Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Mechanisms of mitophagy
Roles of mitophagy in diseases
Small molecules pharmacological...
Conclusion and Outlook
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2023; 13(2):736-766. doi:10.7150/thno.79876 This issue Cite
Review
Cellular mitophagy: Mechanism, roles in diseases and small molecule pharmacological regulation
Yingying Lu1, Zhijia Li1, Shuangqian Zhang1, Tongtong Zhang2,3, Yanjun Liu2,3, ![]() , Lan Zhang1,
, Lan Zhang1, ![]()
1. Sichuan Engineering Research Center for Biomimetic Synthesis of Natural Drugs, School of Life Science and Engineering, Southwest Jiaotong University, Chengdu 610031, China
2. The Center of Gastrointestinal and Minimally Invasive Surgery, Department of General Surgery, The Third People's Hospital of Chengdu, The Affiliated Hospital of Southwest Jiaotong University, Chengdu 610031, China
3. Medical Research Center, The Third People's Hospital of Chengdu, The Affiliated Hospital of Southwest Jiaotong University, Chengdu 610031, China
Received 2022-10-14; Accepted 2022-12-23; Published 2023-1-1
Abstract

Cellular mitophagy means that cells selectively wrap and degrade damaged mitochondria through an autophagy mechanism, thus maintaining mitochondria and intracellular homeostasis. In recent years, mitophagy has received increasing attention as a research hotspot related to the pathogenesis of clinical diseases, such as neurodegenerative diseases, cardiovascular diseases, cancer, metabolic diseases, and so on. It has been found that the regulation of mitophagy may become a new direction for the treatment of some diseases. In addition, numerous small molecule modulators of mitophagy have also been reported, which provides new opportunities to comprehend the procedure and potential of therapeutic development. Taken together, in this review, we summarize current understanding of the mechanism of mitophagy, discuss the roles of mitophagy and its relationship with diseases, introduce the existing small-molecule pharmacological modulators of mitophagy and further highlight the significance of their development.
Keywords: Mitophagy, Mechanism, Diseases, Small molecule modulators
Introduction
Mitochondria, a crucial organelle that serves as the cellular energy hub in all eukaryotic cells, is where cells carry out aerobic respiration and use oxidative phosphorylation to produce ATP [1]. However, mitochondria are easy to be damaged, leading to mitochondrial dysfunction and destruction of cellular homeostasis, which is closely related to the occurrence of a variety of diseases, such as neurodegenerative diseases, cardiovascular diseases, cancer, metabolic diseases, infections and so on [2]. Importantly, the damaged mitochondria need to be immediately separated and selectively removed. Therefore, mitochondrial autophagy is an important way for the body to eliminate dysfunctional mitochondria and maintain the balance of the mitochondrial environment.
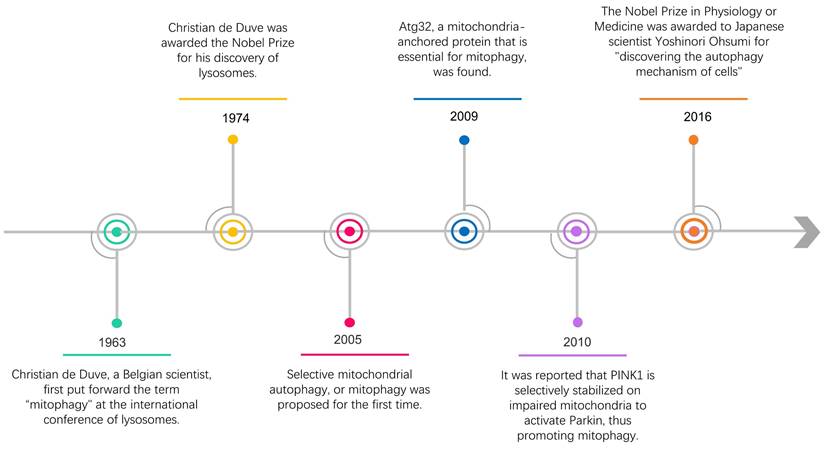
Almost 60 years ago, the term "autophagy" was used for the first time by Christian de Duve, who observed the degradation of mitochondria and other intra-cellular structures in the lysosomes of rat liver [3]. In 2016, the Nobel Prize in Physiology or Medicine was awarded to Japanese scientist Yoshinori Ohsumi for "discovering the autophagy mechanism of cells" [4, 5] (Figure 1). Recently, along with intensive research, the scientific community has gained a deeper insight into the study of autophagy, including selective autophagy such as mitophagy. Within a few years after the word "mitophagy" was initially proposed [6], substantial developments in this field made it possible to identify crucial proteins that specifically mediate mitochondrial breakdown in yeast [7] and mammalian cells [8, 9].
The timeline of some research progress related to mitophagy.
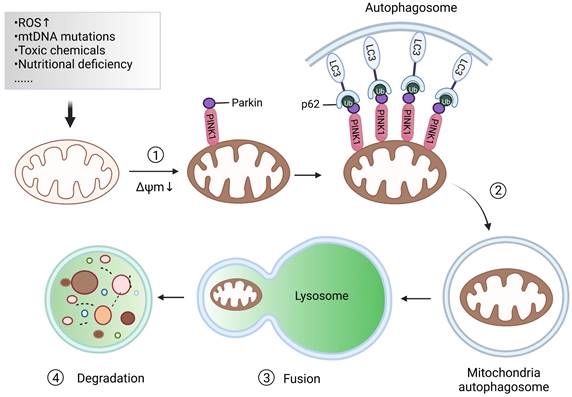
Mitochondrial autophagy, also known as mitophagy, is essential for maintaining mitochondrial and cellular homeostasis [10]. Under the stress of reactive oxygen species (ROS), nutrient deficiency, cell aging and other effects, the mitochondria in cells will show depolarization damage. To maintain the stability of the mitochondrial network and preserve the stability of the intracellular environment, an autophagy mechanism is used by cells to selectively wrap and degrade the damaged or dysfunctional mitochondria within the cells. This procedure is called mitophagy, which is mainly composed of four processes [11]: 1) Damaged mitochondria depolarize and lose membrane potential; Under the action of ROS, nutrient deficiency, cell aging, and other external stimuli, the membrane potential of mitochondria is dissipated, which is the prerequisite for the occurrence of mitophagy. 2) Mitochondria are wrapped by autophagosomes to form mitochondrial autophagosomes; Firstly, the double-membrane phagophores that surround the mitochondria to be degraded begin to initiate and elongate. Subsequently, the phagophore is enclosed in double-membrane vesicles called autophagosomes. 3) Mitochondrial autophagosomes fuse with lysosomes; That is to say, mitochondria can be delivered to the lysosome by autophagosomes for degradation. 4) Mitochondrial contents are degraded by lysosomes. Lysosomal or vacuolar acid hydrolase flows into the autophagosome to degrade mitochondria, and the contents are recycled (Figure 2).
The main processes of mitophagy. When mitochondria are damaged after external stimulation, the damaged mitochondria will depolarize, and the outer membrane potential of the mitochondria will be lost. Subsequently, the autophagosome wraps the mitochondria to become a mitochondrial autophagosome. Lysosomes combine with mitochondrial autophagosomes, thus promoting the degradation of mitochondrial content.
An in-depth analysis of the molecular mechanism of mitophagy is of great help in providing a theoretical basis for further research on the treatment of diseases related to mitophagy dysfunction. Subsequently, we discuss in detail how mitophagy plays a role in some common diseases, indicating the importance of finding new modulators targeting mitophagy. In this review, we also focus on the small molecule pharmacological modulators of mitophagy that have been reported so far, revealing the molecular mechanism of various diseases and helping to develop new drugs targeting mitophagy.
Mechanisms of mitophagy
Since the concept of mitophagy was proposed, research on its mechanisms has received extensive attention. The mechanisms found in current studies can usually be divided into two categories: ubiquitin (Ub)-dependent pathways and Ub-independent pathways. In this section, we will put emphasis on discussing these two categories of mitophagy mechanisms.
Ub-dependent pathways
Ubiquitin-dependent pathways, as the name implies, rely on extensive ubiquitination of mitochondrial surface proteins to promote mitophagy. Among these mechanisms, the PTEN induced putative kinase 1 (PINK1)/Parkin pathway is currently the most widely studied, which is involved in the elimination of damaged mitochondria in mammals [12]. PINK1 is a highly conserved mitochondrial protein encoded by the PARK6 gene and involved in the regulation of many cellular physiological processes, especially the regulation of mitochondrial function [13]. In normal mitochondria, the expression of PINK1 is too low to be detected because it is constantly transferred to the inner membrane of the mitochondria and then removed [14]. However, when the mitochondrial membrane potential (MMP, ΔΨm) is damaged, the path of PINK1 entering the mitochondrial inner membrane is blocked, resulting in PINK1 stably accumulating in the mitochondrial outer membrane. Parkin, a protein encoded by the PARK2 gene, is an E3 ubiquitin ligase, which is responsible for connecting Ub molecules to substrate proteins, and the substrate proteins with Ub tags are recognized by proteasome and then degraded. After mitochondrial damage, the spatial conformation of Parkin changed, and cysteine, which played a catalytic role, was exposed and transformed into an activated E3 ubiquitin ligase [15]. PINK1 interacts with Parkin and jointly regulate the process of mitophagy to maintain mitochondrial quality, whereas loss of either of them will cause mitochondrial damage. Previous research has shown that PINK1 and Parkin are located on the same pathway to protect mitochondria, and Parkin is located downstream of PINK1 [16]. Interestingly, although overexpression of Parkin can restore some of the phenotype of PINK1 deletion, PINK1 cannot compensate for the damage caused by Parkin deletion, which is probably because PINK1 is located in the upstream of Parkin [17].
The damaged mitochondria are commonly characterized by depolarization and a decrease in membrane potential. Recent studies have identified that PINK1 can significantly accumulate on the surface of the mitochondrial membrane with reduced potential, indicating that it may be the main detector of mitochondrial damage [18]. After PINK1 accumulates and stabilizes in the outer membrane of mitochondria (OMM), it activates the E3-Ub ligase Parkin through a mechanism involving phosphorylation of Parkin and its substrate Ub at Ser65 (pSer65-Ub) [19, 20]. Once located on the mitochondria, Parkin will cooperate with PINK1 to amplify the initial signal by modifying the mitochondria with the Ub chain, which is phosphorylated by PINK1 in turn (Figure 3). Sequestosome-1 (p62/ SQSTM1) is an autophagy-associated linker protein that can specifically bridge the ubiquitinated cargos into autophagosomes [21], which is involved in the process of eliminating ubiquitinated proteins and damaged mitochondria as a receptor [22]. Studies have shown that p62, which preferentially locates between adjacent mitochondria in the process of PINK1/Parkin-induced mitophagy, promoted the accumulation of damaged mitochondria through its polybromo 1 (PB1) oligomeric domain [23]. The knockdown of p62 had no effect on the recruitment of Parkin to the mitochondria, but affected the final elimination of damaged mitochondria. The sufficient accumulation of pSer65-Ub on the OMM can trigger the recruitment of autophagy receptors optineurin (OPTN) and nuclear dot protein 52 (NDP52), which can promote the initiation of autophagy close to mitochondria by recruiting autophagy initiation factors such as Unc-51-like kinase 1 (ULK1) [24], Double FYVE-containing protein 1 (DFCP1) and WD repeat domain, phosphoinositide interacting 1 (WIPI1) [25]. Furthermore, OPTN and NDP52 can directly interact with microtubule-associated protein 1 light chain 3 (LC3) through their own LC3 interaction region (LIR) to anchor Ub-labeled mitochondria into autophagy [26, 27] (Figure 3). Importantly, Ub chains assembled by Parkin are also able to recruit autophagy receptors in complexes with a multifunctional kinase called TANK-binding kinase 1 (TBK1) [28], which also directly or indirectly mediates the phosphorylation of all known autophagy receptors. Heo et al. found that TBK1 can directly and specifically phosphorylate Ras-related protein 7A (RAB7A) to promote mitophagy through the PINK1-Parkin pathway [28]. Also, a study conducted by Alban et al. discovered a self-reinforcing positive feedback mechanism that coordinates TBK1-dependent autophagy adaptor phosphorylation with the assembly of ubiquitin chains on mitochondria to facilitate efficient mitophagy [29].
Overview of the mitophagy mechanisms. Mitophagy takes place through many different but interrelated mechanisms, which can usually be divided into Ub-dependent pathways and Ub-independent pathways. Among the Ub-dependent pathways, the PINK1/Parkin pathway is the most common. Besides, a series of mitochondrial autophagic receptors that can directly bind to LC3 without causing extensive ubiquitination are involved in the Ub-independent pathway.
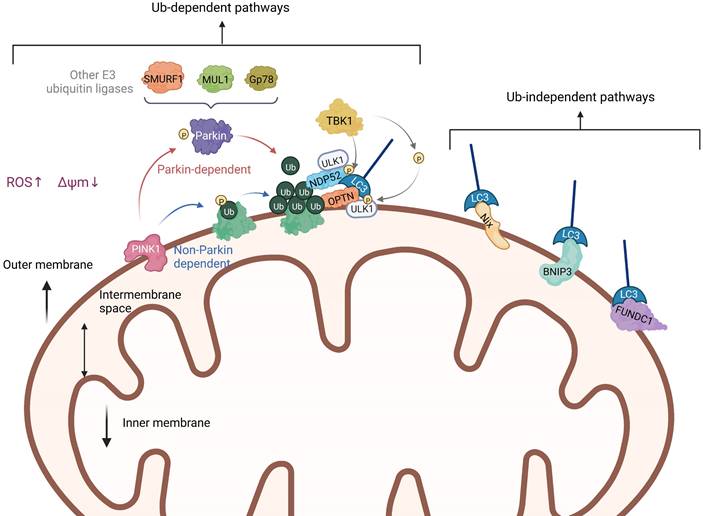
In addition to the PINK1-Parkin pathway, there are ubiquitin-dependent pathways that are not Parkin-dependent. In other words, PINK1 can also directly recruit autophagy receptors OPTN and NDP52 to mitochondria through ubiquitin phosphorylation, and then promote the biogenesis of autophagy [20, 30]. This indicates that OPTN and NDP52 can exist independently of Parkin. However, the presence of Parkin will amplify the signal pathway induced by PINK1 and enhance the process of mitophagy. What's more, besides Parkin, there are also other E3 ubiquitin ligases that can take part in the ubiquitination of mitochondrial proteins and the induction of mitophagy, such as smad ubiquitination regulatory factor-1 (SMURF1) [31], mitochondrial E3 ubiquitin protein ligase 1 (MUL1), Gp78 [32] and so on. It has been reported that SMURF1 promotes mitophagy by recruiting defective mitochondria rather than relying on p62 [33]. In addition, Orvedahl et al. found that SMURF1 controls mitophagy by promoting the transfer of autophagic molecules to the generated autophagosome [31]. Ryoko et al. found that MUL1 can mediate the stability of PINK1 on the mitochondrial membrane, thus inducing mitophagy, but has nothing to do with mitochondrial depolarization [34]. MUL1 deficiency will increase mitofusin2 (Mfn2) activity, which will trigger the first stage of mitochondrial hyperfusion. Over-expression of Mfn2 mimics the defective phenotype of MUL1, along with the expression of protein tyrosine phosphatase interacting protein 51 (PTPIP51), an ER mitochondrial ankyrin, can inhibit Parkin-mediated mitophagy [35]. Some studies have shown that Gp78 regulates mitophagy mainly by inducing mitochondrial disruption and ubiquitination, and the mitophagy induced by Gp78 is independent of Parkin [36].
Ub-independent pathways
Unlike PINK1/Parkin-mediated ubiquitination of mitophagy, there are many proteins containing LIR regions on the OMM, which are receptors for autophagy. They can directly bind to LC3 without ubiquitination, thus initiating mitophagy. In mammals, these receptors mainly include the Nip3-like protein X (NIX)/ BCL2-interacting protein 3 like (BNIP3L) receptor, BCL2-interacting protein 3 (BNIP3) receptor, FUN14 domain containing 1 (FUNDC1) receptor, and others (Figure 3). In this part, we introduce these Ub-independent pathways in detail, namely receptor-mediated mitophagy.
NIX, also known as BNIP3L, was initially reported to be involved in mitochondrial clearance during erythrocyte maturation [9]. The NIX protein can directly bind to LC3 through its BH3 domain and induce mitophagy [37]. It was established that mature red blood cells from mice with NIX deficiency have mitochondria, but NIX deficiency prevents the development of red blood cells, resulting in anemia [38]. Similar to NIX, BNIP3 is also located in OMM and contains a BH3 domain that can bind to LC3. Moreover, they have 56% homology because they both belong to the subfamily of the anti-apoptotic B cell lymphoma-2 (Bcl-2) family, which contains the BH3 domain [39]. When BNIP3 was knocked out, the mitophagy level of neuron cells in mice was significantly reduced under hypoxia. Interestingly, the knock-out of BNIP3 can cause up-regulation of NIX expression, but this up-regulation fails to compensate for the decrease in mitophagy caused by BNIP3 knock-out [40]. Furthermore, BNIP3 deficiency significantly inhibits mitophagy, thus aggravating cell apoptosis and kidney injury [41]. Additionally, FUNDC1 is a mitochondrial outer membrane protein, which can induce Parkin-independent mitophagy in mammalian cells under hypoxia by interacting with LC3 [42]. Although FUNDC1-mediated mitophagy does not depend on Parkin, MARCH5, another mitochondrial E3 ubiquitin ligase, can regulate mitophagy under hypoxia through ubiquitin degradation of FUNDC1 [43]. On the other hand, in the model of cardiac ischemia/reperfusion injury, receptor-interacting serine/threonine-protein kinase 3 (RIPK3) can inhibit FUNDC1-mediated mitophagy and then promote myocardial cell apoptosis, which proves that phosphorylation is involved in regulating FUNDC1-mediated mitophagy [44]. In conclusion, FUNDC1-induced mitophagy is regulated by both ubiquitin degradation and phosphorylation.
