Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Background
Varieties of CRISPR-Cas9 Pooled...
CRISPR-Cas9 Experimental Models...
Application of CRISPR-Cas9...
Technological Advances in...
Discussions and Perspectives
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2022; 12(7):3329-3344. doi:10.7150/thno.71144 This issue Cite
Review
CRISPR-Cas9 library screening approach for anti-cancer drug discovery: overview and perspectives
Yau-Tuen Chan1, Yuanjun Lu1, Junyu Wu1, Cheng Zhang1, Hor-Yue Tan2, Zhao-xiang Bian2, Ning Wang1 ![]() , Yibin Feng1
, Yibin Feng1 ![]()
1. School of Chinese Medicine, The University of Hong Kong.
2. School of Chinese Medicine, Hong Kong Baptist University.
Received 2021-1-17; Accepted 2022-3-31; Published 2022-4-11
Abstract

CRISPR-Cas9 is a Nobel Prize-winning robust gene-editing tool developed in the last decade. This technique enables a stable genetic engineering method with high precision on the genomes of all organisms. The latest advances in the technology include a genome library screening approach, which can detect survival-essential and drug resistance genes via gain or loss of function. The versatile machinery allows genomic screening for gene activation or inhibition, and targets non-coding sequences, such as promoters, miRNAs, and lncRNAs. In this review, we introduce the emerging high-throughput CRISPR-Cas9 library genome screening technology and its working principles to detect survival and drug resistance genes through positive and negative selection. The technology is compared with other existing approaches while focusing on the advantages of its variable applications in anti-cancer drug discovery, including functions and target identification, non-coding RNA information, actions of small molecules, and drug target discoveries. The combination of the CRISPR-Cas9 system with multi-omic platforms represents a dynamic field expected to advance anti-cancer drug discovery and precision medicine in the clinic.
Keywords: CRISPR-Cas9, Library screening, Cancer therapy, Drug discovery, Experimental models
Background
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-associated endonuclease 9 (Cas9) protein was first reported by two Nobel laureates, Charpentier and Doudna, in 2012. It was initially a natural immunity weapon against viral infections in Streptococcus sp. The researchers invented the target DNA cleavage mechanism by integrating CRISPR RNAs (crRNAs) with manually designed trans-activating crRNA (tracrRNA) to form single-guide RNAs (sgRNAs) [1]. Endonuclease Cas9 protein is guided to the target site acting as “scissors” to cleave the DNA, leaving either a double-strand break (DSB), single-strand nick, or mutagenesis [2, 3]. Previous gene-editing technologies, like zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs), require customizable, specific DNA sequence-binding modules fused to the non-specific DNA endonuclease domain. It was advantageous to engineer targeting proteins for editing any DNA sequence with new designs for target sites. Nevertheless, this complex cloning for customized proteins is difficult to be scaled up for whole-genome applications.
Before the advent of CRISPR-Cas9 pooled libraries, RNA interference (RNAi) screens have been widely used. The most applied RNAi pathway is the short hairpin RNAs (shRNAs), which inhibit post-transcriptional levels of mRNAs by inducing endogenous interference via the RNA-induced silencing complex (RISC) [4]. Since RISC acts on cytoplasmic RNAs, it does not affect cell ploidy, DNA tertiary structure, and chromatin conformation. The transduction process is more straightforward than the CRISPR-Cas9 system, as no exogenous sequences coding for endonucleases and transcriptional modifiers are involved. Despite the practical applications of RNAi in many complicated cellular models, its performance in general library screens, especially cancer-related models, has not been satisfactory. The knockdown efficiency is usually unstable and incomplete, making the quality control of the library screens challenging. This problem is most evident in the survival-essential genes [5]. Also, there are reports of persistent off-target activity, minimizing the significance of the screening results [6]. The action of the RNAi machinery is also limited, mainly inside the cytoplasm [7]. Due to its suboptimal efficiency and off-target effect, many shRNAs are usually designed to target a single gene. The nature of shRNAs makes carrying unique barcodes on each backbone impossible. Extra steps are needed to analyze sequencing results after the screening [8]. Hence, more promising library screening approaches are needed, and the CRISPR-Cas9 platform is an excellent tool to fill the gap.
The CRISPR, unlike its ancestors, utilizes a universal Cas9 protein that needs only a guided RNA (gRNA) to match the target. This convenient, rapid, but versatile genome editing technology provides a promising library screen application to identify essential genes for cell survival and drug resistance [9-12] (Table 1). The introduction of the CRISPR-Cas9 system into the library screen application has primarily enhanced the utilization potential of the genome-editing tool. This review focuses on how the genome editing function of CRISPR-Cas9 could be adapted to screen for genes of interest with different experimental model designs in cancer research. We discuss various applications of the pooled library screening approach in anti-cancer drug discovery and the advantages and limitations compared with other current technologies. We also present the latest development of the CRISPR-Cas9 library screening with other omic platforms to provide insights to researchers in this field.
Research Milestones for CRISPR-Cas9 Library Screen Development
| Name of Library | Type of Screening | Type of Cas9 | Study Outline | Model | Research Output | Selection Method | Year [Ref] |
|---|---|---|---|---|---|---|---|
| Customized sgRNA library | CRISPRko | Cas9 | Improving immunotherapy against triple-negative breast cancer | Normal, nude, and immune-competent BALB/c mice | Cop1 as a target on anti-PD-1 treatment | Puromycin | 2022 [55] |
| CHyMErA hgRNA pooled library | Multiplex CRISPRko | SpCas9, LbCas12a | Combinatorial pooled genetic screening method | HEK293T/17 cell line | Establishment of guidelines for combinatorial pooled genetic screens (ChyMErA) | Blasticidin, G418 | 2021 [97] |
| SLALOM | CRISPRko | Cas9 | Method for enzymatic synthesis of sgRNA library | E. coli, zebrafish | Library constructed by a custom sgRNA scaffold sequence | GFP | 2021 [132] |
| PS4 and EMX1 library | CRISPRko | Cas9 variants, SpCas9, SaCas9, SpCas9 HF1, HypaCas9, HiFi Cas9 | Systematic analysis of Cas9 variants in nicking defects | E. coli BL21 | Multiple Cas9s had varied activities and specificities | / | 2021 [51] |
| Customized sgRNA library | CRISPRko | Cas9 | Screening for tumor drivers in human CRC | colorectal cancer organoid, xenografts | TGFBR2 as TSG in CRC | Blasticidin, TGFβ | 2020 [57] |
| Brunello CRISPR library | CRISPRko | Cas9 | Identifying TGFβ-mediated resistance genes in CRC | human small intestine 3D organoid | Tumor-suppressive SWI/SNF chromatin remodeling components | Puromycin, TGFβ | 2020 [56] |
| Customized sgRNAs | CRISPRko | Cas9 variants | Investigating activity and specificity trade-off of Cas9 variants | HEK293T, U-2 OS, K562 cell line | LZ3 Cas9 as high specificity with +1 insertion profile | Puromycin | 2020 [133] |
| Customized sgRNA library | CRISPRko | Inducible Cas9 | Studying the Cas9 toxicity in hPSC | human pluripotent stem cells | Cas9 toxicity was P53/TP53-dependent | Puromycin | 2018 [134] |
| Customized sgRNA library | CRISPRko | hSpCas9 | Screening mutations related to brain tumorigenesis | neoplastic cerebral organoid | Identified three combinations inducing abnormal glial growth | Phenotypic screen | 2018 [58] |
| Brunello CRISPRko | CRISPRko | SpCas9 | Optimizing customized sgRNA libraries in genome-wide screens | A375, HT29, MelJuSo cell line | Knockout, interference, and activation CRISPR libraries were optimized in three tumor cell lines | Vemurafenib, selumetinib, trametinib | 2018 [135] |
| Dolcetto CRISPRi | CRISPRi | ||||||
| Calabrese CRISPRa | CRISPRa | ||||||
| Customized sgRNA library | CRISPRi | dCas9-KRAB-MeCP2 | Testing the effectiveness of modified Cas9 repressor | HEK293T cell line | KRAB-MeCP2 was an improved Cas9 repressor than KRAB | Various | 2018 [45] |
| Big Papi | Multiplex CRISPRko | SpCas9, SaCas9, dCas9-VPR | Using combinatorial genetic screening to explore complex gene networks in cancers | A375, HT29, OVCAR8, 786O, A549, Meljuso cell line | Genetic interactions were identified using two orthologous Cas9 enzymes | Puromycin, survival | 2018 [100] |
| DrugTarget-CDKO library | Multiplex CRISPRko | Cas9 | Screening for gene combinations for drug-resistance | K562, GM12892 cell line | BCL2L1 and MCL1, etc. were responsible for imatinib resistance | Puromycin, ricin | 2017 [19] |
| Customized sgRNA library | CRISPRi, CRISPRa | dCas9KRAB, dCas9p300 | Screening for functions of regulatory regions by parallel LOF and GOF methods | In vitro | Regulatory elements of β-globin locus and HER2 loci were identified | Survival | 2017 [91] |
| Dual-gRNA library | Multiplex CRISPRko | Cas9 | Mapping cancer genetic networks by combinatorial screens | HeLa, 293T, A549 cell line | Numerous therapeutically relevant interactions were identified | Cell growth | 2017 [136] |
| GeCKOv.2 library | CRISPRko | Cas9 | Profiling genes responsible for T-cell therapy resistance | Mel624, A375 cell line | APLNR was identified as the mediator of impaired CD8 T cell | Survival | 2017 [70] |
| GeCKO library | CRISPRko, CRISPRa | SpCas9, dCas9-VP64-KRABMS2 | Optimizing protocol for in vitro CRISPRko and CRISPRa | In vitro models | Guidelines for screening parameters were provided | / | 2017 [18] |
| Customized sgRNA library | Multiplex CRISPRko | Cas9 | Systematic identification of gene and drug combinations | MDA-MB-231, BxPC-3 cell line | KDM4C/BRD4 and KDM6B/BRD4 inhibitors had synergistic efficacy against OC | Zeocin | 2016 [99] |
| PSMB5 tiling library | Point mutagenesis | dCas9-MS2-AID | Characterizing protein functions by point mutagenesis | K562 cell line | Mutations in bortezomib resistance genes were uncovered | Bortezomib, fluorescence | 2016 [75] |
| Customized sgRNA library | CRISPRko | Cas9 | Characterizing functional enhancers in tumor cells | BJ-RAS, MCF-7, T47D, MDA-MD-231 | Several functional enhancer elements mediating TP53 and ESR1 were identified | Survival | 2016 [29] |
| Customized pgRNA library | Paired CRISPRi | Cas9 | Screening for functional lncRNAs regulating cancer cell growth | Huh7.5, 22RV1, HeLa cell line | 51 targets were identified, and 9 of them were validated | Survival | 2016 [42] |
