Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
KEAP1-NRF2 in different...
Cancer Metabolic Reprogramming
TIME
Therapeutic target
Concluding remarks
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2023; 13(2):704-723. doi:10.7150/thno.80184 This issue Cite
Review
Battles against aberrant KEAP1-NRF2 signaling in lung cancer: intertwined metabolic and immune networks
Ke Xu1,*, Jie Ma2,*, Sean R. R. Hall3, Ren-Wang Peng4, Haitang Yang1,*, ![]() , Feng Yao1
, Feng Yao1
1. Department of Thoracic Surgery, Shanghai Chest Hospital, Shanghai Jiao Tong University, Shanghai, 200030, People's Republic of China.
2. Department of Thoracic Surgery, Anhui Chest Hospital, Hefei, 230000, China
3. Wyss Institute for Biologically Inspired Engineering, Harvard University; Boston, MA 02115, USA.
4. Division of General Thoracic Surgery, Department of BioMedical Research (DBMR), Inselspital, Bern University Hospital, University of Bern; Bern, 3010, Switzerland.
*These authors contributed equally to this work.
Received 2022-10-25; Accepted 2022-12-14; Published 2023-1-1
Abstract

The Kelch-like ECH-associated protein 1/nuclear factor erythroid-derived 2-like 2 (KEAP1/NRF2) pathway is well recognized as a key regulator of redox homeostasis, protecting cells from oxidative stress and xenobiotics under physiological circumstances. Cancer cells often hijack this pathway during initiation and progression, with aberrant KEAP1-NRF2 activity predominantly observed in non-small cell lung cancer (NSCLC), suggesting that cell/tissue-of-origin is likely to influence the genetic selection during malignant transformation. Hyperactivation of NRF2 confers a multi-faceted role, and recently, increasing evidence shows that a close interplay between metabolic reprogramming and tumor immunity remodelling contributes to its aggressiveness, treatment resistance (radio-/chemo-/immune-therapy) and susceptibility to metastases. Here, we discuss in detail the special metabolic and immune fitness enabled by KEAP1-NRF2 aberration in NSCLC. Furthermore, we summarize the similarities and differences in the dysregulated KEAP1-NRF2 pathway between two major histo-subtypes of NSCLC, provide mechanistic insights on the poor response to immunotherapy despite their high immunogenicity, and outline evolving strategies to treat this recalcitrant cancer subset. Finally, we integrate bioinformatic analysis of publicly available datasets to illustrate the new partners/effectors in NRF2-addicted cancer cells, which may provide new insights into context-directed treatment.
Keywords: KEAP1-NRF2 signaling, non-small cell lung cancer, metabolic reprogramming, tumor immune microenvironment, bioinformatics, therapeutic vulnerabilities
Introduction
The Kelch-like ECH-associated protein 1 (KEAP1)-nuclear factor erythroid-derived 2-like 2 (NFE2L2) signaling pathway is well characterized by protecting cells from oxidative stress and xenobiotics. Under physiological conditions, KEAP1 binds to NRF2 and promotes its degradation through the ubiquitin-proteasome pathway, acting as an inhibitor of NRF2. Oxidative stress or electrophiles disrupt the KEAP1-NRF2 interaction, resulting in transient activation of NRF2 and subsequent translocation into the nucleus, where it binds to antioxidant response elements (ARE) in the genome. Consequently, transcription factor NRF2 activates several downstream genes including glutamate-cysteine ligase (GCL), heme oxygenase 1 (HO-1), NAD(P)H dehydrogenase (NQO1), glutathione S-transferase (GST) and thioredoxin reductase1 (TrxR1), involved in detoxification and antioxidant function.
NRF2 signaling is also frequently hijacked in various cancer types, most commonly in non-small cell lung cancer (NSCLC) [1, 2]. In cancer, genetic alterations in KEAP1 or NFE2L2 (encoding NRF2) are the most common cause of NRF2 hyperactivation. The mutational landscape and the interaction between KEAP1 and NRF2 have been extensively covered in previous reviews [3-5]. In addition, several other causes of NRF2 hyperactivation in cancer cells have also been described at genomic, transcriptional, and posttranslational levels. Specifically, CUL3 deletion or mutation results in the loss of KEAP1-dependent NRF2 ubiquitylation, causing aberrant NRF2 accumulations [6]. Transcriptional changes, e.g., the loss of RBM47, are associated with KEAP1 mRNA stabilization, and the existence of alternatively spliced NRF2 variants also contributes to NRF2 augmentation [7, 8]. Furthermore, sequestration of KEAP1 by NRF2-competitive-binding proteins including TRIM21, RPRD1A, and FAM129B was found in NRF2-addicted cancer models [9-11]. Thus, it remains to explore the best way to define the NSCLC with high NRF2 addition. NRF2 score-based evaluation might be more feasible [12, 13].
The tumor micro-ecosystem contains cancer, stromal and immune cells, which corporately promote tumorigenesis and disease progression. Of the hallmarks of cancer, metabolism and immune evasion represent the most intertwined partners [14, 15]. Accumulating evidence has revealed that aberrant KEAP1-NRF2 signaling influences both cancer cells and the cancer-associated microenvironment [16, 17], conferring specific metabolic vulnerabilities [18-20] and shaping a unique tumor immune microenvironment (TIME) [16, 21].
In this review, we focus on the role of the aberrant KEAP1-NRF2 signaling pathway in reprogramming tumor metabolism and modulating TIME in NSCLC. We also for the first time summarize the similarities and differences in the dysregulated KEAP1-NRF2 pathway between two major histo-subtypes of NSCLC. We combined literature review and mining of publicly available data to illustrate a more comprehensive landscape of dysregulated KEAP1-NRF2 pathway, which may contribute to a better understanding of the pathogenesis of this special disease and provide new directions for context-dependent treatment.
KEAP1-NRF2 in different histo-subtypes of NSCLC: similarities and differences
NSCLC contains two major histo-subtypes: lung squamous cell carcinoma (LUSC) and adenocarcinoma (LUAD), which originate in different cell types and are related to different risk factors (Figure 1A). The fact that genetic alterations in KEAP1-NFE2L2 preferentially occur in human lung tumors suggests that the cell/tissue-of-origin is likely to influence the genetic selection that drives malignant transformation [22-24].
Similarities and differences of KEAP1-NRF2 aberration in human non-small cell lung cancer. A, Left panel: schematic model showing the common location of lung squamous cell carcinoma (LUSC; central; associated with a smoking history) and adenocarcinoma (LUAD; peripheral). Middle panel: key components of KEAP1-NRF2 pathway. Right panel: genetic alterations of KEAP1, NFE2L2, CUL3 alone or in combination across The Cancer Genome Atlas (TCGA) LUSC and LUAD, respectively. Data were downloaded from the cBioPortal (https://www.cbioportal.org/). B, Oncoprint of genetic alterations of KEAP1, NFE2L2, CUL3 across TCGA LUSC and LUAD. The corresponding patient sex, tumor mutational burden (TMB) and three curated signatures calculating the hypoxia score were also shown, which displayed a different distribution between LUSC and LUAD. Data were downloaded from the cBioPortal (https://www.cbioportal.org/). C, Comparing the transcriptional molecular features between mutated (Mut) (KEAP1, NFE2L2, CUL3) and wildtype (WT) in patient samples of LUSC and LUAD. Nonnegative matrix factorization (NMF) clustering was performed to identify the molecular subtypes. Data were downloaded from the cBioPortal (https://www.cbioportal.org/).
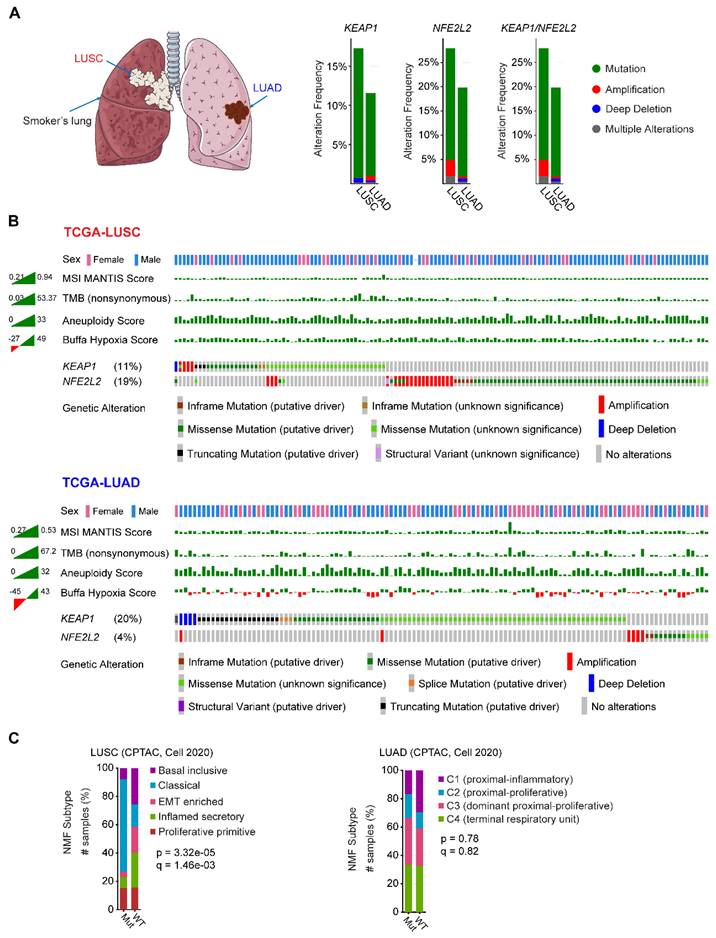
Mining publicly available genomic data from The Cancer Genome Atlas (TCGA) NSCLC cohort revealed that LUSC has a higher proportion of genetic alterations in KEAP1/NFE2L2, particularly NFE2L2, compared with its LUAD counterpart (Figure 1A-B). Furthermore, LUSC harboring KEAP1-NFE2L2 alterations is observed with a higher frequency in male patients, and a differential distribution in the microsatellite instability (MSI), tumor mutational burden (TMB), aneuploidy, and notably, hypoxia scores could be observed between LUSC and LUAD (Figure 1B). Furthermore, non-negative matrix factorization-based clustering analysis of proteomic data also revealed markedly different molecular signatures between these two histo-subtypes (Figure 1C). These observations imply that KEAP1-NFE2L2 alterations confer different biological behaviors in the two subtypes of NSCLC. Supporting this notion, recent multi-omics data of patient samples showed that unlike LUAD [25], KEAP1 mutations did not result in significantly reduced protein expression in LUSC, suggesting the differences in NRF2 pathway dysregulation between NSCLC subtypes [26]. Also, based on genetically engineered mouse models (GEEM), Li et al. showed convincing evidence that there is differential NRF2 and reactive oxygen species (ROS) levels between LUAD and LUSC [27]. Strikingly, ROS functionally modulates LUAD to LUSC transdifferentiation to drive cancer progression and escape therapy. Accordingly, modulation of redox balance by overexpression of Nrf2 or treatment with ROS scavenger N-acetyl cysteine reduced the frequency of squamous tumors. Their data suggested that ROS function as a major driver of lung tumor differentiation between LUAD and LUSC. In addition, Keap1-deficient KrasG12D mouse lung tumors arising from a bronchiolar cell-of-origin, lack pro-tumorigenic macrophages that are observed in tumors originating from alveolar cells [17], further reinforcing the notion that cells-of-origin influence the pathobiology of KEAP1-mutant NSCLC.
