Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. The structure of ATAD2
3. ATAD2 in cancer
4. The molecular basis...
5. Targeting ATAD2 with...
6. Conclusion and perspectives
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2023; 13(2):787-809. doi:10.7150/thno.78840 This issue Cite
Review
ATPase family AAA domain-containing protein 2 (ATAD2): From an epigenetic modulator to cancer therapeutic target
Jiahui Fu1,2#, Jin Zhang3#, Xiya Chen1,3#, Zhiying Liu1,3, Xuetao Yang1, Zhendan He1, Yue Hao3 ![]() , Bo Liu2
, Bo Liu2 ![]() , Dahong Yao1
, Dahong Yao1 ![]()
1. School of Pharmaceutical Sciences, Shenzhen Technology University, Shenzhen, 518118, China.
2. State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University, Chengdu 610041, China.
3. School of Pharmaceutical Sciences, Medical School, Shenzhen University, Shenzhen 518060, China.
#These authors contributed equally to this work.
Received 2022-9-12; Accepted 2022-12-22; Published 2023-1-1
Abstract

ATPase family AAA domain-containing protein 2 (ATAD2) has been widely reported to be a new emerging oncogene that is closely associated with epigenetic modifications in human cancers. As a coactivator of transcription factors, ATAD2 can participate in epigenetic modifications and regulate the expression of downstream oncogenes or tumor suppressors, which may be supported by the enhancer of zeste homologue 2. Moreover, the dominant structure (AAA + ATPase and bromine domains) can make ATAD2 a potential therapeutic target in cancer, and some relevant small-molecule inhibitors, such as GSK8814 and AZ13824374, have also been discovered. Thus, in this review, we focus on summarizing the structural features and biological functions of ATAD2 from an epigenetic modulator to a cancer therapeutic target, and further discuss the existing small-molecule inhibitors targeting ATAD2 to improve potential cancer therapy. Together, these inspiring findings would shed new light on ATAD2 as a promising druggable target in cancer and provide a clue on the development of candidate anticancer drugs.
Keywords: ATPase family AAA domain-containing protein 2 (ATAD2), Epigenetic modification, Therapeutic target, Small-molecule inhibitor, Cancer therapy, Anticancer drug
1. Introduction
AAA ATPase proteins, also known as ATPase proteins, are composed of approximately 200 amino acids and gradually oligomerize into hexamers. This enzyme structure contains ATP binding sites, which can catalyze the hydrolysis of ATP to ADP and phosphate ions with releasing energy [1]. Notably, these energies are essential for organisms to maintain biological functions, such as DNA replication, priming, and remodeling; protein synthesis, modification, and degradation; and transport of nutrients and metabolites [2]. With in-depth research, increasing ATPase family members have been reported [3, 4]. Of them, given the close relationship between ATAD2 and cancer, which has become an increasingly recognized research hotspot [5, 6] (Figure 1).
Timeline illustrating the development of ATAD2 as a potential candidate biomarker and druggable target in cancer therapy.
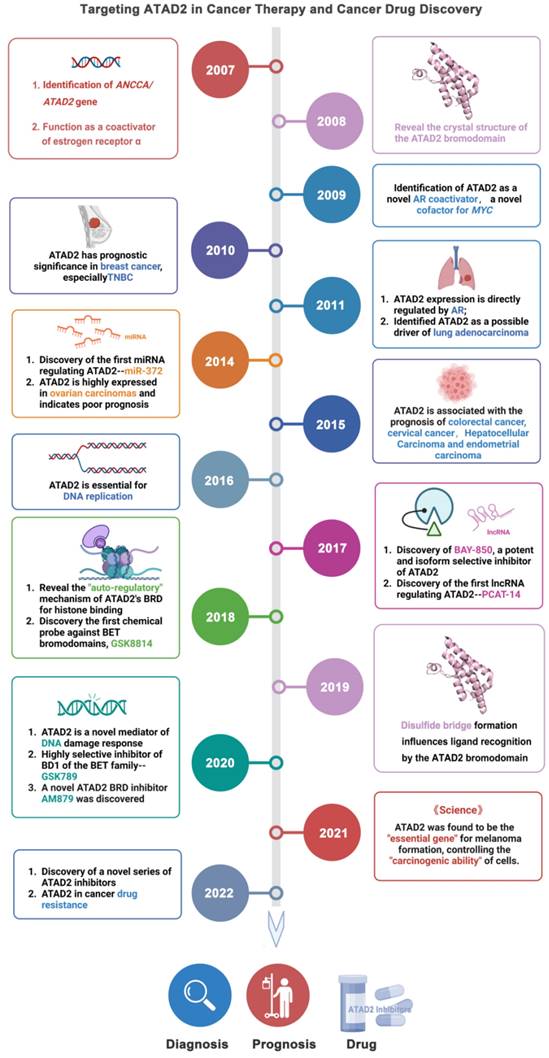
As an epigenetic regulator, ATAD2 is first known for its role in histone modification. In 2007, ANCCA/ATAD2 was first confirmed as a coactivator of estrogen receptor α (ERα). ANCCA binding and hydrolyzing ATP is critical for co-regulating the recruitment of cAMP-responsive element-binding protein (CBP/p300) and histone hyperacetylation on ER target chromatin, providing new insights into the potential mechanisms of ANCCA in breast cancer development [7]. ATAD2 has since been shown to also act as a coactivator of other transcription factors, including c-Myc and the E2F transcription factor 1 (E2F1), E2F transcription factor 2 (E2F2), and E2F transcription factor 3 (E2F3) proteins [8, 9]. In the following decade, studies on ATAD2 became more comprehensive and in-depth, from the upstream factors regulating ATAD2 protein expression (such as miR-372, lnc PACT-14) to the mechanism of action involved in the occurrence and development of breast cancer, lung adenocarcinoma, liver cancer and other diseases [10-13]. In particular, ATAD2 plays an important role in histone modification of tumor cells [14, 15]. For instance, in prostate cancer LNCaP cells, ATAD2 affects the recruitment of histone methylase mixed lineage protein-1 (MLL1) and RNA polymerase II during enhancer of zeste homologue 2 (EZH2) gene transcription. In addition, ATAD2 was confirmed to be closely related to the mRNA and protein levels of the oncogene, EZH2 [16]. EZH2, the main catalytic subunit of polycomb repressive complex 2, catalyzes the trimethylation modification of H3K27 through its SET domain to maintain the silencing state of downstream target genes. Notably, EZH2 is reported to be highly expressed in many human tumors, promoting tumorigenesis and malignant transformation [17, 18]. In prostate cancer, ATAD2 can bind to the promoter of EZH2 together with KDM8 and E2F1 to promote the transcription of EZH2. Therefore, the regulation of EZH2 expression by ATAD2 plays a significant role in the development of cancer [19].
More excitingly, recently it is reported that ATAD2 appears to be a necessary gene for melanoma formation in a melanoma zebrafish model, making ATAD2 a potential cancer therapeutic target. When ATAD2 is knocked out, the cells cannot become cancerous, despite numerous oncogenic and antioncogenic mutations. In contrast, the addition of ATAD2 allows the cells to regain their ability to become cancerous, meaning that ATAD2 is an important factor in melanoma formation [14]. Admittedly, ATAD2 is indeed highly expressed in many cancers and is associated with poor prognosis in cancers including breast, colon and cervical cancer [20, 21]. Over-expressed ATAD2 interacts with various transcription factors and chromatin-modifying proteins in cancer cells and promotes tumor growth by inducing the full expression of genes that promote cell proliferation and inhibit cell apoptosis [22].
Additionally, protein crystal structures of ATAD2 and targeted small-molecule compounds have been deciphered one by one [23-26]. Structurally, the AAA + ATPase domain can mediate ATAD2 polymerization, and mutations in this region will lead to changes in its downstream cascade, such as the regulatory relationship between ATAD2 and ERα [7]. Within the central region of ATAD2, in addition to a functional AAA + ATPase domain, there is also a bromine domain (BRD) near the C-terminus. BRD can specifically recognize peptide chains containing acetylated lysine residues, target ATAD2 protein to activate transcription regions, regulate chromatin structure, and recruit other transcription factors to participate in post-translational modifications of proteins [27]. Currently, a series of compounds regulating BRD have emerged, opening up new prospects for anti-cancer drugs, such as JQ1, AZ13824374 and AM879 [28-30].
Taken together, targeting ATAD2 is a promising strategy for cancer treatment. This review mainly focuses on ATAD2 from the protein structure, biological function, signaling pathway and other aspects to launch a detailed introduction. More importantly, we systematically summarize the design and discovery of small molecule inhibitors that regulate ATAD2, and the characteristics, advantages and disadvantages of these small molecules, which may provide a cornerstone for the development of ATAD2 target drugs for cancer therapy.
2. The structure of ATAD2
The N-terminal acidic domain (NTD, residues 1-403), bipartite domain (AAA-D1, residues 403-690) and AAA-D2 (residues 751-944), AAA + ATPase domain (residues 403-944), bromodomain (BRD, residues 981-1108) and C-terminal domain (CTD, residues 1264-1390), a total of 1390 amino acids, constitute ATAD2. Studies have proven that ATAD2 is evolutionarily conserved across multiple species. Abo1 of S. pombe and lex-1 of C. elegans share the same conserved domain feature with human ATAD2 but differ in the location of the residues in which the domains are located. For example, in C. elegans and S. pombe, the NTD residues are residues 1-371 and 1-288, respectively, and the BRD residues are residues 981-1108 and 755-940 [31] (Figure 2A-B).
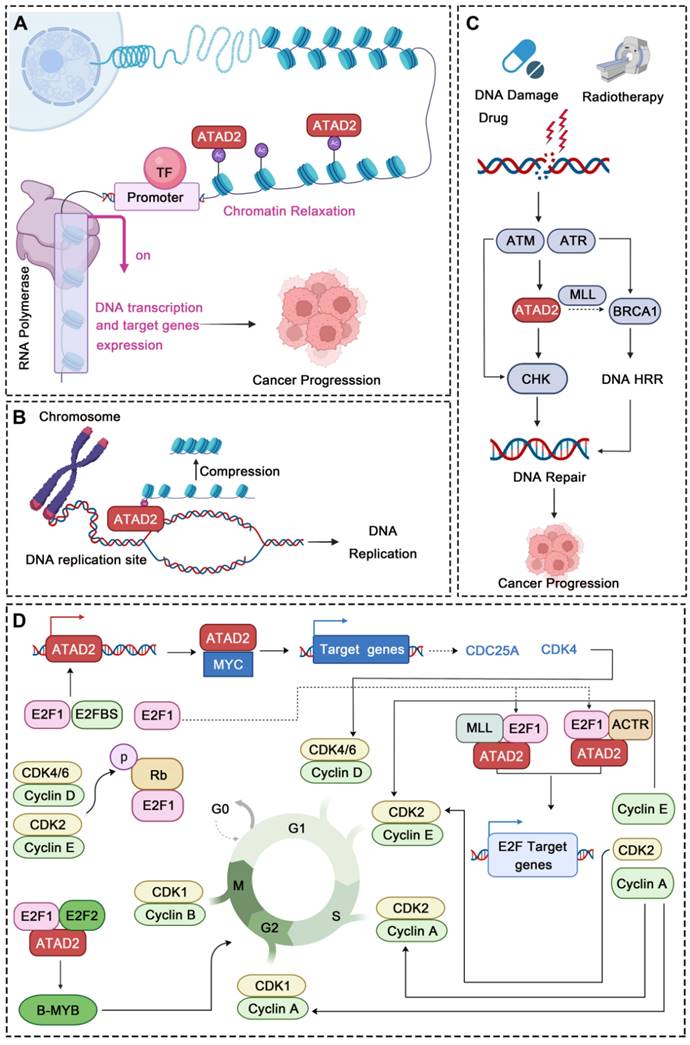
The structure and functions of ATAD2 domains. (A) The domain structure of human ATAD2, C. elegans Lex1, and S. pombe Abo1. NTD, N-terminal acidic domain; AAA-D, AAA+ ATPase domain; BRD, bromodomain; CTD, C-terminal domain. (B) The structure of ATAD2 predicted by AlphaGo [37] and the confidence levels for each domain are also shown. (C) The ATPase domain structure and main function of ATAD2. (D) The bromodomain structure and main function of ATAD2.
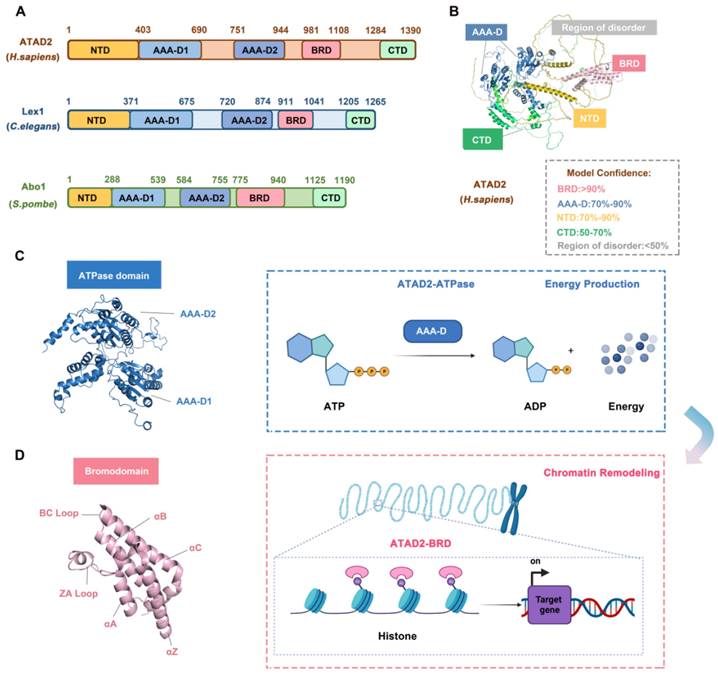
The AAA + ATPase domain is near the NTD module of ATAD2 and consists of a double hexamer consisting of AAA-D1 and AAA-D2, and the former is structurally located on top of the latter. AAA-D1 contains conserved Walker A (GXXXXGKT/S) and B motifs, sensors 1 and 2, and an arginine finger, which is the other conserved domain of ATAD2. The AAA + ATPase domain mediates the oligomerization of proteins to achieve the function of BRD-ATAD2 proteins and is responsible for ATP binding and hydrolysis. Increasing studies have proven that the AAA + ATPase domain mutation will affect its function as an ERα coactivator and interfere with DNA replication [32] (Figure 2C).
