COS-GA/Hep/ACBP complexes.
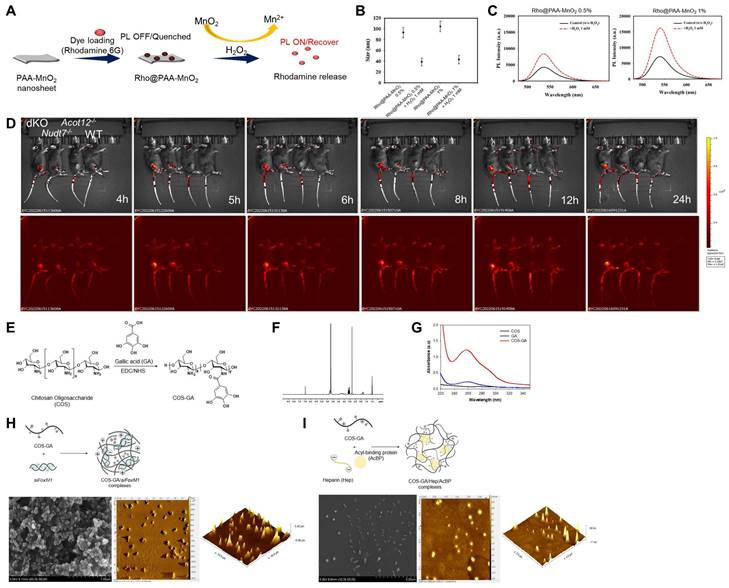
Gallol-containing polymers can be crosslinked in PBS (pH 7.4) by self-oxidation, as previously reported [31]. The morphology of the COS-GA/siFoxM1 complexes after dehydration was examined using scanning electron microscopy (SEM) and atomic force microscopy (AFM). COS-GA/siFoxM1 complexes were well-dispersed spherical nanoparticles. ACBP-loaded nanoparticles were prepared using COS-GA and heparin (Hep). As shown in Figure 7I, the COS-GA/Hep/ACBP complexes were prepared by polyelectrolyte complexation between COS-GA and Hep. The COS-GA/Hep/ACBP complexes were readily dispersed in PBS (pH 7.4).
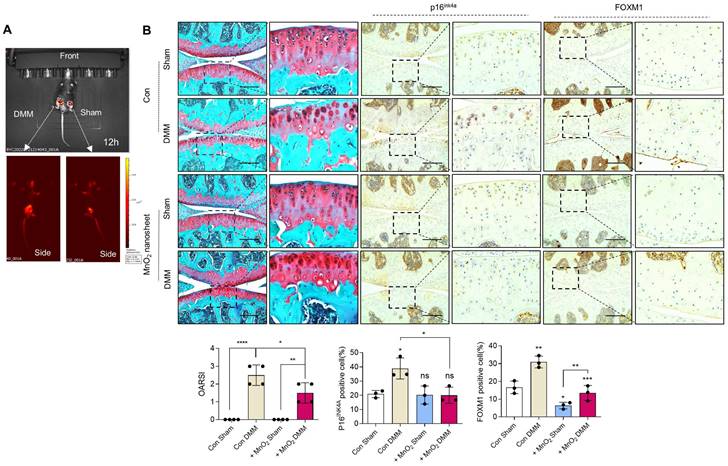
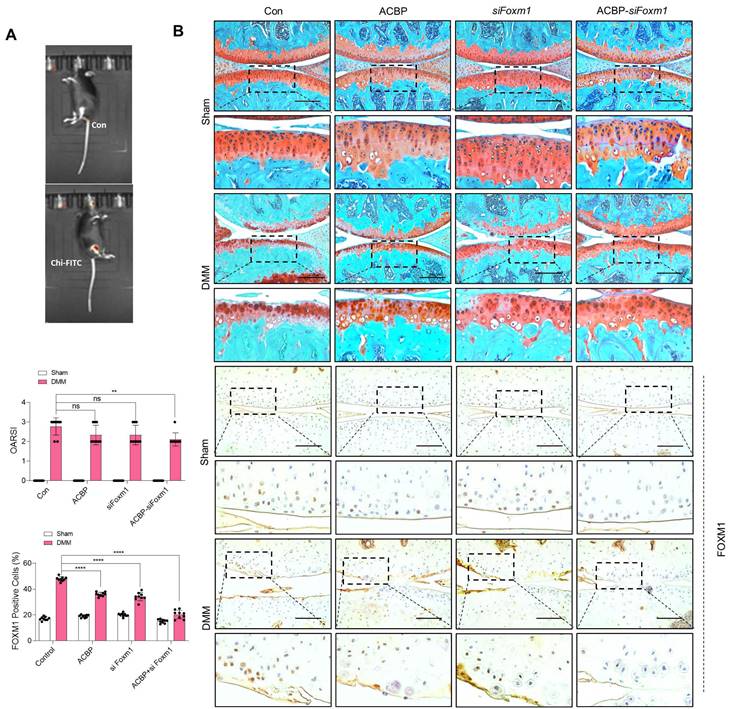
Implantation of PAA-MnO2 nanosheets into the cartilage for eight weeks significantly suppressed DMM-induced cartilage degradation (Figure 8A-B). The expression levels and positive cell numbers of P16ink4a and FOXM1 were significantly decreased by the implantation of the PAA-MnO2 nanosheets into the DMM cartilage. Next, to determine whether capturing accumulated acetyl-CoA and FOXM1 from senescent chondrocytes could suppress OA pathogenesis, we applied heparin-ACBP/COS-GA-siFoxM1 (ACBP-siFoxM1) nanoparticles (Figure 9A-B). Introduction of ACBP-siFoxM1 nanoparticles into the cartilage significantly suppressed DMM-induced cartilage degradation. The most significant inhibition of cartilage degradation was observed after the introduction of the ACBP-siFoxM1 complex.
Delivery of Rho@PAA-MnO2 (MnO2 nanosheet) significantly reduces cartilage degradation. (A) IVIS images of sham and DMM cartilage implanted with senescence sensor. (B) Safranin-O staining in DMM cartilage implanted with MnO2 nanosheet for eight weeks compared to sham cartilage (n = 4 per group). The degree of cartilage degradation is quantified according to the OARSI grade. Scale bar = 200 μm. Immunohistochemistry of P16ink4a and FOXM1 in DMM cartilage (n = 4 per group). Positive cell numbers are indicated by the bar graph. *P < 0.05; **P < 0.01; *** P < 0.001, **** P < 0.0001.
Delivery of heparin-ACBP/COS-GA-siFoxM1 (ACBP-siFoxM1) nanoparticles significantly reduces cartilage degradation. (A) IVIS images of DMM cartilage introduced with ACBP-siFoxM1 nanoparticles compared to sham cartilage. (B) Safranin-O staining in DMM cartilage introduced with ACBP-siFoxM1 nanoparticles for eight weeks compared to sham cartilage (n = 4 per group). The degree of cartilage degradation is quantified according to the OARSI grade. Scale bar = 200 μm. Immunohistochemistry of FOXM1 in DMM cartilage (n = 4 per group). FOXM1-positive-cell numbers are indicated by the bar graph. *P < 0.05; **P < 0.01; *** P < 0.001, **** P < 0.0001.
