Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Structures and functions of...
Neddylation in cancers
Role of neddylation on fibrotic...
Inhibitors of neddylation enzymes
Conclusions and perspectives
Abbreviations
Acknowledgements
References
Author biographies
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2023; 13(14):5017-5056. doi:10.7150/thno.78876 This issue Cite
Review
Targeting cullin neddylation for cancer and fibrotic diseases
Zhang-Xu He1,2#, Wei-guang Yang3#, Dan Zengyangzong2, Ge Gao2, Qian Zhang2, Hong-Min Liu2, Wen Zhao2 ![]() , Li-Ying Ma2,4,5
, Li-Ying Ma2,4,5 ![]()
1. Pharmacy College, Henan University of Chinese Medicine, 450046, Zhengzhou, China.
2. State Key Laboratory of Esophageal Cancer Prevention and Treatment; Key Laboratory of Advanced Pharmaceutical Technology, Ministry of Education of China; School of Pharmaceutical Sciences, Zhengzhou University, Zhengzhou, Henan 450001, China.
3. Children's hospital affiliated of Zhengzhou university; Henan children's hospital; Zhengzhou children's hospital, Henan Zhengzhou 450000, China.
4. China Meheco Topfond Pharmaceutical Co., Zhumadian 463000, China.
5. Key Laboratory of Cardio-cerebrovascular Drug, Henan Province, Zhumadian 463000, China.
#: These authors contribute equally to this work.
*: These senior authors contribute equally to this work.
Received 2022-9-13; Accepted 2023-4-12; Published 2023-9-4
Abstract

Protein neddylation is a post-translational modification, and its best recognized substrates are cullin family proteins, which are the core component of Cullin-RING ligases (CRLs). Given that most neddylation pathway proteins are overactivated in different cancers and fibrotic diseases, targeting neddylation becomes an emerging approach for the treatment of these diseases. To date, numerous neddylation inhibitors have been developed, of which MLN4924 has entered phase I/II/III clinical trials for cancer treatment, such as acute myeloid leukemia, melanoma, lymphoma and solid tumors. Here, we systematically describe the structures and biological functions of the critical enzymes in neddylation, highlight the medicinal chemistry advances in the development of neddylation inhibitors and propose the perspectives concerning targeting neddylation for cancer and fibrotic diseases.
Keywords: Cullin neddylation, Inhibitors, Cancer, Fibrotic diseases
Introduction
Neddylation, a reversible post-translational modification, adds a ubiquitin-like molecule NEDD8 (neuronal precursor cell-expressed developmentally downregulated protein 8) to a lysine residue of targeted substrate proteins through a three-step enzymatic cascade, catalyzed by NEDD8-activating enzyme E1 (NAE), NEDD8-conjugating enzyme E2s (UBC12/UBE2M or UBE2F) and substrate-specific NEDD8-E3 ligases (Figure 1) [1, 2]. Shortly, the mature NEDD8 is firstly adenylated and activated in an ATP-dependent manner by NAE, a heterodimer comprising NAE1/APPBP1 and UBA3/NAEβ. Next, the activated NEDD8 is transmitted to one of two E2s, UBE2F or UBC12 via a trans-thiolation reaction, and then conjugated to its substrate protein through covalent attachment by various substrate-specific NEDD8 E3 ligases, such as RBX1/2 or DCN1-5 [3, 4]. Besides, neddylated substrates could be deneddylated by deneddylases, such as COP9 signalosome and NEDD8 protease 1 [5-9]. The best-identified physiological substrates of neddylation are cullin family proteins of CRLs, including cullin-1, -2, -3, -4A, -4B, -5, -7 and -9. UBC12 mainly interacts with RBX1 to mediate the neddylation of cullin-1, -2, -3, -4A and -4B, while UBE2F is highly selective to the neddylation of RBX2-related cullin-5 [10-12]. Cullin neddylation results in activation of CRLs, which are the biggest family of E3 ubiquitin ligases and control the ubiquitination of up to 20% cellular proteins for targeted degradation via the ubiquitin-proteasome system (UPS) [13-15]. Accumulated evidence clearly demonstrates that many critical proteins (NAE, UBC12, UBE2F, DCN1, RBX1 and RBX2) in neddylation process are overactivated in multiple human diseases, such as cancers [16-22] and fibrotic diseases [23, 24]. Furthermore, the abnormal expression of these enzymes is linked to poor patient prognosis [25], implying targeting neddylation as a promising therapeutic strategy for cancers and fibrotic diseases [26].
The three-step enzymatic reaction for protein neddylation. N8, NEDD8 (neural precursor cell expressed, developmentally down-regulated 8).
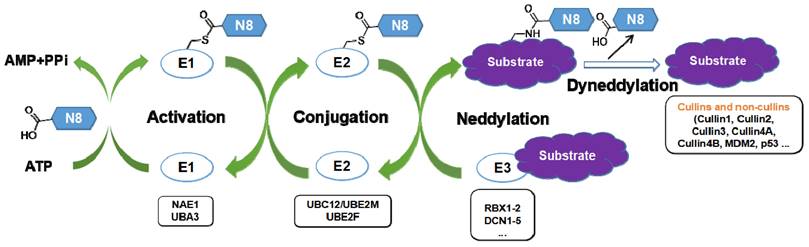
Currently, numerous efforts have been focused on developing novel agents for targeting cullin neddylation. MLN4924 (Pevonedistat), reported in 2009, is a first-in-class NAE inhibitor. It can effectively block neddylation of all cullins, causing the inactivation of CRLs and the accumulation of cancer-related substrates of CRLs [27-30]. Preclinical studies have indicated that MLN4924 possesses robust antitumor activity, which is connected with cell cycle arrest [31, 32], apoptosis [30, 33, 34], senescence [35, 36], autophagy [37], inflammation [38] and angiogenesis [27, 39] in a cell-dependent manner. Given its impressive anticancer effect and well-tolerated toxicity in mice, MLN4924 is currently in many phase I/II/III clinical trials alone or in combination with chemotherapeutic drugs for cancer treatment. In addition to MLN4924, more and more neddylation inhibitors have been identified and show promising anticancer efficacy. Furthermore, the potential roles of neddylation inhibitors on fibrotic diseases are gradually being disclosed [23, 24].
Herein, we are intended to highlight numerous efforts on targeting cullin neddylation for cancer and fibrotic diseases. Specially, we summarize the structures and functions of critical neddylation proteins, the pivotal roles of neddylation in the progression of cancer and fibrotic disease as well as the advances in the development of neddylation inhibitors from a medicinal chemistry perspective. The opportunities and challenges of targeting cullin neddylation for cancer and fibrotic diseases are also discussed.
Structures and functions of critical proteins in neddylation process
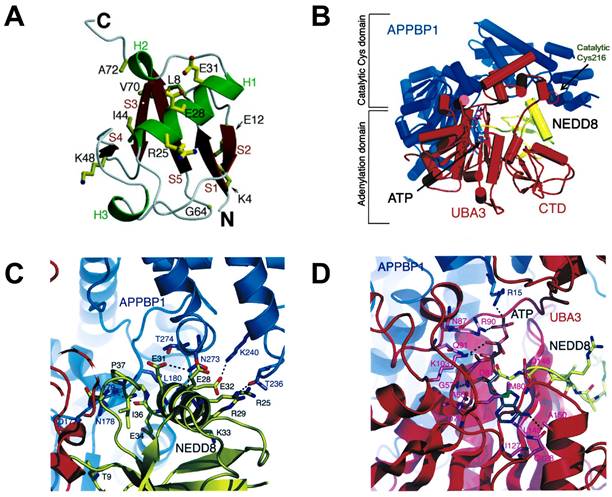
NEDD8, a polypeptide containing 81 amino acid residues, was cloned in mouse brain tissue in 1993. It possesses 80% similarity and 60% identical with ubiquitin and is overexpressed in the nucleus [40, 41]. NEDD8 consists of a globular body containing a five-stranded mixed β sheet (red) and an α helix (green), a flexible C termini and N termini (Figure 2A). Additionally, NEDD8 has an asymmetric distribution of charged residues. One face of NEDD8 is primarily acidic and the other face comprises the Leu8/Ile44/Val70 hydrophobic patch [42, 43]. The structure of APPBP1-UBA3-NEDD8 complex with ATP shows that NAE is composed of three domains: an adenylation domain having ATP binding site is connected via flexible loops to one domain at the C terminus of UBA3 subunit and another domain around the catalytic cysteine (Figure 2B). NEDD8 well fits in the groove of APPBP1-UBA3. The acidic face of NEDD8 in globular domain widely contacts with the catalytic cysteine domain of APPBP1 (Figure 2C). The hydrophobic patch of NEDD8 interacts with the adenylation domain, which is structurally conserved in the enzymatic cascade. Besides, NEDD8's C-terminal tail is inserted within 4A˚ of ATP's α-phosphate and ready for adenylation reaction (Figure 2D) [43, 44].
(A) The overall structure of NEDD8. Reproduced with permission [42]. Copyright 1998, American Society for Biochemistry and Molecular Biology. (B) The structure of the APPBP1-UBA3-NEDD8-ATP complex. (C) The interactions between catalytic cysteine domain of NAE and NEDD8's acidic surface. (D) The interactions between NEDD8's C-terminal tail and ATP of adenylation domain. Reproduced with permission [43]. Copyright 2003, Cell Press.
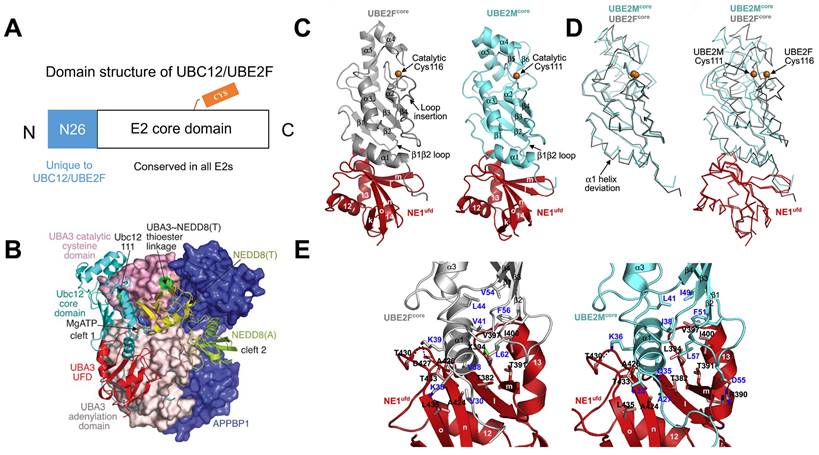
The NEDD8 E2s (UBC12/UBE2M and UBE2F) generally include two regions: (i) a special 26-residue N-terminal extension identified only in UBC12/UBE2M and (ii) an E2 core region, a ∼150-residue domain conserved in all E2s, which possesses E2 catalytic cysteine (Figure 3A). The N-terminal domain can selectively recruit NAE to boost thioester formation between E2s and NEDD8 [45]. In the APPBP1-UBA3~NEDD8(T)-NEDD8(A)-MgATP-UBC12(C111A) complex, the three APPBP1-UBA3 domains pack to form a big groove, which links MgATP, two NEDD8 molecules and UBC12 together (Figure 3B). A crossover loop linking catalytic-cysteine and adenylation domains divides the groove into two parts. The peptide-like extension of UBC12 locates in a groove of UBA3's adenylation domain, and the core domain of UBC12 binds to UBA3's ubiquitin-fold domain (UFD). NE1ufd-UBE2Fcore contains a globular ovoid structure like NE1ufd-UBE2Mcore, with UBE2Fcore indicating a typical E2 catalytic core domain fold (Figure 3C). The superimposition of UBE2Fcore and UBE2Mcore shows different features, such as (i) different orientations of catalyzed cysteine, (ii) a half-turn extension and an offset orientation for UBE2Fcore's N-terminal α1 helix, and (ⅲ) a prolonged C-terminal helix more reminiscent of ubiquitin E2 structures compared with two-stranded β sheet of C terminus in UBE2M (Figure 3D). Further structural analysis displays that NAE binds to different α helix and β1β2 loop sequences of UBE2F and UBE2M. The interactions are surrounded by two hydrophobic clusters between both E2s and NAE (Figure 3E). One cluster consists of NAEufd's Ala424, Ala426, Thr433 and Leu435, UBE2Fcore's Val30 and the hydrophobic moiety of Lys35 from an α helix. These correspond to UBE2Mcore's Ala27 and Leu32 separately. Another cluster comprises NAEufd's Thr382, Thr391, SeMet394, Val397 and Ile400 packing toward UBE2Fcore's Val38, Val41 and Leu44 from an α helix; and Val54, Phe56 and Leu62 from the β1β2 loop. Even though Leu44, Phe56 and Leu62 are conserved between UBE2Mcore and UBE2Fcore, other residues have apparent difference. For example, UBE2Fcore's Val38 forms hydrophobic interactions, but the corresponding UBE2Mcore Gln35 makes a hydrogen bond interaction with E1's Thr382. Also, NAEufd-UBE2Mcore complex is stabilized by many ionic interactions, whereas only one stabilizes NAEufd-UBE2Fcore (Figure 3E) [12, 46-48].
(A) The domain structure of UBC12/UBE2F. (B) Overall structure of the APPBP1-UBA3~NEDD8(T)-NEDD8(A)-MgATP-UBC12(C111A) complex. Reproduced with permission [47]. Copyright 2007, Nature Publishing Group. (C) Overall structure of NAEufd-UBE2Fcore compared to NAEufd-UBC12core. (D) The structural superposition of UBE2Fcore and UBC12core. (E) The interactions between NAEufd and UBE2Fcore, NAEufd and UBC12core separately. Reproduced with permission [12]. Copyright 2009, Elsevier Inc.
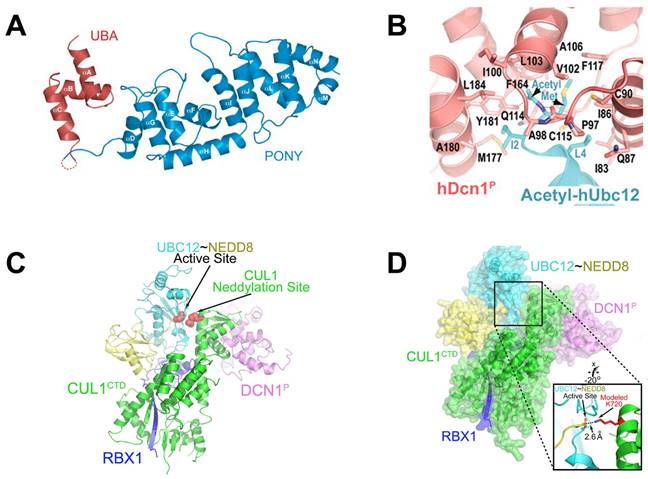
DCN-like proteins, as scaffold-like E3 ligases in neddylation process, do not need cysteine for catalytic function. They can interact with acetylated N-terminus of UBC12/UBE2F and cullin-RBX1 to increase neddylation efficacy [49, 50]. DCN-like proteins include five isoforms (DCN1-5), each of which is composed of an N-terminal ubiquitin associated domain (UBA domain; residues 12-56), a disordered linker (residues 57-69) and a C-terminal domain (PONY domain; residues 70-269) (Figure 4A). Unlike the UBA domain, the PONY domain is mainly responsible for promoting neddylation. The N-terminal acetylation of NEDD8 E2s is usually vital for the activation of DCN-like proteins [49, 51]. The structure of UBC12 complexed with DCN1 shows that interactions are mediated mainly by an N-terminal 12-residue peptide of UBC12 and a well-defined binding groove in DCN1 (Figure 4B) [52]. Additionally, RBX1-UBC12~NEDD8-CUL1-DCN1 structure indicates that oxyester-bonded UBC12~NEDD8 intermediate is steady in complex with RBX1, DCN1P and Cul1CTD (Figure 4C-D). The center of the complex is the active site, in which Cul1 target site and UBC12~NEDD8 covalent linkage are juxtaposed within UBC12's catalytic center. The lysine's ε-amino group of a modelled Cul1 acceptor is ready for ligation at 2.6 A˚ from NEDD8's C-terminal carbon [53].
(A) Ribbon representation of full-length S. cerevisiae DCN1. The PONY and UBA domains are shown in blue and red separately. Reproduced with permission [50]. Copyright 2008, Elsevier Inc. (B) Close-up of UBC12's acetylated N terminus binding to DCN1 in cartoon. Reproduced with permission [52]. Copyright 2011, the American Association for the Advancement of Science. (C) Overall structure of RBX1-UBC12~NEDD8-CUL1-DCN1 complex as cartoon. (D) The surface of RBX1-UBC12~NEDD8-CUL1-DCN1 complex. Reproduced with permission [53]. Copyright 2014, Elsevier Inc.
Neddylation in cancers
Considering that neddylation proteins, including E1, E2s, E3s and neddylation substrates, are overactivated in various cancers and connected with cancer progression, targeting neddylation can be an appealing strategy for cancer treatment [11, 54, 55]. The important roles of neddylation proteins in various cancers are summarized in the current section.
The roles of neddylation in lung cancer
Lung cancer, one of the most common human malignancies, is regarded as a major public health concern around the world [56]. Jia's group found that neddylation pathway was upregulated in both squamous-cell carcinoma and adenocarcinoma of lung, resulting in the increased neddylation of substrates, which induced degradation of tumor suppressors (such as p21 and p27) and promoted carcinogenesis. However, abrogation of neddylation markedly suppressed proliferation, migration, motility and survival of lung cancer cells. Similarly, treated with MLN4924 could also inhibit protein neddylation, inactivate CRL, lead to the accumulation of tumor-related CRL substrates and induce senescence or apoptosis [22]. The treatment of lung cancer metastasis is still challenging, owing to lack of understanding of the potential mechanisms. In 2019, Jia's group recorded the metastatic process in real-time through a whole-mouse imaging system in Lewis lung carcinoma cells [57]. The results showed that MLN4924 effectively blocked the metastatic process, including intravascular survival, extravasation and formation of metastatic colonies, thus inhibiting tumor metastasis. MLN4924 also suppressed the expression of matrix metalloproteinase 9 (MMP9), MMP2 and vimentin, interfered the actin cytoskeleton to weaken invasive potential and then resulted in a DNA damage response, cell cycle arrest as well as apoptosis. Tumor associated macrophages (TAMs) are the most abundant immune cell population infiltrating tumor microenvironment and are beneficial for tumor angiogenesis and metastasis. In addition, tumor-derived chemotactic cytokine ligand 2 (CCL2) is a monocyte-chemotactic protein. Its expression level is positively associated with numbers of TAMs in tumor tissues. Jia et al. discovered that the inactivation of neddylation pathway obviously inhibited the infiltration of TAMs, further suppressing lung cancer metastasis [58]. Specifically, neddylation inactivation inhibited the activity of CRLs and led to the accumulation of its substrate IκBα to suppress nuclear factor-κB (NF-κB) transcriptional activity and CCL2 transactivation, thereby reducing TAMs infiltration.
Jia's group also observed that NEDD8 was upregulated in lung cancer, related to a worse patient overall survival [59]. The knockdown of NEDD8 in lung adenocarcinoma exhibited significantly anticancer effects in vitro and in vivo. Further mechanism research revealed that NEDD8 downregulation caused the accumulation of some tumor-related CRLs substrates through inhibiting their degradation, thereby inducing cell G2 phase arrest, apoptosis and senescence.
Continually, Jia and co-workers indicated that the mRNA expression level of UBC12 in lung cancer tissues was much higher compared with that in normal lung tissues [60]. Besides, the expression of UBC12 protein was not only positively related to the level of global protein neddylation, but also essential for maintaining the malignant phenotypes of lung cancer cells. Downregulation of UBC12 obviously decreased neddylation modification process, while overexpression of UBC12 enhanced protein neddylation. Importantly, UBC12 inhibition could effectively suppress lung cancer cell proliferation in vitro and in vivo. Meanwhile, UBC12 knockdown inhibited the growth of MLN4924-resistant cells by blocking cullin neddylation, leading to the accumulation of CRLs substrates. Mechanically, UBC12 knockdown blocked cullin neddylation, resulted in the inactivation of CRLs and caused the accumulation of tumor-related CRL substrates (p21 and p27) to further induce cell cycle arrest and inhibit the growth of lung cancer cells.
Sun and co-authors reported that UBE2F was upregulated in non-small cell lung cancer (NSCLC), and associated with poor patient survival [61]. UBE2F overexpression contributed to lung cancer cell proliferation in vitro and in vivo, while its downregulation effectively inhibited tumor growth. Additionally, UBE2F coupled with RBX2 to induce Cul5 neddylation, resulting in the activation of CRL5 E3, which further enhanced the pro-apoptotic protein NOXA poly-ubiquitylation through atypical K11 linkage for proteasomal degradation. Thus, by promoting NOXA degradation, UBE2F could exert its growth-stimulating function. Platinum-based chemotherapy remains to be the main way against NSCLC, whereas the increasing occurrence of drug resistance has partly limited the clinical application of platinum-based chemotherapy. Jia's group found that the upregulation of UBE2F was a critical approach for lung cancer cells to escape platinum-induced cell apoptosis, causing insensitivity to platinum-based chemotherapy [62]. Deletion of UBE2F could sensitize lung cancer cells to platinum treatment by increasing the protein levels of pro-apoptotic protein NOXA and subsequently inducing cell apoptosis. Mechanistically, platinum treatment damaged the interaction of UBE2F-RBX1 complex and thus suppressed its degradation. Additionally, the accumulated UBE2F stimulated the neddylation levels and activity of Cul5, in accord with the decreased expression of NOXA, a well-known substrate of CRL5.
The neddylation E2 conjugating enzyme consists of UBC12 and UBE2F, while whether and how both E2s cross-talk with each other is unexplored. Sun and co-workers proposed that UBC12 was a stress-inducible protein, exposed to upregulation by AP-1 and HIF-1, whereas UBE2F was degraded by UBC12-associated E3 ligases. UBC12 served as a neddylation E2 to activate CRL3 Keap1 to maintain UBE2F level upon normal physiological conditions. Upon hypoxia or mitogen stimulation, UBC12 acted as a dual E2 complexed with DJ-1/Parkin to enhance ubiquitylation and degradation of UBE2F, further resulting in CRL5 inactivation, the pro-apoptotic protein NOXA accumulation and the growth inhibition of lung cancer cells [63].
Defective in cullin neddylation 1 (DCN1) is also known as DCUN1D1, DCNL1 or squamous cell carcinoma-related oncogene (SCCRO). It serves as a co-E3 together with RBX1 to promote transfer of NEDD8 from its E2 (UBC12) to cullins, further regulating CRL stability [20]. DCN1 is connected with tumor development and poor outcomes in NSCLC and squamous cell carcinoma (SCC) [64, 65]. Also, DCN1 expression is closely related to tumor stage in patients with NSCLC. It was found in none of the patients with stage I disease, 10% of those with stage II disease and 29% with stage III disease. Moreover, 14 of 16 DCN1 positive patients led to brain metastasis. Besides, knockdown of DCN1 inhibited the growth of 3q26-29-amplified SCC cells [66]. Aberrant expression of circular RNAs (circRNAs) is linked to cancer progression. Liang and co-authors screened circRNA expression of A549 cells, identifying circDCN4, which could inhibit glycolysis and metastasis [67]. Besides, the downregulation of circDCN4 was more common in lymph node metastatic tissues and acted as an important risk factor for the whole survival of lung adenocarcinoma patients.
Cul1 is the scaffold component of SCF (Skp1-Cul1-F-box protein) complexes that control the proteolysis of many proteins associated with cell cycle progression. The abnormal expressions of F-box proteins were found in human cancers, such as lung cancer. Salon et al. reported that the upregulation of Cul1 was observed in 40% (51/128) of all lung tumors in comparison to normal lung tissues, including 75% (18/24), 30% (16/54) and 34% (17/50) of carcinoids, high grade neuroendocrine lung carcinomas and NSCLCs separately [68]. Additionally, high levels of Cul1 protein could protect cells from hyperproliferative signals through cyclin E downregulation. Moreover, neddylated forms of Cul1 were highly expressed in neuroendocrine lung tumors. These results show that alterations of Cul1 neddylation can promote the development of highly aggressive lung tumors.
The roles of neddylation in liver cancer
Liver cancer includes hepatocellular carcinoma (HCC), bile duct cystadenocarcinoma, intrahepatic cholangiocarcinoma (ICC), epitheliod haemangioendothelioma, hepatoblastoma and haemangiosarcoma. In particular, HCC has relatively heterogeneous pathogenesis making it challenging for the existing therapies [69]. The overactivation of neddylation pathway was found in HCC patients with a positive correlation between overall levels of neddylation and poorer prognosis, suggesting that neddylation modification may be a promising therapeutic approach for HCC clinical management [70, 71]. Jia and co-workers disclosed that the effect of MLN4924 on inducing autophagy was related to the CRL inactivation, which was vital for inhibiting the proliferation of liver cancer cells. In addition, MLN4924-induced autophagy was partly linked to inhibition of the mammalian target of rapamycin (mTOR) activity and induction of reactive oxygen species (ROS) stress. Besides, inhibiting autophagy promoted MLN4924-induced apoptosis [72]. Subsequently, Jia and co-authors also examined the clinical relationship and therapeutic potency of targeting neddylation pathway in ICC through analyzing the immunohistochemistry of neddylation proteins in a cohort of 322 cases [16]. The results presented that E1, E2s and global NEDD8 conjugation were overactivated in over 2/3 of human ICC. Besides, MLN4924 treatment obviously suppressed cholangiocarcinoma cell proliferation and tumor growth with synergistic effect when combined with cisplatin.
Jia's group also found that tumor suppressor RhoB was a new substrate of the neddylation-CRL pathway. Ablation of RhoB could decrease the expression of tumor suppressor p21 and p27. Cul2-RBX1 E3 ligase, as the main regulator, functioned for the ubiquitination and degradation of RhoB in liver cancer cells. The overactivation of neddylation-CRL pathway enhanced the degradation of RhoB, thus promoting liver carcinogenesis and tumor progression. However, treated with MLN4924 blocked cullin neddylation, inactivated CRL and caused the accumulation of RhoB, which further induced apoptosis and suppressed proliferation of liver cancer cells [73].
