Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2023; 13(11):3568-3581. doi:10.7150/thno.82535 This issue Cite
Research Paper
The HDAC10 instructs macrophage M2 program via deacetylation of STAT3 and promotes allergic airway inflammation
Yu Zhong1*, Tong Huang1*, Jiewen Huang1*, Jingyun Quan1*, Guomei Su1*, Zhilin Xiong1, Yingying Lv2, Shihai Li1, Xianwen Lai1, Yuanyuan Xiang1, Qu Wang3, Lianxiang Luo3, Xiao Gao1, Yiming Shao4 ![]() , Jing Tang5
, Jing Tang5 ![]() , Tianwen Lai1,2
, Tianwen Lai1,2 ![]()
1. Institute of Respiratory Diseases, Affiliated Hospital of Guangdong Medical University, Zhanjiang 524001, China.
2. Department of Respiratory and Critical Care Medicine, The First Dongguan Affiliated Hospital, Guangdong Medical University, Dongguan 523121, China.
3. The Marine Biomedical Research Institute, Guangdong Medical University; The Marine Biomedical Research Institute of Guangdong Zhanjiang, China.
4. The Intensive Care Unit, The First Dongguan Affiliated Hospital, Guangdong Medical University, Dongguan 523121, China.
5. Department of Anesthesiology, Affiliated Hospital of Guangdong Medical University, Zhanjiang 524001, China.
*These authors contributed equally to this work.
Received 2023-1-10; Accepted 2023-6-10; Published 2023-6-19
Abstract

Background: Perturbation of macrophage homeostasis is one of the key mechanisms of airway inflammation in asthma. However, the exact mechanisms remain poorly understood.
Objectives: We sought to examine the role of histone deacetylase (HDAC) 10 as an epigenetic regulator that governs macrophage M2 program and promotes airway inflammation in asthma, and to elucidate the underlying mechanisms.
Methods: Peripheral blood and airway biopsies were obtained from healthy individuals and asthmatic patients. Asthma was induced by exposure to allergen in mice with myeloid-specific deletion of Hdac10 (Hdac10fl/fl-LysMCre) mice. HDAC10 inhibitor Salvianolic acid B (SAB), STAT3 selective agonist Colivelin, and the specific PI3K/Akt activator 1,3-Dicaffeoylquinic acid (DA) were also used in asthmatic mice. For cell studies, THP1 cells, primary mouse bone marrow derived macrophage (BMDMs) were used and related signaling pathways was investigated.
Results: HDAC10 expression was highly expressed by macrophages and promoted M2 macrophage activation and airway inflammation in asthmatic patients and mice. Hdac10fl/fl-LysMCre mice were protected from airway inflammation in experimental asthma model. Hdac10 deficiency significantly attenuated STAT3 expression and decreased M2 macrophage polarization following allergen exposure. Mechanistically, HDAC10 directly binds STAT3 for deacetylation in macrophages, by which it promotes STAT3 expression and activates the macrophage M2 program. Importantly, we identified SAB as a HDAC10 inhibitor that had protective effects against airway inflammation in mice.
Conclusions: Our results revealed that HDAC10-STAT3 interaction governs macrophage polarization to promote airway inflammation in asthma, implicating HDAC10 as a therapeutic target.
Keywords: HDAC10, STAT3, asthma, airway inflammation, macrophage.
Introduction
Asthma is a common respiratory disease, which characterized by chronic airway inflammation, hyper responsiveness, and airway remodeling. The incidence of asthma increases by 50% every 10 years on average worldwide [1]. However, the mechanisms underlying the pathogenesis of asthma are not completely understood and urgently require in-depth investigation.
Lung macrophages are crucial sentinels in host lung defense and play crucial role in the maintenance of immune regulation, pathogen clearance, and homeostasis [2,3]. The pathogenesis of asthma involves perturbed lung homeostasis response to environmental factors exposure such as house dust mites (HDM), cigarette smoke, and lipopolysaccharide (LPS). Macrophages are classified into two categories: classically activated phenotype (M1) and alternatively activated phenotype (M2) [4]. M1-polarized macrophages are activated under the action of IFN-γ. M2 macrophages are classically activate by IL-4 or IL-13. We, and others, have shown previously that macrophages, particularly M2 macrophages, play an important role in the pathogenesis in asthma, such as bronchial hyperactivity, airway inflammation, and remodeling [5-7]. Although perturbation of macrophage homeostasis is associated with the pathogenesis of asthma, the underlying mechanisms remain elusive. Genes encoding transcription factors of the signal transducer and activator of transcription (STATs) family are involved in the cell activation [8]. STAT3 is associated to the occurrence and development of immune diseases, infectious diseases and tumors. Recent studies have shown that STAT3 is a critical determinant of M2 macrophage polarization and the expression of macrophage M2 markers (Arg1, Fizz1, and Ym1) [9,10]. However, the regulatory mechanisms that repress STAT3 expression, and thereby inhibit macrophage M2 program, remain largely unknown.
Emerging evidence suggests that epigenetic modifications are crucial for the pathogenesis of asthma. Acetylation is a widely occurring epigenetic modification of proteins that are involved in diverse biological processes [11]. The acetylation status is maintained by an intricate balance of histone deacetylase (HDAC) and histone acetyltransferase (HAT) [12]. HDAC10 is a class IIb HDAC that plays critical roles in regulating cellular processes, genomic stability, cancer progression, cells autophagy, and stress response via its epigenetic functions [12-16]. However, whether and how HDAC10 regulates macrophage homeostasis in asthma has not been deciphered.
Here, we found that HDAC10 expression was highly expressed by macrophages and played a critical role in maintaining macrophage homeostasis by controlling M2 macrophage activation through deacetylating STAT3. Mechanistically, HDAC10 directly interacts with STAT3 and targets STAT3 for acetylation, by which it promotes STAT3 expression to increase the macrophage M2 program. Both genetic and pharmacological inhibition of HDAC10 protect against airway inflammation in asthmatic mice, suggesting that the HDAC10/STAT3 axis is a potential target for treatment of asthma.
Results
HDAC10 expression was elevated in asthmatic patients and mice
We first detected HDAC10 expression in the airway biopsies from asthmatic patients (Table S1). HDAC10 was highly expressed in the airway biopsies from asthmatic patients compared with normal controls (Figure 1A-B). Using coimmunostaining of M2 macrophage maker CD206, we found that HDAC10 was mainly localized in M2 macrophages (Figure 1C-D). Using quantitative RT-PCR (qRT-PCR), we next detected the expression of HDAC10 from human normal and asthmatic peripheral blood mononuclear cells (PBMCs) (Table S2). Compared with normal controls, the levels of HDAC10 were also significantly increased in asthmatic patients (Figure 1E).
Analysis of HDAC10 expression in asthmatic patients and mice. (A, B) Representative IHC staining of HDAC10 in the airway from asthmatic patients. Images were captured at ×400 magnification and quantification of IHC was done by using Image J software. (C, D) Representative results for coimmunostaining of HDAC10 and CD206 in the lung sections from patients with asthma. Images were captured at ×400 magnification and quantification of IHC was done by using Image J software. (E) Peripheral blood mononuclear cells (PBMC) were isolated from peripheral blood samples of study subjects. The HDAC10 expression in PBMC was analyzed by using quantitative RT-PCR (qRT-PCR). (F) Schematic illustrating an established house dust mite (HDM)/lipopolysaccharide (LPS)-induced asthma mouse model (n = 6-8 for each group). (G) qRT-PCR analysis of the expression of Hdac10 in mouse lung tissue. Results were normalized to those of the gene encoding β-actin, and relative expression was calculated by the change-in-threshold method. (H, I) Representative IHC images of HDAC10 in lung tissue of Hdac10fl/fl mice following allergen induction. Images were captured at ×400 magnification and quantification of IHC was done by using Image J software. (J, K) Western blot analysis of HDAC10 and macrophage M2 marker Arg1 expression in lung tissues of Hdac10fl/fl mice following allergen induction and quantification was done by using Image J software. (L, M) Results for coimmunostaining of HDAC10 and macrophage M2 marker CD206 in allergen-induced lung sections. Images were captured at ×400 magnification and quantification was done by using Image J software. Data are shown as means ± SEM. ***P < 0.001 and ****P < 0.0001 versus Control (unless otherwise noted) by two-tailed unpaired Student's t test. Data are representative of three independent experiments with similar results (A, C, H, and L) or are from three independent experiments (G and J). See also Figure S1.
