Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2022; 12(2):747-766. doi:10.7150/thno.65828 This issue Cite
Research Paper
Nintedanib enhances the efficacy of PD-L1 blockade by upregulating MHC-I and PD-L1 expression in tumor cells
Jingyao Tu1, Haoran Xu2, Li Ma1, Chunya Li1, Wan Qin1, Xinyi Chen1, Ming Yi1, Li Sun1 ![]() , Bo Liu1
, Bo Liu1 ![]() , Xianglin Yuan1
, Xianglin Yuan1 ![]()
1. Department of Oncology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
2. Department of Orthopedics, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
Received 2021-8-7; Accepted 2021-11-17; Published 2022-1-1
Abstract

Background: Immune checkpoint inhibitors (ICIs), such as programmed cell death protein 1 (PD-1)/programmed death-ligand 1 (PD-L1), have been widely applied in clinical and scientific research. Despite their effective antitumor effects in clinical tumor therapy, most tumors are still resistant to ICIs and long-term benefits are lacking. In addition, tumor patients complicated with interstitial lung disease limit the application of ICI therapy. Therefore, for these cases, there is an urgent need to develop new methods to relieve lung complications and enhance the efficacy of ICI therapy. Nintedanib, a potent triple angiokinase inhibitor approved for the treatment of progressive fibrotic interstitial lung disease. However, its immunotherapy synergy properties and mechanism are still pending further exploration.
Methods: To explore the therapeutic potential of nintedanib and αPD-L1 combination therapy, MC38, LLC, and 4T1 tumor models were used to investigate antitumor and antimetastatic activities in vivo. An idiopathic pulmonary fibrosis-tumor bearing model was used to evaluate the effect of the synergy therapy on tumor model complicated with lung disease. Moreover, RNA-seq, immunohistochemistry, and flow cytometry were utilized to analyze the effect of combination treatment on the tumor microenvironment. The bioactivity following different treatments was determined by western blotting, CCK-8, and flow cytometry.
Results: In this study, nintedanib and αPD-L1 synergy therapy exhibited significant antitumor, antimetastatic and anti-pulmonary fibrosis effects. Both in vitro and in vivo experiments revealed that these effects included promoting vessel normalization, increasing infiltration and activation of immune cells in tumors, enhancing the response of interferon-gamma, and activating the MHC class I-mediated antigen presentation process. Moreover, our results showed an increased expression of PD-L1 and promoted phosphorylation of STAT3 after nintedanib (1 µM) treatment.
Conclusion: The combination of nintedanib and αPD-L1 increased ICI therapy responses, relieved lung complications and further activated the tumor immune microenvironment; thus, exhibiting a notable antitumor effect. Accordingly, the nintedanib synergy strategy is expected to be a promising candidate therapy for tumor patients complicated with interstitial lung disease in clinical practice.
Keywords: Cancer immunotherapy, Nintedanib, PD-L1, MHC class I, Idiopathic pulmonary fibrosis
Introduction
The incidence and mortality rates of malignant tumors continue to rise and show a trend in affecting the younger population. Clinically, immunotherapy is becoming a prevalent approach that is being researched and developed for the treatment of tumors due to its evident therapeutic effects [1-4]. Although immunotherapy can activate the immune system, block immune checkpoint pathways, and overcome immune escape to inhibit tumor growth, there are still many cancer patients who do not obtain long term therapeutic benefits. Among all immune checkpoint inhibitors (ICIs), programmed cell death protein 1 (PD-1)/programmed death-ligand 1 (PD-L1) has attracted the wide attention. However, only a subset of patients benefit from anti-PD-1/PD-L1 treatment, and the overall response rate is relatively low [5, 6]. The effective rate of anti-PD-1/PD-L1, a novel therapy for non-small cell lung cancer (NSCLC), administered alone is approximately 20-30%, while the effective rate of conventional chemotherapy combined with immunotherapy is only 50% [7]. In addition, the effectiveness of anti-PD-1/PD-L1 therapy for colon cancer [8-10] and breast cancer is still under investigation in several clinical trials [11, 12]. Therefore, improving the efficacy of immunotherapy is an urgent problem that needs to be addressed in current oncology treatments.
It is well-established that many intrinsic and extrinsic factors operating within the tumor microenvironment (TME) inhibit the therapeutic effects of ICIs [13, 14]. Intrinsic factors such as the absence of antigenic proteins (low mutational burden), inefficient antigen presentation (downregulation of major histocompatibility complex class I [MHC-I] or transporter associated with antigen processing [TAP]), insensitivity to T cells (oncogenic PD-L1 expression, mutations in the interferon-gamma [IFN-γ] pathway), etc., can contribute to immune escape [13, 15, 16]. Extrinsic factors such as the absence of T cells, inhibitory immune checkpoints (VISTA, LAG-3, TIM-3), immunosuppressive cells (myeloid-derived suppressor cells [MDSCs], Tregs, or type II macrophages), and abnormal tumor extracellular matrix (ECM; abnormal tumor vessel and fibrotic collagen) can create an immunosuppressive environment [13, 15, 17, 18]. The various factors mentioned above would eventually lead to ICI therapy failure. Therefore, dedicated research and strategies to combat these mechanisms that induce immune resistance are key to increasing the efficacy of ICI treatment.
To overcome these obstacles, strategies using synergistic anti-angiogenesis and ICI treatments have been widely utilized in preclinical studies and clinical trials with promising outcomes [19-21]. However, some studies have reported that increased infiltration of immune cells is inversely related to patient overall survival and progression-free survival, which may be attributed to the fact that most of the recruited immune cells after anti-angiogenic therapy were dysfunctional [22]. Therefore, elimination of immunosuppressive TME and activation of immune cells are the primary problems that need to be addressed in anti-angiogenesis treatment combined with ICI therapy.
Indeed, clinical cases have shown that tumor patients complicated with different degrees of interstitial lung disease (ILD; such as chronic obstructive pulmonary diseases, idiopathic pulmonary fibrosis [IPF], ICI therapy-related ILD and radiation-induced lung fibrosis, etc.) are often associated with poor outcomes [23]. However, both ICI and anti-angiogenesis therapy may induce the occurrence or aggravation of a series of ILD [24, 25]. Multiple clinical trials have shown that the incidence of ICI therapy-related ILD is approximately 3.5-14.5% after ICI therapy [26-29]. In particular, the treatment of ICI therapy-related ILD is mainly based on immunosuppressive strategies, which leads to the discontinuation of ICI treatment and tumor progression [24]. Thus, there is an urgent need to develop new methods to relieve lung complications and enhance the efficacy of ICI therapy.
Nintedanib, a potent triple angiokinase inhibitor (VEGFR1/2/3, FGFR1/2/3, and PDGFR α/β) [30], is known to be an excellent anti-angiogenic agent and approved for the treatment of progressive fibrotic interstitial lung disease in 2020 [31-33]. Moreover, recent evidence suggests that nintedanib can increase the infiltration of CD8+ T cells in the tumor environment and inhibit tumor proliferation by inhibiting cancer-associated fibroblasts [34, 35]. Except for alleviating fibrosis in tumor ECM, we hypothesized that nintedanib normalizes distorted vessels in tumor ECM and enhances the ICI efficacy. Moreover, the mechanisms by which nintedanib acts on tumor cells to increase the immune response has not yet been reported.
On the other hand, extensive research has shown that anti-angiogenesis drugs used for tumor treatment may increase the expression of PD-L1 in tumor cells [22, 36-38]. The increased expression of PD-L1 may enhance tumor sensitivity to immunotherapy (anti-PD-1/PD-L1) and improve the prognosis of patients treated with ICIs [39-44]. In summary, we hypothesized that nintedanib combined with anti-PD-L1 (αPD-L1) therapy may promote immunotherapy responses and exert antitumor effects to prevent tumor progression. In this study, the MC38 and LLC tumor-bearing models were used to evaluate the antitumor effect of the synergistic therapy and an IPF-tumor bearing model was used to explore the effect of relieving lung complications. Antimetastatic abilities were verified using the 4T1 model. As for the immune landscape, we explored the difference in tumor immune microenvironment (TIME) after combination treatment with tumor tissue immunohistochemistry (IHC) and flow cytometry. To elucidate the underlying mechanisms, RNA sequencing (RNA-seq) and a series of in vitro experiments were performed. Through this study, we sought to clarify the mechanisms of nintedanib on TIME and explore the antitumor therapeutic potential of nintedanib combined with αPD-L1. This combination may be a potential strategy against immune escape and be applicable in clinical synergy immunotherapy.
Methods
Reagents and materials
Nintedanib was purchased from Selleck Chemicals (Houston, TX, USA). InVivoMab anti-mouse PD-L1 antibody (clone 10F.9G2, Cat#BE0101) was obtained from BioXcell (West Lebanon, NH, USA). Recombinant mouse interferon-gamma (rMuIFN-γ; HY-P7071) and recombinant human interferon-gamma (rHuIFN-γ; HY-P7025) were purchased from MedChem Express (Monmouth Junction, NJ, USA). Dimethyl sulfoxide was purchased from Promoter (Wuhan, China).
Cell lines and culture
The cell lines used in this study were obtained from the Oncology Laboratory, Tongji Hospital, Huazhong University of Science and Technology. MC38 and CT26 (murine colon cancer cells), H1975, A549, H1299, H358, H292, H1703, Hcc827, PC9, and H460 (lung cancer cells) were cultured in RPMI-1640 medium (Hyclone, Logan, UT, USA), and 4T1 (murine breast cancer cells) were cultured in RPMI-1640 medium (Gibco, Grand Island, NY, USA). LLC (murine lung cancer cells), B16-F10 (murine melanoma cells), MCF-7 and 231 (breast cancer cells), and Hct-116 and Sw480 (colon cancer cells) were cultured in Dulbecco's modified Eagle's medium (Hyclone). Cells were cultured in complete medium containing 10% fetal bovine serum (Gibco) and 1% penicillin-streptomycin (Gibco), and incubated with 5% CO2 at 37 °C.
Experimental animals
BALB/c mice, C57BL/6 mice (female, 6-8 weeks, 18-20 g) were obtained from Vital River Laboratories (Beijing, China). The in vivo experiments were performed in accordance with the guidelines of the International Guiding Principles for Animal Research and approved by the Ethics Committee of Huazhong University of Science and Technology. The mice were fed and experiments were conducted under specific pathogen-free grade conditions.
CCK-8 assay
Cells from different cell lines were seeded into 96-well plates (5×103 cells per well) and incubated at 37 °C for 24 h. Different concentrations of nintedanib were added to cells which were then cultured for 24, 48, or 72 h. At the experimental endpoint, Cell Counting Kit-8 (CCK-8; HY-K0301, MedChem Express) was used to evaluate cell viability using absorbance (wavelength of 450 nm) detected by a microplate reader (BioTek, Winooski, VT, USA).