At present, the molecular regulation mechanism of mitophagy is the focus in the research field of mitophagy and cell autophagy. The effective removal of damaged or redundant mitochondria is also the key to maintain the stability of intracellular environment. Abnormal mitophagy is related to a wide array of diseases [45]. Therefore, the research in this field is of great significance to reveal the pathogenesis and prevention mechanism of mitophagy-related diseases, and it is also helpful to find new drug targets as well as promote new drug development and clinical treatment.
Roles of mitophagy in diseases
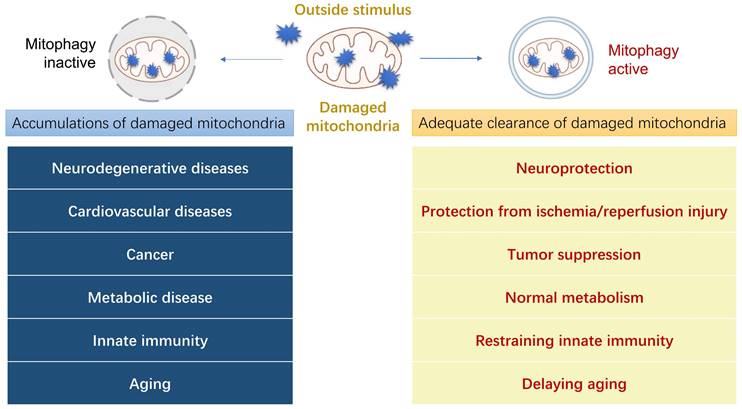
The morphological and functional abnormalities of mitochondria are closely related to the occurrence of various diseases [46]. It is highly probable that the dysfunction of mitophagy has a strong correlation with the pathogenesis of clinical diseases, leading to diseases such as neurodegenerative diseases, cardiovascular diseases, cancer, metabolic diseases, innate immunity, and other diseases (Figure 4). In this section, we shall describe how mitophagy plays a role in several prevalent illnesses and its relationship with their pathogenesis, thus providing a new potential treatment approach for these diseases by regulating mitophagy.
Physiological functions of mitophagy in human diseases. When mitophagy happens normally, it will produce a series of benefits to human body. On the contrary, the accumulation of damaged mitochondria caused by the inactivity of mitophagy will trigger some corresponding diseases.
Neurodegenerative diseases
Neurodegenerative diseases are closely related to age and cannot be cured at present, making it more urgent to deeply study the new pathogenesis and explore effective treatment methods. In neurons, when damaged mitochondria cannot be fully cleared by mitophagy, reactive oxygen species, nitrogen oxides, and other oxidizing substances will be produced. These oxidizing substances can bring about various neurodegenerative diseases, such as Alzheimer's disease (AD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), Huntington's disease (HD), and so on [47]. Although the lesion sites and causes of these diseases vary, neuronal degeneration is their common denominator. Of note, AD and PD mainly occur in the middle-aged and elderly. With the aging of the population, the incidence of AD and PD is increasing. PD is second only to AD in prevalence [48]. However, ALS and HD can occur at different ages. Importantly, impaired mitophagy may give rise to the occurrence or deterioration of a series of such neurodegenerative diseases, making methods targeting mitophagy a viable therapy for these diseases.
AD, the commonest cause of dementia, is a neurodegenerative disorder that has significant implications for both individuals and society. As the largest neurodegenerative disease in the world, its prevalence continues to grow at present because the world population is still aging. Earlier studies have shown that Parkin levels in the brains of AD patients will gradually decrease with the progression of disease, which further infers that there may be a decrease of mitophagy levels in late stages of AD [49]. Additionally, numerous studies have revealed that abnormal mitophagy occurs in AD patients' brains, preventing the damaged mitochondria from being cleared normally. This, in turn, accelerates pathological changes like calcium deposition and Tau protein hyperphosphorylation in AD patients' brains and worsens the onset and course of the disease [50]. Notably, in both amyloid-β (Aβ) and tau C. elegans models of AD, mitophagy induced by urolithin A was demonstrated to restore memory impairment through mechanisms that dependent on PINK1, Parkinson's disease-related-1 (PDR-1) or DAF-16/FOXO-controlled germline-tumor affecting-1 (DCT-1) [50]. At present, it has been proved that the progressive loss of functional nerve cells is also one of the pathological features of AD [51]. For example, it has been reported that obvious neuronal apoptosis may appear in the early stage of AD [52]. At the same time, deficient ATP production due to impaired mitophagy can also enhance the sensitivity of nerve cells to apoptosis-related proteins and accelerate the apoptosis of the cells. Compared with other cells in the brain, neurons are more dependent on ATP provided by oxidative phosphorylation of mitochondria [53]. And dysfunction of mitochondria can directly result in the death of neurons, which affects the functions of neurons in receiving, integrating, transmitting, and conducting information [53]. In brief, abnormal mitophagy induces the increase of AD-related pathological proteins such as the accumulation of Aβ and phosphorylation of Tau protein, increase the apoptosis of nerve cells, and leads to the disorder of brain energy metabolism, thus accelerating the occurrence and development of AD.
At present, there is no drug that can obviously delay or prevent AD, because it is a progressive neurodegenerative disease with multiple factors accumulated. Fortunately, in recent years, there has been evidence that mitophagy inducers can remove the abnormal mitochondria and enhance the ability of mitochondria to resist oxidative stress, namely improve mitochondrial function, thus delaying the course of AD. In the AD mouse model, supplementation of NAD+ precursors (such as nicotinamide) can improve mitochondrial function, reduce the deposition of Aβ and phosphorylation of Tau protein, thus improving the cognitive dysfunction of mice [53]. Therefore, further exploration of the relationship between mitophagy abnormality and AD may provide a new idea for targeted induction of mitophagy regulation and improvement of neuropathological changes in AD, thereby providing a guarantee for clinical selection of treatment strategies for neurodegenerative diseases.
In addition to AD, PD is the second largest neurodegenerative disease in the world, which has no cure method. PD is characterized by the selective death of dopaminergic neurons in the substantia nigra (SN). However, because the etiology of PD is so complex, no theory has been able to fully explain the pathogenesis of PD up until this point. A series of studies have demonstrated that mitochondrial DNA (mtDNA) loss and mitochondrial damage affect individual dopaminergic neurons in PD on a regular basis, and that the autophagy function of mitochondria in the SN and amygdala of PD patients is compromised [54]. Besides, late-stage PD patients can develop dementia with an accumulation of α-synuclein in Lewy bodies [55]. On top of that, the mitochondria in the cells of PD animal model are enlarged and swollen, which may be caused by the wrong folding and aggregation of α-synuclein, eventually culminating in the death of dopaminergic neurons [56]. All of these point to a connection between mitochondrial clearance failure and PD. Due to the identification of two proteins (PINK1 and Parkin) connected to the familial form of PD, the process of mitophagy, which is essential for maintaining neuronal homeostasis, is frequently associated with PD [57].
Studies have found that mutations in either PINK1 or Parkin can lead to the occurrence of genetic PD [20, 58]. The absence of PINK1 or Parkin results in abnormal mitophagy, increased oxidative stress, and the accumulation of large amounts of toxic substances, which cause mitochondrial damage in the mesencephalic SN and eventually culminate in the death of dopaminergic neurons [8, 59]. Therefore, therapeutic interventions aimed at modulating PINK1/Parkin signaling may be effective in treating PD. Notably, nitrosative stress is a significant pathogenic feature in PD [60]. Parkin and PINK1 are S-nitrosylated by nitric oxide, which impairs mitophagy and causes a buildup of damaged mitochondria [61]. According to previous research, Parkin KO animals had abnormal nigrostriatal pathway function, reduced synaptic excitability in spiny neurons, and an increase in extracellular dopamine concentration in the striatum [62]. Parkin was phosphorylated by c-Abl kinase in mouse models of synuclein overexpression, which reduced Parkin's function and caused zinc finger protein 746 (ZNF746) to accumulate as a result. Notably, a number of studies have demonstrated that ZNF746 plays a crucial role in mediating the neurodegeneration caused by synuclein in both dopaminergic and non-dopaminergic neurons [63]. Consequently, the phosphorylation of Parkin is intimately related to the development of PD. Taken together, it is certain that mitochondrial dysfunction caused by mitophagy disorder is an important factor leading to the onset of PD. Therefore, maintaining mitophagy at a normal level by using small molecule modulators will become a promising strategy for the treatment of PD, but this challenging therapeutic strategy still requires further research on the relationship between mitophagy and PD.
Although the incidence of ALS is not as high as that of AD or PD, it is a fatal and incurable disease characterized by selective degeneration of motor neurons, leading to progressive muscle weakness and death due to respiratory failure. It has been reported that inefficient turnover and aggregates of damaged mitochondria might play a role in the disease progression of ALS [64]. Surprisingly, some of the ALS-related genes that have been identified so far, such as OPTN, have a functional connection to mitophagy, particularly the removal of protein aggregates or damaged mitochondria [65]. Olivia et al. found that the multifunctional kinase TBK1 plays an important role in mitophagy. There are nearly one hundred different mutations in TBK1 that are related to ALS and frontotemporal dementia, which are also associated with the mitophagy receptor OPTN [66]. On balance, the autophagy receptor OPTN and its kinase TBK1 are required for the selectivity of phagocytosis of damaged mitochondrial autophagosome. When OPTN is translocated to damaged mitochondria, LC3B is recruited and degraded via lysosomal fusion [67]. The recruitment of p62 to damage mitochondria should be emphasized, however, it was done independently of OPTN and did not lead to the recruitment of LC3. Furthermore, the buildup of damaged or defective mitochondria is believed to be a contributing factor in the case of ALS. To note, ALS has become a public health issue because to current trends in life expectancy. Since this disease manifestly impairs mitochondrial homeostasis, autophagy and mitophagy dysregulation are among the most researched aspects of this pathology. In light of this, it may be suitable to use autophagy and/or mitophagy enhancers in the search for a cure for ALS.
Last but not least, HD is a fatal, incurable, and progressive pure genetic disease characterized clinically by seizures, chorea, cognitive decline, involuntary movement, mental retardation, dystonia, and emotional disorders [68]. Studies have shown that HD is closely related to mitochondrial abnormalities. A typical example is the study on the relationship between mutant Huntington protein (mHtt) and mitochondria. Shirendeb et al. found that mHtt interacted with mitochondrial dynamin-related protein 1 (Drp1), which enhanced the activity of the GTPase Drp1, resulting in excessive fragmentation and abnormal distribution of mitochondria [69]. Recent research has also demonstrated that the progression of HD can start when dead or dying mitochondria fail to be removed [70]. Interestingly, grade-dependent changes in the number and size of mitochondria were observed in HD. Furthermore, Maria et al. showed that the mitochondrial structure of HD-affected neurons was damaged, smaller, and rounder, and the cristae were broken [71]. In addition, research on cellular and animal models of HD shows that both nuclear and mitochondrial DNA are damaged, which further illustrates the correlation between mitophagy damage and the pathogenesis of HD [72].
Collectively, most work on mitophagy in the nervous system focuses on molecular pathogenesis and potential targets for treating nervous system diseases. However, there are still controversies around the target of mitophagy, the exact mechanism of receptor-mediated mitophagy, the differences in research and environmental conditions, whether the disadvantages of disease treatment outweigh the advantages, and how to best solve these problems. The challenge of future research is to design better models to understand the effects of mitophagy, common signal pathway, and mitochondrial defects on the pathological processes of neurodegenerative diseases. Furthermore, modulators that specifically induce or inhibit mitophagy can be used in the model to observe the changes in mitophagy and its relationship with the occurrence and development of diseases. In addition, the results of targeting mitophagy in long-term chronic diseases of the nervous system are still inconclusive, and extensive and rigorous research on short-term acute diseases of the nervous system is lacking. These problems need to be solved in future research.
Cardiovascular diseases
In recent years, with the aging of population worldwide, the incidence and mortality rate of cardiovascular diseases have ranked first. Cardiovascular diseases refer to the diseases of heart and large vessels, which is closely related to the function of myocardial cells and mitochondrial quality control. ROS produced by cell activities and external stimulating factors can cause different degrees of damage to mitochondria, affecting the function and life span of myocardial cells. Therefore, timely and effective removal of aging and damaged mitochondria in myocardial cells plays an essential role in the survival of myocardial cells. In this part, we will discuss the relationship between mitophagy and cardiovascular diseases such as hypertension, atherosclerosis, heart failure, and myocardial ischemia/reperfusion injury respectively, providing new ideas for the clinical treatment of cardiovascular diseases.
Hypertension is a common and frequent systemic disease, which is closely related to cardiovascular diseases, cerebrovascular diseases, and kidney diseases. It is known that enhancing mitophagy helps decrease blood pressure and safeguard the cardiovascular system. Besides, the PARK2 gene, which codes for Parkin, can be the hereditary cause of an elevation in blood pressure. According to Maurizio et al., reactivating mitophagy can lessen the risk of stroke in people with hypertension. They demonstrated that autophagy was reduced, and mitochondrial function was compromised in the brains of spontaneously hypertensive rats (SHR) on a high salt diet [73]. In addition, Chen et al. found that astaxanthin can promote mitophagy and biosynthesis by increasing the expression of PINK1, Parkin, mtDNA and so on, thus reducing vascular remodeling caused by hypertension [74]. However, the specific mechanism by which dysfunctional mitophagy leads to hypertension is still unclear, and further research is definitely needed.
Atherosclerosis is a common and frequently-occurring disease in the cardiovascular system. It is characterized by the formation of atherosclerotic plaques or fibrous plaques in the intima of blood vessels, and is the main cause of coronary heart disease, cerebral infarction, and myocardial infarction [75]. The presence and progression of atherosclerosis are significantly influenced by oxidized low density lipoprotein (ox-LDL), while the level of mitophagy is highly correlated with ox-LDL. Evidence suggests that ox-LDL-induced apoptosis in human vascular smooth muscle cells (VSMC) was reduced by activated mitophagy. What's more, the protective impact of mitophagy on VSMC can be increased by enhancing PINK1 expression [76]. In aortic endothelial cells, ox-LDL can lead to over-expression of nuclear receptor subfamily 4 group A member 1(NR4A1), which contributes to the over-activation of Parkin-mediated mitophagy [77]. Therefore, atherosclerosis is aggravated by the apoptosis of endothelial cells, which is caused by an excessive reduction of mitochondria and insufficient energy supply of cells. Eukaryotic initiation factor 2α (eIF2α) aggravates hyperlipidemia-induced atherosclerotic inflammation by inhibiting the Parkin-induced mitophagy [78]. To sum up, mitophagy plays a key role in the pathogenesis of atherosclerosis. It is expected to slow down or even reverse the progression of atherosclerosis by regulating this process, which may become a new target for the treatment of atherosclerosis.
Heart failure refers to a complex syndrome in which ventricular filling or ejection capacity is impaired due to cardiac dysfunction. Mitophagy is frequently blocked in the initial stages of heart failure. If the degree of mitophagy is appropriately raised, the progress of heart failure can be delayed [79]. On the contrary, mitophagy is frequently overactivated in the late stages of heart failure, which leads to dysfunction of myocardial cells and aggravation of heart failure. At this time, the level of mitophagy should be properly inhibited [80]. Notably, Shires et al. established different models of heart failure and confirmed that the decrease in mitophagy level can aggravate heart injury [81]. Furthermore, other research has demonstrated that mice with PINK1 silenced or knocked out are more prone to reperfusion injury and pressure overload, which can result in cardiac failure [82]. Actually, mitophagy disorder significantly increases ROS level and damages mtDNA, resulting in calcium overload in myocardial cells, inflammatory injury, necrosis, apoptosis, and myocardial fibrosis, all of which promote the occurrence of heart failure [83]. In short, the elucidation of the role of mitophagy in the occurrence of heart failure will not only facilitate a better understanding of the pathogenesis of heart failure, but also provide more potential targets for the treatment of heart failure and have a broad application prospect.
Myocardial ischemia-reperfusion injury (MIRI) is a key problem that must be solved urgently in the treatment of ischemic myocardium. Currently, the main mechanisms of MIRI mainly include ROS, calcium overload, vascular endothelial dysfunction, mitochondrial metabolism disorder, and autophagy, which lead to apoptosis or necrosis of ischemic myocardial cells [84]. The dysfunctional mitochondrial accumulation caused by insufficient mitophagy is harmful to the myocardium. And an appropriate increase in mitophagy, such as by giving therapeutic hypothermia, can alleviate MIRI [85]. On the contrary, according to studies, NIX or BNIP3 is abundantly expressed in ischemic and hypoxic myocardial cells, which stimulates mitophagy. However, high NIX or BNIP3 expression can result in excessive mitochondrial clearance, which alters the energy sources of cells, and instead puts more strain on cardiac cells, producing or exacerbating myocardial ischemia-reperfusion damage [86]. Therefore, the double-sided effect of mitochondria on cardiomyocytes remains to be further studied. As the relationship between the progression of MIRI in each stage and the intensity of mitophagy is still unclear, further research is definitely needed. In the future, ischemia-reperfusion injury can be alleviated and the prognosis of patients can be improved by regulating the intensity of mitophagy.
Accumulating evidence shows that mitophagy plays an irreplaceable role in maintaining the survival and normal function of myocardial cells and large vascular cells, indicating an important role in the onset, progress, and prognosis of cardiovascular diseases. Although the mechanism of mitophagy and its role in diseases have been known to some extent, the specific mechanism still needs further exploration. At the same time, as a research hotspot in recent years, the key proteins in mitophagy regulatory mechanism, such as PINK1, Parkin, BNIP3, and Drp1, are expected to become the targets of molecular therapy for cardiovascular diseases. Consequently, it is of great clinical significance to study the mechanism of mitophagy in cardiovascular diseases at molecular level.