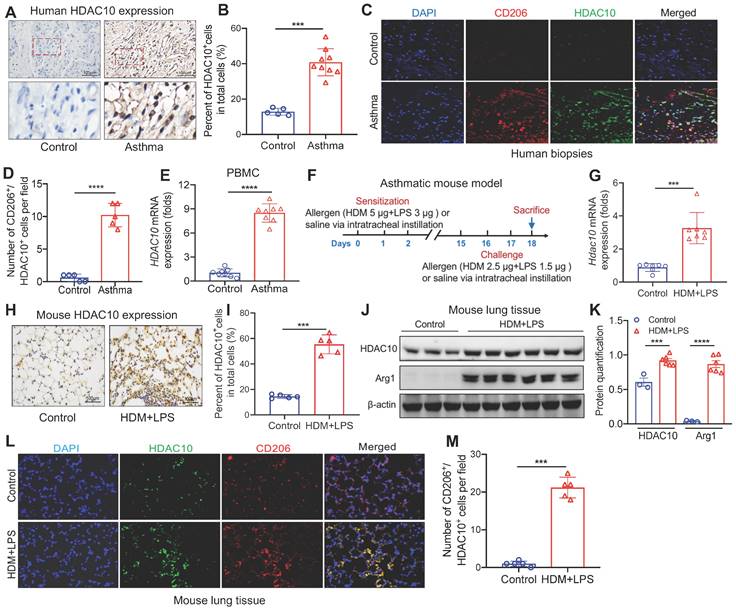
To further determine the role of HDAC10 in the pathogenesis of asthma, we detected HDAC10 expression in the lung tissue in an experimental model of asthma (Figure 1F). As assessed by qRT-PCR, we observed that Hdac10 was significant increased compared with controls (Figure 1G). Those results were confirmed by immunohistochemistry (IHC) and Western blotting (Figure 1H-K). Consistent with human data, HDAC10 abundance was also mainly localized in macrophages in the lung tissue of asthmatic mice (Figure 1L-M). Moreover, we found that HDAC10 expression was induced in the THP1 cells and bone marrow-derived macrophages (BMDMs) in allergic stimuli (Figure S1A-F). Together, these findings suggested that HDAC10 was highly expressed in M2 macrophages and potentially promoted airway inflammation, which also prompted us to further explore the precise role of macrophage HDAC10 in airway inflammation process in asthma.
Hdac10 deficiency protected mice against allergen-induced airway inflammation
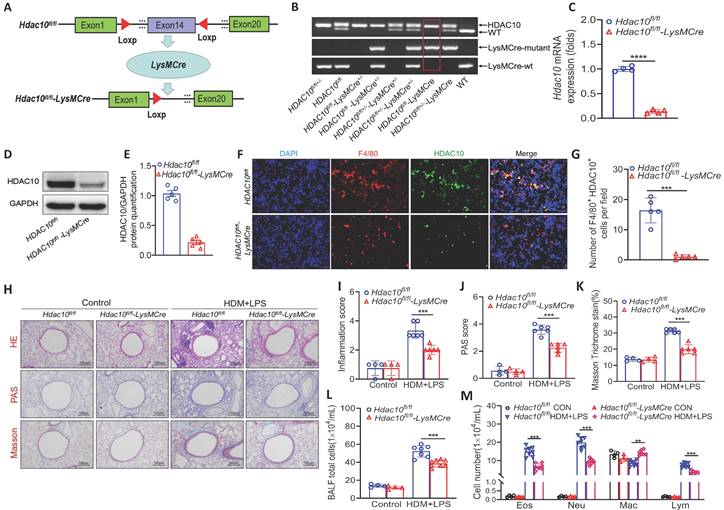
To dissect how HDAC10 exerts its function during allergic airway inflammation, we generated mice with macrophage-specific Hdac10 knocked out (Hdac10fl/fl-LysMCre mice) and their littermates (Hdac10fl/fl mice) (Figure 2A). All mice were genotyped with PCR (Figure 2B). The absence of HDAC10 from bone marrow macrophages was verified by Western blotting and qRT-PCR (Figure 2C-E). The absence of HDAC10 colocalization with macrophages was observed in the lung sections of Hdac10fl/fl-LysMCre mice using immunofluorescence (IF) staining (Figure 2F-G). The Hdac10fl/fl-LysMCre and Hdac10fl/fl mice were exposed to allergen according to an asthma model as described in Figure 1F. Hematoxylin & eosin (HE) staining, periodic acid Schiff (PAS) staining, and peribronchial trichrome (Masson) staining were reduced in HDM/LPS-exposed Hdac10fl/fl-LysMCre mice (Figure 2H-K). The total inflammatory cells, neutrophils, eosinophils, and lymphocytes in bronchoalveolar lavage fluid (BALF) were reduced in allergen-exposed Hdac10fl/fl-LysMCre mice (Figure 2L-M). The mRNA and protein expression of CXCL1 and CXCL2 were lower in the lung tissues and BALF of Hdac10fl/fl-LysMCre mice than in that of Hdac10fl/fl mice exposed to allergen (Figure S2A-F). Consistent with these findings, HDAC10 deficiency suppressed the production of CXCL1 and CXCL2 in allergen-induced BMDMs (Figure S2G-H). Together, our data support that Hdac10 deficiency in macrophages protects mice from allergen-induced airway inflammation.
Hdac10 deficiency protected mice against allergic airway inflammation. (A) Schematic illustrating the genetic approach used to generate macrophages-conditional knockout of Hdac10 (Hdac10fl/fl-LysMCre) mice. (B) Hdac10 deficiency was confirmed by assessing genomic DNA. (C-E) Hdac10 deficiency was assessed in BMDMs from Hdac10fl/fl and Hdac10fl/fl-LysMCre mice using qRT-PCR and Western blotting analysis. Quantification was done by using Image J software. (F, G) Representative result for coimmunostaining of F4/80 and HDAC10 in the lung sections from Hdac10fl/fl and Hdac10fl/fl-LysMCre mice. Images were captured at ×400 magnification and quantification was done by using Image J software. (H-M) Total BAL fluid (BALF) cells, differential cell counts, and histologic analysis of the lung sections were performed with hematoxylin and eosin staining to visualize inflammatory cell recruitment from Hdac10fl/fl and Hdac10fl/fl-LysMCre mice treated with allergen. Images were captured at ×200 magnification and quantification was done by using Image J software. Data are shown as means ± SEM (n = 4-6 mice/group). **P < 0.01 and ***P < 0.001 versus Control (unless otherwise noted) by two-tailed unpaired Student's t test or one-way ANOVA, followed by Tukey's multiple comparisons test. Data are representative of three independent experiments with similar results (F and H) or are from three independent experiments (C, D, L and M). See also Figure S2.
Hdac10 deficiency attenuated M2 macrophage activation
The above results revealed prompted us to further explore the precise role of HDAC10 on M2 macrophage polarization in the airway inflammation process during asthma. The flow cytometry showed that the proportion of M2 macrophages (F4/80+CD206+) in the lung of Hdac10fl/fl-LysMCre mice were lower than that of Hdac10fl/fl mice following allergen exposure (Figure 3A-B). Consistently, Western blotting showed that M2 marker Arg1 was reduced in Hdac10fl/fl-LysMCre mice as compared with the Hdac10fl/fl mice (Figure 3C-D). Moreover, M2 markers Arg1, Fizz1 and Ym1 demonstrated similar results (Figure 3E-G).
