Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results and Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2022; 12(8):3747-3757. doi:10.7150/thno.72559 This issue Cite
Research Paper
Calixarene-modified albumin for stoichiometric delivery of multiple drugs in combination-chemotherapy
Ying Wang1,2#, Zhanzhan Zhang1,2#, Xinzhi Zhao1,2, Lina Xu1,2, Yadan Zheng1,2,4, Hua-Bin Li1,3, Dong-Sheng Guo1,3, Linqi Shi1,2, Yang Liu1,2 ![]()
1. Key Laboratory of Functional Polymer Materials of Ministry of Education, College of Chemistry, Nankai University, Tianjin 300071, China.
2. State Key Laboratory of Medicinal Chemical Biology, Nankai University, Tianjin 300071, China.
3. State Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, China.
4. College of Veterinary Medicine, Northeast Agricultural University, Harbin 150030, China.
#These authors contributed equally to this work.
Received 2022-3-3; Accepted 2022-4-15; Published 2022-5-1
Abstract

Rationale: In combination chemotherapy, the molar ratio of drugs is a critical parameter that determines the synergistic effects. However, most co-delivery vectors are incapable of maintaining the optimal molar ratio of drugs throughout the delivery process. Herein, a calixarene-modified albumin (CaMA), which can co-deliver multiple drugs with precise control of the drug ratio, is presented.
Methods: CaMA was prepared by chemically conjugating multiple sulfonate azocalix[4]arenes (SAC4A) onto the surface of bovine serum albumin (BSA). The precise drug loading and synchronous drug release were measured using fluorescence spectroscopy. Mouse tumor cell 4T1 and 4T1-bearing mice were used to evaluate the combined effects of mitomycin C (MMC) and doxorubicin (DOX) in vitro and in vivo.
Results: With multiple hypoxia-responsive calixarenes conjugated onto a single albumin molecule, CaMA achieved precise drug loading and synchronous release of multiple drugs into the tumor microenvironment. This unique drug loading and release mechanism ensures that CaMA maintains the drug ratio from the initial drug loading to the release site, providing a solid foundation for multi-drug combination therapy with the goal of achieving predictable therapeutic outcomes in vivo. The delivery of the model drug combination MMC and DOX at a prescreened ratio via CaMA achieved significantly enhanced tumor suppression and reduced systemic toxicity.
Conclusions: This stoichiometric delivery feature makes CaMA a powerful tool for the development of combination chemotherapy and personalized medications for cancer treatment.
Keywords: combination chemotherapy, co-delivery, hypoxia-responsive, host-guest interaction, stoichiometric
Introduction
Combination drug therapy is a common practice in cancer treatment to improve therapeutic outcomes by exploiting the different toxicities of multiple drugs [1-7]. In this form of therapy, the molar ratio of the combined drugs is a critical parameter that determines the synergistic effects [8-12]. However, the optimal molar ratio of drugs identified in vitro is difficult to maintain after delivery to the targets in vivo due to the differences in drug biodistribution and pharmacokinetics (PK) [13], which impairs the synergistic effects of the drug combination and hinders its clinical application. To date, several nano-drug delivery systems (nanoDDS), such as polymer micelles [14-16] and liposomes [17], have been investigated for use in the co-delivery of multiple drugs [18-25]. Although these strategies ensure a consistent biodistribution and PK of the loaded drugs [8, 11, 26, 27], precise control of the drug ratio delivered to tumors remains challenging due to the lack of an accurate loading strategy for multiple drugs (physical absorption/embedment is still the major method for drug loading in most co-delivery nanoDDS), as well as the potential for leakage during transportation [23]. Moreover, imprecise loading and drug leakage may lead to “batch-to-batch” variations in preparation and individual differences in drug delivery, resulting in an unpredictable therapeutic efficacy in cancer treatment [28, 29]. To address this issue, covalent conjugation strategies (i.e., drug-to-carrier [13, 30] or drug-to-drug [11, 12, 31, 32]) via stimuli-responsive bonds have been developed [24, 30, 33]. However, the specific groups required for chemical conjugation are not always available for many chemotherapeutic drugs, and tedious synthesis and purification processes limited the fine-tuning of the drug ratio [34]. Therefore, a co-delivery vector that allows for: i) precise loading of multiple drugs, ii) easy tuning of the molar ratio of loaded drugs, and iii) stoichiometric co-delivery of drug combinations to the tumor is in urgent demand for efficient combination chemotherapy.
In the past few decades, macrocyclic molecules (i.e., pillararenes [35-38], cucurbiturils [39, 40], calixarenes [41, 42] and cyclodextrins [43]) have been extensively investigated in drugs delivery to enhance stability, improve solubility, and reduce the side effects of drug [44-47]. These macrocyclic hosts complex with drug molecules through host-guest interactions with a characteristic binding affinity and defined stoichiometry (mostly 1:1) [48, 49]. This unique drug loading mechanism allows for the prediction of the amount of drug loaded into the macrocyclic hosts according to their initial concentrations and characteristic binding affinities, providing a solid foundation for the precise control of drug loading [28, 41]. However, the 1:1 stoichiometry between macrocyclic and drug molecules restricts the simultaneous loading and co-delivery of multi-drug [28]. Therefore, for an efficient macrocyclic-based delivery system for combination chemotherapy, it is essential to develop innovative strategies that can (i) overcome the 1:1 stoichiometric limitation and achieve precise loading of multiple drugs, (ii) effectively target tumor tissues, and (iii) maintain the drug ratio after delivery for optimal synergistic effects of the drug combination.
