Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Characteristics, functions,...
3. Adipose tissue alteration in...
4. Targeting adipose tissue for...
5. Conclusions and Perspectives
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2023; 13(12):3925-3942. doi:10.7150/thno.82911 This issue Cite
Review
New insights into the role of adipocytes in pancreatic cancer progression: paving the way towards novel therapeutic targets
Yu-Chun Lin1, Ya-Chin Hou1,2,3, Hao-Chen Wang1,4, Yan-Shen Shan1,3, ![]()
1. Institute of Clinical Medicine, College of Medicine, National Cheng Kung University, Tainan 704, Taiwan.
2. Department of Clinical Medicine Research Center, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, Tainan 704, Taiwan.
3. Division of General Surgery, Department of Surgery, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, Tainan 704, Taiwan.
4. Medical Imaging Center, Innovation Headquarter, National Cheng Kung University; Tainan 704, Taiwan.
Received 2023-1-25; Accepted 2023-6-21; Published 2023-7-3
Abstract

Pancreatic cancer (PC) remains one of the most lethal malignancies across the world, which is due to delayed diagnosis and resistance to current therapies. The interactions between pancreatic tumor cells and their tumor microenvironment (TME) allow cancer cells to escape from anti-cancer therapies, leading to difficulties in treating PC. With endocrine function and lipid storage capacity, adipose tissue can maintain energy homeostasis. Direct or indirect interaction between adipocytes and PC cells leads to adipocyte dysfunction characterized by morphological change, fat loss, abnormal adipokine secretion, and fibroblast-like transformation. Various adipokines released from dysfunctional adipocytes have been reported to promote proliferation, invasion, metastasis, stemness, and chemoresistance of PC cells via different mechanisms. Additional lipid outflow from adipocytes can be taken into the TME and thus alter the metabolism in PC cells and surrounding stromal cells. Besides, the trans-differentiation potential enables adipocytes to turn into various cell types, which may give rise to an inflammatory response as well as extracellular matrix reorganization to modulate tumor burden. Understanding the molecular basis behind the protumor functions of adipocytes in PC may offer new therapeutic targets.
Keywords: pancreatic cancer, tumor microenvironment, adipocyte dysfunction, fat loss, adipokines, trans-differentiation
1. Introduction
Pancreatic cancer (PC) is one of the most lethal malignancies in the world [1]. Despite advancements in treatments, the 5-year survival rate remains extremely poor. PC is characterized by a highly heterogeneous tumor microenvironment (TME). Apart from PC cells, the TME contains various nonmalignant cell types including vascular endothelial cells, fibroblasts, adipocytes, stellate cells, mesenchymal stem cells (MSCs), and various immune cells. The complex interactions within the pancreatic TME give rise to multiple pathological phenomena, such as chronic inflammation, tissue fibrosis, and even immune escape, which allow PC cells to survive various therapies and disseminate to distant organs. [2-4]. During tumorigenesis, immune cells, particularly macrophages, dendritic cells (DCs), monocytes, and T cells, are recruited to the tumor site to fight against malignant cells through their cytotoxic functions and secretion of pro-inflammatory cytokines. However, chronic inflammation in the TME can impair immune cells or even induce their transformation into a pro-tumor phenotype [5, 6]. Additionally, cancer-derived factors, such as transforming growth factor beta (TGF-β), can activate pancreatic stellate cells (PSCs) and fibroblasts, facilitating the deposition of extracellular matrix (ECM) components and the excessive secretion of cytokines and chemokines [7]. These processes contribute to the development of a desmoplastic TME [8-10]. The hyperplastic stroma in the pancreatic tumor restricts vascularization, creating a physical barrier to anti-tumor reagents and a hypoxic environment that promote cancer development. Besides facilitating tumor progression, the severe cytokine storm generated by the TME can also lead to systemic defects, including adipose dysfunction [11-13].
Adipose tissue regulates energy homeostasis through its lipid storage and endocrine functions [14, 15]. However, pro-inflammatory cytokines derived from the TME can perturb lipid metabolism and adipokine secretion, promoting cancer development in various ways [15]. Previous research has established a strong association between adipose tissue and PC progression, including a higher incidence of tumor metastasis and chemoresistance in obese PC patients [16-19]. Adipose tissue loss and altered circulating lipid profiles are also observed in both PC animal models and clinical studies [20-22]. Additionally, even among PC patients with normal body mass index (BMI) and average blood sugar levels, the presence of peripancreatic fat infiltration portends a high risk of locoregional recurrence and poor overall survival [23-25]. Intrapancreatic fat deposition is also associated with increased levels of pro-inflammatory cytokines in the circulation, which interferes with the immune response and reinforces the development of Kras-driven pancreatic tumorigenesis [26, 27]. These findings identify the significant role of adipose tissue in the development of PC, underscoring the urgent need to better understand the underlying mechanisms for developing therapeutic strategies. In this article, we review recent studies about the molecular mechanisms by which adipose tissue promotes PC development, including nutrient support, endocrine function, and the effects of its plasticity on PC cells with surrounding TME.
2. Characteristics, functions, and development of normal adipose tissue
2.1. Composition of adipose tissue
Adipose tissue is a specialized connective tissue containing two major fractions, lipid-rich adipocytes, and the stromal vascular fraction. Adipocytes store body fat in the form of triglycerides within lipid droplets in their cytoplasm. The stromal vascular fraction consists of various cell types, including endothelial cells, preadipocytes, adipose-derived mesenchymal stem cells (ADSCs), and immune cells. The remarkable heterogeneity of adipose tissue contributes to its multifunctionality and cooperative role in modulating energy balance. Both fractions of adipose tissue are surrounded by the ECM, which serves as a scaffold supporting multiple biological processes, such as cell adhesion, signal transduction, and the formation of new blood vessels [28]. Endothelial cells within the adipose tissue also play a vital role in promoting angiogenesis through their proliferation and migration, ensuring an adequate nutrient supply and guiding immune cells for tissue repair [29, 30]. The tight coordination between the ECM and endothelial cells is closely associated with the expansion of adipocyte precursors and the overall adipose mass, allowing adipose tissue greater expandability and adaptability in response to diverse external stimuli and systemic nutritional status. Apart from non-adipocyte cell types, different categories of adipocytes also exhibit distinct functions in regulating energy homeostasis.
2.2. Function and category of adipocytes
Adipose tissue modulates energy homeostasis by lipid reservation and endocrine secretion [15]. According to functional properties, adipose tissue can be divided into three types, namely white adipose tissue (WAT), beige adipose tissue, and brown adipose tissue (BAT). WAT accounts for the largest proportion of adipose tissue and is characterized by single large lipid droplets in the cytoplasm.
WAT governs metabolic actions in distant organs by releasing numerous adipokines into the circulation [31], while fat storage in WAT is subject to lipid mobilization in response to environmental signals. Nutrients taken up from diet are transported to the liver and converted into lipids through de novo lipogenesis. These newly synthesized fats are then delivered and incorporated into WAT for storage by lipoproteins. To enhance energy storage capacity, adipocytes change through two mechanisms. Firstly, they can incorporate additional lipids content into their lipid droplets leading to an enlargement of their volume, a process known as adipocyte hypertrophy. Secondly, adipocytes can also increase in number by recruiting precursor cells and promoting their proliferation, referred to as hyperplasia. These two processes collectively contribute to the overall growth of adipose tissue mass. In contrast, systemic energy demands, energy deficiency, or catabolic stimulation serve as external stimuli that initiate the process of lipolysis, whereby stored lipid droplets within adipocytes undergo breakdown (Figure 1). Perilipin plays as a gatekeeper in the regulation of lipolysis by forming a complex with CGI-58 on the surface of lipid droplets. When lipolysis is activated, Perilipin undergoes phosphorylation, leading to the dissociation of CGI-58 from the complex. This action facilitates the subsequent binding of CGI-58 to adipose tissue triglyceride lipase (ATGL), enabling the breakdown of triglycerides (TGs) into diglycerides (DGs) and free fatty acids (FFAs). Phosphorylated Perilipin also recruits hormone-sensitive lipase (HSL) to the surface of lipid droplets, working together to break down DGs into FFAs and monoglycerides (MGs). Monoglyceride lipase (MGL) manages the final step in the lipolysis process that breaks down MGs into glycerol and FFAs [32]. Released FFAs can further be plunged into β-oxidation to generate adenosine triphosphate (ATP) for energy production.
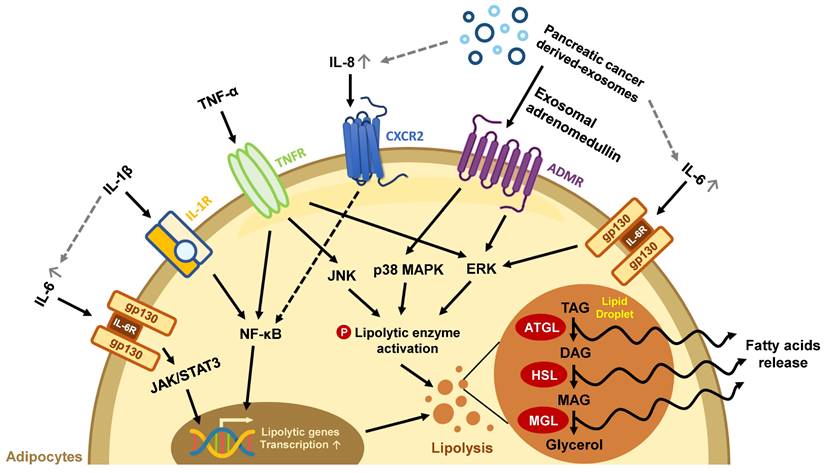
The effects of secretion factors in pancreatic TME on lipid loss in mature adipocytes. Exosomes and pro-inflammatory cytokines released from pancreatic TME trigger excessive lipolysis in mature adipocytes, leading to a decline in lipid storage. The gray dotted line represents the exosome/cytokine involved in IL-6 or IL-8 production.
Unlike WAT, BAT is characterized by the presence of multiple small lipid droplets and high levels of mitochondria. BAT has a substantial contribution to thermoregulation, allowing the body to adjust and maintain its temperature in response to various physical conditions. The high expression of mitochondria uncoupling protein 1 (UCP1) enables BAT to specialize in heat generation. UCP1 acts as a proton pump that balances the ion gradient across the mitochondrial inner membrane during oxidative phosphorylation. Instead of ATP synthesis, this process converts transmembrane potential into heat production [33]. Beige adipocytes residing in WAT originate from the same mesenchymal lineage as WAT, yet harbor a similar phenotype to BAT, possess multilocular lipid droplets, and abundant mitochondria capable of thermogenesis [34]. Several external stimuli such as cold exposure [35], adrenergic stimulation [36], and nutritional manipulation or pharmacological treatments [37, 38], have been demonstrated to induce biogenesis and thermogenesis in beige adipocytes, leading to increased heat production and energy expenditure. Beige adipocytes can also protect against obesity and type 2 diabetes by increasing lipolysis, fatty acid oxidation, glucose uptake, and insulin sensitivity with reduced adiposity [39, 40]. While each of these three types of adipocytes has its unique functions, they are all essential in maintaining energy balance.
2.3. Development of different types of adipocytes
Multipotent MSCs have been suggested as a major source of adipocyte generation, in which numerous critical pathways are involved, such as the TGF-β/bone morphogenic protein (BMP) pathway, Wingless-related integration site (Wnt) signaling, fibroblast growth factors (FGFs) signaling, Hedgehogs signaling, and Notch signaling [41]. The differentiation direction of adipocytes depends on the expression of myogenic Factor-5 (Myf5). Brown adipocytes are derived from the Myf5+ lineage, sharing the same transcriptional profiles as skeletal muscle cells. In contrast, beige and white adipocytes are developed from Myf5- progenitor cells. A transcriptional complex composed of BMP-7 signals, PR domain containing 16 as well as CCAAT/enhancer-binding protein (C/EBP) beta drives Myf5+ precursors into a thermogenic program with co-activation of peroxisome proliferator-activated receptor gamma (PPAR-γ) and PPAR-γ co-activator 1 alpha (PGC-1α), facilitating the formation of brown adipocytes. Partial Myf5- progenitors also undergo a thermogenic process, but they transform into beige-adipocytes with specific genes expression like CD137, transmembrane protein 26, and T-box 1 [34]. The other Myf5- population turns into white pre-adipocytes through BMP2 and BMP4 signaling [42]. PPAR-γ as well as C/EBP family proteins further promote adipogenesis by upregulating lipogenic genes. In the late stage of white adipocyte differentiation, lipid accumulation, and high adipokine secretion, especially leptin and adiponectin can be observed [43]. In addition to de novo differentiation, beige adipocytes can be also transdifferentiated from white adipocytes under certain conditions like cold exposure, β3-adrenergic signals triggered by the sympathetic neuron system, and chronic inflammation in various pathological processes [44]. The white-beige adipocyte transition known as “fat browning” turns the fat storage into the fuel for thermogenesis, leading to energy expenditure and loss of lipid stores. This shows the multidirectional development of adipocytes in response to different environmental stimuli.
3. Adipose tissue alteration in PC development
Exogenous signals and stimuli can greatly affect adipocyte functions and features. During cancer progression, uncontrolled cancer cell proliferation brings high nutrient demands that break normal energy balance [45, 46]; numerous pro-inflammatory cytokines released from the TME trigger catabolic reactions through different pathways in adipose tissue (Figure 1). Inflamed adipose tissue may also undergo morphological transition to other cell types which results in the loss of their original function. In this section, we sum up the findings of how PC induces functional defects in adipose tissue, and how the latter conforms to promote PC growth.