Cancer
Cancer, the abnormal proliferation and differentiation caused by the loss of normal regulation of local tissue cells at the gene level caused by various tumorigenic factors, is one of the main "killers" of human health at present. Studies have found that there are different levels of mitophagy in many cancers compared to normal situations [87], including rectal cancer, lung cancer, and breast cancer, reflecting the close relationship between mitophagy and cancer. Here, we discuss the roles that PINK1, Parkin, BNIP3, NIX, and FUNDC1 play in a wide array of cancers, clarifying the possible mechanisms of mitophagy in cancers.
It is conceivable that mitophagy appears to be important as a tumor-suppressive system because the buildup of defective mitochondria is engaged in carcinogenesis [88]. Lauren et al. discovered that mitophagy inhibits tumor growth by removing malfunctioning mitochondria; otherwise, the abnormal mitochondria may change cells and encourage the development of tumors [89]. According to Xu et al., the Warburg effect, compromised mitophagy, and easier M2 polarization of macrophages are all caused by PINK1 loss in gastric cancer [90]. Furthermore, the occurrence and/or progression of cancer may be promoted by loss of heterozygosity (LOH) observed on chromosomes 6q25-q26, which can inactivate or reduce the expression of Parkin [91]. Interestingly, Parkin is known to influence phosphatase and tensin homolog (PTEN)-mediated glycolytic metabolic control. Previous studies have suggested that Parkin deficiency encourages PTEN degradation, which in turn causes PI3K/AKT signaling to be activated in cancer cells. And cancer cells undergo metabolic remodeling that encourages glycolysis as a result of PI3K/AKT signaling [92]. In some breast cancers, the fragile region of FRA6E containing PARK2 is often absent, which also supports the inhibitory effect of Parkin on tumor cells [93]. As for the receptor of mitophagy, recent studies have shown that the increased level of BNIP3 is related to the poor survival rate of melanoma patients, while the consumption of BNIP3 in B16-F10 melanoma cells will damage the growth of tumors in vivo [94]. High expression of NIX has been found in breast cancer, lung cancer, prostate cancer, cervical cancer, and other tumor cells. Studies have found that NIX-mediated mitophagy can promote pancreatic cancer [95]. In lung cancer, high expression of NIX was also found to be associated with poor prognosis. At the same time, however, it has also been demonstrated that the damaged mitophagy caused by NIX expression deficiency in breast cancer can promote tumor metastasis [96]. In conclusion, NIX may play different roles in different types of tumor cells at different stages, which may be a study hotspot in the future. According to a recent study, hepatocyte-specific FUNDC1 knockouts encourage the tumorigenesis of hepatocellular carcinoma (HCC), but FUNDC1 overexpression in hepatocytes decreases it, indicating that FUNDC1 functions to prevent HCC [97].
All in all, mitophagy, which is crucial for regulating tumor cells, is either directly or indirectly associated with the development of tumors, thus providing a potential therapy for cancer. In malignant tumors, mitophagy involves abnormal activation and proliferation of cancer cells, suggesting that it can exert both an oncogenic effect and a tumor-suppressive effect. The balance between the two effects can determine tumor progression or apoptosis [98]. From this perspective, a new anticancer therapy can be formed, which not only inhibits mitophagy in cancer cells to exert the anti-cancer effect, but also enhances mitophagy in normal cells to remove damaged mitochondria and maintain the stability and normal function of mitochondrial genome to strengthen the anti-cancer effect. Therefore, further understanding of the molecular mechanism of mitophagy signal pathway is expected to provide new ideas for the formulation of clinical anti-tumor treatment strategies, which still needs further efforts by researchers.
Metabolic diseases
Metabolic diseases are diseases caused by metabolic problems, including metabolic disorders. Currently, the most prevalent metabolic diseases include sugar metabolic diseases, lipid metabolic diseases, protein metabolic diseases, and even bone metabolic disorders. With the accelerated pace of life and changes in dietary patterns, the incidence of metabolic diseases such as diabetes and fatty liver is increasing, which has seriously endangered human health. Here, we review several common metabolic diseases that are highly related to abnormal mitophagy, providing a novel treatment strategy for these diseases.
In the past two decades, the prevalence of diabetes has been increasing to a large extent due to the aging society and the rapid increase in the obesity rate. Diabetes can cause damage to multiple organs and systems. And more than half of diabetic patients died of cardiovascular complications. Studies have shown that Parkin-mediated mitophagy can maintain pancreatic islet secretion and avoid the occurrence of type 1 diabetes (T1D) [99]. The symptoms of type 2 diabetes (T2D) include hyperglycemia and insulin resistance (IR). These are invariably linked to mitochondrial damage, probably because high glucose levels boost the production of ROS from mitochondria and the oxidative stress that causes tissue damage [100]. The expression of several mitophagy-related proteins, including NIX, PINK1 and Parkin, increased in early diabetic participants with mild hyperglycemia, but reduced in T2D patients [101]. Interestingly, after Parkin and Atg7 knockout mice were given a high-fat diet for two months, compared to normal control mice, Atg7 and Parkin deficiency reduced mitophagy in myocardial cells, exacerbating T2D-related cardiac lesions [102]. Since obesity raises the risk of T2D, diabetes and obesity are strongly related. As a result, the function that mitophagy serves in them also exhibits similarities. Obesity has also been linked to mitochondrial damage. To note, obesity-induced increases in mitochondrial content are likely due to an accumulation of damaged and fissured mitochondria that cannot be cleared by mitophagy. This is supported by the finding that obese patients have increased skeletal muscle mitochondrial content and decreased mitochondrial biogenesis [103]. Furthermore, obesity is a known risk factor for liver disease. Non-alcoholic fatty liver disease (NAFLD), the largest chronic liver disease in China, is a metabolic stress liver injury that closely related to IR and genetic susceptibility. A significant contributor to the progression of NAFLD to nonalcoholic steatohepatitis is Nod-like receptor protein 3 (NLRP3). According to studies, clearing damaged mitochondria, inhibiting NLRP3 inflammatory corpuscle activation, and promoting liver lipid metabolism all depend on hepatocyte mitophagy [104]. Chen et al. proved that low dose vinyl chloride can aggravate high-fat diet (HFD)-induced liver injury, which can be alleviated by PINK1/Parkin-dependent mitophagy stimulated by Alda-1 [105]. However, at present, the research on mitophagy and liver is still in its infancy, with few studies and inconsistent results. For example, most experimental evidence indicates that the defects of mitophagy contribute to hepatocyte steatosis and liver fibrosis [106], but some experiments also show that mitophagy is involved in the generation and differentiation of fat [107]. Therefore, the relationship between the two needs to be verified by more experiments, so as to clarify how to regulate mitophagy to better treat liver diseases.
On balance, it is a very promising direction to seek new methods to treat metabolic diseases by regulating mitophagy. Also, although some studies have proved that mitophagy is closely related to metabolic diseases, the relationship between them still needs to be much further explored.
Innate immunity
The innate immune response is a rapid response to pathogens or dangerous signals. The accurate activation of it can not only effectively eliminate pathogens, but also avoid excessive inflammation and tissue damage [108]. It is well known that mitochondria are essential organelles for many biological processes, including energy production and immune response. The latest progress in immunology reveals the key role of energy metabolism in innate immune cell function [109]. Therefore, maintaining the integrity and activity of mitochondrial network is a prerequisite for immunity. Here, we review the related molecular mechanism of mitophagy in immune diseases and emphasize its key role in the homeostasis of innate immune system.
Severe trauma or physical injury will lead to tissue tearing and cell damage, which will result in the release of mitochondrial danger-associated molecular pattern (DAMP) molecules (such as mtDNA) into the blood of the host [110]. At this time, the immune system will start to send signals, which will cause systemic inflammatory response syndrome (SIRS), a condition characterized by fever, shortness of breath, hypotension, increased mortality, elevated heart rate, and multiple organ failure. Studies have shown that autophagy protein depletion promotes the accumulation of dysfunctional mitochondria and cytosolic translocation of mtDNA. In other words, the cytoplasmic level of mtDNA and ROS is the key factor of innate immunity activated by the inflammasome [111]. In addition, the NLRP3 inflammasome is capable of detecting mitochondrial dysfunction, which may help to explain why inflammatory illnesses and mitochondrial damage are frequently linked [112]. It is worth noting that mitochondria, as the central signal platform of innate immune response, have a positive feedback loop with the inflammasome. Infection destroys mitochondrial homeostasis, mediates mtDNA release, mtROS overproduction, and then triggers inflammasome stimulation, highlighting the critical role of mitophagy in the innate immune system homeostasis [113]. Furthermore, as mentioned before, the regulation of mitochondrial removal is also influenced by MUL1 and SMURF1, demonstrating the immunosuppressive function of mitophagy in response to noxious stimuli.
In a word, defective removal of damaged mitochondria leads to the activation of inflammatory signaling pathways, which in turn contribute to the development of chronic systemic inflammation and inflammatory diseases. In addition, it has been reported that innate immunity can also be impaired by inducing excessive mitophagy [114]. As mitophagy holds an essential role in the regulation of inflammatory responses, it is promising to develop novel methods targeting mitophagy to treat immune-related diseases.
Other diseases
Apart from those previously discussed, mitophagy has also been demonstrated to be associated with other human diseases. Skeletal muscle atrophy is a major health problem worldwide, especially in the elderly. At present, no treatment can offset the gradual decline of skeletal muscle mass and strength with age, a process termed sarcopenia. However, more and more evidence has proved that the development of sarcopenia is caused by the accumulation of dysfunctional mitochondria, which provides a new window for exploring the treatment of this disease [115]. Moreover, Harmon made the initial suggestion that ROS directly controls the aging process of cells in 1950. Linnane and Fleming both thought that mtDNA mutations and cell aging were caused by an increase in ROS production [116]. However, ROS accumulation is closely related to mitophagy. For example, the knockout of PINK1 and Parkin in human bronchial epithelial cells will accelerate their aging process, accompanied by the accumulation of damaged mitochondria and an increase in ROS production [117]. Besides, adipose-derived stem cells can control metabolic homeostasis and slow the aging process by encouraging mitophagy [118]. Accordingly, mitophagy can remove damaged mitochondria and delay cell aging.
Although many research results to date show that mitophagy plays a key role in the development of some specific diseases, which we have summarized as much as possible here. However, there is still a requirement for more experimental research to explore the relationship between mitophagy and various diseases. We can then further develop new treatment strategies and improve the clinical therapeutic effect of these diseases by regulating mitophagy.
Small molecules pharmacological regulation of mitophagy
Small molecules are important pharmacological tools for dissecting complex biological processes and identifying potential therapeutic interventions. As mentioned above, since mitochondrial dysfunction is associated with many types of diseases, inducing or inhibiting mitophagy is an effective therapeutic intervention strategy. Therefore, it is necessary for us to summarize the current research situation of mitophagy modulators, analyze the existing chemical tools, and discuss their advantages, limitations, as well as current applications. Here, we will review some representative small molecule pharmacological modulators of mitophagy that has been discovered so far. Small molecule mitophagy inducers can be mainly divided into targeting (Table 1) and non-targeting (Table 2), wherein the targeting can be further divided into inducers targeting mitophagy related proteins and inducers targeting non-canonical mitophagy related proteins. As to small molecule mitophagy inhibitors, the relevant study on it is relatively limited up to now, so it will not be involved too much in this section.
Targeting small molecule mitophagy inducers
| Classification | Names | Chemical structure | Target | Mechanisms | Applications | Advantages or limitations | Ref. |
|---|---|---|---|---|---|---|---|
| PINK1 enhancer | Kinetin | 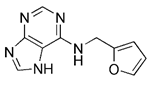 | PINK1 | The precursor of KTP, enhancing the kinase activity of PINK1 | Preventing and treating liver fibrosis | Having no effect on the stability of PINK1 under resting conditions | [119, 120] |
| KTP |  | PINK1 | Enhancing the kinase activity of PINK1 | No application at present | Having higher affinity for PINK1 than the native substrate ATP | [119] | |
| p53 inhibitor | Pifithrin-α |  | p53 | Inhibiting p53 and regulating PARK2 expression | Having neuroprotection effects | Low specificity | [122, 123] |
| Parkinson's toxin | Rotenone |  | Cardiolipin | ROS accumulation and mitochondrial damage, the externalization of cardiolipin | Using as an insecticide, inducing cellular model of PD | High cytotoxicity, inducing PD | [126, 127] |
| MPP+ | 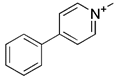 | ERK2 | ROS accumulation and mitochondrial damage, the phosphorylation and accumulation of ERK2 | Inducing cellular model of PD | High cytotoxicity, inducing PD | [129] | |
| 6-OHDA | 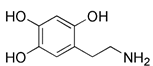 | Cardiolipin, ERK2 | ROS accumulation and mitochondrial damage, ERK2- and Cardiolipin- dependent | Inducing rat and cellular models of PD | High cytotoxicity, inducing PD | [130] | |
| Iron chelators | Phen | 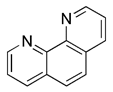 | Drp1 | MMP dissipation and mitochondrial fragmentation, Drp1-dependent | No application at present | Mitochondrial toxicity and respiratory damage | [131] |
| Nrf2 inducers | Sulforaphane | 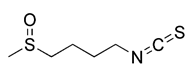 | Keap1 | ROS-dependent mechanism and ERK activation | Anti-cancer, protecting myocardial cells | Low specificity, a host of off-target Effects | [138] |
| PMI | 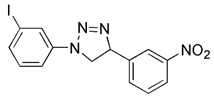 | Keap1 | Inhibiting Keap1 | No application at present | High therapeutic potential, high specificity, further profiling would be beneficial | [142] | |
| Superoxide generator | Sodium selenite | 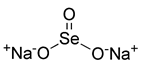 | MUL1 | Activating MUL1, ROS-dependent, down-regulation of MCL-1 | Anti-cancer, anti-inflammation, having radioprotective effects | No toxicity, low cost, selenium supplementation | [143] |