BRD, which is a highly conserved chromatin reader domain, is vital for chromatin modification. Structurally, all BRDs have a unique secondary structure, a left-handed bundle structure of four α-helices (αZ, αA, αB, αC) connected through ZA (αZ-αA) and BC (αB-αC) loops. Functionally, BRD binds and recognizes the acetylated lysine of chromatin, which know as a “reader”, to stimulate the transcription of target genes [33]. BRD-ATAD2 is essential for the interaction of ATAD2 with histones and ATAD2-mediated cancer processes [34]. Of note, the BRD-ATAD2 has a unique tectonic fold, named the “BRD fold” or lysine acetylation (KAc) binding pocket. Within this KAc pocket, some highly conserved residues, such as Asn1064 residues, which are directly involved in mediating histone interactions, are arranged here. Thus, the heterogeneous flexibility of the KAc pocket may affect the binding of ATAD2 to ligands and biological function [35] (Figure 2D).
There is mutual communication between the AAA + ATPase domain and BRD-ATAD2, which is in harmony with the finely regulated functions of ATAD2. First, the ATPase domain utilizes ATP hydrolysis to make bromodomain accessible to acetylated histone tails. Second, multiple bromodomains help to better "capture" proteins. Importantly, when the protein is in a multimeric form or in a high molecular weight complex, the interaction between the AAA + ATPase domain and BRD-ATAD2 improves the binding efficiency of the acetylation site of histone H4 [36].
Therefore, the structure of ATAD2 is the basis of its executive function, and the analysis and study of its structure provide the structural basis and the possibility of drug design for the development of anticancer drug candidates by targeting ATAD2.
3. ATAD2 in cancer
Accumulating evidence has revealed that ATAD2 is involved in the carcinogenesis, proliferation, apoptosis and metastasis of various tumor cells. Importantly, ATAD2 is essential for tumorigenesis in melanoma [14], and overexpression of ATAD2 can facilitate the proliferation of tumor cells and impede apoptosis in a variety of malignancies (Figure 3A-C). For example, overexpression of ATAD2 can activate the mitogen-activated protein kinase (MAPK) pathway to promote the proliferation of ovarian cancer cells [5]. Furthermore, ATAD2 promotes tumor progression as a coactivator of the hormone-induced nuclear receptors ERα and androgen receptor (AR), E2Fs and c-Myc [38]. In lung adenocarcinoma and gastric cancer, ATAD2 could promote cell proliferation via the phosphatidylinositol 3 kinase (PI3K) / AKT pathway and the retinoblastoma tumor suppressor protein (Rb)-E2F-cMyc signaling [39, 40]. While in lung adenocarcinoma, the knockdown of ATAD2 affects glucose metabolism in cancer cells by regulating [18F] FDG uptake and lactate production through the AKT-GLUT1 / HK2 pathway [39]. In addition, silencing ATAD2 also inhibits the growth and colony formation ability of esophageal squamous cell carcinoma (ESCC) cells, and significantly inhibits ESCC tumor growth in vitro [41].
ATAD2 in cancer. ATAD2 is involved in multiple processes of cancer occurrence and development, which mainly include 6 major parts. (A) Promoting carcinogenesis by chromatin remodeling. (B) Facilitating uncontrollable proliferation of cancer cells. (C) Inhibiting cell apoptosis. (D) Accelerating the cancer cell cycle progression. (E) Promoting cancer EMT and metastasis. (F) Potential to predict cancer clinical prognosis and drug response of ATAD2.
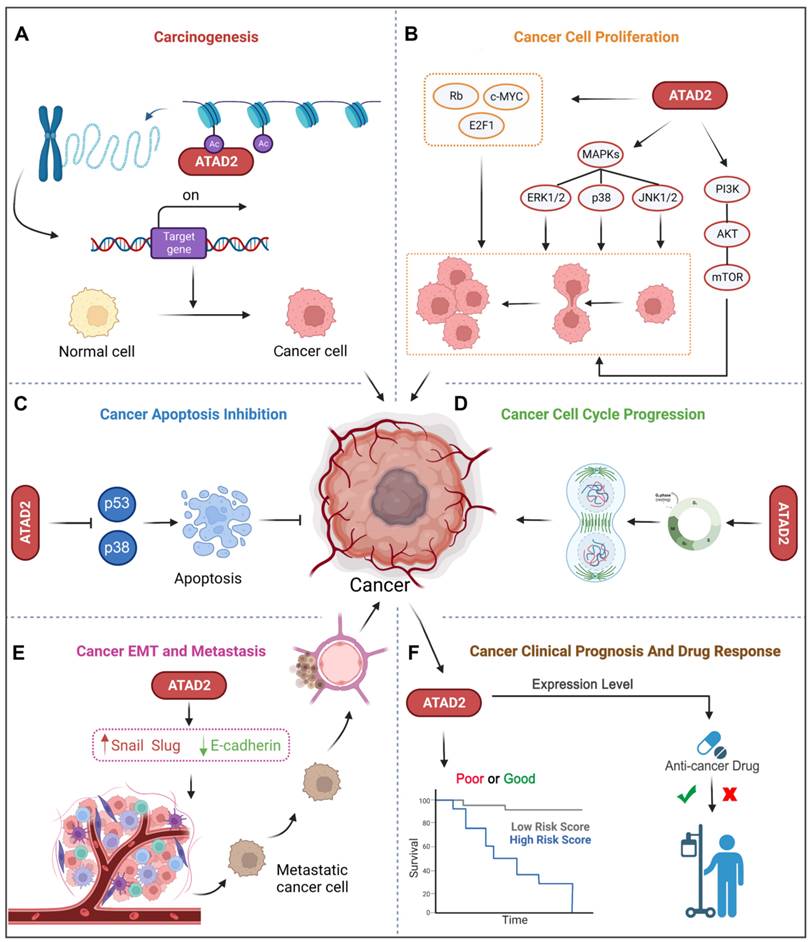
Of note, abnormal regulation of the cell cycle is also a major feature of tumors, and knockdown of the ATAD2 gene leads to decreased expression of cyclin C and cyclin D1 in hepatocellular carcinoma, which is beneficial for cancer treatment [38]. Similarly, the depletion of ATAD2 arrests the cell cycle in G1/S phase and triggers apoptosis through the Rb-E2F1 pathway [42]. And silencing ATAD2 inhibits the proliferation of gastric cancer cells by reducing the expression of the key cell cycle regulator proteins cyclin D1, Rb, E2F1 and cyclin E, thereby arresting the cell cycle in the G1/S phase [40] (Figure 3D).
As we know, metastasis and invasion are one of the malignant behaviors of tumor, and also an important factor leading to high mortality. Studies have shown that the loss of ATAD2 results in decreased protein expression of EMT to impede cancer cell migration and invasion [43]. Silencing ATAD2 inhibited ESCC cell growth and lung metastasis in vivo, and the knockdown of ATAD2 inhibited ESCC migration and invasion in vitro [41]. Additionally, the reduction in ATAD2 could induce a decrease in N-cadherin and Vimentin and an increase in E-cadherin, which is also beneficial for metastasis inhibition [44] (Figure 3E).
In addition to the above-mentioned tumor characteristics, ATAD2 has also been used to evaluate the clinical prognosis of breast cancer (BC), liver cancer, GC and other cancers [39]. Besides, ATAD2 overexpression was significantly associated with poorer overall survival, relapse-free survival, disease-free survival, and disease-specific survival, suggesting an indicative role for ATAD2 in the prognostic significance of human cancers [45]. Some tumor biomarkers (B7-H4, PD-L1 and CMTM6) was notably correlated with ATAD2 in human oral squamous cell carcinoma (OSCC), and it is not surprising that high expression of ATAD2 indicates poor prognosis in OSCC [46]. Furthermore, ATAD2 is also closely related to the cancer drug response, and deletion of ATAD2 has previously been indicated to induce cancer cell apoptosis, both in drug-sensitive and drug-resistant cancer [20]. Remarkably, over-expression of ATAD2 was also significantly associated with tumor size, differentiation, and clinical lymph node metastasis stage and could be used as an independent prognostic factor in GC patients [40] (Figure 3F). Hence, ATAD2 is a promising target for cancer therapy.
4. The molecular basis underlying the regulatory mechanisms of ATAD2
4.1 Upstream regulators
Considering the delicate and complex role of ATAD2 in cells, we first summarized its upstream regulators, including transcriptional regulation, post-transcriptional modification, and protein-protein interactions.
a. miRNAs
MicroRNAs (miRNAs), non-coding RNAs, have the capacity to modulate downstream gene expression by targeting the 3'UTR of target genes [47]. miRNAs negatively regulate gene expression by RNA-induced silencing complex (RISC)-mediated mRNA processing or translational inhibition. Interestingly, studies have demonstrated that a number of miRNAs could regulate the development of different cancer cells by targeting ATAD2 [48]. For instance, miR-520f can restrain the increase in GC cells by targeting the regulation of ATAD2 [49]. MiR-200b-5p inhibits ovarian cancer cell proliferation and promotes apoptosis by targeting ATAD2 and regulating the PI3K/AKT signaling pathway [50]. MiR-372 down-regulates the expression of ATAD2 to inhibit the growth, invasive and migratory capacity of liver cancer cells or renal cell carcinoma cells [13, 51]. Over-expression of miR-186 can reduce the expression of ATAD2, inhibit the Hedgehog signaling pathway and ultimately inhibit the proliferation, migration, invasion and apoptosis of retinoblastoma cells [48]. Besides, miR-302 increases the cisplatin sensitivity of ovarian cancer cells by inhibiting ATAD2 to hinder the invasiveness and EMT in cisplatin-resistant cells [44]. In addition, miR-217 has also been reported to bind to the 3'UTR site, inhibit ATAD2 protein expression and induce apoptosis of pancreatic cancer cells [52]. In summary, miRNAs could directly target and down-regulate the expression of ATAD2 to control cancer progression [53].
b. lncRNAs
Long noncoding RNAs (lncRNAs), with a length of more than 200 nucleotides, are non-translational RNAs that are involved in various cellular processes, including regulation of chromatin structure and transcriptional regulation. Importantly, lncRNAs act as sponges for miRNAs and signal the development of cancer by binding to target genes [47]. PCAT-14 has been reported to promote the proliferation and invasion of cancer cells by inducing miR-372 methylation to increase ATAD2 expression, and then activating the hedgehog signaling pathway in hepatocellular carcinoma [54]. Nuclear paraspeckle assembly transcript 1_2 (NEAT1_2), a common noncoding RNA subtype, can be used as a competitive endogenous RNA of miR-106B-5p to downregulate miR-106B-5p and increase the expression of ATAD2. Therefore, NEAT1_2 knockdown or silencing can inhibit the growth, migration and invasion of thyroid cancer cells [55]. Moreover, MALAT1 promoted the expression of ATAD2 to accelerate cell proliferation, migration, invasion and EMT in Rb by sponging miR-655-3p [43]. LncRNA CRNDE affects CRC cell progression by modulating the miR-126-5p/ATAD2 axis in CRC cells [56]. Thus, lncRNAs also contribute significantly to the regulation of ATAD2 in cancer.
c. Transcription factors and proteins
ATAD2 acts as a cofactor to regulate genes with oncogenic functions together with certain transcription factors, such as c-Myc, E2F, androgen and estrogen receptors [42]. Moreover, ATAD2 has also been identified as a coactivator of ERα, AR, E2Fs, and c-Myc for the promotion of tumor progression [38]. Similarly, ATAD2 can be regulated by a variety of transcription factors or proteins. ATAD2 promoter contains seven potential E2F DNA-binding sites, and E2F1 binds to the regulatory region of the ATAD2 gene to activate its gene transcription [57]. Notably, E2F1 and AR directly target ATAD2 to enhance ATAD2 gene expression at both the mRNA and protein levels [58]. In addition to ERα, AR, and E2Fs, ATAD2 is also a coactivator of c-Myc, enhancing its transcriptional activity [39]. Meanwhile, ATAD2 is a hypoxia-responsive and HIF1α-regulated gene, and HIF1α acts as a transcription factor to mediate the upregulation of ATAD2 in hypoxic SCC by binding to the ATAD2 promoter [59]. Recently, a study found that the transcription factor MYBL2 can bind to the promoter of ATAD2 to promote the expression of ATAD2 to trigger ovarian cancer cell proliferation. Interestingly, when ATAD2 is knocked out, the stability of MYBL2 is also severely reduced as a fed back [60]. Interestingly, ATAD2 can also be directly or indirectly regulated by a variety of proteins. For instance, KDM8 / JMJD5, a histone lysine demethylase/dioxygenase, directly targets ATAD2 to maintain cell survival via AR activation of EZH2 in prostate cancer [19]. Besides, TRIM25 is an oncogenic ubiquitin E3 ligase that feedback regulates ATAD2 expression in a dose-dependent and transcription-independent manner to promote CRC development [61]. Methyltransferase-like 3 (METTL3), a reported m6A “writer”, was found to promote the growth and invasion of osteosarcoma by enhancing the expression of ATAD2, but the regulatory mechanism is unclear [6]. Additionally, the endoplasmic reticulum-associated degradation protein Derlin-1 can upregulate the expression of ATAD2, but the mechanism is also unclear [62]. Notably, ATAD2 recently proved to be a direct target gene of YAP1, whose expression can be enhanced by YAP1 activation. Surprisingly but intriguingly, recruitment of BRD4, TEAD4 and YAP1/TAZ at ATAD2 promoter is interdependent, which can fine-regulate the expression intensity of ATAD2 together [63].