3.1. Impaired energy reservation
3.1.1. Loss of lipid storage during pancreatic tumor progression
Abnormal lipid metabolism in adipocytes has been found in the development of various cancers, including PC [47-49]. Pancreatic tumors are characterized by their massive stroma which gives rise to a hypoxic environment. In this state, PC cells cannot synthesize fatty acids by de novo lipogenesis; instead, they take up exogenous FFAs to maintain lipid homeostasis under hypoxic stress [50]. For example, exosomes secreted from PC cells carry adrenomedullin and can bind to the adrenomedullin receptors on adipocytes, activating lipolysis enzyme HSL via p38 mitogen-activated protein kinases (MAPKs) and extracellular signal‑regulated protein kinase (ERK) 1/2 dependent phosphorylation to turn on lipolysis and release FFAs [51]. PC cells can also cause lipid breakdown directly through cytokine secretion. Numerous pro-inflammatory cytokines such as interleukin (IL)-6, IL-1β, IL-8, and TNF-α in the pancreatic TME have been associated with advanced disease status in clinical studies and can trigger a catabolic response in adipocytes [13]. IL-6 activates its downstream Janus kinase (JAK)/signal transducer and activator of the transcription (STAT) pathway and the ERK pathway to increase lipolytic gene expression and enzyme activity [52]. IL-1β derived from pancreatic tumors stimulates IL-6 production from adipocytes, which aggravates autocrine lipid breakdown in adipocytes [53]. TNF-α activates lipolytic enzymes through stimulating ERK and Jun N-terminal kinase (JNK) kinase activity [54]. Additionally, both IL-1β and TNF-α promote NF-κB nuclear translocation and increase lipolytic gene transcription (Figure 1) [55-57]. PC cells-derived exosomes also enhance the release of inflammatory cytokines IL-6, IL-8, and C-C motif chemokine ligand 2 (CCL2) and promote lipolysis in adipocytes. IL-6 blockade abrogates PC-exosome-induced lipolysis [58], supporting the important role of inflammatory cytokines in lipolysis.
In addition to upregulated lipolysis, WAT may undergo beige adipocyte-transition during chronic inflammation, leading to inefficient energy use through UCP1 expression. IL-6 and parathyroid-hormone-related protein, a hormone frequently upregulated in PC with Kras mutation [59], have been reported to induce fat browning during PC progression [52, 60, 61]. Both lipolysis and fat browning in mature adipocytes increase lipid consumption, leading to the loss of adipocytes' energy storage function.
3.1.2. Energy resources from adipocytes enhance pancreatic tumor development
PC patients with obesity suffer increased tumor burden and faster disease progression compared with those without obesity [62], indicating that enriched lipid depots may exacerbate tumor growth. Another study also mentioned that in KC (fElasCreERT; KrasG12D/+) transgenic mice, a high-fat diet increased the activity of Kras, a substantial oncogenic factor in PC development, while the numbers of both PanIN and PC tumors were increased during the intervention [63, 64]. Furthermore, a high-fat diet enhanced distant metastasis and fat invasion of PC in KPC (Pdx1-Cre; LSL-KrasG12D; Trp53R172H/+) transgenic mice [65], which was also confirmed by direct fatty acid treatment in vitro. The above studies support the idea that lipid resources derived from adipose tissue promote PC progression. Lipids not only serve as substrates for ATP production, but also play central roles in cell membrane construction, signaling transduction, and posttranslational protein modification, all of which are necessary for tumor expansion. [66, 67]. Fatty acids released from adipocytes promote PC development in multiple ways. PC cells treated with primary adipocyte-derived conditioned medium containing oleic, linoleic, and palmitoleic acids, acquired extended lipid storage in their cytoplasm, and demonstrated increased migration and invasion abilities. These pro-tumor effects are positively correlated with the concentration of fatty acids [65]. PC cells take up exogenous FFAs through surface fatty acid translocase-CD36, shutting down this channel with CD36 inhibitors suppresses the enhancement of cancer cell migration by adipocyte condition medium [65]. These FFAs are usually stored as lipid droplets in PC cells to prevent lipotoxicity [49, 68] and reactive oxygen species (ROS) toxicity in hypoxic TME [69].
Besides tumor cells, noncancerous cells in the TME are also affected by abnormal lipid metabolism during cancer progression. PC has a highly immunosuppressive TME where immune cells fail to destroy tumor cells or even transit into a pro-tumor phenotype, exemplified by tumor-associated macrophages (TAMs). Under long-term stimulation with cytokines such as IL-4, IL-10, and IL-13 in the TME, cytotoxic M1 macrophages are polarized into the M2 phenotype with anti-inflammation, pro-angiogenic, and tissue remodeling abilities to promote tumor growth [70, 71]. This polarization process heavily relies on fatty acid oxidation (FAO) for energy supply, which can be supported by extra fatty acids from adipose tissue during tumor progression [72, 73].
CD8+ T lymphocytes recognize specific antigenic peptides presented on the tumor cells by human leukocyte antigen class I and are activated to kill malignant cells through secreting cytotoxic granules or cytokines like interferon and TNF [74]. For prompt energy support, glycolysis is engaged in activated CD8+ T cells [75]. However, glycolytic genes are downregulated in infiltrating CD8+ T cells in a lipid-rich environment [76, 77]. The uptake and accumulation of long-chain fatty acids in the cytoplasm can lead to lipotoxicity and thus cell death of CD8+ T cells [77]. The increased concentration of fatty acids in the TME not only benefits PC cell growth but also disturbs anti-cancer immunity.
Adipocytes can also secrete glutamine to preserve PC cell proliferation in nutrient-poor conditions [78, 79]. PC cells convert glutamine into α-ketoglutarate that can fuel the tricarboxylic acid cycle to turn on multiple biogenesis processes, further indicating the significant role of adipocytes in PC development [78]. Apart from abnormal catabolic reactions of adipose tissue during cancer progression, the extraordinary release of adipose-derived factors also contributes to pancreatic tumor growth.
3.2. Defective endocrine functions
Adipose tissue produces a variety of hormones called adipokines to regulate the metabolic processes throughout the body. However, adipocyte functions can be impaired under pathological conditions. In obesity, adipocytes undergo hypertrophy and hyperplasia, leading to excessive secretion of cytokines and the recruitment of immune cells, which results in low-grade inflammation, and further compromising adipocyte function. This is accompanied by elevated serum levels of leptin, resistin, hepatocyte growth factor (HGF), and TNF-α, along with the decreased release of adiponectin, insulin-like growth factors (IGF), and type two cytokines, which hinder systemic catabolic pathways, insulin sensitivity, and immune regulation [80]. In the context of obesity and cancer, alterations in immune cell populations have been observed. The numbers of M1 macrophages are reduced [81], while inhibitory dendritic cells [82], suppressive T cells [83], and dysfunctional T cells [76] are often found in higher abundance. These observations suggest that secretory factors derived from adipose tissue may play a role in modulating the immune system.
During PC tumorigenesis, the high levels of pro-inflammatory cytokines in the TME bring adipose tissue to an inflammatory state, causing tissue fibrosis and even adipocyte death [15, 84-86]. Moreover, adipocytes stimulated by PC can secrete lipocalin 2 (Lcn2) to facilitate muscle atrophy and fibrosis. The secretion of Lcn2 is involved in the development of cachexia, a condition characterized by severe tissue wasting, which is implicated in the prognosis of PC patients [87, 88]. Above evidence points out the aberrant endocrine function of adipose tissue, which contributes to immune suppression and tumor expansion, thereby potentially impacting patient outcomes. In the following sections, we will discuss the dysregulation of adipokines and their association with PC progression (Figure 2).
Adipokine-related pathways in pancreatic tumorigeneses. Various adipokines are released from inflamed and dysfunctional adipose tissue, triggering different signal pathways involved in the aggressive behaviors of pancreatic tumors, including the NOTCH, JAK/STAT, MAPK, and PI3K/AKT pathways.
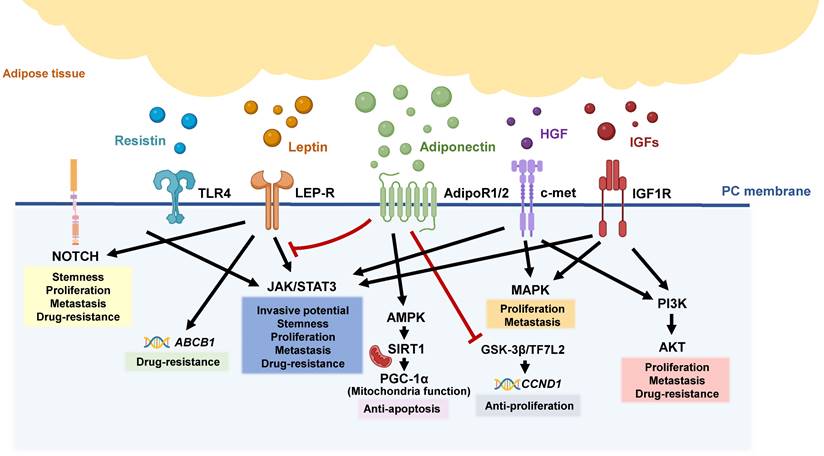
3.2.1. Leptin
Leptin is a key hormone that regulates appetite and food intake, with additional functions involved in immune response and angiogenesis [89, 90]. Even though leptin shows opposite expression patterns in different cancer types, its gene expression is upregulated in response to TNF-α and IL-1 stimulation [91-94], which are elevated in the serum of PC patients and correlated with poor prognosis [95-97]. Leptin facilitates the proliferation of human PC AsPC-1, MIA PaCa-2, and BxPC-3 cells through activation of the JAK2/STAT3 pathway. This pathway also contributes to cancer invasion by producing matrix metalloproteinase (MMP)-13 for tissue remodeling [98, 99]. In addition, leptin promotes the expression of cancer stem cell markers CD24, CD44, and ESA, with increasing tumor-sphere sizes and numbers by a NOTCH-dependent signal [100]. Both STAT3 and NOTCH pathways are vital to gemcitabine-resistance during PC development [101, 102]. Indeed, leptin protects PC cells from the cytotoxicity of 5-FU by inducing drug efflux proteins, including ATP binding cassette subfamily B member 1 (ABCB1). This results in an upregulation of anti-apoptotic proteins, including Bcl-xL, and receptor interacting protein (RIP), while inhibiting the degradation of Poly ADP-ribose Polymerase (PARP) and activation of caspase-3 [100, 103-105]. These findings highlight the important role of leptin in PC progression.
Although the effect of leptin on noncancerous cells within the pancreatic TME is poorly understood, previous research has revealed the roles of leptin in regulating both innate immunity and adaptive immunity. An in vitro study has demonstrated that short-term leptin treatment increases IFN-γ secretion and cytotoxicity of NK cells. However, prolonged leptin stimulation can lead to decreased proliferation and immune function of NK cells [106], indicating the opposite effect of leptin on NK development. Both mature and immature DCs express leptin receptors (LEPR), and leptin signal reception promotes their activation and subsequent release of pro-inflammatory cytokines [107]. Additionally, leptin stimulates macrophage proliferation in a dose-dependent manner [108]. Intriguingly, leptin treatment induces M2-specific surface markers like ARG-1 and Fizz-1 but enhances the production of pro-inflammatory cytokines, including TNF-α, IL-6, IL-1β and CCLs via JAK2/STAT3 mediated pathways [108]. Leptin treatment also induces metabolism toward M1-polarization, with increased lipid accumulation and glycolysis observed in macrophages, underscoring the importance of leptin in immune-metabolic regulation [108, 109]. Regarding adaptive immunity, CD8+ T cells represent the main force against tumor cells. Leptin can also influence the frequency of mitochondrial respiration in CD8+ T cells derived from peripheral lymph nodes near the tumor site, thus enhancing CD8+ T cells' tumor-killing function in a melanoma model [110]. Activation of T cells requires DC-exposed antigens, which can be promoted by leptin as mentioned above, showing that leptin may be a potential immunotherapeutic target during disease progression.
3.2.2. Adiponectin
Adiponectin is the most abundant adipokine secreted by mature adipocytes and possesses insulin-sensitizing and anti-inflammatory abilities [111]. Adiponectin seems to play a dual role in PC progression. Adiponectin treatment triggers glycogen synthase kinase-3 beta (GSK-3β)-dependent β-catenin degradation through activation of its receptor AdipoR1/AdipoR2. Loss of β-catenin restricts the function of its coregulator transcription Factor 7 Like 2, which reduces their downstream target cyclin D1 expression and proliferative attenuation in human PC BxPC-3 and CFPAC-1 cells [112]. This study supports the anti-tumor role of adiponectin. However, another report showed that adiponectin promotes PC progression by inhibiting apoptosis via the activation of AMPK/Sirt1/PGC-1α signaling [113]. Blockade of adiponectin signals through adiporeceptor agonists, which act on both AdipoR1 and AdipoR2, inhibits leptin-mediated STAT3 activation and AMPK activity, ultimately reducing colony formation ability and promoting apoptosis in PC cells [114].
Previous clinical studies have also found that patients with PC have lower plasma adiponectin levels [115], which matches the report showing that adipose tissue expressed less adiponectin under long-term exposure to pro-inflammatory cytokines [116]. However, another clinical study detected a higher level of adiponectin in PC patients [117] while a case-control study identified higher expression levels of adiponectin receptors AdipoR1 and AdipoR2 on pancreatic tumors [118]. These contradictory results may be attributed to the fact that adiponectin levels were measured at only one time point in these clinical studies. The levels and function of adiponectin may be dynamic during PC progression, and a time-lapse study is needed to understand the exact role of adiponectin in PC.