Hdac10 deficiency inhibited the macrophage M2 program. (A, B) Flow cytometry analysis (BD Biosciences) of macrophages derived from lung tissues of Hdac10fl/fl and Hdac10fl/fl-LysMCre mice after allergen induction. Quantification of CD206+ cells was analyzed with FlowJo (TreeStar). (C, D) Western blotting analysis of macrophage M2 marker Arg1 expression in the lung homogenates of Hdac10fl/fl and Hdac10fl/fl-LysMCre mice after allergen induction. Quantification was done by using Image J software. (E-G) qRT-PCR analysis of macrophage M2 markers Arg1, Fizz1 and Ym1 mRNA expression in the lung tissue of Hdac10fl/fl and Hdac10fl/fl-LysMCre mice after allergen exposure. (H, I) Flow cytometry analysis (BD Biosciences) of macrophage M2 marker CD206 expression in BMDMs following IL-4 stimulation. Quantification of CD206+ cells was analyzed with FlowJo (TreeStar). (J-L) Western blotting analysis of HDAC10 and Arg1 expression in the BMDMs after IL-4 induction. Quantification was done by using Image J software. (M-P) qRT-PCR analysis was conducted for Hdac10 mRNA and macrophage M2 markers Arg1, Fizz1, and Ym1 mRNA expression in the BMDMs after IL-4 induction. Data are shown as means ± SEM (n = 4-6 mice/group). **P < 0.01 and ***P < 0.001 versus Control (unless otherwise noted) by two-tailed unpaired Student's t test or one-way ANOVA, followed by Tukey's multiple comparisons test. Data are representative of three independent experiments with similar results (C and J) or are from three independent experiments (A, E-H, and M-P). See also Figure S3.
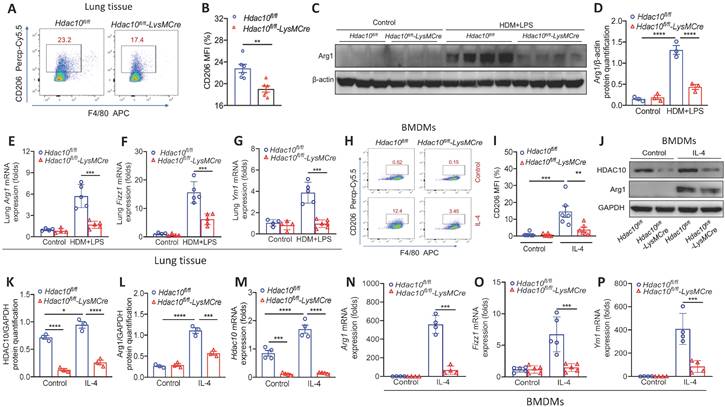
To further dissect the mechanism of Hdac10 regulates the M2 program of macrophages, we generated BMDMs from Hdac10fl/fl and Hdac10fl/fl-LysMCre mice and then stimulated with interleukin-4 (IL-4). The average fluorescence intensity of CD206 in Hdac10fl/fl BMDMs induced by IL-4 was significantly higher than that of Hdac10fl/fl-LysMCre BMDMs (Figure 3H-I). The HDAC10 protein expression was significantly increased in Hdac10fl/fl BMDMs upon IL-4 induction, while Arg 1 was decreased in Hdac10fl/fl-LysMCre BMDMs compared with Hdac10fl/fl BMDMs following IL-4 stimulation (Figure 3J-L). The mRNA expression of Hdac10 and M2 markers Arg1, Fizz1, and Ym1 revealed similar results (Figure 3M-P).
The above data supported the hypothesis that Hdac10 deficiency protected mice against allergen-induced airway inflammation via attenuating the M2 program. To address this assumption, adoptive transfer experiments were performed as described in Methods. Briefly, BMDMs prepared from Hdac10fl/fl and Hdac10fl/fl-LysMCre donor mice and then stimulated with IL-4. The above BMDMs were adoptively transferred into macrophage-depleted wildtype (WT) recipient mice after allergen induction. We found that macrophages were almost undetectable in the lung tissue of mice treat with clodronate liposome (Figure S3A-B). Macrophage-depleted WT mice received Hdac10fl/fl BMDMs treated with IL-4 exhibited higher M2 markers (Arg1 and Ym1) production like Hdac10fl/fl mice following allergen challenge. In contrast, macrophage-depleted WT mice received Hdac10fl/fl-LysMCre BMDMs treated with IL-4 appeared similar to Hdac10fl/fl-LysMCre mice after allergen administration, with lower M2 markers (Arg1 and Ym1) production (Figure S3C-D).
Consistent with these results, HE staining suggested that the adoptive transfer of Hdac10fl/fl BMDMs restored allergen-induced airway inflammation in macrophage-depleted WT mice (Figure S3E-F). As shown in Figure S3G-J, the cytokine secretion remained decreased in allergen-challenged macrophage-depleted WT mice compared with controls after transfer of Hdac10fl/fl-LysMCre BMDMs. Collectively, this data defined a unique function of Hdac10 in macrophages in the regulation of asthmatic airway inflammation.
Depletion of Hdac10 repressed PI3K/Akt signaling
Given the important role of PI3K/Akt signaling played in macrophage M2 polarization induced by IL-4 or IL-13 [17,18], we first assessed the expression of PI3K/Akt signaling in asthmatic patients and mice. The expression of phosphorylated P85 (p-P85) and p-Akt was up-regulated in asthmatic patients and mice (Figure 4A-F). We further found that the levels of p-P85, p-Akt and the Akt downstream effector p-mTOR were decreased in Hdac10fl/fl-LysMCre BMDMs compared with Hdac10fl/fl BMDMs induced by IL-4 stimulation (Figure 4G, Figure S4A-D).
Hdac10 deficiency attenuated allergen-induced PI3K/Akt signaling in macrophages. (A-C) Representative IHC staining of p-Akt and p-P85 in the airway from asthmatic patients. Images were captured at ×400 magnification and quantification of IHC was done by using Image J software. (D-F) Representative IHC staining of p-Akt and p-P85 in the lung tissue from asthmatic mice. Images were captured at ×400 magnification and quantification of IHC was done by using Image J software. (G) Western blotting analysis of p-Akt, p-mTOR and p-P85 expression in the BMDMs from Hdac10fl/fl and Hdac10fl/fl-LysMCre mice after allergen exposure. (H, I) Representative photomicrographs of lung inflammation expression are shown. Images were captured at ×200 magnification and quantification was done by using Image J software. (J-L) The expression of inflammatory cytokines in the lung homogenate of mice was analyzed by using qRT-PCR. (M) qRT-PCR analysis was conducted for macrophage M2 markers Arg1 mRNA expression in the lung homogenate of mice. Data are shown as means ± SEM (n = 4-6 mice/group). ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 versus Control (unless otherwise noted) by two-tailed unpaired Student's t test or one-way ANOVA, followed by Tukey's multiple comparisons test. Data are representative of three independent experiments with similar results (A, D, G, and H) or are from three independent experiments (J-M). See also Figure S4.
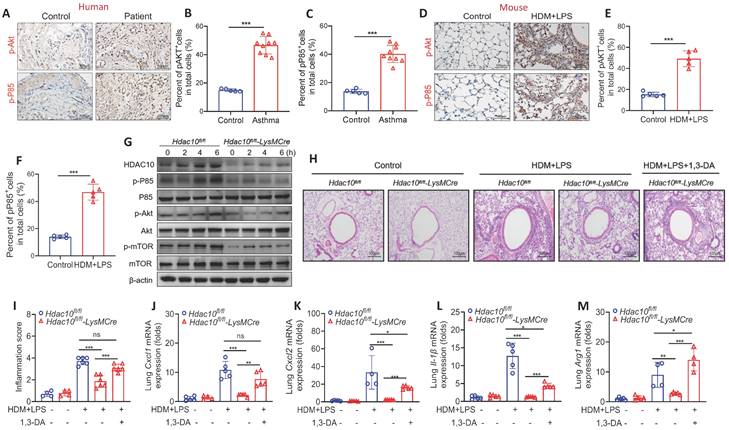
To explore the possible role of PI3K/Akt in mediating the role of HDAC10 in macrophage inflammation, we used the specific PI3K/Akt activator 1,3-Dicaffeoylquinic acid (DA) to activate PI3K/Akt pathway as descried in Figure S4E. Our studies demonstrated that airway inflammation was reduced in allergen-exposed Hdac10fl/fl-LysMCre mice compared with Hdac10fl/fl mice, but 1,3-DA treatment almost completely reversed these phenomena (Figure 4H-I). Consistent with these findings, 1,3-DA treatment significantly rescued the reduced Cxcl1, Cxcl2, IL-1β and Arg1 production in the lung tissue of Hdac10fl/fl-LysMCre mice after allergen stimulation (Figure 4J-M). Collectively, our data support that Hdac10 deficiency repressed PI3K/Akt signaling to attenuate airway inflammation following allergen induction.