Liver metastasis is the main reason of death in patients with uveal melanoma (UM), thus it is urgently required to develop an effective therapy. A recent study has shown that MLN4924 could suppress cancer stem-like cell properties in UM via Slug protein degradation. Additionally, MLN4924 interfered the secretion of NF-kB-mediated VEGF-C and angiogenesis. Besides, neddylation inhibition on proliferation was associated with G2/M phase arrest and activation of ATM-Chk1-Cdc25C DNA damage response. These findings show that neddylation inhibition is beneficial for the treatment of hepatic metastasis of UM [74].
Recently, the potential roles of UBC12 in ICC have been explored by Zhao et al [75]. They found that ICC patients with overexpressed UBC12 showed worse accumulative recurrence and overall survival in comparison to patients with low expression. However, the knockdown of UBC12 caused the decreased viability and colony formation of ICC cell lines, possibly owing to DNA damage responses and apoptosis. Besides, UBC12 silencing inhibited ICC tumor growth, as demonstrated by the decreased size of tumor xenografts infected with the shUBC12 lentivirus. Although knockdown of UBC12 with siRNA can inhibit neddylation pathway and lead to a robust anticancer effect, how to transport siRNA with low toxicity in vivo remains to be solved. Li's group designed a folic acid-modified (poly(lactic-co-glycolic acid))-(thioketal)-(polyethylene glycol) nanomedicine for siRNA delivery [76]. The nanomedicine not only suppressed proliferation and induced apoptosis of liver cancer cells, but also showed favorable therapeutic ability through silencing UBC12, inhibiting neddylation pathway and accumulating tumor-suppressive CRL substrates with low toxicity. More importantly, the nanomedicine exhibited promising potency for multifunctional nano-system applicable in cancer therapy.
RING box protein-1 (RBX1), also named ROC1, is an important RING component of CRL and overactivated in hepatocellular carcinomas connected with poor prognosis. Jia and co-workers found that knockdown of RBX1 suppressed the proliferation of liver cancer cells via inducing autophagy and p21-dependent senescence. Further exploration displayed that RBX1 silencing activated autophagy through suppressing mTOR activity, owing to the accumulation of mTOR-inhibitory protein Deptor, a substrate of CRL. Mechanically, autophagy response on RBX1 silencing was a survival indicator. Additionally, the inhibition of autophagy pathway sensitized cancer cells to apoptosis. The above results not only disclosed RBX1 as an attractive drug target for liver cancer, but also provided a new chance for liver cancer treatment through combining RBX1 inhibitor with autophagy inhibitor [77].
Cul1 acts as a vital scaffold of Skp1-Cul1-F-box protein (SCF) E3 ubiquitin ligase complex that regulates ubiquitination of proteins related to cell cycle, transcription and signal transduction. To explore the role of Cul1 in HCC, Liu et al. determined Cul1 expression by immunohistochemistry using 90 cases HCC tissues and the corresponding adjacent non-cancerous tissues. The results indicated that in comparison to the paired adjacent non-tumor tissues, Cul1 expression was evidently increased in HCC tissues. Also, Cul1 staining was apparently connected with tumor size, TNM stage and histology grade. Besides, the overexpression of Cul1 was a strong independent prognostic indicator of HCC and closely correlated with worse 5-year overall rates in HCC patients. These data demonstrated that Cul1 could be a key prognosis marker for human HCC [78].
Cul4A is also a scaffold protein for E3 ubiquitin ligase and overexpressed in HCC tissues. A negative correlation was observed between Cul4A expression and tumor differentiation grade as well as patient survival, but a positive relationship with venous invasion, lymphatic and hepatocyte proliferation. Additionally, Cul4A knockdown promoted the expression of epithelial marker (E-cadherin), but reduced the steady-state level of mesenchymal marker (Vimentin) as well as EMT-related transcription factors, indicating that Cul4A could promote the development of HCC by stimulating EMT trend. Moreover, Cul4A downregulation suppressed the growth of HCC cells, resulting in the reduction of S-phase and inhibition of Cyclin A and Cyclin B1. The above findings show that Cul4A may be a promising target for HCC treatment [79].
Similarly, Cul4B displays tumor-promoting effects and is noticeably upregulated in different cancers. Yuan et al. observed that overexpression of Cul4B enhanced spontaneous and DEN-induced hepatocarcinogenesis. Additionally, the increased hepatocarcinogenesis in Cul4B transgenic mice could be regulated by the increased proliferation and oxidative stress. Besides, the transgenic mice revealed stronger compensatory proliferation after DEN-induced liver injury, accompanied by the activation of Erk, NF-κB, p38 and Akt. Taken together, Cul4B plays a critical role in hepatocarcinogenesis [69].
Cul7, a non-typical cullin protein comprising approximately 1,700 amino acids, serves as a component of the SCF complex and interacts with RBX1, Fbw8 and Skp1. Zhang et al. found that Cul7 was overexpressed in HCC tumor tissues, particularly in metastatic HCC tumor tissues. In addition, there was an adverse relationship between Cul7 expression and extended survival. Silencing of Cul7 could effectively inhibit invasion, migration and metastatic capacities of liver cancer cells. Besides, upregulation of Cul7 enhanced epithelial-mesenchymal transformation, demonstrated by the increased mesenchymal cell markers (N-cadherin, vimentin and fibronection) and reduced epithelial cell markers (E-cadherin and a-catenin) [80].
The roles of neddylation in leukemia
Acute myelogenous leukemia (AML) is characterized by maturation arrest and hyperproliferation of clonal myeloid precursors. Chemotherapy partly meets the clinical application, whereas patients with AML may relapse because of the occurrence of drug resistance with increased cancer cell ability to rescue DNA damage. Therefore, new therapies are urgently needed [81]. Swords et al. reported that MLN4924 could effectively induce AML cell death and stabilize important NAE targets, further suppressing NF-κB activity, DNA damage and ROS generation. Besides, administration of MLN4924 significantly suppressed AML xenografts growth [33]. Overexpression of microRNA-155 (miR-155) is connected with poor survival in AML. The miR-155 is controlled by NF-κB, the activity of which is partly regulated by the NEDD8-related ubiquitin ligases. Marcucci's group revealed that MLN4924 reduced the binding of NF-κB to miR-155 promoter and downregulated miR-155. Also, overactivation of miR-155 decreased MLN4924-induced antileukemic potency. Moreover, MLN4924 could induce miR-155 expression and prolong the survival of mice xenografted with human leukemic cells [82].
Acute promyelocytic leukemia (APL) is characterized by the arrest of terminal differentiation of myeloid cells into granulocytes. All-trans retinoic acid (ATRA) treatment can clinically relieve APL through inducing granulocytic differentiation and then cell death of differentiated leukemic cells. Wang and co-authors reported that neddylation pathway was involved in the treatment of ATRA for APL, and ATRA treatment in primary APL cells resulted in the degradation of UBA3. Moreover, neddylation inhibition was able to induce differentiation, apoptosis and proliferation suppression [83]. Meanwhile, Liu et al. found that ATRA could decline the expression of NAE, further suppressing neddylation of Cul1 and Cul3 in the APL cell line NB4 [84]. Also, MLN4924 suppressed proliferation and induced apoptosis of APL cells via inhibiting cullin neddylation. Importantly, MLN4924 could promote ATRA-induced differentiation of APL cells by inducing autophagy.
Chronic myeloid leukemia (CML) is involved in the fusion of Abelson oncogene (ABL) with breakpoint cluster region (BCR) gene to encode BCR-ABL, which is the only driving force in pathogenesis of CML. Both resistance to imatinib owing to T315I mutation and leukemia stem cells (LSC) are challenging in patients with CML. Pan and co-workers reported that blocking of neddylation suppressed BCR-ABL point mutations and LSCs, which were responsible for imatinib-resistant recurrences. Biologically, they verified that MLN4924 could prompt CML cell G2/M phase arrest and apoptosis. Besides, MLN4924 inhibited the growth of human CML and LSC cells in CML-bearing mice. These findings show neddylation inhibition as a promising therapeutic strategy for CML with LSC-derived imatinib resistance [85].
Chronic lymphocytic leukemia (CLL) is characterized by the accumulation of neoplastic B cells, resistant to apoptosis. Constitutive and BCR-dependent activation of NF-kB is a vital feature in CLL B cells and forecasts poor survival, but its activation may result in tumor resistance. Therefore, neutralization of NF-kB is an attractive strategy for CLL, which not only has the potential to inhibit the proliferation of CLL cells, but also leads to sensitization to both BCR-targeting agents and conventional chemotherapy. Interestingly, NEDD8 can regulate the activity of CRLs, therefore indirectly controlling NF-κB activity. Danilov's group found that MLN4924 disrupted NF-kB activation and caused Bim expression in CLL cells, further blocking stroma-mediated resistance [86]. Similarly, Godbersen et al. reported that both pulse and continuous exposure to MLN4924 could abolish NF-κB activity and induce apoptosis in CLL B cells, thus circumventing stroma-mediated resistance [87]. NF-κB pathway activity is essential in immune response. In addition, T-cell function is changed in patients with CLL. Danilov's group indicated that MLN4924 abrogated NF-κB signalling in malignant B cells [88]. Besides, T cells treated with MLN4924 recovered NAE activity, and maintained their response to T-cell receptor stimulation and cytotoxic potential, suggesting the immune role of targeting neddylation in lymphoid malignancies and CLL.
The roles of neddylation in glioblastoma
Glioblastoma (GBM) is the most malignant primary brain tumor, with limited treatment options. Jia's group firstly reported that neddylation pathway was overactivated in GBM tumor tissues in comparison to adjacent normal tissues [18, 89]. The upregulation of neddylation pathway was positively related to postoperative recurrence and high-grade disease, while was adversely connected with overall survival. Moreover, treated with MLN4924 significantly suppressed cullin neddylation and inactivated CRL, further inducing senescence, G2 cell cycle arrest and apoptosis. More importantly, inhibition of neddylation by MLN4924 inhibited tumor growth in a xenograft model of human GBM without obvious side effect. Further exploration showed that recurrent GBMs possessed higher neddylation activity than primary GBM [90]. Continually, Han et al. found that NEDD8 E1 (NAE) levels were increased in glioblastoma patients and high NEDD8 levels were connected with poor clinical outcomes. In addition, both patient-derived stem cells and glioblastoma cells were very sensitive to MLN4924, whereas healthy human astrocytes were resistant. Interestingly, MLN4924 could inhibit ERK and AKT phosphorylation in MLN4924 sensitive cells [91].
Programmed death ligand-1 (PDL1) is a central immunological checkpoint ligand molecule, and its overexpression in gliomas leads to the shortening of patient survival and reduction of the antitumor immune response. Filippova and co-workers reported that MLN4924 showed strongly antiproliferative activity against PDGx and glioma cell lines with the IC50 values ranging from 0.2 to 3 uM. Additionally, MLN4924 treatment increased the expression of PDL1 and its main transcriptional enhancer hypoxia-inducible factor 1A (HIF1A) in all glioma cell lines, which might cause potential resistance through evasion of immune surveillance checkpoints. Moreover, the overexpression of PDL1 in glioma cells caused T-cell energy, which could be inhibited by PD1/PDL1 blockage [92]. Continually, Wang's group reported that silencing of neddylation pathway key enzymes strikingly enhanced PDL1 expression in glioblastoma cancer cells. MLN4924 could increase PDL1 mRNA levels by suppressing WD repeat domain-containing 7 E3 ligase activity and Cul1-F-box, and accumulating c-MYC. Additionally, MLN4924 reduced T cell killing via PDL1 induction. However, the inhibition of PDL1 recovered sensitivity of MLN4924-treated glioblastoma cancer cells to T cell killing. To sum up, the combination of PD1/PDL1 inhibition and MLN4924 may be an effective strategy for glioma treatment [93].
Zheng et al. indicated that overexpression of CRL5 components SOCS3, Elongin C, Elongin B and RBX2 forecasted poor prognosis of glioma and all GBM subclasses. In addition, the high protein levels of Cul5 were negatively connected with those of p65/RelA and vHL. Moreover, GBM neovascularization could be enhanced by CRL5-mediated anti-angiogenic vHL protein downregulation. This study shows that CRL5 inhibition may relieve GBM progression [94]. Positive epidermal growth factor receptor (EGFR) immunoreactivity in GBM is used to forecast poor radiation response. Unfortunately, numerous efforts to target EGFR have not been successful because of the molecular heterogeneity of EGFR expression in GBM. Considering that CRL2 can degrade EGFR, Cyclin B1 and HIF-1a, it is valuable to explore the probable role of CRL2 on GBM survival and radiosensitivity. Zheng et al. discovered that expression of Cul2 could forecast GBM development and survival rate. Cul2 protein levels were negatively corrected with those of Cyclin B1, EGFR, HIF-1a and VEGF-A, suggesting that patients with high Cul2 expression level may not develop resistance toward EGFR inhibitors. Besides, elevated Cul2 expression predicted increased radiosensitivity. Together, Cul2 can be regarded as a biomarker in facilitating GBM and radiosensitivity profiling [95].
The roles of neddylation in head and neck squamous cell carcinoma
Head and neck squamous cell carcinoma (HNSCC) arises in the nasal cavity, larynx, pharynx and oral cavity, and shows poor survival rates in advanced stages. Jia's group reported that the expressions of NAE and UBC12 were upregulated in HNSCC tissues than that in normal tissues. Additionally, neddylation inhibition evidently suppressed the abnormal proliferation of HNSCC cells. Mechanistically, MLN4924 could cause the accumulation of CRL ligase substrate c‐Myc that transcriptionally activated pro‐apoptotic protein Noxa, finally resulting in apoptosis of HNSCC [96].
CRL4 with its substrate receptor CDT2 (CRL4-CDT2) is an important regulator of DNA replication via targeting CDT1, p21 and SET8 for ubiquitin-dependent proteolysis. The abnormal enhanced stability of these proteins can lead to DNA re-replication in cells with inactivated CRL4-CDT2, which is harmful to cells. Abbas and co-authors uncovered that CDT2 was upregulated in HNSCC. Its silencing significantly inhibited the growth of human papilloma virus negative (HPV-ve) HNSCC cells mainly via the induction of re-replication. In addition, MLN4924 treatment could induce re-replication, inhibit HNSCC cell proliferation as well as HNSCC xenografts growth. Besides, MLN4924 not only sensitized HNSCC cells to ionizing radiation (IR), but also increased IR-induced inhibition of xenografts in mice. These findings display that induction of re-replication is a new manner to treat radioresistant HNSCC tumors. MLN4924 may act as an adjuvant for IR-based treatments [97].
Chairatvit et al. found that NEDD8 conjugation was upregulated in human oral carcinoma cells. The expression of NEDD8 conjugation was mainly related to the concentration of serum, which also influenced the proliferation of oral squamous cell carcinoma. Importantly, NEDD8 conjugation was necessary for the proliferation of carcinoma cell as the transfection of negative UBC12 could increase proliferation rate [98].
Nasopharyngeal carcinoma (NPC) is one of the most common human malignancies in south China, with a high recurrence rate and treatment resistance. Qian's group reported that both knockdown of NEDD8 by shRNA and MLN4924 treatment significantly inhibited NPC cell proliferation, apoptosis and cycle arrest, whereas overexpression of NEDD8 displayed counter effects. Additionally, the inactivation of NEDD8 increased the sensitivity of NPC cells to cisplatin and radiation. These results reveal that NEDD8 is a novel prognostic marker and MLN4924 may act as an appealing therapeutic target for NPC treatment [21].
14‐3‐3ζ is known as a potential oncogene related to the pathogenesis of HNSCC. Seo and co-workers indicated that knockdown of 14‐3‐3ζ dramatically resulted in senescence phenotypes through p27 accumulations. Cul1 neddylation was reduced via 14‐3‐3ζ silencing. Additionally, 14‐3‐3ζ silencing suppressed the growth of Hep‐2 cells implanted into nude mice. However, downregulation of CSN5, a deneddylase, recovered the senescence induced by 14‐3‐3ζ depletion. In summary, 14‐3‐3ζ silencing caused premature senescence in Hep‐2 laryngeal cancer cells, accompanied by upregulation of p27, which might be correlated with the inactivation of the SCF ubiquitin ligase via the deneddylation of Cul1 [99].
The roles of neddylation in melanoma
Melanoma arises from the melanocyte lineage and is the most lethal form of skin cancer, including four anatomic subtypes (mucosal, cutaneous, uveal, and acral) [100]. Sun's group reported that NAE was upregulated in melanoma cell lines and tissues. The silencing of UBA3, a subunit of NAE, significantly inhibited the proliferation of melanoma M14 cells, led to G0/G1 arrest and apoptosis, suggesting UBA3 as a potential target for melanoma [101, 102]. CDT2 was also overactivated in melanoma. The knockdown of CDT2 could inhibit the proliferation of melanoma cells through a mechanism that was dependent on the stabilization of CRL4CDT2 substrates SET8 and p21. Stimulatingly, MLN4924 revealed strongly anti-melanoma activity by inducing SET8- and p21-dependent senescence and re-replication. These results showed that CRL4-CDT2-SET8/p21 degradation axis could be the main target of MLN4924 in melanoma [103]. Similarly, Wood and co-workers discovered that MLN4924 dramatically inhibited the viability of canine melanoma cells, and induced apoptosis by induction of DNA re-replication and cell senescence [104]. Moreover, most canine melanoma samples displayed sensitivity to MLN4924 of nanomolar concentration, which was related to P21 levels. Continually, Wong et al. evaluated the anticancer efficacy of MLN4924 using melanoma cells and patient-derived tumor xenografts (PDTX). The results indicated that MLN4924 could suppress cell growth (IC50 < 0.3 μM) and induce melanoma cell apoptosis. Also, MLN4924 inhibited tumor growth in melanoma cell xenografts and PDTX [105]. These results presented neddylation inhibition as a promising strategy for melanoma treatment.
The roles of neddylation in colorectal cancer
Colorectal cancer with high morbidity and mortality, is one of the most frequently diagnosed cancers in the world [106]. Gao's group reported that MLN4924 not only suppressed colon cancer cell (HCT116 and HT29) growth, but also induced cell G2/M phase arrest and apoptosis. Additionally, MLN4924 significantly triggered autophagy in HCT116 and HT29 cells through inhibiting PI3K/AKT/mTOR pathway, whereas treated with the autophagy inhibitor 3-MA blocked the effect of MLN4924 on inhibiting colon cancer cell growth and cell death. Moreover, MLN4924 effectively inhibited colon cell proliferation in a xenograft model [107]. Fotouhi and co-authors indicated that both NEDD8 and RBX1 were overexpressed in small intestinal neuroendocrine tumors (SI-NETs) from patients with liver metastasis. MLN4924 treatment displayed potent antiproliferative activity and induced SI-NET cell apoptosis. Besides, proteomics analysis upon neddylation inhibition in two SI-NET cells led to the upregulation of CRL substrates. Furthermore, the inactivation of CRL, by either NEDD8/RBX1 silencing or MLN4924 treatment induced SI-NET cell apoptosis, which was partly rescued by silencing of p27 [108]. NOXA, a BH3-only proapoptotic protein, is overexpressed in colorectal cancer (CRC). Zhang's group found that PRDX1, an antioxidant overexpressed in CRC, could suppress CRC cell apoptosis by downregulating NOXA. Mechanistically, PRDX1 caused NOXA ubiquitination and degradation, which absolutely depended on Cul5 neddylation. Additionally, PRDX1 oligomers could bind to both UBE2F and Cul5, and this tricomplex was important for Cul5 neddylation, as downregulation of PRDX1 could suppress Cul5 neddylation and NOXA degradation [109].
The roles of neddylation in prostate cancer
Prostate cancer (PCa), as the third primary cause of death in men with cancer, is the most common non-cutaneous malignancy. In 2002, Meehan et al. found that the expression of NEDD8 was downregulated in prostate cancer tissues in comparison to normal prostate tissues using proteomics [110]. Subsequently, Guo's group reported that neddylation pathway was critical and targetable in PCa. MLN4924 treatment not only inhibited PCa cell proliferation and clone formation, but also caused DNA damage, G2 phase arrest and apoptosis, which was related to the inactivated CRLs, accompanied by accumulation of tumor-suppressive CRLs substrates [111]. Continually, Rulina and co-workers probed CRLs in PCa cells and found significant plasticity of cells with TMPRSS2-ERG translocation. In addition, CRL suppression by knockdown of RBX1 or chemical inhibition resulted in prostate cancer cell G0/G1 phase arrest that inhibited cell apoptosis. Besides, inhibition of neddylation-dissociated 1 protein (Cand1), cullin-associated targets and androgen receptor (AR) sensitized cancer cells to CRL inhibition [112]. Recently, Balakirev's group analyzed the effect of MLN4924 on spheroids formed by prostate cancer LNCaP and VCaP cells, and found that MLN4924 had diverse effects on the tested cell lines, differing from cell cycle arrest and protective dormancy to apoptosis and senescence [113]. AR and its active variants (AR-Vs) are closely connected with the development and recurrence of PCa. In 2020, Sun's group indicated that both MLN4924 treatment and knockdown of NAE effectively inhibited growth and invasion of PCa cells without significant effects on normal prostate epithelial cells. Additionally, the combination of MLN4924 with castration or AR antagonist displayed more potent anti-prostate cancer activity in vitro and in vivo, compared with monotherapy. Biologically, MLN4924 suppressed the transcription of AR/AR-V7 and its downstream targets, and the expressions of MMP2 and MMP9 [114].
The roles of neddylation in breast cancer
Recently, neddylation inhibition has been reported as an appealing way for breast cancer treatment [115]. Jia and co-authors indicated that neddylation was overactivated in ER-positive breast cancer and forecasted poor prognosis. Treated with MLN4924 downregulated ERα expression in vitro and in vivo. Additionally, MLN4924 single or synergized with fulvestrant dramatically inhibited the growth of ER-positive breast cancer [116]. The overexpression of estrogen-related receptor beta (ERRβ) is positively associated with the improved prognosis and prolonged survival in breast cancer patients. Mishra's group presented that ERRβ was the main substrate of SCF complex. In breast cancer, neddylation could trigger cullin subunits of SCF complex to target ERRβ for further degradation. However, MLN4924 treatment restored ERRβ expression and led to decreased proliferation and migration of breast cancer cells. Besides, the role of ERRβ was not related to the expression of Erα [117].
The roles of neddylation in esophageal cancer
Esophageal squamous cell carcinoma (ESCC) is a primary type of esophageal cancers ranked as the sixth main cause of cancer death worldwide [118]. Jia's group reported that neddylation pathway was overactivated in ESCC and adversely connected with patient survival. Inhibition of neddylation pathway could induce death receptor 5 (DR5)-mediated apoptosis and cause the inhibition of ESCC in vivo. Further studies showed that neddylation inhibition stabilized CRL substrate activating transcription factor 4 (ATF4), which transactivated transcription factor CHOP, resulting in the expression of DR5 to activate caspase-8 and induce extrinsic apoptosis [119]. Continually, Jia and co-workers found that MLN4924-induced neddylation inhibition caused the accumulation of ATF3 to promote prosurvival autophagy by regulating NF-κB-Catalase-ROSATF3 axis in esophageal cancer cells, suggesting inhibition of ATF3-mediated autophagy as a promising strategy to improve neddylation mediated ESCC treatment [120]. Recently, Augoff's group also exhibited that MLN4924 possessed a significantly inhibitory efficacy on the tumor necrosis factor-alpha (TNF-α)-induced activity of MMP9 in ESCC cells, which further inhibited MMP9-dependent cancer cell migration [121]. Wang et al. discovered that UBC12 was upregulated in ESCC tissues and forecasted a poor survival of patients. Knockdown of UBC12 could trigger the tumor-suppressive cellular responses of ESCC cells, and suppress malignant phenotypes of ESCC cells. Besides, downregulation of UBC12 inhibited CRLs and caused the accumulation of CRLs substrates (e.g. p21 and p27), consequently prompting DNA damage, cell cycle arrest, apoptosis or senescence [122].
The roles of neddylation in gastric cancer
Neddylation inhibition is regarded as a promising strategy for gastric cancer (GC) treatment [123]. Zhang and co-authors reported that MLN4924 possessed robust antiproliferative activity against GC cells, accompanied by the accumulation of tumor-suppressor gene P27 and G2/M phase arrest. P27 inactivation in MLN4924-treated cells led to more potent inhibitory activity in vitro and in vivo, which was related to the overproduction of ROS. Additionally, P27 knockdown influenced the expression levels of BH3 family members to enhance the mitochondrial dysfunction and apoptosis. These findings not only uncovered the protective role of P27 in MLN4924-treated GC cells, but also implied that inhibition of MLN4924 with P27 might generate synergistic therapeutic efficacy [124]. Sun's group also found that MLN4924 could suppress Cul1 neddylation, along with the inhibitory proliferation and migration in GC cells. Mechanically, MLN4924 caused the accumulation of numerous CRL substrates to promote DNA damage and induce G2/M phase arrest, senescence as well as autophagy. Besides, MLN4924 could also depressed gastric cancer cell migration by transcriptionally activating E-cadherin and inhibiting MMP-9 [125].
The roles of neddylation in myeloma
In 2012, McMillin et al. reported that MLN4924 inhibited human multiple myeloma (MM) cell proliferation with the EC50 values ranging from 25 to 150 nM. Also, MLN4924 possessed comparable activity toward the proteasome inhibitors bortezomib-resistant ANBL-6 subline and its bortezomib-sensitive parental cells. Moreover, MLN4924 displayed submicromolar activity (EC50 < 0.5 μM) toward CD138+ multiple myeloma cells, and revealed synergistic effect with doxorubicin, bortezomib and dexamethasone on MM.1S cells. More importantly, MLN4924 was well tolerated at doses up to 60 mg/kg 2× daily and could effectively decrease tumor growth in both an orthotopic and a subcutaneous mouse model of multiple myeloma [126]. Continually, Gu and co-workers indicated that the efficacy of MLN4924 on MM cell lines was related to the inhibition of AKT and mTOR pathways via increasing the expression levels of REDD1. The combination of MLN4924 with bortezomib synergistically caused MM cell apoptosis, which overcame the prosurvival effects of growth factors [127].