Although it remains to define the fundamental molecular underpinnings underlying the difference between the two NSCLC histo-subtypes with hyperactivated NRF2, the co-occurring mutations with KEAP1/NFE2L2 genes might provide certain explanations. In LUAD, KEAP1 mutation often co-exists with STK11 and KRAS, whereas NFE2L2 frequently co-occurs with TP53 mutations in LUSC [28]. Co-mutations with KEAP1 and STK11 configure a special metabolically and immunologically addicted phenotype in patients with KRAS-mutant LUAD [29]. Besides, the KEAP1 and STK11 co-mutated LUAD also exhibits a ferroptosis-tolerate behavior, a newly discovered form of programmed cell death [30]. In keeping with these data, clinical evidence shows that in KEAP1-mutant LUAD, co-mutated PBRM1, SMARC4, or STK11 is associated with poor response to immunotherapy as compared with single-mutant or wild-type tumors [31]. As to LUSC, the KEAP1 and TP53 co-mutations have been found in airway basal stem cells, promoting their renewal and expansion characteristics and conferring radiation resistance [32]. While other co-mutations in LUSC are rarely mentioned. Thus, different co-mutated genes might also play a role in contributing to distinct biological behaviors between LUAD and LUSC.
Notably, the majority of preclinical mouse models focus predominantly on LUAD [16, 19, 20, 33, 34], but very few on LUSC [32]. Although the regulation of oxidative stress response has been revealed in both subtypes, whether KEAP1-NFE2L2 alterations hold equal weights in LUSC and LUAD, or whether the features of KEAP1-NFEL2 alterations are governed by the cancer cell-of-origin warrants further investigations.
Cancer Metabolic Reprogramming
Metabolic reprogramming is a hallmark of cancer, sustaining the survival and proliferation of cancer cells by regulating several key metabolic pathways such as glycolysis, glutaminolysis, and lipid metabolism to meet the elevated biosynthetic needs of tumors [35]. Beyond its classical role in regulating oxidative stress, increasing evidence has revealed crosstalk between KEAP1-NRF2 signaling and tumor metabolic reprogramming [18-20], thereby creating targetable metabolic vulnerabilities. More recently, in a systematic study that aims to determine the redox vulnerability of KEAP1-NRF2-mutant NSCLC, CRISPR-Cas9-based screen for antioxidant enzymes led to the identification of multiple hits involved in the pentose phosphate pathway (PPP), the thioredoxin-dependent antioxidant system, and glutathione reductase, as well as mitochondrial superoxide dismutase 2 (SOD2) [36]. This suggests that KEAP1-NRF2 signaling is related to a wide range of metabolic activities.
Glucose metabolism
Aberrant glucose metabolism is a prominent type of metabolic reprogramming in cancer, which not only involves glycolysis that was preferentially used by tumors (Warburg effect) but also other pathways that require glucose, e.g., the PPP that generates pentose phosphates for ribonucleotide synthesis and NADPH for reducing equivalent [37]. Recent studies indicated that KEAP1-NRF2 signaling drives glucose addiction in NSCLC cancer, and cancer cells with KEAP1 inactivation are more vulnerable to glucose deprivation (Figure 2) [18].
Crosstalk between tumor metabolism and immunity in lung cancer with KEAP1-NRF2 aberration. KEAP1-NRF2 aberration plays a pleiotropic role in tumor metabolism, which has a close interplay with tumor immunity
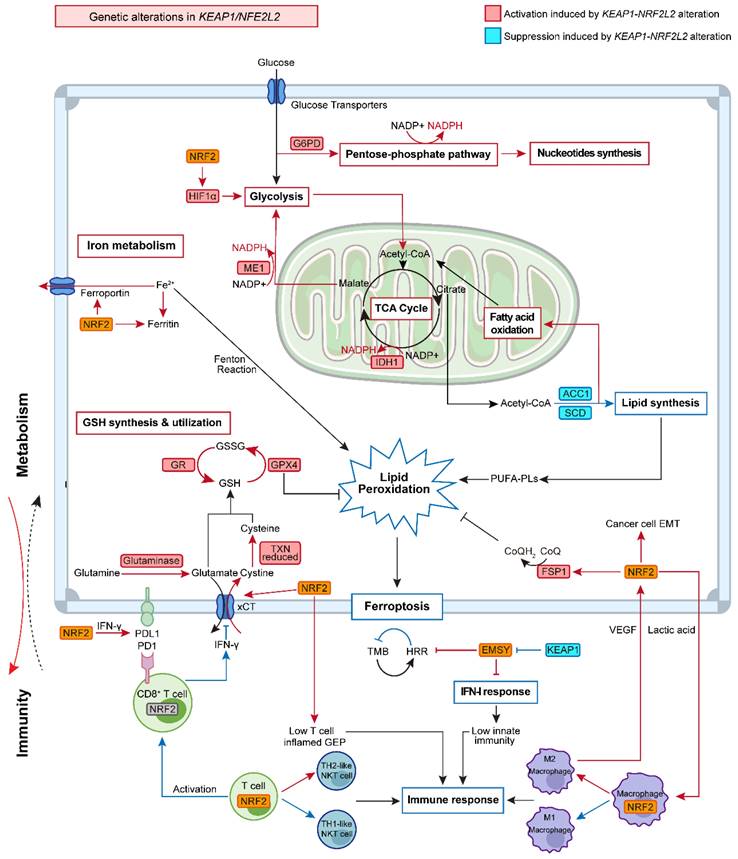
Glycolysis
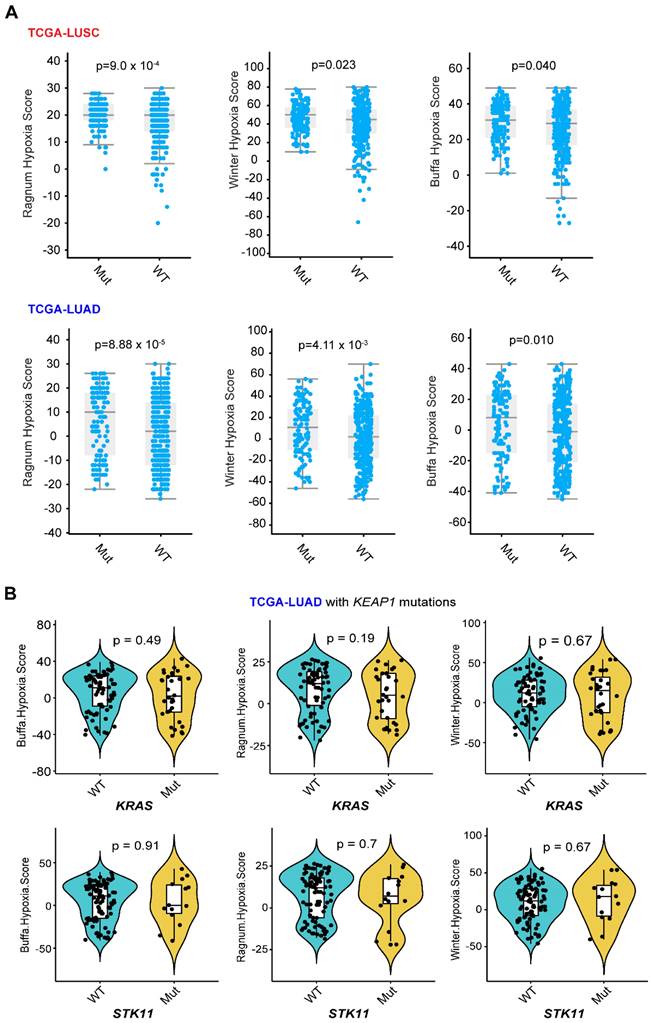
The upregulation of glycolysis in cancer cells was related to hypoxia-inducible factor 1 α (HIF1α), followed by HIF1α-related glycolytic enzyme transcription [38]. Analysis of publicly available data revealed that KEAP1-NRF2 alterations are overall related to a higher hypoxia score based on different algorithms (Figure 3A), and that the common genes co-occurring with KEAP1 mutations in LUAD do not appear to affect their hypoxia status (Figure 3B). Of note, the above data-mining analysis revealed that mutated KEAP1-NRF2 in TCGA LUSC appears to be associated with a markedly higher hypoxia score (Figure 1B), while the biological explanation for this and the reason for the difference between LUSC and LUAD need further investigation.
Different hypoxia index between lung tumor with and without KEAP1-NRF2 aberration. A, Three different curated signatures were used to compare the hypoxia score between mutated (Mut) (KEAP1, NFE2L2, CUL3) and wildtype (WT) in patient samples of TCGA LUSC (lung squamous cell carcinoma) and LUAD (lung adenocarcinoma). Data were downloaded from the cBioPortal (https://www.cbioportal.org/). Of note, the “subtle and marginal significance” between the comparative subgroups might be due to: 1) the presence of high NRF2 activity in the KEAP1-WT lung tumors, as NRF2 activity can be regulated at multiple levels including genetic, transcriptional, and post-transcriptional. As such, NRF2 score [12, 13] might better reflect the activity of NRF2 signaling. B, In LUAD, mutated KEAP1 is commonly co-occurring with KRAS or STK11 mutation. Different algorithms-based hypoxia scores were used to compare the difference between KRAS (upper panel) or STK11 (lower panel) wildtype (WT) and mutant (Mut) tumors under the context of KEAP1 mutation in LUAD. P-value was calculated by Wilcoxon Test.
The hyperactivation of the rate-limiting enzymes of the glycolytic pathway, e.g., hexokinase (HK) and pyruvate kinase (PK) was found in NRF2-addicted cancer [39]. Furthermore, Zhang et al. demonstrated that NRF2 stimulates cancer cell progression by targeting HIF1α to enhance the expression of several key glycolytic genes including hexokinase 2 (HK2), phosphofructokinase-2/fructose-2,6-bisphosphatase 3 (PFKFB3), pyruvate kinase isozymes M2 (PKM2) and lactate dehydrogenase A (LDHA) [40]. Similarly, Lee et al. showed that NRF2-silencing suppressed HIF1α accumulation and subsequently inhibited the expression of glycolysis-associated glucose transporter-1, HK2, pyruvate dehydrogenase kinase-1 (PDK1), and LDHA [41], suggesting the robust regulation of NRF2 on glycolysis is mediated by HIF1α. The interplay between HIF1α and NRF2 has also been revealed in other diseases [42, 43].
PPP and NADPH production
The PPP includes two distinct phases: the oxidative phase with NADPH production and the non-oxidative phase with 5-carbon sugars synthesis. Therefore, the PPP contributes to tumor proliferation not only by producing NADPH to buffer oxidative stress and prevent cells from death, but also by providing materials needed for nucleotide synthesis [44]. The activation of these processes was also found to engage in NRF2-dependent tumor progression and metastasis.
Previous studies have reported that deregulation of the KEAP1-NRF2 signaling pathway promotes cellular proliferation and tumorigenesis in vivo by reprogramming glucose metabolism, with an altered metabolic profile characterized by increased glucose-derived carbon flux towards the PPP and tricarboxylic acid (TCA) cycle [45]. NRF2 can directly regulate the transcription of various enzymes involved in the PPP. Specifically, expression products of genes involved in the oxidative phase (glucose-6-phosphate dehydrogenase [G6PD], 6-phosphogluconate dehydrogenase [6PGD]) and non-oxidative phase (transketolase [TKT], and transaldolase [TALDO1]) and de novo nucleotide synthesis (phosphoribosyl pyrophosphate amidotransferase [PPAT] and methylenetetrahydrofolate dehydrogenase 2 [MTHFD2]) were decreased by the NRF2 knockdown [45]. In addition, sustained activation of NRF2 signaling in cancer cells was also found to indirectly enhance the PPP activation through the inhibition of microRNAs miR-1 and miR-206 expression. Specifically, sustained activation of NRF2 increased redox-sensitive histone deacetylase 4 (HDAC4) expression which attenuated miR-1 and miR-206 expression, leading to activation of PPP genes expression [46].
