Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Statistical analysis
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2023; 13(2):621-638. doi:10.7150/thno.72297 This issue Cite
Research Paper
FAM3C in circulating tumor-derived extracellular vesicles promotes non-small cell lung cancer growth in secondary sites
Win Lwin Thuya1*, Li Ren Kong1,2,4*, Nicholas L. Syn1,11*, Ling-Wen Ding1,3*, Esther Sok Hwee Cheow1, Regina Tong Xin Wong1, Tingting Wang1, Robby Miguel Wen-Jing Goh1, Hongyan Song1, Migara K. Jayasinghe2, Minh TN Le2,4, Jian Cheng Hu4, Wei-Peng Yong1,4,6, Soo-Chin Lee1,4,6, Andrea Li-Ann Wong1,4,6, Gautam Sethi2,4, Huynh The Hung5, Paul Chi-Lui Ho7, Jean-Paul Thiery1,8, Siu Kwan Sze9, Tiannan Guo10,11, Ross A. Soo1,6, Henry Yang1, Yaw Chyn Lim1, Lingzhi Wang1,2,4 ![]() , Boon-Cher Goh1,2,4,6,12
, Boon-Cher Goh1,2,4,6,12 ![]()
1. Cancer Science Institute of Singapore, National University of Singapore, Singapore 117599.
2. Department of Pharmacology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore.
3. Department of Pathology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore.
4. NUS Centre for Cancer Research (N2CR), Yong Loo Lin School of Medicine, National University of Singapore, Singapore.
5. Division of Cellular and Molecular Research, National Cancer Centre, Singapore.
6. Department of Haematology-Oncology, National University Cancer Institute, National University Health System, Singapore.
7. Department of Pharmacy, Faculty of Science, National University of Singapore, Singapore.
8. INSERM Unit 1186, Comprehensive Cancer Center, Institut Gustave Roussy, Villejuif, France.
9. Nanyang Technological University, 60 Nanyang Drive, Singapore 637551, Singapore.
10. Zhejiang Provincial Laboratory of Life Sciences and Biomedicine, Key Laboratory of Structural Biology of Zhejiang Province, School of Life Sciences, Westlake University, China.
11. Institute of Basic Medical Sciences, Westlake Institute for Advanced Study, China.
12. Department of Medicine, Yong Loo Lin School of Medicine, National University of Singapore, Singapore.
*Equal contributions
Received 2022-2-22; Accepted 2022-9-7; Published 2023-1-1
Abstract

Rationale: Metastasis is a complex process with a molecular underpinning that remains unclear. We hypothesize that cargo proteins conducted by extracellular vesicles (EVs) released from tumors may confer growth and metastasis potential on recipient cells. Here, we report that a cytokine-like secreted protein, FAM3C, contributes to late-stage lung tumor progression.
Methods: EV protein profiling was conducted with an unbiased proteomic mass spectrometry analysis on non-small cell lung cancer (NSCLC) and normal lung fibroblast cell lines. Expression of FAM3C was confirmed in a panel of NSCLC cell lines, and correlated to the invasive and metastatic potentials. Functional phenotype of endogenous FAM3C and tumor-derived EVs (TDEs) were further investigated using various biological approaches in RNA and protein levels. Metastasis potential of TDEs secreted by FAM3C-overexpressing carcinoma cells was validated in mouse models.
Results: Transcriptomic meta-analysis of pan-cancer datasets confirmed the overexpression of FAM3C - a gene encoding for interleukin-like EMT inducer (ILEI) - in NSCLC tumors, with strong association with poor patient prognosis and cancer metastasis. Aberrant expression of FAM3C in lung carcinoma cells enhances cellular transformation and promotes distant lung tumor colonization. In addition, higher FAM3C concentrations were detected in EVs extracted from plasma samples of NSCLC patients compared to those of healthy subjects. More importantly, we defined a hitherto-unknown mode of microenvironmental crosstalk involving FAM3C in EVs, whereby the delivery and uptake of FAM3C via TDEs enhances oncogenic signaling - in recipient cells that phenocopies the cell-endogenous overexpression of FAM3C. The oncogenicity transduced by FAM3C is executed via a novel interaction with the Ras-related protein RalA, triggering the downstream activation of the Src/Stat3 signaling cascade.
Conclusions: Our study describes a novel mechanism for FAM3C-driven carcinogenesis and shed light on EV FAM3C as a driver for metastatic lung tumors that could be exploited for cancer therapeutics.
Keywords: FAM3C, non-small cell lung carcinoma, tumor-derived extracellular vesicles, predictive biomarker, tumor metastasis.
Introduction
Metastasis of carcinomas represents the end-product of diverse evolutionary processes including the accumulation of oncogenic mutations [1], acquisition of mesenchymal phenotype [2], as well as co-option of diverse cell types within the tumor microenvironment [3]. Advanced metastases are often associated with cancer mortality [4] and resistance to therapeutic intervention [5], which then undermine the survival gains of targeted and immune-based therapies particularly in non-small cell lung cancer (NSCLC) [6]. NSCLC accounts for the highest cancer mortality worldwide predominantly due to its high metastasis potential. Understanding the molecular mechanisms underpinning these metastatic events and interrupting these pathways is expected to improve clinical outcome among NSCLC patients.
Research on extracellular vesicles (EVs) has gained considerable interest in recent years, and the increased understanding on the pathophysiology of tumor-derived EVs (TDEs) has engendered development of novel diagnostics and therapeutics tools [7]. EVs are comprised of exosomes, ectosomes and microvesicles, etc. While they differ in their cellular origin and biogenesis, EVs are constantly secreted into the extracellular microenvironment and could be detected in biofluids such as plasma and urine [7, 8], and may therefore serve as readily available biomarkers for disease diagnosis, prognosis and prediction of treatment effects [9, 10]. In addition, disease-associated EVs are intracellular communicators that can be transferred to recipient cells through plasma membrane fusion and endocytosis-mediated internalization. The uptake of these vesicles has been proven to be essential for biological processes that range from neurodevelopment [11] to immune defense mechanism [12], and have direct impact on disease severity.
From the perspective of carcinogenesis, TDEs and their cargo molecules are able to reprogram the neoplastic cells and stroma compartment to facilitate tumor development [13-17]. More recently, the discovery that vesicular integrins could determine organ-specific metastasis [18] and facilitate endocytosis of EVs in the recipient cells [19] have placed further emphasis on the biological role of TDEs in reprogramming the tumor microenvironment. This finding challenges the long-standing dogma that autocrine and paracrine signaling are regulated mainly through secreted proteins. We and others have previously articulated a conceptual framework for the mechanisms of EVs-mediated metastasis [7, 20, 21], which involves (i) paracrine and autocrine signaling for the intracellular induction of epithelial-mesenchymal transition (EMT), (ii) distant cell-to-cell communication that facilitate metastatic colonization, and (iii) alteration of the tumor microenvironment to enable escape from anti-tumor immuno-surveillance. While the multifarious roles of TDEs are established in cell-cell communication and cancer progression/metastases, the specific molecules mediating these effects need further elucidation if we are to find therapeutically tractable solutions.
In the present study, by coupling mass-spectrometry-based proteomics and biochemical assays in lung cancer cells validated with patient's plasma samples, we identify FAM3C, a secreted interleukin-like EMT inducer (ILEI), is an oncogenic cargo protein encapsulated within TDEs through which cell-cell growth signals are potentially communicated. FAM3C is a potent inducer of tumor metastasis which regulates EMT through TGF-β signaling [22-24], and is associated with poorer prognosis in several cancer types [17, 25], but its role in NSCLC remains unclear. Functionally, we show that intracellular expression of FAM3C in NSCLC cells not only promotes cancer cell growth and invasion in vitro and in vivo, but extracellular transmission of FAM3C-containing TDEs can also induce aggressive phenotypes in recipient cells resembling those of cell autonomous FAM3C overexpression. Mechanistically, we establish that TDE derived FAM3C interacts with Ras-related protein RalA to trigger oncogenic signals.
Materials and methods
Cell Culture
Lung carcinoma cell lines utilized in this study were purchased from the American Type Culture Collection (ATCC) (Manassas, VA, USA); they include HCC827, H1975, H1650, H226, A549, H1299, SKMES-1, Calu-1, H2172, ChaGo-K, H520, H2170 and growth conditions were detailed in Supplementary Information.
EVs isolation
Cells were cultured in T75 cell culture flasks until 80-90% confluency. The culture media were discarded and the cells were washed with 1x phosphate- buffered saline (PBS) at least 3 times to remove all bovine EVs. Then, the cells were cultured in serum-free RPMI-1640 or MEM supplemented with 1% L-glutamine and 1% penicillin/streptomycin. EVs isolation conditions were detailed in Supplementary Information.
Scanning Electron Microscopy (SEM)
EVs samples were first fixed in 2.5% Glutaraldehyde (Electron Microscopy Services, Hatfield, PA, USA) in 0.1M Sodium Cacodylate buffer, pH 7.4 (Electron Microscopy Services, Hatfield, PA, USA) for 60 min, and rinsed in 0.1M Sodium Cacodylate buffer (3 x 10 min) at 25°C. The process was detailed in Supplementary Information.
Characterization of extracellular vesicles by DLS
The size distribution of EVs isolated from plasma samples were determined using Zetasizer Nano ZS90 (Malvern Instruments Ltd, Malvern, UK) and the light scattering was measured. Each sample was measured at dispersant refractive index of 1.330 and viscosity of 0.8872 cP at 25°C, and the size distribution of each sample was recorded through a sequence of 12×10 s recordings which were repeated three times.
Western blot analysis
Cells were lysed in PIERCE™ RIPA lysis buffer, supplemented with Halt™ protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). The proteins were separated on 8-12% SDS-PAGE gels and the electrophoresed gels were transferred into PVDF membranes. Western blotting conditions were detailed in Supplementary Information.