The roles of neddylation in lymphoma
Smith's group found that MLN4924 treatment in activated B cell-like (ABC) diffuse large B-cell lymphoma (DLBCL) could lead to accumulation of pIκBα, reduction in nuclear p65 content, decrease of NF-κB transcriptional activity and G1 phase arrest, finally causing cell apoptosis. Additionally, in germinalcenter B cell-like (GCB) DLBCL cells, treated with MLN4924 resulted in the increased cellular Cdt-1 and S phase arrest, consistent with cells ongoing DNA re-replication. Besides, MLN4924 treatment in mice with human xenograft tumors of ABC- and GCB-DLBCL suppressed NAE pathway biomarkers and tumor growth [128]. Jia and co-workers reported that MLN4924 blocked protein neddylation, inactivated CRL in lymphoma cells [129]. In addition, MLN4924 inhibited the proliferation of lymphoma cells through inducing G2 phase arrest, along with senescence or apoptosis. Biologically, MLN4924-induced senescence was connected with the expression of tumor suppressor p21/p27, while apoptosis induction on neddylation inhibition was regulated by intrinsic apoptotic signaling pathway.
The roles of neddylation in ovarian cancer
Pan and co-authors reported that CRL4 components were upregulated in human ovarian cancer tissues, and the activity of CRL4CDT2 was related to ovarian cancer cell (OCC) proliferation and survival. MLN4924 treatment could result in DNA damage, cell cycle arrest and apoptosis, and make OCCs more sensitive toward other chemotherapeutic drugs. Silencing of CRL4 components Cul4A, Roc1/2 and DDB1 displayed dramatically inhibitory potency on OCCs similar to MLN4924 treatment, indicating that CRL4 inhibition was beneficial for the efficacy of MLN4924. Besides, CDT2 downregulation mimicked the biological efficacy of MLN4924 and led to accumulation of its substrate CDT1. MLN4924-induced DNA damage and apoptosis in OCCs were partly relieved by CDT1 depletion, implying that CRL4CDT2 inhibition and CDT1 accumulation were related to the genotoxic properties of MLN4924 [130]. Hong et al. found that neddylation inhibition with MLN4924 interrupted PTX-induced microtubule polymerization, and effectively neutralized PTX-mediated antimigration, antiproliferative and apoptotic effects in OCCs. Additionally, disrupting neddylation through silencing of UBC12/UBE2F reduced the anticancer activity of PTX in OCCs. However, in comparison to knockdown of UBC12 or UBE2F alone, downregulation of both NEDD8 E2s simultaneously did not reveal synergistic efficacy on PTX resistance in OCCs. These findings revealed that neddylation potentially regulated the mechanisms of PTX resistance in ovarian cancer [131].
The roles of neddylation in bladder cancer
Bladder cancer (BC) is a common urinary system malignancy, and over 90% of cases of bladder cancer are bladder urothelial carcinoma (UC) [132]. Huang's group explored the effects of MLN4924 on human UC cell lines (NTUB1, T24 and RT4). They found that MLN4924 exerted potent antiproliferative activity, suppressed migration and invasion as well as induced G2M phase arrest and apoptosis, along with activation of Bad, caspase-3/7, PARP and phospho-histone H2A.X, reduction of phospho-Bcl2 level. Importantly, MLN4924 exhibited evidently anticancer activity on xenografted UC tumors in SCID mice without obvious toxicity [133]. Continually, Tian et al. reported that NEDD8 was upregulated in BC tissues and associated with poor clinical outcomes of BC patients via analyzing the Cancer Genome Atlas (TCGA) database. Silencing of NEDD8 caused apoptosis and G2 phase arrest. In addition, NEDD8 inhibition with MLN4924 effectively suppressed proliferation, invasion and migration of BC cells in vitro and inhibited growth and metastasis of tumors in vivo [134].
The roles of neddylation in chondrosarcoma
Chondrosarcoma, as one of the most common primary bone malignancy, usually generates cartilage matrix, which has no response to conventional therapies [135]. MLN4924 revealed significant cytotoxicity against chondrosarcoma cells and caused cell apoptosis by activation of caspase-3/7. In addition, MLN4924 could inhibit cell proliferation through decreasing the phosphorylation of histone H3 to induce G2/M phase arrest. Besides, MLN4924 dramatically suppressed the growth of chondrosarcoma tumors in a xenograft mouse model, accompanied by the stimulation of ER stress-related apoptosis, indicating that neddylation inhibition with MLN4924 was an attractively therapeutic approach for chondrosarcoma [136].
The roles of neddylation in cervical carcinoma
Cervical cancer (CC) is the second most common cancer in women over the world. Huang's group found that MLN4924 suppressed human CC cell proliferation and induced apoptosis, followed by activation of apoptosis-related molecules, interference with cell cycle regulators and Bid phosphorylation interruption. Additionally, MLN4924 resulted in an endoplasmic reticulum stress response (CHOP, ATF-4 and caspase-4 activations) and expression of other cellular stress molecules (c-Jun and JNK activations). Moreover, MLN4924 not only inhibited the growth of CC xenografts in nude mice, but also improved cisplatin-induced growth inhibition of CC xenografts. Together, these results offered a rationale for clinical trials of MLN4924 in CC [137].
The roles of neddylation in pancreatic cancer
Jia's group uncovered that neddylation inhibition with MLN4924 suppressed angiogenesis, leading to the inhibition of tumor growth and metastasis in pancreatic cancer [39]. Mechanically, MLN4924 inhibited CRLs through cullin deneddylation, accompanied by the accumulation of tumor-related substrates of CRLs, which further caused the accumulation of RhoA, DNA damage response, cell cycle arrest and apoptosis. Additionally, the inactivation of CRLs through silencing of RBX1, was similar to antiangiogenic effect of MLN4924.
Based on the relationships between neddylation proteins and various cancers, we can observe that neddylation proteins are overactivated in various cancer types (Table 1), and neddylation inhibition through silencing of single target shows potential antitumor potency. In particular, the effects of neddylation proteins are mainly explored for lung cancer, liver cancer, glioblastoma, HNSCC, colorectal cancer, prostate cancer and ovarian cancer. In addition, although the potential effects of some neddylation proteins like NAE, NEDD8, RBX1 for the occurrence and development of many kinds of cancers have been clarified, the regulatory potency of other neddylation proteins like UBC12, UBE2F, DCN1 etc. in some cancers remains to be further explored. Moreover, even though targeting neddylation is a promising approach for cancers, it is still unknown which protein can be targeted for specific cancer treatment, accompanied by potent activity and low toxicity. Therefore, numerous efforts need to be conducted for investigating the therapeutical effects of neddylation proteins in different cancers, which may be beneficial for precision drug development.
The reported neddylation proteins are overactivated in different cancers.
| Neddylation proteins | Cancer type |
|---|---|
| NAE | lung cancer, liver cancer, leukemia, glioblastoma, HNSCC, melanoma, colorectal cancer, prostate cancer, breast cancer, esophageal cancer, gastric cancer, myeloma, lymphoma, ovarian cancer, bladder cancer, chondrosarcoma, cervical carcinoma, pancreatic cancer |
| NEDD8 | lung cancer, liver cancer, leukemia, glioblastoma, HNSCC, colorectal cancer, prostate cancer, breast cancer, esophageal cancer, gastric cancer, bladder cancer, oral squamous cell carcinoma |
| UBC12 | lung cancer, liver cancer, HNSCC, prostate cancer, breast cancer, esophageal cancer, gastric cancer, ovarian cancer |
| UBE2F | lung cancer, colorectal cancer, ovarian cancer |
| RBX1 | lung cancer, liver cancer, leukemia, melanoma, colorectal cancer, prostate cancer, breast cancer, esophageal cancer, gastric cancer, myeloma, lymphoma, ovarian cancer, bladder cancer |
| RBX2 | lung cancer, leukemia, glioblastoma, colorectal cancer, prostate cancer, ovarian cancer, pancreatic cancer |
| DCN1 | lung cancer, gastric cancer |
| Cul1 | lung cancer, liver cancer, HNSCC, melanoma, colorectal cancer, prostate cancer, breast cancer, gastric cancer |
| Cul2 | glioblastoma |
| Cul4A | liver cancer, melanoma, ovarian cancer |
| Cul4B | liver cancer, melanoma |
| Cul5 | glioblastoma, bowel cancer |
| Cul7 | liver cancer |
Role of neddylation on fibrotic diseases
Fibrotic diseases, such as advanced liver disease, pulmonary fibrosis, heart failure and kidney disease, are a major public health concern in the world. To date, none of examined treatment has been absolutely proved to delay or prevent the progress of fibrotic diseases. Thus, it is imperative to understand the potential pathogenesis of fibrosis in hope of discovering an effective therapeutic approach [138-140]. Recent years, targeting neddylation for the treatment of fibrotic diseases have been obtained more and more attention. In 2017, Imanol et al. firstly reported that neddylation was overactivated in clinical and two mouse models of liver fibrosis, including CCl4 chronic administration and bile duct ligation [24]. Treated with neddylation inhibitor MLN4924 effectively relieved liver injury, apoptosis, inflammation and fibrosis by targeting different hepatic cell types. Our latest report indicated that HZX-960, a DCN1-UBC12 interaction inhibitor, effectively relieved TGFβ-induced liver fibrotic responses, demonstrated by the decrease of collagen I and α-SMA expressions, and the increase of NRF2, HO-1 and NQO1 levels in two hepatic stellate cell lines (LX2 and HSC-T6). Similar effects were observed in CCl4-treated mice after HZX-960 treatment as well [141]. Continually, Wang and co-workers discovered that DCN1-UBC12 interaction inhibitor DI-1859 could protect mice from acetaminophen-induced liver injury, related to the inactivation of CRL3 and the accumulation of its substrate protein NRF2 [142]. Ge's group found that MLN4924 could significantly inhibit the occurrence and development of bleomycin-induced pulmonary fibrosis at the primary inflammatory stage. Mechanically, MLN4924 suppressed neddylation of CRL, resulting in abolishing NF-κB responses, inhibiting MAPK activity and decreasing the secretion of MCP1-induced chemokines and TNF-α-elicited pro-inflammatory cytokines [23]. Neddylation, as an emerging regulatory mechanism of cardiac development and pathological remodeling, is important for heart function and ventricular compaction [143, 144]. Our group observed that DCN1 protein was obviously upregulated in isolated cardiac fibroblasts treated by Angiotensin (Ang) II and in mouse hearts after pressure overload. However, inhibition of DCN1 effectively reversed Ang II-induced cardiac fibroblast activation, which was associated with the inhibition of cullin 3 neddylation and the accumulation of its substrate protein NRF2 [145]. Besides, we also found that treated with DCN1 inhibitor could potentially relieved liver fibrosis in CCl4-induced mouse model [141]. In contrast to the above findings, Lin and co-authors reported that the expression levels of UBE2M and UBE2F were evidently decreased in pancreatic tissues from chronic pancreatitis mice in comparison to control mice [146]. Besides, chronic pancreatitis mice administrated with MLN4924 displayed higher fibrosis-related gene expression, such as α-SMA and TGFβ, compared with the control group. These results reveal that targeting neddylation is a novel and promising therapeutic strategy for fibrotic diseases, which may be organ dependent.
Inhibitors of neddylation enzymes
Although neddylation enzymes are considered as potential targets for cancer treatment, most of them have not been targeted for drug design. Here, we comprehensively summarize the neddylation enzyme inhibitors, including NAE inhibitors, DCN1-UBC12 interaction inhibitors and Cul1/5 inhibitor. Specifically, we cover their clinical applications, biological activities, design strategy, binding modes and structure-activity relationship studies.
NAE inhibitors
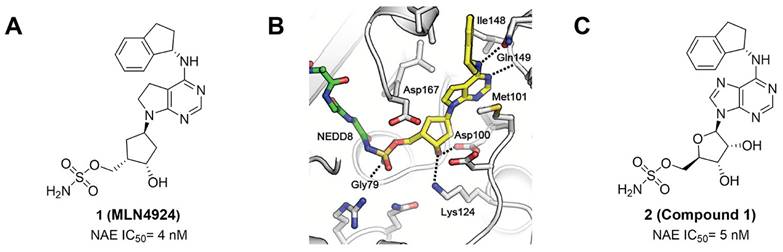
MLN4924 (Pevonedistat), discovered in 2009 through high throughput screening, could selectively bind to the NAE active site to form a covalent NEDD8-MLN4924 adduct that prevented the formation of E2-NEDD8 thioester, therefore potently blocking whole neddylation modification (Figure 5A) [30]. The co-crystal structure of NAE, NEDD8 and MLN4924 shows that there is a covalent NEDD8-MLN4924 adduct, which occupies the same sites as ATP and NEDD8 binding to the adenylation active site (Figure 5B). Especially, we observe several important interactions between MLN4924 and Gly79, Lys124, Asp100, Ile148 as well as Gln149 of NAE. These interactions are similar to those of NAE binding to the adenosine portion of ATP [147]. Considering numerous preclinical data that neddylation inhibition with MLN4924 displayed robust anticancer efficacy with well-tolerated toxicity, several phase I/II/III clinical trials (https://clinicaltrials.gov/) were performed to evaluate the safety, pharmacokinetics (PK), pharmacodynamics and anticancer efficacy of MLN4924 alone or in combinations with chemo-/radiotherapy against patients suffering from AML, MS, melanoma, lymphoma and solid tumors (Table 2). Notably, two phase III trials of the combination of MLN4924 with azacytidine have been launched in patients with AML, MS and CMML. Additionally, MLN4924, as a chemo-/radiosensitizer, can sensitize several types of cancer cells toward chemotherapy agents and radiotherapy with different mechanisms, as shown in Table 3 and 4.
(A) The chemical structure of MLN4924 [30]. (B) The co-crystal structure of NAE (white), NEDD8 (green) and MLN4924 (yellow) (PDB: 3GZN) [147]. Reproduced with permission [147]. Copyright 2010, Elsevier Inc. (C) The chemical structure of molecule 2 [147].
Clinical trials of MLN4924 tested for cancer treatment.
| Identifier | Phase | Cancer type | With combination | Status | Patientnumber | Initial time | Ref. |
|---|---|---|---|---|---|---|---|
| NCT00677170 | I | Solid tumor | Alone | Completed | 62 | 2008,03 | [152] |
| NCT00722488 | I | HM, MM, lymphoma, HL | Alone | Completed | 56 | 2008,07 | [153] |
| NCT00911066 | I | AML, MS, ALL | Azacitidine | Completed | 72 | 2009,03 | [154] |
| NCT01011530 | I | MM | Alone | Completed | 37 | 2009,11 | [155] |
| NCT01415765 | I/II | B-cell lymphoma | Alone | Withdrawn | - | 2011,07 | NR |
| NCT01814826 | I | AML | Azacitidine | Completed | 64 | 2013,03 | [156] |
| NCT01862328 | I | Solid tumor | Docetaxel, Gemcitabine, Carboplatin, Paclitaxel | Completed | 64 | 2013,05 | NR |
| NCT02122770 | I | Solid tumor | Fluconazole, Itraconazole, Docetaxel, Carboplatin, Paclitaxel | Completed | 52 | 2014,04 | NR |
| NCT02782468 | I | AML | Azacitidine | Not yet recruiting | 37 | 2016,03 | NR |
| NCT02610777 | II | MS, CMML, AML | Azacitidine | Not yet recruiting | 120 | 2016,04 | NR |
| NCT03013998 | I/II | AML | Azacitidine | Recruiting | 500 | 2016,11 | NR |
| NCT03057366 | I | Solid tumors | Docetaxel, Carboplatin, Paclitaxel | Completed | 8 | 2017,02 | NR |
| NCT03009240 | I | AML | Decitabine | Recruiting | 30 | 2017,08 | NR |
| NCT03238248 | I | MS, MN | Azacitidine | Recruiting | 71 | 2017,08 | NR |
| NCT03319537 | I/II | Mesothelioma | Pemetrexed, Cisplatin | Recruiting | 42 | 2017,10 | NR |
| NCT03323034 | I | Neoplasm, lymphoma | Temozolomide, Irinotecan | Recruiting | 76 | 2017,11 | NR |
| NCT03268954 | III | MS, CMML, AML | Azacitidine | Recruiting | 450 | 2017,11 | NR |
| NCT03330106 | I | Advanced solid neoplasm | Docetaxel, Carboplatin, Paclitaxel | Recruiting | 45 | 2017,11 | NR |
| NCT03228186 | I | NSCLS | Docetaxel | Recruiting | 37 | 2018,01 | NR |
| NCT03459859 | I | AML, MS | Cytarabine | Recruiting | 18 | 2018,03 | NR |
| NCT03479268 | I | CLL, Non-HL | Ibrutinib | Recruiting | 30 | 2018,03 | NR |
| NCT03386214 | I | Myelofibrosis | Ruxolitinib | Recruiting | 18 | 2018,04 | NR |
| NCT03330821 | I/II | AML | Cytarabine, Idarubicin | Recruiting | 53 | 2018,04 | NR |
| NCT03709576 | II | AML | Azacitidine | Recruiting | 30 | 2018,07 | NR |
| NCT03486314 | I | Advanced solid neoplasm | Rifampin, Docetaxel, Carboplatin, Paclitaxel | Recruiting | 20 | 2018,08 | NR |
| NCT03814005 | I | MS, CMML, AML | Azacitidine | Not yet recruiting | 20 | 2019,02 | NR |
| NCT03862157 | I/II | AML | Azacitidine, Venetoclax | Recruiting | 40 | 2019,02 | NR |
| NCT03349281 | I | ALL | Vincristine, Dexamethasone, PEGasparaginase, Doxorubicin | Recruiting | 18 | 2019,03 | NR |
| NCT03772925 | I | AML, MS | Belinostat | Not yet recruiting | 45 | 2019,03 | NR |
| NCT03745352 | I | AML | Azacitidine | Not yet recruiting | 72 | 2019,03 | NR |
| NCT03813147 | I | AML, MS | Azacitidine, Fludarabine, phosphate, Cytarabine | Not yet recruiting | 23 | 2019,05 | NR |
| NCT03770260 | I | Multiple myeloma | Ixazomib citrate | Not yet recruiting | 54 | 2019,07 | NR |
| NCT04090736 | III | AML | Azacitidine | Recruiting | 466 | 2019,08 | NR |
| NCT03965689 | II | NSCLC | Carboplatin, Paclitaxel | Recruiting | 25 | 2019,09 | NR |
| NCT04175912 | II | Advanced intrahepatic cholangiocarcinoma | Carboplatin, Paclitaxel | Suspended | 52 | 2020,01 | NR |
| NCT04172844 | I | AML | Azacitidine, Venetoclax | Recruiting | 24 | 2020,01 | NR |
| NCT04266795 | II | AML | Venetoclax, Azacitidine | Recruiting | 150 | 2020,10 | NR |
| NCT04712942 | II | MDS, AML | Azacitidine | Recruiting | 102 | 2021,01 | NR |
| NCT04800627 | I/II | dMMR/MSI-H cancers | Pembrolizumab | Recruiting | 39 | 2021,03 | NR |
| NCT04484363 | - | Myelodysplastic syndromes | Azacitidine | Available | - | - | NR |
NR, not reported
MLN4924 serves as a chemosensitizer.
| Drug | Cancer type | Mechanisms | Ref. |
|---|---|---|---|
| Cisplatin | Colon cancer | (1) Suppression of DNA damage-induced FANCD2 monoubiquitination and CHK1 phosphorylation. (2) Increases SUB1/PC4 expression in response to DNA damage, oxidative stress and apoptosis. | [157, 158] |
| Ovarian cancer | (1) Silencing of Cul3. (2) Promotes DNA damage, oxidative stress and expression of the BH3-only protein NBK/BIK. | [159, 160] | |
| Urothelial Carcinoma, | Promotes DNA damage, JNK activation and down-regulation of Bcl-Xl | [161] | |
| Cervical carcinoma, | NR | [162] | |
| Esophageal cancer | Induces apoptosis | [163] | |
| Pancreatic cancer | Promotes DNA damage and the expression of apoptosis-associated proteins through enhancing CDT1, ORC1, p27, p-IκBα and p21 accumulation. | [164] | |
| Triple-negative breast cancer | Increases the DNA damage | [165] | |
| Diffuse large B-cell lymphoma | NR | [166] | |
| Carboplatin | Ovarian cancer | NR | [167] |
| Azacitidine | AML | Inhibits RRM2 expression and induces apoptosis | [168] |
| Cytarabine | AML | (1) MLN4924 promotes cytarabine into the DNA of AML cells. (2) Increases DNA damage. | [169] |
| Belinostat | AML | Disables DNA damage response | [170, 171] |
| All-trans retinoic acid | AML | Induction of apoptosis is related to accumulation of pro-apoptotic proteins, NOXA and c-JUN. | [172] |
| Mitomycin C | Colon cancer, melanoma, lung cancer, osteosarcoma | Induces cell death | [173] |
| Bortezomib | MM | Induces apoptosis | [127] |
| Bendamustine, chlorambucil | CLL | Induces DNA damage, checkpoint activation, cell cycle arrest and apoptosis. | [174] |
| Olaparib | NSCLS | Impairs the DNA repair process | [175] |
| JQ1 | Pancreatic adenocarcinoma | Induces ROS that promotes the increased DNA damage, causing apoptosis. | [176] |
| Gemcitabine | Pancreatic ductal adenocarcinoma | Accumulation of the pro-apoptotic protein NOXA | [177] |
| MK-2206 | Breast cancer | Induces apoptosis | [178] |
| Imatinib | Leukemia | Induces DNA damage and triggers a dramatic shift in the expression of MCL1 and NOXA | [179] |
| Fulvestrant | Breast cancer | Targeting neddylation with MLN4924 can transcriptionally inhibit ER-α expression through SGK1-dependent nuclear export of FOXO3a. | [116] |
| Sorafenib | Hepatocellular carcinoma | Upregulation of CRL/Skp1-Cullin1-F-box E3 ubiquitin ligase substrates p21, p27, Deptor and IκBɑ | [180] |
| Tamoxifen | Breast cancer | Inactivates the FBXW2-MSX2-SOX2 axis | [181] |
MLN4924 serves as a radiosensitizer.
| Cancer type | Mechanisms | Ref. |
|---|---|---|
| Pancreatic cancer | Induces accumulation of CDT1 and WEE1 to trigger DNA damage response and G2/M arrest | [182] |
| Breast cancer | Induction of p21 and apoptosis-related gene, caspase-3 | [183, 184] |
| Colorectal cancer | Accumulation of p27 | [185] |
| Prostate cancer | (1) Accumulation of WEE1/p21/p27. (2) Induction of DNA damage and apoptosis. | [186] |
| Head and neck squamous carcinoma | Induction of re-replication | [97] |
Smith and Petroski et al. disclosed that resistance to MLN4924 in HCT116 cells was correlated with hetereozygous mutations in the NEDD8-binding cleft of NAEβ and the adenosine triphosphate binding pocket [32, 148, 149]. The specific mutations, including A171D, A171T, G201V, E204K, N209K and C324Y, caused decreased drug binding and slower rate of NEDD8-MLN4924 adduct formation. In contrast to MLN4924, compound 2 (Figure 5C), a N6-substituted adenosine sulfamate analog, was reported as a nonselective substrate-assisted inhibitor of canonical E1s and therefore was unsuitable for clinical application [147, 150]. However, the NEDD8-compound 2 adduct bound more tightly to wild-type NAEβ and all mutant NAE enzymes than NEDD8-MLN4924 adduct in HCT116 cells, indicating that compound 2 might be a stronger inhibitor of mutant E1 enzyme in comparison to MLN4924. Continually, Xu et al. found two resistant R-K562MLN and R-U937MLN leukemia cells containing I310N and Y352H mutations in the NAE catalytic subunit UBA3 separately [151]. These mutations led to the decreased efficacy of MLN4924, whereas offering necessary NAE function for leukemia cell growth. Surprisingly, the two MLN4924-resistant cells kept sensitive to compound 2, probably due to its inhibitory effects on other E1 enzymes. Liu and co-workers reported that neddylation inhibition with MLN4924 was effective toward BCR-ABL mutational imatinib-resistant cells, and the self-renewal capacity of leukemia stem cells (LSCs) in CML. Biologically, MLN4924 caused CML cell G2/M phase arrest and apoptosis, regardless of their T315I mutation status in BCR-ABL. In addition, MLN4924 suppressed survival and self-renewal of human CML CD34+ cells, and inhibited LSCs in CML-bearing mice through accumulation of p27kip1 [85].
Given that MLN4924 and compound 2 with similar structures possessed significant difference in their specificity for NAE, Lukkarila and co-workers next investigated the determinants of NAE selectivity based on compound 2 [187]. Compounds with substituents of various size, flexibility and functionality in the purine C6 position were synthesized and evaluated against E1 enzymes NAE, ubiquitin activating enzyme (UAE) and SUMO-activating enzyme (SAE), resulting in compound 3 tethering an n-hexyl group, which showed the strongest binding affinity against NAE (IC50< 10 nM) in tested compounds, and was highly selective for NAE over UAE and SAE (Figure 6). The structure-activity relationships (SARs) indicated that introduction of N-alkyl groups into the purine C6 position was beneficial for NAE specificity, whereas bulky or secondary N-alkyl substituents were not tolerated. The predicted binding modes reveals that the alkyl group of compound 3 well occupies the hydrophobic region formed by Ile148, Gln149, Ile170 and Trp174 (Figure 6A). Besides, analogue 3 exhibited potent antiproliferative activity toward K562 leukemia cells with the IC50 value of 160 nM, slightly weaker than that of MLN4924 (IC50= 108 nM). Based on compound 2, Statsyuk's group developed a pan-E1 inhibitor 4 (Figure 6), which could decrease the expression levels of Ub, NEDD8, SUMO1/2/3 and Ufm1 conjugates in A549 cells. In contrast, compound 5 was reported as a dual inhibitor of ubiquitin- and NEDD8-activating E1 enzymes, and showed modest cytotoxicity against A549 cells with the IC50 value of 2.5 μM (Figure 6). Additionally, compound 5 could effectively induce A549 cell apoptosis, as demonstrated by the detection of cleaved PARP and Annexin V-positive cells [188, 189].