The regulation of PPP by NRF2 was also revealed by systematic investigations. Corroborating this, a metabolism-focused CRISPR screen demonstrated that G6PD was a top dependency in KEAP1-mutant lung tumors [47], which was supported by a more recent work by Jiang et al. [36]. However, whether this dependency is different between KEAP1-mutant and -wildtype tumors remains to be defined. In an independent CRISPR metabolic screen, Zhao et al. revealed that mutational status in the KEAP1-NRF2 pathway influenced the sensitivity of oxidative PPP [48]. Specifically, single knockout of oxidative PPP genes impacted the growth and survival of KEAP1-wildtype HeLa cells more dramatically than KEAP1-mutant lung A549 cells, which was presumably due to the fact that the increased glutathione (GSH) and higher expression of other NADPH-regenerating molecules contribute to the decreased dependence on oxidative PPP flux observed in cells expressing mutated KEAP1. The decreased importance of oxidative PPP for NADPH production and less essentiality of oxidative PPP genes in KEAP1-mutant cancer seem to be at odds with previous work highlighting a dependence on PPP by KEAP1-mutant tumors [36, 45, 47]. The observed differences might be due to transient metabolic reprogramming by CRISPR knockout.
In addition to producing NADPH through modulating the PPP pathway, NRF2 also promotes NADPH production by directly regulating several other metabolic pathways. Specifically, isocitrate dehydrogenase (IDH1 and IDH2) and malic enzyme (ME1, ME2, and ME3), two main enzymes that catalyze isocitrate decarboxylation and the oxidative decarboxylation of malate, respectively, are accompanied by NADPH production [49]. It has been demonstrated that cytosolic ME1 together with mitochondrial IDH2 supports tumor growth and metastasis [50]. Interestingly, NRF2 was found to increase NADPH production by regulating the transcription of ME1 and IDH1 [45].
Taken together, glucose metabolic reprogramming mediated by NRF2 plays a critical role in cancer cell survival and progression not only by providing materials and energy needed for rapid cell proliferation but also by producing NADPH for antioxidant synthesis to overcome oxidant stress.
Amino Acid
Altered amino acid metabolism is another metabolic alteration that occurs in cancer cells for sustaining their uncontrolled proliferation. In addition to directly acting as substrates for protein synthesis, they can also take part in the process of energy generation, nucleoside synthesis, and cellular redox homeostasis maintenance [51]. Recent studies also established a link between altered KEAP1-NRF2 signaling and amino acid metabolism (Figure 2).
GSH synthesis and utilization
GSH, one of the most powerful ROS scavengers, plays a critical role in tumor progression and metastasis and has been confirmed to be modulated by downstream genes of NRF2. The synthesis of GSH requires cysteine, which is converted from cystine uptaken via system Xc- [52]. xCT, a subunit protein of Xc- encoded by SLC7A11, is reported to be regulated by NRF2 [53]. Furthermore, NRF2 also participates in the reduction of cystine to cysteine through transcriptional regulation of thioredoxin (TXN) and thioredoxin reductase 1 (TXNRD1) [54, 55]. After providing sufficient intracellular cysteine, GSH is then synthesized by the consecutive reactions of the two major enzymes including glutamate-cysteine ligase (GCL, a heterodimeric enzyme comprised of GCL catalytic subunit (GCLC) and GCL modifier subunit (GCLM)) and GSH synthetase (GSS) [56]. In addition, NRF2 is also involved in GSH utilization by upregulating enzymes including glutathione reductase (GR), glutathione peroxidase, and glutathione S transferase.
In NSCLC, the upregulation of GSH resulting from constitutive activation of NRF2 contributes to stronger resistance to radiotherapy [32] and chemotherapy [34]. Thus, targeting NRF2/GSH activity may represent a promising strategy to avoid chemoradiotherapy resistance, which has been recently validated [57].
Taken together, the overproduction of GSH regulated by NRF2 inhibits cellular damage by counteracting ROS which maintains redox homeostasis and decreases oxidative damage, contributing to cancer cell survival.
Glutamate
Multi-omics analysis integrating whole-exome sequencing, transcriptomic and metabolic profiling robustly demonstrated that glutamate excretion, cystine uptake, and GSH synthesis represent reproducible features of NRF2-addicted lung cancer cells [58]. Glutamate not only participates in GSH synthesis catalyzed by GCLC and GCLM, but is also exported by xCT coupled with cystine uptake [59]. Mechanistically, NRF2 modulates glutamine metabolism by upregulating glutaminase, which catalyzes glutamine to glutamate [60]. Accordingly, NRF2 knockdown decreased the incorporation of glutamate into GSH, indicating that glutamate production mediated by NRF2 could support GSH synthesis [45]. A combination of CRISPR-Cas9-based genetic screening and metabolomic profiling by Romero et al. showed that KEAP1/NFE2L2-mutant lung cancer relies on increased glutaminolysis, and pharmacological inhibition of glutaminase exerts therapeutic efficacy [19]. LeBoeuf et al. demonstrated that NRF2-mediated secretion of glutamate via activation of xCT suppressed non-essential amino acids (NEAAs) synthesis, which increases the dependency of cancer cells on exogenous NEAAs [61]. Thus, restricting exogenous sources of NEAAs combined with glutaminase inhibition may represent a novel therapeutic strategy in lung tumors with alterations in the KEAP1-NRF2 pathway. Glutamine metabolism mediated by NRF2 activation also contributes to chemoresistance by restraining the assembly of stress granules, and glutaminase inhibitors can play a role in sensitizing cancer cells to chemotherapy [60]. Together, the heavy reliance of NRF2-addicted cancers on glutamate metabolism may present an opportunity for therapeutic targeting in lung cancer.
Tryptophan
In addition to changes in amino acids engaged in cellular redox homeostasis maintenance, NRF2 activation also alters other forms of amino acid metabolism in cancer cells. Tryptophan (Trp) metabolism, a central hub involved in the regulation of immunological and neural functional processes, has been confirmed to be associated with tumor malignancy and immune evasion [62]. Fahrmann et al. confirmed that aberrant NRF2 activation in LUAD alters the Trp metabolism through the kynurenine pathway, contributing to a tumor-promoting, immunosuppressed microenvironment [63]. Specifically, NRF2 upregulates tryptophan-kynurenine enzyme kynureninase (KYNU) and elicits an immunosuppressive microenvironment by efficient activation of T-regulatory cells and increased expression of programmed cell death protein-1 (PD-1) and programmed cell death ligand-1 (PD-L1), eventually leading to poorer survival. This study not only indicates a novel mechanism of NRF2 in Trp metabolism modulation but also provides evidence for the interaction between metabolic reprogramming and immunity remodelling in NRF2-addicted cancers, which will be discussed below in more detail.
Lipid metabolism, iron metabolism and ferroptosis
Emerging evidence highlights the importance of lipid metabolism, which provides energy, signaling molecules, and source material for cell membrane synthesis, as a key regulator in the development of NSCLC [64]. In the glucose-deficient tumor microenvironment (TME), lipid oxidation becomes the main route to generate NADH and FADH2 for ATP production [64, 65]. In the lung, NRF2 regulates several lipases that are involved in degrading triglycerides and phospholipids, providing an important source of lipids [66]. Additionally, previous studies confirmed that NRF2 plays a regulatory role in lipid synthesis by inhibiting two key enzymes, namely, stearoul CoA desaturate 1 (SCD1) and activate acetyl-CoA carboxylase 1 (ACC1). The downregulation of SCD1 and ACC1 results in higher fatty acid oxidation (FAO), which accelerates the oxidation of both long-chain and short-chain fatty acids within the mitochondria [67, 68]. Other genes that regulate FAO include nuclear receptor retinoid X receptor alpha (RXRa), peroxisome proliferator-activated receptor-gamma (PPARg), and peroxisome proliferator-activated receptor delta (PPARδ) were also confirmed to be targeted by NRF2 [69]. These lines of evidence suggest a close relationship between NRF2 and lipid oxidation.
Interestingly, SCD1-catalyzed monounsaturated fatty acids (MUFAs) can replace polyunsaturated fatty acids (PUFAs) in the lipid membrane and decrease the accumulation of lipid peroxides, thus protecting cells against a form of regulated cell death termed ferroptosis that is driven by iron-dependent lipid peroxidation [70, 71]. Disorder of iron metabolism induces an increased level of ferrous iron that interacts with hydrogen peroxide to form an excessive accumulation of reactive species via the Fenton reaction. The excess reactive species will then interact with PUFAs in the cytomembrane to generate excessive lipid peroxides. Previous studies have also demonstrated the critical role of NRF2 in modulating cellular iron metabolism by regulating genes involved in heme synthesis and hemoglobin catabolism [72, 73]. As such, it is not surprising that the KEAP1-NRF2 pathway is involved in affecting this novel cell death cascade.
NRF2 not only upregulates the iron storage protein ferritin (FTL1, FTH1) to decrease labile iron levels but also modulates ferroportin, a transmembrane protein that exports iron out of the cell, which in turn diminishes the accumulation of free iron inside of the cell [60, 61]. Sun et al. found that NRF2 nuclear accumulation activated the transcription of ferritin heavy chain-1 in hepatocellular carcinoma cells [74]. Moreover, the cystine-glutamate transporter system xCT and glutathione peroxidase 4 (GPX4), the two main regulators in the ferroptosis process [71], were also found to be the downstream targets of NRF2 [75]. In agreement, recent work found that cancer cells were able to evade ferroptosis caused by xCT inhibition or GPX4 inhibition via activation of the NRF2-ARE pathway, and the inhibition of NRF2 can reverse the resistance to ferroptotic cell death [13, 76], reinforcing the notion that NRF2 plays a critical role in mediating the resistance to GPX4-dependent ferroptosis.
Recent evidence demonstrates the presence of a GPX4-independent ferroptosis network in cancer, e.g., ferroptosis suppressor protein 1 (FSP1, also known as AIFM2) [77]. Intriguingly, FSP1 was identified as a novel transcriptional target of NRF2 [57]. The ubiquinone (CoQ)-FSP1 axis mediates ferroptosis- and radiation- resistance specifically in lung cancer cells harboring KEAP1 mutations. Pharmacological inhibition of the CoQ-FSP1 axis re-sensitizes KEAP1-deficient lung cancer tumors, which are inherently resistant to radiotherapy, in part, by inducing ferroptosis. This study not only establishes CoQ-FSP1 as a new downstream effector of the KEAP1-NRF2 pathway but also provides an additional therapeutic target for treating KEAP1-mutant lung cancer [57].
In summary, NRF2 is an important transcriptional regulator of anti-ferroptosis genes which prevent lipid peroxidation and the accumulation of free iron. Meanwhile, this regulation also creates new therapeutic vulnerabilities.