| MCL-1 inhibitor | UMI-77 | 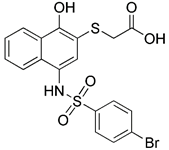 | MCL1 | Inhibiting MCL-1 | Anti-cancer, anti-inflammation | High specificity, independent of apoptosis | [147, 148] |
| USP30 inhibitors | ST-539 | 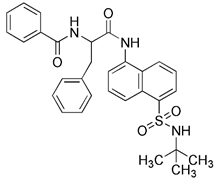 | USP30 | Inhibiting USP30, thus promoting ubiquitination | No application at present | Dependent of PINK1/Parkin activity, low specificity | [151] |
| SIRT1 activators | Resveratrol | 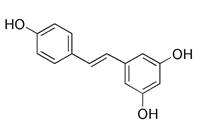 | SIRT1 | Activating SIRT1 directly | Anti-oxidation, anti-cancer, anti-inflammation, protecting cardiovascular system | Low specificity, also inducing general autophagy | [153] |
| Fisetin | 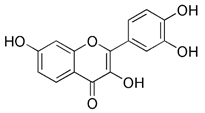 | SIRT1 | Activating SIRT1 directly | Anti-cancer, anti-inflammation, delaying aging | No toxicity and safe; low specificity | [154] | |
| SRT1720 | 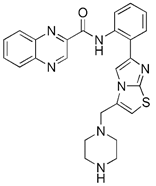 | SIRT1 | Activating SIRT1 directly | Anti-cancer such as bladder cancer, having neuroprotective effects, delaying aging | Having promise in clinical, the actions of it are cell specific. | [155] | |
| Nicotinamide (NAM) | 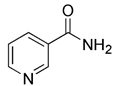 | SIRT1 | The precursor of NAD+, thus activating SIRT1 indirectly | Having neuroprotective effects, protecting skin | Without changing mitochondrial function and membrane potential | [157] | |
| SIRT3 activator | Compound 33c |  | SIRT3 | Activating SIRT3 directly | Anti-cancer such as TNBC | Low specificity, also inducing general autophagy | [161] |
| PARP-1 inhibitor | Olaparib |  | PARP-1 | Inhibiting PARP-1, thus indirectly activating SIRT1 | Anti-cancer such as prostate cancer, pancreatic cancer, ovarian cancer, etc. | Low specificity, inducting autophagy at the same time | [158] |
| Respiratory complex III inhibitor | Antimycin A |  | Respiratory complex III | Increased ROS levels and dissipation of MMP by inhibiting respiratory complex III | Anti-cancer such as oral cancer | Having a relatively limited effect when used alone and is frequently employed in combination with oligomycin A | [162, 163] |
| ATP synthase inhibitor | Oligomycin A |  | ATP synthase | Enhancing the dissipation of MMP by inhibiting ATP synthase | Anti-cancer, anti-bacteria | Having limited effect when used alone | [164] |
Non-targeting small molecule mitophagy inducers
| Classification | Names | Chemical structure | Mechanisms | Applications | Advantages or limitations | Ref. |
|---|---|---|---|---|---|---|
| Protonophores | FCCP | 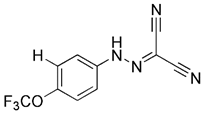 | Proton mediated dissipation of MMP | Having cardioprotective effects | Low specificity, high toxicity, mitochondrial failure or even disappearance | [169] |
| CCCP | 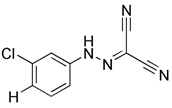 | Proton mediated dissipation of MMP | No application at present | Low specificity, high toxicity, mitochondrial failure or even disappearance | [59] | |
| DNP | 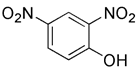 | Proton mediated dissipation of MMP | Using as a weight loss agent | Low specificity, high toxicity, mitochondrial failure or even disappearance | [167] | |
| K+ ionophores | Valinomycin | 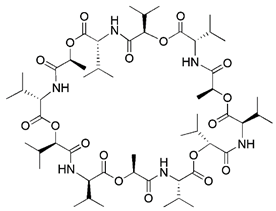 | Dissipation of MMP due to K+ influx | Anti-virus, anti-infection | Activating the PINK1-parkin pathway without altering the pH gradient | [171, 172] |
| Salinomycin | 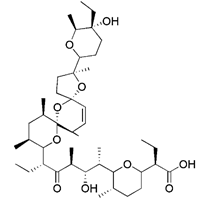 | IMM is hyperpolarized due to K+ outflow and H+ influx | Anti-cancer | Mitochondrial matrix acidification and mitochondrial failure | [173] | |
| Mitochondrial toxins | Diquat | 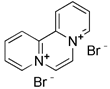 | Overproducing superoxide | Using as a herbicide | High toxicity, chronic neurotoxic effects | [174] |
| Paraquat | 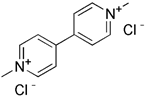 | Mitochondrial depolarization | Using as a pesticide | High toxicity, an acute damage in the lung | [175, 176] | |
| Iron chelators | DFP | 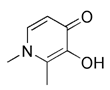 | Inducing iron deficiency, iron-depletion-dependent | Anti-cancer | Mitochondrial toxicity and respiratory damage | [180-182] |
| Metal complexes | Glycosylation zinc (II)-cryptolepine complexes | 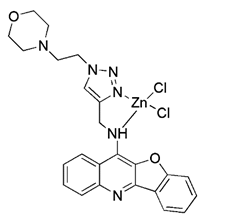 | Blocking ATP production and the respiratory chain | Anti-cancer | Having potential for the development of chemotherapy drugs against cisplatin-resistant cells | [183] |
| Ir-Rhein | 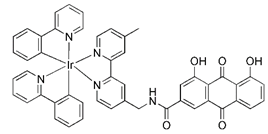 | Inducing ROS elevation and mitochondrial dysfunction | Anti-cancer, overcoming metallodrug resistance | Having superior performance to overcome cisplatin resistance | [184] | |
| Natural products | Urolithin A | 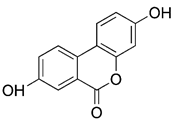 | Dissipation of MMP | Anti-cancer, anti-inflammation, having neuroprotective effects, delaying aging | Easy to obtain, meriting further study | [185-187] |
| Berberine | 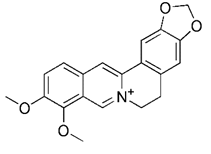 | Upregulating PINK1/Parkin pathway | Anti-cancer, anti-inflammation, lowering blood sugar | Having the potential to treat various disease; side effects such as infant jaundice, and nausea and vomiting | [190] | |
| Curcumin | 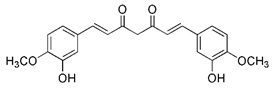 | Through AMPK activation and subsequent TFEB nuclear translocation | Anti-cancer, anti-oxidative stress, anti-inflammation, having neuroprotective effects, treating osteoarthritis | Having promising roles against different diseases; excessive intake may lead to contact dermatitis | [192] | |
| Others | VL-004 | 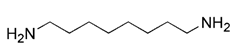 | DCT-1 and PINK1- dependent mitophagy | Extending lifespan, anti-oxidative stress, having neuroprotective effects | Low toxicity, providing a structural template for the development of therapy drugs against diseases related to aging | [193] |
| T-271 | 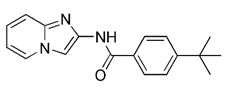 | Parkin-dependent mitophagy | Treating the currently untreatable chronic progressive phases of neurodegenerative diseases | High specificity, few side effects; substantial optimization of the chemical structure is still required | [194] | |
| Flubendazole | 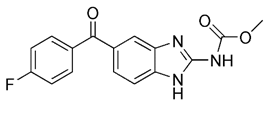 | Inducing DRP1- mediated mitophagy by targeting EVA1A | Anti-cancer, treating spinal cord injury, using as an anthelmintic | Having the potential to be repurposed as a novel anti-tumor agent | [196, 197] |
Targeting small molecules mitophagy inducers
Small molecule inducers targeting mitophagy related proteins
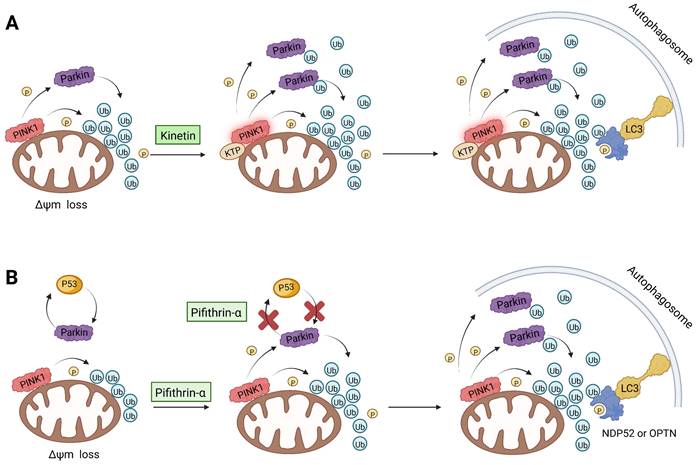
As we mentioned earlier, the most important mechanism of mitophagy is the PINK1-Parkin pathway, so using the activity modulator of this pathway can naturally promote the occurrence of mitophagy. Previous researches have shown that kinetin triphosphate (KTP), as a new substrate, shows higher affinity for PINK1 than the natural substrate ATP [119]. Interestingly, kinetin is the precursor of KTP, and its application to cells will lead to a significant increase in PINK1 activity, resulting in a higher Parkin recruitment level of depolarized mitochondria [119, 120] (Figure 5A). Therefore, kinetin and KTP are essentially PINK1 enhancers, which can promote the biogenesis of mitophagy by enhancing the activity of PINK1. In addition to PINK1 kinase, we can also regulate PINK1-Parkin pathway activity by focusing on Parkin. Previous studies have indicated that the tumor suppressor p53 may inhibit mitophagy by directly interacting with Parkin and preventing it from transferring to mitochondria [121]. To note, pifithrin-α is a small molecule p53 inhibitor that can improve mitophagy by using the feedback loop between Parkin and p53 [122, 123] (Figure 5B). Furthermore, it is reported that pifithrin-α can regulate Parkin expression and thus promote the occurrence of mitophagy [124]. Analogously, various small molecule modulators of Parkin all have the potential to be new inducers of mitophagy.
PINK1-Parkin activity modulators. (A) Kinetin enhanced the activity of PINK1. (B) The p53 inhibitor pifithrin-α promoted the accumulation of Parkin in the mitochondrial outer membrane.
Notably, rotenone, 1-methyl-4-phenylpyridinium (MPP+), and 6-hydroxydopamine (6-OHDA) are all representative and significant Parkinson's toxins, which rely on ROS accumulation and mitochondrial damage to promote mitophagy [125]. The distinction is that rotenone and 6-OHDA can promote the externalization of cardiolipin, which can activate the autophagy mechanism through the direct interaction with LC3 [126, 127] (Figure 6A). Studies have revealed that both 6-OHDA and MPP+ can encourage the phosphorylation of extracellular signal regulated protein kinase 2 (ERK2) and its accumulation in mitochondria, thus promoting mitochondria to enter autophagosomes and accelerate its degradation [128, 129] (Figure 6B). In other words, rotenone induces cardiolipin-dependent mitophagy, whereas MPP+ induces ERK1/2-dependent mitophagy. Intriguingly, the mitophagy induced by 6-OHDA is both cardiolipin and ERK1/2 dependent [130].
The mechanisms of different mitophagy inducers (A and B) Parkinson's toxins (C) Iron chelators and SIRT1 activators (D) Protonophores.
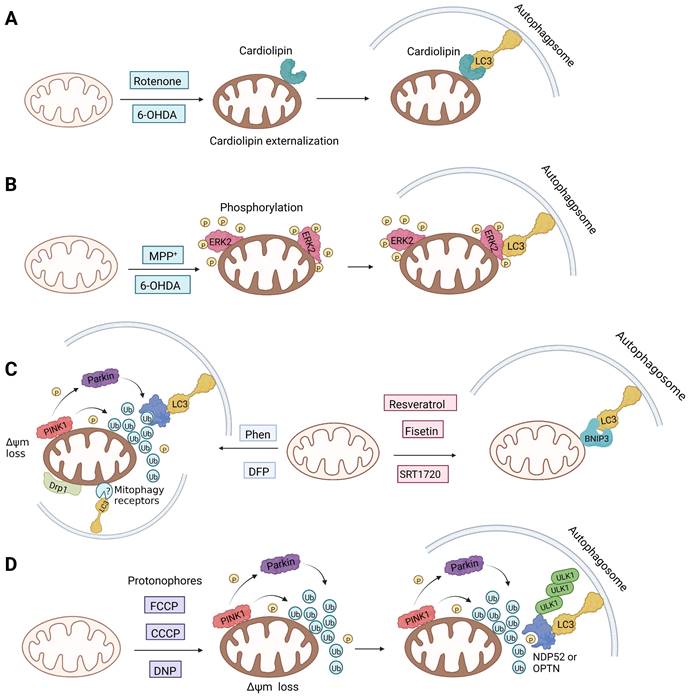
1, 10-phenanthroline (Phen), an iron chelator, can act on Drp1 that is related to mitochondrial motility and induce mitochondrial fragmentation, thereby promoting mitophagy [131]. Although it can mediate MMP dissipation, Park et al. found that Phen-mediated mitophagy was significantly inhibited in Drp1 knockout cells, indicating that Phen induced mitophagy in a Drp1-dependent manner [132]. However, Phen treatment also enhanced autophagy, which makes its specificity for mitochondria low. Furthermore, it has mitochondrial toxicity and can cause respiratory damage. Accordingly, these shortcomings make the use of Phen as mitophagy inducers limited.
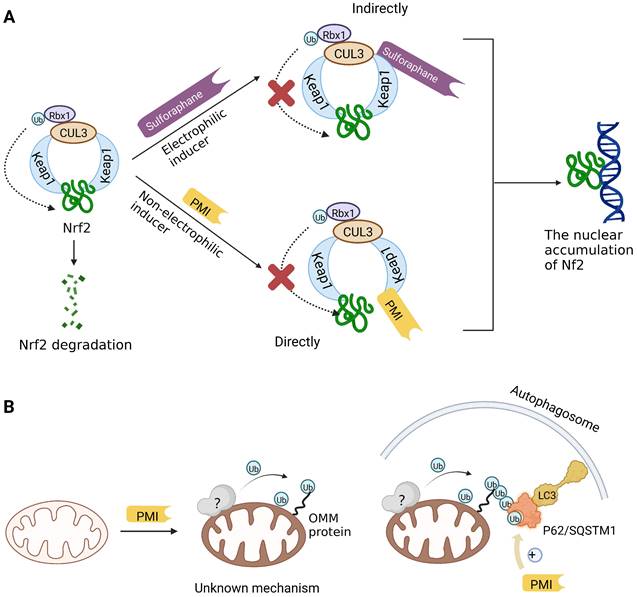
Previous studies have suggested that the nuclear factor erythroid 2-related factor 2 (Nrf2) is a promising target for improving mitochondrial function and health [133]. Numerous advantageous effects on mitochondria are produced when Nrf2 activity is increased genetically or pharmacologically [134]. A number of cell protection genes, including SQSTM1, which is notably connected to selective autophagy, are regulated by the transcription factor Nrf2 [135]. The Keap1-Nrf2 signaling pathway is currently recognized as an important defensive transduction pathway for the body to resist internal and external environmental oxidation and harmful stimulation. Nrf2 is expressed in almost all tissues, and the main factor affecting the function of Nrf2 is Keap1, which is a negative regulator of Nrf2 transcription activity and can regulate Nrf2 transcription level and post-translation level at multiple levels [136]. Although there is growing knowledge of the interaction between Nrf2 and mitochondria, its function in mitophagy has just been investigated lately. Typical Nrf2 activators are electrophiles that interact with the sensor cysteines of Keap1, causing an irreversible conformational shift that prevents Nrf2 from being ubiquitinated [137]. Sulforaphane, a typical example of traditional Nrf2 inducer, has been shown to be a potential therapy for cancer treatment, neurological disorders, diabetic cardiomyopathy, and other conditions [138-140]. Mechanistically, sulforaphane activates mitophagy through a ROS-dependent mechanism and ERK activation [141]. At the same time, it is disappointing that its non-targeted effect and low specificity limit its use. Notably, the newly found p62-mediated mitophagy inducer (PMI), a small molecule that promotes the accumulation of the Nrf2 by targeting the interaction between Nrf2 and Keap1 and inhibiting Keap1, was found to be a promising mitophagy inducer that functions without disrupting the MMP [142]. PMI does not influence general autophagy but instead promotes a targeted autophagic degradation of mitochondria. In addition, compared with traditional inducers of Nrf2, PMI has a unique mode of action, which also highlights its therapeutic potential [142]. Similarly, PMI and Sulforaphane prevent Nrf2 from being degraded by Keap1, causing the nuclear accumulation of Nf2 and activate transcription of genes that control autophagy and mitochondrial function (Figure 7A). However, PMI has the potential to treat diseases characterized by decreased PINK1 and Parkin activity since it may trigger mitophagy in cells lacking a fully functioning PINK1-Parkin pathway (PINK1 knockout or Parkin knockdown). And the specific mechanisms of it still need more research to figure out (Figure 7B).
Mitophagy inducers targeting Nrf2. (A) Sulforaphane (indirectly) and PMI (directly) prevent Nrf2 from being degraded by Keap1, causing the nuclear accumulation of Nf2 which benefit mitochondria. (B) Nrf2 is engaged in controlling mitochondrial activity and gene transcription that activates autophagy. One such gene is p62/SQSTM1, which appears to be crucial for PMI-induced activation of mitophagy.
Sodium selenite has been of special research interest for a long time. Since it causes human glioma cells, instead of astrocytes, to undergo a deadly form of mitophagy, which leads to irreversible cell death in glioma cells [143]. The E3 ubiquitin ligase MUL1 is reportedly activated by sodium selenite in a ROS-dependent manner, which attracts ULK1 to the mitochondria and then facilitates their autophagic degradation [144]. Furthermore, some studies have discovered that down-regulation of the anti-apoptotic protein myeloid cell leukemia-1 (MCL-1) can be caused by sodium selenite, thereby inducing mitochondrial-related apoptosis [145]. Notably, it has been demonstrated that MCL-1 is an LC3-interacting mitophagy receptor [146]. In 2014, a specific inhibitor of MCL-1 protein, UMI-77, was discovered and proved to be effective in inhibiting the growth of pancreatic cancer in vivo and in vitro [147]. Later, in 2020, Cen et al. demonstrated that UMI-77 could interact with LC3A directly to promote mitophagy independent of mitochondrial damage and apoptosis. Meaningfully, they also found that using UMI-77 to target the MCL-1 protein to induce mitophagy may be a promising strategy for the treatment of AD [148].
The ubiquitin-specific peptidase 30 (USP30), a deubiquitinating enzyme found in mitochondria, is famous for its role in the regulation of mitochondrial morphology [149]. By eliminating polyubiquitin chains from damaged mitochondria and hence delaying mitochondrial priming, USP30 combats Parkin-driven mitophagy [150]. Importantly, USP30 knockdown or inhibition is sufficient to restore mitophagy in neuronal cells expressing a dysfunctional Parkin mutant. Consequently, USP30 inhibitors can also stimulate mitophagy by promoting normal ubiquitination. According to recent research, ST-539, a racemic phenylalanine derivative, selectively inhibited the function of the USP30 enzyme in vitro [151]. Overexpression of USP30 prevented the decrease of the translocase of inner mitochondria membrane 23 (TIM23) and the translocase of outer mitochondria membrane 40 (TOM40) levels [149]. However, ST-539 restored the degradation of TIM23 and TOM40, indicating that ST-539 can promote mitophagy by inhibiting USP30. In addition, although ST-539 promotes ubiquitination and mitophagy, PINK1/Parkin activity is imperative for the activity of ST-539 [151]. Disappointingly, inhibition of USP30 may lead to nonspecific events that are not directly related to mitophagy. Therefore, further research is still required to identify the rational use of USP30 inhibition as a treatment strategy for diseases.
Small molecule inducers targeting non-canonical mitophagy related proteins
Apart from targeting mitophagy related proteins, some small molecule that targeted non-mitophagy related proteins can also promote the biogenesis and progress of mitophagy to a high extent.
