Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2022; 12(1):324-339. doi:10.7150/thno.63735 This issue Cite
Research Paper
Tubular epithelial cell-to-macrophage communication forms a negative feedback loop via extracellular vesicle transfer to promote renal inflammation and apoptosis in diabetic nephropathy
Wen-juan Jiang1, Chuan-ting Xu1*, Chang-lin Du1*, Jia-hui Dong1, Song-bing Xu1, Bing-feng Hu1, Rui Feng1, Dan-dan Zang2, Xiao-ming Meng1, Cheng Huang1, Jun Li1, Tao-tao Ma1 ![]()
1. Inflammation and Immune Mediated Diseases Laboratory of Anhui Province, Anhui Institute of Innovative Drugs, School of Pharmacy, Anhui Medical University, Hefei, 230032, China.
2. The Center for Scientific Research of Anhui Medical University, Hefei, 230032, China.
* These authors contributed equally to the first author.
Received 2021-6-10; Accepted 2021-10-30; Published 2022-1-1
Abstract

Background: Macrophage infiltration around lipotoxic tubular epithelial cells (TECs) is a hallmark of diabetic nephropathy (DN). However, how these two types of cells communicate remains obscure. We previously demonstrated that LRG1 was elevated in the process of kidney injury. Here, we demonstrated that macrophage-derived, LRG1-enriched extracellular vesicles (EVs) exacerbated DN.
Methods: We induced an experimental T2DM mouse model with a HFD diet for four months. Renal primary epithelial cells and macrophage-derived EVs were isolated from T2D mice by differential ultracentrifugation. To investigate whether lipotoxic TEC-derived EV (EVe) activate macrophages, mouse bone marrow-derived macrophages (BMDMs) were incubated with EVe. To investigate whether activated macrophage-derived EVs (EVm) induce lipotoxic TEC apoptosis, EVm were cocultured with primary renal tubular epithelial cells. Subsequently, we evaluated the effect of LRG1 in EVe by investigating the apoptosis mechanism.
Results: We demonstrated that incubation of primary TECs of DN or HK-2 mTECs with lysophosphatidyl choline (LPC) increased the release of EVe. Interestingly, TEC-derived EVe activated an inflammatory phenotype in macrophages and induced the release of macrophage-derived EVm. Furthermore, EVm could induce apoptosis in TECs injured by LPC. Importantly, we found that leucine-rich α-2-glycoprotein 1 (LRG1)-enriched EVe activated macrophages via a TGFβR1-dependent process and that tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-enriched EVm induced apoptosis in injured TECs via a death receptor 5 (DR5)-dependent process.
Conclusion: Our findings indicated a novel cell communication mechanism between tubular epithelial cells and macrophages in DN, which could be a potential therapeutic target.
Keywords: diabetic nephropathy, extracellular vesicles, leucine-rich α-2-glycoprotein 1, tumor necrosis factor-related apoptosis-inducing ligand, death receptor 5, tumor necrosis factor α, interleukin
Introduction
Diabetes mellitus is a group of metabolic disorders characterized by high blood sugar levels over a prolonged period [1]. Type 2 diabetes mellitus (T2DM) has the greatest impact and accounts for approximately 90-95% of the global diabetes burden [2]. Complications associated with T2DM include heart attack, kidney failure, unhealed wounds, vision loss, and nerve damage [3, 4]. Furthermore, the rapid global spread of COVID-19 has caused millions to suffer [5-7]. The concurrent global pandemics of COVID-19 and T2DM have resulted in T2DM being the second most common comorbidity of COVID-19 [6].
Tissue stress or dysfunction induces inflammation, which helps tissues adapt to noxious conditions and restore tissue functionality. These processes also include a change in the state of cells from stressed to apoptotic [8]. Diabetic nephropathy (DN), a serious renal complication of T2DM, is characterized by renal tubular epithelial lipoapoptosis (i.e., apoptosis induced by toxic lipid mediators) and inflammatory cell infiltration that is represented, in part, by activated macrophages [9, 10]. However, in DN, it remains unclear how TEC injury promotes macrophage activation and how macrophage activation influences injured TECs and stimulates injured cells to undergo apoptosis to maintain homeostasis. Damaged cells send signals to activate macrophages, which release proinflammatory cytokines and thus stimulate injured cells to undergo apoptosis. This cell communication is an essential component in mammalian development and preservation of homeostasis, ensuring fast and efficient responses to alterations or threats within the environment surrounding host cells. Beyond classical signaling through cell-cell contact and soluble factors, such as cytokines, inflammatory mediators, metabolites, and hormones, intercellular communication also occurs through cellular release of extracellular vesicles (EVs). This mode of communication has the potential to deliver a particularly diverse array of messages to EV-accepting cells at a level beyond that of soluble factor signaling, since EVs may carry a number of bioactive molecules, surface receptors, and genetic information [11, 12].
EVs have been implicated in modulation of the immune system. Moon et al. [13] reported that hyperoxia-induced, lung epithelial cell-derived and caspase-3-enriched EVs activated macrophages and mediated the inflammatory lung responses involved in lung injury. Lv et al. [14] reported that macrophage internalization of exosomes from BSA-treated tubular epithelial cells (TECs) led to an enhanced inflammatory response and macrophage migration, constituting a critical mechanism of albumin-induced tubulointerstitial inflammation. The function of EV is complex. Appiah et al. [15] studied how gut epithelial cell-derived exosomes released in the intestinal luminal space during sepsis affected mucosal inflammation. They found that intestinal epithelial cell (IEC)-derived luminal EVs carried miRNAs that mitigate proinflammatory responses. How these two types of cells, macrophages and TECs, communicate in DN is still unclear.
In the current study, we provided evidence suggesting that lipotoxic TEC-derived EV (EVe) induced the expression and release of proinflammatory cytokines, such as IL-1β and TNF-α, in macrophages and induced the release of macrophage-derived EVm. Furthermore, EVm could induce apoptosis in lipotoxic TECs. Importantly, we found that LRG1-enriched EVe activated macrophages via a TGFβR1-dependent process and that TRAIL-enriched EVm induced apoptosis in lipotoxic TECs via a DR5-dependent process. Taken together, tubular epithelial cell-to-macrophage-to-tubular epithelial cell circulation via EV transfer promoted renal inflammation and apoptosis in DN.
Methods
Experimental reagents
Lysophosphatidylcholine (Sigma, St. Louis, MO) was dissolved as previously described and used for cell culture studies at a concentration of 20 µM[16]. β-actin, DR5, kidney injury molecule-1 (KIM-1) primary antibodies and goat anti-rabbit or anti-mouse immunoglobulin G (IgG) horseradish peroxidase (HRP) secondary antibodies were purchased from Bioss (Beijing, China). Antibodies against TSG101, CD63, ARF6, calnexin, TRAIL, LRG1, and GAPDH were obtained from Abcam (Cambridge, UK). Human recombinant LRG1 and TRAIL were purchased from Abcam (Cambridge, UK). A Cr assay kit was purchased from Njjcbio (Nanjing, China). An ELISA kit for ALB was purchased from Jymbio (Wuhan, China). The Annexin V-FITC/PI Apoptosis Detection Kit was purchased from Bestbio (Shanghai, China). Cytochalasin D was obtained from APExBIO (USA). PKH67 was obtained from Sigma (St. Louis, MO).
Mouse model of T2DM
C57BL/6 mice supplied by the Experimental Animal Center of Anhui Medical University were used to establish the T2DM model. All animal experiments were performed in accordance with the Regulations of the Experimental Animal Administration issued by the State Committee of Science and Technology of China. Efforts were made to minimize the number of animals used and their suffering. Animals were maintained in accordance with the guidance of the Center for Developmental Biology, Anhui Medical University, for the care and use of laboratory animals, and all experiments used protocols approved by the institutions' subcommittees on animal care. C57BL/6 mice were induced by a HFD diet (Xietong Pharmaceutical Bioengineering Co. LTD, Jiangsu, D12492) for four months. The HFD diet ingredients are listed in Supplemental Table 1. Seven days before killing, intraperitoneal injection of streptozotocin (STZ, Sigma Aldrich, St. Louis, MO, USA; 100 mg/kg) freshly dissolved in sodium citrate (0.1 M, pH 4.5) was performed. Diabetes was confirmed by a fasting blood glucose level > 7.8 mmol/L.