TIME
The interplay between tumor metabolism and TIME under NRF2 addiction
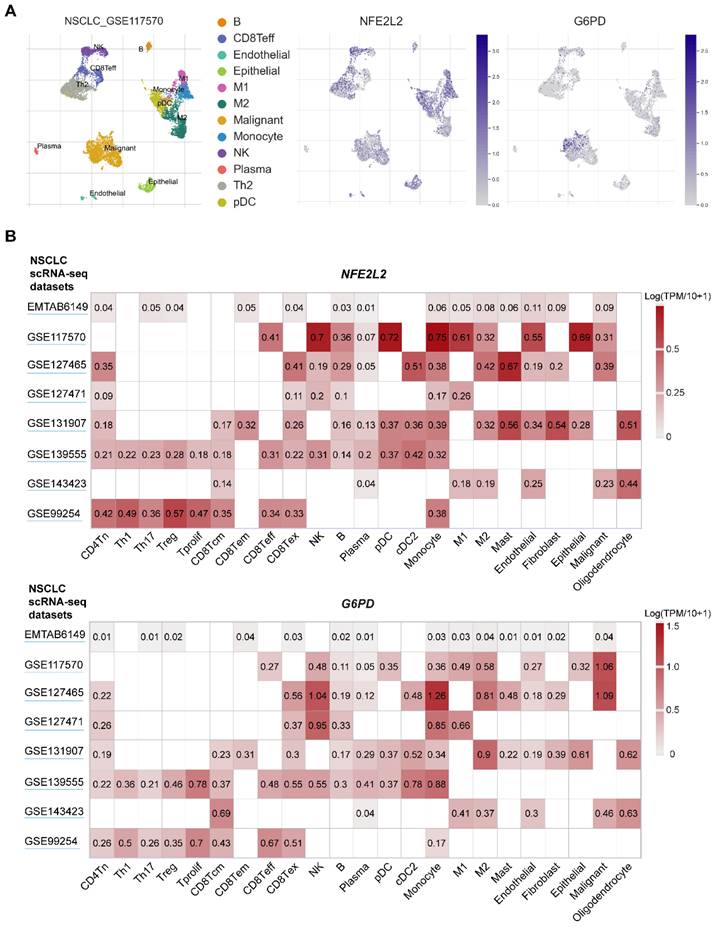
Since genetic mutations are the driving force behind tumor phenotype, e.g., metabolic reprogramming enabled by mutated KEAP1, to support rapid growth and proliferation, thus it is rational to assume that KEAP1-NRF2 mutations in the cancer cell-of-origin promote a metabolic profile that shapes the TIME. As a return, the TIME signals back to the cancer cell, generating a feedback loop such that tumors and TIME co-evolve during progression. Supporting this notion, recent clinical studies demonstrate that the presence of NRF2 activation or KEAP1 mutation in lung cancer is highly predictive of the unresponsiveness to immunotherapy [31, 78]. These observations suggest that lung tumors with dysregulation of NRF2 signaling may be associated with a disordered TIME and a heavily compromised anti-tumor immune response. Mining publicly available single-cell RNA-sequencing (scRNA-seq) data in NSCLC reveals a wide distribution of NFE2L2 and G6PD (a key downstream effector of NRF2) [45, 47] expression across cancer cells, as well as diverse immune cell types, particularly within the myeloid lineage (Figure 4).
scRNA-seq analysis of NRF2 expression across immune cell types within human NSCLC. A, UMAP plots showing the unsupervised clustering of tumor and immune cells from one non-small cell lung cancer (NSCLC) patient cohort. The middle and right panels showing the expression of NFE2L2 and G6PD (one key target gene of NFE2L2) across single cells. Single-cell RNA-seq (scRNA-seq) data were downloaded from the TISCH portal (Tumor Immune Single-cell Hub; http://tisch.comp-genomics.org/home/). B, Heatmap plots showing the distribution of NFE2L2 and G6PD expression across different cell types in NSCLC.
Indeed, emerging evidence highlights that tumor-derived metabolites can modulate the surrounding TIME, which is particularly for glutamine metabolism, a key metabolic molecule engaged in the hyperactive NRF2 signaling [14, 29, 79, 80]. In lung cancer, blocking the metabolism of glutamine to glutamate has pleiotropic effects on the immune system, such as reactivating CD8+T cells [29], enhancing PD-L1 expression [79], and decreasing myeloid-derived suppressor cells (MDSCs) [80]. Of particular interest, glutamine blockade was shown to generate divergent effects on tumors and anti-tumor immune cells [14]. Specifically, limiting glutamine uptake suppresses oxidative and glycolytic metabolism in cancer cells, whereas this markedly enhances oxidative metabolism and promotes a long-lived, highly activated phenotype in effector T cells [14]. The differential reliance on nutrients was further supported by a recent study demonstrating that cancer cells depend on glutamine, whereas immune cells preferentially uptake glucose [81]. As a result, blockage of glutamine utilization using glutamine transport inhibitors shows promise to overcome immunotherapy resistance [14, 29, 79, 80]. Besides, some other forms of metabolism, e.g., ATP/adenosine regulated by ectonucleotidase CD39/CD73 [82], fatty acid [83], and tryptophan [63] also play a critical role in modulating TIME. Together, metabolic reprogramming elicited by hyperactive NRF2 signaling can profoundly affect the surrounding TIME.
Tumor immunogenicity
TMB/MSI
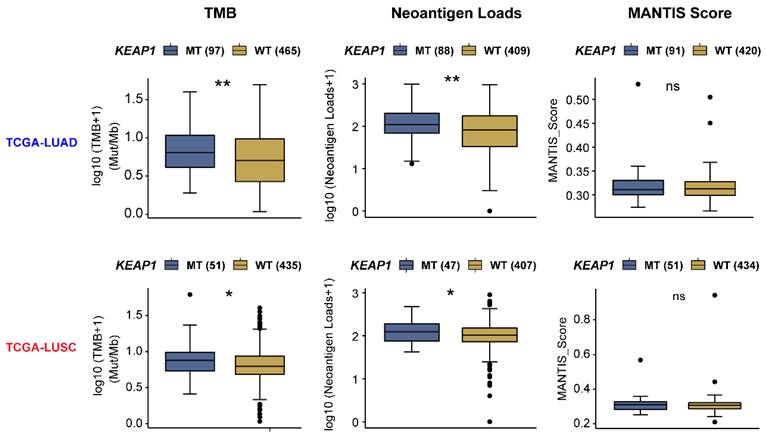
Certain cancer cell-autonomous features of lung tumors, such as TMB, MSI, or neoantigen load, have been well-established to reflect host immunogenicity, and predict a good response to immunotherapy in patients with high expression of these features [84]. Intriguingly, mining the TCGA NSCLC cohort, a significantly higher TMB and neoantigen load were observed in both KEAP1-mutant LUAD and LUSC (Figure 5); however, these features are not associated with a positive response to immunotherapy in this setting. In keeping with these data, clinical evidence shows thatKEAP1 is associated with poor response to immunotherapy despite the high TMB [31]. Thus, the immunogenicity of KEAP1-mutant lung cancer cells is not likely to be the key determinant of the poor immunotherapy response. Conversely, reprogrammed TIME caused by KEAP1 mutation may take the responsibility for this phenomenon.
The association of tumor immunogenicity with the mutant status of the KEAP1-NRF2 pathway in lung cancer. Barplots showing the difference in tumor immunogenicity between KEAP1-mutant (mut) and -wildtype (WT) lung squamous cell carcinoma (LUSC) and adenocarcinoma (LUAD) samples of the TCGA cohort. Data were downloaded from the CAMOIP (Comprehensive Analysis on Multi-Omics of Immunotherapy in Pan-cancer) portal (https://www.camoip.net/).
Mechanistic insights on the observations that KEAP1-mutant lung cancer, despite the presence of high TMB, is associated with immunotherapy resistance remain largely unknown [31]. Recently, Marzio et al. demonstrated that intact KEAP1 targets EMSY for ubiquitin-mediated degradation to regulate homologous recombination repair (HRR) and anti-tumor immunity [21]. In contrast, loss of KEAP1 induces genomic instability due to the defective HRR, resulting in a high TMB through stabilizing EMSY. Critically, accumulated EMSY suppresses the type I interferon response and impairs innate immune signaling, thus promoting cancer immune evasion. Accordingly, activation of the type I interferon response in the TME using a STING agonist results in the engagement of innate and adaptive immune signaling and retards the growth of KEAP1-mutant lung tumors in mouse models. This study not only provides valuable insight into the role of KEAP1 mutation in contributing to high TMB and immune escape but also important evidence for converting cold to hot tumors that are sensitive to immunotherapy. In agreement, Olagnier et al. declared that NRF2 activation represses STING expression by decreasing STING mRNA stability and impairs the responsiveness to STING agonists. Notably, the inhibitory effect of NRF2 on STING seems to be restricted in human cells rather than murine cells [85]. Whether STING agonist represents an effective strategy to treat NRF2-addicted cells in humans warrants further exploration.
PD-L1
Immune checkpoint molecules regulate the balance between activation and suppression during the immune response, and their aberrant expression results in human cancers escaping immune surveillance. PD-1 and its ligand PD-L1 have been identified as immune checkpoint molecules and their interaction results in the immune suppression of T cells and immune evasion of cancer cells [86]. Cancer cells of NSCLC show elevated levels of PD-L1 expression. Although the PD-1/PD-L1 treatment has significantly improved the survival of NSCLC patients, the overall response rate remains unsatisfactory [87].
Interestingly, a recent study identified a new mechanism for the KEAP1-NRF2 pathway in regulating immune checkpoint molecules such as PD-L1 [88]. The cullin (CUL) 3 is a component of Cullin-RING E3 ubiquitin ligase complex (CRLs) which are involved in protein ubiquitylation [89]. The CUL3-speckle-type POZ protein (SPOP) E3 ligase complex has been confirmed to directly ubiquitinate PD-L1, leading to its degradation [90]. Instead, the CUL3-KEAP1 complex was found to regulate the transcription of PD-L1 indirectly [88]. The CUL3-KEAP1 complex stabilizes NRF2 protein, which leads to the upregulation of NRF2-mediated PD-L1 transcription after IFN-γ stimulation. The mechanism mentioned above may provide certain explanations for the findings that KEAP1 mutation or NRF2 activation is associated with resistance to immune checkpoint blockade therapy in lung cancer [31, 78, 91].
T cells
T cells are at the core of adaptive immunity, and CD8+ T cell subsets form the backbone of effective immune checkpoint blockade (ICB) therapy. Ex vivo evidence based on the tumor-infiltrating T cells (TILs) sorted from tumor mass demonstrated a significant upregulation of NRF2 in TILs compared to matched uninvolved T cells. Interestingly, the authors observed that stimulating T cells by TCR activation significantly downregulated NRF2 expression and their target genes [92]. Concordantly, IFN-γ production in NRF2-deficient T cells was dramatically enhanced [92]. As mentioned above, KEAP1-NRF2 signaling plays a critical role in the anti-ferroptosis of cancer cells by regulating multiple target genes involved in cystine-GPX4-dependent and -independent pathways [57, 76]. Interestingly, recent evidence demonstrated that under ICB treatment, CD8+ T cells-derived IFN-γ downregulates the two key subunits (SLC7A11, SLC3A2) of the glutamate-cystine antiporter xCT, thereby promoting tumor ferroptosis [93]. This suggests that in the setting of NSCLC with aberrant KEAP1-NRF2 pathway, increased SLC7A11 expression might empower tumors to evade CD8-IFNγ-induced ferroptosis. In contrast to cancer cells, inhibiting ferroptosis in CD8+ T cells strongly restores their antitumor activity and enables greater antitumor efficacy in combination with ICBs [94].
Furthermore, KEAP1 mutations are associated with a specific T cell-inflamed gene-expression profile (GEP), an emerging biomarker predicting antitumor responses of immunotherapy targeting the PD-1 axis in various tumor types [95]. T cell-inflamed GEP includes 18 genes related to T cell-inflamed TIME [96]. A recent pan-cancer analysis has revealed that tumors with KEAP1 mutations have a poor response to ICB with a low level of T cell-inflamed GEP, and the immunomodulatory effects of KEAP1 mutations appear to be specific to cancer types, especially in LUAD and LUSC [97]. This may lead to tumor adaptation and subsequent immune suppression, allowing the survival and progression of lung cancer [97].
Despite this, there is a reason for caution. Staoh et al. found that NRF2 deficiency created an immunosuppressive microenvironment associated with a higher incidence of lung cancer metastasis following implantation of the mouse Lewis lung carcinoma cell line [98]. In this study, MDSCs, which are immunosuppressive cells, were found to increase in the context of NRF2 deletion. However, an additional study by the same group showed that the increased level of MDSCs and decreased population of the CD8+ T-cells were also observed in the NRF2 wild-type mice bearing tumors [99]. Another study also mentioned that the deletion of KEAP1 promoted tumor metastasis in patients with LUSC [32]. The possible reason for this contradictory result is that NRF2 deficiency increased susceptibility of tumor initiations, whereas NRF2 activation promoted malignant progression at the later stage of cancer [99]. These phenomena indicate a stage-related role of KEAP1-NRF2 signaling in immune modulation during tumor development, which suggests additional mechanisms requiring further investigation. Beyond that, co-mutations with KEAP1 may also play a role. In human LUAD, STK11 is one of the mutated genes that most co-occur with the KEAP1 mutation. Previous evidence also established STK11 as a regulator of ROS [27]. More recently, Sarah et al. investigated the metabolic and immune microenvironment of KRAS-mutant LUAD, based on Keap1 and STK11/Lkb1-mutant mouse models. Surprisingly, they found that increased glutamate abundance was observed in the TME of Lkb1-mutant rather than Keap1-mutant LUAD, which was associated with CD8 T cell activation in response to the immune checkpoint inhibitors targeting PD-1-PD-L1 axis [29]. Combination treatment with the glutaminase inhibitor CB-839 inhibited clonal expansion and activation of CD8 T cells [29], suggesting that glutaminase inhibition negatively impacts CD8 T cells activated by anti-PD-1 immunotherapy [100].