Silent information regulator T1 or Sirtuin 1 (SIRT1) is a deacetylase that regulates a variety of activities by controlling biological processes such as gene expression, DNA repair, metabolism, and mitochondrial function. In terms of mechanisms, SIRT1 may trigger mitophagy by increasing deacetylation of LC3 and then activating LC3 [152] (Figure 6C). The most common activators of SIRT1 are resveratrol [153], fisetin [154], synthetic small molecule SRT1720[155], SRT2104, SRT2379 [156] and so on. They all induce mitophagy by directly activating SIRT1. However, it has recently been reported that nicotinamide (NAM) can also stimulate mitophagy by activating SIRT1 [157]. SIRT1 was discovered to be a NAD+ dependent deacetylase, whereas nicotinamide is the biosynthetic precursor of NAD+, implying that nicotinamide indirectly increases SIRT1 activity by increasing NAD+ [157]. In addition to NAM, the poly (ADP ribose) polymerase-1 (PARP-1) inhibitor, such as Olaparib [158], also functions by indirectly activating SIRT1 [159]. Since PARP-1 is an enzyme that consumes NAD+, blocking it can also raise the NAD+ level in cells, which indirectly activates SIRT1 and then promotes the occurrence of mitophagy. In particular, although the therapeutic effect of SIRT1 activators in in vivo disease models has been fully demonstrated, the low specificity still restricts the use of this kind of small molecule modulator. In addition, Sirtuin-3 (SIRT3) is also an NAD+-dependent protein deacetylase mainly located in mitochondria [160]. As the most important deacetylase in mitochondria, SIRT3 can regulate the acetylation degree of various substrates, thus affecting the malignant progress of tumors. Recently, a new small-molecule SIRT3 activator that could directly bind with SIRT3 called Compound 33c (ADTL-SA1215) was found. It has a general deacetylation activity toward the substrates of SIRT3 and depends on SIRT3 to exert its antiproliferative activity [161]. Notably, studies have suggested that the Compound 33c may inhibit the metastasis and proliferation of triple negative breast cancer (TNBC) cells by promoting SIRT3-regulated mitophagy or autophagy [161]. More crucially, Compound 33c may be a first-in-class selective small molecule SIRT3 activator that will be used to build new anticancer drugs.
There is also a common method of inducing mitophagy, namely respiratory complex III inhibitor and ATP synthase inhibitor. The representative drugs of this class are antimycin A and oligomycin A. By blocking respiratory complex III, antimycin A can increase the production of ROS and decrease MMP [162]. Unfortunately, the decrease in MMP caused by antimycin A is relatively limited, and this decrease is quickly balanced by the reverse hydrolysis activity of ATP synthase [163]. However, oligomycin A is a commonly used inhibitor of ATP synthase [164]. Hence, in order to counteract this compensatory mechanism and simultaneously stimulate higher inner membrane of mitochondria (IMM) depolarization, oligomycin A and antimycin A are frequently employed in combination. Furthermore, compared to mitochondrial toxins, this method is more in line with spontaneous mitophagy and has less toxicity [20].
Non-targeting small molecule mitophagy inducers
There are many non-targeting small molecule inducers of mitophagy, which can induce cell autophagy or other selective autophagy while inducing mitophagy. Consequently, their specificity is relatively low, limiting the possibility of clinical use.
Protonophores
A British academic named Peter Mitchell proposed “the chemical osmosis hypothesis” in 1961 [165]. He claimed that the energy released during electron transfer creates a proton gradient (H+ gradient) across the IMM, which powers ATP synthesis and explains how oxidation and phosphorylation are related. The energy status and operation of mitochondria can be directly measured by MMP. While mitochondrial uncoupler prevents the oxidative phosphorylation of MMP [166], it brings H+ from the membrane gap back to the mitochondria and releases it into the matrix in the form of protonation, thus eliminating the H+ concentration gradient on both sides of the mitochondrial inner membrane. Among chemical uncouplers, according to their structural properties, small molecule compounds can currently be classified as either proton carrier type or ion carrier type, with proton carrier being the most represented. In cell biology studies, these weakly acidic proton (H+) ionophores, also known as protonophores, are frequently utilized as an inducer of mitophagy. Typical protonophores are mainly Carbonyl Cyanide M-Chloro Phenyl Hydrazone (CCCP), Carbonyl Cyanide-P-(Trifluoromethoxy) Phenyl Hydrazone (FCCP), and 2,4-Dinitrophenol (DNP), etc. They have been frequently employed as chemical probes to stimulate mitophagy and investigate potential mitophagy mechanisms. Among them, DNP is the most concerned weakly acid uncoupler, and it is the first pharmacological application of exogenous uncoupler in human [167]. In addition, CCCP, one of the earliest reported drugs to induce mitophagy, has been successfully used to reveal that PD-related protein Parkin is involved in the regulation of mitophagy [59]. Notably, such protonophores induce mitophagy through the same mechanism—proton mediated dissipation of MMP (Figure 6D). Although protonophores are widely used, they still have a host of limitations in inducing mitophagy. For example, 1) the specificity is low, and they act on the entire mitochondrial population rather than a specific subgroup. Moreover, they not only have activity on mitochondrial membranes, but also on other plasma membranes, causing off-target effects. 2) They are highly toxic, and they can even lead to the complete disappearance of mitochondria in the body after long-term treatment. Namely, they have great cytotoxicity. In particular, even when the dose of FCCP is too low to cause MMP dissipation, it is already cytotoxic [168, 169]. 3) This unnatural method, which does not conform to the physiological process, is harmful to mitochondria to a great extent and will lead to mitochondrial failure. Nevertheless, certain research in recent years has discovered some novel mitochondrial protonophore uncouplers with just a few drawbacks, such as BAM15, which does not depolarize the plasma membrane. BAM15 has less cytotoxicity when compared to the uncoupling agent FCCP of equal strength, and it can generate a higher maximum mitochondrial respiratory rate [170].
Mitochondrial toxins
Similar to protonophores, there are various toxins that can activate mitophagy following dissipation of the MMP. One of the typical mitochondrial toxins is K+ ionophores, such as Valinomycin and Salinomycin. Valinomycin is a respiratory chain inhibitor, and its main mechanism of activating mitophagy is to promote the inward flow of K+ and reduce MMP [171, 172]. However, contrary to valinomycin, salinomycin can promote mitochondrial K+ outflow and H+ inflow. In other words, salinomycin causes the IMM to temporarily become hyperpolarized rather than depolarized [173]. As a result, an excessive buildup of H+ in the mitochondrial matrix will cause the acidification that eventually results in mitochondrial failure. Another class of mitochondrial toxins that have been carefully researched includes diquat [174] and paraquat [175, 176]. Diquat cause mitochondrial damage by overproducing superoxide, thus leading to mitophagy [8, 59] Similar to other toxins, paraquat-induced mitophagy is initiated by mitochondrial depolarization and operates through the PINK1-Parkin pathway.
Metal complexes
Metal complexes represent a promising and rapidly evolving area of pharmacotherapy. Since the serendipitous discovery of the antitumor activity of cisplatin, there has been a continuous surge in studies aimed at the development of new cytotoxic metal complexes. One of them is the metal complexes targeting mitochondria, which are able to bypass resistance mechanisms and to (re-) activate cell-death programs. Gou et al. found that some metal complexes targeting mitochondria can initiate mitophagy effectively, including organic phosphine/sulfur salts, quaternary ammonium (QA) salts, peptides, and transition-metal complexes such as guanidinium or bisguanidinium [177]. Chelators are organic molecules possessing specific ligands that have high affinity, which can bind/carry metal ions and play very important roles in living systems [178]. To note, one typical representative of metal complexes targeting mitochondria is iron chelators. The iron chelators can bind free iron ions, resulting in iron deficiency, and this pharmacologically induced iron deficiency can stimulate mitophagy [179]. Current studies have revealed that iron loss caused by iron chelators triggers mitophagy through two or more unknown mechanisms, so further research is required to clarify these mechanisms [180]. As a novel inducer of mitophagy, the iron chelator deferiprone (DFP) was found to increase mitochondrial turnover without causing MMP collapse, but through an iron-depletion-dependent mechanism [179]. Hence, it does not rely on PINK1 or Parkin to play a role, but directly induces mitophagy by inducing iron deficiency [181]. As for its specific mechanism, recent studies have shown that DFP may promote mitophagy through SUMO-specific protease 3 (SENP3)-mediated deSUMOylation of mitochondrial division protein Fission 1 (Fis1) [182]. SUMOylation involves the attachment of a member of the small ubiquitin like modifier (SUMO) family of proteins to lysine residues in target proteins. This type of iron chelator may have therapeutic potential in inducing mitophagy, but iron chelator-induced mitophagy will result in mitochondrial toxicity, which also means respiratory damage, thus limiting the therapeutic application of such medications.
More recently, it was reported that novel glycosylation zinc (II)-cryptolepine complexes perturb mitophagy pathways and thus trigger cancer cell apoptosis and autophagy [183]. Metabolism can help cancer cells escape chemotherapy, and this process mainly involves autophagy and ATP production. In view of this, Wang et al. reported a new ring metallized Ir (III) complex (Ir-Rhein) based on rhein, which can accurately target mitochondria and effectively inhibit metabolic adaptation [184]. The Ir-Rhein complex can cause serious mitochondrial damage, lead to mitophagy and reduce the number of mitochondria, and then restrict the biological energy of mitochondria and glycolysis, eventually resulting in ATP starvation and death. Studies have shown that Ir-Rhein can overcome the resistance of cancer cells to cisplatin, developing a new mitochondrial-related therapy to overcome resistance to metal drugs [184].
All in all, metal complexes have attracted intense interest over recent decades as probes of mitophagy. And the further development of new metal complexes regulating mitophagy is also a very promising direction.
Natural products
Natural products have offered appealing alternatives for disease prevention and treatment, contributing to the development of modern drugs. Urolithin A (UA) is a kind of metabolite of ellagitannin, a polyphenol compound that is readily available and virtually ubiquitous in nature. Common foods rich in ellagic acid and ellagic tannin include pomegranates, raspberries, blueberries, walnuts, etc. [185]. Additionally, UA is a new type of mitophagy enhancer, a dietary, flora-derived metabolite which can enhance muscle strength and endurance as well as mitochondrial and muscle function during aging [186]. UA mainly triggers mitophagy by lowering MMP without interrupting ROS production and the mitochondrial respiratory chain. However, further study is still needed to find the specific mechanism of action. It has been shown that the enhancement of UA on mitophagy can prolong life by maintaining mitochondrial function, and it has a dose-response effect in a certain range [187].
Berberine is the main active ingredient of traditional Chinese medicine Rhizoma Coptidis and Cortex phellodendri. Its most famous function is lowering blood sugar, which has been applied to the treatment of diabetes for thousands of years [188]. In addition, it also has antibacterial, gastric protection, antioxidant, anti-inflammatory, anti-hypertension and other effects [189]. Liu et al. demonstrated that berberine could induce mitophagy and decrease mitochondrial ROS, thereby suppressing influenza virus-triggered NLRP3 inflammasome activation in macrophages [190]. This study also confirmed that berberine can be used as a potential inducer of mitophagy.
Moustapha et al. investigated the effects of curcumin (25 μM, for 24 h) on apoptosis of human hepatoma-derived Huh-7 cells. In these cells, curcumin promote the formation of autophagic vacuoles containing degraded mitochondria and induced autophagy by increasing the expression levels of LC3-II [191]. It was also found that curcumin could promote Parkin-dependent mitophagy through the AMP-activated protein kinase (AMPK)- transcription factor EB (TFEB) signal pathway, thus decrease oxidative stress-induced intestinal barrier injury and mitochondrial damage [192].
Collectively, natural products and their biological functions are currently a subject of great interest in the pharmaceutical industry, and numbers of scientific studies in this field are increasing rapidly. Dissection of the mechanisms underlying natural products that can induce mitophagy is crucial for discovering more potential therapeutic targets and promoting the development and clinical use of natural medicines.
Others
Based on the scaffold of a known mitophagy-promoting agent spermidine (Spd), a family of structurally related compounds were designed and tested recently. Among these compounds, Srivastava et al. found that a prototypic member, 1,8-diaminooctane (VL-004), exceeds Spd in its ability to induce mitophagy and protect against oxidative stress [193]. VL-004 was shown to promote life span and health span in C. elegans and protect against oxidative injury in rodent and human cells. Moreover, Katayama et al. investigated a compound called T-271 as a promising mitophagy enhancer, which was found to act on damaged mitochondria but does not have damage on normal mitochondria [194]. Additionally, Zhen et al. discovered that flubendazole, which was found to have anti-cancer effects via targeting eva-1 homolog A (EVA1A)-modulated autophagy and apoptosis before [195], could induce mitochondrial dysfunction and Drp1-mediated mitophagy by targeting EVA1A in breast cancer [196]. Flubendazole increased DRP1 expression, which leads to the accumulation of PINK1 and subsequent mitochondrial translocation of Parkin, thereby promoting excessive mitophagy [196, 197].
At present, the research findings on mitophagy inducers are relatively complete and new small molecule mitophagy inducers emerge in endlessly. However, although there are many studies on inhibitors of mitophagy, the discovery of small molecule inhibitors targeting mitophagy that has high specificity is still rare, which needs further in-depth research.
Small molecules mitophagy inhibitors
Generally, autophagy inhibitors that have been widely studied can be used to inhibit mitophagy in vitro. However, due to the low specificity of these inhibitors, they can also inhibit general autophagy or autophagy of other organelles besides preventing mitophagy. This also limits the use of these typical autophagy inhibitors to stimulate mitophagy in vivo. Therefore, in this part, we mainly introduce some small molecules that specifically inhibit mitophagy (Table 3).
Small molecule mitophagy inhibitors
| Names | Chemical structure | Mechanisms | Applications | Advantages or limitations | Ref. |
|---|---|---|---|---|---|
| Mdivi-1 | 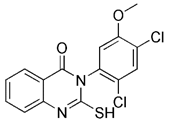 | Inhibiting the activity of Drp1 | Anti-inflammation, anti-oxidative stress, anti-hypertension | Low specificity, protecting cells | [200] |
| Analogue 6 | 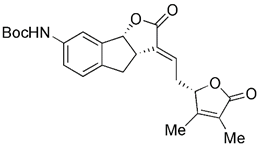 | Increasing the autophagic flux while blocking the autophagosome-lysosome fusion | Anti-cancer such as breast cancer, colorectal cancer, prostate cancer, etc. | Excellent selectivity and potent cytotoxicity against cancer cells | [202] |
| Roflumilast | 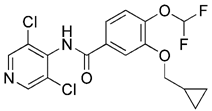 | Inhibiting the expression of phosphor(p)-DRP1 and -PINK1 | Treating chronic obstructive pulmonary disease (COPD), treating cognitive impairment, anti-inflammation | High security, good tolerance, few side effects | [203] |
| IGS2.7 | 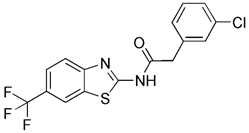 | Inhibiting the expression of ULK1 | Treating ALS | Having no effect on Parkin-dependent mitophagy | [204] |
| QUE | 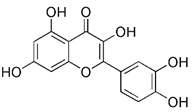 | Inhibiting the expression of p-DRP1 and -PINK1 | Anti-cancer, anti-inflammation, anti-virus, anti-diabetic, anti-oxidative stress | Easy to obtain, low toxicity | [208] |
| Liensinine |  | Inhibiting Drp1-mediated mitochondrial fission, blocking the fusion of mitochondrial autophagosome and lysosomes | Anti-arrhythmia, anti-hypertension, anti-cancer, anti-pulmonary fibrosis | No toxicity, having cardioprotective effects; having potential cardiac side effects | [209, 210] |
Although the application of pharmacological inhibitors of mitophagy has been limited so far, new methods to specifically block mitophagy have been described. Notably, the necessary prerequisite for the initiation of mitophagy is mitochondrial division, which is mediated by the interaction between mitochondrial division protein Fis1 and Drp1 [198]. As Drp1 is the main protein regulating mitochondrial division in mammalian cells [199], the occurrence of mitophagy can be blocked by using compounds that inhibit the activity of Drp1. The mitochondrial division inhibitor 1 (Mdivi-1), an allosteric modulator of Drp1, was discovered by a phenotypic screening procedure in yeast cells [200]. Wu et al. found that Mdivi-1 may alleviate the blood-brain barrier damage and cell death in experimental traumatic brain injury by inhibiting autophagy dysfunction and mitophagy activation [201]. According to a recent study, Yang et al. described the new analogues of strigolactones (SLs) as novel autophagy/mitophagy inhibitors against colorectal cancer cells. Among these analogues, the analogue 6 exerted high specificity against colorectal cancer cells by selectively increasing the autophagic flux while blocking the autophagosome-lysosome fusion, suggesting that it is a novel autophagy/mitophagy inhibitors with high cancer cell specificity [202]. Sun et al. have demonstrated that roflumilast, a phosphodiesterase 4 Inhibitor, was able to reduce the expression of mitophagy regulator proteins Drp1 and PINK1, which shows that roflumilast had the potential to serve as a new inhibitor of mitophagy [203]. Most recently, Maestro et al. identified a small molecule new mitophagy inhibitor named IGS2.7 from the MBC library. Mechanistically, IGS2.7 inhibit the expression of ULK1, thus blocking autophagy and mitophagy. Intriguingly, treating different cellular and in vivo models of ALS with IGS2.7 restores mitophagy to normal levels, indicating a potential and novel therapeutic approach for ALS patients [204]. In addition, besides small molecules, some polypeptides, such as the important Drp1 modulator cell-permeable peptide P110, can block the transport of Drp1 to mitochondria by selectively interfering with its interaction with Fis1 [205]. Furthermore, some peptide inhibitors that can inhibit mitophagy have been found by previous research, such as sequences containing ser13 residues, LIR mimic peptides, a cell permeability probe, and so on [206]. However, although there have been many reports related to the inhibitors of mitophagy hitherto, more research is still needed to developing small molecule pharmacological inhibitors of mitophagy that are highly specific.
Apart from synthetic small molecules inhibitors of mitophagy, some natural products can also inhibit mitophagy. Quercetogetin (QUE) is a polymethoxyflavone found in citrus peels, which has been reported to have different pharmacological effects, such as anticarcinogenic, anti-viral, anti-inflammatory, antioxidant, antithrombogenic, and antiatherogenic effects [207]. Kim et al. found that QUE improved mitochondrial function in human bronchial epithelial cells and protected against cigarette smoke extract (CSE)-induced apoptosis in epithelial cells by inhibiting mitophagy [208]. As a result, QUE may be an option for treating lung diseases brought on by CSE. Liensinine, a major isoquinoline alkaloid, has a wide range of biological activities, including anti-arrhythmia, anti-hypertension, anti-pulmonary fibrosis, and relaxation of vascular smooth muscle, etc [209]. The latest study has revealed that liensinine blocks the fusion of mitochondrial autophagosome and lysosomes by inhibiting the recruitment of RAB7A to lysosomes. Besides downregulating RAB7A, it has also been demonstrated that liensinine could inhibit Drp1-mediated mitochondrial fission [209]. Thus, it is expected to develop into a novel mitophagy inhibitor and be applied to the treatment of various associated diseases [210].