Herein, we developed a calixarene-modified albumin (CaMA) as a stoichiometric co-delivery system for the simultaneous delivery of multiple drugs with a precise control of the drug ratio. As illustrated in Scheme 1, CaMA is prepared by chemically conjugating multiple sulfonate azocalix[4]arenes (SAC4A) onto the surface of bovine serum albumin (BSA). BSA is employed as the structural basis of CaMA due to its nanoscale particle size, stable physicochemical structure, high biocompatibility, and wide availability. SAC4A, which is a stimuli-responsive calixarene that degrades in the hypoxic tumor microenvironment (TME), serves as the functional unit of CaMA to load and control the release of drug molecules. With multiple SAC4As on the surface, CaMA can load multiple drug molecules within one nanostructure. More importantly, more than one type of drug molecule can be loaded into CaMA, and the drug ratio is able to precisely control by adjusting the initial drug concentrations calculated from their characteristic binding affinities. In blood circulation or normal tissue, the strong binding affinity between CaMA and the loaded drugs prevents the drugs from undesired leakage. Upon reaching the TME, the binding affinity decreases as SAC4A degrades, resulting in the rapid and simultaneous release of all drugs. All these characteristics of CaMA, including predictable drug loading ratio, minimal drug leakage during the transportation, enhanced tumor accumulation and simultaneous release of all drugs in response to TME, ensure the stoichiometric delivery of multiple drugs to in vivo target. In this study, doxorubicin (DOX) and mitomycin C (MMC), which are commonly used in clinical chemotherapy, were chosen as the model drug combination. The ratio of DOX to MMC to achieve optimal anti-cancer efficacy was first screened in vitro. By loading DOX and MMC in the pre-screened ratio, CaMA achieved stoichiometric delivery of the drug combination to the tumors, resulting in significantly enhanced tumor suppression and reduced toxicity compared with conventional cocktail therapy. This stoichiometric delivery ability allows CaMA to rapidly translate optimal drug combinations prescreened in vitro into in vivo therapeutic benefits, providing a powerful tool for the development of combination chemotherapies and personalized medications for cancer treatments.
Illustration of CaMA for combination chemotherapy. (A) Illustration of CaMA for stoichiometric co-delivery of drug combinations. (B) Mechanism of SAC4A degradation under hypoxic condition.
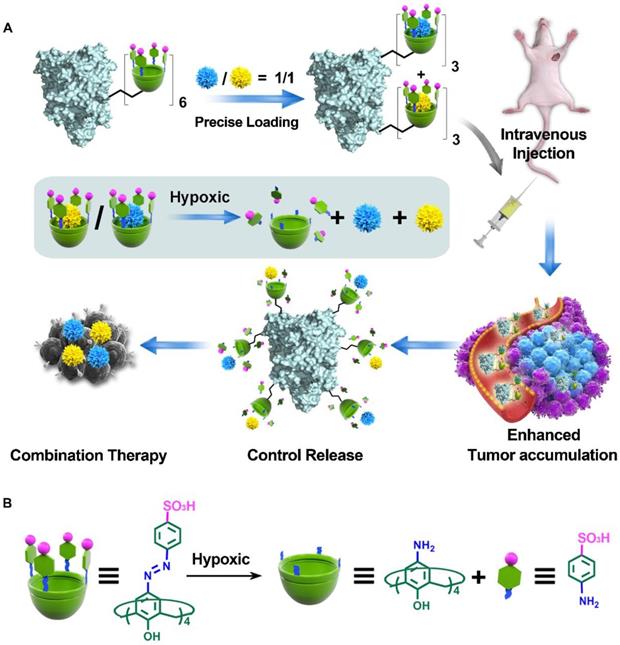
Methods
Preparation of CaMA
For the synthesis of CaMA, SAC4A was first reacted with 1-bromo-2,3-epoxypropane to obtain SAC4A-epoxy. Briefly, 400 mg Na2CO3 (100 mg/mL) and 1-bromo-2,3-epoxypropane (1 g, 7.3 mmol) were added in sequence to a solution of 35 mg SAC4A (28 μmol) in DMF (4 mL) and stirred for 24 h at room temperature. After this reaction, the insoluble Na2CO3 was first removed by centrifugation and unconjugated 1-bromo-2,3-epoxypropane was then removed by precipitation in a large quantity of diethyl ether, following by collecting the precipitation and drying it in vacuo to obtain the SAC4A-epoxy. CaMA was then prepared by conjugating the SAC4A-epoxy onto BSA via a reaction between the amino groups of BSA and the epoxy groups of SAC4A-epoxy. Briefly, 20 mg BSA (0.3 μmol) and 3.75 mg SAC4A-epoxy (3 μmol) were added to 5 mL Na2CO3 buffer (100 mM) and stirred for 24 h at room temperature. The product was isolated by dialyzing against water (MWCO 10000), followed by ultrafiltration (MWCO 30000) and desalination with desalting columns (MWCO 7000). The successful preparation of CaMA was confirmed by MALDI-TOF.
Drug release under cellular hypoxic condition
The hypoxia-triggered drug release behavior of CaMA was observed by CLSM. Briefly, 4T1 cells were first seeded overnight in confocal imaging chambers (1 × 105 cells/well) before treating with CaMA-DOX (8/8 μM). After 6 h's incubation, the culture medium was replaced with fresh medium, and the cells were incubated for another 18 h under hypoxic or normoxic condition, respectively. The cells were then rinsed and stained with DAPI for CLSM image.
Precise drugs loading capability of CaMA
To investigate the capability of CaMA in precisely loading of the drugs, CPT and DOX were employed as the model drugs. The concentration of CaMA (determined by SAC4A) was fixed at 180 μM, the total concentration of CPT and DOX was fixed at 150 μM, and the feeding concentrations of CPT and DOX were calculated according to the expected molar ratio and Ka. Specifically, the concentrations of DOX and CPT were prepared according to the following ratio: 1 : 5 = 21.5 μM : 125 μM, 1 : 2 = 42.5 μM : 100 μM, 1 : 1 = 75 μM : 88.24 μM, 2 : 1 = 100 μM : 58.82 μM and 5 : 1 = 125 μM : 39.41 μM. All mixtures of DOX, CPT and CaMA were shaken for 30 min at room temperature, followed by removing unloaded drugs by ultrafiltration centrifugation (MWCO = 3 kDa). Unloaded DOX and CPT in the filtrate were measured with fluorescence spectrometer at λex = 497 nm and λex = 365 nm.
Synchronous release of loading drugs
To investigate the synchronous release capabilities of CaMA, DOX and CPT were loaded into CaMA at molar ratios ranging from 0.2:1 to 2:1 to form CaMA-DC (0.2:1), CaMA-DC (0.5:1), CaMA-DC (1:1), CaMA-DC (2:1). Specifically, the concentration of CaMA was fixed at 3.2 μM and the total concentration of DOX and CPT was fixed at 3.0 μM, therefore, CaMA-DC (0.2:1) = 0.5 μM : 2.5 μM, CaMA-DC (0.5:1) = 1.0 μM : 2.0 μM, CaMA-DC (1:1) = 1.5 μM : 1.5 μM, CaMA-DC (2:1) = 2.0 μM : 1.0 μM. CaMA-DC at different ratios were then incubated with DT-diaphorase/NADPH and the release of DOX and CPT were continuously monitored during the incubation period.