Hdac10 deficiency suppressed STAT3 to inhibit M2 macrophage program
It has been reported that PI3K/AKT pathway serves as an up-stream participant in the STAT3 activation [19]. STAT3 plays an important role in the activation of immune cells (e.g., T cells, macrophages, eosinophils) and contributes to the development of asthma [20]. Therefore, it promoted us to explore whether Hdac10 deficiency attenuates macrophages M2 program via STAT3 activation. We first compared STAT3 expression in the lung tissue between Hdac10fl/fl and Hdac10fl/fl-LysMCre mice following allergen stimulation. IHC staining showed that STAT3 expression was significantly reduced in lung tissue of Hdac10fl/fl-LysMCre mice compared with Hdac10fl/fl mice exposed to allergen (Figure 5A-B). Consistent with this result, p-STAT3 and STAT3 in Hdac10fl/fl-LysMCre mice was expressed at a much lower level of protein than in the lung tissue of Hdac10fl/fl mice after allergen induction (Figure 5C-E). We further found that STAT3 was also mainly localized in macrophages infiltrated in the lung tissue of mice after allergen induction by F4/80 staining (Figure 5F-G). Furthermore, p-STAT3 and STAT3 expression was obviously increased in allergen-induced Hdac10fl/fl BMDMs, but significantly decreased in allergen-induced Hdac10fl/fl-LysMCre BMDMs (Figure 5H-L). In addition, we expressed a STAT3-TA-luc fusion protein to asses STAT3 transactivation activity by luciferase assay. The activity of STAT3 was significantly augmented in allergen-induced Hdac10fl/fl BMDMs, whereas it was attenuated in allergen-induced Hdac10fl/fl-LysMCre BMDMs (Figure 5M).
Hdac10 deficiency suppressed STAT3 to attenuate M2 program. (A, B) Representative IHC staining of STAT3 in the lung tissue from Hdac10fl/fl and Hdac10fl/fl-LysMCre mice after allergen induction. Images were captured at ×400 magnification and quantification of IHC was done by using Image J software. (C, E) Western blotting analysis of p-STAT3 and STAT3 expression in the lung homogenates of Hdac10fl/fl and Hdac10fl/fl-LysMCre mice after allergen induction. Quantification was done by using Image J software. (F, G) Representative result for coimmunostaining of F4/80 and STAT3 in the lung sections from Hdac10fl/fl and Hdac10fl/fl-LysMCre mice after allergen induction. Images were captured at ×400 magnification and quantification was done by using Image J software. (H, I) Representative result for coimmunostaining of F4/80 and STAT3 in the BMDMs from Hdac10fl/fl and Hdac10fl/fl-LysMCre mice. Images were captured at ×400 magnification and quantification was done by using Image J software. (J-L) Western blotting analysis was conducted for p-STAT3 and STAT3 expression in the BMDMs after allergen stimulation. Quantification was done by using Image J software. (M) BMDMs from Hdac10fl/fl and Hdac10fl/fl-LysMCre mice were transfected with pSTAT3-TA-luc plasmid for 24 h, and then treated with HDM (100 μg/ml) for another 24 h. Luciferase assay was used to detected STAT3-TA-luc fusion protein to asses STAT3 transactivation activity. Data are shown as means ± SEM. **P < 0.01, ***P < 0.001, and ****P < 0.0001 versus Control (unless otherwise noted) by two-tailed unpaired Student's t test or one-way ANOVA, followed by Tukey's multiple comparisons test. Data are representative of three independent experiments with similar results (A, C, F, H and J) or are from three independent experiments (M).
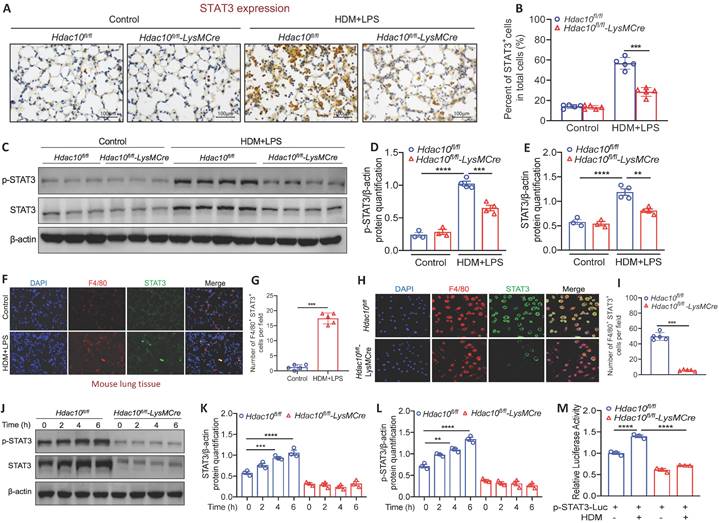
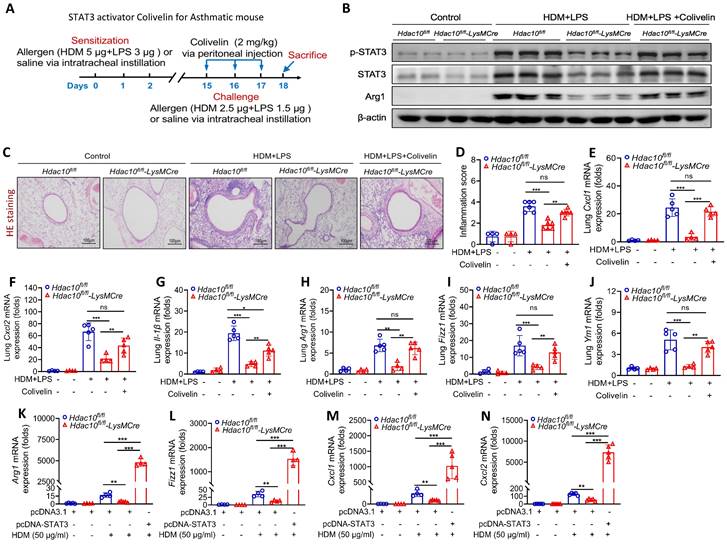
To further investigate whether STAT3 regulate macrophage M2 polarization in this context, STAT3 selective agonist Colivelin was administered to mice as described in Figure 6A. As predicted, p-STAT3, STAT3 and Arg1 expression were significantly lower in lung tissue of Hdac10fl/fl-LysMCre mice compared with Hdac10fl/fl mice exposed to allergen, but Colivelin treatment almost completely reversed these phenomena (Figure 6B, Figure S4F-H). Compared with the control treatment, Colivelin treatment increased the inflammatory cells in airway inflammation (HE staining), inflammatory cytokines, and macrophage M2 markers (Arg1, Fizz1, and Ym1) in lung tissue of Hdac10fl/fl-LysMCre mice exposed to allergen (Figure 6C-J). Consistent with these findings, overexpression of STAT3 promoted allergen-induced inflammatory cytokines, and macrophage M2 markers in allergen-induced Hdac10fl/fl-LysMCre BMDMs (Figure 6K-N). Collectively, our data supported that Hdac10 deficiency suppressed STAT3 to attenuate M2 programming during allergic inflammation.
STAT3 activator Colivelin exacerbated allergic airway inflammation in Hdac10 deficiency mice. (A) Schematic overview of experimental design for STAT3 activator Colivelin for asthmatic mouse. (B) The expression of p-STAT3, STAT3 and Arg1 in the lung homogenate from mice treated with allergen or Colivelin was analyzed by using Western blotting. (C, D) Representative photomicrographs of lung inflammation expression is shown. Images were captured at ×200 magnification and quantification was done by using Image J software. (E-G) The expression of inflammatory cytokines in the lung homogenate of mice was analyzed by using qRT-PCR. (H-J) qRT-PCR analysis was conducted for macrophage M2 markers Arg1, Fizz1, and Ym1 mRNA expression in the lung homogenate of mice. (K-N) BMDMs were transfected with Control or STAT3 plasmid for 24 h and then treated with HDM for another 24 h. Arg1, Fizz1, Cxcl1, and Cxcl2 mRNA expression were assessed by qRT-PCR analysis. Data are shown as means ± SEM (n = 4-6 mice/group). ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 versus Control (unless otherwise noted) by one way ANOVA, followed by Tukey's multiple comparisons test. Data are representative of three independent experiments with similar results (B and C) or are from three independent experiments (E-N). See also Figure S4.