Apart from regulating conventional T cell proliferation and inflammation, the KEAP1-NRF2 pathway also participates in affecting the proliferation and function of innate T cells [101]. Invariant natural killer T (NKT) cell, as the innate lineage of T cell, also plays an important role in immune responses against cancer [102]. There are many subsets of NKT cells that play different immunomodulatory roles by secreting various cell-associated cytokines [103]. For example, TH1-like NKT cell subsets have the potential to activate both tumor-specific T cells and natural killer (NK) cells to eliminate cancer cells, whereas TH2-/T regulatory-like NKT cell subsets exert immunosuppressive effects that facilitate tumor progression and immune escape [102]. Along these lines, Pyaram et al. found an increased frequency of TH2-like NKT cells and a decreased frequency of TH1-like NKT cells in T cells with specific deletion of KEAP1 [101], which creates an immunosuppressive microenvironment for cancer cells in this genetic context.
Altogether, the lung tumors with dysregulated KEAP1-NRF2 pathway appear to have profound effects on the T cells within the TIME, and disrupting this signaling is expected to promote the efficacy of ICB therapy.
Macrophages
Among the immune cells within the TIME, lung macrophages represent the ones that are mostly exposed to a highly oxidative microenvironment, in that redox regulation is fundamental for phagocytic responses of macrophages [104]. The MST1/2-NRF2 axis represents an important protective mechanism for macrophages during an antimicrobial response [105], and a link between NRF2/ERK signaling and ferroptosis is revealed in macrophages [106]. Macrophages are comprised of two major subtypes: proinflammatory M1 macrophages phagocytose tumor cells, while anti-inflammatory M2 macrophages such as tumor‐associated macrophages (TAMs) stimulate tumor growth and invasion [107]. ROS plays a crucial role in promoting macrophage polarization and modulates macrophage immunosuppressive phenotype [108]. In the scRNA-seq dataset of NSCLC, NFE2L2 and its key target gene G6PD are highly expressed, albeit heterogeneously, in M1 and M2 macrophages (Figure 4).
Kobayashi et al. reported that NRF2 plays an anti-inflammatory role by suppressing macrophage inflammatory response by inhibiting pro-inflammatory cytokines expression including IL-6 and IL-1β [109]. Further study suggested that NRF2 contributes to the interaction between cancer cells and macrophages. Activation of NRF2 in cancer cells skews macrophage polarization towards an M2-like phenotype characterized by up-regulation of CD163 and Arg1, and down-regulation of IL-6 and IL-1β. The educated macrophages in turn activate NRF2 with VEGF expression which increases an epithelial-mesenchymal transition in cancer cells [110]. However, direct evidence in the setting of KEAP1-mutant lung cancer is lacking, which warrants further studies.
NK Cells
As mentioned above, previous studies have confirmed that KEAP1-NRF2 pathways influence T cells' proliferation and function. Since many signaling cascades that control NK cell effector function also participate in controlling T cell function [111], it can be reasonably hypothesized that the role of KEAP1-NRF2 pathways is also involved in NK cell regulation. NK cells carry out critical functions in innate immune surveillance in cancer [112]. Deletion of both KEAP1 and PTEN was shown to result in decreasing number of NK cells in mice with lung cancer [16]. Further, activation of NRF2 by the synthetic aromatic organic compound tert-butylhydroquinone (tBHQ) has been found to negatively affect the development and effector functions of NK cells [113].
Paradoxically, a recent study reports that the dysfunction of human NK cells in the TIME is due to suppressed glucose metabolism via lipid peroxidation-associated oxidative stress [114]. Accordingly, expansion of NK cells with IL-21 treatment or an activator of NRF2 antioxidant pathway promotes Warburg-like metabolism with increased glycolysis and reduced oxidative phosphorylation, thereby leading to a markedly improved resistance to the oxidative stressed and nutrient-deprived TME, as well as enhanced in vivo antitumor responses. These findings suggest that activation of the NRF2 antioxidant pathway can restore the metabolic fitness and proper function of NK cells. Whether these observations exist in situ in the context of KEAP1-mutant NSCLC requires further investigations.
Dendritic cells
Dendritic cells (DCs) represent a critical subset of innate immune cells that process and present tumor-derived antigens to prime T-cell immunity. tBHQ mentioned above was also confirmed to affect DCs through NRF2 activation [115]. Specifically, tBHQ significantly attenuates IL-12 expression secreted by DCs, whereas the suppression can be prevented by genetic inhibition of NRF2 expression [115]. In addition, the miR-200 family of microRNAs was also found to participate in the regulation of innate immune response related to DCs [116]. MiR-200a can down-regulate DC maturation and prevents NRF2 ubiquitination caused by KEAP1 overexpression, which eventually reverses the inhibitory effect of DCs on tumors [117]. Taken together, the overactivation of NRF2 contributes to cancer development partly through suppressing DCs function, although detailed mechanistic insights remain to be explored.
Therapeutic target
Given the critical role of the KEAP1-NRF2 pathway in cancer, identifying novel compounds or repurposed drugs that can be used to target this pathway directly or indirectly is of great clinical interest for cancer treatment.
Direct
Small molecule compounds
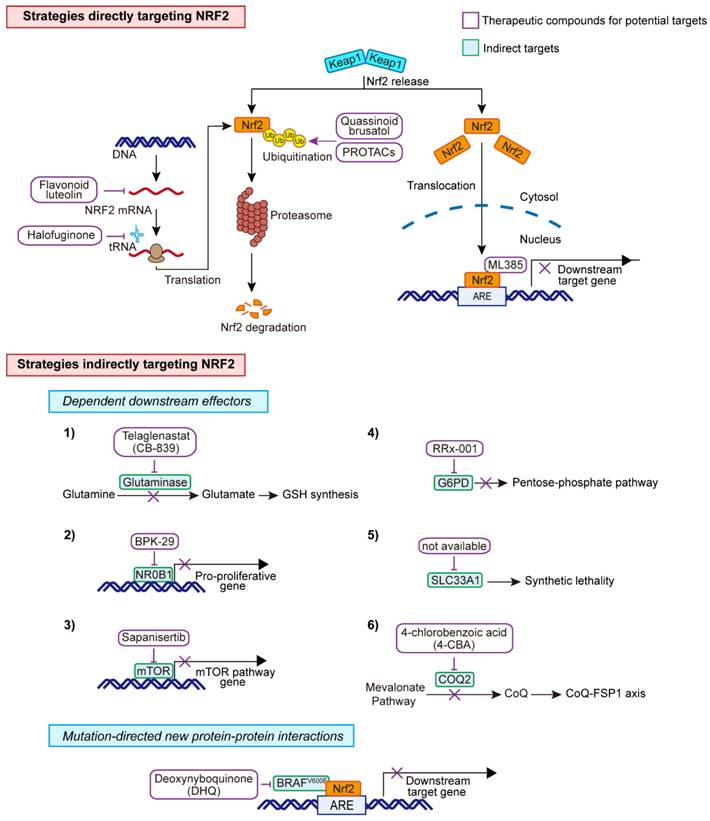
A natural compound, quassinoid brusatol that is extracted from Brucea javanica, was found to stimulate poly-ubiquitination of NRF2, leading to protein degradation [118]. Of note, the inhibitory effect of brusatol on NRF2 is demonstrated to be irrespective of its classical repressor KEAP1. Another NRF2 inhibitor, halofuginone was shown to decrease Nrf2 at the protein level, through suppression prolyl-tRNA synthetase activity, and sensitize the chemotherapy on NRF2-addicted cancer cells [119]. Akin to halofuginone, flavonoid luteolin was reported to accelerate the turnover of NRF2 mRNA, leading to a marked reduction of its mRNA and protein levels and enhanced sensitivity of lung cancer cells to chemotherapeutic agents [120]. Furthermore, in a study screening ∼400,000 small molecules, ML385 was identified as a probe molecule that specifically binds to NRF2 and inhibits the expression of its downstream target gene [121]. Specifically, ML385 binds to Neh1, the Cap 'N' Collar Basic Leucine Zipper (CNC-bZIP) domain of NRF2, and interferes with the binding of the V-Maf Avian Musculoaponeurotic Fibrosarcoma Oncogene Homologue G (MAFG)-NRF2 protein complex to regulatory DNA binding sequences [121].
Targeted protein degradation-PROTACs
NRF2 is typically degraded via the ubiquitination system. Targeted protein degradation is an emerging therapeutic approach to inhibit disease-causing proteins, such as transcription factors, that are historically challenging to target with conventional small molecules. Proteolysis targeting chimeras (PROTACs), which hijack E3 ligases and the ubiquitin-proteasome system (UPS) to selectively degrade the target proteins, represent a new class of promising therapeutic modalities [122] and has shown great promises to target previously undruggable targets. Beyond the KEAP1-Cullins3-RING-box protein 1 (KEAP1-Cul3-RBX1) axis, β-TRCP-S-phase kinase-associated protein 1-Cul1-RBX1 (SKP1-Cul1-RBX1) [123], and HMG-COA reductase degradation 1 homolog (HRD1) [124] have also been uncovered to control the ubiquitination of NRF2. Currently, multiple PROTACs-based or -alternative degrader approaches have been exploited in preclinical and clinical testing [122, 125], which may offer treatment potential in NRF2-addicted.
Indirect
Directly targeting transcription factor NRF2 is still challenging, and efforts are mostly made on the collateral metabolic vulnerabilities rendered by NRF2, as depicted above that KEAP1-NRF2 signaling drives multiple metabolic dependences.
Dependent downstream effectors
Among the candidate targets, inhibition of glutaminase, the enzyme that catalyzes the conversion of glutamine to glutamate, represents the most promising one to treat NRF2-addicted NSCLC [18, 19, 59]. Accordingly, the glutaminase inhibitor telaglenastat (CB-839) is being evaluated in phase II clinical trials, either in combination with chemo-immunotherapy or alone, in NSCLC patients whose tumors harbor KEAP1 or NFE2L2 mutations (NCT04265534 and NCT03872427). Liron et al. did chemical proteomics to identify druggable proteins that are selectively expressed in KEAP1-mutant NSCLC cells. Using this approach, the authors identified NR0B1, a transcriptional regulator that supports KEAP1-mutant NSCLC cells [8]. Importantly, they identified the small compound BPK-29 that disrupts NR0B1 complexes and impairs the anchorage-independent growth of KEAP1-mutant cancer cells.
The dual mTORC1/2 inhibitor sapanisertib is being examined in the advanced setting in NSCLC (NCT02417701 and NCT04250545) based on preclinical evidence showing that NFE2L2 mutations induce mTOR pathway dependency. Other targets including G6PD and endoplasmic reticulum-associated protein SLC33A1 [20] have recently been validated as a new dependency specific in KEAP1-mutant NSCLC. More recently, CoQ-FSP1 axis was also shown to be specific to KEAP1-mutant NSCLC. Clinical evidence has shown that KEAP1-mutant lung tumors are highly refractory to radiotherapy [32]. Inhibition of the CoQ biosynthesis enzyme COQ2 with 4-chlorobenzoic acid (4-CBA) strongly sensitized lung cancer cells to radiotherapy by inducing ferroptosis [57].