| Specialized sgRNA library | CRISPRko, CRISPRi | SpCas9, dCas9-KRAB | Comparing CRISPR with shRNA in lethality screen of tumor cell | RT-112, UM-UC-3 cell line | CRISPR was better than shRNA and CRISPRi based method for identification of essential genes | Puromycin, doxycycline, survival | 2016 [68] |
| Customized sgRNAs | CRISPRko | Cas9 | Investigating amino lipids delivery system of long RNAs | HeLa cell line | Cas9 and DNA editing was sustained 95% | mCherry, luciferase | 2016 [137] |
| Avana, Asiago library | CRISPRko | Cas9 | Optimizing sgRNA design to maximize activity on tumor cell | A375, HT29, MOLM13, BV2 cell line | Rules were set to improve performance | Anti-cancer drugs | 2016 [138] |
| Customized sgRNAs | CRISPRi | dCas9-KRAB, dCas9-VP64 | Generating synthetic transcriptional programs by CRISPR scaffold RNAs | S. cerevisiae, HEK293T cell line | Scaffold RNAs could encode target loci and regulatory actions | mCherry | 2015 [20] |
| Customized sgRNA library | CRISPRko | SpCas9, St1Cas9, SaCas9 | Characterizing various functions of Cas9 derivatives | In vitro | Cas9 variants were modified to recognize alternative PAM sequences | Bacterial | 2015 [139] |
| mGeCKOa library | CRISPRko | lentiCas9-EGFP | Identifying genes responsible for tumor growth and metastasis | KPD cell, mouse NSCLC model | Nf2, Pten, Cdkn2a, Trim72, Fga, miR345, or miR-152 KO accelerated tumor metastasis | Primary tumor growth and metastasis | 2015 [30] |
| Customized sgRNA library | CRISPRa | dCas9-VP64 + MS2-p65-HSF1 (SAM complex) | Screening for BRAF inhibitor resistance genes | A375 cell line | Thirteen genes were identified and validated to confer PLX-4720 resistance | Zeocin, puromycin, PLX-4720 | 2015 [88] |
| Customized sgRNAs | CRISPRko | hSpCas9 | Screening for drug targets on acute myeloid leukemia cells | RN2 cell | Six known targets and 19 additional candidates were identified | GFP/mCherry Competition assay | 2015 [53] |
| TKO library | CRISPRko | Cas9 | Identifying cancer fitness genes | A375, RPE1, GBM, DLD1, HeLa, HCT116 cell line | ANKRD49, ZNF830, CCDC84, and RBM48 were identified and validated as cancer-related fitness gene | Survival | 2015 [5] |
| Customized sgRNAs | CRISPRko, point mutagenesis | Cas9 | Identifying chemical compounds that can modulate gene editing by HDR | Mouse embryonic stem cells, Human iPSC | L755507 and Brefeldin A improved while AZT and TFT decreased HDR efficiency | GFP | 215 [140] |
| Customized sgRNA library | CRISPRko | Cas9 | Developing multiplex CRISPR/Cas9 system | Saccharomyces cerevisiae | HDR could accurately edit up to five loci without many off-target effects | LEU2 selection | 2015 [141] |
| Customized sgRNAs | CRISPRko | Cas9, dCas9 | Mapping of genome-wide dCas9 binding sites in mESC | mouse embryonic stem cells | Complicated off-target binding was observed, models for Cas9 binding were suggested | / | 2014 [25] |
| Genome-scale library | CRISPRi, CRISPRa | dCas9-KRAB, dCas9-SunTag | Optimizing CRISPRi and CRISPRa methodology on cancer cells | K562 cell line | Essential genes, TSG, differentiation regulators, and toxin-sensitive genes were identified | Survival | 2014 [37] |
| Human CRISPR Knockout Pooled Libraries | CRISPRko | hSpCas9 | Screening of resistance genes and essential genes on cancer cells | HL60, KBM7 cell line | TOP2A and CDK6 were responsible for etoposide resistance | 6-thioguianine, etoposide | 2014 [10] |
| Customized sgRNA library | CRISPRko | OCT1-Cas9 | Functional screening by lentiviral library in human cancer cells | HEK293T, HT1080, HeLa cell line | PLXNA1, FZD10, PECR, CD81 and RAB2A were novel targets involving toxin resistance | diphtheria and chimaeric anthrax | 2014 [12] |
| Customized sgRNA library | CRISPRko | piggyBac-hCas9 | LOF screening for drug resistance genes in mESC | Mouse embryonic stem cells | 27 known and four previously unknown genes responsible for drug resistance were identified | C. septicum alpha-toxin, 6-thioguanine | 2014 [11] |
| GeCKO library | CRISPRko | SpCas9 | Identifying essential genes and vemurafenib-resistance genes | A375, HUES62 cell line | NF1, MED12, NF2, CUL3, TADA2B and TADA1 were identified as targets | Vemurafenib | 2014 [9] |
| Evx1 sgRNA library | CRISPRko | SpCas9 | Detecting CRISPR-induced mutations | W9.5 mES cell line | 37 clones were identified that were disrupted | GFP, survival | 2014 [106] |
| Specialized sgRNA library | CRISPRko | SpCas9 | Optimizing design of sgRNA libraries on cancer cells | MOLM13, NB4, TF1, A375 cell line | Sequence features contributed to Cas9 ability of binding DNA | Puromycin, cell surface marker | 2014 [142] |
| CRISPR array | CRISPRko | SpCas9 | Demonstrating potential uses of CRISPR-Cas9 | HEK293FT, N2A cell line | Multiple applications of Cas9 were shown | / | 2013 [2] |
Characteristics of Various CRISPR Screening Technologies
| Type of CRISPR screen | Definition | Applications | Advantages | Disadvantages | Examples |
|---|---|---|---|---|---|
| CRISPRko Screen | Genome-wide irreversible gene ablation by NHEJ or HDR under the action of CRISPR-Cas9, followed by the screening of resulting phenotypic alternations | To detect the loss of fitness in the cell population, such as reduced viability, drug sensitivity, proliferation, and incapability of migration | - Low noise - Convenient for detecting survival essential genes or fitness genes - Higher sensitivity than previous RNAi platforms - Able to modulate nearly the entire genome, including non-coding components | - Low cutting efficiency - Some off-target effects observed - More sgRNAs are required to ensure effectiveness on each target - Heterogeneous and heterozygote knockouts are observed - Cell toxicity may be induced with increased DSBs in the genome | - Hart et al. identified ~2000 fitness genes in five human cancer cell lines, and accurately recapitulated genetic vulnerabilities induced by oncogenes responsive to receptor tyrosine kinases. [5] - Michels et al. screened a pan-cancer TSG library in a CTC organoid model and in vivo xenograft models with TGFβ resistance as a paradigm. [57] - Wang et al. identified E3 ligase Cop1 as a novel immune target modulating the immune microenvironment in an in vivo TNBC model. [55] |
| CRISPRi Screen | Genome-wide reversible gene suppression without perturbating genomic sequence by CRISPR-dCas9, usually adopted with extra regulatory domain, followed by screening of resulting phenotypic alterations | To detect the loss of function in the population; by cooperating with different functional suppression complexes, various precise targets could be obtained | - No perturbation of genetic sequence is needed - No non-target cell toxicity caused - Could interfere precisely with regulatory elements on the genome - Effective in knockdown of lncRNAs | - Vulnerable to sequence variability - Suboptimal in complex TSS regulatory mechanisms - Larger Cas9 complex is needed - Difficult to be packed inside AAV due to its large size, while counterpart lentivirus and adenovirus easily cause host responses | - Gilbert et al. established a robust CRISPRi platform, and screened for essential genes responsible for growth and sensitivity to toxin in K562 cells. [37] - Zhu et al. utilized a paired-guide RNA approach to screen for human lncRNAs regulating human cancer cell growth, and found nine validated hits. [42] - Klann et al. applied the CRISPR-Cas9-based epigenomic regulatory element screening to search for proximal and distal regulatory elements activity. Previously known and unknown elements of β-globin locus and HER2 were identified. [91] |
| CRISPRa Screen | Genome-wide reversible gene activation without perturbating genomic sequence by CRISPR-dCas9, usually adopted with extra regulatory domain, followed by the screening of resulting phenotypic alterations | By regulating the promoter regions, genes or non-coding elements could be activated or overexpressed. A gain-of-function analysis could obtain information on drug resistance genes or essential proteins. | - No perturbation of genetic sequence is needed - Much better and easier than the previous cDNA library overexpression method - More feasible to excite lncRNA expression by regulating the promoter - Robust in vivo activation models | - Vulnerable to sequence variability - Larger Cas9 complex is needed - Difficult to be packed inside AAV due to its large size, while counterpart lentivirus and adenovirus easily cause host responses | Konermann et al. engineered Cas9 activation complex and investigated lincRNA transcripts conferring BRAF inhibitor resistance. [88] - Gilbert et al. established a robust CRISPRa platform, and screened for tumor suppressors and developmental transcription factors in cancer cells [37] - Klann et al. applied the CRISPR-Cas9-based epigenomic regulatory element screening to examine the activity of proximal and distal regulatory elements. Previously known and unknown elements of β-globin locus and HER2 were identified. [91] |
| Point mutagenesis | Multiple base-pair mutations or conversion in regions of interest, without causing indels and frameshifts, followed by functional and structural screens. | By using nickase, this CRISPR-based screening approach could screen for point mutations in oncogenes or TSG, characterize protein functions to assist drug target design, and look for mutagenesis causing drug resistance | - High efficiency - Accuracy in editing targets - Obtain extensive information on SNP in a single run | - Base editing complex is enormous - Long foreign genetic materials are introduced into the cells - Limited efficiency in some genomic structures due to spatial hindrance | - Yu et al. developed CRISPR-mediated HDR machinery that could cause point mutations, and the action of small molecules could enhance the efficiency. [140] - Hess et al. utilized the dCas9 complex with cytidine deaminase attached to create mutagenesis in endogenous targets with limited off-target damage. Known and novel mutations conferring bortezomib resistance were identified. [75] |
Varieties of CRISPR-Cas9 Pooled Libraries
The application of the CRISPR-Cas9 as a gene-editing tool in different research fields has been widely studied, and several reviews on its fundamental principles, variations, and applications in cancer research have been published [13-17]. The pooled library screening approach, using CRISPR-Cas9 based gene-editing tool, has evolved as a powerful way to identify interesting gene mutations through phenotypic changes or viability screens. Currently, customed-designed CRISPR-Cas9 libraries covering the whole genome for essential gene screening or a series of genes with specific phenotypes or cellular functions for target discovery are employed. The experiments are designed not to perturb each cell more than once. The unique genetic change is typically ensured using a viral-packaged sgRNA library at a less than 0.3 to 0.5 low multiplicity of infection (MOI) [18]. In the case of combinatorial screens, gene alterations are performed in multiple (usually two) turns to evaluate combined gene functions [19, 20].
