Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Types of small antibody fragments
Antibody recognition of...
Targeting tumour-associated...
Conclusion
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2023; 13(9):3041-3063. doi:10.7150/thno.80901 This issue Cite
Review
Smaller size packs a stronger punch - Recent advances in small antibody fragments targeting tumour-associated carbohydrate antigens
Sana Khan Khilji1,2, Charlotte Op 't Hoog1,3, David Warschkau1,2, Jost Lühle1,2, Felix Goerdeler1,2, Anika Freitag1,4, Peter H. Seeberger1,2, Oren Moscovitz1 ![]()
1. Department of Biomolecular Systems, Max Planck Institute of Colloids and Interfaces, 14476 Potsdam, Germany.
2. Institute of Chemistry and Biochemistry, Freie Universität Berlin, 14195 Berlin, Germany.
3. Graduate School of Life Sciences, Utrecht University, 3584 CH Utrecht, Netherlands.
4. Institute of Chemistry, University of Potsdam, 14476 Potsdam, Germany.
Received 2022-11-16; Accepted 2023-4-26; Published 2023-5-15
Abstract

Attached to proteins, lipids, or forming long, complex chains, glycans represent the most versatile post-translational modification in nature and surround all human cells. Unique glycan structures are monitored by the immune system and differentiate self from non-self and healthy from malignant cells. Aberrant glycosylations, termed tumour-associated carbohydrate antigens (TACAs), are a hallmark of cancer and are correlated with all aspects of cancer biology. Therefore, TACAs represent attractive targets for monoclonal antibodies for cancer diagnosis and therapy. However, due to the thick and dense glycocalyx as well as the tumour micro-environment, conventional antibodies often suffer from restricted access and limited effectiveness in vivo. To overcome this issue, many small antibody fragments have come forth, showing similar affinity with better efficiency than their full-length counterparts. Here we review small antibody fragments against specific glycans on tumour cells and highlight their advantages over conventional antibodies.
Keywords: fragments, TACAs, cancer, aberrant glycosylation, CAR T-cell, ScFv, nanobody, carbohydrates, tumor-associated carbohydrate antigens, chimeric antigen receptors, single-chain variable fragment, single-chain fragment variable, tumor, Tumour, single domain antibodies, cancer antigens, glycans, immunology, cancer glycans
Introduction
Glycans are the most complex and structurally diverse biomolecules found on all cells and tissues of the human body. Glycosylation is the primary source of microheterogeneity in proteins, and, with few exceptions, all proteins passing through the ER and Golgi during biosynthesis are modified with O- and/or N-linked glycans. As part of the cellular glycocalyx, glycans are involved in various biological processes, including intracellular transport, cell adhesion, cell-cell, and cell-matrix interactions, as well as signalling [1]. Hence, they play a crucial role in human physiology and pathology.
The assembly of glycans can differ considerably from cell to cell. Although at least 4000 human genes are directly and indirectly involved in glycan synthesis, glycans are not directly encoded in the genome. Genetic defects in the glycosylation machinery often tend to be lethal at the embryonic stage, emphasising the vital role of glycans. The dynamic structure and composition of the glycocalyx are affected by various endogenous factors, such as the expression levels of specific glycosyltransferases and pH in the ER and Golgi. Age and environmental factors, such as lifestyle and diet, have also been shown to affect the glycocalyx. Under normal physiological conditions, a cell's glycan characteristics are primarily conserved, and alterations often reflect and result in cancer [1]. These aberrant glycan structures can, therefore, be used as glycan biomarkers for diagnostics as well as specific targeting of the cells that carry them.
Tumour-associated carbohydrate antigens (TACAs) have received increasing attention over the past decades due to their central role in every aspect of cancer progression and the possibility of targeting them specifically using antibodies (Abs). Carbohydrates, however, are generally poorly immunogenic and often elicit a T-cell-independent response, which fails to create immunological memory. Hence, immunisation of animals using glycans primarily leads to the production of low-affinity Immunoglobulin M (IgM), which does not class switch to Immunoglobulin G (IgG) [2]. Thus, vaccine development against TACAs is hampered by the absence of T-cell-mediated immunity, which is critical for active cancer immunotherapy. TACAs are often tolerated by the immune system due to the structural similarity of TACAs with healthy cell glycans [3] and the low expression of TACAs in healthy tissue or during prenatal development [4]. To overcome the poor immunogenicity of glycans in vaccines and therapeutic Ab development, glycans are conjugated to a carrier protein. The generation of glycopeptides in antigen-presenting cells, following their display by the major histocompatibility complex (MHC) system to T-cell receptors, is essential for eliciting a T-cell-dependent immune response and the formation of specific and high-affinity immunoglobulins. To date, production of most anti-glycan monoclonal antibodies (mAbs) involves animal immunisation with glycan-carrier protein conjugates to ensure the essential T-cell-dependent response for the formation of glycan-specific IgGs.
Even though immunotherapy targeting glycans faces several hurdles, it also holds a considerable advantage. One of the major problems in combating cancer is drug resistance. Due to drug-imposed selection pressure in combination with the high mutational rate and clonal expansion, cancer cells often acquire drug resistance genetically. Targeting TACAs reduces the chance of antigen escape tremendously, as their synthesis, in contrast to proteins, is neither linear nor template-driven and involves the coordinated activity of numerous enzymes. Furthermore, TACAs are far more abundant than protein tumour antigens; for instance, the highly expressed protein marker HER-2 has about 106 copies per cell, whereas the TACA Thomsen-Friedenreich (TF) has about 107 copies per cell [5]. TACA expression is often more frequent for given tumour types than protein antigens and identified in a larger percentage of the patients. Targeting TACAs can pave the way to more effective cancer therapies as is demonstrated by the approval of Dinutuximab, a mAb targeting the TACA GD2, for treating neuroblastoma in 2015 [6].
The mAbs used in therapy are typically IgG and are composed of the fragment antigen binding (Fab), and the crystallizable fragment (Fc) (Figure 1A), which interacts with Fc receptors expressed on several immune cells such as natural killer (NK) cells and macrophages, leading to antibody-dependent cellular cytotoxicity (ADCC) or antibody-dependent cell phagocytosis (ADCP). In addition, the Fc fragment can be recognised by components of the complement system and can thereby induce complement-dependent cytotoxicity (CDC). Alternatively, mAbs can be used to deliver a toxic payload in the form of an antibody-drug conjugate (ADC) [7].
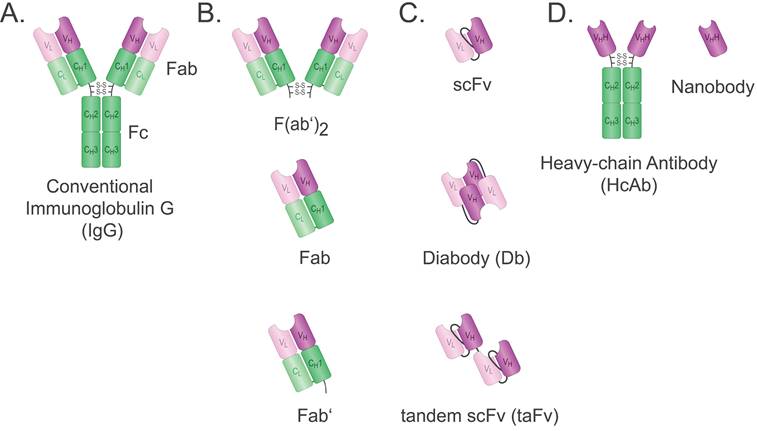
Antibody and antibody fragments. A. The full-length Immunoglobulin G (IgG) consists of two Fab and one Fc fragment. The IgG also contains two heavy (H) and two light (L) chains. Green indicates the constant (C) domains and purple indicates the variable (V) domains. The variable domains of the heavy (VH) and light (VL) chain form the variable fragment (Fv). The Fab and Fc regions are marked. B. Fab: Antigen binding fragment; Fab': Fab with hinge region; F(ab')2: two Fabs joined at the hinge region. C. scFv: single chain variable fragment; diabody: non-covalent scFv dimer; tandem scFv: covalent scFv dimer. D. hcAb: heavy chain only Abs from camelids. Nanobody: variable domain (VHH) of camelid hcAb.
The first mAb for therapeutic use was approved by the U.S Food and Drug Administration (FDA) in 1986. In 2021, the FDA approved the 100th mAb product, highlighting their wide success [8] Nevertheless, their large size and high molecular mass of ~150 kDa, prevents Abs from reaching their full potential in therapeutic efficiency, as penetration into thick and dense tissue, as the tumour microenvironment, is often a limiting factor. In addition, the structure, function, and serum half-life of therapeutic mAbs, as well as non-desired adverse effects in treated human patients are due to Fc fragment interactions with the immune system cells and the N-glycans it carries [9].
To overcome these problems, tremendous efforts have been expended to generate smaller antigen-binding fragments, such as Fab, single chain variable fragment (scFv), and single-domain Abs (Figure 1). Ab fragments are produced through enzymatic cleavage from intact mAbs, or by recombinant protein expression systems such as bacteria, which allow cheaper and faster production in large quantities. The fragments are less immunogenic due to the absence of the Fc fragment, and their size enables better access to dense tissue and contributes to enhanced tumour penetration [10]. Moreover, Ab fragments are readily coupled with different molecules, and their relatively smaller size makes them highly amenable to genetic engineering and the development of multivalent and multispecific tools [11].
However, many recombinant fragments exhibit reduced stability and affinity compared to their parent Ab. Moreover, lying below the glomerular filtration cut-off of approximately 60 kDa, most Ab fragments suffer from fast renal clearance and a half-life ranging from minutes to several hours [12]. A longer circulating time is desirable for cancer therapy to achieve its full therapeutic efficacy through enhanced biodistribution and increased Ab concentration at the target site. Unfortunately, too-small constructs will be excreted through the kidneys before exerting biological effects. Therefore, several strategies are successfully employed to increase the half-life of Ab fragments, including chemical conjugation to polyethylene glycol (PEG) chains or fusion to additional Ab fragments that bind serum proteins such as albumin [13]. Nevertheless, a shorter blood circulation time can also be advantageous for tumour imaging, as it lowers background signal compared with the 2-3 weeks of half-life of canonical Abs [14].
While there have been several comprehensive reviews on full Abs targeting TACAs [15-17], literature regarding Ab fragments targeting TACAs was not reviewed to date. Herein, we provide a comprehensive overview of the achievements and applications of Ab fragments that target specific TACAs. First, we provide a structural overview of the different Ab fragments. Thereafter, we describe specific TACAs, and the fragments that target them. Engineered Ab fragments that contain Fc segments are out of the scope of this review.
Types of small antibody fragments
Fab-based formats
With a weight of ~50 kDa, the Fab is composed of the Ab light chain (VL + CL) covalently bound to the heavy chain VH and CH1 domains via a disulfide bridge securing monovalent and monospecific binding (Figure 1B). Fabs are more stable than other fragments such as their single chain variable fragment (scFv) counterparts, but bigger and, therefore, less efficient in tissue penetration [18]. Fabs can be obtained readily by proteolytic digestion of IgG with papain. Using alternative enzymatic digestion with pepsin that cleaves below the hinge region, F(ab')2 can be obtained. However, this is a time-consuming process as it also requires the production of an Ab first. F(ab')2 is a bivalent Ab format with a molecular weight of ~110 kDa. An antivenom F(ab')2 is used in the clinic and shows a longer half-life compared to the antivenom Fab previously used [19]. No F(ab')2 has been approved by the FDA for cancer therapy to date. It is possible to cleave F(ab')2 into Fab', which is the Fab containing the hinge region. A major advantage of the various Fabs is that linker engineering is not required, which saves time and resources. Fabs are typically conjugated as targeting ligands for therapeutic or diagnostic tools [20].
Fv-based formats
Single chain variable fragment
The scFv was first described by Bird et al. in 1988. It comprises the variable domains of heavy (VH) and light (VL) chains of an Ab, fused together by a linker peptide (Figure 1C) [21]. The linker peptide should be flexible with optimal length and sequence for affinity and thermostability and is often rich in glycine, serine, and hydrophilic residues. The affinity for the antigen is generally comparable to that of the parent Ab [21]. While conventional mAbs usually require a mammalian expression system as well as a balance in expression of the light (L) and heavy (H) chains, the VL and VH domains of an scFv are coded in a single genetic sequence leading to approximately 27 kDa that can be easily expressed in both eukaryotic and prokaryotic systems, hence, making it more cost-effective [22]. Nevertheless, the disadvantages of scFv should also be taken into consideration. The lack of constant domains often results in lower thermostability and a tendency to aggregate. Thus, scFv production may require additional refolding procedures [23]. In addition, scFvs have a short half-life of approximately 0.5-2.0 hours due to their small size. Like Fabs, scFvs are used to generate targeted cancer imaging tracers and drug conjugates [24].
A more recent application of scFvs is their expression as Chimeric Antigen Receptors (CAR) that provide specificity of T cells to their targets but also initiate and determine the strength of their activation [25]. CAR-modified immune cells, mainly cytotoxic T-cells but also natural killer (NK) cells, dendritic cells, and macrophages, are genetically engineered to display antigen-specific scFvs or nanobodies. Upon target binding, additional intracellular CAR domains mediate a cytotoxic response and killing of the cancer cell [25]. To date, the effectiveness of CAR T therapy against solid tumours remains limited, and the six FDA-approved CAR therapies target different haematological malignancies [26]. Nevertheless, extensive efforts are being made to bypass the limitations in targeting solid tumour and generate stable scFv-CARs against protein and glycan targets with optimal affinity to ensure minimal on-target/off-tumour toxicity [25].
Multimeric scFv formats: Diabodies and Tandem scFvs
In addition to the classical monovalent form, scFvs can be readily engineered to form multivalent constructs, which is a viable strategy to improve pharmacokinetic properties. The generated multimers are often bivalent, thus having a molecular weight of 50-60 kDa. The increase in size is accompanied by an increase in serum half-life [27]. The multivalency can either be utilised to increase the functional affinity, termed avidity, or to target different antigens simultaneously. In case of the latter, the additional binding domain can be used to increase serum half-life further by targeting albumin, or to exert biological effects by targeting effectors. Such bivalent Ab constructs are named bispecific Antibodies (BsAbs). There are two main forms of multimeric scFv formats: diabodies and tandem scFvs (taFv) (Figure 1C). The taFv format is formed by covalent linkage of two scFv molecules through a short linker so that they can rotate freely and retain their separate conformations and antigen-binding units [27]. An anti-cancer immune response can be provoked by using an scFv with specificity for cancer cells fused to an scFv with specificity for a type of immune cell. Such a taFv is called a bispecific T-cell engager (BiTE) when a CD3-specific scFv is used to engage T-cells [28].
In diabodies, the two scFvs are not fused together by a linker but instead contain a shorter linker between VH and VL of the same chain forcing the domains to couple with complementary domains of a second scFv. Hence, a rigid conformation is created with two antigen-binding sites. Bispecific diabodies are formed when fusions of variable domains of different Abs (VH-VL' and VH '-VL or VL-VH' and VL '-VH) as scFvs are expressed in the same cell and interact randomly. A mixture of non-functional scFv-homo- and functional scFv-heterodimers follows, of which the latter can be isolated using affinity chromatography [27]. To date, no diabodies are used in the clinic.
Single domain formats
Nanobodies
Three decades ago, the Hamers group at Vrije Universiteit Brussel (VUB) discovered that camelids produce Abs lacking light chains and CH1 domains [29]. The heavy chain-only antibodies (hcAb) bind to their antigen solely through the two VHH domains that constitute the smallest antigen-binding fragments in nature, weighing only ~15 kDa (Figure 1D). The recombinantly expressed VHH domain was termed nanobody (Nb), and as shaped by nature, Nbs are highly stable, durable, and soluble. Moreover, their small size enables Nbs to penetrate narrow cavities and bind epitopes inaccessible to full-size Abs. Additionally, Nbs are easily genetically manipulated to form multivalent and multispecific tools expressed efficiently on a large scale via different expression systems. Due to their structural and functional advantages, using Nbs and Nb-conjugates for in vivo cancer diagnosis and therapeutics is well-studied for a range of intra- and extracellular cancer markers. As a result, five Nb constructs entered (or will soon enter) clinical trials [30], and the first VHH-based chimeric antigen receptors (CAR)-T product was FDA approved in February 2022 for the treatment of relapsed or refractory multiple myeloma [31].
In contrast to the long circulation time of full-length mAbs, Nbs exhibit short circulation time and fast renal clearance (> 2h), which leads to reduced background signals and lower accumulation in nontarget organs. Moreover, fast clearance increases the low tumour-to-blood ratio and reduces the systemic side effects often correlated with mAbs. Thus, Nbs were proven ideal reagents for rapid and specific same-day tumour imaging. Nbs advantages over canonical mAbs for tumour imaging were recently demonstrated in several comparison studies in xenografted mice using fluorescently labelled Nbs against HER2 [10, 32], and epidermal growth factor receptor (EGFR) [33].