Therefore, ATAD2 can be fine-tuning by different regulators to cope with a variety of life processes in cancer, which also provides a theoretical basis for targeting ATAD2 for cancer treatment.
4.2 ATAD2-modulated cancer-associated signaling pathways
As an important epigenetic target, ATAD2 can regulate the expression of various cancer-related genes and participate in all aspects of cancer progression. In this section, we mainly elaborate on the cancer pathways regulated by ATAD2 and further clarify the molecular basis of ATAD2 as a promising candidate cancer target.
4.2.1 ATAD2 in Chromatin Remodeling
As an important epigenetic target, ATAD2 is best known for its role in chromatin remodeling. Of note, chromatin remodeling has been a hot topic in epigenetic research in recent years, and it is also an important issue in cancer biology [66, 67]. Mechanistically, ATAD2 directly binds to chromatin via histone acetylation identification, leading to an increase in chromatin accessibility and histone dynamics, which provide sufficient conditions for the appropriate activity of the highly expressed gene fraction. Additionally, ATAD2 acts as a regulator of chromatin dynamics to maintain specific gene expression programs through histone acetylation-directed chromatin opening [68]. Besides, a structural study of ATAD2 found that BRD-ATAD2 can specifically direct proteins to acetylated histones, and its AAA + ATPase domain can act as a molecular motor in the process of acetylated histones. In this process, the acidic N-terminal region of ATAD2 binds tightly but nonspecifically to chromatin, then the AAA + ATPase domain promotes nucleosome expulsion, and the BRD read the acetylated lysines on histones [69]. Intriguingly, recently a study has found that the D/E repeats sequence of ATAD2 is closely related to its chromatin binding capacity, and the chromatin binding capacity of ATAD2 with D/E repeats knockout is greatly reduced [70], as is the corresponding chromatin remodeling capacity. Undoubtedly, ATAD2 could regulate cancer progression by controlling chromatin remodeling. For instance, a recent study revealed that ATAD2 could promote the formation of melanoma phenotypes through chromatin remodeling. In detail, ATAD2 led to a significant increase in chromatin accessibility and formed a complex with SOX10 that promoted the expression of MAPK-related genes [14]. In hepatocellular carcinoma, clinical value and biological function comprehensive bioinformatics analysis showed that ATAD2 plays an important role in carcinogenesis by interfering with the interaction between chromatin proteins and DNA [38]. In short, ATAD2 can indeed promote cancer progression through chromatin remodeling.
Upstream regulators of ATAD2. (A) Post-transcriptional regulation of ATAD2 expression via miRNAs and long non-coding RNAs. (B) Transcription factors, transcription co-activators, and some proteins can regulate ATAD2 expression or stability.
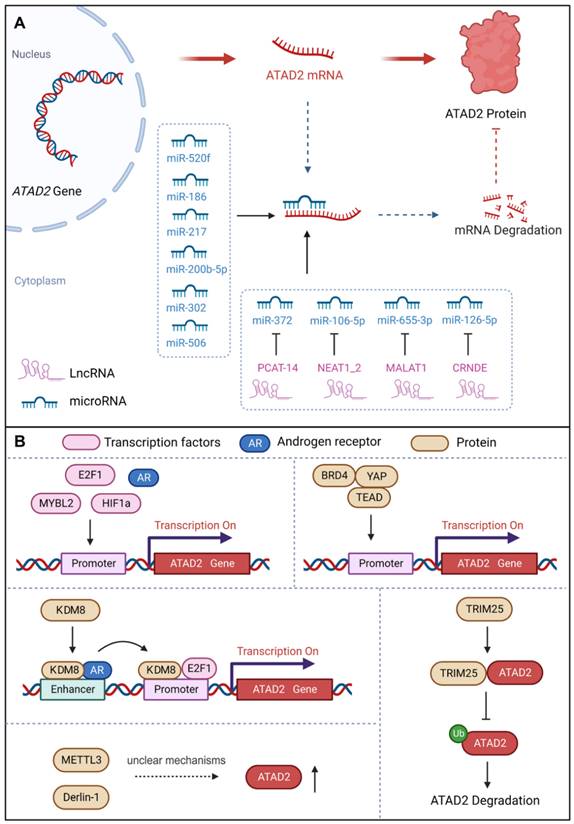
Upstream regulators of ATAD2 in cancer.
| Name | Type | Positive/Negative | Mechanism | Cancer Type | Ref. |
|---|---|---|---|---|---|
| miR-520f | miRNA | - | miR-520f directly targets ATAD2 and downregulates its expression | Gastric carcinoma | [49] |
| miR-200b-5p | miRNA | - | miR-200b-5p directly targets ATAD2 and downregulates its expression | Ovarian cancer | [50] |
| miR-372 | miRNA | - | miR-372 targets ATAD2 and suppresses its expression | Hepatocellular carcinoma | [54] |
| miR-186 | miRNA | - | miR-186 directly targets ATAD2 and downregulates its expression | Retinoblastoma | [48] |
| miR-302 | miRNA | - | miR-302 directly targets ATAD2 and downregulates its expression | Ovarian carcinoma | [44] |
| miR-217 | miRNA | - | miR-217 targets ATAD2 and suppresses its expression | Pancreatic cancer | [52] |
| miR-506 | miRNA | - | miR-506 could target and down-regulate ATAD2 | Lung adenocarcinoma | [53] |
| PCAT-14 | lnc RNA | + | PACT-14 upregulates ATAD2 expression by sponging miR-372 | Hepatocellular carcinoma | [54] |
| NEAT1_2 | lnc RNA | + | NEAT1_2 upregulates ATAD2 expression by sponging miR-106b-5p | Papillary thyroid cancer | [55] |
| miR-106b-5p | miRNA | - | miR-106b-5p directly targets ATAD2 and downregulates its expression | Papillary thyroid cancer | [55] |
| MALAT1 | lnc RNA | + | LncRNA MALAT1 promoted ATAD2 expression via miR-655-3p | Retinoblastoma | [43] |
| miR-655-3p | miRNA | - | miR-655-3p directly targets ATAD2 and downregulates its expression | Retinoblastoma | [43] |
| CRNDE | lnc RNA | + | CRNDE upregulates ATAD2 expression via inhibiting miR-126-5p expression | Colorectal carcinoma | [56] |
| miR-126-5p | miRNA | - | miR-126-5p directly targets ATAD2 and downregulates its expression | Colorectal carcinoma | [56] |
| E2F1 | transcription factor | + | E2F1 directly target ATAD2 by binding to and activating the ATAD2 promoter. | Breast cancer | [64] |
| AR | Androgen receptor | + | Androgens activates ATAD2 via directly interaction of AR with an ARBS within the gene | Prostate cancer | [65] |
| HIF1α | transcription factor | + | HIF1α as the transcription factor mediating upregulation of ATAD2 in hypoxic SCCs | Stomach cancer | [59] |
| MYBL2 | Transcription factor | + | MYBL2 binds the CHR elements in the ATAD2 promoter to promote ATAD2 expression | Ovarian cancer | [60] |
| KDM8/JMJD5 | Protein | + | KDM8 directly targets ATAD2 and up-regulates its expression | Prostate cancer | [19] |
| TRIM25 | Protein | + | TRIM25 promoted ATAD2 expression in a dose-dependent and transcriptional-independent manner | Colorectal cancer | [61] |
| METTL3 | Protein | + | METTL3 up-regulates ATAD2 expression, but the regulatory mechanism is unclear | Osteosarcoma | [6] |
| Derlin-1 | Protein | + | Derlin-1 directly upregulates ATAD2 expression | Breast cancer | [62] |
| YAP1/TAZ | Protein | + | YAP1/TAZ enters the nucleus and binds to the promoter of ATAD2 to promote ATAD2 expression | Head and neck squamous cell carcinoma (HNSCC) | [63] |
4.2.2 ATAD2 in DNA Replication, DNA Damage and Repair
In addition to chromatin remodeling, ATAD2 can also participate in the regulation of DNA replication, DNA damage and repair in cancer. DNA replication occurs in the S phase while ATAD2 is always highly expressed in this phase and directly regulates DNA replication. During this process, ATAD2 is recruited to DNA replication sites through interaction with the di-acetylation mark and binds to newly synthesized histones, which may assist in heterochromatin compression [32]. Furthermore, the newly synthesized histone H4 was incorporated into chromatin during DNA replication at AcK5 and AcK12 sites. Overexpression of ATAD2 can protect this mark from being removed, while HDAC2 competes with ATAD2 for the target acetyl group to fine-tuning the balance of H4 acetylation [71]. In addition, ATAD2 also has a very obvious response to DNA damage and repair (DDR). After DNA damage drug treatment or radiotherapy, ATAD2 can be highly expressed by ATM/ATR pathway. When activated, ATAD2 can accelerate CHK1/2-regulated DNA damage repair signaling and may promote BRCA1 expression by increasing the recruitment of H3K4 methyltransferase MLL1 at the BRCA1 promoter, which may further promote the homologous recombination repair (HRR) of the damaged DNA [15]. For instance, in pancreatic cancer, ATAD2 inhibition increases the sensitivity of pancreatic cancer cells to gemcitabine and radiotherapy. When ATAD2 is inhibited, the DNA damage of the cancer cells is significantly intensified, while the expression of DNA damage repair proteins is significantly down-regulated, further demonstrating the crucial role of ATAD2 in DDR [72]. Thus, due to its important role in DNA replication and DDR, ATAD2 may be a potential candidate target in cancers that are susceptible to DNA damage.
4.2.3 ATAD2 in Cell Cycle Regulation
Strikingly, abnormal cell cycle is also a major feature of cancer, and more and more evidence also shows that ATAD2 plays an important role in the regulation of various stages of cancer cell cycle. In CC and GC, ATAD2 was significantly over-expressed. After ATAD2 knockout, the cancer cells showed significant G1/S cell cycle arrest, which was related to the Rb-regulated cell cycle pathway [40, 73]. Rb signaling is a key signaling point in cell cycle regulation, which is mainly composed of CDKN, D-type cyclins, cyclin-dependent protein kinases (CDK4, CDK6), the Rb pocket protein family (Rb, p107, p130), and the E2F transcription factor family (E2F1-8 and DP1-2). Of them, Cyclin D and CDK4/6 disrupt the Rb-E2F interaction through phosphorylation of Rb proteins, resulting in activation of E2F-regulated gene expression [74]. Cyclin D1 and CDKs can drive G1 to S phase progression by phosphorylating Rb [43]. Interestingly, cyclin D1, E2F1 and Cyclin E proteins all showed a down-regulation trend after ATAD2 knocking out, which further indicated that ATAD2 could regulate cell cycle progression by regulating the expression of key proteins of Rb-E2F1 signaling pathway [40]. Notably, ATAD2 can also regulate transcription factors such as E2F and MYC to control the cell cycle [40]. As to E2F, ATAD2 can interact directly with E2F and bind to H3K14ac via its bromine domain. During the transition from G1 to S, the interaction between ATAD2 and E2F reaches its maximum intensity. In this process, Host cytokine 1 (HCF-1)-MLL histone methyltransferase complex and nuclear hormone receptor coactivator ACTR also contribute to the recruitment of E2F targets by ATAD2 and the assembly of chromosomes [9]. And as to MYC, ATAD2 cooperates with MYC in activating transcription, and it is essential to the efficient transcriptional activation of numerous MYC target genes [64], including key cell cycle regulators, such as CDKs, cyclins, and E2F transcription factors [75]. In addition, B-MYB is a member of the Myb family of highly conserved transcription factors, which not only regulate the cell cycle but are also ubiquitously expressed in strong proliferative cancer cells. High expression of ATAD2 can facilitate the up-regulation of B-MYB [57], while ATAD2 silencing significantly inhibits the expression of B-MYB and some cell cycle proteins, such as cyclin E1, cyclin B1 and cyclin A2 in malignant tumors [76]. Accordingly, ATAD2 can encourage the process of cell cycle through a variety of aspects to promote cancer progression.
4.2.4 ATAD2 in Gonadal Hormone Signaling
As we all know, gonadal hormones are essential for human growth and development. In recent years, more and more studies have illustrated that abnormal gonadal hormone signaling pathway is also an important risk factor for the onset and development of various gonadal hormone-related tumors, especially breast cancer and prostate cancer [77, 78]. Excitingly, ATAD2 has also been identified to be involved in the regulation of gonadal hormone signaling in cancer progression.
