Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
The Blood-Brain Barrier in...
Clinical methods to detect BBB...
Current progress in biomarker...
Conclusion and perspectives
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2022; 12(4):1639-1658. doi:10.7150/thno.68304 This issue Cite
Review
Pathophysiology of blood brain barrier dysfunction during chronic cerebral hypoperfusion in vascular cognitive impairment
Vismitha Rajeev1, David Y. Fann2,3,4, Quynh Nhu Dinh5, Hyun Ah Kim5, T. Michael De Silva5, Mitchell K.P. Lai1, Christopher Li-Hsian Chen1, Grant R. Drummond5, Christopher G. Sobey5, Thiruma V. Arumugam5,6 ![]()
1. Memory Aging and Cognition Centre, Department of Pharmacology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore
2. Department of Biochemistry, Yong Loo Lin School of Medicine, National University of Singapore, Singapore
3. Healthy Longevity Translational Research Program, Yong Loo Lin School of Medicine, National University of Singapore, Singapore
4. Centre for Healthy Longevity, National University Health System (NUHS), Singapore
5. Centre for Cardiovascular Biology and Disease Research, Department of Physiology, Anatomy and Microbiology, School of Life Sciences, La Trobe University, Bundoora, VIC, Australia
6. School of Pharmacy, Sungkyunkwan University, Suwon, Republic of Korea
Received 2021-10-21; Accepted 2022-1-3; Published 2022-1-16
Abstract

The prevalence of cerebrovascular disease increases with age, placing the elderly at a greater lifetime risk for dementia. Vascular cognitive impairment (VCI) encompasses a spectrum of cognitive deficits from mild cognitive impairment to dementia. VCI and its most severe form, vascular dementia (VaD), is becoming a major public health concern worldwide. As growing efforts are being taken to understand VCI and VaD in animal models and humans, the pathogenesis of the disease is being actively explored. It is postulated that chronic cerebral hypoperfusion (CCH) is a major cause of VCI. CCH activates a molecular and cellular injury cascade that leads to breakdown of the blood brain barrier (BBB) and neurodegeneration. The BBB tightly regulates the movement of substances between the blood and the brain, thereby regulating the microenvironment within the brain parenchyma. Here we illustrate how BBB damage is causal in the pathogenesis of VCI through the increased activation of pathways related to excitotoxicity, oxidative stress, inflammation and matrix metalloproteinases that lead to downstream perivascular damage, leukocyte infiltration and white matter changes in the brain. Thus, CCH-induced BBB damage may initiate and contribute to a vicious cycle, resulting in progressive neuropathological changes of VCI in the brain. This review outlines the molecular and cellular mechanisms that govern BBB breakdown during CCH and highlights the clinical evidence in identifying at-risk VCI patients.
Introduction
Vascular cognitive impairment (VCI) covers an entire spectrum of vascular pathologies that contribute to cognitive impairment, from pre-clinical subjective states to the manifestation of a severe state of cognitive decline such as vascular dementia (VaD) [1,2]. Cerebral small vasssel disease (cSVD) is a common age-related condition that drives VCI [3-8]. cSVDs are characterised by pathological processes that affect the structural and functional integrity of small arteries, arterioles, capillaries and venules of the brain [9]. Some of the distinct clinical features of cSVD observed in VCI patients that can be visualised using neuroimaging include lacunar infarcts, enlargement of the perivascular space, cerebral atrophy, microbleeds, microinfarcts, white matter hyperintensities (WMHs) and reduced cerebral blood flow (CBF) [5,6,8,10-13].
The onset of disease manifestation from risk factors are complicated as risk factors lead to multiple layers of damage before reaching the vascular pathology stage in VCI and VaD. The degree of association between the risk factors and the vascular disease is thought to be dependent on age, lifestyle, genetic susceptibility, comorbidity of other diseases and even differential brain reserves as reviewed in detail previously [2,14,15]. Briefly, age has been suggested to be the strongest risk factor for VaD and accounts for many unrecognized vascular changes in the brain. After the age of 65, the risk of developing dementia progressively increases [16]. There are studies on sexual dimorphism that argue that the protective effects of estrogen in women against coronary heart disease account for a lower risk of VaD in females [16-19]. With regards to lifestyle factors, smoking has been identified as significant risk factors for cardiovascular disease, cerebral vascular disease and cognitive decline. Smoking causes vascular endothelial dysfunction and atherosclerotic damage [20-22]. Taken together, these risk factors each contribute to the progression of vascular disease and its unique impact on cognitive impairment. Therefore manipulating some of these risk factors and observing their impact on VCI progression can perhaps provide some pragmatic treatment perspectives to clinicians when identifying vulnerable patients. Understanding the VaD subtypes, lesions and risk factors reflects the complex layers present in people with VaD, and the causes of cognitive impairment are likely to be multifactorial.
Even without the presence of risk factors, vascular aging leads to chronic cerebral hypoperfusion (CCH) that induces phenotypical changes in the brain and therefore makes the brain more vulnerable to disease [23]. This emphasizes the importance of CBF regulation under physiological and pathological conditions. Cerebral blood vessels are responsible for the delivery of many important substances to the brain such as nutrients and oxygen, which is necessary for neuronal oxidative metabolism of energy substrates. Neurons have limited capacity for anaerobic metabolism, thus adequate CBF is critically important for function and viability of neurons [24]. Cerebral hemodynamics play an important role in maintaining coordinated flow responses in the brain as neuronal tissues require tight coordination between neuronal activity and CBF within the brain parenchyma (known as neurovascular coupling). CBF regulation is particularly well-developed in the brain to ensure coordinated flow responses in the brain [25]. The BBB is a relevant component of the microcirculatory response unit during ischemia, as it is involved in hemodynamic regulation of the arterial microcirculatory to allow for increased oxygen extraction from the blood [26]. The endothelial cells are involved in producing several vasoactive mediators such as nitric oxide (NO) that have a significant influence on vascular tone thereby influencing CBF [27]. However, clinical symptoms of hypoperfusion begin to exhibit when the degree of hypoperfusion is higher than the ability of the cerebral vasculature to autoregulate and allow for the supply of oxygen and nutrients to match the metabolic demand of neurons [26,28].
CCH was reported to be a common feature in all subtypes of VCI [29]. In fact, CCH was reported to begin at early stages of VCI and continue till the late demented state of VaD [15,30]. Furthermore, in a severe state of VaD, global CBF reduction in patients was reported to be more extensive than age-matched controls and Alzheimer's Disease (AD) patients [31,32]. VaD patient cohorts reportedly showed decreased CBF to all parts of the brain [33]. Another study reported a 31% decrease in CBF at the frontal cortex and a 39% decrease in CBF at the parietal cortex [34]. These studies implemented the use of various CBF measuring techniques on patients to determine any circulation changes, as explained in detail here [24]. In brief, there are techniques that are able to measure global CBF changes using invasive intravascular measurements such as nitrous oxide (N2O) inhalation and thermodilution. There are many more techniques that are able to measure CBF at a regional and local spatial extent such as positron emission tomography (PET), computed tomography (CT) and dynamic susceptibility contrast magnetic resonance imaging (DSC-MRI), which are all minimally invasive. Other local and regional CBF measurement techniques that are non-invasive and therefore are more popular include arterial spin labelling (ASL), transcranial doppler and thermal diffusion flowmetry (LDF).
Investigations into the pathophysiology of cSVD associated with VCI and VaD revealed endothelial dysfunction and blood-brain barrier (BBB) breakdown in the pathophysiology of cSVD associated with VCI and VaD. In fact, BBB breakdown has been proposed to be a core contributing mechanism in cSVD and associated cognitive decline [35]. There is evidence reporting that BBB leakage precedes the development of WMHs, a diagnostic hallmark of cSVD, in a longitudinal study [36]. BBB damage typically increases with age in normal-appearing white matter, and is more extensive in the white matter of cSVD patients [37]. This suggests that BBB damage leads to cSVD pathology. CCH is often associated with the formation of white matter lesions (WMLs) in the deep structures of the brain [38,39], and accordingly, white matter pathology is commonly observed in clinical cases of VCI.
CCH is also associated with neurodegeneration and varying degrees of cognitive decline within the VCI spectrum [15,29,30,34,40,41]. Decreased cerebral perfusion has been reported to be correlated with dementia severity and is also able to predict which individuals with mild cognitive impairment (MCI) will progress to develop a severe state of dementia [42,43]. Under CCH conditions, there are many mechanisms that are activated chronically in the brain that cause damage to accumulate, and overcome repair processes. These mechanisms include prolonged energy imbalance, oxidative stress, inflammation, endoplasmic reticulum (ER) stress and mitochondrial dysfunction. These mechanisms drive downstream structural changes such as BBB dysfunction, WMLs, glial activation and cell death activation [35,38,39,44]. This suggests that although CCH may not directly cause cognitive deficits, the effect of CCH on cognition is mediated by these mechanistic and structural changes in the brain [10]. Several animal models of VCI have been developed on the basis of CCH being the main driver for cognitive decline, and they have been reported to mimic closely the various aspects of pathology relevant to VCI and which are extensively used as a basis to probe pathophysiological mechanisms related to the disease [45].
Combined with a CCH state, vascular risk factors such as hypertension, stroke and hyperlipidemia frequently exist in VCI patients, contributing to BBB damage, all of which can propagate the hypoperfusive state of the brain [46,47]. Neuropathological studies have reported an increased BBB permeability in other types of dementia including in AD [48-52]; however interestingly, a more extensive BBB permeability was observed among VaD patients compared to AD patients [48]. An increase in BBB permeability was observed with increasing WML in the brain of VaD patients [53,54], supporting the pathological link between BBB permeability and white matter pathology. Hypoperfusion-associated BBB dysfunction may be an early feature in the development of WMLs and cognitive decline, and may play a direct pathogenic role in dementia manifestation. This review discusses the physiological state of the BBB while exploring pathophysiological mechanisms that govern BBB breakdown and the resulting effects of BBB damage such as leukocyte infiltration and WML formation.
The Blood-Brain Barrier in Health and Disease
Blood-Brain Barrier in a Physiological State
Although the term BBB suggests a rigid structure, it is innately semi-permeable and primarily separates the circulating blood and cerebral parenchymal tissue. The BBB surrounds the microvascular architecture, comprising capillaries, arterioles and venules, throughout most central nervous system (CNS) regions [55]. The BBB functions to establish a controlled environment in the cerebral parenchyma by maintaining relatively constant level of hormones, nutrients and water in the brain. This is vital as any fluctuations in the finely-tuned microenvironment of the brain could disrupt CNS homeostasis. The BBB also provides defense against circulating toxins or pathogens that could cause brain infections. Thus, the BBB serves as an important anatomical and biochemical barrier for the CNS. The BBB is comprised of endothelial cells, pericytes, astrocyte end feet and the basement membrane which together, enable these unique structural and functional charactericstics. The close interaction of these cells with the neurons forms the neurovascular unit, which is important in regulating several aspects of brain function, including the regulation of CBF.
Endothelial Cells and Tight Junctions
Endothelial cells are the primary components of the BBB, responsible for the physical barrier between the circulation and the CNS. Compared with peripheral endothelial cell, CNS endothelial cells have specialised characteristics such as the lack of fenestrations and minimal transcytosis activity that greatly restricts vesicle-mediated transcellular movement of solutes. These cells also have an abundant mitochondrial content, which is important for generation of adenoside triphosphate (ATP) during the transport of substances through ATP-dependant pumps [56]. Compared to peripheral endothelial cells, CNS endothelial cells express low levels of leukocyte adhesion molecules, restricting immune cell adhesion and infiltration into the brain parenchyma. Paracellular transport is also restricted by tight junctions (TJs) at the apical end of endothelial cells [57]. TJs are unique to brain endothelial cells and provide a “sealing” capacity for the BBB. The structural and functional properties of TJs are attributed to several proteins that interact and form complexes at the BBB, namely the cytoplasmic zonula occludens (ZO) and the transmembrane proteins (i.e. claudins, occludins, and junctional adhesion molecules (JAMs)) [58]. ZO proteins are scaffold proteins that directly interact with other proteins in the TJs, and maintain their expression at the transendothelial interface of the BBB and in signal transduction and transcriptional modifications [59,60]. Claudin subtypes 1, 3, 5 and 12 are expressed at endothelial TJs, but claudin 5, in particular, makes up a major portion of the BBB [61]. Claudins are involved in the tightening of paracellular junctions between endothelial cells and in the maturation of the BBB [62]. Occludins are the most abundant protein at TJs, and is important in maintaining the transendothelial electrical resistance at the BBB, and in the assembly of Claudin-5 proteins at the TJ interface [63]. In addition to TJs, adherens junctions (AJs) are associated with TJs to mediate their assembly and maintenance. AJs are located on the basolateral end of endothelial cells, and are primarily involved in adhesion of endothelial cells to one another [64]. Vascular endothelial cadherins (VE-cadherins), neural cadherins (N-cadherins) and epithelial cadherins (E-cadherins) are the AJs present in brain endothelial cells, with VE-cadherins the most abundant [65] (Figure 1).
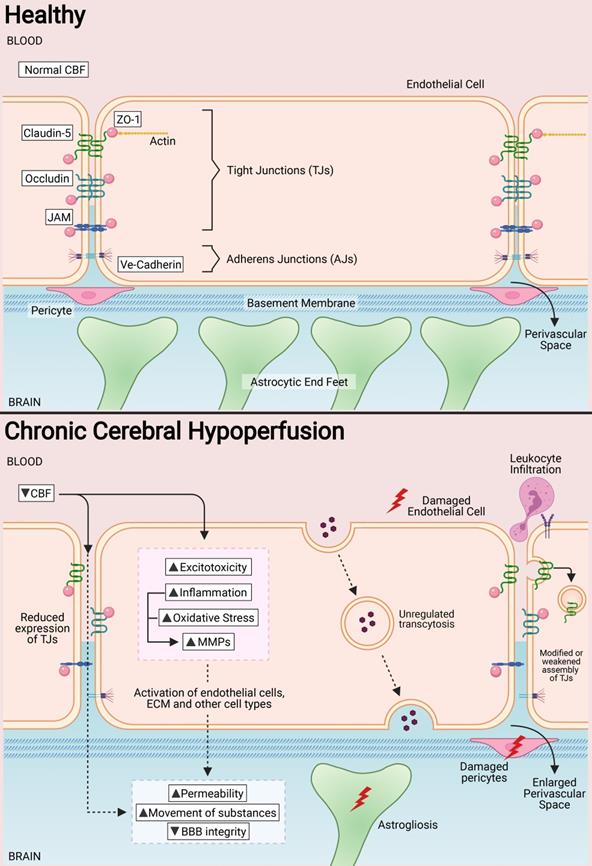
In a healthy state, tight junctions (TJs) zonula occludens (ZO-1), Claudin-5, Occludin and junctional adhesion molecules (JAMs) are involved at the apical end of the endothelial cells. Adherens junctions such as VE-cadherins are involved at the basolateral end of the endothelial cells. Collectively, these proteins provide for a continuous intercellular barrier between the endothelial cells. Pericytes, astrocytic end feet and the basement membrane are associated with the endothelial cells that collectively form the blood brain barrier (BBB) and are involved in the tightly regulated movements of molecules between the blood and the brain. In a chronic cerebral hypoperfused state, the reduced cerebral blood flow (CBF) triggers a myriad of effects on the BBB such as the reduced expression, modification and weakened assembly of the TJ proteins between the endothelial cells. Other effects of a reduced CBF are the induction of increased excitotoxicity, inflammation, oxidative stress and expression of matrix metalloproteinases. Collectively, these effects increase the permeability of the BBB at the endothelial cells and therefore increase the movement of substances between the blood and the brain. Secondary effects of increased BBB permeability include leukocyte infiltration, unregulated transcytosis, damaged pericytes, increased perivascular space and astrogliosis.
