Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusions
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2023; 13(9):2946-2961. doi:10.7150/thno.82889 This issue Cite
Research Paper
Chemogenetic activation of the HPC-mPFC pathway improves cognitive dysfunction in lipopolysaccharide -induced brain injury
Chenglong Ge1,2,3, Wei Chen1,2,3, Lina Zhang1,2,3, Yuhang Ai1,2,3, Yu Zou4, Qianyi Peng1,2,3 ![]()
1. Department of Critical Care Medicine, Xiangya Hospital, Central South University, Changsha, Hunan Province, China, 410008.
2. National Clinical Research Center for Geriatric Disorders, Changsha, Hunan Province, China, 410008.
3. Hunan Provincial Clinical Research Center for Critical Care Medicine, Changsha, Hunan Province, China, 410008.
4. Department of Anesthesia, Xiangya Hospital, Central South University, Changsha, Hunan Province, China, 410008.
Received 2023-1-24; Accepted 2023-5-7; Published 2023-5-11
Abstract

Rationale: Although sepsis-associated encephalopathy (SAE) is a common psychiatric complication in septic patients, the underlying mechanisms remain unclear. Here, we explored the role of the hippocampus (HPC) - medial prefrontal cortex (mPFC) pathway in cognitive dysfunction in lipopolysaccharide-induced brain injury.
Methods: Lipopolysaccharide (LPS, 5 mg/kg, intraperitoneal) was used to induce an animal model of SAE. We first identified neural projections from the HPC to the mPFC via a retrograde tracer and virus expression. The activation viruses (pAAV-CaMKIIα-hM3Dq-mCherry) were injected to assess the effects of specific activation of mPFC excitatory neurons on cognitive tasks and anxiety-related behaviors in the presence of clozapine-N-oxide (CNO). Activation of the HPC-mPFC pathway was evaluated via immunofluorescence staining of c-Fos-positive neurons in mPFC. Western blotting was performed to determine protein levels of synapse- associated factors.
Results: We successfully identified a structural HPC-mPFC connection in C57BL/6 mice. LPS-induced sepsis induces cognitive impairment and anxiety-like behaviors. Chemogenetic activation of the HPC-mPFC pathway improved LPS-induced cognitive dysfunction but not anxiety-like behavior. Inhibition of glutamate receptors abolished the effects of HPC-mPFC activation and blocked activation of the HPC-mPFC pathway. The glutamate receptor-mediated CaMKII/CREB/BDNF/TrKB signaling pathway influenced the role of the HPC-mPFC pathway in sepsis-induced cognitive dysfunction.
Conclusions: HPC-mPFC pathway plays an important role in cognitive dysfunction in lipopolysaccharide-induced brain injury. Specifically, the glutamate receptor-mediated downstream signaling appears to be an important molecular mechanism linking the HPC-mPFC pathway with cognitive dysfunction in SAE.
Keywords: Cognitive dysfunction, Hippocampus, Medial prefrontal cortex, Sepsis-associated encephalopathy, Lipopolysaccharide
Introduction
Sepsis-associated encephalopathy (SAE) is a diffuse form of sepsis-induced cerebral dysfunction that occurs in the absence of direct central nervous system (CNS) infection, structural abnormalities, or other types of encephalopathy [1]. Patients with SAE exhibit varying degrees of cognitive dysfunction, memory loss, and abnormal behavior, etc [2]. However, the underlying pathogenetic mechanisms of cognitive dysfunction in sepsis-induced brain injury have not been well established, and few effective clinical treatments are available. Therefore, elucidating the pathogenesis of sepsis-induced brain injury is an important objective in the search for novel therapeutic targets.
Cognitive function is supported by the coordinated activation of various brain regions, including the hippocampus (HPC) [3], parietal cortices [4], temporal cortices [5], and prefrontal cortex (PFC) [3, 5, 6]. The PFC, particularly the medial prefrontal cortex (mPFC), plays a vital role in social cognition and socio-emotional processing [7, 8]. Studies using human brain imaging and animal neurophysiological recording have suggested that neural activity in the HPC and mPFC is critical for cognitive tasks [9-12]. Numerous studies have reported a direct HPC-mPFC connection, confined to a unidirectional projection from CA1 [13] and the ventral two-thirds of the HPC (vHPC) [14]. The HPC-mPFC pathway has been implicated in spatial encoding as a component of working memory [3, 15], and has also been associated with Alzheimer's disease (AD) [16, 17], depression [18, 19], and neuropathic pain [20]. Working memory defects are a common manifestation of sepsis-induced brain injury. Previous studies have shown that hippocampal dysfunction [21, 22] and prefrontal cortex injury [2, 22] are associated with cognitive dysfunction in sepsis patients. However, to the best of our knowledge, no reports have examined the role of the HPC-mPFC pathway in cognitive dysfunction in sepsis-induced brain injury.
The chemogenetic technology is widely used in remote manipulation of neuronal activity in mice. Designer receptors exclusively activated by designer drugs (DREADD) could be activated specifically by the “designer drug” clozapine N-oxide (CNO). DREADD—namely, mutated human muscarinic receptors (hM3Dq and hM4Di), are engineered G protein coupled receptors (GPCR) which can precisely regulate GPCR signaling pathways [23].
Based on the above-mentioned studies, we hypothesized that the working memory defects in sepsis patients might be associated with dysfunction of the HPC-mPFC pathway, and that activation of this pathway could improve cognitive function in individuals with sepsis-induced brain injury. Previous studies have shown that alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) and N-methyl-D-aspartate receptor (NMDAR) subtypes of glutamate receptors were strongly implicated in hippocampus-dependent spatial learning and memory processes [24, 25]. Further, glutamate neurotransmission has been found to play an important role in the HPC-mPFC pathway, as well as in recognition memory [26]. Accordingly, to determine whether activity of the HPC-mPFC pathway is specifically correlated with that of the postsynaptic glutamate receptor signaling pathway (GRS), and to examine the role of the GRS in SAE, we evaluated the expression of glutamate receptors (AMPAR and NMDAR) and downstream activity of the calmodulin-dependent kinase II (CaMKII)/ cAMP-response element binding protein (CREB)/ brain-derived neurotrophic factor (BDNF)/ tyrosine receptor kinase B (TrKB) signaling pathway.
Materials and Methods
Animals
Adult male C57BL/6 mice (initial weight 18-22 g, 6-8 weeks old) were purchased from Hunan SJA Laboratory Animal Co., Ltd. (Changsha, China). Mice were housed in groups of up to five per cage with libitum access to food and water on a 12/12 h light/dark cycle. All animal experiments were approved by the Animal Care and Use Committee of Central South University (No.2019sydw0240). Mice were handled for 2 min per day for 7 days before the behavioral studies took place. Lipopolysaccharide (LPS, 5 mg/kg, intraperitoneal) was used to induce an animal model of SAE.
Drugs and virus
We used the following compounds in the current study: a lipopolysaccharide (LPS; E. coli 055: B5, L2880; Sigma, USA), Fluoro-Gold (Fluorochrome, LLC, USA), clozapine-N-oxide (CNO; MedChemExpress, USA), D-AP5 (NMDAR competitive antagonist; MedChemExpress, USA), and NBQX (AMDAR inhibitor; MedChemExpress, USA). The doses were as follows: LPS (5 mg/kg dissolved in 0.9% saline, administered via intraperitoneal (i.p.) injection) [27], Fluoro-Gold (4% in 0.9% saline, 0.5 µl/side, administered via microinjection into the mPFC) [28], CNO (1 mM dissolved in 10% DMSO in 0.9% saline, 0.5 µl/side, via microinjection into the mPFC)[29], D-AP5 (1µg/µl dissolved in 10% DMSO in 0.9% saline, 0.5 µl /side, via microinjection into the mPFC) [30], and NBQX (10 nmol/ml dissolved in 10% DMSO in 0.9% saline, 1 µl /side, via microinjection into the mPFC) [31], according to previous studies.
To activate the HPC-mPFC projection pathway, we bilaterally injected an adeno-associated virus (AAV) that expressed hM3Dq, which is an excitatory designer receptor exclusively activated by CNO. The viruses (pAAV-CaMKIIα-hM3Dq-mCherry and pAAV-CaMKIIα-mCherry) used in this study were verified and purchased from OBiO Technology (Shanghai, China) Corp., Ltd.