Western blotting
Tumor cells or tissue proteins were extracted using RIPA buffer (Beyotime, Shanghai, China) containing phenylmethylsulfonyl fluoride and phosphatase inhibitors (Servicebio, Wuhan, China), and the concentration was quantified using a BCA assay kit (Beyotime, Shanghai, China). The protein sample (30 µg) was separated using SDS-PAGE and transferred to a PVDF membrane (Millipore, Burlington, MA, USA). After blocking with 5% BSA for 1 h, the membranes were incubated with the following primary antibodies: GAPDH (1:20000, 60004-1-lg, Proteintech, Wuhan, China), TAP1 (1:800, 11114-1-AP, Proteintech), PD-L1 (1:1000, 17952-1-AP, Proteintech), PD-L1 (1:1000, E1L3N, Cell Signaling Technology, Boston, Massachusetts, USA), STAT3 (1:1000, 10253-2-AP, Proteintech), Phospho-STAT3 (1:1000, D3A7, Cell Signaling Technology), Beta-2-Microglobulin (β2M; 1:1000, 13511-1-AP, Proteintech), Fibronectin (1:1000, 15613-1-AP, Proteintech), E-cadherin (1:1000, 24E10, Cell Signaling Technology), and Vimentin (1:1000, D21H3, Cell Signaling Technology) at 4 °C overnight. The next day, membranes were incubated with secondary antibodies (Promoter, Wuhan, China) and visualized using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific, Waltham, MA, USA). The signal from the blots was detected using the G:BOX Chemi X system (Syngene, Cambridge, UK) and analyzed using ImageJ software.
Real-time PCR (RT-PCR)
Total tumor tissue RNA was extracted using Trizol (NO. 9766, Takara Bio, Shiga, Japan) and reverse transcribed to cDNA using the PrimeScript 1st Strand cDNA Synthesis Kit (NO.6110A, Takara Bio). Gene-specific primers used are listed: Cxcl2, 5'-ACTGCGCTGTCAATGCCTGAAG-3' (forward), 5'-CAGTTAGCCTTGCCTTTGTTCAGTATC-3' (reverse); Cxcl3, 5'-CCACCAACCACCAGGCTACA-3' (forward), 5'-TGGACTTGCCGCTCTTCAGT-3' (reverse); Ccl6, 5'-TTCTTTATCCTTGTGGCTGTCCTTGG‑3' (forward), 5'‑AGGCACCTCTGAACTCTCCGATC‑3' (reverse); Ccl8, 5'-TGCTTCTTTGCCTGCTGCTCATAG‑3' (forward), 5'‑TCTCCATGTACTCACTGACCCACTTC‑3' (reverse); Ccl9, 5'-TCTTCAGATTGCTGCCT GTCCTATAAC‑3' (forward), 5'‑TTGTTTGTAGGTCCGTGGTTGTGAG‑3' (reverse); GAPDH, 5'‑AGGTCGGTGTGAACGGATTTG-3' (forward), 5'‑TGTAGACCATGTAGTTGAGGTCA‑3' (reverse). RT-PCR was performed to detect gene expression levels using an RT-PCR system (7900HT, Applied Biosystems, Waltham, MA, USA). The mRNA expression levels of the target genes were normalized to GAPDH expression levels and analyzed using the 2-ΔΔCt method.
Flow cytometry
Tumor tissue, lymph nodes, and spleen were harvested from the euthanized mice (n = 5 per group). To prepare the tumor cell suspension, tumor tissue was cut into small pieces, ground, and digested with type IV collagenase (Promoter), hyaluronidase, and DNase at 37 °C. The suspensions were filtered using 70 μm nylon cell strainers and treated with RBC lysis buffer. To obtain lymph node and spleen suspensions, tissues were ground and filtered using 70 μm nylon cell strainers (spleen suspension must be processed by RBC lysis). After centrifugation (2000 rpm, 5 min) and resuspension with PBS, cells were stained with APC/Cy7 Fixable Viability Stain 780 (565388, BD, Franklin, NJ, USA), blocked with anti-mouse CD16/32 (101320, BioLegend, San Diego, CA, USA) and stained according to the manufacturer's guidelines. The fluorescent antibodies used are listed as follows: PerCP/Cy5.5-anti-mouse CD11c (560584, BD), PE/Cy7-anti-mouse CD86 (560582, BD), PE-anti-mouse CD45 (553081, BD), FITC-anti-mouse CD8a (553030, BD), BV421-anti-mouse CD4 (562891, BD), BV510-anti-mouse CD3e (563024, BD), Alexa Fluor 674-anti-mouse I-A/I-E (107608, BioLegend), APC-anti-mouse CD274 (124311, BioLegend), PE/Cy7-anti-mouse CD69 (552879, BD), FITC-anti-mouse CD11b (557396, BD), BV421-anti-mouse F4/80 (123131, BioLegend), BV650-anti-mouse CD206 (141723, BioLegend), Alexa Fluor 700-anti-mouse Ly-6G/Ly-6C (557979, BD). Flow cytometry was performed using CytoFLEX LX (Beckman, Brea, CA, USA) and analyzed using FlowJo v10 software.
IFN-γ response analysis
Tumor cells were seeded into 6-well plates (2×105 cells per well) and treated under several conditions (different concentrations of nintedanib and 10 ng/ml rMuIFN-γ) for 24 h. A total of 5×105 cells were obtained, washed with PBS, and stained with fluorescent antibodies: APC-anti-mouse H-2Kd (116619, BioLegend), FITC-anti-mouse H-2Kb (553569, BD), and PE-anti-mouse CD274 (558091, BD). Flow cytometry was performed and analyzed as described above.
RNA‑seq assay
The RNA-seq assay was performed by Novogene (Beijing, China). Briefly, the total RNA of MC38 tumors (minimum of four samples per group) was extracted using Trizol (Takara Bio) and purified for library preparation and sequencing on an Illumina Hiseq platform. For data analysis, differential expression analysis of the four groups was performed using the DESeq2 R package (1.20.0), and genes were considered significantly differentially expressed when the p-value was less than 0.05. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses for differentially expressed genes (DEGs) were performed using the clusterProfiler R package. Immune signatures were designed based on the public lists [45]. In addition, we used the local Gene Set Enrichment Analysis (GSEAtool (http://www.broadinstitute.org/gsea/index.jsp) to independently analyze the GO and KEGG data sets.
Staining and IHC
Isolated lung tissues and tumors were fixed with 4% paraformaldehyde (PFA) at 25 °C for 48 h, dehydrated, and embedded in paraffin wax for further experiments. Hematoxylin-eosin (HE) staining (G1005, Servicebio), Masson's trichrome staining (G1340, Solarbio, Beijing, China) and various IHC assays were performed according to the manufacturer's instructions. The primary antibodies used in the experiments were as follows: anti-CD3 (1:150, ab16669, Abcam, UK), anti-CD8α (1:2000, ab217344, Abcam), and anti-Ki67 (1:200, ab16667, Abcam). Bright-field images (five random fields) were captured using a microscope (EVOS fl auto, Thermo Fisher Scientific, USA). For CD3+ and CD8+ T cell analysis, two blinded pathologists counted tumor center and tumor periphery T cells. In addition, positive Ki67 cells were counted and analyzed using ImageJ software.
Immunofluorescence (IF)
Freshly dissected tumor tissues were fixed with 4% PFA, dehydrated, embedded in paraffin wax, and transferred to the slice. A TUNEL assay staining kit (11684817910, Roche) was used to detect tumor cell apoptosis. Next, according to the manufacturer's protocol, the slices were permeabilized (0.1% Triton X-100), blocked with 10% normal donkey serum (ANT051, antGene, Wuhan, China), and incubated with primary antibodies, CD31 (1:500, ab182981, Abcam), and α-SMA (1:100, AF1032, Affinity) overnight. After incubation with fluorescent secondary antibody and DAPI (C0065, Solarbio), the immunofluorescence images were captured using a microscope (EVOS fl auto, Thermo Fisher Scientific, Waltham, MA, USA) and analyzed using ImageJ software.
Tumor growth inhibition
For in vivo experiments, two different subcutaneous tumor models (MC38 and LLC) were established by subcutaneous inoculation of tumor cells (1×106) into the right flank of C57BL/6 mice. When the volume of the tumor was approximately 100 mm3, the mice (n = 7) were randomly treated with isotype control antibodies (i.p.) and saline (p.o.) as the control group, αPD-L1 (i.p., 10 mg/kg, three times a week, administered three times) as the αPD-L1 group, nintedanib (p.o., 30 mg/kg, 5 days ON, 1 day OFF, administered 10 times) as the nintedanib group, or a combination of αPD-L1 with nintedanib as the combination group. Subsequently, tumor size (length × width2 / 2) and mouse weight were recorded every three days. The endpoint of the animal experiment was when a tumor volume from one mouse was > 2000 mm3 or when natural death of the mice occurred. Finally, the tumor volume, tumor weight, and mouse body weight were measured, and tumor tissues were collected for further experiments.
Survival and tumor growth curves of the tumor-bearing mice were investigated in an independent experiment. Two tumor models were grouped and treated as described above (n = 7). Mouse tumor volumes were recorded until all mice reached the endpoint and were analyzed.
Tumor metastasis inhibition
To validate the antimetastatic effect, a metastatic 4T1 tumor model was established by transplanting 4T1 cells (1×106) into the mammary fat pad of Balb/c mice. When the tumor volume reached 50-80 mm3, the tumor-bearing mice were grouped and treated as mentioned above in the “Tumor growth inhibition” subsection. At the end of the experiment (tumor volume > 2000 mm3), the mice were euthanized. During necropsy, the tumor and lung tissues were weighed and dissected for further investigation. In addition, the lung tissues were fixed with Bouin's fluid to display and count the metastatic nodules.
IPF-tumor bearing model
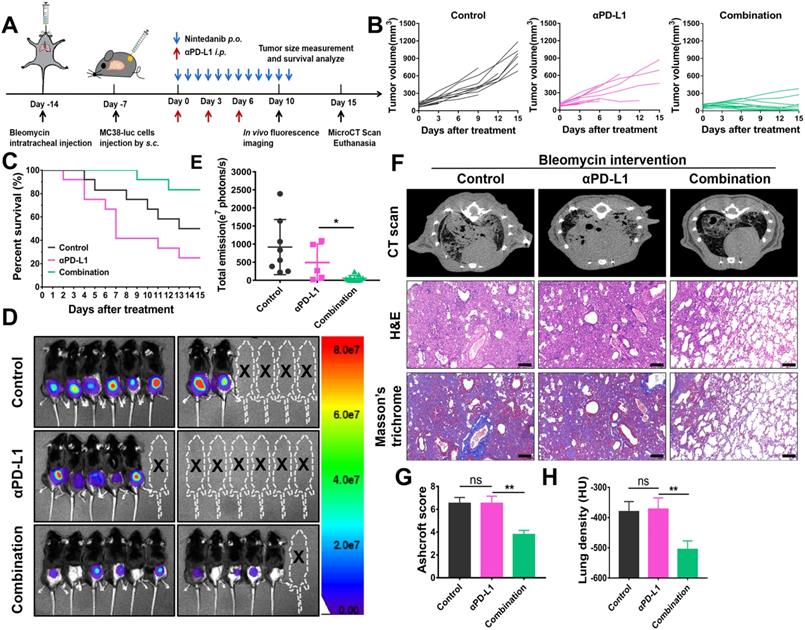
IPF model mice were anesthetized by isoflurane and established by single intratracheal administration of Bleomycin sulfate (1.5 mg/kg, HY-17565, MedChem Express) via microsprayer aerosolizer. After 7 days, MC38-luc tumor cells (1×106) were injected subcutaneously into the right flank of IPF model. When the volume of the tumor was approximately 100 mm3, the mice were divided into control, αPD-L1 and combination group randomly (n = 12) and received corresponding treatment (Figure 8A). Survival of mice were monitored daily. Tumor growth is reflected by volume measurement every three days and in vivo fluorescence imaging on day 10 after treatment. At the endpoint, mice pulmonary fibrosis were evaluated by micro CT scan, HE and Masson's trichrome staining [46].