Cas9 variants have been engineered to adopt different screening strategies to cope with experimental needs and settings. Typical Cas9, which brings about gene knockout (CRISPRko) by inducing error-prone non-homologous end joining (NHEJ) DNA repair or error-free homology-directed repair (HDR), results in irreversible KO indels. In recent developments, variations of Cas9 proteins have emerged (Table 1). Utilizing the Cas9 mutant nickase version (Cas9n) to create nicking of both DNA strands separately by a pair of gRNAs, the genome-editing results in site-specific DSB [21]. This approach maximizes the specificity while maintaining efficiency similar to wild-type Cas9 [22]. The use of catalytically inactive Cas9 (dCas9) could repress target genes by transcriptional inhibition, also known as CRISPR interference (CRISPRi) [23-25]. The dCas9 protein combined with transcriptional repressors, such as KRAB, instead of DNA cleavage, binds to and hinders the target region in the genome, suppressing the RNA polymerase activity and leading to gene repression [1]. On the other hand, the fusion of effector domains e.g. VP64 [26], enables reversible transcriptional activation [18, 24]. The Cas9 complex is guided by sgRNAs but binds to the promoter region of the gene of interest. Another advancement in Cas9 technology is adding a base editor to dCas9 or Cas9n. Deaminase enzymes or adenine base editors (ABEs) can introduce point mutations and enable nucleotide conversion in target sites [27, 28]. After these phenotypic screens, total RNA is extracted, sequenced, and relative levels of all sgRNAs in control and experimental samples are analyzed [18]. Subsequently, the genes of interest are identified by negative or positive selection.
CRISPRko
Compared with the controls, the negative selection screens identify the depleted or reduced levels of sgRNAs in the population. CRISPRko is often used to detect the loss of fitness in the population, such as reduced viability, drug sensitivity, cell proliferation, and incapability of migration (Figure 1). The sgRNA targets could be designed for nearly all genome regions and not just the functional genes, and the non-coding components, such as promoters, enhancers, and miRNAs, could be modulated directly [9, 29, 30]. Pooled screens utilizing typical CRISPR-Cas9 guided by sgRNAs resulted in indels by NHEJs [31], convenient for detecting survival-essential genes or fitness genes with increased sensitivity compared to the previous RNAi platforms [5, 31].
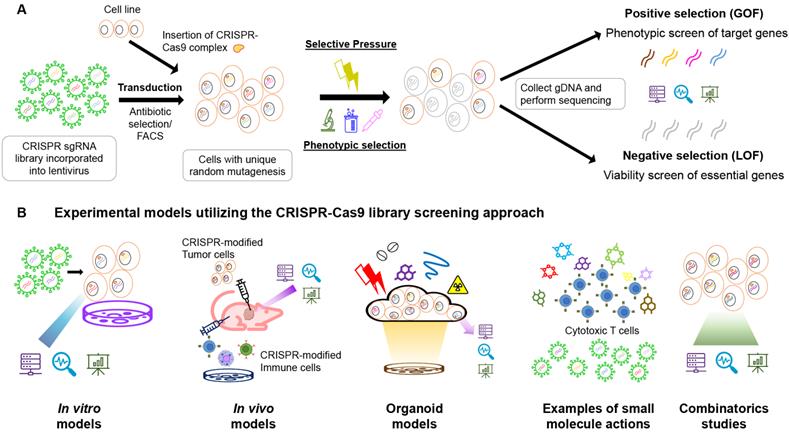
(A) Schematic diagram of the CRISPR-Cas9 library positive and negative selection workflow. The sgRNA library is incorporated into the lentivirus. The library is then transduced into the cell line (with CRISPR-Cas9 complex inserted in the case of the two-plasmid system, or an all-in-one backbone packed inside the lentivirus) with an appropriate MOI (usually less than 0.3-0.5) to ensure unique mutagenesis per cell. The transduced cells were undergone antibiotic selection or FACS to select for successful modifications. The remaining cells are then put under selective pressure or phenotypic selection for desired cell functions. The gDNA of the survived cells are extracted and then subjected to next-generation sequencing to obtain information on the sgRNA presence. The existing amount of sgRNAs are ranked from highest to lowest, and the positive selection is from the top, while the negative selection is from the bottom. (B) Experimental models utilizing the CRISPR-Cas9 library screening approach for anti-cancer drug discovery including in vitro models, in vivo models, organoid models, combinatorics studies and small molecules screenings. In in vitro models, sgRNA libraries are introduced into the cell pool by viral means (usually lentivirus), while the sgRNAs and Cas9 protein complex could be transduced in single or multiple vectors. The foreign genetic materials are often expressed using eukaryotic transposons. In in vivo models searching for target genes in tumours, the cell lines are usually modified in the culture and then injected into animal bodies to form tumour masses. After drug treatments or survival studies, genomic DNAs will be extracted from the tumours and analyzed for the sgRNA appearances using deep sequencing. In some cases, with no handy orthotropic model available, or limited patient-derived xenografts, cultured organoids are ideal models to study for the tissual response of the tumor to the drugs. Selective pressure is applied on the organoid culture with CRISPR-Cas9 library integrated, and the remaining bodies are allowed to grow, and gene mutation pattern is analyzed afterwards. Small molecules are emerging research directions in the study of cancer drug resistance. Large-scale drug screening is comprehended with a CRISPR-Cas9 library screen to detect the potential molecular candidates and genes responsible for drug resistance on the CAR-T therapy. The screening result could be analyzed on both GOF and LOF to look for mutations leading to drug resistance genes and damaged essential proteins. In combinatoric studies, two independent genetic modifications are induced by two sgRNAs. The cells are either exposed to the lentivirus twice for second modifications, or integrating a dual-Cas9 enzyme system with two independent target sites in single transduction. It is possible to induce a knockout and overexpression in the same run simultaneously.
However, the CRISPRko system has low cutting efficiency and off-target effect limitations. The native Cas9 system requires more (around 10) sgRNAs on each target to ensure the effective knockout of a specific gene. Also, despite well-designed sgRNA libraries that could minimize off-target effects [5, 32-34], heterogeneous or heterozygote indels and knockouts remain a concern. When specific mutations fail to induce a stop codon or frameshift, the efficiency is reduced. The sgRNA potency is essential to target the biallelic functional gene mutations [32]. The lowered achievable depletion screening ability could be remedied by sequence-specific barcodes [33, 34]. Intriguingly, the introduction of DSBs on the genome also elevated cell toxicity and jeopardized cell fitness, leading to false-positive selections, which further hinder their applications in combinatorial screens with multiple targets per cell [35, 36].
CRISPRi
The CRISPRi systems integrated with dCas9 reversibly knockdown target genes without perturbating the genome sequence. The loss-of-function screening can be adopted without causing any unpredictable non-target cell toxicity. The sgRNA targets are designed from 50 base pairs before to 300 base pairs after the transcription start site (TSS) with the protospacer length between 18 to 21 base pairs [37]. Homopolymers in the sequence that could substantially hamper the sgRNA activity should be avoided [38]. Since dCas9 directly acts on the homogeneous TSS, and by hindering 23 base pairs on the target genome only [39], CRISPRi could interfere precisely with regulatory elements and non-coding RNAs, including miRNAs and lncRNAs [40, 41]. Especially, lncRNAs could not be easily knocked down with RNAi or CRISPRko as their gene functions are usually silenced by a consequential mutation or multiple indels [10, 42, 43]. The RNAi also has limited efficacy in inhibiting the lncRNAs localized in the nuclei [7].
The CRISPRi system is usually adopted with the most common extra-regulatory domain, the Krüppel-associated box (KRAB), a DNA-binding-dependent transcriptional repressor from the amino terminus of the zinc finger protein 10 [44]. A more recent example of the CRISPRi setup is a dCas9 protein fused with the C-terminal gene-silencing effector domain, KRAB-MeCP2, [45]. This gene suppressor matrix had a more potent effect on most single or multiplex sgRNA targets than the gold standard repressor, dCas9-KRAB. The MeCP2 domain binds to different transcriptional regulators, including DNA methyltransferase and SIN3A-histone deacetylase corepressor complex, further suppressing the target genes. However, the high sensitivity of CRISPRi makes it more vulnerable to sequence variability, for instance, DNA polymorphism in regulatory regions [37]. Furthermore, as the CRISPRi functions by binding to the TSS, this may not be a perfect choice for genes regulated by more than one TSS or multiple genes regulated by a single TSS [36].
CRISPRa
The gain-of-function (GOF) screens are analyzed by positive selection, where the most enriched targets constitute the genes of interest (Figure 1). Before the CRISPRa platform, GOF screens were usually carried out by cDNA library overexpression, which had many drawbacks, such as incomplete coverage, difficulties in design and construction, and lack of endogenous regulations and variability [46, 47]. On the contrary, CRISPRa activates gene expression by targeting promoter regions of the corresponding loci. Based on the targets, only sgRNA libraries are needed and gene transcription is activated endogenously on the proximal promoters. The design and cloning of sgRNAs are easy and inexpensive than previous approaches. The typical CRISPRa involves a Cas9 or dCas9 protein integrated with a transcriptional activator. Tanenbaum et al. employed the SunTag peptide [48], which is a multiple peptide array of epitopes with variable antibody fragments, to recruit VP64 effector protein domains [37]. The VP64-p65-Rta complex, when fused with the dCas9 protein, could also act as a transcriptional activator on endogenous coding and non-coding genes [49]. Another platform developed by Zhang's group incorporated the MS2 sequence onto the sgRNA backbones to recruit two more effector domains, p65 and HSF1. This synergistic activation mediator (SAM) complex, together with the dCas9-VP64 fusion protein, could overexpress the target gene by upregulating transcription. Zalatan et al. integrated CRISPR sgRNAs with scaffolding RNA sequences to allow multidirectional regulations by recruiting protein effectors and epigenetic modifiers into the target region [20]. Various levels of regulation could be achieved by implementing different regulators.
Point Mutagenesis
Besides indel mutations or DSBs leading to frameshifts or nonsense mutations, Cas9n is the emerging version of the CRISPR genetic editing system. Since many genetic diseases arise from point mutations or single-nucleotide polymorphisms, it is necessary to develop a tool that could precisely make single base pair changes. Current CRISPR-Cas9 systems integrated with base editing usually consist of deaminase activity. The cytidine deaminase enzyme can mediate C to T or G to A conversion without causing DSBs or frameshifts. For instance, rat cytidine deaminase enzyme APOBEC1 with a potent conversion activity, when fused with a 16-residue XTEN linker, could extend its effective action range to 4-8 base pairs from the distal end of the protospacer [27]. The efficiency of base editing was higher than 50%. Only 4-6% indels were observed at the target sites in astrocytes compared to the 26-40% indels with the wild-type Cas9 and no evident base repair. However, Cas9n is not yet commonly applied in library screens. Among the few other options available for generating base pair changes, mutation locations could only be determined by site but not the precise base [50]. The mutation frequencies of different bases within the mutation window are different and might be biased when applied to a genome-wide screen [51]. Also, the base editing protein complex is substantial, requiring efficient intracellular delivery tools, and its function may be impaired in some dense genome structures [52].