Combination index analysis of DOX and MMC
A cell viability-based assay was used to determine the combined index (CI) of DOX and MMC. Briefly, different concentrations of DOX and MMC (16, 8, 4, 2, 1, 0.5, 0.25, and 0.125 μM) and combinations of different ratios (4:1, 2:1, 1:1, 1:2, 1:4) were used to treat 4T1 cancer cells, and their cytotoxicity against 4T1 cells was assayed with CCK-8. Afterwards, the CI for DOX and MMC was conducted using CompuSyn software based on the Chou and Talalay method. CI values > 1, = 1, and < 1 indicate antagonistic, additive, and synergistic effects of the drug combination. The best-fit CI values at IC50 were used to show and compare the synergy of drug combination at different ratios.
Anti-tumor efficacy of DOX and MMC
Female BALB/c mice were used to investigate the synergistic anti-tumor effects of MMC and DOX. First, 1×106 4T1 cancer cells were subcutaneously injected into the left mammary fat pad of mice to establish the tumor-bearing mice model. Then the mice were randomly divided into seven groups (n = 6) once the tumor volumes had reached approximately 100 mm3. 200 μL of PBS, CaMA (320 μM), DM (molar ratio 1:1, 150 μM/150 μM), CaMA-DOX (320 μM/300 μM,), CaMA-MMC (320 μM/300 μM), CaMA-DM-med (320 μM/60 μM/240 μM), and CaMA-DM-opt ((320 μM/150 μM/150 μM) were then intravenously injected into the mice every two days for a total of 3 doses. PBS-treated mice were used as negative control. The volume of tumors were consciously monitored for 25 days, and calculated on the basis of the formula: V = (W2 × L)/2, where W represents the shortest dimension and L represents the longest dimension.
Results and Discussion
Structure characterization of CaMA
To prepare CaMA, a hypoxia-responsive macrocyclic host, SAC4A was first synthesized as the functional unit to precisely load and responsively release of drugs [50]. In the design of SAC4A, aminocalix[4]arene (NH2C4A) was used as macrocyclic scaffold. By modifying the upper rim of NH2C4A with 4-sulfobenzenediazonium chloride, SAC4A obtained a cavity to enhance the binding affinity to guests attributed to its deeper cavitand and the formation of salt bridges between the sulfonate and cationic groups of the guest molecules (drugs, probes, etc.). After obtaining SAC4A, CaMA was synthesized by conjugating SAC4A onto BSA. This was achieved by first reacting SAC4A with 1-bromo-2,3-epoxypropane to obtain SAC4A-epoxy, followed by conjugating SAC4A-epoxy onto BSA through a reaction between the amino groups of BSA and the epoxy groups of SAC4A-epoxy (Figure 1A and Figure S1). The successful conjugation of SAC4A onto BSA was confirmed by MALDI-TOF analysis, in which the characteristic peak of CaMA was detected with a molecular weight of 73689.8 Da (Figure 1B). The average conjugated SAC4A molecule per BSA was calculated to be 5.86 according to the MALDI-TOF spectrum. Zeta potential analysis further confirmed the conjugation of SAC4A onto BSA, with the decrease in zeta potential from -15.6 ± 0.21 mV of BSA to -21.2 ± 0.48 mV of CaMA. Dynamic light scattering (DLS) measurements and transmission electron microscopy (TEM) observations indicated that CaMA has a uniform nanomorphology with a diameter of around 6.5 ± 0.78 nm (Figure 1C).
Structural characterization of CaMA. (A) The synthesis of CaMA. (B) MALDI-TOF spectrum of BSA and CaMA. (C) Size distribution and TEM image of CaMA. (D) Fluorescence titration curves of RhB with CaMA and SAC4A fitted according to the 1:1 binding stoichiometry. (E) Fluorescence (FL) recovery ratio of CaMA-SiPcN2 in the presence of biological species in blood, and the fluorescence image of CaMA-SiPcN2 and SiPcN2 in MS (inset).
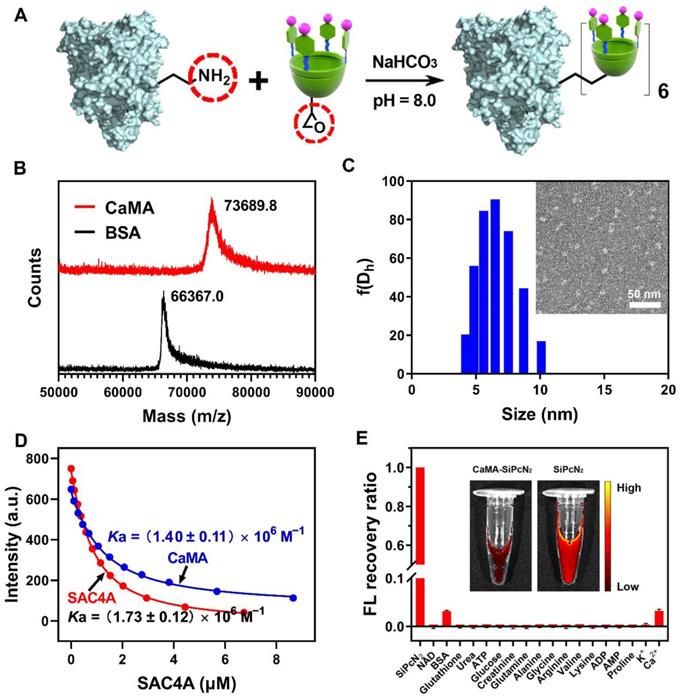
It is necessary for the SAC4A of CaMA to maintain the drug loading capability after chemical conjugation onto BSA for an efficient combination drug therapy. Rhodamine B (RhB), DOX, CPT and MMC were employed as the reporter, and their binding affinity (Ka) to the SAC4A of CaMA and free SAC4A were determined by fluorescence titration. As shown in Table S1, Figure 1D and Figures S2-9, slightly decreased binding affinities to RhB, DOX, CPT and MMC were observed from the SAC4A of CaMA compared to free form of SAC4A, suggesting the uncompromised drug loading capability of the SAC4A after conjugating to BSA and the potential drug loading capability of CaMA. To demonstrate the capability of CaMA as a co-delivery vector for combination drug therapy, common anticancer drugs, including camptothecin (CPT), DOX and MMC were employed and their binding affinities to CaMA were determined. As shown in the results from fluorescence titration (Figures S4, S6 and S8), CaMA exhibited strong binding affinities to CPT ((1.85 ± 0.35) × 105 M-1), DOX ((9.98 ± 0.16) × 106 M-1), and MMC ((8.47 ± 0.71) × 105 M-1) with a 1:1 stoichiometry. This high binding affinity guaranteed the quantitative loading of these drugs. For example, when the concentration of CaMA was 300 μM, the drug loading efficacy of CaMA can be calculated as 98% for DOX, 94% for MMC and 87% for CPT (see Supporting Information for the detailed calculation). Considering that one CaMA possesses multiple SAC4As, the strong binding affinities of these drugs offer CaMA great potential for achieving the precise loading of drug combinations by control the initial drug concentrations calculated from their characteristic binding affinities.