The effects of nintedanib combined with αPD-L1 on the IPF-tumor model. (A) Model establishment and treatment schedule of IPF-tumor model. (B) Individual tumor volume curves for the IPF-tumor model. (C) Survival curves of the IPF -tumor model. (D) In vivo fluorescence imaging and (E) its quantitative analysis on day 10. (F) The mirco CT scan, HE and Masson's trichrome staining of mice lungs. (G) CT lung density quantification and (H) lung Ashcroft score. The scale bar is 200 µm.
In vivo fluorescence imaging
Mice were injected by D-Luciferin (115144-35-9, GoldBio, USA) and anesthetized with pentobarbital sodium (50 mg/kg). Bioluminescence was detected by IVIS 200 Xenogen system (IVIS Spectrum; Perkin Elmer, Waltham, USA).
Micro CT scan
Mice were anesthetized with pentobarbital sodium and set in the prone position. Thorax computed tomography (CT) was scanned and reconstructed by micro-CT scanner (SkyScan 1176, Bruker, Billerica, MA, USA). The IPF model lung density was quantified as Hounsfield units (HU) used in clinical CT scanners.
Safety evaluation
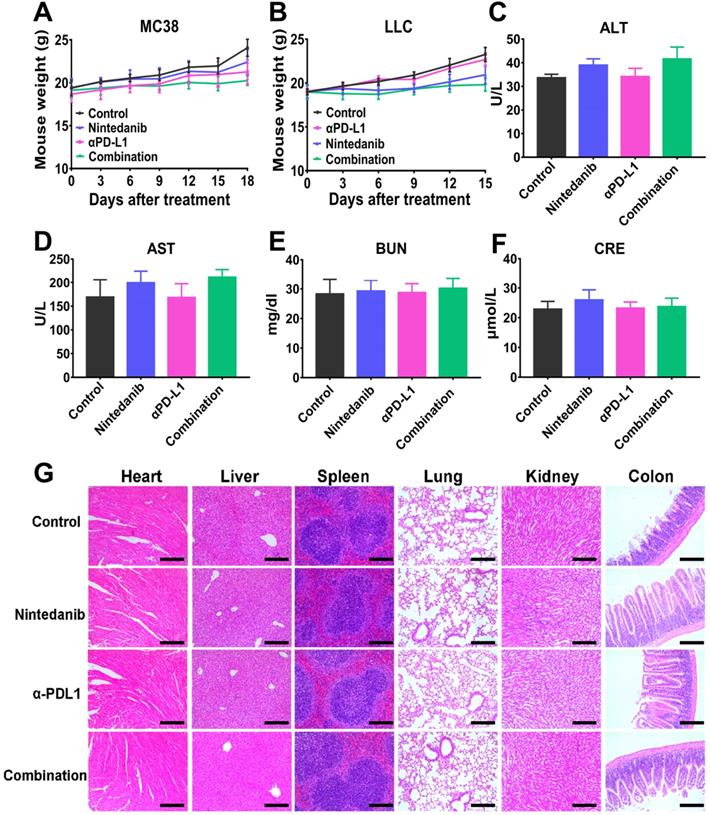
At the end of the animal experiment, the main organs (heart, liver, lung, colon, spleen, and kidney) and serum of the euthanized mice were collected for further investigation. Major organs were fixed with 4% PFA, embedded in paraffin, and sectioned for HE staining analysis to evaluate histopathological damage. Mouse serum was collected for blood biochemistry evaluation (alanine aminotransferase [ALT], aspartate aminotransferase [AST], blood urea nitrogen [BUN], and creatinine [CRE]) and analyzed using a Chemray 240 Automatic Biochemical Analyzer (Rayto Science, Shenzhen, China).
Statistical analyses
All statistical analysis and statistical graph generation were performed using GraphPad Prism 8, and the data are presented as mean ± standard deviation. Student's t-test was performed to analyze data between two experimental groups, while multiple group comparisons were performed using one-way analysis of variance (ANOVA). Statistical significance was shown as *p < 0.05, **p < 0.01, ***p < 0.001, ****p< 0.0001.
Results
Antitumor ability of nintedanib combined with αPD-L1 in vivo
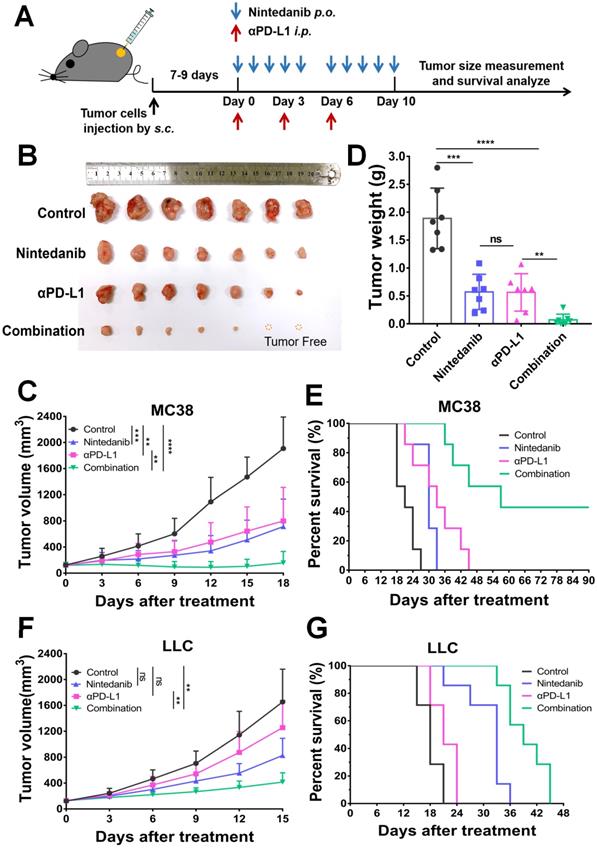
The antitumor effect of nintedanib combined with αPD-L1 was evaluated in both the MC38 and LLC tumor models. After subcutaneous injection of tumor cells for 7-9 days (tumor volume around 100 mm3), mice were grouped and treated as shown in Figure 1A (“Tumor growth inhibition” subsection of the Methods). Although the nintedanib and αPD-L1 groups partially suppressed tumor growth in the MC38 tumor model, the combination therapy exhibited a notable antitumor effect, with some tumors (2/7) showing complete regression (Figure 1B). Individual tumor growth curves are shown in Figure S1A. The tumor volume curves and tumor weight also reflected the tumor suppression effect of the combination therapy (Figure 1C-D). Furthermore, the combination group showed prolonged overall survival compared to the other groups (Figure 1E). Individual tumor growth curves are shown in Figure S1B. In the LLC tumor model, αPD-L1 monotherapy did not slow tumor progression and reduce tumor burden, which may be due to the weak response of LLC tumors to immune checkpoint therapy (Figure 1F). However, αPD-L1 combined with nintedanib effectively inhibited tumor growth and prolonged overall survival of the mice compared to monotherapy (Figure 1F-G, and Figure S1C), demonstrating the enhanced immunotherapy effect of nintedanib.
The antitumor effect of nintedanib combined with α-programmed death-ligand 1 (αPD-L1) in vivo. (A) Treatment schedule of tumor-bearing mice. (B) Images of isolated tumors from MC38 tumor-bearing mice. (C) Growth curves and (D) weight of tumors from MC38 tumor-bearing mice. (E) Survival curves of MC38 tumor-bearing mice. (F) Tumor growth curves and (G) survival curves of LLC tumor-bearing mice.
Nintedanib promotes tumor cell apoptosis and tumor blood vessel normalization
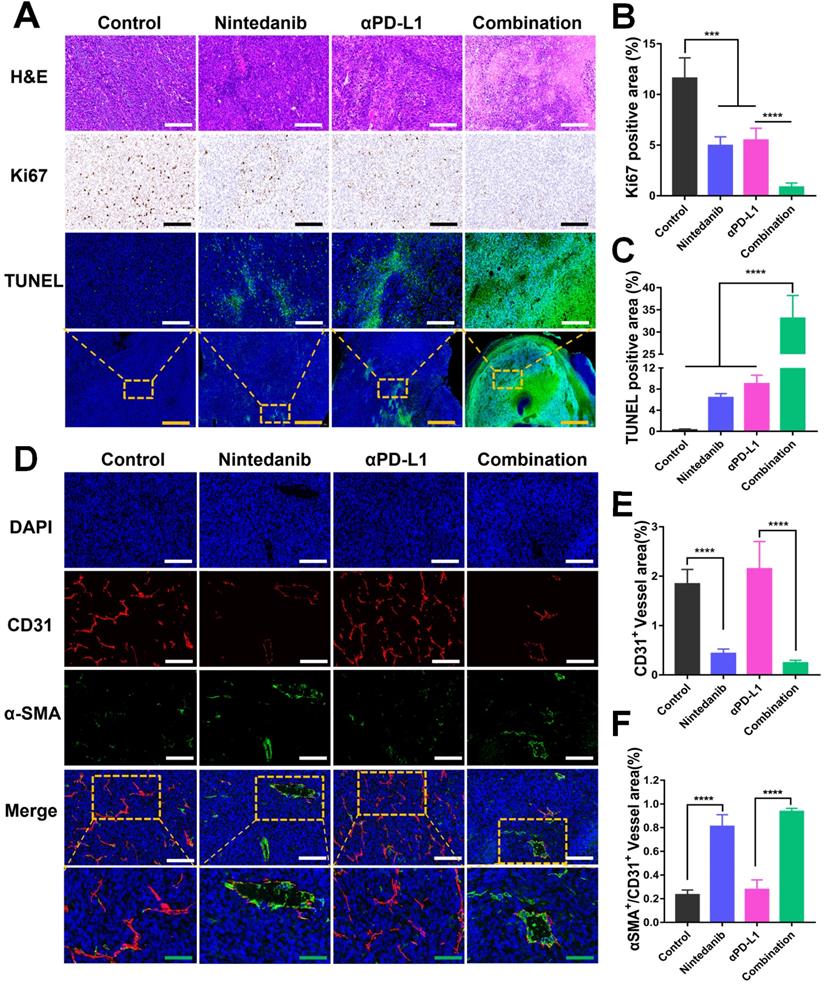
Tumors isolated from the MC38 and LLC models that received different treatments were collected for further investigation. As shown in Figure 2A-C, the promotion of tumor apoptosis and inhibition of tumor proliferation were confirmed with HE, Ki67, and TUNEL staining. Relative to the control, nintedanib, and αPD-L1 groups, the combination group showed the highest necrotic and apoptotic activity and the largest population of TUNEL-positive cells, while Ki67 had the lowest level of expression in this group. A biomarker of angiogenesis, CD31, used to monitor vessel density was detected in tumor tissues by IF analysis. The mean vessel density in tumors was notably reduced after nintedanib administration (Figure 2D-E, and Figure S2). In contrast, α-SMA+/CD31+ (a sign of tumor vessel normalization), which reflects perfused functional tumor vessels, was markedly increased (Figure 2D-F, and Figure S2). From this it can be concluded that dying tumor cells and functional blood vessel normalization may contribute to the infiltration and activation of immune cells.