(A) The predicted binding modes of compound 3 (green) with NAE (white) [187]. Reproduced with permission [187]. Copyright 2011, American Chemical Society. (B) The chemical structures of MLN4924 analogues 3-7 [188-191].
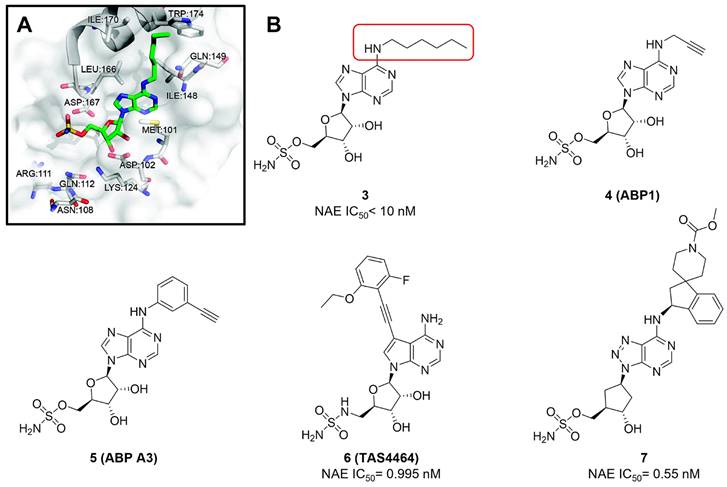
From library screening and structure-based design, compound TAS4464 (6) was identified and displayed more potent and more selective against NAE than MLN4924 (Figure 6) [190]. Compound 6 could effectively inhibit cullin neddylation, further inducing the accumulation of CRL substrates. Additionally, treated with compound 6 showed strongly and broadly antiproliferative activity toward cancer cell lines as well as patient-derived tumor cells, and revealed a wider therapeutic index in comparison to MLN4924. Meanwhile, Ohkubo's group reported that compound 6 could cause growth arrest and cell death in tested 14 multiple myeloma cell lines [192]. It also impaired the activities of NF‐κB transcription factors p65 and RelB and reduced the expression of NF‐κB target genes, indicating that derivative 6 inhibited both the canonical and non-canonical NF‐κB pathways. Moreover, compound 6 synergistically improved the anticancer efficacy of MM chemotherapies bortezomib, daratumumab, elotuzumab and dexamethasone. Considering these favorable preclinical findings, a phase 1 study was conducted to explore the efficacy of compound 6 in patients with advanced/metastatic solid tumors [193]. The results indicated that abnormal liver function test (LFT) changes and gastrointestinal effects were the most common adverse events (AEs). Dose-limiting toxicities with 56-mg/m2 weekly dosing occurred in 1/5 patients; five patients possessed grade ≥ 2 abnormal LFT changes. These severe AEs terminated the applications of compound 6 in clinical trial.
Recently, a structural hopping strategy was performed for optimizing the deazapurine skeleton and the solvent interaction region of MLN4924, providing molecule 7 tethering a pyrimidotriazole scaffold (Figure 6) [191]. Compound 7 was a potent NAE and UAE dual inhibitor with the IC50 values of 0.55, 66.84 nM separately. Additionally, derivative 7 possessed robust antiproliferative activity against ten tested cancer cell lines with submicromolar activity. Besides, analogue 7 displayed higher plasma concentrations and better PK properties in mice compared to its parental compound MLN4924. Mechanically, treated with compound 7 effectively suppressed the neddylation pathway and increased CRL substrates. Importantly, molecule 7 revealed potent antitumor potency and a good safety profile in MV-4-11 and HCT-116 xenograft models.
With the aim of identification of novel covalent NAE inhibitors, Zhang et al. conducted a combined virtual screen strategy through using both structure-based pharmacophore modelling and covalent docking, resulting in the discovery of sulfamoyl-substituted compounds 8-10 with the IC50 values of 1.06, 9.18, 19.71 μM separately (Figure 7A) [194]. The docking studies exhibits that the sulfamoyl moiety of three molecules can form covalent bonds with Gly79 of NAE separately (Figure 7B-D), similar to the binding modes of MLN4924. In particular, compound 8 presented potent in vitro anticancer activity toward Bel-7402, Caco-2 and MCF-7 cells with the IC50 values ranging from 12.3 to 29.5 μM. Taken together, these novel NAE inhibitors can be regarded as hit compounds for further optimization.
(A) The chemical structures of sulfamoyl-substituted analogues 8-10. (B-D) The corresponding binding modes of compounds 8-10 with NAE, respectively [194]. Reproduced with permission [194]. Copyright 2014, American Chemical Society.
Compound 11 was found as a new NAE inhibitor via virtual screening using a chemical library of natural product and natural product-like structures (Figure 8) [195]. It revealed modest binding activity on NAE with the IC50 value of 20 μM. Treated with compound 11 could effectively inhibit NAE activity in colorectal adenocarcinoma Caco-2 cells with the concentration of 25 μM. In contrast to MLN4924, molecule 11 cannot form a covalent adduct with NEDD8. Similarly, Leung et al. discovered the dipeptide-conjugated deoxyvasicinone analogue 12-1 as a noncovalent NAE inhibitor through virtual screening of about 90,000 molecules from the ZINC database of natural products (Figure 8) [196]. Densitometry analysis indicated that analogue 12-1 significantly suppressed NAE activity with the IC50 value of 0.8 μM. Also, compound 12-1 was highly selective for NAE over analogous E1 enzymes UAE and SAE at the concentration of 12.5 μM. Besides, derivative 12-1 showed moderate antiproliferative activity against Caco-2 cells (IC50= 10 μM, 48 h), which might be partly associated with the suppression of NAE activity, further causing the accumulation of CRL substrates like p27kip1. Encouraged by the efficacy of compound 12-1, Leung's group used the pharmacophore screening to further identify analogues 12-2 and 12-3 as more potent NAE inhibitors with the IC50 values of 0.39, 0.27 μM, respectively [197]. 12-2 and 12-3 showed similarly biological profiles compared with molecule 12-1. Compounds 13 and 14 (Figure 8), as naturally occurring chalcone discovered in the Kava plant, were new NAE inhibitors and suppressed NEDD8 to both Cul1 and UBE2M in prostate cancer cells and UBE2M neddylation [198, 199]. Molecular docking study and the cellular thermal shift assay revealed that both compounds could directly bind to the NAE regulatory subunit APP-BP1, which was responsible for anti-prostate cancer activity. Mechanically, they interacted with APP-BP1, leading to the deneddylation of Cul1 and UBE2M, the decreased activity of SCFSKP2 complex and SKP2 ubiquitination and degradation, as well as the activation of caspase mediated apoptotic pathway. Gartanin (15), a 4-prenylated xanthone, was identified from the purple mangosteen fruit, and showed potential enzymatic activity on NAE with the predicted IC50 value of 0.735 μM (Figure 8) [200]. In-depth mechanism research indicated that compound 15 could deprive NEDD8 from UBC12 and Cul1, which changed the specific SCF ubiquitin ligases and finally resulted in cell growth inhibition and autophagy.
Zhong et al. designed and synthesized a novel class of rhodium(III) complexes as NAE inhibitors, resulting in molecule 16 (R1, R2, R3= H) with the IC50 value of 1.5 μM (Figure 9) [201]. The SARs showed that replacement of the hydrogen at R1, R2 and R3 with other substituents caused weaker inhibitory activity. Also, the metal center played a critical role for recognizing protein binding site. Derivative 16 exerted potent antitumor effects on Caco-2 cells with the IC50 value of 0.3 μM, ten folds stronger than that of MLN4924 in a parallel experiment, which might be related to the inhibition of CRL substrate degradation, increase of p27 protein level and reduction of NF-kB pathway. Continually, Zhong and co-authors reported the rhodium(III) complex [Rh(phq)2(MOPIP)]+ (17) as a potent and selective ATP-competitive NAE inhibitor with the IC50 value of 0.1 μM using an AlphaScreen assay, about 15 folds more active in comparison to compound 16 (Figure 9) [202]. In addition, compound 17 could inhibit Caco-2 cell proliferation with the IC50 value of 4.3 μM. The preliminary SARs study indicated that the 1H-imidazo[4,5-f][1,10]phenanthroline moiety and methoxy group in compound 17 were beneficial for binding affinity. Besides, changing the Rh(III) center of 17 to Ir(III) dramatically decreased NAE inhibitory activity. Furthermore, complex 17 could block the degradation of CRL substrates NRF2 and CDT1, partly linked to NAE inhibition.
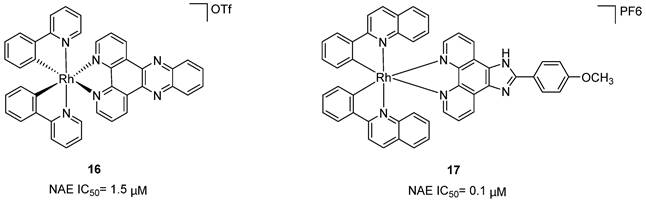
Through the structure-based virtual screening using an FDA-approved drug library of around 3000 molecules, Zhong et al. identified piperacillin (18) as an effective NAE inhibitor with the IC50 value of 1 μM (Figure 10A) [203]. Compound 18 exhibited highly selective for NAE over SAE and ubiquitin E2 enzyme UBC10 in Caco-2 cells. Also, treated with 18 caused the accumulation of CRL substrate p27kip1. The docking study presents that the binding pose of molecule 18 in NAE overlaps markedly with that of ATP (Figure 10B), indicating that 18 may serve as a reversible ATP-competitive inhibitor of NAE. Mitoxantrone (19) was also reported as an ATP-competitive reversible inhibitor of NAE. It could inhibit NAE-mediated UBC12-NEDD8 conjugation in Caco-2 cells with the IC50 value of 1.3 μM (Figure 10C) [204]. In addition, compound 19 displayed higher selectivity against NAE over SAE and UAE. The molecular docking study reveals that the hydroxyl group of 19 makes a hydrogen bond interaction with Thr103 (Figure 10D). One of the carbonyl groups acts as a hydrogen bond acceptor with Gln112, whereas the other carbonyl group interacts with the sidechain of Lys307. Besides, one of the alkyl chains of derivative 19 extends out the solvent region, while the other is located in the binding pocket. Biologically, analogue 19 induced Caco-2 cell apoptosis via regulation of cullin-dependent substrates.
(A) and (C) The chemical structures of FDA-approved drugs 18-19. (B) The binding modes of compound 18 (yellow) with NAE (green). Reproduced with permission [203]. Copyright 2014, Elsevier Masson SAS. (D) The binding modes of compound 19 (yellow) with NAE (green) [203, 204]. Reproduced with permission [204]. Copyright 2018, Elsevier Masson SAS.
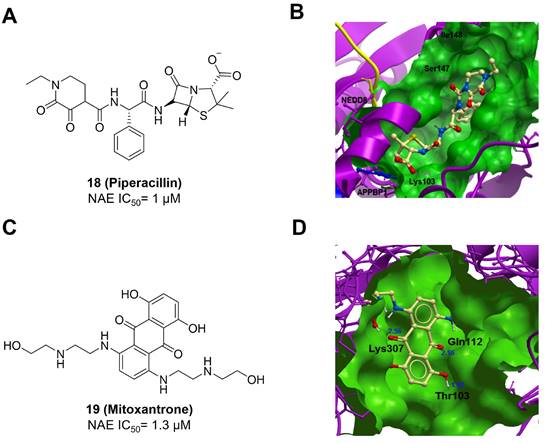
To discover new NAE inhibitors, Jia's group screened 1331 approved drugs by evaluating the Cul1-NEDD8 inhibition, yielding the hit compound candesartan cilexetil (20) with the IC50 value of 16.43 μM (Figure 11) [205]. Compound 20 bearing a benzimidazole moiety could obviously inhibit A549 cell proliferation with the IC50 value of 63.93 μM in CCK-8 cell viability assay and induce A549 cell apoptosis. Besides, molecule 20 not only suppressed neddylation pathway through determining the formations of Cul1-NEDD8 and UBC12-NEDD8 adducts, but ATP-competitively inhibited neddylation modification. The antitumor activity in vivo presented that molecule 20-treated with 60 or 30 mg/kg group effectively inhibited the growth of A549 xenograft tumors. To overcome the defect of structurally easy hydrolysis and enhance the antitumor efficacy of derivative 20, further structural modifications were performed via focusing on the region A, B and C of compound 20, providing 42 new derivatives with a benzimidazole skeleton [206]. The SARs indicated that the benzimidazole and tetrazole groups were beneficial for neddylation inhibitory activities. The introduction of (cyclo) alkyl, benzoalicyclic and heterocyclic groups into region C caused decreased activities, whereas the replacement of phenyl groups improved biological activities. Among synthesized compounds, molecule 21 (Figure 11) possessed the strongest neddylation inhibitory activity with the IC50 value of 5.51 μM, about 3 folds more active than compound 20. Additionally, analogue 21 showed potent antiproliferative activity toward A549 cells with the inhibition rate of 83.9% at 100 μM. Besides, molecule 21 could induce A549 cell apoptosis and senescence. Notably, compound 21 not only inhibited the growth of A549 xenograft tumors on administration of intraperitoneal injection (30 mg/kg), but also displayed potent anticancer potency via oral administration (60 mg/kg).
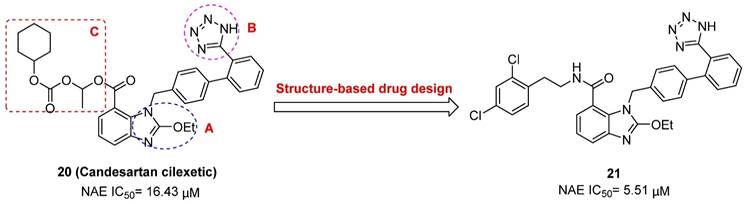
Lu and co-workers performed structure-based virtual screening of a compound library comprising 50,000 molecules, resulting in the identification of compound 22 (Figure 12) as a selective and reversible NAE inhibitor [207]. Next, a dose-dependent decrease of the intensity of UBC12-NEDD8 bands was determined after compound 22 treatment. The results indicated that treated with molecule 22 with the concentration of 3.33 μM evidently suppressed the formation of UBC12-NEDD8. Additionally, derivative 22 exerted moderate in vitro anticancer effects on Caco-2, SK-OV-3, K562, AGS, and BXPC-3 cell lines with the GI90 values of 12.7, 13.2, 8.98, 14.9, and 26.6 μM separately. Besides, analogue 22 blocked neddylation and decreased the degradation of related substrates, leading to cell apoptosis. Furthermore, compound 22 effectively inhibited tumor growth in the nude mice xenograft model without obvious toxicity in the zebrafish model.
The chemical structures and design strategy of chromen-based NAE inhibitors 22-24 [207-209].
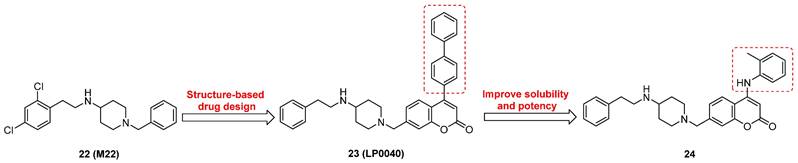
Based on compound 22, a new class of 4-substituted 2H-chromen-2-one analogues were designed and synthesized through mainly focusing on the 4-position of 2H-chromen-2-one [208]. In comparison to molecule 22, most synthesized compounds displayed increased antiproliferative activity toward A549, K562, HCT116, SKOV-3, BxPC-3 and AGS cells with micromolar activity. The SARs studies disclosed that 4-hydrophobic substituents could benefit inhibitory activity, while 4-hydrophilic substituents decreased activity. Additionally, introduction of ethynyl groups into 4-position of 2H-chromen-2-one contributed to the enhancement of the antiproliferative activity. Especially, compound 23 (Figure 12) bearing a biphenyl group indicated the strongest biological activity against AGS cells (IC50= 0.76 μM) and low toxicity toward normal HEK293T, HUVEC and L-02 cells with the LC50 values of 12.93, 10.69, 13.54 μM separately. Derivative 23 was found as a NAE/UAE dual inhibitor and could inhibit the formation of UBC12-NEDD8 at the concentration of 0.37 μM. Besides, compound 23 effectively induced AGS cell G1 phase arrest and apoptosis.
Continually, to improve water solubility of compound 23, Lu et al. introduced various aliphatic amino or aromatic amino into the 4-position of 2H-chromen-2-one, offering a new class of 2H-chromen-2-one analogues [209]. In particular, compound 24 (Figure 12) exerted the strongest cytotoxicity on BxPC-3 cells with the IC50 value of 0.17 μM, more potent than compound 23 (IC50= 2.8 μM), and also indicated low toxicity toward normal cells. Compared to analogue 23, compound 24 tethering a 4-aromatic amino group revealed better solubility at 340.73 μg/mL and its Log P value was 1.66, implying that it possessed a reasonable druglike property. Additionally, molecule 24 was a promising ATP-dependent NAE inhibitor and inhibited the formation of UBC12-NEDD8 at 0.56 μM. Moreover, derivative 24 effectively induced BxPC-3 cell apoptosis and played a synergistic effect with bortezomib on BxPC-3 cell growth inhibition.
Miao's group conducted a target-based virtual screening to discover new NAE inhibitors, leading to compound 25 featuring a benzothiazole skeleton (Figure 13A) [210]. Derivative 25 possessed strongly antiproliferative activity on HCT116 and U-2OS cells with the IC50 values of 2.12, 3.19 μM, respectively. Next, two round structural optimizations were performed via focusing on the non-hydrogen bonding sides of compound 25, yielding compound 26, which displayed significantly increased cytotoxicity toward HCT116 and U-2OS cells with the IC50 values of 0.1, 1.22 μM separately. The docking study indicates that both compounds make hydrogen bond interactions with Asp100, Ile148, Gln149 and Asp167 (Figure 13B-C). Biologically, derivative 26 could effectively suppress NEDD8 protein and subsequently elevate the UBC12 level.
(A) The chemical structures of benzothiazole derivatives 25-26. (B) Docking modes of analogues 25 (B) and 26 (C) with NAE [210]. Reproduced with permission [210]. Copyright 2017, Elsevier Masson SAS.
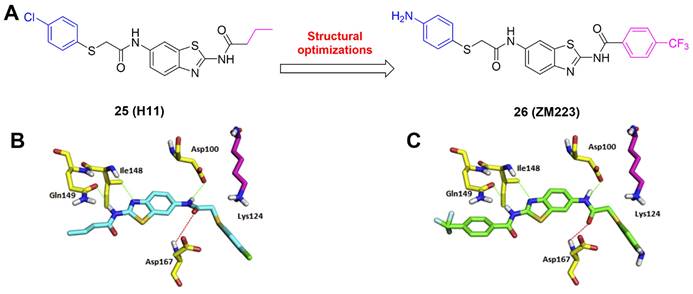
Zhang's group screened their in-house structurally diverse molecular library, providing triazine-based compound 27 as a hit for NAE inhibition (Figure 14A) [211]. Further modifications were focused on the 3-position of triazine moiety in 27 via introducing various aromatic groups and indole groups. Afterwards, molecule 28 featuring an indole group was obtained and displayed modest anticancer activity toward MGC803, PC3, MCF-7 cells with the IC50 values of 8.22, 9.28, 9.64 μM separately. Compound 28 evidently inhibited the activity of NAE, demonstrated by the decrease of UBC12-NEDD8 conjugation in MGC803 cells with the EC50 value of 3.56 μM. The molecular docking indicates that derivative 28 forms hydrogen bonds with Lys124, Ile148 and Gly165, respectively (Figure 14B). Additionally, the indole ring makes strongly electrostatic interactions with the surrounding residues Gly79, Arg111, Gln112 and Asn317, and hydrophobic interactions with Leu80, Thr203, Ile310 and Ala312. Besides, analogue 28 caused MGC803 cell G2/M phase arrest and induced apoptosis. Continually, Zhang's group synthesized a new array of indole-based compounds mainly through the modifications of rings A and B (Figure 14C) [212]. Particularly, compound 29 exhibited the strongest biological activity toward MGC803, PC3, EC109 cells with the IC50 values of 1.59, 3.56, 14.52 μM separately. Also, molecule 29 interacted with NAE regulatory subunit to suppress UBC12 neddylation in MGC803 cells. The docking study discloses that compound 29 well occupies the active cavity of NAE (Figure 14D). Hydrogen bond interactions are formed between 4-methoxyl-phenyl group and Ile148, 3,4,5-trimethoxyl-phenyl group and Serl68. The 5-methoxy-indole group also makes hydrogen bond interactions with Leu80 and Lys124. Mechanism research indicated that analogue 29 not only resulted in MGC803 cell G2/M phase arrest, but also inhibited neddylation and MAPK pathways, further leading to activated extrinsic and intrinsic apoptosis pathways.
(A) The chemical structures and design strategy of indole-based NAE inhibitors 27-28. (B) Docking modes of compound 28 with NAE. Reproduced with permission [211]. Copyright 2020, Elsevier Ltd. (C) The chemical structure of indole-based NAE inhibitor 29. (D) Docking modes of compound 29 with NAE [211, 212]. Reproduced with permission [212]. Copyright 2020, Elsevier Inc.
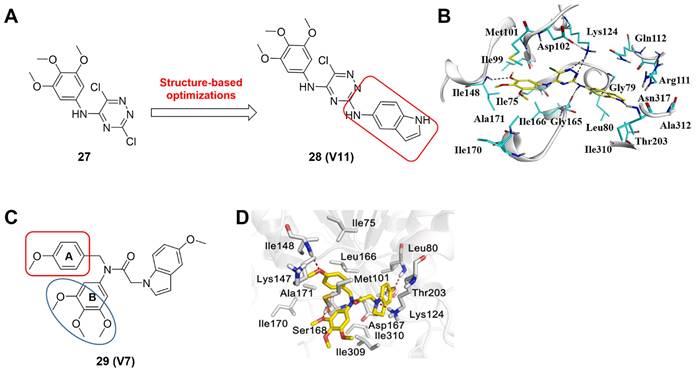
In 2021, Sun's group identified the NAE inhibitor 30 (HA-1141) through virtual screening and further modifications, which significantly suppressed neddylation of cullins 1-5 (Figure 15A) [213]. The docking study shows that compound 30 well occupies the active pocket consisting of amino acids Gln 442, Thr405, Thr403, Asn410, Leu 456 and Thr454 (Figure 15B). Especially, derivative 30 forms two critical hydrogen bonds with Thr403 and Thr405, respectively. Additionally, a halogen bond is found between Gln442 and a fluorine atom in compound 30. In contrast to MLN4924, compound 30 could effectively inhibit the growth of lung cancer cells in vitro and tumor growth in vivo xenograft lung cancer models via suppressing neddylation and triggering ER stress.
(A) The chemical structure of urea derivative-based NAE inhibitor 30. (B) Docking modes of compound 30 with NAE [213]. Reproduced with permission [213]. Copyright 2021, Elsevier B.V.
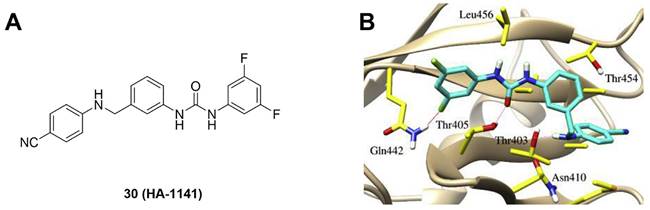
Considering that neddylation affecting many pathological processes, NAE activators also have been reported recent years. In 2020, Fu et al. found a new series of amide derivatives, indicating potent antiproliferative activity toward MGC803, MCF-7 and PC-3 cell lines [214]. In particular, molecule 31 displayed the strongest effects on inhibiting MGC803 growth with the IC50 value of 94 nM (Figure 16). Further evaluation revealed that compound 31 not only caused MGC803 cell arrest at G2/M phase, but induced MGC803 cell apoptosis through intrinsic and extrinsic pathways. Further mechanism study showed that compound 31 could activate NAE1-UBC12-Cullin1 neddylation through binding NAE protein straightly, leading to the degradation of anti-apoptosis protein IAPs. Besides, compound 31 could significantly suppress tumor growth in xenograft models without noticeable toxicity in vivo. Compound 31 was the first molecule reported for activating NAE. Continually, their group developed a new class of 1,2,4-triazine-dithiocarbamate derivatives as anti-cancer agents in 2021, of which the representative compound 32 (Figure 16) possessed the most potent cytotoxicity on MGC-803, PC-3 and EC-109 cell lines with the IC50 values of 2.35, 5.71 and 10.1 μM separately, more active compared to the positive control 5-Fu [215]. In addition, compound 32 could enhance the neddylation of MGC-803 and HGC-27 cell lines through activating NAE, causing the degradation of anti-apoptosis proteins c-IAP and YAP/TAZ, which further induced cell growth inhibition and apoptosis. These findings indicates that the development of NAE activators may be also a new strategy for cancer treatment.
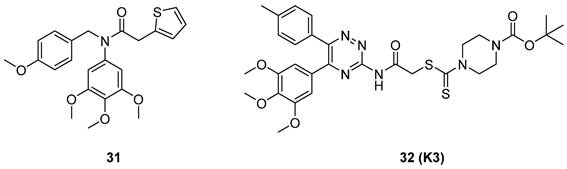
Overall, NAE modulators of different structural types have been reported, including N6-substituted adenosine sulfamate derivatives, sulfamoyl-substituted derivatives, flavonoid derivatives, chalcone derivatives, 4-prenylated xanthone derivatives, rhodium (III) complexes, beta-lactam derivatives, anthracenedione derivatives, benzimidazole derivatives, 2H-chromen-2-one derivatives, benzothiazole derivatives, indole derivatives and urea derivatives (Table 5). These modulators were developed mainly through high throughput virtual screening, structure-based drug design, structural hopping strategy, pharmacophore screening, structure-based virtual screening and drug repurposing. Generally, these reported NAE inhibitors revealed potent anticancer activity. Compared with other NAE inhibitors, the most N6-substituted adenosine sulfamate derivatives could covalently target NAE active site, and indicated stronger NAE inhibitory activity with low nanomolar level (Figure 5-6). However, due to the hetereozygous mutations (A171D, A171T, G201V, E204K, N209K and C324Y) in the NEDD8-binding cleft of NAEβ and the adenosine triphosphate binding pocket, these adenosine sulfamate-based NAE inhibitors partly occurred to resistance in some cancer cells. The development of new NAE inhibitors targeting those mutations may be a method for overcoming resistance. It is worth noting that Rh(III) complex 16-17 also showed obvious NAE binding activity. The specific structural features of the octahedral coordination geometry of the Rh(III) complex 16-17 made them form beneficial interactions with NAE, probably further resulting in enhanced activity and selectivity toward NAE over other closed-related E1 enzyme [201, 202]. In addition to NAE inhibitors, Zhang's findings concerning NAE activators delivered us a new prospect for activating neddylation pathway in cancer treatment [214, 215]. Besides, although numerous clinical trials are undergoing for exploring the anticancer effects of NAE inhibitor MLN4924, it is unclear whether NAE is an effective target for diseases clinically. Thus, more potent and selective NAE inhibitors with low toxicity and novel chemotypes remain to be urgently needed for investigating the clinical efficacy of targeting NAE.