Proteomics sample processing and analysis
Briefly, extracted EV proteins were separated in gel digestion, following tryptic digestion into peptides which were subsequently analyzed by Linear Trap Quadrupole Fourier Transform (LTQ-FT) mass spectrometry. The sample processing and analysis were detailed in Supplementary Information.
RT-qPCR analysis
Reverse transcription-quantitative polymerase chain reaction was performed to quantify and validate RNA sequencing results, using GAPDH as a loading control. For primers used, see Table S3.
Immunohistochemistry (IHC) staining and scoring
The NSCLC tissue microarray comprising 45 cases of NSCLC tissues, 47 cases of tumor adjacent normal tissues and 8 cases of normal lung tissues was obtained from US Biomax, Inc. IHC conditions and description of the scoring criteria for FAM3C expression were detailed in Supplementary Information.
FAM3C plasmid constructs and lentiviruses transduction
Human FAM3C MISSION shRNA Lentiviral Transduction Particles and MISSION TRC2 Control Vector pLKO-puro TurboGFP shRNA were obtained from Sigma. The sequence for FAM3C shRNA was CCGGCTTGGTGTGTGCATGAGTATTCTCGAGAATACTCATGCACACACCAAGTTTTTG. Lentiviruses transduction procedure was detailed in Supplementary Information.
Recombinant human FAM3C protein treatment
Recombinant human FAM3C protein was purchased from MyBiosource (Catalog #: MBS204141). NSCLC cells including H2170, A549 and SKMES-1 were treated with 10 ng/mL FAM3C recombinant protein for 48 hours before experimental analyses.
In vitro invasion assay
Cell invasion capability was performed using a Corning® BioCoat™ Matrigel Invasion Chambers (#354480). Cell invasion procedure was detailed in Supplementary Information.
In vitro cell migration assay
The cells were cultured until they reached 90-95% confluence. A scratch was created through the monolayer in each well using a 300ul pipette tip. Monolayer was washed with 1 x PBS and media was changed daily. Cell movement into the wound area was monitored and photographed at 0 hour and 24 hours using Zeiss Axio Vert A1 inverted microscope (Carl Zeiss Microscopy GmbH, Oberkochen, Germany).
Anchorage-dependent and -independent colony formation assays
For anchorage-dependent assay, human lung carcinoma (SKMES-1, A549 and H2170) control, FAM3C-overexpressed and shFAM3C-knockdown cells were seeded at a density of 5,000 cells/well in 6 well plates. The anchorage-dependent and independent colony assays were detailed in Supplementary Information.
Extracellular vesicles uptake by NSCLC cells
The fluorescent reagent Exo-Green (System Biosciences) was utilized to label the protein component of H2170 lung carcinoma cell-derived EVs. The process was detailed in Supplementary Information.
Duolink Proximity Ligation in situ Assay (P-LISA)
For Duolink in situ, all incubations were performed in a 37 °C humidity chamber. Cells were plated onto glass coverslips and fixed with 4% paraformaldehyde at room temperature for 30 min. PLA staining was conducted according to the manufacturer's protocol (Duolink, #DUO92101). Preparations were mounted with prolong gold antifade reagent with DAPI (Life Technologies). PLA images were analyzed using ImageJ software (NIH), expressed with the mean PLA/cell intensity ± SEM.
Phosphokinase antibody array
Human phosphokinase antibody array kits were obtained from R&D Systems® to analyze the phosphorylation profiles of kinases according to manufacturer's instructions (https://www.rndsystems.com/resources/technical/assays-analytes-represented-human-phospho-kinase-array-kit).
RNA sequencing analysis
Total RNA was isolated using Qiagen RNeasy Kit and sequenced using Illumina Hiseq4000. Differently expressed genes [with a threshold log2 (fold change) ≥ 0.5] were analyzed using Gene Set enrichment analysis (GSEA) and ConsensusPathDB for pathway enrichment analysis.
TCGA data of NSCLC
The TCGA pan cancer RNA sequencing data were analyzed using Gene Expression Profiling Interactive Analysis (GEPIA). The expression level of FAM3C in different sets of lung cancer patient cohorts was retrieved and analyzed using GEO gene expression database.
In vivo lung tumor colonization assay
The animal experiments were approved by ethics board from the SingHealth Institutional Animal Care and Use Committee (IACUC) at the National Cancer Centre Singapore and Singapore General Hospital. All mice were maintained according to the "Guide for the Care and Use of Laboratory Animals" published by National Institute of Health, USA. They were provided with sterilized food and water ad libitum, and housed in negative pressure isolators with 12h light/dark cycles.
Metastatic ability of FAM3C was determined by tail vein injection of Control-Vector cells, FAM3C knockdown and over-expressed A549 cells into mice. 1×106 cells/100 µl PBS were injected into tail vein of the respective mice. After 2 months, the mice were euthanized; and their lungs were harvested, fixed in buffered formalin and stained with Bouin's solution. After that, the lungs were destained prior to processing into paraffin blocks and 4µm thick sections prepared for IHC staining.
For determination of metastasis potential of tumor-derived EVs in mice, 10 μg of control A549 and FAM3C-overexpressing A549 cells-derived EVs were injected intravenously via the tail vein into mice twice a week for two weeks prior to primary cells administration. At the beginning of week 3, the mice were inoculated with A549 primary cells together with tumor-derived EVs via tail vein injection. EVs were injected subsequently twice a week for an additional 5 weeks. Mice were sacrificed and lung organs were harvested to investigate the efficiency of tumor-derived EVs to induce tumour cell engraftment. Lungs were fixed in formalin, processed into paraffin blocks and tissue sections were stained for pan-cytokeratin and FAM3C expression as described above.
Statistical analysis
All data analysis was conducted using Graph Pad Prism 9 software (GraphPad Software, La Jolla, CA, USA). All results are reported as mean ± SEM. Statistical differences between two groups were evaluated using two-sided paired Student's t-test, while comparison between multiple groups was conducted using one-way ANOVA. Differences with a p value < 0.05 were considered statistically significant. The non-parametric one-tailed Mann-Whitney U test was carried out for the mouse lung tumor nodule analyses.
Results
FAM3C is expressed in TDEs released from NSCLC tumors and its expression correlates with poor prognosis
Our central hypothesis is that EV proteins may drive oncogenic signaling within the tumor microenvironment. Proteomic quantification was performed on EVs derived from a panel of NSCLC cell lines (HCC827, H1650, and H520) and fibroblast cell line using label-free LTQ-FT mass spectrometry. EVs were purified using differential centrifugation followed by ultracentrifugation - an established method for EVs isolation (Figure S1A). To verify the characteristics of isolated EVs, we showed - by dynamic light scattering analysis - that isolated EVs formed a bell-shaped size distribution profile with average size of 130.4 nm (Figure S1B). The purity of the extracted EVs were validated with the universal EVs markers (TSG101, CD63, and CD9), without the contamination of the cis-Golgi matrix protein (GM130) and marker for endoplasmic reticulum (Calnexin) (Figure S1C).
From the purified EVs (Figure S1C), we identified a total of 2,658 distinct proteins in EVs shed by NSCLC cells (HCC827, H1650 and H520) and IMR90 cells at a false discovery rate (FDR) of 1%, with 1,369 proteins found to be specific to NSCLC (Figure S1D, left). Among the EV proteins detected in the three NSCLC cell lines (2,371), 709 proteins were commonly shared by all cancer cell lines (Figure S1D, right). Among the top 20 differentially expressed proteins with a fold-change of > 6 (Figure S1E, Table S1), we identified FAM3C, a small secreted protein with strong EMT inducing property. To investigate the clinical relevance of this finding, we successfully purified and validated EVs from plasma samples of NSCLC patients and healthy individuals (Figure S2A), and measured EV FAM3C expression by immunoblotting in 84 plasma samples (Figure 1E) and ELISA in 213 plasma samples (Figure 1F), demonstrating significantly higher FAM3C in cancer patients. More importantly, higher FAM3C concentrations in the EVs extracted from patient plasma was associated with more advanced stages of NSCLC (Figure S2B). In addition, the other top hits identified from the cell line-derived EVs (ANXA4, RPS9, RPLP1, and PSME3) were detected in plasma EVs as well (Figure S2C-F), thus validating the robustness of our proteomic approach. Given the known role of FAM3C in promoting EMT in a few cancers, we hypothesized that FAM3C carried on TDEs could promote proliferation of NSCLC at secondary sites.
FAM3C expression is associated with poorer prognosis in lung carcinomas. (A) Analysis of FAM3C mRNA expression in 11 different types of human cancer (TCGA PanCancer Atlas Studies from cBioPortal database). Red, tumor samples; green, normal tissue. BRCA, breast carcinoma; LUAD, lung adenocarcinoma, LUSC, lung squamous cell carcinoma; COAD, colon adenocarcinoma; PRAD, prostate adenocarcinoma; STAD, stomach adenocarcinoma; LIHC, liver hepatocellular carcinoma; ESCA, esophageal carcinoma; UCS, uterine carcinosarcoma; THCA, thyroid carcinoma; BLCA, bladder carcinoma. (B) FAM3C mRNA expressions in five independent lung carcinoma datasets: GSE10072, GSE18842, GSE19804, GSE31210 and GSE19188. LUAD, lung adenocarcinoma; NSCLC, non-small cell lung carcinoma. (C) Kaplan-Meier survival plot showing prognosis of patients stratified by FAM3C expression (Data retrieved from gene expression profiling interactive analysis). (D) NSCLC tissue microarray with 45 cases of NSCLC and 47 cases of tumor adjacent normal tissue including 8 cases of normal lung tissues (US Biomax). The representative images of FAM3C expression in normal lung tissues, LUAD and LUSC (left panel). Scale bar, 100 μM. Box plot showing the histoscore for FAM3C expression in TMA for adenocarcinoma and squamous cell carcinomas (right panel). (E) Quantitative analysis by Western blot for FAM3C expression in plasma EVs in NSCLC patients (n=42) and healthy controls (n=42). The heatmap in the bottom right panel represents relative density of the Western blot bands of FAM3C normalized to the densitometry values of internal exosomal marker, TSG101 compared with control (p value <0.0001). (F) Scatter plot of EV FAM3C concentrations in plasma samples based on the ELISA results of the NSCLC group (n=102) and healthy control group (n=111).