Cell lines and culture
HK-2 and mTECs, kindly provided by Prof. Huiyao Lan, were cultured in DME/F-12 (HyClone, Logan, UT, USA) supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum (Merck Millipore, Darmstadt, Germany) at 37 °C in a humidified incubator under 5% CO2. THP-1 cells were cultured in RPMI-1640 (HyClone, Logan, UT, USA) supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum (Merck Millipore, Darmstadt, Germany) at 37 °C in a humidified incubator under 5% CO2. For all experiments, THP-1 monocytes were differentiated into macrophages using phorbol 12-myristate 13-acetate (5 ng/mL).
Primary cell isolation and culture
Bone marrow-derived macrophages (BMDMs) were isolated from wild-type mice as previously described [17]. Briefly, bone marrow was flushed from dissected mouse tibias and femurs with sterile PBS. Cells were resuspended and plated in RPMI-1640 (Gibco, Carlsbad, CA) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 U/mL penicillin, 100 μg/mL streptomycin, and M-CSF (10 ng/mL) (Novoprotein, Shanghai, China) conditioned media. After 3 days, the medium was replaced with the same medium without M-CSF. BMDMs were used for experiments after a differentiation period of 7 days. For real-time quantitative PCR analysis, 3 × 105 cells were plated per well into 12-well plates. The next day, BMDMs were treated with EV in RPMI 1640 medium for 8 h before RNA isolation.
Extracellular vesicle isolation
Renal primary epithelial cells and macrophage-derived EVs isolated from T2D mice were collected over a 24 h time period in EV-depleted complete medium, which was prepared by overnight centrifugation at 100,000 × g at 4 °C. Unless stated otherwise, EVs were isolated from cell culture medium by differential ultracentrifugation using a modified version of a protocol by Thery et al. [17]. The collected medium was depleted of cells and cell debris by consecutive, low-speed centrifugation (2,000 × g for 15 min and 16,000 × g for 20 min). The supernatants obtained were carefully collected and centrifuged for 90 min at 100,000 × g at 4 °C. Pellets from this centrifugation step were washed in PBS, pooled, and centrifuged again for 90 min at 100,000 × g at 4 °C. The obtained pellets were resuspended in lysis buffer (see below), PBS solution or RPMI-1640 medium, depending on subsequent experiments. EV solutions intended for cell treatment were sterile filtered through a 0.22 µm syringe filter. Resuspended EVs were either used for subsequent analysis or aliquoted and stored at -80 °C. For isolation of EVs derived from primary cells, a commercially available ExoQuick-TC exosome precipitation solution kit from System Biosciences (Palo Alto, CA) was utilized.
Electron microscopy
Isolated EVs were fixed in 2% paraformaldehyde in 0.1 M phosphate buffer overnight at 4 °C. The samples were then placed on a Formvar-carbon-coated grid and air dried for 20 min. After being rinsed with PBS, grids were transferred to 1% glutaraldehyde for 5 min and washed with distilled water. The grids were first contrasted with uranyl-oxalate solution and then contrasted and embedded in a mixture of 4% uranylacetate and 2% methylcellulose (1:9 ratio). The grids were air dried and visualized with a JEOL 1400 electron microscope (JEOL USA, Peabody, MA) at 80 kV. For immunogold staining, grids were blocked with 10% FBS for 20 min followed by overnight incubation at 4 °C with primary anti-TRAIL and anti-LRG1 antibodies diluted 1:20 in blocking solution overnight. Next, grids were incubated with secondary antibody for 1 h. In negative control samples, primary antibody was omitted. Samples were then labeled with protein A-10-nm gold for 1 h. The grids were contrasted and embedded with a mixture of 4% uranyl acetate and 2% methylcellulose (1:9 ratio) and observed as described above.
Nanoparticle tracking analysis
The concentration and size distribution of isolated EVs were assessed by nanoparticle tracking analysis (NTA) using a NanoSight NS300 instrument (NanoSight, Amesbury, UK). EV samples were diluted with PBS at a range of concentrations between 4 × 108 and 8 × 108 particles per milliliter in a total volume of 1 milliliter. Each sample was continuously run through a flow-cell top plate set up to 23.3 °C using a syringe pump at a rate of 25 µL/min. At least three videos lasting 30 seconds documenting Brownian motion of nanoparticles were recorded and at least 1000 completed tracks were analyzed by NanoSight software (NTA 2.3.5).
Cell EV uptake
HK-2-derived purified EVs were labeled with the green fluorescent dye PKH67 (Sigma) and washed with 100,000 × g spinning. Differentiated THP-1 macrophages were then incubated with EVs (1010/ml) for 1 h (with or without 10 μM cytochalasin D pre-Hirsova et al. treatment), and their cellular internalization was observed using an LSM880 confocal microscope (Carl Zeiss, Jena, Germany). HK-2 cells were incubated with THP-1-derived purified EVs as mentioned above.
Flow cytometry
The extent of programmed cell death was detected by flow cytometry (CytoFlex, Beckman Coulter, USA) using an AV-FITC/PI apoptosis detection kit (Bestbio, Shanghai, China). To evaluate the apoptosis level of HK-2 and mTECs, the attached and supernatant cells were stained with 5 µl of Annexin V-FITC and 10 µl of PI in the dark, detected by flow cytometry, and analyzed using CytExpert 2.1 software (CytExpert, Beckman Coulter, USA).
Assay of caspase-3 activity
The activity of caspase-3 was measured by using a caspase-3 activity kit (Bestbio, Shanghai China) according to the manufacturer's instructions. Assays were performed on 96-well microtiter plates. Ten microliters of protein extract, 90 μl of reaction buffer, and 10 μl of caspase substrate were added in turn. Then, the protein extracts were incubated at 37 °C for 2-3 h. Samples were measured with Multiskan MK3 at an absorbance of 405 nm.
Transient transfection
A small interfering RNA (siRNA) was used to silence TGFβ-R1 in THP-1 cells. Cells transfected with scramble siRNA were used as controls. Cells were grown in 60-mm dishes and transiently transfected with SmartPool siRNA (5 nM, Hanbio) using LipoFiter 3.0 (Hanbio). Experiments were performed 48 h after transfection.
Acridine orange-ethidium bromide staining
The apoptotic morphology of treated cells was detected and distinguished by acridine orange-ethidium bromide (AO/EB) fluorescent staining. The staining method was performed by introducing a mixture of fluorescent dyes, acridine orange and ethidium bromide at a 1:1 ratio to treated cells. HK-2 cells and mTECs (2 × 103 cells/well) were seeded in chamber slides. The adherent cells were washed with 200 μl of PBS, and 2 μl of the dye mixture containing ethidium bromide (100 mg/ml) and acridine orange (100 mg/ml) in a 1:1 ratio was placed on each well of the chamber slide. Chamber slides were examined immediately under a fluorescence microscope (Carl Zeiss Axio Vert. A1, Jena, Germany).
Creatinine assay kits and albumin ELISA kits
Twenty-four-hour urine samples were collected one day before the sacrifice of mice. The concentrations of creatinine (Cr) in urine from C57BL/6 T2D mice were determined by Cr assay kits according to the manufacturer's instructions. The concentration of albumin (ALB) in urine from T2D mice was measured via ELISA using a commercialized protocol.
Histopathology
Renal tissues of mice were fixed in 4% paraformaldehyde for 24 h immediately following killing, processed for histological examination according to a conventional method, and stained with PAS, Oil Red O, H&E and F4/80. The slides were scored in a blinded manner and deidentified.
TUNEL assay
Renal cell apoptosis was examined by TUNEL assay using the One step TUNEL Apoptosis Assay Kit from Beyotime Biotechnology (Beyotime, Jiangsu, China). Briefly, cells were fixed with 4% paraformaldehyde in PBS and then exposed to the TUNEL reaction mixture containing TM red-labeled dUTP. Finally, samples were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). TUNEL-positive nuclei were identified by fluorescence microscopy.
Western blot
Whole extracts were separated by 10 or 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), transferred to a polyvinylidene difluoride membrane, and incubated with primary antibodies against LRG1/TRAIL/TSG101/ARF6/CD63/calnexin/DR5/β-actin/GAPDH. The membranes were then washed in TBS-Tween 20 and incubated with the corresponding secondary antibodies. After extensive washing in TBS-Tween 20, protein bands were visualized with an ECL chemiluminescent kit (ECL-plus; Thermo Fisher Scientific, Pittsburgh, PA, USA).