Experimental design
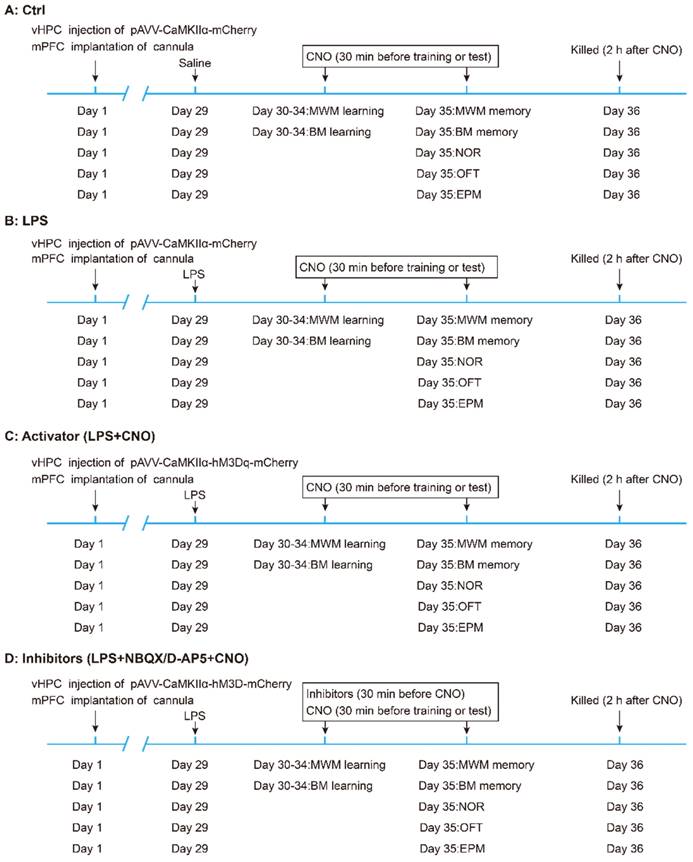
The experimental design of the study is summarized in Figure 1. Mice were randomly divided into five groups: (A) the control (Ctrl) group (n = 30, 6 mice for each behavioral test). (B) the LPS group (n = 30, 6 mice for each behavioral test). (C) the LPS+CNO (Activator) group (n = 30, 6 mice for each behavioral test). (D) LPS+ NBQX/D-AP5+CNO (Inhibitors) groups (two sub-groups, n=30/group, with 6 mice for each behavioral test). The sepsis model was induced via an i.p. LPS injection at a dose of 5 mg/kg [27, 32].
Flow chart of experimental design. Mice were randomly divided into five groups: (A) the control (Ctrl) group (n = 30, 6 mice for each behavioral test). (B) the LPS group (n = 30, 6 mice for each behavioral test). (C) the LPS+CNO (Activator) group (n = 30, 6 mice for each behavioral test). (D) LPS+NBQX/D-AP5+CNO (Inhibitors) groups (two sub-groups, n=30/group; with 6 mice for each behavioral test). vHPC, ventral hippocampus; mPFC, medial prefrontal cortex; CNO, clozapine-N-oxide; MWM, Morris water maze; BM, Barnes maze; NOR New object recognition; OFT Open field test; EPM, Elevated plus maze.
Retrograde tracing
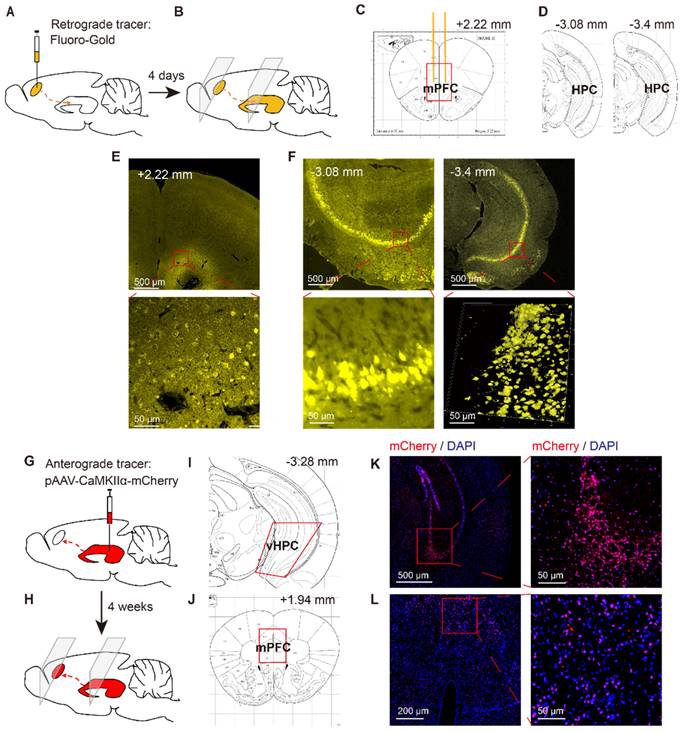
To verify the structure of neurons projecting to the mPFC, another 6 mice were anesthetized via sodium pentobarbital (60 mg/kg, i.p.) and placed in a stereotaxic frame, where they received bilateral injections of the retrograde tracer Fluoro-Gold (4% in saline, 0.5 µl/side) into the mPFC (1.8 mm anterior, 0.4 mm lateral, 1.4 mm ventral) [3] via microinjection needles (33 G). About 4 days after this injection, the mice were anaesthetized via isoflurane inhalation and killed by decapitation. The brain tissue was removed and frozen, and the frozen brain sections (10 µm) were prepared for tracing via Fluoro-Gold in the mPFC and vHPC (3.1 mm posterior, 3.0 mm lateral, 3.9 mm ventral) [3] (Figure 2A~F).
Identification of HPC-mPFC structural connection in C57BL/6 mice. (A-F) Retrograde tracing: (A) Retrograde fluorescent tracer (FluoroGold) was injected into medial prefrontal cortex (mPFC); (B) after 4 days, the FluoroGold was mainly shown in ventral hippocampus (vHPC) by coronal sections; (C, D) Representative figures showing the location of the mPFC and vHPC; (E) Representative images showing the FluoroGold in the mPFC; (F) FluoroGold microinjected into the mPFC labeled cells in the vHPC. (G-L) Anterograde tracing: (G) Microinjection of pAAV-CaMKIIα-mCherry into the vHPC; (H) after 4 weeks, the mCherry was mainly shown in ventral mPFC by coronal sections; (I, J) Representative figures showing the location of the mPFC and vHPC; (K) A representative image showing expression of mCherry in the vHPC; (L) Four weeks after viral microinjection into the vHPC, mCherry was detected in the mPFC.
Stereotaxic microinjection and cannula implantation
Mice were deeply anesthetized with sodium pentobarbital (60 mg/kg, i.p.) and placed into a stereotactic device (RWD Life Sciences, Shenzhen, China). We bilaterally injected 0.5 µl pAAV-CaMKIIα-mCherry (negative control) or pAAV-CaMKIIα-hM3Dq-mCherry into the vHPC (3.1 mm posterior, 3.0 mm lateral, 3.9 mm ventral; 0.5 µl/side) using Hamilton syringes with 33 G needles (Hamilton Reno, NV, USA). To enhance virus expression in the vHPC, we microinjected AAV into a pair of adjacent coordinates for the vHPC (anteroposterior (AP) -3.30, mediolateral (ML) ± 3.30, dorsoventral (DV) -3.60) [33]. The viruses were injected using a 33 G needle at a rate of 0.1 µl/min, and the needles were left in place for an additional 5 min to allow for sufficient diffusion.
Bilateral cannulas (OD 0.48 mm, ID 0.34 mm) were implanted 1 mm above the mPFC (AP 1.80, ML ±0.40, DV -1.40) [3]. The cannulas were secured to the skull using two stainless-steel screws and dental cement, and 33 G internal cannulas were inserted into the bilateral cannulas to prevent infection and clogging. About 4 weeks after the AVV injection, CNO (0.5 µl/side) or inhibitors were infused bilaterally into the cannulas for more than 1 min using a microinjection needle (33 G) that extended 1 mm beyond the tip of the cannula prior to each trial (including 5 training sessions and 1 testing session). The needles were left in place for over 1 min to allow for adequate drug diffusion.
Behavioral tests
Behavioral tests began 24 h after the LPS infusion. All behavioral experiments were conducted between 09:00 and 17:00, and each animal was only used in one experimental group.