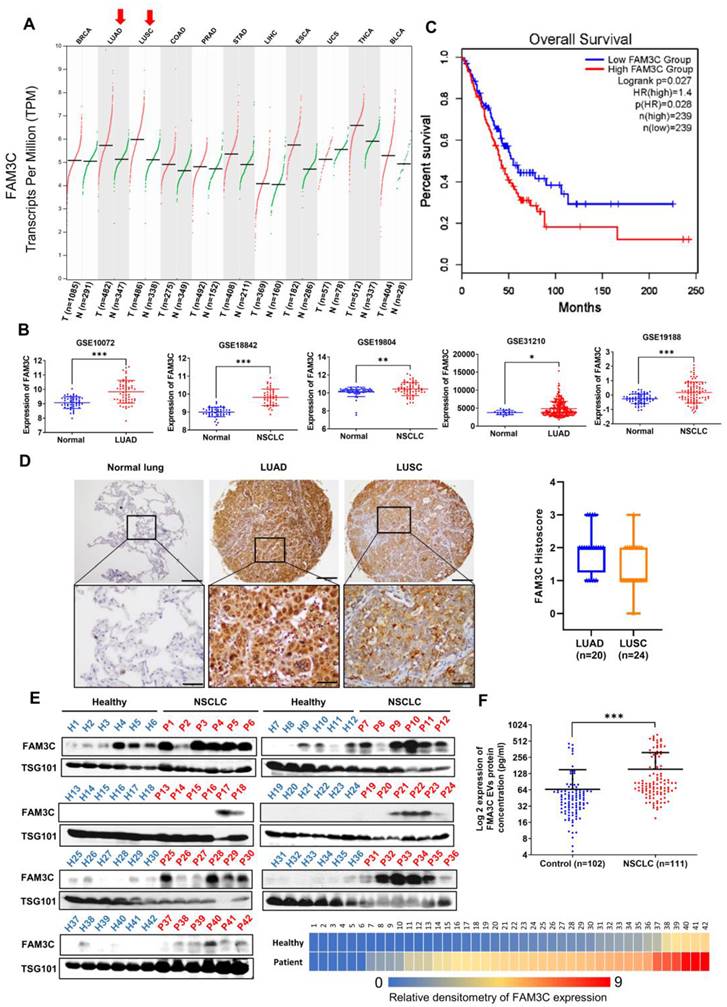
To unravel the clinical significance of FAM3C in carcinomas, we began by delineating its clinicopathological and molecular correlation by leveraging on publicly available transcriptomic datasets as well as immunohistochemical evaluation of FAM3C expression in tissue microarrays. Notably, FAM3C expression is elevated in malignant lung tissues (lung adenocarcinoma, LUAD; and lung squamous cell carcinoma, LUSC) compared to the non-malignant tissues based on The Cancer Genome Atlas (TCGA) database (Figure 1A). Furthermore, significantly higher mRNA expression of FAM3C in carcinoma tissues was observed as compared to the normal lung counterparts based on five independent lung carcinoma datasets (Figure 1B). Using one-stage meta-analysis on patient survival data from these datasets, we found high FAM3C mRNA expression (stratified at 239 for both high and low median) to be correlated with poorer prognosis (marginal HR = 1.40; 95% CI: 1.04-1.89; p = 0.028) (Figure 1C), thereby suggesting its pertinence in NSCLC.
To confirm the specificity of FAM3C in tumor cells, its expression profile across normal lung tissues and early to late stage carcinomas was assessed in a scRNA-Seq readout of 44 LUAD patients [26]. Of note, FAM3C is scarcely detected in non-transformed epithelial tissues and lymph nodes (Figure S3A-B). In contrast, FAM3C is significantly elevated in the epithelial compartment of the cancerous tissues (Figure S3C), with an enrichment of FAM3C-positive populations in the lymph node and brain metastases (Figure S3D-E). By focusing on the epithelial compartments of the primary tumors and metastases, there was an increase of FAM3C expression in most of the tumor clusters of the metastases (Figure S3F-G). Collectively, these analyses suggested that FAM3C is likely of tumor origin and may play a significant role in the disease progression of lung carcinoma metastasis.
Semi-quantitative measurement of FAM3C using immunohistochemical staining was performed on 45 cases of NSCLC, 47 cases of tumor-adjacent normal tissue, and 8 cases of normal lung tissue (Table S2). Microscopic analyses of the tissue histology revealed that FAM3C is predominantly localized in the cytosol, with strong expression detected in the cancer cells (Figure 1D). Interestingly, all normal lung parenchyma showed negative staining (n=0/10, 0.0%), while 17 of 47 of tumor-adjacent “normal” tissue (36.2%), and 44 of 45 of NSCLC samples (97.2%) were stained positive (Pearson's χ2 = 50.28, p=1.2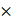 10-11). FAM3C expression was detected in both LUAD and LUSC (Figure 1D, left), although more variable staining intensities were seen within the LUSC subtype (Figure 1D, right). Collectively, both transcriptomic and immunohistochemical data confirmed the specificity of FAM3C in lung tumors and it is associated with poorer prognosis.
10-11). FAM3C expression was detected in both LUAD and LUSC (Figure 1D, left), although more variable staining intensities were seen within the LUSC subtype (Figure 1D, right). Collectively, both transcriptomic and immunohistochemical data confirmed the specificity of FAM3C in lung tumors and it is associated with poorer prognosis.
Aberrant FAM3C promotes NSCLC cell motility and anchorage-independent growth
Endogenous FAM3C expression has been reported to drive metastatic outgrowth via EMT induction in breast and gastric carcinomas [22, 24, 27]. However, its pathophysiologic role in NSCLC has not been investigated in depth. Here, we screened FAM3C protein expression in 12 NSCLC cell lines. FAM3C expression was low in the normal lung fibroblast (IMR90) cell line. In contrast, most of adenocarcinoma or squamous cell lines showed high FAM3C content (Figure S4A), which is consistent with the scRNA-Seq database (Figure S3A). To delineate the role of FAM3C in NSCLC, we modulated its expression in the FAM3C-high SKMES-1 (using shRNA-mediated knockdown), FAM3C-low H2170 (using plasmid-based overexpression), and FAM3C-intermediate A549 cell lines (knockdown or overexpression). In agreement with previous reports from others, our data demonstrated that FAM3C is a strong inducer of cell motility and malignancy. FAM3C-knockdown (FAM3C_kd) in SKMES-1 effectively increased E-cadherin expression and reduced vimentin (Figure S4B) - both quintessential EMT-related markers and reduced cell migration (Figure S4C), invasion (Figure S4D), and clonogenicity (Figure S4E). Reciprocally, FAM3C-overexpression (FAM3C_ox) in H2170 suppressed E-cadherin while induced vimentin, and concomitantly induced invasiveness, migration and anchorage-independent growth (Figure S4B-E, middle). In consonance with experiments with SKMES-1 and H2170 cells, knockdown and stable overexpression of FAM3C in A549 was accompanied by downregulation and upregulation of E-cadherin and vimentin respectively (Figure S4B, right). Modulating FAM3C levels in A549 cells had significant effect in cell motility and were mirrored by statistically significant changes in anchorage-dependent and anchorage-independent tumorigenesis both in terms of colony counts and size of colonies (Figure S4C-E, right). Furthermore, FAM3C oncogenic activity was confirmed via rescue experiments in FAM3C_kd A549 cells by co-incubation with recombinant FAM3C protein (Figure 2A-B). Taken together, the afore set of observations convincingly established the role of FAM3C in imparting NSCLC cells with pro-invasive and tumorigenic properties.
FAM3C impacts invasiveness of lung carcinoma cells. (A) A549 FAM3C knockdown cells (FAM3C_kd) were treated with human recombinant FAM3C (rFAM3C) protein and expression of FAM3C and EMT markers (E-cadherin and Vimentin) were analyzed by Western blot analysis. (B) The effect of recombinant FAM3C on cell motility was examined by Matrigel invasion and wound healing assays. The data presented were from three independent experiments, each with three replicates and are represented as mean ± SEM. *** p < 0.001. (C-E) FAM3C promotes lung colonization in vivo. (C) A549 parental cells (Ctrl), A549-FAM3C knockdown (FAM3C_kd) cells, and A549-FAM3C overexpressed (FAM3C_ox) cells were injected intravenously into five nude mice per cell type. Mice were sacrificed and analyzed for the presence of lung metastasis. (D) Macroscopic images of Bouin's stained lungs from five individual mice from each group. Visible lung tumor colonies appear as yellowish white spots on the lung surface. (E) Representative images of whole lung sections stained with cytokeratin (CK). Bright red clusters are A549 lung colonization. Counts for CK-positive nodules were tabulated as median ± interquartile range (n = 5). (F) (upper panels) Representative images of FAM3C expression (brown) in the A549 Ctrl cells, FAM3C_kd and FAM3C_ox transduced cells, red arrows indicate FAM3C-positive A549 Ctrl cells; (bottom panels) FAM3C (brown)-Ki-67 (red) double staining of lung sections of A549 Ctrl, FAM3C_kd and FAM3C_ox tumors. (G) Representative images of A549 Ctrl lung tumor section stained with cytokeratin (red). Box, 40X magnification of indicated area.