3.2.3. Resistin
Resistin is originally secreted from adipose tissue and takes part in obesity, inflammation, and insulin resistance. A clinical study indicated that patients with resistin-positive pancreatic tumors displayed shorter relapse-free survival times [119, 120]. After binding to adenylyl cyclase‐associated protein 1 and toll‐like receptor 4 (TLR4), resistin activates STAT3 to promote gemcitabine-resistance via modulating cell cycle from arresting S phase enter into G2-M phase in MIA PaCa‐2 and SW1990 cells [120]. These results suggest that resistin released from adipose tissue may support the survival of cancer cells.
3.2.4. Hepatocyte growth factor (HGF)
HGF has been well known to promote epithelial cell proliferation and cell motility [121]. HGF produced by adipocytes can interacts with its receptors c-mesenchymal-epithelial transition factor (c-Met) on PC cells to promote the proliferation and migration of Panc02 and TGP-47 murine cells through activation of downstream MAPK, PI3K/AKT, and STAT3 signaling [122-124]. c-Met upregulation and elevated circulating HGF have been found in locally advanced PC and are associated with poor clinical outcomes [125]. HGF/c-Met activation also provides chemoprotection against gemcitabine during KrasG12D/+-driven pancreatic intraepithelial neoplasia development in vivo [126]. HGF secreted from PSCs has been demonstrated to mediate tissue remodeling, tumor invasion, and metastasis in PC. This effect can be further exacerbated by the excessive release of HGF from dysfunctional adipocytes.
3.2.5. Insulin-like growth factors (IGFs)
IGFs have two isoforms, IGF1 and IGF2, and are produced by the liver and adipose tissue. IGFs participate in adipocyte differentiation and blood glucose regulation, and they have been reported as cancer risk factors in various tumor types [127]. During PC progression, inflammation, and insulin resistance promote IGF secretion from adipose tissue [128, 129]. IGF-1 protects PC cells from gemcitabine and Nab-Paclitaxel through activating AKT, which can be diminished by specific antibody blockade with their receptor IGF-1R [130]. IGF-1R signaling can also promote the proliferation and metastasis of HPAC and PANC-1 cells through the PI3K/PTEN/AKT, MAPK, and JAK/STAT pathways [131-133]. Inhibition of AKT is able to decrease IGF-1R expression and reduce PC cell growth [134].
3.2.6. Proinflammatory cytokines
Normally, resident macrophages in adipose tissue produce anti-inflammatory factors to maintain an appropriate environment for regular adipose tissue function. However, during cancer progression, systemic inflammation activates macrophages in adipose tissue, leading to the secretion of inflammatory cytokines from adipocytes, which in turn results in immune cell infiltration, intensifying the immune response in adipose tissue [135, 136]. Among these cytokines, IL-1β, IL-6, and TNF-α are well-known to mediate PC progression by promoting desmoplasia, proliferation, metastasis, immunosuppression, and chemoresistance [137-140]. A previous study on secretome has shown that omental adipocytes secrete IL-1β, IL-6, and TGF-β1 into pancreatic the TME, promoting the growth, metastasis, and chemoresistance of PC cells [141]. Another study has demonstrated that CXCL14 secreted from thermogenetically activated beige/brown adipocytes promotes the recruitment of macrophages and M2 polarization in high-fat diet-induced obese animal models, indicating that the browning of adipocytes may also contribute to immunosuppression in PC [142]. These studies offer clues that adipose tissue enhances systemic inflammation response by secreting a variety of pro-inflammatory cytokines, which promote cancer growth and survival and can even intensify fat wasting via autocrine effects. This creates a vicious circle that impairs adipose tissue function and exacerbates the pathological processes.
3.3. Adipocyte plasticity in PC development
Numerous signal pathways that sustain the normal function of adipocytes are interfered with by stimulation of cytokines derived from the pancreatic TME or adipose tissue itself, leading to altered expression of adipogenic and metabolic genes. Over time, adipocytes gradually lose their specific characteristics acquired during their development and undergo phenotypic changes, transitioning into progenitor-like cell types or directly trans-differentiating into pro-tumor fibroblasts. In this section, we will discuss the phenotypic changes observed in adipocytes following their interaction with PC cells and elucidate how these dedifferentiated adipocytes contribute to the progression of PC (Figure 3).
Interaction between pancreatic tumors and adjacent adipose tissue promotes adipose tissue dysfunction and cancer progression. Dysfunctional adipocytes support PC growth through various secreting factors. Long-term stimulation of pro-inflammatory cytokines also transforms white adipocytes into beige adipocytes, MSC-like progenitor cells, or CAF-like cell types that participate in ECM remodeling in PC.
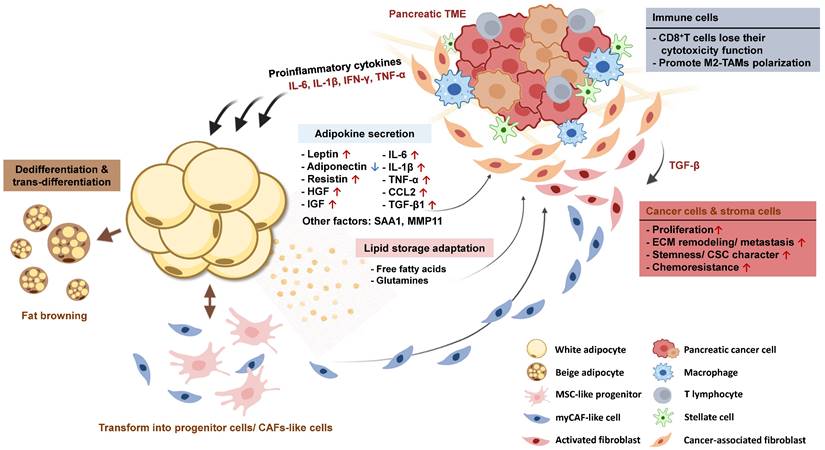
3.3.1. Morphological change and impaired function of adipocytes during PC progression
During the progression of PC, adipocytes undergo distinct morphological changes. When interacting with PC cells, adipocytes lose their lipid stores and transit into fibroblast-like cells [143, 144]. Gene expression profiling studies have revealed that co-culturing adipocytes with PANC-1 cells resulted in a significant downregulation of genes associated with adipogenesis, lipogenesis, and TG storage in mature adipocytes. Additionally, in the presence of PC cells, mature adipocytes lose their specific markers including adiponectin, leptin, resistin, and fatty acid-binding protein 4 (FABP4), while acquiring fibroblast markers like α-smooth muscle actin (α-SMA), S100 Calcium Binding Protein A4 (S100A4), FSP-1, plasminogen activator inhibitor-1, MMP-9 and MMP-11 [143, 144]. These findings suggest that PC cells can induce fibrogenic changes in adipocytes.
In addition to adipocytes, skeletal and smooth muscle in patients of PDAC exhibit increased collagen content, which has been associated with lymph node metastasis [88, 145, 146]. This muscle fibrosis appears to be influenced by the function of TGF-β, which is known to be released by TME and dysfunctional adipose tissue. The collaborative effect of fibrosis in both adipose tissue and muscle, along with the enhanced ECM deposition, creates a favorable environment for the expansion of PC.
3.3.2. Mechanism involved in adipocyte dedifferentiation
In the absence of adipogenic gene expression, particularly PPAR-γ, adipocytes undergo a fibrotic pathway leading to cellular reprogramming, which transforms adipocytes into preadipocytes, mesenchymal progenitor cells, or myofibroblasts [147]. The Wnt pathway is one of the important mechanisms in mediating adipocyte dedifferentiation. IL-6 and TGF-β derived from the PC TME induce Wnt5a expression in mature adipocytes, triggering the downstream signaling involving c-Jun/activator protein-1 (AP-1) complex for the transcription of genes associated with fibroblast transformation and MMP proteins [148, 149]. In addition to adopting fibroblast phenotype, adipocytes can transit to acquire stem cell characteristics. After being co-cultured with MIA PaCa-2 cells, mature adipocytes exhibited significant induction of genes associated with pluripotent stem cells, such as Oct3/4, Klf4, c-Myc, and Sox2 [149]. Another study has also suggested that dedifferentiated fat cells possess a multilineage potential to differentiate into various cell types within the mesenchymal lineage, including adipocytes, chondrocytes, and osteoblasts, under appropriate stimulation [150].
Collectively, the stimulation of pro-inflammatory cytokines or other factors secreted from the pancreatic TME can trigger the trans-differentiation of adipocytes into mesenchymal lineage cells or fibroblast-like cells. Adipocytes in close contact with cancer cells may progressively transit into cancer-associated fibroblasts (CAFs), which contribute to fibrosis and tissue remodeling within the TME and further intensify the chemoresistance and metastatic potential of PC.
3.4. Interaction between adipose tissue and pancreatic TME
Obesity and other metabolic syndromes have been linked to the progression of PC. In addition to the influence of remote subcutaneous fat, fat tissue adjacent to the tumor also plays a vital role in cancer development. In the KPC transgenic mouse model, a high-fat diet significantly increases the percentages of intra-tumoral adipocytes and the incidence of distant metastasis compared with a standard diet [65]. Clinical research has depicted that PC tumors with peripancreatic fat infiltration are typically associated with advanced cancer stage, locoregional recurrence, and reduced overall survival [24]. Increased fat accumulation in acinar cells also leads to the extra release of pro-inflammatory cytokines, which recruit immune cells and modulate immune responses in the pancreatic TME [26, 27]. In this section, we will focus on how adjacent adipose tissue interacts with the TME to exacerbate PC aggressiveness.
3.4.1. The effect of mature adipocytes on PC progression
Conditioned medium (CM) collected from omental fat promotes the proliferation, invasion, migration, and gemcitabine resistance of PC cells [65]. The results from xenograft nude mouse models of PC have shown that the CM of adipocytes had a higher angiogenic potential [141]. In addition, the secretome analysis of omental CM identified 157 cancer progression-associated proteins, with IL-6, IL-1β, and TGF-β as the key upstream mediators. ANGPTL4 and SPP1, encoding angiopoietin-like 4 and osteopontin respectively, were found to be upregulated in CM-treated PC cells. Both are ECM-associated glycoproteins involved in stroma structure remodeling for tumor metastasis [141, 151]. These findings support that an adipose-rich environment may boost PC development.
Besides the uni-directional signal from adipocytes, the crosstalk between adipocytes and PC cells also brings benefits for tumor growth. Previous studies have shown that adipocytes increase serum amyloid A1 (SAA1) expression in PC, which mediates PC cell migration by upregulating metastasis-related proteins N-cadherin, Snail, and Slug [144]. SAA1 expression is clinically associated with a poor prognosis of PC and plays an important role in 5-Fluorouracil (5-FU) resistance and stroma formation [144, 152]. Because SAA1 has been demonstrated to activate NF-κB and proinflammatory genes [153, 154], it may further facilitate the inflammation response in the TME. These findings suggest the noticeable role of adipocyte-induced SAA1 in PC tumor progression.
The interaction between adipocytes and PC cells also enhances the expression of MMP-11 in PC cells, which has been reported to inhibit apoptosis and is capable of degrading ECM components for tumor invasion or migration [155]. MMP-11 accelerates the cell cycle of PC cells by upregulating cyclin-dependent kinase 4 and cyclin D1 [156]. Of note, adipocytes can also secrete MMP-11 to affect tumor growth when they are nearby due to peripancreatic fat infiltration or PC metastasis into adipose tissue [157]. Not only mature adipocytes but also the resident progenitors in adipose tissue, contribute to PC development.
3.4.2. The role of adipose tissue-derived stem cells in PC
Adipose tissue-derived stem cells (ADSCs) are mesenchymal stem cells that stay in the stromal vascular fraction of adipose tissue and are capable of self-renewal as well as multipotent differentiation. ADSCs near the tumor site can easily interact with surrounding cancer cells. ADSCs excrete stromal cell-derived factor-1 (SDF-1) into the interstitial space and turn on CXC chemokine receptor 4 (CXCR4) signaling in PC cells, consequently triggering the proliferation, invasion, and migration of PC [158]. SDF-1/CXCR4 signaling has also been reported to regulate apoptosis in cancer cells and participates in tumor angiogenesis by recruiting endothelial progenitor cells [159-161]. Extracellular vesicles originating from ADSCs have been found to carry the long non-coding RNA (lncRNA) NEAT1, which promotes PC progression by facilitating proliferation, upregulating stemness-related genes, and enhancing resistance to gemcitabine. These effects are achieved through the suppression of miR-491-5p expression. Moreover, the elevated expression of the EMT-related protein Snail, along with the downregulation of suppressor of cytokine signaling (SOCS) proteins, which act as negative regulators of the JAK/STAT3 signaling pathway, can further contribute to the progression of PC [162]. However, there is also conflicting evidence about the role of ADSCs in cancer progression. A previous study reported that conditioned medium collected from primary ADSCs induced cell cycle delay by inhibiting CDK4 and cyclinD1, simultaneously promoting necrosis of PC cells. These effects ultimately lead to a reduction in tumor volume in a Capan-1 subcutaneous injection model [163].
