Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2023; 13(12):4042-4058. doi:10.7150/thno.84388 This issue Cite
Research Paper
HMGB1-mediated elevation of KLF7 facilitates hepatocellular carcinoma progression and metastasis through upregulating TLR4 and PTK2
Weibo Feng1#, Jie Chen1#, Wenjie Huang2,3#, Guodong Wang1#, Xilang Chen1#, Lili Duan1, Yue Yin1, Xiaoping Chen3, Bixiang Zhang3, Mengyu Sun2, Xiangyuan Luo2, Yongzhan Nie1, Daiming Fan1 ![]() , Kaichun Wu1
, Kaichun Wu1 ![]() , Limin Xia1,2
, Limin Xia1,2 ![]()
1. State Key Laboratory of Holistic Integrative Management of Gastrointestinal Cancers and National Clinical Research Center for Digestive Diseases, Xijing Hospital of Digestive Diseases, Fourth Military Medical University, Xi'an 710032, Shaanxi Province, China.
2. Department of Gastroenterology, Institute of Liver and Gastrointestinal Diseases, Hubei Key Laboratory of Hepato-Pancreato-Biliary Diseases, Tongji Hospital of Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, Hubei Province, China.
3. Hubei Key Laboratory of Hepato-Pancreato-Biliary Diseases; Hepatic Surgery Center, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology; Clinical Medicine Research Center for Hepatic Surgery of Hubei Province; Key Laboratory of Organ Transplantation, Ministry of Education and Ministry of Public Health, Wuhan, Hubei, 430030, China.
#These authors contributed equally to this work.
Received 2023-3-17; Accepted 2023-7-5; Published 2023-7-14
Abstract

Background: Metastasis is a major cause of HCC-related deaths with no effective pharmacotherapies. Chronic inflammation promotes HCC dissemination, however, its underlying mechanisms are not fully understood. Here, we investigated the role of Krüppel-like factor 7 (KLF7) in inflammation-provoked HCC metastasis and proposed therapeutic strategies for KLF7-positive patients.
Methods: The expression of KLF7 in human HCC specimens were examined by immunohistochemistry and quantitative real-time PCR. The luciferase reporter assays and chromatin immunoprecipitation assays were conducted to explore the transcriptional regulation related to KLF7. Orthotopic xenograft models and DEN/CCl4-induced HCC models were established to evaluate HCC progression and metastasis.
Results: KLF7 overexpression promotes HCC metastasis through transactivating toll-like receptor 4 (TLR4) and protein tyrosine kinase 2 (PTK2) expression. High mobility group box 1 (HMGB1) upregulates KLF7 expression through the TLR4/advanced glycosylation end-product specific receptor (RAGE)-PI3K-AKT-NF-κB pathway, forming an HMGB1-KLF7-TLR4 positive feedback loop. The HMGB1-KLF7-TLR4/PTK2 axis is gradually activated during the progression of inflammation-HCC transition. Genetic depletion of KLF7 impedes HMGB1-mediated HCC progression and metastasis. The combined application of TLR4 inhibitor TAK-242 and PTK2 inhibitor defactinib alleviates HCC progression and metastasis induced by the HMGB1-KLF7 axis. In human HCCs, KLF7 expression is positively correlated with cytoplasmic HMGB1, p-p65, TLR4, and PTK2 levels, and patients positively co-expressing HMGB1/KLF7, p-p65/KLF7, KLF7/TLR4 or KLF7/PTK2 exhibit the worst prognosis.
Conclusions: HMGB1-induced KLF7 overexpression facilitates HCC progression and metastasis by upregulating TLR4 and PTK2. Genetic ablation of KLF7 via AAV gene therapy and combined blockade of TLR4 and PTK2 represents promising therapy strategies for KLF7-positive HCC patients.
Keywords: krüppel-like factor 7, toll-like receptor 4, protein tyrosine kinase 2, high mobility group box 1, hepatocellular carcinoma
Introduction
Hepatocellular carcinoma (HCC) is a lethal malignancy with a dismal prognosis, which remains a global health challenge lacking effective intervention [1]. Although the evolution of systemic therapies has enriched therapeutic drugs for unresectable HCC, patients commonly receive limited benefits owing to the low objective response rate (ORR) and acquired resistance [2]. High genetic heterogeneity of HCC is considered the primary cause of treatment failure, and combination therapies have been proposed to overcome this obstacle [3]. Recently, the combination of atezolizumab and bevacizumab has become the first-line treatment for HCC instead of sorafenib. Several ongoing clinical trials of combination therapy also show encouraging results in improving HCC prognosis, indicating the exciting prospect of combination strategies in future HCC treatment [1]. Currently, early recurrence and distant metastasis remain major hurdles in the clinical management of HCC. Hence, it is necessary to further elucidate the molecular mechanisms underlying HCC metastasis and propose feasible combination strategies for certain HCC subpopulations based on specific molecular signatures.
Dysregulated transcription factors that shape the aberrant cancer transcriptome are significant drivers of tumor progression and metastasis, thus representing a unique class of therapeutic targets [4]. Krüppel-like factors (KLFs), a family of zinc finger-containing transcription factors with 17 members in humans, are involved in the onset and progression of human cancers by regulating various cancerous phenotypes [5]. We profiled the expression patterns of KLF genes in paired HCC tissues and identified KLF7 as the most upregulated gene that significantly promoted HCC cell migration and proliferation. KLF7 is a well-defined oncogenic factor and an unfavorable prognostic biomarker in pancreatic ductal adenocarcinoma (PDAC), endometrial cancer and glioma [6-8]. A recent study reported that KLF7 is up-regulated in human HCC and enhances tumor proliferation and invasion [9]. Nevertheless, the functional role of KLF7 in HCC metastasis remains elusive, and further exploration of its oncogenic mechanisms in HCC may lead to new translational discoveries in HCC diagnosis and treatment.
The feedforward cycle of inflammation and hepatocyte death plays a critical role in the pathogenesis of chronic liver disease and HCC [10]. HMGB1, a non-histone chromatin-associated protein, can be passively released from necrotic cells or actively secreted from immunocompetent cells during liver injury and inflammation [11]. As a typical damage-associated molecular pattern (DAMP), extracellular HMGB1 transmits danger signals to neighboring cells by interacting with its classical receptors, including Toll-like receptors 2/4/9 (TLR2/4/9) and receptor for advanced glycation end products (RAGE) [12]. Numerous studies have suggested that inflammation-provoked HMGB1 release promotes HCC initiation, progression and metastasis via multiple mechanisms [13-15]. Here, we found that extracellular HMGB1 induced KLF7 upregulation in HCC cells and explored the functional role of KLF7 in HMGB1-mediated HCC development and metastasis.
In this study, we reported that HMGB1-induced KLF7 overexpression facilitated HCC development and metastasis through upregulating TLR4 and protein tyrosine kinase 2 (PTK2). Adeno-associated virus 8 (AAV8)-mediated KLF7 knockout in hepatocytes or combination of TLR4 inhibitor TAK-242 and PTK2 antagonist defactinb effectively suppressed HMGB1-KLF7 axis-driven HCC development and metastasis.
Materials and Methods
Establishment of Orthotopic HCC Models
Male BALB/C nude mice (5 weeks old) were fed under standard conditions and cared for in accordance with the institutional guidelines for animal care. For orthotopic implantation, 50 µL PBS/Matrigel mixture with 2×106 HCC cells was injected into the left liver lobe of mice via an 8 mm epigastric incision under anesthesia. In vivo liver tumor development and metastasis were monitored using bioluminescence. To detect in vivo signals, mice were intraperitoneally injected with D-luciferin (Xenogen, Hopkinton, MA, USA) at a dose of 100 mg/kg, and bioluminescence was recorded using an IVIS 100 Imaging System (Xenogen). The survival of the mice was recorded daily. After 9 weeks, the mice were sacrificed and their lungs were dissected for standard histological examination. These animal experiments were approved by the Committee on the Use of Live Animals in Teaching and Research (CULATR), Fourth Military Medical University.
DEN/CCl4-induced HCC models
For chemically induced HCC, male C57BL/6 mice were intraperitoneally injected with DEN (25mg/kg) at week 2 postpartum and subsequently injected weekly with CCl4 (0.5 mL/kg, i.p., diluted with corn oil) from 4-week-old to 15-week-old. Mice were sacrificed at 34 weeks for statistical analysis or raised until death for survival analysis. All animal experiments were approved by the Committee on the Use of Live Animals in Teaching and Research (CULATR), Fourth Military Medical University.
Additional materials and methods are available in the online supplementary materials.
Results
KLF7 upregulation predicts poor clinical outcomes and high metastatic potentials in human HCC
Dysregulated KLF genes contribute to cancer development and metastasis [5]. To identify dysregulated KLFs in HCC, we analyzed the expression profiles of KLF genes in HCC samples. Compared to para-tumor specimens, the mRNA levels of KLF5, KLF7, KLF8, and KLF13 were elevated, whereas those of KLF4, KLF6, and KLF10 were reduced in HCC specimens. The expressions of KLF2, KLF3, KLF9, KLF11, KLF12, KLF15, and KLF16 did not differ between adjacent non-tumor tissues and HCC tissues. Moreover, KLF1 and KLF17 were undetectable in HCC tissues (Figure S1). To preliminarily explore the function of the upregulated KLFs in HCC malignant behavior, we individually knocked down the four KLFs in MHCC97H cells and performed in vitro assays. Notably, KLF7 exhibited the largest fold change among the elevated KLFs and KLF7 downregulation significantly inhibited HCC cell migration, invasion, and proliferation (Figure S2). Based on these findings, we focused on KLF7 in further studies.