Estrogen Receptor
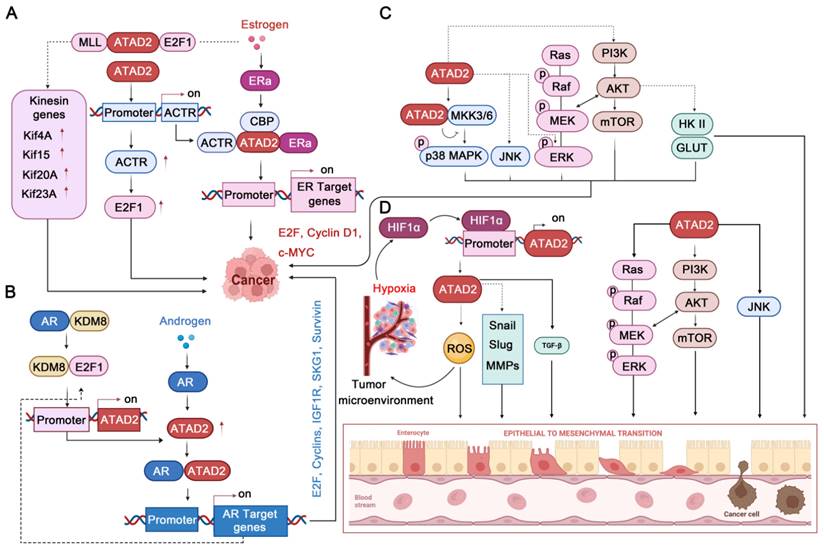
ATAD2 is a transcriptional co-regulator of ER, which can affect tumorigenesis by enhancing the transcription of ER target genes [7, 9]. Upon estrogen stimulation, ATAD2 is selectively recruited to the promoters of ERα target genes, such as cyclin D1, c-Myc and E2F1, which are required for estrogen-induced cancer cell proliferation. Intriguingly, ATAD2 does not occupy the promoters in ERα target genes but promotes the recruitment or assembly of the ERα-CBP complex at the chromatin, which is crucial for histone modification [7]. In addition, ATAD2 participates in regulating the E2-dependent recruitment of E2F and MLL1 at the promoters of the kinesin genes (such as Kif4A, Kif15, Kif20A, and Kif23), promoting their expression. Of note, the promotion of kinesin expression mediated by ATAD2-E2F1-MLL1 is required for BC cell proliferation and survival and is associated with the prognosis of BC patients [12]. Besides, ATAD2 can control the photo-oncogene ACTR/A1B1 expression by recruiting to its promoter [79], while ACTR can interact with estrogen-bound ER to strongly co-activate the transcription of ER target genes [80]. Additionally, ACTR can directly interact with E2F1 and then locates in the promoter of E2F target genes to promote their expression [81]. It is worth noting that the ACTR-E2F pathway can promote the proliferation of BC even without the stimulation of estrogen and the participation of ER [81]. Therefore, the ATAD2-ACTR-E2F pathway may still have potential targeting value in the treatment of ER-negative BC.
Androgen Receptor
Similar to ER, androgen receptor (AR) is mainly activated by the androgen steroid hormones testosterone and 5α-dihydrotestosterone (5α-DHT), which is the major factor promoting the proliferation of prostate cancer [82]. Transcriptional activation or repression mediated by AR, as a member of the nuclear hormone receptor superfamily, requires several co-regulators, including ATAD2 [83]. It has been found that inhibition of ATAD2 strongly impedes the proliferation of prostate cancer cells and leads to apoptosis enhancement. In addition, overexpression of ATAD2 enhances the expression of a multitude of AR target genes, such as IGF1R, IRS-2, SGK1, E2F1, Cyclin A2, Cyclin D3, and Survivin. Mechanistically, ATAD2 directly interacts with the DBD-hinge region of AR via its NH2 terminus to facilitate its transcriptional activity and enhance androgen-stimulated gene expression [83]. Likewise, ATAD2 can be also regulated by the E2F1 transcription factor, and E2F1 interacts with AR on the ATAD2 promoter through a chromatin loop to activate transcription upon androgen stimulation [65]. Interestingly, in the case of androgen deficiency, KDM8 could interact with AR to enhance the expression of androgen-responsive genes, such as ATAD2 and EZH2. Impressively, EZH2 can also be activated by AR via ATAD2 binding to its promoter [19]. Therefore, ATAD2 can assist the expression of androgen receptor target genes from multiple paths.
ATAD2 regulates chromatin remodeling, DNA Replication, DNA Damage and Repair, and cell cycle. (A) ATAD2 promotes chromatin remodeling by binding to the acetylation site of histones. This binding leads to chromatin relaxation and promotes DNA transcription and gene expression. (B) ATAD2 regulates DNA replication through interaction with the di-acetylation mark and binds to newly synthesized histones, which may assist in heterochromatin compression. (C) ATAD2 enhances the DDR response via activation of the CHK1/2 and BRCA1. (D) ATAD2 facilitates the cell cycle progression mainly through MYC, b-MYB, and E2F activation. In this process, the activation of CDKs and cyclins is also regulated by ATAD2.
Together, ATAD2 acts as a transcriptional co-activator of ER and AR, regulating the expression of ER/AR-targeted genes to control cancer cell proliferation and survival [84], even without gonadal hormone stimulation. Therefore, it is not difficult to speculate that in cancer with negative or positive gonadal hormone receptors, ATAD2 may both have strong potential targeting value.
4.2.5 ATAD2 in MAPKs and PI3K/AKT/mTOR Pathways
As well-known cancer-related family proteins, MAPK cascades and AKT signaling play important roles in a variety of cellular processes. Notably, MAPK and PI3K/AKT/mammalian target of rapamycin (mTOR) signaling have a close relationship, and the complex network they constitute is of great significance in cancer cell proliferation, angiogenesis, metastasis, metabolic changes, and response to anticancer drugs [85-87]. Excitingly, ATAD2 is also involved in regulating this complex network that governs the fate of cancer. In ovarian cancer, ATAD2 deletion attenuates the MAPK pathways by decreasing the phosphorylation levels of JNK1/2 and ERK1/2 MAPK [5]. Likewise, ATAD2 inhibition by miR-200b impedes the protein expression levels of PI3K and p-Akt, thereby inhibiting the proliferation and apoptosis of ovarian cancer cells. Conversely, highly expressed ATAD2 protein can promote the protein expression levels of PI3K and p-Akt to promote ovarian cancer progression [50]. In HCC, ATAD2 deletion could activate p38 MAPK to activate apoptosis. Mechanistically, ATAD2 directly interacts with MKK3/6, which is the upstream regulator of p38 MAPK [88]. Similarly, ATAD2 can also regulate the proliferation, tumorigenicity, and migration of cancer cells through PI3K/AKT pathway in lung adenocarcinoma [39, 53]. In this process, ATAD2 significantly positively upregulates the expression of p-AKT, GLUT1, and HK2, which also contribute to glucose uptake and metabolism [39]. In addition, ATAD2 inhibition by miR-506 can improve lung adenocarcinoma's sensitivity to cisplatin-based hyperthermia by inhibiting the ATAD2-PI3K-AKT pathway [53]. Encouragingly, a recent study found that AM879, an inhibitor of ATAD2, blocks the PI3K-AKT-mTOR pathway through ATAD2 inhibition, thereby facilitating breast cancer cell apoptosis and autophagy-associated cell death [89]. Unfortunately, but still hopeful, although the involvement of ATAD2 in the regulation of MAPKs and AKT signaling pathways is well established, the exact mechanisms are still in their infancy.
4.2.6 ATAD2 in HIF1α-mediated Hypoxia Signaling and EMT Pathways
In cancer, another major issue that cannot be ignored is cancer microenvironment and cancer metastasis [90, 91]. Of note, the hypoxic microenvironment is necessary for the development of solid tumors and contributes to tumor EMT, angiogenesis, and metastasis [92]. EMT is a biological process in which epithelial cells are transformed into cells with a mesenchymal phenotype by a specific procedure. In epithelial-derived malignancies, the migration and invasion abilities of tumor cells are closely related to EMT [93]. Intriguingly, ATAD2 has also been found to contribute significantly to cancer response to hypoxia and EMT. In stomach cancer and lung cancer, ATAD2 and HIF-1 α always highly expressed [59, 94]. Mechanistically, ATAD2 is a hypoxia-responsive gene, and its promoter contains an HIF1α binding site (HBS) and HIF1α ancillary site. Under hypoxic conditions, HIF1α binds to the ATAD2 promoter, and the expression of ATAD2 is encouraged, which further promotes the cancer proliferation and migration process [59]. In lung cancer, HIF-1α activates ATAD2 to enhance cancer development, which may further facilitate mitochondrial ROS production that contributes to enhancing cancer cell stemness [94]. As to EMT, the most striking sign is the upregulation of N-cadherin and Vimentin protein expression and downregulation of E-cadherin [43]. Notably, inhibition of ATAD2 can inhibit EMT by reducing the expression of EMT regulators such as slug and snail in OSCC [46]. In addition, ATAD2 silencing inhibits EMT through upregulation of E-cadherin and downregulation of vimentin in CRC cells, thereby preventing tumor invasion and migration [38]. Besides, the activation of PI3K-AKT-mTOR and MAPKs signaling by ATAD2 may contribute to EMT. Interestingly, the TGF-β1 cascade activation by ATAD2 may also contribute to the EMT process [41]. It is worth noting that the underlying mechanism of how ATAD2 regulates these key EMT regulators have not yet been fully revealed (TGF-β1-SMAD3 may contribute a lot), but ATAD2 does contribute significantly to cancer progression by regulating HIF1α-mediated hypoxia signaling pathways and EMT pathways.
4.2.7 ATAD2 in Other Cancer-associated Signaling Pathways
Despite the research history of ATAD2 is not long, with the deepening of research, ATAD2 has also been found to be involved in many other cancer related pathways, including hedgehog (HH), TGF-β1, YAP/Hippo, p53 and NF-κB signaling pathways.
ATAD2 in gonadal hormone MAPK, and PI3K/AKT/mTOR, HIF1α-mediated hypoxia, and EMT signaling pathways. (A-B) ATAD2 plays an important role in gonadal hormone signaling by accelerating ER/AR target gene expression. (C) ATAD2 activates p38-MAPK, JNK1/2, and ERK1/2 MAPK to regulate cancer cell progression. In addition, ATAD2 activates PI3K-AKT-mTOR signaling to stimulate glucose metabolism and cancer progression. (D) HIF1α promotes ATAD2 expression to enhance EMT through up-regulating EMT-related genes, such as Snail, Slug, MMPs. Besides, ATAD2-regulated ROS production, ERK1/2, AKT and JNK1/2, and TGF-β pathways also contribute to enhancing EMT.
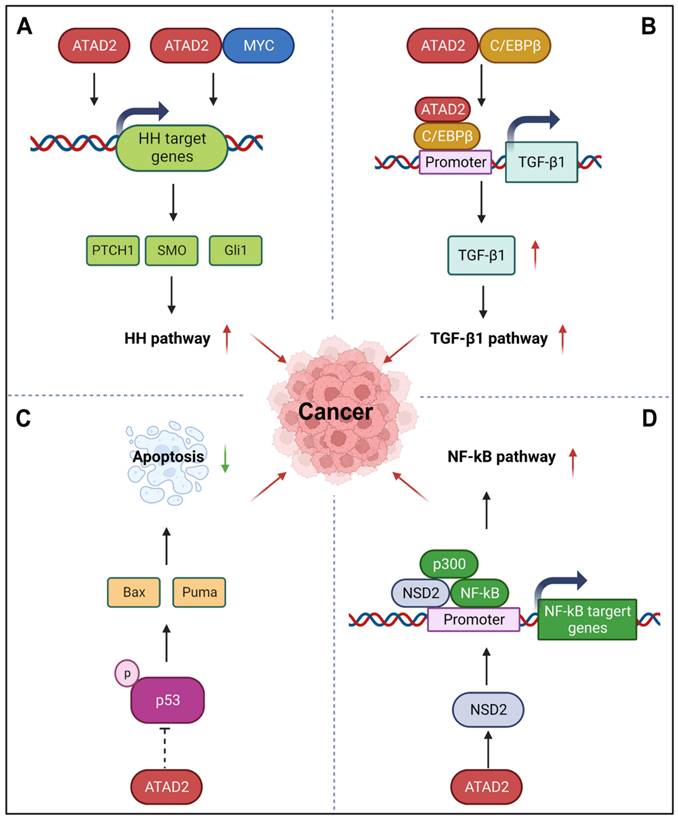
HH signaling pathway is a highly conserved evolutionary pathway that involves various stages of carcinogenesis [95]. Interestingly, ATAD2 can regulate the HH pathway to promote cancer development, angiogenesis, invasion, and metastasis. For instance, silencing ATAD2 can impede the HH signaling pathway by reducing the expression of Gli1, SMO, and PTCH1 [48]. Additionally, ATAD2 and MYC can co-regulate the expression of the HH pathway-related genes SMO and Gli1 through MYC-interacting zinc finger protein 1 (MIZ1) and accelerate the activation of the Hedgehog pathway [96]. For example, miR-186 can inhibit HH pathway by impairing the expression of ATAD2, which is beneficial to the inhibition of angiogenesis of retinoblastoma [48]. Similar to the HH pathway, TGF-β1 signaling is also an important developmental signaling pathway that controls cell fate [97]. While ATAD2 directly interacts with C/EBPβ, the transcriptional activator of TGF-β1, to promote its nuclear translocation and directly bind to the promoter of TGF-β1 to activate TGF-β1 expression. Then, the activation of ATAD2-TGF-β-SMAD3 signaling may further enhance the EMT of ESCC [41]. YAP/Hippo signaling is another key pathway that controls organ development, its incontrollable is closely related to the occurrence and development of cancer [98]. Recently, ATAD2 was found to be directly regulated by YAP1, which contributes to the tumorigenesis of head and neck squamous cell carcinomas. During this process, YAP, TAZ, and TEAD form transcription complexes and recruit BRD4 to promote the open state of chromatin and promote the transcriptional activation of ATAD2 [63]. Furthermore, p53, as the most well-known tumor suppressor protein, its important role in cancer that needs not to be explained. Importantly, ATAD2 has also been reported to be involved in the p53-regulated apoptotic cascade. In the presence of wild-type p53, silencing of ATAD2 induces the phosphorylation activation of p53 in HCC cells, as well as the activation of a variety of pro-apoptotic proteins, including Puma, Bax, and so on [88]. Unfortunately, the exact regulatory mechanisms of ATAD2 on p53 remain to be fully elucidated. Besides p53, NF-κB signaling pathway also participates in the regulation of apoptosis. Meanwhile, it is also known for its role in the cellular response to inflammation and immunity [99]. Hopefully, ATAD2 may regulate NF-κB signaling through its target protein histone methyltransferase NSD2. As a co-activator of NF-κB, NSD2 directly binds to NF-κB and is recruited to the NF-κB target gene promoter. In addition, co-activator p300 is also involved in this process, which further activates the NF-κB signaling [100].