A summary of the reported NAE modulator.
| Compound ID | Structure | Year reported | Biological functions | Reference |
|---|---|---|---|---|
| 1 (MLN4924) |  | 2009 | A selective and covalent NAE inhibitor (IC50= 4 nM) The detailed biological functions of MLN4924 in cancers were introduced in the part 3 of this manuscript. | [30] |
| 2 (compound 1) |  | 2010 | An equipotent inhibitor of NAE, UAE, and SAE-dependent transthiolation reactions with IC50 values of 5 nM | [147, 150] |
| 3 | 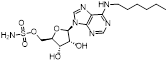 | 2011 | A selective and covalent NAE inhibitor (IC50 < 10 nM) Potent antiproliferative activity toward K562 leukemia cells (IC50= 160 nM) | [187] |
| 4 (ABP1) |  | 2013 | ABPQ, a nonselective and covalent E1 inhibitor, could decrease the expression levels of Ub, NEDD8, SUMO1/2/3 and Ufm1 conjugates in A549 cells. | [188] |
| 5 (ABP A3) |  | 2015 | ABP A3, a dual inhibitor of Ub and NAE inhibitor, possessed potent anti-cancer activity toward A549 cells (IC50= 2.5 μM), and could induce A549 cell apoptosis. | [189] |
| 6 (TAS4464) |  | 2019 | A selective and covalent NAE inhibitor (IC50= 0.955 nM) TAS4464 showed extensively anticancer activity, and revealed a wider therapeutic index than MLN4924. However, owing to the potential hepatic and gastrointestinal toxicity in the phase 1 study, TAS4464 was terminated in the subsequent clinical evaluation. | [190, 192, 193] |
| 7 |  | 2021 | A potent NAE and UAE dual inhibitor with the IC50 values of 0.55, 66.84 nM separately. molecule 7 revealed potent antitumor potency and a good safety profile in MV-4-11 and HCT-116 xenograft models. | [191] |
| 8 (LZ3) |  | 2014 | A covalent NAE inhibitor (IC50= 1.06 μM), Caco-2 cells (IC50= 12.3 μM) | [194] |
| 9 (LZ6) | 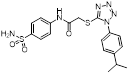 | 2014 | A covalent NAE inhibitor (IC50= 9.18 μM) | [194] |
| 10 (LZ7) | 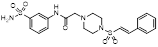 | 2014 | A covalent NAE inhibitor (IC50= 19.71 μM) | [194] |
| 11 (6,6''-biapigenin) | 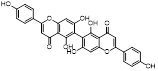 | 2011 | A non-covalent NAE inhibitor (IC50= 20 μM), Caco-2 cells (IC50= 25 μM) | [195] |
| 12-1 | 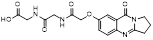 | 2012 | A selective and non-covalent NAE inhibitor (IC50= 0.8 μM), Caco-2 cells (IC50= 10 μM) | [196] |
| 12-2 |  | 2015 | A selective and non-covalent NAE inhibitor (IC50= 0.39 μM) | [197] |
| 12-3 | 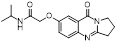 | 2015 | A selective and non-covalent NAE inhibitor (IC50= 0.27 μM) | [197] |
| 13 (Flavokawain A) | 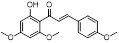 | 2015 | A non-covalent NAE inhibitor (IC50= 5 μM) Flavokawain A inhibited the formation of high-grade prostatic intra-epithelial neoplasia lesions and prostate adenocarcinomas. | [199] |
| 14 (Flavokawain B) | 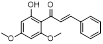 | 2019 | A non-covalent NAE inhibitor (IC50= 11 μM) Flavokawain B's inhibitory activity toward prostate cancer cells is owing to FKB's binding to the NAE regulatory subunit APP-BP1. | [198] |
| 15 (Gartanin) | 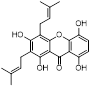 | 2019 | A non-covalent NAE inhibitor (IC50= 0.735 μM) Gartanin effectively inhibited the growth of prostate cancer lines through autophagy initiation. | [200] |
| 16 |  | 2012 | A non-covalent NAE inhibitor (IC50= 1.5 μM), Caco-2 cells (IC50= 0.3 μM) Compound 16 suppressed CRL substrate degradation and NF-kB activation in Caco-2 cells. | [201] |
| 17 |  | 2016 | A non-covalent NAE inhibitor (IC50= 0.1 μM), Caco-2 cells (IC50= 4.3 μM) Complex 17 indicated promising effect on the treatment of inflammatory bowel disease. | [202] |
| 18 (Piperacillin) |  | 2014 | A selective and non-covalent NAE inhibitor (IC50= 1 μM) Compound 18 could effectively inhibit the degradation of NAE downstream protein substrate p27kip1. | [203] |
| 19 (Mitoxantrone) |  | 2018 | A selective and non-covalent NAE inhibitor (IC50= 1 μM) Analogue 19 could induce Caco-2 cell apoptosis through regulation of cullin-dependent substrates p53. | [204] |
| 20 (Candesartan cilexetic) | 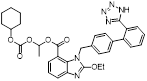 | 2020 | A non-covalent NAE inhibitor (IC50= 16.43 μM), A549 cells (IC50= 63.93 μM) Compound 20-treated with 60 or 30 mg/kg group effectively inhibited the growth of A549 xenograft tumors. | [205] |
| 21 | 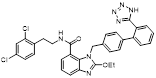 | 2021 | A non-covalent NAE inhibitor (IC50= 5.51 μM), A549 cells (Inhibition rate= 83.9%, 100μM) Compound 21 not only could induce A549 cell apoptosis and senescence, but also inhibit the growth of A549 xenograft tumors. | [206] |
| 23 (LP0040) | 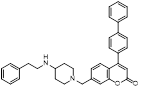 | 2018 | A NAE/UAE dual inhibitor (IC50= 0.37 μM), AGS cells (IC50= 0.76 μM) Compound 23 effectively induced AGS cell G1 phase arrest and apoptosis. | [208] |
| 24 | 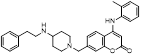 | 2020 | A non-covalent NAE inhibitor (IC50= 0.56 μM), BxPC-3 cells (IC50= 0.17 μM) Derivative 24 induced BxPC-3 cell apoptosis and played a synergistic effect with bortezomib on BxPC-3 cell growth inhibition. | [209] |
| 26 (ZM223) |  | 2017 | A non-covalent NAE inhibitor, HCT116 cells (IC50= 0.1 μM) U-2OS cells (IC50= 1.22 μM) | [210] |
| 28 (V11) | 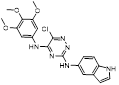 | 2020 | Compound 28 evidently inhibited the activity of NAE in MGC803 cells (EC50= 3.56 μM). Molecule 28 displayed modest anticancer activity toward MGC803, PC3, MCF-7 cells with the IC50 values of 8.22, 9.28, 9.64 μM separately. | [211] |
| 29 (V7) | 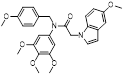 | 2021 | A non-covalent NAE inhibitor Compound 29 exhibited potent biological activity toward MGC803, PC3 and EC109 cells with the IC50 values of 1.59, 3.56, 14.52 μM. Induction of MGC803 cell G2/M phase arrest and apoptosis | [212] |
| 30 (HA-1141) |  | 2021 | A non-covalent NAE inhibitor Compound 30 possessed the dual activities of blocking neddylation and triggering ER stress, leading to growth suppression of cancer cells. | [213] |
| 31 | 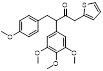 | 2020 | An NAE activator Induction of MGC803 cell G2/M phase arrest and apoptosis Compound 31 could activate NAE1-UBC12-Cullin1 neddylation through binding NAE straightly, leading to the degradation of anti-apoptosis protein IAPs. | [214] |
| 32 (K3) | 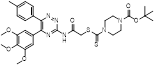 | 2021 | An NAE activator Compound 32 displayed potent cytotoxicity on MGC-803, PC-3 and EC-109 cell lines with the IC50 values of 2.35, 5.71 and 10.1 μM, separately. Induction of MGC-803 and HGC-27 cell growth inhibition and apoptosis | [215] |
Inhibitors targeting DCN1-UBC12 interaction
Among the five DCN isoforms, human DCN1 is highly amplified in several tumors and essential in tumor formation, maintenance and regulation of neddylation pathway [216, 217]. Biologically, DCN1, a scaffold-like co-E3 ligase, promotes the formation of multi-protein complex, while UBC12 facilitates cullin neddylation by recruiting DCN1. Targeting DCN1-UBC12 interaction could selectively regulate cullin neddylation. Structurally, the interaction is mainly mediated by a well-defined binding groove in DCN1 and an N-terminal 12-residue of UBC12 peptide. The groove in DCN1 shows four hydrophobic hotspots, including P1, P2, P3 and P4, which cover those regions occupied by Met1, Ile2 and Leu4 residues of UBC12 separately (Figure 17A-B). These hotspots appear to be amenable for the design of DCN1-UBC12 interaction inhibitors [218].
(A) The crystal structure of DCN1 complexed with UBC12 peptide (PDB: 3TDU). (B) Hydrophobic hotspots (purple mesh) at the UBC12 peptide binding site. (C) The co-crystal structure of DCN1 complexed with DI-591 (PDB: 5UFI). (D) The chemical structures and design strategy of peptidomimetic compounds 33-35 [218]. Reproduced with permission [218]. Copyright 2017, Nature Publishing Group.
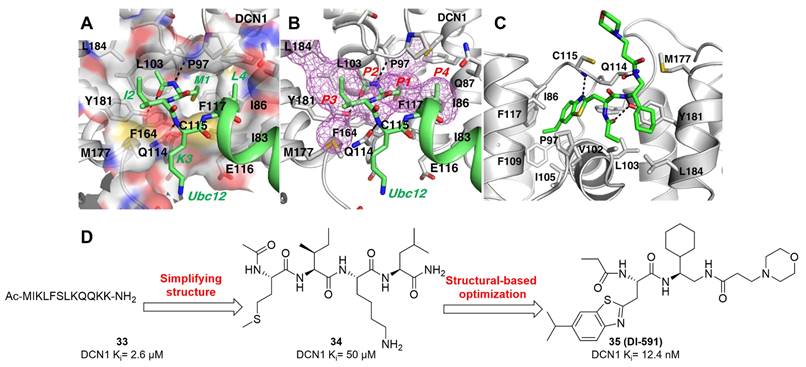
Based on the observed DCN1-UBC12 interaction, Wang's group determined the 12-residue UBC12 peptide 33 binding to DCN1 with the Ki value of 2.6 µM (Figure 17D). Next, they tried to shorten the peptide 33, leading to hit 34 (Ki= 50 µM) with a smaller molecular weight (MW) for further optimizations [218]. Eventually, compound 35 (DI-591) was provided with the Ki value of 12.4 nM and MW of 586, and had excellent aqueous solubility. Compound 35 displayed potent binding affinity to DCN1 and 2 with the Kd values of 21.9, 11.2 nM separately, but no inhibitory activity against DCN3-5 at concentration up to 10 µM. The co-crystal structure shows that molecule 35 binds to DCN1 protein with similar binding modes with UBC12 (Figure 17B-C). The bicyclic aromatic group of compound 35 is located in a deep hydrophobic pocket surrounded by I86, F89, C90, F109, F117, A106, A111 and F164. The methyl group of propionyl group in analogue 35 fits well in the hydrophobic pocket defined by L103, L184, Y181, A106 and V102. Additionally, the propionyl group makes hydrogen bond interactions with P97 and Y181. Meanwhile, the amide in the soluble portion of derivative 35 forms a hydrogen bond with Q114. Besides, the cyclohexyl group is half buried in a hydrophobic pocket surrounded by M177, A180, Y181 and L184. Mechanism study indicated that compound 35 could selectively inhibit neddylation of Cul3, resulting in the accumulation of NRF2 protein. In contrast to MLN4924, compound 35 did not exert significantly antiproliferative activity.
As a parallel work to develop compound 35 (DI-591), tetrapeptide 36 (Ki= 3.6 µM) also was used as a starting point for in-depth optimizations based on the predicted binding modes (Figure 18A), aiming to better target the four pockets (P1, P2, P3 and P4) of DCN1 protein [219]. The structural modifications were mainly centered on the marked 5 positions in compound 36, further producing molecule 37 with the KD value of 6.9 nM (Figure 18B). Compound 37 had an aqueous solubility of 54 μM in PBS at pH = 7.4. In addition, molecule 37 could target DCN1 protein and inhibit DCN1-UBC12 interaction in H2170 cells. Similar to DI-591, derivative 37 selectively suppressed neddylation of cullin 3, causing the accumulation of NRF2 protein, a substrate of CRL3 in H2170 and SK-MES-1 cells.
(A) Predicted binding modes of derivative 36 with DCN1. (B) The chemical structures of peptidomimetic compounds 36-37 [219]. Reproduced with permission [219]. Copyright 2018, American Chemical Society.
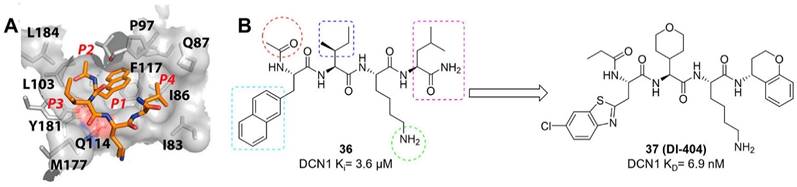
By analyzing the co-crystal structure of DI-591 complexed with DCN1, Wang's group found that the sulfur atom of Cys115 in DCN1 was located 4.1 Å from the α carbon of the 3-morpholinopropanamide moiety in DI-591 (Figure 19B), which might be a chance for designing covalent DCN1 inhibitors [142]. Therefore, a novel class of DI-591 analogues containing various Michael acceptor groups were synthesized and evaluated toward the inhibition of cullin 3 neddylation. In particular, compound 38 (Figure 19A) selectively suppressed cullin 3 neddylation at concentrations of 0.3 nM, about 1000 times stronger than DI-591 in four tested cancer cell lines. From the mass spectrometry analysis, 100% of DCN1 protein was observed to form a complex with compound 38 within 10 min incubation. The co-crystal structure shows that the electron density between Cys115 and molecule 38 verifies the covalent nature of this complex, and the dimethylamino group in molecule 38 is absent (Figure 19C-D). Molecule 38 could specifically target DCN1 over DCN3, although DCN3 has a Cys140 in its binding site corresponding to Cys115 in DCN1. More importantly, they found that pretreated with compound 38 could effectively protect mice from acetaminophen-induced liver damage.
(A) The chemical structures and design strategy of covalent inhibitor 38. (B) and (C) The co-crystal structures of DCN1 complexed with peptidomimetic compounds 35 and 38 (PDB: 6XOO), respectively. (D) The covalent adduct of peptidomimetic compound 38 with DCN1 [142]. Reproduced with permission [142]. Copyright 2021, Nature Publishing Group.
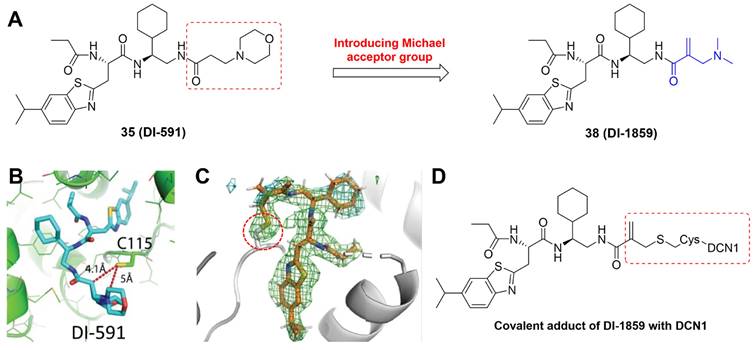
In 2017, Guy's group reported the screening of a library containing 601,194 unique small molecules using TR-FRET assay, leading to the piperidinyl urea derivative 39 (NAcM-HIT) as a potential DCN1 inhibitor with the IC50 value of 7 μM (Figure 20) [220]. After analyzing the co-crystal structure, they found that the N-acetyl and Leu pockets in DCN1 were not occupied by compound 39, which was regarded as an approach to improve the binding affinity (Figure 20A). Thus, the structural optimizations were performed to target the N-acetyl and Leu pockets of DCN1, yielding molecule 40 (Figure 20) with the IC50 value of 50 nM, over 100 folds more active than compound 39. Besides, analogue 40 displayed better solubility and permeability [221]. Unfortunately, compound 40 was quickly metabolized in microsomal models (CLint= 170 mL/min/kg). Therefore, their continuing optimization efforts were centered on improving the clearance of molecule 40, through (1) analyzing the physical drivers for bioavailability, and (2) determining metabolic and toxic liabilities related to the scaffold. Subsequently, compound 41 (NAcM-OPT) was provided (Figure 20), which not only possessed favorable biochemical activity (IC50= 60 nM), but was significantly more stable (CLint= 25 mL/min/kg). Also, derivative 41 was orally bioavailable and well tolerated in mice [222]. Based on the co-crystal structure compound 41 with DCN1, Guy and co-authors then developed a covalent inhibitor 42 (NAcM-COV) with more potent binding affinity, whose allylamide group could form a covalent bond with Cys115 of DCN1 (Figure 20). Both compounds 41 and 42 could effectively interrupt DCN1-UBC12 interaction, leading to the deceased neddylation of cullin-1/3 in DCN1-overexpressed HCC95 cells, and inhibited anchorage-independent growth of HCC95 cells [220].
The chemical structures and design strategy of piperidinyl urea derivatives 39-42. (A) The co-crystal structure overlay of 39 (orange) with DCN1 and UBC12NAc (magenta) with DCN1 (PDB: 5V83 and 3TDU), indicating the critical regions targeted for optimizations [220-222]. Reproduced with permission [221]. Copyright 2018, American Chemical Society.
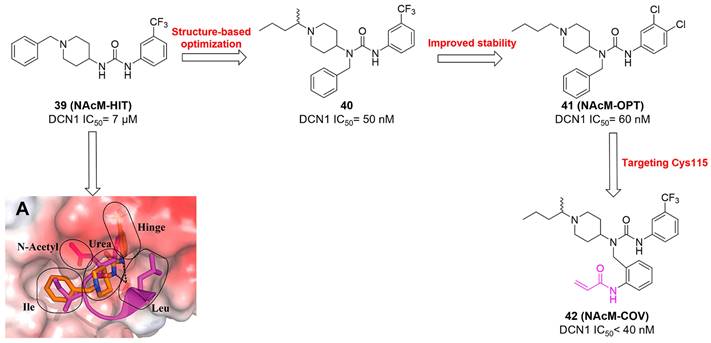
From the above high-throughput screening results, Guy and co-workers also discovered a pyrazolo-pyridone derivative 43 (Figure 21A), which targeted DCN1-UBC12 interaction with the IC50 value of 5.1 μM [223]. Compared with the piperidinyl urea derivatives, such as NAcM-OPT, these pyrazolo-pyridone compounds have a high degree of three-dimensional structure that could be beneficial for increasing activity, selectivity and solubility. The co-crystal structure indicates that molecule 43 positions the central pyrazolo-pyridone ring closer to the N-acetyl pocket, which offers a chance for targeting the N-acetyl pocket (Figure 21B). Therefore, the structure-based optimizations were carried out via firstly defining the core pharmacophore, followed by identifying the optimal substituents to occupy the key subpockets. Subsequently, analogue 44 (Figure 21A) was generated and showed about 25 folds more potent activity relative to the initial compound 43. The co-crystal structure of 44 with DCN1 displays that the ethyl group on pyrazolo-pyridone ring well occupies the N-acetyl pocket and pushes the ligand deeper into the Leu pocket (Figure 21C), which is mainly responsible for the improved potency. Considering that compound 44 possessed poor oral bioavailability and relatively rapid clearance, Guy et al. performed further medicinal chemistry optimizations to enhance its bioavailability, yielding derivative 45 (Figure 21A) with the IC50 value of 0.1 μM, more potent than 44 [224]. Derivative 45 revealed increased aqueous solubility (71 μM), oxidative metabolic stability (MLM Clint< 10 mL/(min kg)), oral bioavailability (92%) and maintained minimal clearance (plasma ClIV = 1.2 L/(kg h)). The SARs showed that replacement of the methyl pyrazole in 44 with ionizable polar substituents significantly improved aqueous solubility without undesirably influencing the activity. The introduction of CF3 group resulted in the electronic and steric deactivation of aryl ring, thus inhibiting oxidative metabolism in microsomal and murine model. Compound 45 possessed stronger thermal stabilization of DCN1 protein in comparison to 44. Additionally, molecule 45 evidently suppressed anchorage-independent colony formation of HCC95 cells, indicating the main expected phenotypic cellular response. In summary, derivative 45 with potent activity and good oral bioavailability, could be used for exploring the acute pharmacologic effects of DCN1-UBC12 interaction in murine models.
(A) The chemical structures and design strategy of pyrazolo-pyridone derivatives 43-45. (B) The co-crystal structure overlay of 43 (orange) with DCN1 and UBC12NAc (blue) with DCN1 (PDB: 6P5W and 3TDU). Reproduced with permission [223]. Copyright 2019, American Chemical Society. (C) The co-crystal structure of 44 with DCN1 (PDB: 6P5V). Reproduced with permission [223]. Copyright 2019, American Chemical Society. (D) The co-crystal structure of 45 with DCN1 (PDB: 7KWA) [223, 224]. Reproduced with permission [224]. Copyright 2021, American Chemical Society.
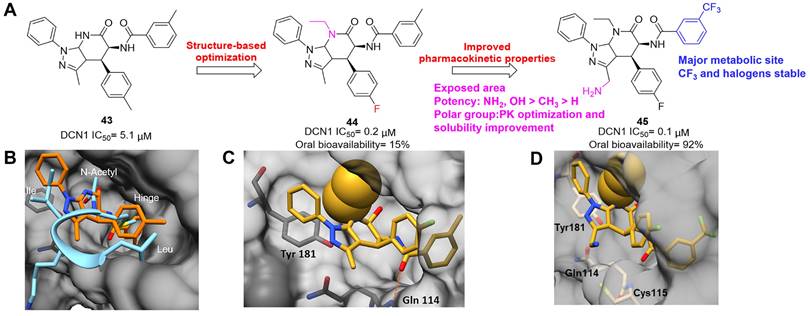
To find hit compounds targeting DCN1-UBC12 interaction, Wang et al. screened ∼15 000 molecules with 5000 diverse scaffolds using HTRF assay, providing the triazolo[1,5-a]pyrimidine-based hit 46 with the IC50 value of 5.82 μM (Figure 22A) [225]. Next, structure-based optimizations were carried out based on hit 46, producing a novel class of triazolo[1,5-a]pyrimidine derivatives, of which compound 47 revealed the strongest binding affinity toward DCN1 protein (IC50= 11 nM) in a reversible manner and possessed high selectivity on DCN1 over BTK, CDK and EGFR kinases. The predicted binding modes of 47 with DCN1 shows that the 4-Cl phenyl group is deeply inserted into the hydrophobic pocket, defined by Ile86, Phe89, Val102, Ile105, Ala106, Phe109, Ala111, Phe117 and Phe164, and has π-π stacking interactions with Phe89, Phe109, Phe117 and Phe164 separately (Figure 22B). The 5-methylpyrimidine group is located in the hydrophobic pocket, defined by Ala180, Tyr181 and Leu184. Additionally, the pyrimidine ring possesses an arene-H electrostatic interaction with Ala98. Pro97 has an arene-H electrostatic interaction with the tetrazole ring. Besides, the N,N-dimethyl group of 47 is observed to form hydrophobic interactions with pocket surrounded by Ile83 and Ile86. These interactions may be responsible for the efficacy of compound 47. In MGC-803 and KYSE70 cells, molecule 47 selectively suppressed the neddylation of cullin-1/3 over other cullin members, accompanied by the accumulation of their downstream proteins p21, p27 and NRF2. Unfortunately, derivative 47 was metabolized rapidly (T1/2< 5 min) and possessed high clearance in human, rat and dog liver microsomes.
(A) The chemical structures and design strategy of triazolo[1,5-a]pyrimidine derivatives 46-47. (B) The predicted binding modes of compound 47 with DCN1 [225]. Reproduced with permission [225]. Copyright 2019, American Chemical Society.
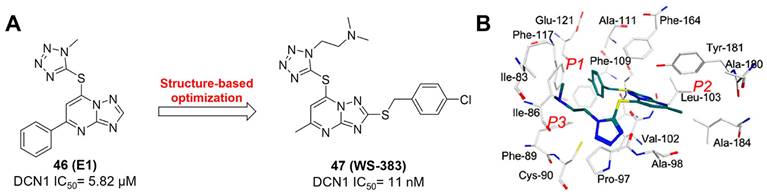
Meanwhile, our group designed and synthesized a novel series of 5‑cyano-6-phenyl-pyrimidine derivatives based on the hit 48 (IC50= 1.2 μM) that was identified via screening our in-house structurally different molecular library (ca. 1000 compounds), using both FP and HTRF assays (Figure 23A) [226]. Among synthesized compounds, molecule 49 disclosed the most potent activity against DCN1-UBC12 interaction with the IC50 value of 15 nM, about 80 folds more effective than 48. Besides, 49 revealed high selectivity on DCN1-2 over DCN3-5. The docking modes indicate that the sulfur atom connecting the thiazole ring makes a hydrogen bond interaction with the carbonyl group of Pro97 with a distance of 3.55 Å (Figure 23B). The nitrogen atom of cyan group makes a hydrogen bond with the hydroxyl group of Tyr181 with a distance of 2.09 Å. Moreover, the methylbenzene group is extended to the hydrophobic pocket defined by Ile86, Phe89, Val102, Leu103, Ala106 and Phe117, and has an additional arene-H interaction with Phe164. The propargyl group in compound 49 is located in the hydrophobic region surrounded by Ile83, Ile86, Gln87 and Cys115. Preliminary mechanism research indicated that derivative 49 specifically suppressed DCN1-UBC12 interaction, causing the reduction of neddylation of cullin-3 and the accumulation of NRF2 as well as its downstream proteins HO1 and NQO1 in H1975 and PC9 cells.