Discussion
Cellular senescence, a state of stable cell cycle arrest, is a controlled process that occurs in diverse biological processes [32]. However, the persistence and accumulation of senescent cells can impair diverse cellular functions and induce age-related diseases such as OA. Senescent chondrocytes accumulate with age and increase in number in human OA cartilage compared with normal cartilage [4]. Senescent chondrocytes localize near osteoarthritic lesions of the cartilage, but not in intact areas of the cartilage, indicating a positive connection between chondrocyte senescence and OA pathogenesis [4]. Chondrocyte senescence is a common molecular mechanism that drives or promotes OA development. SASP, a robust proinflammatory secretome, can cause an imbalance in cartilage homeostasis and alter the structure and function of cells and tissues in the joint [4,33]. Expression of the SASP factors, Mmp-13, IL-6, and IL-1β stimulates inflammation and degradation of cartilage, which are the hallmarks of OA. Recently, molecular mechanisms underlying the contribution of cell senescence to the initiation and progression of OA have been reported. Oxidative stress may play a major role in the association between chondrocyte senescence and OA development [34-36]. Increased levels of intracellular ROS in the cartilage contribute to aging changes in cells and tissues by causing oxidative damage to proteins, lipids, and DNA, resulting in chondrocyte senescence, apoptosis, and cartilage matrix loss. Furthermore, increased oxidative stress in senescent chondrocytes alters mitochondrial mass, potential, and morphology; accumulates dysfunctional mitochondria; and stimulates inflammatory and matrix catabolic responses in chondrocytes [37,38]. However, many other questions, such as other factors that induce chondrocyte senescence or other underlying mechanisms that determine articular degeneration that causes osteoarthritis, remain to be answered. In the present study, we observed a high number of senescent cells with peroxisomal dysfunction. Double-knockout mice lacking Acot12 and Nudt7 showed stimulated chondrocyte senescence along with upregulation of inflammatory and cartilage catabolic factors and downregulation of cartilage anabolic factors, resulting in the development and progression of OA pathogenesis.