Real-time reverse transcriptase-PCR
Total RNA was collected from kidney tissues, THP-1 cells and BMDMs using TRIzol reagent (Invitrogen). First-strand cDNA was synthesized using a ThermoScript RT-PCR synthesis kit (Fermentas, Pittsburgh, PA, USA) according to the manufacturer's instructions. Real-time quantitative PCR analyses for mRNA were performed using ThermoScript RT-qPCR kits in an ABI Prizm step-one plus real-time PCR System (Applied Biosystems, Foster City, CA, USA). The products were used as templates for amplification using SYBR Green PCR amplification reagent (Qiagen, Valencia, CA, USA) and gene-specific primers. Relative expression levels were calculated according to the standard 2-ΔΔCt method. The forward and reverse primers used for PCR are listed in Table S1.
Statistical analysis
Data are expressed as the means ± SEM and represent at least three independent experiments. Differences between two groups were compared using the two-tailed Student's t-test. Differences between multiple groups were compared using one-way analysis of variance followed by Student's t-test. Differences were considered significant at P < 0.05. All analyses were performed using GraphPad Prism 5.0 software (San Diego, CA).
Results
Lipotoxicity induces the release of tubular epithelial cell (TEC)-derived EVe in vivo and in vitro
To study lipotoxicity in diabetic nephropathy (DN), a mouse model of diabetes was successfully induced by a high-fat diet (HFD) in C57BL/6 mice, as demonstrated by an equal level of increased blood glucose (Figure 1A). Periodic acid-Schiff (PAS) staining showed that glomerular mesangial expansion (GME) was increased in the DN mouse group compared to the vehicle group (Figure 1B). Furthermore, DN mice developed significant urinary albumin (ALB) and urinary creatinine (Cr) at 18 weeks after HFD induction (Figure 1C - D). Immunoblot analysis indicated that KIM-1, a kidney injury marker, was significantly upregulated in the DN group (Figure 1E). Lipid accumulation occurred in tubule cells compared to vehicle, as assessed by Oil Red O (ORO) staining (Figure 1F).
Lipotoxicity induces the release of tubular epithelial cell (TEC)-derived EVe in vivo and in vitro. (A) Blood glucose assay. (B) Kidney tissues stained with periodic acid-Schiff (PAS). (C) Urinary albumin (ALB) assay. (D) Urinary creatinine (Cr) assay. (E) Expression of KIM-1 was detected by western blot. (F) Kidney tissues stained with Oil Red O (ORO). (G) Primary TEC-derived EVe of DN representative image with nanoparticle tracking analysis (NTA). (H) Transmission electron photomicrographs of primary TEC-derived EVe of DN. (I) The expression of TSG101, ARF6, CD63 and calnexin in primary TEC-derived EVe from DN patients was detected by western blot. Similar results were obtained in 3 independent experiments with 10 mice per group.
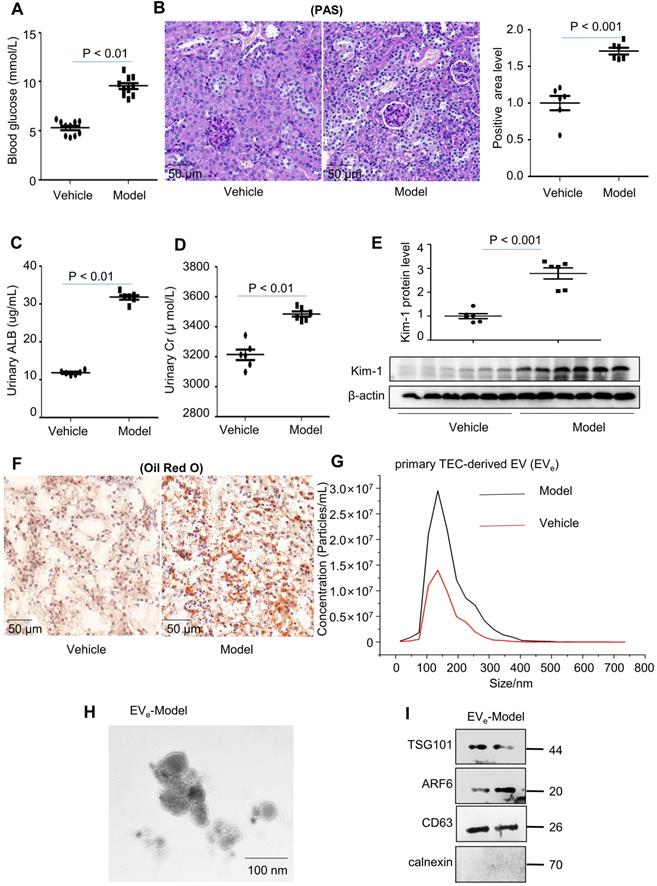
Primary tubular epithelial cell-derived EVe from DN mice were increased by approximately 2-fold, as quantified via nanoparticle tracking analysis (NTA) (Figure 1G). The size characteristics of the released EVe were confirmed by electron microscopy (Figure 1H). Immunoblot analysis indicated that isolated EVe were positive for tumor susceptibility gene 101 (TSG101), adenosine diphosphateribosyltion factor 6 (ARF6) and CD63 and were negative for the endoplasmic reticulum marker calnexin (Figure 1I).
To explore the effect of lipotoxicity on TEC-derived EVe release in vitro, we established treatment conditions that did not induce cell death in HK-2 cells and mTECs to avoid the collection of apoptotic bodies. A 6-hour treatment with LPC did not induce apoptosis but increased injury in these cells, and thus they were used for our experiments (Supplementary Figure S1A - B). We then examined the damage from lipids to renal tubular epithelial cells with lysophosphatidyl choline (LPC) to stimulate kidney tubular epithelial cells (HK-2) and mouse kidney tubular epithelial cells (mTECs). As shown in Supplementary Figure S1C - D, the protein level of KIM-1, an established biomarker for renal proximal tubule injury, was increased dramatically after 20 μmol/L LPC stimulation.
Upon LPC treatment, released EVe were isolated from the cell culture medium and quantified via nanoparticle tracking analysis (NTA). Over a 6-hour incubation period, LPC induced an approximately 2-fold increase in the release of EVe in HK-2 cells (Supplementary Figure S2A). The size characteristics of the released EVe were confirmed by electron microscopy (Supplementary Figure S2B). Immunoblot analysis indicated that isolated EVe were positive for TSG101, ARF6 and CD63 (Supplementary Figure S2C). Similar results were confirmed in mTECs (Supplementary Figure S2D - F).
This suggested that lipotoxicity induced the release of tubular epithelial cell (TEC)-derived EVe in vivo and in vitro.
Lipotoxic TEC-derived EVe activate macrophages
Inflammation is a central contributor to kidney pathology during DN. Characterization of DN mice was confirmed by infiltration of macrophages detected by immunohistochemical analysis. As shown in Figure 2A, the infiltration of macrophages was clearly upregulated in DN mice.
Lipotoxic TEC-derived EVe activates macrophages. (A) Immunohistochemical staining of F4/80 in kidney tissues. Scale bar, 50 μM; magnification, 20 ×. (B) Mouse bone marrow-derived macrophages (BMDMs) were incubated with purified EVe (1010/mL) isolated from DN mouse-derived TECs. (C) The levels of CXCL12, TNF-α, and IL-1β in BMDMs treated with primed TEC-derived EVe from DN mice were analyzed by quantitative real-time PCR. (D) The levels of CXCL12, TNF-α, and IL-1β in BMDMs treated with lipotoxic mTEC-derived EVe were analyzed by quantitative real-time PCR. (E) The levels of CXCL12, TNF-α, and IL-1β in THP-1 cells treated with lipotoxic HK-2-derived EVe were analyzed by quantitative real-time PCR. (F) The levels of CXCL12, TNF-α, and IL-1β in THP-1 cells treated with lipotoxic HK-2-derived EVe and boiled lipotoxic HK-2-derived EVe analyzed by quantitative real-time PCR. Similar results were obtained in 3 independent experiments or in triplicate culture assays.