In this section, we review the present small molecules inhibitors of mitophagy, including synthetic and natural compounds. Also, we summarized some mitophagy inhibitors with great potential, such as the polypeptides mentioned above, to bring inspiration for the further development of mitophagy inhibitors. Of note, more research is still needed to find highly specific autophagy inhibitors for mitochondria.
Conclusion and Outlook
In recent years, there has been a longstanding interest in research on mitophagy and the pathogenesis of clinical diseases. It has been found that regulating mitophagy may become a new direction for the treatment of some diseases. By deeply dissecting the molecular mechanism of mitophagy, it may provide a theoretical basis for further research on the treatment of these diseases. In many diseases, the significance and central correlation of mitophagy have been fully confirmed. Mitophagy is needed to control metabolic homeostasis or remove damaged and unnecessary mitochondria, which prevents mitochondrial dysfunction and subsequent molecular events leading to disease development, such as oxidative stress. In other words, mitophagy modulators, particularly activators or inducers, may have great therapeutic potential in diseases. Accordingly, it is imperative to continue researching how diseases are impacted by mitophagy. To thoroughly explore the potential mitophagy regulatory factors, which are not only limited to proteins but also non-coding RNA, the use of some omics techniques, such as proteomics, transcriptomics, metabolomics or various modification omics, as well as single cell sequencing technology, is of particular importance. In order to lay the groundwork for the discovery of pharmacological small molecules targeting mitophagy, we need to further investigate the functions of these mitophagy-regulating factors in diseases.
Although compared to other fields, our understanding of the mitophagy pathway has advanced significantly, it is plainly weak in translating mechanistic research into therapeutically effective drugs. Nowadays, the traditional method of triggering mitophagy in vitro is to induce the collapse of MMP and the stability of PINK1 by chemical methods. Up to now, the vast majority of mitophagy inducers have been essentially mitochondrial toxins or mitochondrial uncouplers, which have numerous limitations, such as low specificity, high toxicity, and respiratory damage. More importantly, the clinical efficacy of mitophagy modulators has not yet been fully defined. According to the most recent research, it is noteworthy that the original protocols based on acute mitochondrial depolarization brought on by non-targeting substances (like FCCP) have been gradually refined into "milder" strategies (like antimycin A and oligomycin) intended to promote mitophagy in a way that is more in line with physiological requirements. Consequently, it is necessary to design new pharmacological methods to manipulate mitophagy and develop drugs to selectively activate mitophagy without interfering with other organelles. With the help of new detection, this process of finding novel modulators can be based on phenotypic screening, rational drug design based on specific target proteins, or accidental discovery based on natural products. In addition, artificial intelligence (AI) technology can also be used to boost drug discovery. Better utilization of modern assay technologies will contribute a lot to the future research into and validation of various medicines. For example, recently, a research team conducted virtual drug screening through AI technology and successfully found two natural small molecules, kaempferol and emodin, with obvious mitophagy induction, which indicated their potential for treatment of AD[211]. It is worth noting that in this study, researchers also compared this model with other methods to determine the accuracy of this model in identifying mitophagy inducers. The results show that the AI model used in this research is the most accurate, and the compounds found by other methods can't induce mitophagy in neurons. Moreover, the hit rate of the AI model used is significantly higher than that of high-throughput screening, and it is superior to other machine learning, quantitative structure-activity relationships (QSAR), and computer-aided methods.
Small molecules are splendid tools for studying and validating the therapeutic implications of mitophagy because they are easy to manage, act rapidly, and are mostly reversible [212]. However, one of the main challenges is to find small molecules that selectively act on mitophagy. Therefore, while designing better mitophagy modulators, targeting of selective mitophagy pathways should be pursued, which may open up a wider therapeutic window. Just as how to accurately and reasonably apply autophagy modulators is still a difficult problem for these small molecule medicines to enter clinical application, the accurate application of existing mitophagy modulators will also be a big challenge in the future. Therefore, there are a series of "points for attention" that need to be considered when assessing chemical probes targeting mitophagy. It is crucial to make sure that the small molecules we choose to use conform to the recommended guidelines for developing tool compounds and chemical probes. Fortunately, the number of these chemicals is continuously increasing. Of note, in the process of small molecule drug delivery or drug release, some nanomaterials can be used to increase mitochondrial targeting and reduce toxicity, such as Titanium dioxide nanoparticles (TiO2-NPs) [213], superparamagnetic iron oxide nanoparticles (SPIO-NPs) [214], zinc oxide nanoparticles (ZnO-NPs) [215], and mesoporous silica nanoparticles (MSNPs) [216]. Using nanomaterials to promote the transport of small molecules drug targeting mitochondria is also a potential and effective method to reduce the off-target effects of some small molecule modulators. In the future, still a lot of work remains to do to discover novel nanomaterials of all kinds targeting mitophagy. And how to exert the advantages of nanotechnology in drug delivery through reasonable design, construct ideal nanocarrier and dosage form, and develop nano-drugs with high-efficiency loading, tumor-specific enrichment and controlled release are the key problems that are waiting to be solved.
In summary, this paper reviews the mechanism of mitophagy, its roles in diseases, and its small molecule pharmacological regulation. It hopes to attract people's attention to new and specific mitophagy modulators and promote the discovery of the next generation of chemical probes and best methods for clinical use. As our understanding of mitophagy deepens, we will be able to select more challenging, but possibly more specific targets. In this way, a host of diseases related to dysfunctional mitophagy will harvest promising potential therapies.
Abbreviations
ROS: reactive oxygen species; Ub: ubiquitin; PINK1: PTEN induced putative kinase 1; MMP/ΔΨm: mitochondrial membrane potential; OMM: outer membrane of mitochondria; p62/SQSTM1: sequestosome-1; PB1: polybromo 1; OPTN: optineurin; NDP52: nuclear dot protein 52; ULK1: Unc-51-like kinase 1; DFCP1: Double FYVE-containing protein 1; WIPI1: WD repeat domain, phosphoinositide interacting 1; LC3: microtubule-associated protein 1 light chain 3; LIR: LC3 interaction region; TBK1: TANK-binding kinase 1; RAB7A: Ras-related protein 7A; SMURF1: smad ubiquitination regulatory factor-1; MUL1: mitochondrial E3 ubiquitin protein ligase 1; Mfn2: mitofusin2; PTPIP51: protein tyrosine phosphatase interacting protein 51; NIX: Nip3-like protein X; BNIP3L: BCL2-interacting protein 3 like; BNIP3: BCL2-interacting protein 3; FUNDC1: FUN14 domain containing 1; Bcl-2: B cell lymphoma-2; RIPK3: receptor-interacting serine/threonine-protein kinase 3; AD: Alzheimer's disease; PD: Parkinson's disease; ALS: Amyotrophic lateral sclerosis; HD: Huntington's disease; Aβ: amyloid-β; PDR-1: Parkinson's disease-related-1; DCT-1: DAF-16/FOXO-controlled germline-tumor affecting-1; SN: substantia nigra; mtDNA: mitochondrial DNA; ZNF746: zinc finger protein 746; mHtt: mutant Huntington protein; Drp1: dynamin-related protein 1; SHR: spontaneously hypertensive rats; ox-LDL: oxidized low density lipoprotein; VSMC: vascular smooth muscle cells; NR4A1: nuclear receptor subfamily 4 group A member 1; eIF2α: eukaryotic initiation factor 2α; MIRI: Myocardial ischemia-reperfusion injury; LOH: loss of heterozygosity; PTEN: phosphatase and tensin homolog; HCC: Hepatocellular carcinoma; T1D: type 1 diabetes; T2D: type 2 diabetes; IR: insulin resistance; NAFLD: Non-alcoholic fatty liver disease; NLRP3: Nod-like receptor protein 3; HFD: high-fat diet; DAMP: danger-associated molecular pattern; SIRS: systemic inflammatory response syndrome; KTP: kinetin triphosphate; MPP+: 1-methyl-4-phenylpyridinium; 6-OHDA: 6-hydroxydopamine; ERK2: extracellular signal regulated protein kinase 2; Phen: 1, 10-phenanthroline; Nrf2: Nuclear factor erythroid 2-related factor 2; PMI: p62-mediated mitophagy inducer; MCL-1: myeloid cell leukemia-1; USP30: ubiquitin-specific peptidase 30; TIM23: translocase of inner mitochondria membrane 23; TOM40: translocase of outer mitochondria membrane 40; SIRT1: Silent information regulator T1 / Sirtuin 1; NAM: nicotinamide; PARP-1: poly (ADP ribose) polymerase-1; SIRT3: Sirtuin-3; TNBC: triple negative breast cancer; IMM: inner membrane of mitochondria; CCCP: Carbonyl Cyanide M-Chloro Phenyl Hydrazone; FCCP: Carbonyl Cyanide-P-(Trifluoromethoxy) Phenyl Hydrazone; DNP: 2,4-Dinitrophenol; QA: quaternary ammonium; DFP: deferiprone; SENP3: SUMO-specific protease 3; Fis1: Fission 1; SUMO: small ubiquitin like modifier; UA: Urolithin A; AMPK: AMP-activated protein kinase; TFEB: transcription factor EB; Spd: spermidine; VL-004: 1,8-diaminooctane; EVA1A: eva-1 homolog A; Mdivi-1: mitochondrial division inhibitor 1; SLs: strigolactones; QUE: Quercetogetin; CSE: cigarette smoke extract; AI: artificial intelligence; QSAR: quantitative structure-activity relationships; TiO2-NPs: titanium dioxide nanoparticles; SPIO-NPs: superparamagnetic iron oxide nanoparticles; ZnO-NPs: zinc oxide nanoparticles; MSNPs: mesoporous silica nanoparticles; COPD: chronic obstructive pulmonary disease.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (Grant No. 22277102, Grant No. 82170887) and Fundamental Research Funds for the Central Universities (Grant No. 2682022ZTPY032).
Author Contributions
L. Zhang and Y. Lu conceived and wrote this review. Y. Lu, Z. Li and S. Zhang searched and analyzed the literature. L. Zhang and Y. Liu reviewed and edited this review. Y. Lu, Z. Li and T. Zhang modified this review. All authors have read and approved the final review manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145-59
2. Doblado L, Lueck C, Rey C, Samhan-Arias AK, Prieto I, Stacchiotti A. et al. Mitophagy in Human Diseases. Int J Mol Sci. 2021 22
3. Deter RL, De Duve C. Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J Cell Biol. 1967;33:437-49
4. Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol. 2009;10:458-67
5. Ohsumi Y. Historical landmarks of autophagy research. Cell Res. 2014;24:9-23
6. Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005;8:3-5
7. Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell. 2009;17:87-97
8. Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J. et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298
9. Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M. et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454:232-5
10. Onishi M, Yamano K, Sato M, Matsuda N, Okamoto K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021;40:e104705
11. Xu Y, Shen J, Ran Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy. 2020;16:3-17
12. Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013;20:31-42
13. Wang N, Zhu P, Huang R, Wang C, Sun L, Lan B. et al. PINK1: The guard of mitochondria. Life Sci. 2020;259:118247
14. Deas E, Plun-Favreau H, Gandhi S, Desmond H, Kjaer S, Loh SH. et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet. 2011;20:867-79
15. Riley BE, Lougheed JC, Callaway K, Velasquez M, Brecht E, Nguyen L. et al. Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat Commun. 2013;4:1982
16. Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway: a mitochondrial quality control system? J Bioenerg Biomembr. 2009;41:499-503
17. Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH. et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162-6
18. Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA. et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211-21
19. Kazlauskaite A, Martínez-Torres RJ, Wilkie S, Kumar A, Peltier J, Gonzalez A. et al. Binding to serine 65-phosphorylated ubiquitin primes Parkin for optimal PINK1-dependent phosphorylation and activation. EMBO Rep. 2015;16:939-54
20. Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL. et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309-14
21. Nguyen TD, Shaid S, Vakhrusheva O, Koschade SE, Klann K, Thölken M. et al. Loss of the selective autophagy receptor p62 impairs murine myeloid leukemia progression and mitophagy. Blood. 2019;133:168-79
22. Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ. et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119-31
23. Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010;6:1090-106
24. Vargas JNS, Wang C, Bunker E, Hao L, Maric D, Schiavo G. et al. Spatiotemporal Control of ULK1 Activation by NDP52 and TBK1 during Selective Autophagy. Mol Cell. 2019;74:347-62.e6
25. Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013;14:759-74
26. Qiu Y, Wang J, Li H, Yang B, Wang J, He Q. et al. Emerging views of OPTN (optineurin) function in the autophagic process associated with disease. Autophagy. 2022;18:73-85
27. Padman BS, Nguyen TN, Uoselis L, Skulsuppaisarn M, Nguyen LK, Lazarou M. LC3/GABARAPs drive ubiquitin-independent recruitment of Optineurin and NDP52 to amplify mitophagy. Nat Commun. 2019;10:408
28. Heo JM, Ordureau A, Swarup S, Paulo JA, Shen K, Sabatini DM. et al. RAB7A phosphorylation by TBK1 promotes mitophagy via the PINK-PARKIN pathway. Sci Adv. 2018;4:eaav0443
29. Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol Cell. 2015;60:7-20
30. Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P. et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A. 2016;113:4039-44
31. Orvedahl A, Sumpter R Jr, Xiao G, Ng A, Zou Z, Tang Y. et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature. 2011;480:113-7
32. Mukherjee R, Chakrabarti O. Ubiquitin-mediated regulation of the E3 ligase GP78 by MGRN1 in trans affects mitochondrial homeostasis. J Cell Sci. 2016;129:757-73
33. Dagar N, Kale A, Steiger S, Anders HJ, Gaikwad AB. Receptor-mediated mitophagy: An emerging therapeutic target in acute kidney injury. Mitochondrion. 2022;66:82-91
34. Igarashi R, Yamashita SI, Yamashita T, Inoue K, Fukuda T, Fukuchi T. et al. Gemcitabine induces Parkin-independent mitophagy through mitochondrial-resident E3 ligase MUL1-mediated stabilization of PINK1. Sci Rep. 2020;10:1465
35. Puri R, Cheng XT, Lin MY, Huang N, Sheng ZH. Mul1 restrains Parkin-mediated mitophagy in mature neurons by maintaining ER-mitochondrial contacts. Nat Commun. 2019;10:3645
36. Fu M, St-Pierre P, Shankar J, Wang PT, Joshi B, Nabi IR. Regulation of mitophagy by the Gp78 E3 ubiquitin ligase. Mol Biol Cell. 2013;24:1153-62
37. Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A. et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11:45-51
38. Yuan Y, Zheng Y, Zhang X, Chen Y, Wu X, Wu J. et al. BNIP3L/NIX-mediated mitophagy protects against ischemic brain injury independent of PARK2. Autophagy. 2017;13:1754-66
39. Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC. et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A. 2007;104:19500-5
40. Shi RY, Zhu SH, Li V, Gibson SB, Xu XS, Kong JM. BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci Ther. 2014;20:1045-55