HDAC10 directly interacted with STAT3 and targeted STAT3 for deacetylation
To investigate the mechanisms by which HDAC10 regulates STAT3 in asthma, we have therefore completed a series of experiments. Immunofluorescence assay indicated that HDAC10 and STAT3 were expressed in both the nucleus and cytoplasm, but mainly located in the nucleus of THP1 cells (Figure 7A). To determine whether HDAC10 and STAT3 physically interact, co-immunoprecipitation (Co-IP) analysis were performed. HDAC10 and STAT3 coimmunoprecipitated with each other and that allergen could up-regulate HDAC10 and STAT3 complex (Figure 7B-C). We further determined whether HDAC10 was able to interact with STAT3 exogenously. The exogenous HDAC10 and STAT3 complex was observed in THP1 cells (Figure 7D-E). Moreover, the direct interaction of HDAC10 and STAT3 was verified by glutathione S-transferase (GST) pull-down (Figure 7F).
HDAC10 directly interacted with STAT3 and targeted STAT3 for deacetylation. (A) Confocal microscopy of the location of endogenous HDAC10 (Red) and STAT3 (Green) in THP1 cells treated with HDM for 24 h. DAPI, DNA binding dye. Images were captured at ×400 magnification. (B, C) Immunoblot (IB) analysis of endogenous HDAC10 or STAT3 in THP1 cells infected with HDM, assessed before (input) or after IP with IgG (control) or antibody to HDAC10 or STAT3. (D, E) IB analysis of exogenous HDAC10 or STAT3 in THP1 cells transfected with HA-tagged STAT3 alone or together with Flag-tagged HDAC10, assessed before (input) or after IP with antibody to HA or Flag. (F) IB analysis of HDAC10 and STAT3 interaction in a GST pull-down assay. (G) IB analysis of STAT3 acetylation (Ack) in THP1 cells transfected with HA-tagged STAT3 alone or together with histone deacetylase inhibitor Trichostatin A (TSA), assessed before (input) or after IP with antibody to STAT3 and Ack. (H) THP1 cells were transfected with Control siRNA or Hdac10 siRNA for 24 h and then treated with HDM (100 μg/ml) for 24 h, and assessed before (input) or after IP with antibody to STAT3 and Ack. (I) THP1 cells were transfected with pcDNA3.1 or pcDNA-HDAC10 plasmid for 24 h and then treated with HDM (100 μg/ml) for 24 h, and assessed before (input) or after IP with antibody to STAT3 and Ack. Data are shown as means ± SEM. Data are representative of three independent experiments with similar results.
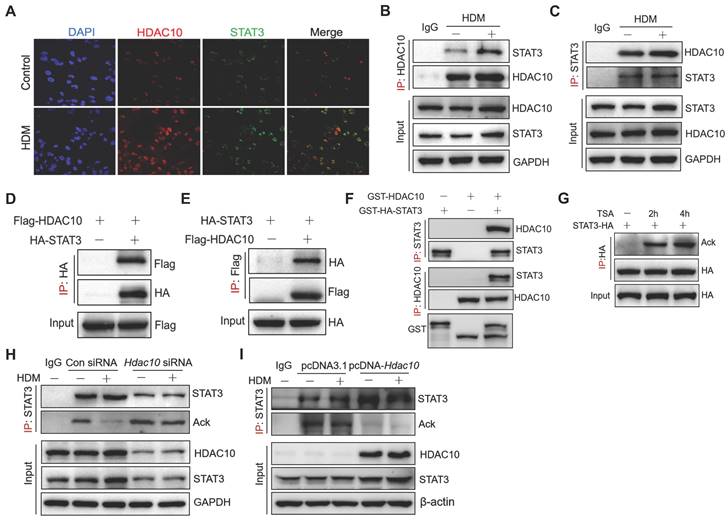
Given that HDAC10 is a deacetylase, we investigated whether HDAC10 targets STAT3 for deacetylation. STAT3 was overexpressed in THP1 cells using HA-STAT3 plasmid and then treated with histone deacetylase inhibitor trichostatin A (TSA) for 2 h or 4 h. We immunoprecipitated HA-STAT3 with anti-HA and found that STAT3 was indeed acetylated, and its acetylation was enhanced after treatment with TSA (Figure 7G). To further illustrate the involvement of HDAC10 in STAT3 acetylation modification, Hdac10 was knocked-down using a siRNA approach or was overexpressed using HDAC10 plasmid in THP-1 cells and then treated with allergen for 24 h. We found that HDM treatment decreased STAT3 acetylation in THP1 cells, but was inhibited by Hdac10 knockdown using siRNA (Figure 7H). In contrast, HDAC10 overexpression in THP1 cells using HDAC10 plasmid significantly reduced STAT3 acetylation compared with the control cells exposed to allergen (Figure 7I). Collectively, this finding suggested that HDAC10 directly interacted with STAT3 and targeted STAT3 for deacetylation, which contributes to promote M2 program during allergic inflammation.
HDAC10 inhibitor treatment attenuated allergic airway inflammation
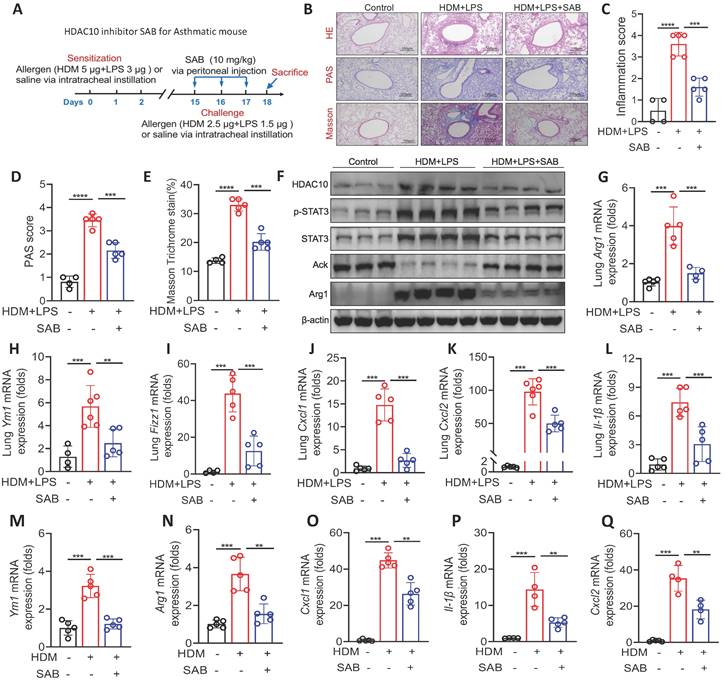
Our data indicated that HDAC10 inhibition might represent a valuable therapeutic approach for asthma. Therefore, we identified a HDAC10 inhibitor using a molecular docking screening strategy as described in Methods. The docking scores of 5 molecules and HDAC10 structure and other basic information are shown in Table S3. To further screen compounds with good target inhibition activity, we used the protein structure of HDAC10 as the target for fine molecular docking with compound Salvianolic acid B (SAB). SAB, one of the main active ingredients of Salvia miltiorrhiza, has anti-inflammatory effects and a beneficial effect on cardiovascular diseases, cancers, and liver fibrosis [21-23]. The interaction between HDAC10 and SAB were analyzed in detail. The interaction diagram of compound SAB with HDAC10 (Figure S4I) and the three-dimensional binding pattern diagram (Figure S4J) showed that the compound forms hydrogen bond interaction with Gly-145, Asp-174, Arg-198, Glu-276, Pro-273, Glu-274, suggesting that they are closely combined.
We further characterized the specificity of the HDAC10 inhibitor SAB in our study. BMDMs were isolated from WT mice and treated with HDM (100 μg/ml) and/or SAB (20 μM) for 24 h. The expression of HDACs (HDAC1-11) in BMDMs were analyzed using qRT-PCR. We found that the levels of Hdac5, Hdac6, Hdac8, Hdac9, and Hdac10 were significantly increased in BMDMs treated with HDM compared with the control group, but only Hdac10 was significantly decreased in BMDMs treated with HDM plus SAB (Figure S4K). We then evaluated the role of SAB in experimental mouse model as described in Figure 8A. HE staining, PAS staining, and Masson staining data demonstrated that SAB treatment attenuated allergen-induced airway inflammation, mucus secretion, and collagen deposition (Figure 8B-E). The expression of HDAC10, p-STAT3, STAT3, and Arg1 in the lung homogenates was obviously decreased by SAB administration compared with normal saline administration (Figure 8F, Figure S4L). SAB treatment also inhibited macrophage M2 markers (Arg1, Fizz1 and Ym1) and inflammatory cytokines in the lung homogenate of mice (Figure 8G-L). Consistent with animal data, SAB treatment inhibited macrophage M2 markers (Arg1, Fizz1, and Ym1) and inflammatory cytokines in HDM-induced BMDMs (Figure 8M-Q). Collectively, these findings suggested that inhibition of HDAC10 with SAB treatment may be of potential value in the treatment of airway inflammation in asthma.