In contrast to mice and humanised mAbs, Nbs are similar to human VH sequences, enabling repeated treatment cycles with a lower risk of anti-drug Ab development [34]. Although lacking an FC domain and the ability to engage and activate immune effector functions, Nbs coupled to immunotoxin, drugs, or radio-isotope probes can kill cancer cells, block tumour growth, and prolong the survival time of xenografted mice [11]. Moreover, cargo-free Nbs can also inhibit tumour growth by blocking immune-suppressing molecules at the tumour microenvironment or breaking the inhibitory immune checkpoint axis. Nevertheless, in most comparison studies, full-length mAbs with a longer circulation time and the ability to initiate the range of Ab-dependent anti-cancer immune responses still obtain a better anti-cancer therapeutic outcome [35]. Yet, despite their obvious limitations for cancer therapy compared to mAbs, several Nbs still demonstrated a comparable, and in some cases, better activity than their full-size counterparts. For example, radioimmunotherapy using anti-CD20 Nbs in hCD20pos B16 tumour-bearing mice significantly prolonged their median survival and was as effective as treatment with the radiolabelled mAb [36]. In addition, a unique bispecific nanobody against the immune system suppressing molecules PD-L1 and CXCR4 was superior in inhibiting tumour growth compared with anti-PD-L1 mAb in pancreatic cancer xenografted mice model [37].
Furthermore, due to their structural robustness, Nbs are gaining increased attention as chimeric antigen receptors (CARs) and are investigated in pre-clinical and clinical studies against solid and non-solid tumours [38]. Interestingly, in addition to their advantages as CARs, CAR T cells engineered to secrete Nbs in the tumour microenvironment demonstrate enhanced persistence and improved anti-tumoral response compared with conventional CAR T cells [39]. Currently, no Nb-based CAR T-cell targeting a TACA has been described. Moreover, obtaining purely anti-glycan Nbs is still challenging [40]. Interestingly, we recently reported the development of a specific Nb against Globo-H, a glycosphingolipid expressed in many solid tumours. Using alpaca immunisation with synthetic glycans, we generated a unique Nb that binds specifically to Globo-H, albeit with a relatively low affinity [41].
Antibody recognition of carbohydrate structures
Our current understanding of how mAbs recognise specific glycan structures has advanced significantly in recent years. It has been found that recognition of particular glycan epitopes by Abs is governed by the precise arrangements of the sugar residues and their linkage, as well as the size and shape of the overall glycan structure. Naturally, higher specificity and lower cross-reactivity of mAbs are dictated by the exact binding epitope along the glycan chain and the number of monosaccharides involved in the interaction. Thus, synthetic glycan array characterisation is crucial to elucidate the specificity and cross-reactivity of anti-glycan Abs. Unfortunately, most existing mAbs cross-react with additional glycan structures, and the number of monospecific anti-glycan Abs is still low. However, currently available three-dimensional structures of Ab-glycan complexes provide valuable information and demonstrate the lack of a universal rule for the binding modes of glycan-targeting Abs and bound glycan conformations. Thus, mAbs recognise distinct glycan epitopes through diverse binding mechanisms. They can exhibit no notable conformational changes upon glycan binding [42] but also undergo an induced-fit conformational change that creates a binding pocket to occupy a portion of the glycan in an end-on insertion [43] or a parallel configuration mode [44]. Some mAbs form an extensive glycan-binding paratope that spans the entire VL-VH interface, engaging glycans through all six CDRs [45]. On the other hand, other Abs rely on only a portion of their CDRs, predominantly those of the VH chain, for glycan binding [46], and some interact exclusively via the VL chain [47]. In addition, the bound glycans can adopt an anchored, perpendicular, or parallel orientation to accommodate the Ab binding cavity. Interestingly, recent work on anti-CA19.9 Abs demonstrated that despite the high flexibility of glycan structures, Abs with different binding modes still recognise a similar distinct low-energy conformer [42].
Targeting tumour-associated carbohydrate antigens
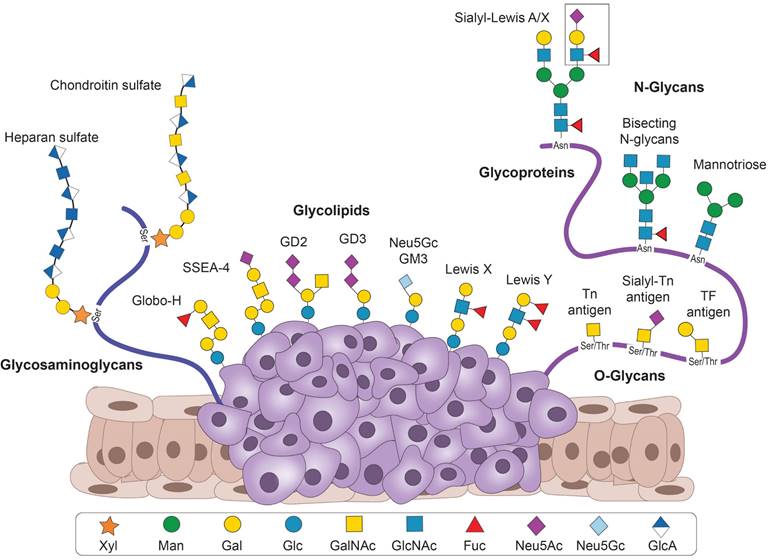
Small Ab fragments against TACAs have been developed for various applications and target TACAs from several groups (Figure 2). These include 1. Glycosphingolipids from the ganglio-series (GD2, GD3, N-glycolyl GM3), and the globo-series (Globo-H and SSEA-4); 2. Blood group-related Lewis antigens (Lewis Y, Lewis X, sialyl-Lewis X, and sialyl-Lewis A); 3. Aberrant N-glycans or the truncated O-glycans (Thomsen-Freidenreich (TF), Thomsen-nouveau (Tn) and sialyl-Tn (sTn antigens); and 4. several cancer-related glycosaminoglycans. We describe below the antigens, the Ab fragments targeting them, and their applications.
The structures of the TACAs targeted by small antibody fragments described in this review. Glycosaminoglycans are shown on the left, TACAs carried by lipids as glycolipids and glycosphingolipids in the middle, and cancer-associated N-and O- glycans carried by proteins (glycoproteins) on the right. Lewis antigens can be present as glycosphingolipids and on glycoproteins.
Glycosphingolipids
Glycosphingolipids (GSLs) are cell surface glycolipids that can be classified according to their core structure as the ganglio-series, the globo-series, and the lacto-series. Each ganglioside series is found in a specific cell type or tissue type and may contribute to adhesion or signalling characteristics of cells [48].
Gangliosides
Gangliosides are a GSL subclass, characterised by the presence of one or more sialic acid residues. They interact with phospholipids, cholesterol, and transmembrane proteins and play an essential role in cell adhesion and signalling. Gangliosides are particularly important for the development of the nervous system [49]. While most types of gangliosides are widely expressed across tissues, making the majority unsuitable for targeted therapy, some subtypes have limited expression in healthy tissues compared to cancer tissue. Aberrant expression of gangliosides is often detected in tumours of the central or peripheral nervous system (neuroectodermal origin). Aberrant gangliosides that have been targeted using small Ab fragments include GD2, GD3 and N-glycolyl GM3.
Disialoganglioside GD2
Disialoganglioside GD2 (GalNAcβ1-4(Neu5Acα2-8Neu5Acα2-3)Galβ1-4Glcβ1Cer) is a TACA found in a wide variety of paediatric and adult malignancies, including neuroblastomas, retinoblastomas, gliomas, osteosarcoma, Ewing sarcoma, rhabdomyosarcoma, small cell lung cancer, melanoma, breast cancer, bladder cancer, and soft tissue sarcoma [50]. The U.S. National Cancer Institute ranked GD2 at position 12 out of 75 potential targets based on various aspects such as therapeutic potential, expression level, and specificity [51]. GD2 expression in normal tissues is restricted to the central nervous system, peripheral sensory nerves, and skin melanocytes. It presents a promising immunotherapeutic target, especially in case of neuroblastomas, as more than 95% cases of neuroblastomas are GD2-positive. Therefore, mAbs against GD2 have been actively developed in recent years. Currently, the only FDA-approved immunotherapeutic drugs on the market targeting GD2 are Dinutuximab (Unituxin/ch14.18) and Naxitamab (Danyelza/hu3F8), both mouse/human chimeric IgG1 Abs. The Abs are effective in patients, but one of the major side effects is acute pain during administration due to activation of the complement system [52]. Therapeutics involving small Ab fragments can overcome this problem due to the absence of Fc-mediated complement activation. Small Ab fragments have been developed from ch14.18 (IgG3), its humanised derivative hu14.18K322A, as well as the class-switched 14G2a (IgG2a). Furthermore, fragments have been generated from hu3F8 (IgG3), 5F11 (IgM), 7A4 (IgG3) and ME361 (IgG2a) Abs. Finally, a Nb targeting GD2 generated by camel immunisation has been recently patented (ID: CN110551218B).
I. Anti-GD2 Fabs
The Fabs of 14.G2a, 7A4, and ME361 have been obtained and tested for several applications. Bernhard et al. chemically conjugated an anti-CD3 Fab' to ME361 Fab'. The Fab'-based BsAb could specifically mediate lysis of various human GD2-positive melanoma and glioma cell lines in vitro by engaging T-cells [53]. Later it was shown by Doronin et al. that ME361 Fab by itself can inhibit proliferation and induce apoptosis in GD2-expressing mice lymphoma EL4 cells [54]. Michon et al. chemically conjugated an anti-FcγRI Fab' to 7A4 Fab'. This Fab'-based BsAb (7A4 bis 22) engages macrophages instead of T-cells, which was also successful in binding human neuroblastoma cells in vitro as well as grafted in mice [55]. The Fab' fragment obtained from 14.G2a was used to produce GD2-targeting liposomes carrying various types of therapeutic agents. Cargo included antisense oligonucleotides against proto-oncogene c-myb, which showed long-term survival (more than 120 days) of mice xenografted with human neuroblastoma [56], chemotherapeutic doxorubicin, which selectively inhibited growth and dissemination of human neuroblastoma in mice [57], and siRNA, which knocked down the gene for anaplastic lymphoma kinase (ALK) in neuroblastoma xenografted in mice, causing cell growth arrest, apoptosis, and prolonged survival [58].
II. Anti-GD2 scFvs
ScFvs have been generated from 5F11, ch14.18, 14.G2a, hu3F8, KM8138 and 7A4. ScFvs are reported in the application of immunotoxins, pre-targeted radioimmunotherapy (PRIT), BiTEs and CAR T-cells.
Two generated immunotoxins include 14.18-scFv fused to Pseudomonas exotoxin A [59] and 5F11-scFv linked to Diphtheria toxin A [60] that have shown specific cytotoxicity in neuroblastoma cell lines. However, the immunogenicity of foreign toxins may pose a problem that may be overcome by various approaches such as PEGylation, patient treatment with immunosuppressive drugs, and protein engineering of toxins to modify and/or eliminate immune cell epitopes have been suggested [61].
GD2-targeting scFvs have also been used in radioimmunotherapy. The 5F11-scFv was labelled with streptavidin for multistep delivery of radiolabelled biotin in PRIT [62]. First, 5F11-scFv-streptavidin is administered, followed by a chemical clearing agent that removes unbound protein. Subsequently, radiolabelled biotin is added, hence, separating the targeting agent and cytotoxic agent. The tumour-to-blood ratio achieved by this system is almost 60-fold higher in mice compared to radio-iodinated anti-GD2 mAb 3F8 that is currently in phase II clinical trials (NCT00445966). Unfortunately, translation to the clinic is difficult due to the immunogenicity of streptavidin. Nevertheless, a multistep approach employing an scFv to reduce the size and, therefore, clearance time, is promising. Although some efforts have been made to reduce the immunogenicity of streptavidin, other scFv-targeted systems have attracted attention [63].
Incorporating 5F11-scFv into a BiTE can efficiently inhibit melanoma and neuroblastoma xenograft growth [64]. A novel platform with human transcription factor HNF1α-derived dimerisation tag enhanced the potency and efficacy of the BiTE by increasing the avidity and serum half-life in vivo [65]. The hu3F8-scFv has a 13-fold higher affinity (KD = 19 nM) for GD2 compared to 5F11-scFv (KD = 250 nM). Therefore, the group exchanged the anti-GD2 scFv in the construct to assess the effect on the antitumour response. The BiTE holding hu3F8-scFv showed 5000-fold higher potency. This increase in affinity showed improved T-cell activation in vitro and better tumour growth inhibition in neuroblastoma and melanoma xenograft models [66].
Hu3F8-scFv has also been engineered to carry a BTX binding peptide that strongly interacts with anti-bungarotoxin (BTX). BTX, on the other hand, was covalently attached to a polymer including doxorubicin [67]. While doxorubicin is a popular cytotoxic agent against chemo-sensitive tumours like neuroblastomas, its therapeutic efficacy is limited by dose-dependent toxicity to bone marrow and heart tissue [68]. Doxorubicin covalently attached to the scFv directly showed lower binding than the Hu3F8-scFv-BTX-Doxorubicin complex and, therefore, 10x and ~3x lower IC50 values against murine lymphoma and human neuroblastoma cells, respectively [67].
Another scFv from murine 3F8 was used to produce GD2-targeted hexanoyl chitosan-based nanoparticles by N-(ß-maleimidopropionyl)succinimide (BMPS) conjugation. The Fab of ME361 was used to compare the BMPS with the carbodiimide (CDI) conjugation method. EL-4 mouse lymphoma cells were used to show that BMPS conjugated constructs exhibited improved binding. The 3F8 scFv showed better binding than ME361 Fab. However, the anti-GD2 fragment-nanoparticle complex was mildly cytotoxic in both cases as cell viability decreased only to about 70% after 72 hours of incubation possibly due to a lack of internalisation of the complex on the cell surface that may be required for cell death induction [69].
The scFv derived from 14.18 was used to generate mono-, di-, and tetra-scFvs by site-directed PEGylation. The di-scFv and tetra-scFv exhibited highest binding efficiency, while tetra-scFvs were better GD2 binders than the di-scFv in a direct enzyme-linked immunosorbent assay (ELISA). The tetra-scFv were also the best binders for the GD2-positive IMR-32 and EL-4 cell lines, while none of the scFv constructs showed binding to the GD2-negative NGP-127 cell line. The cytotoxic effect of tetra-scFv on IMR-32 cells was again higher than the other constructs and comparable to that of the full-length chimeric 14.18 Ab. In addition, in the EL-4 syngeneic mice model, the accumulation of tetra-scFv after 24 h was significantly higher compared to the monomeric scFv and even better than the full-length 14.18 Ab. This highlights the significance and advantage of multimerisation of small Ab fragments for better therapeutic characteristics [70].
More than two decades ago, the 7A4 Ab was used to generate an scFv. The binding was tested in an ELISA and cross-reactivity with other closely related gangliosides was included. ScFv 7A4 retains its target specificity with weak cross-reactivity to GD3, as is seen for the full Ab [71]. Later, scFv 7A4 was incorporated into a CAR and expressed in T-cells. The CAR T-cells were, however, not functional. In the same report, scFv 14.G2a was also tested, which showed neuroblastoma-specific cell lysis. However, functionality decreased over time without nonspecific stimulation of transduced cells, and antigenic stimulation of the chimeric receptor alone could not sustain proliferation [72]. Many reports followed this GD2-targeting CAR T-cell since. Sujjitjoon et al. generated T-cells with fourth generation CAR moieties. For this, the scFv derived from mAb hu3F8 was used, and GD2 expressed on retinoblastomas was targeted. The CAR T-cells were effective in killing retinoblastomas, but extended exposure to the T-cells diminished their effect on the retinoblastomas. Immune evasion mechanism by retinoblastomas was also studied. This was the first study to show a CAR T-cell strategy targeting retinoblastomas [73].