In addition to the impact of secreted factors from ADSCs on PC growth, the inherent pluripotency of ADSCs also enables them to undergo phenotypic changes that promote PC development. When exposed to external stimuli, ADSCs can differentiate into various cell types. Specifically, TGF-β released by pancreatic tumors activates ADSCs, leading to the downregulation of S100A4 expression and their subsequent transformation into CAF-like cells. These transformed cells acquire an enhanced ability to produce ECM components that support PC cell migration [164]. Distinct co‐culture conditions also determine the fate of ADSCs. When ADSCs come directly in contact with Capan‐1 cells, they differentiate into myoblastic CAFs (myCAFs) and inflammatory CAFs (iCAFs). When ADSCs were co-cultured with PC indirectly through the trans-well system, the iCAF phenotype was induced [165]. iCAFs with high expression of IL6, CXCL1, and LIF genes are characterized by a secretory phenotype and are able to stimulate STAT3 activation in PC cells. myCAFs with high ACTA2 (α-SMA) and TPM1 expression are usually adjacent to cancer cells and contribute to ECM production [165-167].
Given the high potential of ADSCs transiting into pro-tumor CAFs, targeting the factors and mechanisms involved in this process can potentially suppress the development and progression of PC. A single-cell analysis from patients' samples reveals the gene expression pattern during ADSCs-CAFs conversion. APOD is the representative marker of the ADSCs population, together with other genes like CFD, CXCL12, MGP, and PTGDS expressed predominantly. During transformation, SFRP4, a Wnt pathway regulator, and RARRES1, which is known for regulating the proliferation and differentiation of ADSCs were both highly expressed. At the same time, the levels of PLA2G2A proteins were elevated during this transition process. PLA2G2A participates in the hydrolysis of phospholipids, leading to the production of FFAs that provide additional energy resources for cancer metastasis. At the end of the transition, ADSCs lose the expression of SFRP4 and RARRES1 and turn into COL11A1-expressing CAFs [168]. This time-scale experiment identifies the essential regulators during ADSC-CAF transition, inhibition of this pathway may be able to reduce CAF populations and their related desmoplasia.
4. Targeting adipose tissue for pancreatic cancer treatment
Emerging evidence underscores the vital role of adipose tissue in the development of PC. There is increasing interest in exploring innovative approaches to target dysfunctional adipose tissue, intending to reduce the aggressiveness of tumors and overcome treatment resistance in PC. Strategies such as blocking catabolic pathways, regulating the production of specific molecules known as adipokines and their downstream effects, or preventing the transformation of adipocytes show great promise in this regard. In this section, we provide a summary of the current applications of anti-pancreatic tumor reagents that target different functions of adipose tissue and their impact on the development of PC (Table 1). By understanding the role of adipose tissue in PC progression, novel therapeutic approaches can be explored to effectively counteract this disease.
Inhibition of the interaction between PC and adipose tissue by different mechanisms.
| Inhibiting adipokines downstream signal pathways | |||||
|---|---|---|---|---|---|
| Inhibitor name | Major molecular target | Experiment model | Results | Notes | References |
| LPrA2 (Leptin receptor antagonist) | Leptin-Notch signal | MIA PaCa-2 xenografts in Nude CD1 nu/nu mice | Delayed tumor onset Reduced tumor volume Decreased cancer stem cell marker expression | LPrA2 was delivered by iron oxide nanoparticles | [100] |
| DAPT (γ-Secretase Inhibitor) | Notch signal | Human pancreatic cancer cell lines, BxPC-3, PANC-1, and MIA PaCa-2 | Deceased number and size of tumor spheres Enhanced 5-FU sensitivity through increasing caspase-3 activity (apoptosis induction) | DAPT+5-FU | [100], [104] |
| RO4929097 (γ-Secretase Inhibitor) | Notch signal | Human tumor-sphere implant in NOD/SCID mice | Reduced cancer proliferation and tumor size Declined cancer stem cell population (ESA+/ CD44+/ CD24+) | RO4929097+Gemcitabine | [173] |
| PF-03084014 (γ-Secretase Inhibitor) | Notch signal | Patient-derived PC xenografts in athymic nude mice | Enhanced anti-tumor effect of gemcitabine Preventing lung and liver metastasis Suppressed tumor proliferation Reduced angiogenesis in pancreatic tumor | PF-03084014+Gemcitabine | [102] |
| AZD1480 | JAK/STAT3 signal | PANC-1 xenografts in Fox1-nu/nu mice | Reduced tumor size Improved mice survival Depleted desmoplastic stroma Enhanced gemcitabine delivery in tumor | AZD1480+Gemcitabine | [101] |
| Rilotumumab (human anti-HGF mAba)/ Compound A (Blocking c-met activation) | HGF-c-Met signal | AsPC-1 orthotopically inoculates in BALBc nu/nu mice | Increased gemcitabine-induced cancer cell apoptosis Reduced tumor size and metastasis events Decreased the expression of stemness markers | A triple combination treatment of Rilotumumab, compound A, and Gemcitabine shows the highest anti-cancer effect. | [123] |
| Galunisertib | TGF-β/Smad2 signal | PANC-1 human PC cells | Reduction in the population of CD25+FOXP3+ Treg cells Reduced stroma by suppressing the proliferation of epithelial cells | Transgenic mice with MT-TGFBR2DN or globally Tgfbr1+/- deficiencyb exhibit improved stromal fibrosis and restored CD8+ T cell cytotoxicity during PC development. | [181] |
| 2G8 (TGFβR2 inhibitor) | TGF-β/Smad2/4 signal | Transgenic model KICc mice and KPCd mice | Decreased IL-6 secretion from stroma Promoting M1-like macrophages, NK cells, and cytotoxic CD8+ T cell activation | 2G8 treatment shows decreased tumor proliferation and improved survival only in TGFβR2-deficient pancreatic tumors. | [182] |
| Tocilizumab (anti-IL-6 receptor antibody) | IL-6/STAT3 signal | Colo357 and PancTuI cells orthotopic xenografts in SCID/bg mice | Reduced tumor weight and proliferation Decreased local recurrence and distant metastasis | The combination treatment of Tocilizumab and Gemcitabine did not show a significant additive effect. | [184] |
| Etanercept/ Infliximab | TNF-α signal | Colo357, BxPC-3, and PancTuI cells orthotopic xenografts in SCID/bg mice | Reduced tumor volume and liver metastasis | Etanercept and Infliximab have been plunged into clinical trials of PC treatment but failed to enhance Gemcitabine efficiency. | [96, 195, 196] |
| Anakinra (human IL-1 receptor antagonist) | IL-1/NF-κB signal | AsPC-1 orthotopic xenograft in athymic nude mice | Reduced tumor size through suppressing proliferation Enhanced Gemcitabine sensitivity | The combination of Gemcitabine and Anakinra reduced the migration and induced apoptosis in PC cells. | [185] |
| Suppressing PC progression through metabolic function | |||||
| Etomoxir | Inhibition of carnitine palmitoyl transferase 1A (CPT1)/ Fatty acid oxidation | Panc02 syngeneic model with subcutaneously implantation into C57BL/6 mouse | Combination treatment of Etomoxir and anti-VEGF significantly reduced proliferation and enhanced apoptosis in PC | The inhibitory effect on PC proliferation only displays on combination treatment of Etomoxir and anti-VEGF treatment. | [190] |
| Trimetazidine | Fatty acid oxidation | MIA PaCa-2 and SNU-324 human PC cells | Trimetazidine treatment reduced ATP production and PC proliferation | PC produces ATP through FAO more than glycolysis. | [191] |
| Targeting adipocyte dedifferentiation | |||||
| Anti-Wnt5a antibody/SFRP-5 | Wnt/β-catenin Wnt5a/c-Jun/AP-1 | In vitro study of MIA PaCa-2 PC cells with 3T3-L1 adipocytes | Inhibition of the Wnt5a signal partially sustains adipocyte morphology and lipid storage | MIA PaCa-2 cells induced the transition of adipocytes into fibroblast-like phenotype. | [149] |
amAb: monoclonal antibody; bMT-TGFB2DN: TGF-β suppression through expressing dominant-negative TGFBR2 in epithelial cells; MT, metallothionein 1 promoter, allows gene modification in tissue that rich in cation, including pancreas, gastrointestinal tract, and liver. cKIC mice: mice with genetical engineer in KrasLSL-G12D/+; Cdkn2aflox/flox; Ptf1aCre/+.d KPC mice with genetical engineer in KrasLSL-G12D/+; Trp53LSL-R172H/+; Ptf1aCre/+ mice.
4.1. Receptor/downstream signaling blockade
Adipocytes upregulate pro-survival and pro-metastasis signaling in PC cells, promoting their growth through direct contact or secretion of adipokines. Numerous in vitro and in vivo studies have revealed that targeting these pathways could successfully reduce PC growth.
Clinical studies have demonstrated the correlation between high serum leptin and the risk of PC [169, 170]. The protumor functions of leptins include promoting PC proliferation, tumor sphere formation, cell cycle progression, and chemoresistance through JAK/STAT or NOTCH signaling pathways [100, 103-105]. The activation of Notch signaling by γ-secretase, which cleaves the Notch 1 intracellular domain, has been associated with increased proliferation, migration, invasion, and drug resistance in PC cells [171, 172]. Blockade of the Notch1 pathway with a leptin receptor antagonist or γ-secretase inhibitor enhances tumor sensitivity to gemcitabine and 5-FU and reduces pancreatic tumor size [100, 102, 104, 173]. The JAK/STAT pathway participates in tumorigenesis and regulates the expression of programmed death ligand-1 (PD-L1) that suppress the immune response in PC cells [174-176]. STAT3 inhibition by a competitive small-molecule inhibitor of JAK1/2 kinase, such as AZD1480, sensitizes PC cells to gemcitabine [101]. Therefore, targeting leptin and its downstream signaling pathways represent a conceivable strategy for suppressing pancreatic tumorigenesis.
SAA1 secreted from dedifferentiated adipocytes has been implicated in promoting migration and chemoresistance to 5-FU in PC cells [144]. Studies conducted in a glioblastoma model have demonstrated that SAA1 provides chemo-protection by activating AKT and downstream anti-apoptotic proteins such as BCL-2 and BAX [177]. The expression of SAA1 is regulated by H3K27 acetylation on its enhancer region, and transcription factors NF-κB and STAT3 also bind to the SAA1 promoter region. Treatment with JQ1 and mivebresib, inhibitors of the BET bromodomain involved in lysine acetylation, has been shown to effectively reduce the expression of SAA1 [178]. These findings suggest that targeting SAA1 through epigenetic modification in dedifferentiated adipocytes represents a potential therapeutic strategy for cancer.
Adipocyte-derived HGF enhances PC cell growth and gemcitabine-resistance through its interaction with the c-Met receptor. The HGF/c-Met pathway is upregulated in PC and is involved in the interaction with stromal PSCs [126]. Blockade of the HGF/c-Met signaling axis by either the ligand inhibitor rilotumumab or the receptor agonist Compound A, in combination with gemcitabine, has been found to significantly reduce pancreatic tumor size and metastasis [123]. A similar inhibitory effect has also been observed during the early development of PC in orthotopic mouse models [179], suggesting that targeting HGF/c-Met signaling to abolish the communication between PC cells and PSCs provides a promising approach for PC treatment.
Proinflammatory cytokines originating from both the pancreatic TME and inflamed adipose tissue facilitate tumor progression. Targeting the signal pathways of these cytokines is crucial for PC treatment. TGF-β exhibits distinct roles in both pro-tumorigenic and tumor-suppressive processes during PC progression, depending on the specific tumor stage and microenvironment. It exerts suppressive effects on tumor-induced inflammation and early carcinogenesis [180]. However, its downstream signaling pathway also inhibits the activation of CD8+ T cells [181]. Various strategies have been employed to disrupt TGF-β signaling, including RNA interference (RNAi), neutralizing antibodies, and small molecule inhibitors, which have shown promising results in animal models and clinical trials with improved survival rates [181-183]. In addition to TGF-β, proinflammatory cytokines such as TNF-α, IL-6, and IL-1β released from both the PC TME and inflamed adipose tissue have been found to cause adipose wasting. Blocking of these cytokines and their associated signaling pathway has demonstrated efficacy in slowing PC development and preserving adipose function. For instance, inhibition of the IL-6/STAT3 signal pathway using anti-IL-6 receptor antibodies has been successfully in reducing tumor growth, local recurrence, and distant metastasis [184]. Similarly, the blockade of TNF-α signaling has shown anti-cancer effects, mainly by reducing tumor volume and inhibiting liver metastasis [96]. Furthermore, suppression of the IL-1β pathway has been shown to preserve gemcitabine-mediated cancer cell apoptosis in AsPC-1 orthotopic xenograft models [185].
4.2. Modulation of metabolic functions in TME components
Overactivated lipolysis induced by proinflammatory cytokines during PC progression leads to the release of FFAs into the circulation or surrounding tumor area. These FFAs can be taken up and accumulated in the pancreatic TME, which not only serves as a source of nutrients for tumor growth but also induces CD8+ T cell dysfunction and M2 macrophage polarization that impair anti-tumor immunity. To combat these effects, it is crucial to suppress PC-induced fat wasting and reduce lipid uptake by the TME. Inhibiting the uptake of lipids by targeting scavenger receptor CD36 through genetic depletion, gene silencing, or irreversible inhibitor sulfosuccinimidyl oleate (SSO) treatment has been demonstrated to maintain immune system functions, prevent gemcitabine resistance, and inhibit PC metastasis [72, 77, 186-188].