Morris water maze
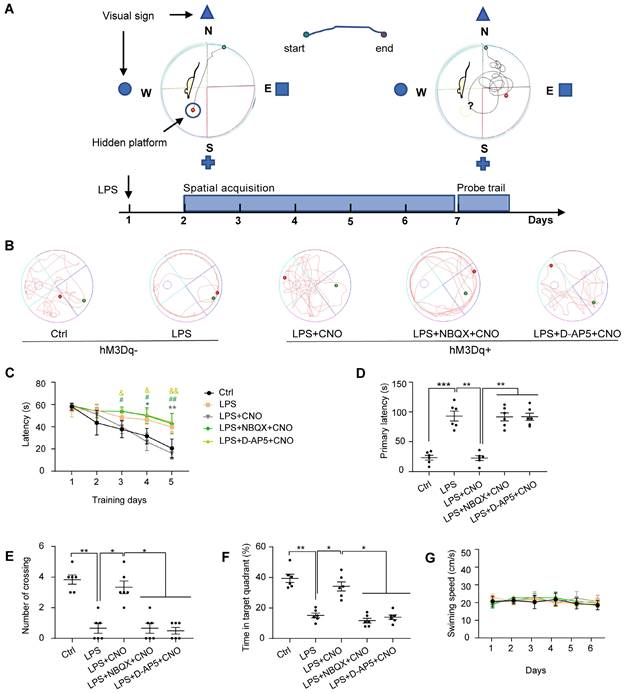
The Morris water maze (MWM) consisted of a circular plastic tank (1.2 m in diameter) filled with water (19-22 °C) and a platform (10 cm in diameter) submerged 1 cm below the surface of the water, as described previously [34]. A white food color additive was added to the water to make it opaque and to obscure the platform. The MWM tank was divided into four equal quadrants: northeast (NE), southeast (SE), southwest (SW), and northwest (NW) (Figure 4A). There were visual cues in the room for the mice to use to determine what quadrant they were in. A video camera was positioned above the center of the tank to record the mouse movement traces. We used the MWM to assess the spatial learning and memory abilities of the mice. For the spatial acquisition task, mice were trained over five consecutive days (four trials per day with a 30-s inter-trial interval). CNO (0.5 µl/side) was injected 30 min before trails. In inhibitors group, NBQX (1 µl/side) and D-AP5 (0.5 µl/side) were administrated 30 min before a CNO injection. The mice were gently placed into the water starting at one of four different quadrants (the same quadrant was used in each subsequent trial) and allowed 1 min to find the hidden platform. Each mouse was guided to the platform if it failed to locate the position after 60 s. After the last training session, a probe trial was performed to evaluate spatial memory. In this trial, the platform was removed, and the mice were placed in the water at the same point as in the acquisition trials and allowed to explore freely for 120 s. The escape latency during spatial acquisition, primary latency to the target site, number of platform crossings, time spent in the target quadrant, and swimming speeds and paths in the probe trial were recorded and analyzed using Smart 3.0 software (Figure 4).
The performance of the mice in the Morris water maze test. (A) Experimental protocol of the Morris water maze (MWM). (B) Representative swimming paths during probe trial. (C) Activation of HPC-mPFC by CNO improves the LPS-induced learning deficits (*: CNO+LPS versus LPS; #: LPS+NBQX+CNO versus LPS; &: LPS+D-AP5+CNO versus LPS). (D-F) Activation of HPC-mPFC may ameliorate the LPS-induced spatial memory deficit, but the effect was blocked by glutamate receptor inhibitors (D-AP5 and NBQX). (G) Movement is unaffected. Data were presented as mean ± SEM. n=6/group. * p < 0.05, **: p < 0.01.
Barnes maze
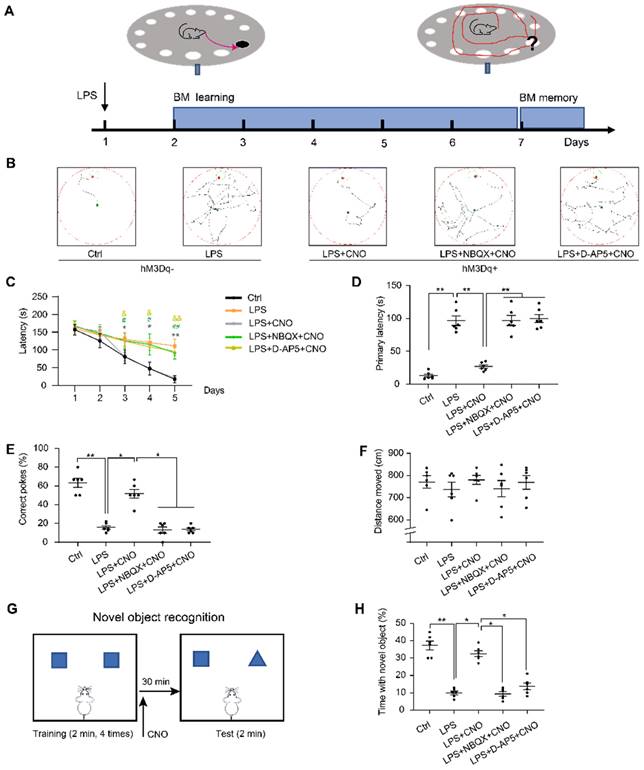
Barnes maze (BM) consisted of a circular table (92 cm diameter, 105 cm high) with 20 holes (19 empty holes and 1 target hole with escape box) located around the perimeter. As a reference to reach the target hole, we used different cues around the room (black square, yellow triangle, red diamond, and green circle). To prompt mice to complete BM test, bright lights were hung above the center of the maze as negative reinforcement. CNO (0.5 µl/side) was injected 30 min before trails. In inhibitors group, NBQX (1 µl/side) and D-AP5 (0.5 µl/side) were administrated 30 min before a CNO injection. The mice were placed in a cylindrical container at the center of the table for 10 s before each trial. During the learning phase, the mice were allowed to freely explore the target hole for 180 s per trial (3 trials per day) for 5 consecutive days. If a mouse was unable to find the target hole, it was guided towards that location and kept in the escape box for 60 s. After each trial, 75% alcohol was used to wipe and clean the maze to reduce the influence of residual odors on the subsequent test. On day 6, the escape box was removed, and the mice were allowed to freely search the maze for 120 s. During the learning trials, the latencies to the target hole were recorded. During the memory trial, the primary latency to the escape hole, percentage of correct pokes, and total distance moved were recorded and analyzed (Figure 5A~F).
The performance of the mice in the Barns maze and new object recognition test. (A) Experimental protocol of the Barns maze (BM). (B) Representative searching paths during probe trial. (C) Chemogenetic activation of the HPC-mPFC pathway improves the LPS-induced learning deficits (*: CNO+LPS versus LPS; #: LPS+NBQX+CNO versus LPS; &: LPS+D-AP5+CNO versus LPS). (D-E) Activation of HPC-mPFC may improve the LPS-induced spatial memory deficit, but the effect was blocked by glutamate receptor inhibitors (D-AP5 and NBQX). (F) Motor function was not affected. (G) Experimental protocol for CNO injections using the new object recognition (NOR). (H) LPS group showed significantly worse recognition memory than controls, while activation of HPC-mPFC signaling pathway may improve the memory (LPS versus Ctrl; LPS+CNO versus LPS). This effect of HPC-mPFC was blocked by the glutamate receptor inhibitors (LPS+NBQX/D-AP5+CNO versus LPS+CNO). Data were presented as mean ± SEM. n=6/group. * p < 0.05, ** p < 0.01.
New object recognition (NOR)
We used a new object recognition (NOR) task to assess recognition memory in the subject mice, as described previously [35, 36]. Mice were placed in an empty box (50 × 50 × 50 cm) for 10 min to enable habituation 24 h before testing. Each mouse completed 5 trials (2 minutes each). In the first four trials, the mice were placed inside the box where they were able to explore two identical objects. To reduce odor cues, the arenas and objects were carefully cleaned with 70% alcohol between the trials. On the fifth trial, mice received an injection of CNO (0.5 µl/side) 30 min before the NOR test. In inhibitors group, NBQX (1 µl/side) and D-AP5 (0.5 µl/side) were administrated 30 min before a CNO injection. For the NOR test, one of objects was replaced by a new object. The percentage of time spent exploring the new was calculated (Figure 5G~H).
Elevated plus maze test
The elevated plus maze (EPM) is commonly used to assess anxiety-related behavior. Here, we conducted the EPM in accordance with a previously described protocol [37]. The EPM in this study had two open arms and two enclosed arms, connected by a central platform (5 × 5 cm). Thirty min after a CNO injection, mice were placed on the central platform facing an open arm and allowed to freely explore for 5 min. In inhibitors group, NBQX (1 µl/side) and D-AP5 (0.5 µl/side) were administrated 30 min before a CNO injection. The number of entries to the arms and the time spent on the arms were separately measured (Figure 6A~E).
Chemogenetic activation of the HPC-mPFC pathway do not improve anxiety-related behaviors in LPS-induced sepsis-associated encephalopathy. (A-E) Activation of HPC-mPFC pathway did not alter LPS-induced anxiety-related behavior in the elevated plus maze (EPM) tests. (A) Representative searching paths on EPM. There were no significant differences in open arm entries (B), closed arm entries (C), time with open arm (D), and time with close arm (E) among LPS, Activator, and Inhibitors groups. (F-H) Activation of HPC-mPFC pathway did not improve LPS-induced anxiety-related behavior in the open field test (OFT). (F) Representative searching paths on OFT. (G) No significant difference was observed in time with center zone among LPS, Activator, and Inhibitors groups. (H) The distance moved in OFT (P > 0.05). Data were presented as mean ± SEM. n=6/group. * p < 0.05, ** p < 0.01.