The HDAC10 inhibitor Salvianolic acid B (SAB) prevented allergen-induced airway inflammation. (A) Schematic overview of experimental design for (B-Q) in an asthma mouse model. (B-E) Representative photomicrographs of HE staining, PAS staining, and Masson staining in the lung tissue of mice are shown. Images were captured at ×200 magnification and quantification was done by using Image J software. (F) The expression of HDAC10, STAT3, and Arg1 in the lung homogenate of mice was analyzed by using Western blotting. (G-I) qRT-PCR analysis was conducted for macrophage M2 markers Arg1, Fizz1, and Ym1 mRNA expression in the lung homogenate of mice. (J-L) The expression of inflammatory cytokines in the lung homogenate of mice was analyzed by using qRT-PCR. (M, N) The mRNA expression of Arg1 and Ym1 in BMDMs was conducted by qRT-PCR analysis. (O-Q) The levels of inflammatory cytokines in BMDMs was conducted by qRT-PCR analysis. Data are shown as means ± SEM (n = 4-6 mice/group). **P < 0.01, ***P < 0.001, and ****P < 0.0001 versus Control (unless otherwise noted) by one-way ANOVA, followed by Tukey's multiple comparisons test. Data are representative of three independent experiments with similar results (B and F) or are from three independent experiments (G-Q). See also Figure S4.
Discussion
In this study, we performed experiments in patients and animals to investigate the role of histone deacetylase HDAC10 on the pathogenesis of asthma. We found that HDAC10 expression was elevated in human biopsies, mouse models, and macrophages in response to allergen. Mice with a myeloid cell-specific Hdac10 deficiency attenuated airway inflammation in asthmatic mice. We conjectured that HDAC10 directly interacts with STAT3 for its deacetylation in macrophages. The complex then promotes macrophage M2 polarization via PI3K/Akt signaling pathway. Importantly, we identified SAB as a HDAC10 inhibitor that effectively suppressed airway inflammation in asthmatic mice. Upregulation of HDAC10 was positively associated with disease severity of asthma. Collectively, our study disclosed a previously unknown HDAC10-STAT3 axis that facilitates macrophage M2 polarization and led to airway inflammation in asthma, implicating HDAC10 as a therapeutic target.
Epigenetic regulation plays a critical role in the effects of environmental factors in the pathogenesis of asthma [24]. HDAC10 is a histone deacetylase that regulates melanogenesis in ovarian cancer patients [25]. Oehme et al. demonstrated that HDAC10 protects cancer cells from cytotoxic agents [16]. The expression of HDAC10 is inhibited in human renal cell carcinoma (RCC) cells, suggesting that HDAC10 is an independent predictor of the prognosis of RCC, and activating HDAC10 expression may be a new therapeutic strategy for advanced RCC [26]. However, whether and how HDAC10 regulates the pathogenesis of asthma have not been reported. Here, we found that HDAC10 expression was elevated in asthmatic patients and mice. Hdac10fl/fl-LysMCre mice exhibited significantly reduced airway inflammation, cytokine production, and mucus production compared with Hdac10fl/fl mice exposed to allergens. Previous studies had shown that PI3K/Akt signaling participates in physiological functions such as regulating the expression of various inflammatory genes, DNA damage repair, and aging [18,19]. PI3K/Akt signaling is also plays a role in macrophage M2 program [19]. We found that the specific PI3K/Akt activator 1,3-DA significantly rescued the reduced airway inflammation and macrophage M2 program in Hdac10fl/fl-LysMCre mice after allergen stimulation. Specifically, the macrophage depletion and adoptive transfer experiments confirmed that reduced M2 macrophages protected against allergen-induced airway inflammation. Moreover, we identified SAB as a HDAC10 inhibitor that effectively suppressed airway inflammation in asthmatic mice.
STAT3 plays a critical role in determinant of polarization of the alternatively activated M2 macrophages [20]. In our study, STAT3 expression was decreased in Hdac10 deficient BMDMs. Consistent with in vitro experiments, STAT3 expression was significantly reduced in M2 macrophages of Hdac10fl/fl-LysMCre mice compared with Hdac10fl/fl mice exposed to allergen. Furthermore, compared with the control treatment, STAT3 selective agonist Colivelin treatment increased airway inflammation, lung inflammatory cytokines, and macrophage M2 markers (Arg1, Fizz1, and Ym1) in Hdac10fl/fl-LysMCre mice exposed to allergen. These results indicated that Hdac10 deficiency suppressed STAT3 to attenuate macrophage M2 program during allergic inflammation. However, the mechanism of HDM in observed intracellular signaling mechanisms like HDAC10/STAT3 expression and activation remains unknown and needs further investigation.
Acetylation plays a critical regulatory role in protein-protein interactions. Acetylation of histones causes the relaxation of chromatin structure as a necessary but not sufficient condition for gene transcription [27]. Deacetylation is also linked directly to gene transcription. H4K16 deacetylation is required for regulating transcription activation and promotes regional gene expression [28]. STAT3 is involved in the pathology of asthma [20]. However, the regulatory mechanisms remain unclear. To the best of our current knowledge, no studies have addressed whether and how HDAC10 regulates STAT3 expression and promotes macrophage M2 program in asthma. We discovered that HDAC10 directly interacted with STAT3 and targeted STAT3 for deacetylation. The complex was then promoted macrophage M2 polarization via PI3K/Akt signaling pathway. However, the more detailed mechanisms of STAT3 deacetylation that regulates STAT3 transcriptional activity still need to be further studied.
The present study does have some limitations. First, the Hdac10fl/fl-LysMCre mice were conditionally knocked out for HDAC10 in macrophages (CKO). HDAC10 is expressed in various types of cells, such as epithelial cells, endothelial cells and T cells [29,30]. Further experiments are needed to investigate the effect of other cells on HDAC10 in the regulation of BMDMs and the role of HDAC10 in other cells. Second, although SAB has the ability to inhibit HDAC10 activity and transcription, the precise mechanisms underlying the impact of SAB on HDAC10 expression and activity remain elusive and warrant further investigation. Therefore, caution should be exercised when interpreting the role of SAB in asthma due to this limitation.
In conclusion, we have shown that HDAC10 was highly expressed in macrophages and Hdac10 deficiency protected mice against allergen-induced airway inflammation via PI3K/Akt signaling. We conjectured that HDAC10 directly interacts with STAT3 and targeted STAT3 for deacetylation to enhance an M2 program. Moreover, HDAC10 inhibitor treatment attenuated allergic airway inflammation. These data provided insight into the relation of epigenetic regulators with STAT3 in macrophage M2 program and supplied important clues for a potential strategy against airway inflammation in asthma.
Materials and methods
Human samples
The diagnosis of asthma was based on the Global Initiative on Asthma (GINA) guidelines [31]. Asthmatic patients between 18 and 65 years old were included. Exclusion criteria were combined respiratory diseases other than asthma such as chronic obstructive pulmonary disease (COPD), and lung cancer. Patients who had lung nodules and underwent surgery were used as normal control. Bronchial biopsies were obtained from patients undergoing bronchoscopy for diagnostic purposes. Peripheral blood mononuclear cells (PBMC) were isolated from peripheral blood samples of study subjects using density gradient centrifugation according to manufacturer instructions. Clinical information is summarized in Table S1 and S2. The study protocols were approved by the Medical Ethics Committee of Affiliated Hospital of Guangdong Medical University (PJKT2022-079). Written informed consent was obtained from all participants.
Animal studies
Hdac10fl/fl mice were generated using the CRISPR-Cas9 system by the Cyagen Biosciences Inc. (Suzhou, China). Two loxP sequences were inserted in the introns flanked with the exon 2 and exon 14 of HDAC10 as described in Figure 2A. The LysMCre mice were a generous gift from Dr. G. Feng (University of California at San Diego, CA, USA). Hdac10fl/fl-LysMCre mice were generated by crossing the LysMCre mice with Hdac10fl/fl mice for specific deletion of Hdac10 in macrophages. All experimental procedures were approved by the Guangdong Medical University's Animal Ethical Committee.