Deregulation of organelle interactions between the endoplasmic reticulum, mitochondria, endosomes, and cellular metabolites contributes to cellular senescence and is associated with aging [39]. Mitochondria are closely involved in cellular senescence [4]. Mitochondrial dysfunction/alteration-associated senescence produces the SASP, and mitochondria in senescent hepatocytes have an impaired capacity for fatty acid oxidation, leading to increased hepatic lipid deposition, a typical pathophysiological characteristic of non-alcoholic fatty liver disease [40,41]. In this study, we found that the dysfunction of peroxisomes induced by NUDT7 and ACOT12 deficiency also induced cellular senescence in the cartilage through the accumulation of acetyl-CoA. Lipotoxicity caused by the accumulation of fatty acids damages mitochondrial structure and function and induces a rapid increase in ROS levels, thereby contributing to cellular senescence [42]. Here, exposure of iMACs to acetyl-CoA induced the SASP. However, the relevance of peroxisomal dysfunction or acetyl-CoA accumulation induced by peroxisomal dysregulation in cellular senescence has not yet been elucidated. Furthermore, the key regulatory mechanisms and molecules responsible for this process have not yet been fully investigated. Here, we found that in chondrocytes, acetyl-CoA accumulated due to a deficiency of NUDT7 and ACOT12 induces cellular senescence via upregulation of FoxM1, a member of the forkhead superfamily of transcription factors. FOXM1 is a crucial regulator of many biological responses and dysregulation of FOXM1 is known to significantly contribute tumorigenesis and cancer progression via modulation of cell proliferation, invasion and metastasis, cancer stem cell properties, genomic instability, and cellular metabolism [43]. In particular, FOXM1 controls a network of genes necessary for the G2-M transition during cell division, suggesting its involvement in cell proliferation [44]. FOXM1 is overexpressed in various human malignancies, including prostate, breast, lung, colon, stomach, liver, and kidney cancer, via the activation of Ras and cyclin D1 and FOXM1-depletion is sufficient to decrease carcinogenesis by stimulating apoptosis [45-47]. Unlike in cancer cells, FOXM1 is known to have the opposite effect in chondrocytes. The level of FOXM1 is significantly upregulated in the articular tissues of patients with OA, as well as in chondrocytes after stimulation with IL-1β, a key regulator of cartilage destruction in OA [48]. In contrast to cancer cells, the knockdown of FoxM1 in IL-1 β-induced chondrocytes improved cell viability and attenuated inflammatory responses. In this study, we found that the upregulation of FOXM1 induced by a deficiency of NUDT7 and ACOT12 was responsible for the stimulation of chondrocyte senescence during OA pathogenesis. Taken together, in this study, we found that peroxisomal dysfunction induced by NUDT7 and ACOT12 deficiency stimulated cartilage degradation by activating chondrocyte senescence through the upregulation of FOXM1 via acetyl-CoA accumulation. Furthermore, the implantation of MnO2 nanosheets to scavenge ROS generated by senescent chondrocytes or delivery of siFoxM1 conjugated with ACBP significantly suppressed chondrocyte senescence and cartilage degradation during OA pathogenesis.
Materials and Methods
Ethical approval
All animal studies were approved by Wonkwang University Animal Care and Use Committee and performed in compliance with the institutional guidelines (#WKU21-36, #WKU22-40, #WKU22-41, #WKU22-42). Human articular cartilage specimens were obtained from patients who underwent total knee arthroplasty. Human cartilage tissue collection was approved by the Human Subjects Committee of Wonkwang University Hospital, and the studies were performed in compliance with the institutional guidelines (WKUH 201605-HRBR-041). Written informed consent was obtained from all adult patients or at least one guardian of each patient before the start of the experiment.