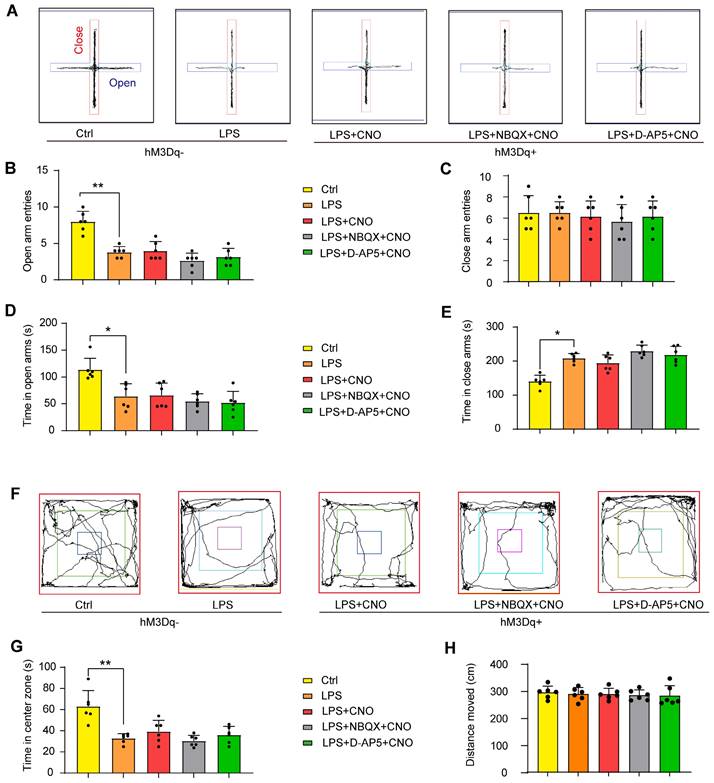
Open field test
At 30 min post-CNO injection, mice were subjected to an open field test (OFT) as a way to estimate anxiety-related behavior. In inhibitors group, NBQX (1 µl/side) and D-AP5 (0.5 µl/side) were administrated 30 min before a CNO injection. The mice were gently placed in the corner of a plastic chamber (40 × 40 × 40 cm) and allowed to move freely for 5 min. The time spent in the center area (25 × 25 cm) and total distance traveled in this region were recorded (Figure 6F~H).
Immunofluorescent staining and image analysis
Immunofluorescent staining was performed as described in a previous study [29]. At 120 min after a microinjection of CNO, mice were anesthetized with 1% sodium pentobarbital (100 mg/kg, i.p.) and perfused with 0.9 % saline followed by 4 % paraformaldehyde. In inhibitors group, NBQX (1 µl/side) and D-AP5 (0.5 µl/side) were administrated 30 min before a CNO injection. Subsequently, the brains were rapidly removed and fixed overnight in 4% paraformaldehyde, and then sectioned coronally with a sliding microtome into frozen sections (10 µm thick) at the vHPC and mPFC level. For the c-Fos experiment, the tissue sections were washed 3 × 10 min in phosphate-buffered saline (PBS) followed by a 50 min incubation period with blocking buffer (3% BSA in PBS, 0.1% Triton X100). The sections were then incubated at 4°C overnight with the following primary antibodies, diluted using a commercial antibody diluent (NCM Biotech, China): anti-c-Fos (1:500, ab208942, Abcam, USA) and anti-mCherry (1:500, ab183628, Abcam, USA). On the following day, the sections were washed 3 times with PBS and incubated with the corresponding secondary antibodies diluted in PBS (1:500) for 60 min. The secondary antibodies included Alexa Fluor 488 (ab150113, Abcam, USA) and Alexa Fluor 594 (ab150080, Abcam, USA). After washing 3 times with PBS, sections were counterstained with 4′,6-Diamidino-2-phenylindole (DAPI) and observed using a fluorescence microscope. Images were captured using a confocal microscope. The number of c-Fos positive cells was counted using Image J software. For the Fluoro-Gold experiment, tissue sections were directly observed and photographed using ultraviolet light (380 nm excitation wavelength, 420 nm absorption wavelength) under a fluorescence confocal microscope.
Western blotting
The brain regions containing the HPC and mPFC were carefully isolated using ophthalmic micro-tweezers and stored at -80 °C until use. The proteins were separated via SDS polyacrylamide gel electrophoresis (SDS-PAGE) and probed with appropriate antibodies against NR2A (1:500), NR2B (1:500), NR1 (1:1000), TrKB (1:1000), GluA1 (1:1000), GluA2 (1:1000), CaMKII (1:1000), CREB (1:1000), pCREB (1:500), BDNF (1:1000), and GAPDH (1:5000). All of these antibodies were purchased from Bioworld Technology (Bioworld, Louis Park, MN, USA). Blotting was detected with secondary antibodies and visualized via chemiluminescence using an enhanced chemiluminescence (Omni-ECL) detection reagents kit (Epizyme Biotechnology, Shanghai, China). The protein bands were quantitatively analyzed using Image J software.
Statistical analyses
Data were presented as mean ± standard error of the mean and analyzed using GraphPad Prism 9.0 software (GraphPad software, San Diego, CA, USA). Behavioral tests, c-Fos results, and WB values were analyzed using one-way analysis of variance (one-way ANOVA). Time series data were analyzed with repeated measures two-way ANOVA. Following a D'Agostino-Pearson's test, determining the normality of the distributions, multiple comparison between the groups was performed using post hoc Tukey-Kramer method. P < 0.05 was considered statistically significant.
Results
Identification of a structural HPC-mPFC connection in C57BL/6 mice
To verify the neural projections from the HPC to mPFC, a retrograde fluorescent tracer (Fluoro-Gold) was microinjected into the mPFC of C57BL/6 mice (Figure 2A, C, E). After 4 days, the Fluoro-Gold tracing was predominantly visible in the HPC area, as viewed in serial coronal sections (Figure 2B, D, F). To further examine direct monosynaptic inputs from the HPC to the mPFC via anterograde tracing, we microinjected pAVV-CaMKIIα-mCherry into the vHPC and visualized the neural terminals in the mPFC (Figure 2G, I, K). After 4 weeks, we observed extensive expression of mCherry (red) in the mPFC (Figure 2H, J, l).
Chemogenetic activation of the HPC-mPFC pathway
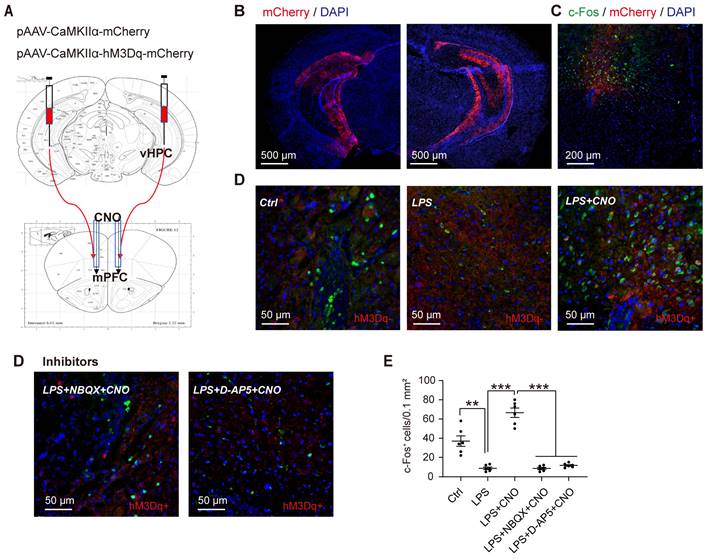
To selectively activate the HPC-mPFC pathway, we used DREADD-based chemogenetic tools [38]. Specifically, we microinjected the pAVV-CaMKIIα-hM3Dq-mCherry expressing excitatory DREADD hM3Dq into the HPC, and then 4 weeks later, microinjected CNO (1 mM, 0.5 µl/side) via the cannula implanted into the mPFC (Figure 3A-C). To verify whether CNO activates the HPC-mPFC pathway, we employed immunofluorescent double labeling of mCherry and c-Fos (a marker of neuronal activation), enabling us to visualize the expression of c-Fos and mCherry in the mPFC (Figure 3C-D). Compared with the control group, mice that received LPS exhibited a reduced number of c-Fos-positive neurons in the PFC, while the microinjection of CNO into the PFC of septic mice expressing hM3Dq increased c-Fos expression (Figure 3D: LPS+CNO vs. LPS, p < 0.001). Our results demonstrate that the HPC-mPFC pathway can be activated by a CNO injection.
Activation of the HPC-mPFC pathway. (A) Representative figure showing chemogenetic activation of the HPC-mPFC pathway. (B) Representative images showing expression of hM3Dq-mCherry and -mCherry in the HPC. (C) Representative image showing c-Fos and mCherry expression in the mPFC. (D) Representative images showing c-Fos expression in the mPFC of different groups. (E) Chemogenetic activation of the HPC-mPFC pathway increased the number of c-Fos-positive cells in the mPFC of septic mice (LPS+CNO vs. LPS, *** P<0.001); Compared with the Ctrl group, the expression of c-Fos is significantly lower in LPS group; Compared with the LPS+CNO group, the expressions of c-Fos were inhibited in inhibitors (D-AP5, NBQX) group. Data were presented as mean ± SEM. n=5/group. ** p < 0.01, *** p < 0.001.
LPS-induced sepsis induces cognitive impairment and anxiety-like behaviors
First, to test the effect of LPS-induced sepsis (LPS 5 mg/kg, i.p.) on cognitive function in mice, we performed a battery of behavioral tests associated with spatial learning and memory: the MWM, BM, and NOR. Representative swimming trajectories are shown in Figure 4B. In the spatial acquisition phase of the MWM, septic mice were much slower to learn to find the hidden platform compared with the Ctrl group (Figure 4C). During the memory test (probe trial) on day 6, in which the platform was removed, septic mice had a longer escape latency, fewer instances of crossing the area where the platform had been located, and spent less time in the target quadrant compared with mice in the Ctrl group (Figure 4D-F). We observed no differences in motor function in terms of swimming speed (Figure 4G).