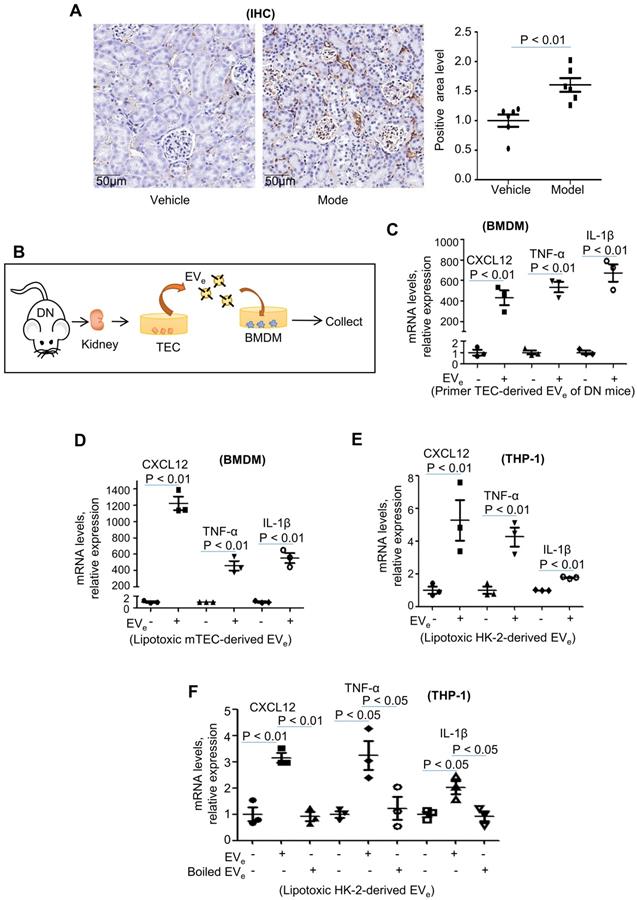
Interestingly, mouse bone marrow-derived macrophages (BMDMs) were incubated with primary tubular epithelial cell-derived EVs (EVe, 1010/ml) from DN mice (Figure 2B), and EVe significantly increased CXCL12, TNF-α and IL-1β mRNA expression (Figure 2C). Similarly, incubation of BMDMs with LPC-treated mTEC-derived EVe (1010/ml) (Figure 2D) or incubation of THP-1 cells with LPC-treated HK-2-derived EVe (1010/ml) (Figure 2E) also increased TNF-α, IL-1β, and CXCL12 mRNA expression.
To demonstrate that an EV-unrelated fraction was not responsible for macrophage activation, we destroyed EVs by boiling and observed that this fraction abolished their stimulatory activity (Figure 2F).
This suggested that lipotoxicity-induced EVe promote proinflammatory responses.
LRG1-enriched EVe-mediated macrophage activation is TGFβR1-dependent
Our previous study revealed that LRG1 was highly elevated upon renal damage [18]. Interestingly, immunogold labeling and electron microscopy showed that LRG1 was found in mouse primary TEC-derived EVe (Figure 3A). Similar results were also found in lipotoxic HK-2-derived EVe (Figure 3B). Moreover, western blot analysis showed a significant increase in LRG1 expression in lipotoxic HK-2-derived EVe and lipotoxic mTEC-derived EVe (Figure 3C). In addition, THP-1 cells treated with recombinant human LRG1 (rhLRG1) clearly upregulated the expression of CXCL12, TNF-α and IL-1β (Figure 3D - E). The results indicated that LRG1 induced the expression of cytokines.
LRG1-enriched EVe-mediated macrophage activation is TGFβR1-dependent. (A) Representative transmission electron photomicrographs immunogold-labeled with an anti-LRG1 antibody of primary TEC-derived EVe of DN. Scale bar: 100 nm. (B) Representative transmission electron photomicrographs immunogold-labeled with an anti-LRG1 antibody of lipotoxic HK-2-derived EVe. Scale bar: 100 nm. (C) Expression of LRG1 in HK-2-derived EVe and mTEC-derived EVe treated with LPC was detected by western blot. (D) Effects of various concentrations of rhLRG1 on TNF-α mRNA levels in THP-1 cells analyzed by quantitative real-time PCR. (E) Effects of rhLRG1 (10 ng/mL) on CXCL12, TNF-α and IL-1β mRNA levels in THP-1 cells analyzed by quantitative real-time PCR. (F) THP-1 cells were differentiated using PMA treatment for 48 h. Thereafter, THP-1 cells were pretreated with cytochalasin D (1 μM) followed by 2 hours of incubation with EVe (1010/mL) isolated from LPC-treated HK-2 cells. Prior to this incubation, EVe were labeled using the fluorescent dye PKH67. EVe uptake by macrophages was visualized using confocal microscopy and quantified. (G) Differentiated macrophage-like THP-1 cells were pretreated with cytochalasin D (1 μM) for 1 hour followed by 2 hours of incubation with lipotoxic HK-2-derived EVe (1010/mL) for 2 hours. Effect of cytochalasin D on CXCL12, TNF-α and IL-1β mRNA levels in THP-1 cells analyzed by quantitative real-time PCR. (H) The efficiency of TGFβR1 knockdown in THP-1 cells analyzed by quantitative real-time PCR. (I) CXCL12, TNF-α and IL-1β mRNA levels in THP-1 cells treated with siRNA-TGFβR1 analyzed by quantitative real-time PCR. Similar results were obtained in 3 independent experiments or in triplicate culture assays.
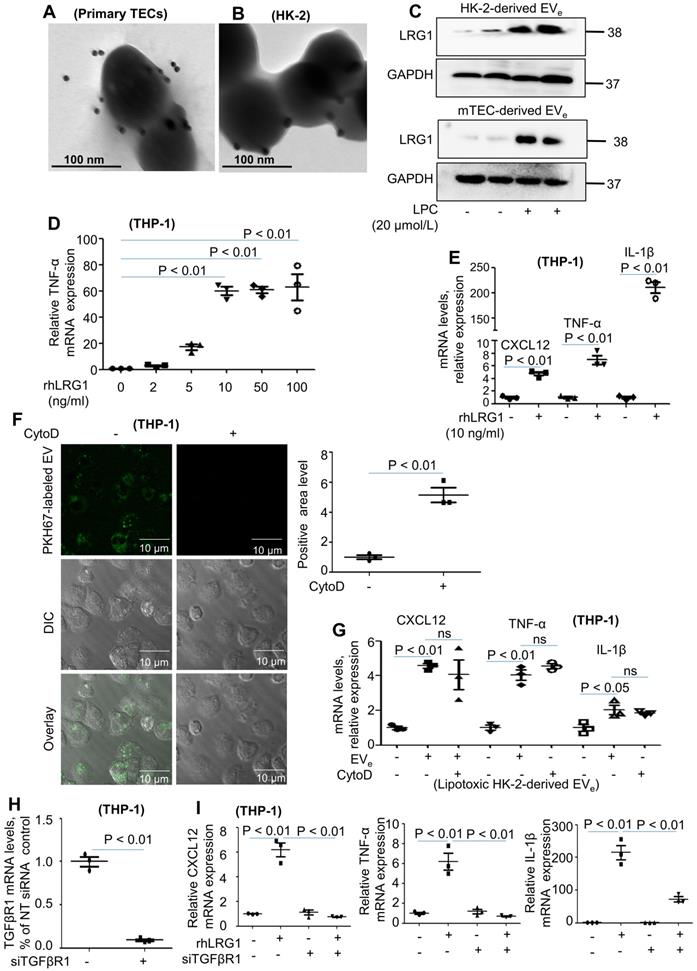
We then examined whether phagocytosis was required for macrophage activation by EVe. Cytochalasin D, a pharmacologic inhibitor of phagocytosis, efficiently inhibited phagocytosis of fluorescently labeled EVe but had no effect on macrophage activation (Figure 3F - G). These data suggested that EVe-macrophage interactions at the level of the plasma membrane were likely responsible for proinflammatory macrophage activation.
Although LRG1 binds directly to TGFβ receptor 1 (TGFβR1) [19], it is not known whether LRG1-enriched EVe could promote the activation of macrophages via TGFβR1. To determine whether TGFβR1 regulated LRG1-enriched, EVe-induced macrophage activation, we then treated THP-1 cells with siRNA-TGFβR1 for 24 hours (Figure 3H). PCR analysis indicated that knockdown of TGFβR1 significantly inhibited the expression of TNF-α, IL-1β, and CXCL12 (Figure 3I).
Taken together, lipotoxic TEC-derived, LRG1-enriched EVe promoted the activation of macrophages in a TGFβR1-dependent manner.