High-throughput screens
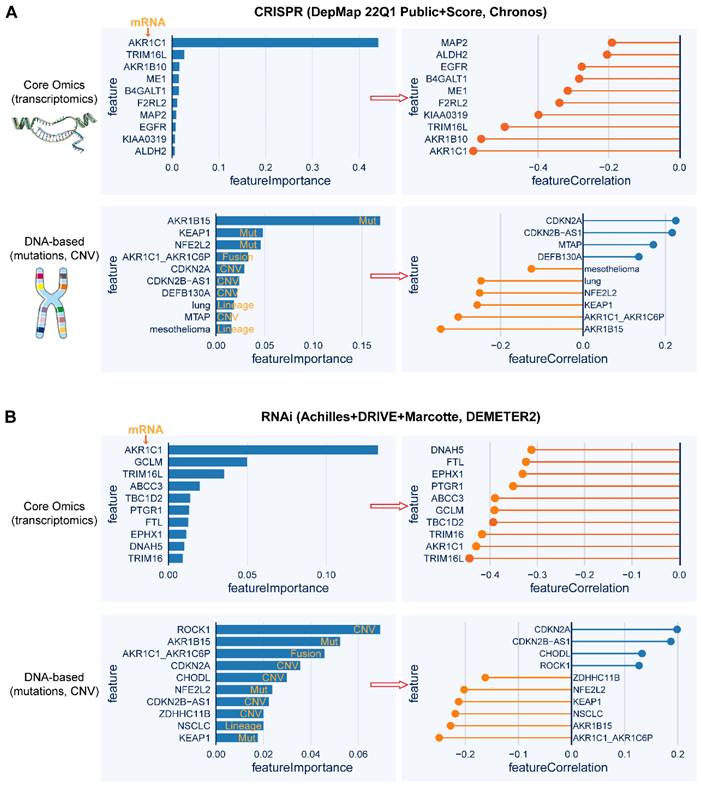
Quantitative high-throughput screens can robustly facilitate the identification of genes whose expression or mutation is correlated with the degree of addiction to NRF2 function. Based on the publicly available datasets of CRISPR-Cas9 and RNA interference (RNAi) screening data, the top 10 genes whose mRNA level or genetic mutational status are tightly correlated (positively or negatively) with NFE2L2 knockout or knockdown, respectively (Figure 6A-B). Specifically, in the CRISPR-Cas9 screen, AKR1C1 expression represents the strongest feature that predicts the sensitivity to NFE2L2 knockout, and strikingly, a genetic mutation in the AKR1B15 exhibits higher predictive power than KEAP1 and NFE2L2 mutations. The predictive ability of AKR1C1 expression and mutational status of KEAP1 and NFE2L2 is also validated in the RNAi screen data. Interestingly, CDKN2A amplification is predicted to resist NFE2L2 knockout or knockdown. These high-throughput analyses might provide a further understanding of the pathogenesis of NRF2-addictive cancer while also uncovering new therapeutic targets.
Whole-genome screens reveal potential targeted dependency in KEAP1-mutant cancer cells. A, B, Top 10 essential genes whose mRNA level (upper panel) or genetic mutation (lower panel) is correlated with the dependence score of NFE2L2, based on CRISPR (A)- and RNAi (B)-based whole-genome screen (https://depmap.org/portal/). In the left panels of A and B, the random forest machine learning strategy was used to identify the most important molecular features related to NFE2L2 dependence. Right panels showing the Pearson correlation of the indicated genes with NFE2L2 dependence score. The negative correlation indicates that genes whose higher expression or mutation signifies higher sensitivity to knockout or knockdown of NFE2L2.
Mutation-directed new protein-protein interactions (PPI)
Mutations-driven loss or gain of PPIs illustrates another dimension of molecular underpinnings that enable tumorigenesis and cancer progression. In a recent study that aims to identify mutations-driven loss or gain of PPIs, the authors systematically performed the comparative analysis of wildtype and the paired mutant PPI profiles, uncovering an unappreciated function of mutated BRAF as a direct regulator of the KEAP1-mediated redox pathway through KEAP1-NRF2-ARE signaling, as overexpression of BRAFV600E, but not its wildtype counterpart, significantly stabilizes NRF2 protein and increases the transcriptional activity of ARE reporter [126]. As a result, BRAFV600E-mutant cells are specifically vulnerable to quinone compounds such as deoxynyboquinone (DNQ). This example illustrates the potential of mutation-guided PPI to uncover oncogenic pathways as a promising strategy for pathobiological and therapeutic exploration, which is highly relevant for KEAP1-mutant lung tumors. The therapeutic strategies are summarized in Figure 7.
Therapeutic strategies for KEAP1-mutant cancer cells. Direct and indirect targets for the aberrant KEAP1-NRF2 pathway.
Concluding remarks
Aberrant KEAP1-NRF2 signaling in NSCLC has both cancer cell-autonomous and non-autonomous effects, driving unique metabolic and immunological states, which suggests that combined targeting the metabolic vulnerabilities of cancer cells and disrupting their close interaction with the TIME would provide a more effective approach for this disease. Given the critical role of TIME in empowering NRF2-addicted NSCLC, new compounds or repurposed drugs that treat lung tumors with aberrant KEAP1-NRF2 signaling pathway should be tested in an intact immune system. Additionally, it is noteworthy that in preclinical mouse models, more attention should be paid to KEAP1-mutant LUSC, given that a higher incidence of mutations in KEAP1-NRF2 pathway occurs in LUSC, and more importantly, cell-of-origin in the lung might also matter in this genetic background. PROTACs and other alternative approaches show great promise in directly targeting the “undruggable” transcription factors, although their applications have not been achieved in the KEAP1-mutant setting. Other evolving strategies, e.g., mutations-driven loss or gain of PPIs, are also powerful to increase understanding of the KEAP1-NRF2 pathobiology, which is critical toward uncovering new therapeutic vulnerabilities of this daunting disease.
Abbreviations
4-CBA: 4-chlorobenzoic acid; 6PGD: 6-phosphogluconate dehydrogenase; ACC1: acetyl-CoA carboxylase 1; ARE: antioxidant response elements; CNC-bZIP: Cap 'N' Collar Basic Leucine Zipper; CoQ: ubiquinone; CRLs: cullin-RING E3 ubiquitin ligase complex; CUL: cullin; DCs: dendritic cells; DNQ: deoxynyboquinone; EpRE: electrophile-responsive element; FAO: fatty acid oxidation; FSP1: ferroptosis suppressor protein 1; G6PD: glucose-6-phosphate dehydrogenase; GCL: glutamate-cysteine ligase; GCL: glutamate-cysteine ligase; GCLC: GCL catalytic subunit; GCLM: GCL modifier subunit; GEEM: genetically engineered mouse models; GEP: gene-expression profile; GPX4: glutathione peroxidase 4; GR: glutathione reductase; GSH: glutathione; GSS: GSH synthetase; GST: glutathione S-transferase; HIF1α: hypoxia-inducible factor 1 α; HK: hexokinase; HO-1: heme oxygenase 1; HRD1: HMG-COA reductase degradation 1 homolog; HRR: homologous recombination repair; ICB: immune checkpoint blockade; IDH: isocitrate dehydrogenase; KEAP1: kelch-like ECH-associated protein 1; KEAP1-Cul3-RBX1: KEAP1-Cullins3-RING-box protein 1; LDHA: lactate dehydrogenase A; LUAD: lung adenocarcinoma; LUSC: lung squamous cell carcinoma; MAFG: V-Maf Avian Musculoaponeurotic Fibrosarcoma Oncogene Homologue G; MDSCs: myeloid-derived suppressor cells; ME: malic enzyme; MSI: microsatellite instability; MTHFD2: methylenetetrahydrofolate dehydrogenase 2; MUFAs: monounsaturated fatty acids; NEAAs: non-essential amino acids; NFE2L2: nuclear factor erythroid-derived 2-like 2; NK: natural killer; NKT: natural killer T; NQO1: NAD(P)H dehydrogenase; NSCLC: non-small cell lung cancer; PD-1: programmed cell death protein-1; PDK1: pyruvate dehydrogenase kinase-1; PD-L1: programmed cell death ligand-1; PFKFB3: phosphofructokinase-2/fructose-2,6-bisphosphatase 3; PK: pyruvate kinase; PKM2: pyruvate kinase isozymes M2; PPARg: peroxisome proliferator-activated receptor-gamma; PPARδ: peroxisome proliferator-activated receptor delta; PPAT: phosphoribosyl pyrophosphate amidotransferase; PPI: mutation-directed new protein-protein interactions; PPP: pentose phosphate pathway; PROTACs: proteolysis targeting chimeras; PUFAs: polyunsaturated fatty acids; RNAi: RNA interference; ROS: reactive oxygen species; RXRa: receptor retinoid X receptor alpha; SCD1: stearoul CoA desaturate 1; scRNA-seq: single-cell RNA-sequencing; SKP1-Cul1-RBX1: β-TRCP-S-phase kinase-associated protein 1-Cul1-RBX1; SOD2: mitochondrial superoxide dismutase 2; SPOP: speckle-type POZ protein; TALDO1: transaldolase; TAMs: tumor‐associated macrophages; tBHQ: tert-butylhydroquinone; TCA: tricarboxylic acid; TCGA: The Cancer Genome Atlas; TILs: infiltrating T cells; TIME: tumor immune microenvironment; TKT: transketolase; TME: tumor microenvironment; TMB: tumor mutational burden; TrxR1: thioredoxin reductase1; TXN: thioredoxin; UPS: ubiquitin-proteasome system.
Acknowledgements
This study used the databases from TCGA Program, and the authors acknowledge the efforts of the corresponding institutes. The interpretation and reporting of these data are the sole responsibility of the authors. Graphical abstract in this article was created with BioRender.com.
Author Contributions
Review concept and design: Ke Xu and Haitang Yang
Bioinformatics mining and analysis: Ke Xu, Jie Ma and Haitang Yang
Drafting of the manuscript and figures: Ke Xu and Haitang Yang
Critical revision: Jie Ma, Sean R. R. Hall and Ren-Wang Peng
Study supervision: Feng Yao, and Haitang Yang
All authors contributed to the manuscript for important intellectual content and approved the submission.