We then used the BM, which is considered to be less stressful than the MWM, to test spatial reference memory. As with the MWM, the LPS group spent longer exploring the target hole than the Ctrl group during the five consecutive learning days (Figure 5C). During the memory test on day 6, the LPS group had a longer primary latency and fewer correct pokes than the Ctrl group (Figure 5D, E). There were no significant differences in the distance moved between the two groups (Figure 5F). Figure 5B shows representative movement traces.
We administered the NOR (Figure 5G~H) to assess cognitive memory ability. We found that the septic mice explored the new object for less time than the mice in the Ctrl group (Figure 5H). These findings suggest that LPS-induced sepsis induces cognitive and spatial memory impairment in mice.
We used the EPM and OFT to assess anxiety-like behaviors. Representative animal traces are shown in Figure 6A, F. In the EPM, the septic mice displayed fewer open-arm entries for both arms, and spent less time in the open arms compared with the mice in the Ctrl group (Figure 6B, D). Similarly, in the OFT, the septic mice spent less time than the control mice in the central area (Figure 6G). There was no obvious difference in the distance moved between the groups (Figure 6H). These results indicate that LPS-induced sepsis induces anxiety-like behaviors.
Chemogenetic activation of the HPC-mPFC pathway improved LPS-induced cognitive dysfunction but not anxiety-like behavior
To investigate the role of the HPC-mPFC pathway in SAE, we first measured the effect of HPC-mPFC pathway activation on performance in the MWM, BM, and NOR. In the spatial acquisition phase of the MWM, the LPS+CNO group took less time to find the hidden platform than the LPS group (Figure 4C: * LPS+CNO vs. LPS). During the memory test (probe trial), the LPS+CNO group had a shorter escape latency, higher number of crossings of the region that had contained the platform, and spent more time in the target quadrant compared with the LPS group (Figure 4D-F). There were no significant differences in exercise ability in terms of swimming speed between the groups (Figure 4G). Representative swimming traces are shown in Figure 4B. Similarly, during the learning phase in the BM test, mice in the LPS+CNO group spent less time than the septic mice exploring the target hole (Figure 5C: *LPS+CNO vs. LPS). During the BM memory test, mice in the LPS+CNO group also had a shorter primary latency and more correct pokes than the LPS group (Figure 5D, E). The distance moved was not significantly different between the groups (Figure 5F). Figure 5B shows representative movement traces. In the NOR task (Figure 5G~H), the mice in the LPS+CNO group spent more time exploring the new object compared with the LPS group (Figure 5H). These behavioral data indicate that chemogenetic activation of the HPC-mPFC pathway improved LPS-induced cognitive dysfunction.
We also measured the effects of HPC-mPFC pathway activation on performance in the EPM and OFT. However, we found no significant differences in behavioral performance between the LPS+CNO and LPS group (Figure 6).
Inhibition of glutamate receptors abolished the effects of HPC-mPFC activation and blocked activation of the HPC-mPFC pathway
To learn how glutamate receptors and associated downstream signaling pathways interact with the HPC-mPFC pathway, we assessed behavioral performance and c-Fos expression via microinjection of a glutamate receptor inhibitor 30 min before a CNO injection in the mPFC. In terms of the MWM, BM, and NOR tests, the mice in all of the inhibitor groups performed significantly worse than those in the Activator group on tests of spatial memory and cognitive function (Figure 4, Figure 5: LPS+NBQX/D-AP5+CNO vs. LPS+CNO). These data indicate that inhibitors of glutamate receptors abolished the effects of HPC-mPFC activation and blocked the activation of the HPC-mPFC pathway.
The glutamate receptor-mediated CaMKII/CREB/BDNF/TrKB signaling pathway influenced the role of the HPC-mPFC pathway in sepsis-induced cognitive dysfunction
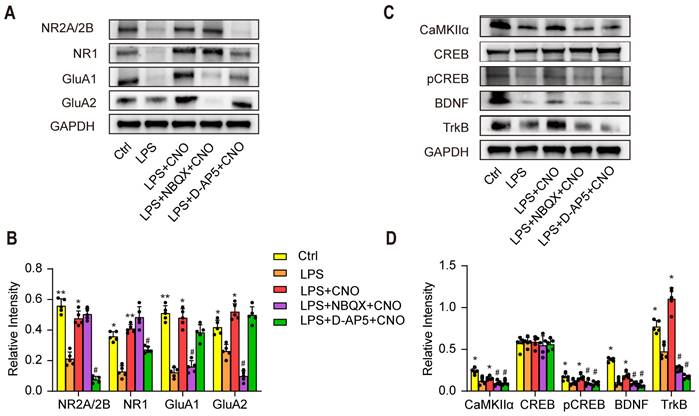
To further explore the molecular mechanisms by which the HPC-mPFC pathway regulates cognitive dysfunction in sepsis-induced brain injury, we used WB to examine the expression levels of AMPAR (GluA1, GluA2), NMDAR (NR1, NR2A/2B), and downstream signaling molecules including CaMKII, pCREB/CREB, BDNF, and TrKB in the mPFC (Figure 7A~D). We found that the levels of AMPAR (GluA1, GluA2), NMDAR (NR1, NR2A/2B), CaMKII, pCREB/CREB, BDNF, and TrKB increased significantly after activation of the HPC-mPFC pathway (Figure 7 A~D; * p < 0.05, ** p < 0.01), and that obstructing glutamate receptors via NBQX (AMPAR inhibitor) or D-AP5 (NMDAR inhibitor) abolished this increase (Figure 7A~B, C~D: # P < 0.05, ## P < 0.01). These data suggest that the glutamate receptor-mediated CaMKII/CREB/BDNF/TrKB signaling pathway influenced the role of the HPC-mPFC pathway in sepsis-induced cognitive dysfunction.
The glutamate receptor-mediated CaMKII/CREB/BDNF/TrKB signaling pathway influenced the role of the HPC-mPFC pathway in sepsis-induced cognitive dysfunction. (A-B) LPS reduces levels of glutamate receptors (AMPAR subunit GluA1/2, NMDAR subunit NR2A/2B and NR1) in mPFC, while activation of HPC-mPFC abolishes the decrease. AMDAR inhibitor (NBQX) and NMDAR inhibitor (D-AP5) effectively reduce the express of AMDAR (GluA1/2) and DMDAR (NR2A/2B and NR1) (* LPS+CNO versus LPS, # LPS+NBQX/D-AP5+CNO versus LPS+CNO). (C-D) NBQX and D-AP5 effectively reduce the express of CaMKIIα, pCREB/CREB, BDNF, and TrkB in mPFC, while activation of HPC-mPFC prevents the decrease (* LPS+CNO versus LPS, # LPS+NBQX/D-AP5+CNO versus LPS+CNO). Data were presented as mean ± SEM. n=5/group. * p < 0.05, ** p < 0.01, # p<0.05, ## p<0.01.
Discussion
Sepsis has a high mortality rate, and thus remains a major public health problem worldwide. How, the mechanisms by which sepsis affects cognitive function are unclear. DREADD-based chemogenetic technology has become increasingly popular in the field of neuroscience [38]. It can be used to identify neurocircuits associated with behavior, cognition, and emotions in animals ranging from flies to nonhuman primates [38]. Here, we performed a projection-specific manipulation to examine the role of the HPC-mPFC pathway in cognitive dysfunction in septic mice. Our study has shown that activation of the HPC-mPFC pathway improved cognitive function in normal mice. This result is consistent with the previous reports revealing that HPC-PFC input promote spatial memory [3, 15]. Moreover, we also demonstrated that chemogenetic activation of the HPC-mPFC pathway improved cognitive function in septic mice. This suggests that abnormal function of the HPC-mPFC pathway is implicated in the pathogenesis of sepsis-induced brain injury. These observations indicated that the HPC-mPFC pathway played an important role in cognitive dysfunction induced by sepsis. Alternatively, the glutamate receptor inhibitors did not alter behavior in both control mice (non-LPS) as well as in LPS treated mice expressing pAAV-CaMKIIα-mCherry. This might be partly explained by that glutamate receptor inhibitors had no effect on resting potential in normal mice [39]. Separately, as shown by other studies, we also found that AMPAR and DMDAR were down-regulated significantly after LPS injection [40]. This could probably explain why glutamate receptor inhibitors did not alter behavior in LPS treated mice expressing pAAV-CaMKIIα-mCherry. Although there still were some studies mentioned that injection of D-AP5 in the hippocampus might affect partially cognitive function, these injection sites were done directly in the hippocampus [41, 42]. Few studies have reported the role of D-AP5 or NBQX injection in mPFC on cognitive function. Furthermore, our studies show that the effect of HPC-mPFC activity in improving cognitive function in septic mice could be blocked by glutamate receptor (AMPAR or DMDAR) inhibitors. This suggested that the effects of HPC-mPFC signaling pathway may be regulated by glutamate receptor. Impairment of the HPC-mPFC pathway via LPS reduced levels of postsynaptic glutamate receptors (AMPAR and NMPAR), inhibited activity in the CaMKII/CREB/BDNF/TrKB signaling pathway, and diminished spatial cognitive function in mice (Figure 7). Therefore, the glutamate receptor-mediated CaMKII/CREB/BDNF/TrKB signaling pathway appears to modulate the role of the HPC-mPFC pathway in LPS-induced cognitive dysfunction. Our data reveal a novel mechanism by which the HPC-mPFC pathway is involved in sepsis-related cognitive dysfunction, thus providing new evidence for therapeutic targets for SAE.