Human OA specimens
Human articular cartilage specimens were obtained from patients undergoing total knee replacement surgery and classified into relatively healthy (non-OA) and severely damaged (OA) regions. Human articular cartilage specimens were cut into pieces and some pieces were immediately fixed in 10% neutral buffered formalin.
Animals
Nudt7-/- and Acot12-/- mice were generated using TALEN- or RGEN-induced mutations, as previously described [25,26]. dKO mice were generated by crossbreeding of Nudt7-/- mice with Acot12-/- mice. Wild-type C57BL/6N mice were purchased from Samtako BioKorea, Inc. (Osan, Korea). All mice were housed at 22 ± 1℃ with 12 h light/dark cycles and a relative humidity of 50 ± 5% with food and water available ad libitum. DMM was performed in eight-week-old mice and knee joints were analyzed eight weeks after surgery. The degree of cartilage degradation was scored from 0 to 6 using the Osteoarthritis Research Society International (OARSI) scoring system.
Alizarin red/Alcian blue staining for a newborn skeleton
Postnatal 0 pups were euthanized, skinned, and eviscerated including eyes and fat. After fixation in 95% ethanol for 24 h, specimens were submerged in Alcian blue solution overnight at room temperature (RT). Specimens were destained in 95% ethanol and transferred to 2% KOH for cleaning the tissue. The mice were soaked in alizarin red staining solution for 24 h at RT to counterstain the bone. After clearing tissues in 20% glycerol/1% KOH, 50% glycerol/1% KOH, and 80% glycerol/1% KOH for 24 h each, samples were stored in 100% glycerol.
Histological analysis
Cartilage was fixed in 10% neutral buffered formalin and decalcified in 14% EDTA (pH 7.4). The tissues were embedded in paraffin and sectioned to a thickness of 5 μm. Safranin O staining was performed after rehydration, and the Mankin's score was measured. For immunohistochemistry, antigen retrieval using 0.01 M sodium citrate buffer (pH 6.0) was performed and sections were blocked with normal horse serum. Sections were incubated overnight with the following primary antibodies: C1,2C (1:100, 50-1035, IBEX Pharmaceuticals, Canada), FOXM1 (1:100, sc-376471, Santa Cruz, USA), MMP13 (1:200, 3533, BioVision, USA), Aggrecan Neoepitope (1:200, 100-74350, NOVUS, USA), and P16INK4A (1:100, ab189034, abcam, UK). After incubation with a peroxidase-conjugated secondary antibody, detection with the ImmPACT DAB Substrate (Vector Laboratories, USA) was performed.
iMACs culture
The iMACs were isolated from postnatal day 5 pups using a previously published method [25]. iMACs were cultured with Dulbecco's modified Eagle's medium (DMEM; Gibco) with 10% fetal bovine serum (FBS; Gibco, USA) and 100 units/ml of penicillin and streptomycin (Gibco) at 37℃ supplied with 5% of CO2. pcDNA-FoxM1 and/or siFoxM1 were transfected for 24 h following with or without 5 ng/ml IL-1β for an additional 24 h. 50 μM acetyl-CoA or 25, 50, and 100 μM sodium acetate was treated for 24 h.
Primary cell cultures
Mesenchymal cells (at a density of 2 × 107 cells/ml) were derived from the limb bud of E9 to E12.5 mouse embryo and micromass cultured in Ham's F-12 medium containing 10% FBS, 100 units/ml of penicillin and streptomycin.
Analysis of cell differentiation and pre-cartilage condensation
Alcian blue bound to sulfated glycosaminoglycans was extracted with 6 M guanidine-HCl and quantified by measuring the absorbance of the extract at 600 nm. Cultures were incubated with 100 μg/ml biotinylated peanut agglutinin (Sigma-Aldrich, USA) and visualized with the VECTASTAIN ABC and DAB substrate solution kit.
Microarray using mouse chondrocytes
Mouse whole-transcript expression array analysis was performed using the Affymetrix GeneChip Whole Transcript PLUS reagent. cDNA was synthesized using the GeneChip Whole Transcript Amplification kit. Sense cDNA was then fragmented, biotin-labeled with TdT using the GeneChip WT Terminal labeling kit, and hybridized to the Affymetrix GeneChip Mouse 2.0 ST Array for 16 h at 45 °C. The hybridized array was washed, stained on a GeneChip Fluidics Station 450, and scanned using a GGCS3000 Scanner (Affymetrix, USA). Signal values were computed using the Affymetrix GeneChip Command Console software.