Image analysis of tumor tissue stained with different stains. (A) Images of tumor tissue stained with hematoxylin-eosin (HE), Ki67, and TUNEL. Quantitative analysis of the (B) Ki67 positive area and (C) TUNEL positive area. (D) Immunofluorescence (IF) images of CD31 and α-SMA in LLC tumor tissue. The quantitative analysis of the (E) CD31+ vessel area and (F) α-SMA+/ CD31+ vessel area. The green scale bar is 100 µm, the white and black scale bar is 200 µm, and the yellow scale bar is 1000 µm.
Nintedanib increases immune cell infiltration and reshapes the TIME
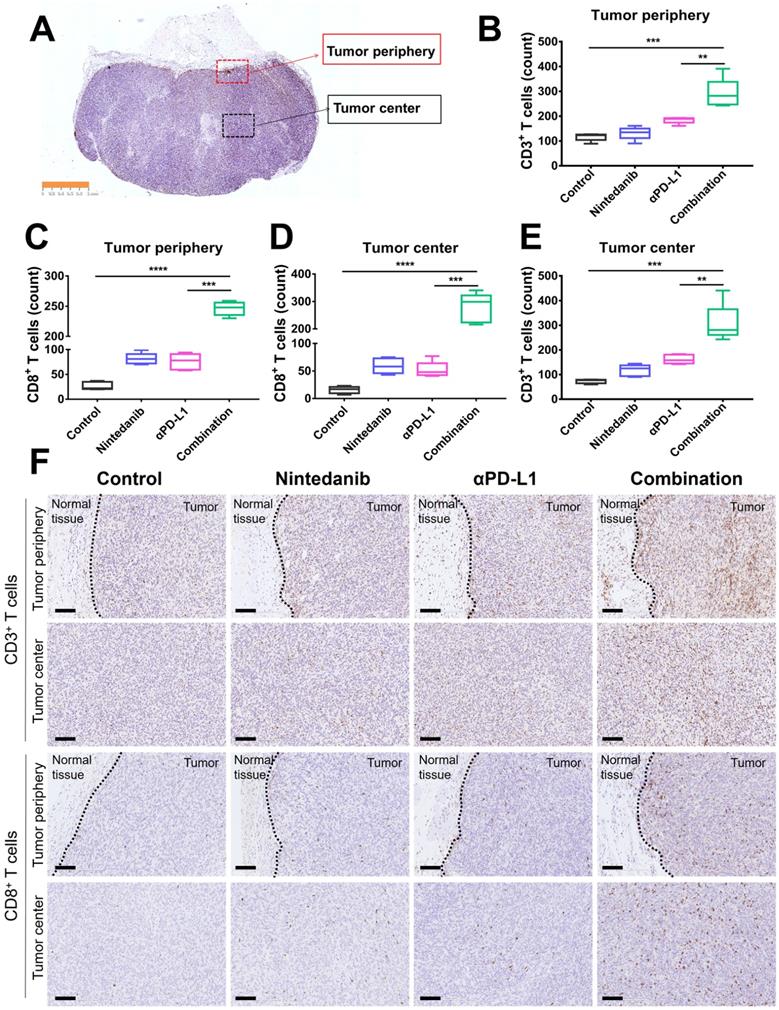
To further determine the immune landscape differences of synergistic nintedanib and αPD-L1 therapy, IHC of CD3+ and CD8+ T cells in MC38 tumors was performed. CD3+ and CD8+ T cells in the tumor center and tumor periphery were counted and analyzed (Figure 3A). IHC images (Figure 3F) and quantification (Figure 3B-E) prove that combination treatment remarkably facilitated the infiltration of CD3+ and CD8+ T cells into both the tumor center and the tumor periphery.
Immunohistochemistry (IHC) analysis of CD3+ T cells and CD8+ T cells in MC38 tumor tissues. (A) Schematic diagram of the tumor center and periphery. Quantitative analysis of (B) CD3+ T cells and (C) CD8+ T cells in the tumor periphery. Quantitative analysis of (D) CD8+ T cells and (E) CD3+ T cells in the tumor center. (F) IHC images of CD3+ T cells and CD8+ T cells in the tumor periphery and center. The scale bar is 100 µm.
The tumor tissue, spleen, and tumor-draining lymph node (TDLN) of the MC38 and LLC models were collected to explore the immune landscape using flow cytometry. In the MC38 model, combination treatment significantly promoted CD8+ T cell infiltration and activation in the tumor tissue (Figure 4A-D). In addition to T cells, the combination treatment also increased the density of dendritic cells (DCs) and promoted their activation simultaneously (Figure 4E-H). Nintedanib alone partially enhanced the infiltration and activation of immune cells compared to the control group. As for changes in the TDLN and spleen immune microenvironment, after combination treatment, we found that the density of CD8+ T cells also increased notably, while the DC number was similar to that of the monotherapy group but more than that of the control group (Figure 4I-N). Due to their recruitment in tumor tissue, the number of DCs after combination treatment did not further increase in the TDLN and spleen compared to nintedanib or αPD-L1 monotherapy. Moreover, PD-L1 levels in tumor cells were increased after nintedanib alone and the combination treatment (Figure 4O).
Flow cytometry analysis of the immune microenvironment in MC38 tumor tissue, tumor-draining lymph node (TDLN), and spleen. Representative images of flow cytometry of (A) CD8+ T cells (gated on CD45+ cells), (C) CD69+ T cells (gated on CD3+CD8+ cells), (E) CD11c+MHCII+ cells (gated on CD45+ cells), and (G) CD11c+CD86+ cells (gated on CD45+ cells). Quantitative analysis of the proportion of (B) CD8+ cells in CD45+ cells, (D) CD3+CD8+CD69+ cells in lymphocytes, (F) CD11c+MHCII+ cells, and (H) CD11c+CD86+ cells in tumors. Representative images of flow cytometry of (I) CD3+CD8+ T cells (gated on CD45+ cells) and quantitative analysis of the proportion of (J) CD3+CD8+ T cells and (K) CD11c+MHCII+ cells in the TDLN. Representative images of flow cytometry of (L) CD8+ T cells (gated on CD45+CD3+ cells) and quantitative analysis of the proportion of (M) CD3+CD8+ T cells and (N) CD11c+MHCII+ cells in the spleen. Representative images of flow cytometry of (O) the PD-L1 level (gated on CD45- cells) and quantitative analysis of PD-L1 mean fluorescence intensity (MFI) in tumors.
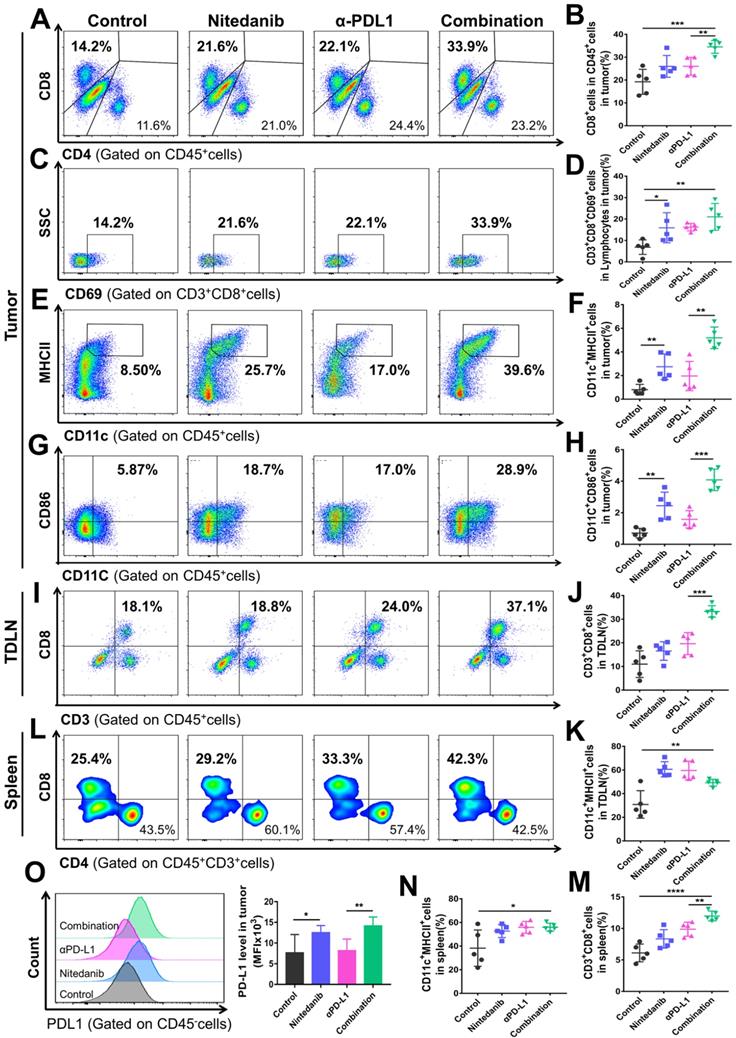
A similar tendency was observed in the LLC model flow cytometry results. After combination treatment, the TIME not only showed an enhancement in total lymphocyte cells, CD45+ cells, CD8+ T cells, and DC infiltration and activation (Figure S3A-F and S3K-M), but also a notable reduction in immunosuppressive MDSCs (Figure S3G-H). Although there was no significant difference in the total number of tumor-associated macrophages (TAMs), the combination group regulated the polarization of macrophages and increased the ratio of M1-like/M2-like macrophages (M1/M2; Figure S3I-J, N). In addition, increased CD8+ T cell density and activation was also detected in the LLC model TDLN and spleen (Figure S4). Our results collectively demonstrated that the combination of nintedanib and αPD-L1 could effectively promote T cell and DC infiltration and activation, while eliminating the immunosuppressive environment (MDSCs and TAMs ratio), thus enhancing the antitumor immune response.