The expression of TACAs on healthy tissues, albeit low, can cause off-target effects in conventional CAR T-cell therapy. CAR T-cells targeting large tumours may unpredictably proliferate and cause adverse effects like cytokine storms and tumour lysis syndrome in patients. Mitwasi et al. used a novel CAR T-cell (UniCAR) approach to target GD2. Unlike conventional CAR T-cells, UniCAR T-cells are not targeted towards a cancer cell surface epitope directly. Instead, they bind to a peptide epitope (so-called target module (TM)) that is not only specific for the cancer cell surface epitope but also modulates the UniCAR T-cells in a switch on/off manner as the UniCAR T-cells would target the cancer cell only in their presence. In this case, the TM comprised the scFv 14.G2a fused with the UniCAR epitope. Three GD2-specific TMs were used to show target-specific and -dependent activation of UniCAR T-cells, secretion of pro-inflammatory cytokines, and tumour cell lysis for Ewing sarcoma and neuroblastoma in vitro. TM enrichment and rapid elimination at the tumour site were studied via PET imaging in vivo in a neuroblastoma mouse model to confirm the TMs fulfilled all prerequisites [74]. As a proof of concept, the same group used NK-92 cells as an alternative to the CAR T-cells with the same UniCAR strategy to target GD2 on neuroblastomas in vitro and in vivo. NK cells boast a lower risk of toxicities. In addition to using scFv 14.G2a fused with the UniCAR epitope, they studied varying half-life of TMs, by also generating an IgG4-Fc-fused scFv 14.G2a and UniCAR epitope. Specific killing of GD2-expressing cells in vitro and in vivo was observed, along with increased production of interferon-γ. With the help of radiolabelled proteins, it was demonstrated that the IgG4 Fc region and homodimerisation of the TM molecule increased the in vivo half-life dramatically [75].
Previously, first-generation CAR NK92 cells were also generated using scFv ch14.18 targeting GD2 on neuroblastoma, melanoma, and breast cancer cells. Specific targeting and cytotoxicity against neuroblastoma cells initially resistant to parental NK-92 was established. Targeting was blocked by GD2-specific mAb or an anti-idiotypic mAb occupying the CAR's cell recognition domain. Additionally, enhanced cytotoxicity of CAR NK-92 targeting GD2 was found against primary neuroblastoma cells as well as GD2-expressing melanoma and breast cancer cells, offering potential clinical utility of retargeted effector cells [76]. Several GD2-targeting CAR T-cells reached clinical trials and the results of three phase I clinical trials for children with neuroblastoma were published. First-generation CAR T-cells comprising autologous activated T-cells and Epstein Barr-virus specific T-cells were both modified to express a CAR construct containing the scFv 14.G2a (NCT00085930). The treatment did not induce neurotoxicity and lead to complete remission in 3/19 patients, which has persisted for 2 patients for at least four years [77]. A third generation CAR T containing scFv 14.G2a and an inducible caspase 9 suicide switch was tested in neuroblastoma patients (NCT01822652). Patients were divided in three cohorts: CAR T alone, CAR T with preparative lymphodepletion and CAR T with lymphodepletion and a PD-1 inhibitor. The lymphodepletion resulted in increased CAR T expansion. PD-1 inhibition did, however, not result in enhanced expansion or persistence. The treatments were safe and lead to a modest antitumour activity after six weeks [78].
1RG-CART, a second-generation CAR with scFv KM8138 was tested in children with relapsed/refractory neuroblastoma (NCT02761915). Three out of six patients receiving high doses (≥108/m2 CAR T-cells) in combination with preparative lymphodepletion showed antitumour activity in sites of disease, without on-target off-tumour neurotoxicity. Nonetheless, the effect was transient and modifications to enhance T-cell persistence are required [79]. In another study, the fourth generation 4SCAR-GD2 containing the scFv hu3F8 and an inducible caspase 9 suicide switch was tested (NCT02765243). Tumour growth was delayed, and patients' survival was prolonged without neurotoxicity. Nevertheless, the 4SCAR-GD2 T-cells expanded when the disease relapsed, indicating other factors at play determining treatment efficacy [80]. Clinical trials based on GD2-targeting CAR T-cells are listed in Table 1.
GD2-targeting CAR T-cells in clinical trials.
| Identifier | Phase | scFv | Generation | Disease | Age | (Estimated) completion |
|---|---|---|---|---|---|---|
| NCT01460901 | I | 14.G2a | 1 | Neuroblastoma | 1.5-17 | January 2015 |
| NCT02107963 | I | 14.G2a | 3 | GD2+ solid tumour | 1-35 | January 2017 |
| NCT02919046 | I/II | 14.G2a | 3 | Neuroblastoma | 1-14 | September 2020 |
| NCT02992210 | I/II | 4 | GD2+ solid tumour | 1-65 | December 2020 | |
| NCT03356795 | I/II | Cervical cancer | 18-70 | December 2020 | ||
| NCT02761915 | I | KM8138 | 2 | Neuroblastoma | 1+ | August 2021 |
| NCT02173093 | I | 4 | Neuroblastoma | 1-29 | December 2022 | |
| NCT02765243 | I | Hu3F8 | 4 | Neuroblastoma | 1-14 | December 2022 |
| NCT03423992 | I | Glioma | 18-70 | January 2023 | ||
| NCT00085930 | I | 14.G2a | 1 | Neuroblastoma | < 21 | June 2023 |
| NCT04637503 | I/II | 4 | Neuroblastoma | 1-65 | December 2023 | |
| NCT03356782 | I/II | hu3F8 | 4 | Sarcoma, osteoid Sarcoma and Ewing Sarcoma | 1-75 | December 2023 |
| NCT04433221 | I/II | Sarcoma Osteoid Sarcoma Ewing Sarcoma | 1-75 | December 2023 | ||
| NCT04430595 | I/II | 4 | Breast cancer | 18-75 | December 2023 | |
| NCT04429438 | I/II | 4 | B cell lymphoma | 0.5-75 | December 2023 | |
| NCT04539366 | I | 14.G2a | 3 | Neuroblastoma and osteosarcoma | <35 | August 2024 |
| NCT05438368 | I/II | (bispecific) | GD2+ and CD70+ solid tumour | 1-75 | June 2026 | |
| NCT05437315 | I/II | 4 (bispecific) | GD2+ and PSMA+ solid tumour | 1-75 | June 2026 | |
| NCT05437328 | I/II | 4 (bispecific) | GD2+ and CD56+ solid tumour | 1-75 | June 2026 | |
| NCT03373097 | I/II | 14.G2a | 3 | GD2+ solid tumour | 1-25 | December 2027 |
| NCT01822652 | I | 14.G2a | 3 | Neuroblastoma | All | October 2030 |
| NCT03294954 | I | 14.G2a | 2 (NK cells) | Neuroblastoma | 1-21 | August 2034 |
| NCT01953900 | I | 14.G2a | 3 | Neuroblastoma and sarcoma | All | October 2034 |
| NCT05298995 | I | 3 | GD2+ CNS tumour | 0.5-30 | May 2037 | |
| NCT03635632 | I | 4 | GD2+ solid tumour | 1-74 | December 2038 | |
| NCT04099797 | I | 14.G2a | 4 | GD2+ brain tumours | 1-21 | February 2038 |
| NCT03721068 | I | 14.G2a | 4 | Neuroblastoma and osteosarcoma | 1.5-18 | June 2039 |
| NCT04196413 | I | 14.G2a | Diffuse intrinsic pontine glioma and spinal diffuse glioma | 2-30 | July 2042 |
Disialoganglioside GD3
Another highly relevant ganglioside TACA is GD3 (Neu5Acα2-8Neu5Acα2-3Galβ1-4Glcβ1Cer), which, like GD2, is found in cancers of neuroectodermal origin [81]. Its normal expression pattern is mainly restricted to high levels in embryonic neural stem cells [82] and low levels in melanocytes, retinal pigment cells and the central nervous system (CNS) [83, 84]. GD3 is highly overexpressed in melanoma, astrocytoma, medulloblastoma, meningioma, neuroblastoma, lung, and breast cancer, while 9-O-acetyl-GD3 is overexpressed in lymphoblasts of childhood acute lymphoblastic leukaemia [81]. The use of a small Ab fragment targeting GD3 for cancer therapy is limited to the application of the scFv from MB3.6 murine mAb in CAR. A first-generation CAR [85] and later a second-generation CAR was created [86]. Following treatment with the second-generation CAR in combination with IL-2, a remission rate of 50% was observed in a subcutaneous melanoma mouse model. In addition, the CAR T-cells could substantially reduce the tumour burden in a genetic mouse model of tuberous sclerosis complex (TSC). TSC is a genetic disease that causes benign tumours to grow. The tumours mostly affect the brain, skin, kidneys, heart, and lungs [87]. To date, there is no cure for TSC, rendering anti-GD3 CAR T a valuable therapeutic option.
Kotlan et al. reported a method that can aid in biomarker identification and simultaneously generate human-derived scFvs against an antigen. Tumour infiltrating B (TIL-B) cells were isolated from metastatic melanoma, and following RT-PCR, the variable domains were cloned in the scFv format to generate an scFv library. Following phage display and binding evaluation to melanoma cells, binders were found for GD3 and its O-acetylated form. The presented method can provide means to obtain tumour-specific Abs of human origin. The study provides evidence for a two-way regulation mechanism between TIL-B cells and tumour cells and opens a way to potentially new cancer treatment strategies [88].
N-Glycolylneuraminic acid GM3
The N-Acetylneuraminic acid (NeuAcGM3) ganglioside is normally detected in normal human tissues. However, N-glycolylneuraminic acid (NeuGcGM3) ganglioside is a TACA found in various tumours, including lung cancer, breast cancer, neuroblastoma, pancreatic cancer, and gastrointestinal tumour [89]. As healthy cells are incapable of synthesising Neu5Gc due to a deletion in the CMP-NeuAc hydroxylase gene, the current hypothesis to explain the presence of NeuGcGM3 on malignant cells is attributed to incorporation from dietary sources due to altered metabolism and hypoxia [89]. Due to the low expression in normal tissue, NeuGcGM3 is an attractive therapeutic target candidate. However, a close structural homologue, N-acetyl GM3 is widely expressed in the body and only differs from N-glycolyl GM3 by a single oxygen atom. The murine mAb 14F7 targets NeuGcGM3 specifically with no cross-reactivity with NeuAcGM3 [90]. Therefore, Roque-Navarro et al. generated Fabs of 14F7 and showed, that while 14F7-Fab does not induce cytotoxicity in murine lymphocytic leukaemia, F(ab')2 retains the induction capacity of the full-length Ab, possibly due to cross-linking of carbohydrate epitopes. The novel mechanism of NeuGcGM3-mediated 14F7-induced cell death accompanied by cellular swelling, membrane lesion formation, and cytoskeleton activation, suggested a complement-independent phenomenon, and demonstrated the therapeutic usefulness of NeuGcGM3 for Ab-based cancer immunotherapy [91]. The generation of an scFv from 14F7 by fusing the original hybridoma variable regions did not yield a functional Ab fragment. Light-chain shuffling trying various combinations of the original 14F7 VH region with various naïve VL regions followed by phage display yielded various scFvs with affinities ranging from KD = 15-38 nM, thus retaining the affinity and specificity of 14F7 (KD = 25±4 nM) [92].
Globo family
The Globo family shares the core structure Galα1-4Galβ1-4Glcβ-Cer, and the TACAs in this family include Stage Specific Antigen 3 ((SSEA-3) also called GB5), Globo-H (SSEA-3b), and SSEA-4. The last two have received the greatest attention as candidate targets. Globo-H and SSEA-4 are both synthesised from SSEA-3, and differ only by their terminal moiety, which is fucose or sialic acid, respectively [93].
Globo-H
Globo-H (Fucα1-2Galβ1-3GalNAcβ1-3Galα1-4Galβ1-4Glcβ1-Cer) is overexpressed in a wide variety of epithelial cancers, including breast, gastric, lung, ovarian, endometrial, pancreatic, and prostate cancers. Globo-H has been shown to promote immunosuppression, angiogenesis, and metastasis of tumours [15]. During normal development, Globo-H is abundantly expressed in undifferentiated embryonic stem cells (ESCs) and is lost after differentiation [94]. Later, in normal differentiated tissue, it is only moderately expressed in apical epithelial cells at the lumen borders of glandular tissue, which are inaccessible to the immune system [15]. Hence, Globo-H is a promising candidate for immunotherapies. Most research regarding Globo-H therapeutics is focused on the development of active immunotherapy, however, one mAb targeting Globo-H, OBI-888, is currently being tested in phase I/II clinical trials for locally advanced or metastatic solid tumours with sufficient target expression (NCT03573544). In addition, OBI-999, an ADC composed of a Globo-H-specific mAb and the anti-mitotic agent monomethyl auristatin E, is also being tested in phase I/II clinical trials (NCT04084366). So far, only one small Ab fragment was described against Globo-H, a Nb termed GH46 that we produced through immunisation of an alpaca with synthetic Globo-H conjugated to a carrier protein [41]. Although highly specific to its target, the monovalent GH46 exhibited a relatively low affinity to Globo-H. Nevertheless, a trivalent construct of the Nb showed a nine-fold improved affinity (apparent KD from 18 ± 8 μM to 2 ± 1 μM) through improved avidity on breast cancer cells [41].
SSEA-4
Like Globo-H, SSEA-4 (Siaα2-3Galβ1-3GalNAcβ1-3Galα1-4Galβ14Glcβ1-1Cer) is expressed in ESCs. Previously, it was believed that the expression is strictly limited to the embryonic stage, and that expression in adults is limited to malignant tissues. Several examples of such malignancies include malignant glioma cells, breast cancer, ovarian cancer, teratocarcinoma, lung cancer, and prostate cancer. In cancer cells, SSEA-4 promotes invasion and metastasis by mediating loss of the cell-cell interactions and the gain of a migratory phenotype. SSEA-4-expressing cancer cells often display high levels of stem cell-specific markers [93]. Therefore, SSEA-4 seemed an excellent TACA to target for anti-cancer therapy. However, SSEA-4 is in fact expressed in healthy adult tissues, namely in the mesenchymal stem cell population in bone marrow [95] and in germ cells in testis [96], and ovary [97]. Probably owing to this, so far, no agents targeting SSEA-4 have entered clinical trials. Nevertheless, efforts are being made towards developing SSEA-4-targeted CAR T-cell therapy. Pfeifer et al. generated a second-generation CAR using an scFv derived from the SSEA-4-specific mAb REA101 (Miltenyi Biotec). The CAR T-cells could inhibit triple-negative breast cancer cells in vitro and in vivo in a xenograft mouse model. However, on-target/off-tumour toxicity was observed, due to the co-targeting of hematopoietic multipotent progenitor cells in the bone marrow and pluripotent epithelial cells in the lungs expressing SSEA-4 [98, 99]. On the other hand, Lin et al. generated a second-generation CAR with the scFv of a newly developed humanised murine mAb hMC48. The CAR T-cells could inhibit pancreatic cancer cells in vitro and in vivo in a xenograft mouse model without inducing any toxicity [100].
Lewis blood antigens
The blood type is determined by the specific oligosaccharide structures found on various cells, such as red blood cells, platelets, leucocytes, plasma proteins, certain tissues, and various cell surface enzymes. These glycans also exist in soluble form in body secretions such as breast milk, seminal fluid, saliva, sweat, gastric secretions, urine as well as amniotic fluid, and their composition is defined by the expression of specific glycosyltransferase enzymes. Since the discovery of blood antigens in the early 20th century, over 30 blood group systems have been described [101]. One of these systems concerns the Lewis blood antigens. The Lewis antigens can be carried on GSLs, as well as N- and O-glycans [102]. The antigens are primarily involved in cell adhesion and cell signalling during embryogenesis and later development. Aberrant expression of Lewis antigens correlates with malignant transformation and poor prognosis. Therefore, they constitute an attractive class of TACAs. The Lewis antigens are all structurally related, in which three monosaccharide units are present: N-acetylglucosamine, galactose, and fucose. The different glycosidic bonds can give rise to different Lewis antigens (Galβ1-3GlcNAc in type I and Galβ1-4GlcNAc in type II Lewis antigens). The TACAs that have been targeted using small Ab fragments include Lewis Y (LeY) and Lewis X (LeX), as well as the sialylated Lewis antigens sialyl-Lewis A (sLeA) and sialyl-Lewis X (sLeX) [103].
Lewis Y
LeY (CD174) is a difucosylated tetrasaccharide (Fucα1-2Galβ1-4Fucα1 -3GlcNAc). In healthy tissue, LeY is mainly found in several epithelial tissues and weakly on granulocytes. Overexpression of LeY has been reported for breast, gastrointestinal, pancreatic, ovarian, and prostate cancers, as well as hepatocellular carcinoma, and non-small-cell lung carcinoma [103]. In addition, the upregulated expression of LeY on endothelial cells contributes to tumour vascularisation and leucocyte recruitment to inflamed tissue [104].