Other BBB Components
Pericytes are involved in the regulation of BBB homeostasis by maintaining low levels of transcytosis at endothelial cells [66]. Pericytes are also important within the pericyte-endothelial interactions for modulating CBF within capillaries [67], and in maintaining the structural integrity of the BBB as they modulate the basement membrane and TJ structure and function [68]. In addition, pericytes are involved in fine-tuning inflammatory responses in the brain by controlling leukocyte infiltration [69]. The position of pericytes around endothelial cells restricts the migration of leukocytes into brain parenchymal tissue. Leukocyte infiltration increases when there is pericyte gap enlargement, increased expression of adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1) and secretion of migration inhibitory factor (MIF) in response to inflammatory signals [70,71]. Astrocytes are also essential for the maintence of the BBB. Mature astrocytes are able to extend their end feet processes to completely ensheath the brain microvasculature. These astrocytic end feet processes also extend out to the neuronal circuity, making astrocytes a central link between the microvasculature and neuronal network. Astrocytes can sense the metabolic demands of neurons, and relay signals to the vasculature to coordinate CBF changes to a focal brain area [72]. Moreover, astrocytes function to provide barrier properties onto endothelial cells in the brain [73]. The vascular basement membrane surrounding the BBB is a component of the extracellular matrix produced by mature endothelial cells [74]. Common constituents of the basement membrane include specific laminin isoforms, type IV collagens and proteoglycans. The basement membrane and its protein components provide structural support to anchor the components of the BBB tightly together, while allowing for interactions between various cell types during signal processing [75]. In a mouse model of multiple sclerosis, deletion of the laminin α2 within the vascular basement membrane results in increased infiltration of T lymphocytes into the brain [76]. Another study reported haemorrhage in deep brain regions and impaired vascular smooth muscle cell function following deletion of astrocytic laminin α2 [77]. These studies validate that the composition of the vascular basement membrane is important in the integrity and function of the BBB.
BBB in Chronic Cerebral Hypoperfusion (CCH)
Pathological states in the CNS have been associated with BBB dysfunction. The BBB is intrinsically dynamic, hence it has important functional implications during development, the physiological aging process and age-associated neurodegenerative diseases and cognitive performance. Therefore, it is unsurprising that increased BBB permeability is observed in VaD patients [10,52,78-83]. An increased BBB permeability allows for uncontrolled movement of substances between the blood and brain parenchyma, which may include entry of immune cells and pathogens into the brain. Impairment of the BBB can be further aggravated when endothelial cells and other components of the neurovascular unit, such as pericytes, astrocytes and basement membranes, are affected. During CCH, the anatomical and functional properties of TJs at the endothelial cell interface can be modulated by changes in the expression of TJ proteins during hypoperfusion. In fact, increased BBB permeability in VaD has been reported to occur with neuronal loss and white matter degeneration during disease progression [84]. BBB damage generally propagates through various molecular cascades of events due to CCH. These mechanisms include, but are not limited to excitotoxicity, oxidative stress, inflammation and activation of matrix metalloproteinases (MMPs). Concurrently, BBB damage induces a series of downstream events that contribute to secondary brain injury, such as perivascular damage, astrogliosis, leukocyte infiltration and WML formation (Figure 1).
Primary Pathological Mechanisms Underlying BBB Damage During CCH
Excitotoxicity
CCH induces cellular energy imbalance and consequently, a state of excitotoxicity in the CNS [75,85-87]. Excitotoxicity is defined as the state of increased activation of neurons in response to increased levels of extracellular glutamate and subsequent calcium ion (Ca2+) influx [88]. High levels of glutamate release during excitotoxicity cause a persistent activation of N-methyl-d-aspartate acid (NMDA) receptors, ∝-amino-3-hydroxy-5-methylisoxazole propionic acid (AMPA) receptors, and metabotropic glutamate receptors (mGluRs) on neurons during CCH. Consistent with neuronal excitotoxicity, the released glutamate activates glutamate receptors on other brain cells such as endothelial cells, astrocytes and microglia.
Endothelial cells express NMDA receptors, and activation of these receptors during excitotoxicity results in calcium signaling and downstream NO production that promotes vascular permeability in the brain [89-91] (Figure 2). Persistent activation of glutamate receptors at the BBB during CCH may promote increased movement of Ca2+ into the endothelial cytosol, which induces disruption of metabolism, mitochondrial dysfunction, activation of proteases and phospholipases, and production of free radicals that can collectively contribute to membrane damage, vascular cell death and disruption of BBB integrity [92,93]. Another consequence of excitotoxicity is the modulation of ion channels and transporters at the BBB. Ion exchangers that carry ions such as sodium, potassium, chloride and calcium, and membrane transporters such as ATP-binding cassette (ABC) transporters are responsible for the maintenance of physiological and metabolic homeostasis, as well as ionic balance at the BBB. Increased vesicular transport across endothelial cells within a leaky BBB contributes to vasogenic edema. High Ca2+ levels in the cytosol can also lead to increased phosphorylation and activation of myosin light chain kinase (MLCK) [94]. Activated MLCK leads to the contraction of actin in the cytosol and relocation of TJ proteins between endothelial cells [95].
Chronic cerebral hypoperfusion or reduced cerebral blood flow (CBF) results in various mechanisms triggered in endothelial and neighbouring cells of the neurovascular unit including excitotoxicity. Excitotoxicity is induced in endothelial cells in response to a decreased glucose intake. Abnormally high levels of cytosolic Ca2+ induces the disruption of metabolism, mitochondrial dysfunction, activation of proteases and phospholipases, and production of reactive oxidative species (ROS) that collectively contribute to cell membrane damage and vascular cell death leading to disruption of BBB integrity. Astrocytes of the BBB are adversely affected by excitotoxins, as the interaction between astrocytes and the extracellular matrix (ECM) is rapidly disrupted. Astrocytic dysfunction or astrogliosis coupled with increased transendothelial pinocytosis contributes mostly to BBB breakdown after excitotoxicity.
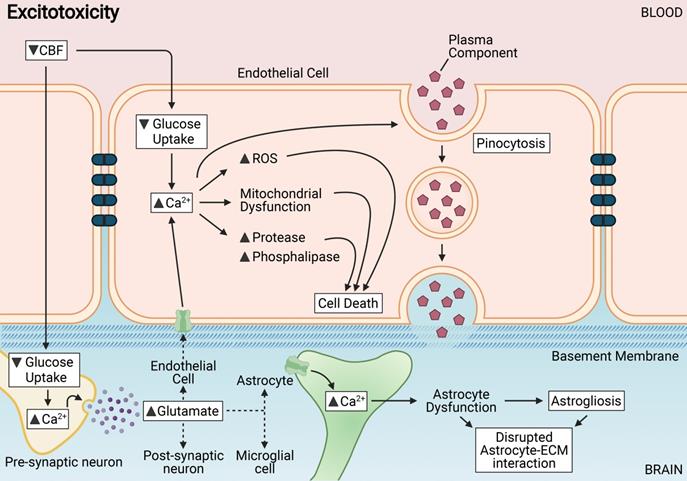
Astrocytes of the BBB have been shown to be adversely affected by persistent and excessive glutamate release, as astrocyte interaction with the extracellular matrix is rapidly disrupted [96]. Endothelial cells have been reported to in fact be highly resistant to primary glutaminergic stimuli and prevent the passive transport of glutamate into the brain parenchyma [97]. Glutamate has, interestingly, no direct effect on the expression of TJ proteins between endothelial cells, but is involved in the trigger of pinocytosis - a transendothelial transport process [98]. Thus, astrocytic dysfunction coupled with increased transendothelial pinocytosis contributes mostly to BBB breakdown during excitotoxicity.
Microglia are resident immune cells that play an essential function in immune surveillance, host defense against injury and tissue repair in the brain. CCH-induced excitotoxic stimuli have been reported to alter microglial function and can contribute to the pathogenesis of chronic CNS diseases [99]. Upon excessive and persistent glutamate release, activated microglia initiate several responses that elevate signaling pathways related to neurotransmitter homeostasis, ATP energy utilization and macromolecular synthesis in the brain [100]. In light of the importance of microglia in the brain, dysregulation of microglia activity may severely hamper its capacity for combating inflammatory challenges and tissue repair in the brain [101]. Overactivated microglia can contribute to neurotoxicity by becoming a source for glutamate release in the brain further modulating the pathogenesis of BBB disruption during CCH.
Interest in targeting excitotoxicity expanded following its implication in the pathogenesis of ischemic brain injury but there are little evidence in clinical trials that prove its efficacy [102]. However, there has been a gain of interest in this field in recent years that suggests that this knowledge should be leveraged to develop neuroprotective treatment against VaD. Glutamate antagonists against hypoperfusion have been reported to fail in clinical testing [103,104]. It is also noted that pan blockade of NMDA receptors could lead to anti-excitotoxic efficacy but widespread loss of fast synaptic excitation would not be well tolerated, therefore admistration of these drugs at the correct time is cruicial and may bring back these drugs into active consideration in clinical trials [102]. A combination approach of other drugs with anti-excitotoxic efficacy may be a more favourable approach.
Oxidative Stress
Hypoxia leads to increased production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) in the brain microvascular endothelium, thus leading to oxidative stress [105]. Mechanistically, hypoxia leads to a coordinated downregulation of metabolic utilization of ATP and ultimately a bioenergetic collapse. The cellular production of ATP is usually due to a decrease in activity of the electron transport chain in the mitochondria [106]. Complex IV is the main enzyme in the electron transport chain (ETC) that utilizes oxygen and couples it to the generation of ATP. During low oxygen levels, the electron transport chain is the major site for ROS production [107]. The three main sources of oxidative stress in the vasculature are the nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, mitochondria and cyclooxygenases (COX) [108]. While there are several ROS species such as peroxides, superoxides, hydroxyl radicals, many studies have suggested that superoxide plays a key role in BBB damage [109-112]. During CNS injury, the cellular defense systems are weakened, and ROS species will lead to the oxidation of lipids, proteins, and nucleic acids that alter endothelial cell function, therefore leading to BBB dysfunction [113]. Patients with VaD have reported increased levels of malondialdehyde, a marker of lipid peroxidation, as compared to controls, and even higher than those reported in AD patients [114].
NADPH oxidases have been reported to be involved in the progression of VaD states [115-117]. Angiotensin II is an important inducer of NADPH oxidase-modulated superoxide production and endothelial dysfunction within the BBB [118]. Angiotensin II has also been implicated in cognitive impairment and dementia [119]. Mitochondria are abundant within endothelial cells and these organelles are activated by NADPH oxidases to generate ROS. COX was shown to increase ROS production in endothelial cells through pro-inflammatory responses [120]. ROS may affect BBB integrity by modifying the interface of TJ proteins and inducing a decrease in E-cadherin levels, which are important for BBB maintenance [121] (Figure 3).
Oxidative Stress induces endothelial cell dysfunction. Increased reactive oxygen species (ROS) and reactive nitrogen species (RNS) from sources such as NADPH oxidases, cyclooxygenases and mitochondria. Increased oxidative stress can modify TJs directly, cause endothelial dysfunction, and activate MMPs.
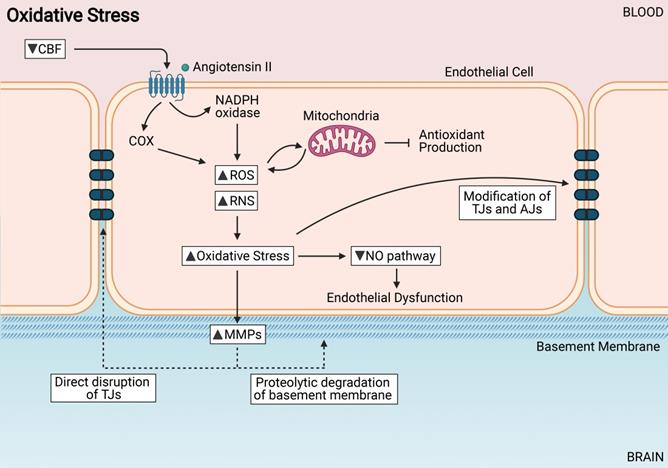
An increase in ROS in the endothelial cells leads to an impairment of NO signaling [122]. In physiological conditions, NO is a vasodilator controlling CBF; it is also a neurotransmitter that regulates vascular smooth muscle cells; it regulates Ca2+ levels in the brain therefore protecting neurons from excitotoxic damage; and is important in preserving and maintaining learning and memory function [123]. NO bioavailibility is decreased in endothelial cells during CCH as it can react with superoxide (O2-) to form peroxynitrite (OONO-), causing extensive oxidative stress, endothelial dysfunction and gradual BBB impairment [120] (Figure 3). Another pathway that increases oxidative stress in the BBB is through the upregulation of the receptor for advanced glycation end products (RAGE). Upregulation of RAGE results in the activation of downstream nuclear factor kappa beta (NF-κB) pathways which eventually lead to the dysregulation of the BBB via reduction in the TJ proteins between endothelial cells and thus increasing BBB permeability [124-126]. This pathway might play an important role in the pathogenesis of BBB breakdown during CCH.
Given the association between increased oxidative stress during CCH and increased BBB permeability, preventing or inherently reversing BBB breakdown through antioxidant therapies may be a target for the treatment of VaD as described here [127]. Antioxidant therapy such as ocimum sanctum (OS) has been reported to reduce lipid peroxidation after CCH was induced in rats [128]. Another popular antioxidant treatment is resveratrol, which reported a reduced lipid peroxidation and restored glutathione levels in rat brains following CCH [129]. Scavengers of ROS such as edaravone have been reported to ameliorate oxidative stress under CCH and also rescue white matter integrity and memory function in rats [130]. Another antioxidant therapy Chotosan enhanced antioxidative pathways in rats subjected to CCH, reversing ROS production and ameliorate memory impairment [131]. There are no trials for antioxidant therapies in VaD to date, although there are completed and ongoing clinical trials for AD [132]. There may therefore be potential in repurposing some of the antioxidant therapies that have had clinical success into VaD patients to examine their effects.