Hence, the relevant signal network with ATAD2 as the core plays an important role in various pathways that cancer depends on for survival. Undoubtedly, ATAD2 has an important hopeful targeting value in cancer therapy, and these biological processes regulated by ATAD2 are the molecular basis for ATAD2 to become a promising candidate drug target in cancer therapy.
ATAD2 in other signaling pathways in cancer. (A) ATAD2 can stimulate or co-regulate the expression of some key HH pathway genes with MYC to activate the HH pathway. (B) ATAD2 promotes the expression of TGF-β1 via interacting with C/EBPβ, the transcriptional activator of TGF-β1, thus enhancing the TGF-β1 pathway. (C) ATAD2 inhibits the activation of p53 to suppress cancer cell apoptosis. (D) ATAD2 may activate the NF-κB signaling through the activation of NSD2, which is a co-activator of NF-κB.
5. Targeting ATAD2 with pharmacological small-molecule compounds in cancer therapy
Given the comprehensive cancer-related functions of ATAD2, disrupting the ATAD2-acetyl-lysine interactions has emerged as a potential therapeutic target in cancer, and a lot of pioneering efforts have been made to develop ATAD2 inhibitors from both pharmaceutical and academic settings.
5.1 The discovery of fragment hits for ATAD2 Bromodomain
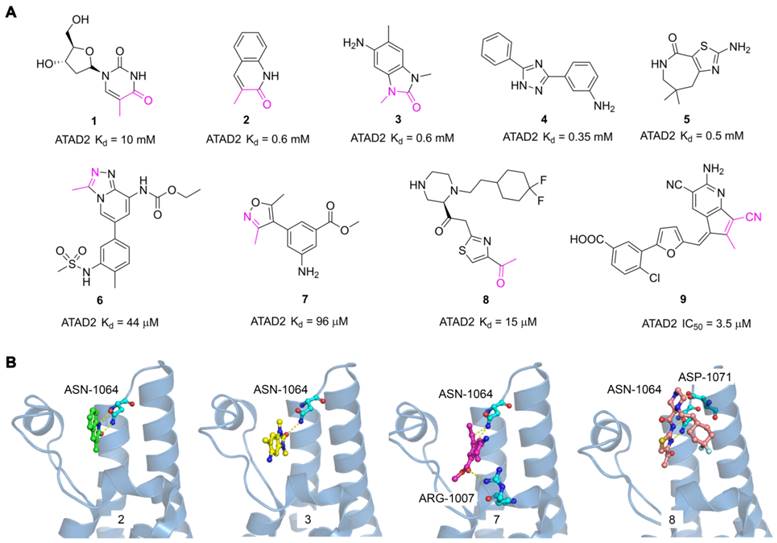
The discovery of hits with potential activity is crucial for the development of targeted drugs. In 2014, Apirat Chaikuad et al. first discovered nine novel hits with weak affinity for ATAD2 by crystallographic fragment screening, including thymidine (1) and 3-methylquinolin-2(1H)-one (2) with Kd value of 10 mM and 0.6 mM respectively [101]. The crystal structures of fragments showed the endyllamide group of 3-methylquinolin-2(1H)-one (2) initiated two conservative hydrogen bonds with the ASN1064 residue which is the crucial active center to adopt the acetyl-lysine of substrates. Furthermore, the methyl group in the adjacent position occupied a tight hydrophobic space, which enhanced the affinity to ATAD2. 3-methylquinolin-2(1H)-one (2) is a novel acetyl-lysine mimetic ligand, offering a promising chemical starting point for the development of more potent ATAD2 inhibitors. Another fragment-based approach was performed to discovery new ATAD2 fragment hits, the results revealed three hits (Compound 3, 4, 5) were found by NMR spectroscopy. According to the crystal structure of 3 with ATAD2, a conserved hydrogen interaction was formed between the carbonyl group and ASN1064 residue and the methyl group played an important role in the simulation of the acetyl group [102]. To screen new ATAD2 inhibitors, structure-guided studies were conducted by John E. Ladbury from the MD Anderson Cancer Center. They elucidated an unexpected conformational change of the conserved ASN1064 residue plays an important role in driving the peptide-binding conformation remodeling. The methyltriazolium- and dimethylisoxazole-containing ligands (6, 7) were identified as potential ATAD2 inhibitors by imitating acetyl group [103]. The conformational selection of the ATAD2 bromodomain was explored through the synergistic combination of molecular dynamics and protein crystallography. The gatekeepers Ile1074, Val1008, and Val1013 residues played an essential role in assuming multiple orientations for different ligands. It is a novel strategy to discover potent and selective ATAD2 inhibitors by coupling the ZA-loop rigidification and the rearrangement of the hydrophobic side chains. Based on the strategy, a potent ATAD2 inhibitor (8) was screened with a Kd value of 15 μM [104]. The linker of acid amides formed two hydrogen bonds with ASN1064, and the piperazine group initiated a hydrogen interaction with ASP1071 located at the BC loop, which provided a promising scaffold to further optimize selective ATAD2 inhibitors. Based on the resolution of binding modes of ligands and ATAD2 bromodomain, they adopted a structure-based virtual screening strategy to discover a novel ATAD2 inhibitor (9, AM879) with an IC50 value of 3565 nM. AM879 significantly inhibited breast cancer cell proliferation with an IC50 value of 6.0 μM and induced significant apoptosis and autophagy via PI3K-AKT-mTOR signaling in MDA-MB-231 cells [89]. Collectively, although these fragment hits and leads did not show a very strong affinity for the ATAD2 bromodomain, they promoted the understanding of the ligand and receptor binding modes, which lays a key foundation for the discovery of highly selective novel targeting ATAD2 inhibitors.
5.2 Azepines derivatives
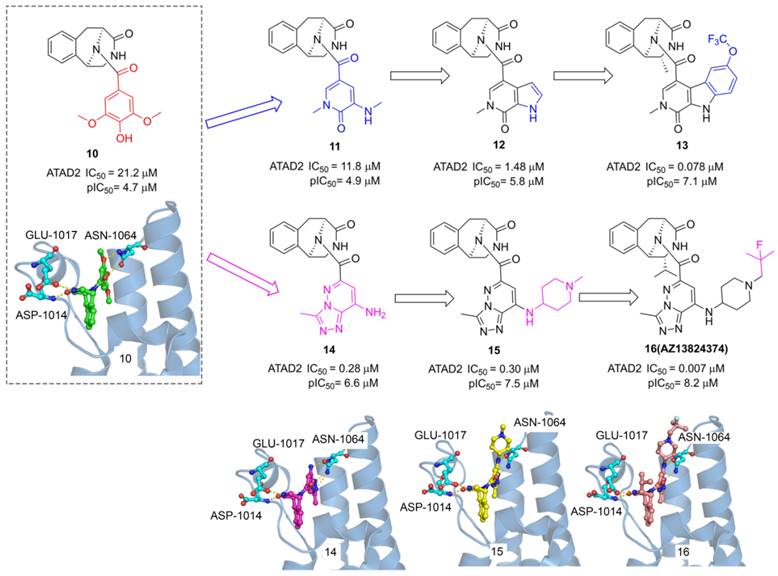
ATAD2, as an epigenetic bromodomain-containing oncology target overexpressed in many cancers, has attracted widespread attention among pharmaceutical companies. The researchers from AstraZeneca conducted a high throughput screen by TR-FRET assay, a tetrahydroisoquinoline derivative (10) with IC50 value of 21.2 μM was screened from the AstraZeneca screening collection including 1.8 M samples [29]. The X-ray structure showed 6 occupied in the acetyl-lysine binding site of ATAD2, two key hydrogen interactions were observed between the phenol and Asn1064, and two additional hydrogen interactions were formed between the tetrahydroisoquinoline group and GLU1017, ASP1014 from ZA loop associated with the selectivity for ATAD2. To improve the affinity for active Asn1064, the phenol group of 10 was replaced by a pyridone warhead inspired by the previous fragment hit (2). Derivative 11 presented a significantly improved potency with an IC50 value of 11.8 μM. More pyridine derivatives were designed and synthesized, such as pyrrolopyridinone derivative (12, IC50 value of 1.48 μM). Compound 13 showed the most potent against ATAD2 in this subseries with an IC50 value of 0.078 μM, which combines a 2R-methyl with an OCF3 substituent on the pyridoindolone ring. As expected, the carbonyl of pyridone formed a hydrogen bond with Asn1064 and the methyl group occupied the hydrophobic pocket. Furthermore, the phenol group of 10 was replaced by triazolopyridazine, a classical acetyl group mimics, producing a new series of TAD2 inhibitors. 3-methyl-[1,2,4]triazolo[4,3-b]pyridazin-8-amine derivative (14) played a significantly more potent affinity against ATAD2 than its pyrrolopyridinone analog 11. The triazolopyridazine group initiated two conserved hydrogen bonds with ASN1064 and additional two hydrogen bonds were preserved between tetrahydroisoquinoline group and GLU1017, ASP1014 residues. Next, 1-methylpiperidin-4-amine moiety was introduced to product 15 with a similar potency as 14. The 2-fluoro-2-methyl-propyl-piperidine derivative (16, AZ13824347) showed the most potent activity with an IC50 of 7 nM, and antiproliferatory activity with a pIC50 of 6.9. Due to the good potency, selectivity, and permeability of AZ13824347, AstraZeneca is conducting a comprehensive pharmaceutical study on AZ13824347, expected to enter into clinical trials.
The reported fragment hits for ATAD2 Bromodomain and some representative X-ray structures of ligands and ATAD2 Bromodomain, including 2 (PDB code 5A5O), 3 (PDB code 4TYL), 7 (PDB code 4TTE), and 8 (PDB code 6HIE).
5.3 3-methylquinolinone derivatives
The 3-methylquinolin-2(1H)-one (2) was an important fragment hit to optimize potent selective ATAD2 inhibitors which caused the extensive interest of GlaxoSmithKline researchers. Firstly, N,N-dimethylethane-1,2-diamine group was incorporated into the 8th site of quinolinone core to yield 17 with pIC50 of 3.3 μM. The endyllamide group formed two conserved hydrogen bonds with ASN1064, and the NH moiety also initiated a hydrogen interaction with ASN1064 enhancing the affinity. The methyl group occupied the hydrophobic pocket and the N, N-dimethylethane-1,2-diamine side chain interacts with ASP1071 by a hydrogen bond. The side chain was replaced with piperidin-4-amine group to product 18 which presents an improved affinity with a pIC50 of 4.9 μM. To optimize the selectivity for ATAD2, 3-methoxypyridine group was incorporated into the 5th of 3-methylquinolinone to form a hydrogen bond with ASP1014 located in the ZA loop, 19 presented a significant affinity for ATAD2 with pIC50 of 1.3 μM [105]. Based on the analysis of 18 and ATAD2, a potential hydrophobic pocket closing to the RVF shelf of the ZA loop was found. A cyclic sulfone group was incorporated into the 3'-site of piperidine to yield 20 with pIC50 of 5.6 μM, initiating additional three hydrogen bonds with ARG1077 and ARG1007. Based upon the structure of 19, 3-methylpyridine group was also induced the 5th of 3-methylquinolinone, 21 showed the most potent affinity with IC50 of 130 nM and 100-fold BET selectivity [106]. On this basis of 21, the cyclic sulfone group was replaced by the CF2 group to yield 23 (GSK8814) which was identified as the first reported low-nanomolar, selective, and cell-permeable chemical probe for ATAD2 Bromodomain [107]. To make further efforts, the researchers hypothesized that constrained bridged piperidine derivatives would show greater selectivity over the BET bromodomains. The piperidine was replaced with tropane yield 22 which is a potent ATAD2 inhibitor with IC50 of 100 nM and has fairly good BRD4 selectivity [25].