C-fos is a transcription factors typically expressed in the nucleus [43]. However, our present study shows that the c-fos staining in Figure 3 is largely cytoplasmic. The c-Fos level is very low without stimulation and induced rapidly at the transcriptional level by a number of stimuli. Research has shown that c-fos proteins left the nucleus for the cytoplasm when proliferation was stopped by the high density of culture cells or by serum starvation in cultures [44]. The fact that the cFos protein would be present in cytoplasm for at least a few hours after the onset of activation has made identification of the stimulated neurons possible [43]. This led us to speculate that c-fos could rapidly translocate from nuclear to cytoplasm after CNO-induced activation. This point needs further experimental confirmation.
Both the LPS model and cecal ligation and puncture (CLP) model of SAE have been used to create short- and long-term behavioral impairments, including changes in non-aversive (MWM, OFT, EPM, NOR) and aversive memory (fear conditioning) [45]. Compared with the CLP model, cognitive dysfunction can be assessed earlier after the initiation of LPS-induced SAE because movement is less limited after the procedure [45]. In the present study, we comprehensively examined the early impact of LPS-induced sepsis on cognitive and anxiety-related behavioral parameters in mice. We found that LPS-induced sepsis not only impaired cognitive function, but also altered affective behaviors. These behavioral results are partially consistent with previous reports [32, 46, 47]. However, one previous study did not find evidence of both impaired memory and affective behavior in the MWM, NOR, and OFT one month following the injection of LPS (5 mg/kg) [36], and cognitive dysfunction was intact 30 days after the administration of LPS (5 mg/kg) for 7 consecutive days in one study [48]. In addition to the LPS dosage and mouse strain, factors including mouse sex and age, the length of the interval between the model induction and test, the timing of the training and test, etc., may influence both the degree of cognitive impairment and the results of behavioral tests [45].
Many therapeutic approaches have been used to prevent sepsis-induced cognitive impairment in mice. These include acetylcholinesterase inhibitors [49], erythropoietin [32], ACE inhibitors [50], morphine [51], the benzodiazepine agonist MRK-016[52], the NMDA antagonist MK-801 [53], and others. Among these strategies, anti-oxidative and anti-inflammatory treatments were the most frequently used, which demonstrates the importance of oxidative stress and inflammatory pathways on the pathogenesis of SAE. Non-pharmacological methods including physical exercise [54] and environmental rehabilitation [55] have also been used to address cognitive impairment after sepsis. However, the neurological pathways associated with SAE have not been previously reported. HPC projections to the mPFC are thought to be associated with memory [3, 56] and anxiety regulation [57]. Further, the HPC-mPFC pathway is important for encoding task-relevant location cues [3]. Here, we verified that the vHPC projects to the mPFC at the structural level in C57BL/6 mice (Figure 2). Then, we observed the function of this projection in terms of alterations in memory and anxiety-associated behaviors. By selectively regulating the mPFC-vHPC projection via chemogenetics, we found that the HPC-mPFC pathway regulates spatial memory, but not anxiety-associated behaviors in SAE mice. This has not been previously reported, and thus needs to be confirmed by further studies.
Ionotropic glutamate receptors (iGluR) are the main mediators of excitatory synaptic transmission in the brain [58]. NMDAR-dependent plasticity in the HPC is vital for episodic-like memory. Further, the activation of NMDA receptors is key for memory encoding, and can trigger memory formation [59]. The results showed that the expressions of glutamatergic receptor were down-regulated significantly after LPS injection. This is consistent with previous results reported by Wu et al [40, 60]. In their study, they found that a significant decrease in GluR1 and NMDA receptor (NR1) after four consecutive days of LPS injection. Regarding glutamatergic receptors, previous reports have indicated that alterations of NMDAR subunits mediate synaptic plasticity impairment in the prefrontal cortex (PFC) and hippocampus (HPC) of LPS-induced septic mice [61, 62]. NMDARs, comprising NR1, NR2A and 2B, and NR3 subunits, have different features and functions [63]. Regarding neuronal development and memory consolidation in mice, some studies have revealed that the NR2A/2B subunits of NMDA mediate synaptic plasticity and learning ability [64, 65]. Jonatan et al [66] have demonstrated that downregulation of NR2A/2B subunits is associated with cognitive impairment in a pristane-induced lupus BALB/c mice. Our study showed that pharmacological blockade of NMDA receptors reversed the effects of activation mainly on NR2A/B expression while AMPA receptor antagonists block the effects on AMPAR subunit GluA1/2 expression. This suggests that the NR2A/B and GluA1/2 subunits play a crucial role in neuroinflammation-related cognitive impairment.
AMPARs enable information transfer via NMDAR-dependent synaptic plasticity, specifically, by alleviating voltage-dependent channel blocking through Mg2+, allowing postsynaptic Ca2+ entry, and enhancing excitatory synaptic transmission [67]. In some synapses, AMPARs can also directly mediate Ca2+ influx, thus enabling the regulation of postsynaptic plasticity [67]. A Ca2+ influx can contribute to the activation of CaMKII, which induces long term potentiation and enhances learning and memory [68]. CaMKII phosphorylates the transcription factor CREB, and converts it into active form p-CREB [69]. CREB activation results in BDNF transcription in the hippocampus [70]. As an important molecule involved in memory, BDNF binds to the TrkB receptor, thus promoting excitatory signaling and long-term potentiation [71]. In the present study, we detected the expression of both AMPAR and NMDAR, and their downstream signaling molecules, including CaMKII, CREB, pCREB, BDNF, and TrKB in the mPFC. Our data showed that the effect of HPC-mPFC in improving cognitive function could be blocked by both glutamate receptor (AMPAR or DMDAR) inhibitors. The levels of glutamate receptors (AMPAR or DMDAR) and CaMKII/CREB/BDNF/TrKB were reduced by LPS, but increased by the activation of the HPC-mPFC pathway. The increased expression of both AMPAR and NMDAR activated the downstream CaMKII/CREB/BDNF/TrKB signaling pathway after targeted HPC-mPFC activation. These results indicate that the glutamate receptor modulates the role of the HPC-mPFC pathway in sepsis-induced cognitive dysfunction.
This study represents an innovative exploration in the field of SAE research, as previous studies had not paid sufficient attention to the relationship between the HPC-PFC pathway and SAE. Therefore, this experiment fills a critical gap in our understanding. Given the high mortality rates associated with sepsis and SAE being one of its most common complications, severely affecting both survival outcomes and quality of life for septic patients, this experiment emphasizes clinical needs and cutting-edge global issues. By adopting various techniques such as behavioral studies, chemical genetics, anterograde and retrograde tracing, etc., this study provides the first evidence from a neural projection pathway level that HPC-PFC pathway plays a vital role in mediating cognitive impairment in SAE. As a result, it proposes new insights into the pathogenesis of SAE and potential therapeutic targets that are of great significance and innovation.
Our study has some limitations. First, CNO can be reverse metabolized to clozapine which itself is biologically active and can produce off-target effects. CNO has very good drug-like properties with rapid CNS diffusion and distribution, and has at least a 60 min residence in vivo in mice [38]. To avoid the off-target effects, on the one hand, intracranial injection was used to prolong residence time of CNO in brain; on the other hand, time of behavioral test per mouse was controlled within half an hour as promptly as possible. Second, some studies revealed sex differences in acute immune response and neuro-inflammatory response after exposure to LPS and possible mechanisms involved in the enduring alterations in behavior and brain function following LPS treatment [72-74]. Given that all studies were conducted in male mice, our findings may not be generalizable. Besides, the effect of inhibiting the HPC-mPFC on learning and memory by chemogenetic approach has not been tested. Thus, this study can merely demonstrate the role of glutamate receptors in modulating spatial memory deficits induced by sepsis via HPC-mPFC pathway, without specifying its causal link. It is crucial to conduct more in-depth studies, including inhibitor experiments, in the future to further investigate the specific mechanisms and causal relationships of HPC-mPFC pathway in cognitive impairment caused by sepsis.
Conclusions
In summary, we identified a prominent excitatory connection between the HPC and mPFC, and revealed the vital role of the HPC-mPFC pathway in improving cognitive dysfunction after LPS-induced brain injury. Further, the glutamate receptor appears to be an important molecular mechanism linking the HPC-mPFC pathway with cognitive dysfunction in SAE.