CRISPR-Cas9 Experimental Models for the Discovery and Development of Anti-cancer Drugs
The CRISPR library screening method is one of the most adaptable research methods for forward candidate gene identification from the phenotype-to-genotype approach. Various applications of CRISPR-Cas9 library screens for the anti-cancer drug target discovery are discussed below (Figure 1) (Table 1).
In vitro Models
The CRISPR system from the bacteria was adapted as an effective genetic editing tool in 2013, and subsequently, the first genetic screens using the CRISPR-Cas9 system were reported [9-12]. Cell lines stably expressing the Cas9 protein were established, and vectors coding the sgRNA were delivered through lentivirus. Also, a different method integrating the Cas9 protein-coding sequence with the corresponding individual sgRNA segment into the same lentiviral backbone was chosen by Shalem et al. [9]. The single lentiviral vector carrying the Cas9 protein, sgRNA, and the selection marker makes this platform more accessible to any cell line of interest without establishing the Cas9-expressing cell line. Koike-Yusa et al. used the piggyBac transposon to carry and express Cas9 [11], while promoters like doxycycline [10] and OCT1 [12] were also employed. The efficiency of the null mutagenesis was greatly enhanced when sgRNAs were targeted directly into the coding exons of important protein domains [53].
In vivo Models
In vivo models are established to study the effect of target genes in the tumor tissue or on the microenvironment, facilitate screening effectiveness, and study the primary tumor growth or metastasis. The most common method to establish a tumor model with library screens is by culturing the engineered cells in vitro and then transplanting them into the animals [30]. For instance, the NSCLC cell line KPD was transduced with the Cas9 protein and a GFP marker to ensure the homogeneity of the cell line and assist in monitoring tumorigenesis and metastasis. The library-transduced cells were injected into the mice after culturing for one week. Genomic DNA was extracted from the tissues of interest, and then deep sequencing was performed to analyze target genes.
Furthermore, CRISPR-Cas9 with sgRNAs were transduced into TCR-expressing CD8+ T cells to investigate the target genes of long-lived effector T cells [54]. The modified T cells were then adoptively transferred into mice bearing B16 melanoma. The sgRNA library was analyzed seven days after the transfer, in which 218 genes were significantly depleted. REGNASE-1 was the most highly increased gene, the functions of which in effector T cells and the anti-tumor immunity were validated. A secondary genome-scale CRISPR library screen was performed to identify the mechanism involved in the REGNASE-1 pathway. BATF was significantly increased in the REGNASE-1-null cells and therefore was the key target of REGNASE-1. By enhancing BATF function and metabolism, REGNASE-1-deficient CD8+ cells were reprogrammed and exhibited an improved response to adoptive cell therapy.
Wang et al. used a CRISPRko library containing over 4,500 genes related to tumor development [55]. The CRISPR library was transduced into membrane-bound ovalbumin-expressing 4T1 cells, an approach to enhance cellular immune response. Cells with 200-fold sgRNAs were transplanted into the fat pads of normal, nude, and immune-competent (pre-vaccinated with ovalbumin) BALB/c mice. The sgRNA abundance distributions at different levels of host immunity were compared and analyzed. Eventually, E3 ubiquitin ligase Cop1 was identified as a regulator of the macrophage chemoattractant, and its inhibition resulted in increased sensitivity to anti-PD1 treatment and prolonged survival.
In general, the CRISPR library screening approach identified potential key genes in the anti-tumor immunity, but the mechanisms of downstream gene regulation require further characterization. Also, it is currently challenging to search for effective therapeutic drugs against the target as it is not a commonly studied gene due to the randomness of the discovery process.
Organoids
Some cancers like brain tumors cannot be studied in vivo easily due to the absence of a good orthotopic model. Genetically engineered mouse models are a choice, but the genetic features of mice are limited, and the cost is high. Patient-derived xenografts are also not ideal for studying tumor initiation and drug screening. Also, although easy, the use of cancer cell lines is not appropriate for investigations involving cell differentiation, cancer stem cells, and the influence of the tumor microenvironment. In this context, in vitro organoid cultures for tumor modeling represent a recent advance often used in drug discovery. However, the CRISPR-based genome editing platform has not been integrated into the organoid models due to technical limitations, such as the need for large cell numbers, heterogeneous growth rates, and low survival rates. Nevertheless, a recent study utilized the CRISPR screen in human intestinal organoids to identify TGF-β resistance genes [56]. The investigators performed CRISPR screening with a single organoid sequencing analysis approach, where each surviving clone was individually amplified with different barcoded primers. Since the organoids were grown from single clones, the noise from heterogenous growth rates could be removed entirely.
In another study, Michels et al. devised an optimized protocol to apply CRISPR-Cas9 screening in the 3D colorectal cancer (CRC) organoid system [57]. They selected TGF-β sensitivity as a positive selection phenotypic trait as this pathway contains several mutated tumor suppressor genes in CRC. The gRNA against TGF-β receptor-2 (TGFBR2) was used as a positive control. The MOI for infecting organoids was titrated by transducing two TGFBR2 gRNA vectors containing GFP or DsRed reporters to be <1. With TGF-β selection, strong signals were observed in 68% of the cells, while a small 5.7% proportion was double-integrated. The pooled library was then transduced to the human colon organoid stably expressing Cas9; cultures were collected after 2 days, 2 weeks, and TGF-β selection for 6.5 weeks, and sgRNAs were analyzed. The group also xenotransplanted the modified organoids into mice models, showing the diverse utilization of CRISPR library screens.
In the studies of brain tumorigenesis, a 3D neoplastic cerebral organoid model was adapted to recapitulate tumor formation [58]. Human embryonic stem cells were used to generate embryoid bodies on ultra-low attachment plates and then induced to form neuroepithelial tissues on matrigel. Subsequently, brain tumors were initiated by the transfected plasmids. CRISPR-Cas9 library provided the tumor-suppressor gene mutation, while Sleeping Beauty transposon was inserted for oncogene amplification. The expanded cells were collected, and the gene mutation pattern was analyzed. This model was instrumental in identifying the mutation driver assemblies where the organoids were initiated from the patient-collected pluripotent stem cells.
Actions of Small Molecules
In the drug discovery process, identifying the cellular targets of the candidate molecules is valuable information. Immunotherapies have improved remarkably in treating multiple cancers, in which chimeric antigen receptor (CAR) T-cell therapy is prominent on B-cell neoplasms. However, primary and acquired resistance is a hurdle [59], and small-molecule inhibitors affect the modulated immune cells. A large-scale drug sensitivity screen was conducted with a genome-scale CRISPR-Cas9 library screen to select potential candidates for enhancing the CAR-T efficacy [60]. More than 500 chemical compounds were screened on cytotoxic T cells for function and downstream signaling, and a CRISPR screen was used to investigate the genes responsible for impaired cytotoxicity of therapeutic CAR-T cells. Screening results identified that SMAC mimetics potently sensitized malignant B cells to CAR-T cytotoxicity, and the library screen characterized the mechanism of action of the molecule. The pharmacological effect was mediated through the RIPK1 pathway, indicating the involvement of programmed cell deaths, including necroptosis [61]. Hence, the combination of the small molecule profiling and the CRISPR library screen results in a fast and systematic selection of effective compounds with known genetic mechanisms of action. Furthermore, the CRISPR-Cas9 library screen for genome-wide mutations provides a fast and effective way to search for the possible drug resistance genetic variations. Neggers et al. demonstrated the beneficial approach of creating localized genetic variations on the gene screening [62]. The mutagenesis in candidate genes leading to drug resistance was indicative of gain-of-function screen, while mutations leading to essential protein knockout represent loss-of-function screen. In brief, using CRISPR-Cas9 pooled library is favorable in screening small molecule actions.
Application of CRISPR-Cas9 Library in Target Discovery and Development of Anti-cancer Drugs
Essential Genes for Survival
Essential genes or fitness genes are the genes that must be present in surviving cells, and their perturbation would negatively affect cell survival and proliferation [63]. A common feature of the cancer genome is chromosomal perturbation, causing lesions in driver genes, passenger genes, and other essential genes [64]. Of these, essential genes offer potential therapeutic windows as drug targets in the tumor [65-67]. Hart et al. improved screening methods for essential genes named as the TKO library screen [5]. The TKO library identified more survival essential genes than the first-generation GeCKO screen. The improvements in the sgRNA library design included removing the strong-biased uridines in the last positions of the sgRNA, sequences with high (>70%) or low (<45%) GC contents, and gRNA with more than one potential off-target site on the genome. Up to 12 sgRNAs were designed for each target gene, and 176,500 sgRNAs were targeted on 17,611 coding genes. As discussed in the study, the distribution of the fold-changes on essential genes should not shift remarkably on the graph compared with non-essential genes. In this case, the expression level of the corresponding sgRNAs could be meaningful as a reference of common core fitness genes or cell-dependent pathway-specific essential genes.
The efficiency of CRISPRi and CRISPRko for screening survival-essential genes was studied by Evers et al. The results showed that CRISPRko technology was the most suitable CRISPR-Cas9 platform in lethality screens [68]. The various sgRNAs among the same target genes appeared to be consistent. The lowest false-discovery rate among all types of library screens makes it the most promising technology in studying fitness genes [69]. Notably, the number of breaks present on the genome by CRISPR-Cas9 editing resulted in an anti-proliferative response unrelated to the gene targets [35]. This unpredicted cell toxicity must be handled carefully in the studies of cancer essential genes, as tumor cells tend to have high copy-number alterations in the genome. In summary, the CRISPR-Cas9 library screen aids the identification and characterization of essential genes in tumor cells.
Identification of Druggable Target Genes
One major obstacle in pharmacological research is the identification of de novo druggable targets. The CRISPR-Cas9 library screens can be adopted in different models to facilitate drug discovery. Patel et al. developed the 2CT-CRISPR assay system to confirm the critical genes responsible for evasion from the effector T cell response [70]. Human CD8+ T cells were isolated and then engineered to be specifically targeted to the HLA-class-1 antigen only. Melanoma cells were transduced with the CRISPRko GeCKOv.2 library and then exposed to the effector T cells. The change in sgRNA levels revealed genes directly related to MHC class 1 antigen processing and elevated unreported genes, such as APLNR, which was later validated for its anti-tumor response of T cells via JAK-STAT signaling.