We next profiled the protein levels of KLF7 in two independent HCC cohorts using immunohistochemical (IHC) staining. KLF7 was predominantly localized in the nucleus and showed markedly elevated intensity in HCC tissues compared to para-cancer tissues (Figure 1A). In our HCC cohorts, KLF7-positive patients showed poorer OS and higher relapse probabilities than KLF7-negative patients (Figure 1B). Moreover, KLF7-positive patients exhibited a higher incidence of absent tumor encapsulation, microvascular invasion, poor tumor differentiation, and advanced tumor-node-metastasis (TNM) stage (Table S1). Positive KLF7 expression was also an independent predictor of poor OS and high recurrence in HCC patients, according to multivariate analysis (Table S2). HCC samples had higher KLF7 mRNA expression than adjacent non-tumor samples and normal livers (Figure 1C), and HCC patients who had ever undergone recurrence or metastasis displayed elevated KLF7 expression compared to those without recurrence or metastasis (Figure 1D). The protein levels of KLF7 were also markedly higher in metastatic HCC tissues than in primary tumors (Figure 1E), which was supported by online transcriptome data (Figure 1F). These findings collectively suggested that KLF7 elevation may be a key driver of HCC progression, especially in HCC metastasis.
KLF7 overexpression indicates poor prognosis in HCC patients and promotes HCC metastasis. (A) Representative IHC images and IHC scores of KLF7 staining in para-cancerous nontumor specimens and HCC specimens in two independent microarrays. (B) Kaplan-Meier analysis of the correlation of KLF7 expression with overall survival and recurrence in two independent HCC cohorts. (C) Relative KLF7 mRNA expression in 20 normal liver samples and 50 pairs of para-cancer nontumor and HCC tissues. (D) The mRNA expression of KLF7 in primary HCC samples from patients without or with recurrence (left) and metastasis (right). (E) Representative IHC pictures and IHC scores of KLF7 staining in adjacent nontumorous samples, HCC samples, and matched metastatic HCC samples. (F) The gene expression of KLF7 in primary live tumors (n = 806) and liver cancer metastatic tissues (n = 24) was analyzed. Data from the TNMplot database (https://tnmplot.com/analysis/). (G) The protein levels of KLF7 in human HCC cell lines (left). KLF7 upregulation and knockdown in indicated HCC cells after lentiviral transfection were confirmed by immunoblotting (right). (H) The motility of indicated HCC cell lines with KLF7 expression changes were detected by transwell assays. (I-M) Effect of KLF7 overexpression on HCC metastasis were evaluated by orthotopic HCC models. (I) Representative bioluminescent images of hepatic tumors and incidence of pulmonary metastases. (J) Dynamic intensity of bioluminescent signals of liver tumors. (K) Number of lung metastatic nodules. (L) Overall survival time of nude mice. (M) Representative H&E staining of lung tissues. *P < 0.05. Abbreviations: IHC, Immunohistochemical; H&E, hematoxylin and eosin.
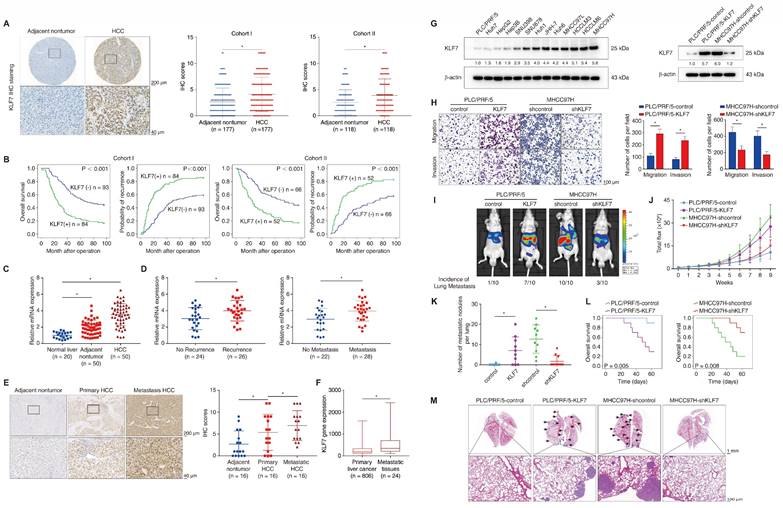
KLF7 overexpression promotes HCC metastasis
KLF7 expression was examined in different HCC cell lines (Figure 1G, left). PLC/PRF/5 cells with low endogenous KLF7 expression and low metastatic capabilities and MHCC97H cells with high endogenous KLF7 expression and high metastatic capabilities were chosen for gain and loss-of-function studies (Figure 1G, right). KLF7 upregulation enhanced the migration and invasion of PLC/PRF/5 cells, whereas KLF7 knockdown impaired the mobility of MHCC97H cells (Figure 1H). KLF7 overexpression also decreased epithelial marker E-cadherin expression and increased mesenchymal marker vimentin expression in HCC cells, and vice versa (Figure S3). Orthotopic HCC models further revealed that mice implanted with PLC/PRF/5-KLF7 cells exhibited higher pulmonary metastasis and shorter OS than those in the control group. In contrast, mice from the MHCC97H-shKLF7 group showed lower pulmonary metastasis rates and prolonged OS than those from the MHCC97H-shcontrol group (Figure 1I-M). These observations demonstrated that KLF7 promotes HCC metastasis.
KLF7 upregulates TLR4 and PTK2 in HCC cells
To investigate the molecular mechanism underlying KLF7-mediated HCC metastasis, we compared transcriptome changes between PLC/PRF/5-control and PLC/PRF/5-KLF7 cells using a Human Liver cancer RT2 Profiler PCR Array. Fourteen of the eighty-four genes were upregulated (fold change > 2.0) after KLF7 overexpression (Figure 2A, Table S3). Of particular interest were TLR4 and PTK2, which were ranked as the two most upregulated genes. Considering that both TLR4 and PTK2 are upregulated in HCC tissues and play critical roles in promoting HCC metastasis [16, 17], we further explored whether they are downstream effectors in KLF7-mediated HCC metastasis. KLF7 overexpression upregulated TLR4 and PTK2 expression, whereas KLF7 knockdown decreased TLR4 and PTK2 levels in HCC cells (Figure 2B). The luciferase activities of TLR4 and PTK2 promoters were also increased in KLF7-overexpressing cells, indicating that TLR4 and PTK2 were transcriptionally activated by KLF7 in HCC cells (Figure 2C).
KLF7 fosters HCC metastasis through transcriptionally upregulating TLR4 and PTK2 expression. (A) Differentially-expressed genes between PLC/PRF/5-KLF7 cells and PLC/PRF/5-control cells were detected using A human liver cancer PCR array. (B) The mRNA and protein levels of TLR4 and PTK2 in PLC/PRF/5 cells with KLF7 overexpression or in MHCC97H cells with KLF7 knockdown. (C) Relative luciferase activities of TLR4 and PTK2 promotor reporter plasmids in PLC/PRF/5 cells co-transfected with pCMV-KLF7 or pCMV-Taq. (D-E) Serially truncated/mutated TLR4 or PTK2 promotor constructs were co-transfected with pCMV-KLF7 into PLC/PRF/5 cells for testing luciferase activities. (F-G) ChIP assays showed KLF7 directly bound to the TLR4 and PTK2 promoters in HCC cells and HCC specimens. (H) TLR4 and PTK2 knockdown in KLF7-overexpressing PLC/PRF/5 cells and upregulation in MHCC97H cells with KLF7 knockdown were confirmed by western blot. (I) The migratory and invasive capacities of the indicated HCC cell lines were evaluated by transwell assays. (J-N) In vivo metastatic experiments revealed that TLR4 and PTK2 upregulation was essential for KLF7-fostered HCC metastasis. (J) Representative bioluminescent pictures of liver tumors and rate of lung metastasis. (K) Bioluminescent signals of liver tumors were dynamically monitored. (L) Number of lung-colonizing nodules. (M) Typical H&E pictures of metastatic lung nodules. (N) Overall survival of different groups. *P < 0.05.
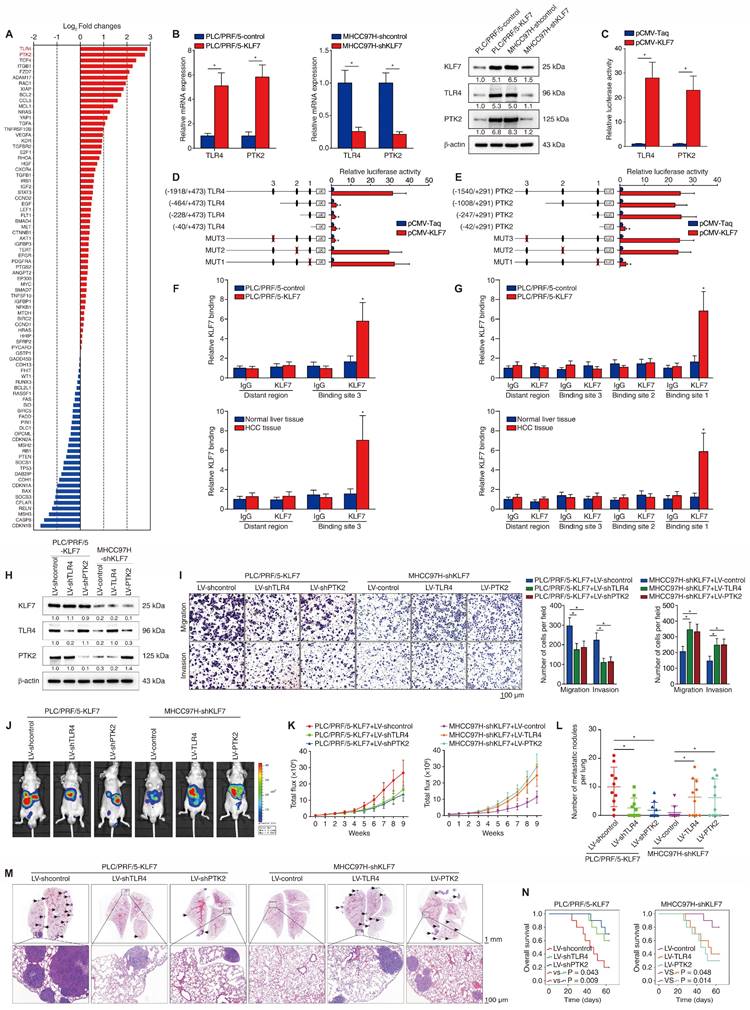
Through sequence analysis, we identified multiple putative KLF7-binding motifs in both the TLR4 promoter (n = 3) and the PTK2 promoter (n =3) (Figure S4, S5). Serially truncated or mutated TLR4 promoter reporter plasmids were constructed to explore the functions of these motifs in the transcriptional regulation of TLR4. Deletion of the fragment from -1918 to -464 base pairs (bp) significantly impaired KLF7-induced TLR4 promoter activation. Consistently, mutation of putative KLF7-binding motif 3 within this region also abolished KLF7-activated TLR4 promoter activity (Figure 2D). Likewise, we used the same approach to characterize KLF7-dependent cis elements in the PTK2 promoter and identified that the putative KLF7-binding motif 1 in the -247bp ~ -42bp fragment was required for KLF7-induced PTK2 promoter transactivation (Figure 2E). A chromatin immunoprecipitation (ChIP) assay further validated that KLF7 directly bound to the TLR4 and PTK2 promoters in both HCC cells and human HCC samples (Figure 2F-G). These results demonstrated that KLF7 transcriptionally upregulates TLR4 and PTK2 in human HCC cells.
