Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Biology of Aabs with relevance...
Exchange of immunoglobulins...
Amyloid-β Aabs
Microtubule protein tau Aabs
Neurofilament Aabs
α-Synuclein Aabs
Conclusion and future directions
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2022; 12(7):3045-3056. doi:10.7150/thno.72126 This issue Cite
Review
Autoantibodies targeting neuronal proteins as biomarkers for neurodegenerative diseases
Gabriela Kocurova1, Jan Ricny1, Saak V. Ovsepian2 ![]()
1. Experimental Neurobiology Program, National Institute of Mental Health, Klecany, Czech Republic.
2. Faculty of Science and Engineering, University of Greenwich London, Chatham Maritime, Kent, ME4 4TB, United Kingdom.
Received 2022-2-17; Accepted 2022-3-9; Published 2022-3-28
Abstract

Neurodegenerative diseases (NDDs) are associated with the accumulation of a range of misfolded proteins across the central nervous system and related autoimmune responses, including the generation of antibodies and the activation of immune cells. Both innate and adaptive immunity become mobilized, leading to cellular and humoral effects. The role of humoral immunity in disease onset and progression remains to be elucidated with rising evidence suggestive of positive (protection, repair) and negative (injury, toxicity) outcomes. In this study, we review advances in research of neuron-targeting autoantibodies in the most prevalent NDDs. We discuss their biological origin, molecular diversity and changes in the course of diseases, consider their relevance to the initiation and progression of pathology as well as diagnostic and prognostic significance. It is suggested that the emerging autoimmune aspects of NDDs not only could facilitate the early detection but also might help to elucidate previously unknown facets of pathobiology with relevance to the development of precision medicine.
Keywords: Fluid biomarkers, autoimmunity, dementia, differential diagnosis, immunoglobins
Introduction
Neurodegenerative diseases (NDDs) are chronic incurable disorders of the Central Nervous System (CNS) characterized by a progressive decline of synaptic functions and irreversible neuronal loss, with devastating personal impact and overwhelming socio-economical costs. With aging as the main risk factor, the most prevalent NDDs such as Alzheimer's disease (AD), Parkinson's disease (PD), Dementia with Lewy bodies (DLB), Frontotemporal Lobar Neurodegeneration (FTLD), Amyotrophic Lateral Sclerosis (ALS), and Vascular Dementia (VD) are currently on the rise [1, 2]. Despite the considerable symptomatic overlap, NDDs are viewed as independent entities affecting specific functional systems of the CNS and manifesting via a set of distinctive symptoms and histopathological characteristics [3-7].
Amongst shared features of NDDs, deposition of misfolded proteins and fragments across CNS, neuroinflammation, dysregulation of glutamatergic signaling, oxidative stress with cytotoxic effects are the most prominent, contributing to neurological and psychiatric symptoms with behavioral impairments. Disruption of neuronal activity, synaptic transmission, and plasticity mechanisms are thought to be caused primarily by the accumulation of aggregation-prone toxic amyloid proteins in the CNS and dysregulation of Ca2+ homeostasis [8-13]. Due to the alleged causal role and differential prevalence in various NDDs, amyloid proteins and their fragments accumulating in the brain and cerebrospinal fluid (CSF) prompted much interest as biomarkers for diagnosis, patient stratification, and monitoring the disease progression [14-18]. The routine use of CNS tissue and CSF-based assays, however, is hampered by invasive procedures they involve with significant related health risks.
Currently, there is a major unmet need for low-cost, non-invasive, and reliable methods for the early detection of CNS diseases. With advances in sensing technologies, it is expected that new approaches will be developed to facilitate the accurate diagnosis of NDDs and timely interventions [17, 19, 20] (Fig. 1). Over recent years, autoantibodies (Aabs) have generated much interest as putative biomarkers for NDDs [21-23]. The abundance of Aabs in CSF and blood with their specific reaction to a range of neuronal proteins have been explicitly shown in preclinical studies as well as clinical reports involving patients [24-27]. As emerges from this review, while major progress has been made in the analysis and characterization of Aab response in NDDs, the field is far from maturity, with numerous outstanding issues impeding the effective translation of Aabs-based approaches in diagnostic laboratories and clinical practice.
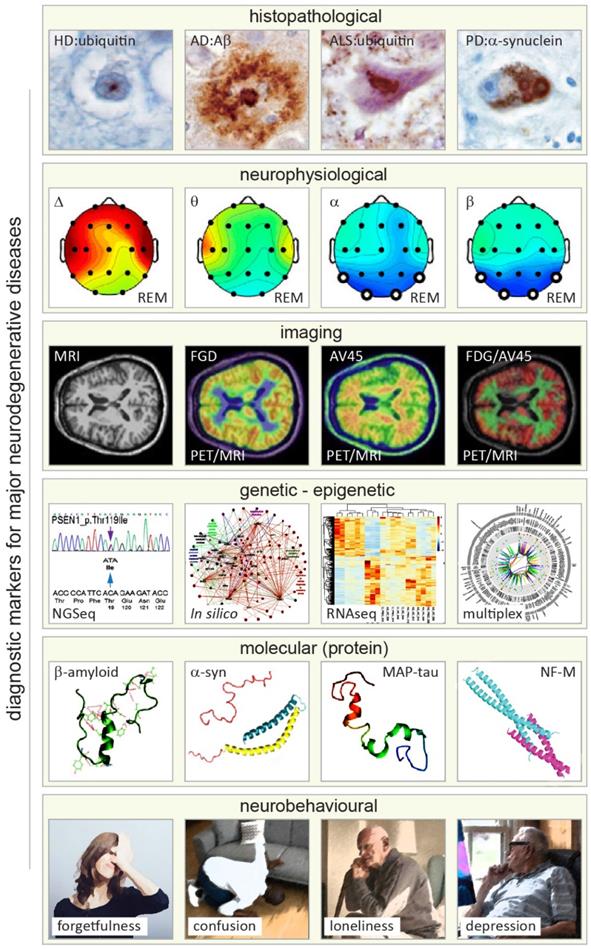
Primary approaches and readouts used for diagnosis of neurodegenerative diseases (NDDs). From top to bottom. First row: histopathological hallmarks of Huntington's, Alzheimer's, Lou Gehring's (known also as amyotrophic lateral sclerosis, ALS), and Parkinson's diseases shown in brain autopsy staining exemplifying deposition of distinguishing amyloid proteins (amyloid lesions, left to right). Adapted with permission from [142]. Second row: neurophysiological readouts (electroencephalographic (EEG) maps) illustrating the distribution of neural dynamics and activity across various brain structures and areas in Alzheimer's disease with reference to changes in four major types of EEG activity (Δ, θ, α and β bands) in rapid eye movement (REM) phase of sleep (left to right). Adapted with permission from [143]. Third row: magnetic and nuclear brain imaging (magnetic resonance imaging, MRI and positron emission tomography, PET) with various contrasts for detecting NDD-related changes in metabolic activity of the brain (Fluorodeoxyglucose, FDG) and amyloid distribution (Florpiramine F18; AV-45) targeting amyloid plaques, and hybrid MIR/PET, and dual FDG/AV-45 PET imaging modes (left to right). Adapted with permission from [144]. Fourth row: primary genomic, transcriptomic, and bioinformatics (in silico) methods applied for diagnosis of NDDs analyzing genetic and epigenetic alterations (left to right). Adapted with permission from [145-147]. Fifth row: 3D structure of four principal neuronal proteins enriched in amyloid deposits of the most prevalent NDDs (left to right). Note that for illustration purposes, the Ca2+ binding C-terminal domain of a-synuclein is truncated (pink). Adapted with permission from [148-151]. Sixth row: major neurobehavioral symptoms of NDDs (exemplified by symptoms of Alzheimer's disease), which can vary between NDD conditions (Illustrations modified from iflScience.com).
Biology of Aabs with relevance to NDDs
Antibodies (Ab) are large Y-shaped proteins used by the immune system for recognizing and neutralizing foreign materials, through activating the complement system and phagocytosis. Abs are generated by two types of B lymphocytes: B1 and B2. B2 cells produce Abs in follicles of secondary lymphatic organs, which in their majority are regular proteins derived after specific antigenic stimulation [28]. Some of these Abs may be directed against “auto” antigens, including those released from damaged and degenerating cells [29, 30]. Up until now, the role of Aabs produced by B2 cells remained unclear, with emerging data suggesting their homeostatic effects. Unlike, Abs generated by B1 cells are typically poly-reactive and can be produced in the absence of extrinsic antigens (i.e., bacteria, viruses, fungi) or self-antigens. The latter account for ~5% of the whole Ab pool of blood. Because of their broad reactivity, Abs of B1 cells play a key role in wide-ranging first-line defense against infections and foreign proteins. For the same reason, a small portion of these Abs could demonstrate auto-reactivity, i.e. qualify as natural Aabs [31]. In this way, B1 cells can play an important role in the clearance of cellular debris and removal of apoptotic tissue, protecting host organisms from toxic waste.