41. Lin Q, Li S, Jiang N, Jin H, Shao X, Zhu X. et al. Inhibiting NLRP3 inflammasome attenuates apoptosis in contrast-induced acute kidney injury through the upregulation of HIF1A and BNIP3-mediated mitophagy. Autophagy. 2021;17:2975-90
42. Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P. et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177-85
43. Chen Z, Liu L, Cheng Q, Li Y, Wu H, Zhang W. et al. Mitochondrial E3 ligase MARCH5 regulates FUNDC1 to fine-tune hypoxic mitophagy. EMBO Rep. 2017;18:495-509
44. Zhou H, Zhu P, Guo J, Hu N, Wang S, Li D. et al. Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol. 2017;13:498-507
45. Lisanti MP, Martinez-Outschoorn UE, Chiavarina B, Pavlides S, Whitaker-Menezes D, Tsirigos A. et al. Understanding the "lethal" drivers of tumor-stroma co-evolution: emerging role(s) for hypoxia, oxidative stress and autophagy/mitophagy in the tumor micro-environment. Cancer Biol Ther. 2010;10:537-42
46. Perez Ortiz JM, Swerdlow RH. Mitochondrial dysfunction in Alzheimer's disease: Role in pathogenesis and novel therapeutic opportunities. Br J Pharmacol. 2019;176:3489-507
47. Shefa U, Jeong NY, Song IO, Chung HJ, Kim D, Jung J. et al. Mitophagy links oxidative stress conditions and neurodegenerative diseases. Neural Regen Res. 2019;14:749-56
48. Luo F, Sandhu AF, Rungratanawanich W, Williams GE, Akbar M, Zhou S. et al. Melatonin and Autophagy in Aging-Related Neurodegenerative Diseases. Int J Mol Sci. 2020 21
49. Ye X, Sun X, Starovoytov V, Cai Q. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer's disease patient brains. Hum Mol Genet. 2015;24:2938-51
50. Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B. et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer's disease. Nat Neurosci. 2019;22:401-12
51. Fang EF, Scheibye-Knudsen M, Chua KF, Mattson MP, Croteau DL, Bohr VA. Nuclear DNA damage signalling to mitochondria in ageing. Nat Rev Mol Cell Biol. 2016;17:308-21
52. Zhao S, Zhao J, Zhang T, Guo C. Increased apoptosis in the platelets of patients with Alzheimer's disease and amnestic mild cognitive impairment. Clin Neurol Neurosurg. 2016;143:46-50
53. Magistretti PJ, Allaman I. A cellular perspective on brain energy metabolism and functional imaging. Neuron. 2015;86:883-901
54. Wager K, Russell C. Mitophagy and neurodegeneration: the zebrafish model system. Autophagy. 2013;9:1693-709
55. Lee VM, Trojanowski JQ. Mechanisms of Parkinson's disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron. 2006;52:33-8
56. Grünewald A, Kumar KR, Sue CM. New insights into the complex role of mitochondria in Parkinson's disease. Prog Neurobiol. 2019;177:73-93
57. Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9-14
58. Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S. et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605-8
59. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795-803
60. Oh CK, Sultan A, Platzer J, Dolatabadi N, Soldner F, McClatchy DB. et al. S-Nitrosylation of PINK1 Attenuates PINK1/Parkin-Dependent Mitophagy in hiPSC-Based Parkinson's Disease Models. Cell Rep. 2017;21:2171-82
61. Rizza S, Cardaci S, Montagna C, Di Giacomo G, De Zio D, Bordi M. et al. S-nitrosylation drives cell senescence and aging in mammals by controlling mitochondrial dynamics and mitophagy. Proc Natl Acad Sci U S A. 2018;115:E3388-e97
62. Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A. et al. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem. 2003;278:43628-35
63. Brahmachari S, Lee S, Kim S, Yuan C, Karuppagounder SS, Ge P. et al. Parkin interacting substrate zinc finger protein 746 is a pathological mediator in Parkinson's disease. Brain. 2019;142:2380-401
64. Moore AS, Holzbaur EL. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci U S A. 2016;113:E3349-58
65. Moore AS, Holzbaur EL. Spatiotemporal dynamics of autophagy receptors in selective mitophagy. Autophagy. 2016;12:1956-7
66. Harding O, Evans CS, Ye J, Cheung J, Maniatis T, Holzbaur ELF. ALS- and FTD-associated missense mutations in TBK1 differentially disrupt mitophagy. Proc Natl Acad Sci U S A. 2021 118
67. Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A. 2014;111:E4439-48
68. Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, Wild EJ, Saft C, Barker RA. et al. Targeting Huntingtin Expression in Patients with Huntington's Disease. N Engl J Med. 2019;380:2307-16
69. Shirendeb UP, Calkins MJ, Manczak M, Anekonda V, Dufour B, McBride JL. et al. Mutant huntingtin's interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington's disease. Hum Mol Genet. 2012;21:406-20
70. Franco-Iborra S, Plaza-Zabala A, Montpeyo M, Sebastian D, Vila M, Martinez-Vicente M. Mutant HTT (huntingtin) impairs mitophagy in a cellular model of Huntington disease. Autophagy. 2021;17:672-89
71. Manczak M, Reddy PH. Mitochondrial division inhibitor 1 protects against mutant huntingtin-induced abnormal mitochondrial dynamics and neuronal damage in Huntington's disease. Hum Mol Genet. 2015;24:7308-25
72. Siddiqui A, Rivera-Sánchez S, Castro Mdel R, Acevedo-Torres K, Rane A, Torres-Ramos CA. et al. Mitochondrial DNA damage is associated with reduced mitochondrial bioenergetics in Huntington's disease. Free Radic Biol Med. 2012;53:1478-88
73. Forte M, Bianchi F, Cotugno M, Marchitti S, De Falco E, Raffa S. et al. Pharmacological restoration of autophagy reduces hypertension-related stroke occurrence. Autophagy. 2020;16:1468-81
74. Chen Y, Li S, Guo Y, Yu H, Bao Y, Xin X. et al. Astaxanthin Attenuates Hypertensive Vascular Remodeling by Protecting Vascular Smooth Muscle Cells from Oxidative Stress-Induced Mitochondrial Dysfunction. Oxid Med Cell Longev. 2020;2020:4629189
75. Poznyak AV, Nikiforov NG, Wu WK, Kirichenko TV, Orekhov AN. Autophagy and Mitophagy as Essential Components of Atherosclerosis. Cells. 2021 10
76. Docherty CK, Carswell A, Friel E, Mercer JR. Impaired mitochondrial respiration in human carotid plaque atherosclerosis: A potential role for Pink1 in vascular smooth muscle cell energetics. Atherosclerosis. 2018;268:1-11
77. Morciano G, Patergnani S, Bonora M, Pedriali G, Tarocco A, Bouhamida E. et al. Mitophagy in Cardiovascular Diseases. J Clin Med. 2020 9
78. Humeau J, Leduc M, Cerrato G, Loos F, Kepp O, Kroemer G. Phosphorylation of eukaryotic initiation factor-2α (eIF2α) in autophagy. Cell Death Dis. 2020;11:433
79. Lam CK, Zhao W, Liu GS, Cai WF, Gardner G, Adly G. et al. HAX-1 regulates cyclophilin-D levels and mitochondria permeability transition pore in the heart. Proc Natl Acad Sci U S A. 2015;112:E6466-75
80. Wang B, Nie J, Wu L, Hu Y, Wen Z, Dong L. et al. AMPKα2 Protects Against the Development of Heart Failure by Enhancing Mitophagy via PINK1 Phosphorylation. Circ Res. 2018;122:712-29
81. Shires SE, Gustafsson Å B. Mitophagy and heart failure. J Mol Med (Berl). 2015;93:253-62
82. Chang X, Zhang W, Zhao Z, Ma C, Zhang T, Meng Q. et al. Regulation of Mitochondrial Quality Control by Natural Drugs in the Treatment of Cardiovascular Diseases: Potential and Advantages. Front Cell Dev Biol. 2020;8:616139
83. van der Pol A, van Gilst WH, Voors AA, van der Meer P. Treating oxidative stress in heart failure: past, present and future. Eur J Heart Fail. 2019;21:425-35
84. Xiao Y, Chen W, Zhong Z, Ding L, Bai H, Chen H. et al. Electroacupuncture preconditioning attenuates myocardial ischemia-reperfusion injury by inhibiting mitophagy mediated by the mTORC1-ULK1-FUNDC1 pathway. Biomed Pharmacother. 2020;127:110148
85. Marek-Iannucci S, Thomas A, Hou J, Crupi A, Sin J, Taylor DJ. et al. Myocardial hypothermia increases autophagic flux, mitochondrial mass and myocardial function after ischemia-reperfusion injury. Sci Rep. 2019;9:10001
86. Zhou H, Ma Q, Zhu P, Ren J, Reiter RJ, Chen Y. Protective role of melatonin in cardiac ischemia-reperfusion injury: From pathogenesis to targeted therapy. J Pineal Res. 2018 64
87. Ferro F, Servais S, Besson P, Roger S, Dumas JF, Brisson L. Autophagy and mitophagy in cancer metabolic remodelling. Semin Cell Dev Biol. 2020;98:129-38
88. Vara-Perez M, Felipe-Abrio B, Agostinis P. Mitophagy in Cancer: A Tale of Adaptation. Cells. 2019 8
89. Drake LE, Springer MZ, Poole LP, Kim CJ, Macleod KF. Expanding perspectives on the significance of mitophagy in cancer. Semin Cancer Biol. 2017;47:110-24
90. Xu Y, Lu J, Tang Y, Xie W, Zhang H, Wang B. et al. PINK1 deficiency in gastric cancer compromises mitophagy, promotes the Warburg effect, and facilitates M2 polarization of macrophages. Cancer Lett. 2022;529:19-36
91. Cesari R, Martin ES, Calin GA, Pentimalli F, Bichi R, McAdams H. et al. Parkin, a gene implicated in autosomal recessive juvenile parkinsonism, is a candidate tumor suppressor gene on chromosome 6q25-q27. Proc Natl Acad Sci U S A. 2003;100:5956-61
92. Gupta A, Anjomani-Virmouni S, Koundouros N, Dimitriadi M, Choo-Wing R, Valle A. et al. PARK2 Depletion Connects Energy and Oxidative Stress to PI3K/Akt Activation via PTEN S-Nitrosylation. Mol Cell. 2017;65:999-1013.e7
93. Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y. et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486:395-9
94. Vara-Pérez M, Rossi M, Van den Haute C, Maes H, Sassano ML, Venkataramani V. et al. BNIP3 promotes HIF-1α-driven melanoma growth by curbing intracellular iron homeostasis. EMBO J. 2021;40:e106214
95. Humpton TJ, Alagesan B, DeNicola GM, Lu D, Yordanov GN, Leonhardt CS. et al. Oncogenic KRAS Induces NIX-Mediated Mitophagy to Promote Pancreatic Cancer. Cancer Discov. 2019;9:1268-87
96. Sowter HM, Ferguson M, Pym C, Watson P, Fox SB, Han C. et al. Expression of the cell death genes BNip3 and NIX in ductal carcinoma in situ of the breast; correlation of BNip3 levels with necrosis and grade. J Pathol. 2003;201:573-80
97. Li W, Li Y, Siraj S, Jin H, Fan Y, Yang X. et al. FUN14 Domain-Containing 1-Mediated Mitophagy Suppresses Hepatocarcinogenesis by Inhibition of Inflammasome Activation in Mice. Hepatology. 2019;69:604-21
98. Mizushima N, Levine B. Autophagy in Human Diseases. N Engl J Med. 2020;383:1564-76
99. Glick D, Zhang W, Beaton M, Marsboom G, Gruber M, Simon MC. et al. BNip3 regulates mitochondrial function and lipid metabolism in the liver. Mol Cell Biol. 2012;32:2570-84
100. Rovira-Llopis S, Bañuls C, Diaz-Morales N, Hernandez-Mijares A, Rocha M, Victor VM. Mitochondrial dynamics in type 2 diabetes: Pathophysiological implications. Redox Biol. 2017;11:637-45
101. Scheele C, Nielsen AR, Walden TB, Sewell DA, Fischer CP, Brogan RJ. et al. Altered regulation of the PINK1 locus: a link between type 2 diabetes and neurodegeneration? FASEB J. 2007;21:3653-65
102. Rabinovich-Nikitin I, Dhingra R, Kirshenbaum LA. Activation of Mitophagy in High-Fat Diet-Induced Diabetic Cardiomyopathy. Circ Res. 2019;124:1288-90
103. Rosa-Caldwell ME, Lee DE, Brown JL, Brown LA, Perry RA Jr, Greene ES. et al. Moderate physical activity promotes basal hepatic autophagy in diet-induced obese mice. Appl Physiol Nutr Metab. 2017;42:148-56
104. Eid N, Ito Y, Otsuki Y. Triggering of Parkin Mitochondrial Translocation in Mitophagy: Implications for Liver Diseases. Front Pharmacol. 2016;7:100
105. Chen L, Lang AL, Poff GD, Ding WX, Beier JI. Vinyl chloride-induced interaction of nonalcoholic and toxicant-associated steatohepatitis: Protection by the ALDH2 activator Alda-1. Redox Biol. 2019;24:101205
106. Zhou H, Du W, Li Y, Shi C, Hu N, Ma S. et al. Effects of melatonin on fatty liver disease: The role of NR4A1/DNA-PKcs/p53 pathway, mitochondrial fission, and mitophagy. J Pineal Res. 2018 64
107. Zhou T, Chang L, Luo Y, Zhou Y, Zhang J. Mst1 inhibition attenuates non-alcoholic fatty liver disease via reversing Parkin-related mitophagy. Redox Biol. 2019;21:101120
108. Zhang Q, Cao X. Epigenetic Remodeling in Innate Immunity and Inflammation. Annu Rev Immunol. 2021;39:279-311
109. Gkikas I, Palikaras K, Tavernarakis N. The Role of Mitophagy in Innate Immunity. Front Immunol. 2018;9:1283
110. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W. et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104-7
111. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC. et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222-30
112. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221-5
113. Yu J, Nagasu H, Murakami T, Hoang H, Broderick L, Hoffman HM. et al. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc Natl Acad Sci U S A. 2014;111:15514-9
114. Wang R, Zhu Y, Ren C, Yang S, Tian S, Chen H. et al. Influenza A virus protein PB1-F2 impairs innate immunity by inducing mitophagy. Autophagy. 2021;17:496-511
115. Giovarelli M, Zecchini S, Martini E, Garrè M, Barozzi S, Ripolone M. et al. Drp1 overexpression induces desmin disassembling and drives kinesin-1 activation promoting mitochondrial trafficking in skeletal muscle. Cell Death Differ. 2020;27:2383-401
116. Linnane AW, Marzuki S, Ozawa T, Tanaka M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet. 1989;1:642-5
117. Ito S, Araya J, Kurita Y, Kobayashi K, Takasaka N, Yoshida M. et al. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy. 2015;11:547-59
118. Lv M, Zhang S, Jiang B, Cao S, Dong Y, Cao L. et al. Adipose-derived stem cells regulate metabolic homeostasis and delay aging by promoting mitophagy. FASEB J. 2021;35:e21709
119. Hertz NT, Berthet A, Sos ML, Thorn KS, Burlingame AL, Nakamura K. et al. A neo-substrate that amplifies catalytic activity of parkinson's-disease-related kinase PINK1. Cell. 2013;154:737-47
120. Osgerby L, Lai YC, Thornton PJ, Amalfitano J, Le Duff CS, Jabeen I. et al. Kinetin Riboside and Its ProTides Activate the Parkinson's Disease Associated PTEN-Induced Putative Kinase 1 (PINK1) Independent of Mitochondrial Depolarization. J Med Chem. 2017;60:3518-24
121. Zhang F, Peng W, Zhang J, Dong W, Wu J, Wang T. et al. P53 and Parkin co-regulate mitophagy in bone marrow mesenchymal stem cells to promote the repair of early steroid-induced osteonecrosis of the femoral head. Cell Death Dis. 2020;11:42
122. Zhang C, Lin M, Wu R, Wang X, Yang B, Levine AJ. et al. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc Natl Acad Sci U S A. 2011;108:16259-64
123. Garbern JC, Helman A, Sereda R, Sarikhani M, Ahmed A, Escalante GO. et al. Inhibition of mTOR Signaling Enhances Maturation of Cardiomyocytes Derived From Human-Induced Pluripotent Stem Cells via p53-Induced Quiescence. Circulation. 2020;141:285-300
124. Karim MR, Liao EE, Kim J, Meints J, Martinez HM, Pletnikova O. et al. α-Synucleinopathy associated c-Abl activation causes p53-dependent autophagy impairment. Mol Neurodegener. 2020;15:27
125. Borsche M, Pereira SL, Klein C, Grünewald A. Mitochondria and Parkinson's Disease: Clinical, Molecular, and Translational Aspects. J Parkinsons Dis. 2021;11:45-60
126. Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA. et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15:1197-205
127. Wang H, Dou S, Zhu J, Shao Z, Wang C, Xu X. et al. Ghrelin protects against rotenone-induced cytotoxicity: Involvement of mitophagy and the AMPK/SIRT1/PGC1α pathway. Neuropeptides. 2021;87:102134
128. Dagda RK, Zhu J, Kulich SM, Chu CT. Mitochondrially localized ERK2 regulates mitophagy and autophagic cell stress: implications for Parkinson's disease. Autophagy. 2008;4:770-82
129. Li C, Zhang Y, Liu R, Mai Y. Ramelteon ameliorated 1-methyl-4-phenylpyridinium (MPP+)-induced neurotoxicity in neuronal cells in a mitochondrial-dependent pathway. Bioengineered. 2021;12:4868-77
130. Urano Y, Mori C, Fuji A, Konno K, Yamamoto T, Yashirogi S. et al. 6-Hydroxydopamine induces secretion of PARK7/DJ-1 via autophagy-based unconventional secretory pathway. Autophagy. 2018;14:1943-58
131. Park SJ, Shin JH, Kim ES, Jo YK, Kim JH, Hwang JJ. et al. Mitochondrial fragmentation caused by phenanthroline promotes mitophagy. FEBS Lett. 2012;586:4303-10
132. Simula L, Campanella M, Campello S. Targeting Drp1 and mitochondrial fission for therapeutic immune modulation. Pharmacol Res. 2019;146:104317