As a non-covalent co-delivery vector, it is essential for CaMA to avoid undesired payload leakage during blood circulation. For the verification, the major species existing in blood were employed to challenging the integrity the CaMA. A fluorescent probe, silicon (IV) phthalocyanine (SiPcN2), was loaded into CaMA to form CaMA-SiPcN2 (Figure S10), then the major species in blood were utilized to challenge the stability of CaMA-SiPcN2. Almost no fluorescence recovery was observed from CaMA-SiPcN2 after incubation with these biological species (Figure 1E), indicating the robustness of CaMA-SiPcN2 under physiological conditions. Furthermore, CaMA-SiPcN2 also maintained high stability in mouse serum (MS), in which negligible fluorescence signals were observed from CaMA-SiPcN2 in the presence of MS (Figure 1E, inset). A similar result was also observed from a similar stability study with CaMA-DOX complex (Figure S11). The anti-interference ability should be attributed to the strong host-guest interaction between CaMA and the guest molecules. This important feature of CaMA effectively avoids undesired drug leakage during blood circulation, thereby endowing CaMA with great potential for the stoichiometric co-delivery of multiple drugs to tumor.
Hypoxia-responsive degradation of CaMA
The ability to precisely confine drug release to only the tumor site is another requirement for CaMA in order to improve therapeutic outcomes with reduced side effects. To this end, SAC4A of CaMA was designed to be reduced to NH2C4A by the reductase present in the hypoxic TME, thereby significantly reducing its binding affinity to the loaded drugs and releasing them. To demonstrate the hypoxia-responsive capability of CaMA, sodium dithionite (SDT) was employed as a chemical mimic of reductase and incubated with CaMA. The absorbance was continuously monitored at 420 using a UV-vis spectrometer. The absorbance at 420 nm declined sharply and disappeared within 10 min after the addition of SDT (Figure 2A), indicating a rapid and complete reduction of the azo groups. Similar studies confirmed that CaMA could also be reduced by DT-diaphorase with NAD phosphate (NADPH) under hypoxic condition (Figure S12). Next, the binding affinities of CaMA to the drug molecules before and after reduction were studied using DOX as guest molecule. The binding affinity between DOX and reduced CaMA (BSA-NH2C4A) (Ka < 1000 M-1) was four orders of magnitude lower than that of CaMA (Ka = (9.98 ± 0.16) × 106 M-1) (Figures 2B, S4 and S13). This significantly reduced binding affinity indicates the potential of CaMA releasing loaded drugs after reduction. To demonstrate, CaMA-DOX was treated with different concentrations of SDT and the fluorescence intensity was recorded using a fluorescence spectrometer. The fluorescence intensity of DOX was gradually recovered as the concentration of SDT increased (Figure 2C), indicating the loaded drug was released from CaMA under hypoxic TME. The release behaviors of each drug also confirmed that CaMA was able to achieve effective drug release under hypoxic TME (Figure S14). Further studies by challenging CaMA-DOX with other factors commonly present in inflammatory tissues, including low pH (pH = 6.5), elevated levels of ATP (100 μM) and redox species (10 mM GSH, 100 μM H2O2), resulted in negligible fluorescence recovery (Figure S15), suggesting that the drug release of CaMA can only be triggered by the reductases. This unique controllable release mechanism of CaMA not only optimizes the bioavailability of the drugs, but also provides the necessary prerequisites for the stoichiometric co-delivery of multiple drugs to tumor.
Hypoxia-responsive degradation of CaMA. (A) Absorbance changes for CaMA (5 µM) at 420 nm after adding SDT (2 µM), and the UV spectra of CaMA before and after SDT addition (inset). (B) Fluorescence titration curves of CaMA and BSA-NH2C4A with DOX, fitted according to the 1:1 binding stoichiometry. (C) Fluorescence spectra of CaMA-DOX after incubation with different concentrations of SDT (0-600 µM) in PBS for 30 min. λex = 497 nm. (D) Cell viabilities of the DOX and CaMA-DOX treated cells under different conditions. 'Nor' stands for normoxic condition, 'Hyp' stands for hypoxic condition.