In addition to blocking lipid influx, disturbing the metabolism of PC also has a significant impact on tumor growth. Pancreatic tumors with high ATGL levels show increased desmoplasia [189], while PC implantation in adipose tissue switches its metabolism from glycolysis to FAO [190], showing the significant role of fatty acid metabolism in PC progression. Fatty acid oxidation inhibition with trimetazidine reduced ATP production in human PC cell lines remarkably [191]. Targeting FAO by inhibiting the transport of fatty acids into mitochondria with etomoxir has been shown to reduce cell proliferation and promote apoptosis in PC cells under treatment with an anti-angiogenesis reagent.
On the other hand, targeting the upstream regulator of lipolysis is an effective way to prevent fat wasting during PC progression. Administration of anit-IL-6 receptor antibodies or genetic knockout of IL-6 prohibits lipid storage loss from adipose tissue [52, 192]. Treatment with inhibitors against JAK, a downstream kinase of IL-6, prevented adipose tissue loss and prolonged the lifespan of tumor-bearing mice [193]. This evidence highlights the importance of blocking lipid breakdown or cellular uptake as a critical strategy for suppressing tumor growth. However, it remains uncertain whether the observed antitumor effects in these studies are solely attributed to the inhibition of fat-wasting, as the pathways involved in lipolysis are not exclusive to adipose tissue.
4.3. Inhibition of adipocyte dedifferentiation
Mature adipocytes can transit into CAF-like cells under chronic cytokine stimulation to exacerbate desmoplasia, metastasis, and chemo-resistance of PC. It is known that adipocyte dedifferentiation initiates activation of Notch signaling to cut down fatty acid synthesis and PPAR-γ activity that sustains adipogenesis. Wnt5a signaling induced by TGF-β and IL-6 promotes MMPs production and the expression of fibrotic genes in the adipocytes. Treatment with rosiglitazone, a synthetic ligand for PPAR-γ, has successfully reversed the dedifferentiation of adipocytes [148]. Similarly, treatment with IWP2, an inhibitor of Wnt ligand secretion, has been shown to preserve the normal morphology of mature adipocytes. Additionally, the use of Wnt-5a neutralizing antibodies as well as a Wnt antagonist, such as secreted frizzled related-protein 5, has also been found to decelerate the transition of adipocytes [149]. A study conducted on mouse models of breast cancer revealed a significant reduction in tumor growth when dedifferentiated adipocytes were absent [194]. These findings provide compelling evidence that targeting the plasticity of adipocyte differentiation holds promise for suppressing the progression of PC.
5. Conclusions and Perspectives
As the significance of lipid metabolism in tumor progression becomes more prominent, an increasing number of studies are now focusing on the role of adipose tissue in the development of PC, especially in obese subjects. Growing evidence suggests a strong connection between adipose tissue and aggressive PC, characterized by enhanced metastasis and chemoresistance. Long-term exposure to proinflammatory cytokines secreted from the pancreatic TME triggers the release of excessive lipid resources and abnormal adipokines from dysfunctional adipose tissue, providing a supportive environment for cancer cell proliferation and the establishment of a tumor-promoting TME. The aberrant transition of mature adipocytes into a cell type exhibiting myCAF characteristics gives rise to severe desmoplasia in the pancreatic TME. Cytokines and growth factors produced by these cells further stimulate the proliferation, invasion, migration, and chemoprotection of PC. Thus, disrupting the communication between adipose tissue and PC cells holds great promise for improving the efficacy of antineoplastic treatments. Indeed, in vivo studies have demonstrated significant advancements in anti-pancreatic tumor progression and improving survival by employing this approach. Combining targeted anti-adipokine reagents or neutralizing antibodies with chemotherapy drugs has proven successful in overcoming chemoresistance mediated by dysfunctional adipocytes. These findings provide valuable insights and a hopeful outlook for overcoming cancer drug resistance in clinical treatment. They offer valuable guidance for future therapeutic strategies, paving the way for innovative directions in the field of cancer treatment. However, there remains a need to better understand the impact of adipose tissue on pancreatic tumor expansion and the mechanisms underlying the transition of adipocytes.
Most studies investigating the involvement of adipose tissue in PC progression have utilized obesity as their disease model. However, emerging evidence suggests that adipose tissue adjacent to the pancreas can promote locoregional recurrence and result in unfavorable clinical outcomes, even in the absence of metabolic disorders. In animal models of cachexia, the loss and remodeling of adipose tissue have been observed during PC development, showing that while obesity can be a contributing factor, it is not a necessary condition for disease progression. Chronic inflammation induced by adipose tissue itself during obesity creates a metabolic imbalance and unhealthy immune response that may not accurately reflect the conditions in patients without metabolic diseases. Therefore, further investigation is needed to understand the complicated communication between adipose tissue and PC. Additionally, different adipose tissue depots exhibit heterogeneous responses to physiological stimulation. For instance, epididymal and mesenteric adipose tissue display a higher inflammation status compared to subcutaneous depots during tumor progression. This indicates that the distance between adipose tissue and the tumor may result in varying degrees of impact. While most studies have focused on peritoneal fat in PC analyses, it is important to consider that proinflammatory cytokines released from TME may influence all fat depots through the systemic circulation. Exploring whether other adipose tissue depots exhibit similar pro-tumor effects as peritoneal fat is an interesting area for future investigations. Moreover, limited attention has been given to the direct effects of adipokines on immune cells rather than cancer cells in PC models. The immune escape continues to pose a significant challenge in the treatment of PC, and exploring the potential role of adipokines in the development of treatment resistance in PC cells could yield valuable insights for overcoming this difficulty. Future research focusing on the direct impact of adipokines on immune cells may uncover novel strategies to enhance immune responses and improve treatment outcomes in PC.
Nowadays, increasing evidence reveal the connection between adipose tissue and the clinical outcomes of PC, highlighting adipose tissue as a promising target for therapeutic interventions. However, it is important to note that most intervention methods targeting adipose-tumor interactions have primarily been tested in vitro. The lack of in vivo evaluation hinders better assessment of their efficacy and safety, which is essential before clinical translation. Although the journey towards the clinical use and approval of these therapeutics may be long, acquiring a more comprehensive understanding of the molecular mechanisms through which adipose tissue promotes pancreatic tumors and associated wasting symptoms will undoubtedly contribute to the development of more effective anti-cancer strategies.
Abbreviations
5-FU: 5-fluorouracil; ABCB1: ATP binding cassette subfamily B member 1; ADSCs: adipose-derived mesenchymal stem cells; AMPK: AMP activated protein kinase; AP-1: activator protein-1; ATGL: adipose triglyceride lipase; ATP: adenosine triphosphate; BAT: brown adipose tissue; BMI: body mass index; BMP: bone morphogenic protein; C/EBP: CCAAT-enhancer-binding protein; CAFs: cancer-associated fibroblast; CC: cancer cachexia; CCL-2: C-C motif chemokine ligand 2; CM: conditioned medium; c-Met: c-mesenchymal-epithelial transition factor; CXCR4: CXC chemokine receptor 4; DCs: dendritic cells; DGs: diglycerides; ECM: extracellular matrix; FABP4: fatty acid binding protein 4; FAO: fatty acid oxidation; FFAs: free fatty acids; FGFs: fibroblast growth factors; GSK-3β: glycogen synthase kinase-3 beta; HGF: hepatocyte growth factor; HSL: hormone-sensitive lipase; iCAFs: inflammatory CAF; IFN-γ: interferon gamma; IGFs: insulin-like growth factors; IL: interleukin; JAK: janus kinase; JNK: jun N-terminal kinase; KC: fElasCreERT;KrasG12D/+; KPC: Pdx1-Cre;LSL-KrasG12D/+;Trp53R172H/+; Lcn2: lipocalin 2; LEPR: leptin receptors; lncRNA: long non-coding RNA; MAPKs: mitogen-activated protein kinases; MGL: monoglyceride lipase; MMP: matrix metalloproteinase; MSCs: mesenchymal stem cells; myCAFs: myoblastic CAFs; Myf5: myogenic factor-5; NF-κB: nuclear factor kappa B; NK cells: nature killer cells; PARP: poly ADP-ribose polymerase; PC: pancreatic cancer; PGC-1α: PPAR-γ co-activator 1 alpha; PI3K: phosphatidylinositol 3-kinase; PPAR-γ: peroxisome proliferator-activated receptor gamma; PSCs: pancreatic stellate cells; RIP: receptor interacting protein; RNAi: RNA interference; ROS: reactive oxygen species; S100A4: S100 calcium binding protein A4; SAA1: serum amyloid A1; SDF-1: stromal cell-derived factor-1; SOCS: suppressor of cytokine signaling; STAT: signal transducer and activator of transcription; TAMs: tumor-associated macrophages; TG: triglycerides; TGF-β: transforming growth factor beta; TLR4: toll‐like receptor 4; TME: tumor microenvironment; TNF-α: tumor necrosis factor alpha; UCP1: uncoupling protein 1; WAT: white adipose tissue; Wnt: wingless-related integration site; α-SMA: alpha-smooth muscle actin.
Acknowledgements
We thank core labs of Clinical Medicine Research Center of NCKUH for the technical support.
Author Contributions
Conceptualization, Y.-C.L., Y.-C.H., and Y.-S.S.; visualization, Y.-C.L., and Y.-C.H.; writing—original draft preparation, Y.-C.L.; writing—review and editing, Y.-C.H., H.-C.W., and Y.-S.S.; Funding acquisition, Y.-S.S. All authors have read and agreed to the published version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209-49
2. Ho WJ, Jaffee EM, Zheng L. The tumour microenvironment in pancreatic cancer—clinical challenges and opportunities. Nat Rev Clin Oncol. 2020;17:527-40
3. Ren B, Cui M, Yang G, Wang H, Feng M, You L. et al. Tumor microenvironment participates in metastasis of pancreatic cancer. Mol Cancer. 2018;17:1-15
4. Wang S, Li Y, Xing C, Ding C, Zhang H, Chen L. et al. Tumor microenvironment in chemoresistance, metastasis and immunotherapy of pancreatic cancer. Am J Cancer Res. 2020;10:1937-53
5. Lee HS, Leem G, Kang H, Jo JH, Chung MJ, Jang SJ. et al. Peripheral natural killer cell activity is associated with poor clinical outcomes in pancreatic ductal adenocarcinoma. J Clin Gastroenterol Hepatol. 2021;36:516-22
6. Marcon F, Zuo J, Pearce H, Nicol S, Margielewska-Davies S, Farhat M. et al. NK cells in pancreatic cancer demonstrate impaired cytotoxicity and a regulatory IL-10 phenotype. Oncoimmunology. 2020;9:1845424
7. Bachem MG, Zhou Z, Zhou S, Siech M. Role of stellate cells in pancreatic fibrogenesis associated with acute and chronic pancreatitis. J Clin Gastroenterol Hepatol. 2006;21(Suppl 3):S92-6
8. McCarroll JA, Naim S, Sharbeen G, Russia N, Lee J, Kavallaris M. et al. Role of pancreatic stellate cells in chemoresistance in pancreatic cancer. Front Physiol. 2014;5:141
9. Wang H-C, Lin Y-L, Hsu C-C, Chao Y-J, Hou Y-C, Chiu T-J. et al. Pancreatic stellate cells activated by mutant KRAS-mediated PAI-1 upregulation foster pancreatic cancer progression via IL-8. Theranostics. 2019;9:7168-83