As natural immunoglobulins (Ig), Aabs occur in three isotypes: IgM, IgG, and IgA. IgM recognizes and binds post-apoptotic antigens and markers of cell senescence [32-34]. Although IgM is mainly produced by CD5+ B1 cells, in limited amounts it can be also generated by B2 cells [35]. Of all three immunoglobulins, IgM is the most abundant and of the lowest-affinity, whereas the amount of IgG and IgA are lower and vary considerably, with both showing higher immunoreactivity and specificity, as compared to IgM [36]. It is noteworthy that IgM producing B2 cells respond poorly to receptor-mediated activation and rarely undergo affinity maturation. They may, however, undergo a class switching to generate high-affinity pathogenic IgG [37]. Despite constant negative selection or targeted inactivation of self-reactive B lymphocytes in bone marrow, their positive selection can also occur. This process may lead to the emergence of immune cells producing Ab reacting to surface proteins of intact neurons and other brain cells, as well as peptides and proteins released after their pathological breakdown [38-40]. Such autoimmune reaction has been implicated in autoimmune response-related psychosis and schizophrenia [41, 42] as well as neural autoantibody-associated dementias (NABD) with signs of axonal degeneration [43, 44]. Quantitative analysis and profiling of Aabs targeting neuronal proteins, may, therefore, provide specific and instructive information on the onset, mechanisms, and severity of brain pathology.
Exchange of immunoglobulins between peripheral circulation and CNS
CNS is considered immunologically privileged with very limited exposure to antigens and restricted infiltration of Abs taking place under physiological conditions. This is due to physical and molecular barriers at the blood-CNS interface (known as blood-brain barriers, BBB) and elaborate system of meningeal lymphatic vessels (mLVs) which control the concentration and isoforms of Abs entering the CNS and guide immune cells out to cervical lymph nodes [45]. The latter is known to be the main site for presenting neuronal antigens to B lymphocytes and stimulating Ab production [46, 47]. Nonetheless, there is growing evidence for a quantitative correlation of Aabs of the CFS and blood (serum), with their concentration in the CSF significantly lower than that in the peripheral circulation [48, 49]. These findings suggest that under physiological conditions, a limited quantity of immunoglobulins can infiltrate the CNS from the peripheral circulation.
Many disorders affecting CNS, including NDDs, are associated with the disintegration of BBB, which may lead to an out-of-control outflow of neuronal and glial proteins with activation of autoimmune response [50, 51]. Accordingly, a variety of Aabs target neuronal and glial proteins, and their changes can be detected in the blood and CSF of patients with NDDs. Amongst these, Aabs specific to neurofilament heavy subunit, tubulin, glial fibrillary acidic protein, S100b protein, tau, β-amyloid peptide, α-synuclein, myelin basic protein (MBP), and heparan sulfate proteoglycan are most extensively studied [52-56]. Despite low amounts of Aabs in CSF, considerable evidence suggests their biological effects. In AD, for instance, Aabs may play dual, pathogenic, and protective roles, with levels of Ig recognizing self-antigens (protein tau, Aβ-amyloid peptide) correlating with specific disease stages and associated comorbidities.
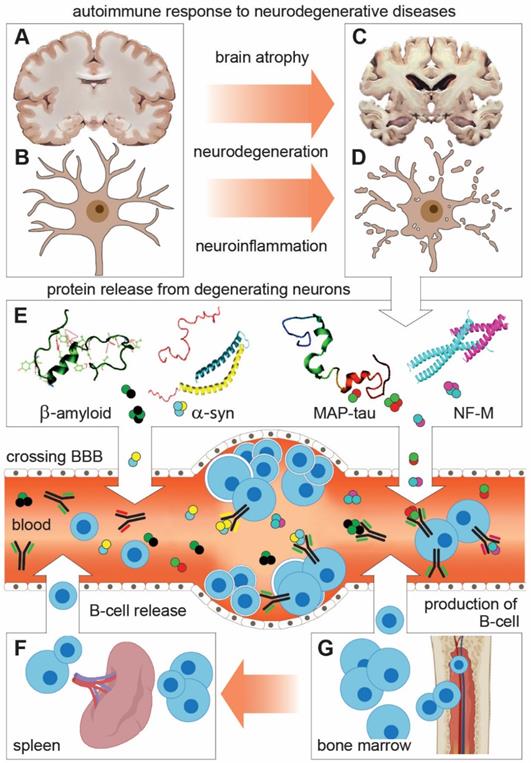
Schematic representation of shared features and mechanisms of autoimmune response to neuronal proteins in neurodegenerative diseases (NDDs). (A-D) Degeneration of neurons leads to brain atrophy and the release of neuronal proteins and their fragments (autoantigens) in the interstitial space and CSF. (E) From there, autoantigens cross BBB to enter the peripheral circulation (Blood and lymphatic system) where they encounter and activate B lymphocytes to produce and release neuronal protein-specific autoantibodies (Aabs). This reaction underlies changes in Aabs profile and activity in peripheral circulation, which can be taken as indicative (biomarker) for NDDs. (F and G) B lymphocytes are produced in the bone marrow to be released directly in blood or stored in the spleen and released in blood as part of the immunogenic response.
Amyloid-β Aabs
Gaskin et al. presented the first evidence for Aβ Aabs in the peripheral circulation of AD patients [57]. This was followed by reports of Ig in serum of healthy and AD patients [58-65] in free form as well as in complex with Aβ, with complexation affecting the sensitivity of detection methods. In studies with dissociation of Aβ-Ab complex, the amount of detected Aβ was less variable [66]. Most of Aβ42 and Aβ40 Aab studies showed a lower titer of unbound Ig in sera of AD patients as compared to healthy controls [59, 64, 67-69], with some reports also showing no difference [58] (Table 1). The lower levels of Aβ Aabs in serum of AD suggest the reduced passage of Aβ to blood, which could accelerate its accumulation in CNS and development of fibrillary deposits. Using ELISA for monomeric Aβ42 and aggregated soluble Aβ, Nath et al. found that titers of Aβ42 Aabs in serum of AD patients are higher as compared to patients with multiple sclerosis (MS) and HIV encephalitis. Comparative analysis showed that a significant fraction of Aβ Aabs in redox-treated serum peptides of clinical AD was reactive to Aβ oligomers, which were also reduced as compared to that in age-matched healthy controls. This observation infers that Aβ oligomer can leak from the CNS to plasma, supporting the potential usefulness of Aβ oligomer immunotherapy [68]. Of note, in AD, the titre of monomeric Aβ1-42 Aabs in serum was lower than that for aggregated Aβ1-42, a finding implying that the immune response to Aβ targets specific conformational epitopes, which have higher immunogenicity in Aβ aggregates [63]. Another report comparing serum IgG against Aβ1-42 mono- and oligomers in AD, MCI, and cognitively normal controls (10/group) with subtraction of polyvalent antibodies binding and dissociating Ab-Ig complexes did not find differences between the three groups [61]. In contrast, analysis of the level of Aabs reactive to Aβ25-35 oligomers in serum showed their increase in AD patients as compared to controls [70]. This short peptide is regarded as the main toxic domain of Aβ [71]. Interestingly, longitudinal studies of Aabs changes in AD showed that the levels of Aabs to aggregated Aβ variants in sera increase during the mild to moderate phase of the disease but decline with the progression of the pathology into the severe phase [70].
A summary table of Aβ Aabs values in NDD patients versus controls
| Directionality | Index change | Diagnosis | Material | Method | Aβ, variant | Reference |
|---|---|---|---|---|---|---|
| Increase | 1.33; 1.41 | AD | Serum | ELISA | Mono- Agg- | [63] |
| No change | 1.04 | AD | Serum | ELISA | - | [58] |
| Decrease | 0.53 | AD | Serum | IP | - | [67] |
| Decrease | 0.69 | AD | CSF | ELISA | - | [59] |
| Increase | 10.2; 47.5 | AD<5y; AD>15y | Serum | ELISA | Oligo- | [152] |
| Increase | 40; 5; 160; 30 | AD short, long (stages) | Serum | ELISA | - | [70] |
| Increase | 1.05 - 1.27 | AD, mild, severe | Serum | ELISA | Mono- Oligo- | [61] |
| Increase | 2.2 | AD | Serum | ELISA | - | [66] |
| No change | 0.95 | AD | Plasma | ELISA | - | [60] |
| Decrease | 0.41 | AD | Serum | ELISA | - | [65] |
| Increase | 1.23 | AD | Serum | ELISA | - | [74] |
| Decrease | 0.71 | AD | Serum | ELISA | Oligo- | [68] |
| Decrease | 0.69 | AD | Serum | ELISA | - | [153] |
| Decrease | 0.51 | AD | Serum | ELISA | - | [69] |
| No change | 1.0 | AD | Plasma | TAPIR | - | [154] |
| No change | 1.0 | AD | Plasma | Pep. microarray | Oligo - | [48] |
| Decrease | 0.88 | AD | Serum | ELISA | - | [155] |
| Increase | 1.36-1.69 | AD | Serum | ELISA | - | [66, 72] |
| No change, | 0.96; 1.0 | PD, PDND | Serum, CSF | ELISA | - | [137, 156] |
| Increase | 3.68 | AD | Plasma | EIA/RIA | - | [62] |
| Decrease | 0.63 | AD | Serum | ELISA | Fragments | [64] |
| No change | 1.01 | VD | CSF | ELISA | - | [137] |
| Increase | 1.35; 1.14 | DLB/PD; AD/FTD | CSF | ELISA | - | [137] |
Using an affinity purification approach, Mruthinti et al. found a higher titer of IgG binding Aβ42 peptide in plasma of AD [62]. This observation agrees with the results of the earlier report with the use of acidic dissociation to measure bound and unbound antibodies, showing that their levels in AD exceeded that of age-matched controls [66, 72]. It is important to note that exposure to low pH can cause partial denaturation of Aabs which can lead to increased reactivity [73]. Other tests using Aabs specific to Aβ (21-37), and monoclonal mouse 6E10 antibody (mAb 6E10) that binds to Aβ (3-8) were also able to detect Aβ-IgG complexes in serum and CSF, which were more prevalent in AD patients. In combined analytical assays with clinical tests in AD patients, the titer of immune complexes in CSF and serum negatively correlated with the cognitive performance of subjects [74]. A recent random-effect meta-analysis containing 30 case-control studies with a total of 2901 individuals (1311 and 1590, AD and healthy subjects, respectively) demonstrated an increase of Aβ IgG in the blood of AD, whereas IgM was lower in these subjects as compared to healthy. In the same report, assessments of CSF Aβ Aabs in AD against healthy showed no difference, while meta-regression analyses suggestive of measurable sex-related effects [75]. Overall, although many studies advocate the diagnostic relevance of Aβ Aabs changes in the CSF and serum of AD, the results are controversial, calling for further research with careful stratification of subjects and the use of standardized methods.