Inflammation
Although the brain has historically been considered to be an immune-privileged site, it is now understood that the CNS and the immune system are well connected and inflammation is involved in vascular and tissue remodelling after injury. With the reduction of CBF, an ischemic cascade consisting of a complex series of events is initiated that ultimately lead to activation of various brain cells to induce their death. The ischemic cascade triggers expression of pro-inflammatory cytokines, chemokines, and inflammatory mediators by various brain cells. Excitotoxicity, oxidative stress, microvascular injury, BBB dysfunction and secondary inflammation can exacerbate hypoperfusion injury in the brain and lead to permanent cerebral damage. In VaD, inflammatory genes and pathways are upregulated during disease progression [133], and so there has been increased interest in translational research to mitigate brain injury following CCH.
The cerebral vasculature is an important site of neuroinflammation and the BBB becomes compromised under inflammatory conditions [134]. Inflammation can affect the primary component of the BBB - the endothelial cells - through a number of different mechanisms. These include formation of membrane abnormalities that can compromise the integrity of the BBB via increased fenestrations; increased damage to the mitochondria that can prevent regulated transcytotic diffusion across the BBB and activation of apoptotic signals that can lead to endothelial cell death [135]. Generally, pro-inflammatory cytokines and chemokines activate specific signalling cascades that increase BBB permeability at the endothelial cell interface through weakening of the assembly and expression levels of TJ proteins [136-142]. These pro-inflammatory cytokines also increase expression of cell adhesion molecules on the endothelial cell surface of the BBB which increases leukocyte infiltration [143-150]. It was also shown that CCH-induced pro-inflammatory cytokines can activate Rho-kinase signalling within endothelial cells [151]. The Rho-kinase pathway is involved in the internalisation of TJ proteins via endocytosis that can directly influence the disruption and permeability of the BBB [152]. During chronic inflammatory states such as in VaD, the internalised TJ proteins may not be recycled to the plasma membrane, but are instead directed to the lysosome for degradation, leading to permanent BBB dysfunction [153].
In addition, microglia plays a central role in orchestrating neuroinflammation in the brain. Activated microglia have a pro-inflammatory phenotype that contributes to BBB damage by activating downstream inflammatory mediators such as interleukin-1 beta (IL-1β), tumour necrosis factor (TNF), interleukin-6 (IL-6), and monocyte chemoattractant protein-1 (MCP-1) [154,155]. These cytokines released by activated microglial cells can directly and indirectly contribute to BBB damage [156,157]. In particular, it was reported that necroptosis, a regulated mode of inflammatory cell death, was the underlying mechanism responsible for endothelial cell death contributing to BBB disruption [154] (Figure 4).
Inflammation is induced via the release of pro-inflammatory cytokines and chemokines. This release can activate various cell types including endothelial cells. These cytokines can directly damage the BBB, and increase permeability at the endothelial cell interface via either weakening assembly or endocytosis of tight junctions (TJs). They can also activate apoptotic pathways; unregulated transcytosis across the endothelial cells and also occur in response to inflammation. MMPs are activated by inflammation and can affect ECM and TJ disruption. Leukocyte infiltration is a downstream effect of increased cell adhesion pathways during inflammation. ECM edema can also cause other downstream effects such as white matter lesions (WMLs) and perivascular damage.
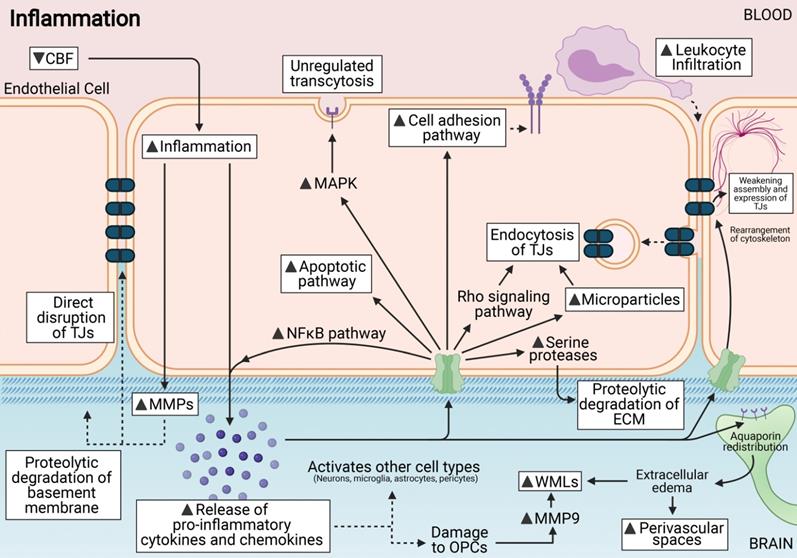
Here we summarize various cytokines and chemokines formed during CCH and discuss the role of each in BBB breakdown. Inflammatory cytokines such as IL-1β, IL-6, interleukin-17 (IL-17), interferon gamma (IFN-γ), TNF and C-C motif Chemokine Ligand 2 (CCL2) have been reported to be increased during CCH [158-161]. This suggests that CCH-induced inflammatory responses in various cell types may play an important role in neurovascular pathology.
Interleukin-1β (IL-1β)
In response to CNS injury, IL-1β expression has been reported to be elevated in various CNS diseases. In humans, IL-1β was elevated in the plasma of VaD human patients and in the cortex, hippocampus, striatum and microglial lysates in mouse models of VaD [158,162,163]. IL-1β can be produced by various cell types including microglia, astrocytes, infiltrating leukocytes, neurons and oligodendrocytes, and its primary effects are exerted mainly on microglia, astrocytes and endothelial cells, all of which contribute to BBB integrity [164,165]. IL-1β induces strong inflammatory effects on endothelial cells, which induces BBB leakiness via reduced expression of TJ protein ZO-1 [166]. In particular, IL-1β is produced by the inflammasome - a caspase-1 activating molecular platform expressed in various cell types [163]. Activation of caspase-1 in the brain increased adhesion and transmigration of peripheral immune cells across the BBB [167]. Increased inflammasome-mediated pyroptosis in damaged endothelial cells also contributes to BBB insult [168]. Additionally, IL-1β has a positive feedback mechanism where it can trigger an increase in its own expression in the brain [169], thus promoting increased leukocyte infiltration and exacerbating the inflammatory response in the brain [166].
Interleukin-6 (IL-6)
IL-6 has both pro-inflammatory and anti-inflammatory properties. Increased levels of IL-6 have been reported in the cerebrospinal fluid of VaD patients when compared to AD or cerebrovascular disease patients [159]. It has been reported that high levels of IL-6 in the brain reduces occludin and claudin-5 expression between the endothelial cells [121,137]. Therefore, this reduction in TJ proteins can weaken BBB integrity during CCH.
Interleukin-17 (IL-17)
IL-17 is a pro-inflammatory cytokine secreted by T cells to initiate neutrophil recruitment. IL-17 primarily exerts neuroinflammatory properties, but also promotes oxidative stress by inducing NADPH oxidase [170]. IL-17 is also a potent mediator of secondary chemokine release, activation of endothelial cells, and recruitment of other immune cells [171]. Consistent with these findings, in a mouse model of ischemic stroke, IL-17 exacerbated inflammatory responses following ischemia, increasing infarct size and worsening neurological outcomes [172]. Serum IL-17 was reported to be higher in VaD patients than in control patients [173]. Specifically, at the BBB, IL-17 has been reported to decrease occludin and claudin 5 expression between endothelial cells, leading to BBB leakage [174]. Thus, increased IL-17 in VaD brains is likely to also reduce expression of TJ proteins and BBB integrity [173].
Tumour Necrosis Factor (TNF)
TNF is a multifunctional cytokine secreted by several cell types including macrophages, natural killer cells, and lymphocytes, and plays a key role in the initiation and perpetuation of cerebral inflammation. Activation of TNF production has been implicated in various diseases including VaD and AD [143,175]. Exogenous TNF administration increased BBB permeability in control mice and in ischemic states [176]. At the BBB, inceased TNF expression in endothelial cells result in a rearrangement of the cytoskeleton within the cytoplasm, thus disrupting TJ protein assembly and basement membrane [177-180]. TNF also activates NF-κB and mitogen-activated protein kinase (MAPK) signaling pathways, whose major targets include the increased transcription of other pro-inflammatory cytokines and apoptosis related genes, all contributing to the activation and dysfuntion of endothelial cells [181-184]. TNF upregulates expression of toll-like receptors (TLR2, TLR3, TLR4 and TLR6) in various cell types including endothelial cells. In particular, activation of TLR2 at endothelial cells increases BBB permeability via disruption of TJ proteins occludin and claudin-5 [185]. Peripheral levels of TNF are reported to be involved in BBB disruption at the endothelial interface as increased serum TNF levels downregulate occludin expression, thereby inducing BBB disruption in a mouse model of liver failure [138]. Peripheral TNF levels have been reported to be increased in VaD patients compared to control subjects [186,187].
Interferon gamma (IFN-γ)
IFN-γ is a cytokine that plays an important role in both innate and adaptive immunity. It is secreted by activated T cells and natural killer cells and functions as a primary activator of macrophages. TNF and IFN-γ are cytokines that act synergistically in response to inflammation during pathology. BBB endothelial cells express TNF and IFN-γ receptors, and TNF itself has been reported to independently increase IFN-γ receptors on endothelial cells [188]. These cytokines bind to their receptors on endothelial cells to induce various signaling cascades including the recruitment of extracellular signal-regulated kinases (Erk1/2) [188]. TNF and IFN-γ synergistically increase chemokine secretion in CNS inflammation [189], and also stimulate the release of microparticles, cell membrane-derived particles generated by budding from the endothelial cell surface [190]. These microparticles export TJs and AJs away from the transendothelial surface following cytokine stimulation, thus contributing to the loss of the barrier property of the BBB [190].
Chemokine Ligand 2 (CCL2)
Chemokine CCL2 is the main effector of monocyte transmigration during CNS inflammation and is associated with endothelial dysfunction [191]. CCL2 participates in increasing BBB permeability during monocyte infiltration into the brain parenchyma [192]. CCL2 contributes to BBB opening by the dysregulation of the AJs between the endothelial cells [140]. AJs play a role in maintaining mechanical strength and stability of endothelial cells, thus with increased CCL2 expression their dysregulation can lead to increased BBB permeability. Consistent with this, in older adults, CCL2 is reported to be increased in the brain, and is associated with longitudinal decline in memory [193].
Based on many lines of evidence as highlighted above, inflammation is a major pathophysiological mechanism in BBB injury. This evidence highlights the potential importance of looking at anti-inflammatory therapies to combat CCH associated BBB injury. The use of non-steroidal-anti-inflammatory drugs (NSAID) and statins have been reported to reduce BBB damage through suppressing inflammation, particularly within the hippocampus [194]. Morin, a bioflavonoid, had been reported to attenuate BBB disruption after cerebral hypoperfusion via its anti-inflammatory properties [195]. Glucocorticoid steroids have been reported to increase the tightness of the BBB [196]. These steroids influence TJ protein expression between endothelial cells, transendothelial electrical resistance and ZO-1 expression [197]. A major limitation with employing steroids as a potential therapeutic intervention is determining the concentration, and the form of the steroids administered, as administration is highly dependent on age, gender, health, diet and the source of the steroids [198]. However, to date, there are no novel treatment agents with disease-modifying activity against VaD. There have, however, been many observational studies that indicate the potential of NSAIDs in limiting the progression of dementia as a whole [199].
Matrix Metalloproteinases (MMPs)
MMPs are zinc-dependent enzymes responsible for the proteolytic degradation of the extracellular matrix in the brain. In pathological states such as hypoxia and CCH, MMPs are activated by oxidative stress and inflammation in endothelial cells [153,200]. MMP substrates include fibronectin and laminins within the basal lamina, which provide a structural scaffold for endothelial cells in the BBB [75]. Although the basement membrane is the main substrate of MMPs, TJ proteins are also MMP substrates. Occludin, claudin-5 and ZO-1 proteins are degraded by MMP2 and MMP9 during CCH [153,201,202]. Thus, MMPs are robustly and directly involved in disruption of BBB structural integrity and hence its permeability. Activated microglia are the main source of MMP activation in the brain during CCH. In the hypoxic state of VaD, microglial MMP2 and MMP9 are increased [203], and are involved in the degradation of the basement membrane and TJ proteins of the BBB [204]. Though MMP2 and MMP9 enzymes are both upregulated during CCH, MMP2 expression in the microglia and vascular endothelium of white matter contributes more extensively to the remodeling of white matter myelin [205].
Upstream of the MMP pathway is the activator known as cyclophilin A (CypA). The CypA protein has multiple functions including protein folding, protein trafficking, assembly and cell signalling, and is activated by oxidative stress and inflammation [206]. CypA is activated in various human diseases including dementia, and the primary mechanism through which it exerts its inflammatory effects is through inducing an imbalance in the MMP levels in the brain and altering collagen turnover and extracellular remodelling, thus affecting vascular structure [207-209]. Changes in cell-matrix attachment sites (integrins) and their topographical localization may modulate arterial structure. Specifically at the BBB, both intracellular and extracellular CypA contribute to inflammation by promoting expression of adhesion molecules and enhancing T cell responses [210].
In this context, recent years have witnessed tremendous advancements in the field of MMP research, with a special emphasis for neuroprotection at the site of the BBB. Since MMPs have been implicated in the pathology of BBB insult, there has been much interest in the use of MMP inhibitors to treat diseases, and hence improve BBB integrity. Over the past decade, there has been better understanding of MMP activity and its influence on the BBB, and specific unique MMP inhibitors have been designed. Until recently, these MMP inhibitors have not been used in clinical trials, as MMP inhibitors have been designed to have desirable selectivity and improved pharmacokinetics [211]. However, the reluctance of using MMP inhibitors still exists as the same MMP is able to exert both beneficial and non-beneficial roles, and also have differing roles in different organ systems [212].
Pathological Secondary Effects Underlying BBB Damage During CCH
Perivascular Damage
A consequence of BBB damage is an increased size of perivascular spaces, a common observation in VaD. Perivascular spaces are part of an intricate fluid clearance system in the brain, and its dysfunction in disease results in accumulation of harmful waste products in the brain [213]. BBB damage includes the damage of pericytes. Although a direct link of pericytes to VaD is not clear, studies have shown that pericyte damage is associated with CCH, white matter damage, neuronal loss and cognitive impairment [214].