RNA sequencing using human chondrocytes
RNA sequencing was performed using an Illumina HiSeq4000 instrument, libraries were quantified using quantitative real-time PCR (qPCR), and their quality was checked using an Agilent Technologies 2100 Bioanalyzer. Raw data were calculated as fragments per kilobase of transcript per million mapped reads (FPKM) for each sample using Cufflinks software. Data were logarithmically transformed and normalized using the quantile normalization method.
RT-PCR
Total RNA was isolated using RNAiso Plus (Takara, Japan) and reverse transcribed using 5X RT Master Mix (Takara). Quantitative PCR was performed using AMPIGENE qPCR Green Mix Hi-ROX (Enzo, USA) on an ABI StepOnePlus instrument (Applied Biosystems, USA) and normalized to the 18S rRNA expression level. The qRT-PCR primer sequences used in this study are listed in Supplementary Table 1.
TUNEL assay
An in situ cell death detection kit (Roche, Switzerland) was used with deparaffinized cartilage sections according to the manufacturer's instructions. Images were acquired by fluorescence microscopy after nuclear staining with 4′,6-diamidino-2-phenylindole (DAPI).
Cell apoptosis and proliferation assay
Cell apoptosis was analyzed using the Muse Annexin V & Dead Cell Kit (MCH100105, Luminex,USA), and cell proliferation was measured using the Quick Cell Proliferation Colorimetric Assay Kit (K301, Biovision) with a Muse Cell Analyzer (Merck Millipore, USA), following the manufacturer's instructions.
SA-β-gal staining
Cellular senescence was imaged using a Senescence β-Galactosidase Staining Kit (9860, Cell Signaling Technology, USA) following the manufacturer's instructions.
Neutral lipid staining
Fixed cells or mouse cartilage were stained with BODIPY493/503 (Thermo Fisher Scientific, USA) for 20 min and mounted with DAPI mounting medium (Vector Laboratories). Fluorescence images were acquired using the cartilage images.
Preparation of chitosan-FITC/acetyl-CoA complexes
A chitosan-FITC stock solution (1 mg/ml chitosan-FITC in 0.05 N HCl) was diluted 10-fold with pH 7.4 PBS and 10 µL of acetyl-CoA (50 mM) were added. After reacting for 6 h to ensure the formation of chitosan-FITC/acetyl-CoA complexes, the final product was purified by dialysis (MWCO:12-14 kDa, SpectraPor, USA) against DDW for 1 d.
Synthesis of manganese oxide nanoparticle-decorated polyacrylic acid (PAA-MnO2)
PAA (Mw: 400.000, Sigma-Aldrich) was dissolved in 50 ml dH2O and a solution of potassium permanganate (KMnO4, Sigma-Aldrich) (75 mg in 10 ml dH2O). Then, 45 ml of 2-(N-morpholino) ethanesulfonic acid (MES, Sigma-Aldrich) (0.1 M, pH 6.0) was added slowly and allowed to react at RT for 4 h until the solution color gradually changed from purple to brown. After 4 h, the solution was dialyzed (MWCO: 3.5 kDa) against dH2O and freeze-dried.
Synthesis of Rhodamine 6G-loaded PAA-MnO2 (Rho@PAA-MnO2)
Fluorescence dye Rhodamine 6G was loaded onto PAA-MnO2 based on the weight ratio of Rhodamine 6G to PAA-MnO2 (0.5% and 1%). Briefly, 1 g of PAA-MnO2 was dissolved in 20 ml dH2O and 5 mg of Rhodamine 6G was added to the PAA-MnO2 solution. After reacting for 12 h with stirring at RT, the solution was dialyzed against dH2O (MWCO 1 kDa) for 6 h and freeze-dried to obtain Rho@PAA-MnO2 (0.5%).
Synthesis of gallic acid-grafted chitosan oligosaccharide (COS-GA)
COS-GAs were synthesized using an EDC coupling agent. Briefly, 1 g of COS (Mn 5 kDa, TCI, Japan) was dissolved in 100 ml dH2O. Then, 0.49 g of GA (Sigma-Aldrich), 0.34 g of 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC, Tokyo, Japan), and 0.34 g of N-hydroxysuccinimide (NHS, Sigma-Aldrich) were added dropwise to the COS solution and reacted for 12 h at a pH of 5.0. The product was dialyzed against HCl solution (pH 2; MWCO: 1 kDa) for 4 h and freeze-dried.