Nintedanib combined with αPD-L1 further activates immune-related pathways
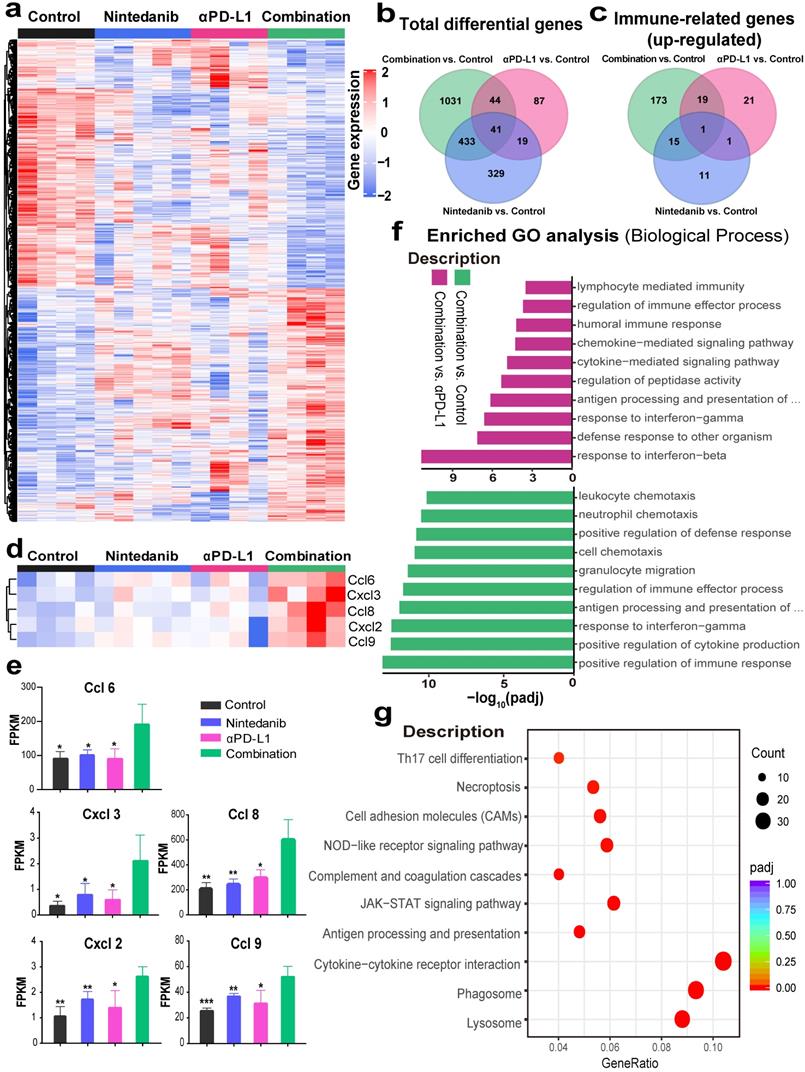
To investigate the mechanisms of the enhancement of the antitumor effects and the increase of immune cell infiltration after synergistic treatment, RNA-seq in MC38 model tumor tissues was performed. DEG analysis revealed that 1549, 291, and 349 genes were differentially expressed in the control, nintedanib, and αPD-L1 groups, respectively, relative to the combination group (Figure 5A). The total DEGs are also presented intuitively in a Venn diagram (Figure 5B). Among the top 100 immune-related GO terms union for the DEGs, the combination group showed obvious upregulation of immune-related genes compared to other therapies (Figure 5C). A detailed pathway and gene list are shown in Table S1. Among these, some immune-chemokine-related genes, such as Ccl6, Ccl8, Ccl9, Cxcl2, and Cxcl3, were significantly upregulated after combination treatment (Figure 5D-E), which was verified with RT-PCR (Figure S5). The functional classification and enrichment of the RNA-seq results was performed using GO and KEGG analysis. The top 10 significantly (p < 0.05) enriched immune-related GO terms for the DEGs in the combination group compared to the control or αPD-L1 groups are shown in Figure 5F and Table S2. Combination therapy activated immune-related pathways (such as response to IFN-γ, antigen processing and presentation of peptide antigen via MHC-I, etc.) to a larger degree than αPD-L1 monotherapy. In addition, KEGG analysis of the combination and control groups also reflected the activation of the immune system (such as JAK-STAT signaling pathway, antigen processing and presentation, and cytokine-cytokine receptor interaction, etc.) in the combination group (Figure 5G). The RNA-seq results may provide further evidence for nintedanib improving the antitumor effects of ICI therapy and increasing immune cell infiltration in the transcriptomic level.
RNA sequencing (RNA-seq) to explore changes in the immune microenvironment of MC38 tumors after different treatments. (A) Heat map of genes that were significantly differentially expressed (p < 0.05). (B) Venn diagram of total differentially expressed genes (DEGs). (C) Venn diagram of the top 100 immune-related Gene Ontology (GO) terms union for the DEGs. (D) The expression levels of CcL6, Ccl8, Ccl9, Cxcl2 and Cxcl3, and their (E) fragments per kilobase million (FPKM) quantification. (F) The top 10 significantly enriched immune-related GO terms. (G) The Kyoto Encyclopedia of Genes and Genomes (KEGG) dot plot (combination vs. control group).
Nintedanib enhances IFN-γ response and the MHC-I-mediated antigen presentation process
To determine the effects of different treatments on tumor cells, we screened and analyzed immune-related signature gene sets that directly affect immune response. As shown in Figure 6A-D, enriched genes and signature scores of IFN-γ and IFN-α response were increased after combination treatment. A detailed list of the enriched genes is presented in Tables S3 and S4. Additionally, GSEA also showed that genes in the combination group were significantly enriched in “RESPONSE TO INTERFERON-GAMMA” (Figure 6E) and “ANTIGEN PROCESSING AND PRESENTATION” (Figure 6F). Among the genes enriched in the latter category were H2-K1, H2-T24, H2-T23, H2-T22, H2-Q7, TAP 1/2 and β2M, which are closely related to MHC-I, a key player in antigen processing and presentation. Based on the screening and experimental results mentioned above, we hypothesized that nintedanib could improve the efficacy of immunotherapy by enhancing the IFN-γ response and upregulating MHC-I expression in tumor cells.
Analysis of mechanisms related to immune activation. (A) Heat map of the expression of signature genes of interferon-gamma (IFN-γ) response (B) quantified by signature score. (C) Heat map of the expression of signature genes of IFN-α response (D) quantified by signature score. Gene Set Enrichment Analysis (GSEA) plot and gene sets for (E) RESPONSE TO INTERFERON-GAMMA and (F) ANTIGEN PROCESSING AND PRESENTATION. Flow cytometry analysis of (G) major histocompatibility complex class I (MHC-I) and (H) PD-L1 expression after different treatments in MC38 cells. The MHC-I expression levels were evaluated by MFI in (I) MC38, (J) LLC, (K) 4T1, and (O) B16F10 cells. The PD-L1 expression levels were evaluated by MFI in (L) MC38, (M) LLC, (N) 4T1, and (P) B16F10 cells. (Q) Representative western blot images of p-STAT3/STAT3, transporter associated with antigen processing 1 (TAP1), PD-L1 and beta-2-microglobulin (β2M).
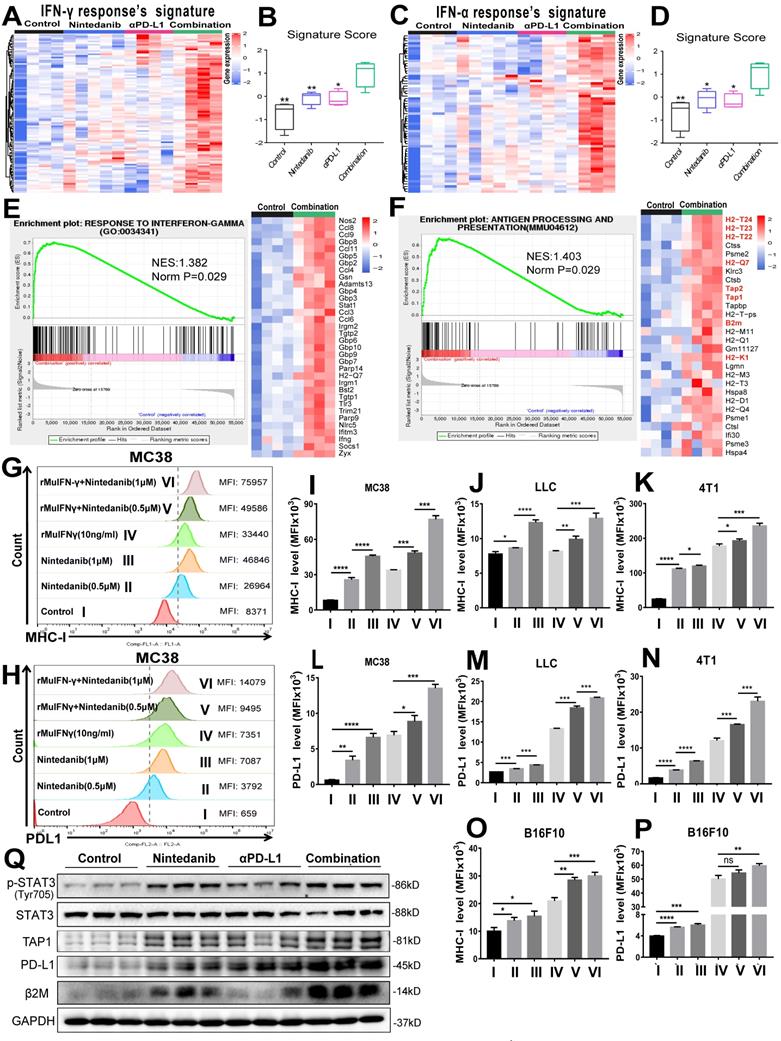
To test our hypothesis, flow cytometry analysis of different tumor cell lines was performed (Figure S6A). Although rMuIFN-γ treatment partially increased MHC-I, rMuIFN-γ + nintedanib (0.5 μM or 1 μM) markedly upregulated MHC-I expression, which was seen with an increase in mean fluorescence intensity (MFI; Figure 6G, I-K, O and Figure S6B). In response to rMuIFN-γ - a stimulating factor that activates PD-L1 expression - PD-L1 expression was upregulated to a certain degree. However, PD-L1 expression was further upregulated when IFN-γ and nintedanib were combined (Figure 6H, I-N, P and Figure S6B). These results indicated that nintedanib enhanced the tumor cell response to IFN-γ, thus upregulating MHC-I and PD-L1 expression. However, in the CT26 cells (Figure S6C-D), this synergistic treatment only increased PD-L1 expression and not MHC-I expression.
At the protein level, the upregulation of p-STAT3, TAP1, β2M (MHC-I-related protein) and PD-L1 were also observed in MC38 tumor tissues (Figure 6Q). In in vitro experiments, a low concentration of nintedanib (1 μM) can promote STAT3 phosphorylation in different tumor cell lines (Figure S7A). Similarly, PD-L1 and β2M were upregulated in tumor cells under nintedanib (1 μM) treatment, while TAP1 had no obvious effect (Figure S7B). In addition, nintedanib combined with IFN-γ could further upregulate TAP1, PD-L1 and β2M expression compared with a single intervention (Figure S7C). IFN-γ exerted its action in TME mainly by regulating the STAT1 pathway [47, 48]. Therefore, nintedanib combined with IFN-γ further activated downstream protein expression which may be attributed to additional STAT3 pathway activation.