(A and D) The chemical structures of DCN1 inhibitors 48-52. (B) The predicted binding modes of 5‑cyano-6-phenyl-pyrimidine derivative 49 (blue) with DCN1 protein (white) [226]. Reproduced with permission [226]. Copyright 2019, American Chemical Society. (C) The predicted binding modes of compound 50 (blue) with DCN1 protein (green) [145]. Reproduced with permission [145]. Copyright 2022, American Chemical Society. (E) The predicted binding modes of compound 52 (blue) with DCN1 protein (white) [227]. Reproduced with permission [227]. Copyright 2021, Elsevier Masson SAS.
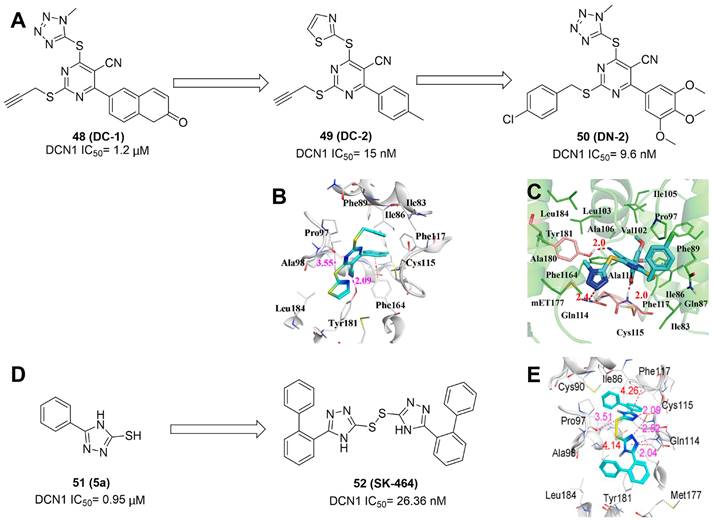
Based on the docking modes of DCN1 inhibitor 49, we conducted further structural optimizations and SARs using several drug design strategies, including structure-based drug design and bioisosterism etc., yielding 77 novel 2-thiopyrimidine derivatives as DCN1 inhibitors [145]. In comparison to molecule 49, the new synthesized compound 50 (IC50 = 9.55 ± 0.98 nM) possessed stronger inhibitory effect, higher selectivity toward DCN-like proteins and weaker toxicity against cardiac fibroblasts (CFs) (Figure 23A). Molecular docking (Figure 23C), biolayer interferometry, NAE and co-immunoprecipitation assays confirmed that compound 50 could specifically target DCN1 and interfere DCN1-UBC12 interaction. More importantly, we, for the first time, reported that compound 50 could effectively reduce Ang Ⅱ-induced CFs activation, suggesting the potential anti-cardiac fibrotic effect through targeting DCN1.
In addition to hit 48, we also found the phenyltriazole thiol-based hit 51 as a potential DCN1 inhibitor with the IC50 value of 0.95 μM (Figure 23D) [227]. Subsequently, structure-based optimizations were performed based on 51, generating a new array of phenyltriazole thiol analogues. In particular, compound 52 exhibited the strongest biological activity against DCN1-UBC12 interaction with the IC50 value of 26.36 nM. In addition, compound 52 disclosed high selectivity and specificity for DCN1 and 2. The predicted binding modes of 52 complexed with DCN1 indicate that in the inside of the pocket, two hydrogen bonds are formed between the nitrogen of triazole ring and the amino group of Cys115, the sulfur of triazole ring and the carbonyl group of Pro97 (Figure 23E). Moreover, the triazole ring makes an arene-H interaction with Ile86, and the diphenyl is extended to the hydrophobic pocked defined by Ile86, Phe89, Cys90, Leu93, Ile105, Ala106, Phe109 and Phe117. In the outside of the pocket, the nitrogen atoms of the triazole make two hydrogen bond interactions with Gln114 and Cys115, respectively. The diphenyl forms hydrophobic interactions with Ala98, Leu103, Met177, Ala180, Tyr181 and Leu184. Further exploration showed that compound 52 could bind to DCN1, block the neddylation of cullin-3 and suppress KYSE70 and H2170 cell migration and invasion.
Taken together, since 2017, DCN1 inhibitors with various chemotypes have been identified, including peptidomimetic derivatives, piperidinyl urea derivatives, pyrazolo-pyridone derivatives, triazolo[1,5-a]pyrimidine derivatives, 5‑cyano-6-phenyl-pyrimidine derivatives and phenyltriazole thiol-based derivatives. These DCN1 inhibitors were found mainly via structure-based drug design, high throughput virtual screening and screening in-house structurally different molecular library. The most reported DCN1 inhibitors showed robust binding affinity with the IC50 values ranging from 1 to 200 nmol/L. Covalent DCN1 inhibitors seemed to be more potent than non-covalent DCN1 inhibitors. Owing to the high homology between DCN1 and DCN2, all the reported DCN1 inhibitors were highly selective toward DCN1-2 over DCN3-5. Interestingly, the structural differences of DCN1 inhibitors may result in their diverse effects on inhibiting cullin3 or/and cullin1 neddylation. In addition, many DCN1 inhibitors were lack of favorable pharmacokinetic and physicochemical properties. Besides, DCN1 was initially regarded as a promising anti-cancer therapeutical target, while the anti-cancer effects of DCN1 inhibitors were subsequently found to be weak. Considering the NRF2, an antioxidant transcription factor, is a typical substrate of cullin3 [10], DCN1 inhibitors could play extensive efficacy for the treatment of diseases linked to over-production of reactive oxygen species (ROS). Especially, the latest findings indicated that DCN1 inhibitors exhibited effective potency on relieving acetaminophen-induced liver damage and AngⅡ -induced cardiac fibrosis, which was related with the inhibition of cullin3 neddylation and its substrate NRF2 activation [142, 145]. All in all, more potent and selective DCN1 inhibitors with drug-like properties are urgently required, aiming to further investigate their therapeutical potential for human diseases and DCN1 protein's potentially biological roles.
A summary of the reported DCN1 inhibitors.
| Inhibitors | Year reported | DCN1 (IC50) | Half-life | Biology | Reference |
|---|---|---|---|---|---|
| 35 (DI-591) | 2017 | 12 nmol/L | - | A non-covalent DCN1 inhibitor High selectivity toward DCN1-2 over DCN3-5 Selective inhibition of cullin 3 neddylation | [218] |
| 37 (DI-404) | 2018 | 6.9 nmol/L | - | A non-covalent DCN1 inhibitor Selective inhibition of cullin 3 neddylation | [219] |
| 38 (DI-1859) | 2021 | < 1 nmol/L | - | A covalent DCN1 inhibitor High selectivity toward DCN1-2 over DCN3-5 Compound 38 selectively suppressed cullin 3 neddylation at concentrations of 0.3 nM, about 1000 times stronger than DI-591. Also, compound 38 effectively protected mice from acetaminophen-induced liver damage. | [142] |
| 40 (7) | 2018 | 50 nmol/L | iv: 0.14 h | A non-covalent DCN1 inhibitor High selectivity toward DCN1-2 over DCN3-5 Compound 40 not only had favorable solubility and permeability, but also could selectively reduce steady-state levels of neddylated cullin1 and cullin3. | [222] |
| 41 (OPT) | 2018 | 60 nmol/L | iv: 3.6 h, po: 4.2 h | A non-covalent DCN1 inhibitor High selectivity toward DCN1-2 over DCN3-5 Selective inhibition of cullin 1 and cullin 3 neddylation | [220-222] |
| 42 (COV) | 2018 | < 40 nmol/L | - | A covalent DCN1 inhibitor High selectivity toward DCN1-2 over DCN3-5 Selective inhibition of cullin 1 and cullin 3 neddylation | [220] |
| 44 (27) | 2019 | 200 nmol/L | iv: 1.2 h | A non-covalent DCN1 inhibitor High selectivity toward DCN1-2 over DCN3-5 Selective inhibition of cullin 1 and cullin 3 neddylation | [223] |
| 45 (40) | 2021 | 100 nmol/L | iv: 7.2 h, po: 9.5 h | A non-covalent DCN1 inhibitor High selectivity toward DCN1-2 over DCN3-5 Selective inhibition of cullin 1 and cullin 3 neddylation | [224] |
| 47 (WS-383) | 2019 | 11 nmol/L | < 5 min | A non-covalent DCN1 inhibitor Selective inhibition of cullin 1 and cullin 3 neddylation | [225] |
| 49 (DC-2) | 2019 | 15 nmol/L | < 5 min | A non-covalent DCN1 inhibitor High selectivity toward DCN1-2 over DCN3-5 Selective inhibition of cullin 3 neddylation | [226] |
| 50 (DN-2) | 2022 | 9.55 nmol/L | < 5 min | A non-covalent DCN1 inhibitor High selectivity toward DCN1-2 over DCN3-5 Selective inhibition of cullin 3 neddylation Potential anti-cardiac fibrotic effect | [145] |
| 52 (SK-464) | 2021 | 26 nmol/L | iv: 0.63 h | A non-covalent DCN1 inhibitor High selectivity toward DCN1-2 over DCN3-5 Compound 52 selectively blocked the neddylation of cullin 3 and suppressed KYSE70 and H2170 cell migration and invasion. | [227] |
Cullin 1/5 inhibitors
To discover potent and novel inhibitors targeting Cul5 neddylation, Sun's group screened a library of 17,000 molecules using an AlphaScreen-based assay, yielding the natural 53 (gossypol) as a potent inhibitor of cullin neddylation (Figure 24) [228, 229]. Molecule 53 effectively inhibited the neddylation of Cul5 and Cul1 with the IC50 values of 4.3 and 3.4 μM, respectively. Besides, it could straightly bind to SAG-Cul5 and RBX1-Cul1 complexes in multiple cancer cells, accompanied by accumulation of Cul5 substrate NOXA and Cul1 substrate MCL1. Interestingly, the combination of compound 53 and MCL1 inhibitor S63845 displayed a synergistic anticancer effect in multiple human cancer cells.
The chemical structure of Cul1/5 inhibitor Gossypol [228].
UBE2F inhibitors
In 2022, Sun's research group, for the first time, reported an azaindole-based UBE2F inhibitor HA-9104, which was obtained through a structure-based virtual screen and multiple rounds of chemistry-based optimization [230]. HA-9104 could effectively bind to UBE2F protein and decrease its expression, resulting in the inhibition of cullin-5 neddylation. In addition, the inhibition of cullin-5 neddylation inactivated CRL-5 activity, further causing the accumulation of NOXA, a substrate of CRL-5, to induce apoptosis. Besides, the 7-azaindole group of HA-9104 may form a DNA adduct to cause DNA damage and G2/M arrest. Furthermore, HA-9104 displayed potent antiproliferative activity against lung cancer H1650, H2170, H358 cells with the IC50 values of 5.260, 1.765, 4.377 μΜ separately and possessed radiosensitization in both in vitro cell culture and in vivo xenograft tumor models. Taken together, HA-9104, a novel inhibitor of UBE2F-CRL5 axis, showed highly anti-cancer activity alone or in combination with radiation, and remained to be needed further optimization and development.
The chemical structure of UBE2F inhibitor HA-9104 [230].
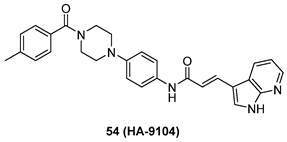
UBC12 inhibitors
In 2022, Chen and co-authors, for the first time, disclosed an UBC12 inhibitor arctigenin via screening a natural product library with 32 compounds [231]. Arctigenin could significantly decrease the level of NEDD8-conjugated UBC12, further resulting in the reduction of the neddylation levels of cullin 1, cullin 3 and cullin 4. Additionally, treated with arctigenin dose-dependently suppressed the growth of UBC12 overexpressed PC9, H1299 and HGC27 cells with IC50 values of 9.7, 17.2 and 9.5 μM separately. Further mechanism studies indicated that arctigenin obviously inhibited cullin neddylation in gastric cancer cells, causing the accumulation of cullin downstream substrate PDCD4 that was a tumor suppressive protein and could induce cell death. Besides, the suppression of UBC12 and cullin neddylation was beneficial for regulating protein stability by arctigenin.
The chemical structure of UBC12 inhibitor arctigenin [231].
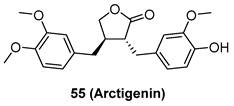
A summary of targeting neddylation through inhibiting various proteins of this pathway.
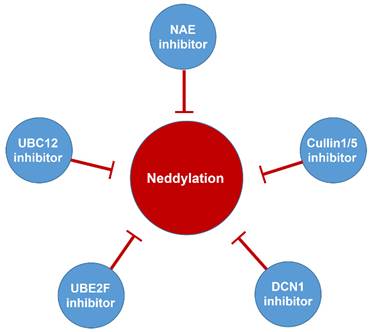
Conclusions and perspectives
The overactivated neddylation proteins and CRL components play pivotal roles on cancer progression and confer poor overall survival, indicating that targeting neddylation is a promising strategy for cancer treatment. However, there is challenging to identify which of neddylation proteins probably possess the strongest therapeutic effects when being targeted, and how to develop effective and selective drugs that can inhibit these proteins without side effects.
NAE is regarded as a promising therapeutic target for human diseases, and its inhibitor MLN4924 is undergoing many phase I/Ⅱ/III trials for cancer treatment. However, treated with MLN4924 in phase I trials unexpectedly led to modest response [152-154], the emergence of resistance owing to heterozygous mutations in UBA3 [32, 148, 149, 151], and liver toxicity [152]. The discovery of small molecules targeting NAE mutations might be an effective way to reduce resistance. In addition, some NAE inhibitors also displayed significantly inhibitory activity against other E1, like UAE and SAE, which may cause inevitable side effects. Therefore, it was necessary to find the differences between the active sites of these E1 proteins to better design selective NAE inhibitors. Besides, numerous clinical trials and preclinical studies disclosed that MLN4924 could be regarded as a sensitizer in the combination with chemo- or radiotherapy, which exerted stronger efficacy for a variety of chemo-/radio-resistant cancers (Table 2-4). Especially, two phase III trials of MLN4924 in combination with azacytidine have been launched for the treatment of AML, MS and CMML. These findings show that targeting NAE is a meaningful strategy for cancers, and more potent and specific NAE inhibitors with low toxicity are still desired.
In addition to NAE inhibitors, many potent DCN1-UBC12 interaction inhibitors were reported and could selectively inhibit neddylation of cullin 3 and/or cullin 1, leading to more specifically explore the pharmacological roles of individual CRL. Although DCN1 was a promising therapeutical target for the treatment of diseases, DCN1 inhibitors displayed limited anticancer potency in vitro. Also, the effects of DCN1 inhibitors at the cellular level remain moderate, indicating more efforts needed to further enhance their cellular efficacy. Of note, compared to non-covalent DCN1 inhibitors like DI-591, covalent DCN1 inhibitors like DI-1859 seemed to have stronger cellular potency, suggesting that the development of covalent DCN1 inhibitors may have more important value [142]. Moreover, given that the antioxidant transcription factor NRF2 is a typical substrate of cullin3 [10], DCN1 inhibitors probably play vital role for the treatment of diseases related to over-production of ROS. In particular, the recent findings disclosed that DCN1 inhibitors possessed evident potency on reducing acetaminophen-induced liver damage and AngⅡ-induced cardiac fibrosis, connected with the inhibition of cullin3 neddylation and its substrate NRF2 activation [142, 145]. Besides, considering that neddylation inhibition well sensitize chemo-/radio-resistant cancer cells [159, 167, 172], DCN1 inhibitors can be used to investigate their potency on relieving resistance in the future. Anyhow, more powerful and selective DCN1 inhibitors with favorable drug-like properties should be developed in the future, aiming to further explore the potential functions of DCN1 protein in human diseases.
Overall, the development of neddylation inhibitors remains in infancy and it is highly desired to develop more active agents selectively targeting neddylation E2s (UBC12 or UBE2F), E3s (such as RBX1 or RBX2) or E3-substrates for cancer treatment. Considering the potential anti-cancer potency of NAE activators [214, 215]. the design of modulators activating neddylation proteins may be also a new therapeutical strategy for human diseases. In addition, neddylation protein degradation may be an effective approach for treating diseases caused by neddylation protein mutation or activation. Until now, no structures of neddylation protein in complex with PROTAC-based degraders have been reported, more efforts are needed to solve the crystal structures of these proteins to guide the rational degrader design. The application of highly accurate protein structure prediction technologies, such as alphafold [232], may be an important opportunity to predict the crystal structures unresolved protein in neddylation pathway. Furthermore, trivalent and dual-targeting PROTACs might also have therapeutic potential for treating neddylation dysregulated human diseases.
Finally, we want to highlight the critical roles of targeting neddylation for fibrotic diseases. Although important involvement of neddylation, the therapeutic roles of targeting neddylation pathway or specific enzymes of this pathway in fibrotic diseases remain to be further explored.
Abbreviations
ABL: Abelson oncogene
AEs: Adverse events
AML: Acute myelogenous leukemia
Ang II: Angiotensin II
AP-1: Activating protein-1
APL: Acute promyelocytic leukemia
APPBP1: Amyloid beta precursor protein binding protein 1
AR: Androgen receptor
AR-Vs: AR and its active variants
ATF4: Activating transcription factor 4
ATP: Adenosine-triphosphate
ATRA: All-trans retinoic acid
BCR: Breakpoint cluster region
BTK: Bruton'styrosinekinase
Cand1: Neddylation-dissociated 1 protein
CC: Cervical cancer
CCL2: Chemotactic cytokine ligand 2
CDK: Cyclin-dependent kinases
CLL: Chronic lymphocytic leukemia
CML: Chronic myeloid leukemia
CRC: Colorectal cancer
CRLs: Cullin-RING ligases
DCN1: Defective in cullin neddylation 1
DLBCL: Diffuse large B-cell lymphoma
DR5: Death receptor 5
EGFR: Epidermal growth factor receptor
Erk: Extracellular regulated protein kinases
ERRβ: Estrogen-related receptor beta
ESCC: Esophageal squamous cell carcinoma
FP: Fluorescence polarization
GBM: Glioblastoma
GC: Gastric cancer
GCB: Germinalcenter B cell-like
HCC: Hepatocellular carcinoma
HIF-1: Hypoxia-inducible factor 1
HIF1A: Hypoxia-inducible factor 1A
HNSCC: Head and neck squamous cell carcinoma
HTRF: Homogeneous time resolved fluorescence
ICC: Intrahepatic cholangiocarcinoma
IR: Ionizing radiation
LFT: Liver function test
LSC: Leukemia stem cells
MAPK: Mitogen-activated protein kinase
miR-155: Microrna-155
MM: Multiple myeloma
MMP9: Matrix metalloproteinase 9
mTOR: Mammalian target of rapamycin
MW: Molecular weight
NAE: NEDD8-activating enzyme E1
NEDD8: Neuronal precursor cell-expressed developmentally downregulated protein 8
NF-κB: Nuclear factor-κB
NPC: Nasopharyngeal carcinoma
NSCLC: Non-small cell lung cancer
OCC: Ovarian cancer cell
PCa: Prostate cancer
PDL1: Programmed death ligand-1
PDTX: Patient-derived tumor xenografts
PK: Pharmacokinetics
RBX1: RING-box protein 1
RBX2: RING-box protein 2
ROS: Reactive oxygen species
SAE: SUMO-activating enzyme
SARs: Structure-activity relationships
SCC: Squamous cell carcinoma
SCF: Skp1-Cul1-F-box protein
SI-NETs: Small intestinal neuroendocrine tumors
Skp1: S-phase kinase associated protein 1
TAMs: Tumor associated macrophages
TCGA: Cancer Genome Atlas database
TGFβ: Transforming growth factor
TNF-α: Tumor necrosis factor-alpha
TR-FRET: Time-resolved fluorescence resonance energy transfer
UAE: Ubiquitin activating enzyme
UBA3: Ubiquitin-like modifier activating enzyme 3
UBC12: NEDD8 conjugating enzyme
UBE2F: Ubiquitin-conjugating enzyme E2F
UC: Urothelial carcinoma
UM: Uveal melanoma
UPS: Ubiquitin-proteasome system
α-SMA: Α-smooth muscle actin
Acknowledgements
Funding
This work was supported by China Postdoctoral Science Foundation (2022M721060 by H. ZX, 2020M672249 and 2022T150465 by M. L-Y,); National Natural Science Foundation of China (82270290 by Z. W); Natural Science Foundation of Henan Province (No. 212300410392 by M. L-Y).
Author contributions
Zhang-Xu He and Wei-guang Yang drafted this review and designed the figures and tables; Dan Zengyangzong, Ge Gao and Qian Zhang collected the related references and gave some valuable suggestions; Hong-Min Liu provided guidance and revised this manuscript; Wen Zhao and Li-Ying Ma provided guidance, revised this manuscript, conceived the final approval of the version to be submitted and obtained funding supports. All authors read and approved the final manuscript.