Lipotoxic TEC-derived, LRG1-enriched EVe induce the release of macrophage-derived EVm
Interestingly, we found that lipotoxic TEC-derived, LRG1-enriched EVe not only activated macrophages but also induced the release of macrophage-derived EVm. Primary TEC-derived EVe from DN mice induced an approximately 4-fold increase in the release of macrophage-derived EVm in BMDMs, as quantified by NTA in Figure 4A. The size characteristics of the released EVm were confirmed by electron microscopy (Figure 4B). Immunoblot analysis indicated that isolated EVm were positive for TSG101, ARF6 and CD63 (Figure 4C). Furthermore, analysis using NTA, western blot and electron microscopy showed that recombinant human LRG1 (rhLRG1) also induced an approximately 2-fold increase in the release of macrophage-derived EVm in THP-1 cells (Figure 4D - F).
Lipotoxic TEC-derived, LRG1-enriched EVe induce the release of macrophage-derived EVm. (A) Representative images of BMDM-derived EVm treated with primary TEC-derived EVe from DN with nanoparticle tracking analysis (NTA). (B) Transmission electron photomicrographs of BMDM-derived EVm treated with primary TEC-derived EVe of DN. (C) Expression of TSG101, ARF6 and CD63 in BMDM-derived EVm treated with primary TEC-derived EVe from DN patients was detected by western blot. (D) Representative images of THP-1-derived EVm treated with rhLRG1 with nanoparticle tracking analysis (NTA). (E) Transmission electron photomicrographs of THP-1-derived EVm treated with rhLRG1. (F) The expression of TSG101, ARF6 and CD63 in THP-1-derived EVm treated with rhLRG1 was detected by western blot. Similar results were obtained in 3 independent experiments or in triplicate culture assays.
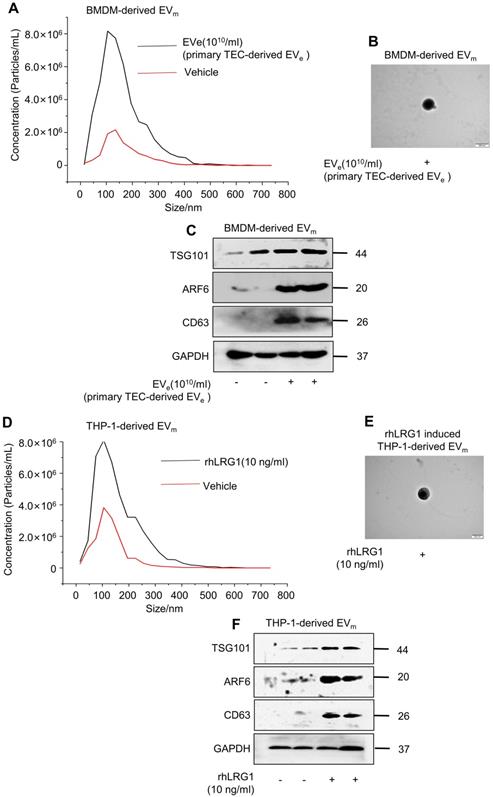
All of the above data supported the conclusion that lipotoxic TEC-derived, LRG1-enriched EVe induced the activation of macrophages and promoted the release of macrophage-derived EVm.
Activated macrophage-derived EVm induce lipotoxic TEC apoptosis
Available evidence suggests that apoptosis of tubular epithelial cells is one of the characteristics of DN. Numerous TUNEL-positive cells were observed in DN mice in contrast with the vehicle group (Figure 5A).
Activated macrophage-derived EVm induce lipotoxic TEC apoptosis. (A) Representative images of TUNEL staining in various groups. Scale bar, 50 μm. (B) Tubular epithelial cells of DN were incubated with purified EVm (1010/mL) isolated from DN mice-derived macrophages. (C) Flow cytometry analysis of primary TECs in each group. (D) Flow cytometry analysis of HK-2 in each group. (E) Flow cytometry analysis of mTEC in each group. (F) The activity of caspase-3 in HK-2 cells induced by THP-1-derived EVm. (G) The activity of caspase-3 in mTECs induced by BMDM-derived EVm. Similar results were obtained in 3 independent experiments or in triplicate culture assays.
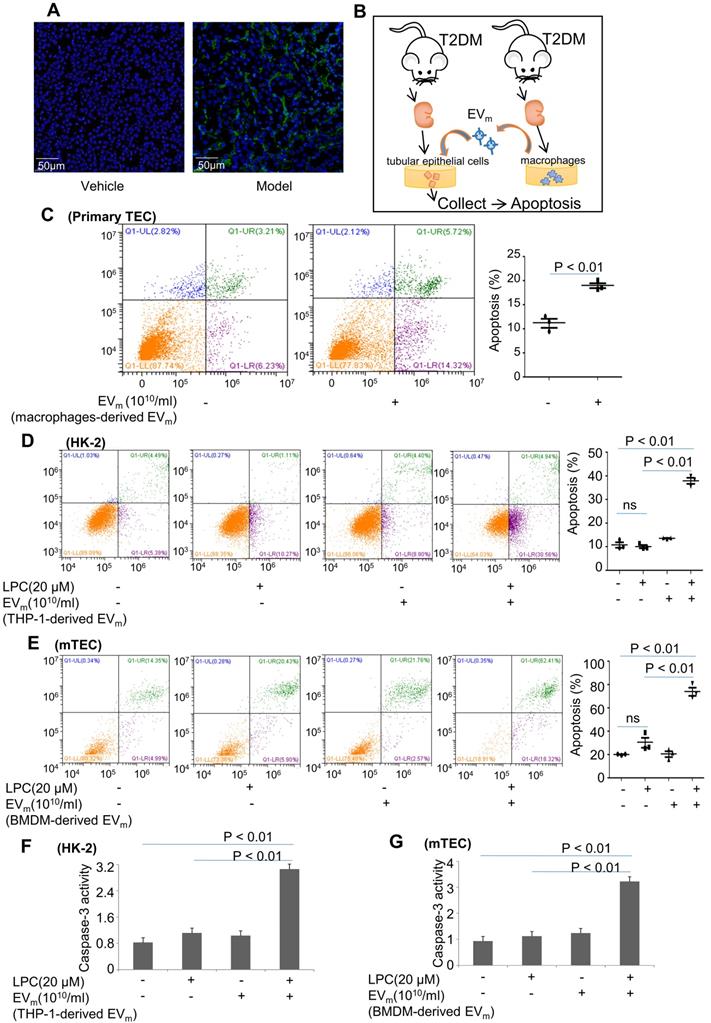
To confirm the role of EVm in apoptosis of tubular epithelial cells in DN, primary renal macrophage-derived EVm were cocultured with primary renal tubular epithelial cells (Figure 5B). Over an 8-hour incubation period, primary mouse renal macrophage-derived EVm significantly promoted apoptosis in primary renal tubular epithelial cells (Figure 5C).
Next, we treated HK-2 cells with LPC and then cocultured them with macrophage-derived EVm in THP-1 cells. As illustrated in Figure 5D, in the presence of EVm, LPC-pretreated HK-2 cells exhibited a much higher level of apoptosis, whereas LPC treatment alone in the absence of EVm did not affect the basal level of apoptosis in HK-2 cells. Similar results revealed that activated macrophage-derived EVm in BMDMs induced lipotoxic mTEC apoptosis (Figure 5E). Interestingly, the activity of caspase-3 in lipotoxic mTECs or HK-2 cells was increased by macrophage-derived EVm (Figure 5F- G).
Activated macrophage-derived EVm induces tubular epithelial cell apoptosis in a TRAIL-DR5-dependent manner
We then examined whether phagocytosis is required for tubular epithelial cell apoptosis by EVm. Cytochalasin D had no effect on HK-2 apoptosis (Figure 6A - B). These data suggested that EVm-tubular epithelial cell interaction at the level of the plasma membrane was likely responsible for tubular epithelial cell apoptosis.
Activated macrophage-derived EVm induce tubular epithelial cell apoptosis in a TRAIL-DR5-dependent manner. (A) HK-2 cells were pretreated with cytochalasin D (1 μM) followed by 2 hours of incubation with THP-1-derived EVm (1010/mL). Prior to this incubation, EVm were labeled using the fluorescent dye PKH67. EVm uptake by HK-2 cells was visualized using confocal microscopy and quantified. (B) Flow cytometry analysis of HK-2 in each group. (C) The expression of DR5 in primary TECs was detected by western blot. (D) The expression of DR5 in HK-2 cells treated with LPC was detected by western blot. Similar results were obtained in 3 independent experiments or in triplicate culture assays.