Preparation and characterization of COS-GA/siFoxM1 complexes
Polyelectrolyte complexes of siFoxM1 were formed using COS-GAs. Briefly, 4.5 mg of COS-GA was dissolved in 724 μl of PBS buffer (pH 7.4), and 50 μl of 20 nM siFoxM1 solution was prepared as a stock solution. siFoxM1 was complexed with COS-GA at an N/P ratio of 10 at 4°C for 15 min.
Preparation and characterization of COS-GA/heparin/ACBP complexes
To prepare the COS-GA/heparin/ACBP complexes, 1 μl of 2 μg/μl Acyl-CoA-binding protein (MyBioSource, USA) and 2 μl of 2.5 mg/ml heparin (Sigma-Aldrich) were mixed for 30 min. Then, 2 μl of COS-GA solution was added and the resulting solution reacted for 3 h.
Statistical analyses
Data are presented as the means ± SD from at least three experiments. The results were analyzed using the Student's t-test and one- or two-way analysis of variance (ANOVA) using GraphPad Prism. Significance was defined as * P < 0.05, ** P < 0.01, ***P < 0.001, and ****P < 0.0001.
Supplementary Material
Supplementary figures and table.
Acknowledgements
We thank former and current laboratory members for their technical support and helpful comments.
Funding
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean Government (MSIT) (2022R1A4A1031259 and 2021R1I1A3041149), and by the Alchemist Project of the Korea Evaluation Institute of Industrial Technology (KEIT 20018560, NTIS 1415180625) funded by the Ministry of Trade, Industry & Energy (MOTIE) to E-J J.
Author contributions
Conceptualization: E-J J; Methodology: JSS, EHK, AIR, SAK; Investigation: JSS, EHK, JHY, DHK, E-J J, JHR, SYP; Visualization: JSS, EHK, JHY, DHK; Funding acquisition: E-J J; Project administration: E-J J, JHR, SYP; Supervision: E-J J, JHR; Writing-original draft: E-J J, EHK, SYP; Writing-review & editing: E-J J, JHR, SYP.
Data and materials availability
All data are available in the main text or the supplementary materials.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Martel-Pelletier J, Barr AJ, Cicuttini FM, Conaghan PG, Cooper C, Goldring MB. et al. Osteoarthritis. Nat Rev Dis Primers. 2016;2:16072
2. L J Sandell, T Aigner. Articular cartilage and changes in arthritis. an introduction: cell biology of osteoarthritis. Arthritis Res. 2001;3:107-13
3. Kolasinski SL, Neogi T, Hochberg MC, Oatis C, Guyatt G, Block J. et al. 2019 American college of rheumatology/arthritis foundation guideline for the management of osteoarthritis of the hand, hip, and knee. Arthritis Care Res. 2020;72:149-62
4. Price JS, Waters JG, Darrah C, Pennington C, Edwards DR, Donell ST. et al. The role of chondrocyte senescence in osteoarthritis. Aging Cell. 2002;1:57-65
5. Rim YA, Nam Y, Ju JH. The role of chondrocyte hypertrophy and senescence in osteoarthritis initiation and progression. Int J Mol Sci. 2020;21:2358
6. Ashraf S, Cha BH, Kim JS, Ahn J, Han I, Park H. et al. Regulation of senescence associated signaling mechanisms in chondrocytes for cartilage tissue regeneration. Osteoarthritis Cartilage. 2016;24:196-205
7. Cao H, Chen M, Cui X, Liu Y, Liu Y, Deng S. et al. Cell-free osteoarthritis treatment with sustained-release of chondrocyte-targeting exosomes from umbilical cord-derived mesenchymal stem cells to rejuvenate aging chondrocytes. ACS Nano. 2023;17:13358-76
8. Bolduc JA, Collins JA, Loeser RF. Reactive oxygen species, aging and articular cartilage homeostasis. Free Radic Biol Med. 2019;132:73-82
9. Liu Y, Zhang Z, Li T, Xu H, Zhang H. Senescence in osteoarthritis: from mechanism to potential treatment. Arthritis Res Ther. 2022;24:174