Abbreviations
SAE: sepsis-associated encephalopathy; CNS: central nervous system; LPS: lipopolysaccharide; HPC: hippocampus; vHPC: ventral hippocampus; PFC: prefrontal cortex; mPFC: medial prefrontal cortex; CNO: clozapine-N-oxide; DREADD: designer receptors exclusively activated by designer drugs; GPCR: G protein coupled receptors; GRS: glutamate receptor signaling pathway; AMPAR: alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; NMDAR: N-methyl-D-aspartate receptor; MWM: Morris water maze; BM: Barnes maze; NOR: new object recognition; OFT: open field test; EPM: elevated plus maze; CaMKII: calmodulin-dependent kinase II; CREB: cAMP-response element binding protein; BDNF: brain-derived neurotrophic factor; TrKB: tyrosine receptor kinase B; AAV: adeno-associated virus.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81974285), the National Natural Science Foundation of China (81901264), the Natural Science Foundation of Hunan Province (2020JJ5918), the Natural Science Foundation of Hunan Province (2020JJ5959), China.
Author contributions
Chenglong Ge performed the experiments and wrote the manuscript. Qianyi Peng designed the research. Yu Zou and Wei Chen collected the data. Yuhang Ai and Lina Zhang reviewed the manuscript. All the authors reviewed and approved the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gofton TE, Young GB. Sepsis-associated encephalopathy. Nat Rev Neurol. 2012;8:557-66
2. Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. Jama. 2010;304:1787-94
3. Spellman T, Rigotti M, Ahmari SE, Fusi S, Gogos JA, Gordon JA. Hippocampal-prefrontal input supports spatial encoding in working memory. Nature. 2015;522:309-14
4. Mackey WE, Curtis CE. Distinct contributions by frontal and parietal cortices support working memory. Sci Rep. 2017;7:6188
5. Rissman J, Gazzaley A, D'Esposito M. Dynamic adjustments in prefrontal, hippocampal, and inferior temporal interactions with increasing visual working memory load. Cerebral cortex (New York, NY: 1991). 2008;18:1618-29
6. Constantinidis C, Procyk E. The primate working memory networks. Cogn Affect Behav Neurosci. 2004;4:444-65
7. Nocjar C, Alex KD, Sonneborn A, Abbas AI, Roth BL, Pehek EA. Serotonin-2C and -2a receptor co-expression on cells in the rat medial prefrontal cortex. Neuroscience. 2015;297:22-37
8. Morgan MA, Romanski LM, LeDoux JE. Extinction of emotional learning: contribution of medial prefrontal cortex. Neurosci Lett. 1993;163:109-13
9. Baeg EH, Kim YB, Huh K, Mook-Jung I, Kim HT, Jung MW. Dynamics of population code for working memory in the prefrontal cortex. Neuron. 2003;40:177-88
10. Floresco SB, Seamans JK, Phillips AG. Selective roles for hippocampal, prefrontal cortical, and ventral striatal circuits in radial-arm maze tasks with or without a delay. J Neurosci. 1997;17:1880-90
11. Schon K, Atri A, Hasselmo ME, Tricarico MD, LoPresti ML, Stern CE. Scopolamine reduces persistent activity related to long-term encoding in the parahippocampal gyrus during delayed matching in humans. J Neurosci. 2005;25:9112-23
12. Jung MW, Qin Y, McNaughton BL, Barnes CA. Firing characteristics of deep layer neurons in prefrontal cortex in rats performing spatial working memory tasks. Cerebral cortex (New York, NY: 1991). 1998;8:437-50
13. Jay TM, Witter MP. Distribution of hippocampal CA1 and subicular efferents in the prefrontal cortex of the rat studied by means of anterograde transport of Phaseolus vulgaris-leucoagglutinin. J Comp Neurol. 1991;313:574-86
14. Oh SW, Harris JA, Ng L, Winslow B, Cain N, Mihalas S. et al. A mesoscale connectome of the mouse brain. Nature. 2014;508:207-14
15. Tamura M, Spellman TJ, Rosen AM, Gogos JA, Gordon JA. Hippocampal-prefrontal theta-gamma coupling during performance of a spatial working memory task. Nat Commun. 2017;8:2182
16. Xu F, Ono M, Ito T, Uchiumi O, Wang F, Zhang Y. et al. Remodeling of projections from ventral hippocampus to prefrontal cortex in Alzheimer's mice. J Comp Neurol. 2021;529:1486-98
17. Bazzigaluppi P, Beckett TL, Koletar MM, Lai AY, Joo IL, Brown ME. et al. Early-stage attenuation of phase-amplitude coupling in the hippocampus and medial prefrontal cortex in a transgenic rat model of Alzheimer's disease. J Neurochem. 2018;144:669-79
18. Zheng C, Zhang T. Synaptic plasticity-related neural oscillations on hippocampus-prefrontal cortex pathway in depression. Neuroscience. 2015;292:170-80
19. Banagozar Mohammadi A, Torbati M, Farajdokht F, Sadigh-Eteghad S, Fazljou SMB, Vatandoust SM. et al. Sericin alleviates restraint stress induced depressive- and anxiety-like behaviors via modulation of oxidative stress, neuroinflammation and apoptosis in the prefrontal cortex and hippocampus. Brain Res. 2019;1715:47-56
20. Cardoso-Cruz H, Lima D, Galhardo V. Impaired spatial memory performance in a rat model of neuropathic pain is associated with reduced hippocampus-prefrontal cortex connectivity. J Neurosci. 2013;33:2465-80
21. Liu L, Xie K, Chen H, Dong X, Li Y, Yu Y. et al. Inhalation of hydrogen gas attenuates brain injury in mice with cecal ligation and puncture via inhibiting neuroinflammation, oxidative stress and neuronal apoptosis. Brain Res. 2014;1589:78-92
22. Heming N, Mazeraud A, Verdonk F, Bozza FA, Chrétien F, Sharshar T. Neuroanatomy of sepsis-associated encephalopathy. Crit care (London, England). 2017;21:65
23. Gomez JL, Bonaventura J, Lesniak W, Mathews WB, Sysa-Shah P, Rodriguez LA. et al. Chemogenetics revealed: DREADD occupancy and activation via converted clozapine. Science (New York, NY). 2017;357:503-7
24. Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361-79
25. Lee YS, Silva AJ. The molecular and cellular biology of enhanced cognition. Nat Rev Neurosci. 2009;10:126-40
26. Barker GR, Warburton EC. Object-in-place associative recognition memory depends on glutamate receptor neurotransmission within two defined hippocampal-cortical circuits: a critical role for AMPA and NMDA receptors in the hippocampus, perirhinal, and prefrontal cortices. Cerebral cortex (New York, NY: 1991). 2015;25:472-81
27. Luo RY, Luo C, Zhong F, Shen WY, Li H, Hu ZL. et al. ProBDNF promotes sepsis-associated encephalopathy in mice by dampening the immune activity of meningeal CD4(+) T cells. J Neuroinflammation. 2020;17:169
28. Worth AA, Shoop R, Tye K, Feetham CH, D'Agostino G, Dodd GT. et al. The cytokine GDF15 signals through a population of brainstem cholecystokinin neurons to mediate anorectic signalling. eLife. 2020 9
29. Liu J, Wu R, Johnson B, Vu J, Bass C, Li JX. The Claustrum-Prefrontal Cortex Pathway Regulates Impulsive-Like Behavior. J Neurosci. 2019;39:10071-80
30. Alijanpour S, Zarrindast MR. Potentiation of morphine-induced antinociception by harmaline: involvement of μ-opioid and ventral tegmental area NMDA receptors. Psychopharmacology. 2020;237:557-70
31. Yu H, Li M, Zhou D, Lv D, Liao Q, Lou Z. et al. Vesicular glutamate transporter 1 (VGLUT1)-mediated glutamate release and membrane GluA1 activation is involved in the rapid antidepressant-like effects of scopolamine in mice. Neuropharmacology. 2018;131:209-22
32. Gao R, Tang YH, Tong JH, Yang JJ, Ji MH, Zhu SH. Systemic Lipopolysaccharide Administration-Induced Cognitive Impairments are Reversed by Erythropoietin Treatment in Mice. Inflammation. 2015;38:1949-58
33. Padilla-Coreano N, Bolkan SS, Pierce GM, Blackman DR, Hardin WD, Garcia-Garcia AL. et al. Direct Ventral Hippocampal-Prefrontal Input Is Required for Anxiety-Related Neural Activity and Behavior. Neuron. 2016;89:857-66
34. Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1:848-58
35. Tuscher JJ, Taxier LR, Fortress AM, Frick KM. Chemogenetic inactivation of the dorsal hippocampus and medial prefrontal cortex, individually and concurrently, impairs object recognition and spatial memory consolidation in female mice. Neurobiol Learn Mem. 2018;156:103-16
36. Anderson ST, Commins S, Moynagh PN, Coogan AN. Lipopolysaccharide-induced sepsis induces long-lasting affective changes in the mouse. Brain Behav Immun. 2015;43:98-109
37. Walf AA, Frye CA. The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nat Protoc. 2007;2:322-8
38. Zhu H, Aryal DK, Olsen RH, Urban DJ, Swearingen A, Forbes S. et al. Cre-dependent DREADD (Designer Receptors Exclusively Activated by Designer Drugs) mice. Genesis (New York, NY: 2000). 2016;54:439-46
39. Greenhill SD, Jones RS. Simultaneous estimation of global background synaptic inhibition and excitation from membrane potential fluctuations in layer III neurons of the rat entorhinal cortex in vitro. Neuroscience. 2007;147:884-92
40. Wu L, Zhang T, Chen K, Lu C, Liu XF, Zhou JL. et al. Rapid antidepressant-like effect of Fructus Aurantii depends on cAMP-response element binding protein/Brain-derived neurotrophic facto by mediating synaptic transmission. Phytother Res2021; 35: 404-14.