Previously, we explored PD-L1 protein levels in various human cancer cell lines (Figure S7D) and low levels of PD-L1 were found in colon cancer cell lines and breast cancer cell lines, which may cause αPD-L1 therapy resistance. Thus, nintedanib may be an ideal complementary therapy for these tumor immunotherapies. Additionally, there were no obvious cytotoxicity of nintedanib on the viability of different tumor cells and human umbilical vein endothelial cells (HUVECs) at the experimental concentration (≤ 1 μM) within 24 h, except for the H1703 cell line (Figure S8 and S9). Apart from a series of positive effects on the TME, nintedanib increased the PD-L1 level and enhanced the αPD-L1 response, thereby exerting an excellent antitumor effect.
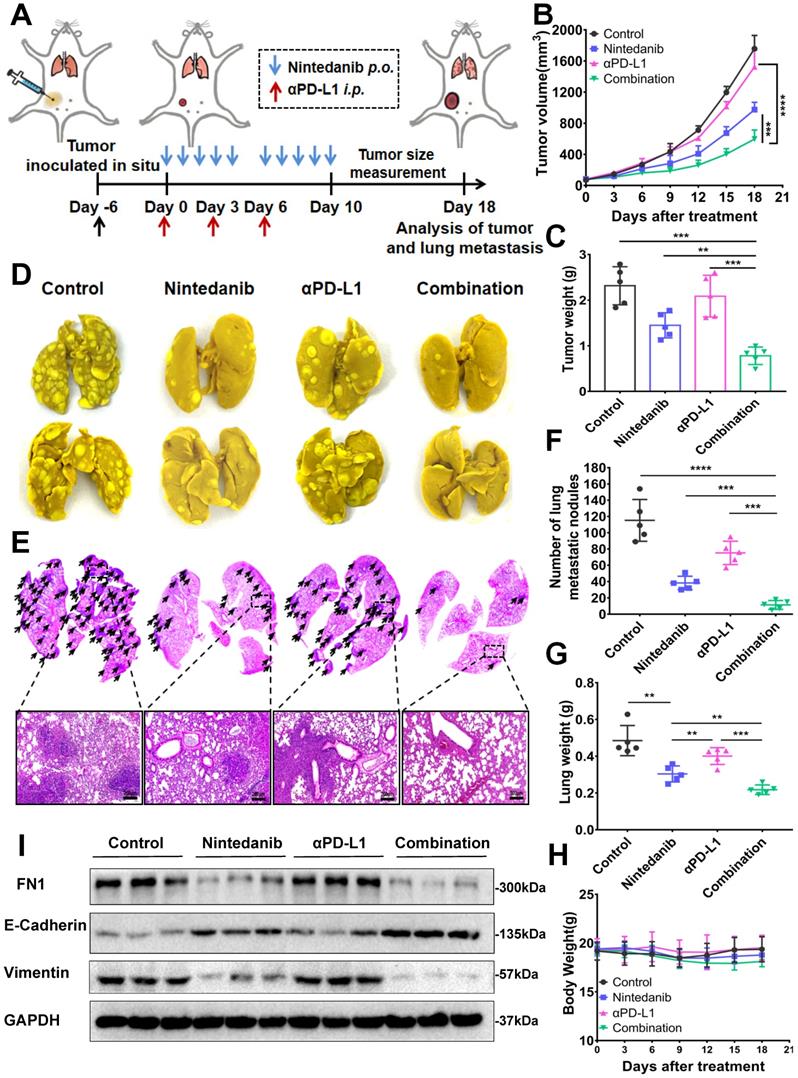
Nintedanib inhibits tumor metastasis
Nintedanib, which enhanced the efficacy of immunotherapy, also inhibited tumor metastasis in a metastatic 4T1 tumor model. Establishment of the metastatic 4T1 tumor model (Figure S10A), and the medication method is shown in Figure 7A. The combination group notably inhibited the growth of 4T1 tumors compared to the effect on tumor growth in the other groups (Figure 7B-C, and Figure S10B-C). As presented in Figure 7D-G and Figure S10D-E, there were obvious metastatic nodules in the lung tissue of the control group and the αPD-L1 group, whereas the nintedanib-treated group had fewer nodules, suggesting that it potentially has antitumor and antimetastatic effects. Moreover, the combination of nintedanib and αPD-L1 demonstrated further anti-invasive capabilities without increasing the physical burden (Figure 7H). The mechanism by which nintedanib inhibits tumor metastasis may be related to the reversal of the epithelial-mesenchymal transition (EMT) in the tumor microenvironment. Mesenchymal protein (Vimentin, FN) levels increased while epithelial protein (E-cadherin) levels were decreased in 4T1 tumor tissue (Figure 7I). In addition, in vitro experiments, similar EMT reversal trend could be observed in different tumor cells after nintedanib treatment (Figure S10F). The metastatic tumor signature EMT tendency was reversed after nintedanib treatment, which may contribute to increased immune cell infiltration and enhanced antitumor effects and metastasis inhibition [49, 50]. These results demonstrate that there are more options for the treatment of metastatic tumors.
The antimetastatic effect of nintedanib combined with αPD-L1. (A) Treatment schedule of 4T1 tumor-bearing mice. (B) Growth curves and (C) weight of tumors from 4T1 tumor-bearing mice. (D) Images and (E) HE staining of lung tissues and lung metastatic nodules (black arrows are lung metastatic nodules). (F) Count of lung metastatic nodules. (G) Lung and (H) body weight of 4T1 models. (I) Representative western blot of epithelial-mesenchymal transition (EMT) related indicators. The scale bar is 200 µm.
Nintedanib alleviates pulmonary fibrosis and increases the efficacy of immunotherapy in IPF-tumor model
To mimic tumor patients complicated with lung disease, IPF-tumor bearing model was established and administered as shown in Figure 8A. Under IPF conditions, combination therapy significantly inhibits tumor growth compared to other treatments (Figure 8B, 8D-E). Although combination therapy effectively improves the survival rate of the model, mice receiving αPD-L1 alone had higher mortality than those in the control group (Figure 8C-D). As shown in lung tissue staining and CT scan results, lungs of mice in the control and αPD-L1 groups had evident pulmonary consolidation, whereas nintedanib application notably reduced lung fibrosis (Figure 8F-H). These findings suggest that the combination of nintedanib and αPD-L1 may be applied as a potential treatment for tumor patients complicated with ILD.
Nintedanib combined with αPD-L1 is found safe
Although the previously presented results showed that nintedanib combined with αPD-L1 enhanced the efficacy of immunotherapy and antitumor effects, biosafety of this synergy therapy is still a primary concern in clinical practice. At the end of the animal experiment, mouse weights were recorded, and there was no significant weight loss in the treated mice compared to untreated mice, indicating that nintedanib was well tolerated in vivo (Figure 9A-B). The serum of the euthanized mice was also sampled for the measurement of blood biochemical parameters. As regular indicators of organ function, ALT, AST, BUN, and CRE were found to be within the normal range in this study (Figure 9C-F). Finally, there were no obvious pathological sites in HE staining of the main organs (Figure 9G). These results further demonstrate the safety in clinical application of this synergistic therapy.
The safety evaluation of different treatments. Mouse weight in the (A) MC38 and (B) LLC models. The measurement of blood biochemistry parameters including (C) alanine aminotransferase (ALT), (D) aspartate aminotransferase (AST), (E) Blood urea nitrogen (BUN), and (F) Creatinine (CRE). (G) HE staining of major organs. The scale bar is 200 µm.
Discussion
At present, although ICI therapy has been one of the optimal choices in partial tumor treatment cases, prolonging unprecedented survival in some patients, initial or acquired immunotherapy-related resistance in many cancers limits its effect and application [13, 18, 51]. Based on extensive clinical research cases, only a small proportion of patients achieve a long-term and beneficial response, while immunotherapy resistance is an unfortunate experience for most patients. To solve this problem, multiagent cancer immunotherapy combination regimens have been widely recognized and explored, thus broadening the immunotherapy-responding population [52, 53]. We are the first to report that nintedanib and αPD-L1 combination treatment not only increased the infiltration and activation of immune cells in tumor tissues through normalization of tumor blood vessels, but also upregulated the expression of PD-L1 and activated the MHC-I-mediated antigen processing and presentation process, thus improving the efficacy of immunotherapy and overcoming partial ICI resistance (Figure 10).
Schematic illustration of nintedanib effects in the tumor microenvironment (TME).
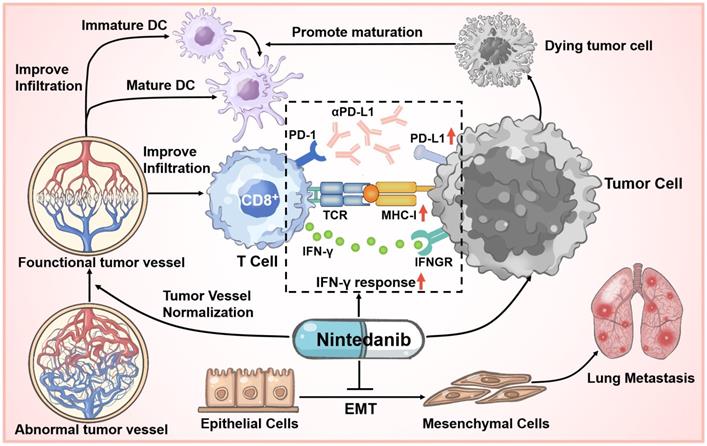
Currently, to explore effective antitumor mechanisms, various immune-related signaling pathways, such as the cGAS-STING pathway have been widely studied [54, 55]. However, in our previous work, RNA-seq and pre-experiment results did not show that nintedanib therapy was related to the effects of these pathways. Despite this, it is undeniable that the combination of nintedanib and αPD-L1 exerted remarkable antitumor effects in multiple murine tumor-bearing models, and HE staining and TUNEL staining also showed this tendency. In a metastatic 4T1 tumor model, nintedanib treatment significantly reduced the metastatic nodules in the lung, which may be caused by inhibition of the EMT process [56, 57], reflecting its antimetastatic potential. Second, the distorted tumor blood vessel architecture and function normalization contributed to inhibition of tumor growth, recruitment of immune cells, improvement of tumor hypoxia, and transport of drugs [17, 19, 58]. As an indicator of perfused functional vessels, the ratio of α-SMA/CD31 increased, while the number of tumor vessels was obviously reduced after nintedanib treatment, indicating tumor ECM vessel normalization [59]. As for changes in the immune landscape, tumor tissue IHC and flow cytometry results reflected a substantial increase in the infiltration and activation of CD8+ T cells and promotion of DC cell maturation after combination treatment. This TME enhancement may be related to tumor vessel normalization and the induction of tumor cell apoptosis.