FAM3C promotes lung carcinoma cell colonization in vivo
Having shown that FAM3C promotes EMT and cancer aggressiveness in vitro, we wonder if changes in intracellular FAM3C translates to any appreciable effects in vivo. We established mouse models of lung colonization through tail vein injection of parental A549 (Ctrl) cells or the modified counterparts with FAM3C knockdown (FAM3C_kd) and overexpression (FAM3C_ox) (Figure 2C). Resected lungs were washed and stained in Bouin's solution, and both microscopy and macroscopy analyses revealed gross disparities in intrapulmonary tumor burden of mice that were positively correlated to FAM3C status. Tumor nodules appeared as yellowish white spots on the lungs of mice injected with A549 parental and FAM3C_kd cells (Figure 2D). In contrast, A549 FAM3C_ox cells have colonized the whole lung, as demonstrated by the enlarged lung size and the yellowish white coloration of the entire lung (Figure 2D). Pan-cytokeratin staining (red) revealed the presence of tumor nodules in the lungs of mice injected with A549 parental and FAM3C_kd cells (Figure 2E); however, the number of nodules in the A549 FAM3C_kd group was significantly fewer than that of the parental cell group (Figure 2E, right panel). The nodules in the lungs of mice injected with A549 FAM3C_ox cells cannot be counted because they were extremely aggressive and had colonized and defaced the entire lung parenchyma of the host animals.
We next validated FAM3C expression in the parental and transduced cell lines; and their respective lung tumors (Figure 2F upper panels). While only a small proportion of the A549 Ctrl cells show weak FAM3C expression (red arrows), strong FAM3C expression was detected in ~70% of the A549 FAM3C_ox cells. As expected, the cytokeratin positive FAM3C_kd derived tumor nodules exhibit negligible FAM3C expression (Figure S5A; Figure 2F middle panels). In contrast, the FAM3C expression in the FAM3C_ox derived lung tumor nodules were more intense than that of A549 parental cell origin (Figure 2F).
While some tumor nodules or part of some nodules in the lung sections appear to be tightly packed and intensely stained (Figure 2G, #); the rest exhibit an open and lacey appearance with less intense cytokeratin staining (Figure 2G, *). Further examination at high magnification show that darkly stained nodules are made up small cancer cells with strong cytoplasmic and membrane cytokeratin staining (1, Figure 2G and Figure S5A), while the cells in lighter nodules often appeared thinly spread and elongated (2, Figure 2G and Figure S5A). More intriguingly, positive nuclear Ki67 staining (red), indicating the presence of proliferating cells, is consistently found within the clusters of small cells (Figure 2F, 1 Figure 5B) but rarely amongst the larger spread cells (Figure 2F, 3 Figure S5B). FAM3C expression in the A549 parental and FAM3C_ox tumor nodules appear to be unipolar or perinulcear in these small tightly packed carcinoma cells (Figure 2F upper panels; 1, Figure S5B, red arrows) and as punctate cytoplasmic positivity proximal to the nuclei in the larger spread carcinoma cells (2, Figure S5B, black arrows).
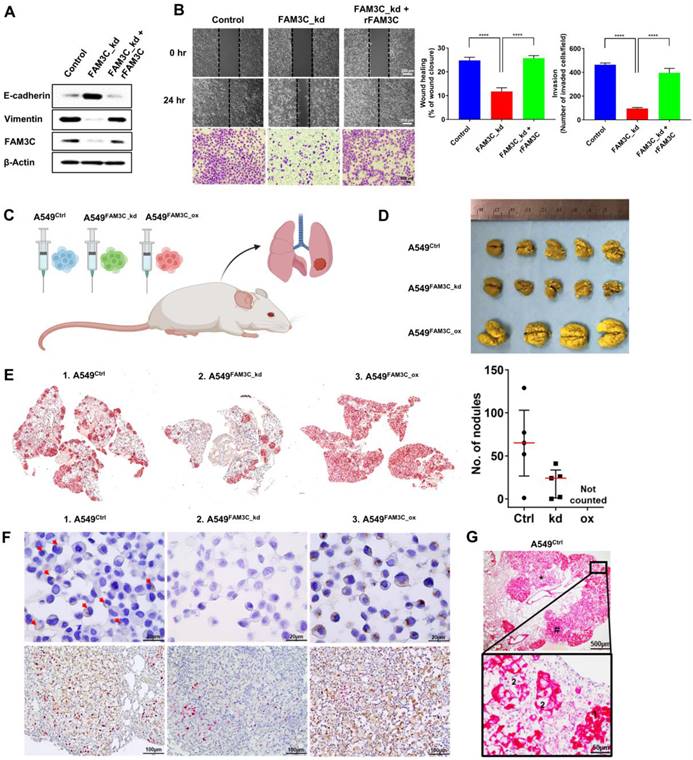
Transcriptomic and molecular perturbations in FAM3C overexpressed cells. RNA-Seq analysis of FAM3C_ox cells versus control cells. (A) Volcano plot of log2 (fold change) and -log10 (adjusted p-value) of all identified genes between control and FAM3C-overexpressing cells. (B) The top 10 enriched disease ontology (left panel) and KEGG pathway (right panel). (C) GSEA analysis of significantly enriched genes in FAM3C-overexpressing cells vs the control cells. NES, normalized enrichment score; FDR, false discovery rate. The red color scale indicates the positive and blue color indicates the negative correlation. (D) Histograms plotting the gene targets within the significantly enriched GSEA pathways. (E) Key targets genes from the RNA-Seq were validated using quantitative PCR (qPCR) analysis. (F-H) Kinome profiling of FAM3C_ox cells. (F) Human phospho-kinase array analysis was performed in H2170 and FAM3C_ox cells. Red box, blue box, green box and orange box indicates the phosphoproteins of Src, ERK1/2, Akt (T308) and STAT3 (Y705) respectively. (G) Bar graph represents the fold change expression of each phosphokinase proteins relative to control cells. (H) Western blot analysis of the indicated targets in H2170 and FAM3C_ox cells. Beta actin was used as a loading control.
Taken together these observations suggest that the small tightly pack cells are the newly divided and actively proliferating cells; and the thinly spread and elongated cells are cells that appear to have lost the ability to divide. At this juncture, we are unclear of the reason for this loss. As this observed change in cell morphology and proliferation potential are common features in the A549 parental/Ctrl (Figure 2G), FAM3C_kd (Figure S5A) and FAM3C_ox (not shown) lung sections, we can conclude that this is an inherent characteristic of the A549 lung cancer cell line independent of the cells' FAM3C expression status. In contrast, the aggressiveness of the FAM3C_ox cancer cells in near complete colonization the host lungs compared to the A549 ctrl and FAM3C_kd cells further supports our hypothesis that FAM3C is a potent inducer of cancer metastasis and disease progression.
TDEs mediate cell-to-cell transfer of FAM3C protein and increase migration and invasion
In recent years, compelling evidences have indicated that TDEs may serve as paracrine and autocrine messengers within the tumor microenvironment to induce tumor drug resistance [15], metastasis [13, 14, 16, 17], and malignant growth [28]. While FAM3C was first characterized as a secreted protein, we showed here that FAM3C was also enriched in TDEs released by the panel of NSCLC cell lines (Figure 3A), with strong concordance to the endogenous protein expression (Figure S4A). Accordingly, we postulate here that FAM3C-containing EVs may promote intercellular crosstalk within the tumor microenvironment to drive tumor malignancy and metastases. By Here, we proposed to investigate whether (i) the secreted EVs can be internalized by neighboring neoplastic cells, (ii) if their uptake is sufficient to confer invasive cue in recipient cells, and (iii) whether TDEs secreted by carcinoma cells could hasten disease progression in vivo. To probe the first two questions, EVs shed by FAM3C-overexpressed H2170 and A549 cells (EVsFAM3C) and their respective control cells (EVsControl) were isolated. The particle size of the isolated EVs were in the range of 30-100 nm (Figure 3B, left and right), and the purity was confirmed by the strong EV surface markers (Alix, Tsg101 and CD63) relative to cell lysates (Figure 3B, middle) (Figure S6B). On the contrary, two EV negative (-) markers (GM130 and Calnexin) were highly expressed in cell lysates but undetectable in EVs. We showed that EV FAM3C protein was protected by proteinase k treatment; whereas disruption of the surface membrane of EV led to degradation of FAM3C, suggesting that EV encapsulation could enhance the stability of the cargo molecules (Figure S6C). Next, the EVs isolated from H2170 and A549 control (EVsControl) and FAM3C-overexpressed (EVsFAM3C) cells were incubated in their parental cells (Figure 3C) (Figure S6E), and the cellular uptake was confirmed by probing for intracellular FAM3C expression (Figure 3D). Moreover, direct imaging of H2170 cells cultured with fluorescent-labelled EVsFAM3C showed punctate fluorescence signals (Figure 3E), proving that successful uptake of EVs may facilitate transfer of cargo molecules. Next, we asked whether the internalization of EVsFAM3C elicits metastatic properties in recipient cells as compared to EVsControl treatment. We observed a more efficient wound closure and invasion in H2170 and A549 cells incubated with EVsFAM3C as compared to EVsControl (Figure 3F-G) (Figure S6F-G).
Tumor-derived EVs containing FAM3C promote invasiveness of lung carcinoma cells in vitro. (A) Western blot analysis of FAM3C expression in cell lines-derived EVs isolated from 12 NSCLC cell lines compared to normal lung fibroblast (NLF) cell line (IMR90). The histogram represents expression levels of FAM3C in EVs derived from normal lung fibroblast (black), LUAD (blue) and LUSC (orange) after normalization against TSG101. (B) Morphological characterization of EVs were performed by scanning electron microscopy at 60k magnification (SEM). Scale bar, 1 μM (left panel). Western blot analysis of three positive (+) EVs markers (Alix, Tsg101 and CD63), and two EV negative (-) markers (GM130 and Calnexin) in cell lysates and EVs isolated from the culture media of the respective cell lines (middle panel). GAPDH served as loading control. Size distribution of H2170 cell-derived EVs was assessed by the Zetasizer Nano ZS90 (right panel). (C) Experimental set up for EVs uptake by lung carcinoma cells. The EVs isolated from H2170 control (EVsControl) and FAM3C-overexpressed (EVsFAM3C) cells were added into culture medium of parental H2170 cells to evaluate for metastasis potential. (D) Western blot analysis of EV markers (CD9 and Alix) and FAM3C expression in cell lysates and EVs isolated from the culture media of the H2170 control and overexpressed FAM3C cells (left panel). Western blot analysis of FAM3C expression in H2170 cells treated with PBS, 25 μg/mL of EVsControl and 25 μg/mL of EVsFAM3C (right panel). (E) Bright field and fluorescent images of cells 6 hours post EVs administration. H2170 cells were treated either with PBS, 25 μg/mL of EVsControl or 25 μg/mL of EVsFAM3C, and labelled with Exo-Green. Representative images were shown. Scale bar: 100 μM. (F-G) Effects of PBS, EVsControl or EVsFAM3C are assessed with wound healing (top) and Matrigel invasion assays (bottom) in H2170 cells after treatment with 25 μg/mL of respective EVs. (F) Representative images are shown. (G) Data are presented as mean of three independent experiments ± SEM. * p < 0.05, ** p < 0.01 and *** p < 0.001.