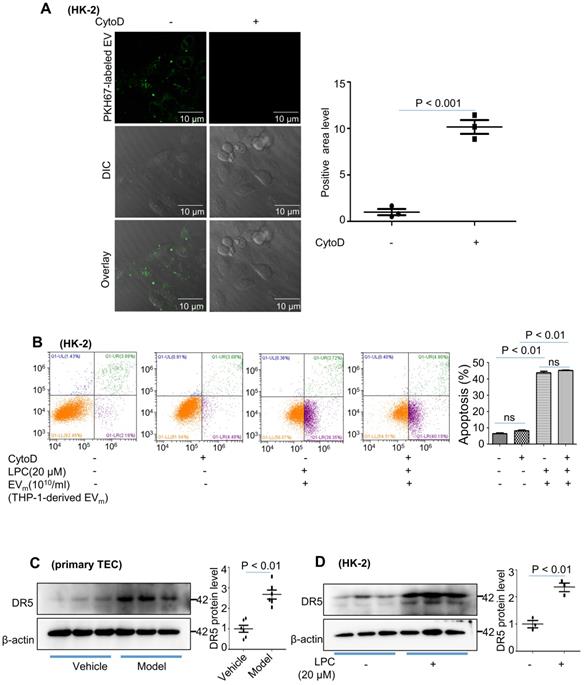
Lorz and colleagues reported that tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced cell death played an important role in the progression of diabetic nephropathy [20]. Recent evidence has shown that TRAIL-R2 (DR5) is the protein that is most strongly associated with kidney function decline. We examined the expression of DR5 in primary tubular epithelial cells and found that DR5 was significantly upregulated in the DN group (Figure 6C). Similar results were revealed in HK-2 cells after LPC treatment (Figure 6D).
Macrophage infiltration and TEC apoptosis are hallmarks of DN. However, how these two types of cells communicate remains obscure. As illustrated in Figure 7A, primary tubular epithelial cell-derived EVe induced a much higher level of TRAIL, the ligand of DR5, in BMDMs. Similar results were observed in mTEC-derived EVe-induced BMDMs (Figure 7B). Interestingly, the expression of TRAIL in macrophage-derived EVm of DN mice was observed by western blot and immunogold labeling (Figure 7C - D). Furthermore, primary tubular epithelial cell-derived EVe- or LPC-treated mTEC-derived EVe induced a much higher level of TRAIL in BMDM-derived EVm (Figure 7E - F). Importantly, rhLRG1 also increased the mRNA level of TRAIL in BMDMs (Figure 7g) and increased the protein level of TRAIL in BMDM-derived EVm (Figure 7H). To gain greater insight regarding TRAIL-induced apoptosis of lipotoxic TECs, recombinant human TRAIL (rhTRAIL) was used. Concomitant stimulation of HK-2 cells with TRAIL and LPC significantly increased apoptosis (Figure 7I).
Activated macrophage-derived EVm induces tubular epithelial cell apoptosis in a TRAIL-DR5-dependent manner. (A) Effect of primary TEC-derived EVe on TRAIL mRNA levels in BMDMs analyzed by quantitative real-time PCR. (B) Effect of mTEC-derived EVs on TRAIL mRNA levels in BMDMs analyzed by quantitative real-time PCR. (C) The levels of CXCL12, TNF-α, IL-1β, TRAIL and calnexin in macrophage-derived EVm of DN mice analyzed by western blot. (D) Representative transmission electron photomicrographs immunogold-labeled with an anti-TRAIL antibody of macrophage-derived, TRAIL-enriched EVm of DN mice. Scale bar: 100 nm. (E) The expression of TRAIL in BMDM-derived EVm treated with primary TEC-derived EVe from DN mice was detected by western blot. (F) The expression of TRAIL in BMDM-derived EVm treated with mTEC-derived EVe from DN mice was detected by western blot. (G) Effect of rhLRG1 (10 ng/mL) on TRAIL mRNA levels in BMDMs analyzed by quantitative real-time PCR. (H) Effect of rhLRG1 (10 ng/mL) on TRAIL protein levels in BMDMs analyzed by western blot. (I) Flow cytometry analysis of HK-2 in each group. Similar results were obtained in 3 independent experiments or in triplicate culture assays.
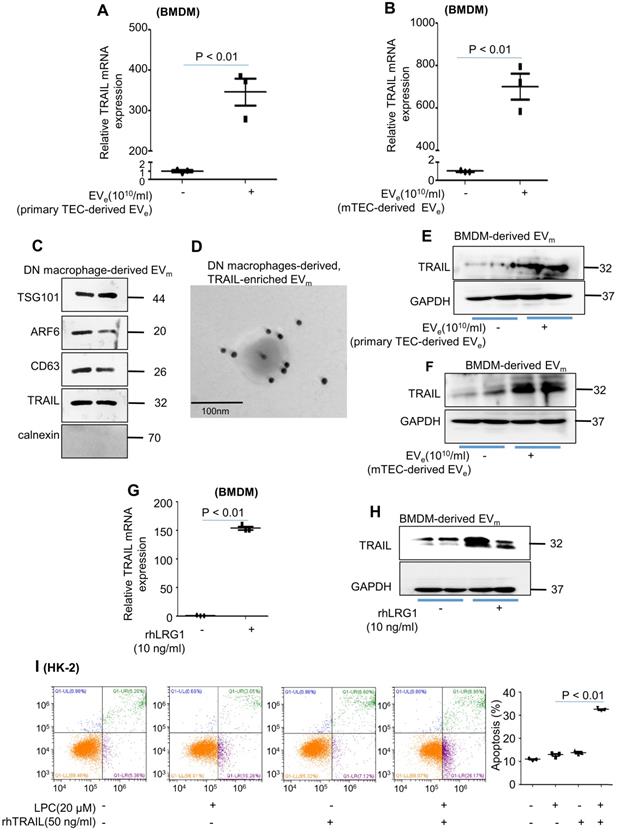
Taken together, our results indicated that TRAIL-enriched EVm from activated macrophages induced lipotoxic TEC apoptosis via a DR5 proapoptotic signaling pathway.
Discussion
Diabetic nephropathy (DN), whose pathogenesis is complex, is increasing in incidence and is now the number one cause of end-stage renal disease in the industrialized world [21]. Recent studies have confirmed that apoptosis contributes to diabetic nephropathy (DN) [22, 23]. Tojo et al. [24] reported that tubular dysfunction may partly explain microalbuminuria in early-stage diabetic nephropathy. Kumar et al. [25] also found apoptosis in all five DN biopsy specimens examined, either in epithelial cells of the proximal or distal tubules or in endothelial cells or interstitial cells. Mou et al. [26] further demonstrated that apoptosis of renal tubular epithelial cells may participate in the development and progression of DN in rats. The potential therapeutic mechanisms of PGE1 and AGEI, at least in part, inhibited the apoptosis of renal tubular epithelial cells. In our study, similar results demonstrated that the apoptosis of proximal tubular cells was increased in a DN mouse model. Cell death via apoptosis is an active response of cells to alter microenvironments [27]; however, the mechanism of renal tubular epithelial cell apoptosis is obscure.
Renal lipotoxicity due to derangement in lipid metabolism may be a pathogenic mechanism leading to diabetic nephropathy and renal dysfunction [28]. Ectopic fat deposition (EFD) in the kidney has been shown to play a causal role in diabetic nephropathy [29]. Opazo-Rios et al. [30] found that serum lipid abnormalities and renal ectopic lipid accumulation were associated with the development of kidney diseases, particularly diabetic nephropathy. Xu et al. [31] also reported that the manipulation of lipid metabolism might act as a promising therapeutic intervention for diabetic nephropathy. Our study found that LPC promoted proximal tubular cell injury. However, the process by which lipotoxicity promotes the progression of diabetic nephropathy remains obscure.
It has been confirmed that diabetes is an inflammatory disease caused by metabolic disorders. Bohle et al. [32] found that infiltration of inflammatory cells was detected by immunostaining in renal biopsy specimens from 488 patients with DN. Inflammation is a complex biological response that is essential for eliminating microbial pathogens and repairing tissues after injury. Macrophages have a crucial role in the regulation of both innate and adaptive immune responses in DN [33, 34]. Chow et al. [35] reported that kidney macrophages were found to increase with the duration of diabetes, and their numbers correlated with the severity of renal injury and dysfunction. We also found massive macrophage infiltration in the current study. This new pathogenic perspective of DN as an inflammatory condition leads to novel horizons [36]. Targeting inflammatory pathways could possibly be a component of the strategies to prevent and control diabetes and related complications [37]. However, these processes and mechanisms of activation of macrophages, playing active defense roles and signals transmitted between cells in DN, remain obscure.