5.4. Imidazolidine-2,4-dione and furane derivatives
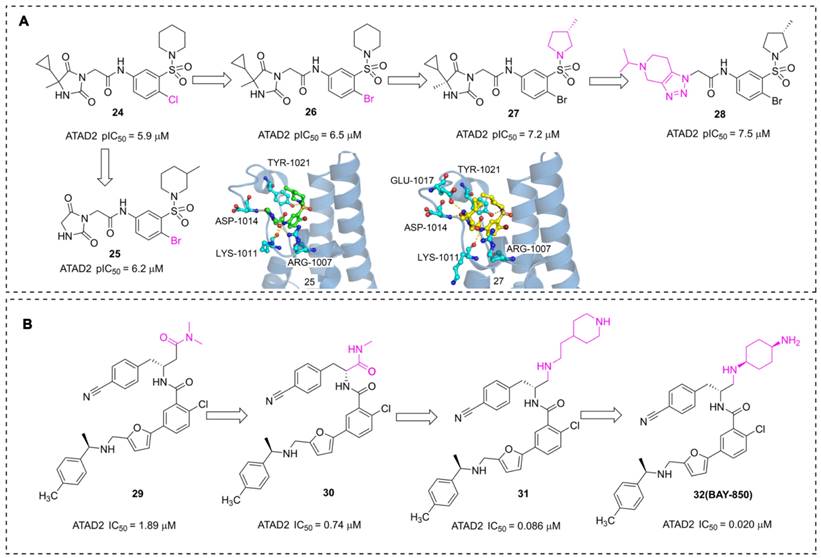
In addition to the acetyl-mimetic agent, imidazolidine-2,4-dione and furane derivatives were also developed as potent ATAD2 inhibitors with atypical binding modes. The researchers from AstraZeneca conducted a high throughput screen by TR-FRET assay, NMR, and SPR, 16 hits were obtained from the 1.7 million collections [108]. An imidazolidine-2,4-dione derivative (24) presented the most potent affinity for ATAD2 with pIC50 of 5.9 μM. The Br substituted (26) displayed an improved affinity with pIC50 of 6.5 μM. Further, the 5-cyclopropyl-5-methylimidazolidine-2,4-dione group was replaced with imidazolidine-2,4-dione to yield 25. Unfortunately, the activity of 25 showed no significant improvement. According to the X-ray structure of 25 and ATAD2, the sulfonyl group initiated a hydrogen bond with TYR1021, and three hydrogen interactions were observed between imidazolidine-2,4-dione group and ARG1007, LYS1011 residues. No interaction was observed between the ligand and ASN1064, which is a significant difference from the common ATAD2 inhibitors. These findings suggested that imidazolidine-2,4-dione derivatives bind to ATAD2 with a novel binding mode. Additional two derivatives were designed and synthesized [109], 27, an (S)-3-methylpyrrolidine substituted, presented an improved activity with pIC50 of 7.2 μM. The sulfonyl group initiated a hydrogen bond with TYR1021, and four hydrogen interactions were observed between the side chain and residues GLU1017, ASP1014, LYS1011, and ARG1007. The 5-cyclopropyl-5-methylimidazolidine-2,4-dione group was substituted with 5-isopropyl-4,5,6,7-tetrahydro-[1,2,3] triazolo[4,5-c]pyridine to yield 28, and 28 showed the most potent affinity for ATAD2 with pIC50 of 7.5 μM.
The chemistry structures of compounds 10-16 and X-ray structures of ligands and ATAD2 Bromodomain, including 10 (PDB code 7Q6U), 14 (PDB code 7Q6V), 15 (PDB code 7Q6W), and 16 (PDB code 7Q6T).
The chemistry structures of compounds 17-23 and X-ray structures of ligands and ATAD2 Bromodomain, including 17 (PDB code 5A5P), 19 (PDB code 5A5R), 20 (PDB code 5A82), and 23 (PDB code 5lj0).
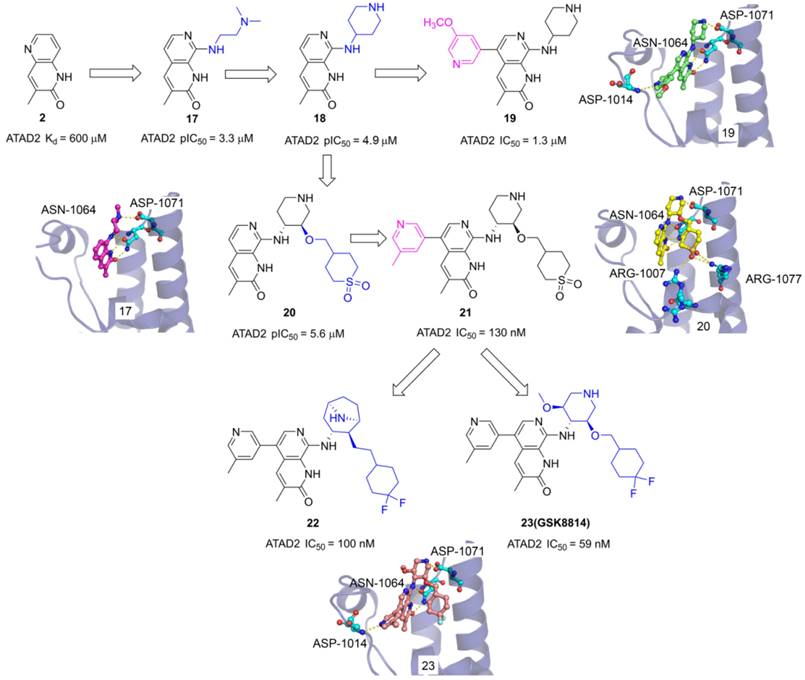
Pharmaceutical giant Bayer has also made significant efforts to develop novel ATAD2 inhibitors as innovative anti-tumor drugs. A series of furane derivatives were discovered by a DNA-encoded library screen, 29 displayed an IC50 value of 1.89 μM against ATAD2. A shorter formylamine derivative (30) presented slightly increased inhibitory activity of ATAD2 with an IC50 value of 0.74 μM. A 2-(piperidin-4-yl)ethan-1-amine moiety was incorporated to product 31, displaying a potent inhibitory potency of ATAD2 with an IC50 value of 0.086 μM [110]. The cyclohexane-1,4-diamine substituted (32, BAY-850) showed the most potent inhibitory activity with an IC50 value of 0.02 μM. BAY-850 is widely used as an isoform-selective ATAD2 chemical probe to explore the biological function of ATAD2 because of its potent activity against ATAD2 [24]. However, the binding mode of BAY-850 and ATAD2 has not yet been resolved, some researchers put forward the hypothesis that BAY-850 could specifically induce ATAD2 bromodomain dimerization and prevented interactions with acetylated histones, but more solid evidence needs to provide.
5.2 AAA + ATPase domain
As a candidate drug target, in addition to the abovementioned BRD region, ATAD2 also exists as an important domain responsible for ATP binding, hydrolysis, and polymerization, ---AAA + ATPase domain. The importance of AAA + ATPase domain for estrogen-stimulated-mediated gene expression, as well as efficient binding to chromatin, has been demonstrated by specific mutations. For instance, studies have shown that mutations in the AAA + ATPase domain, which is responsible for mediating the multimerization of ATAD2, affect the recruitment and/or occupancy of CBP at the promoter of Erα [7]. Despite the biological functional importance of the AAA + ATPase domain, small molecules developed to target it still need to be further explored, possibly because the spatial structure of the AAA + ATPase domain of ATAD2 has not been resolved and because of the high homology with other AAA + ATPase family members. To date, small-molecule compounds targeting AAA + ATPase family members are gradually being reported. Dynein, an AAA + ATPase, is responsible for powering all microtubule-negative-oriented movements within eukaryotic cells, which plays a crucial role in a variety of cellular functions [111]. In a study of Caenorhabditis elegans, deficiency of either light or heavy chains of dynein at the nuclear envelope inhibited physiological apoptosis in phagocytic defective mutants, involving the CED-4/Apaf1 protein [112]. A novel aminothiazol inhibitor of cytoplasmic dynein 1/2, Dynarrestin, blocks smooth-mediated Hedgehog signaling by transmembrane receptors [113]. Furthermore, another AAA + ATPase family member, p97, has been greatly developed in recent years. Accumulating studies have found that the expression of p97 is upregulated in a variety of cancers and is closely related to the poor prognosis of diseases such as gastric cancer and liver cancer [114-116]. NMS-873, a potent allosteric inhibitor of p97, activates unfolded protein responses, interferes with autophagy and promotes cell death in CRC HCT-116 cells [117].
The chemistry structures of compounds 24-32 and X-ray structures of ligands and ATAD2 Bromodomain, including 25 (PDB code 6s55), 27 (PDB code 6YB4).
In short, the AAA + ATPase domain still has a long way to go as an alternative strategy for small-molecule compounds to target ATAD2. It is hoped that the abovementioned successful examples of developing targeted dynein and p97 will provide new inspiration for the design of compounds targeting ATAD2.
6. Conclusion and perspectives
Up to present, the involvement of ATAD2 in epigenetic modifications that promote cancer cell proliferation and its clinical significance in diagnosis and prognosis has been continuously revealed, making it an emerging and promising cancer therapeutic target. For instance, ATAD2 is highly expressed in breast cancer, and its expression level is remarkably correlated with tumor grade [22]. In addition, inhibition of ATAD2 expression can effectively inhibit the proliferation of prostate cancer cells and induce apoptosis [27]. To further evaluate the potential of ATAD2 as an anticancer target, it is essential to understand its biological role in cancer. In this thesis, we describe in detail the structure of ATAD2, which is mainly composed of BRD and AAA + ATPase domains, and its important biological roles in cancer, including participating in epigenetic modifications and regulating downstream oncogene expression and signaling pathways. Notably, ATAD2 functions as a coactivator of epigenetic readers or transcription factors in the Rb/E2F-CMYc pathway, PI3K/AKT/mTOR, and other signaling pathways. Finally, we summarized small-molecule inhibitors targeting BRD in ATAD2, such as GSK8814, AZ13824374, and BAY-850. Taken together, elaborating on the abovementioned contents will not only help to deepen the understanding of ATAD2 in cancer development but also provide an unprecedented opportunity for the development of anticancer drugs with new targets.
Anticancer therapy targeting ATAD2 is not a simple subject; thus far, there are still many problems and challenges to be solved. One of the best problems is the design of potent novel small molecule inhibitors targeting ATAD2. The BRD domain in ATAD2 is a promising target. Nevertheless, unlike the classical BRD4 domain, the BRD in ATAD2 replaces the WPF (Trp-Pro-Phe) motif with the RVF motif (Arg-Val-Phe) which is more flexible and hydrophilic, resulting in more challenging to engage in productive interactions when designing ligands. Another distinctive Arg1077 in ATAD2 in place of BRD4 BD1 residue Met149, leading to a more hydrophilic binding site in ATAD2. Moreover, the ZA loop of ATAD2 is two residues shorter than that of BRD4, contributing further to the relative solvent exposure and hydrophilicity of its KAc site [103, 107, 118, 119]. Similarly, the AAA + ATPase domain also faces challenges in drug discovery. For example, the lack of a detailed spatial crystal structure of this domain and its high similarity to other AAA + ATPase family members implies that a series of adverse reactions such as poor selectivity and side effects may occur. Second, the reported small-molecule inhibitors, such as GSK8814 and AM879, are still in their infancy, and the anticancer mechanism and pharmacodynamic studies should be further explored to pave a credible path for them to become effective anticancer drugs.
In conclusion, the development of targeted ATAD2 inhibitors is a daunting and creative task that will require the combined efforts of basic pharmacology, structural biology, and proteomics to usher in an unprecedented anticancer drug era. Increasing pharmaceutical giants, such as AstraZeneca, Bayer, and GlaxoSmithKline, have entered the development of ATAD2-targeted drugs, and the clinical application prospect of ATAD2-targeted small-molecule inhibitors is promising.
Abbreviations
5α-dihydrotestosterone, 5α-DHT; Androgen receptor, AR; ATPase family AAA domain-containing protein 2, ATAD2; Breast cancer, BC; Bromine domain, BRD; cAMP-responsive element-binding protein, CBP/p300; C-Jun N-terminal kinase 1/2, JNK1/2; Colorectal cancer, CRC; C-terminal domain, CTD; Cyclin-dependent protein kinases, CDKs; DNA damage and repair, DDR; E2F transcription factor 1, E2F1; E2F transcription factor 2, E2F2; E2F transcription factor 3, E2F3; Enhancer of zeste homologue 2, EZH2; Epithelial-mesenchymal transition, EMT; Esophageal squamous cell carcinoma, ESCC; Estrogen receptor α, ERα; Extracellular signal-regulated kinase 1/2, ERK1/2; Fluorodeoxyglucose, FDG; Gastric cancer, GC; Glucose transporter type 1, GLUT1; Head and neck squamous cell carcinoma, HNSCC; Hedgehog, HH; Hepatocellular carcinoma, HCC; Hexokinase 2, HK2; HIF1α binding site, HBS; Histone deacetylases 2, HDAC2; Homologous recombination repair, HRR; Host cytokine 1, HCF-1; Hypoxia-inducible factor 1, HIF1; Long noncoding RNAs, lncRNAs; Lysine acetylation, KAc; Mammalian target of rapamycin, mTOR; MAP Kinase Kinase 3, MKK3/6; Methyltransferase-like 3, METTL3; MicroRNAs, miRNAs; Mitogen-activated protein kinase, MAPK; Mixed lineage protein-1, MLL1; MYC-interacting zinc finger protein 1, MIZ1; N-terminal acidic domain, NTD; Nuclear paraspeckle assembly transcript 1_2, NEAT1_2; Oral squamous cell carcinoma, OSCC; Patched 1, PTCH1; Phosphatidylinositol 3 kinase, PI3K; Retinoblastoma tumor suppressor protein, Rb; RNA-induced silencing complex, RISC; Smoothened, SMO; Stomach cancer cells, SCC.
Acknowledgements
This work was supported in part by National Natural Science Foundation of China (Grant No. 82003580, Grant No. 82172649 and Grant No. 82173666), Foundation Committee of basic and applied basic research of Guangdong Province (2019B1515120029 and 2019A1515110482), as well as Shenzhen science and technology research and development funds (Grant No. JCYJ20210324094612035, JCYJ20190808171803553, and 2022071718149001). We also thank some materials in the graphical abstract and figures that are produced by Servier Medical Art (https://smart.servier.com) and BioRender (https://biorender.com). We also thank Dr. Yuqian Zhao for her help in the inquiry of the drug preclinical study. We also thank the Language Editing Services of Elsevier (Serial number: LE-247622-27C189AB0929).