41. Inglis J, Martin SJ, Morris RG. Upstairs/downstairs revisited: spatial pretraining-inducedrescue of normal spatial learning during selective blockade of hippocampal N-methyl-d-aspartate receptors. Eur. J. Neurosci. 2013;37:718-27
42. Steele RJ, Morris RG. Delay-dependent impairment of a matching-to-place task with chronic and intrahippocampal infusion of the NMDA-antagonist D-AP5. Hippocampus. 1999;9:118-36
43. Hoffman GE, Smith MS, Verbalis JG. c-Fos and related immediate early gene products as markers of activity in neuroendocrine systems. Front Neuroendocrinol. 1993;14:173-213
44. Vriz S, Lemaitre JM, Leibovici M, Thierry N, Méchali M. Comparative analysis of the intracellular localization of c-Myc, c-Fos, and replicative proteins during cell cycle progression. Mol Cell Biol. 1992;12:3548-55
45. Savi FF, de Oliveira A, de Medeiros GF, Bozza FA, Michels M, Sharshar T. et al. What animal models can tell us about long-term cognitive dysfunction following sepsis: A systematic review. Neurosci Biobehav Rev. 2021;124:386-404
46. Zhu SZ, Huang WP, Huang LQ, Han YL, Han QP, Zhu GF. et al. Huperzine A protects sepsis associated encephalopathy by promoting the deficient cholinergic nervous function. Neurosci Lett. 2016;631:70-8
47. Yamanaka D, Kawano T, Nishigaki A, Aoyama B, Tateiwa H, Shigematsu-Locatelli M. et al. Preventive effects of dexmedetomidine on the development of cognitive dysfunction following systemic inflammation in aged rats. J Anesth. 2017;31:25-35
48. Bian Y, Zhao X, Li M, Zeng S, Zhao B. Various roles of astrocytes during recovery from repeated exposure to different doses of lipopolysaccharide. Behav Brain Res. 2013;253:253-61
49. Gamal M, Moawad J, Rashed L, El-Eraky W, Saleh D, Lehmann C. et al. Evaluation of the effects of Eserine and JWH-133 on brain dysfunction associated with experimental endotoxemia. J Neuroimmunol. 2015;281:9-16
50. Abareshi A, Hosseini M, Beheshti F, Norouzi F, Khazaei M, Sadeghnia HR. et al. The effects of captopril on lipopolysaccharide induced learning and memory impairments and the brain cytokine levels and oxidative damage in rats. Life Sci. 2016;167:46-56
51. Rostami F, Oryan S, Ahmadiani A, Dargahi L. Morphine preconditioning protects against LPS-induced neuroinflammation and memory deficit. J Mol Neurosci. 2012;48:22-34
52. Eimerbrink MJ, White JD, Pendry RJ, Hodges SL, Sadler LN, Wiles JD. et al. Administration of the inverse benzodiazepine agonist MRK-016 rescues acquisition and memory consolidation following peripheral administration of bacterial endotoxin. Behav Brain Res. 2015;288:50-3
53. Cassol OJ Jr, Comim CM, Constantino LS, Rosa DV, Mango LA, Stertz L. et al. Acute low dose of MK-801 prevents memory deficits without altering hippocampal DARPP-32 expression and BDNF levels in sepsis survivor rats. J Neuroimmunol. 2011;230:48-51
54. Wu CW, Chen YC, Yu L, Chen HI, Jen CJ, Huang AM. et al. Treadmill exercise counteracts the suppressive effects of peripheral lipopolysaccharide on hippocampal neurogenesis and learning and memory. J Neurochem. 2007;103:2471-81
55. Kawano T, Morikawa A, Imori S, Waki S, Tamura T, Yamanaka D. et al. Preventive effects of multisensory rehabilitation on development of cognitive dysfunction following systemic inflammation in aged rats. J Anesth. 2014;28:780-4
56. Phillips ML, Robinson HA, Pozzo-Miller L. Ventral hippocampal projections to the medial prefrontal cortex regulate social memory. eLife. 2019 8
57. Schoenfeld TJ, Kloth AD, Hsueh B, Runkle MB, Kane GA, Wang SS. et al. Gap junctions in the ventral hippocampal-medial prefrontal pathway are involved in anxiety regulation. J Neurosci. 2014;34:15679-88
58. Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK. et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405-96
59. Morris RG. NMDA receptors and memory encoding. Neuropharmacology. 2013;74:32-40
60. Wu Y, Wei Z, Li Y, Wei C, Li Y, Cheng P. et al. Perturbation of Ephrin Receptor Signaling and Glutamatergic Transmission in the Hypothalamus in Depression Using Proteomics Integrated With Metabolomics. Front Neurosci. 2019;13:1359
61. Walker AK, Budac DP, Bisulco S, Lee AW, Smith RA, Beenders B. et al. NMDA receptor blockade by ketamine abrogates lipopolysaccharide-induced depressive-like behavior in C57BL/6J mice. Neuropsychopharmacology. 2013;38:1609-16
62. Francija E, Petrovic Z, Brkic Z, Mitic M, Radulovic J, Adzic M. Disruption of the NMDA receptor GluN2A subunit abolishes inflammation-induced depression. Behav Brain Res. 2019;359:550-9
63. Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature. 1994;368:144-7
64. Brigman JL, Feyder M, Saksida LM, Bussey TJ, Mishina M, Holmes A. Impaired discrimination learning in mice lacking the NMDA receptor NR2A subunit. Learn Mem (Cold Spring Harbor, NY). 2008;15:50-4
65. von Engelhardt J, Doganci B, Jensen V, Hvalby Ø, Göngrich C, Taylor A. et al. Contribution of hippocampal and extra-hippocampal NR2B-containing NMDA receptors to performance on spatial learning tasks. Neuron. 2008;60:846-60
66. Luciano-Jaramillo J, Sandoval-García F, Vázquez-Del Mercado M, Gutiérrez-Mercado YK, Navarro-Hernández RE, Martínez-García EA. et al. Downregulation of hippocampal NR2A/2B subunits related to cognitive impairment in a pristane-induced lupus BALB/c mice. PloS one. 2019;14:e0217190
67. Greger IH, Watson JF, Cull-Candy SG. Structural and Functional Architecture of AMPA-Type Glutamate Receptors and Their Auxiliary Proteins. Neuron. 2017;94:713-30
68. Lisman J, Yasuda R, Raghavachari S. Mechanisms of CaMKII action in long-term potentiation. Nat Rev Neurosci. 2012;13:169-82
69. Yan X, Liu J, Ye Z, Huang J, He F, Xiao W. et al. CaMKII-Mediated CREB Phosphorylation Is Involved in Ca2+-Induced BDNF mRNA Transcription and Neurite Outgrowth Promoted by Electrical Stimulation. PloS one. 2016;11:e0162784
70. Navabpour S, Rezayof A, Ghasemzadeh Z. Activation of VTA/CeA/mPFC cannabinoid CB1 receptors induced conditioned drug effects via interacting with hippocampal CAMKII-CREB-BDNF signaling pathway in rats. Eur J Pharmacol. 2021;909:174417
71. Bekinschtein P, Cammarota M, Medina JH. BDNF and memory processing. Neuropharmacology. 2014 76 Pt C: 677-83
72. Cai KC, van Mil S, Murray E, Mallet JF, Matar C, Ismail N. Age and sex differences in immune response following LPS treatment in mice. Brain Behav Immun. 2016;58:327-37
73. Murtaj V, Belloli S, Di Grigoli G, Pannese M, Ballarini E, Rodriguez-Menendez V. et al. Age and Sex Influence the Neuro-inflammatory Response to a Peripheral Acute LPS Challenge. Front Aging Neurosci. 2019;11:299
74. Chistyakov DV, Azbukina NV, Astakhova AA, Goriainov SV, Chistyakov VV, Sergeeva MG. Sex-Mediated Differences in LPS Induced Alterations of TNFα, IL-10 Expression, and Prostaglandin Synthesis in Primary Astrocytes. Int J Mol Sci. 2018 19
Author contact
![]() Corresponding author: Qianyi Peng. Email: PQYicuedu.cn or 405905edu.cn
Corresponding author: Qianyi Peng. Email: PQYicuedu.cn or 405905edu.cn
Tel: +8615974252010 (Mobile). Fax: +86073184327074. Address: No. 87 Xiang-Ya Road, Changsha, Hunan Province, China. Code: 410008.