Funding
This research was supported by the National Natural Science Foundation of China (82072570 to F.Y; 82202925 to H.Y); Excellent talent program of Shanghai Chest Hospital (2021YF1028 to F.Y); Basic Foundation Program for Youth of Shanghai Chest Hospital (2021YNJCQ2 to H.Y), and sponsored by Shanghai Pujiang Program (22PJD068 to H.Y) and Shanghai Medicine and Health Development Foundation (to H.Y).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC. et al. Oncogenic signaling pathways in the cancer genome atlas. Cell. 2018;173:321-37 e10
2. Saleh MM, Scheffler M, Merkelbach-Bruse S, Scheel AH, Ulmer B, Wolf J. et al. Comprehensive analysis of TP53 and KEAP1 mutations and their impact on survival in localized- and advanced-stage NSCLC. J Thorac Oncol. 2022;17:76-88
3. Kansanen E, Kuosmanen SM, Leinonen H, Levonen AL. The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol. 2013;1:45-9
4. Wu WL, Papagiannakopoulos T. The pleiotropic role of the KEAP1/NRF2 pathway in cancer. Annu Rev Cancer Biol. 2020;4:413-35
5. Liu S, Pi J, Zhang Q. Signal amplification in the KEAP1-NRF2-ARE antioxidant response pathway. Redox Biol. 2022;54:102389
6. Ooi A, Dykema K, Ansari A, Petillo D, Snider J, Kahnoski R. et al. CUL3 and NRF2 mutations confer an NRF2 activation phenotype in a sporadic form of papillary renal cell carcinoma. Cancer Res. 2013;73:2044-51
7. Goldstein LD, Lee J, Gnad F, Klijn C, Schaub A, Reeder J. et al. Recurrent loss of NFE2L2 exon 2 is a mechanism for Nrf2 pathway activation in human cancers. Cell Rep. 2016;16:2605-17
8. Bar-Peled L, Kemper EK, Suciu RM, Vinogradova EV, Backus KM, Horning BD. et al. Chemical proteomics identifies druggable vulnerabilities in a genetically defined cancer. Cell. 2017;171:696-709.e23
9. Cheng KC, Lin RJ, Cheng JY, Wang SH, Yu JC, Wu JC. et al. FAM129B, an antioxidative protein, reduces chemosensitivity by competing with Nrf2 for Keap1 binding. EBioMedicine. 2019;45:25-38
10. Feng X, Jiang T, Yang C, Pang S, Ding Z, Hu H. et al. RPRD1A stabilizes NRF2 and aggravates HCC progression through competing with p62 for TRIM21 binding. Cell Death Dis. 2021;13:6
11. Pan JA, Sun Y, Jiang YP, Bott AJ, Jaber N, Dou Z. et al. TRIM21 ubiquitylates SQSTM1/p62 and suppresses protein sequestration to regulate redox homeostasis. Mol Cell. 2016;61:720-33
12. Okazaki K, Anzawa H, Liu Z, Ota N, Kitamura H, Onodera Y. et al. Enhancer remodeling promotes tumor-initiating activity in NRF2-activated non-small cell lung cancers. Nat Commun. 2020;11:5911
13. Takahashi N, Cho P, Selfors LM, Kuiken HJ, Kaul R, Fujiwara T. et al. 3D culture models with CRISPR screens reveal hyperactive NRF2 as a prerequisite for spheroid formation via regulation of proliferation and ferroptosis. Mol Cell. 2020;80:828-44 e6
14. Leone RD, Zhao L, Englert JM, Sun IM, Oh MH, Sun IH. et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science. 2019;366:1013-21
15. DeBerardinis RJ. Tumor microenvironment, metabolism, and immunotherapy. N Engl J Med. 2020;382:869-71
16. Best SA, De Souza DP, Kersbergen A, Policheni AN, Dayalan S, Tull D. et al. Synergy between the KEAP1/NRF2 and PI3K pathways drives non-small-cell lung cancer with an altered immune microenvironment. Cell Metab. 2018;27:935-43.e4
17. Best SA, Ding S, Kersbergen A, Dong X, Song JY, Xie Y. et al. Distinct initiating events underpin the immune and metabolic heterogeneity of KRAS-mutant lung adenocarcinoma. Nat Commun. 2019;10:4190
18. Koppula P, Olszewski K, Zhang Y, Kondiparthi L, Liu X, Lei G. et al. KEAP1 deficiency drives glucose dependency and sensitizes lung cancer cells and tumors to GLUT inhibition. iScience. 2021;24:102649
19. Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE. et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med. 2017;23:1362-8
20. Romero R, Sanchez-Rivera FJ, Westcott PMK, Mercer KL, Bhutkar A, Muir A. et al. Keap1 mutation renders lung adenocarcinomas dependent on Slc33a1. Nat Cancer. 2020;1:589-602
21. Marzio A, Kurz E, Sahni JM, Di Feo G, Puccini J, Jiang S. et al. EMSY inhibits homologous recombination repair and the interferon response, promoting lung cancer immune evasion. Cell. 2022;185:169-83.e19
22. Ferone G, Lee MC, Sage J, Berns A. Cells of origin of lung cancers: lessons from mouse studies. Genes Dev. 2020;34:1017-32
23. Sutherland KD, Song JY, Kwon MC, Proost N, Zevenhoven J, Berns A. Multiple cells-of-origin of mutant K-Ras-induced mouse lung adenocarcinoma. Proc Natl Acad Sci U S A. 2014;111:4952-7
24. Schneider G, Schmidt-Supprian M, Rad R, Saur D. Tissue-specific tumorigenesis: context matters. Nat Rev Cancer. 2017;17:239-53
25. Gillette MA, Satpathy S, Cao S, Dhanasekaran SM, Vasaikar SV, Krug K. et al. Proteogenomic characterization reveals therapeutic vulnerabilities in lung adenocarcinoma. Cell. 2020;182:200-25 e35
26. Satpathy S, Krug K, Jean Beltran PM, Savage SR, Petralia F, Kumar-Sinha C. et al. A proteogenomic portrait of lung squamous cell carcinoma. Cell. 2021;184:4348-71 e40
27. Li F, Han X, Li F, Wang R, Wang H, Gao Y. et al. LKB1 inactivation elicits a redox imbalance to modulate non-small cell lung cancer plasticity and therapeutic response. Cancer Cell. 2015;27:698-711
28. Scalera S, Mazzotta M, Cortile C, Krasniqi E, De Maria R, Cappuzzo F. et al. KEAP1-mutant NSCLC: the catastrophic failure of a cell-protecting hub. J Thorac Oncol. 2022;17:751-7
29. Best SA, Gubser PM, Sethumadhavan S, Kersbergen A, Negrón Abril YL, Goldford J. et al. Glutaminase inhibition impairs CD8 T cell activation in STK11-/Lkb1-deficient lung cancer. Cell Metab. 2022;34:874-87.e6
30. Wohlhieter CA, Richards AL, Uddin F, Hulton CH, Quintanal-Villalonga À, Martin A. et al. Concurrent mutations in STK11 and KEAP1 promote ferroptosis protection and SCD1 dependence in lung cancer. Cell Rep. 2020;33:108444
31. Marinelli D, Mazzotta M, Scalera S, Terrenato I, Sperati F, D'Ambrosio L. et al. KEAP1-driven co-mutations in lung adenocarcinoma unresponsive to immunotherapy despite high tumor mutational burden. Ann Oncol. 2020;31:1746-54
32. Jeong Y, Hoang NT, Lovejoy A, Stehr H, Newman AM, Gentles AJ. et al. Role of KEAP1/NRF2 and TP53 mutations in lung squamous cell carcinoma development and radiation resistance. Cancer Discov. 2017;7:86-101
33. Galan-Cobo A, Sitthideatphaiboon P, Qu X, Poteete A, Pisegna MA, Tong P. et al. LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence in KRAS-mutant lung adenocarcinoma. Cancer Res. 2019;79:3251-67
34. Goeman F, De Nicola F, Scalera S, Sperati F, Gallo E, Ciuffreda L. et al. Mutations in the KEAP1-NFE2L2 pathway define a molecular subset of rapidly progressing lung adenocarcinoma. J Thorac Oncol. 2019;14:1924-34
35. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. 2020 368
36. Jiang C, Ward NP, Prieto-Farigua N, Kang YP, Thalakola A, Teng M. et al. A CRISPR screen identifies redox vulnerabilities for KEAP1/NRF2 mutant non-small cell lung cancer. Redox Biol. 2022;54:102358
37. Bose S, Zhang C, Le A. Glucose metabolism in cancer: the Warburg effect and beyond. Adv Exp Med Biol. 2021;1311:3-15
38. Lu H, Forbes RA, Verma A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J Biol Chem. 2002;277:23111-5
39. Fu J, Xiong Z, Huang C, Li J, Yang W, Han Y. et al. Hyperactivity of the transcription factor Nrf2 causes metabolic reprogramming in mouse esophagus. J Biol Chem. 2019;294:327-40
40. Zhang HS, Du GY, Zhang ZG, Zhou Z, Sun HL, Yu XY. et al. NRF2 facilitates breast cancer cell growth via HIF1ɑ-mediated metabolic reprogramming. Int J Biochem Cell Biol. 2018;95:85-92
41. Lee S, Hallis SP, Jung KA, Ryu D, Kwak MK. Impairment of HIF-1α-mediated metabolic adaption by NRF2-silencing in breast cancer cells. Redox Biol. 2019;24:101210
42. Hamada S, Matsumoto R, Masamune A. HIF-1 and NRF2; key molecules for malignant phenotypes of pancreatic cancer. Cancers (Basel). 2022 14
43. Potteti HR, Noone PM, Tamatam CR, Ankireddy A, Noel S, Rabb H. et al. Nrf2 mediates hypoxia-inducible HIF1α activation in kidney tubular epithelial cells. Am J Physiol Renal Physiol. 2021;320:F464-f74
44. Ju HQ, Lin JF, Tian T, Xie D, Xu RH. NADPH homeostasis in cancer: functions, mechanisms and therapeutic implications. Signal Transduct Target Ther. 2020;5:231
45. Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H. et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012;22:66-79
46. Singh A, Happel C, Manna SK, Acquaah-Mensah G, Carrerero J, Kumar S. et al. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J Clin Invest. 2013;123:2921-34
47. Ding H, Chen Z, Wu K, Huang SM, Wu WL, LeBoeuf SE. et al. Activation of the NRF2 antioxidant program sensitizes tumors to G6PD inhibition. Sci Adv. 2021;7:eabk1023
48. Zhao D, Badur MG, Luebeck J, Magaña JH, Birmingham A, Sasik R. et al. Combinatorial CRISPR-Cas9 metabolic screens reveal critical redox control points dependent on the KEAP1-NRF2 regulatory axis. Mol Cell. 2018;69:699-708.e7
49. Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 2014;510:298-302
50. Shao C, Lu W, Du Y, Yan W, Bao Q, Tian Y. et al. Cytosolic ME1 integrated with mitochondrial IDH2 supports tumor growth and metastasis. Redox Biol. 2020;36:101685
51. Vettore L, Westbrook RL, Tennant DA. New aspects of amino acid metabolism in cancer. Br J Cancer. 2020;122:150-6
52. Lo M, Wang YZ, Gout PW. The x(c)- cystine/glutamate antiporter: a potential target for therapy of cancer and other diseases. J Cell Physiol. 2008;215:593-602
53. Hu K, Li K, Lv J, Feng J, Chen J, Wu H. et al. Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. J Clin Invest. 2020;130:1752-66
54. Hawkes HJ, Karlenius TC, Tonissen KF. Regulation of the human thioredoxin gene promoter and its key substrates: a study of functional and putative regulatory elements. Biochim Biophys Acta. 2014;1840:303-14
55. Malhotra D, Portales-Casamar E, Singh A, Srivastava S, Arenillas D, Happel C. et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010;38:5718-34
56. Tonelli C, Chio IIC, Tuveson DA. Transcriptional regulation by Nrf2. Antioxid Redox Signal. 2018;29:1727-45
57. Koppula P, Lei G, Zhang Y, Yan Y, Mao C, Kondiparthi L. et al. A targetable CoQ-FSP1 axis drives ferroptosis- and radiation-resistance in KEAP1 inactive lung cancers. Nat Commun. 2022;13:2206
58. Saigusa D, Motoike IN, Saito S, Zorzi M, Aoki Y, Kitamura H. et al. Impacts of NRF2 activation in non-small-cell lung cancer cell lines on extracellular metabolites. Cancer Sci. 2020;111:667-78
59. Sayin VI, LeBoeuf SE, Singh SX, Davidson SM, Biancur D, Guzelhan BS. et al. Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. Elife. 2017 6
60. Mukhopadhyay S, Goswami D, Adiseshaiah PP, Burgan W, Yi M, Guerin TM. et al. Undermining glutaminolysis bolsters chemotherapy while NRF2 promotes chemoresistance in KRAS-driven pancreatic cancers. Cancer Res. 2020;80:1630-43
61. LeBoeuf SE, Wu WL, Karakousi TR, Karadal B, Jackson SR, Davidson SM. et al. Activation of oxidative stress response in cancer generates a druggable dependency on exogenous non-essential amino acids. Cell Metab. 2020;31:339-50.e4
62. Platten M, Nollen EAA, Röhrig UF, Fallarino F, Opitz CA. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat Rev Drug Discov. 2019;18:379-401