10. Maneiro E, Martín MA, De Andres MC, López-Armada MJ, Fernández-Sueiro JL, Hoyo P. et al. Mitochondrial respiratory activity is altered in osteoarthritic human articular chondrocytes. Arthritis Rheum. 2003;48:700-8
11. Wu X, Liyanage C, Plan M, Stark T, McCubbin T, Barrero RA. et al. Dysregulated energy metabolism impairs chondrocyte function in osteoarthritis. Osteoarthritis Cartilage. 2023;31:613-26
12. Yang X, Chen W, Zhao X, Chen L, Li W, Ran J. et al. Pyruvate kinase M2 modulates the glycolysis of chondrocyte and extracellular matrix in osteoarthritis. DNA Cell Biol. 2018;37:271-7
13. Adams SB, Setton LA, Kensicki E, Bolognesi MP, Toth AP, Nettles DL. Global metabolic profiling of human osteoarthritic synovium. Osteoarthritis Cartilage. 2012;20:64-7
14. Kim S, Hwang J, Kim J, Ahn JK, Cha HS, Kim KH. Metabolite profiles of synovial fluid change with the radiographic severity of knee osteoarthritis. Joint Bone Spine. 2017;84:605-10
15. Shen J, Wang C, Li D, Xu T, Myers J, Ashton JM. et al. DNA methyltransferase 3b regulates articular cartilage homeostasis by altering metabolism. JCI Insight. 2017;2:e93612
16. Loef M, Schoones JW, Kloppenburg M, Ioan-Facsinay A. Fatty acids and osteoarthritis: different types, different effects. Joint Bone Spine. 2019;86:451-8
17. Abshirini M, Ilesanmi-Oyelere BL, Kruger MC. Potential modulatory mechanisms of action by long-chain polyunsaturated fatty acids on bone cell and chondrocyte metabolism. Prog Lipid Res. 2021;83:101113
18. Ruiz-Romero C, Calamia V, Mateos J, Carreira V, Martínez-Gomariz M, Fernández M. et al. Mitochondrial dysregulation of osteoarthritic human articular chondrocytes analyzed by proteomics: a decrease in mitochondrial superoxide dismutase points to a redox imbalance. Mol Cell Proteomics. 2009;8:172-89
19. Jiang N, Xing B, Peng R, Shang J, Wu B, Xiao P. et al. Inhibition of Cpt1a alleviates oxidative stress-induced chondrocyte senescence via regulating mitochondrial dysfunction and activating mitophagy. Mech Ageing Dev. 2022;205:111688
20. Lee SW, Rho JH, Lee SY, Chung WT, Oh YJ, Kim JH. et al. Dietary fat-associated osteoarthritic chondrocytes gain resistance to lipotoxicity through PKCK2/STAMP2/FSP27. Bone Res. 2018;6:20
21. Bastiaansen-Jenniskens YM, Siawash M, van de Lest CHA, Verhaar JAN, Kloppenburg M, Zuurmond A-M. et al. Monounsaturated and saturated, but not n-6 polyunsaturated fatty acids decrease cartilage destruction under inflammatory conditions: a preliminary study. Cartilage. 2013;4:321-8
22. Alvarez-Garcia O, Rogers NH, Smith RG, Lotz MK. Palmitate has proapoptotic and proinflammatory effects on articular cartilage and synergizes with interleukin-1. Arthritis Rheumatol. 2014;66:1779-88
23. Tan L, Harper LR, Armstrong A, Carlson CS, Yammani RR. Dietary saturated fatty acid palmitate promotes cartilage lesions and activates the unfolded protein response pathway in mouse knee joints. PLoS One. 2021;16:e0247237
24. Song J, Kang YH, Yoon S, Chun CH, Jin EJ. HIF-1α: CRAT:miR-144-3p axis dysregulation promotes osteoarthritis chondrocyte apoptosis and VLCFA accumulation. Oncotarget. 2017;8:69351-61
25. Song J, Baek IJ, Chun CH, Jin EJ. Dysregulation of the NUDT7-PGAM1 axis is responsible for chondrocyte death during osteoarthritis pathogenesis. Nat Commun. 2018;9:3427
26. Park S, Baek IJ, Ryu JH, Chun CH, Jin EJ. PPARα-ACOT12 axis is responsible for maintaining cartilage homeostasis through modulating de novo lipogenesis. Nat Commun. 2022;13:3
27. Li J, Zhang H, Han Y, Hu Y, Geng Z, Su J. Targeted and responsive biomaterials in osteoarthritis. Theranostics. 2023;13:931-54
28. Zhang H, Wu S, Chen W, Hu Y, Geng Z, Su J. Bone/cartilage targeted hydrogel: strategies and applications. Bioact Mater. 2023;23:156-169