10. Jin G, Hong W, Guo Y, Bai Y, Chen B. Molecular mechanism of pancreatic stellate cells activation in chronic pancreatitis and pancreatic cancer. J Cancer. 2020;11:1505-15
11. Kordes M, Larsson L, Engstrand L, Löhr J-M. Pancreatic cancer cachexia: three dimensions of a complex syndrome. Br J Cancer. 2021;124:1623-36
12. Ni J, Zhang L. Cancer cachexia: Definition, staging, and emerging treatments. Cancer Manag Res. 2020;12:5597-605
13. Hou Y-C, Wang C-J, Chao Y-J, Chen H-Y, Wang H-C, Tung H-L. et al. Elevated serum interleukin-8 level correlates with cancer-related cachexia and sarcopenia: an indicator for pancreatic cancer outcomes. J Clin Med. 2018;7:502
14. Choe SS, Huh JY, Hwang IJ, Kim JI, Kim JB. Adipose tissue remodeling: its role in energy metabolism and metabolic disorders. Front Endocrinol (Lausanne). 2016;7:30
15. Vegiopoulos A, Rohm M, Herzig S. Adipose tissue: between the extremes. EMBO J. 2017;36:1999-2017
16. Incio J, Liu H, Suboj P, Chin SM, Chen IX, Pinter M. et al. Obesity-induced inflammation and desmoplasia promote pancreatic cancer progression and resistance to chemotherapy. Cancer Discov. 2016;6:852-69
17. Xu M, Jung X, Hines OJ, Eibl G, Chen Y. Obesity and pancreatic cancer: overview of epidemiology and potential prevention by weight loss. Pancreas. 2018;47:158-62
18. Bhaskaran K, Douglas I, Forbes H, dos-Santos-Silva I, Leon DA, Smeeth L. Body-mass index and risk of 22 specific cancers: a population-based cohort study of 5· 24 million UK adults. Lancet. 2014;384:755-65
19. Ligibel JA, Alfano CM, Courneya KS, Demark-Wahnefried W, Burger RA, Chlebowski RT. et al. American Society of Clinical Oncology position statement on obesity and cancer. J Clin Oncol. 2014;32:3568-74
20. Riccardi DMdR, Das Neves RX, de Matos-Neto EM, Camargo RG, Lima JDCC, Radloff K. et al. Plasma lipid profile and systemic inflammation in patients with cancer cachexia. Front Nutr. 2020;7:4
21. Mormile R. Total serum cholesterol and pancreatic cancer risk: what is the link? Pathol Oncol Res. 2020;26:1361
22. Fonseca GWPd, Farkas J, Dora E, von Haehling S, Lainscak M. Cancer cachexia and related metabolic dysfunction. Int J Mol Sci. 2020;21:2321
23. Takahashi M, Hori M, Ishigamori R, Mutoh M, Imai T, Nakagama H. Fatty pancreas: A possible risk factor for pancreatic cancer in animals and humans. Cancer Sci. 2018;109:3013-23
24. Jamieson NB, Foulis AK, Oien KA, Dickson EJ, Imrie CW, Carter R. et al. Peripancreatic fat invasion is an independent predictor of poor outcome following pancreaticoduodenectomy for pancreatic ductal adenocarcinoma. Journal of Gastrointestinal Surgery. 2011;15:512-24
25. Hori M, Takahashi M, Hiraoka N, Yamaji T, Mutoh M, Ishigamori R. et al. Association of pancreatic fatty infiltration with pancreatic ductal adenocarcinoma. Clin Transl Gastroenterol. 2014;5:e53
26. Singh RG, Nguyen NN, Cervantes A, Ramos GCA, Cho J, Petrov MS. Associations between intra-pancreatic fat deposition and circulating levels of cytokines. Cytokine. 2019;120:107-14
27. Eibl G, Rozengurt E. KRAS, YAP, and obesity in pancreatic cancer: A signaling network with multiple loops. Semin Cancer Biol. 2019;54:50-62
28. Bourgeois C, Gorwood J, Barrail-Tran A, Lagathu C, Capeau J, Desjardins D. et al. Specific biological features of adipose tissue, and their impact on HIV persistence. Front Microbiol. 2019;10:2837
29. Corvera S, Solivan-Rivera J, Yang Loureiro Z. Angiogenesis in adipose tissue and obesity. Angiogenesis. 2022;25:439-53
30. Herold J, Kalucka J. Angiogenesis in adipose tissue: the interplay between adipose and endothelial cells. Front Physiol. 2021;11:624903
31. Chait A, den Hartigh LJ. Adipose tissue distribution, inflammation and its metabolic consequences, including diabetes and cardiovascular disease. Front Cardiovasc Med. 2020;7:22
32. Duncan RE, Ahmadian M, Jaworski K, Sarkadi-Nagy E, Sul HS. Regulation of lipolysis in adipocytes. Annu Rev Nutr. 2007;27:79-101
33. Jastroch M. Uncoupling protein 1 controls reactive oxygen species in brown adipose tissue. Proceedings of the National Academy of Sciences. 2017;114:7744-6
34. Shan T, Liang X, Bi P, Zhang P, Liu W, Kuang S. Distinct populations of adipogenic and myogenic Myf5-lineage progenitors in white adipose tissues. J Lipid Res. 2013;54:2214-24
35. Berry DC, Jiang Y, Graff JM. Mouse strains to study cold-inducible beige progenitors and beige adipocyte formation and function. Nat Commun. 2016;7:10184
36. Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84:277-359
37. Petrovic N, Walden TB, Shabalina IG, Timmons JA, Cannon B, Nedergaard J. Chronic peroxisome proliferator-activated receptor γ (PPARγ) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J Biol Chem. 2010;285:7153-64
38. Merlin J, Evans BA, Dehvari N, Sato M, Bengtsson T, Hutchinson DS. Could burning fat start with a brite spark? Pharmacological and nutritional ways to promote thermogenesis. Mol Nutr Food Res. 2016;60:18-42
39. Singh AM, Zhang L, Avery J, Yin A, Du Y, Wang H. et al. Human beige adipocytes for drug discovery and cell therapy in metabolic diseases. Nat Commun. 2020;11:2758
40. Cheng L, Wang J, Dai H, Duan Y, An Y, Shi L. et al. Brown and beige adipose tissue: a novel therapeutic strategy for obesity and type 2 diabetes mellitus. Adipocyte. 2021;10:48-65
41. Chen Q, Shou P, Zheng C, Jiang M, Cao G, Yang Q. et al. Fate decision of mesenchymal stem cells: adipocytes or osteoblasts? Cell Death Differ. 2016;23:1128-39
42. Reddy NL, Tan BK, Barber TM, Randeva HS. Brown adipose tissue: endocrine determinants of function and therapeutic manipulation as a novel treatment strategy for obesity. BMC Obes. 2014;1:1-12
43. Madsen MS, Siersbæk R, Boergesen M, Nielsen R, Mandrup S. Peroxisome proliferator-activated receptor γ and C/EBPα synergistically activate key metabolic adipocyte genes by assisted loading. Membr Cell Biol. 2014;34:939-54
44. Mulya A, Kirwan JP. Brown and beige adipose tissue: therapy for obesity and its comorbidities? Endocrinol Metab Clin North Am. 2016;45:605-21
45. García-Jiménez C, Goding CR. Starvation and pseudo-starvation as drivers of cancer metastasis through translation reprogramming. Cell Metab. 2019;29:254-67
46. Frühbeck G, Méndez-Giménez L, Fernández-Formoso J-A, Fernández S, Rodriguez A. Regulation of adipocyte lipolysis. Nutr Res Rev. 2014;27:63-93
47. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
48. Koundouros N, Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J Cancer. 2020;122:4-22
49. Sunami Y, Rebelo A, Kleeff J. Lipid metabolism and lipid droplets in pancreatic cancer and stellate cells. Cancers (Basel). 2018;10:3
50. Hao X, Ren Y, Feng M, Wang Q, Wang Y. Metabolic reprogramming due to hypoxia in pancreatic cancer: Implications for tumor formation, immunity, and more. Biomed Pharmacother. 2021;141:111798
51. Sagar G, Sah RP, Javeed N, Dutta SK, Smyrk TC, Lau JS. et al. Pathogenesis of pancreatic cancer exosome-induced lipolysis in adipose tissue. Gut. 2016;65:1165-74
52. Han J, Meng Q, Shen L, Wu G. Interleukin-6 induces fat loss in cancer cachexia by promoting white adipose tissue lipolysis and browning. Lipids Health Dis. 2018;17:1-8
53. Uno T, He J, Usui I, Kanatani Y, Bukhari A, Fujisaka S. et al. Long-term interleukin-1α treatment inhibits insulin signaling via IL-6 production and SOCS3 expression in 3T3-L1 adipocytes. Horm Metab Res. 2008;40:8-12
54. Ryden M, Arner P. Tumour necrosis factor-α in human adipose tissue-from signalling mechanisms to clinical implications. J Intern Med. 2007;262:431-8
55. Grant RW, Stephens JM. Fat in flames: influence of cytokines and pattern recognition receptors on adipocyte lipolysis. Am J Physiol Endocrinol Metab. 2015;309:E205-13
56. Gao D, Madi M, Ding C, Fok M, Steele T, Ford C. et al. Interleukin-1β mediates macrophage-induced impairment of insulin signaling in human primary adipocytes. Am J Physiol Endocrinol Metab. 2014;307:E289-304
57. Jurga L, Elisabet ANm, Andrea D, Lennart B, Erik Ns, Dominique L. et al. NF-κB is important for TNF-α-induced lipolysis in human adipocytes. J Lipid Res. 2007;48:1069-77
58. Wang S, Xu M, Xiao X, Wang L, Sun Z, Guan M. et al. Pancreatic cancer cell exosomes induce lipidomics changes in adipocytes. Adipocyte. 2022;11:346-55
59. Pitarresi JR, Norgard RJ, Chiarella AM, Suzuki K, Bakir B, Sahu V. et al. PTHrP drives pancreatic cancer growth and metastasis and reveals a new therapeutic vulnerability. Cancer Discov. 2021;11:1774-91
60. Petruzzelli M, Schweiger M, Schreiber R, Campos-Olivas R, Tsoli M, Allen J. et al. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab. 2014;20:433-47
61. Kir S, White JP, Kleiner S, Kazak L, Cohen P, Baracos VE. et al. Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature. 2014;513:100-4
62. Chung KM, Singh J, Lawres L, Dorans KJ, Garcia C, Burkhardt DB. et al. Endocrine-exocrine signaling drives obesity-associated pancreatic ductal adenocarcinoma. Cell. 2020;181:832-47
63. Philip B, Roland CL, Daniluk J, Liu Y, Chatterjee D, Gomez SB. et al. A high-fat diet activates oncogenic Kras and COX2 to induce development of pancreatic ductal adenocarcinoma in mice. Gastroenterology. 2013;145:1449-58
64. Zhu L, Ji J, Ma J, Wang D, Liu M, Du JX. et al. Differential effects of dietary macronutrients on the development of oncogenic KRAS-mediated pancreatic ductal adenocarcinoma. Cancers (Basel). 2022;14:2723
65. Okumura T, Ohuchida K, Sada M, Abe T, Endo S, Koikawa K. et al. Extra-pancreatic invasion induces lipolytic and fibrotic changes in the adipose microenvironment, with released fatty acids enhancing the invasiveness of pancreatic cancer cells. Oncotarget. 2017;8:18280-95
66. Luo X, Cheng C, Tan Z, Li N, Tang M, Yang L. et al. Emerging roles of lipid metabolism in cancer metastasis. Mol Cancer. 2017;16:1-10
67. Swierczynski J, Hebanowska A, Sledzinski T. Role of abnormal lipid metabolism in development, progression, diagnosis and therapy of pancreatic cancer. World J Gastroenterol. 2014;20:2279-303
68. Munir R, Lisec J, Swinnen JV, Zaidi N. Lipid metabolism in cancer cells under metabolic stress. Br J Cancer. 2019;120:1090-8
69. Bensaad K, Favaro E, Lewis CA, Peck B, Lord S, Collins JM. et al. Fatty acid uptake and lipid storage induced by HIF-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014;9:349-65
70. Chen Y, Song Y, Du W, Gong L, Chang H, Zou Z. Tumor-associated macrophages: an accomplice in solid tumor progression. J Biomed Sci. 2019;26:1-13
71. Belgiovine C, Digifico E, Anfray C, Ummarino A, Torres Andón F. Targeting tumor-associated macrophages in anti-cancer therapies: convincing the traitors to do the right thing. J Clin Med. 2020;9:3226
72. Su P, Wang Q, Bi E, Ma X, Liu L, Yang M. et al. Enhanced lipid accumulation and metabolism are required for the differentiation and activation of tumor-associated macrophages. Cancer Res. 2020;80:1438-50
73. Batista-Gonzalez A, Vidal R, Criollo A, Carreño LJ. New insights on the role of lipid metabolism in the metabolic reprogramming of macrophages. Front Immunol. 2020;10:2993
74. Durgeau A, Virk Y, Corgnac S, Mami-Chouaib F. Recent advances in targeting CD8 T-cell immunity for more effective cancer immunotherapy. Front Immunol. 2018;9:14
75. Menk AV, Scharping NE, Moreci RS, Zeng X, Guy C, Salvatore S. et al. Early TCR signaling induces rapid aerobic glycolysis enabling distinct acute T cell effector functions. Cell Rep. 2018;22:1509-21
76. Ringel AE, Drijvers JM, Baker GJ, Catozzi A, García-Cañaveras JC, Gassaway BM. et al. Obesity shapes metabolism in the tumor microenvironment to suppress anti-tumor immunity. Cell. 2020;183:1848-66
77. Manzo T, Prentice BM, Anderson KG, Raman A, Schalck A, Codreanu GS. et al. Accumulation of long-chain fatty acids in the tumor microenvironment drives dysfunction in intrapancreatic CD8+ T cells. J Exp Med. 2020;217:e20191920
78. Bott AJ, Shen J, Tonelli C, Zhan L, Sivaram N, Jiang Y-P. et al. Glutamine anabolism plays a critical role in pancreatic cancer by coupling carbon and nitrogen metabolism. Cell Rep. 2019;29:1287-98