I. Lewis Y-targeting immunotoxins
Since the early 1990s, efforts have been made to generate LeY-targeting therapies using small Ab fragments. The older therapeutics that were primarily developed were immunotoxins. The first immunotoxin targeting LeY using a small Ab fragment is LMB-7. It comprises of an scFv derived from the murine mAb B3 and a truncated form of Pseudomonas exotoxin A, which inhibits protein synthesis. Later, a disulfide stabilised version was generated and was called LMB-9. Both immunotoxins showed a binding affinity of KD = ~1.5 nM [105] and were tested in phase I clinical trials. LMB-7 was tested in patients with leptomeningeal metastases (NCT00003020) and LMB-9 in patients with advanced colon, breast, non-small cell lung, bladder, pancreatic, or ovarian cancer (NCT00005858). Patients experienced renal and gastrointestinal toxicities, due to the expression of LeY on normal gastric cells and some tubular cells in the kidney. The latter could be prevented by blocking acid secretion in the stomach, however renal toxicity remained dose limiting. Therefore, no significant anti-tumour activity could be observed [106].
A similar immunotoxin was generated by conjugating a truncated Pseudomonas exotoxin to Ab fragments derived from the mouse-human chimeric IgG BR96. First, the Fab formats (Fab', and F(ab')2) were compared to the full-length IgG immunotoxin conjugates. Although internalisation was similar, the Fab' immunotoxin was considerably less cytotoxic than F(ab')2 and IgG. This was believed to be a result of loss of avidity through the monovalent nature of Fab' [107]. Nevertheless, later the toxin was genetically fused to an scFv derived from BR96 such that the construct was specific for LeY, although with 5-fold less binding compared to the mAb BR96. Interestingly, the scFv-based toxin was 4-fold more potent than the intact IgG-immunotoxin construct on LeY-positive MCF-7 cells [108]. The scFv immunotoxin is approximately one-third of the size of the IgG immunotoxin, resulting in four times shorter blood half-life time and enhanced tumour penetration in breast and lung xenograft mice models [109]. BR96 scFv-PE40 requires no chemical conjugation and can be produced entirely in bacteria relatively rapidly and inexpensively [108]. BR96 scFv-PE40 was named SGN-10 and entered phase I clinical trials for patients with different types of LeY-positive metastatic carcinomas. About one-third of the patients showed no signs of disease progression, indicating a favourable outcome. However, the treatment was hampered by human antitoxin Ab (HATA) and toxicity responses due to the expression of LeY on gastrointestinal epithelial cells [110].
Harmful HATA responses can be bypassed using human toxins. A generated immunotoxin (KD = ~0.45 µM) composed of an scFv derived from the LeY-specific mAb B1 and tumour necrosis factor α (TNF-α), a protein that can induce cell death upon binding to cell surface receptors, was specifically cytotoxic in vitro and in vivo in TNF-α-sensitive MCF-7 human breast cancer cells xenograft mice [111].
II. Lewis Y-specific multimers
To generate enhanced anti-LeY fragments, Rheinnecker et al. developed an scFv derived from MSL5 (murine anti-LeY IgM). Self-assembling multimers were generated by fusing the scFv sequence to either an artificial dimerisation domain or the tetramerisation domain of the human transcription factor p53. The monomeric scFv showed specific but weak binding to LeY on surface plasmon resonance (SPR). In contrast, the dimeric and the tetrameric scFv exhibited an eight-fold and a twenty-fold increase in binding through increased avidity [112]. Power et al. generated a diabody against LeY by altering the linker length between VH and VL in an scFv. By using a five-residue linker in the scFv derived from humanised mAb hu3S193, the generated construct was pushed to form a non-covalent bivalent diabody. The diabody with a molecular weight of 54 kDa demonstrated similar binding activity as the parent mAb on SPR as well as in MCF-7 cell binding assays. After radiolabelling the diabody, rapid tumour uptake and fast blood clearance were observed in an MCF-7 xenograft mouse model. The LeY-specific diabody proved to be a promising candidate for tumour imaging [113]. The 111In-radiolabelled C-functionalised trans-cyclohexyl diethylenetriaminepentaacetic acid chelated diabody was compared to F(ab')2 (generated with pepsin digest of mAb hu3S193) in terms of affinity and biodistribution in breast carcinoma xenograft mice. The affinity of the diabody was approximately 10-fold lower than the intact mAb. The F(ab′)2 displayed a somewhat higher affinity than the diabody, as well as increased stability. The results demonstrate that the diabody is more effective as a diagnostic imaging tool, whereas F(ab')2 is better for tumour targeting [114]. Obtaining multimers by varying the linker length was explored to generate other multimers. The scFvs a linker that is extended by three residues length formed scFv dimers, whereas decreasing the length or completely removing the linker formed scFv triabodies and tetrabodies. Expressed in Escherichia coli, all scFv formats gave active protein [115]. The affinity of the hu3S193 multimers expressed as Ka was 158 nM, which is slightly higher, compared to hu3S193 mAb. On the other hand, the affinity of hu3S193 F(ab′)2 was 4.31 µM, which is approximately 10-fold lower. Further experiments showed maximum tumour uptake for the scFv trimer/tetramer mixture within six hours and for the F(ab')2 within 24 hours in MCF-7 xenografted mice. Despite the tumour targeting, however, high in vitro instability and renal uptake prevented the scFv multimer mixture from being used in radioimmunotherapy and imaging [116].
III. Lewis Y-targeting CAR T-cells
Targeting LeY with CAR T-cells was first described for CAR targeting LeY used an scFv derived from the MluC1 mAb as antigen-binding domain. The scFv was linked to the γ-chain of the Fc receptor and transduced to cytotoxic T-cells. Even though the binding ability of the soluble scFv was low, seven of thirteen genetically engineered cytotoxic T lymphocyte clones inhibited the growth of LeY-positive cells in vitro [117]. More advanced, second-generation CAR T-cells were generated using the scFv derived for the first time from a humanised mAb, hu3S193, targeting LeY. The engineered T-cells inhibited subcutaneous human ovarian OVCAR-3 tumours, which are recognised as a difficult model to treat, in xenograft mice. Normal tissue cells expressing lower amounts of antigen were not harmed by T-cells that were redirected towards them. The antitumour response was not inhibited in presence of excess numbers of normal LeY+ cells [118]. Based on these findings, the tolerability and potential therapeutic effect of anti-LeY CAR T-cells were examined in a phase I clinical trial with four patients suffering from acute myeloid leukaemia. While CAR T-cell infusions were generally tolerated well and the CAR T-cells accumulated and persisted in the bone marrow and the spleen, they failed to impair tumour progression in patients [119]. Since LeY has generally higher expression on epithelial tumours, another phase I clinical study is currently assessing the tolerability and potential anti-tumour activity of anti-LeY CAR T-cells in patients with advanced solid tumours (NCT03851146).
Lewis X and sialyl-Lewis X
LeX (SSEA-1/CD15) is a trisaccharide (Galβ1-4(Fucα1-3)GlcNAc) that is involved in embryonic development, particularly of the central nervous system (CNS). In addition, it is a typical antigen found on myeloid cells, while also expressed in normal tissues of the stomach, colon, salivary glands, kidneys, bladder, epididymis, uterus, cervix, and medulla, and weakly in several other tissues. LeX is overexpressed on various types of cancer cells, including both haematological malignancies (Hodgkin's lymphoma) and solid tumours (colorectal, thyroid, urological, lung, breast, and oral cancer, as well as hepatocellular carcinoma and glioma). Moreover, it is a ligand for selectins and thereby mediates adhesion to endothelial cells and transendothelial migration. Overexpression of LeX on cancer cells has a pivotal role in metastasis [103].
Sialylation of LeX forms the tetrasaccharide sLeX (NeuAc-Galβ1-4(Fucα1-3)GlcNAc). sLeX is expressed on neutrophils and monocytes, allowing for extravasation to sites of inflammation via their interaction with selectins [103]. In contrast to LeX, sLeX can also be present in serum in soluble form, where it can compete for interactions with selectins and is, therefore, correlated with favourable prognosis [120]. However, overexpression on the cancer cell surface is correlated with poor prognosis. sLeX overexpression has been reported in gastrointestinal, bladder, prostate, lung, and breast cancer [103].
ScFvs are generally obtained from murine sources. To obtain completely human scFvs targeting LeX and sLeX, a phage-display library derived from the peripheral blood lymphocytes of 20 patients of various cancer types to select and isolate scFvs specific to the carbohydrate antigens was generated. At least four different scFv genes could be obtained that have KD values ranging from 0.11 nM to 0.62 nM, which are comparable to the affinities of the mAbs. The isolated scFvs showed specificity for LeX and sLeX on pancreatic adenocarcinoma cells. This was the first time the technique was employed to generate human Ab fragments against TACAs directly [121]. In another study, the two human scFvs, LeX1 and sLeX10, selected from the n-CoDeR phage display library and targeting LeX and sLeX, respectively, were shown to bind rapidly and specifically to their antigens on SPR. LeX1 and sLeX10 showed relatively low KD values of 35 ± 7 μM for LeX and 26 ± 7 μM for sLeX, respectively [122].
Sialyl-Lewis A
sLeA (CA19.9/CD43) is a tetrasaccharide (Neu5Acα2-3Galβ1-3(Fucα1-4)GlcNAc) that is normally expressed in low levels on fibroblasts and on luminal and glandular epithelium [103]. However, sLeA expression is particularly elevated in gastrointestinal cancers. Notably, sLeA is the only FDA-approved biomarker for pancreatic cancer [123]. Disialyl-Lewis A (LeA), sLeA with an additional sialic acid, is commonly expressed on non-malignant epithelial cells and is a ligand for immunosuppressive receptors. During malignant transformation, the overexpression of sLeA originates from the incomplete synthesis of LeA. Hence, the aberrant expression contributes to cancer progression in two ways: the loss of LeA contributes to tumour-promoting inflammation, and gain of sLeA allows binding to selectins, like LeX and sLeX [124].
F(ab')2 prepared from the murine SA23.2 IgM mAb bound more strongly to sLeA-positive colon adenocarcinomas, pancreatic, stomach, and lung cancers, and showed much less unspecific binding than the original IgM [125]. As a follow-up, BsF(ab')2 from SA23.2 IgM and anti-carcinoembryonic antigen (CEA) IgM was generated via disulfide bond exchange. The BsF(ab')2 had almost the same affinity (KD = 0.17 μM) as the parent F(ab')2 construct (KD = 0.18 μM). While the size and low solubility of an IgM restrict its number of applications, the BsF(ab')2 format might be a useful immunotherapy and -diagnosis tool [126]. A radiolabelled diabody from the murine mAb 1116-NS-19-9, with an apparent affinity of KD < 3 nM, can be conjugated to nanoparticles and was suitable for PET imaging in a pancreatic cell xenograft mouse model [127]. The diabody was conjugated to liposomal nanoparticles that specifically target sLeA-expressing pancreatic cells in vitro as well as xenografted in mice, indicating the potential of the diabody to deliver targeted treatment [128]. Human 5B1 (MVT-5873), another mAb against sLeA is currently being tested in phase I clinical trials in patients with advanced pancreatic cancer or other sLeA positive malignancies (NCT02672917). Borenstein-Katz et al. used sequences of both mAbs, 5B1 and 1116-NS-19-9, to generate scFvs using yeast surface display. These were used in return to generate a mutated version of 1116-NS-19-9 with 10-fold increased affinity and computer-assisted sequence optimisation [42]. An scFv derived from 5B1 was used to develop a sLeA-targeting CAR T-cell therapy. The 5B1-based CAR T-cells were tested in vivo in an orthotopic pancreatic cancer mouse model in combination with low-dose radiation. The radiation was used to sensitise cancer cells to activated CAR T-cells. Remarkably, sLeA- cancer cells were also eliminated upon immune activation by sLeA+ cancer cells. Thus, the therapy is promising for targeting heterogenous solid tumours [129].
Glycoproteins
Truncated O-glycans
Aberrant O-glycan structures in mucins and other glycoproteins are abundantly found in many carcinomas and associated with cancer aggressiveness as well as poor prognosis. Their expression directly affects oncogenic parameters such as cancer cell proliferation, motility, and invasion. O-glycan TACAs include the Thomsen-nouveau antigen (Tn or CD175, GalNAcα-O-Ser/Thr) and its sialylated structure sTn (CD175s/CA72-4), as well as Thomsen-Friedenreich antigen (TF/T antigen or CD176, Galβ1-3GalNAcα-O-Ser/Thr) with its mono- and di-sialylated forms [130]. Truncated O-glycans are among the most prevalent TACAs and have been a target for mAb development for almost 40 years. Although considered poorly immunogenic [131], various Abs and Ab fragments with different affinities and specificities were developed against Tn, sTn, and TF antigens over the years.
Thomsen-nouveau antigen
The Tn antigen is expressed in many human carcinomas such as breast, prostate, bladder, lung, colon, and stomach. Anti-Tn mAbs have shown promising results in pre-clinical trials. However, low binding specificities and a broad tissue distribution of the Tn antigen still prevent their testing in clinical trials [131]. Most scFvs against the Tn antigen were adapted from successful mAbs that show specific Tn antigen-binding properties and tumour cell growth inhibition. The Tn antigen, consisting of GalNAc, is too small for stable binding by Abs, and therefore Tn-antigens are generally recognised as two (Tn2) or three (Tn3) consecutive Tn-antigens or the Tn-antigen in combination with the peptide backbone [132]. Hence, Sakai et al. used synthetic Tn3 for panning and screening of a phage library to generate scFvs. The generated scFvs, 4E10, and 4G2 bound Tn3 with KD values of 0.37 μM and 0.14 μM, respectively [133]. Other scFvs generated against Tn include 83D4-scFv [134], G2-D11 (Tn2) (KD = 13.3 nM - 23.4 nM), sH1 [135], MLS128 scFv (KD Tn2 = 5.9 μM; KD Tn3 = 1.1 μM) [136], H5, G2-H7 [137], as well as 3-9 and 3-18 [138]. None of the described scFvs have been incorporated into Tn-targeted therapeutics. Many Tn/sTn-mucin 1 Abs have been generated that recognise not only the glycan, but a glycopeptide epitope. Efforts towards targeted therapies using such epitopes fall outside the scope of this review.
Sialyl-Thomsen-nouveau antigen
Sialylation of the Tn antigen to form sTn is due to overexpression of the ST6GalNAc I enzyme, and its aberrant localisation in the Golgi. Sialyl Tn has been reported to be overexpressed in breast, gastric, pancreatic, ovarian, and bladder cancer [130]. The first-generation of anti-sTn mAbs was developed in the 1980s using a variety of sTn-expressing immunogens. A comparison of Abs such as B72.3, TKH2, HB-STn1, and MLS102 gave conflicting results as they differ significantly in binding among different sTn-expressing cancer tissues [139]. The development of glycan arrays contributed to defining sTn specificity for mAbs as, L2A5 [140], and the humanised 2G12-2B2-L0H3 [141].
Several sTn-specific scFvs were adapted from B72.3, the first mAb developed against sTn [142, 143] on tumour-associated glycoprotein 72 (TAG-72) [144] using enriched membrane fractions of a breast carcinoma biopsy as immunogen [145]. Muraro et al. generated CC49 using purified TAG-72 isolated from colon carcinoma xenografted mice as immunogen. One of the purification steps for TAG-72 included affinity purification with B72.3 [146]. Different formats including intact radiolabelled IgG, Fab, F(ab')2 and scFv were generated, and then compared their efficacy against colon carcinoma xenograft extracts in vitro and xenografted tumours in vivo in mice and rhesus monkeys [147]. The mAb CC49 and its derived F(ab')2 displayed similar relative dissociation constant, whereas for the scFv and the Fab' they were 8-fold and 7.4-fold lower, respectively. The radiolabelled scFv tracer was tested successfully in a clinical trial in five patients with metastasising colorectal carcinoma using single photon emission computed tomography (SPECT) and whole-body imaging. The tracer was cleared from the blood with a biphasic clearance t1/2 of 30 minutes and 10.5 hours. The tumours were visualised in all patients in both primary and metastatic lesions. Although the image quality was suboptimal, the administration was safe and allowed imaging on the same day [148]. In a similar preclinical study, the Colcher group generated Ab fragments derived from CC49, including scFv, diabody, Fab' and F(ab')2, and radiolabelled them. The scFv monomer had a 25 amino acid-helical linker, which allowed for non-covalent dimerisation to the diabody. The KA for the mAb CC49, dimeric scFv and monomeric scFv were 1.7, 1.99, and 0.52 nM by Scatchard analysis and 11.4, 446, and 150 nM, respectively, by BlAcore analysis. Thereafter, pharmacokinetic, biodistribution, and tumour targeting characteristics were compared in colon as well as pancreatic cancer xenograft mice. The stable scFv dimer demonstrated a two-fold higher tumour uptake than the monomeric scFv and Fab' as well as longer retention time and, therefore, was found to have the ideal characteristics for therapeutic applications [149]. Following this, the group also showed that a tetravalent CC49 scFv format improved avidity and biological half-life. The KA value for the tetravalent and mAb were similar, 0.102 and 0.114 nM respectively. In addition, the KA were 4-fold higher than its divalent scFv (275 nM) [150]. A CC49-derived diabody was PEGylated and radiolabelled for tumour positron emission tomography (PET) imaging. The construct, PEG-AVP0458, was tested in vivo in colon cancer xenograft mice [151] and in clinical trials in patients with relapsed/metastatic prostate or ovarian cancer. This is the first-in-human clinical trial of a diabody, demonstrating the safety and feasibility of using diabodies for tumour imaging [152]. Yang et al. generated a tetravalent scFv using CC49 and fused it to streptavidin for pre-targeting of Jurkat leukemia cells in vitro and in xenografted mice. The biotin-functionalised nanoparticles could then target the tetravalent scFv and, through it, the leukemia cells. Compared to nanoparticles only, the targeting of TAG-72 carrying leukemia cells was much higher with this strategy and presented a potential way for drug delivery to cancer cells [153]. Roberge et al. also constructed an scFv of CC49 for therapeutic application. The VH and VL sequences were mutated to enhance stability, and the resulting scFv was fused to β-lactamase (BLA). BLA can be used as an enzyme to catalyse the activation of prodrugs in a treatment called antibody-dependent enzyme prodrug therapy (ADEPT) [154]. The fusion protein named TAB2.5 was tested in combination with the prodrug GC-Mel in colorectal cancer xenograft mice. Cleavage of GC-Mel by BLA releases the chemotherapeutic drug melphalan. The treatment was effective, but additional optimisations of TAB2.5, the dosing regimen, and the mouse model were proposed [155]. Chan et al. generated a different type of CC49 scFv drug conjugate by fusing the scFv to the extracellular cytotoxic domain of CD178 (FasL). The fusion boasted 30.000-fold higher cytotoxicity compared to soluble FasL in vitro and cured mice with intraperitoneally implanted lymphoma cells [156].