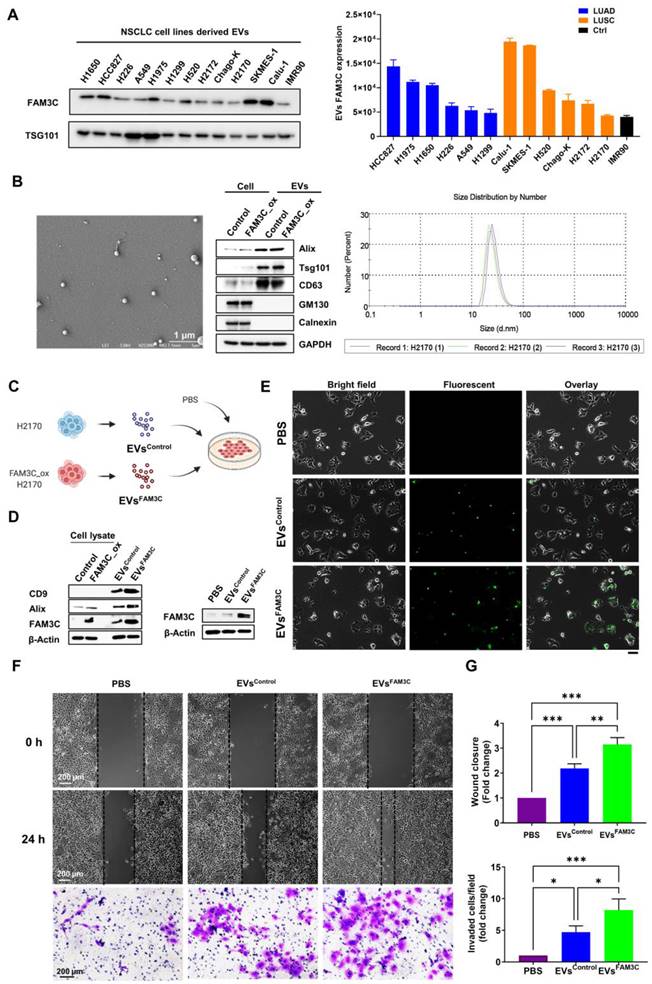
As a secreted protein, FAM3C is reported to bind to the leukemia inhibitory factor receptor (LIFR) to drive oncogenic signaling [29]. As such, we further investigated the effect of LIFR on FAM3C intracellular uptake. The results of our study indicated that there was no association of LIFR with FAM3C cellular expression in NSCLC (Figure S9A-B). While rFAM3C and EVs enhanced pro-migratory and pro-invasive cue of both in control and LIFR knockdown cells (Figure S9C-E), the EVs exhibited greater effect than rFAM3C. Taken together, our data strongly indicates that TDEs secreted by FAM3C-overexpressing NSCLC cells can indeed evoke biochemical and phenotypic changes in recipient cells.
FAM3C containing TDEs promote distant colony formation
Having demonstrated the effect of TDEs in inducing cell motility in vitro, we next explored their role in lung tumor colonization in vivo. To achieve this, we performed lung colonization studies using the tail vein injection model. Mice were randomly assigned to receive a 7-weeks' regimen of twice-weekly intravenous infusion of either PBS (negative control), EVsControl or EVsFAM3C. As summarized in Figure 4A, the mice were first primed with TDEs for 2 weeks prior to a one-time tail vein injection of A549 cells mixed with either PBS, EVsControl or EVsFAM3C. The animals continued to receive biweekly instillation of PBS or the appropriate EVs for another 5 weeks and then were sacrificed at the end of week 7. As characterized previously in Figure 2E, A549 cells steadily colonized the lung and formed large tumor nodules with open lacy appearance in all three treatment groups (Figure 4B). In this lung colonization model, the established A549 tumor nodules have the propensity to slowly seed new aggregates of carcinoma cells within the normal lung parenchyma (N) as seen in the PBS treated control group (Figure 4B, upper left). In comparison with mice exposed to PBS, animals treated with EVsControl and EVsFAM3C had respective ~2-fold and 4-fold increase in the number of tumor aggregates that appear as cell clusters with high pan-cadherin staining (Figure 4B, bottom right). This is concordant with recent reports on the role of TDEs in lung metastasis [18, 19, 28], that TDEs can communicate cell to cell metastatic cues. These lung tumor aggregates were found to express FAM3C (brown) and Ki67 positive cells (red) (Figure 4C), providing further evidence that FAM3C is an important mediator of carcinoma cell invasion and engraftment.
Tumor-derived EVs containing FAM3C trigger aggressiveness of lung carcinoma cells in vivo. (A) Tail vein injection of A549 lung carcinoma cells in mouse model after 2 weeks pre-educated with PBS, EVsControl or EVsFAM3C. Animals were sacrificed after week 7. (B) Representative images of cytokeratin (red) stained tissues. Tumor nodules are stained pink with lacy appearance. Black circle, small tumor cell aggregates highlighted as dark red clusters; N=normal lung parenchyma. Counts for tumor aggregates were tabulated as median ± interquartile range (n=4 for PBS, n=5 for EVsControl and EVsFAM3C). (C) Representative images of lung sections stained for FAM3C (brown) and Ki67 (red) positivity. Original images were captured at 20x magnification; box showed cropped image of the indicated area prior to image size reduction.
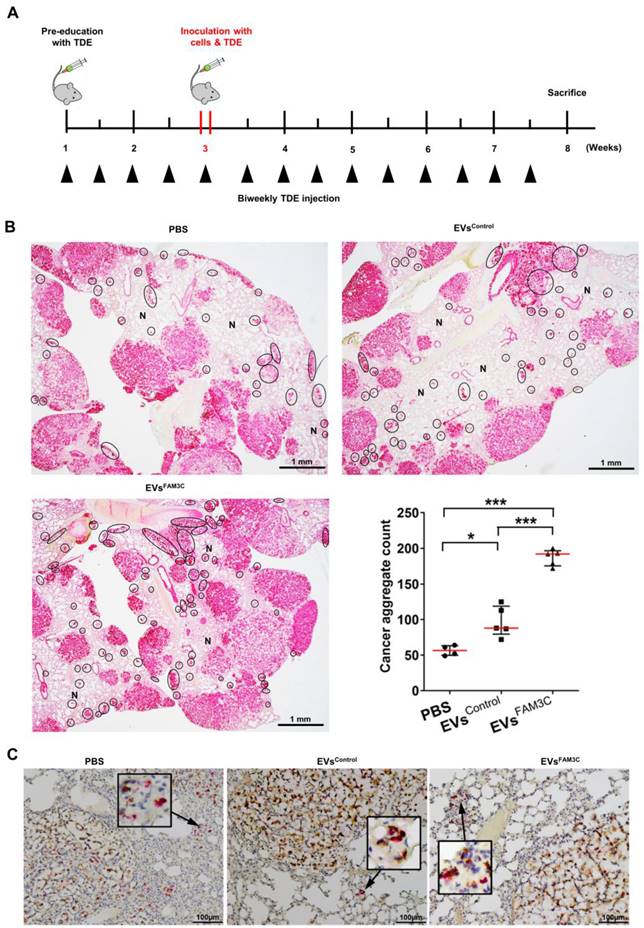
FAM3C induces transcriptomic activation of oncogenic signaling
We next sought to elucidate the molecular mechanisms underpinning FAM3C function. RNA-Seq analyses identified 4210 upregulated genes and 4702 downregulated genes in FAM3C_ox A549 cell lines compared to control cells (Figure 5A). Unsurprisingly, the transcriptomic perturbations evoked by FAM3C-overexpression were found to closely resemble curated gene signatures relevant to lung carcinoma, breast carcinoma, and prostate carcinoma, based on enrichment analyses using the Human Disease Ontology (DO) (Figure 5B, left panel). These differentially-expressed genes were also found to be overrepresented in KEGG-pathway ontologies related to focal adhesion and lysosome (Figure 5B, right panel), while gene-set enrichment analyses (GSEA) in the Molecular Signatures Database (MSigDB) further demonstrated significant overrepresentation of input genes in the hallmark gene set (identifier: M2572) for epithelial-to-mesenchymal transition (FDR q-value = 0), and two curated gene sets related to KRAS signaling (identifier: M2448; FDR q-value = 0.002) and tumor cell motility and invasiveness (identifier: M2572; FDR q-value = 0.09) (Figure 5C-D).
Selected genes involved in these GSEA pathways had been selected for independent validation using real-time quantitative PCR (Figure 5E). Notably, integrin beta 3 (ITGB3) was significantly elevated in FAM3C_ox cells. It has been previously reported that ITGB3 plays an important role in facilitating endocytosis-mediated uptake of EVs into recipient cells, and is critical for colonization of carcinoma cells in the lungs [19]. The positive correlation of FAM3C and ITGB3 implicates a possible feedforward mechanism of FAM3C in favor of EVs uptake through promoting integrin internalization. Our transcriptomic analysis revealed that genes involved in the JAK-STAT3 pathway (IL6, JAK1) and KRAS signaling were upregulated in the FAM3C_ox cells (Figure 5D-E), thereby suggesting a possible involvement of FAM3C in signal transduction cascades. To examine this, we harnessed a human phosphokinase array to detect simultaneous perturbations in kinase activities. In H2170 cells, FAM3C overexpression were found to enhance the activation of oncogenic phosphoinositide-dependent signaling, as indicated by elevated levels of phosphorylated ERK1/2, MEK1/2, Akt, Src and Stat3 (Tyr705) (Figure 5F-G). These phosphorylation events were validated independently via immunoblotting (Figure 5H). Though these multi-omic analyses are meant to be hypothesis generating and nondefinitive, collectively the results suggest that FAM3C is involved in intracellular kinase signaling potentially through activation of Stat3 signaling.