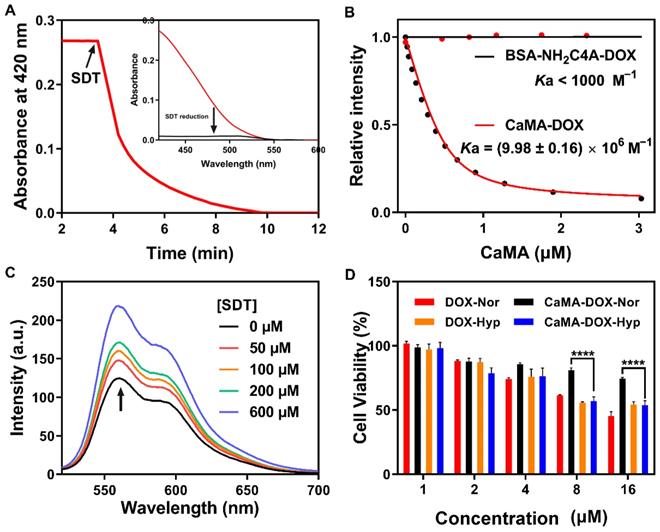
Next, the capability for controlled drug release was further verified with cell-based studies. For this demonstration, CaMA-DOX was first incubated with mouse breast cancer cells (4T1) under normoxic and hypoxic conditions, and the release of DOX was observed with confocal laser scanning microscopy (CLSM). An obvious fluorescence signal of DOX (red) was observed from the CaMA-DOX treated cells under hypoxic condition rather than under normoxic condition (Figure S16), indicating the hypoxia-responsive release of DOX from CaMA-DOX. The cell viability assay further confirmed this result (Figure 2D), in which significantly lower cell viability was observed from the CaMA-DOX treated cells under hypoxic condition than those under normoxic condition. The significantly different cytotoxicity of CaMA-DOX under different conditions demonstrated the capability of CaMA in optimizing the anti-tumor effect and reducing the undesired toxicity of the loaded drugs, which is essential for CaMA to achieve optimal therapeutic outcomes against tumor without obvious side effects.
Precise drug loading and synchronous drug release capability of CaMA
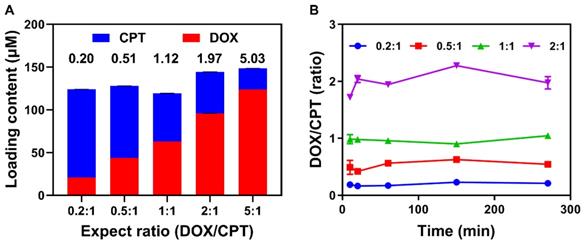
For efficient combination chemotherapy, the precise loading of drug combinations in vitro and the synchronous release of loaded drugs in tumor are critical for CaMA to ensure that the optimal drug ratio is maintained in the tumor. To investigate the precise drug-loading capability of CaMA, CPT and DOX were chosen as drug combinations. Five combinations of DOX and CPT with different expected ratios (0.2:1, 0.5:1, 1:1, 2:1 and 5:1) with a fixed total concentration of 150 μM were loaded into CaMA (denoted as CaMA-DC, details in the Supporting Information). The initial concentrations of DOX and CPT were calculated on the basis of their characteristic binding affinities. Drug loading was achieved by mixing CaMA, DOX, and CPT in pre-calculated concentrations, and the unloaded drugs were separated by filtration (MWCO = 3 kDa). To calculate the drug-loading content, the concentrations of the unloaded CPT and DOX were measured with fluorescence spectrometer. The molar ratio of DOX into CPT loaded in CaMA was almost the same as expected (0.2 vs 0.20, 0.5 vs 0.51, 1.0 vs 1.12, 2.0 vs 1.97, 5.0 vs 5.03) (Figure 3A). These results indicated the excellent capability of CaMA in precise loading of multiple drugs. More importantly, the molar ratio of the loaded drugs could be easily tuned by altering their feeding concentrations. Next, we studied the synchronous release of CaMA. CaMA-DCs at different ratios (DOX to CPT, 0.2:1, 0.5:1, 1:1 and 2:1) were incubated with DT-diaphorase/NADPH and monitored the fluorescence intensities of CPT and DOX continuously. The release of CPT and DOX increased gradually within the observed 240 min (Figures S17-18), indicating that DOX and CPT were effectively released from CaMA after reduction. Notably, the molar ratio of DOX to CPT released from CaMA-DC for each group maintained almost unchanged during the drug release and was consistent with the initial loading ratio (Figure 3B), suggesting the synchronous release of the multiple drugs and the ability to maintain the drug ratio after release.
Precise drug loading and synchronous drug release capability of CaMA. (A) Loading contents and molar ratio of CPT and DOX in CaMA for different ratios. (B) The molar ratios of DOX to CPT released from CaMA-DC (DOX to CPT, 0.2:1, 0.5:1, 1:1 and 2:1) with the time of DT-diaphorase and NADPH added in PBS under hypoxic condition.
Tumor-targeting capability of CaMA
Subsequently, we studied the potential of CaMA for tumor-targeted delivery of drug combinations. In this study, SiPcN2 was employed as a fluorescence probe and loaded into CaMA to obtain CaMA-SiPcN2. Upon reaching the tumor tissues, SiPcN2 was released from CaMA-SiPcN2, leading to the recovery in fluorescence. For the demonstration, 4T1-bearing mice were administrated with CaMA-SiPcN2 and free SiPcN2 via tail-vein. At 48 and 72 h post-injection, tumors and major organs were collected for ex vivo observation. As shown in Figures S19-20, the tumors were clearly delineated by CaMA-SiPcN2 at 48 h post-injection, suggesting that SiPcN2 was successfully released from CaMA-SiPcN2. Moreover, the fluorescence intensity of the tumor from CaMA-SiPcN2 treated mice was significantly higher than free SiPcN2 treated, indicating the ability of CaMA to deliver the loaded drug to tumor tissues.