TLR4 and PTK2 are essential for KLF7-provoked HCC metastasis
Previous studies have demonstrated that both TLR4 and PTK2 play important roles in promoting HCC metastasis [16, 17]. To investigate whether TLR4 and PTK2 function in KLF7-mediated HCC metastasis, we downregulated TLR4 or PTK2 expression in KLF7-overexpressing PLC/PRF/5 cells and upregulated TLR4 or PTK2 in MHCC97H cells with KLF7 knockdown (Figure 2H). TLR4 or PTK2 knockdown attenuated the migration and invasion of PLC/PRF/5-KLF7 cells, whereas TLR4 or PTK2 upregulation largely rescued the mobility defects of MHCC97H-shKLF7 cells (Figure 2I). In vivo metastatic assays demonstrated that TLR4 or PTK2 knockdown in PLC/PRF/5-KLF7 cells dramatically reduced pulmonary metastatic loads while extending the OS time of nude mice. Reciprocally, upregulation of TLR4 or PTK2 rescued the decline in pulmonary metastasis and reduced OS time in mice implanted with MHCC97H-shKLF7 cells (Figure 2J-N). Additionally, the orthotopic HCC models showed that the impact of knockdown or overexpression of TLR4 or PTK2 on HCC metastasis was less significant in the KLF7 non-manipulated state than in the KLF7 manipulated state, further demonstrating that the TLR4 and PTK2 effects are downstream of KLF7 (Figure S6). These findings indicated that TLR4 and PTK2 are essential downstream effectors of KLF7-mediated HCC metastasis.
KLF7 expression positively correlated with TLR4 and PTK2 expression in human HCC specimens
IHC staining was performed to evaluate the clinical relevance of KLF7, TLR4, and PTK2 in HCC cohorts. Representative IHC staining of KLF7, TLR4, and PTK2 in HCC tissues and adjacent non-tumor tissues is presented (Figure 3A). Correlation analysis indicated that KLF7 expression was positively associated with TLR4 and PTK2 expression in both cohorts (Figure 3B). Positive TLR4 or PTK2 expression was accompanied by incomplete tumor encapsulation, microvascular invasion, poor differentiation, advanced TNM stage, poor OS, and high recurrence rates in our cohorts (Figure 3C-D, Table S4, S5). Notably, the subgroups of HCC patients co-expressing KLF7/TLR4 or KLF7/PTK2 displayed the worst prognosis in our cohorts (Figure 3E-F). Online bioinformatics analysis further validated these clinical observations. In TCGA LIHC samples, positive correlations were observed between KLF7 and TLR4 or PTK2 expression, and HCC patients with high levels of KLF7/TLR4 or KLF7/PTK2 co-expression exhibited an obviously poorer OS than patients with low levels (Figure S7A -B).
KLF7 expression is positively correlated with TLR4 and PTK2 expression in human HCC. (A) Representative IHC images of KLF7, TLR4 and PTK2 staining in adjacent nontumorous and HCC tissues. (B) The correlations between KLF7 expression and TLR4 or PTK2 expression in two HCC cohorts. (C, D) The prognostic significance of TLR4 or PTK2 expression on overall survival and recurrence rates in two HCC cohorts. (E, F) The correlation of KLF7/TLR4 or KLF7/PTK2 co-expression with overall survival and recurrence rates in two HCC cohorts. *P < 0.05.
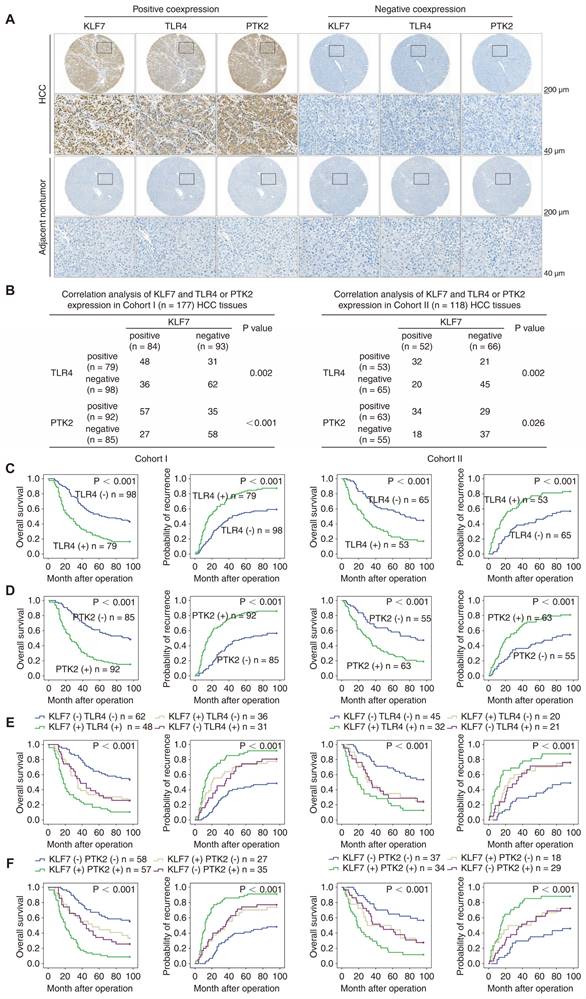
To evaluate the pro-metastatic properties of KLF7, TLR4, and PTK2 in clinical samples, we compared their expression levels in 20 pairs of primary and metastatic HCC tissues. KLF7, TLR4, and PTK2 levels were markedly elevated in metastatic HCC tissues compared to those in primary HCC and adjacent non-neoplastic tissues (Figure S8). Correspondingly, by employing gene signature analysis in online LIHC databases, we found that the mean expression of the gene set composed of KLF7, TLR4 and PTK2 was elevated in liver cancer tissues compared to normal livers, and further increased in liver cancer metastatic tissues (Figure S9A). High levels of this gene set predicted much poorer survival in HCC samples (Figure S9B). These clinical observations collectively supported the conclusion that KLF7 promotes HCC metastasis by upregulating TLR4 and PTK2.
Extracellular HMGB1 upregulates KLF7 expression through the TLR4/RAGE-PI3K-AKT-NF-κB signaling pathway
The upstream mechanism of KLF7 dysregulation in HCC cells still remains unclear. Chronic inflammation is a recognized hallmark of HCC and contributes to HCC metastasis [18]. Inflammatory factors, including cytokines, damage-associated molecular patterns (DAMPs) and gut-derived pathogen-associated molecular patterns (PAMPs), are vital pro-metastatic signals in the HCC tumor microenvironment (TME) that can aberrantly activate pro-metastatic genes and pathways within HCC cells [18, 19]. To determine whether proinflammatory factors triggered KLF7 overexpression in HCC cells, PLC/PRF/5 and Huh7 cells with low endogenous KLF7 expression were stimulated with a panel of inflammatory factors (IL-1β, IL-6, IL-17A, IL-8, TNF-α, TGF-β, LPS and HMGB1) enriched under chronic liver inflammation [19, 20]. In both cell lines, HMGB1 was the most powerful inducer of KLF7 overexpression among all factors (Figure 4A). HMGB1 treatment induced KLF7 expression in a dose-dependent manner and significantly enhanced luciferase activity of KLF7 promoter (Figure 4B-C). These observations suggested that extracellular HMGB1 upregulates KLF7 in HCC cells.