Microtubule protein tau Aabs
Microtubule-associated protein tau, which in AD becomes hyperphosphorylated (p-tau), is the main constituent of neurofibrillary tangles. An increase in the level of p-tau in the brain and CSF has been considered as one of the key biomarkers of AD [76-78]. The presence of tau-reactive IgG and IgM have been reported in CSF and sera of AD patients, as well as in healthy controls [52, 53, 79-83] (Table 2). Tau Aabs were shown in various immunoglobulin (IVIG) products from large cohorts of healthy donors [81, 84-86] as well as in children [82] suggesting that they are unlikely to be harmful [82, 84] and may have some physiological role. Bartos and co-workers observed lower levels of tau-reactive Aabs in serum of AD patients as compared to controls, with titers declining further with the progression of the pathology [52]. Considerable evidence suggests that anti-tau antibodies can infiltrate the CNS through impaired BBB to bind neurofibrillary tangles [87, 88] as well as intracellular tau deposits. The intracellular interaction may happen similar to the binding of paraneoplastic Ab to nuclear or cytoplasmic elements [89, 90]. This process may interfere with the cytoskeletal functions, aggravating the disease process. It is interesting to note that reduction of tau Aabs in serum was also reported in PD [91]. On the other hand, Rosenmann et al. have reported higher levels of IgM class Abs against p-tau in AD [83]. This was, however, a pilot study using low number of samples, hence, the results warrant independent verification. Klaver et al. tested the binding of IgG and IgM from AD, MCI, and control subjects to p-tau and tau, using as antigens 196-207 tau peptide, as well as full-length variants (tau and p-tau at Serine-199 and Serine-202). Authors found specific antibodies to both p-tau and tau in most subjects, regardless of cognitive status, with increased specific IgG binding to p-tau (an increase in the p-tau IgG ratio) detected in MCI subjects as compared to AD patients and healthy controls [80].
A summary table of tau and neurofilament Aabs values in NDD patients versus controls
| Directionality | Index change | Diagnosis | Material | Method | MAP tau, variants | Authors/Year |
|---|---|---|---|---|---|---|
| No change | 0.83, 1.0 | MCI, AD | Serum | ELISA | IgM non p- tau | [80] |
| No change | 0.95, 0.66 | MCI, AD | Serum | ELISA | IgM p-tau | [80] |
| Increase | 1.7, 1.02 | MCI, AD | Serum | ELISA | IgG p-tau | [80] |
| Increase | 1.7 | AD | Intrathec. synth. | ELISA | - | [79] |
| Decrease | 0.80 | AD | Serum | ELISA | - | [52] |
| Decrease | 0.45, 0.68 | MCI | Serum, CSF | ELISA | - | [157] |
| Increase | 2.0 | AD | Serum | ELISA | p-tau | [83] |
| Increase | 2.5 | MS | Intrathec. synth. | ELISA | - | [158] |
| Increase | 1.95 | PD vs PDND | Serum | ELISA | - | [91] |
| Increase | 2.2 | AD | Intrathec. synth. | ELISA | NF-H | [79] |
| Decrease | 0.62 | AD | Serum | ELISA | NF-H | [52] |
| No change | 1 | AD | Intrathec. synth. | ELISA | NF-L | [79] |
| No change | 1 | AD | Serum | ELISA | NF-L | [52] |
Using circulating IgGs, it was shown that they can recognize modified tau variants, which differ in their characteristics [81, 85]. These observations suggest that despite modifications of tau protein by aggregation, formation of paired helical filaments (PHFs), phosphorylation, and polymerization [92-96], they are still recognized by specific Aabs. Preliminary data from our laboratory (PI, Dr. Ricny) showed that tau Ab from the serum of AD patients interact equally with both, recombinant and natural monomeric tau derived from brain homogenates [97]. On the contrary, antibodies isolated from IVIG and pooled from the plasma of healthy controls showed stronger reactivity with recombinant tau fragment (155-421 aa) and with aggregated forms [81, 85]. Notwithstanding considerable research, currently, there is a lack of consensus if tau Aabs levels are altered in the peripheral circulation of AD and MCI patients. It is important to note that the results of clinical trials with anti-Aβ antibodies leading to the removal of amyloid plaques suggest that neurofibrillary tangle pathology is secondary to the build-up of amyloid deposits, and the reversal of tau pathology might be important in the onset of clinical benefits with cognitive improvements [98]. Currently, it is unclear if anti-tau Aabs have any protective role or can influence the formation of NF tangles, which in turn could influence the spread of tau pathology and cognitive functions.
Neurofilament Aabs
Neurofilaments (NFs) belong to a family of intermediate filaments with their diameter (~10 nm) falling between two other cytoskeletal polymers, i.e., microtubules (~25 nm) and actin (~6 nm). Based on their gene sequence and structural characteristics, NFs are divided into six types (I-VI) [99, 100]. Adult neurons in CNS are enriched with pan-neuronal type IV NFs (i.e. NF triplet proteins light, middle and heavy, NF-L, NF-M, NF-H, and α-internexin), while peripheral neurons express NF triplet proteins with type III IF peripherin [101, 102]. NFs are integral structural elements of synapses, enriched especially at postsynaptic sites of glutamatergic synapses, with their impairments disrupting synaptic plasticity and memory formation in animal models, and implicated in several NDDs and neuropsychiatric conditions [103].
NF deposits were found to co-localize with tau tangles in brains affected by AD [104] as well as within LBs of dopaminergic cells in PD [105] and dystrophic neurites of ALS motor neurons [106]. Increased levels of NF in the blood and CSF infers axonal injury, which can result as a part of normal brain aging and pathological processes, such as autoimmune diseases, inflammation, vascular and traumatic disorders of the CNS and PNS [101, 107, 108] (Table 2). Higher NF levels in peripheral circulation have been reported in association with neuronal damage caused by several NDDs [17, 109-111]. Although the ubiquitous presence of NFs in the CNS and their release in CSF and blood in various NDDs rule out their utility for differential diagnosis, increase in NF levels and reactive Ab provide a sensitive means for detecting the onset as well as the progression of neuronal degeneration. Fialova et al. used anti-NF Aab profiling to monitor disease progression in patients diagnosed with early MS and clinically isolated syndrome [112]. In addition to distinguishing various phases of MS (i.e. relapsing-remitting), the approach showed potential for detecting secondary progressive phases of the pathology related to continuous spillage of NF. It is important to note that, like Aβ, tau, and α-syn Aabs, NF Aabs can be detected in the serum and CSF of not only diseased but also healthy individuals [112, 113] (Table 2). Under certain conditions, NF Aabs seem to contribute to the pathogenesis of several NDDs and can aggravate the disease process in AD patients [114-117].
Soussan et al. compared NF Aab profiles in serum of AD patients and healthy controls. Unlike controls showing equal binding for different isoforms of NF-H (bovine ventral root and dorsal root NF-H) without changing their specificity during aging, in AD, the levels of Aabs against ventral root cholinergic NF-H was higher than those directed against dorsal root NF-H. The phosphoserine content analysis of NFs showed its higher levels in ventral as compared to that of dorsal root NF-H, with Aabs from AD patients binding more effectively phosphorylated epitopes, which show higher prevalence in ventral root NF-H [118]. Of note, serum levels of NF-H Aabs in AD patients were lower as compared to healthy controls, while the levels of NF-L Aabs remained unaltered [52]. Moreover, AD patients had elevated intrathecal synthesis of tau and NF-H Aabs [79] while patients with multi-infarct dementia showed higher titers of NF-H IgG as compared to the serum of healthy controls. In the context of the current discussion, it is important to note that the prevalence of sub-classes of NF Aabs varies in different neuropsychiatric diseases, which might be due to the immunogenicity of different NF sub-classes (NF-H/NF-M/NF-L) resulting from various modifications of the protein and epitope sites, including phosphorylation of the C-terminal domain of NF, which might impact the pathogenicity of NF Aabs [115, 118].