Antitumor efficacy of CaMA-DM
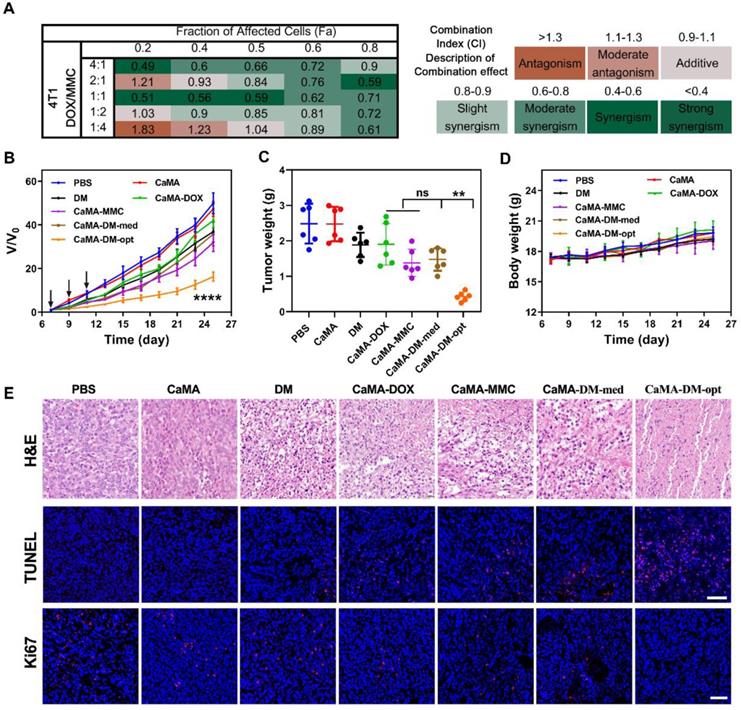
Next, we evaluated the general applicability of CaMA as a co-delivery vector for effective combination chemotherapy. DOX and MMC were chosen as the drug combination owing to their non-overlapping cytotoxicity. Before applying to animals, the optimal ratio for the highest drug synergy of DOX and MMC on 4T1 cells was determined to be 1:1 with a cell-based assay on the basis of Chou-Talalay's method [51, 52] (Figure 4A). DOX and MMC at the optimal ratio (1:1) and the mediocre ratio (1:4) were then loaded into CaMA to form CaMA-DM-opt and CaMA-DM-med, followed by intravenously injecting into 4T1-bearing mice. PBS, CaMA (no drug), the mixture of DOX and MMC at ratio of 1:1 (DM), CaMA-DOX and CaMA-MMC were employed as the control and comparative groups for further investigation. The frequency of administration was once every two days for three doses (days 7, 9, and 11). The body weight and tumor volume were continuously monitored for 25 days. CaMA-DM-opt-treated mice exhibited the most effective tumor suppression than all other treatments (Figure 4B-C). In contrast, no significant difference in tumor suppression was observed between the groups treated with CaMA-DM-med, CaMA-DOX, and CaMA-MMC. These results are in keeping with the results from in vitro experiments, suggesting that CaMA can effectively maintain the molar ratio of drug combination throughout the delivery process. Additionally, the mice body weights in each group remained essentially unchanged within observed 25 days (Figure 4D). Further analysis of histopathological morphology of major organs (Figure S21) indicated that no obvious abnormalities or damages had been caused by CaMA-DM treatment. And a safety assessment by blood routine (Figure S22) indicated that no obvious inflammation was caused by CaMA. For the further investigation of the antitumor effects of CaMA-DM, the tumors were collected and sliced for hematoxylin-eosin (H&E), in situ terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), and Ki67 staining. As shown in the H&E image (Figure 4E), the tumor sections from the mice treated with CaMA-DM-opt exhibited classical apoptotic features, while negligible apoptosis signals were observed in other groups. Further staining with TUNEL and Ki67 also confirmed the same result, i.e., obvious apoptosis and significantly inhibited tumor proliferation were observed in tumor sections from the mice treated with CaMA-DM-opt. Considering that tumor suppression is closely related to the molar ratio of drug combinations, CaMA achieved to effectively translate the optimal molar ratio of the drug combination screened in vitro into in vivo tumor suppressions, providing a powerful tool for the development of safe and efficient combination chemotherapy and personalized medicine.
In vivo antitumor efficacy of CaMA-DM. (A) Combination index of DOX and MMC in 4T1 cells and corresponding color bar. (B) Growth curves of tumor from mice administrated with PBS, DM, CaMA, CaMA-DOX, CaMA-MMC, CaMA-DM-med and CaMA-DM-opt. (C) The tumor weights of mice from each group. (D) The changes in body weight of mice from each group. (E) H&E, TUNEL and Ki67 staining results of tumors from each group. Data are represented as mean ± s.e.m. (n = 6). The significant levels are **P < 0.01, and ****P < 0.0001, analyzed by one-way ANOVA for (c) and two-way ANOVA for (b) with a Tukey's test.
Conclusions
In conclusion, we developed CaMA that achieves precise loading and stoichiometric delivery of multiple drugs for efficient combination chemotherapy. With multiple SAC4As on the surface, CaMA can load multiple drug molecules into one nanostructure through host-guest interactions. More importantly, the drug ratio is able to precisely control by adjusting the initial drug concentrations calculated from their characteristic binding affinities. Upon reaching the tumor, SAC4A of CaMA was degraded, allowing for the simultaneous release of all drugs. By employing DOX and MMC as a combination example, delivery of this drug combination at the pre-screened and optimal ratio via CaMA significantly enhanced tumor suppression with reduced toxicity compared with a traditional cocktail therapy. This stoichiometric delivery ability allows CaMA to rapidly translate optimal drug combinations pre-screened in vitro into in vivo tumor suppression, providing a powerful tool for the development of combination chemotherapies and personalized medications for cancer treatments.
Abbreviations
CaMA: calixarene-modified albumin; MMC: mitomycin C; DOX: doxorubicin; PK: pharmacokinetics; nanoDDS: nano-drug delivery systems; BSA: bovine serum albumin; TME: tumor microenvironment; SAC4A: sulfonate azocalix[4]arene; NH2C4A: aminocalix[4]arene; DLS: dynamic light scattering; TEM: transmission electron microscopy; RhB: rhodamine B; CPT: camptothecin; MS: mouse serum; SiPcN2: silicon (IV) phthalocyanine; SDT: sodium dithionite; CLSM: confocal laser scanning microscope; H&E: hematoxylin-eosin; TUNEL: in situ terminal deoxynucleotidyl transferase dUTP nick end labeling.
Supplementary Material
Supplementary materials and methods, figures and table.
Acknowledgements
This work was supported by National Key Research and Development Programs of China (2018YFA0209700), National Natural Science Foundation of China (NSFC; Grant No. 22077073 and U20A20259), Frontiers Science Center for New Organic Matter (63181206), and Fundamental Research Funds for the Central Universities (Nankai University; Grant No. 63206015).