HMGB1 induces KLF7 expression through the TLR4/RAGE-PI3K-AKT-NF-κB signaling pathway. (A) PLC/PRF/5 and Huh7 cells were incubated with a panel of inflammatory factors for 24 hours, and KLF7 expression was then examined. (B) Western blotting and real-time PCR analysis of KLF7 expression in PLC/PRF/5 and Huh7 cells after HMGB1 treatment at a series of gradient doses for 24 hours. (C) The luciferase reporter activity of the KLF7 promoter was measured in PLC/PRF/5 and Huh7 cells treated with HMGB1. (D) PLC/PRF/5 cells were transfected with LV-shTLR4, LV-shRAGE or a combination before HMGB1 treatment, KLF7, TLR4 and RAGE expression were then detected (left). TLR4 and RAGE inhibitors were applied in PLC/PRF/5 cells under HMGB1 treatment, and KLF7 expression was then examined (right). (E) PLC/PRF/5 cells were transfected with serially truncated or mutated KLF7 promoter constructs and treated with or without HMGB1. Luciferase reporter activities were then detected. (F-G) PLC/PRF/5 cells were treated with NF-κB inhibitor BAY 11-7082 (F) or transfected with LV-shp65 (G) before HMGB1 stimulation. KLF7 expression and KLF7 promoter activity were measured by western blotting, real-time PCR and luciferase reporter assays. (H) PLC/PRF/5 cells were precultured with inhibitors specific to PI3K, JNK, ERK and p38 before HMGB1 treatment. KLF7 expression as well as the levels of phosphorylated and total AKT, JNK, ERK, p38 and p65 were then detected. (I) A ChIP assay was conducted to examine the relative enrichment of p65 on the KLF7 promoter when PLC/PRF/5 cells were treated with HMGB1 and inhibitors of PI3K, JNK, ERK and p38.
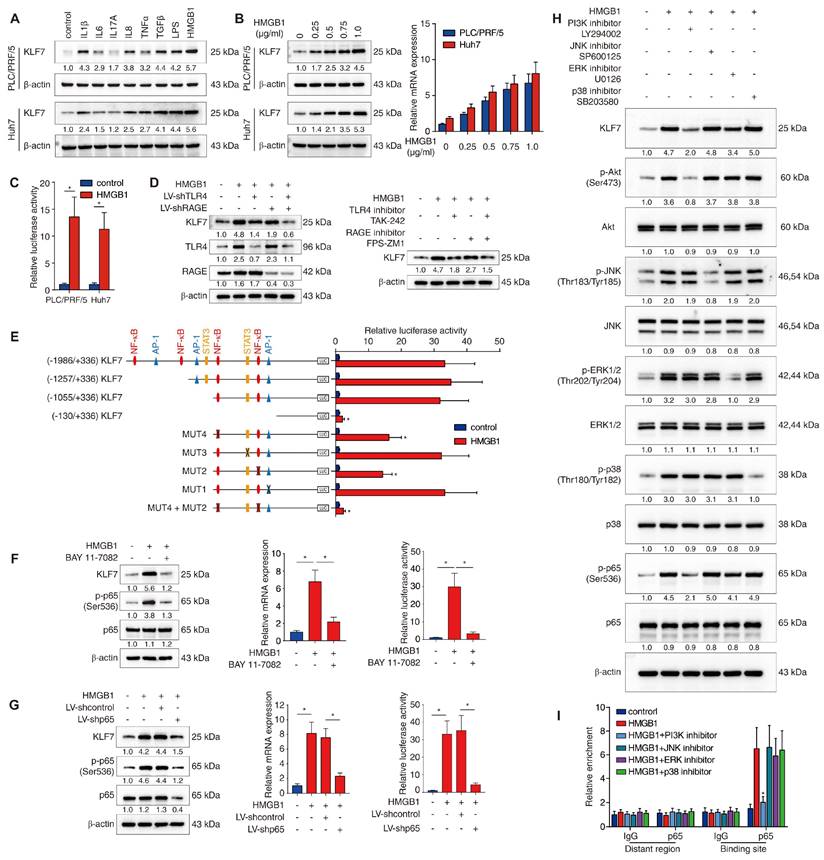
TLR4 and RAGE function as primary HMGB1 receptors and participate in HMGB1-mediated HCC metastasis [15, 21]. To explore which receptor is required for HMGB1-induced KLF7 expression, PLC/PRF/5 cells were transfected with TLR4 knockdown, RAGE knockdown, or combined knockdown before HMGB1 treatment. Combined silencing of TLR4 and RAGE dramatically abolished HMGB1-induced KLF7 expression, whereas a single knockdown showed only a modest inhibitory effect (Figure 4D, left). Consistently, the combined blockade of TLR4 and RAGE showed more potent inhibition than a single drug treatment (Figure 4D, right). These data proved that both TLR4 and RAGE are required for HMGB1-induced KLF7 upregulation.
To identify the cis-regulatory elements responsible for HMGB1-dependent KLF7 expression, we analyzed the KLF7 promoter and discovered multiple putative binding motifs of transcription factors downstream of TLR4/RAGE signaling (Figure S10). Next, we generated various luciferase reporter constructs containing truncated or mutated KLF7 promoters and transfected them into PLC/PRF/5 cells. The absence of the -1055bp ~ -130bp region significantly decreased HMGB1-dependent KLF7 promoter activity (Figure 4E). Co-mutation of the two NF-κB binding sites within this region largely impaired HMGB1-dependent KLF7 promoter activity, whereas disruption of binding sites of other transcription factors had little effect (Figure 4E). Subsequently, PLC/PRF/5 cells were treated with p65 knockdown or pretreated with an NF-κB inhibitor prior HMGB1 administration. These operations inhibited NF-κB phosphorylation and reduced HMGB1-induced KLF7 overexpression and HMGB1-dependent KLF7 promoter activity (Figure 4F-G). TLR4/RAGE signaling activates intracellular PI3K, JNK, ERK, and p38 pathways [22, 23]. To identify which signaling pathway mediated HMGB1-dependent NF-κB activation, PLC/PRF/5 cells were treated with signaling-specific inhibitors. The PI3K inhibitor largely abolished, while the ERK inhibitor slightly lowered HMGB1-induced NF-κB phosphorylation and KLF7 overexpression (Figure 4H). Furthermore, a ChIP assay validated that PI3K inhibitor treatment significantly mitigated the binding of NF-κB to the KLF7 promoter, while JNK, ERK, and P38 inhibitors had no such obvious effect (Figure 4I). Above all, these findings indicated that extracellular HMGB1 upregulates KLF7 expression through the TLR4/RAGE-PI3K-AKT-NF-κB signaling pathway in HCC cells.
KLF7 is crucial for HMGB1-induced HCC metastasis
The pivotal role of HMGB1 in promoting HCC metastasis has been identified [15]. Considering that KLF7 is induced by HMGB1 and facilitates HCC metastasis, we investigated its role in HMGB1-mediated HCC metastasis. HMGB1 was used to stimulate PLC/PRF/5 and Huh7 cells, with or without KLF7 knockdown (Figure 5A). HMGB1 treatment markedly enhanced the migration and invasion of PLC/PRF/5 and Huh7 cells, whereas KLF7 downregulation largely reversed these changes (Figure 5B). Consistently, HMGB1 upregulation augmented the migration and invasion of PLC/PRF/5 and Huh7 cells, whereas KLF7 knockdown mostly abolished HMGB1-enhanced migration and invasion of HCC cells (Figure 5C-D). Moreover, nude mice implanted with PLC/PRF/5-HMGB1 or Huh7-HMGB1 cells exhibited higher pulmonary metastasis and poorer OS than their control groups, whereas KLF7 knockdown significantly reversed these HMGB1-dependent phenotypes (Figure 5E-I, Figure 5J-5N). These data indicated that KLF7 elevation is crucial for HMGB1-induced HCC metastasis.
KLF7 is essential for HMGB1-induced HCC metastasis. (A) PLC/PRF/5 cells and Huh7 with or without KLF7 knockdown were treated with HMGB1, and KLF7 expression was examined by Western blotting. (B) Transwell analysis of the mobility of indicated PLC/PRF/5 cells and Huh7 cells. (C) Lentivirus infection was used to upregulate HMGB1 in PLC/PRF/5 and Huh7 cells, and silence KLF7 in HMGB1-overexpressing PLC/PRF/5 and Huh7 cells. The protein levels of cytoplasmic HMGB1, total HMGB1 and KLF7 were examined. (D) The migration and invasion of PLC/PRF/5-HMGB1 cells and Huh7-HMGB1 cells with or without KLF7 knockdown were assessed by transwell assays. (E-I) Orthotopic HCC models shown that KLF7 knockdown abrogated HMGB1-dependent HCC metastasis. The nude mice were injected with the indicated PLC/PRF/5 cells in the livers. (E) Typical bioluminescent images of hepatic tumors in different groups. (F) Bioluminescent signals of liver tumors. (G) Number of metastatic nodules in lung. (H) Representative H&E images of metastases in lung tissues. (I) Overall survival time of nude mice. (J-N) Orthotopic HCC models shown that KLF7 downregulation inhibited HMGB1-mediated HCC metastasis. The nude mice were injected with the indicated Huh7cells in the livers. (J) Typical bioluminescent images of hepatic tumors in different groups. (K) Bioluminescent signals of liver tumors. (L) Number of metastatic nodules in lung. (M) Representative H&E images of metastases in lung tissues. (N) Overall survival time of nude mice. *P < 0.05.