α-Synuclein Aabs
Accumulation of insoluble and misfolded α-syn in neurons leads to synaptic failure with the build-up of fibrils constituting Lewy bodies (LB) and neurites of DLB and PD [4, 119]. Based on the localization and clinical signs of LB, the Newcastle-McKeith criteria distinguishes three main forms: (1) brain stem predominant form, affecting IX-X motor nucleus, locus coeruleus, and substantia nigra (2) limbic form affecting the amygdala, trans-entorhinal cortex, and cingulate cortex, and (3) neocortical form targeting frontal, temporal and parietal areas [120, 121]. Considerable data suggest that α-syn upregulation alone can lead to synaptic pathology and set on the formation of LB, even with retained physiological conformation [122]. Like Aβ and tau protein, pathological increase in α-syn is associated with local immune reaction in the brain as well as systemic response. In the PD brain, for instance, aggregates of α-syn in substantia nigra co-localize with deposits of IgG [123], indicating that α-syn build-up induces local Aab response. Of note, exogenously applied monoclonal antibodies to α-syn can alter the rate of protein aggregates in cellular models and animal studies of PD [124-126], inferring that α-syn Aabs may influence the onset and progression of the disease [127, 128].
The results of the analysis of α-syn Aabs in the blood of PD patients and comparison with controls vary considerably (Table 3). While some reports found α-syn Aabs titers unaltered [129-131], others showed significant changes. Besong-Agbo and co-workers [132], for instance, report lower α-syn Aabs levels in sera of PD compared to healthy controls and AD patients. Another small cohort study divided patients into two groups (1) with ≤5-years and (2) ≥10-years PD and described higher levels of α-syn Aabs in sera of both PD patient groups compared to healthy controls. Interestingly, the antibody activity in the second group of patients gradually declined over time, implying that the auto-immune response can be regulated throughout the disease process [128]. Similar findings were reported by other studies of α-syn Aabs in PD sera [133] and plasma [134]. The level of α‐syn Aabs and changes appears to be gender-dependent, with PD and healthy men showing typically higher titers than women [135]. In addition to the blood, alterations of α-syn were investigated in the CSF. Akhtar, Horvath et al [134, 135] have found higher CSF Aabs levels in PD, unlike Heinzel et al [129] reporting no differences from healthy controls. α-syn Aabs levels were also reported to be increased in DLB, and to a lesser extent in AD [136, 137] (Table 3). Finally, a significant rise of α-syn Aabs was found in behavioral variant FTD (bvFTD) patients, where serum levels of α-syn Aabs were significantly higher compared to PD patients [91]. Overall, from the autoimmune point of view, the response of Aabs to α-syn varies widely across several NDDs and can be influenced by multiple factors, including the stage of diseases, its severity, patient gender, and others.
A summary table of α-synuclein Aabs values in NDD patients versus controls
| Directionality | Index change | Diagnosis | Material | Method | α-syn variant | Authors/Year |
|---|---|---|---|---|---|---|
| Decrease | 0.90, 0.91 | VD, AD/FTD | CSF | ELISA | - | [137] |
| Increase | 1.27 | DLB/PD | CSF | ELISA | - | [137] |
| Increase | 1.53 | PD | CSF | ELISA | - | [135] |
| Decrease | 0.94, 0.69 | AD, PD | Serum | ELISA | - | [132] |
| No change | 0.63 | PD | Serum | ELISA | - | [130] |
| No change | 0.61, 0.81 | PD | Serum, CSF | ELISA | - | [129] |
| No change | 0.82 | PD | Serum | ELISA | - | [131] |
| Increase | 16.2, 4.0;4.0, 2.0 | PD<5y PD>10y | Serum | ELISA | Mono- Oligo, - | [159] |
| Increase | 1.39, 1.3;1.29, 1.2 | PD mild,moderate | Serum,CSF | ELISA | - | [134] |
| No change | 1.1, 0.9 | PD, PDND | Serum, CSF | ELISA | - | [129, 137] |
| Increase | 1.3 - 3.7 | PD | Serum | EIS | - | [160] |
| Increase | 2.5 - 6.0 | PD | Serum | ELISA | Mono- | [133] |
| Conditional | - | PD | Serum | WB | - | [161] |
| Decrease in HA Abs | 1.37 | PD | Plasma | ELISA | - | [162] |
| Increase | 2.5 | PD | Serum | ELISA | - | [163] |
| Increase | 6.3 - 10.7 | PD | Serum | ELISA | - | [164] |
| Increase | 1.32 | PD | Serum | EIS | - | [165] |
| No change | 1 | PD, PDND | Serum | ELISA | - | [91] |
Conclusion and future directions
Despite two decades of in-depth research and major progress in developing biomarkers for CNS disorders, the definitive diagnosis of NDDs remains a major challenge. The current diagnostic gold standard - positron emission tomography (PET) - has low sensitivity and is of limited availability, due to high costs and requirements for specialized infrastructure and skilled staff, as well as potential health risks related to the use of radioactive tracers. Substantial drawbacks are also associated with the use of CNS tissue as well as CSF-based assays involving biopsies and lumbar puncture, which necessitates invasive procedures and related major health risks. The emerging Aabs based blood tests seem to offer a specific, rapid, and affordable approach for diagnosis of NDD without major risks and adverse effects. Nevertheless, significant challenges and questions remain, which impede their effective translation and widespread clinical use, calling for further research and optimization in clinical trials. One of the key difficulties is imposed by the discovery of significant amounts of neuronal Aabs in the peripheral circulation in healthy subjects, inferring their potential physiological role, and questing the specificity of selected Aabs for a particular NDD. Another major challenge is imposed by the results of comparative studies, which demonstrate considerable variations of Aabs levels in the peripheral circulation of NDDs that frequently deviate from changes in Aabs titers in CSF. These observations also substantiate the highly complex nature of the immune response to NDDs and underscore the potential shortcomings of utilized detection methods. Together with numerous conflicting reports and outstanding methodological issues, the above-listed considerations call for revision and improvements of sample preparation and standardization of sensing methods. They also highlight the need for more stringent stratification of target groups and profiling of Aabs, to ensure accurate and specific detection and quantification of Aabs. In this context, the use of genetic methods is especially warranted, given the causative and predisposing effects of specific genes in NDDs. Because of the association of NDD with genetic alterations (e.g., AopE4) [138, 139], the latter might also influence the level and activity of Aabs as biomarkers. Importantly, the emerging inconsistent data highlight numerous outstanding biological questions, which require careful analysis and interpretation. For instance, the cellular origin, induction mechanisms, and potential significance of the physiological presence of Aabs in peripheral circulation remain to be elucidated. Likewise, it must be shown if higher levels of oligomer Aabs as compared to monomers, and changes in their ratio, is of any diagnostic or biological importance under normal and disease conditions. Finally, major outstanding questions remain in the basic neurobiology of neurodegenerative diseases, with important, previously unknown, mechanisms regulating the production, processing, and secretion of amyloid peptides reported recently [140, 141]. As binding of Aabs can influence the propensity of α-syn, tau, or Aβ42 for aggregation in fibrillary deposits, alterations in titers of oligomer-specific Aabs might influence the onset of amyloid depositions as well as the pathological spread of misfolded proteins throughout the CNS. Whether this is the case or not remains to be demonstrated. Addressing these and many other technical challenges and scientific questions underscored throughout this study warrants further preclinical research and clinical trials, with the view of improving the diagnostic and therapeutic utility of Aabs in the foreseeable future.
Acknowledgements
This study was supported by the project Sustainability for the National Institute of Mental Health (NIMH, IN: 00023752) under grant number LO1611, from the Ministry of Education, Youth and Sports of the Czech Republic under the NPU I program.