RalA is a functional interacting partner of FAM3C
Though FAM3C is known to induce metastasis, the signaling pathway mediating this is unclear. As FAM3C lacks a functional kinase domain, it is likely that interaction with other signaling molecules mediate this effect. Accordingly, we sought to identify potential interacting partners of FAM3C by employing the GPS-Prot bioinformatics database (GPS-Prot: Data Visualization for Protein-Protein Interactions (gpsprot.org)) (Figure S10). The Ras-related proto-oncogene A (RalA), a GTPase normally involved in regulating cell motility, vesicular trafficking, and endo/exocytosis [30-39], was identified as a plausible binding partner of FAM3C. Activation of RalA through phosphorylation at the C-terminal Ser194 [35] residue is integral in Ras-mediated tumorigenesis, invasion, and metastasis [38, 39]. We confirmed that these cell-biological effects are applicable to lung carcinoma, as overexpressing RalA in H2170 lung carcinoma cells increased Src and Stat3 phosphorylation (Figure S7A). In addition, RalA-overexpressing cells (RalA_ox) phenocopied those of FAM3C_ox with enhancement in their migration abilities, invasiveness, and in vitro malignant transformation (Figure S7B-E).
The notion that FAM3C is a novel bona fide binding partner of RalA was supported by multiple converging lines of evidence: First, we observed in immunoblot assays that FAM3C expression correlated with levels of total and especially phosphorylated RalA (Ser194) across an array of lung carcinoma cell lines (Figure 6A, left). This was further corroborated by the reciprocal observations that H2170 FAM3C_ox cells expressed higher p-RalA (Figure 6B), whereas FAM3C silencing in four FAM3C highly expressed lung cancer cell lines ablated expression of RalA phosphorylation (Figure 6A, right). Second, protein-protein interaction between FAM3C and RalA was confirmed by biochemical co-immunoprecipitation (Co-IP) assays through the overexpression of HA-tagged RalA and FLAG-tagged FAM3C either alone or simultaneously in HEK293T cells. As shown in Figure 6C (top panel), both HA and RalA were detected in the FLAG-FAM3C and HA-RalA co-transfected complex by immunoblot analysis after immunoprecipitation using anti-FLAG M2 magnetic beads. To dispel the notion that FAM3C interacts with RalA only at artificially high levels of protein expression, we repeated the experiment with FAM3C-high SKMES-1 lung carcinoma cells and confirmed the endogenous interaction of RalA and FAM3C (Figure 6C, bottom panel). Third, to further reinforce the findings from Co-IP assays, we also utilized an in situ Duolink proximity ligation assay (PLA) and confirmed the proximal co-localization of FAM3C with RalA in A549 parental and FAM3C_ox lung carcinoma cells (Figure 6D-E). In contrast, there was reduced PLA signals in the FAM3C_kd counterpart, suggesting a clear association between FAM3C and RalA. Collectively, the compatible findings provide consistent evidence that RalA is a functional interacting partner of oncogenic FAM3C.
RalA is an interacting partner of FAM3C. (A) Differential expression of FAM3C and RalA in normal fibroblast IMR90 and NSCLC cell lines by Western blot (left panel). Western blot analysis of phosphorylated and total RalA expression in control and siRNA FAM3C knockdown NSCLC cells including HCC827, H1975, SKMES1 and Calu-1 (right panel). (B) Western blot analysis of phosphorylated and total RalA expression in control and FAM3C overexpressed H2170 cells. (C) Co-Immunoprecipitation (Co-IP) assay showing interaction of FAM3C and RalA. HEK293T cells were transfected with FLAG-tagged FAM3C and HA-tagged RalA plasmids. FLAG-FAM3C complex were pulled down by anti-FLAG beads and immunoblot with FAM3C and RalA antibodies (top panel). Immunoprecipitation of endogenous FAM3C protein was performed using anti-FAM3C antibody in SKMES-1 cells and immunoblot with FAM3C and RalA antibodies (bottom panel). (D) Microscopic images of FAM3C/ RalA staining by Duolink proximity ligation assay (PLA) in A549 cells. Red signals indicating proximity of FAM3C and RalA. DAPI was used to counterstain nucleus. (E) The PLA signals were quantified and expressed as number of signals per cell ± SEM (n > 3). (F) Effect of the RalA inhibitor BQU57 on protein expression of FAM3C and the phosphorylation of RalA and Stat3 in SKMES-1 FAM3C-knockdown cells. (G, H) Representative images are shown. Scale bar: 200 μM. Effect of BQU57 treatment on the anchorage-independent growth and invasion of SKMES-1 cells. SKMES-1 parental and FAM3C-knockdown cells were seeded in Matrigel invasion chambers (G) and soft agar (H) containing various concentrations of BQU57 drug with their corresponding bar graphs enumerating on the right panels. The bar graphs showed normalized data of BQU57 treatment to their corresponding controls. All data represented mean as ± SEM, ns: not significant, * p < 0.5, p < 0.01 and *** p < 0.001.
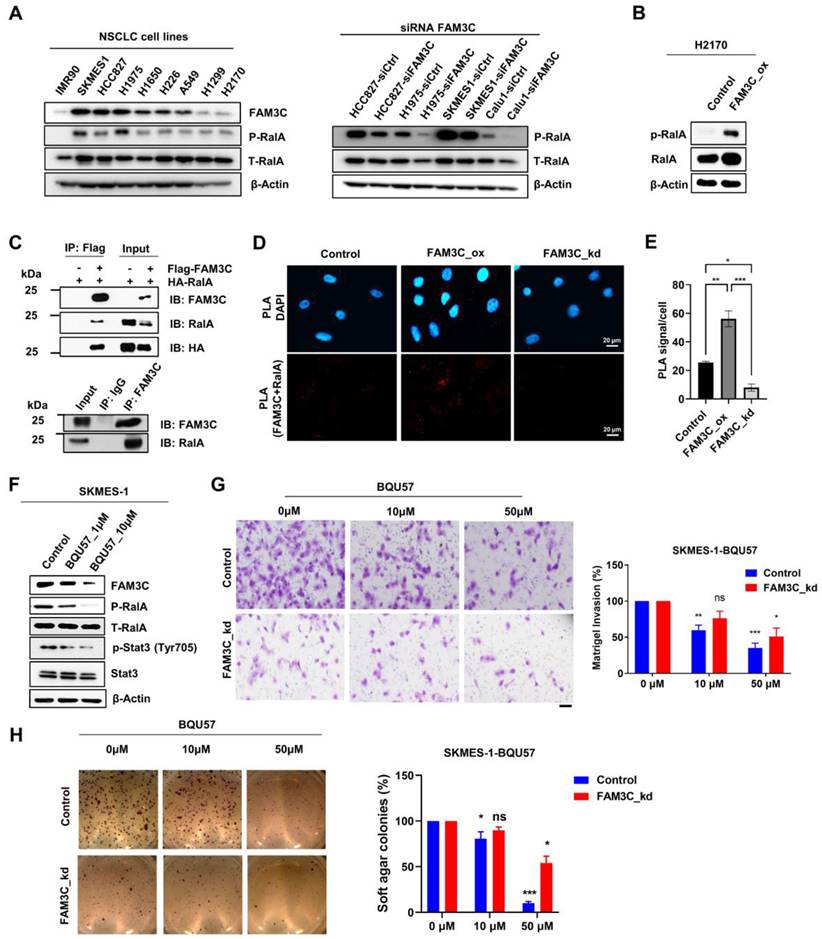
We next explored strategies in targeting FAM3C-driven cancers. While there is no specific antagonist for FAM3C, we proposed to exploit RalA inhibition. By generating stable RalA-silenced A549 cells (Figure S8A), we showed that reduced expression of RalA could suppressed the oncogenicity associated with FAM3C (Figure S8B-D). Interestingly, the effect of recombinant FAM3C on cell migration and invasion was suppressed in shRalA cell line (Figure S8C-D), thereby confirming the dependency of FAM3C on RalA. Accordingly, we wondered if RalA inhibition may exert anti-tumor effect in FAM3C-driven cells by testing the efficacy of BQU57, a covalent small molecule inhibitor of RalA [32, 40]. Immunoblotting analysis demonstrated that BQU57 dose-dependently reduced the expressions of both p-RalA and FAM3C with concomitant suppression of Stat3 phosphorylation, which is downstream effector of FAM3C (Figure 6F). Functionally, BQU57 exhibited dose-dependent inhibitory effects on the oncogenicity in SKMES-1 control cells, as demonstrated by the reduced cellular invasiveness (Figure 6G) and anchorage-independent growth (Figure 6H).
Discussion
As a secreted protein, FAM3C is released into the extracellular milieu through unconventional mechanisms of protein secretion such as autophagy-dependent secretion [41] and plasminogen-urokinase plasminogen activator receptor-regulated secretion [42]. Notwithstanding, the precise cell-biological and molecular crosstalk through which FAM3C acts has remained poorly defined. As FAM3C was found to exhibit an unexpected β-β-α fold [43] and also undergo covalent dimerization [44], it is believed to represent a structurally distinct and previously-unknown class of signaling molecules that is evolutionarily-unrelated [23] to classical four-helix cytokines. This motivated us to elucidate its role as a functional cargo molecule of carcinoma EVs. Several pertinent EVs mediated pathways have already been established in the metastatic cascade. Our current work adds to this metastatic paradigm in NSCLC, demonstrating that FAM3C carried in EVs of NSCLC overexpressing the protein promulgates distant colony formation at least in the lungs.