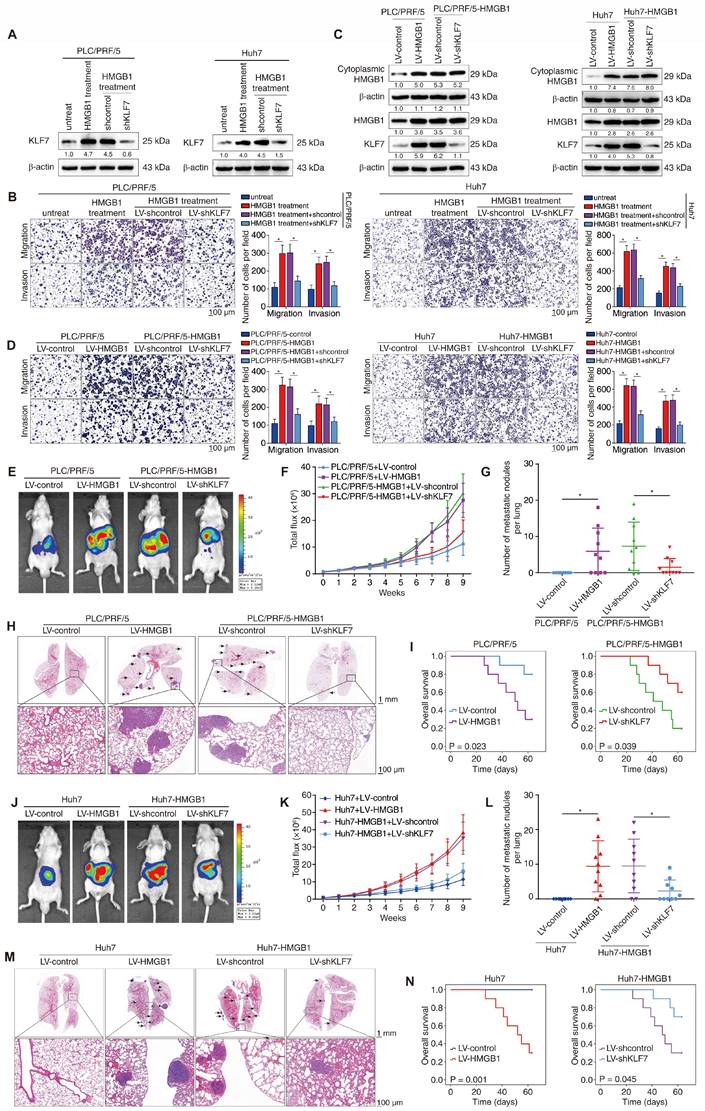
KLF7 expression was positively associated with cytoplasmic HMGB1 and phosphorylated NF-κB p65 levels in human HCC specimens
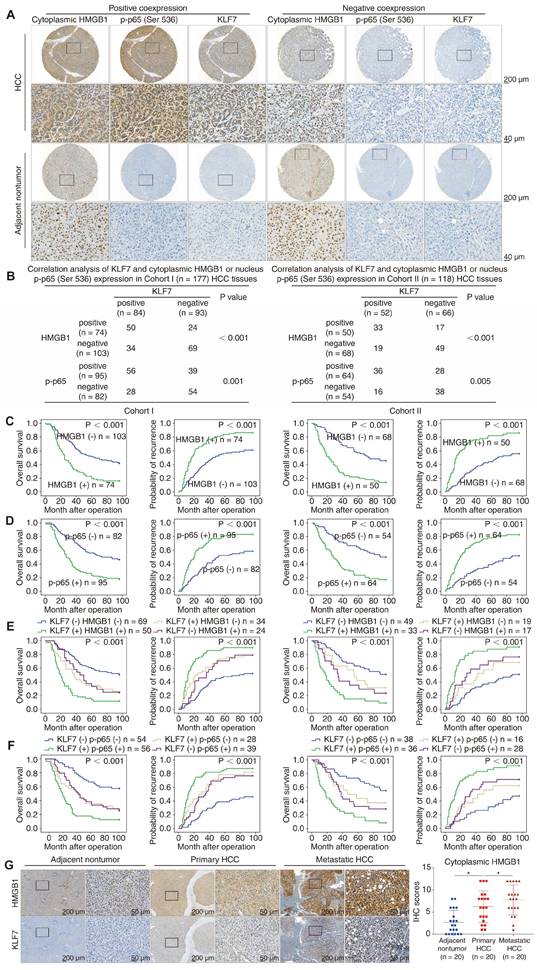
To confirm the clinical significance of HMGB1, p-p65 (Ser536), and KLF7 in HCC tissues, we profiled their expression using IHC staining in our HCC cohorts. Representative IHC images of HMGB1, p-p65(Ser536), and KLF7 expression are shown (Figure 6A). While para-cancerous non-tumor tissues always displayed nuclear HMGB1 immunoreactivity, positive cytoplasmic expression of HMGB1 was detected in nearly half of the analyzed HCC tissues. Compelling data demonstrated that HMGB1 can translocate from the nuclear to the cytoplasm and then secrete into the extracellular context as a danger alarm in inflammatory/stress hepatocytes [11]. Thus, cytoplasmic HMGB1 levels can be used as a sign to reflect HMGB1 secretion levels in HCC tissues [24, 25]. In our HCC cohorts, KLF7 expression positively correlated with cytoplasmic HMGB1 and nucleus p-p65 levels (Figure 6B). HCC patients with cytoplasmic HMGB1 or nucleus p-p65 upregulation displayed poor prognosis and aggressive pathological parameters (Figure 6C-D, Tables S6, S7). Furthermore, patients with either HMGB1/KLF7 or p-p65/KLF7 positive co-expression had the poorest prognosis in our cohorts (Figure 6E-F). Data from the TCGA LIHC cohort also confirmed that KLF7 expression was positively correlated with HMGB1 and RELA expression in HCC samples, and HCC patients highly co-expressing HMGB1/KLF7 or RELA/KLF7 had significantly poorer OS, especially in Asian subpopulations (Figure S7C-D). Furthermore, we detected the expression levels of HMGB1 and KLF7 by IHC staining in 20 pairs of primary and metastatic HCC tissues. The staining of HMGB1 in metastatic HCC tissues was primarily localized in the cytoplasm and was more intense than in primary HCC tissues, which was in accordance with the KLF7 expression pattern (Figure 6G).
KLF7 expression is positively correlated with the expression levels of cytoplasmic HMGB1 and nucleus p-p65 in human HCC tissues. (A) Representative IHC staining of HMGB1, p-p65(Ser536) and KLF7 expression in HCC tissues and para-cancer nontumor specimens. (B) Correlation analysis of KLF7 expression and cytoplasmic HMGB1 or nucleus p-p65 (Ser536) expression in two independent HCC cohorts. (C-D) Overall survival and recurrence rates of HCC patients with positive or negative cytoplasmic HMGB1 (C) or p-p65 (D) in two independent cohorts. (E-F) The correlation of KLF7/cytoplasmic HMGB1 (E) or KLF7/p-p65 (F) co-expression and overall survival and recurrence rates in our HCC cohorts. (G) Representative IHC staining of HMGB1 and KLF7 expression in para-cancerous nontumor samples, HCC specimens, and metastatic HCC tissues. IHC scores of and HMGB1 were presented. *P < 0.05.
Hepatocyte-specific knockout of Klf7 impedes DEN/CCl4-induced HCC development and metastasis
The DEN/CCl4-induced HCC model can imitate the process of hepatic inflammation-cirrhosis-HCC sequence and is suitable for studying inflammation-mediated HCC progression and metastasis [26]. Previous studies have reported that cytoplasmic translocation and secretion of HMGB1, as well as phosphorylation of NF-κB p65, were markedly upregulated in mouse livers following chronic diethyl nitrosamine (DEN) or carbon tetrachloride (CCl4) treatment [13, 27, 28], we thus wondered whether the HMGB1-p-p65-KLF7-TLR4/PTK2 pathway was activated in the DEN/CCl4-induced HCC model. By performing immunoblotting at different time points during DEN/CCl4 treatment, we observed that the cytoplasmic translocation of HMGB1 and the levels of p-p65, KLF7, TLR4 and PTK2 were gradually upregulated during HCC initiation and progression. (Figure 7B). IHC assays further confirmed that the HMGB1-p-p65-KLF7-TLR4/PTK2 pathway was more highly activated in DEN/CCl4-induced HCC tissues than in normal livers (Figure 7C-D).
Hepatocyte-specific knockout of Klf7 impedes DEN/CCl4-induced HCC development and metastasis. (A) Schematic diagram of DEN/CCl4-induced HCC model. (B) The levels of cytoplasmic HMGB1, p-p65, p65, KLF7, TLR4, and PTK2 in the livers of control mice or in mice livers from the DEN/CCl4-treated group at the indicated time points were detected by immunostaining. (C-D) IHC scores of the levels of HMGB1, p-p65, KLF7, TLR4, and PTK2 in mice livers from the control group and in HCC tissues from the DEN/CCl4 group at 34 weeks (C). Representative IHC staining of each group was shown (D). (E) The schematic showed the time point of AAV8-TBG-null or AAV8-TBG-Cre injection during DEN/CCl4 treatment. (F) The schematic showed the mechanism by which Klf7 was knockout in hepatocytes by TBG-Cre recombinase in the Klf7fl/fl mice. (G) Expression of KLF7 in hepatocytes and non-parenchymal cells of AAV8-TBG-null and AAV8-TBG-Cre groups at the indicated time points after AAV8 injection. (H) Representative pictures of whole-liver morphology, H&E of liver sections, and H&E of lung metastases from the AAV8-TBG-null and AAV8-TBG-Cre groups at 34 weeks. (I) Liver weight/body weight ratio, tumor number, largest tumor size, and the number of pulmonary metastatic nodules in each group were statically analyzed at 34 weeks (n = 10 per group). (J) 15 mice in each group were raised until natural death for survival analysis. Survival rates of Klf7fl/fl mice treated with AAV8-TBG-null or AAV8-TBG-Cre were shown (n = 15 per group). (K) The protein levels of cytoplasmic HMGB1, p-p65, KLF7, TLR4, and PTK2 in the liver tumors from the AAV8-TBG-null group and the AAV8-TBG-Cre group at 34 weeks were examined by immunostaining. *P < 0.05.