79. Meyer KA, Neeley CK, Baker NA, Washabaugh AR, Flesher CG, Nelson BS. et al. Adipocytes promote pancreatic cancer cell proliferation via glutamine transfer. Biochem Biophys Rep. 2016;7:144-9
80. Taylor EB. The complex role of adipokines in obesity, inflammation, and autoimmunity. Clinical Science. 2021;135:731-52
81. Singh A, Mayengbam SS, Yaduvanshi H, Wani MR, Bhat MK. Obesity programs macrophages to support cancer progression. Cancer Res. 2022;82:4303-12
82. James BR, Tomanek-Chalkley A, Askeland EJ, Kucaba T, Griffith TS, Norian LA. Diet-induced obesity alters dendritic cell function in the presence and absence of tumor growth. J Immunol. 2012;189:1311-21
83. Wogsland CE, Lien HE, Pedersen L, Hanjra P, Grondal SM, Brekken RA. et al. High dimensional immunotyping of the obese tumor microenvironment reveals model specific adaptation. Dis Model Mech. 2020;14:dmm048977
84. Silvério R, Lira FS, Oyama LM, do Nascimento CMO, Otoch JP, Alcântara PS. et al. Lipases and lipid droplet-associated protein expression in subcutaneous white adipose tissue of cachectic patients with cancer. Lipids Health Dis. 2017;16:1-11
85. Brocco D, Florio R, De Lellis L, Veschi S, Grassadonia A, Tinari N. et al. The role of dysfunctional adipose tissue in pancreatic cancer: A molecular perspective. Cancers (Basel). 2020;12:1849
86. Lindhorst A, Raulien N, Wieghofer P, Eilers J, Rossi F, Bechmann I. et al. Adipocyte death triggers a pro-inflammatory response and induces metabolic activation of resident macrophages. Cell Death Dis. 2021;12:1-15
87. Lemecha M, Chalise JP, Takamuku Y, Zhang G, Yamakawa T, Larson G. et al. Lcn2 mediates adipocyte-muscle-tumor communication and hypothermia in pancreatic cancer cachexia. Mol Metab. 2022;66:101612
88. Judge SM, Nosacka RL, Delitto D, Gerber MH, Cameron ME, Trevino JG. et al. Skeletal muscle fibrosis in pancreatic cancer patients with respect to survival. JNCI Cancer Spectr. 2018;2:pky043
89. Procaccini C, La Rocca C, Carbone F, De Rosa V, Galgani M, Matarese G. Leptin as immune mediator: Interaction between neuroendocrine and immune system. Dev Comp Immunol. 2017;66:120-9
90. Yu F, Fu R, Liu L, Wang X, Wu T, Shen W. et al. Leptin-induced angiogenesis of EA. Hy926 endothelial cells via the Akt and Wnt signaling pathways in vitro and in vivo. Front Pharmacol. 2019;10:1275
91. Barone I, Giordano C, Bonofiglio D, Andò S, Catalano S. Leptin, obesity and breast cancer: progress to understanding the molecular connections. Curr Opin Pharmacol. 2016;31:83-9
92. Demiray G, Değirmencioğlu S, Uğurlu E, Yaren A. Effects of serum leptin and resistin levels on cancer cachexia in patients with advanced-stage non-small cell lung cancer. Clin Med Insights Oncol. 2017;11:1179554917690144
93. Słomian GJ, Nowak D, Buczkowska M, Głogowska-Gruszka A, Słomian SP, Roczniak W. et al. The role of adiponectin and leptin in the treatment of ovarian cancer patients. Endokrynol Pol. 2019;70:57-63
94. Liu C, Feng X, Li Q, Wang Y, Li Q, Hua M. Adiponectin, TNF-α and inflammatory cytokines and risk of type 2 diabetes: a systematic review and meta-analysis. Cytokine. 2016;86:100-9
95. Karayiannakis AJ, Syrigos KN, Polychronidis A, Pitiakoudis M, Bounovas A, Simopoulos K. Serum levels of tumor necrosis factor-alpha and nutritional status in pancreatic cancer patients. Anticancer Res. 2001;21:1355-8
96. Egberts J-H, Cloosters V, Noack A, Schniewind B, Thon L, Klose S. et al. Anti-tumor necrosis factor therapy inhibits pancreatic tumor growth and metastasis. Cancer Res. 2008;68:1443-50
97. Mitsunaga S, Ikeda M, Shimizu S, Ohno I, Furuse J, Inagaki M. et al. Serum levels of IL-6 and IL-1 β can predict the efficacy of gemcitabine in patients with advanced pancreatic cancer. Br J Cancer. 2013;108:2063-9
98. Mendonsa AM, Chalfant MC, Gorden LD, VanSaun MN. Modulation of the leptin receptor mediates tumor growth and migration of pancreatic cancer cells. PLoS One. 2015;10:e0126686
99. Fan Y, Gan Y, Shen Y, Cai X, Song Y, Zhao F. et al. Leptin signaling enhances cell invasion and promotes the metastasis of human pancreatic cancer via increasing MMP-13 production. Oncotarget. 2015;6:16120-34
100. Harbuzariu A, Rampoldi A, Daley-Brown DS, Candelaria P, Harmon TL, Lipsey CC. et al. Leptin-Notch signaling axis is involved in pancreatic cancer progression. Oncotarget. 2017;8:7740-52
101. Nagathihalli NS, Castellanos JA, Shi C, Beesetty Y, Reyzer ML, Caprioli R. et al. Signal transducer and activator of transcription 3, mediated remodeling of the tumor microenvironment results in enhanced tumor drug delivery in a mouse model of pancreatic cancer. Gastroenterology. 2015;149:1932-43
102. Yabuuchi S, Pai SG, Campbell NR, De Wilde RF, De Oliveira E, Korangath P. et al. Notch signaling pathway targeted therapy suppresses tumor progression and metastatic spread in pancreatic cancer. Cancer Lett. 2013;335:41-51
103. Mantho CIT, Harbuzariu A, Gonzalez-Perez RR. Histone deacetylases, microRNA and leptin crosstalk in pancreatic cancer. World J Clin Oncol. 2017;8:178-89
104. Harbuzariu A, Gonzalez-Perez RR. Leptin-Notch axis impairs 5-fluorouracil effects on pancreatic cancer. Oncotarget. 2018;9:18239-53
105. Paz-Filho G, Mastronardi C, Franco CB, Wang KB, Wong M-L, Licinio J. Leptin: molecular mechanisms, systemic pro-inflammatory effects, and clinical implications. Arq Bras Endocrinol Metabol. 2012;56:597-607
106. Wrann CD, Laue T, Hübner L, Kuhlmann S, Jacobs R, Goudeva L. et al. Short-term and long-term leptin exposure differentially affect human natural killer cell immune functions. Am J Physiol Endocrinol Metab. 2012;302:E108-16
107. Al-Hassi H, Bernardo D, Murugananthan A, Mann E, English N, Jones A. et al. A mechanistic role for leptin in human dendritic cell migration: differences between ileum and colon in health and Crohn's disease. Mucosal Immunol. 2013;6:751-61
108. Monteiro L, Pereira JAdS, Palhinha L, Moraes-Vieira PMM. Leptin in the regulation of the immunometabolism of adipose tissue-macrophages. J Leukoc Biol. 2019;106:703-16
109. Monteiro LdB, Prodonoff JS, Favero de Aguiar C, Correa-da-Silva F, Castoldi A, Bakker NvT. et al. Leptin signaling suppression in macrophages improves immunometabolic outcomes in obesity. Diabetes. 2022;71:1546-61
110. Harjes U. Leptin boosts T cell function in tumours. Nat Rev Cancer. 2019;19:607
111. Maeda N, Funahashi T, Matsuzawa Y, Shimomura I. Adiponectin, a unique adipocyte-derived factor beyond hormones. Atherosclerosis. 2020;292:1-9
112. Jiang J, Fan Y, Zhang W, Shen Y, Liu T, Yao M. et al. Adiponectin suppresses human pancreatic cancer growth through attenuating the β-catenin signaling pathway. Int J Biol Sci. 2019;15:253-64
113. Huang B, Cheng X, Wang D, Peng M, Xue Z, Da Y. et al. Adiponectin promotes pancreatic cancer progression by inhibiting apoptosis via the activation of AMPK/Sirt1/PGC-1α signaling. Oncotarget. 2014;5:4732-45
114. Messaggio F, Mendonsa AM, Castellanos J, Nagathihalli NS, Gorden L, Merchant NB. et al. Adiponectin receptor agonists inhibit leptin induced pSTAT3 and in vivo pancreatic tumor growth. Oncotarget. 2017;8:85378-91
115. Bao Y, Giovannucci EL, Kraft P, Stampfer MJ, Ogino S, Ma J. et al. A prospective study of plasma adiponectin and pancreatic cancer risk in five US cohorts. J Natl Cancer Inst. 2013;105:95-103
116. Simons PJ, van den Pangaart PS, Aerts JM, Boon L. Pro-inflammatory delipidizing cytokines reduce adiponectin secretion from human adipocytes without affecting adiponectin oligomerization. J Endocrinol. 2007;192:289-99
117. Gasiorowska A, Talar-Wojnarowska R, Kaczka A, Borkowska A, Czupryniak L, Małecka-Panas E. Subclinical inflammation and endothelial dysfunction in patients with chronic pancreatitis and newly diagnosed pancreatic cancer. Dig Dis Sci. 2016;61:1121-9
118. Dalamaga M, Migdalis I, Fargnoli JL, Papadavid E, Bloom E, Mitsiades N. et al. Pancreatic cancer expresses adiponectin receptors and is associated with hypoleptinemia and hyperadiponectinemia: a case-control study. Cancer Causes Control. 2009;20:625-33
119. Jiang C-Y, Wang W, Yin Y-L, Yuan Z-R, Wang L-B. Expression of the adipocytokine resistin and its association with the clinicopathological features and prognosis of pancreatic ductal adenocarcinoma. Oncol Lett. 2012;4:960-4
120. Zhang M, Yan L, Wang GJ, Jin R. Resistin effects on pancreatic cancer progression and chemoresistance are mediated through its receptors CAP1 and TLR4. J Cell Physiol. 2019;234:9457-66
121. Kato T. Biological roles of hepatocyte growth factor-Met signaling from genetically modified animals. Biomed Rep. 2017;7:495-503
122. Ziegler KM, Considine RV, True E, Swartz-Basile DA, Pitt HA, Zyromski NJ. Adipocytes enhance murine pancreatic cancer growth via a hepatocyte growth factor (HGF)-mediated mechanism. Int J Surg. 2016;28:179-84
123. Xu Z, Pang TC, Liu AC, Pothula SP, Mekapogu AR, Perera CJ. et al. Targeting the HGF/c-MET pathway in advanced pancreatic cancer: A key element of treatment that limits primary tumour growth and eliminates metastasis. Br J Cancer. 2020;122:1486-95
124. Matsumura A, Kubota T, Taiyoh H, Fujiwara H, Okamoto K, Ichikawa D. et al. HGF regulates VEGF expression via the c-Met receptor downstream pathways, PI3K/Akt, MAPK and STAT3, in CT26 murine cells. Int J Oncol. 2013;42:535-42
125. Yu J, Ohuchida K, Mizumoto K, Ishikawa N, Ogura Y, Yamada D. et al. Overexpression of c-met in the early stage of pancreatic carcinogenesis; altered expression is not sufficient for progression from chronic pancreatitis to pancreatic cancer. World J Gastroenterol. 2006;12:3878-82
126. Noguchi K, Konno M, Eguchi H, Kawamoto K, Mukai R, Nishida N. et al. c-Met affects gemcitabine resistance during carcinogenesis in a mouse model of pancreatic cancer. Oncol Lett. 2018;16:1892-8
127. Hua H, Kong Q, Yin J, Zhang J, Jiang Y. Insulin-like growth factor receptor signaling in tumorigenesis and drug resistance: a challenge for cancer therapy. J Hematol Oncol. 2020;13:1-17
128. Lashinger LM, Harrison LM, Rasmussen AJ, Logsdon CD, Fischer SM, McArthur MJ. et al. Dietary energy balance modulation of Kras-and Ink4a/Arf+/--driven pancreatic cancer: The role of insulin-like growth factor-1. Cancer Prev Res (Phila). 2013;6:1046-55
129. Deng J, Guo Y, Du J, Gu J, Kong L, Tao B. et al. The intricate crosstalk between insulin and pancreatic ductal adenocarcinoma: A review from clinical to molecular. Front Cell Dev Biol. 2022;10:844028
130. Camblin AJ, Pace EA, Adams S, Curley MD, Rimkunas V, Nie L. et al. Dual inhibition of IGF-1R and ErbB3 enhances the activity of gemcitabine and nab-paclitaxel in preclinical models of pancreatic cancer. Clin Cancer Res. 2018;24:2873-85
131. Subramani R, Lopez-Valdez R, Arumugam A, Nandy S, Boopalan T, Lakshmanaswamy R. Targeting insulin-like growth factor 1 receptor inhibits pancreatic cancer growth and metastasis. PLoS One. 2014;9:e97016
132. Jaquish DV, Yu PT, Shields DJ, French RP, Maruyama KP, Niessen S. et al. IGF1-R signals through the RON receptor to mediate pancreatic cancer cell migration. Carcinogenesis. 2011;32:1151-6
133. Ma J, Sawai H, Matsuo Y, Ochi N, Yasuda A, Takahashi H. et al. IGF-1 mediates PTEN suppression and enhances cell invasion and proliferation via activation of the IGF-1/PI3K/Akt signaling pathway in pancreatic cancer cells. J Surg Res. 2010;160:90-101
134. Trajkovic-Arsic M, Kalideris E, Siveke JT. The role of insulin and IGF system in pancreatic cancer. J Mol Endocrinol. 2013;50:R67-74
135. Corrêa LH, Corrêa R, Farinasso CM, de Sant'Ana Dourado LP, Magalhães KG. Adipocytes and macrophages interplay in the orchestration of tumor microenvironment: new implications in cancer progression. Front Immunol. 2017;8:1129
136. Wu Q, Li B, Li J, Sun S, Yuan J, Sun S. Cancer-associated adipocytes as immunomodulators in cancer. Biomark Res. 2021;9:1-21