An scFv derived from humanised and mutationally improved 3E8 counterpart of CC49 (KD = ~12 nM) was conjugated with IR800 to generate a tool for optical surgical navigation (OSN), which can help surgeons to resect tumours more accurately. The generated OSN agent was validated in a human orthotopic colon adenocarcinoma mouse model [157]. The same group later engineered the scFv linker to optimise various properties. The best construct, 3E8.G4S with a low apparent KD of 3.6 nM, resulted in the formation of various oligomeric states: 47% dimer, 42% trimer, and 11% tetramer. In addition, 3E8.G4S was shown to be an effective delivery vehicle in PET imaging of TAG-72-positive colorectal tumours in xenograft mice [158].
Anti-sTn scFvs have also been used as CARs in CAR T-cell therapy. Murad et al. reported a significant reduction in peritoneal ovarian tumour growth and extended overall survival of mice treated with sTn-binding second-generation CAR T-cells based on CC49. However, in early recurring tumours, reduction in expression of TAG-72 was observed, explaining decrease in T-cell persistence [159]. On the other hand, results from a long-term first-generation CAR T-cell scFv huCC49 clinical trial against metastatic colorectal cancer proved disappointing. Although proven as relatively safe with minimal on target/off tumour toxicity, no clinical responses were observed [160]. The reason of failure could be that the huCC49 scFv has 23- to 30-fold lower affinity compared to murine CC49, and, therefore, compromised the efficacy. A second-generation CAR T with a deimmunised version of murine CC49 scFv with high and specific tumour uptake and lower immunogenicity was generated. The CAR T-cells carried an additional binding domain specific for the tumour antigen CD47, albeit without signalling domains, to facilitate enhanced binding and avoid targeting of healthy cells. The dual CAR T-cells were effective in human ovarian xenograft mice [161].
Sharifzadeh et al. generated a CAR T-cell using an anti-TAG-72 Nb as binding domain fused to human CH3 and CH2 with two hinge regions. The CAR T-cell was effective in in vitro co-culture assays with colon adenocarcinoma and breast cancer cells. However, while 13 Nbs were initially generated and shown to bind to TAG-72 with nanomolar affinity, they were hypothesised to target different epitopes of TAG-72. Therefore, it is unclear whether the CAR T-cells bind a glycan, peptide, or glycopeptide epitope [162]. In another attempt to generate sTn-targeting CAR T-cells, an scFv derived from L2A5 was used to redirect novel engineered T-cells to tumour cells in the UniCAR T strategy. The authors reported effective eradication of malignant sTn-expressing cells both in vitro in metastatic breast and bladder carcinoma as well as in a mouse model [163]. To extend the half-life of the TM, which was initially composed of the scFv L2A5 and the UniCAR epitope only, the hinge region and Fc domain of the IgG4 Ab were added on to the UniCAR epitope. In comparison to the previous smaller-sized anti-sTn TM, the specific binding, functionality, and efficiency of the construct were then assessed on metastatic breast and bladder carcinoma in vitro and breast carcinoma in vivo assays. The construct bound with a KD value of approximately 4 nM in comparison to 57 and 75 nM determined for the former construct. Efficient activation and redirection of UniCAR T-cells to cancer cells in a target-specific and TM-dependent manner as well as the secretion of proinflammatory cytokine and cell lysis were observed. The prolonged serum half-life of the anti-sTn-IgG4 TM presents a promising strategy for retargeting of UniCAR T-cells [164].
Currently, a phase I clinical trial with TAG-72 CAR T-cell therapy is being set up for patients with platinum-resistant epithelial ovarian cancer (NCT05225363).
Thomsen-Friedenreich antigen
The TF antigen is expressed during foetal development and in most human carcinomas. Although located in several healthy tissues, the epitope is masked by its elongated glycosylation that is truncated during malignant transformation. The TF antigen plays a critical role in metastasis through its binding to galectin on endothelial and hepatocyte cells [165].
The first TF-specific scFvs were produced via phage display selection where they focused on creating multimers based on scFvs with shorter linkers. The tetramer displayed the best binding in ELISA and SPR assays, yielding a KD value of approximately 88 nM [166]. They further tested the trimeric and tetrameric scFv constructs for radioimmunotargeting in a breast cancer mouse model and observed fast renal excretion and reduced kidney uptake for an scFv tetramer [167]. As a validation of their previously described strategy [133], Matsumoto-Takasaki et al. also employed phage display screening to identify human scFvs against TF antigen. Pure scFv binds to the TF antigen in ELISA and SPR assays [168]. ScFvs expressed in Drosophila showed specific binding. Furthermore, using STD-NMR, it was shown that the generated scFv 1E8 binds the terminal non-reducing end galactose unit of the TF-antigen [169]. In a follow up study, the scFvs were expressed in E. coli and purified from inclusion bodies and refolded. The scFvs were active and 1E8 (KD = ~83 nM) showed higher affinity than 1E6 (KD = ~1.3 µM) [170]. A TF-specific scFv was discovered by screening phage display libraries derived from TF-antigen immunised mice. The scFv showed binding to TF-positive leukaemia, colorectal, and gastric cancer cells, as well as inhibited adhesion of colorectal and gastric cancer cells to endothelial cells and hepatocytes in vitro [171].
N-glycans
The overexpression of tumour-associated N-glycans can serve as a marker for certain cancers, including most ovarian cancer subtypes [172]. For example, the generation of human scFvs against tumour-specific glycoforms containing bisecting N-glycans of the ECM protein Periostin via yeast display led to functional fragments, of which one (scFvC9) was validated for its tumour specificity in ovarian cancer cells in vitro and xenografted in mice [173]. Such N-glycosylation-specific Ab fragments may be novel agents in cancer diagnosis and therapy. Another N-linked TACA is mannotriose, a trisaccharide composed of the three mannose moieties on the core base of canonical N-glycans. In normal cells, core mannose residues are masked by terminal glycosylation, whereas in cancer cells, they can become exposed due to aberrant glycosylation [174]. Therefore, scFvs against mannotriose were developed as a putative therapeutic tool and tested on breast cancer cells along with anti-Tn scFv [136] in vitro. However, the scFvs (1A4, 1G4 and 5A3 with KD values of 2.5, 1.8 and 3.8 µM, respectively) did not inhibit breast cancer cell growth, and the potential application for cancer therapy still needs to be developed [175].
Proteoglycans
Proteoglycans are major components of the extracellular matrix (ECM) in animal tissues and are composed of a protein part and a glycosaminoglycan (GAG) moiety. GAGs are long linear repeats of disaccharide units that consist mainly of a uronic acid and an amino sugar. An important structural feature of GAGs is the extensive N- and O-sulfation, which plays vital roles in cell-cell and cell-ECM interactions due to its negative charge. Consequently, alterations in sulfation patterns often have adverse effects and are associated with various disorders, including several cancer types [176]. Based on the identity of the disaccharide repeats, GAGs are divided into four groups: heparin/heparan sulfate, chondroitin/dermatan sulfate, keratan sulfate, and hyaluronan. Despite the important roles GAGs play in numerous signalling pathways, Abs and Ab fragments targeting specific GAG epitopes are scarce. This is mainly due to the enormous complexity of the sulfation pattern along the GAGs backbone chain, referred to as the “sulfation code”. The high structural complexity of sulfate groups on the disaccharide repeats prevents the development of molecular tools that target specific GAG epitopes. Therefore, nearly all anti-GAG Abs lack selectivity among the various structures of GAGs and, hence, have broader targets, such as heparan sulfate or chondroitin sulfate. Another limiting factor is access to pure, structurally defined GAGs oligosaccharides for Ab generation and characterisation, as isolation from natural sources is highly challenging, and chemical synthesis is generally slow, costly, and low yielding. Therefore, the importance of GAGs demands new, sulfation-specific Abs that would help to better understand GAGs' biology and the structure-to function of specific sulfation patterns [177].
Heparan sulfate
A predominant class of proteoglycans is heparan sulfates (HS), which are involved in a variety of biological processes, for instance, growth factor signalling and cell adhesion [177]. Changes in the degree and pattern of sulfation of HS are associated with breast, ovarian, colorectal, and gastric cancers, among others. It has been suggested as a biomarker for breast cancer [176]. Aberrant sulfation of HS has been shown to negatively affect the survival of patients with head and neck squamous cell carcinoma [178]. Due to the low immunogenicity of HS, conventional methods to generate anti-HS Abs are very challenging. However, that problem has been overcome by generating HS-specific Ab fragments using human phage display library [179]. Kuppevelt et al. generated scFvs using a synthetic human phage display library against HS from bovine kidney and became the first group to generate scFvs against polysaccharides [180]. The obtained scFvs had KD values of 0.12 and 0.15 µM. The anti-HS scFv library was used to study HS distribution in muscle basal lamina of various species (including human, mouse, and rat) and found the scFv binding to be strongly sulfation dependent. HS differences were found between the neural, synaptic, and extrasynaptic basal laminae, and changes in distribution during muscle development were time- and region-dependent both in vitro (mouse-derived skeletal muscle and mutant Chinese hamster ovary cells) and in vivo (mouse and rat model). The study underlines the importance of HS in skeleton muscular and neural development [181]. Further, by using HS from synthetic and native sources and anti-HS scFvs that bind distinct HS sulfation patterns, the group identified 15 varying HS domains with specific tissue distribution, indicating a tightly regulated topological distribution [182]. Anti-heparin scFvs were also identified and characterised as well as investigated for inhibition of heparin anticoagulant effect. The scFvs could bind heparin on mast cells in human lung cryosection. Some scFvs exclusively bound to heparin, while others also bound to structurally related HS and chondroitin sulfate [183]. Notably, most Ab fragments against HS exhibit a similar affinity to heparin. Because medicinal heparin can have various severe side effects, including heparin-induced thrombocytopenia, agents are needed to block the anticoagulant effect if necessary [184]. Therefore, scFvs providing the required inhibitory effect on anticoagulation could be promising candidates [183]. Even though defined HS structures were used in the studies, the oligosaccharide sequence was largely unknown. Therefore, the group identified a specific scFv (MW3G3) against acharan sulfate, which is a HS-similar snail GAG and has a repeating disaccharide structure of α-d-N-acetylglucosaminyl-2-O-sulfo-α-l-iduronic acid (GlcNAc-IdoA2S) residues. The scFv was then used to identify HS oligosaccharides containing the disaccharide repeats and study their distribution in rat organs [185]. Using another scFv (RB4CD12) that recognises highly sulfated domains (6S-domains) of HS, it could be shown that HS S-domains play a significant role in lung and ovarian cancer progression [186-188]. Recently, one of the well characterised scFvs from the initial synthetic phage display library, HS4C3, which binds strongly to the 3-O-sulfated motif in HS and weakly to any N-sulfated, 2-O- and 6-O-sulfated hexa- to octasaccharide fragment [189], was also used as a template, containing a heparan-binding consensus site in CDR3 to establish another scFv library with a large number of scFvs against HS only differing in the consensus region [190].
Sulfation-specific scFvs, D4A4 and D6B10, were obtained against syndecan-1, an HS proteoglycan associated with multiple myeloma (MM) cells. Both scFvs pulled down syndecan-1 from MM cell lysates as well as competed for binding to cells. D4A4 and D6B10 scFvs may provide help reading changes in sulfation patterns of HS during myeloma tumour progression [191]. Another scFv, HS20, was selected from a phage display library against glypican-3, which is often highly expressed in hepatocellular carcinoma and correlates with poor prognosis. The scFv was, however, converted to a full IgG that showed binding to a Wnt binding domain in the HS structure and inhibited Wnt signalling, suppressing hepatocellular carcinoma cell growth in vitro and in xenografted mice [192].
Chondroitin and Dermatan sulfate
Chondroitin sulfate (CS) plays an essential role in skeletal development and is responsible for 67-97% of total GAG content of bone. As coreceptors in various signalling cascades, the sulfation degree and pattern can greatly affect skeletal development. The sulfation changes can also be associated with cancers such as breast, ovarian, colorectal, prostate, and gastric cancers, among others [176]. Therefore, investigating CS-specific Ab fragments is highly relevant regarding both cancer diagnosis and therapy. With the phage display technique that had previously been used for HS [180], Smetsers et al. used a synthetic human scFv library for the selection of Ab fragments against CS. The scFvs IO3D9, IO3H10, and IO3H12 possess high specificity for CS, whereas IO4C2 also shows cross-reactivity with heparin. While all reacted to CS-A, CS-C, and CS-E, IO3D9 showed higher specificity towards CS-E, which is highly sulfated compared to CS-A and CS-C. Expression and localisation of CS were studied on rat organs and melanoma metastases in situ. These may be valuable tools in detecting alterations in CS patterns in both healthy and malignant tissues [193]. E-units on CS-E and dermatan sulfate (DS) are involved in metastasis of Lewis lung carcinoma cells. The scFv GD3G7 recognises E-units, GlcAβ1-3GalNAc(4S,6S), where 4S and 6S stand for 4-O- and 6-O-sulfate, respectively. Using GD3G7, it was shown that the presence of the E-unit was crucial for the metastatic potential and that the addition of GD3G7 and CS-E decasaccharide fraction (minimal epitope of the scFv) could strongly inhibit metastasis [194]. GD3G7 was also used to test 148 ovarian tumours including benign and malignant for the presence of 4,6-disulfated CS motifs, potentially an ovarian cancer biomarker. 4,6-disulfated CS had significantly higher expression in malignant tumour, and its expression correlated with serous subtype, high tumour grade, advanced FIGO-stage and high CA-125 levels [195]. The scFv GD3A10 was selected in the biopanning against embryonic GAGs as a source of cancer antigen. It specifically targeted the GAG moieties of CS associated with ovarian cancer [196]. In the next study, the binding specificity of scFvs GD3A11 to malignant ovarian tumours was confirmed in large-scale studies (N = 359) on human patient tissues. Unlike with GD3A10, the expression of highly sulfated CS showed no correlation to tumour grade, FIGO stage, and the use chemotherapy. For aggressive ovarian cancer, however, high expression independently predicted poor prognosis [197].
The two scFvs, GD3A12 and LKN1, were also generated against 4/2,4 di-O-sulfated DS, which was found to be expressed in rat thymus and spleen. In ovarian tumours, DS expression was high in the stromal parts, and occasionally on tumour cells. These scFvs may help study the function, expression, and localisation of DS in healthy tissue as well as cancer [198, 199]. GAGs and their sulfation moieties play an important role in the ECM and aberrant alterations including increased sulfation of CS have been proposed as potential cancer biomarkers [176].
Conclusion
Due to their boasted thermo- and chemostability as well as their small size, Ab fragments have been investigated as therapeutics against several diseases targeting different glycan antigens. Access to cryptic epitopes that are out of reach for conventional Abs, including the heavily concentrated tumour microenvironment, as well as high stability of small fragments are major advantages. Moreover, the lack of a Fc portion reduces the risk of side effects as immune effectors such as the complement-dependent cytotoxicity will not be activated.