The CRISPR library screen can quickly and efficiently discover novel drug targets. For example, CMTM6, a previously uncharacterized PD-L1-related protein, was discovered as a potential therapeutic target in the anti-tumor immunotherapy using a similar approach by the CRISPR library screen [71]. Also, ferroptosis is a vital cell death mechanism in studying anti-cancer therapies. Using the CRISPR-Cas9 suppression screen, cytochrome P450 oxidoreductase (POR) was essential for ferroptotic cell death in cancer [72]. POR knockout induces ferroptosis in various cell lines and is critical for lipid peroxidation. It is a potential druggable target for developing anti-tumor therapies using the ferroptosis approach. In another study applying the CRISPR-Cas9 suppressor screen, peroxisomes were shown to contribute to ferroptosis in renal and ovarian carcinoma cell lines. The results also indicated that inhibition of specific peroxisome component genes lowered the vulnerability of cells to ferroptosis induced by peroxisomes through the synthesis of polyunsaturated ether phospholipids, which could be an essential mediator in the anti-cancer strategy [73].
Drug Resistance Genes
Mutations are a common cause of acquired drug resistance [74]. The information on the resistance-contributing mutations could facilitate the development of detection methods, diagnosis standards, prognosis references, and drug target discovery and design. For example, the drug target of the proteasome inhibitor bortezomib, PSMB5, was mutagenized by a library of 143 sgRNAs incorporating a dCas9-MS2-AIDΔ mutagenesis complex in the human erythroleukemic K562 cell line [75]. After transduction and drug selection, mutation frequencies were quantified at every single base pair. The results were validated by separate editing that demonstrated five sites of powerful resistance-induced mutations against bortezomib. Another study investigated the resistance mechanism against immunotherapy in melanoma [76] by transducing a library of 9,872 sgRNAs targeting 2,368 genes into the stable Cas9-expressing B16 cell line. The cells were then transplanted into wild-type and TCRa-/- mice. Apart from the well-known immune evasion PD-L1 and CD47 markers, levels of IFNγ signaling pathway genes targeted by sgRNAs were significantly increased in the immunotherapy group. By analyzing the most depleted genes in the treatment group, PTPN2 was identified to be the cause for resistance to immunotherapy since sgRNAs targeting this gene sensitized the response to immunotherapy. Ptpn2-null tumors were less resistant to the treatment, while tumor suppression was not observed in the TCRa-/- mice. Likewise, a CRISPR-Cas9 screen was performed on an AML-derived cell line MOLM-13 to study the resistance to BCL2 inhibitor venetoclax [77]. The cell line was first transduced to express Cas9 and then infected with sgRNA-carrying lentivirus. Cells were selected by puromycin for five days and then followed by venetoclax for 14 days. Sequencing results showed that sgRNAs of TP53, BAX, and proteins in the apoptosis pathway or mitochondrial homeostasis, such as PMAIP1, were enriched most significantly, and their inactivation contributed to drug resistance.
Non-coding RNAs
With the advances in whole-genome analysis technology, the role of non-coding mutations contributing to tumor development was uncovered. Known mutations in the non-coding regions of the genome can also drive cancer by altering gene expression, transcriptional, post-transcriptional, and epigenetic regulation, regulatory elements, chromatin structure, and regulatory non-coding RNAs [78-80]. The non-coding elements in the genome affect the expression of oncogenes and tumor suppressor genes via direct or indirect actions [81-83]. As discussed in the previous sections, small-scaled indel cuts or base-pair substitutions by the Cas9 suppression system are not likely to inhibit or elevate the function of the non-coding elements, especially the long non-coding RNAs (lncRNAs). Therefore, the CRISPR-Cas9 system had to be improved for effective functional modulation of non-coding RNAs.
A high-throughput genomic deletion method was proposed in 2016 involving a paired-guide RNA (pgRNA) library [42]. The pgRNA strategy is a one-step approach to target two cutting sites for the Cas9 protein with a gap as long as 23kb between the two sites [84]. Increasing the number of sgRNA pairs could further enhance the targeting efficiency. It was also suggested that the dual sgRNA system could improve the Cas9-mediated targeting modification efficiency in vivo with transmittable on-target and off-target mutations [85]. The pgRNA library screening was validated to be specific and more effective than the individual CRISPR-Cas9 knockout, providing another potent tool for studying genome-wide lncRNAs. Although the dual sgRNA system can delete a large fragment of DNA on the site, the targets must be carefully designed to avoid overlapping with other functional non-coding elements in the genome, such as enhancers and miRNAs, or disrupting the introns of other coding genes [86]. Also, the screening results provide no information on the molecular mechanism of lncRNAs, requiring further research to understand the downstream interactions [87]. The pgRNA library approach can detect loss-of-function, but it is tedious to design and set up and not suitable for gain-of-function screens.
On the other hand, the CRISPRi and CRISPRa approaches are more feasible to disrupt or excite lncRNA expression. The dCas9 protein and a suppression or activation domain could readily modulate gene expression, including the lncRNA level [88]. Zhang's group applied the dCas9-VP64 protein with an MS2-p65-HSF1 fusion protein to form a SAM complex, which could upregulate coding genes, intragenic non-coding RNAs, and activate multiple genes concurrently. The activation targets depend on the design of the sgRNA library; for example, more than 10,000 lncRNA TSS were shown to be targeted in a melanoma cell line [89]. The screen was performed on the malignant melanoma cell line A375 with BRAF inhibitor vemurafenib as the selection pressure. Sixteen novel target sgRNAs, which were not previously reported, were identified using enrichment ranking analysis. One selected candidate EMICERI, which activated the neighboring genes in a dose-dependent manner, was validated.
Recently, the possibility of targeting sgRNAs on splice sites was suggested. Cutting sites of CRISPR were designed to be within 50-75 bp of the 5' splice donor or the 3' splice acceptor sites flanking the intron sequences. This method of gene perturbation has advantages over CRISPRi and CRISPRa in terms of specificity. The phenotypic effects on target sites in the proximity of essential coding genes have been controversial due to uncertain hits on the precise target or regulatory elements of the corresponding neighbouring gene [41]. Therefore, the splice site targeting approach can lower the false positive hits in the library screen as the cutting sites are not next to any coding genes. However, this strategy could only effectively target trans-acting lncRNAs. Therefore, targeting the splice sites prior to the promoters is feasible but not ideal [90].
Besides the non-coding RNAs, the regulatory elements at the epigenomic level are also essential features in oncogene regulation. The CRISPR-Cas9-based epigenomic regulatory element screening (CERES) [91] utilizes dCas9KRAB and dCas9p300 proteins to suppress or activate DNase I hypersensitive sites (DHS) [92] by the sgRNA library. Results showed that although the gRNAs did not commonly bring gene expression to more than two-fold change, they were validated by further experiments to confirm the modest regulatory actions. Another study by Fulco et al. also reported a similar screen by CRISPR-dCas9KRAB suppression [40]. Instead of focusing on DHS, they applied the gRNA targets throughout the genome. Results suggested complicated relationships between genes and enhancers, including multiple genes regulated by one enhancer or more than one enhancer controlling a single gene. There was also evidence of enhancers competing with neighbouring promoters in gene regulation.
With the customized design of sgRNA libraries, complex transcription networks and non-coding regulatory elements on specific or arbitrary genes could be mapped and illustrated. The architecture of the sgRNA library for screening non-coding elements requires extra attention, as there have been reports of off-target effects in the Cas9 library and CRIPSRi/a studies [93]. The problem shall be ameliorated by improving sgRNA design strategies [94], producing new variants of Cas9 protein [95], as well as engineering the adaptation requirement of the sgRNAs to CRISPR complex [96].
Combinatorial Studies
Many biological processes, especially in tumors, are controlled by multiple regulatory genes. A combinatorial genetic screen approach for CRISPR-Cas9 library screens has been adopted to identify the complex associations and interactions between various oncogenes or metabolic functions. The multiplex gene targeting system can reveal potential roles of uncharacterized transcriptomes and functions of untranslated regions [97].
The multiplex genetic modification can be attained by transducing sgRNAs targeting two loci in the same cell. It is also achievable by using CRISPR array encoding for more than one targeting spacers [2, 98]. A more advanced design to overcome the problem of multiple transductions would be using the CombiGEM technology [99]. This strategy was used to build a multiplex gRNA library by ligating restriction-enzyme-digested sgRNAs into compatible overhangs on the backbones. The effect was tested on ovarian cancer cells, and two sets of drug-target combinations were identified as therapeutic candidates.
Furthermore, combination therapies are commonly used to overcome drug resistance in cancers; however, direct screening for possible combinations is not feasible. Han et al. examined a functional genetic interaction (GI) map using a massive parallel pairwise gene knockout [19]. More than 21,000 pairs of drug targets were mapped against each other using statistical scoring, and the corresponding lethal drug combinations were identified. More personalized targeted therapies are feasible with the help of this systematic GI network.
Also, a group from the Broad Institute, USA, has reported an innovative methodology in this research field [100]. Genetically editing two independent sites became feasible. The researchers could genetically edit two independent sites by ingeniously using two unique Cas9 enzymes in the same transduction. In addition to the typical SpCas9 (isolated from Streptococcus pyogenes), they picked the orthogonal SaCas9 protein from Staphylococcus aureus. A different set of sgRNAs matching with the SaCas9 was designed with machine learning, and the dual CRISPR system was transduced into different cell lines. This system enabled a simultaneous knockout and overexpression mediated by different CRISPR-Cas9 proteins.
Technological Advances in CRISPR-Cas9 Screening
Publicly Accessible Repositories and In Silico Studies
Despite numerous benefits of CRISPR library research, it may not be accessible or affordable for some laboratories. However, ready-to-use sgRNA libraries on some non-profit repository platforms are available to scientists for research purposes with an acknowledgment to the contributor. Another option is to perform secondary analysis on prior CRISPR-Cas9 library screening data to obtain more valuable information. The Bayesian Analysis of Gene EssentiaLity (BAGEL) is a machine learning method for studying gene knockout screening data [101]. It offers a greater sensitivity but shorter runtime to identify more fitness genes in library screens. The methodology is easily executed in Python, and therefore it could be widely applied in every laboratory. In addition, sequencing data from multiple CRISPR screens could be analyzed together. A total of 31 library screens against 27 drugs using one cell line were integrated to search for genes responsible for sensitivity or resistance to genotoxic agents [102]. Previously unknown DNA repair elements and undetected mechanisms of drug action were discovered from the dataset and demonstrated a valuable learning analysis approach. A project Score (https://score.depmap.sanger.ac.uk/) collects CRISPR-Cas9 whole-genome drop-out screens data across all cell lines to develop a human cancer cell model collection [103]. Until July 2021, there were 914 cell lines from 25 tissues collected in the database. The unbiased and systematic framework determines context-specific and human core fitness genes [104]. The datasets are analyzed in parallel with the patient genomic data to obtain prioritized candidate cancer drug targets. The Biological General Repository for Interaction Datasets (BioGRID) Open Repository of CRISPR Screens (ORCS) (https://orcs.thebiogrid.org/) is an open repository to store CRISPR screens with comprehensive data. It currently holds 1422 CRISPR screens from 719 cell lines [105], and the sgRNA sequence data were re-formatted and collated to be easily accessible.