Deng et al. [38] found that exosome-like vesicles (ELVs) released by adipose tissue can act as a mode of communication between adipose tissues and macrophages. Li et al. [39] reported that the HIF-1alpha-dependent release of miRNA-23a-enriched exosomes from hypoxic tubular epithelial cells activated macrophages to promote tubulointerstitial inflammation. Interestingly, we also found that lipotoxicity-induced EVe promote proinflammatory responses. This suggested that lipotoxic TEC-derived EVe activated macrophages in DN. In our previous study, leucine-rich alpha-2-glycoprotein 1 (LRG1) was highly elevated upon renal damage [18]. Interestingly, LRG1 was also found in lipotoxic TEC-derived EVe. LRG1 can promote completely different cellular processes, including survival, proliferation, migration, and metastasis [40, 41]. LRG1 is an innovative biomarker for inflammation and angiogenesis. Liu et al. [42] found that LRG1 deletion caused impaired immune cell infiltration. Hong et al. [19] identified LRG1 as a potential novel pathogenic mediator that promoted diabetic kidney disease progression by enhancing TGF-beta-induced angiogenesis. In our study, it was also observed that stimulation of TGF-βR1 with recombinant LRG1- or LRG1-enriched EVe activated macrophages toward a proinflammatory phenotype. All of the above data supported the conclusion that lipotoxic TEC-derived, LRG1-enriched EVe induced the activation of macrophages.
Interestingly, we found that lipotoxic TEC-derived, LRG1-enriched EVe not only activated macrophages but also induced the release of macrophage-derived EVm. Next, we found that activated macrophage-derived EVm induced lipotoxic TEC apoptosis in DN. To explore the mechanism, we tested the level of DR5 and found that DR5 was significantly upregulated in the DN group and in LPC-treated HK-2 cells. Similarly, Hirsova et al. [43] also reported that lipids stimulated the expression of DR5. TRAIL has been studied extensively as a death ligand and initiator of the apoptotic signaling cascade via its cognate receptor DR5 [44]. Ke et al. [45] reported that TRAIL was naturally delivered as a membrane-bound form by EVs and was highly efficient for apoptosis induction. Thereafter, we focused on the potential role of macrophage-derived EVm carrying TRAIL as contributors to TEC apoptosis. It was demonstrated that lipotoxic TEC-derived, LRG1-enriched EVe promoted the level of TRAIL expression not only in macrophages but also in macrophage-derived EVm. Herein, we found that stimulation of TRAIL receptor (DR5) with recombinant TRAIL or macrophage-derived TRAIL-enriched EVm promoted TEC apoptosis.
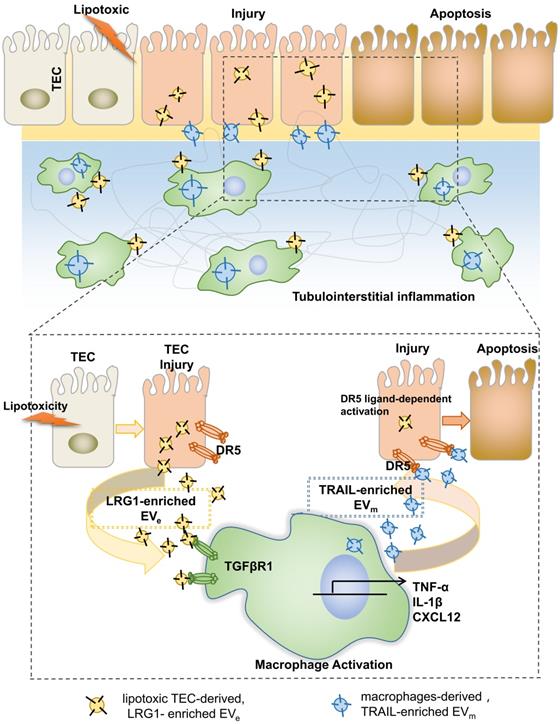
In summary, on the basis of our findings, we proposed a working model in Figure 8. During DN progression, LRG1-enriched, lipotoxic TEC-derived EVe activated macrophages by a TGFβR1-dependent pathway and subsequently upregulated the expression of many inflammatory genes, thereby inducing DN inflammation and injury. Meanwhile, LRG1/TGFβR1 signaling also elevated TRAIL expression in macrophages, and TRAIL-enriched, macrophage-derived EVm promoted apoptosis of TECs, which acted as a feedback loop in DN. Thus, our findings indicated a novel cell communication mechanism between tubular epithelial cells and macrophages in DN, which could be a potential therapeutic target.
Tubular epithelial cell-to-macrophage forms a feedback loop via extracellular vesicle transfer promotes renal inflammation and apoptosis in diabetic nephropathy.
Abbreviations
T2DM: Type 2 diabetes mellitus; EV: extracellular vesicles; TECs: tubular epithelial cells; DN: diabetic nephropathy; LRG1: leucine-rich α-2-glycoprotein 1; DR5: death receptor 5; TNFRSF10B: factor receptor superfamily member 10b; TRAIL: tumor necrosis factor-related apoptosis-inducing ligand; LPC: lysophosphatidyl choline; HK-2: human kidney tubular epithelial cells; mTEC: mouse kidney tubular epithelial cells; NTA: nanoparticle tracking analysis; TSG101: tumor susceptibility gene 101; ARF6: adenosine diphosphateribosylation factor 6; BG: blood glucose; NLs: neutral lipids; ORO: Oil Red O; PAS: periodic acid-Schiff; GME: glomerular mesangial expansion; EPCAM: mouse primary epithelial cell adhesion molecule; TNF-α: tumor necrosis factor α; IL: interleukin; CXCL: chemokine (X-C motif) ligand; Cyto D: Cytochalasin D.
Supplementary Material
Supplementary figures and table.
Acknowledgements
This work was supported by funding from the National Science Foundation of China (U19A2001), Institute of translational medicine (2021zhyx-B06), University Synergy Innovation Program of Anhui Province (GXXT-2019-045, GXXT-2020-063, GXXT-2020-025) and Natural Science Foundation of Anhui Province (2008085MH273). The authors thank the Center for Scientific Research of Anhui Medical University for valuable help in our experiment.