Author contributions
Dahong Yao, Bo Liu and Jin Zhang conceived the project and supervised the project. Dahong Yao, Jiahui Fu and Jin Zhang wrote the manuscript. Zhiying Liu and Xuetao Yang summed up the literature and the compounds in the manuscript. Xiya Chen and Zhiying Liu was involved in chemical structure drawing. Jin Zhang proofread the structure and figures. Zhendan He, Bo Liu and Yue Hao polished the manuscript. All authors approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jessop M, Felix J, Gutsche I. AAA+ ATPases: structural insertions under the magnifying glass. Curr Opin Struct Biol. 2021;66:119-28
2. Clapier CR, Iwasa J, Cairns BR, Peterson CL. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat Rev Mol Cell Biol. 2017;18:407-22
3. Prattes M, Grishkovskaya I, Hodirnau VV, Rossler I, Klein I, Hetzmannseder C. et al. Structural basis for inhibition of the AAA-ATPase Drg1 by diazaborine. Nat Commun. 2021;12:3483
4. Li S, Hsieh KY, Su SC, Pintilie GD, Zhang K, Chang CI. Molecular basis for ATPase-powered substrate translocation by the Lon AAA+ protease. J Biol Chem. 2021;297:101239
5. Liu Q, Liu H, Li L, Dong X, Ru X, Fan X. et al. ATAD2 predicts poor outcomes in patients with ovarian cancer and is a marker of proliferation. Int J Oncol. 2020;56:219-31
6. Zhou L, Yang C, Zhang N, Zhang X, Zhao T, Yu J. Silencing METTL3 inhibits the proliferation and invasion of osteosarcoma by regulating ATAD2. Biomed Pharmacother. 2020;125:109964
7. Zou JX, Revenko AS, Li LB, Gemo AT, Chen HW. ANCCA, an estrogen-regulated AAA+ ATPase coactivator for ERalpha, is required for coregulator occupancy and chromatin modification. Proc Natl Acad Sci U S A. 2007;104:18067-72
8. Logotheti S, Marquardt S, Gupta SK, Richter C, Edelhauser BAH, Engelmann D. et al. LncRNA-SLC16A1-AS1 induces metabolic reprogramming during Bladder Cancer progression as target and co-activator of E2F1. Theranostics. 2020;10:9620-43
9. Revenko AS, Kalashnikova EV, Gemo AT, Zou JX, Chen HW. Chromatin loading of E2F-MLL complex by cancer-associated coregulator ANCCA via reading a specific histone mark. Mol Cell Biol. 2010;30:5260-72
10. Wang D, Pan Y, Hao T, Chen Y, Qiu S, Chen L. et al. Clinical and Prognostic Significance of ANCCA in Squamous Cell Lung Carcinoma Patients. Arch Med Res. 2016;47:89-95
11. Hwang HW, Ha SY, Bang H, Park CK. ATAD2 as a Poor Prognostic Marker for Hepatocellular Carcinoma after Curative Resection. Cancer Res Treat. 2015;47:853-61
12. Zou JX, Duan Z, Wang J, Sokolov A, Xu J, Chen CZ. et al. Kinesin family deregulation coordinated by bromodomain protein ANCCA and histone methyltransferase MLL for breast cancer cell growth, survival, and tamoxifen resistance. Mol Cancer Res. 2014;12:539-49
13. Wu G, Liu H, He H, Wang Y, Lu X, Yu Y. et al. miR-372 down-regulates the oncogene ATAD2 to influence hepatocellular carcinoma proliferation and metastasis. BMC Cancer. 2014;14:107
14. Baggiolini A, Callahan SJ, Montal E, Weiss JM, Trieu T, Tagore MM. et al. Developmental chromatin programs determine oncogenic competence in melanoma. Science. 2021;373:eabc1048
15. Duan Z, Andrews NP, Chen CZ, Fan M, Wang J, Shen J. et al. Targeting bromodomain protein ANCCA/ATAD2 enhances the efficacy of DNA-damaging chemotherapy agents and radiation. Oncol Rep. 2020;43:318-27
16. Duan Z, Zou JX, Yang P, Wang Y, Borowsky AD, Gao AC. et al. Developmental and androgenic regulation of chromatin regulators EZH2 and ANCCA/ATAD2 in the prostate Via MLL histone methylase complex. Prostate. 2013;73:455-66
17. Wen Y, Hou Y, Yi X, Sun S, Guo J, He X. et al. EZH2 activates CHK1 signaling to promote ovarian cancer chemoresistance by maintaining the properties of cancer stem cells. Theranostics. 2021;11:1795-813
18. Duan R, Du W, Guo W. EZH2: a novel target for cancer treatment. J Hematol Oncol. 2020;13:104
19. Wang HJ, Pochampalli M, Wang LY, Zou JX, Li PS, Hsu SC. et al. KDM8/JMJD5 as a dual coactivator of AR and PKM2 integrates AR/EZH2 network and tumor metabolism in CRPC. Oncogene. 2019;38:17-32
20. Liu N, Funasaka K, Obayashi T, Miyahara R, Hirooka Y, Goto H. et al. ATAD2 is associated with malignant characteristics of pancreatic cancer cells. Oncol Lett. 2019;17:3489-94
21. Caron C, Lestrat C, Marsal S, Escoffier E, Curtet S, Virolle V. et al. Functional characterization of ATAD2 as a new cancer/testis factor and a predictor of poor prognosis in breast and lung cancers. Oncogene. 2010;29:5171-81
22. Kalashnikova EV, Revenko AS, Gemo AT, Andrews NP, Tepper CG, Zou JX. et al. ANCCA/ATAD2 overexpression identifies breast cancer patients with poor prognosis, acting to drive proliferation and survival of triple-negative cells through control of B-Myb and EZH2. Cancer Res. 2010;70:9402-12
23. Gay JC, Eckenroth BE, Evans CM, Langini C, Carlson S, Lloyd JT. et al. Disulfide bridge formation influences ligand recognition by the ATAD2 bromodomain. Proteins. 2019;87:157-67
24. Fernandez-Montalvan AE, Berger M, Kuropka B, Koo SJ, Badock V, Weiske J. et al. Isoform-Selective ATAD2 Chemical Probe with Novel Chemical Structure and Unusual Mode of Action. ACS Chem Biol. 2017;12:2730-6
25. Bamborough P, Chung CW, Furze RC, Grandi P, Michon AM, Watson RJ. et al. Aiming to Miss a Moving Target: Bromo and Extra Terminal Domain (BET) Selectivity in Constrained ATAD2 Inhibitors. J Med Chem. 2018;61:8321-36
26. Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D. et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214-31
27. Evans CM, Phillips M, Malone KL, Tonelli M, Cornilescu G, Cornilescu C. et al. Coordination of Di-Acetylated Histone Ligands by the ATAD2 Bromodomain. Int J Mol Sci. 2021 22
28. Wang X, Yang Y, Ren D, Xia Y, He W, Wu Q. et al. JQ1, a bromodomain inhibitor, suppresses Th17 effectors by blocking p300-mediated acetylation of RORgammat. Br J Pharmacol. 2020;177:2959-73
29. Winter-Holt JJ, Bardelle C, Chiarparin E, Dale IL, Davey PRJ, Davies NL. et al. Discovery of a Potent and Selective ATAD2 Bromodomain Inhibitor with Antiproliferative Activity in Breast Cancer Models. J Med Chem. 2022;65:3306-31
30. Yao D, Li C, Jiang J, Huang J, Wang J, He Z. et al. Design, synthesis and biological evaluation of novel HDAC inhibitors with improved pharmacokinetic profile in breast cancer. Eur J Med Chem. 2020;205:112648
31. Nayak A, Dutta M, Roychowdhury A. Emerging oncogene ATAD2: Signaling cascades and therapeutic initiatives. Life Sci. 2021;276:119322
32. Koo SJ, Fernandez-Montalvan AE, Badock V, Ott CJ, Holton SJ, von Ahsen O. et al. ATAD2 is an epigenetic reader of newly synthesized histone marks during DNA replication. Oncotarget. 2016;7:70323-35
33. Lloyd JT, Glass KC. Biological function and histone recognition of family IV bromodomain-containing proteins. J Cell Physiol. 2018;233:1877-86
34. Cattaneo M, Morozumi Y, Perazza D, Boussouar F, Jamshidikia M, Rousseaux S. et al. Lessons from yeast on emerging roles of the ATAD2 protein family in gene regulation and genome organization. Mol Cells. 2014;37:851-6
35. Zhou Y, Hussain M, Kuang G, Zhang J, Tu Y. Mechanistic insights into peptide and ligand binding of the ATAD2-bromodomain via atomistic simulations disclosing a role of induced fit and conformational selection. Phys Chem Chem Phys. 2018;20:23222-32
36. Boussouar F, Jamshidikia M, Morozumi Y, Rousseaux S, Khochbin S. Malignant genome reprogramming by ATAD2. Biochim Biophys Acta. 2013;1829:1010-4
37. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O. et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583-9
38. Meng X, Wang L, Zhu B, Zhang J, Guo S, Li Q. et al. Integrated Bioinformatics Analysis of the Clinical Value and Biological Function of ATAD2 in Hepatocellular Carcinoma. Biomed Res Int. 2020;2020:8657468
39. Sun T, Du B, Diao Y, Li X, Chen S, Li Y. ATAD2 expression increases [18F]Fluorodeoxyglucose uptake value in lung adenocarcinoma via AKT-GLUT1/HK2 pathway. BMB Rep. 2019;52:457-62
40. Zhou X, Ji H, Ye D, Li H, Liu F, Li H. et al. Knockdown of ATAD2 Inhibits Proliferation and Tumorigenicity Through the Rb-E2F1 Pathway and Serves as a Novel Prognostic Indicator in Gastric Cancer. Cancer Manag Res. 2020;12:337-51
41. Cao LJ, Zhang YJ, Dong SQ, Li XZ, Tong XT, Chen D. et al. ATAD2 interacts with C/EBPbeta to promote esophageal squamous cell carcinoma metastasis via TGF-beta1/Smad3 signaling. J Exp Clin Cancer Res. 2021;40:109
42. Wang JH, Yu TT, Li Y, Hao YP, Han L, Xu KY. et al. Silence of ATAD2 inhibits proliferation of colorectal carcinoma via the Rb-E2F1 signaling. Eur Rev Med Pharmacol Sci. 2020;24:6055-63
43. Zhao Y, Wang Z, Gao M, Wang X, Feng H, Cui Y. et al. lncRNA MALAT1 regulated ATAD2 to facilitate retinoblastoma progression via miR-655-3p. Open Med (Wars). 2021;16:931-43
44. Ge T, Liu T, Guo L, Chen Z, Lou G. MicroRNA-302 represses epithelial-mesenchymal transition and cisplatin resistance by regulating ATAD2 in ovarian carcinoma. Exp Cell Res. 2020;396:112241
45. Han HJ, Huang QY, Huang LJ, Chang F, Diao QZ. Prognostic value of ATPase family, AAA+ domain containing 2 expression in human cancers: A systematic review and meta-analysis. Medicine (Baltimore). 2019;98:e17180
46. Wang XL, Wang S, Wu ZZ, Yang QC, Li H, Xiong HG. et al. Overexpression of ATAD2 indicates Poor Prognosis in Oral Squamous Cell Carcinoma. Int J Med Sci. 2020;17:1598-609
47. Zhao R, Fu J, Zhu L, Chen Y, Liu B. Designing strategies of small-molecule compounds for modulating non-coding RNAs in cancer therapy. J Hematol Oncol. 2022;15:14
48. Wu S, Han M, Zhang C. Overexpression of microRNA-186 inhibits angiogenesis in retinoblastoma via the Hedgehog signaling pathway by targeting ATAD2. J Cell Physiol. 2019;234:19059-72
49. Hong S, Bi M, Chen S, Zhao P, Li B, Sun D. et al. MicroRNA-520f suppresses growth of gastric carcinoma cells by target ATPase family AAA domain-containing protein 2 (ATAD2). Neoplasma. 2016;63:873-9
50. Wang AQ, Lv M, Xu YH, Xie PM, Dong YY. MiR-200b-5p inhibits proliferation of ovarian cancer cells by targeting ATAD2 and regulating PI3K/AKT signaling pathway. Eur Rev Med Pharmacol Sci. 2020;24:9860-8
51. Ji S, Su X, Zhang H, Han Z, Zhao Y, Liu Q. MicroRNA-372 functions as a tumor suppressor in cell invasion, migration and epithelial-mesenchymal transition by targeting ATAD2 in renal cell carcinoma. Oncol Lett. 2019;17:2400-8
52. Dutta M, Das B, Mohapatra D, Behera P, Senapati S, Roychowdhury A. MicroRNA-217 modulates pancreatic cancer progression via targeting ATAD2. Life Sci. 2022;301:120592
53. Zhang K, Sun X, Sun W, Wang M, Han F. Exosomal microRNA-506 inhibits biological activity of lung adenocarcinoma cells and increases sensitivity to cisplatin-based hyperthermia. Cell Signal. 2022;100:110469
54. Wang Y, Hu Y, Wu G, Yang Y, Tang Y, Zhang W. et al. Long noncoding RNA PCAT-14 induces proliferation and invasion by hepatocellular carcinoma cells by inducing methylation of miR-372. Oncotarget. 2017;8:34429-41
55. Sun W, Lan X, Zhang H, Wang Z, Dong W, He L. et al. NEAT1_2 functions as a competing endogenous RNA to regulate ATAD2 expression by sponging microRNA-106b-5p in papillary thyroid cancer. Cell Death Dis. 2018;9:380
56. Liu C, Hou J, Shan F, Wang L, Lu H, Ren T. Long Non-Coding RNA CRNDE Promotes Colorectal Carcinoma Cell Progression and Paclitaxel Resistance by Regulating miR-126-5p/ATAD2 Axis. Onco Targets Ther. 2020;13:4931-42