High expression of FAM3C is associated with poorer survival outcomes in multiple malignancies. This is explained by its role in promoting metastases; in pancreatic carcinoma for example, FAM3C is associated with hepatic metastases [17]. Recently, its overexpression is reported to be regulated by copy number gains in carcinomas [25]. Here, we present several lines of evidence to support the notion that FAM3C is prominently involved in colonization of lung carcinoma. First, comprehensive analyses on TCGA pan-cancer transcriptome revealed the high expression of FAM3C both in lung adenocarcinoma and squamous cell carcinoma. Second, distribution of FAM3C transcript is heterogenous across various cell types [26], and significant enrichments of FAM3C-positive populations are observed in lymph node and brain metastases of lung carcinoma patients. Third, protein-based analyses on tumor specimens and circulating EVs isolated from lung carcinoma donors detected an increasing level of FAM3C that is specifically associated with lung carcinoma progression. Fourth, we have demonstrated that delivery of FAM3C in lung carcinoma cells - either by ectopic gene expression or direct EVs uptake - significantly induced in vitro cell motility and in vivo lung colonization. Given these distinct lines of evidences, aberrant FAM3C expression is likely to represent a prognostic biomarker for predicting the risk of developing metastatic disease, and opens an opportunity to investigate RalA inhibitors as potential targeted therapies.
FAM3C appears to be overexpressed across NSCLC and is not associated with common driver oncogenes in lung carcinomas such as EGFR mutations. It is significant that FAM3C is present in both the primary tumor as well as the secretome as part of the circulatory EVs. This suggests that NSCLC utilizes vesicular FAM3C to prepare secondary or distant tissues for colonization. In this context, FAM3C overexpression was associated with expression of ITGB3, a known cell-cell and cell matrix adhesion molecule, raising the possibility that vesicular FAM3C may induce ITGB3 expression to promote uptake into recipient cells, thus forming the foundation for the establishment of distant colonies. Our tail vein mouse models provide strong evidence for this with development of higher density of lung colonization in mice primed with TDEs enriched with FAM3C. These experiments support the oncogenic role of FAM3C not only through mediating intracellular signaling events, but also in disseminating this stimulatory signal to distantcarcinoma cells through the vesicular cell communication circuit, which culminates in colony formation in distant tissues (Figure 7).
Schematic diagram of the potential role of EVs-derived FAM3C in lung tumor invasion and distant colonization. EVs-derived FAM3C activates the phosphorylation of RalA (Ser194) in lung carcinoma cells. RalA GTPase and subsequently activates the phosphorylation of SRC and STAT3 (Tyr705) in the downstream signaling pathway. This oncogenic activity induces cellular transformation and distant colonization in the recipient cells.
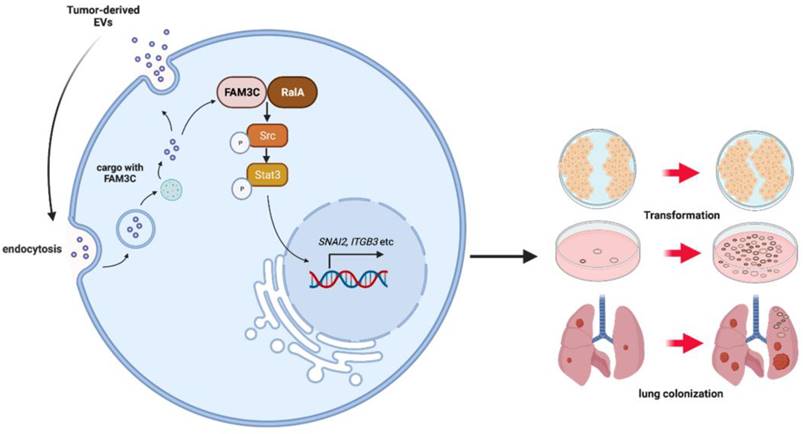
It is pertinent to unravel the cell signaling mechanisms contributory to this invasiveness phenotype since FAM3C has physiological functions in lipid and glucose metabolism [45], osteogenic differentiation [46], retinal laminar formation [47] and brain amyloid peptide formation [48]. Consequently, direct inhibition of FAM3C may result in undesirable off-target effects. Though FAM3C can mediate carcinoma phenotype through various pathways including RAS, Jak/Stat3, and TGFβ, our work gives credence to its stimulation of migration and invasiveness through association with RalA, a Ras GTPase that is activated in many carcinomas, through downstream signaling pathways that include Src, JNK, NF-kB, cyclin D and Aurora A that may lead to cellular transformation. Despite so, it remains uncertain how RalA signaling is activated in carcinomas apart from its canonical upstream RalGEF effectors. As RalA co-precipitates with FAM3C, levels of both proteins are concordant, RalA silencing reduces FAM3C levels and abrogates FAM3C mediated cell migration, RalA signaling via direct interaction with FAM3C is likely, making RalA inhibition a strong candidate for disruption of TDE carried FAM3C effects.
The TGF-β signaling plays a prominent role in EMT and metastasis [49]. While FAM3C acts as a downstream effector of TGFβ receptor [23, 24], it is notable that aberrant FAM3C expression is able to induce mesenchymal phenotype independently of TGF-β through the activation of MAPK/Erk pathway [22, 23]. Given that the lack of correlation between FAM3C expression and its known receptor LIFR in lung cancer, it signifies the need to elucidate the underlying mechanisms of its signal transduction. As depicted in our model, cells incubated with TDEs isolated from FAM3C-overexpresssed cells are found to transduce pro-invasive cues, likely via a novel RalA-FAM3C axis that engender the downstream activation of MAPK and Src pathways. These observations recapitulate previous observations on the pro-metastatic cues of secreted FAM3C [50], and raise the possibility that EVs secreted from primary tumors are essential to the tumor-microenvironment interaction. RalA GTPase functions downstream of RAS as one of its oncogenic pathways and RalA is active in the GTP bound form. Recently, inhibitors of RalA have been developed including BQU57 that binds preferentially to the allosteric site of inactive RalA-GDP, preventing it from being activated and bind to its downstream signaling partner RalBP1. This inhibition of FAM3C by inhibition of RalA may avoid interrupting its physiological function and yet disrupting its gain of function oncogenicity.
While our in vitro findings show that uptake of FAM3C through EVs could regulate tumor localization, the mechanism of EV transmission remains unclear. The upregulation of ITGB3 transcript in FAM3C overexpressed cells suggests a direct impact on endocytosis. In addition, RalA regulates EVs processing through medication by exocyst complex proteins SEC5 and EXO84. In this context, inhibition of RalA would impair EVs trafficking and release, potentially impacting on vesicular FAM3C-mediated carcinoma cell invasiveness. Yet, this hypothesis is unexplored due to the limitation of our animal models that restricts the examination of intercellular transfer of FAM3C. As a follow up study, we intend to label TDEs with carboxyfluorescein succinimidyl ester (CFSE) for live tracking of TDE uptake within the tumor compartment. Further studies are warranted to explore the underlying mechanisms of RalA-FAM3C interaction, binding sites and protein stability, etc.
In summary, our current study describes a novel mechanism for FAM3C-driven carcinogenesis which is not relevant to expression level of LIFR. Though our study was pertinent in lung carcinoma, it may be relevant for other malignancies that highly express FAM3C, especially in EVs of cancer patients. It would be important to extend the investigation into these carcinomas to determine the role of FAM3C. Stratification of patients based on high FAM3C expression in plasma EVs may pave the path for developing strategies against widespread tumor colonization.
Supplementary Material
Supplementary methods, figures and tables.
Acknowledgements
We thank Dr Hae-Ock Lee from Catholic University of Korea for the usage of the scRNA-Seq analysis platform (http://ureca-singlecell.kr).
Funding
This research is supported by the National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centres of Excellence initiative. This work was also funded by the Singapore Ministry of Health's National Medical Research Council (NMRC/CSA-SI/0006/2016), the National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centres of Excellence initiatives (Boon-Cher Goh); the Singapore Ministry of Health's National Medical Research Council (NMRC/CNIG/1146/2016, Lingzhi Wang), NCIS-N2CR Seed Grant (Boon-Cher Goh, Lingzhi Wang) and the Singapore Ministry of Health's National Medical Research Council under its NCIS Centre Grant (NMRC/CG/012/2013, Boon-Cher Goh, Li-Ren Kong).
Author contributions
Project conception: B.C.G., L.W., H.Y., R.A.S., P.C.H., T.G. and S.K.S.; Experimental design: W.L.T., N.S, L.R.K, L.D., and E.S.C.; Experimental work: W.L.T., R.T.W., T.W., H.S., Y.C.L. and H.T.H.; Data analysis: H.Y., W.L.T., N.S, L.R.K, Y.C.L., R.M.W.G., and L.D; Writing original draft: W.L.T., N.S, L.R.K, L.W., and B.C.G; Writing review and editing, L.R.K, J.C.H., W.P.Y., S.C.L, A.L.W., M.T.L., G.S., R.M.W.G., P.C.H., M.K.J. and J.T.; All authors reviewed the manuscript.
Availability of data and material
FAM3C expression elevated in malignant lung tissues is from The Cancer Genome Atlas (TCGA) database. RNAseq sequencing data has been deposited onto public database.
Ethics approval and consent to participate
The animal experiments were approved by ethics board from the SingHealth Institutional Animal Care and Use Committee (IACUC, 2019/SHS/1458).