Allergic asthma mouse model
Allergic asthma mouse model was established according to we and others previously described [32-34]. Briefly, six to eight-week-old mice were sensitized by intratracheal instillation of 5 μg HDM plus 3 μg LPS in 50 μl normal saline (NS) on days 0, 1, and 2. On days 15, 16 and 17 after the initial sensitization, the mice were challenged with 2.5 μg HDM plus 1.5 μg LPS in 50 μl NS using intratracheal instillation. Mice were administered with same volume of normal saline served as controls. The HDAC10 inhibitor (Salvianolic acid B, SAB), specific PI3K/Akt activator 1,3-Dicaffeoylquinic acid (DA) or STAT3 agonist (Colivelin) was injected intraperitoneally into the mice 2 h before the 2.5 μg HDM plus 1.5 μg LPS intratracheal instillation. The mice were sacrificed under anesthesia 24 h after the final challenge.
Histological and immunohistochemical analysis
Lung tissue sections were subjected to hematoxylin and eosin (HE) staining, Masson's trichrome staining (Masson), and Periodic Acid-Schiff (PAS) staining. For immunostaining, the frozen sections were incubated with antibodies against CD206, STAT3, HDAC10, and F4/80, followed by staining with Alexa Fluor 594-labeled anti-mouse/rabbit or Alexa Fluor 488-conjugated anti-rabbit/mouse antibodies, respectively. All slides were examined in a random blinded fashion by two independent investigators. At least 10 bronchioles were counted on each slide and the data used for statistical analysis.
Quantitative RT-PCR (qRT-PCR)
Total RNA was isolated from the cells and mice lung tissues using Trizol reagent (Invitrogen, Carlsbad, CA, USA) and was reverse-transcribed into cDNA using the PrimeScript™RT reagent kit (Takara, Beijing, China). cDNA was used for RT-PCR amplification using TB Green™ Premix Ex Taq™ (Takara, Beijing, China). The expression of individual genes was normalized to the expression of β-actin. The primers used to amplify each target gene were shown in Table S4.
Culture and treatment of BMDMs
BMDMs were isolated and cultured as we previously reported [7]. Briefly, BMDMs were collected from six-to eight-week-old male mice. Red blood cells (RBCs) Lysing Buffer (Tbdscience.com) was used to remove red blood cells. The remaining cells were incubated in DMEM containing antibiotics, 10% fetal bovine serum (FBS), and 10 ng/ml recombinant mouse M-CSF (Novoprotein, Catalog #0331488) for 7 days to promote differentiate bone marrow-derived macrophages. After 7 days, the differentiated macrophages were cocultured with IL-4 (10 ng/ml) or HDM (0, 25, 50, 75, 100 μg/ml) at the indicated time points.
Dual-luciferase reporter assay
Hdac10fl/fl and Hdac10fl/fl-LysMCre BMDMs transfected with Renilla TK luciferase reporter vectors, luciferase reporter vectors and pSTAT3-TA-luc plasmid for 24 h, and then treated with HDM (100 μg/ml) for another 24 h. The cells were collected for luciferase activity assays using the Dual-Luciferase Reporter Assay System kit (Promega) according to the manufacturer's instructions.
Macrophage depletion and adoptive transfer studies
According to previous study [18], WT mice were injected with Clodronate liposomes (200 μl) or equal volume liposomes through the caudal vein for two consecutive days. Mice were sacrificed 24 h after intravenous injection to evaluate the macrophage depletion efficiency. For adoptive transfer studies, the Hdac10fl/fl and Hdac10fl/fl-LysMCre naive BMDMs were first stimulated with IL-4 (10 ng/ml) or equal volume of NS for 12 h to induce M2 macrophage polarization, and then transferred into the lungs of liposome-treated WT mice at a density of 1×106 cells per mouse (50 μl) on days 16 and 17 of allergen induction as described above through intratracheal instillation. The mice were sacrificed 24 h after adoptive transplantation for analysis.
Western blotting
Total proteins were extracted from lung tissues and cells using RIPA, and the concentration was detected by BCA protein assay kit according to the manufacturer's protocol (Beyotime, Shanghai, China). Equal amounts of protein were separated via New Semet Express-cast PAGE color gel electrophoresis and electrophoresed onto PVDF membranes. The membranes were blocked with 5% skim milk or 5% BSA for 1 h, and incubated with primary antibodies incubated at 4 °C overnight. After washed with 1×TBST solution for 3 times, the PVDF membrane was incubated with the corresponding secondary antibody for 1 h. Then the PVDF membrane was washed with 1×TBST solution 3 times again, and the exposure was analyzed and sorted out.
Co-immunoprecipitation (Co-IP)
After Total proteins of cells were extracted with cell lysis buffer, the protein concentration was determined by the BCA method and 1mg protein was taken for subsequent immunoprecipitation. Each sample was incubated with 1 µg antibody overnight at 4 °C, then incubation with 30 μl of Protein G Beads at 4 °C for 2 h. The immunocomplexes were washed 5 times with IP lysis buffer, then resuspended with 2×SDS Sample Buffer for Western blotting.
GST pull-down
GST-HDAC10 and GST-HA-STAT3 plasmids were constructed. The GST fusion protein was expressed in E. coli BL21 cells and purified according to manufacturer's instructions using the GST-Tag Protein Purification Kit (Beyotime). GST-HDAC10 and GST-HA-STAT3 proteins were mixed in equal amounts using the BCA kit to quantify the protein concentration, and then the corresponding primary antibody was added and incubated at 4 °C overnight. On the second day, each sample was added with the same amount of Protein G Beads and incubated at 4 °C for 1~2 h. After washing, western blotting was performed with anti-HDAC10 or anti-STAT3 antibody.
ELISA
Mouse CXCL1 and mouse CXCL2 in BALF and lung homogenate of mice were measured using ELISA kit (Elabscience). Assays were according to the manufacturer's protocol.
Flow cytometry
The cultured naïve BMDMs were stimulated by IL-4 (10 ng/ml) for 12 h. The cells were stained with anti-mouse CD206-PerCP/Cy5.5 and F4/80-APC. All staining processes were performed according to recommended protocols. The staining was detected by flow cytometry (BD FACSCantoⅡ) as previously described [35], and the data were analyzed using Flow Jo v10 (Becton Dickinson).
Molecular docking
The molecular docking program consists of two parts. First, in order to rapidly screen out molecules with good target binding activity, we first combined the traditional Chinese medicine database with HDAC10 (PDB; 5TD7) to perform rapid virtual filtering. In the next step, the compound Salvianolic acid B with the highest scoring data was matched with the target carefully, and the sites were selected as Glu 272, Asp 177 and Gln 302. Finally, we show the docking scoring table and the combination model diagram [36,37].
Reagents and antibodies
Please see the Table S5.
Statistical analysis
Unless stated otherwise, data are presented as means ± SEM. The data were analyzed using GraphPad Prism (San Diego, CA, version 8.0). Two experimental groups were compared using Mann-Whitney U-test or Student's t test. For comparisons between more than two groups, an ordinary one-way analysis of variance (ANOVA) followed by Tukey's post hoc test. Categorical variables were tested using Chi-square. A P value of < 0.05 was considered statistically significant.
Abbreviations
HDAC10: Atopic dermatitis; STAT3: Expression quantitative trait locus; BMDMs: Formaldehyde-assisted isolation of regulatory elements; SAB: Salvianolic acid B; DA: 1,3-Dicaffeoylquinic acid; HDM: house dust mites; LPS: lipopolysaccharide; PBMCs: peripheral blood mononuclear cells.
Supplementary Material
Supplementary tables.
Supplementary figures.
Acknowledgements
This research was supported by the Guangdong Basic and Applied Basic Research Foundation (2020B1515020004), National Natural Science Foundation of China (82170030; 82072208), Guangdong Provincial Key Laboratory of Autophagy and Major Chronic Noncommunicable Diseases (2022B1212030003).
Author contributions
Y.Z., T.H., J.W.H., and J.Y.Q. conducted experiments, data analysis. G.M.S., Z.L.X., S.H.L., X.W.L., Y.Y.X., and Q.W. assisted with the experiments and data analysis. Y.Y.L. collected clinical samples. Y.M.S., X.G., and L.X.L. analyzed/interpreted results. T.W.L., J.T., and Y.M.S. conceived, designed, and supervised the whole study. T.W.L. and Y.Z. wrote the manuscript. All authors read and approved the final manuscript.