Synergistic Usage of OMICs Platforms with CRISPR Library Screens
Several other cutting-edge molecular biology techniques were adapted together with the genetic editing system to enhance data analysis and depth of the results from the CRISPR library screen. Next-generation sequencing is widely applied in most library screens to determine the remaining sgRNA levels in the selected population compared with the control/wild-type setup. The unique barcode for the sgRNA targets is usually added to the PCR primers and embedded next to the Cas9 target site [106]. Recent advances have enabled the integration of single-cell transcriptomics with library screens to map the genetic information that regulates cellular phenotypes [107-109]. CRISPR library screen could also be combined with metabolomic analyses, demonstrating resistance to several therapies due to one gene perturbation and identifying the potential target of a metabolic dependency [110]. Also, Wang et al. utilized integrative omics platforms including proteome, phosphoproteome, and transcriptome to analyze and identify various master regulators [111]. CRISPR library screen was subsequently carried out to validate downstream transcription factors and crucial metabolic pathways. A combination of bioinformatics and structural biology is expected to further refine the CRISPR-Cas9 system and facilitate more effective and accurate screening strategies.
Modified Systems for Enhanced Effectiveness of Genome Engineering
Despite its high efficiency, the unexpected off-target effects of the CRISPR-Cas9-based gene-editing technique limit its wide adoption in clinical studies. Current strategies to reduce undesirable editing include specific directed delivery of CRISPR complex, modification of Cas9, and sgRNA engineering [112]. In most settings of pooled library screens, Cas9 endonucleases are modified in different ways to accommodate various experimental needs while not sacrificing accuracy. Two typical Cas9 orthologs are SpCas9 and SaCas9. There are many SpCas9 variants for high-fidelity experiments. SpCas9-HF1, eSpCas9, HypaCas9, and SuperFi-Cas9 are examples of structure-guided engineered proteins in which amino acid residues that contact with the DNA strands are modified [95, 113-115]. Another approach for obtaining improved variants is random mutagenesis and end-point selection, examples of which include evoCas9, Sniper-Cas9, and xCas9 [96, 112, 116]. SaCas9, on the other hand, has a much smaller molecular size than SpCas9 and, due to its easy packaging into viral vectors, is more commonly used in animal models. SaCas9-HF, efSaCas9, and KKH-SaCas9 are alternatives to wild-type SaCas9 for reduced off-target activities while preserving on-target efficiency [117-119].
dCas9 proteins fused with the effector domain are usually adopted for epigenome editing and transcriptional modulation. Furthermore, dual-vector adeno-associated virus (AAV) systems [120], smaller Cas9 orthologues [121, 122], and truncated regulatory elements [122, 123] have been developed to cope with the complicated structure of the editing complex. Although these systems facilitate the integration of epigenetic and transcriptional modulations for in vivo applications, many obstacles hinder the in vivo genetic modification. Delivery methods such as lentivirus and adenovirus can incorporate large packages. Lentivirus is commonly used in pooled library screens on cell lines or primary cells, while adenovirus is usually adopted in vaccine production or immune therapy research. Most current cancer-related in vivo studies involving CRISPR-Cas9 technology transplant modified cell lines because viruses are highly immunogenic in animals [124, 125]. AAV systems resulted in less severe host response and cellular damage but elicited the humoral response and T cell activation [126, 127]. The in vivo CRISPR-Cas9-based genetic modification has not been optimized yet, and nor has the robustness of pooled library screens. The usage of CRISPR-Cas9 library screens in animal models is still in its infancy, and further research is required to enhance the accuracy, potency, and safety.
There are alternatives to Cas9 proteins, such as Cas12, Cas13, Cas14 and Casɸ, but this discussion would be beyond the scope of this review.
Discussions and Perspectives
The CRISPR-Cas9 genetic editing system is one of the most significant breakthroughs of molecular biology in the twenty-first century. The library screening approach is a remarkable consequence derived from its original function as a ge nome perturbation tool. With machine learning and computing tools, designing a genome-scale sgRNA library with thousands of targets is not a fantasy anymore. Artificial intelligence can now predict if sgRNAs are functional for the CRISPR-Cas9 system [128-131], making it easier to obtain more sophisticated and efficient sgRNA libraries focusing on specific areas of interest rather than screening the entire genome. A well-designed, precise sgRNA library could provide large-scale experimental results for identifying gene functions where the genetic information is obtained from phenotypic selections. Anti-cancer drug discovery is a complicated process that involves the entire spectrum, from determining the genetic mutation causes to identifying the potential drug targets. The research had been like finding a needle in the haystack until the invention of the CRISPR library screen. The pre-clinical stage can be completed faster and more systematically using in vitro cell experiments and/or in vivo animal models. The CRISPR genetic engineering system enables rapid, versatile, and accurate genome editing. The combination of CRISPR/Cas9 gene-editing tool and omics technologies has enabled genome-wide genetic screen and mutagenesis analysis, revealing the underlying causes of many genetic problems and functions of non-coding elements in the genome. The ultimate goal of eliminating and/or effectively treating cancers appears to be within reach. Oncology research and genome editing have been placed on a fast track, providing hope that soon we would be able to edit our genomes for curing multiple diseases. Along with the exciting and exploding research outputs, we must consider the possible ethical issues that may arise from this ground-breaking advancement. CRISPR technology to modify genetic information in human embryos is prohibited now, and society has not consented to the concept of genetically engineered babies. Also, the potential off-target effect must be eliminated to ensure patient safety for the clinical application of this technology. Modified genes in adoptively transferred cells may lead to inadvertent detrimental results in humans. Therefore, it is imperative to safeguard technological advances for the benefit of humanity.
Acknowledgements
Funding
This research was partially supported by the Research Council of the University of Hong Kong (project codes: 104004092 and 104004460), Wong's donation (project code: 200006276), a donation from the Gaia Family Trust of New Zealand (project code: 200007008), the Research Grants Committee (RGC) of Hong Kong, HKSAR (Project Codes: 740608, 766211, 17152116 and 17121419), Health and Medical Research Fund (Project code: 15162961 and 16172751), Enhanced new staff start-up fund (Project code: 204610519) and Pre-emptive retention fund (Project code: 202007002).
Author contributions
YTC drafted the manuscript. YL, JW, CZ, HYT and ZXB commented and revised the manuscript. NW and YF designed, revised, and finalized the manuscript. All authors have read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science. 2012;337:816-21
2. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N. et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819-23
3. Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE. et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823-6
4. Liu Q, Rand TA, Kalidas S, Du F, Kim H-E, Smith DP. et al. R2D2, a Bridge Between the Initiation and Effector Steps of the Drosophila RNAi Pathway. Science. 2003;301:1921-5
5. Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G. et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell. 2015;163:12
6. Jackson AL, Linsley PS. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat Rev Drug Discov. 2010;9:57-67
7. Zeng YAN, Cullen BR. RNA interference in human cells is restricted to the cytoplasm. RNA. 2002;8:855-60
8. Fellmann C, Lowe SW. Stable RNA interference rules for silencing. Nat Cell Biol. 2014;16:10-8
9. Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS. et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84-7
10. Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic Screens in Human Cells Using the CRISPR-Cas9 System. Science. 2014;343:80-4
11. Koike-Yusa H, Li Y, Tan E-P, Velasco-Herrera MDC, Yusa K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol. 2014;32:267-73
12. Zhou Y, Zhu S, Cai C, Yuan P, Li C, Huang Y. et al. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature. 2014;509:487-91
13. Dominguez AA, Lim WA, Qi LS. Beyond editing: repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat Rev Mol Cell Biol. 2016;17:11
14. Komor AC, Badran AH, Liu DR. CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell. 2017;168:20-36
15. Rees HA, Liu DR. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet. 2018;19:770-88
16. Li HY, Yang Y, Hong WQ, Huang MY, Wu M, Zhao X. Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Signal Transduct Target Ther. 2020;5:23
17. Zhang H, Qin C, An C, Zheng X, Wen S, Chen W. et al. Application of the CRISPR/Cas9-based gene editing technique in basic research, diagnosis, and therapy of cancer. Mol Cancer. 2021;20:126
18. Joung J, Konermann S, Gootenberg JS, Abudayyeh OO, Platt RJ, Brigham MD. et al. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat Protoc. 2017;12:828-63
19. Han K, Jeng EE, Hess GT, Morgens DW, Li A, Bassik MC. Synergistic drug combinations for cancer identified in a CRISPR screen for pairwise genetic interactions. Nat Biotechnol. 2017;35:463-74
20. Zalatan Jesse G, Lee Michael E, Almeida R, Gilbert Luke A, Whitehead Evan H, La Russa M. et al. Engineering Complex Synthetic Transcriptional Programs with CRISPR RNA Scaffolds. Cell. 2015;160:339-50
21. Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S. et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol. 2013;31:833-8
22. Ran FA, Hsu Patrick D, Lin C-Y, Gootenberg Jonathan S, Konermann S, Trevino AE. et al. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell. 2013;154:1380-9
23. Qi Lei S, Larson Matthew H, Gilbert Luke A, Doudna Jennifer A, Weissman Jonathan S, Arkin Adam P. et al. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell. 2013;152:1173-83
24. Gilbert Luke A, Larson Matthew H, Morsut L, Liu Z, Brar Gloria A, Torres Sandra E. et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell. 2013;154:442-51
25. Wu X, Scott DA, Kriz AJ, Chiu AC, Hsu PD, Dadon DB. et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat Biotechnol. 2014;32:670-6
26. Konermann S, Brigham MD, Trevino AE, Hsu PD, Heidenreich M, Le C. et al. Optical control of mammalian endogenous transcription and epigenetic states. Nature. 2013;500:472-6
27. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420-4
28. Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI. et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017;551:464-71
29. Korkmaz G, Lopes R, Ugalde AP, Nevedomskaya E, Han R, Myacheva K. et al. Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nat Biotechnol. 2016;34:192-8
30. Chen S, Sanjana Neville E, Zheng K, Shalem O, Lee K, Shi X. et al. Genome-wide CRISPR Screen in a Mouse Model of Tumor Growth and Metastasis. Cell. 2015;160:1246-60
31. Henkel L, Rauscher B, Schmitt B, Winter J, Boutros M. Genome-scale CRISPR screening at high sensitivity with an empirically designed sgRNA library. Bmc Biology. 2020;18:21
32. Yuen G, Khan FJ, Gao S, Stommel JM, Batchelor E, Wu X. et al. CRISPR/Cas9-mediated gene knockout is insensitive to target copy number but is dependent on guide RNA potency and Cas9/sgRNA threshold expression level. Nucleic Acids Res. 2017;45:12039-53
33. Boettcher M, Covarrubias S, Biton A, Blau J, Wang HP, Zaitlen N. et al. Tracing cellular heterogeneity in pooled genetic screens via multi-level barcoding. BMC Genomics. 2019;20:9
34. Iida M, Suzuki M, Sakane Y, Nishide H, Uchiyama I, Yamamoto T. et al. A simple and practical workflow for genotyping of CRISPR-Cas9-based knockout phenotypes using multiplexed amplicon sequencing. Genes Cells. 2020;25:498-509
35. Aguirre AJ, Meyers RM, Weir BA, Vazquez F, Zhang C-Z, Ben-David U. et al. Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov. 2016;6:914-29
36. Rosenbluh J, Xu H, Harrington W, Gill S, Wang XX, Vazquez F. et al. Complementary information derived from CRISPR Cas9 mediated gene deletion and suppression. Nat Commun. 2017;8:8
37. Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH. et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell. 2014;159:647-61
38. Radzisheuskaya A, Shlyueva D, Müller I, Helin K. Optimizing sgRNA position markedly improves the efficiency of CRISPR/dCas9-mediated transcriptional repression. Nucleic Acids Res. 2016;44:e141-e
39. Nishimasu H, Cong L, Yan Winston X, Ran FA, Zetsche B, Li Y. et al. Crystal Structure of Staphylococcus aureus Cas9. Cell. 2015;162:1113-26
40. Fulco CP, Munschauer M, Anyoha R, Munson G, Grossman SR, Perez EM. et al. Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science. 2016;354:769-73
41. Liu SJ, Horlbeck MA, Cho SW, Birk HS, Malatesta M, He D. et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science. 2017;355:eaah7111
42. Zhu S, Li W, Liu J, Chen C-H, Liao Q, Xu P. et al. Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR-Cas9 library. Nat Biotechnol. 2016;34:1279-86
43. Paralkar Vikram R, Taborda Cristian C, Huang P, Yao Y, Kossenkov Andrew V, Prasad R. et al. Unlinking an lncRNA from Its Associated cis Element. Mol Cell. 2016;62:104-10
44. Groner AC, Meylan S, Ciuffi A, Zangger N, Ambrosini G, Dénervaud N. et al. KRAB-zinc finger proteins and KAP1 can mediate long-range transcriptional repression through heterochromatin spreading. PLoS genetics. 2010;6:e1000869
45. Yeo NC, Chavez A, Lance-Byrne A, Chan Y, Menn D, Milanova D. et al. An enhanced CRISPR repressor for targeted mammalian gene regulation. Nat Methods. 2018;15:611-6
46. Lavaissiere L, Jia S, Nishiyama M, de la Monte S, Stern AM, Wands JR. et al. Overexpression of human aspartyl(asparaginyl)beta-hydroxylase in hepatocellular carcinoma and cholangiocarcinoma. J Clin Invest. 1996;98:1313-23
47. Pritsker M, Ford NR, Jenq HT, Lemischka IR. Genomewide gain-of-function genetic screen identifies functionally active genes in mouse embryonic stem cells. Proc Natl Acad Sci U S A. 2006;103:6946-51
48. Tanenbaum Marvin E, Gilbert Luke A, Qi Lei S, Weissman Jonathan S, Vale Ronald D. A Protein-Tagging System for Signal Amplification in Gene Expression and Fluorescence Imaging. Cell. 2014;159:635-46
49. Chavez A, Scheiman J, Vora S, Pruitt BW, Tuttle M, P R Iyer E. et al. Highly efficient Cas9-mediated transcriptional programming. Nat Methods. 2015;12:326-8
50. Kim YB, Komor AC, Levy JM, Packer MS, Zhao KT, Liu DR. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat Biotechnol. 2017;35:371-6
51. Murugan K, Suresh SK, Seetharam AS, Severin AJ, Sashital DG. Systematic in vitro specificity profiling reveals nicking defects in natural and engineered CRISPR-Cas9 variants. Nucleic Acids Res. 2021;49:4037-53
52. Liu Y, Zhou C, Huang S, Dang L, Wei Y, He J. et al. A Cas-embedding strategy for minimizing off-target effects of DNA base editors. Nat Commun. 2020;11:6073
53. Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, Vakoc CR. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol. 2015;33:661-7
54. Wei J, Long L, Zheng W, Dhungana Y, Lim SA, Guy C. et al. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature. 2019;576:471-6
55. Wang X, Tokheim C, Gu SS, Wang B, Tang Q, Li Y. et al. In vivo CRISPR screens identify the E3 ligase Cop1 as a modulator of macrophage infiltration and cancer immunotherapy target. Cell. 2021;184:5357-74.e22
56. Ringel T, Frey N, Ringnalda F, Janjuha S, Cherkaoui S, Butz S. et al. Genome-Scale CRISPR Screening in Human Intestinal Organoids Identifies Drivers of TGFβ; Resistance. Cell Stem Cell. 2020;26:431-40.e8
57. Michels BE, Mosa MH, Streibl BI, Zhan T, Menche C, Abou-El-Ardat K. et al. Pooled In vitro and In vivo CRISPR-Cas9 Screening Identifies Tumor Suppressors in Human Colon Organoids. Cell Stem Cell. 2020;26:782-92.e7
58. Bian S, Repic M, Guo Z, Kavirayani A, Burkard T, Bagley JA. et al. Genetically engineered cerebral organoids model brain tumor formation. Nat Methods. 2018;15:631-9
59. June CH, Sadelain M. Chimeric Antigen Receptor Therapy. N Engl J Med. 2018;379:64-73
60. Dufva O, Koski J, Maliniemi P, Ianevski A, Klievink J, Leitner J. et al. Integrated drug profiling and CRISPR screening identify essential pathways for CAR T-cell cytotoxicity. Blood. 2020;135:597-609
61. McComb S, Aguadé-Gorgorió J, Harder L, Marovca B, Cario G, Eckert C. et al. Activation of concurrent apoptosis and necroptosis by SMAC mimetics for the treatment of refractory and relapsed ALL. Sci Transl Med. 2016;8:339ra70-ra70
62. Neggers JE, Kwanten B, Dierckx T, Noguchi H, Voet A, Bral L. et al. Target identification of small molecules using large-scale CRISPR-Cas mutagenesis scanning of essential genes. Nat Commun. 2018;9:502
63. Hart T, Brown KR, Sircoulomb F, Rottapel R, Moffat J. Measuring error rates in genomic perturbation screens: gold standards for human functional genomics. Mol Syst Biol. 2014;10:733
64. Muller FL, Aquilanti EA, DePinho RA. Collateral Lethality: A New Therapeutic Strategy in Oncology. Trends in Cancer. 2015;1:161-73
65. Pertesi M, Ekdahl L, Palm A, Johnsson E, Järvstråt L, Wihlborg A-K. et al. Essential genes shape cancer genomes through linear limitation of homozygous deletions. Commun Biol. 2019;2:262
66. Liu Y, Zhang X, Han C, Wan G, Huang X, Ivan C. et al. TP53 loss creates therapeutic vulnerability in colorectal cancer. Nature. 2015;520:697-701
67. Aksoy BA, Demir E, Babur Ö, Wang W, Jing X, Schultz N. et al. Prediction of individualized therapeutic vulnerabilities in cancer from genomic profiles. Bioinformatics. 2014;30:2051-9
68. Evers B, Jastrzebski K, Heijmans JPM, Grernrum W, Beijersbergen RL, Bernards R. CRISPR knockout screening outperforms shRNA and CRISPRi in identifying essential genes. Nat Biotechnol. 2016;34:631-3
69. Munoz DM, Cassiani PJ, Li L, Billy E, Korn JM, Jones MD. et al. CRISPR Screens Provide a Comprehensive Assessment of Cancer Vulnerabilities but Generate False-Positive Hits for Highly Amplified Genomic Regions. Cancer Discov. 2016;6:900-13
70. Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M. et al. Identification of essential genes for cancer immunotherapy. Nature. 2017;548:537-42
71. Burr ML, Sparbier CE, Chan Y-C, Williamson JC, Woods K, Beavis PA. et al. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature. 2017;549:101-5
72. Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W. et al. Cytochrome P450 oxidoreductase contributes tophospholipid peroxidation in ferroptosis. Nat Chem Biol. 2020;16:302-9
73. Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P. et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature. 2020;585:603-8
74. Aleksakhina SN, Kashyap A, Imyanitov EN. Mechanisms of acquired tumor drug resistance. Biochim Biophys Acta Rev Cancer. 2019;1872:188310
75. Hess GT, Frésard L, Han K, Lee CH, Li A, Cimprich KA. et al. Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat Methods. 2016;13:1036-42
76. Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC. et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature. 2017;547:413-8
77. Nechiporuk T, Kurtz SE, Nikolova O, Liu T, Jones CL, D'Alessandro A. et al. The TP53 Apoptotic Network Is a Primary Mediator of Resistance to BCL2 Inhibition in AML Cells. Cancer Discov. 2019;9:910-25
78. Elliott K, Larsson E. Non-coding driver mutations in human cancer. Nat Rev Cancer. 2021;21:500-9
79. Yan H, Bu P. Non-coding RNA in cancer. Essays Biochem. 2021;65:625-39
80. Jiang W, Pan S, Chen X, Wang ZW, Zhu X. The role of lncRNAs and circRNAs in the PD-1/PD-L1 pathway in cancer immunotherapy. Mol Cancer. 2021;20:116