29. Sahranavard M, Sarkari S, Safavi S, Ghorbani F. Three-dimensional bio-printing of decellularized extracellular matrix-based bio-inks for cartilage regeneration: a systematic review. Biomater Transl. 2022;3:105-15
30. Pinho E, Soares G, Henriques M. Cyclodextrin modulation of gallic acid in vitro antibacterial activity. J Incl Phenom Macrocycl Chem. 2015;81:205-14
31. Lee JS, Cho JH, An S, Shin J, Choi S, Jeon EJ. et al. In situ self-cross-linkable, long-term stable hyaluronic acid filler by gallol autoxidation for tissue augmentation and wrinkle correction. Chem Mater. 2019;31:9614-24
32. Di Micco R, Krizhanovsky V, Baker D, d'Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. 2021;22:75-95
33. Coryell PR, Diekman BO, Loeser RF. Mechanisms and therapeutic implications of cellular senescence in osteoarthritis. Nat Rev Rheumatol. 2021;17:47-57
34. Yudoh K, Nguyen van T, Nakamura H, Hongo-Masuko K, Kato T, Nishioka K. Potential involvement of oxidative stress in cartilage senescence and development of osteoarthritis: oxidative stress induces chondrocyte telomere instability and downregulation of chondrocyte function. Arthritis Res Ther. 2005;7:R380-91
35. Brandl A, Hartmann A, Bechmann V, Graf B, Nerlich M, Angele P. Oxidative stress induces senescence in chondrocytes. J Orthop Res. 2011;29:1114-20
36. Yagi M, Endo K, Komori K, Sekiya I. Comparison of the effects of oxidative and inflammatory stresses on rat chondrocyte senescence. Sci Rep. 2023;13:7697
37. Shang J, Lin N, Peng R, Jiang N, Wu B, Xing B. et al. Inhibition of Klf10 attenuates oxidative stress-induced senescence of chondrocytes via modulating mitophagy. Molecules. 2023;28:924
38. Kim S, Lee K, Choi YS, Ku J, Kim H, Kharbash R. et al. Mitochondrial double-stranded RNAs govern the stress response in chondrocytes to promote osteoarthritis development. Cell Rep. 2022;40:111178
39. Huang J, Meng P, Wang C, Zhang Y, Zhou L. The relevance of organelle interactions in cellular senescence. Theranostics. 2022;12:2445-64
40. Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A. et al. Cellular senescence drives age-dependent hepatic steatosis. Nat Commun. 2017;8:15691
41. Dabravolski SA, Bezsonov EE, Orekhov AN. The role of mitochondria dysfunction and hepatic senescence in NAFLD development and progression. Biomed Pharmacother. 2021;142:112041
42. Hamsanathan S, Gurkar AU. Lipids as regulators of cellular senescence. Front Physiol. 2022;13:796850
43. Kalathil D, John S, Nair AS. FOXM1 and cancer: faulty cellular signaling derails homeostasis. Front Oncol. 2021;10:626836
44. Fischer M, Schade AE, Branigan TB, Müller GA, DeCaprio JA. Coordinating gene expression during the cell cycle. Trends Biochem Sci. 2022;47:1009-22
45. Cai Y, Balli D, Ustiyan V, Fulford L, Hiller A, Misetic V. et al. Foxm1 expression in prostate epithelial cells is essential for prostate carcinogenesis. J Biol Chem. 2013;288:22527-41
46. Wang IC, Ustiyan V, Zhang Y, Cai Y, Kalin TV, Kalinichenko VV. Foxm1 transcription factor is required for the initiation of lung tumorigenesis by oncogenic KrasG12D. Oncogene. 2014;33:5391-6
47. Anders L, Ke N, Hydbring P, Choi YJ, Widlund HR, Chick JM. et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011;20:620-34
48. Zeng R-M, Lu X-H, Lin J, Hu J, Rong Z-J, Xu W-C. et al. Knockdown of FOXM1 attenuates inflammatory response in human osteoarthritis chondrocytes. Int Immunopharmacol. 2019;68:74-80
Author contact
![]() Corresponding author: Email: jineunjungac.kr.
Corresponding author: Email: jineunjungac.kr.