Leukocyte Infiltration
The absence of leukocytes in a healthy brain is due to restriction of leukocyte infiltration into the CNS by the BBB [215]. However, loss in BBB integrity during CCH can lead to uncontrolled movement of substances, resulting in glial activation and an altered extracellular environment around neurons. During CCH, a hypoxia-mediated inflammatory response triggers leukocyte infiltration through increased expression of pro-inflammatory chemokines and cytokines. Recruitment of these inflammatory cells into the microvasculature further compromises vessel perfusion, contributing to a persistent state of neuroinflammation in CCH [216].
Activation of endothelial cells in response to inflammation initiates a coordinated and sequential recruitment of inflammatory cells into the cerebral vasculature, including populations of neutrophils and mononuclear leukocytes such as lymphocytes, monocytes, macrophages and dendritic cells, that have been extensively described [216,217]. Briefly, adhesion molecules such as ICAM, vascular adhesion molecule (VCAM), selectins and leukocyte integrins are commonly expressed on endothelial cells to aid leukocyte infiltration. Selectins are involved in the tethering and rolling interactions of endothelial and leukocyte recruitment by binding to fucosylated oligosaccharide ligands on circulating leukocytes [218]. ICAMs and VCAMs allow for the arrest of leukocytes, and a subsequently tighter adhesion of leukocytes onto the endothelial cells [219]. Integrins expressed on leukocytes allow for the transmigration between endothelial cells into parenchymal tissue [220]. Therefore, increased expression of adhesion molecules on endothelial cells collectively accelerates leukocyte infiltration into the brain [216].
At endothelial cells, increased expression of chemokines, cytokines and P-selectin collectively activates the NF-κB pathway to mediate increased expression and recruitment of adhesion molecules on cell surface membranes [221,222]. TNF expression decreases BBB integrity via upregulation of ICAM and selectins on endothelial cells [223]. Activated leukocytes contribute to a chronic inflammatory state of CCH through further release of pro-inflammatory cytokines, forming a positive feedback loop of endothelial activation that perpetuates BBB damage. Leukocytes recruited into the brain also produce ROS, which exacerbates brain injury from immune cell infiltration by activating the NF-κB pathway.
Pericytes also play a role in leukocyte infiltration into the brain in CCH-mediated inflammation. Pericytes coordinate the navigation of infiltrated cells and regulate the type of cells infiltrating the brain by controlling the opening size of intra-pericyte gaps [70,71,224,225]. This controlled relaxation and opening of pericytic gaps is coordinated by Rho and its downstream Rho-assocated forming protein kinase (RhoA/ROCK) signaling pathway [225]. In addition, the basement membrane has also been recently reported to mediate leukocyte extravasation into the brain [76]; it was reported that there was a limited ability for T cells to enter the CNS due to the presence of laminin 5 expressed on endothelial cells, therefore potentially reducing disease susceptibility and severity.
Endothelial inflammation is associated with excessive activated platelets. Platelets are part of the body's defence system when there is excessive bleeding. The presences of activated platelets in circulation induce leukocyte adhesion on activated endothelial cells and promote inflammation in the brain [221,226,227]. This is followed by increased transcription and expression of adhesion receptors that mediate the docking and extravasation of leukocytes into the brain parenchyma under pathological conditions [228]. In VCI, vascular abnormalities such as disrupted microvascular integrity and microbleeds may trigger platelet activation [229]. The uncontrolled activation of platelets in VaD can result in chronic inflammatory reactions that can mediate endothelial cell stress. Besides having a key role in inflammation, platelets play an important role in hemostasis, whereby they initiate clotting following endothelial disruption or tissue injury. Platelets release substances such as thromboxane A2 that can deposit in the walls of cerebral blood vessels, which ultimately translates into vascular degeneration [230]. Platelets have gained much interest in recent years as they respond rapidly to environmental changes and potentially be a valid cellular tool to study pathogenesis of VaD.
In summary, increased BBB permeability allows infiltration of peripheral leukocytes. Activated leukocytes can further increase BBB permeability by exacerbating the inflammatory response via disruption of TJ proteins between the endothelial cells, modifying basement membrane proteins, activating pericytes and platelets. Yet, in contrast to their involvement in BBB damage, extravasation of immune cells in the brain increases migration of protective anti-inflammatory cells into the brain that may contribute to the repair of the BBB [231]. This emphasizes the need to identify the precise molecular and cellular pathways involved in BBB-induced leukocyte infiltration into the brain during CCH.
White Matter Damage
WMLs are associated with cognitive dysfunction in VaD patients and involve injury to oligodendrocytes. The pathogenesis of WMLs is associated with ischemic conditions in the brain [232-234]. In fact, many studies emphasize the role of damaged BBB in the induction and progression of WMLs [235-237]. A rodent model of CCH underlines the importance of an intact BBB in WML formation as it suggests that BBB damage is the starting point for WML formation [47]. Following white matter injury, oligodendrocyte precursor cells (OPC) proliferate and differentiate into mature cells in an attempt to restore damaged white matter. However, the proliferation capacity of OPCs is limited as CCH-induced pro-inflammatory cytokines disrupt the ability of vascular endothelial cells to support OPCs [238]. OPCs rapidly respond to early white matter damage and express MMP-9 endopeptidases, which further aggravates BBB disruption [239]. A higher leakage volume at BBB dysfunction areas was shown to be associated with aggravation of lesion severity in the white matter [37,240,241].
Another contributor of WMLs during hypoperfused and inflammatory states is the redistribution of the water channel aquaporin-4 (AQP4). AQP4 is abundantly expressed on astrocytic end feet process, contributing to the polarity of astrocytic cells and BBB integrity [242,243]. CCH-driven redistribution of AQP4 mediates osmotically-driven water transport into the brain parenchyma, resulting in edema within the white matter regions, and hence WML formation [244,245]. This fluid accumulation appears as white matter hyperintensities on magnetic resonance imaging (MRI), which is one of the diagnostic criteria in identifying VaD patients.
Clinical methods to detect BBB breakdown
Several techniques can measure BBB permeability in humans (Figure 5). The ratio of cerebrospinal fluid (CSF) and serum levels of plasma protein albumin correlates with BBB permeability. Serum albumin is usually not synthesized within the CNS, and so when detected in CSF fluid it is indicative of increased BBB permeability [246]. Another method is serum analysis of CNS proteins such as serum S100β. S100β is not usually synthesised outside the CNS, hence its detection in the serum correlates with BBB permeability [247]. Consistent with this phenomenon, elevated serum S100β levels have been reported in VaD patients [248]. Similar to S100β, CSF protein transthyretin is extravasated when BBB is damaged [249]. While transthyretin has not been studied in the context of VaD, CSF transthyretin levels have been implicated in other dementias with vascular contributions [250]. However, biochemical assays involving CSF sampling is invasive which represents a major limitation for detection assays.
Clinical Detection Method for BBB Breakdown. In patients, there are three main ways that BBB breakdown is measured. The first detection method is via neuroimaging techniques. The most sensitive and commonly used method is T1-weighted dynamic contrast-enhanced MRI (DCE-MRI), to image for low molecular weight paramagnetic tracers in the blood. Positron emission tomography (PET) imaging visualises the movement of radiolabelled tracers injected such as gallium. However, these methods are quite invasive in nature. A non-invasive MRI method known as diffusion-prepared arterial spin labelling (DP-ASL) is an effective technique to measures subtle dysfunctions associated with altered water exchange rates across the BBB. Another detection method of BBB permeability is through the measurement of fluid biomarkers. While cerebral spinal fluid (CSF) albumin and transthyretin have been studied to be effective markers of BBB permeability, their invasive nature proves to be ineffective. Hence blood biomarkers Albumin and S100b have gained much interest lately in measuring BBB breakdown.
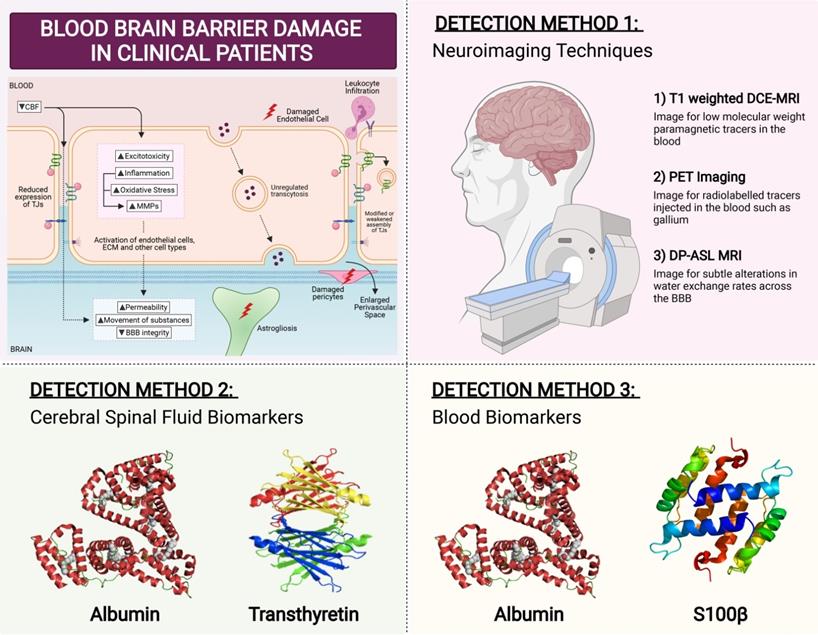
Brain imaging techniques can be used to assess the functional integrity of the BBB (Figure 5). The most sensitive and commonly used method is T1-weighted dynamic contrast-enhanced MRI (DCE-MRI), to image for low molecular weight paramagnetic tracers in the blood [251]. DCE-MRI has been employed to detect BBB breakdown in VaD patients [252]. In preclinical models of vascular diseases, PET imaging of BBB breakdown has also been reported, albeit in limited numbers [253]. PET imaging visualises the movement of radiolabelled tracers injected such as gallium [254]. There are many disadvantages in using neuroimaging including its high cost, slow-processing time, and high invasiveness. However, perhaps the greatest limitation is that only advanced stages of BBB dysfunction can be detected, as neuroimaging is not sensitive enough to detect small changes in the BBB [255]. In a continuum of disease progression such as occurs in VCI, it is important to identify BBB breakdown at early stages when BBB changes are relatively minor. This may be an important factor whereby BBB disruption could serve as a predictor for identification of at-risk dementia patients, so as to administer treatment options as early as possible.
A potentially effective measure of BBB dysfunction is through the use of a non-invasive MRI method known as diffusion-prepared arterial spin labelling (DP-ASL). This method measures subtle dysfunctions associated with altered water exchange rates across the BBB. Mapping the water exchange rate across the BBB without contrast imaging and invasiveness has been reported to have good reproducibility and has been proposed to be used as an imaging marker for cSVD and its associated cognitive impairment [256]. The potential use of this method in clinics as a surrogate imaging biomarker for VCI and early dementia may help in identifying at-risk patients by modulating BBB damage early before the leakage of large molecules across the BBB occurs during more advanced stages of dementia (Figure 5).
Current progress in biomarker profiling of BBB dysfunction
Diagnostics spanning molecular biomarkers and neuroimaging techniques are important for indicating the presence of disease. Biomarkers specific for a particular disease would be ideal, yet they could also be broader in nature; for example, biomarkers for BBB dysfunction. One candidate for measuring BBB leakage is through radiolabelling of albumin protein and tracking its movement across the BBB. The invasive process of albumin tracing further limits its use in both clinical and research studies. Less invasive, yet effective biomarkers are necessary in the determination of BBB dysfunction. BBB dysfunction leads to parenchymal proteins moving into the circulation; therefore, parenchymal proteins may potentially be used as novel biomarkers to detect BBB dysfunction [257].
As our understanding of CCH improves, the ultimate goal of biomarker profiling for VCI and VaD will be to refine the biomarkers to deliver a disease-specific indicator. While these molecular markers have been correlated to BBB dysfunction, they have been reported to arise in various settings such as ischemic stroke, AD, traumatic brain injury and schizophrenia, rendering these markers unspecific to VCI [257-260]. Increased BBB permeability is associated with increased brain pathology during CCH. Hence, there is a growing interest for potential therapeutic strategies to be introduced to target BBB damage and ultimately prevent the onset and progression of CCH-induced neuropathological features. Detailed understanding of the mediators of BBB disruption is key to finding potential treatments to prevent or reverse BBB breakdown in VaD. However a BBB dysfunction-related biomarker with specificity and sensitivity for VCI or VaD has not been reported to date. Prospective studies to evaluate the relationship between biomarkers of BBB dysfunction and VCI are likely to be an important area of study.
Conclusion and perspectives
The BBB is an important barrier that maintains and regulates the brain microenvironment to allow for proper neuronal functioning. In the context of vascular cognitive disorders, the brain experiences a state of CCH, where the cells do not receive sufficient oxygen and nutrients. The brain cells then initiate a pathophysiological process through various mechanisms. At the BBB interface, abnormal changes in endothelial cells directly leads to impairment of the BBB, further reducing CBF in the brain. Damages to pericytes, astrocytes and surrounding microglia further aggravate CBF regulation, and induce further injury at the BBB. This review has elucidated the various known pathological drivers of BBB dysfunction and secondary injuries downstream. While there is indeed progress being made in the field regarding BBB dysfunction during CCH, many questions remain unanswered. For instance, which is the main pathway governing dynamic regulation of the BBB in response to CCH? Are there preferential areas of BBB breakdown occurring throughout the brain that account for differential impact of cognitive loss in humans? Research geared towards answering these questions is necessary to understand the mechanisms underlying CCH. Future studies in the field of VCI should target ways to reduce small vessel endothelial damage to prevent BBB damage and brain injury. These studies may include, improvement of lifestyle factors such as diet and exercise. Additional studies are needed to determine whether BBB dysfunction can predict 'at risk' white matter, and whether treatment targeting BBB dysfunction can reverse WML and cognitive impairment. Therapeutic interventions that promote the health of the BBB may provide opportunities for the brain to control the course of VCI development.
Acknowledgements
All figures in this article were created using BioRender.
Funding
This work was supported by the National Medical Research Council Research Grants (NMRC-CBRG-0102/2016; NMRC/CSA-SI/007/2016 and NMRC/OFIRG/0036/2017), Singapore.