Ab fragments can be manipulated into being functional in many ways. By selecting a specific format, the pharmacokinetics of the targeting moiety can be adapted for the application. In general, increasing size leads to increased half-life. For imaging purposes, the half-life should be short, whereas it should be longer for therapeutic purposes to exert biological effects. Increasing size decreases tumour penetration. Hence, each fragment has advantages and disadvantages regarding different applications. Prolonging their circulation half-life can be accomplished by PEGylation, fusion with an albumin-binding Ab fragment or multimerisation [200]. Increasing valency can aid in improving the overall affinity for the targets through avidity.
TACA-targeting scFvs have been researched the most compared to any other Ab fragment. The widespread applications such as imaging agents, drug conjugates, BiTES, and CAR-T cells allow for easy multimerisation in the form of taFv and diabodies. The small Ab fragments targeting TACAs that reached clinical trials mostly represent scFvs incorporated in CAR T cells for LeY, sTn, and particularly for GD2. Other treatment modalities still face problems due to toxicity or immunogenicity. Nonetheless, small Ab fragments are particularly suitable for tumour imaging, illustrated by numerous reports of pharmacokinetic properties of fragments in murine xenograft models. The sTn-specific PEGylated and radiolabelled diabody PEG-AVP0458 was the first diabody to reach clinical trials [152]. It was successfully used in PET imaging, emphasising the suitability of using recombinant anti-TACA Ab fragments in cancer diagnosis. The Nb format has been gaining increasing interest since it is more stable and easier to produce and functionalise. Recently published Nbs against Globo-H [41], and GD2 (patent ID: CN110551218B) indicate the ability to produce even smaller antibody fragments targeting TACAs to further innovate cancer diagnosis and therapy.
Acknowledgements
Generous financial support by the Max Planck Society is gratefully acknowledged. This work was supported by the IMPRS on Multiscale Bio-Systems (A.F.), and Deutsche Forschungsgemeinschaft (RTG2046 for F.G. and J.L.). Lastly, the authors wish to thank Ms. Arbel Moscovitz for her beautiful graphical abstract.
Competing Interests
O.M. and P.H.S. are co-founders of Tacalyx GmbH, where P.H.S. is a boardmember. O.M. and P.H.S. have a significant financial interest in the company and field a patent for the Nbs mentioned in the manuscript (EP3799881A1).
References
1. Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M. et al. Essentials of Glycobiology. 4 ed. New York: Cold Spring Harbor Laboratory Press. 2022
2. Heimburg-Molinaro J, Lum M, Vijay G, Jain M, Almogren A, Rittenhouse-Olson K. Cancer vaccines and carbohydrate epitopes. Vaccine. 2011;29:8802-26
3. Livingston PO. Approaches to augmenting the immunogenicity of melanoma gangliosides: from whole melanoma cells to ganglioside-KLH conjugate vaccines. Immunol Rev. 1995;145:147-66
4. Zinkernagel RM. Localization dose and time of antigens determine immune reactivity. Semin Immunol. 2000;12:163-71
5. Goletz S, Cao Y, Danielczyk A, Ravn P, Schoeber U, Karsten U. Thomsen-friedenreich antigen: The ́hiddeń tumor antigen. Adv Exp Med Biol. 2002;535:147-62
6. Dhillon S. Dinutuximab: first global approval. Drugs. 2015:923-7
7. Zahavi D, Weiner L. Monoclonal Antibodies in Cancer Therapy. Antibodies (Basel). 2020
8. Mullard A. FDA approves 100th monoclonal antibody product. Nat Rev Drug Discov. 2021;20:491-5
9. Bates A, Power CA. David vs. Goliath: The Structure, Function, and Clinical Prospects of Antibody Fragments. Antibodies (Basel). 2019;8:28
10. Debie P, Lafont C, Defrise M, Hansen I, van Willigen DM, van Leeuwen FWB. et al. Size and affinity kinetics of nanobodies influence targeting and penetration of solid tumours. J Control Release. 2020;317:34-42
11. Yang EY, Shah K. Nanobodies: Next Generation of Cancer Diagnostics and Therapeutics. Front Oncol. 2020 10
12. Schaller J, Gerber S, Kämpfer U, Lejon S, Trachsel C. Human Blood Plasma Proteins. England: John Wiley & Sons. 2008
13. Adams R, Griffin L, Compson JE, Jairaj M, Baker T, Ceska T. et al. Extending the half-life of a fab fragment through generation of a humanized anti-human serum albumin Fv domain: An investigation into the correlation between affinity and serum half-life. MAbs. 2016;8:1336-46
14. Kontermann RE. Half-life extended biotherapeutics. Expert Opin Biol Ther. 2016;16:903-15
15. Berois N, Pittini A, Osinaga E. Targeting Tumor Glycans for Cancer Therapy: Successes, Limitations, and Perspectives. Cancers (Basel). 2022;14:645
16. Sorieul C, Papi F, Carboni F, Pecetta S, Phogat S, Adamo R. Recent advances and future perspectives on carbohydrate-based cancer vaccines and therapeutics. Pharmacology Ther. 2022;235:108158
17. Soliman C, Yuriev E, Ramsland PA. Antibody recognition of aberrant glycosylation on the surface of cancer cells. Curr Opin Struct Biol. 2017;44:1-8
18. Li Z, Krippendorff BF, Sharma S, Walz AC, Lavé T, Shah DK. Influence of molecular size on tissue distribution of antibody fragments. MAbs. 2016;8:113-9
19. Wilson BZ, Bahadir A, Andrews M, Karpen J, Winkler G, Smelski G. et al. Initial Experience with F(ab')2 Antivenom Compared with Fab Antivenom for Rattlesnake Envenomations Reported to a single poison center during 2019. Toxicon. 2022;209:10-7
20. Richards DA, Maruani A, Chudasama V. Antibody fragments as nanoparticle targeting ligands: a step in the right direction. Chem Sci. 2017;8:63-77
21. Bird RE, Hardman K. D. Jacobson JW, Johnson S, Kaufman BM, Lee SM, Lee T, et al. Single-chain antigen-binding proteins. Science. 1988;242:423-6
22. Spadiut O, Capone S, Krainer F, Glieder A, Herwig C. Microbials for the production of monoclonal antibodies and antibody fragments. Trends Biotechnol. 2014;32:54-60
23. Nieba L, A H, Krebber C, Plückthun A. Disrupting the hydrophobic patches at the antibody variable/constant domain interface: improved in vivo folding and physical characterization of an engineered scFv fragment. Protein Eng. 1997;10:435-44
24. Weisser NE, Hall JC. Applications of single-chain variable fragment antibodies in therapeutics and diagnostics. Biotechnol Adv. 2009;27:502-20
25. Duan Y, Chen R, Huang Y, Meng X, Chen J, Liao C. et al. Tuning the ignition of CAR: optimizing the affinity of scFv to improve CAR-T therapy. Cell Mol Life Sci. 2022;79:14
26. Car T Cells: Engineering immune cells to treat cancer-National Cancer Institute 2013. Revised 10 March 2022. https://www.cancer.gov/about-cancer/treatment/research/car-t-cells#:~:text=CAR%20T%2Dcell%20therapy%3A%20A%20%22living%20drug%22&text=They%20are%20made%20by%20collecting,the%20surface%20of%20cancer%20cells
27. Hornig N, Färber-Schwarz A. Production of Bispecific Antibodies: Diabodies and Tandem scFv. Methods Mol Biol. 2012;907:713-27
28. Suurs FV, Lub-de Hooge MN, de Vries EG, de Groot DJA. A review of bispecific antibodies and antibody constructs in oncology and clinical challenges. Pharmacol Ther. 2019;201:103-19
29. Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hammers C, Songa EB. et al. Naturally occurring antibodies devoid of light chains. Nature. 1993;363:446-8
30. Muyldermans S. Applications of Nanobodies. Annu Rev Anim Biosci. 2021;9:401-21
31. Arbabi-Ghahroudi M. Camelid Single-Domain Antibodies: Promises and Challenges as Lifesaving Treatments. Int J Mol Sci. 2022;23:5009
32. Kijanka M, Warnders F-J, El Khattabi M, Lub-de Hooge M, van Dam GM, Ntziachristos V. et al. Rapid optical imaging of human breast tumour xenografts using anti-HER2 VHHs site-directly conjugated to IRDye 800CW for image-guided surgery. Eur J Nucl Med Mol Imaging. 2013;40:1718-29
33. Oliveira S, Van Dongen GA, Walsum MS-v, Roovers RC, Stam JC, Mali W. et al. Rapid visualization of human tumor xenografts through optical imaging with a near-infrared fluorescent anti-epidermal growth factor receptor nanobody. Mol Imaging. 2012;11:33-46
34. D'Huyvetter M, Xavier C, Caveliers V, Lahoutte T, Muyldermans S, Devoogdt N. Radiolabeled nanobodies as theranostic tools in targeted radionuclide therapy of cancer. Expert Opin Drug Deliv. 2014;11:1939-54
35. D'Huyvetter M, De Vos J, Xavier C, Pruszynski M, Sterckx YGJ, Massa S. et al. 131I-labeled anti-HER2 camelid sdAb as a theranostic tool in cancer treatment. Clin Cancer Res. 2017;23:6616-28
36. Krasniqi A, D'Huyvetter M, Xavier C, der Jeught KV, Muyldermans S, Der Heyden JV. et al. Theranostic radiolabeled anti-CD20 sdAb for targeted radionuclide therapy of non-hodgkin lymphoma. Mol Cancer Ther. 2017;16:2828-39
37. Hao S, Xu S, Li L, Li Y, Zhao M, Chen J. et al. Tumour inhibitory activity on pancreatic cancer by bispecific nanobody targeting PD-L1 and CXCR4. BMC Cancer. 2022;22:1092
38. Maali A, Gholizadeh M, Feghhi-Najafabadi S, Noei A, Seyed-Motahari SS, Mansoori S. et al. Nanobodies in cell-mediated immunotherapy: On the road to fight cancer. Front Immunol. 2023;14:1012841
39. Kozani PS, Naseri A, Mirarefin SMJ, Salem F, Nikbakht M, Bakhshi SE. et al. Nanobody-based CAR-T cells for cancer immunotherapy. Biomark Res. 2022;10:24
40. Henry KA, Hussack G, Kumaran J, Gilbert M, MacKenzie CR, Sulea T. et al. Role of the non-hypervariable FR3 D-E loop in single-domain antibody recognition of haptens and carbohydrates. J Mol Recognit. 2019;32:e2805
41. Khilji SK, Goerdeler F, Frensemeier K, Warschkau D, Lühle J, Fandi Z. et al. Generation of glycan-specific nanobodies. Cell Chem Biol. 2022;29:1353-61
42. Borenstein-Katz A, Warszawski S, Amon R, Eilon M, Cohen-Dvashi H, Ben-Arye SL. et al. Biomolecular recognition of the glycan neoantigen CA19-9 by distinct antibodies. J Mol Biol. 2021;433:167099
43. Horwacik I, Golik P, Grudnik P, Kolinski M, Zdzalik M, Rokita H. et al. Structural basis of GD2 ganglioside and mimetic peptide recognition by 14G2a antibody. Mol Cell Proteomics. 2015;14:2577-90
44. Soliman C, Chua JX, Vankemmelbeke M, McIntosh RS, Guy AJ, Spendlove I. et al. The terminal sialic acid of stage-specific embryonic antigen-4 has a crucial role in binding to a cancer-targeting antibody. J Biol Chem. 2020;295:1009-20
45. Soliman C, Walduck AK, Yuriev E, Richards JS, Cywes-Bentley C, Pier GB. et al. Structural basis for antibody targeting of the broadly expressed microbial polysaccharide poly- N-acetylglucosamine. J Biol Chem. 2018;293:5079-89
46. Ramsland PA, Farrugia W, Bradford TM, Hogarth PM, Scott AM. Structural Convergence of Antibody Binding of Carbohydrate Determinants in Lewis Y Tumor Antigens. J Mol Biol. 2004;340:809-18
47. Yu ED, Girardi E, Wang J, Mac T-T, Karl O, Van Calenbergh S. et al. Structural basis for the recognition of C20: 2-αGalCer by the invariant natural killer T cell receptor-like antibody L363. J Biol Chem. 2012;287:1269-78
48. Hakomori S. Structure, organization, and function of glycosphingolipids in membrane. Curr Opin Hematol. 2003;10:16-24
49. Ledeen R, Wu G. Gangliosides of the nervous system. Methods Mol Biol. 2018;1804:19-55
50. Nazha B, Inal C, Owonikoko TK. Disialoganglioside GD2 expression in solid tumors and role as a target for cancer therapy. Front Oncol. 2020;10:1000
51. Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT. et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15:5323-37
52. Sorkin L, Yu A, Junger H, Doom C. Antibody directed against GD2 produces mechanical allodynia, but not thermal hyperalgesia when administered systemically or intrathecally despite its dependence on capsaicin sensitive afferents. Brain Res. 2002;930:67-74
53. Bernhard H, Karbach J, Büschenfelde KHMZ, Strittmatter W, Knuth A. Induction of tumor-cell lysis by bi-specific antibody recognizing ganglioside GD2 and T-cell antigen CD3. Int J Cancer. 1993;55:465-70
54. Doronin I, Kholodenko I, Molotkovskaya I, Kholodenko R. Preparation of Fab-fragments of GD2-specific antibodies and analysis of their antitumor activity in vitro. Bull Exp Biol Med. 2013;154:658-63
55. Michon J, Perdereau B, Brixy F, Moutel S, Fridman W-H, Teillaud J-L. In vivo targeting of human neuroblastoma xenograft by anti-GD2/anti-FcγRI (CD64) bispecific antibody. Eur J Cancer. 1995;31A:631-6
56. Brignole C, Pastorino F, Marimpietri D, Pagnan G, Pistorio A, Allen TM. et al. Immune cell-mediated antitumor activities of GD2-targeted liposomal c-myb antisense oligonucleotides containing CpG motifs. J Natl Cancer Inst. 2004;96:1171-80
57. Pastorino F, Brignole C, Marimpietri D, Sapra P, Moase EH, Allen TM. et al. Doxorubicin-loaded Fab′ fragments of anti-disialoganglioside immunoliposomes selectively inhibit the growth and dissemination of human neuroblastoma in nude mice. Cancer Res. 2003;63:86-92
58. Di Paolo D, Ambrogio C, Pastorino F, Brignole C, Martinengo C, Carosio R. et al. Selective therapeutic targeting of the anaplastic lymphoma kinase with liposomal siRNA induces apoptosis and inhibits angiogenesis in neuroblastoma. Mol Ther. 2011;19:2201-12
59. Tur MK, Sasse S, Stocker M, Djabelkhir K, Huhn M, Matthey B. et al. An anti-GD2 single chain Fv selected by phage display and fused to Pseudomonas exotoxin A develops specific cytotoxic activity against neuroblastoma derived cell lines. Int J Mol Med. 2001;8:579-84
60. Thomas PB, Delatte SJ, Sutphin A, Frankel AE, Tagge EP. Effective targeted cytotoxicity of neuroblastoma cells. J Pediatr Surg. 2002;37:539-44
61. Akbari B, Farajnia S, Ahdi Khosroshahi S, Safari F, Yousefi M, Dariushnejad H. et al. Immunotoxins in cancer therapy: Review and update. Int Rev Immunol. 2017;36:207-19
62. Cheung N-KV, Modak S, Lin Y, Guo H, Zanzonico P, Chung J. et al. Single-chain Fv-streptavidin substantially improved therapeutic index in multistep targeting directed at disialoganglioside GD2. J Nucl Med. 2004;45:867-77
63. Yumura K, Ui M, Doi H, Hamakubo T, Kodama T, Tsumoto K. et al. Mutations for decreasing the immunogenicity and maintaining the function of core streptavidin. Protein Sci. 2013;22:213-21
64. Cheng M, Ahmed M, Xu H, Cheung NKV. Structural design of disialoganglioside GD2 and CD3-bispecific antibodies to redirect T cells for tumor therapy. Int J Cancer. 2015;136:476-86