Consent for publication
We declared that this manuscript is original, has not been published before, and is not currently being considered for publication elsewhere.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Rabut G, Peter M. Function and regulation of protein neddylation. EMBO reports. 2008;9:969-76
2. Zheng YC, Guo YJ, Wang B, Wang C, Mamun MAA, Gao Y. et al. Targeting neddylation E2s: a novel therapeutic strategy in cancer. J Hematol Oncol. 2021;14:57
3. Soucy TA, Smith PG, Rolfe M. Targeting NEDD8-activated cullin-ring ligases for the treatment of cancer. Clin Cancer Res. 2009;15:3912-6
4. Watson IR, Irwin MS, Ohh M. NEDD8 pathways in cancer, sine quibus non. Cancer Cell. 2011;19:168-76
5. Mendoza HM, Shen LN, Botting C, Lewis A, Chen J, Ink B. et al. NEDP1, a highly conserved cysteine protease that deneddylates cullins. J Biol Chem. 2003;278:25637-43
6. Lyapina S, Cope G, Shevchenko A, Serino G, Tsuge T, Zhou CS. et al. Promotion of NEDD8-Cul1 conjugate cleavage by COP9 signalosome. Science. 2001;292:1382-5
7. Min KW, Kwon MJ, Park HS, Park Y, Yoon SK, Yoon JB. CAND1 enhances deneddylation of Cul1 by COP9 signalosome. Biochem Biophys Res Commun. 2005;334:867-74
8. Cavadini S, Fischer ES, Bunker RD, Potenza A, Lingaraju GM, Goldie KN. et al. Cullin-RING ubiquitin E3 ligase regulation by the COP9 signalosome. Nature. 2016;531:598-603
9. Faull SV, Lau AMC, Martens C, Ahdash Z, Hansen K, Yebenes H. et al. Structural basis of cullin 2 ring E3 ligase regulation by the COP9 signalosome. Nat Commun. 2019;10:3814
10. Zhao Y, Morgan MA, Sun Y. Targeting neddylation pathways to inactivate cullin-ring ligases for anticancer therapy. Antioxid Redox Signal. 2014;21:2383-400
11. Duncan K, Schafer G, Vava A, Parker M, Zerbini LF. Targeting neddylation in cancer therapy. Future Oncol. 2012;8:1461-70
12. Huang DT, Ayrault O, Hunt HW, Taherbhoy AM, Duda DM, Scott DC. et al. E2-ring expansion of the NEDD8 cascade confers specificity to cullin modification. Mol Cell. 2009;33:483-95
13. Bulatov E, Ciulli A. Targeting cullin-ring E3 ubiquitin ligases for drug discovery: structure, assembly and small-molecule modulation. Biochem J. 2015;467:365-86
14. Wu S, Yu L. Targeting cullin-ring ligases for cancer treatment: rationales, advances and therapeutic implications. Cytotechnology. 2016;68:1-8
15. Zhou L, Jia L. Targeting protein neddylation for cancer therapy. Adv Exp Med Biol. 2020;1217:297-315
16. Gao Q, Yu GY, Shi JY, Li LH, Zhang WJ, Wang ZC. et al. Neddylation pathway is up-regulated in human intrahepatic cholangiocarcinoma and serves as a potential therapeutic target. Oncotarget. 2014;5:7820-32
17. Jia L, Soengas MS, Sun Y. ROC1/RBX1 E3 ubiquitin ligase silencing suppresses tumor cell growth via sequential induction of G2-M arrest, apoptosis, and senescence. Cancer Res. 2009;69:4974-82
18. Hua W, Li C, Yang Z, Li L, Jiang Y, Yu G. et al. Suppression of glioblastoma by targeting the overactivated protein neddylation pathway. Neuro Oncol. 2015;17:1333-43
19. Ma T, Shi T, Huang J, Wu L, Hu F, He P. et al. DCUN1D3, a novel UVC-responsive gene that is involved in cell cycle progression and cell growth. Cancer Sci. 2008;99:2128-35
20. Huang G, Kaufman AJ, Ramanathan Y, Singh B. SCCRO (DCUN1D1) promotes nuclear translocation and assembly of the neddylation E3 complex. J Biol Chem. 2011;286:10297-304
21. Xie P, Yang JP, Cao Y, Peng LX, Zheng LS, Sun R. et al. Promoting tumorigenesis in nasopharyngeal carcinoma, NEDD8 serves as a potential theranostic target. Cell Death Dis. 2017;8:e2834
22. Li L, Wang M, Yu G, Chen P, Li H, Wei D. et al. Overactivated neddylation pathway as a therapeutic target in lung cancer. J Natl Cancer I. 2014;106:083
23. Deng Q, Zhang JJ, Gao YQ, She XF, Wang YC, Wang YL. et al. MLN4924 protects against bleomycin-induced pulmonary fibrosis by inhibiting the early inflammatory process. Am J Transl Res. 2017;9:1810-21
24. Zubiete-Franco I, Fernandez-Tussy P, Barbier-Torres L, Simon J, Fernandez-Ramos D, Lopitz-Otsoa F. et al. Deregulated neddylation in liver fibrosis. Hepatology. 2017;65:694-709
25. Xie P, Zhang M, He S, Lu K, Chen Y, Xing G. et al. The covalent modifier nedd8 is critical for the activation of smurf1 ubiquitin ligase in tumorigenesis. Nat Commun. 2014;5:3733
26. Ying J, Zhang M, Qiu X, Lu Y. Targeting the neddylation pathway in cells as a potential therapeutic approach for diseases. Cancer Chemother Pharmacol. 2018;81:797-808
27. Shi CS, Kuo KL, Lin WC, Chen MS, Liu SH, Liao SM. et al. Neddylation inhibitor, MLN4924 suppresses angiogenesis in huvecs and solid cancers: in vitro and in vivo study. Am J Cancer Res. 2020;10:953-64
28. Mao H, Tang Z, Li H, Sun B, Tan M, Fan S. et al. Neddylation inhibitor MLN4924 suppresses cilia formation by modulating AKT1. Protein Cell. 2019;10:726-44
29. Zhou X, Tan M, Nyati MK, Zhao Y, Wang G, Sun Y. Blockage of neddylation modification stimulates tumor sphere formation in vitro and stem cell differentiation and wound healing in vivo. PNAS. 2016;113:E2935-44
30. Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S. et al. An inhibitor of nedd8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732-6
31. Milhollen MA, Narayanan U, Soucy TA, Veiby PO, Smith PG, Amidon B. Inhibition of nedd8-activating enzyme induces rereplication and apoptosis in human tumor cells consistent with deregulating CDT1 turnover. Cancer Res. 2011;71:3042-51
32. Nawrocki ST, Griffin P, Kelly KR, Carew JS. MLN4924: a novel first-in-class inhibitor of NEDD8-activating enzyme for cancer therapy. Expert Opin Investig Drugs. 2012;21:1563-73
33. Swords RT, Kelly KR, Smith PG, Garnsey JJ, Mahalingam D, Medina E. et al. Inhibition of nedd8-activating enzyme: a novel approach for the treatment of acute myeloid leukemia. Blood. 2010;115:3796-800
34. Liu X, Jiang Y, Wu J, Zhang W, Liang Y, Jia L. et al. NEDD8-activating enzyme inhibitor, MLN4924 (Pevonedistat) induces NOXA-dependent apoptosis through up-regulation of ATF-4. Biochem Bioph Res Co. 2017;488:1-5
35. Lin JJ, Milhollen MA, Smith PG, Narayanan U, Dutta A. NEDD8-targeting drug MLN4924 elicits DNA rereplication by stabilizing Cdt1 in S phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. Cancer Res. 2010;70:10310-20
36. Jia L, Li H, Sun Y. Induction of p21-dependent senescence by an NAE inhibitor, MLN4924, as a mechanism of growth suppression. Neoplasia. 2011;13:561-9
37. Luo Z, Pan Y, Jeong LS, Liu J, Jia L. Inactivation of the cullin (CUL)-ring E3 ligase by the nedd8-activating enzyme inhibitor MLN4924 triggers protective autophagy in cancer cells. Autophagy. 2012;8:1677-9
38. Mao H, Sun Y. Neddylation-independent activities of MLN4924. Adv Exp Med Biol. 2020;1217:363-72
39. Yao WT, Wu JF, Yu GY, Wang R, Wang K, Li LH. et al. Suppression of tumor angiogenesis by targeting the protein neddylation pathway. Cell Death Dis. 2014;5:1059
40. Soucy TA, Dick LR, Smith PG, Milhollen MA, Brownell JE. The nedd8 conjugation pathway and its relevance in cancer biology and therapy. Genes Cancer. 2010;1:708-16
41. Tan KL, Pezzella F. Inhibition of nedd8 and FAT10 ligase activities through the degrading enzyme nedd8 ultimate buster 1: a potential anticancer approach. Oncol Lett. 2016;12:4287-96
42. Whitby FG, Xia G, Pickart CM, Hill CP. Crystal structure of the human ubiquitin-like protein nedd8 and interactions with ubiquitin pathway enzymes. J Bio Chem. 1998;273:34983-91
43. Walden H, Podgorski MS, Huang DT, Miller DW, Howard RJ, Minor DL. et al. The structure of the APPBP1-UBA3-NEDD8-ATP complex reveals the basis for selective ubiquitin-like protein activation by an E1. Mol Cell. 2003;12:1427-37
44. Walden H, Podgorski MS, Schulman BA. Insights into the ubiquitin transfer cascade from the structure of the activating enzyme for nedd8. Nature. 2003;422:330-4
45. Huang DT, Miller DW, Mathew R, Cassell R, Holton JM, Roussel MF. et al. A unique E1-E2 interaction required for optimal conjugation of the ubiquitin-like protein NEDD8. Nat Struct Mol Biol. 2004;11:927-35
46. Huang DT, Zhuang M, Ayrault O, Schulman BA. Identification of conjugation specificity determinants unmasks vestigial preference for ubiquitin within the NEDD8 E2. Nat Struct Mol Biol. 2008;15:280-7
47. Huang DT, Hunt HW, Zhuang M, Ohi MD, Holton JM, Schulman BA. Basis for a ubiquitin-like protein thioester switch toggling E1-E2 affinity. Nature. 2007;445:394-8
48. Huang DT, Paydar A, Zhuang M, Waddell MB, Holton JM, Schulman BA. Structural basis for recruitment of Ubc12 by an E2 binding domain in NEDD8's E1. Mol Cell. 2005;17:341-50
49. Monda JK, Scott DC, Miller DJ, Lydeard J, King D, Harper JW. et al. Structural conservation of distinctive N-terminal acetylation-dependent interactions across a family of mammalian NEDD8 ligation enzymes. Structure. 2013;21:42-53
50. Kurz T, Chou YC, Willems AR, Meyer-Schaller N, Hecht ML, Tyers M. et al. DCN1 functions as a scaffold-type E3 ligase for cullin neddylation. Mol Cell. 2008;29:23-35
51. Burgie ES, Bingman CA, Makino S, Wesenberg GE, Pan X, Fox BG. et al. Structural architecture of galdieria sulphuraria DCN1L. Proteins. 2011;79:1329-36
52. Scott DC, Monda JK, Bennett EJ, Harper JW, Schulman BA. N-terminal acetylation acts as an avidity enhancer within an interconnected multiprotein complex. Science. 2011;334:674-8
53. Scott DC, Sviderskiy VO, Monda JK, Lydeard JR, Cho SE, Harper JW. et al. Structure of a ring E3 trapped in action reveals ligation mechanism for the ubiquitin-like protein NEDD8. Cell. 2014;157:1671-84
54. Zhao YC, Sun Y. Cullin-RING ligases as attractive anti-cancer targets. Curr Pharm Design. 2013;19:3215-25
55. Zhou L, Zhang W, Sun Y, Jia L. Protein neddylation and its alterations in human cancers for targeted therapy. Cell Signal. 2018;44:92-102
56. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7-34
57. Jiang Y, Liang Y, Li L, Zhou L, Cheng W, Yang X. et al. Targeting neddylation inhibits intravascular survival and extravasation of cancer cells to prevent lung-cancer metastasis. Cell Biol Toxicol. 2019;35:233-45
58. Zhou L, Jiang Y, Liu X, Li L, Yang X, Dong C. et al. Promotion of tumor-associated macrophages infiltration by elevated neddylation pathway via NF-kappaB-CCL2 signaling in lung cancer. Oncogene. 2019;38:5792-804
59. Zhou Q, Zheng Y, Sun Y. Neddylation regulation of mitochondrial structure and functions. Cell Biosci. 2021;11:55
60. Li L, Kang J, Zhang W, Cai L, Wang S, Liang Y. et al. Validation of NEDD8-conjugating enzyme UBC12 as a new therapeutic target in lung cancer. EBioMedicine. 2019;45:81-91
61. Zhou W, Xu J, Li H, Xu M, Chen ZJ, Wei W. et al. Neddylation E2 UBE2F promotes the survival of lung cancer cells by activating CRL5 to degrade NOXA via the K11 linkage. Clin Cancer Res. 2017;23:1104-16
62. Zhou L, Zhu J, Chen W, Jiang Y, Hu T, Wang Y. et al. Induction of NEDD8-conjugating enzyme E2 UBE2F by platinum protects lung cancer cells from apoptosis and confers to platinum-insensitivity. Cell Death Dis. 2020;11:975
63. Zhou W, Xu J, Tan M, Li H, Li H, Wei W. et al. UBE2M Is a stress-inducible dual E2 for neddylation and ubiquitylation that promotes targeted degradation of UBE2F. Mol Cell. 2018;70:1008-24
64. Yoo J, Lee SH, Lym KI, Park SY, Yang SH, Yoo CY. et al. Immunohistochemical expression of DCUN1D1 in non-small cell lung carcinoma: its relation to brain metastasis. Cancer Res Treat. 2012;44:57-62
65. Huang G, Singh B. Coamplification and cooperation: toward identifying biologically relevant oncogenes. Clin Cancer Res. 2013;19:5549-51
66. Wang J, Qian J, Hoeksema MD, Zou Y, Espinosa AV, Rahman SM. et al. Integrative genomics analysis identifies candidate drivers at 3q26-29 amplicon in squamous cell carcinoma of the lung. Clin Cancer Res. 2013;19:5580-90
67. Liang Y, Wang H, Chen B, Mao Q, Xia W, Zhang T. et al. circDCUN1D4 suppresses tumor metastasis and glycolysis in lung adenocarcinoma by stabilizing TXNIP expression. Mol Ther Nucleic Acids. 2021;23:355-68
68. Salon C, Brambilla E, Brambilla C, Lantuejoul S, Gazzeri S, Eymin B. Altered pattern of Cul1 protein expression and neddylation in human lung tumours: relationships with CAND1 and cyclin E protein levels. J Pathol. 2007;213:303-10
69. Yuan JP, Jiang BC, Zhang AZ, Qian YY, Tan HN, Gao JG. et al. Accelerated hepatocellular carcinoma development in Cul4B transgenic mice. Oncotarget. 2015;6:15209-21
70. Delgado TC, Barbier-Torres L, Zubiete-Franco I, Lopitz-Otsoa F, Varela-Rey M, Fernandez-Ramos D. et al. Neddylation, a novel paradigm in liver cancer. Transl Gastroenterol Hepatol. 2018;3:37
71. Yu J, Huang WL, Xu QG, Zhang L, Sun SH, Zhou WP. et al. Overactivated neddylation pathway in human hepatocellular carcinoma. Cancer Med. 2018;7:3363-72
72. Luo Z, Yu G, Lee HW, Li L, Wang L, Yang D. et al. The nedd8-activating enzyme inhibitor MLN4924 induces autophagy and apoptosis to suppress liver cancer cell growth. Cancer Res. 2012;72:3360-71
73. Xu J, Li L, Yu G, Ying W, Gao Q, Zhang W. et al. The neddylation-cullin 2-RBX1 E3 ligase axis targets tumor suppressor RhoB for degradation in liver cancer. Mol Cell Proteomics. 2015;14:499-509
74. Jin Y, Zhang P, Wang Y, Jin B, Zhou J, Zhang J. et al. Neddylation blockade diminishes hepatic metastasis by dampening cancer stem-like cells and angiogenesis in uveal melanoma. Clin Cancer Res. 2018;24:3741-54
75. Zhao B, Gao C, Shi D, Mao J, Zhao J, Guo L. et al. Knockdown of nedd8 conjugating enzyme UBE2M suppresses the proliferation and induces the apoptosis of intrahepatic cholangiocarcinoma cells. Oncol Rep. 2019;42:2670-9
76. Zhang Z, Heng Y, Cheng W, Pan Y, Ni S, Li H. Reactive oxygen species (ROS)-response nanomedicine through knocking down a novel therapeutic target NEDD8-conjugating enzyme UBC12 (UBE2M) in the treatment of liver cancer. Mater Design. 2021;204:109648
77. Yang D, Li L, Liu H, Wu L, Luo Z, Li H. et al. Induction of autophagy and senescence by knockdown of ROC1 E3 ubiquitin ligase to suppress the growth of liver cancer cells. Cell Death Differ. 2013;20:235-47
78. Liu W, Wang Y, Zhang CL, Huang BS, Bai J, Tian LJ. Cullin1 is up-regulated and associated with poor patients' survival in hepatocellular carcinoma. Int J Clin Exp Pathol. 2015;8:4001-7
79. Pan Y, Wang B, Yang X, Bai F, Xu Q, Li X. et al. Cul4A facilitates hepatocarcinogenesis by promoting cell cycle progression and epithelial-mesenchymal transition. Sci Rep. 2015;5:17006
80. Zhang D, Yang G, Li X, Xu C, Ge H. Inhibition of liver carcinoma cell invasion and metastasis by knockdown of cullin7 in vitro and in vivo. Oncol Res. 2016;23:171-81
81. Lai QY, He YZ, Peng XW, Zhou X, Liang D, Wang L. Histone deacetylase 1 induced by neddylation inhibition contributes to drug resistance in acute myelogenous leukemia. Cell Commun and Signal. 2019;17:86
82. Khalife J, Radomska HS, Santhanam R, Huang X, Neviani P, Saultz J. et al. Pharmacological targeting of miR-155 via the NEDD8-activating enzyme inhibitor MLN4924 (Pevonedistat) in FLT3-ITD acute myeloid leukemia. Leukemia. 2015;29:1981-92
83. Cao H, Xie J, Guo L, Han K, Pei Y, Li X. et al. All-trans retinoic acid induces autophagic degradation of ubiquitin-like modifier activating enzyme 3 in acute promyelocytic leukemia cells. Leuk Lymphoma. 2018;59:1222-30
84. Liu S, Wan J, Kong Y, Zhang Y, Wan L, Zhang Z. Inhibition of CRL-NEDD8 pathway as a new approach to enhance ATRA-induced differentiation of acute promyelocytic leukemia cells. Int J Med Sci. 2018;15:674-81
85. Liu C, Nie D, Li J, Du X, Lu Y, Li Y. et al. Antitumor effects of blocking protein neddylation in T315I-BCR-ABL leukemia cells and leukemia stem cells. Cancer Res. 2018;78:1522-36
86. Godbersen JC, Humphries LA, Danilova OV, Kebbekus PE, Brown JR, Eastman A. et al. The nedd8-activating enzyme inhibitor MLN4924 thwarts microenvironment-driven NF-kappaB activation and induces apoptosis in chronic lymphocytic leukemia B cells. Clin Cancer Res. 2014;20:1576-89
87. Godbersen JC, Paiva C, Danilova OV, Berger A, Brown JR, Danilov AV. Targeting neddylation effectively antagonizes nuclear factor-kappaB in chronic lymphocytic leukemia B-cells. Leuk Lymphoma. 2015;56:1566-9
88. Best S, Lam V, Liu T, Bruss N, Kittai A, Danilova OV. et al. Immunomodulatory effects of pevonedistat, a NEDD8-activating enzyme inhibitor, in chronic lymphocytic leukemia-derived T cells. Leukemia. 2021;35:156-68
89. Cao Y, Zhou H, Chen X, Li Y, Hu J, Zhou G. et al. Recent insight into the role of ring-finger E3 ligases in glioma. Biochem Soc Trans. 2021;49:519-29
90. Mansouri S, Zadeh G. Neddylation in glioblastomas. Neuro-Oncology. 2015;17:1305-6
91. Han S, Shin H, Oh JW, Oh YJ, Her NG, Nam DH. The protein neddylation inhibitor MLN4924 suppresses patient-derived glioblastoma cells via inhibition of ERK and AKT signaling. Cancers. 2019;11:1849
92. Filippova N, Yang X, An Z, Nabors LB, Pereboeva L. Blocking PD1/PDL1 interactions together with MLN4924 therapy is a potential strategy for glioma treatment. J Cancer Sci Ther. 2018;10:190-7
93. Zhou S, Zhao X, Yang Z, Yang R, Chen C, Zhao K. et al. Neddylation inhibition upregulates PD-L1 expression and enhances the efficacy of immune checkpoint blockade in glioblastoma. Int J Cancer. 2019;145:763-74
94. Zheng SH, Li ZH. Identification of a cullin5-ring E3 ligase transcriptome signature in glioblastoma multiforme. Aging. 2020;12:17380-92
95. Zheng S, Wu Y, Li Z. Integrating cullin2-ring E3 ligase as a potential biomarker for glioblastoma multiforme prognosis and radiosensitivity profiling. Radiother Oncol. 2021;154:36-44
96. Zhang W, Liang Y, Li L, Wang X, Yan Z, Dong C. et al. The nedd8-activating enzyme inhibitor MLN4924 (TAK-924/Pevonedistat) induces apoptosis via c-Myc-Noxa axis in head and neck squamous cell carcinoma. Cell Prolif. 2019;52:12536
97. Vanderdys V, Allak A, Guessous F, Benamar M, Read PW, Jameson MJ. et al. The neddylation inhibitor pevonedistat (MLN4924) suppresses and radiosensitizes head and neck squamous carcinoma cells and tumors. Mol Cancer Ther. 2018;17:368-80
98. Chairatvit K, Ngamkitidechakul C. Control of cell proliferation via elevated NEDD8 conjugation in oral squamous cell carcinoma. Mol Cell Bio. 2007;306:163-9
99. Seo SB, Baek JY, Lim JH, Jin X, Lee MY, Lee JH. 14-3-3zeta targeting induced senescence in Hep-2 laryngeal cancer cell through deneddylation of cullin1 in the skp1-cullin-F-box protein complex. Cell Prolif. 2019;52:12654
100. Darp R, Ceol C. Making a melanoma: molecular and cellular changes underlying melanoma initiation. Pigment Cell Melanoma Res. 2021;34:280-7
101. Cheng F, Chen H, Zhang L, Li RH, Liu Y, Sun JF. Inhibition of the NEDD8 conjugation pathway by shRNA to UBA3, the subunit of the NEDD8-activating enzyme, suppresses the growth of melanoma cells. Asian Pac J Cancer Prev. 2012;13:57-62
102. Cheng F, He R, Zhang L, Li H, Zhang W, Ji X. et al. Expression of neddylation-related proteins in melanoma cell lines and the effect of neddylation on melanoma proliferation. Oncol Lett. 2014;7:1645-50
103. Benamar M, Guessous F, Du K, Corbett P, Obeid J, Gioeli D. et al. Inactivation of the CRL4-CDT2-SET8/p21 ubiquitylation and degradation axis underlies the therapeutic efficacy of pevonedistat in melanoma. EBioMedicine. 2016;10:85-100
104. Wood EA, Lu Z, Jia S, Assumpcao A, Van Hesteren MA, Huelsmeyer MK. et al. Pevonedistat targeted therapy inhibits canine melanoma cell growth through induction of DNA re-replication and senescence. Vet Comp Oncol. 2020;18:269-80
105. Wong KM, Micel LN, Selby HM, Tan AC, Pitts TM, Bagby SM. et al. Targeting the protein ubiquitination machinery in melanoma by the NEDD8-activating enzyme inhibitor pevonedistat (MLN4924). Invest New Drugs. 2016;35:11-25
106. Brenner H, Schrotz-King P, Holleczek B, Katalinic A, Hoffmeister M. Declining bowel cancer incidence and mortality in germany: an analysis of time trends in the first ten years after the introduction of screening colonoscopy. Dtsch Arztebl Int. 2016;113:101-6
107. Lv Y, Li B, Han K, Xiao Y, Yu X, Ma Y. et al. The nedd8-activating enzyme inhibitor MLN4924 suppresses colon cancer cell growthviatriggering autophagy. Korean J Physiol Pha. 2018;22:617-25
108. Fotouhi O, Kjellin H, Juhlin CC, Pan Y, Vesterlund M, Ghaderi M. et al. Proteomics identifies neddylation as a potential therapy target in small intestinal neuroendocrine tumors. Oncogene. 2019;38:6881-97
109. Xu S, Ma Y, Tong Q, Yang J, Liu J, Wang Y. et al. Cullin-5 neddylation-mediated NOXA degradation is enhanced by PRDX1 oligomers in colorectal cancer. Cell Death Dis. 2021;12:265
110. Meehan KL, Holland JW, Dawkins HJ. Proteomic analysis of normal and malignant prostate tissue to identify novel proteins lost in cancer. Prostate. 2002;50:54-63
111. Wang X, Li L, Liang Y, Li C, Zhao H, Ye D. et al. Targeting the neddylation pathway to suppress the growth of prostate cancer cells: therapeutic implication for the men's cancer. Biomed Res Int. 2014;2014:974309
112. Rulina AV, Mittler F, Obeid P, Gerbaud S, Guyon L, Sulpice E. et al. Distinct outcomes of CRL-nedd8 pathway inhibition reveal cancer cell plasticity. Cell Death Dis. 2016;7:2505
113. Mittler F, Obeid P, Rulina AV, Haguet V, Gidrol X, Balakirev MY. High-content monitoring of drug effects in a 3D spheroid model. Front Oncol. 2017;7:293
114. Zhou X, Han S, Wilder-Romans K, Sun GY, Zhu H, Liu X. et al. Neddylation inactivation represses androgen receptor transcription and inhibits growth, survival and invasion of prostate cancer cells. Neoplasia. 2020;22:192-202
115. Fuentes-Antras J, Alcaraz-Sanabria AL, Morafraile EC, Noblejas-Lopez MDM, Galan-Moya EM, Baliu-Pique M. et al. Mapping of genomic vulnerabilities in the post-translational ubiquitination, sumoylation and neddylation machinery in breast cancer. Cancers. 2021;13:833
116. Jia X, Li C, Li L, Liu X, Zhou L, Zhang W. et al. Neddylation inactivation facilitates FOXO3a nuclear export to suppress estrogen receptor transcription and improve fulvestrant sensitivity. Clin Cancer Res. 2019;25:3658-72
117. Naik SK, Lam EW, Parija M, Prakash S, Jiramongkol Y, Adhya AK. et al. Neddylation negatively regulates ERRbeta expression to promote breast cancer tumorigenesis and progression. Cell Death Dis. 2020;11:703
118. Abnet CC, Arnold M, Wei WQ. Epidemiology of esophageal squamous cell carcinoma. Gastroenterology. 2018;154:360-73
119. Chen P, Hu T, Liang Y, Li P, Chen X, Zhang J. et al. Neddylation inhibition activates the extrinsic apoptosis pathway through ATF4-CHOP-DR5 axis in human esophageal cancer cells. Clin Cancer Res. 2016;22:4145-57
120. Liang Y, Jiang Y, Jin X, Chen P, Heng Y, Cai L. et al. Neddylation inhibition activates the protective autophagy through NF-κB-catalase-ATF3 Axis in human esophageal cancer cells. Cell Commun Signal. 2020;18:72
121. Hryniewicz-Jankowska A, Wierzbicki J, Tabola R, Stach K, Sossey-Alaoui K, Augoff K. The effect of neddylation inhibition on inflammation-induced MMP9 gene expression in esophageal squamous cell carcinoma. Int J Mol Sci. 2021;22:1716
122. Wang S, Xian J, Li L, Jiang Y, Liu Y, Cai L. et al. NEDD8-conjugating enzyme UBC12 as a novel therapeutic target in esophageal squamous cell carcinoma. Signal Transduct Target Ther. 2020;5:123
123. Song Q, Feng S, Peng W, Li A, Ma T, Yu B. et al. Cullin-ring ligases as promising targets for gastric carcinoma treatment. Pharmacol Res. 2021:105493.