Author Contributions
VR, DYF led the writing of the manuscript and devised all the figures. QND, HAK, TMDS, MKPL and GRD edited the manuscript. TVA, CGS and CLHC supervised the writing and co-edited the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Román GC, Sachdev P, Royall DR, Bullock RA, Orgogozo J-M, López-Pousa S. et al. Vascular cognitive disorder: a new diagnostic category updating vascular cognitive impairment and vascular dementia. J Neurol Sci. 2004;226:81-7
2. van der Flier WM, Skoog I, Schneider JA, Pantoni L, Mok V, Chen CLH. et al. Vascular cognitive impairment. Nat Rev Dis Prim. 2018;4:1-16
3. Akinyemi R, Mukaetova-Ladinska E, Attems J, Ihara M, Kalaria R. Vascular Risk Factors and Neurodegeneration in Ageing Related Dementias: Alzheimer's Disease and Vascular Dementia. Curr Alzheimer Res. 2013;10:642-53
4. Cheng Y-W, Chiu M-J, Chen Y-F, Cheng T-W, Lai Y-M, Chen T-F. The contribution of vascular risk factors in neurodegenerative disorders: from mild cognitive impairment to Alzheimer's disease. Alzheimer's Res Ther. 2020;12:1-10
5. Gyanwali B, Shaik M, Tan B, Venketasubramanian N, Chen C, Hilal S. Risk Factors for and Clinical Relevance of Incident and Progression of Cerebral Small Vessel Disease Markers in an Asian Memory Clinic Population. J Alzheimers Dis. 2019;67:1209-19
6. Hilal S, Mok V, Youn YC, Wong A, Ikram MK, Chen CLH. Prevalence, risk factors and consequences of cerebral small vessel diseases: Data from three Asian countries. J Neurol Neurosurg Psychiatry. 2017;88:669-74
7. Skrobot OA, Black SE, Chen C, DeCarli C, Erkinjuntti T, Ford GA. et al. Progress toward standardized diagnosis of vascular cognitive impairment: Guidelines from the Vascular Impairment of Cognition Classification Consensus Study. Alzheimer's Dement. 2018;14:280-92
8. Xu X, Chan YH, Chan QL, Gyanwali B, Hilal S, Tan BY. et al. Global cerebrovascular burden and long-term clinical outcomes in Asian elderly across the spectrum of cognitive impairment. Int Psychogeriatr. 2018;30:1355-63
9. Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010;9:689-701
10. Duncombe J, Kitamura A, Hase Y, Ihara M, Kalaria RN, Horsburgh K. Chronic cerebral hypoperfusion: a key mechanism leading to vascular cognitive impairment and dementia. Closing the translational gap between rodent models and human vascular cognitive impairment and dementia. Clin Sci. 2017;131:2451-68
11. Gorelick PB, Scuteri A, Black SE, DeCarli C, Greenberg SM, Iadecola C. et al. Vascular Contributions to Cognitive Impairment and Dementia. Stroke. 2011;42:2672-713
12. Schmidt R, Seiler S, Loitfelder M. Longitudinal change of small-vessel disease-related brain abnormalities. J Cereb Blood Flow Metab. 2015;36:26-39
13. Wardlaw JM, Smith C, Dichgans M. Mechanisms of sporadic cerebral small vessel disease: insights from neuroimaging. Lancet Neurol. 2013;12:483-97
14. Chui HC. Subcortical ischemic vascular dementia. Neurol Clin. 2007;25:717-vi
15. Iadecola C. The Pathobiology of Vascular Dementia. Neuron. 2013;80:844-66
16. Corrada M, Brookmeyer R, Berlau D, Paganini-Hill A, Kawas C. Prevalence of dementia after age 90 - Results from The 90+Study. Neurology. 2008;71:337-43
17. Stampfer MJ, Colditz GA, Willett WC, Manson JE, Rosner B, Speizer FE. et al. Postmenopausal Estrogen Therapy and Cardiovascular Disease. N Engl J Med. 1991;325:756-62
18. Gorelick PB. Risk Factors for Vascular Dementia and Alzheimer Disease. Stroke. 2004;35:2620-2
19. Gannon OJ, Robison LS, Custozzo AJ, Zuloaga KL. Sex differences in risk factors for vascular contributions to cognitive impairment & dementia. Neurochem Int. 2019;127:38-55
20. Bakhru A, Erlinger TP. Smoking cessation and cardiovascular disease risk factors: results from the Third National Health and Nutrition Examination Survey. PLoS Med. 2005;2:e160
21. Anstey K, Sanden C, Salim A, O'Kearney R. Smoking as a Risk Factor for Dementia and Cognitive Decline: A Meta-Analysis of Prospective Studies. Am J Epidemiol. 2007;166:367-78
22. Gordon P, Flanagan P. Smoking: A risk factor for vascular disease. J Vasc Nurs. 2016;34:79-86
23. Ungvari Z, Kaley G, de Cabo R, Sonntag WE, Csiszar A. Mechanisms of vascular aging: new perspectives. J Gerontol A Biol Sci Med Sci. 2010;65:1028-41
24. Fantini S, Sassaroli A, Tgavalekos KT, Kornbluth J. Cerebral blood flow and autoregulation: current measurement techniques and prospects for noninvasive optical methods. Neurophotonics. 2016;3:31411
25. Kulik T, Kusano Y, Aronhime S, Sandler AL, Winn HR. Regulation of cerebral vasculature in normal and ischemic brain. Neuropharmacology. 2008;55:281-8
26. Kunz A, Iadecola C. Cerebral vascular dysregulation in the ischemic brain. Handb Clin Neurol. 2008;92:283-305
27. Lok J, Gupta P, Guo S, Kim WJ, Whalen MJ, van Leyen K. et al. Cell-cell Signaling in the Neurovascular Unit. Neurochem Res. 2007;32:2032-45
28. Cipolla MJ. The Cerebral Circulation. San Rafael (CA): Morgan & Claypool Life Sciences; Cereb Circ. 2009:185-7
29. Venkat P, Chopp M, Chen J. Models and mechanisms of vascular dementia. Exp Neurol. 2015;272:97-108
30. Ruitenberg A, Heijer T den, Bakker SLM, Swieten JC van, Koudstaal PJ, Hofman A. et al. Cerebral hypoperfusion and clinical onset of dementia: The Rotterdam study. Ann Neurol. 2005;57:789-94
31. Sabayan B, Jansen S, Oleksik AM, van Osch MJP, van Buchem MA, van Vliet P. et al. Cerebrovascular hemodynamics in Alzheimer's disease and vascular dementia: A meta-analysis of transcranial Doppler studies. Ageing Res Rev. 2012;11:271-7
32. Scheel P, Puls I, Becker G, Schöning M. Volume reduction in cerebral blood flow in patients with vascular dementia. Lancet. 1999;354:2137
33. Kawamura J, Meyer JS, Terayama Y, Weathers S. Leukoaraiosis correlates with cerebral hypoperfusion in vascular dementia. Stroke. 1991;22:609-14
34. Schuff N, Matsumoto S, Kmiecik J, Studholme C, Du A, Ezekiel F. et al. Cerebral blood flow in ischemic vascular dementia and Alzheimer's disease, measured by arterial spin-labeling magnetic resonance imaging. Alzheimer's Dement. 2009;5:454-62
35. Wardlaw JM, Makin SJ, Hernández MCV, Armitage PA, Heye AK, Chappell FM. et al. Blood-brain barrier failure as a core mechanism in cerebral small vessel disease and dementia: evidence from a cohort study. Alzheimer's Dement. 2017;13:634-43
36. Muñoz Maniega S, Chappell FM, Valdés Hernández MC, Armitage PA, Makin SD, Heye AK. et al. Integrity of normal-appearing white matter: Influence of age, visible lesion burden and hypertension in patients with small-vessel disease. J Cereb Blood Flow Metab. 2017;37:644-56
37. Zhang CE, Wong SM, Uiterwijk R, Backes WH, Jansen JFA, Jeukens CRLPN. et al. Blood-brain barrier leakage in relation to white matter hyperintensity volume and cognition in small vessel disease and normal aging. Brain Imaging Behav. 2018;13:389-95
38. O'Sullivan M, Lythgoe DJ, Pereira AC, Summers PE, Jarosz JM, Williams SCR. et al. Patterns of cerebral blood flow reduction in patients with ischemic leukoaraiosis. Neurology. 2002;59:321-6
39. Vernooij MW, Lugt A van der, Ikram MA, Wielopolski PA, Vrooman HA, Hofman A. et al. Total Cerebral Blood Flow and Total Brain Perfusion in the General Population: The Rotterdam Scan Study. J Cereb Blood Flow Metab. 2007;28:412-9
40. Deng Y, Wang L, Sun X, Liu L, Zhu M, Wang C. et al. Association Between Cerebral Hypoperfusion and Cognitive Impairment in Patients With Chronic Vertebra-Basilar Stenosis. Front Psychiatry. 2018;9:455
41. Safouris A, Hambye A-S, Sculier C, Papageorgiou SG, Vasdekis SN, Gazagnes M-D. et al. Chronic Brain Hypoperfusion due to Multi-Vessel Extracranial Atherosclerotic Disease: A Potentially Reversible Cause of Cognitive Impairment. J Alzheimer's Dis. 2015;43:23-7
42. Chao L, Buckley S, Kornak J, Schuff N, Madison C, Yaffe K. et al. ASL perfusion MRI predicts cognitive decline and conversion from MCI to dementia. Alzheimer Dis Assoc Disord. 2010;24:19-27
43. Alsop DC, Dai W, Grossman M, Detre JA. Arterial spin labeling blood flow MRI: its role in the early characterization of Alzheimer's disease. J Alzheimers Dis. 2010;20:871-80
44. Shabir O, Berwick J, Francis S. Neurovascular dysfunction in vascular dementia, Alzhiemers and atherosclerosis. BMC Neurosci. 2018 19
45. Hainsworth A, Allan S, Boltze J, Cunningham C, Farris C, Head E. et al. Translational models for vascular cognitive impairment: A review including larger species. BMC Med. 2017 15
46. Poggesi A, Pasi M, Pescini F, Pantoni L, Inzitari D. Circulating biologic markers of endothelial dysfunction in cerebral small vessel disease: A review. J Cereb Blood Flow Metab. 2015;36:72-94
47. Huang J, Li J, Feng C, Huang X, Wong L, Liu X. et al. Blood-Brain Barrier Damage as the Starting Point of Leukoaraiosis Caused by Cerebral Chronic Hypoperfusion and Its Involved Mechanisms: Effect of Agrin and Aquaporin-4. Biomed Res Int. 2018:1-10
48. Farrall AJ, Wardlaw JM. Blood brain barrier: ageing and microvascular disease-systematic review and meta-analysis. J Neurol Sci. 2009;283:261
49. Rosenberg GA. Blood-Brain Barrier Permeability in Aging and Alzheimer's Disease. J Prev Alzheimer's Dis. 2014;1:138-9
50. Haar H, Burgmans S, Jansen J, van Osch M, van Buchem M, Muller M. et al. Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology. 2016;31:152244
51. Skillbäck T, Delsing L, Synnergren J, Mattsson-Carlgren N, Janelidze S, Nägga K. et al. CSF/serum albumin ratio in dementias: a cross-sectional study on 1861 patients. Neurobiol Aging. 2017 59
52. Nation DA, Sweeney MD, Montagne A, Sagare AP, D'Orazio LM, Pachicano M. et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med. 2019;25:270-6
53. Wallin A, Sjögren M, Edman Å, Blennow K, Regland B. Symptoms, Vascular Risk Factors and Blood-Brain Barrier Function in Relation to CT White-Matter Changes in Dementia. Eur Neurol. 2000;44:229-35
54. Hanyu H, Asano T, Tanaka Y, Iwamoto T, Takasaki M, Abe K. Increased Blood-Brain Barrier Permeability in White Matter Lesions of Binswanger's Disease Evaluated by Contrast-Enhanced MRI. Dement Geriatr Cogn Disord. 2002;14:1-6
55. Wilhelm I, Nyúl-Tóth Á, Suciu M, Hermenean A, Krizbai IA. Heterogeneity of the blood-brain barrier. Tissue Barriers. 2016 4
56. Chai J. Roles of SRF in Endothelial Cells During Hypoxia. Res Dir Tumor Angiogenes. 2013
57. Berndt P, Winkler L, Cording J, Breitkreuz-Korff O, Rex A, Dithmer S. et al. Tight junction proteins at the blood-brain barrier: far more than claudin-5. Cell Mol Life Sci. 2019;76:1987-2002
58. Haseloff RF, Dithmer S, Winkler L, Wolburg H, Blasig IE. Transmembrane proteins of the tight junctions at the blood-brain barrier: Structural and functional aspects. Semin Cell Dev Biol. 2015;38:16-25
59. Bauer H, Zweimueller-Mayer J, Steinbacher P, Lametschwandtner A, Bauer HC. The dual role of zonula occludens (ZO) proteins. J Biomed Biotechnol. 2010. 2010
60. Tornavaca O, Chia M, Dufton N, Almagro LO, Conway DE, Randi AM. et al. ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation. J Cell Biol. 2015;208:821-38
61. Ohtsuki S, Yamaguchi H, Katsukura Y, Asashima T, Terasaki T. mRNA expression levels of tight junction protein genes in mouse brain capillary endothelial cells highly purified by magnetic cell sorting. J Neurochem. 2008;104:147-54
62. Krause G, Winkler L, Mueller SL, Haseloff RF, Piontek J, Blasig IE. Structure and function of claudins. Biochim Biophys Acta. 2008;1778:631-45
63. Feldman GJ, Mullin JM, Ryan MP. Occludin: Structure, function and regulation. Adv Drug Deliv Rev. 2005;57:883-917
64. Taddei A, Giampietro C, Conti A, Orsenigo F, Breviario F, Pirazzoli V. et al. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol. 2008;10:923-34
65. Bazzoni G, Dejana E. Endothelial Cell-to-Cell Junctions: Molecular Organization and Role in Vascular Homeostasis. Physiol Rev. 2004;84:869-901
66. Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C. et al. Pericytes regulate the blood-brain barrier. Nat. 2010;468:557-61
67. Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA. et al. Capillary pericytes regulate cerebral blood flow in health and disease. Nat. 2014;508:55-60
68. Bergers G, Song S. The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol. 2005;7:452-64
69. Rudziak P, Ellis CG, Kowalewska PM. Role and molecular mechanisms of pericytes in regulation of leukocyte diapedesis in inflamed tissues. Mediators Inflamm. 2019. 2019