63. Fahrmann JF, Tanaka I, Irajizad E, Mao X, Dennison JB, Murage E. et al. Mutational activation of the NRF2 pathway upregulates kynureninase resulting in tumor immunosuppression and poor outcome in lung adenocarcinoma. Cancers (Basel). 2022 14
64. Koundouros N, Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J Cancer. 2020;122:4-22
65. Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer. 2013;13:227-32
66. Paek J, Lo JY, Narasimhan SD, Nguyen TN, Glover-Cutter K, Robida-Stubbs S. et al. Mitochondrial SKN-1/Nrf mediates a conserved starvation response. Cell Metab. 2012;16:526-37
67. Dobrzyn P, Dobrzyn A, Miyazaki M, Cohen P, Asilmaz E, Hardie DG. et al. Stearoyl-CoA desaturase 1 deficiency increases fatty acid oxidation by activating AMP-activated protein kinase in liver. Proc Natl Acad Sci U S A. 2004;101:6409-14
68. Kim C-W, Addy C, Kusunoki J, Anderson NN, Deja S, Fu X. et al. Acetyl CoA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: a bedside to bench investigation. Cell Metab. 2017;26:394-406 e6
69. Bowman BM, Montgomery SA, Schrank TP, Simon JM, Ptacek TS, Tamir TY. et al. A conditional mouse expressing an activating mutation in NRF2 displays hyperplasia of the upper gastrointestinal tract and decreased white adipose tissue. J Pathol. 2020;252:125-37
70. Das UN. Saturated fatty acids, MUFAs and PUFAs regulate ferroptosis. Cell Chem Biol. 2019;26:309-11
71. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060-72
72. Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, Cook JL. Nrf2, a Cap'n'Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem. 1999;274:26071-8
73. Campbell MR, Karaca M, Adamski KN, Chorley BN, Wang X, Bell DA. Novel hematopoietic target genes in the NRF2-mediated transcriptional pathway. Oxid Med Cell Longev. 2013;2013:120305
74. Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R. et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63:173-84
75. Habib E, Linher-Melville K, Lin HX, Singh G. Expression of xCT and activity of system xc(-) are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biol. 2015;5:33-42
76. Shin D, Kim EH, Lee J, Roh JL. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic Biol Med. 2018;129:454-62
77. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH. et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688-92
78. Scalera S, Mazzotta M, Corleone G, Sperati F, Terrenato I, Krasniqi E. et al. KEAP1 and TP53 frame genomic, evolutionary, and immunologic subtypes of lung adenocarcinoma with different sensitivity to immunotherapy. J Thorac Oncol. 2021;16:2065-77
79. Byun JK, Park M, Lee S, Yun JW, Lee J, Kim JS. et al. Inhibition of glutamine utilization synergizes with immune checkpoint inhibitor to promote antitumor immunity. Mol Cell. 2020;80:592-606 e8
80. Oh MH, Sun IH, Zhao L, Leone RD, Sun IM, Xu W. et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J Clin Invest. 2020;130:3865-84
81. Reinfeld BI, Madden MZ, Wolf MM, Chytil A, Bader JE, Patterson AR. et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature. 2021;593:282-8
82. Yang H, Yao F, Davis PF, Tan ST, Hall SRR. CD73, Tumor plasticity and immune evasion in solid cancers. Cancers (Basel). 2021 13
83. Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu X. et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature. 2017;543:252-6
84. Bagchi S, Yuan R, Engleman EG. Immune checkpoint inhibitors for the treatment of cancer: clinical impact and mechanisms of response and resistance. Annu Rev Pathol. 2021;16:223-49
85. Olagnier D, Brandtoft AM, Gunderstofte C, Villadsen NL, Krapp C, Thielke AL. et al. Nrf2 negatively regulates STING indicating a link between antiviral sensing and metabolic reprogramming. Nat Commun. 2018;9:3506
86. Kornepati AVR, Vadlamudi RK, Curiel TJ. Programmed death ligand 1 signals in cancer cells. Nat Rev Cancer. 2022;22:174-89
87. Niu M, Yi M, Li N, Luo S, Wu K. Predictive biomarkers of anti-PD-1/PD-L1 therapy in NSCLC. Exp Hematol Oncol. 2021;10:18
88. Papalexi E, Mimitou EP, Butler AW, Foster S, Bracken B, Mauck WM 3rd. et al. Characterizing the molecular regulation of inhibitory immune checkpoints with multimodal single-cell screens. Nat Genet. 2021;53:322-31
89. Dubiel D, Bintig W, Kähne T, Dubiel W, Naumann M. Cul3 neddylation is crucial for gradual lipid droplet formation during adipogenesis. Biochim Biophys Acta Mol Cell Res. 2017;1864:1405-12
90. Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT. et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature. 2018;553:91-5
91. Singh A, Daemen A, Nickles D, Jeon SM, Foreman O, Sudini K. et al. NRF2 activation promotes aggressive lung cancer and associates with poor clinical outcomes. Clin Cancer Res. 2021;27:877-88
92. JO Y, Lee B, Joo M, Hong C. Nrf2 expression is upregulated in tumor infiltrating T cells and induces T cell anergy. J Immunol. 2016;196:143.15-15
93. Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK. et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270-4
94. Ma X, Xiao L, Liu L, Ye L, Su P, Bi E. et al. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab. 2021;33:1001-12 e5
95. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR. et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127:2930-40
96. Ott PA, Bang YJ, Piha-Paul SA, Razak ARA, Bennouna J, Soria JC. et al. T-cell-inflamed gene-expression profile, programmed death ligand 1 expression, and tumor mutational burden predict efficacy in patients treated with pembrolizumab across 20 cancers: KEYNOTE-028. J Clin Oncol. 2019;37:318-27
97. Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J. et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science. 2018 362
98. Satoh H, Moriguchi T, Taguchi K, Takai J, Maher JM, Suzuki T. et al. Nrf2-deficiency creates a responsive microenvironment for metastasis to the lung. Carcinogenesis. 2010;31:1833-43
99. Satoh H, Moriguchi T, Takai J, Ebina M, Yamamoto M. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Res. 2013;73:4158-68
100. Best SA, Gubser PM, Sethumadhavan S, Kersbergen A, Negron Abril YL, Goldford J. et al. Glutaminase inhibition impairs CD8 T cell activation in STK11-/Lkb1-deficient lung cancer. Cell Metab. 2022
101. Pyaram K, Kumar A, Kim YH, Noel S, Reddy SP, Rabb H. et al. Keap1-Nrf2 system plays an important role in invariant natural killer T cell development and homeostasis. Cell Rep. 2019;27:699-707.e4
102. Krijgsman D, Hokland M, Kuppen PJK. The role of natural killer T cells in cancer-a phenotypical and functional approach. Front Immunol. 2018;9:367
103. Brennan PJ, Brigl M, Brenner MB. Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat Rev Immunol. 2013;13:101-17
104. Herb M, Schramm M. Functions of ROS in macrophages and antimicrobial immunity. Antioxidants (Basel). 2021 10
105. Wang P, Geng J, Gao J, Zhao H, Li J, Shi Y. et al. Macrophage achieves self-protection against oxidative stress-induced ageing through the Mst-Nrf2 axis. Nat Commun. 2019;10:755
106. Liu N, Liang Y, Wei T, Zou L, Huang X, Kong L. et al. The role of ferroptosis mediated by NRF2/ERK-regulated ferritinophagy in CdTe QDs-induced inflammation in macrophage. J Hazard Mater. 2022;436:129043
107. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. 2019;19:369-82
108. Roux C, Jafari SM, Shinde R, Duncan G, Cescon DW, Silvester J. et al. Reactive oxygen species modulate macrophage immunosuppressive phenotype through the up-regulation of PD-L1. Proc Natl Acad Sci U S A. 2019;116:4326-35
109. Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H. et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun. 2016;7:11624
110. Feng R, Morine Y, Ikemoto T, Imura S, Iwahashi S, Saito Y. et al. Nrf2 activation drive macrophages polarization and cancer cell epithelial-mesenchymal transition during interaction. Cell Commun Signal. 2018;16:54
111. Sun JC. Transcriptional control of NK cells. Curr Top Microbiol Immunol. 2016;395:1-36
112. Maskalenko NA, Zhigarev D, Campbell KS. Harnessing natural killer cells for cancer immunotherapy: dispatching the first responders. Nat Rev Drug Discov. 2022;21:559-77
113. Boss AP, Freeborn RA, Duriancik DM, Kennedy RC, Gardner EM, Rockwell CE. The Nrf2 activator tBHQ inhibits the activation of primary murine natural killer cells. Food Chem Toxicol. 2018;121:231-6
114. Poznanski SM, Singh K, Ritchie TM, Aguiar JA, Fan IY, Portillo AL. et al. Metabolic flexibility determines human NK cell functional fate in the tumor microenvironment. Cell Metab. 2021;33:1205-20 e5
115. Macoch M, Morzadec C, Génard R, Pallardy M, Kerdine-Römer S, Fardel O. et al. Nrf2-dependent repression of interleukin-12 expression in human dendritic cells exposed to inorganic arsenic. Free Radic Biol Med. 2015;88:381-90
116. Wendlandt EB, Graff JW, Gioannini TL, McCaffrey AP, Wilson ME. The role of microRNAs miR-200b and miR-200c in TLR4 signaling and NF-κB activation. Innate Immun. 2012;18:846-55
117. Liu QL, Zhang J, Liu X, Gao JY. Role of growth hormone in maturation and activation of dendritic cells via miR-200a and the Keap1/Nrf2 pathway. Cell Prolif. 2015;48:573-81
118. Olayanju A, Copple IM, Bryan HK, Edge GT, Sison RL, Wong MW. et al. Brusatol provokes a rapid and transient inhibition of Nrf2 signaling and sensitizes mammalian cells to chemical toxicity-implications for therapeutic targeting of Nrf2. Free Radic Biol Med. 2015;78:202-12
119. Tsuchida K, Tsujita T, Hayashi M, Ojima A, Keleku-Lukwete N, Katsuoka F. et al. Halofuginone enhances the chemo-sensitivity of cancer cells by suppressing NRF2 accumulation. Free Radic Biol Med. 2017;103:236-47
120. Tang X, Wang H, Fan L, Wu X, Xin A, Ren H. et al. Luteolin inhibits Nrf2 leading to negative regulation of the Nrf2/ARE pathway and sensitization of human lung carcinoma A549 cells to therapeutic drugs. Free Radic Biol Med. 2011;50:1599-609
121. Singh A, Venkannagari S, Oh KH, Zhang YQ, Rohde JM, Liu L. et al. Small molecule inhibitor of NRF2 selectively intervenes therapeutic resistance in KEAP1-deficient NSCLC tumors. ACS Chem Biol. 2016;11:3214-25
122. Bekes M, Langley DR, Crews CM. PROTAC targeted protein degraders: the past is prologue. Nat Rev Drug Discov. 2022;21:181-200
123. Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, Cuadrado A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol. 2011;31:1121-33
124. Wu T, Zhao F, Gao B, Tan C, Yagishita N, Nakajima T. et al. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014;28:708-22
125. Alabi SB, Crews CM. Major advances in targeted protein degradation: PROTACs, LYTACs, and MADTACs. J Biol Chem. 2021;296:100647
126. Mo X, Niu Q, Ivanov AA, Tsang YH, Tang C, Shu C. et al. Systematic discovery of mutation-directed neo-protein-protein interactions in cancer. Cell. 2022;185:1974-85.e12
Author contact
![]() Corresponding author: Haitang Yang (haitang.yangedu.cn, +86 18217015189), Feng Yao (yaofengedu.cn, +86 13636354837), Department of Thoracic Surgery, Shanghai Chest Hospital, Shanghai Jiao Tong University. West Huaihai 241, 200030, Shanghai, People's Republic of China.
Corresponding author: Haitang Yang (haitang.yangedu.cn, +86 18217015189), Feng Yao (yaofengedu.cn, +86 13636354837), Department of Thoracic Surgery, Shanghai Chest Hospital, Shanghai Jiao Tong University. West Huaihai 241, 200030, Shanghai, People's Republic of China.