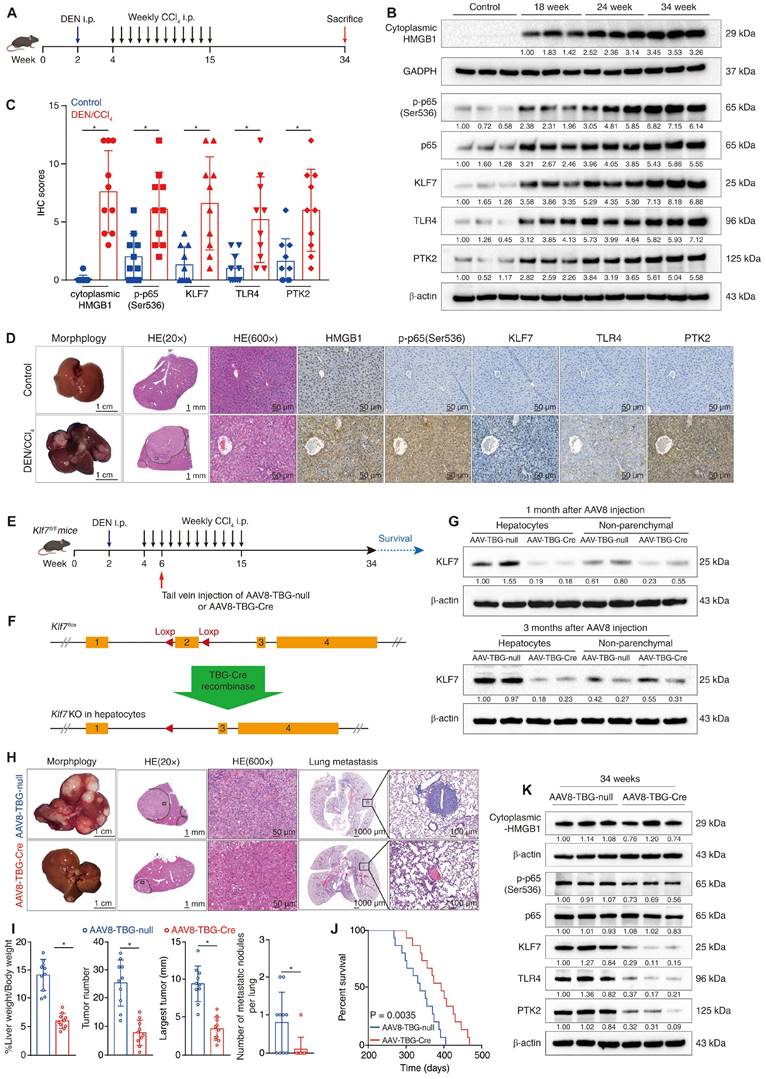
To explore the role of KLF7 in HMGB1-mediated HCC development and metastasis, we performed hepatocyte-specific knockout of Klf7 in Klf7fl/fl mice via tail vein injection of AAV8-TBG-Cre vectors in the early stage of DEN/CCl4 treatment (Figure 7E-F). The effectiveness of Klf7 knockout in the hepatocytes of Klf7fl/fl mice was verified using western blotting (Figure 7G). Surprisingly, mice in the Klf7 knockout group developed smaller hepatic tumors with fewer nodules and less pulmonary metastasis, while exhibiting a much longer survival time (Figure 7H-J). Hepatic tumors from the Klf7 knockout group also showed low levels of KLF7, TLR4, and PTK2 (Figure 7K). These data proved that DEN/CCl4-induced HMGB1 release promotes HCC progression by activating the KLF7-TLR4/PTK2 axis in hepatocytes, and that hepatocyte-specific Klf7 knockout significantly impedes HMGB1-induced HCC development and metastasis.
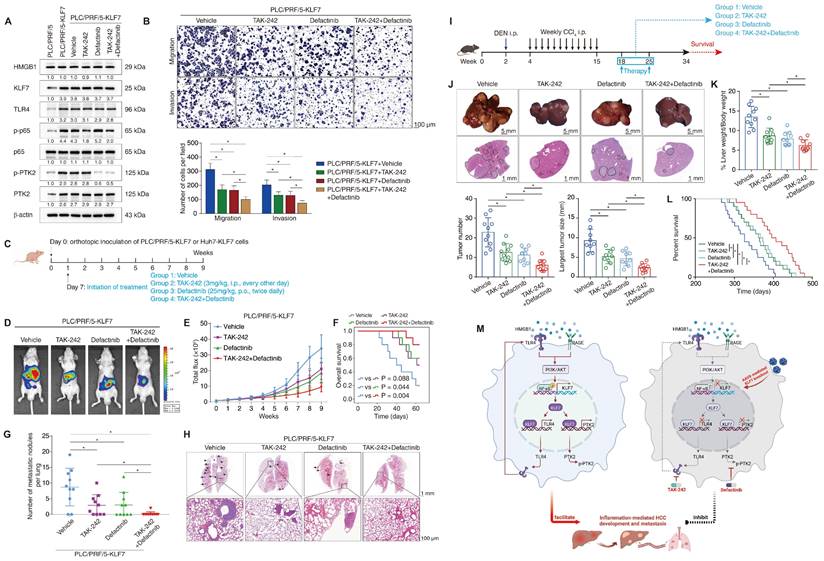
Combined administration of TLR4 inhibitor TAK-242 and PTK2 inhibitor defactinib significantly alleviates KLF7-mediated HCC development and metastasis
Given that TLR4 and PTK2 are downstream effectors of the HMGB1-KLF7 axis, we explored whether the pharmacological inhibition of TLR4 and PTK2 could impede KLF7-mediated HCC development and metastasis. The small-molecule TLR4 inhibitor TAK-242 and PTK2 inhibitor defactinib attracted our attention because of their verified safety and efficacy profiles [29, 30]. PLC/PRF/5 and Huh7 cells stably overexpressing KLF7 were treated with TAK-242, defactinib, or a combination of both drugs. The efficacy of TAK-242 and defactinib was verified by decreased levels of p-p65 and p-PTK2, respectively (Figure 8A, Figure S11A). TAK-242 or defactinib alone partly while their combination potently inhibited the migration and invasion of PLC/PRF/5-KLF7 and Huh7-KLF7 cells (Figure 8B, Figure S11B). Consistently, the combination of TAK-242 and defactinib significantly alleviated pulmonary metastatic burden and extended OS time compared to the single-agent groups in nude mice implanted with PLC/PRF/5-KLF7 or Huh7-KLF7 cells (Figure 8D-H, Figure S11C-G). We examined the therapeutic effects of our combination strategy in the DEN/CCl4-induced HCC model. Combination therapy with TAK-242 and defactinib markedly suppressed HCC development and extended mouse survival time, while single-drug treatment showed moderate efficacy (Figure 8I-L).
Combined administration of TLR4 and PTK2 inhibitors suppresses KLF7-mediated HCC development and metastasis. (A) PLC/PRF/5-KLF7 cells were incubated with vehicle, TAK-242, defactinib, or a combination of both, then the protein levels of HMGB1, KLF7, TLR4, p-p65, p65, p-PTK2 and PTK2 were detected. (B) Transwell analysis of the mobility of PLC/PRF/5-KLF7 cells treated with TAK-242 alone, defactinib alone, or a combination of both. (C) The diagram of drug treatment. Nude mice were transplanted with HCC cells and randomly divided into four groups. Then, vehicle, TAK-242, defactinib, or combined administration were applied to different groups, respectively. (D-H) Combined blockade of TLR4 and PTK2 alleviated KLF7-mediated HCC metastasis. (D) Representative bioluminescent pictures of liver tumors in nude mice. (E) Intensity of bioluminescent signals of liver tumors. (F) Overall survival of mice. (G)The amount of pulmonary metastatic nodules. (H) Representative pictures of H&E staining of lung tissues. (I) Schematic diagram of DEN/CCl4 treatment and drug regimens. Mice were randomly divided into four groups at 18 weeks. Then, vehicle, TAK-242, defactinib, or combined application was applied to each group, respectively. (J) Typical images of whole-liver morphology and H&E of liver sections from different groups at 34 weeks. (K) 10 mice in each group were sacrificed at 34 weeks for statistical analysis. The analysis of liver weight/body weight ratio, tumor number, and largest tumor size in each group were shown (n = 10 per group). (L) 20 mice in each group were raised until natural death for survival analysis. The survival rates of mice in different groups were shown (n = 20 per group). (M) A schematic diagram depicting the function of the HMGB1-KLF7-TLR4/PTK2 pathway in inflammation-mediated HCC development and metastasis. Inflammation-provoked HMGB1 release upregulates KLF7 expression via the TLR4/RAGE-PI3K-AKT-NF-κB pathway within HCC cells. Furthermore, KLF7 promotes HCC development and metastasis by transcriptionally upregulating TLR4 and PTK2 expression, forming an HMGB1-KLF7-TLR4 positive feedback loop. Hepatocyte-specific knockout of Klf7 via AAV8 gene therapy, or combined administration of TLR4 inhibitor TAK-242 and PTK2 inhibitor defactinib significantly impedes HCC development and metastasis mediated by the HMGB1-KLF7 axis. *P < 0.05.