Consent for publication
This original work has not been submitted to other journals for consideration, and all authors have approved the manuscript and agree with its submission to Theranostics.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Turajlic S, Swanton C. Metastasis as an evolutionary process. Science. 2016;352(6282):169-75
2. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871-90
3. Talmadge JE, Fidler IJ. AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res. 2010;70(14):5649-69
4. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331(6024):1559-64
5. Bhang HE, Ruddy DA, Krishnamurthy Radhakrishna V, Caushi JX, Zhao R, Hims MM. et al. Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nat Med. 2015;21(5):440-8
6. Yuan M, Huang LL, Chen JH, Wu J, Xu Q. The emerging treatment landscape of targeted therapy in non-small-cell lung cancer. Signal Transduct Target Ther. 2019;4:61
7. van Niel G, D'Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19(4):213-228
8. Kalluri R, LeBleu VS. The biology. Science. 2020 367(6478)
9. Moon PG, Lee JE, Cho YE, Lee SJ, Jung JH, Chae YS. et al. Identification of Developmental Endothelial Locus-1 on Circulating Extracellular Vesicles as a Novel Biomarker for Early Breast Cancer Detection. Clin Cancer Res. 2016;22(7):1757-66
10. Melo SA, Luecke LB, Kahlert C, Fernandez AF, Gammon ST, Kaye J. et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015;523(7559):177-82
11. Chan JC, Morgan CP, Adrian Leu N, Shetty A, Cisse YM, Nugent BM. et al. Reproductive tract extracellular vesicles are sufficient to transmit intergenerational stress and program neurodevelopment. Nat Commun. 2020;11(1):1499
12. Toda H, Diaz-Varela M, Segui-Barber J, Roobsoong W, Baro B, Garcia-Silva S. et al. Plasma-derived extracellular vesicles from Plasmodium vivax patients signal spleen fibroblasts via NF-kB facilitating parasite cytoadherence. Nat Commun. 2020;11(1):2761
13. Yang E, Wang X, Gong Z, Yu M, Wu H, Zhang D. Exosome-mediated metabolic reprogramming: the emerging role in tumor microenvironment remodeling and its influence on cancer progression. Signal Transduct Target Ther. 2020;5(1):242
14. Jiang K, Dong C, Yin Z, Li R, Mao J, Wang C. et al. Exosome-derived ENO1 regulates integrin alpha6beta4 expression and promotes hepatocellular carcinoma growth and metastasis. Cell Death Dis. 2020;11(11):972
15. Xu J, Ji L, Liang Y, Wan Z, Zheng W, Song X. et al. CircRNA-SORE mediates sorafenib resistance in hepatocellular carcinoma by stabilizing YBX1. Signal Transduct Target Ther. 2020;5(1):298
16. Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G. et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18(6):883-91
17. Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK. et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015;17(6):816-26
18. Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M. et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527(7578):329-35
19. Fuentes P, Sesé M, Guijarro PJ, Emperador M, Sánchez-Redondo S, Peinado H. et al. ITGB3-mediated uptake of small extracellular vesicles facilitates intercellular communication in breast cancer cells. Nat Commun. 2020;11(1):4261
20. Syn N, Wang L, Sethi G, Thiery JP, Goh BC. Exosome-Mediated Metastasis: From Epithelial-Mesenchymal Transition to Escape from Immunosurveillance. Trends Pharmacol Sci. 2016;37(7):606-617
21. Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020 367(6478)
22. Waerner T, Alacakaptan M, Tamir I, Oberauer R, Gal A, Brabletz T. et al. ILEI: a cytokine essential for EMT, tumor formation, and late events in metastasis in epithelial cells. Cancer Cell. 2006;10(3):227-39
23. Jansson AM, Csiszar A, Maier J, Nystrom AC, Ax E, Johansson P. et al. The interleukin-like epithelial-mesenchymal transition inducer ILEI exhibits a non-interleukin-like fold and is active as a domain-swapped dimer. J Biol Chem. 2017;292(37):15501-15511
24. Yang W, Feng B, Meng Y, Wang J, Geng B, Cui Q. et al. FAM3C-YY1 axis is essential for TGFβ-promoted proliferation and migration of human breast cancer MDA-MB-231 cells via the activation of HSF1. J Cell Mol Med. 2019;23(5):3464-3475
25. Schmidt U, Heller G, Timelthaler G, Heffeter P, Somodi Z, Schweifer N. et al. The FAM3C locus that encodes interleukin-like EMT inducer (ILEI) is frequently co-amplified in MET-amplified cancers and contributes to invasiveness. J Exp Clin Cancer Res. 2021;40(1):69
26. Kim N, Kim HK, Lee K, Hong Y, Cho JH, Choi JW. et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat Commun. 2020;11(1):2285
27. Shi M, Duan G, Nie S, Shen S, Zou X. Elevated FAM3C promotes cell epithelial- mesenchymal transition and cell migration in gastric cancer. Onco Targets Ther. 2018;11:8491-8505
28. Jiang K, Dong C, Yin Z, Li R, Mao J, Wang C. et al. Exosome-derived ENO1 regulates integrin α6β4 expression and promotes hepatocellular carcinoma growth and metastasis. Cell Death Dis. 2020;11(11):972
29. Woosley AN, Dalton AC, Hussey GS, Howley BV, Mohanty BK, Grelet S. et al. TGFbeta promotes breast cancer stem cell self-renewal through an ILEI/LIFR signaling axis. Oncogene. 2019;38(20):3794-3811
30. Neel NF, Martin TD, Stratford JK, Zand TP, Reiner DJ, Der CJ. The RalGEF-Ral Effector Signaling Network: The Road Less Traveled for Anti-Ras Drug Discovery. Genes Cancer. 2011;2(3):275-87
31. Martin TD, Mitin N, Cox AD, Yeh JJ, Der CJ. Phosphorylation by protein kinase Calpha regulates RalB small GTPase protein activation, subcellular localization, and effector utilization. J Biol Chem. 2012;287(18):14827-36
32. Falsetti SC, Wang DA, Peng H, Carrico D, Cox AD, Der CJ. et al. Geranylgeranyltransferase I inhibitors target RalB to inhibit anchorage-dependent growth and induce apoptosis and RalA to inhibit anchorage-independent growth. Mol Cell Biol. 2007;27(22):8003-14
33. Kinsella BT, Erdman RA, Maltese WA. Carboxyl-terminal isoprenylation of ras-related GTP-binding proteins encoded by rac1, rac2, and ralA. J Biol Chem. 1991;266(15):9786-94
34. Shipitsin M, Feig LA. RalA but not RalB enhances polarized delivery of membrane proteins to the basolateral surface of epithelial cells. Mol Cell Biol. 2004;24(13):5746-56
35. Lim KH, Brady DC, Kashatus DF, Ancrile BB, Der CJ, Cox AD. et al. Aurora-A phosphorylates, activates, and relocalizes the small GTPase RalA. Mol Cell Biol. 2010;30(2):508-23
36. Lim KH, O'Hayer K, Adam SJ, Kendall SD, Campbell PM, Der CJ. et al. Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells. Curr Biol. 2006;16(24):2385-94
37. Chien Y, White MA. RAL GTPases are linchpin modulators of human tumour-cell proliferation and survival. EMBO Rep. 2003;4(8):p. 800-6
38. Lim KH, Baines AT, Fiordalisi JJ, Shipitsin M, Feig LA, Cox AD. et al. Activation of RalA is critical for Ras-induced tumorigenesis of human cells. Cancer Cell. 2005;7(6):533-45
39. Bodemann BO, White MA. Ral GTPases and cancer: linchpin support of the tumorigenic platform. Nat Rev Cancer. 2008;8(2):133-40
40. Yan C, Liu D, Li L, Wempe MF, Guin S, Khanna M. et al. Discovery and characterization of small molecules that target the GTPase Ral. Nature. 2014;515(7527):443-7
41. Kraya AA, Piao S, Xu X, Zhang G, Herlyn M, Gimotty P. et al. Identification of secreted proteins that reflect autophagy dynamics within tumor cells. Autophagy. 2015;11(1):60-74
42. Csiszar A, Kutay B, Wirth S, Schmidt U, Macho-Maschler S, Schreiber M. et al. Interleukin-like epithelial-to-mesenchymal transition inducer activity is controlled by proteolytic processing and plasminogen-urokinase plasminogen activator receptor system-regulated secretion during breast cancer progression. Breast Cancer Res. 2014;16(5):433
43. Johansson P, Bernstrom J, Gorman T, Oster L, Backstrom S, Schweikart F. et al. FAM3B PANDER and FAM3C ILEI represent a distinct class of signaling molecules with a non-cytokine-like fold. Structure. 2013;21(2):306-13
44. Kral M, Klimek C, Kutay B, Timelthaler G, Lendl T, Neuditschko B. et al. Covalent dimerization of interleukin-like epithelial-to-mesenchymal transition (EMT) inducer (ILEI) facilitates EMT, invasion, and late aspects of metastasis. FEBS J. 2017;284(20):3484-3505
45. Chen Z, Ding L, Yang W, Wang J, Chen L, Chang Y. et al. Hepatic Activation of the FAM3C-HSF1-CaM Pathway Attenuates Hyperglycemia of Obese Diabetic Mice. Diabetes. 2017;66(5):1185-1197
46. Bendre A, Buki KG, Maatta JA. Fam3c modulates osteogenic differentiation by down-regulating Runx2. Differentiation. 2017;93:50-57
47. Katahira T, Nakagiri S, Terada K, Furukawa T. Secreted factor FAM3C (ILEI) is involved in retinal laminar formation. Biochem Biophys Res Commun. 2010;392(3):301-6
48. Hasegawa H, Liu L, Tooyama I, Murayama S, Nishimura M. The FAM3 superfamily member ILEI ameliorates Alzheimer's disease-like pathology by destabilizing the penultimate amyloid-beta precursor. Nat Commun. 2014;5:3917
49. Padua D, Massague J. Roles of TGFbeta in metastasis. Cell Res. 2009;19(1):89-102
50. Halberg N, Sengelaub CA, Navrazhina K, Molina H, Uryu K, Tavazoie SF. PITPNC1 Recruits RAB1B to the Golgi Network to Drive Malignant Secretion. Cancer Cell. 2016;29(3):339-353
Author contact
![]() Corresponding author: Boon-Cher Goh (phcgbcedu.sg); Lingzhi Wang (csiwledu.sg).
Corresponding author: Boon-Cher Goh (phcgbcedu.sg); Lingzhi Wang (csiwledu.sg).