Moreover, the MC38 model tumor RNA-seq revealed that several immune responses were activated and enhanced after combination treatment, including improved IFN-γ response and MHC-I-mediated antigen processing and presentation. IFN-γ acting on tumor cells can upregulate MHC-I and PD-L1 expression [39, 60, 61]. To simulate this process, different tumor cells received IFN-γ, nintedanib, or a combination treatment and the expression of MHC-I (β2M, H-2kb and H-2kd), TAP1 and PD-L1 was analyzed by western blot or flow cytometry. We found that combination treatment further upregulated MHC-I, TAP1 and PD-L1 expression compared to IFN-γ administered alone. Some studies have reported that STAT3 pathway activation may contribute to increase PD-L1, TAP1 and MHC-I levels [39, 62, 63]. In addition, based on extensive research, the effect of nintedanib on the STAT3 pathway mainly depends on the concentration and cell responsiveness [64-66]. Herein, our in vitro results demonstrated that a low concentration of nintedanib (1 μM) can promote STAT3 phosphorylation and downstream protein expression (PD-L1 and β2M) in different tumor cell lines (A549, MC38 and LLC) and gradually suppress these pathways at high doses (5 μM). This proved that nintedanib enhanced the IFN-γ response and the increased of MHC-I expression may be attributed to additional STAT3 pathway activation (Figure S7A-C). These results suggest that a low concentration of nintedanib may contribute to a better synergy when combined with ICIs. Regardless of whether nintedanib was used alone or in combination, its upregulation effect on PD-L1 has been verified in a variety of tumor cell lines. Recently, increasing evidence has shown that PD-L1 expression levels in tumor cells might affect the clinical response to anti-PD-1/PD-L1 therapies, and PD-L1 (+) patients have a better response rate to ICIs and better overall survival [39-44]. The effect of nintedanib on tumor cells effectively increased the immune response of αPD-L1 and reshaped the TIME. Thus, nintedanib is an important complement to ICI resistance improvement.
On the other hand, one of major difficulties in tumor immunotherapy is complications in other organs [67]. Among these, ILD limited ICI application and severely reduced the prognosis and survival of cancer patients [23, 24]. Nintedanib, one of the most effective treatments for ILD, may be a valuable strategy for tumor patients complicated with ILD. Indeed, multiple clinical trials have shown that nintedanib combined with chemotherapy is well tolerated and has clinical benefits [68-70]. Moreover, some patients who experienced disease progression after prior ICI therapy responded partially to the nintedanib combination strategy [71]. Based on our work, nintedanib combined with αPD-L1 exerted notable antitumor and anti-pulmonary fibrosis effects and showed satisfactory safety in a IPF-tumor model. However, the αPD-L1 group had a higher mortality outcome than the control, which may have been caused by an aggravation in inflammation and fibrosis after ICI application [72, 73]. There were no obvious differences in pulmonary fibrosis between αPD-L1 and the control group at the endpoint of experiment, possibly due to mice with severe pulmonary fibrosis dying earlier (Figure 8C and 8F-H). These encouraging results show that nintedanib has the potential to alleviate pulmonary fibrosis, increase the efficacy of immunotherapy and overcome ICI resistance and side effects. Our results provide convincing evidence to support the advancement of this synergistic therapy to clinical trials. To translate our results into practical clinical applications, further clinical evaluation is necessary to determine the efficacy and safety of nintedanib and αPD-L1 combination therapy. Therefore, our team is preparing to launch a Phase II clinical trial of nintedanib combined with αPD-L1.
However, there are some limitations to this study. Herein, we suggest that nintedanib and αPD-L1 combination treatment improves the immune response by enhancing the tumor cell IFN-γ response and increasing PD-L1 and MHC-I levels. Further investigation into specific targets, regulatory relationships that influence these pathways, and studies on whether nintedanib is suitable for other combination regimens, such as radiotherapy, are necessary. Due to the upregulation of PD-L1 and MHC-I [74], radiotherapy with nintedanib and αPD-L1 synergy therapy may be a valuable combination worthy of further study. Moreover, the efficacy of the combined treatment was weaker against the CT26 cell line, which is a pMMR (MSS), responding poorly to immunotherapy compared to the efficacy against other tumor cells [75, 76]. Unfortunately, these results imply that patients with MSS tumors, in clinical settings, may only partially benefit from this synergistic therapy. Despite this, it is undeniable that nintedanib and αPD-L1 combination treatment exerts excellent antitumor effects and overcomes partial ICI therapy resistance in multiple cancers.
Conclusions
We found, with a series of in vivo and in vitro experiments, that nintedanib and αPD-L1 combination treatment exhibited significant antitumor and antimetastatic effects. Further investigation of these effects revealed that nintedanib could promote tumor vascular normalization and increase T cell and DC infiltration and activation while reshaping the TIME (enhanced IFN-γ responses and MHC-I-mediated antigen presentation). Moreover, as the upregulation of PD-L1 expression increased the tumor cell response to αPD-L1, nintedanib combined with αPD-L1 further exhibited its immune activation effects; thus, enhancing the efficacy of ICI therapy. Based on these encouraging results, nintedanib and αPD-L1 synergy therapy may provide a potential strategy for the treatment of tumor patients with immunotherapy resistance and ILD.
Abbreviations
ALT: alanine aminotransferase; ANOVA: analysis of variance; AST: aspartate aminotransferase; BUN: blood urea nitrogen; CRE: creatinine; DCs: dendritic cells; DEGs: differentially expressed genes; EMT: epithelial-mesenchymal transition; ECM: extra cellular matrix; GO: gene ontology; GSEA: gene set enrichment analysis; HE: hematoxylin-eosin; HUVECs: human umbilical vein endothelial cells; ICIs: immune checkpoint inhibitors; ILD: interstitial lung diseases; IF: immunofluorescence; IHC: immunohistochemistry; IFN-γ: interferon-gamma; IPF: idiopathic pulmonary fibrosis; KEGG: kyoto encyclopedia of genes and genomes; MHC-I: major histocompatibility complex class I; MDSCs: myeloid-derived suppressor cells; NSCLC: non-small cell lung cancer; PD-1: programmed cell death protein 1; PD-L1: programmed death-ligand 1; RT-PCR: Real-time PCR; rMuIFN-γ: recombinant mouse interferon-gamma; rHuIFN-γ: recombinant human interferon-gamma; RNA-seq: RNA sequencing; TAP: Transporter associated with antigen processing; TIME: Tumor immune microenvironment; TME: Tumor microenvironment; TAMs: tumor-associated macrophages; TDLN: tumor-draining lymph node; β2M: beta-2-microglobulin.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant number: 81773370) and the Radiotherapy and Protection Engineering Center Innovation Capacity Building Project of Hubei Province (grant number: 2018-420114-35-03-071705). In addition, we thank the Medical Subcenter of HUST Analytical & Testing Center in data acquisition.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kruger S, Ilmer M, Kobold S, Cadilha BL, Endres S, Ormanns S. et al. Advances in cancer immunotherapy 2019 - latest trends. J Exp Clin Cancer Res. 2019;38:268
2. Tan S, Li D, Zhu X. Cancer immunotherapy: pros, cons and beyond. Biomed Pharmacother. 2020;124:109821
3. Zhao Z, Zheng L, Chen W, Weng W, Song J, Ji J. Delivery strategies of cancer immunotherapy: recent advances and future perspectives. J Hematol Oncol. 2019;12:126
4. Szeto GL, Finley SD. Integrative approaches to cancer immunotherapy. Trends Cancer. 2019;5:400-10
5. Yi M, Jiao D, Xu H, Liu Q, Zhao W, Han X. et al. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol Cancer. 2018;17:129
6. Akinleye A, Rasool Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J Hematol Oncol. 2019;12:92
7. Cho JH. Immunotherapy for non-small-cell lung cancer: current status and future obstacles. Immune Netw. 2017;17:378-91
8. O'Neil BH, Wallmark JM, Lorente D, Elez E, Raimbourg J, Gomez-Roca C. et al. Safety and antitumor activity of the anti-PD-1 antibody pembrolizumab in patients with advanced colorectal carcinoma. PLoS One. 2017;12:e0189848
9. Chen EX, Jonker DJ, Loree JM, Kennecke HF, Berry SR, Couture F. et al. Effect of combined immune checkpoint inhibition vs best supportive care alone in patients with advanced colorectal cancer: the canadian cancer trials group CO.26 study. JAMA Oncol. 2020;6:831-8
10. Lau D, Kalaitzaki E, Church DN, Pandha H, Tomlinson I, Annels N. et al. Rationale and design of the POLEM trial: avelumab plus fluoropyrimidine-based chemotherapy as adjuvant treatment for stage III mismatch repair deficient or POLE exonuclease domain mutant colon cancer: a phase III randomised study. ESMO Open. 2020;5:e000638
11. Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H. et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018;379:2108-21
12. Mittendorf EA, Zhang H, Barrios CH, Saji S, Jung KH, Hegg R. et al. Neoadjuvant atezolizumab in combination with sequential nab-paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy in patients with early-stage triple-negative breast cancer (IMpassion031): a randomised, double-blind, phase 3 trial. Lancet. 2020;396:1090-100
13. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707-23
14. Pitt JM, Vetizou M, Daillere R, Roberti MP, Yamazaki T, Routy B. et al. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and -extrinsic factors. Immunity. 2016;44:1255-69
15. Hegde PS, Chen DS. Top 10 challenges in cancer immunotherapy. Immunity. 2020;52:17-35
16. Bagchi S, Yuan R, Engleman EG. Immune checkpoint inhibitors for the treatment of cancer: clinical impact and mechanisms of response and resistance. Annu Rev Pathol. 2021;16:223-49
17. Munn LL, Jain RK. Vascular regulation of antitumor immunity. Science. 2019;365:544-545
18. Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. 2018;118:9-16
19. Yi M, Jiao D, Qin S, Chu Q, Wu K, Li A. Synergistic effect of immune checkpoint blockade and anti-angiogenesis in cancer treatment. Mol Cancer. 2019;18:60
20. Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S. et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun. 2016;7:12624
21. Chen B, Gao A, Tu B, Wang Y, Yu X, Wang Y. et al. Metabolic modulation via mTOR pathway and anti-angiogenesis remodels tumor microenvironment using PD-L1-targeting codelivery. Biomaterials. 2020;255:120187