Discussion
Transcriptional factors are vital integrators of input oncogenic signals and represent the central nodes regulating tumor biology in HCC [31]. Genomic and transcriptomic data on human HCC revealed that many transcription factors are dysregulated in aggressive HCC subtypes, suggesting a promising prospect for translating dysfunctional transcription factors into HCC stratification and therapy [31]. Here, we identified KLF7 elevation as an independent prognostic predictor of HCC and highlighted its vital role in HCC progression and metastasis. As a transcription factor, KLF7 exerts cancer-promoting effects through transcriptional activation of oncogenic genes in human cancers [6, 8]. Our study demonstrated that KLF7 transcriptionally upregulates TLR4 and PTK2 expression in HCC cells. TLR4 is a transmembrane pattern-recognition receptor that recognizes both PAMPs and DAMPs and senses danger signals [32]. TLR4 is overexpressed in HCC tissues and predicts poor survival and early recurrence [16, 33]. The TME of HCC is commonly enriched with a variety of TLR4 ligands such as gut-derived lipopolysaccharide (LPS), dying or stressed cell-released HMGB1/heat shock protein 70 (HSP70), and hepatitis B virus (HBV) antigens [32]. These ligands persistently stimulate HCC cells overexpressing TLR4, ultimately leading to hyperactivation of TLR4 signaling. Activated TLR4 signaling subsequently induces hepatocarcinogenesis and promotes HCC growth, stemness, EMT and metastasis [16, 33, 34]. PTK2, also known as focal adhesion kinase (FAK), is a non-receptor tyrosine kinase downstream of multiple extracellular signals such as growth factors, integrins, and cytokines [35]. PTK2 is upregulated and hyperphosphorylated in multiple tumors and contributes to poor prognosis [35]. In human HCC, PTK2 expression is elevated and serves as an independent prognostic factor [36]. PTK2 overexpression significantly accelerates hepatocarcinogenesis and enhances HCC growth, invasion, metastasis, and self-renewal [17, 37-39]. Thus, TLR4 and PTK2 are vital regulators of HCC progression and metastasis, respectively. Our study found that KLF7 promotes HCC progression and metastasis by transcriptionally upregulating TLR4 and PTK2. In HCC samples, KLF7 expression was positively correlated with TLR4 and PTK2 expression, and patients positively co-expressing KLF7/TLR4 or KLF7/PTK2 exhibited the worst prognosis.
As a typical inflammation-induced cancer, HCC generally has an inflammatory TME enriched with cytokines, PAMPs, and DAMPs, which are vital pro-tumorigenic and pro-metastatic signals for HCC cells [18]. In this study, we found that inflammatory factors triggered KLF7 expression in HCC cells and identified HMGB1 as a potent inducer of KLF7 upregulation. HMGB1, a typical DAMP, can be translocated from the nucleus to the cytoplasm and subsequently secreted during cell injury, HBV X protein stimulation, hypoxia, sustained p53 activation, etc. [13, 24, 40], which leads to HMGB1 accumulation in the extracellular milieu of chronic liver diseases and HCC [11]. Extracellular HMGB1 ulteriorly facilitates HCC initiation, growth, and metastasis by interacting with cell-surface TLR4/RAGE receptors and endosome-membrane TLR9 receptor [14, 15, 40]. Immunostaining of HMGB1 in HCC specimens is primarily observed in the cytoplasm of carcinoma cells and is stronger than in para-tumors [41]. High cytoplasmic HMGB1 levels are positively associated with poor prognosis and aggressive pathological parameters in HCC patients [25, 41]. Therefore, extracellular HMGB1 is an inflammation-related oncogenic factor in human HCC. Our study revealed that extracellular HMGB1 induced KLF7 elevation in HCC cells through the TLR4/RAGE-PI3K-AKT-NFκB signaling pathway. Genetic depletion of KLF7 significantly inhibited HMGB1-mediated HCC progression and metastasis. In the HCC cohorts, KLF7 expression was positively associated with cytoplasmic HMGB1 intensity, and positive HMGB1/KLF7 co-occurrence predicted the worst prognosis. Moreover, high KLF7 expression predicted inferior OS in HMGB1-high HCC patients. In conclusion, our data identified a novel HMGB1-KLF7 axis involved in inflammation-induced HCC progression and metastasis.
To pharmacologically inhibit the HMGB1-KLF7 axis in HCC, we focused on small-molecule inhibitors targeting its downstream effectors, TLR4 and PTK2. TAK-242 (resatorvid) is a selective small-molecule TLR4 inhibitor that interferes with interactions between the TLR4 intracellular domain and its adaptor molecules [29]. An attempt to use TAK-242 for treating human sepsis has reached phase Ⅲ trials, confirming the safety of this drug [29]. Recently, TAK-242 alone or in combination with other drugs, has shown anti-tumor effects in preclinical models of colorectal cancer, breast cancer, glioma, and skin cancer [42-45]. Defactinib (VS-6063) is an orally bioavailable PTK2 inhibitor that suppresses Y397 phosphorylation of PTK2 via an ATP-competitive mechanism [30]. Several completed clinical trials on solid tumors have proven the tolerable safety profile and modest efficacy of defactinib [30]. Moreover, defactinib monotherapy or co-administration with other agents has shown exciting therapeutic efficacy in preclinical models of ovarian cancer, PDAC, and EGFR-mutant non-small-cell lung cancer [46-48]. This study found that the combined administration of TAK-242 and defactinib effectively inhibited KLF7-mediated HCC progression and metastasis, as well as prolonged OS in preclinical HCC mouse models. Gene signature analysis further revealed that the KLF7-TLR4/PTK2 axis is activated in HCC tissues and contributes to HCC metastasis and poor prognosis. These findings indicate an excellent application prospect for translating our combination strategy into treating the KLF7-positive HCC subpopulation.
In conclusion, extracellular HMGB1 induced KLF7 expression via the TLR4/RAGE-PI3K-AKT-NF-κB pathway in HCC cells. KLF7 was critical for HMGB1-mediated HCC progression and metastasis. The HMGB1-KLF7 axis facilitated HCC progression and metastasis by upregulating TLR4 and PTK2 expression, forming an HMGB1-KLF7-TLR4 positive feedback loop. Combined administration of the TLR4 inhibitor TAK-242 and PTK2 inhibitor defactinib or genetic ablation of KLF7 in hepatocytes significantly impeded the HMGB1-KLF7 axis-mediated HCC progression and metastasis, which represented promising therapeutic strategies for KLF7-positive HCC patients.
Abbreviations
HCC, hepatocellular carcinoma; KLF7, krüppel-like factor 7; TLR4, toll-like receptor 4; PTK2, protein tyrosine kinase 2; HMGB1, high mobility group box 1; RAGE, advanced glycosylation end-product specific receptor; RELA, RELA proto-oncogene, NF-κB subunit; DAMPs, damage-associated molecular patterns; PAMPs, pathogen-associated molecular patterns; AAV8, adeno-associated virus 8; H&E, Hematoxylin and eosin; IHC, immunohistochemistry; mRNA, messenger RNA; shRNA, short hairpin RNA; ChIP, chromatin immunoprecipitation analysis; BLI, bioluminescent imaging; TCGA, The Cancer Genome of Atlas; LIHC, liver hepatocellular carcinoma; OS, overall survival; RFS, relapse-free survival; DFS, disease-free survival; HR, hazard ratio.
Supplementary Material
Supplementary materials and methods, figures and tables.
Acknowledgements
This research was supported by grants from the National Natural Science Foundation of China No. 82273374 (K.W.), No. 82273310 (L.X.), No. 82173313 (W.H.), No. 82103303 (G.W.), the Natural Science Foundation of Hubei Province 2022CFA016 (L.X.), and the Basic Research Support Program of Huazhong University of Science and Technology 2023BR038 (L.X.).
Author Contributions
Weibo Feng, Jie Chen, Wenjie Huang, Guodong Wang and Xilang Chen contributed equally to this work. Weibo Feng performed experiments. Jie Chen, Wenjie Huang, Guodong Wang, Xilang Chen and Yue Yin assisted with immunohistochemical staining, HE staining and animal experiments. Lili Duan assisted in performing the bioinformatics analysis. Mengyu Sun and Xiangyuan Luo assisted in collecting tissues samples and performing clinical statistical analysis. Xiaoping Chen, Bixiang Zhang, Yongzhan Nie, Daiming Fan, and Kaichun Wu conceived of the experiments and reviewed the manuscript. Weibo Feng, Limin Xia, Kaichun Wu, and Daiming Fan designed the study and prepared the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S. et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7:6
2. Finn RS, Zhu AX. Evolution of Systemic Therapy for Hepatocellular Carcinoma. Hepatology. 2021;73(Suppl 1):150-7
3. Chen S, Cao Q, Wen W, Wang H. Targeted therapy for hepatocellular carcinoma: Challenges and opportunities. Cancer Lett. 2019;460:1-9
4. Bushweller JH. Targeting transcription factors in cancer - from undruggable to reality. Nat Rev Cancer. 2019;19:611-24
5. Tetreault MP, Yang Y, Katz JP. Kruppel-like factors in cancer. Nat Rev Cancer. 2013;13:701-13
6. Gupta R, Malvi P, Parajuli KR, Janostiak R, Bugide S, Cai G. et al. KLF7 promotes pancreatic cancer growth and metastasis by up-regulating ISG expression and maintaining Golgi complex integrity. Proc Natl Acad Sci U S A. 2020;117:12341-51
7. De Donato M, Babini G, Mozzetti S, Buttarelli M, Ciucci A, Arduini G. et al. KLF7: a new candidate biomarker and therapeutic target for high-grade serous ovarian cancer. J Exp Clin Cancer Res. 2020;39:265