Author Contributions
G.K., J.R., and S.V.O. designed the study; G.K., J.R., and S.V.O. wrote and revised the manuscript; G.K. and S.V.O. prepared the illustrations and tables. All authors have read and agreed to the final submission version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hou YJ, Dan XL, Babbar M, Wei Y, Hasselbalch SG, Croteau DL. et al. Ageing as a risk factor for neurodegenerative disease. Nature Reviews Neurology. 2019;15:565-81
2. Feigin VL, Nichols E, Alam T, Bannick MS, Beghi E, Blake N. et al. Global, regional, and national burden of neurological disorders, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18:459-80
3. Focus on neurodegenerative disease. Nat Neurosci. 2018; 21: 1293.
4. Goedert M, Jakes R, Spillantini MG. The Synucleinopathies: Twenty Years On. J Parkinsons Dis. 2017;7:S51-S69
5. Koikkalainen J, Rhodius-Meester H, Tolonen A, Barkhof F, Tijms B, Lemstra AW. et al. Differential diagnosis of neurodegenerative diseases using structural MRI data. Neuroimage Clin. 2016;11:435-49
6. Marsh AP. Molecular mechanisms of proteinopathies across neurodegenerative disease: a review. Neurol Res Pract. 2019;1:35
7. Saxena S, Caroni P. Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron. 2011;71:35-48
8. Camandola S, Mattson MP. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017;36:1474-92
9. Ovsepian SV, O'Leary VB, Zaborszky L, Ntziachristos V, Dolly JO. Synaptic vesicle cycle and amyloid beta: Biting the hand that feeds. Alzheimers Dement. 2018;14:502-13
10. Ovsepian SV, O'Leary VB, Zaborszky L, Ntziachristos V, Dolly JO. Amyloid Plaques of Alzheimer's Disease as Hotspots of Glutamatergic Activity. Neuroscientist. 2019;25:288-97
11. Wishart TM, Parson SH, Gillingwater TH. Synaptic vulnerability in neurodegenerative disease. J Neuropathol Exp Neurol. 2006;65:733-9
12. Henstridge CM, Pickett E, Spires-Jones TL. Synaptic pathology: A shared mechanism in neurological disease. Ageing Res Rev. 2016;28:72-84
13. Alzheimer's Association Calcium Hypothesis W. Calcium Hypothesis of Alzheimer's disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement. 2017;13:178-82 e17
14. Cummings J. The Role of Biomarkers in Alzheimer's Disease Drug Development. Adv Exp Med Biol. 2019;1118:29-61
15. Polanco JC, Li C, Bodea LG, Martinez-Marmol R, Meunier FA, Gotz J. Amyloid-beta and tau complexity - towards improved biomarkers and targeted therapies. Nat Rev Neurol. 2018;14:22-39
16. Simren J, Ashton NJ, Blennow K, Zetterberg H. An update on fluid biomarkers for neurodegenerative diseases: recent success and challenges ahead. Curr Opin Neurobiol. 2020;61:29-39
17. Zetterberg H, Burnham SC. Blood-based molecular biomarkers for Alzheimer's disease. Mol Brain. 2019;12:26
18. Zetterberg H, Schott JM. Biomarkers for Alzheimer's disease beyond amyloid and tau. Nat Med. 2019;25:201-3
19. O'Bryant SE, Mielke MM, Rissman RA, Lista S, Vanderstichele H, Zetterberg H. et al. Blood-based biomarkers in Alzheimer disease: Current state of the science and a novel collaborative paradigm for advancing from discovery to clinic. Alzheimers Dement. 2017;13:45-58
20. Ehrenberg AJ, Khatun A, Coomans E, Betts MJ, Capraro F, Thijssen EH. et al. Relevance of biomarkers across different neurodegenerative diseases. Alzheimers Res Ther. 2020;12:56
21. Hampel H, O'Bryant SE, Molinuevo JL, Zetterberg H, Masters CL, Lista S. et al. Blood-based biomarkers for Alzheimer disease: mapping the road to the clinic. Nat Rev Neurol. 2018;14:639-52
22. Pruss H. Autoantibodies in neurological disease. Nat Rev Immunol. 2021;21:798-813
23. Obrocki P, Khatun A, Ness D, Senkevich K, Hanrieder J, Capraro F. et al. Perspectives in fluid biomarkers in neurodegeneration from the 2019 biomarkers in neurodegenerative diseases course-a joint PhD student course at University College London and University of Gothenburg. Alzheimers Res Ther. 2020;12:20
24. DeMarshall CA, Han M, Nagele EP, Sarkar A, Acharya NK, Godsey G. et al. Potential utility of autoantibodies as blood-based biomarkers for early detection and diagnosis of Parkinson's disease. Immunol Lett. 2015;168:80-8
25. Nagele E, Han M, DeMarshall C, Belinka B, Nagele R. Diagnosis of Alzheimer's Disease Based on Disease-Specific Autoantibody Profiles in Human Sera. PLoS One. 2011;6:e23112
26. Nagele EP, Han M, Acharya NK, DeMarshall C, Kosciuk MC, Nagele RG. Natural IgG autoantibodies are abundant and ubiquitous in human sera, and their number is influenced by age, gender, and disease. PLoS One. 2013;8:e60726
27. Reddy MM, Wilson R, Wilson J, Connell S, Gocke A, Hynan L. et al. Identification of Candidate IgG Antibody Biomarkers for Alzheimer's Disease Through Screening of Synthetic Combinatorial Libraries. Cell. 2011;144:132-42
28. Hoffman W, Lakkis FG, Chalasani G. B Cells, Antibodies, and More. Clin J Am Soc Nephrol. 2016;11:137-54
29. Neiman M, Hellström C, Just D, Mattsson C, Fagerberg L, Schuppe-Koistinen I. et al. Individual and stable autoantibody repertoires in healthy individuals. Autoimmunity. 2019;52:1-11
30. Jain RW, Yong VW. B cells in central nervous system disease: diversity, locations and pathophysiology. Nat Rev Immunol. 2021
31. Coutinho A, Kazatchkine MD, Avrameas S. Natural autoantibodies. Curr Opin Immunol. 1995;7:812-8
32. Hardy RR, Hayakawa K. Development of B cells producing natural autoantibodies to thymocytes and senescent erythrocytes. Springer Semin Immunopathol. 2005;26:363-75
33. Madi A, Hecht I, Bransburg-Zabary S, Merbl Y, Pick A, Zucker-Toledano M. et al. Organization of the autoantibody repertoire in healthy newborns and adults revealed by system level informatics of antigen microarray data. Proc Natl Acad Sci U S A. 2009;106:14484-9
34. Mouthon L, Haury M, Lacroix-Desmazes S, Barreau C, Coutinho A, Kazatchkine MD. Analysis of the normal human IgG antibody repertoire. Evidence that IgG autoantibodies of healthy adults recognize a limited and conserved set of protein antigens in homologous tissues. J Immunol. 1995;154:5769-78
35. Zhou ZH, Notkins AL. Polyreactive antigen-binding B (PAB+) cells are widely distributed and the PAB+ population consists of both B-1+ and B-1- phenotypes. Clin Exp Immunol. 2004;137:88-100
36. Sigounas G, Kolaitis N, Monell-Torrens E, Notkins AL. Polyreactive IgM antibodies in the circulation are masked by antigen binding. J Clin Immunol. 1994;14:375-81
37. Baumgarth N, Tung JW, Herzenberg LA. Inherent specificities in natural antibodies: a key to immune defense against pathogen invasion. Springer Semin Immunopathol. 2005;26:347-62
38. Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don't see). Nat Rev Immunol. 2014;14:377-91
39. Cano RLE LH. Introduction to T and B lymphocytes. In: Anaya JM SY, Rojas-Villarraga A, et al. Autoimmunity: From Bench to Bedside. Bogota (Colombia): El Rosario University Press. 2013
40. Giannoccaro MP, Gastaldi M, Rizzo G, Jacobson L, Vacchiano V, Perini G. et al. Antibodies to neuronal surface antigens in patients with a clinical diagnosis of neurodegenerative disorder. Brain, behavior, and immunity. 2021;96:106-12
41. Jezequel J, Johansson EM, Leboyer M, Groc L. Pathogenicity of Antibodies against NMDA Receptor: Molecular Insights into Autoimmune Psychosis. Trends Neurosci. 2018;41:502-11
42. Jorratt P, Hoschl C, Ovsepian SV. Endogenous antagonists of N-methyl-d-aspartate receptor in schizophrenia. Alzheimers Dement. 2021;17:888-905
43. Hansen N. Current Nosology of Neural Autoantibody-Associated Dementia. Front Aging Neurosci. 2021;13:711195
44. Hansen N, Juhl AL, Grenzer IM, Hirschel S, Teegen B, Fitzner D. et al. Cerebrospinal Fluid Total Tau Protein Correlates With Longitudinal, Progressing Cognitive Dysfunction in Anti-Neural Autoantibody-Associated Dementia and Alzheimer's Dementia: A Case-Control Study. Front Immunol. 2022
45. Sun BL, Wang LH, Yang T, Sun JY, Mao LL, Yang MF. et al. Lymphatic drainage system of the brain: A novel target for intervention of neurological diseases. Prog Neurobiol. 2018;163-164:118-43