Author Contributions
Y.W., Z.Z.Z., D.S.G, and Y.L. designed the project. Y.W., Z.Z.Z., L.N.X., Y.D.Z., and H.B.L. carried out chemical experiments. Y.W. performed cell experiments. Y.W., Z.Z.Z. and X.Z.Z. performed the biological experiments in vivo. Y.W., Z.Z.Z., L.Q.S. and Y.L. contributed to writing of this manuscript with input from the other authors.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Webster RM. Combination therapies in oncology. Nat Rev Drug Discov. 2016;15:81-2
2. Rationalizing combination therapies. Nat Med. 2017; 23: 1113.
3. Pusuluri A, Krishnan V, Wu D, Shields CWt, Wang LW, Mitragotri S. Role of synergy and immunostimulation in design of chemotherapy combinations: An analysis of doxorubicin and camptothecin. Bioeng Transl Med. 2019;4:e10129
4. Chen C, Tao R, Ding D, Kong D, Fan A, Wang Z. et al. Ratiometric co-delivery of multiple chemodrugs in a single nanocarrier. Eur J Pharm Sci. 2017;107:16-23
5. Miao L, Guo S, Zhang J, Kim WY, Huang L. Nanoparticles with Precise Ratiometric Co-Loading and Co-Delivery of Gemcitabine Monophosphate and Cisplatin for Treatment of Bladder Cancer. Adv Funct Mater. 2014;24:6601-11
6. Na D, Chae J, Cho S-Y, Kang W, Lee A, Min S. et al. Predictive biomarkers for 5-fluorouracil and oxaliplatin-based chemotherapy in gastric cancers via profiling of patient-derived xenografts. Nat Commun. 2021;12:4840
7. Liu Y, Ma X, Zhu Y, Lv X, Wang P, Feng L. pH-responsive nanomedicine co-encapsulated with Erlotinib and chlorin e6 can enable effective treatment of triple negative breast cancer via reprogramming tumor vasculature. Chem Eng J. 2022 135305
8. Meng F, Wang J, Ping Q, Yeo Y. Camouflaging Nanoparticles for Ratiometric Delivery of Therapeutic Combinations. Nano Lett. 2019;19:1479-87
9. Pan J, Rostamizadeh K, Filipczak N, Torchilin VP. Polymeric Co-Delivery Systems in Cancer Treatment: An Overview on Component Drugs' Dosage Ratio Effect. Molecules. 2019;24:1035
10. Jia J, Zhu F, Ma X, Cao ZW, Li YX, Chen YZ. Mechanisms of drug combinations: interaction and network perspectives. Nat Rev Drug Discov. 2009;8:111-28
11. Feng C, Zhang H, Chen J, Wang S, Xin Y, Qu Y. et al. Ratiometric co-encapsulation and co-delivery of doxorubicin and paclitaxel by tumor-targeted lipodisks for combination therapy of breast cancer. Int J Pharm. 2019;560:191-204
12. Goldman A, Kulkarni A, Kohandel M, Pandey P, Rao P, Natarajan SK. et al. Rationally Designed 2-in-1 Nanoparticles Can Overcome Adaptive Resistance in Cancer. ACS Nano. 2016;10:5823-34
13. Yang T, Yu S, Liu L, Sun Y, Lan Y, Ma X. et al. Dual polymeric prodrug co-assembled nanoparticles with precise ratiometric co-delivery of cisplatin and metformin for lung cancer chemoimmunotherapy. Biomater Sci. 2020;8:5698-714
14. Zhao D, Wu J, Li C, Zhang H, Li Z, Luan Y. Precise ratiometric loading of PTX and DOX based on redox-sensitive mixed micelles for cancer therapy. Colloid Surface B. 2017;155:51-60
15. Yin Q, Pan A, Chen B, Wang Z, Tang M, Yan Y. et al. Quantitative imaging of intracellular nanoparticle exposure enables prediction of nanotherapeutic efficacy. Nat Commun. 2021;12:2385
16. Duan X, Xiao J, Yin Q, Zhang Z, Yu H, Mao S. et al. Smart pH-Sensitive and TemporalControlled Polymeric Micelles for Effective Combination Therapy of Doxorubicin and Disulfiram. ACS Nano. 2013:5858-69
17. Yang Z, Zhao L, Wang X, He Z, Wang Y. Ratiometric Delivery of Mitoxantrone and Berberine Co-encapsulated Liposomes to Improve Antitumor Efficiency and Decrease Cardiac Toxicity. AAPS PharmSciTech. 2021;22:46
18. Aryal S, Hu CM, Zhang L. Polymeric nanoparticles with precise ratiometric control over drug loading for combination therapy. Mol Pharmaceut. 2011;8:1401-7
19. Wan X, Min Y, Bludau H, Keith A, Sheiko SS, Jordan R. et al. Drug Combination Synergy in Worm-like Polymeric Micelles Improves Treatment Outcome for Small Cell and Non-Small Cell Lung Cancer. ACS Nano. 2018;12:2426-39
20. Chen Y, Zhang W, Huang Y, Gao F, Sha X, Fang X. Pluronic-based functional polymeric mixed micelles for co-delivery of doxorubicin and paclitaxel to multidrug resistant tumor. Int J Pharm. 2015;488:44-58
21. Hu Q, Sun W, Wang C, Gu Z. Recent advances of cocktail chemotherapy by combination drug delivery systems. Adv Drug Deliv Rev. 2016;98:19-34
22. Ruiz-Esparza GU, Wu S, Segura-Ibarra V, Cara FE, Evans KW, Milosevic M. et al. Polymer Nanoparticles Encased in a Cyclodextrin Complex Shell for Potential Site- and Sequence-Specific Drug Release. Adv Funct Mater. 2014;24:4753-61
23. Kumar S, Singhal A, Narang U, Mishra S, Kumari P. Recent Progresses in Organic-Inorganic Nano Technological Platforms for Cancer Therapeutics. Curr Med Chem. 2020;27:6015-56
24. Zhou F, Wang P, Peng Y, Zhang P, Huang Q, Sun W. et al. Molecular Engineering-Based Aptamer-Drug Conjugates with Accurate Tunability of Drug Ratios for Drug Combination Targeted Cancer Therapy. Angew Chem Int Ed Engl. 2019;58:11661-5
25. He Y, Cong C, Liu Z, Li X, Zhu R, Gao D. Stealth surface driven accumulation of “Trojan Horse” for tumor hypoxia relief in combination with targeted cancer therapy. Chem Eng J. 2019 378