Data and materials availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The data that support the findings of this study are available from the corresponding author upon reasonable request.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hammad H, Lambrecht BN. The basic immunology of asthma. Cell. 2021;184:1469-85
2. Aegerter H, Lambrecht BN, Jakubzick CV. Biology of lung macrophages in health and disease. Immunity. 2022;55:1564-80
3. Joshi N, Walter JM, Misharin AV. Alveolar Macrophages. Cell Immunol. 2018;330:86-90
4. Sharma N, Akkoyunlu M, Rabin RL. Macrophages-common culprit in obesity and asthma. Allergy. 2018;73:1196-205
5. Lechner A, Henkel FDR, Hartung F, Bohnacker S, Alessandrini F, Gubernatorova EO. et al. Macrophages acquire a TNF-dependent inflammatory memory in allergic asthma. J Allergy Clin Immunol. 2022;149:2078-90
6. Han X, Huang S, Xue P, Fu J, Liu L, Zhang C. et al. LncRNA PTPRE-AS1 modulates M2 macrophage activation and inflammatory diseases by epigenetic promotion of PTPRE. Sci Adv. 2019;5:eaax9230
7. Lai T, Su G, Wu D, Chen Z, Chen Y, Yi H. et al. Myeloid-specific SIRT1 deletion exacerbates airway inflammatory response in a mouse model of allergic asthma. Aging (Albany NY). 2021;13:15479-90
8. Ihle JN. The Stat family in cytokine signaling. Curr Opin Cell Biol. 2001;13:211-7
9. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14:736-46
10. Bi C, Fu Y, Li B. Brain-derived neurotrophic factor alleviates diabetes mellitus-accelerated atherosclerosis by promoting M2 polarization of macrophages through repressing the STAT3 pathway. Cell Signal. 2020;70:109569
11. Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol. 2014;15:536-50
12. Cosío BG, Mann B, Ito K, Jazrawi E, Barnes PJ, Chung KF. et al. Histone acetylase and deacetylase activity in alveolar macrophages and blood mononocytes in asthma. Am J Respir Crit Care Med. 2004;170:141-7
13. Li Y, Zhang X, Zhu S, Dejene EA, Peng W, Sepulveda A. et al. HDAC10 Regulates Cancer Stem-Like Cell Properties in KRAS-Driven Lung Adenocarcinoma. Cancer Res. 2020;80:3265-78
14. Bugide S, Gupta R, Green MR, Wajapeyee N. EZH2 inhibits NK cell-mediated antitumor immunity by suppressing CXCL10 expression in an HDAC10-dependent manner. Proc Natl Acad Sci U S A. 2021 118
15. Tong JJ, Liu J, Bertos NR, Yang XJ. Identification of HDAC10, a novel class II human histone deacetylase containing a leucine-rich domain. Nucleic Acids Res. 2002;30:1114-23
16. Oehme I, Linke JP, Böck BC, Milde T, Lodrini M, Hartenstein B. et al. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc Natl Acad Sci U S A. 2013;110:E2592-601
17. Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt Signaling Pathway in Macrophage Activation and M1/M2 Polarization. J Immunol. 2017;198:1006-14
18. Wang Y, Zhang L, Wu GR, Zhou Q, Yue H, Rao LZ. et al. MBD2 serves as a viable target against pulmonary fibrosis by inhibiting macrophage M2 program. Sci Adv. 2021 7
19. Li J, Diao B, Guo S, Huang X, Yang C, Feng Z. et al. VSIG4 inhibits proinflammatory macrophage activation by reprogramming mitochondrial pyruvate metabolism. Nat Commun. 2017;8:1322
20. Simeone-Penney MC, Severgnini M, Tu P, Homer RJ, Mariani TJ, Cohn L. et al. Airway epithelial STAT3 is required for allergic inflammation in a murine model of asthma. J Immunol. 2007;178:6191-9
21. Li CL, Liu B, Wang ZY, Xie F, Qiao W, Cheng J. et al. Salvianolic acid B improves myocardial function in diabetic cardiomyopathy by suppressing IGFBP3. J Mol Cell Cardiol. 2020;139:98-112
22. Wang D, Lu X, Wang E, Shi L, Ma C, Tan X. Salvianolic acid B attenuates oxidative stress-induced injuries in enterocytes by activating Akt/GSK3β signaling and preserving mitochondrial function. Eur J Pharmacol. 2021;909:174408
23. Ko YS, Jin H, Park SW, Kim HJ. Salvianolic acid B protects against oxLDL-induced endothelial dysfunction under high-glucose conditions by downregulating ROCK1-mediated mitophagy and apoptosis. Biochem Pharmacol. 2020;174:113815
24. DeVries A, Vercelli D. Epigenetic Mechanisms in Asthma. Ann Am Thorac Soc. 2016;13(Suppl 1):S48-50
25. Islam MM, Banerjee T, Packard CZ, Kotian S, Selvendiran K, Cohn DE. et al. HDAC10 as a potential therapeutic target in ovarian cancer. Gynecol Oncol. 2017;144:613-20
26. Fan W, Huang J, Xiao H. Histone deacetylase 10 suppresses proliferation and invasion by inhibiting the phosphorylation of β-catenin and serves as an independent prognostic factor for human clear cell renal cell carcinoma. Int J Clin Exp Med. 2015;8:3734-42
27. Gallinari P, Di Marco S, Jones P, Pallaoro M, Steinkühler C. HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics. Cell Res. 2007;17:195-211
28. Ray A, Khan P, Nag Chaudhuri R. Deacetylation of H4 lysine16 affects acetylation of lysine residues in histone H3 and H4 and promotes transcription of constitutive genes. Epigenetics. 2021;16:597-617
29. Su Y, Tan R, Sun M, Yuan L, Ruiz M, Dupuis J. et al. MiR-1249 on Endothelial Extracellular Vesicles Mediates Cigarette Smoke-Induced Pulmonary Hypertension by Inhibiting HDAC10 (Histone Deacetylase 10)-NFκB (Nuclear Factor κB)-CaSR (Calcium-Sensing Receptor) Cascade. Hypertension. 2022;79:2721-32
30. Ran X, Ao Z, Olukitibi T, Yao X. Characterization of the Role of Host Cellular Factor Histone Deacetylase 10 during HIV-1 Replication. Viruses. 2019 12
31. Reddel HK, Bacharier LB, Bateman ED, Brightling CE, Brusselle GG, Buhl R. et al. Global Initiative for Asthma Strategy 2021: executive summary and rationale for key changes. Eur Respir J. 2022 59
32. Lai T, Wu M, Zhang C, Che L, Xu F, Wang Y. et al. HDAC2 attenuates airway inflammation by suppressing IL-17A production in HDM-challenged mice. Am J Physiol Lung Cell Mol Physiol. 2019;316:L269-l79
33. Kwak DW, Park D, Kim JH. Leukotriene B(4) receptors play critical roles in house dust mites-induced neutrophilic airway inflammation and IL-17 production. Biochem Biophys Res Commun. 2021;534:646-52
34. Qian G, Jiang W, Zou B, Feng J, Cheng X, Gu J. et al. LPS inactivation by a host lipase allows lung epithelial cell sensitization for allergic asthma. J Exp Med. 2018;215:2397-412
35. Uba AI, Yelekçi K. Crystallographic structure versus homology model: a case study of molecular dynamics simulation of human and zebrafish histone deacetylase 10. J Biomol Struct Dyn. 2020;38:4397-406
36. Géraldy M, Morgen M, Sehr P, Steimbach RR, Moi D, Ridinger J. et al. Selective Inhibition of Histone Deacetylase 10: Hydrogen Bonding to the Gatekeeper Residue is Implicated. J Med Chem. 2019;62:4426-43
37. Ibrahim Uba A, Yelekçi K. Homology modeling of human histone deacetylase 10 and design of potential selective inhibitors. J Biomol Struct Dyn. 2019;37:3627-36
Author contact
![]() Corresponding author: laitianwen2011com (T.W.L.), tanglitangjingcom (J.T.), symedu.cn (Y.M.S.).
Corresponding author: laitianwen2011com (T.W.L.), tanglitangjingcom (J.T.), symedu.cn (Y.M.S.).