46. Ahn JH, Cho H, Kim J-H, Kim SH, Ham J-S, Park I. et al. Meningeal lymphatic vessels at the skull base drain cerebrospinal fluid. Nature. 2019;572:62-6
47. Hu X, Deng Q, Ma L, Li Q, Chen Y, Liao Y. et al. Meningeal lymphatic vessels regulate brain tumor drainage and immunity. Cell Res. 2020;30:229-43
48. Britschgi M, Olin CE, Johns HT, Takeda-Uchimura Y, LeMieux MC, Rufibach K. et al. Neuroprotective natural antibodies to assemblies of amyloidogenic peptides decrease with normal aging and advancing Alzheimer's disease. Proc Natl Acad Sci U S A. 2009;106:12145-50
49. Kheirkhah R, DeMarshall C, Sieber F, Oh E, Nagele RG. The origin and nature of the complex autoantibody profile in cerebrospinal fluid. Brain Behav Immun Health. 2020;2:100032
50. Sweeney MD, Ayyadurai S, Zlokovic BV. Pericytes of the neurovascular unit: key functions and signaling pathways. Nat Neurosci. 2016;19:771-83
51. Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol Rev. 2019;99:21-78
52. Bartos A, Fialová L, Švarcová J. Lower Serum Antibodies Against Tau Protein and Heavy Neurofilament in Alzheimer's Disease. J Alzheimers Dis. 2018;64:751-60
53. Terryberry JW, Thor G, Peter JB. Autoantibodies in Neurodegenerative Diseases: Antigen-Specific Frequencies and Intrathecal Analysis. Neurobiol Aging. 2017;19:205-16
54. Avrameas S, Dighiero G, Lymberi P, Guilbert B. Studies on natural antibodies and autoantibodies. Ann Immunol (Paris). 1983;134d:103-13
55. Poletaev AB, Morozov SG, Gnedenko BB, Zlunikin VM, Korzhenevskey DA. Serum anti-S100b, anti-GFAP and anti-NGF autoantibodies of IgG class in healthy persons and patients with mental and neurological disorders. Autoimmunity. 2000;32:33-8
56. Park H, Kim M, Kim HJ, Lee Y, Seo Y, Pham CD. et al. Heparan sulfate proteoglycans (HSPGs) and chondroitin sulfate proteoglycans (CSPGs) function as endocytic receptors for an internalizing anti-nucleic acid antibody. Sci Rep. 2017;7:14373
57. Gaskin F, Finley J, Fang Q, Xu S, Fu SM. Human antibodies reactive with beta-amyloid protein in Alzheimer's disease. J Exp Med. 1993;177:1181-6
58. Baril L, Nicolas L, Croisile B, Crozier P, Hessler C, Sassolas A. et al. Immune response to Aβ-peptides in peripheral blood from patients with Alzheimer's disease and control subjects. Neurosci Lett. 2004;355:226-30
59. Du Y, Dodel R, Hampel H, Buerger K, Lin S, Eastwood B. et al. Reduced levels of amyloid beta-peptide antibody in Alzheimer disease. Neurology. 2001;57:801-5
60. Hyman BT, Smith C, Buldyrev I, Whelan C, Brown H, Tang M-X. et al. Autoantibodies to amyloid-β and Alzheimer's disease. Ann Neurol. 2001;49:808-10
61. Klaver AC, Coffey MP, Smith LM, Bennett DA, Finke JM, Dang L. et al. ELISA measurement of specific non-antigen-bound antibodies to Aβ1-42 monomer and soluble oligomers in sera from Alzheimer's disease, mild cognitively impaired, and noncognitively impaired subjects. J Neuroinflammation. 2011;8:93 -
62. Mruthinti S, Buccafusco JJ, Hill WD, Waller JL, Jackson TW, Zamrini EY. et al. Autoimmunity in Alzheimer's disease: increased levels of circulating IgGs binding Aβ and RAGE peptides. Neurobiol Aging. 2004 25
63. Nath A, Hall E, Tuzova M, Dobbs M, Jones M, Anderson C. et al. Autoantibodies to amyloid β-peptide (Aβ) are increased in Alzheimer's disease patients and Aβ antibodies can enhance Aβ neurotoxicity. Neuromol Med. 2003;3:29-39
64. Qu B-X, Gong Y, Moore C, Fu M, German DC, Chang L-Y. et al. Beta-Amyloid Auto-antibodies are reduced in Alzheimer's disease. J Neuroimmunol. 2014;274:168-73
65. Weksler ME, Relkin N, Turkenich R, LaRusse S, Zhou L, Szabo P. Patients with Alzheimer disease have lower levels of serum anti-amyloid peptide antibodies than healthy elderly individuals. Exp Gerontol. 2002 37
66. Gustaw KA, Garrett MR, Lee H-g, Castellani RJ, Zagorski MG, Prakasam A. et al. Antigen-antibody dissociation in Alzheimer disease: a novel approach to diagnosis. J Neurochem. 2008;106:1350-6
67. Brettschneider S, Morgenthaler NG, Teipel SJ, Fischer-Schulz C, Bürger K, Dodel R. et al. Decreased serum amyloid β1-42 autoantibody levels in Alzheimer's disease, determined by a newly developed immuno-precipitation assay with radiolabeled amyloid β1-42 peptide. Biol Psychiatry. 2005;57:813-6
68. Moir RD, Tseitlin KA, Soscia S, Hyman BT, Irizarry MC, Tanzi RE. Autoantibodies to Redox-modified Oligomeric Aβ Are Attenuated in the Plasma of Alzheimer's Disease Patients. J Biol Chem. 2005;280:17458-63
69. Song MS, Mook-Jung I, Lee HJ, Min JY, Park MH. Serum Anti-Amyloid-β Antibodies and Alzheimer's Disease in Elderly Korean Patients. J Int Med Res. 2007;35:301-6
70. Gruden MA, Davidova TB, Mališauskas M, Sewell RDE, Voskresenskaya NI, Wilhelm K. et al. Differential neuroimmune markers to the onset of Alzheimer's disease neurodegeneration and dementia: Autoantibodies to Aβ(25-35) oligomers, S100b and neurotransmitters. J Neuroimmunol. 2007;186:181-92
71. Millucci L, Ghezzi L, Bernardini G, Santucci A. Conformations and biological activities of amyloid beta peptide 25-35. Curr Protein Pept Sci. 2010;11:54-67
72. Gustaw-Rothenberg KA, Siedlak SL, Bonda DJ, Lerner A, Tabaton M, Perry G. et al. Dissociated amyloid-beta antibody levels as a serum biomarker for the progression of Alzheimer's disease: a population-based study. Exp Gerontol. 2010;45:47-52
73. McMahon MJ, O'Kennedy R. Polyreactivity as an acquired artefact, rather than a physiologic property, of antibodies: evidence that monoreactive antibodies may gain the ability to bind to multiple antigens after exposure to low pH. J Immunol Methods. 2000;241:1-10
74. Maftei M, Thurm F, Schnack C, Tumani H, Otto M, Elbert T. et al. Increased Levels of Antigen-Bound β-Amyloid Autoantibodies in Serum and Cerebrospinal Fluid of Alzheimer's Disease Patients. PLoS One. 2013;8:e68996
75. Li X-W, Li X-X, Liu Q-S, Cheng Y. Blood and Cerebrospinal Fluid Autoantibody to Aβ Levels in Patients with Alzheimer's Disease: a Meta-Analysis Study. J Mol Neurosci. 2020;70:1208-15
76. Itoh N, Arai H, Urakami K, Ishiguro K, Ohno H, Hampel H. et al. Large-scale, multicenter study of cerebrospinal fluid tau protein phosphorylated at serine 199 for the antemortem diagnosis of Alzheimer's disease. Ann Neurol. 2001;50:150-6
77. Kolarova M, Garcia-Sierra F, Bartos A, Ricny J, Ripova D. Structure and pathology of tau protein in Alzheimer disease. Int J Alzheimers Dis. 2012;2012:731526
78. Skillbäck T, Farahmand BY, Rosén C, Mattsson N, Nägga K, Kilander L. et al. Cerebrospinal fluid tau and amyloid-β1-42 in patients with dementia. Brain. 2015;138:2716-31
79. Bartos A, Fialová L, Švarcová J, Ripova D. Patients with Alzheimer disease have elevated intrathecal synthesis of antibodies against tau protein and heavy neurofilament. J Neuroimmunol. 2012;252:100-5
80. Klaver AC, Coffey MP, Bennett DA, Loeffler DA. Specific serum antibody binding to phosphorylated and non-phosphorylated tau in non-cognitively impaired, mildly cognitively impaired, and Alzheimer's disease subjects: an exploratory study. Transl Neurodegener. 2017 6
81. Krestova M, Hromadkova L, Bilkova Z, Bartos A, Ricny J. Characterization of Isolated Tau-Reactive Antibodies From the Ivig Product, Plasma of Patients with Alzheimer's Disease and Cognitively Normal Individuals. J Neuroimmunol. 2017;313:16-24
82. Kuhn I, Rogosch T, Schindler TI, Tackenberg B, Zemlin M, Maier RF. et al. Serum titers of autoantibodies against α-synuclein and tau in child- and adulthood. J Neuroimmunol. 2018;315:33-9
83. Rosenmann H, Meiner Z, Geylis V, Abramsky O, Steinitz M. Detection of circulating antibodies against tau protein in its unphosphorylated and in its neurofibrillary tangles-related phosphorylated state in Alzheimer's disease and healthy subjects. Neurosci Lett. 2006;410:90-3
84. Smith LM, Coffey MP, Klaver AC, Loeffler DA. Intravenous immunoglobulin products contain specific antibodies to recombinant human tau protein. Int Immunopharmacol. 2013;16:424-8