22. Liu XD, Hoang A, Zhou L, Kalra S, Yetil A, Sun M. et al. Resistance to antiangiogenic therapy is associated with an immunosuppressive tumor microenvironment in metastatic renal cell carcinoma. Cancer Immunol Res. 2015;3:1017-29
23. Barczi E, Nagy T, Starobinski L, Kolonics-Farkas A, Eszes N, Bohacs A. et al. Impact of interstitial lung disease and simultaneous lung cancer on therapeutic possibilities and survival. Thorac Cancer. 2020;11:1911-7
24. Kalisz KR, Ramaiya NH, Laukamp KR, Gupta A. Immune checkpoint inhibitor therapy-related pneumonitis: patterns and management. Radiographics. 2019;39:1923-37
25. Lind JS, Senan S, Smit EF. Pulmonary toxicity after bevacizumab and concurrent thoracic radiotherapy observed in a phase I study for inoperable stage III non-small-cell lung cancer. J Clin Oncol. 2012;30:e104-8
26. Delaunay M, Cadranel J, Lusque A, Meyer N, Gounant V, Moro-Sibilot D. et al. Immune-checkpoint inhibitors associated with interstitial lung disease in cancer patients. Eur Respir J. 2017;50:1750050
27. Yamagata A, Yokoyama T, Fukuda Y, Ishida T. Impact of interstitial lung disease associated with immune checkpoint inhibitors on prognosis in patients with non-small-cell lung cancer. Cancer Chemother Pharmacol. 2021;87:251-8
28. Suzuki Y, Karayama M, Uto T, Fujii M, Matsui T, Asada K. et al. Assessment of immune-related interstitial lung disease in patients with NSCLC treated with immune checkpoint inhibitors: a multicenter prospective study. J Thorac Oncol. 2020;15:1317-27
29. Atchley WT, Alvarez C, Saxena-Beem S, Schwartz TA, Ishizawar RC, Patel KP. et al. Immune checkpoint inhibitor-related pneumonitis in lung cancer: real-world incidence, risk factors, and management practices across six health-care centers in North Carolina. Chest. 2021;160:731-42
30. Hilberg F, Roth GJ, Krssak M, Kautschitsch S, Sommergruber W, Tontsch-Grunt U. et al. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008;68:4774-82
31. Spagnolo P, Kropski JA, Jones MG, Lee JS, Rossi G, Karampitsakos T. et al. Idiopathic pulmonary fibrosis: disease mechanisms and drug development. Pharmacol Ther. 2021;222:107798
32. Sgalla G, Franciosa C, Simonetti J, Richeldi L. Pamrevlumab for the treatment of idiopathic pulmonary fibrosis. Expert Opin Investig Drugs. 2020;29:771-7
33. Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y. et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med. 2019;381:1718-27
34. Kato R, Haratani K, Hayashi H, Sakai K, Sakai H, Kawakami H. et al. Nintedanib promotes antitumour immunity and shows antitumour activity in combination with PD-1 blockade in mice: potential role of cancer-associated fibroblasts. Br J Cancer. 2021;124:914-24
35. Yamanaka T, Harimoto N, Yokobori T, Muranushi R, Hoshino K, Hagiwara K. et al. Nintedanib inhibits intrahepatic cholangiocarcinoma aggressiveness via suppression of cytokines extracted from activated cancer-associated fibroblasts. Br J Cancer. 2020;122:986-94
36. Ramjiawan RR, Griffioen AW, Duda DG. Anti-angiogenesis for cancer revisited: Is there a role for combinations with immunotherapy? Angiogenesis. 2017;20:185-204
37. Chen Y, Ramjiawan RR, Reiberger T, Ng MR, Hato T, Huang Y. et al. CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology. 2015;61:1591-602
38. Chen Y, Liu YC, Sung YC, Ramjiawan RR, Lin TT, Chang CC. et al. Overcoming sorafenib evasion in hepatocellular carcinoma using CXCR4-targeted nanoparticles to co-deliver MEK-inhibitors. Sci Rep. 2017;7:44123
39. Zhang J, Dang F, Ren J, Wei W. Biochemical aspects of PD-L1 regulation in cancer immunotherapy. Trends Biochem Sci. 2018;43:1014-32
40. Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019;19:133-50
41. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF. et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443-54
42. Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A. et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387:1909-20
43. Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A. et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375:1823-33
44. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP. et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018-28
45. Lan Y, Zhang D, Xu C, Hance KW, Marelli B, Qi J. et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-β. Sci Transl Med. 2018;10:eaan5488
46. Hubner RH, Gitter W, El Mokhtari NE, Mathiak M, Both M, Bolte H. et al. Standardized quantification of pulmonary fibrosis in histological samples. Biotechniques. 2008;44:507-11 14-7
47. Zhou F. Molecular mechanisms of IFN-gamma to up-regulate MHC class I antigen processing and presentation. Int Rev Immunol. 2009;28:239-60
48. Hu X, Ivashkiv LB. Cross-regulation of signaling pathways by interferon-gamma: implications for immune responses and autoimmune diseases. Immunity. 2009;31:539-50
49. Lu W, Kang Y. Epithelial-mesenchymal plasticity in cancer progression and metastasis. Dev Cell. 2019;49:361-74
50. Horn LA, Fousek K, Palena C. Tumor plasticity and resistance to immunotherapy. Trends Cancer. 2020;6:432-41
51. Schoenfeld AJ, Hellmann MD. Acquired resistance to immune checkpoint inhibitors. Cancer Cell. 2020;37:443-55
52. Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. 2019;18:197-218
53. Murciano-Goroff YR, Warner AB, Wolchok JD. The future of cancer immunotherapy: microenvironment-targeting combinations. Cell Res. 2020;30:507-19
54. Wang Y, Luo J, Alu A, Han X, Wei Y, Wei X. cGAS-STING pathway in cancer biotherapy. Mol Cancer. 2020;19:136
55. Zhang X, Bai XC, Chen ZJ. Structures and mechanisms in the cGAS-STING innate immunity pathway. Immunity. 2020;53:43-53
56. Na TY, Schecterson L, Mendonsa AM, Gumbiner BM. The functional activity of E-cadherin controls tumor cell metastasis at multiple steps. Proc Natl Acad Sci U S A. 2020;117:5931-7
57. Wang WD, Shang Y, Li Y, Chen SZ. Honokiol inhibits breast cancer cell metastasis by blocking EMT through modulation of Snail/Slug protein translation. Acta Pharmacol Sin. 2019;40:1219-27
58. Sung YC, Jin PR, Chu LA, Hsu FF, Wang MR, Chang CC. et al. Delivery of nitric oxide with a nanocarrier promotes tumour vessel normalization and potentiates anti-cancer therapies. Nat Nanotechnol. 2019;14:1160-9
59. Huang N, Liu Y, Fang Y, Zheng S, Wu J, Wang M. et al. Gold nanoparticles induce tumor vessel normalization and impair metastasis by inhibiting endothelial Smad2/3 signaling. ACS Nano. 2020;14:7940-58
60. Cha JH, Chan LC, Li CW, Hsu JL, Hung MC. Mechanisms controlling PD-L1 expression in cancer. Mol Cell. 2019;76:359-70
61. Stifter K, Krieger J, Ruths L, Gout J, Mulaw M, Lechel A. et al. IFN-γ treatment protocol for MHC-I(lo)/PD-L1(+) pancreatic tumor cells selectively restores their TAP-mediated presentation competence and CD8 T-cell priming potential. J Immunother Cancer. 2020;8:e000692
62. Liu X, Wu XP, Zhu XL, Li T, Liu Y. IRG1 increases MHC class I level in macrophages through STAT-TAP1 axis depending on NADPH oxidase mediated reactive oxygen species. Int Immunopharmacol. 2017;48:76-83
63. Luo M, Wang F, Zhang H, To KKW, Wu S, Chen Z. et al. Mitomycin C enhanced the efficacy of PD-L1 blockade in non-small cell lung cancer. Signal Transduct Target Ther. 2020;5:141
64. Shochet GE, Pomerantz A, Shitrit D, Bardenstein-Wald B, Ask K, Surber M. et al. Galectin-3 levels are elevated following nintedanib treatment. Ther Adv Chronic Dis. 2020;11:2040622320968412
65. Bosch-Barrera J, Verdura S, Ruffinelli JC, Carcereny E, Sais E, Cuyas E. et al. Silibinin suppresses tumor cell-intrinsic resistance to nintedanib and enhances its clinical activity in lung cancer. Cancers (Basel). 2021;13:4168
66. Tai WT, Shiau CW, Li YS, Chang CW, Huang JW, Hsueh TT. et al. Nintedanib (BIBF-1120) inhibits hepatocellular carcinoma growth independent of angiokinase activity. J Hepatol. 2014;61:89-97
67. Kennedy LB, Salama AKS. A review of cancer immunotherapy toxicity. CA Cancer J Clin. 2020;70:86-104
68. du Bois A, Kristensen G, Ray-Coquard I, Reuss A, Pignata S, Colombo N. et al. Standard first-line chemotherapy with or without nintedanib for advanced ovarian cancer (AGO-OVAR 12): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2016;17:78-89
69. de Braud F, Cascinu S, Spitaleri G, Pilz K, Clementi L, Liu D. et al. A phase I, dose-escalation study of volasertib combined with nintedanib in advanced solid tumors. Ann Oncol. 2015;26:2341-6
70. Eisen T, Loembé AB, Shparyk Y, MacLeod N, Jones RJ, Mazurkiewicz M. et al. A randomised, phase II study of nintedanib or sunitinib in previously untreated patients with advanced renal cell cancer: 3-year results. Br J Cancer. 2015;113:1140-7
71. Puri S, Niyongere S, Chatwal MS, Boyle TA, Gray JE. Phase I/II study of nivolumab and ipilimumab combined with nintedanib in advanced NSCLC. J Clin Oncol. 2018;36:TPS9112-TPS
72. Ni K, Liu M, Zheng J, Wen L, Chen Q, Xiang Z. et al. PD-1/PD-L1 pathway mediates the alleviation of pulmonary fibrosis by human mesenchymal stem cells in humanized mice. Am J Respir Cell Mol Biol. 2018;58:684-95
73. Habiel DM, Espindola MS, Kitson C, Azzara AV, Coelho AL, Stripp B. et al. Characterization of CD28(null) T cells in idiopathic pulmonary fibrosis. Mucosal Immunol. 2019;12:212-22
74. Sharabi AB, Lim M, DeWeese TL, Drake CG. Radiation and checkpoint blockade immunotherapy: radiosensitisation and potential mechanisms of synergy. Lancet Oncol. 2015;16:e498-509
75. Ooki A, Shinozaki E, Yamaguchi K. Immunotherapy in colorectal cancer: current and future strategies. J Anus Rectum Colon. 2021;5:11-24
76. Efremova M, Rieder D, Klepsch V, Charoentong P, Finotello F, Hackl H. et al. Targeting immune checkpoints potentiates immunoediting and changes the dynamics of tumor evolution. Nat Commun. 2018;9:32
Author contact
![]() Corresponding authors: Xianglin Yuan, Department of Oncology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 1095 Jiefang Avenue, Wuhan 430030, Hubei Province, China. E-mail: yuanxianglinedu.cn; Bo Liu, Department of Oncology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 1095 Jiefang Avenue, Wuhan 430030, Hubei Province, China. E-mail: boliu888com; Li Sun, Department of Oncology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 1095 Jiefang Avenue, Wuhan 430030, Hubei Province, China. E-mail: litchisuncom.
Corresponding authors: Xianglin Yuan, Department of Oncology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 1095 Jiefang Avenue, Wuhan 430030, Hubei Province, China. E-mail: yuanxianglinedu.cn; Bo Liu, Department of Oncology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 1095 Jiefang Avenue, Wuhan 430030, Hubei Province, China. E-mail: boliu888com; Li Sun, Department of Oncology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 1095 Jiefang Avenue, Wuhan 430030, Hubei Province, China. E-mail: litchisuncom.