70. Proebstl D, Voisin M-B, Woodfin A, Whiteford J, D'Acquisto F, Jones GE. et al. Pericytes support neutrophil subendothelial cell crawling and breaching of venular walls in vivo. J Exp Med. 2012;209:1219-34
71. Stark K, Eckart A, Haidari S, Tirniceriu A, Lorenz M, von Brühl M-L. et al. Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and “instruct” them with pattern-recognition and motility programs. Nat Immunol. 2012;14:41-51
72. Howarth C. The contribution of astrocytes to the regulation of cerebral blood flow. Front Neurosci. Frontiers. 2014;8:103
73. Ramsauer M, Krause D, Dermietzel R. Angiogenesis of the blood-brain barrier in vitro and the function of cerebral pericytes. FASEB J. 2002;16:1274-6
74. Thomsen MS, Birkelund S, Burkhart A, Stensballe A, Moos T. Synthesis and deposition of basement membrane proteins by primary brain capillary endothelial cells in a murine model of the blood-brain barrier. J Neurochem. 2017;140:741-54
75. Daneman R, Prat A. The Blood-Brain Barrier. Cold Spring Harb Perspect Biol. 2015;7:a020412
76. Wu C, Ivars F, Anderson P, Hallmann R, Vestweber D, Nilsson P. et al. Endothelial basement membrane laminin α5 selectively inhibits T lymphocyte extravasation into the brain. Nat Med. 2009;15:519-27
77. Chen Z-L, Yao Y, Norris EH, Kruyer A, Jno-Charles O, Akhmerov A. et al. Ablation of astrocytic laminin impairs vascular smooth muscle cell function and leads to hemorrhagic stroke. J Cell Biol. 2013;202:381-95
78. Al Ahmad A, Gassmann M, Ogunshola OO. Involvement of oxidative stress in hypoxia-induced blood-brain barrier breakdown. Microvasc Res. 2012;84:222-5
79. Candelario-Jalil E, Thompson J, Taheri S, Grossetete M, Adair JC, Edmonds E. et al. Matrix Metalloproteinases Are Associated With Increased Blood-Brain Barrier Opening in Vascular Cognitive Impairment. Stroke. 2011;42:1345-50
80. Erhardt EB, Pesko JC, Prestopnik J, Thompson J, Caprihan A, Rosenberg GA. Biomarkers identify the Binswanger type of vascular cognitive impairment. J Cereb Blood Flow Metab. 2018;39:1602-12
81. Ueno M, Tomimoto H, Akiguchi I, Wakita H, Sakamoto H. Blood-Brain Barrier Disruption in White Matter Lesions in a Rat Model of Chronic Cerebral Hypoperfusion. J Cereb Blood Flow Metab. 2016;22:97-104
82. Wardlaw JM, Doubal F, Armitage P, Chappell F, Carpenter T, Maniega SM. et al. Lacunar stroke is associated with diffuse blood-brain barrier dysfunction. Ann Neurol. 2009;65:194-202
83. Wardlaw JM. Blood-brain barrier and cerebral small vessel disease. J Neurol Sci. 2010;299:66-71
84. Toth P, Tarantini S, Csiszar A, Ungvari Z. Functional vascular contributions to cognitive impairment and dementia: mechanisms and consequences of cerebral autoregulatory dysfunction, endothelial impairment, and neurovascular uncoupling in aging. Am J Physiol Heart Circ Physiol. 2017;312:H1-20
85. Dong X, Wang Y, Qin Z. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin. 2009;30:379-87
86. Ganzella M, de Oliveira EDA, Comassetto DD, Cechetti F, Cereser VH, Moreira JD. et al. Effects of chronic guanosine treatment on hippocampal damage and cognitive impairment of rats submitted to chronic cerebral hypoperfusion. Neurol Sci. 2011;33:985-97
87. Marosi M, Fuzik J, Nagy D, Rákos G, Kis Z, Vécsei L. et al. Oxaloacetate restores the long-term potentiation impaired in rat hippocampus CA1 region by 2-vessel occlusion. Eur J Pharmacol. 2009;604:51-7
88. Velasco M, Rojas-Quintero J, Chávez-Castillo M, Rojas M, Bautista J, Martínez MS. et al. Excitotoxicity: An Organized Crime at The Cellular Level. J Neurol Neurosci. 2017;8:193
89. Muir KW. Glutamate-based therapeutic approaches: clinical trials with NMDA antagonists. Curr Opin Pharmacol. 2006;6:53-60
90. Vazana U, Veksler R, Pell GS, Prager O, Fassler M, Chassidim Y. et al. Glutamate-Mediated Blood-Brain Barrier Opening: Implications for Neuroprotection and Drug Delivery. J Neurosci. 2016;36:7727-39
91. Xhima K, Weber-Adrian D, Silburt J. Glutamate Induces Blood-Brain Barrier Permeability through Activation of N-Methyl-D-Aspartate Receptors. J Neurosci. 2016;36:12296-8
92. András IE, Deli MA, Veszelka S, Hayashi K, Hennig B, Toborek M. The NMDA and AMPA/KA Receptors are Involved in Glutamate-Induced Alterations of Occludin Expression and Phosphorylation in Brain Endothelial Cells. J Cereb Blood Flow Metab. 2007;27:1431-43
93. Collard CD, Park KA, Montalto MC, Alapati S, Buras JA, Stahl GL. et al. Neutrophil-derived Glutamate Regulates Vascular Endothelial Barrier Function. J Biol Chem. 2002;277:14801-11
94. Wang J, Sun L, Si Y-F, Li B-M. Overexpression of actin-depolymerizing factor blocks oxidized low-density lipoprotein-induced mouse brain microvascular endothelial cell barrier dysfunction. Mol Cell Biochem. 2012;371:1-8
95. Rigor RR, Shen Q, Pivetti CD, Wu MH, Yuan SY. Myosin Light Chain Kinase Signaling in Endothelial Barrier Dysfunction. Med Res Rev. 2013;33:911-33
96. Milner R, Hung S, Wang X, Berg GI, Spatz M, Zoppo GJ del. Responses of Endothelial Cell and Astrocyte Matrix-Integrin Receptors to Ischemia Mimic Those Observed in the Neurovascular Unit. Stroke. 2008;39:191-7
97. Domoki F, Kis B, Gáspár T, Bari F, Busija DW. Cerebromicrovascular endothelial cells are resistant to l-glutamate. Am J Physiol Regul Integr Comp Physiol. 2008;295:1099-108
98. Yan BC, Xu P, Gao M, Wang J, Jiang D, Zhu X. et al. Changes in the Blood-Brain Barrier Function Are Associated With Hippocampal Neuron Death in a Kainic Acid Mouse Model of Epilepsy. Front Neurol. 2018;9:775
99. Bordeleau M, ElAli A, Rivest S, Bordeleau M, ElAli A, Rivest S. Severe chronic cerebral hypoperfusion induces microglial dysfunction leading to memory loss in APPswe/PS1 mice. Oncotarget. 2016;7:11864-80
100. Domercq M, Vázquez-Villoldo N, Matute C. Erratum: Neurotransmitter signaling in the pathophysiology of microglia. Front Cell Neurosci. Frontiers. 2013;7:107
101. Liu B, Wang K, Gao H-M, Mandavilli B, Wang J-Y, Hong J-S. Molecular consequences of activated microglia in the brain: overactivation induces apoptosis. J Neurochem. 2001;77:182-9
102. Choi DW. Excitotoxicity: Still Hammering the Ischemic Brain in 2020. Front Neurosci. 2020;14:579953
103. Hoyte L, Barber PA, Buchan A, Hill M. The Rise and Fall of NMDA Antagonists for Ischemic Stroke. Curr Mol Med. 2004;4:131-6
104. Lai TW, Zhang S, Wang YT. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog Neurobiol. 2014;115:157-88
105. Chen R, Lai UH, Zhu L, Singh A, Ahmed M, Forsyth NR. Reactive Oxygen Species Formation in the Brain at Different Oxygen Levels: The Role of Hypoxia Inducible Factors. Front Cell Dev Biol. Frontiers. 2018;0:132
106. Wheaton WW, Chandel NS. Hypoxia. 2. Hypoxia regulates cellular metabolism. Am J Physiol Cell Physiol. 2011;300:C385-93
107. Li W, Yang S. Targeting oxidative stress for the treatment of ischemic stroke: Upstream and downstream therapeutic strategies. Brain Circ. 2016;2:153-63
108. Lee MY, Griendling KK. Redox Signaling, Vascular Function, and Hypertension. Antioxid Redox Signal. 2008;10:1045-59
109. Kahles T, Luedike P, Endres M, Galla H-J, Steinmetz H, Busse R. et al. NADPH Oxidase Plays a Central Role in Blood-Brain Barrier Damage in Experimental Stroke. Stroke. 2007;38:3000-6
110. Lee HS, Namkoong K, Kim DH, Kim KJ, Cheong YH, Kim SS. et al. Hydrogen peroxide-induced alterations of tight junction proteins in bovine brain microvascular endothelial cells. Microvasc Res. 2004;68:231-8
111. Maier CM, Hsieh L, Crandall T, Narasimhan P, Chan PH. A new approach for the investigation of reperfusion-related brain injury. Biochem Soc Trans. 2006;34:1366-9
112. Schreibelt G, Musters RJP, Reijerkerk A, Groot LR de, Pol SMA van der, Hendrikx EML. et al. Lipoic Acid Affects Cellular Migration into the Central Nervous System and Stabilizes Blood-Brain Barrier Integrity. J Immunol. 2006;177:2630-7
113. Lewén A, Matz P, Chan P. Free radical pathways in CNS Injury. J Neurotrauma. 2000;17:871-90
114. Gustaw Rothenberg K, Kowalczuk K, Stryjecka-Zimmer M. Lipids' peroxidation markers in Alzheimer's disease and vascular dementia. Geriatr Gerontol Int. 2010;10:161-6
115. Choi D-H, Lee J. A Mini-Review of the NADPH Oxidases in Vascular Dementia: Correlation with NOXs and Risk Factors for VaD. Int J Mol Sci. 2017;18:2500
116. ChoiDong-Hee LeeKyoung-Hee, KimJi-Hye SeoJu-Ha, Young K Young S. et al. NADPH Oxidase 1, a Novel Molecular Source of ROS in Hippocampal Neuronal Death in Vascular Dementia. Antioxid Redox Signal. 2014;21:533-50
117. Toyama K, Koibuchi N, Uekawa K, Hasegawa Y, Kataoka K, Katayama T. et al. Apoptosis Signal-Regulating Kinase 1 Is a Novel Target Molecule for Cognitive Impairment Induced by Chronic Cerebral Hypoperfusion. Arterioscler Thromb Vasc Biol. 2014;34:616-25
118. Chrissobolis S, Banfi B, Sobey CG, Faraci FM. Role of Nox isoforms in angiotensin II-induced oxidative stress and endothelial dysfunction in brain. J Appl Physiol. 2012;113:184-91
119. Takeda S, Sato N, Takeuchi D, Kurinami H, Shinohara M, Niisato K. et al. Angiotensin Receptor Blocker Prevented β-Amyloid-Induced Cognitive Impairment Associated With Recovery of Neurovascular Coupling. Hypertension. 2009;54:1345-52
120. Wang F, Cao Y, Ma L, Pei H, Rausch WD, Li H. Dysfunction of Cerebrovascular Endothelial Cells: Prelude to Vascular Dementia. Front Aging Neurosci. 2018;10:376
121. Rochfort KD, Collins LE, Murphy RP, Cummins PM. Downregulation of Blood-Brain Barrier Phenotype by Proinflammatory Cytokines Involves NADPH Oxidase-Dependent ROS Generation: Consequences for Interendothelial Adherens and Tight Junctions. PLoS One. 2014;9:e101815
122. Förstermann U. Nitric oxide and oxidative stress in vascular disease. Pflügers Arch - Eur J Physiol. 2010;459:923-39
123. Lourenço CF, Ledo A, Barbosa RM, Laranjinha J. Neurovascular-neuroenergetic coupling axis in the brain: master regulation by nitric oxide and consequences in aging and neurodegeneration. Free Radic Biol Med. 2017;108:668-82
124. Yang H, Wang W, Jia L, Qin W, Hou T, Wu Q. et al. The Effect of Chronic Cerebral Hypoperfusion on Blood-Brain Barrier Permeability in a Transgenic Alzheimer's Disease Mouse Model (PS1V97L). J Alzheimer's Dis. 2020;74:261-75
125. Deane R, Yan S, Submamaryan R, LaRue B, Jovanovic S, Hogg E. et al. RAGE mediates amyloid-peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907-13
126. Kook S-Y, Hong HS, Moon M, Ha CM, Chang S, Mook-Jung I. Aβ1-42 RAGE Interaction Disrupts Tight Junctions of the Blood-Brain Barrier Via Ca2+-Calcineurin Signaling. J Neurosci. 2012;32:8845-8854
127. Kuang H, Zhou Z-F, Zhu Y-G, Wan Z-K, Yang M-W, Hong F-F. et al. Pharmacological Treatment of Vascular Dementia: A Molecular Mechanism Perspective. Aging Dis. 2021;12:308
128. Yanpallewar S, Rai S, Kumar M, Acharya S. Evaluation of antioxidant and neuroprotective effect of Ocimum sanctum on transient cerebral ischemia and long-term cerebral hypoperfusion. Pharmacol Biochem Behav. 2004;79:155-64
129. Ozacmak V, Sayan-Ozacmak H, Barut F. Chronic treatment with resveratrol, a natural polyphenol found in grapes, alleviates oxidative stress and apoptotic cell death in ovariectomized female rats subjected to chronic cerebral hypoperfusion. Nutr Neurosci. 2015 19
130. Miyamoto N, Maki T, Pham L-DD, Hayakawa K, Seo JH, Mandeville ET. et al. Oxidative Stress Interferes With White Matter Renewal After Prolonged Cerebral Hypoperfusion in Mice. Stroke. 2013;44:3516-21
131. Jiang P, Chen L, Sun J, Li J, Xu J, Liu W. et al. Chotosan ameliorates cognitive impairment and hippocampus neuronal loss in experimental vascular dementia via activating the Nrf2-mediated antioxidant pathway. J Pharmacol Sci. 2018 139
132. Mecocci P, Polidori MC. Antioxidant clinical trials in mild cognitive impairment and Alzheimer's disease. Biochim Biophys Acta. 2012;1822:631-8
133. Chen Y, Liu Q, Liu J, Wei P, Li B, Wang N. et al. Revealing the Modular Similarities and Differences Among Alzheimer's Disease, Vascular Dementia, and Parkinson's Disease in Genomic Networks. NeuroMolecular Med. 2021:1-14
134. Zhou T, Zhao L, Zhan R, He Q, Tong Y, Tian X. et al. Blood-brain barrier dysfunction in mice induced by lipopolysaccharide is attenuated by dapsone. Biochem Biophys Res Commun. 2014;453:419-24