81. Chen X, Ma Q, Shang Z, Niu Y. Super-enhancer in prostate cancer: transcriptional disorders and therapeutic targets. NPJ Precis Oncol. 2020;4:31
82. Ganem NS, Ben-Asher N, Lamm AT. In cancer, A-to-I RNA editing can be the driver, the passenger, or the mechanic. Drug Resist Updat. 2017;32:16-22
83. Diederichs S, Bartsch L, Berkmann JC, Fröse K, Heitmann J, Hoppe C. et al. The dark matter of the cancer genome: aberrations in regulatory elements, untranslated regions, splice sites, non-coding RNA and synonymous mutations. EMBO Mol Med. 2016;8:442-57
84. Han J, Zhang J, Chen L, Shen B, Zhou J, Hu B. et al. Efficient in vivo deletion of a large imprinted lncRNA by CRISPR/Cas9. RNA Biology. 2014;11:829-35
85. Zhou J, Wang J, Shen B, Chen L, Su Y, Yang J. et al. Dual sgRNAs facilitate CRISPR/Cas9-mediated mouse genome targeting. The FEBS Journal. 2014;281:1717-25
86. Louro R, Smirnova AS, Verjovski-Almeida S. Long intronic noncoding RNA transcription: Expression noise or expression choice? Genomics. 2009;93:291-8
87. Rinn JL, Chang HY. Genome Regulation by Long Noncoding RNAs. Annu Rev Biochem. 2012;81:145-66
88. Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C. et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583-U332
89. Joung J, Engreitz JM, Konermann S, Abudayyeh OO, Verdine VK, Aguet F. et al. Genome-scale activation screen identifies a lncRNA locus regulating a gene neighbourhood. Nature. 2017;548:343-6
90. Engreitz JM, Haines JE, Perez EM, Munson G, Chen J, Kane M. et al. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature. 2016;539:452-5
91. Klann TS, Black JB, Chellappan M, Safi A, Song L, Hilton IB. et al. CRISPR-Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nat Biotechnol. 2017;35:561-8
92. Sabo PJ, Kuehn MS, Thurman R, Johnson BE, Johnson EM, Cao H. et al. Genome-scale mapping of DNase I sensitivity in vivo using tiling DNA microarrays. Nat Methods. 2006;3:511-8
93. Tycko J, Wainberg M, Marinov GK, Ursu O, Hess GT, Ego BK. et al. Mitigation of off-target toxicity in CRISPR-Cas9 screens for essential non-coding elements. Nat Commun. 2019;10:4063
94. Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32:279-84
95. Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84-8
96. Hu JH, Miller SM, Geurts MH, Tang W, Chen L, Sun N. et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 2018;556:57-63
97. Aregger M, Xing K, Gonatopoulos-Pournatzis T. Application of CHyMErA Cas9-Cas12a combinatorial genome-editing platform for genetic interaction mapping and gene fragment deletion screening. Nat Protoc. 2021;16:4722-65
98. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N. et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 2013;339:819-23
99. Wong ASL, Choi GCG, Cui CH, Pregernig G, Milani P, Adam M. et al. Multiplexed barcoded CRISPR-Cas9 screening enabled by CombiGEM. Proc Natl Acad Sci U S A. 2016;113:2544-9
100. Najm FJ, Strand C, Donovan KF, Hegde M, Sanson KR, Vaimberg EW. et al. Orthologous CRISPR-Cas9 enzymes for combinatorial genetic screens. Nat Biotechnol. 2018;36:179-89
101. Hart T, Moffat J. BAGEL: a computational framework for identifying essential genes from pooled library screens. BMC Bioinformatics. 2016;17:7
102. Olivieri M, Cho T, Álvarez-Quilón A, Li K, Schellenberg MJ, Zimmermann M. et al. A Genetic Map of the Response to DNA Damage in Human Cells. Cell. 2020;182:481-96.e21
103. Dwane L, Behan FM, Gonçalves E, Lightfoot H, Yang W, van der Meer D. et al. Project Score database: a resource for investigating cancer cell dependencies and prioritizing therapeutic targets. Nucleic Acids Res. 2020;49:D1365-D72
104. Behan FM, Iorio F, Picco G, Gonçalves E, Beaver CM, Migliardi G. et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature. 2019;568:511-6
105. Oughtred R, Rust J, Chang C, Breitkreutz B-J, Stark C, Willems A. et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Science. 2021;30:187-200
106. Bell CC, Magor GW, Gillinder KR, Perkins AC. A high-throughput screening strategy for detecting CRISPR-Cas9 induced mutations using next-generation sequencing. BMC Genomics. 2014;15:1002
107. Gasperini M, Hill AJ, McFaline-Figueroa JL, Martin B, Kim S, Zhang MD. et al. A Genome-wide Framework for Mapping Gene Regulation via Cellular Genetic Screens. Cell. 2019;176:377-90.e19
108. McFaline-Figueroa JL, Hill AJ, Qiu X, Jackson D, Shendure J, Trapnell C. A pooled single-cell genetic screen identifies regulatory checkpoints in the continuum of the epithelial-to-mesenchymal transition. Nat Genet. 2019;51:1389-98
109. Replogle JM, Norman TM, Xu A, Hussmann JA, Chen J, Cogan JZ. et al. Combinatorial single-cell CRISPR screens by direct guide RNA capture and targeted sequencing. Nat Biotechnol. 2020;38:954-61
110. Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE. et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med. 2017;23:1362-8
111. Wang H, Diaz AK, Shaw TI, Li Y, Niu M, Cho J-H. et al. Deep multiomics profiling of brain tumors identifies signaling networks downstream of cancer driver genes. Nat Commun. 2019;10:3718
112. Casini A, Olivieri M, Petris G, Montagna C, Reginato G, Maule G. et al. A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nat Biotechnol. 2018;36:265-71
113. Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z. et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490-5
114. Chen JS, Dagdas YS, Kleinstiver BP, Welch MM, Sousa AA, Harrington LB. et al. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature. 2017;550:407-10
115. Bravo JPK, Liu M-S, Hibshman GN, Dangerfield TL, Jung K, McCool RS. et al. Structural basis for mismatch surveillance by CRISPR-Cas9. Nature. 2022
116. Lee JK, Jeong E, Lee J, Jung M, Shin E, Kim Y-h. et al. Directed evolution of CRISPR-Cas9 to increase its specificity. Nat Commun. 2018;9:3048
117. Tan Y, Chu AHY, Bao S, Hoang DA, Kebede FT, Xiong W. et al. Rationally engineered Staphylococcus aureus Cas9 nucleases with high genome-wide specificity. Proc Natl Acad Sci U S A. 2019;116:20969-76
118. Xie H, Ge X, Yang F, Wang B, Li S, Duan J. et al. High-fidelity SaCas9 identified by directional screening in human cells. PLOS Biology. 2020;18:e3000747
119. Kleinstiver BP, Prew MS, Tsai SQ, Nguyen NT, Topkar VV, Zheng Z. et al. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat Biotechnol. 2015;33:1293-8
120. Thakore PI, Kwon JB, Nelson CE, Rouse DC, Gemberling MP, Oliver ML. et al. RNA-guided transcriptional silencing in vivo with S. aureus CRISPR-Cas9 repressors. Nat Commun. 2018;9:1674
121. Kim E, Koo T, Park SW, Kim D, Kim K, Cho H-Y. et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat Commun. 2017;8:14500
122. Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ. et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186-91
123. Moreno AM, Fu X, Zhu J, Katrekar D, Shih Y-RV, Marlett J. et al. In situ Gene Therapy via AAV-CRISPR-Cas9-Mediated Targeted Gene Regulation. Mol Ther. 2018;26:1818-27
124. Wang D, Mou H, Li S, Li Y, Hough S, Tran K. et al. Adenovirus-mediated somatic genome editing of Pten by CRISPR/Cas9 in mouse liver in spite of Cas9-specific immune responses. Hum Gene Ther. 2015;26:432-42
125. Annoni A, Gregori S, Naldini L, Cantore A. Modulation of immune responses in lentiviral vector-mediated gene transfer. Cell Immunol. 2019;342:103802
126. Chew WL, Tabebordbar M, Cheng JKW, Mali P, Wu EY, Ng AHM. et al. A multifunctional AAV-CRISPR-Cas9 and its host response. Nat Methods. 2016;13:868-74
127. Mays LE, Wilson JM. The Complex and Evolving Story of T cell Activation to AAV Vector-encoded Transgene Products. Mol Ther. 2011;19:16-27
128. Xue L, Tang B, Chen W, Luo J. Prediction of CRISPR sgRNA Activity Using a Deep Convolutional Neural Network. J Chem Inf Model. 2019;59:615-24
129. Erard N, Knott SRV, Hannon GJ. A CRISPR Resource for Individual, Combinatorial, or Multiplexed Gene Knockout. Mol Cell. 2017;67:348-54.e4
130. Guo J, Wang T, Guan C, Liu B, Luo C, Xie Z. et al. Improved sgRNA design in bacteria via genome-wide activity profiling. Nucleic Acids Res. 2018;46:7052-69
131. Jing R, Li Y, Xue L, Liu F, Li M, Luo J. autoBioSeqpy: A Deep Learning Tool for the Classification of Biological Sequences. J Chem Inf Model. 2020;60:3755-64
132. Yates JD, Russell RC, Barton NJ, Yost HJ, Hill JT. A simple and rapid method for enzymatic synthesis of CRISPR-Cas9 sgRNA libraries. Nucleic Acids Res. 2021
133. Schmid-Burgk JL, Gao L, Li D, Gardner Z, Strecker J, Lash B. et al. Highly Parallel Profiling of Cas9 Variant Specificity. Mol Cell. 2020;78:794-800.e8
134. Ihry RJ, Worringer KA, Salick MR, Frias E, Ho D, Theriault K. et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat Med. 2018;24:939-46
135. Sanson KR, Hanna RE, Hegde M, Donovan KF, Strand C, Sullender ME. et al. Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat Commun. 2018;9:15
136. Shen JP, Zhao D, Sasik R, Luebeck J, Birmingham A, Bojorquez-Gomez A. et al. Combinatorial CRISPR-Cas9 screens for de novo mapping of genetic interactions. Nat Methods. 2017;14:573-6
137. Miller JB, Zhang S, Kos P, Xiong H, Zhou K, Perelman SS. et al. Non-Viral CRISPR/Cas Gene Editing In vitro and In vivo Enabled by Synthetic Nanoparticle Co-Delivery of Cas9 mRNA and sgRNA. Angew Chem Int Ed Engl. 2017;56:1059-63
138. Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF. et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34:184 -+
139. Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z. et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015;523:481-5
140. Yu C, Liu Y, Ma T, Liu K, Xu S, Zhang Y. et al. Small Molecules Enhance CRISPR Genome Editing in Pluripotent Stem Cells. Cell Stem Cell. 2015;16:142-7
141. Jakočiūnas T, Bonde I, Herrgård M, Harrison SJ, Kristensen M, Pedersen LE. et al. Multiplex metabolic pathway engineering using CRISPR/Cas9 in Saccharomyces cerevisiae. Metab Eng. 2015;28:213-22
142. Doench JG, Hartenian E, Graham DB, Tothova Z, Hegde M, Smith I. et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotechnol. 2014;32:1262-7
Author contact
![]() Corresponding authors: Dr. Ning Wang (E-mail: ckwanghk); Prof Yibin Feng (E-mail: yfenghk).
Corresponding authors: Dr. Ning Wang (E-mail: ckwanghk); Prof Yibin Feng (E-mail: yfenghk).