137. Zhao X, Fan W, Xu Z, Chen H, He Y, Yang G. et al. Inhibiting tumor necrosis factor-alpha diminishes desmoplasia and inflammation to overcome chemoresistance in pancreatic ductal adenocarcinoma. Oncotarget. 2016;7:81110-22
138. van Duijneveldt G, Griffin MD, Putoczki TL. Emerging roles for the IL-6 family of cytokines in pancreatic cancer. Clinical science. 2020;134:2091-115
139. Thomas H. IL-6 drives niche formation in pancreatic cancer liver metastasis. Nat Rev Gastroenterol Hepatol. 2019;16:263
140. Takahashi R, Macchini M, Sunagawa M, Jiang Z, Tanaka T, Valenti G. et al. Interleukin-1β-induced pancreatitis promotes pancreatic ductal adenocarcinoma via B lymphocyte-mediated immune suppression. Gut. 2021;70:330-41
141. Feygenzon V, Loewenstein S, Lubezky N, Pasmanic-Chor M, Sher O, Klausner JM. et al. Unique cellular interactions between pancreatic cancer cells and the omentum. PLoS One. 2017;12:e0179862
142. Cereijo R, Gavaldà-Navarro A, Cairó M, Quesada-López T, Villarroya J, Morón-Ros S. et al. CXCL14, a brown adipokine that mediates brown-fat-to-macrophage communication in thermogenic adaptation. Cell Metab. 2018;28:750-63
143. Cai Z, Liang Y, Xing C, Wang H, Hu P, Li J. et al. Cancer-associated adipocytes exhibit distinct phenotypes and facilitate tumor progression in pancreatic cancer. Oncol Rep. 2019;42:2537-49
144. Takehara M, Sato Y, Kimura T, Noda K, Miyamoto H, Fujino Y. et al. Cancer-associated adipocytes promote pancreatic cancer progression through SAA1 expression. Cancer Sci. 2020;111:2883-94
145. Vaes RD, Welbers TT, van Dijk DP, Rennspiess D, zur Hausen A, Damink SWO. et al. Intestinal smooth muscle aberrations in pancreatic cancer patients with sarcopenia. JCSM Rapid Commun. 2021;4:187-96
146. Washington TA, Schrems ER, Haynie WS, Rosa-Caldwell ME, Brown JL, Saling L. et al. Development of skeletal muscle fibrosis in a rodent model of cancer cachexia. Cell Biochem Funct. 2023;41:478-89
147. Marangoni RG, Korman B, Varga J. Adipocytic progenitor cells give rise to pathogenic myofibroblasts: Adipocyte-to-mesenchymal transition and its emerging role in fibrosis in multiple organs. Curr Rheumatol Rep. 2020;22:1-8
148. Bi P, Yue F, Karki A, Castro B, Wirbisky SE, Wang C. et al. Notch activation drives adipocyte dedifferentiation and tumorigenic transformation in mice. J Exp Med. 2016;213:2019-37
149. Zoico E, Darra E, Rizzatti V, Budui S, Franceschetti G, Mazzali G. et al. Adipocytes WNT5a mediated dedifferentiation: a possible target in pancreatic cancer microenvironment. Oncotarget. 2016;7:20223-35
150. Matsumoto T, Kano K, Kondo D, Fukuda N, Iribe Y, Tanaka N. et al. Mature adipocyte-derived dedifferentiated fat cells exhibit multilineage potential. J Cell Physiol. 2008;215:210-22
151. Zhao H, Chen Q, Alam A, Cui J, Suen KC, Soo AP. et al. The role of osteopontin in the progression of solid organ tumour. Cell Death Dis. 2018;9:1-15
152. Djurec M, Graña O, Lee A, Troulé K, Espinet E, Cabras L. et al. Saa3 is a key mediator of the protumorigenic properties of cancer-associated fibroblasts in pancreatic tumors. Proc Natl Acad Sci U S A. 2018;115:E1147-56
153. Ignacio RMC, Gibbs CR, Kim S, Lee E-S, Adunyah SE, Son D-S. Serum amyloid A predisposes inflammatory tumor microenvironment in triple negative breast cancer. Oncotarget. 2019;10:511-26
154. Jijon HB, Madsen KL, Walker JW, Allard B, Jobin C. Serum amyloid A activates NF-κB and proinflammatory gene expression in human and murine intestinal epithelial cells. Eur J Immunol. 2005;35:718-26
155. Zhang X, Huang S, Guo J, Zhou L, You L, Zhang T. et al. Insights into the distinct roles of MMP-11 in tumor biology and future therapeutics. Int J Oncol. 2016;48:1783-93
156. Zhang X, Lu J, Zhou L, You L, Liang Z, Guo J. et al. Matrix metalloproteinase 11 as a novel tumor promoter and diagnostic and prognostic biomarker for pancreatic ductal adenocarcinoma. Pancreas. 2020;49:812-21
157. Motrescu ER, Rio M-C. Cancer cells, adipocytes and matrix metalloproteinase 11: a vicious tumor progression cycle. Biol Chem. 2008;389:1037-41
158. Ji S, Cao J, Zhang Q, Li Y, Yan Y, Yu F. Adipose tissue-derived stem cells promote pancreatic cancer cell proliferation and invasion. Braz J Med Biol Res. 2013;46:758-64
159. Petit I, Jin D, Rafii S. The SDF-1-CXCR4 signaling pathway: a molecular hub modulating neo-angiogenesis. Trends Immunol. 2007;28:299-307
160. Tseng D, Vasquez-Medrano D, Brown J. Targeting SDF-1/CXCR4 to inhibit tumour vasculature for treatment of glioblastomas. Br J Cancer. 2011;104:1805-9
161. Cun Y, Diao B, Zhang Z, Wang G, Yu J, Ma L. et al. Role of the stromal cell derived factor-1 in the biological functions of endothelial progenitor cells and its underlying mechanisms. Exp Ther Med. 2021;21:39
162. Wu R, Su Z, Zhao L, Pei R, Ding Y, Li D. et al. Extracellular vesicle-loaded oncogenic lncRNA NEAT1 from adipose-derived mesenchymal stem cells confers gemcitabine resistance in pancreatic cancer via miR-491-5p/Snail/SOCS3 axis. Stem Cells Int. 2023;2023:6510571
163. Cousin B, Ravet E, Poglio S, De Toni F, Bertuzzi M, Lulka H. et al. Adult stromal cells derived from human adipose tissue provoke pancreatic cancer cell death both in vitro and in vivo. PLoS One. 2009;4:e6278
164. Okumura T, Ohuchida K, Kibe S, Iwamoto C, Ando Y, Takesue S. et al. Adipose tissue-derived stromal cells are sources of cancer-associated fibroblasts and enhance tumor progression by dense collagen matrix. Int J Cancer. 2019;144:1401-13
165. Miyazaki Y, Oda T, Mori N, Kida YS. Adipose-derived mesenchymal stem cells differentiate into pancreatic cancer-associated fibroblasts in vitro. FEBS Open Bio. 2020;10:2268-81
166. Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M. et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. 2017;214:579-96
167. Vaish U, Jain T, Are AC, Dudeja V. Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma: An Update on Heterogeneity and Therapeutic Targeting. Int J Mol Sci. 2021;22:13408
168. Zhu K, Cai L, Cui C, de Los Toyos JR, Anastassiou D. Single-cell analysis reveals the pan-cancer invasiveness-associated transition of adipose-derived stromal cells into COL11A1-expressing cancer-associated fibroblasts. PLoS Comput Biol. 2021;17:e1009228
169. Stolzenberg-Solomon RZ, Newton CC, Silverman DT, Pollak M, Nogueira LM, Weinstein SJ. et al. Circulating leptin and risk of pancreatic cancer: a pooled analysis from 3 cohorts. Am J Epidemiol. 2015;182:187-97
170. Babic A, Bao Y, Qian ZR, Yuan C, Giovannucci EL, Aschard H. et al. Pancreatic cancer risk associated with prediagnostic plasma levels of leptin and leptin receptor genetic polymorphisms. Cancer Res. 2016;76:7160-7
171. Tang Y, Tang Y, Cheng Y-s. miR-34a inhibits pancreatic cancer progression through Snail1-mediated epithelial-mesenchymal transition and the Notch signaling pathway. Sci Rep. 2017;7:1-11
172. Lin J, Xu Z, Xie J, Deng X, Jiang L, Chen H. et al. Oncogene APOL1 promotes proliferation and inhibits apoptosis via activating NOTCH1 signaling pathway in pancreatic cancer. Cell Death Dis. 2021;12:1-11
173. Abel EV, Kim EJ, Wu J, Hynes M, Bednar F, Proctor E. et al. The Notch pathway is important in maintaining the cancer stem cell population in pancreatic cancer. PLoS One. 2014;9:e91983
174. Palagani V, Bozko P, El Khatib M, Belahmer H, Giese N, Sipos B. et al. Combined inhibition of Notch and JAK/STAT is superior to monotherapies and impairs pancreatic cancer progression. Carcinogenesis. 2014;35:859-66
175. Ishikawa T, Okayama T, Oka K, Mizushima K, Yasuda T, Sakamoto N. et al. The JAK/STAT pathway is involved in the upregulation of PD-L1 expression in pancreatic cancer cell lines. Oncol Rep. 2017;37:1545-54
176. Corcoran RB, Contino G, Deshpande V, Tzatsos A, Conrad C, Benes CH. et al. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res. 2011;71:5020-9
177. Zhang H, Xu Y, Deng G, Yuan F, Tan Y, Gao L. et al. SAA1 knockdown promotes the apoptosis of glioblastoma cells via downregulation of AKT signaling. J Cancer. 2021;12:2756-67
178. Yasukawa Y, Hattori N, Iida N, Takeshima H, Maeda M, Kiyono T. et al. SAA1 is upregulated in gastric cancer-associated fibroblasts possibly by its enhancer activation. Carcinogenesis. 2021;42:180-9
179. Pothula SP, Xu Z, Goldstein D, Merrett N, Pirola RC, Wilson JS. et al. Targeting the HGF/c-MET pathway: stromal remodelling in pancreatic cancer. Oncotarget. 2017;8:76722-39
180. Shen W, Tao G-q, Zhang Y, Cai B, Sun J, Tian Z-q. TGF-β in pancreatic cancer initiation and progression: Two sides of the same coin. Cell Biosci. 2017;7:1-7
181. Principe DR, DeCant B, Mascariñas E, Wayne EA, Diaz AM, Akagi N. et al. TGFβ signaling in the pancreatic tumor microenvironment promotes fibrosis and immune evasion to facilitate tumorigenesis. Cancer Res. 2016;76:2525-39
182. Huang H, Zhang Y, Gallegos V, Sorrelle N, Zaid MM, Toombs J. et al. Targeting TGFβR2-mutant tumors exposes vulnerabilities to stromal TGFβ blockade in pancreatic cancer. EMBO Mol Med. 2019;11:e10515
183. Alvarez MA, Freitas JP, Hussain SM, Glazer ES. TGF-β inhibitors in metastatic pancreatic ductal adenocarcinoma. Int J Gastrointest Cancer. 2019;50:207-13
184. Goumas FA, Holmer R, Egberts JH, Gontarewicz A, Heneweer C, Geisen U. et al. Inhibition of IL-6 signaling significantly reduces primary tumor growth and recurrencies in orthotopic xenograft models of pancreatic cancer. Int J Cancer. 2015;137:1035-46
185. Zhuang Z, Ju H-Q, Aguilar M, Gocho T, Li H, Iida T. et al. IL1 receptor antagonist inhibits pancreatic cancer growth by abrogating NF-κB activation. Clin Cancer Res. 2016;22:1432-44
186. Xu S, Chaudhary O, Rodríguez-Morales P, Sun X, Chen D, Zappasodi R. et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8+ T cells in tumors. Immunity. 2021;54:1561-77
187. Kubo M, Gotoh K, Eguchi H, Kobayashi S, Iwagami Y, Tomimaru Y. et al. Impact of CD36 on chemoresistance in pancreatic ductal adenocarcinoma. Ann Surg Oncol. 2020;27:610-9
188. Tanase C, Gheorghisan-Galateanu A-A, Popescu ID, Mihai S, Codrici E, Albulescu R. et al. CD36 and CD97 in pancreatic cancer versus other malignancies. Int J Mol Sci. 2020;21:5656
189. Grace SA, Meeks MW, Chen Y, Cornwell M, Ding X, Hou P. et al. Adipose triglyceride lipase (ATGL) expression is associated with adiposity and tumor stromal proliferation in patients with pancreatic ductal adenocarcinoma. Anticancer Res. 2017;37:699-703
190. Iwamoto H, Abe M, Yang Y, Cui D, Seki T, Nakamura M. et al. Cancer lipid metabolism confers antiangiogenic drug resistance. Cell Metab. 2018;28:104-17
191. Lee J-S, Oh S-J, Choi H-J, Kang JH, Lee S-H, Ha JS. et al. ATP production relies on fatty acid oxidation rather than glycolysis in pancreatic ductal adenocarcinoma. Cancers (Basel). 2020;12:2477
192. Rupert JE, Narasimhan A, Jengelley DH, Jiang Y, Liu J, Au E. et al. Tumor-derived IL-6 and trans-signaling among tumor, fat, and muscle mediate pancreatic cancer cachexia. J Exp Med. 2021;218:e20190450
193. Arora G, Gupta A, Guo T, Gandhi A, Laine A, Ahn C. et al. JAK inhibitors suppress colon cancer cachexia-associated anorexia and adipose sasting in mice. JCSM Rapid Commun. 2020;3:115-28
194. Li Y, Mao AS, Seo BR, Zhao X, Gupta SK, Chen M. et al. Compression-induced dedifferentiation of adipocytes promotes tumor progression. Sci Adv. 2020;6:eaax5611
195. Wu C, Fernandez SA, Criswell T, Chidiac T, Guttridge D, Villalona-Calero M. et al. Disrupting cytokine signaling in pancreatic cancer: A phase I/II study of etanercept in combination with gemcitabine in patients with advanced disease. Pancreas. 2013;42:813-8
196. Wiedenmann B, Malfertheiner P, Friess H, Ritch P, Arseneau J, Mantovani G. et al. A multicenter, phase II study of infliximab plus gemcitabine in pancreatic cancer cachexia. J Support Oncol. 2008;6:18-25
Author contact
![]() Corresponding author: ysshanncku.edu.tw, Tel.: +886-6-2353535 (ext. 3105); Fax: +886-6-2766676
Corresponding author: ysshanncku.edu.tw, Tel.: +886-6-2353535 (ext. 3105); Fax: +886-6-2766676