65. Ahmed M, Cheng M, Cheung IY, Cheung N-KV. Human derived dimerization tag enhances tumor killing potency of a T-cell engaging bispecific antibody. Oncoimmunology. 2015;4:e989776
66. Cheng M, Santich BH, Xu H, Ahmed M, Huse M, Cheung N-KV. Successful engineering of a highly potent single-chain variable-fragment (scFv) bispecific antibody to target disialoganglioside (GD2) positive tumors. Oncoimmunology. 2016;5:e1168557
67. Pola R, Král V, Filippov SK, Kaberov L, Etrych T, Sieglová I. et al. Polymer Cancerostatics Targeted by Recombinant Antibody Fragments to GD2-Positive Tumor Cells. Biomacromolecules. 2019;20:412-21
68. Niethammer D, Handgretinger R. Clinical strategies for the treatment of neuroblastoma. Eur J Cancer. 1995;31A:568-71
69. Zubareva A, Boyko A, Kholodenko I, Rozov F, Larina M, Aliev T. et al. Chitosan nanoparticles targeted to the tumor-associated ganglioside GD2. Russ J Bioorg Chem. 2016;42:532-45
70. Kholodenko IV, Kalinovsky DV, Svirshchevskaya EV, Doronin II, Konovalova MV, Kibardin AV. et al. Multimerization through pegylation improves pharmacokinetic properties of scFv fragments of GD2-specific antibodies. Molecules. 2019;24:3835
71. Moutel S, Birkle S, Laurence V, Michon J, Fridman WH, Aubry J. et al. Generation and characterization of a mouse single-chain antibody fragment specific for disialoganglioside (GD2). Hybridoma. 1997;16:335-46
72. Rossig C, Bollard CM, Nuchtern JG, Merchant DA, Brenner MK. Targeting of GD2-positive tumor cells by human T lymphocytes engineered to express chimeric T-cell receptor genes. Int J Cancer. 2001;94:228-36
73. Sujjitjoon J, Sayour E, Tsao S-T, Uiprasertkul M, Sanpakit K, Buaboonnam J. et al. GD2-specific chimeric antigen receptor-modified T cells targeting retinoblastoma-assessing tumor and T cell interaction. Transl Oncol. 2021;14:100971
74. Mitwasi N, Feldmann A, Bergmann R, Berndt N, Arndt C, Koristka S. et al. Development of novel target modules for retargeting of UniCAR T cells to GD2 positive tumor cells. Oncotarget. 2017;8:108584-603
75. Mitwasi N, Feldmann A, Arndt C, Koristka S, Berndt N, Jureczek J. et al. “UniCAR”-modified off-the-shelf NK-92 cells for targeting of GD2-expressing tumour cells. Sci Rep. 2020;10:2141
76. Esser R, Müller T, Stefes D, Kloess S, Seidel D, Gillies SD. et al. NK cells engineered to express a GD2 -specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J Cell Mol Med. 2012;16:569-81
77. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD. et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050-6
78. Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B. et al. CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol Ther. 2017;25:2214-24
79. Straathof K, Flutter B, Wallace R, Jain N, Loka T, Depani S. et al. Antitumor activity without on-target off-tumor toxicity of GD2-chimeric antigen receptor T cells in patients with neuroblastoma. Sci Transl Med. 2020;12:eabd6169
80. Yang L, Ma X, Liu Y-C, Zhao W, Yu L, Qin M. et al. Chimeric antigen receptor 4SCAR-GD2-modified T cells targeting high-risk and recurrent neuroblastoma: a phase II multi-center trial in China. Blood. 2017;130:3335
81. Bobowski M, Cazet A, Steenackers A, Delannoy P. Role of complex gangliosides in cancer progression. In: Rauter P, Lindhorst TK, editors. Carbohydrate Chemistry. Cambridge, UK: Royal Society of Chemistry. 2011 p. 1-20
82. Yanagisawa M, Yoshimura S, Yu RK. Expression of GD2 and GD3 gangliosides in human embryonic neural stem cells. ASN Neuro. 2011;3:e00054
83. Goldman JE, Reynolds R. A reappraisal of ganglioside GD3 expression in the CNS. Glia. 1996;16:291-5
84. Urmacher C, Cordon-Cardo C, Houghton AN. Tissue distribution of GD3 ganglioside detected by mouse monoclonal antibody R24. Am J Dermatopathol. 1989;11:577-81
85. Yun C, Nolan K, Beecham E, Reisfeld R, Junghans P. Targeting of T lymphocytes to melanoma cells through chimeric anti-GD3 immunoglobulin T-cell receptors. Neoplasia. 2000;2:449-59
86. Lo AS, Ma Q, Liu DL, Junghans RP. Anti-GD3 chimeric sFv-CD28/T-cell receptor zeta designer T cells for treatment of metastatic melanoma and other neuroectodermal tumors. Clin Cancer Res. 2010;16:2769-80
87. Thomas A, Sumughan S, Dellacecca ER, Shivde RS, Lancki N, Mukhatayev Z. et al. Benign tumors in TSC are amenable to treatment by GD3 CAR T cells in mice. JCI insight. 2021;6:e152014
88. Kotlan B, Horvath S, Eles K, Plotar VK, Naszados G, Czirbesz K. et al. Tumor-Associated Disialylated Glycosphingolipid Antigen-Revealing Antibodies Found in Melanoma Patients' Immunoglobulin Repertoire Suggest a Two-Direction Regulation Mechanism Between Immune B Cells and the Tumor. Front Immunol. 2019;10:650
89. Uskent N, Ayla S, Molinas Mandel N, Ozkan M, Teomete M, Baloglu H. et al. Prognostic Significance of Tumor Tissue NeuGcGM3 Ganglioside Expression in Patients Receiving Racotumomab Immunotherapy. J Oncol. 2020;2020:1360431
90. Carr A, Mullet A, Mazorra Z, Vázquez AM, Alfonso M, Mesa C. et al. A mouse IgG1 monoclonal antibody specific for N-glycolyl GM3 ganglioside recognized breast and melanoma tumors. Hybridoma. 2000;19:241-7
91. Roque-Navarro L, Chakrabandhu K, de Leon J, Rodríguez S, Toledo C, Carr A. et al. Anti-ganglioside antibody-induced tumor cell death by loss of membrane integrity. Mol Cancer Ther. 2008;7:2033-41
92. Rojas G, Talavera A, Munoz Y, Rengifo E, Krengel U, Ångström J. et al. Light-chain shuffling results in successful phage display selection of functional prokaryotic-expressed antibody fragments to N-glycolyl GM3 ganglioside. J Immunol Methods. 2004;293:71-83
93. Sigal DS, Hermel DJ, Hsu P, Pearce T. The role of Globo H and SSEA-4 in the development and progression of cancer, and their potential as therapeutic targets. Future Oncol. 2021;18:117-34
94. Ho M-Y, Yu AL, Yu J. Glycosphingolipid dynamics in human embryonic stem cell and cancer: their characterization and biomedical implications. Glycoconj J. 2017;34:765-77
95. Gang EJ, Bosnakovski D, Figueiredo CA, Visser JW, Perlingeiro RC. SSEA-4 identifies mesenchymal stem cells from bone marrow. Blood. 2007;109:1743-51
96. Izadyar F, Wong J, Maki C, Pacchiarotti J, Ramos T, Howerton K. et al. Identification and characterization of repopulating spermatogonial stem cells from the adult human testis. Hum Reprod. 2011;26:1296-306
97. Virant-Klun I, Skutella T, Hren M, Gruden K, Cvjeticanin B, Vogler A. et al. Isolation of small SSEA-4-positive putative stem cells from the ovarian surface epithelium of adult human ovaries by two different methods. BioMed Res Int. 2013;2013:690415
98. Pfeifer R, Lock D, Aloia A, Bosio A, Kaiser A, Hardt O. et al. Sialyl Glycolipid Stage-Specific Embryonic Antigen 4 (SSEA4)-A Novel Target for CAR T Cell Therapy of Solid Cancers. Mol Ther. 2016;24:259
99. Luo C, Wang P, He S, Zhu J, Shi Y, Wang J. Progress and Prospect of Immunotherapy for Triple-Negative Breast Cancer. Front Oncol. 2022;12:919072
100. Lin C-W, Wang Y-J, Lai T-Y, Hsu T-L, Han S-Y, Wu H-C. et al. Homogeneous antibody and CAR-T cells with improved effector functions targeting SSEA-4 glycan on pancreatic cancer. Proc Natl Acad Sci U S A. 2021;118:e2114774118
101. Ewald DR, Sumner SC. Blood type biochemistry and human disease. Wiley Interdiscip Rev Syst Biol Med. 2016;8:517-35
102. Stanley P, Wuhrer M, Lauc G, Stowell SR, Cummings RD. Structures common to different glycans. In: Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, et al. Essentials of Glycobiology 4th edition. New York: Cold Spring Harbor. 2022
103. Blanas A, Sahasrabudhe NM, Rodríguez E, van Kooyk Y, van Vliet SJ. Fucosylated Antigens in Cancer: An Alliance toward Tumor Progression, Metastasis, and Resistance to Chemotherapy. Front Oncol. 2018;8:39
104. Moehler TM, Sauer S, Witzel M, Andrulis M, Garcia-Vallejo JJ, Grobholz R. et al. Involvement of α 1-2-fucosyltransferase I (FUT1) and surface-expressed lewisy (CD174) in first endothelial cell-cell contacts during angiogenesis. J Cell Physiol. 2008;215:27-36
105. Reiter Y, Pastan I. Recombinant Fv immunotoxins and Fv fragments as novel agents for cancer therapy and diagnosis. Trends Biotechnol. 1998;16:513-20
106. Pastan I, Hassan R, FitzGerald DJ, Kreitman RJ. Immunotoxin therapy of cancer. Nat Rev Cancer. 2006;6:559-65
107. Siegall CB, Gawlak SL, Chin JJ, Zoeckler ME, Kadow KF, Brown JP. et al. Cytotoxicity of chimeric (human-murine) monoclonal antibody BR96 IgG, F (ab') 2, and Fab'conjugated to Pseudomonas exotoxin. Bioconjug Chem. 1992;3:302-7
108. Friedman PN, McAndrew SJ, Gawlak SL, Chace D, Trail PA, Brown JP. et al. BR96 sFv-PE40, a potent single-chain immunotoxin that selectively kills carcinoma cells. Cancer Res. 1993;53:334-9
109. Friedman P, Chace D, Trail P, Siegall C. Antitumor activity of the single-chain immunotoxin BR96 sFv-PE40 against established breast and lung tumor xenografts. J Immunol. 1993;150:3054-61
110. Posey JA, Khazaeli MB, Bookman MA, Nowrouzi A, Grizzle WE, Thornton J. et al. A phase I trial of the single-chain immunotoxin SGN-10 (BR96 sFv-PE40) in patients with advanced solid tumors. Clin Cancer Res. 2002;8:3092-9
111. Scherf U, Benhar I, Webber KO, Pastan I, Brinkmann U. Cytotoxic and antitumor activity of a recombinant tumor necrosis factor-B1(Fv) fusion protein on LeY antigen-expressing human cancer cells. Clin Cancer Res. 1996;2:1523-31
112. Rheinnecker M, Hardt C, Ilag LL, Kufer P, Gruber R, Hoess A. et al. Multivalent antibody fragments with high functional affinity for a tumor-associated carbohydrate antigen. J Immunol. 1996;157:2989-97
113. Power BE, Caine JM, Burns JE, Shapira DR, Hattarki MK, Tahtis K. et al. Construction, expression and characterisation of a single-chain diabody derived from a humanised anti-Lewis Y cancer targeting antibody using a heat-inducible bacterial secretion vector. Cancer Immunol Immunother. 2001;50:241-50
114. Tahtis K, Lee F-T, Smyth FE, Power BE, Renner C, Brechbiel MW. et al. Biodistribution properties of 111Indium-labeled C-functionalized trans-cyclohexyl diethylenetriaminepentaacetic acid humanized 3S193 diabody and F (ab′) 2 constructs in a breast carcinoma xenograft model. Clin Cancer Res. 2001;7:1061-72
115. Power BE, Doughty L, Shapira DR, Burns JE, Bayly AM, Caine JM. et al. Noncovalent scFv multimers of tumor-targeting anti-Lewisy hu3S193 humanized antibody. Protein Sci. 2003;12:734-47
116. Kelly MP, Lee F-T, Tahtis K, Power BE, Smyth FE, Brechbiel MW. et al. Tumor targeting by a multivalent single-chain Fv (scFv) anti-Lewis Y antibody construct. Cancer Biother Radiopharm. 2008;23:411-23
117. Mezzanzanica D, Canevari S, Mazzoni A, Figini M, Colnaghi MI, Waks T. et al. Transfer of chimeric receptor gene made of variable regions of tumor-specific antibody confers anticarbohydrate specificity on T cells. Cancer Gene Ther. 1998;5:401-7
118. Westwood JA, Smyth MJ, Teng MW, Moeller M, Trapani JA, Scott AM. et al. Adoptive transfer of T cells modified with a humanized chimeric receptor gene inhibits growth of Lewis-Y-expressing tumors in mice. Proc Natl Acad Sci U S A. 2005;102:19051-6
119. Ritchie DS, Neeson PJ, Khot A, Peinert S, Tai T, Tainton K. et al. Persistence and efficacy of second generation CAR T Cell against the LeY Antigen in acute myeloid leukemia. Mol Ther. 2013;21:2122-9
120. Paganuzzi M, Bobbio B, Marroni P, Filiberti R, Secco GB, Grossi CE. Prognostic role of serum sialyl Lewisx (CD15s) in colorectal cancer. Oncology. 2003;65:52-9
121. Mao S, Gao C, Lo CH, Wirsching P, Wong CH, Janda KD. Phage-display library selection of high-affinity human single-chain antibodies to tumor-associated carbohydrate antigens sialyl Lewisx and Lewisx. Proc Natl Acad Sci U S A. 1999;96:6953-8
122. Johansson R, Ohlin M, Jansson B, Ohlson S. Transiently binding antibody fragments against Lewis x and sialyl-Lewis x. J Immunol Methods. 2006;312:20-6
123. Zhang X, Shi S, Zhang B, Ni Q, Yu X, Xu J. Circulating biomarkers for early diagnosis of pancreatic cancer: facts and hopes. Am J Cancer Res. 2018;8:332-53
124. Miyazaki K, Ohmori K, Izawa M, Koike T, Kumamoto K, Furukawa K. et al. Loss of disialyl Lewis(a), the ligand for lymphocyte inhibitory receptor sialic acid-binding immunoglobulin-like lectin-7 (Siglec-7) associated with increased sialyl Lewis(a) expression on human colon cancers. Cancer Res. 2004;64:4498-505
125. Morimoto K, Inouye K. Flow cytometric analysis of sialyl Lewis A antigen on human cancer cells by using F(ab')2μ fragments prepared from a mouse IgM monoclonal antibody. Cytotechnology. 1997;24:219-26
126. Morimoto K, Inouye K. Method for the preparation of bispecific F(ab')2mu fragments from mouse monoclonal antibodies of the immunoglobulin M class and characterization of the fragments. J Immunol Methods. 1999;224:43-50
127. Girgis MD, Kenanova V, Olafsen T, McCabe KE, Wu AM, Tomlinson JS. Anti-CA19-9 Diabody as a PET Imaging Probe for Pancreas Cancer. J Surg Res. 2011;170:169-78
128. Girgis MD, Federman N, Rochefort MM, McCabe KE, Wu AM, Nagy JO. et al. An engineered anti-CA19-9 cys-diabody for positron emission tomography imaging of pancreatic cancer and targeting of polymerized liposomal nanoparticles. J Surg Res. 2013;185:45-55
129. DeSelm C, Palomba ML, Yahalom J, Hamieh M, Eyquem J, Rajasekhar VK. et al. Low-Dose Radiation Conditioning Enables CAR T Cells to Mitigate Antigen Escape. Mol Ther. 2018;26:2542-52
130. Pinho SS, Reis CA. Glycosylation in cancer: Mechanisms and clinical implications. Nat Rev Cancer. 2015;15:540-55
131. Loureiro LR, Carrascal MA, Barbas A, Ramalho JS, Novo C, Delannoy P. et al. Challenges in antibody development against Tn and sialyl-Tn antigens. Biomolecules. 2015;5:1783-809
132. Fujita-Yamaguchi Y. Production of Single-Chain Variable-Fragments against Carbohydrate Antigens. Antibodies (Basel). 2014;3:155-68
133. Sakai K, Yuasa N, Tsukamoto K, Takasaki-Matsumoto A, Yajima Y, Sato R. et al. Isolation and characterization of antibodies against three consecutive Tn-antigen clusters from a phage library displaying human single-chain variable fragments. J Biochem. 2010;147:809-17
134. Babino A, Pritsch O, Oppezzo P, Du Pasquier R, Roseto A, Osinaga E. et al. Molecular cloning of a monoclonal anti-tumor antibody specific for the Tn antigen and expression of an active single-chain Fv fragment. Hybridoma. 1997;16:317-24