135. Cardoso FL, Kittel Á, Veszelka S, Palmela I, Tóth A, Brites D. et al. Exposure to Lipopolysaccharide and/or Unconjugated Bilirubin Impair the Integrity and Function of Brain Microvascular Endothelial Cells. PLoS One. 2012;7:e35919
136. Ahmad S, Khan SA, Kindelin A, Mohseni T, Bhatia K, Hoda MN. et al. Acetyl-11-keto-β-boswellic acid (AKBA) Attenuates Oxidative Stress, Inflammation, Complement Activation and Cell Death in Brain Endothelial Cells Following OGD/Reperfusion. NeuroMolecular Med. 2019;21:505-16
137. Cohen SS, Min M, Cummings EE, Chen X, Sadowska GB, Sharma S. et al. Effects of Interleukin-6 on the Expression of Tight Junction Proteins in Isolated Cerebral Microvessels from Yearling and Adult Sheep. Neuroimmunomodulation. 2013;20:264-73
138. Lv S, Song H-L, Zhou Y, Li L-X, Cui W, Wang W. et al. Tumour necrosis factor-α affects blood-brain barrier permeability and tight junction-associated occludin in acute liver failure. Liver Int. 2010;30:1198-210
139. Rahman MT, Ghosh C, Hossain M, Linfield D, Rezaee F, Janigro D. et al. IFN-γ, IL-17A, or zonulin rapidly increase the permeability of the blood-brain and small intestinal epithelial barriers: Relevance for neuro-inflammatory diseases. Biochem Biophys Res Commun. 2018;507:274-9
140. Roberts TK, Eugenin EA, Lopez L, Romero IA, Weksler BB, Couraud P-O. et al. CCL2 disrupts the adherens junction: implications for neuroinflammation. Lab Investig. 2012;92:1213-33
141. Wang Y, Jin S, Sonobe Y, Cheng Y, Horiuchi H, Parajuli B. et al. Interleukin-1β Induces Blood-Brain Barrier Disruption by Downregulating Sonic Hedgehog in Astrocytes. PLoS One. 2014;9:e110024
142. Yang J, Wang K, Hu T, Wang G, Wang W, Zhang J. Vitamin D3 Supplement Attenuates Blood-Brain Barrier Disruption and Cognitive Impairments in a Rat Model of Traumatic Brain Injury. NeuroMolecular Med. 2021:1-9
143. Chua XY, Chai YL, Chew WS, Chong JR, Ang HL, Xiang P. et al. Immunomodulatory sphingosine-1-phosphates as plasma biomarkers of Alzheimer's disease and vascular cognitive impairment. Alzheimer's Res Ther. 2020;12:1-12
144. Kallmann BA, Hummel V, Lindenlaub T, Ruprecht K, Toyka K V, Rieckmann P. Cytokine-induced modulation of cellular adhesion to human cerebral endothelial cells is mediated by soluble vascular cell adhesion molecule-1. Brain. 2000;123:687-97
145. Page S, Raut S, Al-Ahmad A. Oxygen-Glucose Deprivation/Reoxygenation-Induced Barrier Disruption at the Human Blood-Brain Barrier is Partially Mediated Through the HIF-1 Pathway. NeuroMolecular Med. 2019;21:414-31
146. Stamatovic SM, Shakui P, Keep RF, Moore BB, Kunkel SL, Rooijen N Van. et al. Monocyte Chemoattractant Protein-1 Regulation of Blood-Brain Barrier Permeability. J Cereb Blood Flow Metab. 2005;25:593-606
147. Stamatovic SM, Dimitrijevic OB, Keep RF, Andjelkovic A V. Protein Kinase Cα-RhoA Cross-talk in CCL2-induced Alterations in Brain Endothelial Permeability. J Biol Chem. 2006;281:8379-88
148. Yao Y, Tsirka SE. Truncation of monocyte chemoattractant protein 1 by plasmin promotes blood-brain barrier disruption. J Cell Sci. 2011;124:1486-95
149. Jiao H, Wang Z, Liu Y, Wang P, Xue Y. Specific Role of Tight Junction Proteins Claudin-5, Occludin, and ZO-1 of the Blood-Brain Barrier in a Focal Cerebral Ischemic Insult. J Mol Neurosci. 2011;44:130-9
150. Yarlagadda A, Alfson E, Clayton AH. The Blood Brain Barrier and the Role of Cytokines in Neuropsychiatry. Psychiatry (Edgmont). 2009;6:18
151. González-Mariscal L, Tapia R, Chamorro D. Crosstalk of tight junction components with signaling pathways. Biochim Biophys Acta. 2008;1778:729-56
152. Utech M, Mennigen R, Bruewer M. Endocytosis and recycling of tight junction proteins in inflammation. J Biomed Biotechnol. 2010. 2010
153. Stamatovic SM, Johnson AM, Keep RF, Andjelkovic A V. Junctional proteins of the blood-brain barrier: New insights into function and dysfunction. Tissue Barriers. 2016;4:e1154641
154. Chen A-Q, Fang Z, Chen X-L, Yang S, Zhou Y-F, Mao L. et al. Microglia-derived TNF-α mediates endothelial necroptosis aggravating blood brain-barrier disruption after ischemic stroke. Cell Death Dis. 2019;10:1-18
155. Davalos D, Kyu Ryu J, Merlini M, Baeten KM, Le Moan N, Petersen MA. et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun. 2012;3:1-15
156. Nishioku T, Matsumoto J, Dohgu S, Sumi N, Miyao K, Takata F. et al. Tumor Necrosis Factor-α Mediates the Blood-Brain Barrier Dysfunction Induced by Activated Microglia in Mouse Brain Microvascular Endothelial Cells. J Pharmacol Sci. 2010;112:251-4
157. Sumi N, Nishioku T, Takata F, Matsumoto J, Watanabe T, Shuto H. et al. Lipopolysaccharide-Activated Microglia Induce Dysfunction of the Blood-Brain Barrier in Rat Microvascular Endothelial Cells Co-Cultured with Microglia. Cell Mol Neurobiol. 2009;30:247-53
158. Matsuyama H, Shindo A, Shimada T, Yata K, Wakita H, Takahashi R. et al. Chronic cerebral hypoperfusion activates AIM2 and NLRP3 inflammasome. Brain Res. 2020;1736:146779
159. Wada-Isoe K, Wakutani Y, Urakami K, Nakashima K. Elevated interleukin-6 levels in cerebrospinal fluid of vascular dementia patients. Acta Neurol Scand. 2004;110:124-7
160. Miyanohara J, Kakae M, Nagayasu K, Nakagawa T, Mori Y, Arai K. et al. TRPM2 Channel Aggravates CNS Inflammation and Cognitive Impairment via Activation of Microglia in Chronic Cerebral Hypoperfusion. J Neurosci. 2018;38:3520-33
161. Hou X, Liang X, Chen J-F, Zheng J. Ecto-5′-nucleotidase (CD73) is involved in chronic cerebral hypoperfusion-induced white matter lesions and cognitive impairment by regulating glial cell activation and pro-inflammatory cytokines. Neuroscience. 2015;297:118-26
162. Malaguarnera L, Motta M, Rosa M Di, Anzaldi M, Malaguarnera M. Interleukin-18 and transforming growth factor-beta1 plasma levels in Alzheimer's disease and vascular dementia. Neuropathology. 2006;26:307-12
163. Poh L, Fann DY, Wong P, Lim HM, Foo SL, Kang S-W. et al. AIM2 inflammasome mediates hallmark neuropathological alterations and cognitive impairment in a mouse model of vascular dementia. Mol Psychiatry. 2020:1-17
164. Basu A, Krady JK, Levison SW. Interleukin-1: A master regulator of neuroinflammation. J Neurosci Res. 2004;78:151-6
165. Gibson RM, Rothwell NJ, Le Feuvre RA. CNS injury: the role of the cytokine IL-1. Vet J. 2004;168:230-7
166. Labus J, Häckel S, Lucka L, Danker K. Interleukin-1β induces an inflammatory response and the breakdown of the endothelial cell layer in an improved human THBMEC-based in vitro blood-brain barrier model. J Neurosci Methods. 2014;228:35-45
167. Israelov H, Ravid O, Atrakchi D, Rand D, Elhaik S, Bresler Y. et al. Caspase-1 has a critical role in blood-brain barrier injury and its inhibition contributes to multifaceted repair. J Neuroinflammation. 2020;17:1-19
168. Ge X, Li W, Huang S, Yin Z, Xu X, Chen F. et al. The pathological role of NLRs and AIM2 inflammasome-mediated pyroptosis in damaged blood-brain barrier after traumatic brain injury. Brain Res. 2018;1697:10-20
169. Shaftel SS, Griffin WST, O'Banion MK. The role of interleukin-1 in neuroinflammation and Alzheimer disease: an evolving perspective. J Neuroinflammation. 2008;5:1-12
170. Huppert J, Closhen D, Croxford A, White R, Kulig P, Pietrowski E. et al. Cellular mechanisms of IL-17-induced blood-brain barrier disruption. FASEB J. 2010;24:1023-34
171. Thom V, Schmid S, Gelderblom M, Hackbusch R, Kolster M, Schuster S. et al. IL-17 production by CSF lymphocytes as a biomarker for cerebral vasculitis. Neurol Neuroimmunol Neuroinflammation. 2016;3:e214
172. Gelderblom M, Weymar A, Bernreuther C, Velden J, Arunachalam P, Steinbach K. et al. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood. 2012;120:3793-802
173. Hamdan A, Melconian A, Adhia A, Alhaidary A. The level of IL-1alpha, IL-10 and IL-17A in Alzheimer's disease patients: Comparative study. Baghdad Sci J. 2014;11:1486-92
174. Setiadi AF, Abbas AR, Jeet S, Wong K, Bischof A, Peng I. et al. IL-17A is associated with the breakdown of the blood-brain barrier in relapsing-remitting multiple sclerosis. J Neuroimmunol. 2019;332:147-54
175. Swardfager W, Lanctôt K, Rothenburg L, Wong A, Cappell J, Herrmann N. A Meta-Analysis of Cytokines in Alzheimer's Disease. Biol Psychiatry. 2010;68:930-41
176. Tsuge M, Yasui K, Ichiyawa T, Saito Y, Nagaoka Y, Yashiro M. et al. Increase of tumor necrosis factor-α in the blood induces early activation of matrix metalloproteinase-9 in the brain. Microbiol Immunol. 2010;54:417-24
177. Coelho-Santos V, Leitão RA, Cardoso FL, Palmela I, Rito M, Barbosa M. et al. The TNF-α/Nf-κB Signaling Pathway has a Key Role in Methamphetamine-Induced Blood-Brain Barrier Dysfunction. J Cereb Blood Flow Metab. 2015;35:1260-71
178. Poritz LS, Garver KI, Tilberg AF, Koltun WA. Tumor necrosis factor alpha disrupts tight junction assembly1,2. J Surg Res. 2004;116:14-8
179. Rosenberg GA. Matrix metalloproteinases in neuroinflammation. Glia. 2002;39:279-91
180. Sharony R, Yu P-J, Park J, Galloway AC, Mignatti P, Pintucci G. Protein targets of inflammatory serine proteases and cardiovascular disease. J Inflamm. 2010;7:1-17
181. Pasparakis M. Regulation of tissue homeostasis by NF-κB signalling: implications for inflammatory diseases. Nat Rev Immunol. 2009;9:778-88
182. Miller F, Fenart L, Landry V, Coisne C, Cecchelli R, Dehouck M-P. et al. The MAP kinase pathway mediates transcytosis induced by TNF-α in an in vitro blood-brain barrier model. Eur J Neurosci. 2005;22:835-44
183. Förster C, Burek M, Romero IA, Weksler B, Couraud P-O, Drenckhahn D. Differential effects of hydrocortisone and TNFα on tight junction proteins in an in vitro model of the human blood-brain barrier. J Physiol. 2008;586:1937-49
184. Franzén B, Duvefelt K, Jonsson C, Engelhardt B, Ottervald J, Wickman M. et al. Gene and protein expression profiling of human cerebral endothelial cells activated with tumor necrosis factor-α. Mol Brain Res. 2003;115:130-46
185. Nagyoszi P, Wilhelm I, Farkas AE, Fazakas C, Dung NTK, Haskó J. et al. Expression and regulation of toll-like receptors in cerebral endothelial cells. Neurochem Int. 2010;57:556-64
186. Álvarez A. Neuroimmunotrophic regulation in vascular dementia: Serum TNF-alpha and IGF-I. J Neurol Sci. 2009;283:249
187. Tisato V, Rimondi E, Brombo G, Volpato S, Zurlo A, Zauli G. et al. Serum Soluble Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand Levels in Older Subjects with Dementia and Mild Cognitive Impairment. Dement Geriatr Cogn Disord. 2016;41:273-80
188. Lombardi A, Cantini G, Mello T, Francalanci M, Gelmini S, Cosmi L. et al. Molecular mechanisms underlying the pro-inflammatory synergistic effect of tumor necrosis factor α and interferon γ in human microvascular endothelium. Eur J Cell Biol. 2009;88:731-42
189. Chui R, Dorovini-Zis K. Regulation of CCL2 and CCL3 expression in human brain endothelial cells by cytokines and lipopolysaccharide. J Neuroinflammation. 2010;7:1-12
190. Yun JW, Barzegar M, Boyer CJ, Minagar A, Couraud P-O, Alexander JS. Brain Endothelial Cells Release Apical and Basolateral Microparticles in Response to Inflammatory Cytokine Stimulation: Relevance to Neuroinflammatory Stress? Front Immunol. 2019;0:1455
191. Maus U, Henning S, Wenschuh H, Mayer K, Seeger W, Lohmeyer J. Role of endothelial MCP-1 in monocyte adhesion to inflamed human endothelium under physiological flow. Am J Physiol Heart Circ Physiol. 2002;283:2584-91
192. Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic A V. Effects of the Chemokine CCL2 on Blood-Brain Barrier Permeability during Ischemia-Reperfusion Injury. J Cereb Blood Flow Metab. 2005;26:797-810
193. Bettcher BM, Neuhaus J, Wynn MJ, Elahi FM, Casaletto KB, Saloner R. et al. Increases in a Pro-inflammatory Chemokine, MCP-1, Are Related to Decreases in Memory Over Time. Front Aging Neurosci. Frontiers. 2019;0:25
194. Pallebage-Gamarallage M, Lam V, Takechi R, Galloway S, Clark K, Mamo J. Restoration of dietary-fat induced blood-brain barrier dysfunction by anti-inflammatory lipid-modulating agents. Lipids Health Dis. 2012;11:117
195. Khamchai S, Chumboatong W, Hata J, Tocharus C, Suksamrarn A, Tocharus J. Morin protects the blood-brain barrier integrity against cerebral ischemia reperfusion through anti-inflammatory actions in rats. Sci Rep. 2020;10:13379
196. Salvador E, Shityakov S, Förster C. Glucocorticoids and endothelial cell barrier function. Cell Tissue Res. 2014;355:597-605