124. Zhang Q, Hou D, Luo Z, Chen P, Lv B, Wu L. et al. The novel protective role of P27 in MLN4924-treated gastric cancer cells. Cell Death Dis. 2015;6:e1867
125. Lan H, Tang Z, Jin H, Sun Y. Neddylation inhibitor MLN4924 suppresses growth and migration of human gastric cancer cells. Sci Rep. 2016;6:24218
126. McMillin DW, Jacobs HM, Delmore JE, Buon L, Hunter ZR, Monrose V. et al. Molecular and cellular effects of NEDD8-activating enzyme inhibition in myeloma. Mol Cancer Ther. 2012;11:942-51
127. Gu Y, Kaufman JL, Bernal L, Torre C, Matulis SM, Harvey RD. et al. MLN4924, an NAE inhibitor, suppresses AKT and mTOR signaling via upregulation of REDD1 in human myeloma cells. Blood. 2014;123:3269-76
128. Milhollen MA, Traore T, Adams-Duffy J, Thomas MP, Berger AJ, Dang L. et al. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-κB-dependent lymphoma. Blood. 2010;116:1515-23
129. Wang Y, Luo Z, Pan Y, Wang W, Zhou X, Jeong LS. et al. Targeting protein neddylation with an NEDD8-activating enzyme inhibitor MLN4924 induced apoptosis or senescence in human lymphoma cells. Cancer Biol Ther. 2015;16:420-9
130. Pan WW, Zhou JJ, Yu C, Xu Y, Guo LJ, Zhang HY. et al. Ubiquitin E3 ligase CRL4(CDT2/DCAF2) as a potential chemotherapeutic target for ovarian surface epithelial cancer. J Biol Chem. 2013;288:29680-91
131. Hong X, Li S, Li W, Xie M, Wei Z, Guo H. et al. Disruption of protein neddylation with MLN4924 attenuates paclitaxel-induced apoptosis and microtubule polymerization in ovarian cancer cells. Biochem Bioph Res Co. 2019;508:986-90
132. Jemal A, Siegel R, Xu JQ, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277-300
133. Kuo KL, Ho IL, Shi CS, Wu JT, Lin WC, Tsai YC. et al. MLN4924, a novel protein neddylation inhibitor, suppresses proliferation and migration of human urothelial carcinoma: In vitro and in vivo studies. Cancer Lett. 2015;363:127-36
134. Tian DW, Wu ZL, Jiang LM, Gao J, Wu CL, Hu HL. Neural precursor cell expressed, developmentally downregulated 8 promotes tumor progression and predicts poor prognosis of patients with bladder cancer. Cancer Sci. 2019;110:458-67
135. Mery B, Espenel S, Guy JB, Rancoule C, Vallard A, Aloy MT. et al. Biological aspects of chondrosarcoma: Leaps and hurdles. Crit Rev Oncol Hematol. 2018;126:32-6
136. Wu MH, Lee CY, Huang TJ, Huang KY, Tang CH, Liu SH. et al. MLN4924, a protein neddylation inhibitor, suppresses the growth of human chondrosarcoma through inhibiting cell proliferation and inducing endoplasmic reticulum stress-related apoptosis. Int J Mol Sci. 2018;20:72
137. Lin WC, Kuo KL, Shi CS, Wu JT, Hsieh JT, Chang HC. et al. MLN4924, a novel nedd8-activating enzyme inhibitor, exhibits antitumor activity and enhances cisplatin-induced cytotoxicity in human cervical carcinoma: in vitro and in vivo study. Am J Cancer Res. 2015;5:3350-62
138. Gu C, Shi X, Dang X, Chen J, Chen C, Chen Y. et al. Identification of common genes and pathways in eight fibrosis diseases. Front Genet. 2020;11:627396
139. Sun C, Zhang H, Liu X. Emerging role of CCN family proteins in fibrosis. J Cell Physiol. 2021;236:4195-206
140. Lachiondo-Ortega S, Mercado-Gómez M, Serrano-Maciá M, Lopitz-Otsoa F, Salas-Villalobos T. B, Varela-Rey M, et al. Ubiquitin-like post-translational modifications (Ubl-PTMs): small peptides with huge impact in liver fibrosis. Cells. 2019;8:1575
141. Zhou WJ, Dong GJ, Ge G. et al. Evaluation of HZX-960, a novel DCN1-UBC12 interaction inhibitor as a potential antifibrotic compound for liver fibrosis. Biochem Cell Biol. 2022 doi 10.1139/bcb-2021-0585
142. Zhou H, Lu J, Chinnaswamy K, Stuckey JA, Liu L, McEachern D. et al. Selective inhibition of cullin 3 neddylation through covalent targeting DCN1 protects mice from acetaminophen-induced liver toxicity. Nat Commun. 2021;12:2621
143. Kandala S, Kim IM, Su HB. Neddylation and deneddylation in cardiac biology. Am J Cardiovasc Dis. 2014;4:140-58
144. Zou J, Ma W, Li J, Littlejohn R, Zhou H, Kim IM. et al. Neddylation mediates ventricular chamber maturation through repression of hippo signaling. PNAS. 2018;115:4101-10
145. He ZX, An Q, Wei B, Zhou WJ, Wei BF, Gong YP. et al. Discovery of potent and selective 2-(benzylthio)pyrimidine-based DCN1-UBC12 inhibitors for anticardiac fibrotic effects. J Med Chem. 2022;62:163-95
146. Lin Y, Chen Y, Feng W, Hua R, Zhang J, Huo Y. et al. Neddylation pathway alleviates chronic pancreatitis by reducing HIF1alpha-CCL5-dependent macrophage infiltration. Cell Death Dis. 2021;12:273
147. Brownell JE, Sintchak MD, Gavin JM, Liao H, Bruzzese FJ, Bump NJ. et al. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol Cell. 2010;37:102-11
148. Milhollen MA, Thomas MP, Narayanan U, Traore T, Riceberg J, Amidon BS. et al. Treatment-emergent mutations in NAEbeta confer resistance to the NEDD8-activating enzyme inhibitor MLN4924. Cancer Cell. 2012;21:388-401
149. Toth JI, Yang L, Dahl R, Petroski MD. A gatekeeper residue for NEDD8-activating enzyme inhibition by MLN4924. Cell Rep. 2012;1:309-16
150. Chen JJ, Tsu CA, Gavin JM, Milhollen MA, Bruzzese FJ, Mallender WD. et al. Mechanistic studies of substrate-assisted inhibition of ubiquitin-activating enzyme by adenosine sulfamate analogues. J Biol Chem. 2011;286:40867-77
151. Gires O, Xu GW, Toth JI, da Silva SR, Paiva SL, Lukkarila JL. et al. Mutations in UBA3 confer resistance to the NEDD8-activating enzyme inhibitor MLN4924 in human leukemic cells. PLoS One. 2014;9:93530
152. Sarantopoulos J, Shapiro GI, Cohen RB, Clark JW, Kauh JS, Weiss GJ. et al. Phase I study of the investigational NEDD8-activating enzyme inhibitor Pevonedistat (TAK-924/MLN4924) in patients with advanced solid tumors. Clin Cancer Res. 2016;22:847-57
153. Shah JJ, Jakubowiak AJ, O'Connor OA, Orlowski RZ, Harvey RD, Smith MR. et al. Phase I study of the novel investigational NEDD8-activating enzyme inhibitor Pevonedistat (MLN4924) in patients with relapsed/refractory multiple myeloma or lymphoma. Clin Cancer Res. 2016;22:34-43
154. Swords RT, Erba HP, DeAngelo DJ, Bixby DL, Altman JK, Maris M. et al. Pevonedistat (MLN4924), a first-in-Class NEDD8-activating enzyme inhibitor, in patients with acute myeloid leukaemia and myelodysplastic syndromes: a phase 1 study. Br J Haematol. 2015;169:534-43
155. Bhatia S, Pavlick AC, Boasberg P, Thompson JA, Mulligan G, Pickard MD. et al. A phase I study of the investigational NEDD8-activating enzyme inhibitor pevonedistat (TAK-924/MLN4924) in patients with metastatic melanoma. Invest New Drugs. 2016;34:439-49
156. Swords RT, Coutre S, Maris MB, Zeidner JF, Foran JM, Cruz J. et al. Pevonedistat, a first-in-class NEDD8-activating enzyme inhibitor, combined with azacitidine in patients with AML. Blood. 2018;131:1415-24
157. Kee Y, Huang M, Chang S, Moreau LA, Park E, Smith PG. et al. Inhibition of the nedd8 system sensitizes cells to DNA interstrand cross-linking agents. Mol Cancer Res. 2012;10:369-77
158. Megger DA, Abou-Eid S, Zulch B, Sitek B. Systematic analysis of synergistic proteome modulations in a drug combination of cisplatin and MLN4924. Mol Omics. 2018;14:450-7
159. Jazaeri AA, Shibata E, Park J, Bryant JL, Conaway MR, Modesitt SC. et al. Overcoming platinum resistance in preclinical models of ovarian cancer using the neddylation inhibitor MLN4924. Mol Cancer Ther. 2013;12:1958-67
160. Nawrocki ST, Kelly KR, Smith PG, Espitia CM, Possemato A, Beausoleil SA. et al. Disrupting protein neddylation with MLN4924 is a novel strategy to target cisplatin resistance in ovarian cancer. Clin Cancer Res. 2013;19:3577-90
161. Ho IL, Kuo KL, Liu SH, Chang HC, Hsieh JT, Wu JT. et al. MLN4924 synergistically enhances cisplatin-induced cytotoxicity via JNK and bcl-xL pathways in human urothelial carcinoma. Sci Rep. 2015;5:16948
162. Lin WC, Kuo KL, Shi CS, Wu JT, Hsieh JT, Chang HC. et al. MLN4924, a novel nedd8-activating enzyme inhibitor, exhibits antitumor activity and enhances cisplatin-induced cytotoxicity in human cervical carcinoma: in vitro and in vivo study. Am J Cancer Res. 2015;5:3350-62
163. Lin S, Shang Z, Li S, Gao P, Zhang Y, Hou S. et al. Neddylation inhibitor MLN4924 induces G2 cell cycle arrest, DNA damage and sensitizes esophageal squamous cell carcinoma cells to cisplatin. Oncol Lett. 2018;15:2583-9
164. Zeng Y, Iv YS, Pan QH, Zhou YG, Li H. An overactive neddylation pathway serves as a therapeutic target and MLN4924 enhances the anticancer activity of cisplatin in pancreatic cancer. Oncol Lett. 2019;18:2724-32
165. Misra S, Zhang XL, Wani NA, Sizemore S, Ray A. Both BRCA1-wild type and-mutant triple-negative breast cancers show sensitivity to the NAE inhibitor MLN4924 which is enhanced upon MLN4924 and cisplatin combination treatment. Oncotarget. 2020;11:784-800
166. Torka P, Mavis C, Kothari S, Belliotti S, Gu J, Sundaram S. et al. Pevonedistat, a NEDD8-activating enzyme inhibitor, induces apoptosis and augments efficacy of chemotherapy and small molecule inhibitors in pre-clinical models of diffuse large B-cell lymphoma. EJHaem. 2020;1:122-32
167. de Sousa GF, Lima Mde A, Custodio DF, Freitas VM, Monteiro G. Chemogenomic study of carboplatin in saccharomyces cerevisiae: inhibition of the neddylation process overcomes cellular resistance mediated by HuR and cullin proteins. PLoS One. 2015;10:0145377
168. Visconte V, Nawrocki ST, Espitia CM, Kelly KR, Possemato A, Beausoleil SA. et al. Comprehensive quantitative proteomic profiling of the pharmacodynamic changes induced by MLN4924 in acute myeloid leukemia cells establishes rationale for its combination with azacitidine. Leukemia. 2016;30:1190-4
169. Nawrocki ST, Kelly KR, Smith PG, Keaton M, Carraway H, Sekeres MA. et al. The NEDD8-activating enzyme inhibitor MLN4924 disrupts nucleotide metabolism and augments the efficacy of cytarabine. Clin Cancer Res. 2015;21:439-47
170. Moskowitz AJ. Bleomycin for older patients: less is more. Blood. 2016;127:2167-8
171. Zhou L, Chen S, Zhang Y, Kmieciak M, Leng Y, Li L. et al. The NAE inhibitor pevonedistat interacts with the HDAC inhibitor belinostat to target AML cells by disrupting the DDR. Blood. 2016;127:2219-30
172. El-Deiry WS, Tan M, Li Y, Yang R, Xi N, Sun Y. Inactivation of SAG E3 ubiquitin ligase blocks embryonic stem cell differentiation and sensitizes leukemia cells to retinoid acid. PLoS One. 2011;6:27726
173. Garcia K, Blank JL, Bouck DC, Liu XJ, Sappal DS, Hather G. et al. Nedd8-activating enzyme inhibitor MLN4924 provides synergy with mitomycin C through interactions with ATR, BRCA1/BRCA2, and chromatin dynamics pathways. Mol Cancer Ther. 2014;13:1625-35
174. Paiva C, Godbersen JC, Berger A, Brown JR, Danilov AV. Targeting neddylation induces DNA damage and checkpoint activation and sensitizes chronic lymphocytic leukemia B cells to alkylating agents. Cell Death Dis. 2015;6:1807
175. Guo ZP, Hu YC, Xie Y, Jin F, Song ZQ, Liu XD. et al. MLN4924 suppresses the BRCA1 complex and synergizes with PARP inhibition in NSCLC cells. Biochem Bioph Res Co. 2017;483:223-9
176. Langdon CG, Platt JT, Means RE, Iyidogan P, Mamillapalli R, Klein M. et al. Combinatorial screening of pancreatic adenocarcinoma reveals sensitivity to drug combinations including bromodomain inhibitor plus neddylation inhibitor. Mol Cancer Ther. 2017;16:1041-53
177. Li H, Zhou W, Li L, Wu J, Liu X, Zhao L. et al. Inhibition of neddylation modification sensitizes pancreatic cancer cells to gemcitabine. Neoplasia. 2017;19:509-18
178. Chen X, Cui D, Bi Y, Shu J, Xiong X, Zhao Y. AKT inhibitor MK-2206 sensitizes breast cancer cells to MLN4924, a first-in-class NEDD8-activating enzyme (NAE) inhibitor. Cell Cycle. 2018;17:2069-79
179. Bahjat M, de Wilde G, van Dam T, Maas C, Bloedjes T, Bende RJ. et al. The NEDD8-activating enzyme inhibitor MLN4924 induces DNA damage in Ph+ leukemia and sensitizes for ABL kinase inhibitors. Cell Cycle. 2019;18:2307-22
180. Yang Z, Zhang J, Lin X, Wu D, Li G, Zhong C. et al. Inhibition of neddylation modification by MLN4924 sensitizes hepatocellular carcinoma cells to sorafenib. Oncol Rep. 2019;41:3257-69
181. Yin Y, Xie CM, Li H, Tan M, Chen G, Schiff R. et al. The FBXW2-MSX2-SOX2 axis regulates stem cell property and drug resistance of cancer cells. PNAS. 2019;116:20528-38
182. Wei D, Li H, Yu J, Sebolt JT, Zhao L, Lawrence TS. et al. Radiosensitization of human pancreatic cancer cells by MLN4924, an investigational NEDD8-activating enzyme inhibitor. Cancer Res. 2012;72:282-93
183. El-Deiry WS, Yang D, Tan M, Wang G, Sun Y. The p21-dependent radiosensitization of human breast cancer cells by MLN4924, an investigational inhibitor of NEDD8 activating enzyme. PLoS One. 2012;7:34079
184. Oladghaffari M, Shabestani Monfared A, Farajollahi A, Baradaran B, Mohammadi M, Shanehbandi D. et al. MLN4924 and 2DG combined treatment enhances the efficiency of radiotherapy in breast cancer cells. Int J Radiat Biol. 2017;93:590-9
185. Wan J, Zhu J, Li G, Zhang Z. Radiosensitization of human colorectal cancer cells by MLN4924. Technol Cancer Res T. 2016;15:527-34
186. Wang XF, Zhang WJ, Yan Z, Liang YP, Li LH, Yu XL. et al. Radiosensitization by the investigational NEDD8-activating enzyme inhibitor MLN4924 (pevonedistat) in hormone-resistant prostate cancer cells. Oncotarget. 2016;7:38380-91
187. Lukkarila JL, da Silva SR, Ali M, Shahani VM, Xu GW, Berman J. et al. Identification of NAE inhibitors exhibiting potent activity in leukemia cells: exploring the structural determinants of NAE specificity. ACS Med Chem Lett. 2011;2:577-82
188. An H, Statsyuk AV. Development of activity-based probes for ubiquitin and ubiquitin-like protein signaling pathways. J Am Chem Soc. 2013;135:16948-62
189. An H, Statsyuk AV. An inhibitor of ubiquitin conjugation and aggresome formation. Chem Sci. 2015;6:5235-45
190. Yoshimura C, Muraoka H, Ochiiwa H, Tsuji S, Hashimoto A, Kazuno H. et al. TAS4464, a highly potent and selective inhibitor of NEDD8-activating enzyme, suppresses Neddylation and shows antitumor activity in diverse cancer models. Mol Cancer Ther. 2019;18:1205-16
191. Xiong C, Zhou L, Tan J, Song S, Bao X, Zhang N. et al. Development of potent NEDD8-activating enzyme inhibitors bearing a pyrimidotriazole scaffold. J Med Chem. 2021;64:6161-78
192. Muraoka H, Yoshimura C, Kawabata R, Tsuji S, Hashimoto A, Ochiiwa H. et al. Activity of TAS4464, a novel NEDD8 activating enzyme E1 inhibitor, against multiple myeloma via inactivation of nuclear factor κB pathways. Cancer Sci. 2019;110:3802-10
193. Yamamoto N, Shimizu T, Yonemori K, Kitano S, Kondo S, Iwasa S. et al. A first-in-human, phase 1 study of the NEDD8 activating enzyme E1 inhibitor TAS4464 in patients with advanced solid tumors. Invest New Drugs. 2021;39:1036-46
194. Zhang S, Tan J, Lai Z, Li Y, Pang J, Xiao J. et al. Effective virtual screening strategy toward covalent ligands: identification of novel NEDD8-activating enzyme inhibitors. J Chem Inf Model. 2014;54:1785-97
195. Leung CH, Chan DS, Yang H, Abagyan R, Lee SM, Zhu GY. et al. A natural product-like inhibitor of NEDD8-activating enzyme. Chem Commun. 2011;47:2511-3
196. Zhong HJ, Ma VPY, Cheng Z, Chan DSH, He HZ, Leung KH. et al. Discovery of a natural product inhibitor targeting protein neddylation by structure-based virtual screening. Biochimie. 2012;94:2457-60
197. Zhong HJ, Leung KH, Lin S, Chan DS, Han QB, Chan SL. et al. Discovery of deoxyvasicinone derivatives as inhibitors of NEDD8-activating enzyme. Methods. 2015;71:71-6
198. Li X, Pham V, Tippin M, Fu D, Rendon R, Song L. et al. Flavokawain B targets protein neddylation for enhancing the anti-prostate cancer effect of Bortezomib via Skp2 degradation. Cell Commun Signal. 2019;17:25
199. Li XS, Yokoyama NN, Zhang SY, Ding LN, Liu HM, Lilly MB. et al. Flavokawain A induces deneddylation and Skp2 degradation leading to inhibition of tumorigenesis and cancer progression in the TRAMP transgenic mouse model. Oncotarget. 2015;6:41809-24
200. Pham V, Rendon R, Le VX, Tippin M, Fu DJ, Le TH. et al. Gartanin is a novel neddylation inhibitor for induction of Skp2 degradation, FBXW2 expression, and autophagy. Mol Carcinogen. 2019;59:193-201
201. Zhong HJ, Yang H, Chan DS, Leung CH, Wang HM, Ma DL. A metal-based inhibitor of NEDD8-activating enzyme. PLoS One. 2012;7:49574
202. Zhong HJ, Wang W, Kang TS, Yan H, Yang Y, Xu L. et al. A rhodium(III) complex as an inhibitor of neural precursor cell expressed, developmentally down-regulated 8-activating enzyme with in vivo activity against inflammatory bowel disease. J Med Chem. 2016;60:497-503
203. Zhong HJ, Liu LJ, Chan DS, Wang HM, Chan PW, Ma DL. et al. Structure-based repurposing of FDA-approved drugs as inhibitors of NEDD8-activating enzyme. Biochimie. 2014;102:211-5
204. Wu KJ, Zhong HJ, Li G, Liu C, Wang HD, Ma DL. et al. Structure-based identification of a NEDD8-activating enzyme inhibitor via drug repurposing. Eur J Med Chem. 2018;143:1021-7
205. Ni S, Chen X, Yu Q, Xu Y, Hu Z, Zhang J. et al. Discovery of candesartan cilexetic as a novel neddylation inhibitor for suppressing tumor growth. Eur J Med Chem. 2020;185:111848
206. Chen X, Yang X, Mao F, Wei J, Xu Y, Li B. et al. Development of novel benzimidazole-derived neddylation inhibitors for suppressing tumor growth invitro and invivo. Eur J Med Chem. 2021;210:112964
207. Lu P, Liu X, Yuan X, He M, Wang Y, Zhang Q. et al. Discovery of a novel NEDD8 activating enzyme inhibitor with piperidin-4-amine scaffold by structure-based virtual screening. ACS Chem Biol. 2016;11:1901-7
208. Lu P, Guo Y, Zhu L, Xia Y, Zhong Y, Wang Y. A novel NAE/UAE dual inhibitor LP0040 blocks neddylation and ubiquitination leading to growth inhibition and apoptosis of cancer cells. Eur J Med Chem. 2018;154:294-304
209. Zhu L, Lu P, Gong L, Lu C, Li M, Wang Y. Design, synthesis, and biological evaluation of 4-amino substituted 2Hchromen- 2-one derivatives as an NEDD8 activating enzyme inhibitor in pancreatic cancer cells. Med Chem. 2020;16:969-83
210. Ma H, Zhuang C, Xu X, Li J, Wang J, Min X. et al. Discovery of benzothiazole derivatives as novel non-sulfamide NEDD8 activating enzyme inhibitors by target-based virtual screening. Eur J Med Chem. 2017;133:174-83
211. Song J, Cui XX, Wu BW, Li D, Wang SH, Shi L. et al. Discovery of 1,2,4-triazine-based derivatives as novel neddylation inhibitors and anticancer activity studies against gastric cancer MGC-803 cells. Bioorg Med Chem Lett. 2020;30:126791
212. Fu DJ, Cui XX, Zhu T, Zhang YB, Hu YY, Zhang LR. et al. Discovery of novel indole derivatives that inhibit neddylation and MAPK pathways against gastric cancer MGC803 cells. Bioorg Chem. 2021;107:104634
213. Li Y, Wang C, Xu T, Pan P, Yu Q, Xu L. et al. Discovery of a small molecule inhibitor of cullin neddylation that triggers ER stress to induce autophagy. Acta Pharm Sin B. 2021;11:3567-84
214. Fu DJ, Song J, Zhu T, Pang XJ, Wang SH, Zhang YB. et al. Discovery of novel tertiary amide derivatives as NEDDylation pathway activators to inhibit the tumor progression in vitro and in vivo. Eur J Med Chem. 2020;192:112153
215. Song J, Liu Y, Yuan XY, Liu WB, Li YR, Yu GX. et al. Discovery of 1,2,4-triazine dithiocarbamate derivatives as NEDDylation agonists to inhibit gastric cancers. Eur J Med Chem. 2021;225:113801
216. Yu Q, Jiang Y, Sun Y. Anticancer drug discovery by targeting cullin neddylation. Acta Pharm Sin B. 2019;10:746-65
217. Fang Y, Yu L, Liao GC, Yu B. Targeting the DCN1-UBC12 protein-protein interaction for selective modulation of cullin substrates. Curr Top Med Chem. 2018;18:1423-5
218. Zhou H, Lu J, Liu L, Bernard D, Yang CY, Fernandez-Salas E. et al. A potent small-molecule inhibitor of the DCN1-UBC12 interaction that selectively blocks cullin 3 neddylation. Nat Commun. 2017;8:1150
219. Zhou H, Zhou W, Zhou B, Liu L, Chern TR, Chinnaswamy K. et al. High-affinity peptidomimetic inhibitors of the DCN1-UBC12 protein-protein interaction. J Med Chem. 2018;61:1934-50
220. Scott DC, Hammill JT, Min J, Rhee DY, Connelly M, Sviderskiy VO. et al. Blocking an N-terminal acetylation-dependent protein interaction inhibits an E3 ligase. Nat Chem Biol. 2017;13:850-7
221. Hammill JT, Scott DC, Min J, Connelly MC, Holbrook G, Zhu F. et al. Piperidinyl ureas chemically control defective in cullin neddylation 1 (DCN1)-mediated cullin neddylation. J Med Chem. 2018;61:2680-93
222. Hammill JT, Bhasin D, Scott DC, Min J, Chen Y, Lu Y. et al. Discovery of an orally bioavailable inhibitor of defective in cullin neddylation 1 (DCN1)-mediated cullin neddylation. J Med Chem. 2018;61:2694-706
223. Kim HS, Hammill JT, Scott DC, Chen Y, Min J, Rector J. et al. Discovery of novel pyrazolo-pyridone DCN1 inhibitors controlling cullin neddylation. J Med Chem. 2019;62:8429-42
224. Kim HS, Hammill JT, Scott DC, Chen Y, Rice AL, Pistel W. et al. Improvement of oral bioavailability of pyrazolo-pyridone inhibitors of the interaction of DCN1/2 and UBE2M. J Med Chem. 2021;64:5850-62
225. Wang S, Zhao L, Shi XJ, Ding L, Yang L, Wang ZZ. et al. Development of highly potent, selective, and cellular active triazolo[1,5- a]pyrimidine-based inhibitors targeting the DCN1-UBC12 protein-protein interaction. J Med Chem. 2019;62:2772-97
226. Zhou W, Ma L, Ding L, Guo Q, He Z, Yang J. et al. Potent 5-cyano-6-phenyl-pyrimidin-based derivatives targeting DCN1-UBE2M interaction. J Med Chem. 2019;62:5382-403
227. Zhou W, Xu C, Dong G, Qiao H, Yang J, Liu H. et al. Development of phenyltriazole thiol-based derivatives as highly potent inhibitors of DCN1-UBC12 interaction. Eur J Med Chem. 2021;217:113326
228. Yu Q, Hu Z, Shen Y, Jiang Y, Pan P, Hou T. et al. Gossypol inhibits cullin neddylation by targeting SAG-Cul5 and RBX1-Cul1 complexes. Neoplasia. 2020;22:179-91
229. Yu Q, Sun Y. Targeting protein neddylation to inactivate cullin-ring ligases by gossypol: a lucky hit or a new start? Drug Des Devel Ther. 2021;15:1-8
230. Xu T, Ma Q, Li Y, Yu Q, Pan P, Zheng Y. et al. A small molecule inhibitor of the UBE2F-CRL5 axis induces apoptosis and radiosensitization in lung cancer. Signal Transduct Target Ther. 2022;7:354
231. Chen YF, Liu RZ, Ying WW, Yang YN, Xiang SF, Shao XJ. et al. Arctigenin impairs UBC12 enzyme activity and cullin neddylation to attenuate cancer cells. Acta Pharmacol Sin. 2023;44:661-669
232. Callaway E. Deepmind's AI predicts structures for a vast trove of proteins. Nature. 2021;595:635
Author biographies
Li-Ying Ma received his PhD degree from the Department of Chemistry of Zhengzhou University (China, 2011-2015). He was subsequently appointed as an assistant professor at Zhengzhou University, and thereafter promoted to associate professor in Medicinal Chemistry, School of Pharmaceutical Sciences. His research interest is the discovery and development of novel small‐molecule therapeutics for cancer treatment. To date, Dr. Ma has published over 30 peer reviewed papers in drug discovery and related area.
Wen Zhao received her B.S. in Henan Medical University and Ph.D. in Beijing Medical University. She used to work in Dr. Evangelia G. Kranias' lab as a postdoctoral fellow and research associate in University of Cincinnati College of Medicine, Cincinnati, Ohio, in 1999-2011. She was appointed a tenured professor and PI for new drug discovery in anticancer and cardiovascular diseases in Zhengzhou University School of Pharmaceutical Sciences in 2011. To date, Dr. Zhao has published over 56 peer reviewed papers in drug discovery and related area.
![]() Corresponding author: Zhao Wen, Ph.D, zhaowen100com and Ma Li-Ying, Ph.D, maliyingedu.cn.
Corresponding author: Zhao Wen, Ph.D, zhaowen100com and Ma Li-Ying, Ph.D, maliyingedu.cn.