135. Persson N, Stuhr-Hansen N, Risinger C, Mereiter S, Polónia A, Polom K. et al. Epitope mapping of a new anti-Tn antibody detecting gastric cancer cells. Glycobiology. 2017;27:635-45
136. Yuasa N, Ogawa H, Koizumi T, Tsukamoto K, Matsumoto-Takasaki A, Asanuma H. et al. Construction and expression of anti-Tn-antigen-specific single-chain antibody genes from hybridoma producing MLS128 monoclonal antibody. J Biochem. 2012;151:371-81
137. Persson N, Jansson B, Stuhr-Hansen N, Kovács A, Welinder C, Danielsson L. et al. A combinatory antibody-antigen microarray assay for high-content screening of single-chain fragment variable clones from recombinant libraries. PLoS One. 2016;11:e0168761
138. Kubota T, Matsushita T, Niwa R, Kumagai I, Nakamura K. Novel anti-Tn single-chain Fv-Fc fusion proteins derived from immunized phage library and antibody Fc domain. Anticancer Res. 2010;30:3397-405
139. Julien S, Videira PA, Delannoy P. Sialyl-Tn in cancer: (How) did we miss the target? Biomolecules. 2012;2:435-66
140. Loureiro LR, Sousa DP, Ferreira D, Chai W, Lima L, Pereira C. et al. Novel monoclonal antibody L2A5 specifically targeting sialyl-Tn and short glycans terminated by alpha-2-6 sialic acids. Sci Rep. 2018;8:12196
141. Eavarone DA, Al-Alem L, Lugovskoy A, Prendergast JM, Nazer RI, Stein JN. et al. Humanized anti-Sialyl-Tn antibodies for the treatment of ovarian carcinoma. PLoS One. 2018;13:e0201314
142. Kjeldsen T, Clausen H, Hirohashi S, Ogawa T, Iijima H, Hakomori Si. Preparation and characterization of monoclonal antibodies directed to the tumor-associated O-linked sialosyl-2--6 alpha-N-acetylgalactosaminyl (sialosyl-Tn) epitope. Cancer Res. 1988;48:2214-20
143. Reddish MA, Jackson L, Koganty RR, Qiu D, Hong W, Longenecker BM. Specificities of anti-sialyl-Tn and anti-Tn monoclonal antibodies generated using novel clustered synthetic glycopeptide epitopes. Glycoconj J. 1997;14:549-60
144. Thor A, Gorstein F, Ohuchi N, Szpak CA, Johnston WW, Schlom J. Tumor-associated glycoprotein (TAG-72) in ovarian carcinomas defined by monoclonal antibody B72.3. J Natl Cancer Inst. 1986;76:995-1006
145. Colcher D, Hand PH, Nuti M, Schlom J. A spectrum of monoclonal antibodies reactive with human mammary tumor cells. Proc Natl Acad Sci U S A. 1981;78:3199-203
146. Muraro R, Kuroki M, Wunderlich D, Poole DJ, Colcher D, Thor A. et al. Generation and characterization of B72.3 second generation monoclonal antibodies reactive with the tumor-associated glycoprotein 72 antigen. Cancer Res. 1988;48:4588-96
147. Milenic DE, Yokota T, Filpula DR, Finkelman MA, Dodd SW, Wood JF. et al. Construction, binding properties, metabolism, and tumor targeting of a single-chain Fv derived from the pancarcinoma monoclonal antibody CC49. Cancer Res. 1991;51:6363-71
148. Larson SM, El-Shirbiny AM, Divgi CR, Sgouros G, Finn RD, Tschmelitsch J. et al. Single chain antigen binding protein (sFv CC49): first human studies in colorectal carcinoma metastatic to liver. Cancer. 1997;80:2458-68
149. Pavlinkova G, Beresford GW, Booth BJ, Batra SK, Colcher D. Pharmacokinetics and biodistribution of engineered single-chain antibody constructs of MAb CC49 in colon carcinoma xenografts. J Nucl Med. 1999;40:1536-46
150. Goel A, Colcher D, Baranowska-Kortylewicz J, Augustine S, Booth BJ, Pavlinkova G. et al. Genetically engineered tetravalent single-chain Fv of the pancarcinoma monoclonal antibody CC49: improved biodistribution and potential for therapeutic application. Cancer Res. 2000;60:6964-71
151. Scott A, Lee F, Bogdal R, Kocovski P, Rigopoulos A, Cao D. et al. Characterisation of 124I radiolabelled anti-TAG72 pegylated diabody AVP0458. J Nucl Med. 2012;53:114
152. Scott AM, Akhurst T, Lee FT, Ciprotti M, Davis ID, Weickhardt AJ. et al. First clinical study of a pegylated diabody (124)I-labeled PEG-AVP0458 in patients with tumor-associated glycoprotein 72 positive cancers. Theranostics. 2020;10:11404-15
153. Yang Q, Parker CL, Lin Y, Press OW, Park SI, Lai SK. Pretargeting with bispecific fusion proteins facilitates delivery of nanoparticles to tumor cells with distinct surface antigens. J Control Release. 2017;255:73-80
154. Roberge M, Estabrook M, Basler J, Chin R, Gualfetti P, Liu A. et al. Construction and optimization of a CC49-based scFv-beta-lactamase fusion protein for ADEPT. Protein Eng Des Sel. 2006;19:141-5
155. Alderson RF, Toki BE, Roberge M, Geng W, Basler J, Chin R. et al. Characterization of a CC49-based single-chain fragment-beta-lactamase fusion protein for antibody-directed enzyme prodrug therapy (ADEPT). Bioconjug Chem. 2006;17:410-8
156. Chan DV, Sharma R, Ju CY, Roffler SR, Ju ST. A recombinant scFv-FasLext as a targeting cytotoxic agent against human Jurkat-Ras cancer. J Biomed Sci. 2013;20:16
157. Gong L, Ding H, Long NE, Sullivan BJ, Martin EW Jr, Magliery TJ. et al. A 3E8.scFv.Cys-IR800 Conjugate Targeting TAG-72 in an Orthotopic Colorectal Cancer Model. Mol Imaging Biol. 2018;20:47-54
158. Long NE, Sullivan BJ, Ding H, Doll S, Ryan MA, Hitchcock CL. et al. Linker engineering in anti-TAG-72 antibody fragments optimizes biophysical properties, serum half-life, and high-specificity tumor imaging. J Biol Chem. 2018;293:9030-40
159. Murad JP, Kozlowska AK, Lee HJ, Ramamurthy M, Chang WC, Yazaki P. et al. Effective targeting of TAG72+peritoneal ovarian tumors via regional delivery of CAR-engineered T cells. Front Immunol. 2018;9:2268
160. Hege KM, Bergsland EK, Fisher GA, Nemunaitis JJ, Warren RS, McArthur JG. et al. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J Immunother Cancer. 2017;5:22
161. Shu R, Evtimov VJ, Hammett MV, Nguyen NN, Zhuang J, Hudson PJ. et al. Engineered CAR-T cells targeting TAG-72 and CD47 in ovarian cancer. Mol Ther Oncolytics. 2021;20:325-41
162. Sharifzadeh Z, Rahbarizadeh F, Shokrgozar MA, Ahmadvand D, Mahboudi F, Jamnani FR. et al. Genetically engineered T cells bearing chimeric nanoconstructed receptors harboring TAG-72-specific camelid single domain antibodies as targeting agents. Cancer Lett. 2013;334:237-44
163. Loureiro LR, Feldmann A, Bergmann R, Koristka S, Berndt N, Arndt C. et al. Development of a novel target module redirecting UniCAR T cells to Sialyl Tn-expressing tumor cells. Blood Cancer J. 2018;8:81
164. Loureiro LR, Feldmann A, Bergmann R, Koristka S, Berndt N, Máthé D. et al. Extended half-life target module for sustainable UniCAR T-cell treatment of STn-expressing cancers. J Exp Clin Cancer Res. 2020;39:77
165. Yu LG. The oncofetal Thomsen-Friedenreich carbohydrate antigen in cancer progression. Glycoconj J. 2007;24:411-20
166. Ravn P, Danielczyk A, Bak Jensen K, Kristensen P, Astrup Christensen P, Larsen M. et al. Multivalent scFv display of phagemid repertoires for the selection of carbohydrate-specific antibodies and its application to the Thomsen-Friedenreich antigen. J Mol Biol. 2004;343:985-96
167. Ravn P, Stahn R, Danielczyk A, Faulstich D, Karsten U, Goletz S. The Thomsen-Friedenreich disaccharide as antigen for in vivo tumor targeting with multivalent scFvs. Cancer Immunol Immunother. 2007;56:1345-57
168. Matsumoto-Takasaki A, Horie J, Sakai K, Furui Y, Sato R, Kawakami H. et al. Isolation and characterization of anti-T-antigen single chain antibodies from a phage library. Biosci Trends. 2009;3:87-95
169. Yuasa N, Koyama T, Subedi GP, Yamaguchi Y, Matsushita M, Fujita-Yamaguchi Y. Expression and structural characterization of anti-T-antigen single-chain antibodies (scFvs) and analysis of their binding to T-antigen by surface plasmon resonance and NMR spectroscopy. J Biochem. 2013;154:521-9
170. Yuasa N, Koyama T, Fujita-Yamaguchi Y. Purification and refolding of anti-T-antigen single chain antibodies (scFvs) expressed in Escherichia coli as inclusion bodies. Biosci Trends. 2014;8:24-31
171. Liu J, Yi B, Zhang Z, Cao Y. CD176 single-chain variable antibody fragment inhibits the adhesion of cancer cells to endothelial cells and hepatocytes. Front Med. 2016;10:204-11
172. Tian Y, Zhang H. Characterization of disease-associated N-linked glycoproteins. Proteomics. 2013 p. 504-11
173. Lu Z, Kamat K, Johnson BP, Yin CC, Scholler N, Abbott KL. Generation of a Fully Human scFv that binds Tumor-Specific Glycoforms. Sci Rep. 2019;9:5101
174. Durand G, Seta N. Protein glycosylation and diseases: blood and urinary oligosaccharides as markers for diagnosis and therapeutic monitoring. Clin Chem. 2000;46:795-805
175. Matsumoto-Takasaki A, Yuasa N, Katagiri D, Koyama T, Sakai K, Zamri N. et al. Characterization of three different single chain antibodies recognizing non-reducing terminal mannose residues expressed in Escherichia coli by an inducible T7 expression system. J Biochem. 2011;150:439-50
176. Soares da Costa D, Reis RL, Pashkuleva I. Sulfation of Glycosaminoglycans and Its Implications in Human Health and Disorders. Annu Rev Biomed Eng. 2017;19:1-26
177. Sterner E, Flanagan N, Gildersleeve JC. Perspectives on Anti-Glycan Antibodies Gleaned from Development of a Community Resource Database. ACS Chem Biol. 2016;11:1773-83
178. Yang Y, Ahn J, Raghunathan R, Kallakury BV, Davidson B, Kennedy ZB. et al. Expression of the Extracellular Sulfatase SULF2 Affects Survival of Head and Neck Squamous Cell Carcinoma Patients. Front Oncol. 2020;10:582827
179. Kim H, Ho M. Isolation of Antibodies to Heparan Sulfate on Glypicans by Phage Display. Curr Protoc Protein Sci. 2018;94:e66
180. van Kuppevelt TH, Dennissen MA, van Venrooij WJ, Hoet RM, Veerkamp JH. Generation and application of type-specific anti-heparan sulfate antibodies using phage display technology. Further evidence for heparan sulfate heterogeneity in the kidney. J Biol Chem. 1998;273:12960-6
181. Jenniskens GJ, Oosterhof A, Brandwijk R, Veerkamp JH, Van Kuppevelt TH. Heparan sulfate heterogeneity in skeletal muscle basal lamina: Demonstration by phage display-derived antibodies. J Neurosci. 2000;20:4099-111
182. Dennissen MA, Jenniskens GJ, Pieffers M, Versteeg EM, Petitou M, Veerkamp JH. et al. Large, tissue-regulated domain diversity of heparan sulfates demonstrated by phage display antibodies. J Biol Chem. 2002;277:10982-6
183. van de Westerlo EM, Smetsers TF, Dennissen MA, Linhardt RJ, Veerkamp JH, van Muijen GN. et al. Human single chain antibodies against heparin: selection, characterization, and effect on coagulation. Blood. 2002;99:2427-33
184. Jang IK, Hursting MJ. When heparins promote thrombosis review of heparin-induced thrombocytopenia. Circulation. 2005 p. 2671-83
185. ten Dam GB, van de Westerlo EM, Smetsers TF, Willemse M, van Muijen GN, Merry CL. et al. Detection of 2-O-sulfated iduronate and N-acetylglucosamine units in heparan sulfate by an antibody selected against acharan sulfate (IdoA2S-GlcNAc)n. J Biol Chem. 2004;279:38346-52
186. Iwahashi N, Ikezaki M, Nishikawa T, Namba N, Ohgita T, Saito H. et al. Sulfated glycosaminoglycans mediate prion-like behavior of p53 aggregates. Proc Natl Acad Sci U S A. 2020;117:33225-34
187. Lemjabbar-Alaoui H, van Zante A, Singer MS, Xue Q, Wang YQ, Tsay D. et al. Sulf-2, a heparan sulfate endosulfatase, promotes human lung carcinogenesis. Oncogene. 2010;29:635-46
188. Hossain MM, Hosono-Fukao T, Tang R, Sugaya N, van Kuppevelt TH, Jenniskens GJ. et al. Direct detection of HSulf-1 and HSulf-2 activities on extracellular heparan sulfate and their inhibition by PI-88. Glycobiology. 2010;20:175-86
189. Ten Dam GB, Kurup S, van de Westerlo EM, Versteeg EM, Lindahl U, Spillmann D. et al. 3-O-sulfated oligosaccharide structures are recognized by anti-heparan sulfate antibody HS4C3. J Biol Chem. 2006;281:4654-62
190. Damen LAA, van de Westerlo EMA, Versteeg EMM, van Wessel T, Daamen WF, van Kuppevelt TH. Construction and evaluation of an antibody phage display library targeting heparan sulfate. Glycoconj J. 2020;37:445-55
191. Delcommenne M, Klingemann HG. Detection and characterization of syndecan-1-associated heparan sulfate 6-O-sulfated motifs overexpressed in multiple myeloma cells using single chain antibody variable fragments. Hum Antibodies. 2012;21:29-40
192. Gao W, Kim H, Feng M, Phung Y, Xavier CP, Rubin JS. et al. Inactivation of Wnt signaling by a human antibody that recognizes the heparan sulfate chains of glypican-3 for liver cancer therapy. Hepatology. 2014;60:576-87
193. Smetsers TF, van de Westerlo EM, ten Dam GB, Overes IM, Schalkwijk J, van Muijen GN. et al. Human single-chain antibodies reactive with native chondroitin sulfate detect chondroitin sulfate alterations in melanoma and psoriasis. J Invest Dermatol. 2004;122:707-16
194. Li F, Ten Dam GB, Murugan S, Yamada S, Hashiguchi T, Mizumoto S. et al. Involvement of highly sulfated chondroitin sulfate in the metastasis of the Lewis lung carcinoma cells. J Biol Chem. 2008;283:34294-304
195. Vallen MJ, Massuger LF, ten Dam GB, Bulten J, van Kuppevelt TH. Highly sulfated chondroitin sulfates, a novel class of prognostic biomarkers in ovarian cancer tissue. Gynecol Oncol. 2012;127:202-9
196. Vallen MJE, Van Tilborg AAG, Tesselaar MH, Ten Dam GB, Bulten J, Van Kuppevelt TH. et al. Novel single-chain antibody GD3A10 defines a chondroitin sulfate biomarker for ovarian cancer. Biomark Med. 2014;8:699-711
197. van der Steen SC, van Tilborg AA, Vallen MJ, Bulten J, van Kuppevelt TH, Massuger LF. Prognostic significance of highly sulfated chondroitin sulfates in ovarian cancer defined by the single chain antibody GD3A11. Gynecol Oncol. 2016;140:527-36
198. Lensen JF, Wijnhoven TJ, Kuik LH, Versteeg EM, Hafmans T, Rops AL. et al. Selection and characterization of a unique phage display-derived antibody against dermatan sulfate. Matrix Biol. 2006;25:457-61
199. Ten Dam GB, Yamada S, Kobayashi F, Purushothaman A, van de Westerlo EM, Bulten J. et al. Dermatan sulfate domains defined by the novel antibody GD3A12, in normal tissues and ovarian adenocarcinomas. Histochem Cell Biol. 2009;132:117-27
200. Zaman R, Islam RA, Ibnat N, Othman I, Zaini A, Lee CY. et al. Current strategies in extending half-lives of therapeutic proteins. J Control Release. 2019;301:176-89
Author contact
![]() Corresponding author: oren.moscovitzmpg.de.
Corresponding author: oren.moscovitzmpg.de.