26. Yu Z, Zhou P, Pan W, Li N, Tang B. A biomimetic nanoreactor for synergistic chemiexcited photodynamic therapy and starvation therapy against tumor metastasis. Nat Commun. 2018;9:5044
27. Cheng Z, Cheng Y, Chen Q, Li M, Wang J, Liu H. et al. Self-assembly of pentapeptides into morphology-adaptable nanomedicines for enhanced combinatorial chemo-photodynamic therapy. Nano Today. 2020;33:100878
28. Zhang Z, Yue YX, Xu L, Wang Y, Geng WC, Li JJ. et al. Macrocyclic-Amphiphile-Based Self-Assembled Nanoparticles for Ratiometric Delivery of Therapeutic Combinations to Tumors. Adv Mater. 2021;33:e2007719
29. Kolishetti N, Dhar S, Valencia PM, Lin LQ, Karnik R, Lippard SJ. et al. Engineering of self-assembled nanoparticle platform for precisely controlled combination drug therapy. Proc Natl Acad Sci U S A. 2010;107:17939-44
30. Greco F, Vicent MJ. Combination therapy: Opportunities and challenges for polymer-drug conjugates as anticancer nanomedicines. Adv Drug Deliv Rev. 2009;61:1203-13
31. Zhu L, Guo Y, Qian Q, Yan D, Li Y, Zhu X. et al. Carrier-Free Delivery of Precise Drug-Chemogene Conjugates for Synergistic Treatment of Drug-Resistant Cancer. Angew Chem Int Ed Engl. 2020;59:17944-50
32. Choi J, Shim MK, Yang S, Hwang HS, Cho H, Kim J. et al. Visible-Light-Triggered Prodrug Nanoparticles Combine Chemotherapy and Photodynamic Therapy to Potentiate Checkpoint Blockade Cancer Immunotherapy. ACS Nano. 2021 15, 12086-12098
33. Pusuluri A, Krishnan V, Lensch V, Sarode A, Bunyan E, Vogus DR. et al. Treating Tumors at Low Drug Doses Using an Aptamer-Peptide Synergistic Drug Conjugate. Angew Chem Int Ed Engl. 2019;58:1437-41
34. Chen H, Zeng X, Tham HP, Phua SZF, Cheng W, Zeng W. et al. NIR-Light-Activated Combination Therapy with a Precise Ratio of Photosensitizer and Prodrug Using a Host-Guest Strategy. Angew Chem Int Ed Engl. 2019;58:7641-6
35. Tian X, Zuo M, Niu P, Velmurugan K, Wang K, Zhao Y. et al. Orthogonal Design of a Water-Soluble meso-Tetraphenylethene-Functionalized Pillar[5]arene with Aggregation-Induced Emission Property and Its Therapeutic Application. ACS Appl Mater Interfaces. 2021;13:37466-74
36. Hu XY, Gao L, Mosel S, Ehlers M, Zellermann E, Jiang H. et al. From Supramolecular Vesicles to Micelles: Controllable Construction of Tumor-Targeting Nanocarriers Based on Host-Guest Interaction between a Pillar[5]arene-Based Prodrug and a RGD-Sulfonate Guest. Small. 2018;14:e1803952
37. Xia L, Tian J, Yue T, Cao H, Chu J, Cai H. et al. Pillar[5]arene-Based Acid-Triggered Supramolecular Porphyrin Photosensitizer for Combating Bacterial Infections and Biofilm Dispersion. Adv Healthc Mater. 2022;11:e2102015
38. Guo S, Huang Q, Chen Y, Wei J, Zheng J, Wang L. et al. Synthesis and Bioactivity of Guanidinium-Functionalized Pillar[5]arene as a Biofilm Disruptor. Angew Chem Int Ed Engl. 2021;60:618-23
39. Wang H, Xue KF, Yang Y, Hu H, Xu JF, Zhang X. In situ Hypoxia-Induced Supramolecular Perylene Diimide Radical Anions in Tumors for Photothermal Therapy with Improved Specificity. J Am Chem Soc. 2022;144:2360-7
40. Sun C, Wang Z, Yue L, Huang Q, Cheng Q, Wang R. Supramolecular Induction of Mitochondrial Aggregation and Fusion. J Am Chem Soc. 2020;142:16523-7
41. Zhang TX, Zhang ZZ, Yue YX, Hu XY, Huang F, Shi L. et al. A General Hypoxia-Responsive Molecular Container for Tumor-Targeted Therapy. Adv Mater. 2020;32:e1908435
42. Zheng Z, Yu H, Geng WC, Hu XY, Wang YY, Li Z. et al. Guanidinocalix[5]arene for sensitive fluorescence detection and magnetic removal of perfluorinated pollutants. Nat Commun. 2019;10:5762
43. Yang K, Yu G, Yang Z, Yue L, Zhang X, Sun C. et al. Supramolecular Polymerization-Induced Nanoassemblies for Self-Augmented Cascade Chemotherapy and Chemodynamic Therapy of Tumor. Angew Chem Int Ed Engl. 2021;60:17570-8
44. Webber MJ, Appel EA, Meijer EW, Langer R. Supramolecular biomaterials. Nat Mater. 2016;15:13-26
45. Cui H, Xu B. Supramolecular medicine. Chem Soc Rev. 2017;46:6430-2
46. Zhou J, Yu G, Huang F. Supramolecular chemotherapy based on host-guest molecular recognition: a novel strategy in the battle against cancer with a bright future. Chem Soc Rev. 2017;46:7021-53
47. Wang H, Wu H, Yi Y, Xue K-F, Xu J-F, Wang H. et al. Self-Motivated Supramolecular Combination Chemotherapy for Overcoming Drug Resistance Based on Acid-Activated Competition of Host-Guest Interactions. CCS Chem. 2021:1413-25
48. Webber MJ, Langer R. Drug delivery by supramolecular design. Chem Soc Rev. 2017;46:6600-20
49. Geng WC, Sessler JL, Guo DS. Supramolecular prodrugs based on host-guest interactions. Chem Soc Rev. 2020;49:2303-15
50. Zhang TX, Li JJ, Li HB, Guo DS. Deep Cavitand Calixarene-Solubilized Fullerene as a Potential Photodynamic Agent. Front Chem. 2021;9:710808
51. Chou T-C. Drug combination against xenograft tumors in nude mice: Experimental design, execution, and computerized simulation of synergism and antagonism. Cancer Res. 2008;68(9_Supplement):4183
52. Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440-6
Author contact
![]() Corresponding author: Yang Liu (E-mail: yliuedu.cn).
Corresponding author: Yang Liu (E-mail: yliuedu.cn).