8. Guan F, Kang Z, Zhang JT, Xue NN, Yin H, Wang L. et al. KLF7 promotes polyamine biosynthesis and glioma development through transcriptionally activating ASL. Biochem Biophys Res Commun. 2019;514:51-7
9. Guo Y, Chai B, Jia J, Yang M, Li Y, Zhang R. et al. KLF7/VPS35 axis contributes to hepatocellular carcinoma progression through CCDC85C-activated beta-catenin pathway. Cell Biosci. 2021;11:73
10. Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol. 2013;59:583-94
11. Gaskell H, Ge X, Nieto N. High-Mobility Group Box-1 and Liver Disease. Hepatol Commun. 2018;2:1005-20
12. Wang S, Zhang Y. HMGB1 in inflammation and cancer. J Hematol Oncol. 2020;13:116
13. Yan HX, Wu HP, Zhang HL, Ashton C, Tong C, Wu H. et al. p53 promotes inflammation-associated hepatocarcinogenesis by inducing HMGB1 release. J Hepatol. 2013;59:762-8
14. Chen R, Zhu S, Fan XG, Wang H, Lotze MT, Zeh HJ 3rd. et al. High mobility group protein B1 controls liver cancer initiation through yes-associated protein -dependent aerobic glycolysis. Hepatology. 2018;67:1823-41
15. Yan W, Chang Y, Liang X, Cardinal JS, Huang H, Thorne SH. et al. High-mobility group box 1 activates caspase-1 and promotes hepatocellular carcinoma invasiveness and metastases. Hepatology. 2012;55:1863-75
16. Liu WT, Jing YY, Yu GF, Han ZP, Yu DD, Fan QM. et al. Toll like receptor 4 facilitates invasion and migration as a cancer stem cell marker in hepatocellular carcinoma. Cancer Lett. 2015;358:136-43
17. Chen JS, Huang XH, Wang Q, Chen XL, Fu XH, Tan HX. et al. FAK is involved in invasion and metastasis of hepatocellular carcinoma. Clin Exp Metastasis. 2010;27:71-82
18. Yu LX, Ling Y, Wang HY. Role of nonresolving inflammation in hepatocellular carcinoma development and progression. NPJ Precis Oncol. 2018;2:6
19. Ringelhan M, Pfister D, O'Connor T, Pikarsky E, Heikenwalder M. The immunology of hepatocellular carcinoma. Nat Immunol. 2018;19:222-32
20. Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology. 2015;61:1066-79
21. Yang H, Wang H, Andersson U. Targeting Inflammation Driven by HMGB1. Front Immunol. 2020;11:484
22. Yang Y, Lv J, Jiang S, Ma Z, Wang D, Hu W. et al. The emerging role of Toll-like receptor 4 in myocardial inflammation. Cell Death Dis. 2016;7:e2234
23. Hudson BI, Lippman ME. Targeting RAGE Signaling in Inflammatory Disease. Annu Rev Med. 2018;69:349-64
24. Chen S, Dong Z, Yang P, Wang X, Jin G, Yu H. et al. Hepatitis B virus X protein stimulates high mobility group box 1 secretion and enhances hepatocellular carcinoma metastasis. Cancer Lett. 2017;394:22-32
25. Li J, Ren H, Wang J, Zhang P, Shi X. Extracellular HMGB1 promotes CD44 expression in hepatocellular carcinoma via regulating miR-21. Aging (Albany NY). 2021;13:8380-95
26. Brown ZJ, Heinrich B, Greten TF. Mouse models of hepatocellular carcinoma: an overview and highlights for immunotherapy research. Nat Rev Gastroenterol Hepatol. 2018;15:536-54
27. Ge X, Arriazu E, Magdaleno F, Antoine DJ, Dela Cruz R, Theise N. et al. High Mobility Group Box-1 Drives Fibrosis Progression Signaling via the Receptor for Advanced Glycation End Products in Mice. Hepatology. 2018;68:2380-404
28. Xu X, Lei Y, Chen L, Zhou H, Liu H, Jiang J. et al. Phosphorylation of NF-kappaBp65 drives inflammation-mediated hepatocellular carcinogenesis and is a novel therapeutic target. J Exp Clin Cancer Res. 2021;40:253
29. Rice TW, Wheeler AP, Bernard GR, Vincent JL, Angus DC, Aikawa N. et al. A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit Care Med. 2010;38:1685-94
30. Lu Y, Sun H. Progress in the Development of Small Molecular Inhibitors of Focal Adhesion Kinase (FAK). J Med Chem. 2020;63:14382-403
31. Malz M, Pinna F, Schirmacher P, Breuhahn K. Transcriptional regulators in hepatocarcinogenesis-key integrators of malignant transformation. J Hepatol. 2012;57:186-95
32. Sepehri Z, Kiani Z, Kohan F, Alavian SM, Ghavami S. Toll like receptor 4 and hepatocellular carcinoma; A systematic review. Life Sci. 2017;179:80-7
33. Jing YY, Han ZP, Sun K, Zhang SS, Hou J, Liu Y. et al. Toll-like receptor 4 signaling promotes epithelial-mesenchymal transition in human hepatocellular carcinoma induced by lipopolysaccharide. BMC Med. 2012;10:98
34. Dapito DH, Mencin A, Gwak GY, Pradere JP, Jang MK, Mederacke I. et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell. 2012;21:504-16
35. Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer. 2014;14:598-610
36. Fujii T, Koshikawa K, Nomoto S, Okochi O, Kaneko T, Inoue S. et al. Focal adhesion kinase is overexpressed in hepatocellular carcinoma and can be served as an independent prognostic factor. J Hepatol. 2004;41:104-11
37. Shang N, Wang H, Bank T, Perera A, Joyce C, Kuffel G. et al. Focal Adhesion Kinase and beta-Catenin Cooperate to Induce Hepatocellular Carcinoma. Hepatology. 2019;70:1631-45
38. Fan Z, Duan J, Wang L, Xiao S, Li L, Yan X. et al. PTK2 promotes cancer stem cell traits in hepatocellular carcinoma by activating Wnt/beta-catenin signaling. Cancer Lett. 2019;450:132-43
39. Gnani D, Romito I, Artuso S, Chierici M, De Stefanis C, Panera N. et al. Focal adhesion kinase depletion reduces human hepatocellular carcinoma growth by repressing enhancer of zeste homolog 2. Cell Death Differ. 2017;24:889-902
40. Liu Y, Yan W, Tohme S, Chen M, Fu Y, Tian D. et al. Hypoxia induced HMGB1 and mitochondrial DNA interactions mediate tumor growth in hepatocellular carcinoma through Toll-like receptor 9. J Hepatol. 2015;63:114-21
41. Liu F, Zhang Y, Peng Z, Gao H, Xu L, Chen M. High expression of high mobility group box 1 (hmgb1) predicts poor prognosis for hepatocellular carcinoma after curative hepatectomy. J Transl Med. 2012;10:135
42. Song W, Tiruthani K, Wang Y, Shen L, Hu M, Dorosheva O. et al. Trapping of Lipopolysaccharide to Promote Immunotherapy against Colorectal Cancer and Attenuate Liver Metastasis. Adv Mater. 2018;30:e1805007
43. Jiang Y, Zhou J, Luo P, Gao H, Ma Y, Chen YS. et al. Prosaposin promotes the proliferation and tumorigenesis of glioma through toll-like receptor 4 (TLR4)-mediated NF-kappaB signaling pathway. EBioMedicine. 2018;37:78-90
44. Sherwani MA, Abdelgawad A, Chung M, Ibrahim S, Eraslan M, Elmets CA. et al. Toll-Like Receptor-4 Antagonist Enhances the Repair of Ultraviolet Radiation-Induced DNA Damage and Augments Anti-Tumor Immune Responses in Mice. Cancers (Basel). 2021 13
45. Zandi Z, Kashani B, Bashash D, Poursani EM, Mousavi SA, Chahardoli B. et al. The anticancer effect of the TLR4 inhibition using TAK-242 (resatorvid) either as a single agent or in combination with chemotherapy: A novel therapeutic potential for breast cancer. J Cell Biochem. 2020;121:1623-34
46. Kang Y, Hu W, Ivan C, Dalton HJ, Miyake T, Pecot CV. et al. Role of focal adhesion kinase in regulating YB-1-mediated paclitaxel resistance in ovarian cancer. J Natl Cancer Inst. 2013;105:1485-95
47. Le Large TYS, Bijlsma MF, El Hassouni B, Mantini G, Lagerweij T, Henneman AA. et al. Focal adhesion kinase inhibition synergizes with nab-paclitaxel to target pancreatic ductal adenocarcinoma. J Exp Clin Cancer Res. 2021;40:91
48. Fu Y, Zhang Y, Lei Z, Liu T, Cai T, Wang A. et al. Abnormally activated OPN/integrin alphaVbeta3/FAK signalling is responsible for EGFR-TKI resistance in EGFR mutant non-small-cell lung cancer. J Hematol Oncol. 2020;13:169
Author contact
![]() Corresponding authors: Dr. Limin Xia, Department of Gastroenterology, Institute of Liver and Gastrointestinal Diseases, Hubei Key Laboratory of Hepato-Pancreato-Biliary Diseases, Tongji Hospital of Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, Hubei Province, China; Phone: 86 27 6937 8507; Fax: 86 27 8366 2832; Email: xialimintjmu.edu.cn. Dr. Kaichun Wu, State Key Laboratory of Holistic Integrative Management of Gastrointestinal Cancers and National Clinical Research Center for Digestive Diseases, Xijing Hospital of Digestive Diseases, Fourth Military Medical University, Xi'an 710032, Shaanxi Province, China; Email: kaicwuedu.cn. Dr. Daiming Fan, State Key Laboratory of Holistic Integrative Management of Gastrointestinal Cancers and National Clinical Research Center for Digestive Diseases, Xijing Hospital of Digestive Diseases, Fourth Military Medical University, Xi'an 710032, Shaanxi Province, China; Email: fandaimedu.cn.
Corresponding authors: Dr. Limin Xia, Department of Gastroenterology, Institute of Liver and Gastrointestinal Diseases, Hubei Key Laboratory of Hepato-Pancreato-Biliary Diseases, Tongji Hospital of Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, Hubei Province, China; Phone: 86 27 6937 8507; Fax: 86 27 8366 2832; Email: xialimintjmu.edu.cn. Dr. Kaichun Wu, State Key Laboratory of Holistic Integrative Management of Gastrointestinal Cancers and National Clinical Research Center for Digestive Diseases, Xijing Hospital of Digestive Diseases, Fourth Military Medical University, Xi'an 710032, Shaanxi Province, China; Email: kaicwuedu.cn. Dr. Daiming Fan, State Key Laboratory of Holistic Integrative Management of Gastrointestinal Cancers and National Clinical Research Center for Digestive Diseases, Xijing Hospital of Digestive Diseases, Fourth Military Medical University, Xi'an 710032, Shaanxi Province, China; Email: fandaimedu.cn.