85. Hromadkova L, Kolarova M, Jankovicova B, Bartos A, Ricny J, Bilkova Z. et al. Identification and characterization of natural antibodies against tau protein in an intravenous immunoglobulin product. J Neuroimmunol. 2015;289:121-9
86. Smith LM, Coffey MP, Loeffler DA. Specific binding of intravenous immunoglobulin products to tau peptide fragments. Int Immunopharmacol. 2014;21:279-82
87. Levin EC, Acharya NK, Han M, Zavareh SB, Sedeyn JC, Venkataraman V. et al. Brain-reactive autoantibodies are nearly ubiquitous in human sera and may be linked to pathology in the context of blood-brain barrier breakdown. Brain Res. 2010;1345:221-32
88. D'Andrea MR. Evidence that immunoglobulin-positive neurons in Alzheimer's disease are dying via the classical antibody-dependent complement pathway. Am J Alzheimers Dis Other Demen. 2005;20:144-50
89. Bartos A, Stourac P, Rusina R, Sejdová M, Velenská Z. [Paraneoplastic cerebellar degeneration associated with ovarian cancer: anti-Yo immunoreactivity in autoptic cerebellum and ovarian carcinoma]. Nervenarzt. 2002;73:995-8
90. Graus F, Saiz A, Dalmau J. Antibodies and neuronal autoimmune disorders of the CNS. J Neurol. 2010;257:509-17
91. Kronimus Y, Albus A, Balzer-Geldsetzer M, Straub S, Semler E, Otto M. et al. Naturally Occurring Autoantibodies against Tau Protein Are Reduced in Parkinson's Disease Dementia. PLoS One. 2016;11:e0164953
92. Abraha A, Ghoshal N, Gamblin TC, Cryns V, Berry RW, Kuret J. et al. C-terminal inhibition of tau assembly in vitro and in Alzheimer's disease. J Cell Sci. 2000 113 Pt 21: 3737-45
93. Augustinack JC, Schneider A, Mandelkow EM, Hyman BT. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol. 2002;103:26-35
94. Berry RW, Abraha A, Lagalwar S, LaPointe N, Gamblin TC, Cryns VL. et al. Inhibition of tau polymerization by its carboxy-terminal caspase cleavage fragment. Biochemistry. 2003;42:8325-31
95. Binder LI, Guillozet-Bongaarts AL, Garcia-Sierra F, Berry RW. Tau, tangles, and Alzheimer's disease. Biochim Biophys Acta Mol Basis Dis. 2005;1739:216-23
96. García-Sierra F, Ghoshal N, Quinn B, Berry RW, Binder LI. Conformational changes and truncation of tau protein during tangle evolution in Alzheimer's disease. J Alzheimers Dis. 2003;5:65-77
97. Hromadkova L, Ovsepian SV. Tau-Reactive Endogenous Antibodies: Origin, Functionality, and Implications for the Pathophysiology of Alzheimer's Disease. J Immunol Res. 2019;2019:7406810
98. Karran E, De Strooper B. The amyloid hypothesis in Alzheimer disease: new insights from new therapeutics. Nat Rev Drug Discov. 2022
99. Gafson AR, Barthélemy NR, Bomont P, Carare RO, Durham HD, Julien JP. et al. Neurofilaments: neurobiological foundations for biomarker applications. Brain. 2020;143:1975-98
100. Perrot R, Berges R, Bocquet A, Eyer J. Review of the multiple aspects of neurofilament functions, and their possible contribution to neurodegeneration. Molecular neurobiology. 2008;38:27-65
101. Khalil M, Teunissen CE, Otto M, Piehl F, Sormani MP, Gattringer T. et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol. 2018;14:577-89
102. Trojanowski JQ, Walkenstein N, Lee VM. Expression of neurofilament subunits in neurons of the central and peripheral nervous system: an immunohistochemical study with monoclonal antibodies. J Neurosci. 1986;6:650-60
103. Yuan A, Nixon RA. Specialized roles of neurofilament proteins in synapses: Relevance to neuropsychiatric disorders. Brain Res Bull. 2016;126:334-46
104. Ishii T, Haga S, Tokutake S. Presence of neurofilament protein in Alzheimer's neurofibrillary tangles (ANT). An immunofluorescent study. Acta Neuropathol. 1979;48:105-12
105. Goldman JE, Yen SH, Chiu FC, Peress NS. Lewy bodies of Parkinson's disease contain neurofilament antigens. Science. 1983;221:1082-4
106. Delisle MB, Carpenter S. Neurofibrillary axonal swellings and amyotrophic lateral sclerosis. J Neurol Sci. 1984;63:241-50
107. Norgren N, Rosengren L, Stigbrand T. Elevated neurofilament levels in neurological diseases. Brain Res. 2003;987:25-31
108. Petzold A. Neurofilament phosphoforms: surrogate markers for axonal injury, degeneration and loss. J Neurol Sci. 2005;233:183-98
109. Gordon BA. Neurofilaments in disease: what do we know? Curr Opin Neurobiol. 2020;61:105-15
110. Hansson O, Janelidze S, Hall S, Magdalinou N, Lees AJ, Andreasson U. et al. Blood-based NfL: A biomarker for differential diagnosis of parkinsonian disorder. Neurology. 2017;88:930-7
111. Ashton NJ, Janelidze S, Al Khleifat A, Leuzy A, van der Ende EL, Karikari TK. et al. A multicentre validation study of the diagnostic value of plasma neurofilament light. Nat Commun. 2021;12:3400
112. Fialová L, Bartos A, Svarcová J, Zimova D, Kotoucova J, Malbohan I. Serum and cerebrospinal fluid light neurofilaments and antibodies against them in clinically isolated syndrome and multiple sclerosis. J Neuroimmunol. 2013;262:113-20
113. Ehling R, Lutterotti A, Wanschitz J, Khalil M, Gneiss C, Deisenhammer F. et al. Increased frequencies of serum antibodies to neurofilament light in patients with primary chronic progressive multiple sclerosis. Mult Scler. 2004;10:601-6
114. Lu XY, Chen XX, Huang LD, Zhu CQ, Gu YY, Ye S. Anti-alpha-internexin autoantibody from neuropsychiatric lupus induce cognitive damage via inhibiting axonal elongation and promote neuron apoptosis. PLoS One. 2010;5:e11124
115. Oron L, Dubovik V, Perlman M, Novitsky L, Michaelson DM. Model Studies of the Role of Anti-Neurofilament Antibodies in Neurodegeneration in Alzheimer's Disease. Boston, MA: Birkhäuser Boston. 1994 p. 395-401
116. Oron L, Dubovik V, Novitsky L, Eilam D, Michaelson DM. Animal model and in vitro studies of anti neurofilament antibodies mediated neurodegeneration in Alzheimer's disease. J Neural Transm Suppl. 1997;49:77-84
117. Stubbs EB Jr, Lawlor MW, Richards MP, Siddiqui K, Fisher MA, Bhoopalam N. et al. Anti-neurofilament antibodies in neuropathy with monoclonal gammopathy of undetermined significance produce experimental motor nerve conduction block. Acta Neuropathol. 2003;105:109-16
118. Soussan L, Tchernakov K, Bachar-Lavi O, Yuvan T, Wertman E, Michaelson DM. Antibodies to different isoforms of the heavy neurofilament protein (NF-H) in normal aging and Alzheimer's disease. Molecular neurobiology. 1994;9:83-91
119. Braak H, Tredici KD, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197-211
120. McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor JP, Weintraub D. et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology. 2017;89:88-100
121. Attems J, Toledo JB, Walker L, Gelpi E, Gentleman S, Halliday G. et al. Neuropathological consensus criteria for the evaluation of Lewy pathology in post-mortem brains: a multi-centre study. Acta Neuropathol. 2021;141:159-72
122. Oczkowska A, Kozubski W, Lianeri M, Dorszewska J. Mutations in PRKN and SNCA Genes Important for the Progress of Parkinson's Disease. Curr Genomics. 2013;14:502-17
123. Orr CF, Rowe DB, Mizuno Y, Mori H, Halliday GM. A possible role for humoral immunity in the pathogenesis of Parkinson's disease. Brain. 2005;128:2665-74
124. Games D, Valera E, Spencer B, Rockenstein E, Mante M, Adame A. et al. Reducing C-Terminal-Truncated Alpha-Synuclein by Immunotherapy Attenuates Neurodegeneration and Propagation in Parkinson's Disease-Like Models. J Neurosci. 2014;34:9441-54
125. Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M. et al. Effects of alpha-synuclein immunization in a mouse model of Parkinson's disease. Neuron. 2005;46:857-68
126. Tran HT, Chung CH-Y, Iba M, Zhang B, Trojanowski JQ, Luk KC. et al. Α-synuclein immunotherapy blocks uptake and templated propagation of misfolded α-synuclein and neurodegeneration. Cell Rep. 2014;7:2054-65
127. Monahan AJ, Warren M, Carvey PM. Neuroinflammation and Peripheral Immune Infiltration in Parkinson's Disease: An Autoimmune Hypothesis. Cell Transplant. 2008;17:363-72