133. Dinkova-Kostova AT, Abramov AY. The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med. 2015;88:179-88
134. Tsushima M, Liu J, Hirao W, Yamazaki H, Tomita H, Itoh K. Emerging evidence for crosstalk between Nrf2 and mitochondria in physiological homeostasis and in heart disease. Arch Pharm Res. 2020;43:286-96
135. Jain A, Lamark T, Sjøttem E, Larsen KB, Awuh JA, Øvervatn A. et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285:22576-91
136. Lu MC, Ji JA, Jiang ZY, You QD. The Keap1-Nrf2-ARE Pathway As a Potential Preventive and Therapeutic Target: An Update. Med Res Rev. 2016;36:924-63
137. Bar-Peled L, Kemper EK, Suciu RM, Vinogradova EV, Backus KM, Horning BD. et al. Chemical Proteomics Identifies Druggable Vulnerabilities in a Genetically Defined Cancer. Cell. 2017;171:696-709.e23
138. Russo M, Spagnuolo C, Russo GL, Skalicka-Woźniak K, Daglia M, Sobarzo-Sánchez E. et al. Nrf2 targeting by sulforaphane: A potential therapy for cancer treatment. Crit Rev Food Sci Nutr. 2018;58:1391-405
139. Uddin MS, Mamun AA, Jakaria M, Thangapandiyan S, Ahmad J, Rahman MA. et al. Emerging promise of sulforaphane-mediated Nrf2 signaling cascade against neurological disorders. Sci Total Environ. 2020;707:135624
140. Gu J, Cheng Y, Wu H, Kong L, Wang S, Xu Z. et al. Metallothionein Is Downstream of Nrf2 and Partially Mediates Sulforaphane Prevention of Diabetic Cardiomyopathy. Diabetes. 2017;66:529-42
141. Jo C, Kim S, Cho SJ, Choi KJ, Yun SM, Koh YH. et al. Sulforaphane induces autophagy through ERK activation in neuronal cells. FEBS Lett. 2014;588:3081-8
142. East DA, Fagiani F, Crosby J, Georgakopoulos ND, Bertrand H, Schaap M. et al. PMI: a ΔΨm independent pharmacological regulator of mitophagy. Chem Biol. 2014;21:1585-96
143. Kim EH, Sohn S, Kwon HJ, Kim SU, Kim MJ, Lee SJ. et al. Sodium selenite induces superoxide-mediated mitochondrial damage and subsequent autophagic cell death in malignant glioma cells. Cancer Res. 2007;67:6314-24
144. Li J, Qi W, Chen G, Feng D, Liu J, Ma B. et al. Mitochondrial outer-membrane E3 ligase MUL1 ubiquitinates ULK1 and regulates selenite-induced mitophagy. Autophagy. 2015;11:1216-29
145. Shi K, Jiang Q, Li Z, Shan L, Li F, An J. et al. Sodium selenite alters microtubule assembly and induces apoptosis in vitro and in vivo. J Hematol Oncol. 2013;6:7
146. Liu T, Lam V, Thieme E, Sun D, Wang X, Xu F. et al. Pharmacologic Targeting of Mcl-1 Induces Mitochondrial Dysfunction and Apoptosis in B-Cell Lymphoma Cells in a TP53- and BAX-Dependent Manner. Clin Cancer Res. 2021;27:4910-22
147. Abulwerdi F, Liao C, Liu M, Azmi AS, Aboukameel A, Mady AS. et al. A novel small-molecule inhibitor of mcl-1 blocks pancreatic cancer growth in vitro and in vivo. Mol Cancer Ther. 2014;13:565-75
148. Cen X, Chen Y, Xu X, Wu R, He F, Zhao Q. et al. Pharmacological targeting of MCL-1 promotes mitophagy and improves disease pathologies in an Alzheimer's disease mouse model. Nat Commun. 2020;11:5731
149. Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q. et al. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. 2014;510:370-5
150. Cunningham CN, Baughman JM, Phu L, Tea JS, Yu C, Coons M. et al. USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat Cell Biol. 2015;17:160-9
151. Luo H, Krigman J, Zhang R, Yang M, Sun N. Pharmacological inhibition of USP30 activates tissue-specific mitophagy. Acta Physiol. 2021;232:e13666
152. Huang R, Xu Y, Wan W, Shou X, Qian J, You Z. et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell. 2015;57:456-66
153. Yang X, Jiang T, Wang Y, Guo L. The Role and Mechanism of SIRT1 in Resveratrol-regulated Osteoblast Autophagy in Osteoporosis Rats. Sci Rep. 2019;9:18424
154. Zheng W, Feng Z, You S, Zhang H, Tao Z, Wang Q. et al. Fisetin inhibits IL-1β-induced inflammatory response in human osteoarthritis chondrocytes through activating SIRT1 and attenuates the progression of osteoarthritis in mice. Int Immunopharmacol. 2017;45:135-47
155. Tan P, Wang M, Zhong A, Wang Y, Du J, Wang J. et al. SRT1720 inhibits the growth of bladder cancer in organoids and murine models through the SIRT1-HIF axis. Oncogene. 2021;40:6081-92
156. Villalba JM, Alcaín FJ. Sirtuin activators and inhibitors. Biofactors. 2012;38:349-59
157. Hwang ES, Song SB. Nicotinamide is an inhibitor of SIRT1 in vitro, but can be a stimulator in cells. Cell Mol Life Sci. 2017;74:3347-62
158. Arun B, Akar U, Gutierrez-Barrera AM, Hortobagyi GN, Ozpolat B. The PARP inhibitor AZD2281 (Olaparib) induces autophagy/mitophagy in BRCA1 and BRCA2 mutant breast cancer cells. Int J Oncol. 2015;47:262-8
159. Fang EF, Scheibye-Knudsen M, Brace LE, Kassahun H, SenGupta T, Nilsen H. et al. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell. 2014;157:882-96
160. Diao Z, Ji Q, Wu Z, Zhang W, Cai Y, Wang Z. et al. SIRT3 consolidates heterochromatin and counteracts senescence. Nucleic Acids Res. 2021;49:4203-19
161. Zhang J, Zou L, Shi D, Liu J, Zhang J, Zhao R. et al. Structure-Guided Design of a Small-Molecule Activator of Sirtuin-3 that Modulates Autophagy in Triple Negative Breast Cancer. J Med Chem. 2021;64:14192-216
162. Barzegari A, Nouri M, Gueguen V, Saeedi N, Pavon-Djavid G, Omidi Y. Mitochondria-targeted antioxidant mito-TEMPO alleviate oxidative stress induced by antimycin A in human mesenchymal stem cells. J Cell Physiol. 2020;235:5628-36
163. Ivanes F, Faccenda D, Gatliff J, Ahmed AA, Cocco S, Cheng CH. et al. The compound BTB06584 is an IF1 -dependent selective inhibitor of the mitochondrial F1 Fo-ATPase. Br J Pharmacol. 2014;171:4193-206
164. Lin X, Wen Y, Li M, Chen Z, Guo J, Song Y. et al. A new strain of Streptomyces avermitilis produces high yield of oligomycin A with potent anti-tumor activity on human cancer cell lines in vitro. Appl Microbiol Biotechnol. 2009;81:839-45
165. Harvey WR. Voltage coupling of primary H+ V-ATPases to secondary Na+- or K+-dependent transporters. J Exp Biol. 2009;212:1620-9
166. Valmas N, Zuryn S, Ebert PR. Mitochondrial uncouplers act synergistically with the fumigant phosphine to disrupt mitochondrial membrane potential and cause cell death. Toxicology. 2008;252:33-9
167. Shrestha R, Johnson E, Byrne FL. Exploring the therapeutic potential of mitochondrial uncouplers in cancer. Mol Metab. 2021;51:101222
168. Tsiper MV, Sturgis J, Avramova LV, Parakh S, Fatig R, Juan-García A. et al. Differential mitochondrial toxicity screening and multi-parametric data analysis. PLoS One. 2012;7:e45226
169. Brennan JP, Berry RG, Baghai M, Duchen MR, Shattock MJ. FCCP is cardioprotective at concentrations that cause mitochondrial oxidation without detectable depolarisation. Cardiovasc Res. 2006;72:322-30
170. Kenwood BM, Weaver JL, Bajwa A, Poon IK, Byrne FL, Murrow BA. et al. Identification of a novel mitochondrial uncoupler that does not depolarize the plasma membrane. Mol Metab. 2014;3:114-23
171. Xiong X, Li S, Han TL, Zhou F, Zhang X, Tian M. et al. Study of mitophagy and ATP-related metabolomics based on β-amyloid levels in Alzheimer's disease. Exp Cell Res. 2020;396:112266
172. Zhang D, Ma Z, Chen H, Lu Y, Chen X. Valinomycin as a potential antiviral agent against coronaviruses: A review. Biomed J. 2020;43:414-23
173. Managò A, Leanza L, Carraretto L, Sassi N, Grancara S, Quintana-Cabrera R. et al. Early effects of the antineoplastic agent salinomycin on mitochondrial function. Cell Death Dis. 2015;6:e1930
174. Cao S, Wu H, Wang C, Zhang Q, Jiao L, Lin F. et al. Diquat-induced oxidative stress increases intestinal permeability, impairs mitochondrial function, and triggers mitophagy in piglets. J Anim Sci. 2018;96:1795-805
175. Lee IJ, Chao CY, Yang YC, Cheng JJ, Huang CL, Chiou CT. et al. Huang Lian Jie Du Tang attenuates paraquat-induced mitophagy in human SH-SY5Y cells: A traditional decoction with a novel therapeutic potential in treating Parkinson's disease. Biomed Pharmacother. 2021;134:111170
176. Dinis-Oliveira RJ, Duarte JA, Sánchez-Navarro A, Remião F, Bastos ML, Carvalho F. Paraquat poisonings: mechanisms of lung toxicity, clinical features, and treatment. Crit Rev Toxicol. 2008;38:13-71
177. Guo X, Yang N, Ji W, Zhang H, Dong X, Zhou Z. et al. Mito-Bomb: Targeting Mitochondria for Cancer Therapy. Adv Mater. 2021;33:e2007778
178. Kontoghiorghes GJ. Advances on Chelation and Chelator Metal Complexes in Medicine. Int J Mol Sci. 2020 21
179. Allen GF, Toth R, James J, Ganley IG. Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep. 2013;14:1127-35
180. Hara Y, Yanatori I, Tanaka A, Kishi F, Lemasters JJ, Nishina S. et al. Iron loss triggers mitophagy through induction of mitochondrial ferritin. EMBO Rep. 2020;21:e50202
181. Fiorillo M, Tóth F, Brindisi M, Sotgia F, Lisanti MP. Deferiprone (DFP) Targets Cancer Stem Cell (CSC) Propagation by Inhibiting Mitochondrial Metabolism and Inducing ROS Production. Cells. 2020 9
182. Wilkinson KA, Guo C. Iron chelation promotes mitophagy through SENP3-mediated deSUMOylation of FIS1. Autophagy. 2022;18:1743-5
183. Zhou Z, Du LQ, Huang XM, Zhu LG, Wei QC, Qin QP. et al. Novel glycosylation zinc(II)-cryptolepine complexes perturb mitophagy pathways and trigger cancer cell apoptosis and autophagy in SK-OV-3/DDP cells. Eur J Med Chem. 2022;243:114743
184. Wang MM, Xu FJ, Su Y, Geng Y, Qian XT, Xue XL. et al. A New Strategy to Fight Metallodrug Resistance: Mitochondria-Relevant Treatment through Mitophagy to Inhibit Metabolic Adaptations of Cancer Cells. Angew Chem Int Ed Engl. 2022;61:e202203843
185. Singh A, D'Amico D, Andreux PA, Fouassier AM, Blanco-Bose W, Evans M. et al. Urolithin A improves muscle strength, exercise performance, and biomarkers of mitochondrial health in a randomized trial in middle-aged adults. Cell Rep Med. 2022;3:100633
186. Ryu D, Mouchiroud L, Andreux PA, Katsyuba E, Moullan N, Nicolet-Dit-Félix AA. et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med. 2016;22:879-88
187. Andreux PA, Blanco-Bose W, Ryu D, Burdet F, Ibberson M, Aebischer P. et al. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat Metab. 2019;1:595-603
188. Tian X, Liu F, Li Z, Lin Y, Liu H, Hu P. et al. Enhanced Anti-diabetic Effect of Berberine Combined With Timosaponin B2 in Goto-Kakizaki Rats, Associated With Increased Variety and Exposure of Effective Substances Through Intestinal Absorption. Front Pharmacol. 2019;10:19
189. Wang K, Feng X, Chai L, Cao S, Qiu F. The metabolism of berberine and its contribution to the pharmacological effects. Drug Metab Rev. 2017;49:139-57
190. Liu H, You L, Wu J, Zhao M, Guo R, Zhang H. et al. Berberine suppresses influenza virus-triggered NLRP3 inflammasome activation in macrophages by inducing mitophagy and decreasing mitochondrial ROS. J Leukoc Biol. 2020;108:253-66
191. Moustapha A, Pérétout PA, Rainey NE, Sureau F, Geze M, Petit JM. et al. Curcumin induces crosstalk between autophagy and apoptosis mediated by calcium release from the endoplasmic reticulum, lysosomal destabilization and mitochondrial events. Cell Death Discov. 2015;1:15017
192. Cao S, Wang C, Yan J, Li X, Wen J, Hu C. Curcumin ameliorates oxidative stress-induced intestinal barrier injury and mitochondrial damage by promoting Parkin dependent mitophagy through AMPK-TFEB signal pathway. Free Radic Biol Med. 2020;147:8-22
193. Srivastava V, Zelmanovich V, Shukla V, Abergel R, Cohen I, Ben-Sasson SA. et al. Distinct designer diamines promote mitophagy, and thereby enhance healthspan in C. elegans and protect human cells against oxidative damage. Autophagy. 2022:1-31
194. Katayama H, Hama H, Nagasawa K, Kurokawa H, Sugiyama M, Ando R. et al. Visualizing and Modulating Mitophagy for Therapeutic Studies of Neurodegeneration. Cell. 2020;181:1176-87.e16
195. Zhen Y, Zhao R, Wang M, Jiang X, Gao F, Fu L. et al. Flubendazole elicits anti-cancer effects via targeting EVA1A-modulated autophagy and apoptosis in Triple-negative Breast Cancer. Theranostics. 2020;10:8080-97
196. Zhen Y, Yuan Z, Zhang J, Chen Y, Fu Y, Liu Y. et al. Flubendazole induces mitochondrial dysfunction and DRP1-mediated mitophagy by targeting EVA1A in breast cancer. Cell Death Dis. 2022;13:375
197. Yu CG, Bondada V, Ghoshal S, Singh R, Pistilli CK, Dayaram K. et al. Repositioning Flubendazole for Spinal Cord Injury. J Neurotrauma. 2019;36:2618-30
198. Kageyama Y, Hoshijima M, Seo K, Bedja D, Sysa-Shah P, Andrabi SA. et al. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J. 2014;33:2798-813
199. Han H, Tan J, Wang R, Wan H, He Y, Yan X. et al. PINK1 phosphorylates Drp1(S616) to regulate mitophagy-independent mitochondrial dynamics. EMBO Rep. 2020;21:e48686
200. Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T. et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193-204
201. Wu Q, Gao C, Wang H, Zhang X, Li Q, Gu Z. et al. Mdivi-1 alleviates blood-brain barrier disruption and cell death in experimental traumatic brain injury by mitigating autophagy dysfunction and mitophagy activation. Int J Biochem Cell Biol. 2018;94:44-55
202. Yang ST, Fan JB, Liu TT, Ning S, Xu JH, Zhou YJ. et al. Development of Strigolactones as Novel Autophagy/Mitophagy Inhibitors against Colorectal Cancer Cells by Blocking the Autophagosome-Lysosome Fusion. J Med Chem. 2022
203. Kyung SY, Kim YJ, Son ES, Jeong SH, Park JW. The Phosphodiesterase 4 Inhibitor Roflumilast Protects against Cigarette Smoke Extract-Induced Mitophagy-Dependent Cell Death in Epithelial Cells. Tuberc Respir Dis. 2018;81:138-47
204. Maestro I, de la Ballina LR, Porras G, Corrochano S, De Lago E, Simonsen A. et al. Discovery of Mitophagy Inhibitors with Therapeutic Potential in Different Familial Amyotrophic Lateral Sclerosis Mutations. Int J Mol Sci. 2022 23
205. Qi X, Qvit N, Su YC, Mochly-Rosen D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J Cell Sci. 2013;126:789-802
206. Chen G, Han Z, Feng D, Chen Y, Chen L, Wu H. et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell. 2014;54:362-77
207. Gattuso G, Barreca D, Gargiulli C, Leuzzi U, Caristi C. Flavonoid composition of Citrus juices. Molecules. 2007;12:1641-73
208. Son ES, Kim SH, Ryter SW, Yeo EJ, Kyung SY, Kim YJ. et al. Quercetogetin protects against cigarette smoke extract-induced apoptosis in epithelial cells by inhibiting mitophagy. Toxicol In Vitro. 2018;48:170-8
209. Liang X, Wang S, Wang L, Ceylan AF, Ren J, Zhang Y. Mitophagy inhibitor liensinine suppresses doxorubicin-induced cardiotoxicity through inhibition of Drp1-mediated maladaptive mitochondrial fission. Pharmacol Res. 2020;157:104846
210. Zhou J, Li G, Zheng Y, Shen HM, Hu X, Ming QL. et al. A novel autophagy/mitophagy inhibitor liensinine sensitizes breast cancer cells to chemotherapy through DNM1L-mediated mitochondrial fission. Autophagy. 2015;11:1259-79
211. Xie C, Zhuang XX, Niu Z, Ai R, Lautrup S, Zheng S. et al. Amelioration of Alzheimer's disease pathology by mitophagy inducers identified via machine learning and a cross-species workflow. Nat Biomed Eng. 2022;6:76-93
212. Workman P, Collins I. Probing the probes: fitness factors for small molecule tools. Chem Biol. 2010;17:561-77
213. Hou J, Wang L, Wang C, Zhang S, Liu H, Li S. et al. Toxicity and mechanisms of action of titanium dioxide nanoparticles in living organisms. J Environ Sci. 2019;75:40-53
214. He C, Jiang S, Yao H, Zhang L, Yang C, Jiang S. et al. High-content analysis for mitophagy response to nanoparticles: A potential sensitive biomarker for nanosafety assessment. Nanomedicine. 2019;15:59-69
215. Wang J, Gao S, Wang S, Xu Z, Wei L. Zinc oxide nanoparticles induce toxicity in CAL 27 oral cancer cell lines by activating PINK1/Parkin-mediated mitophagy. Int J Nanomedicine. 2018;13:3441-50
216. Orlando A, Cazzaniga E, Tringali M, Gullo F, Becchetti A, Minniti S. et al. Mesoporous silica nanoparticles trigger mitophagy in endothelial cells and perturb neuronal network activity in a size- and time-dependent manner. Int J Nanomedicine. 2017;12:3547-59
Author contact
![]() Corresponding authors: Yanjun Liu, E-mail: liuyanjun_001com; Lan Zhang, E-mail: zhanglanx_9com
Corresponding authors: Yanjun Liu, E-mail: liuyanjun_001com; Lan Zhang, E-mail: zhanglanx_9com