197. Buse P, Woo PL, Alexander DB, Reza A, Firestone GL. Glucocorticoid-induced Functional Polarity of Growth Factor Responsiveness Regulates Tight Junction Dynamics in Transformed Mammary Epithelial Tumor Cells. J Biol Chem. 1995;270:28223-7
198. Witt K, Sandoval K. Steroids and the Blood-Brain Barrier. Adv Pharmacol. 2014;71C:361-90
199. Ali MM, Ghouri RG, Ans AH, Akbar A, Toheed A. Recommendations for Anti-inflammatory Treatments in Alzheimer's Disease: A Comprehensive Review of the Literature. Cureus. 2019;11:e4620-e4620
200. Lenglet S, Montecucco F, Mach F, Schaller K, Gasche Y, Copin J-C. Analysis of the expression of nine secreted matrix metalloproteinases and their endogenous inhibitors in the brain of mice subjected to ischaemic stroke. Thromb Haemost. 2017;112:363-78
201. Romanitan MO, Popescu BO, Winblad B, Bajenaru OA, Bogdanovic N. Occludin is overexpressed in Alzheimer's disease and vascular dementia. J Cell Mol Med. 2007;11:569-79
202. Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix Metalloproteinase-Mediated Disruption of Tight Junction Proteins in Cerebral Vessels is Reversed by Synthetic Matrix Metalloproteinase Inhibitor in Focal Ischemia in Rat. J Cereb Blood Flow Metab. 2006;27:697-709
203. Rosenberg GA, Sullivan N, Esiri MM. White Matter Damage Is Associated With Matrix Metalloproteinases in Vascular Dementia. Stroke. 2001;32:1162-7
204. Truettner JS, Alonso OF, Dietrich WD. Influence of Therapeutic Hypothermia on Matrix Metalloproteinase Activity after Traumatic Brain Injury in Rats. J Cereb Blood Flow Metab. 2005;25:1505-16
205. Ihara M, Tomimoto H, Kinoshita M, Oh J, Noda M, Wakita H. et al. Chronic Cerebral Hypoperfusion Induces MMP-2 but Not MMP-9 Expression in the Microglia and Vascular Endothelium of White Matter. J Cereb Blood Flow Metab. 2016;21:828-34
206. Nigro P, Pompilio G, Capogrossi MC. Cyclophilin A: a key player for human disease. Cell Death Dis. 2013;4:e888-e888
207. Melo A, Monteiro L, Lima RMF, De Oliveira DM, De Cerqueira MD, El-Bachá RS. Oxidative stress in neurodegenerative diseases: Mechanisms and therapeutic perspectives. Oxid Med Cell Longev. 2011;2011:467180
208. Nigro P, Satoh K, O'Dell MR, Soe NN, Cui Z, Mohan A. et al. Cyclophilin A is an inflammatory mediator that promotes atherosclerosis in apolipoprotein E-deficient mice. J Exp Med. 2011;208:53-66
209. Satoh K, Matoba T, Suzuki J, O'Dell MR, Nigro P, Cui Z. et al. Cyclophilin A Mediates Vascular Remodeling by Promoting Inflammation and Vascular Smooth Muscle Cell Proliferation. Circulation. 2008;117:3088-98
210. Satoh K, Nigro P, Berk BC. Oxidative Stress and Vascular Smooth Muscle Cell Growth: A Mechanistic Linkage by Cyclophilin A. Antioxid Redox Signal. 2010;12:675-82
211. Fields GB. The Rebirth of Matrix Metalloproteinase Inhibitors: Moving Beyond the Dogma. Cells. 2019;8:984
212. Vandenbroucke R, Libert C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat Rev Drug Discov. 2014 13
213. Brown R, Benveniste H, Black SE, Charpak S, Dichgans M, Joutel A. et al. Understanding the role of the perivascular space in cerebral small vessel disease. Cardiovasc Res. 2018;114:1462-73
214. Montagne A, Nikolakopoulou AM, Zhao Z, Sagare AP, Si G, Lazic D. et al. Pericyte degeneration causes white matter dysfunction in the mouse central nervous system. Nat Med. 2018;24:326-37
215. Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS immune privilege: hiding in plain sight. Immunol Rev. 2006;213:48-65
216. Yilmaz G, Granger DN. Leukocyte Recruitment and Ischemic Brain Injury. NeuroMolecular Med. 2009;12:193-204
217. Ala A, Dhillon AP, Hodgson HJ. Role of cell adhesion molecules in leukocyte recruitment in the liver and gut. Int J Exp Pathol. 2003;84:1-16
218. Kappelmayer J, Nagy B. The interaction of selectins and PSGL-1 as a key component in thrombus formation and cancer progression. Biomed Res Int. 2017:1-18
219. Muller WA. Leukocyte-Endothelial Cell Interactions in the Inflammatory Response. Lab Investig. 2002;82:521-34
220. Shaw SK, Ma S, Kim MB, Rao RM, Hartman CU, Froio RM. et al. Coordinated Redistribution of Leukocyte LFA-1 and Endothelial Cell ICAM-1 Accompany Neutrophil Transmigration. J Exp Med. 2004;200:1571-80
221. Dole VS, Bergmeier W, Mitchell HA, Eichenberger SC, Wagner DD. Activated platelets induce Weibel-Palade-body secretion and leukocyte rolling in vivo: role of P-selectin. Blood. 2005;106:2334-9
222. Jin AY, Tuor UI, Rushforth D, Kaur J, Muller RN, Petterson JL. et al. Reduced blood brain barrier breakdown in P-selectin deficient mice following transient ischemic stroke: a future therapeutic target for treatment of stroke. BMC Neurosci. 2010;11:1-9
223. Abadier M, Jahromi NH, Alves LC, Boscacci R, Vestweber D, Barnum S. et al. Cell surface levels of endothelial ICAM-1 influence the transcellular or paracellular T-cell diapedesis across the blood-brain barrier. Eur J Immunol. 2015;45:1043-58
224. Kovac A, Erickson MA, Banks WA. Brain microvascular pericytes are immunoactive in culture: cytokine, chemokine, nitric oxide, and LRP-1 expression in response to lipopolysaccharide. J Neuroinflammation. 2011;8:1-9
225. Wang S, Cao C, Chen Z, Bankaitis V, Tzima E, Sheibani N. et al. Pericytes Regulate Vascular Basement Membrane Remodeling and Govern Neutrophil Extravasation during Inflammation. PLoS One. 2012;7:e45499
226. Theilmeier G, Lenaerts T, Remacle C, Collen D, Vermylen J, Hoylaerts MF. Circulating Activated Platelets Assist THP-1 Monocytoid/Endothelial Cell Interaction Under Shear Stress. Blood. 1999;94:2725-34
227. Hundelshausen P, Weber K, Huo Y, Proudfoot A, Nelson P, Ley K. et al. RANTES Deposition by Platelets Triggers Monocyte Arrest on Inflamed and Atherosclerotic Endothelium. Circulation. 2001 103
228. Gawaz M, Brand K, Dickfeld T, Pogatsa-Murray G, Page S, Bogner C. et al. Platelets induce alterations of chemotactic and adhesive properties of endothelial cells mediated through an interleukin-1-dependent mechanism. Implications for atherogenesis. Atherosclerosis. 2000;148:75-85
229. Stellos K, Katsiki N, Tatsidou P, Bigalke B, Laske C. Association of Platelet Activation with Vascular Cognitive Impairment: Implications in Dementia Development? Curr Vasc Pharmacol. 2012 12
230. Kniewallner K, Foidl B, Humpel C. Platelets isolated from an Alzheimer mouse damage healthy cortical vessels and cause inflammation in an organotypic ex vivo brain slice model. Sci Rep. 2018 8
231. Jian Z, Liu R, Zhu X, Smerin D, Zhong Y, Gu L. et al. The Involvement and Therapy Target of Immune Cells After Ischemic Stroke. Front Immunol. 2019;0:2167
232. Bastian C, Day J, Politano S, Quinn J, Brunet S, Baltan S. Preserving Mitochondrial Structure and Motility Promotes Recovery of White Matter After Ischemia. NeuroMolecular Med. 2019;21:484-92
233. Cupini LM, Diomedi M, Placidi F, Silvestrini M, Giacomini P. Cerebrovascular Reactivity and Subcortical Infarctions. Arch Neurol. 2001;58:577-81
234. Terborg C, Gora F, Weiller C, Röther J. Reduced Vasomotor Reactivity in Cerebral Microangiopathy. Stroke. 2000;31:924-9
235. Simpson JE, Wharton SB, Cooper J, Gelsthorpe C, Baxter L, Forster G. et al. Alterations of the blood-brain barrier in cerebral white matter lesions in the ageing brain. Neurosci Lett. 2010;486:246-51
236. Narantuya D, Nagai A, Sheikh AM, Wakabayashi K, Shiota Y, Watanabe T. et al. Microglia transplantation attenuates white matter injury in rat chronic ischemia model via matrix metalloproteinase-2 inhibition. Brain Res. 2010;1316:145-52
237. Schreiber S, Bueche CZ, Garz C, Braun H. Blood brain barrier breakdown as the starting point of cerebral small vessel disease? - New insights from a rat model. Exp Transl Stroke Med. 2013;5:1-8
238. Arai K, Lo EH. An Oligovascular Niche: Cerebral Endothelial Cells Promote the Survival and Proliferation of Oligodendrocyte Precursor Cells. J Neurosci. 2009;29:4351-5
239. Seo JH, Miyamoto N, Hayakawa K, Pham L-DD, Maki T, Ayata C. et al. Oligodendrocyte precursors induce early blood-brain barrier opening after white matter injury. J Clin Invest. 2013;123:782-6
240. Li Y, Li M, Zuo L, Shi Q, Qin W, Yang L. et al. Compromised Blood-Brain Barrier Integrity Is Associated With Total Magnetic Resonance Imaging Burden of Cerebral Small Vessel Disease. Front Neurol. 2018;9:221
241. Young VG, Halliday GM, Kril JJ. Neuropathologic correlates of white matter hyperintensities. Neurology. 2008;71:804-11
242. Momenabadi S, Vafaei AA, Bandegi AR, Zahedi-Khorasani M, Mazaheri Z, Vakili A. Oxytocin Reduces Brain Injury and Maintains Blood-Brain Barrier Integrity After Ischemic Stroke in Mice. NeuroMolecular Med. 2020;22:557-71
243. Nicchia GP, Nico B, Camassa LMA, Mola MG, Loh N, Dermietzel R. et al. The role of aquaporin-4 in the blood-brain barrier development and integrity: Studies in animal and cell culture models. Neuroscience. 2004;129:935-44
244. Vella J, Zammit C, Di Giovanni G, Muscat R, Valentino M. The central role of aquaporins in the pathophysiology of ischemic stroke. Front Cell Neurosci. 2015;0:108
245. Wolburg H, Noell S, Fallier-Becker P, MacK AF, Wolburg-Buchholz K. The disturbed blood-brain barrier in human glioblastoma. Mol Aspects Med. 2012;33:579-89
246. Akaishi T, Narikawa K, Suzuki Y, Mitsuzawa S, Tsukita K, Kuroda H. et al. Importance of the quotient of albumin, quotient of immunoglobulin G and Reibergram in inflammatory neurological disorders with disease-specific patterns of blood-brain barrier permeability. Neurol Clin Neurosci. 2015;3:94-100
247. Kapural M, Krizanac-Bengez L, Barnett G, Perl J, Masaryk T, Apollo D. et al. Serum S-100β as a possible marker of blood-brain barrier disruption. Brain Res. 2002;940:102-4
248. Shi S, Wang G, Zhang K, Zhang Z, Liang K, Li K. et al. Expression of S100β protein in patients with vascular dementia after basal ganglia hemorrhage and its clinical significance. Exp Ther Med. 2017;13:1917-21
249. Marchi N, Fazio V, Cucullo L, Kight K, Masaryk T, Barnett G. et al. Serum Transthyretin Monomer as a Possible Marker of Blood-to-CSF Barrier Disruption. J Neurosci. 2003;23:1949-55
250. Gloeckner SF, Zerr I. Transthyretin cerebrospinal fluid levels in Alzheimer's disease - is this a biomarker for disease severity? Eur Neurol Rev. 2009;4:17-9
251. Cramer SP, Simonsen H, Frederiksen JL, Rostrup E, Larsson HBW. Abnormal blood-brain barrier permeability in normal appearing white matter in multiple sclerosis investigated by MRI. NeuroImage Clin. 2014;4:182-9
252. Raja R, Rosenberg GA, Caprihan A. MRI measurements of Blood-Brain Barrier function in dementia: A review of recent studies. Neuropharmacology. 2018;134:259-71
253. Giridharan V V, Barichello T, Selvaraj S. Molecular Imaging of Blood-Brain Barrier Permeability in Preclinical Models Using PET and SPECT. Neuromethods. 2019;142:329-42
254. Iannotti F, Fieschi C, Alfano B, Picozzi P, Mansi L, Pozzilli C. et al. Simplified, Noninvasive PET Measurement of Blood-Brain Barrier Permeability. J Comput Assist Tomogr. 1987;11:390-7
255. Cuenod CA, Balvay D. Perfusion and vascular permeability: Basic concepts and measurement in DCE-CT and DCE-MRI. Diagn Interv Imaging. 2013;94:1187-204
256. Shao X, Ma SJ, Casey M, D'Orazio L, Ringman JM, Wang DJJ. Mapping water exchange across the blood-brain barrier using 3D diffusion-prepared arterial spin labeled perfusion MRI. Magn Reson Med. 2019;81:3065-79
257. Li Pan R, Qi Z Liu KJ. Current progress in searching for clinically useful biomarkers of blood-brain barrier damage following cerebral ischemia. Brain Circ. 2018;4:145
258. Anderson RE, Hansson L-O, Nilsson O, Dijlai-Merzoug R, Settergren G. High Serum S100B Levels for Trauma Patients without Head Injuries. Neurosurgery. 2001;48:1255-60
259. Chaves ML, Camozzato AL, Ferreira ED, Piazenski I, Kochhann R, Dall'Igna O. et al. Serum levels of S100B and NSE proteins in Alzheimer's disease patients. J Neuroinflammation. 2010;7:1-7
260. Sun B, Liu H, Nie S. S100B protein in serum is elevated after global cerebral ischemic injury. World J Emerg Med. 2013;4:165
Author contact
![]() Corresponding author: Thiruma V. Arumugam, Department of Physiology, Anatomy and Microbiology, La Trobe University, Bundoora, VIC, Australia. E-mail: g.arumugamedu.au
Corresponding author: Thiruma V. Arumugam, Department of Physiology, Anatomy and Microbiology, La Trobe University, Bundoora, VIC, Australia. E-mail: g.arumugamedu.au