128. Gruden MA, Sewell RDE, Yanamandra K, Davidova TV, Kucheryanu VG, Bocharov EV. et al. Immunoprotection against toxic biomarkers is retained during Parkinson's disease progression. J Neuroimmunol. 2011;233:221-7
129. Heinzel S, Gold M, Deuschle C, Bernhard F, Maetzler W, Berg D. et al. Naturally occurring alpha-synuclein autoantibodies in Parkinson's disease: sources of (error) variance in biomarker assays. PLoS One. 2014;9:e114566
130. Smith LM, Schiess MC, Coffey MP, Klaver AC, Loeffler DA. α-Synuclein and Anti-α-Synuclein Antibodies in Parkinson's Disease, Atypical Parkinson Syndromes, REM Sleep Behavior Disorder, and Healthy Controls. PLoS One. 2012;7:e52285
131. Woulfe JM, Duke R, Middeldorp JM, Stevens S, Vervoort M, Hashimoto M. et al. Absence of elevated anti-alpha-synuclein and anti-EBV latent membrane protein antibodies in PD. Neurology. 2002;58:1435-6
132. Besong-Agbo D, Wolf E, Jessen F, Oechsner M, Hametner E, Poewe W. et al. Naturally occurring α-synuclein autoantibody levels are lower in patients with Parkinson disease. Neurology. 2013;80:169-75
133. Yanamandra K, Gruden MA, Casaite V, Meskys R, Forsgren L, Morozova-Roche LA. α-Synuclein Reactive Antibodies as Diagnostic Biomarkers in Blood Sera of Parkinson's Disease Patients. PLoS One. 2011;6:e18513
134. Horvath I, Iashchishyn IA, Forsgren L, Morozova-Roche LA. Immunochemical Detection of α-Synuclein Autoantibodies in Parkinson's Disease: Correlation between Plasma and Cerebrospinal Fluid Levels. ACS Chem Neurosci. 2017;8:1170-6
135. Akhtar RS, Licata JP, Luk KC, Shaw LM, Trojanowski JQ, Lee VMY. Measurements of auto-antibodies to α-synuclein in the serum and cerebral spinal fluids of patients with Parkinson's disease. J Neurochem. 2018;145:489-503
136. Koehler NKU, Stransky E, Shing M, Gaertner S, Meyer M, Schreitmüller B. et al. Altered Serum IgG Levels to α-Synuclein in Dementia with Lewy Bodies and Alzheimer's Disease. PLoS One. 2013;8:e64649
137. Maetzler W, Berg D, Synofzik M, Brockmann K, Godau J, Melms A. et al. Autoantibodies against amyloid and glial-derived antigens are increased in serum and cerebrospinal fluid of Lewy body-associated dementias. J Alzheimers Dis. 2011;26:171-9
138. Benson GS, Bauer C, Hausner L, Couturier S, Lewczuk P, Peters O. et al. Don't forget about tau: the effects of ApoE4 genotype on Alzheimer's disease cerebrospinal fluid biomarkers in subjects with mild cognitive impairment-data from the Dementia Competence Network. J Neural Transm (Vienna). 2022
139. Konijnenberg E, Tijms BM, Gobom J, Dobricic V, Bos I, Vos S. et al. APOE epsilon4 genotype-dependent cerebrospinal fluid proteomic signatures in Alzheimer's disease. Alzheimers Res Ther. 2020;12:65
140. Klafki HW, Wirths O, Mollenhauer B, Liepold T, Rieper P, Esselmann H. et al. Detection and quantification of Abeta-3-40 (APP669-711) in cerebrospinal fluid. J Neurochem. 2022;160:578-89
141. Willem M, Tahirovic S, Busche MA, Ovsepian SV, Chafai M, Kootar S. et al. eta-Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature. 2015;526:443-7
142. Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(Suppl):S10-7
143. D'Atri A, Scarpelli S, Gorgoni M, Truglia I, Lauri G, Cordone S. et al. EEG alterations during wake and sleep in mild cognitive impairment and Alzheimer's disease. iScience. 2021;24:102386
144. Thientunyakit T, Sethanandha C, Muangpaisan W, Chawalparit O, Arunrungvichian K, Siriprapa T. et al. Relationships between amyloid levels, glucose metabolism, morphologic changes in the brain and clinical status of patients with Alzheimer's disease. Ann Nucl Med. 2020;34:337-48
145. Guennewig B, Lim J, Marshall L, McCorkindale AN, Paasila PJ, Patrick E. et al. Defining early changes in Alzheimer's disease from RNA sequencing of brain regions differentially affected by pathology. Sci Rep. 2021;11:4865
146. Giau VV, Bagyinszky E, Yang YS, Youn YC, An SSA, Kim SY. Genetic analyses of early-onset Alzheimer's disease using next generation sequencing. Sci Rep. 2019;9:8368
147. Dourlen P, Kilinc D, Malmanche N, Chapuis J, Lambert JC. The new genetic landscape of Alzheimer's disease: from amyloid cascade to genetically driven synaptic failure hypothesis? Acta Neuropathol. 2019;138:221-36
148. Herrmann H, Aebi U. Intermediate Filaments: Structure and Assembly. Cold Spring Harb Perspect Biol. 2016 8
149. Kadavath H, Jaremko M, Jaremko L, Zweckstetter M. Structure of Tau(267-312) bound to Microtubules. PDB Entry - 2MZ7. 2015
150. Ulmer TS, Bax A, Cole NB, Nussbaum RL. Structure and dynamics of micelle-bound human alpha-synuclein. J Biol Chem. 2005;280:9595-603
151. Vivekanandan S, Brender JR, Lee SY, Ramamoorthy A. A partially folded structure of amyloid-beta(1-40) in an aqueous environment. Biochem Biophys Res Commun. 2011;411:312-6
152. Gruden MA, Davudova TB, Malisauskas M, Zamotin VV, Sewell RD, Voskresenskaya NI. et al. Autoimmune responses to amyloid structures of Abeta(25-35) peptide and human lysozyme in the serum of patients with progressive Alzheimer's disease. Dement Geriatr Cogn Disord. 2004;18:165-71
153. Jianping L, Zhibing Y, Wei Q, Zhikai C, Jie X, Jinbiao L. Low avidity and level of serum anti-Abeta antibodies in Alzheimer disease. Alzheimer Dis Assoc Disord. 2006;20:127-32
154. Xu W, Kawarabayashi T, Matsubara E, Deguchi K, Murakami T, Harigaya Y. et al. Plasma antibodies to Abeta40 and Abeta42 in patients with Alzheimer's disease and normal controls. Brain Res. 2008;1219:169-79
155. Sohn JH, So JO, Hong HJ, Kim JW, Na DR, Kim M. et al. Identification of autoantibody against beta-amyloid peptide in the serum of elderly. Front Biosci (Landmark Ed). 2009;14:3879-83
156. Maetzler W, Stapf AK, Schulte C, Hauser A-K, Lerche S, Wurster I. et al. Serum and cerebrospinal fluid uric acid levels in lewy body disorders: associations with disease occurrence and amyloid-β pathway. J Alzheimers Dis. 2011;27:119-26
157. Krestova M, Ricny J, Bartos A. Changes in concentrations of tau-reactive antibodies are dependent on sex in Alzheimer's disease patients. J Neuroimmunol. 2018;322:1-8
158. Fialová L, Bartos A, Švarcová J, Malbohan I. Increased Intrathecal High-Avidity Anti-Tau Antibodies in Patients with Multiple Sclerosis. PLoS One. 2011 6
159. Gruden MA, Yanamandra K, Kucheryanu VG, Bocharova OR, Sherstnev VV, Morozova-Roche LA. et al. Correlation between protective immunity to α-synuclein aggregates, oxidative stress and inflammation. Neuroimmunomodulation. 2012;19:334-42
160. Bryan T, Luo X, Forsgren L, Morozova-Roche LA, Davis JJ. The robust electrochemical detection of a Parkinson's disease marker in whole blood sera. Chem Sci. 2012;3:3468-73
161. Papachroni KK, Ninkina N, Papapanagiotou A, Hadjigeorgiou GM, Xiromerisiou G, Papadimitriou A. et al. Autoantibodies to alpha-synuclein in inherited Parkinson's disease. J Neurochem. 2007;101:749-56
162. Brudek T, Winge K, Folke J, Christensen S, Fog K, Pakkenberg B. et al. Autoimmune antibody decline in Parkinson's disease and Multiple System Atrophy; a step towards immunotherapeutic strategies. Mol Neurodegener. 2017;12:44
163. Caggiu E, Paulus K, Arru G, Piredda R, Sechi GP, Sechi LA. Humoral cross reactivity between α-synuclein and herpes simplex-1 epitope in Parkinson's disease, a triggering role in the disease? J Neuroimmunol. 2016;291:110-4
164. Shalash A, Salama M, Makar M, Roushdy T, Elrassas HH, Mohamed W. et al. Elevated Serum α-Synuclein Autoantibodies in Patients with Parkinson's Disease Relative to Alzheimer's Disease and Controls. Front Neurol. 2017;8:720
165. Xu L, Qi X, Duan S, Xie Y, Ren X, Chen G. et al. MicroRNAs: potential biomarkers for disease diagnosis. Biomed Mater Eng. 2014;24:3917-25
Author contact
![]() Corresponding author: Saak V. Ovsepian, Faculty of Science and Engineering, University of Greenwich London, Chatham Maritime, Kent, ME4 4TB, United Kingdom; E-mail: s.v.ovsepianac.uk.
Corresponding author: Saak V. Ovsepian, Faculty of Science and Engineering, University of Greenwich London, Chatham Maritime, Kent, ME4 4TB, United Kingdom; E-mail: s.v.ovsepianac.uk.