57. Krakstad C, Tangen IL, Hoivik EA, Halle MK, Berg A, Werner HM. et al. ATAD2 overexpression links to enrichment of B-MYB-translational signatures and development of aggressive endometrial carcinoma. Oncotarget. 2015;6:28440-52
58. Hussain M, Zhou Y, Song Y, Hameed HMA, Jiang H, Tu Y. et al. ATAD2 in cancer: a pharmacologically challenging but tractable target. Expert Opin Ther Targets. 2018;22:85-96
59. Nayak A, Roy AD, Rout N, Singh SP, Bhattacharyya A, Roychowdhury A. HIF1alpha-dependent upregulation of ATAD2 promotes proliferation and migration of stomach cancer cells in response to hypoxia. Biochem Biophys Res Commun. 2020;523:916-23
60. Liu Q, Liu H, Huang X, Fan X, Xiao Z, Yan R. et al. A targetable MYBL2-ATAD2 axis governs cell proliferation in ovarian cancer. Cancer Gene Ther. 2022
61. Tong Y, Li J, Peng M, Qian Q, Shi W, Chen Z. et al. ATAD2 drives colorectal cancer progression by regulating TRIM25 expression via a positive feedback loop with E2F transcriptional factors. Biochem Biophys Res Commun. 2022;594:146-52
62. Liu Y, Wang Z, Liu H, Wang X, Zhang Z, Xiao B. et al. Derlin-1 functions as a growth promoter in breast cancer. Biol Chem. 2020;401:377-87
63. Chen N, Golczer G, Ghose S, Lin B, Langenbucher A, Webb J. et al. YAP1 maintains active chromatin state in head and neck squamous cell carcinomas that promotes tumorigenesis through cooperation with BRD4. Cell Rep. 2022;39:110970
64. Ciro M, Prosperini E, Quarto M, Grazini U, Walfridsson J, McBlane F. et al. ATAD2 is a novel cofactor for MYC, overexpressed and amplified in aggressive tumors. Cancer Res. 2009;69:8491-8
65. Altintas DM, Shukla MS, Goutte-Gattat D, Angelov D, Rouault JP, Dimitrov S. et al. Direct cooperation between androgen receptor and E2F1 reveals a common regulation mechanism for androgen-responsive genes in prostate cells. Mol Endocrinol. 2012;26:1531-41
66. Ahmad K, Henikoff S, Ramachandran S. Managing the Steady State Chromatin Landscape by Nucleosome Dynamics. Annu Rev Biochem. 2022;91:183-95
67. Belk JA, Yao W, Ly N, Freitas KA, Chen YT, Shi Q. et al. Genome-wide CRISPR screens of T cell exhaustion identify chromatin remodeling factors that limit T cell persistence. Cancer Cell. 2022;40:768-86 e7
68. Urbanucci A, Barfeld SJ, Kytola V, Itkonen HM, Coleman IM, Vodak D. et al. Androgen Receptor Deregulation Drives Bromodomain-Mediated Chromatin Alterations in Prostate Cancer. Cell Rep. 2017;19:2045-59
69. Morozumi Y, Boussouar F, Tan M, Chaikuad A, Jamshidikia M, Colak G. et al. Atad2 is a generalist facilitator of chromatin dynamics in embryonic stem cells. J Mol Cell Biol. 2016;8:349-62
70. Shukla S, Lazarchuk P, Pavlova MN, Sidorova JM. Genome-wide survey of D/E repeats in human proteins uncovers their instability and aids in identifying their role in the chromatin regulator ATAD2. iScience. 2022;25:105464
71. Lazarchuk P, Hernandez-Villanueva J, Pavlova MN, Federation A, MacCoss M, Sidorova JM. Mutual Balance of Histone Deacetylases 1 and 2 and the Acetyl Reader ATAD2 Regulates the Level of Acetylation of Histone H4 on Nascent Chromatin of Human Cells. Mol Cell Biol. 2020 40
72. Dutta M, Mohapatra D, Mohapatra AP, Senapati S, Roychowdhury A. ATAD2 suppression enhances the combinatorial effect of gemcitabine and radiation in pancreatic cancer cells. Biochem Biophys Res Commun. 2022;635:179-86
73. Yao D, Li C, Rajoka MSR, He Z, Huang J, Wang J. et al. P21-Activated Kinase 1: Emerging biological functions and potential therapeutic targets in Cancer. Theranostics. 2020;10:9741-66
74. Knudsen ES, Wang JY. Targeting the RB-pathway in cancer therapy. Clin Cancer Res. 2010;16:1094-9
75. Bretones G, Delgado MD, Leon J. Myc and cell cycle control. Biochim Biophys Acta. 2015;1849:506-16
76. Jin Y, Qi G, Chen G, Wang C, Fan X. Association between B-Myb proto-oncogene and the development of malignant tumors. Oncol Lett. 2021;21:166
77. Desai K, McManus JM, Sharifi N. Hormonal Therapy for Prostate Cancer. Endocr Rev. 2021;42:354-73
78. Waks AG, Winer EP. Breast Cancer Treatment: A Review. JAMA. 2019;321:288-300
79. Hsia EY, Kalashnikova EV, Revenko AS, Zou JX, Borowsky AD, Chen HW. Deregulated E2F and the AAA+ coregulator ANCCA drive proto-oncogene ACTR/AIB1 overexpression in breast cancer. Mol Cancer Res. 2010;8:183-93
80. Yuan Y, Liao L, Tulis DA, Xu J. Steroid receptor coactivator-3 is required for inhibition of neointima formation by estrogen. Circulation. 2002;105:2653-9
81. Louie MC, Zou JX, Rabinovich A, Chen HW. ACTR/AIB1 functions as an E2F1 coactivator to promote breast cancer cell proliferation and antiestrogen resistance. Mol Cell Biol. 2004;24:5157-71
82. Snaterse G, Visser JA, Arlt W, Hofland J. Circulating steroid hormone variations throughout different stages of prostate cancer. Endocr Relat Cancer. 2017;24:R403-R20
83. Zou JX, Guo L, Revenko AS, Tepper CG, Gemo AT, Kung HJ. et al. Androgen-induced coactivator ANCCA mediates specific androgen receptor signaling in prostate cancer. Cancer Res. 2009;69:3339-46
84. Hajduczok G, Chapleau MW, Abboud FM. Rheoreceptors in the carotid sinus of dog. Proc Natl Acad Sci U S A. 1988;85:7399-403
85. Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y, Hu LL. ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med. 2020;19:1997-2007
86. Li ZL, Zhang HL, Huang Y, Huang JH, Sun P, Zhou NN. et al. Autophagy deficiency promotes triple-negative breast cancer resistance to T cell-mediated cytotoxicity by blocking tenascin-C degradation. Nat Commun. 2020;11:3806
87. Endo M, Yamamoto H, Setsu N, Kohashi K, Takahashi Y, Ishii T. et al. Prognostic significance of AKT/mTOR and MAPK pathways and antitumor effect of mTOR inhibitor in NF1-related and sporadic malignant peripheral nerve sheath tumors. Clin Cancer Res. 2013;19:450-61
88. Lu WJ, Chua MS, So SK. Suppression of ATAD2 inhibits hepatocellular carcinoma progression through activation of p53- and p38-mediated apoptotic signaling. Oncotarget. 2015;6:41722-35
89. Yao D, Zhang J, Wang J, Pan D, He Z. Discovery of novel ATAD2 bromodomain inhibitors that trigger apoptosis and autophagy in breast cells by structure-based virtual screening. J Enzyme Inhib Med Chem. 2020;35:713-25
90. Ganesh K, Massague J. Targeting metastatic cancer. Nat Med. 2021;27:34-44
91. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423-37
92. Wang M, Zhao X, Zhu D, Liu T, Liang X, Liu F. et al. HIF-1alpha promoted vasculogenic mimicry formation in hepatocellular carcinoma through LOXL2 up-regulation in hypoxic tumor microenvironment. J Exp Clin Cancer Res. 2017;36:60
93. Mittal V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu Rev Pathol. 2018;13:395-412
94. Hao S, Li F, Jiang P, Gao J. Effect of chronic intermittent hypoxia-induced HIF-1alpha/ATAD2 expression on lung cancer stemness. Cell Mol Biol Lett. 2022;27:44
95. Skoda AM, Simovic D, Karin V, Kardum V, Vranic S, Serman L. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn J Basic Med Sci. 2018;18:8-20
96. Wu G, Lu X, Wang Y, He H, Meng X, Xia S. et al. Epigenetic high regulation of ATAD2 regulates the Hh pathway in human hepatocellular carcinoma. Int J Oncol. 2014;45:351-61
97. Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12:325-38
98. Dey A, Varelas X, Guan KL. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat Rev Drug Discov. 2020;19:480-94
99. Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344-62
100. Yang P, Guo L, Duan ZJ, Tepper CG, Xue L, Chen X. et al. Histone methyltransferase NSD2/MMSET mediates constitutive NF-kappaB signaling for cancer cell proliferation, survival, and tumor growth via a feed-forward loop. Mol Cell Biol. 2012;32:3121-31
101. Chaikuad A, Petros AM, Fedorov O, Xu J, Knapp S. Structure-based approaches towards identification of fragments for the low-druggability ATAD2 bromodomain. Medchemcomm. 2014;5:1843-8
102. Harner MJ, Chauder BA, Phan J, Fesik SW. Fragment-based screening of the bromodomain of ATAD2. J Med Chem. 2014;57:9687-92
103. Poncet-Montange G, Zhan Y, Bardenhagen JP, Petrocchi A, Leo E, Shi X. et al. Observed bromodomain flexibility reveals histone peptide- and small molecule ligand-compatible forms of ATAD2. Biochem J. 2015;466:337-46
104. Dolbois A, Batiste L, Wiedmer L, Dong J, Brutsch M, Huang D. et al. Hitting a Moving Target: Simulation and Crystallography Study of ATAD2 Bromodomain Blockers. ACS Med Chem Lett. 2020;11:1573-80
105. Demont EH, Chung CW, Furze RC, Grandi P, Michon AM, Wellaway C. et al. Fragment-Based Discovery of Low-Micromolar ATAD2 Bromodomain Inhibitors. J Med Chem. 2015;58:5649-73
106. Bamborough P, Chung CW, Furze RC, Grandi P, Michon AM, Sheppard RJ. et al. Structure-Based Optimization of Naphthyridones into Potent ATAD2 Bromodomain Inhibitors. J Med Chem. 2015;58:6151-78
107. Bamborough P, Chung CW, Demont EH, Furze RC, Bannister AJ, Che KH. et al. A Chemical Probe for the ATAD2 Bromodomain. Angew Chem Int Ed Engl. 2016;55:11382-6
108. Bamborough P, Chung CW, Demont EH, Bridges AM, Craggs PD, Dixon DP. et al. A Qualified Success: Discovery of a New Series of ATAD2 Bromodomain Inhibitors with a Novel Binding Mode Using High-Throughput Screening and Hit Qualification. J Med Chem. 2019;62:7506-25
109. Lucas SCC, Atkinson SJ, Bamborough P, Barnett H, Chung CW, Gordon L. et al. Optimization of Potent ATAD2 and CECR2 Bromodomain Inhibitors with an Atypical Binding Mode. J Med Chem. 2020;63:5212-41
110. Berger M, Huebner J, Ter Laak A, Gorjannacz M, Fernandez-Montalvan AE, Rodeschini V. et al. Furan derivatives as inhibitors of ATAD2 and their preparation. Bayer Pharma Aktiengesellschaft, Germany. 2017 p. 248pp
111. Bhabha G, Johnson GT, Schroeder CM, Vale RD. How Dynein Moves Along Microtubules. Trends Biochem Sci. 2016;41:94-105
112. Harders RH, Morthorst TH, Lande AD, Hesselager MO, Mandrup OA, Bendixen E. et al. Dynein links engulfment and execution of apoptosis via CED-4/Apaf1 in C. elegans. Cell Death Dis. 2018;9:1012
113. Hoing S, Yeh TY, Baumann M, Martinez NE, Habenberger P, Kremer L. et al. Dynarrestin, a Novel Inhibitor of Cytoplasmic Dynein. Cell Chem Biol. 2018;25:357-69 e6
114. Costantini S, Capone F, Polo A, Bagnara P, Budillon A. Valosin-Containing Protein (VCP)/p97: A Prognostic Biomarker and Therapeutic Target in Cancer. Int J Mol Sci. 2021 22
115. Yamamoto S, Tomita Y, Hoshida Y, Takiguchi S, Fujiwara Y, Yasuda T. et al. Expression level of valosin-containing protein is strongly associated with progression and prognosis of gastric carcinoma. J Clin Oncol. 2003;21:2537-44
116. Pu Z, Duda DG, Zhu Y, Pei S, Wang X, Huang Y. et al. VCP interaction with HMGB1 promotes hepatocellular carcinoma progression by activating the PI3K/AKT/mTOR pathway. J Transl Med. 2022;20:212
117. Magnaghi P, D'Alessio R, Valsasina B, Avanzi N, Rizzi S, Asa D. et al. Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat Chem Biol. 2013;9:548-56
118. Chung CW, Coste H, White JH, Mirguet O, Wilde J, Gosmini RL. et al. Discovery and characterization of small molecule inhibitors of the BET family bromodomains. J Med Chem. 2011;54:3827-38
119. Vidler LR, Brown N, Knapp S, Hoelder S. Druggability analysis and structural classification of bromodomain acetyl-lysine binding sites. J Med Chem. 2012;55:7346-59
Author contact
![]() Corresponding authors: E-mail addresses: yuehaoedu.cn (Yue Hao); liubo2400com (Bo Liu), or yaodahongedu.cn (Dahong Yao). Tel./Fax. (+86)-28-85164063.
Corresponding authors: E-mail addresses: yuehaoedu.cn (Yue Hao); liubo2400com (Bo Liu), or yaodahongedu.cn (Dahong Yao). Tel./Fax. (+86)-28-85164063.