Author Contributions
W.-J.J. performed the cell experiments, analyzed the data and wrote the manuscript. T.-T. M and C.H. designed, supervised and wrote the manuscript. C.-T.X. provided a series of experimental instructions, and S.-B.X. and B.-F.H. performed the animal experiments. C.-L.D. and J.-H.D. helped with the experiments. F.R. and D.-D.Z. contributed new reagents or analytic tools. X.-M. M and J.L. designed the experiments. All authors approved the final version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Diabetes Canada Clinical Practice Guidelines Expert C, Punthakee Z, Goldenberg R, Katz P. Definition, Classification and Diagnosis of Diabetes, Prediabetes and Metabolic Syndrome. Canadian journal of diabetes. 2018;42(Suppl 1):S10-S5
2. Classification and Diagnosis of Diabetes. Standards of Medical Care in Diabetes-2021. Diabetes Care. 2021;44:S15-S33
3. Jia G, Whaley-Connell A, Sowers JR. Diabetic cardiomyopathy: a hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia. 2018;61:21-8
4. Saedi E, Gheini MR, Faiz F, Arami MA. Diabetes mellitus and cognitive impairments. World J Diabetes. 2016;7:412-22
5. Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y. et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus-Infected Pneumonia. N Engl J Med. 2020;382:1199-207
6. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z. et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054-62
7. Chan JF, Yuan S, Kok KH, To KK, Chu H, Yang J. et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 2020;395:514-23
8. Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428-35
9. Krausgruber T, Fortelny N, Fife-Gernedl V, Senekowitsch M, Schuster LC, Lercher A. et al. Structural cells are key regulators of organ-specific immune responses. Nature. 2020;583:296-302
10. Schleimer RP, Kato A, Kern R, Kuperman D, Avila PC. Epithelium: at the interface of innate and adaptive immune responses. J Allergy Clin Immunol. 2007;120:1279-84
11. Pitt JM, Kroemer G, Zitvogel L. Extracellular vesicles: masters of intercellular communication and potential clinical interventions. J Clin Invest. 2016;126:1139-43
12. Quaglia M, Dellepiane S, Guglielmetti G, Merlotti G, Castellano G, Cantaluppi V. Extracellular Vesicles as Mediators of Cellular Crosstalk Between Immune System and Kidney Graft. Front Immunol. 2020;11:74
13. Moon HG, Cao Y, Yang J, Lee JH, Choi HS, Jin Y. Lung epithelial cell-derived extracellular vesicles activate macrophage-mediated inflammatory responses via ROCK1 pathway. Cell Death Dis. 2015;6:e2016
14. Lv LL, Feng Y, Wen Y, Wu WJ, Ni HF, Li ZL. et al. Exosomal CCL2 from Tubular Epithelial Cells Is Critical for Albumin-Induced Tubulointerstitial Inflammation. J Am Soc Nephrol. 2018;29:919-35
15. Appiah MG, Park EJ, Darkwah S, Kawamoto E, Akama Y, Gaowa A. et al. Intestinal Epithelium-Derived Luminally Released Extracellular Vesicles in Sepsis Exhibit the Ability to Suppress TNF-a and IL-17A Expression in Mucosal Inflammation. Int J Mol Sci. 2020 21
16. Kakisaka K, Cazanave SC, Fingas CD, Guicciardi ME, Bronk SF, Werneburg NW. et al. Mechanisms of lysophosphatidylcholine-induced hepatocyte lipoapoptosis. Am J Physiol Gastrointest Liver Physiol. 2012;302:G77-84
17. Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006 Chapter 3: Unit 3 22
18. Jiang W, Ma T, Zhang C, Tang X, Xu Q, Meng X. Identification of urinary candidate biomarkers of cisplatin-induced nephrotoxicity in patients with carcinoma. J Proteomics. 2020;210:103533
19. Hong Q, Zhang L, Fu J, Verghese DA, Chauhan K, Nadkarni GN. et al. LRG1 Promotes Diabetic Kidney Disease Progression by Enhancing TGF-beta-Induced Angiogenesis. J Am Soc Nephrol. 2019;30:546-62
20. Lorz C, Benito-Martin A, Boucherot A, Ucero AC, Rastaldi MP, Henger A. et al. The death ligand TRAIL in diabetic nephropathy. J Am Soc Nephrol. 2008;19:904-14
21. Qi C, Mao X, Zhang Z, Wu H. Classification and Differential Diagnosis of Diabetic Nephropathy. J Diabetes Res. 2017;2017:8637138
22. Sonoki K, Iwase M, Sasaki N, Ohdo S, Higuchi S, Matsuyama N. et al. Relations of lysophosphatidylcholine in low-density lipoprotein with serum lipoprotein-associated phospholipase A2, paraoxonase and homocysteine thiolactonase activities in patients with type 2 diabetes mellitus. Diabetes Res Clin Pract. 2009;86:117-23
23. Balusu S, Van Wonterghem E, De Rycke R, Raemdonck K, Stremersch S, Gevaert K. et al. Identification of a novel mechanism of blood-brain communication during peripheral inflammation via choroid plexus-derived extracellular vesicles. EMBO Mol Med. 2016;8:1162-83
24. Tojo A, Onozato ML, Ha H, Kurihara H, Sakai T, Goto A. et al. Reduced albumin reabsorption in the proximal tubule of early-stage diabetic rats. Histochem Cell Biol. 2001;116:269-76
25. Kumar D, Robertson S, Burns KD. Evidence of apoptosis in human diabetic kidney. Mol Cell Biochem. 2004;259:67-70
26. Martinez-Lorenzo MJ, Anel A, Gamen S, Monle n I, Lasierra P, Larrad L. et al. Activated human T cells release bioactive Fas ligand and APO2 ligand in microvesicles. J Immunol. 1999;163:1274-81
27. De Miguel D, Gallego-Lleyda A, Anel A, Martinez-Lostao L. Liposome-bound TRAIL induces superior DR5 clustering and enhanced DISC recruitment in histiocytic lymphoma U937 cells. Leuk Res. 2015;39:657-66
28. Thongnak L, Pongchaidecha A, Lungkaphin A. Renal Lipid Metabolism and Lipotoxicity in Diabetes. Am J Med Sci. 2020;359:84-99
29. Chen X, Han Y, Gao P, Yang M, Xiao L, Xiong X. et al. Disulfide-bond A oxidoreductase-like protein protects against ectopic fat deposition and lipid-related kidney damage in diabetic nephropathy. Kidney Int. 2019;95:880-95
30. Opazo-Rios L, Mas S, Marin-Royo G, Mezzano S, Gomez-Guerrero C, Moreno JA. et al. Lipotoxicity and Diabetic Nephropathy: Novel Mechanistic Insights and Therapeutic Opportunities. Int J Mol Sci. 2020 21
31. Xu Y, Huang J, Xin W, Chen L, Zhao X, Lv Z. et al. Lipid accumulation is ahead of epithelial-to-mesenchymal transition and therapeutic intervention by acetyl-CoA carboxylase 2 silence in diabetic nephropathy. Metabolism. 2014;63:716-26
32. Bohle A, Wehrmann M, Bogenschutz O, Batz C, Muller CA, Muller GA. The pathogenesis of chronic renal failure in diabetic nephropathy. Investigation of 488 cases of diabetic glomerulosclerosis. Pathol Res Pract. 1991;187:251-9
33. Kimball A, Schaller M, Joshi A, Davis FM, denDekker A, Boniakowski A. et al. Ly6C(Hi) Blood Monocyte/Macrophage Drive Chronic Inflammation and Impair Wound Healing in Diabetes Mellitus. Arterioscler Thromb Vasc Biol. 2018;38:1102-14
34. Wang Y, Cui J, Liu M, Shao Y, Dong X. Schisandrin C attenuates renal damage in diabetic nephropathy by regulating macrophage polarization. Am J Transl Res. 2021;13:210-22
35. Chow FY, Nikolic-Paterson DJ, Atkins RC, Tesch GH. Macrophages in streptozotocin-induced diabetic nephropathy: potential role in renal fibrosis. Nephrol Dial Transplant. 2004;19:2987-96
36. Perez-Morales RE, Del Pino MD, Valdivielso JM, Ortiz A, Mora-Fernandez C, Navarro-Gonzalez JF. Inflammation in Diabetic Kidney Disease. Nephron. 2019;143:12-6
37. Lontchi-Yimagou E, Sobngwi E, Matsha TE, Kengne AP. Diabetes mellitus and inflammation. Curr Diab Rep. 2013;13:435-44
38. Deng ZB, Poliakov A, Hardy RW, Clements R, Liu C, Liu Y. et al. Adipose tissue exosome-like vesicles mediate activation of macrophage-induced insulin resistance. Diabetes. 2009;58:2498-505
39. Li ZL, Lv LL, Tang TT, Wang B, Feng Y, Zhou LT. et al. HIF-1alpha inducing exosomal microRNA-23a expression mediates the cross-talk between tubular epithelial cells and macrophages in tubulointerstitial inflammation. Kidney Int. 2019;95:388-404
40. Gao Y, Xie Z, Ho C, Wang J, Li Q, Zhang Y. et al. LRG1 Promotes Keratinocyte Migration and Wound Repair through Regulation of HIF-1alpha Stability. J Invest Dermatol. 2020;140:455-64 e8
41. Su Q, Lv XW, Sun YH, Ye ZL, Kong BH, Qin ZB. MicroRNA-494 Inhibits the LRG1 Expression to Induce Proliferation and Migration of VECs in Rats following Myocardial Infarction. Mol Ther Nucleic Acids. 2019;18:110-22
42. Liu C, Teo MHY, Pek SLT, Wu X, Leong ML, Tay HM. et al. A Multifunctional Role of Leucine-Rich alpha-2-Glycoprotein 1 in Cutaneous Wound Healing Under Normal and Diabetic Conditions. Diabetes. 2020;69:2467-80
43. Hirsova P, Ibrahim SH, Krishnan A, Verma VK, Bronk SF, Werneburg NW. et al. Lipid-Induced Signaling Causes Release of Inflammatory Extracellular Vesicles From Hepatocytes. Gastroenterology. 2016;150:956-67
44. Yuan X, Gajan A, Chu Q, Xiong H, Wu K, Wu GS. Developing TRAIL/TRAIL death receptor-based cancer therapies. Cancer Metastasis Rev. 2018;37:733-48
45. Ke C, Hou H, Li J, Su K, Huang C, Lin Y. et al. Extracellular Vesicle Delivery of TRAIL Eradicates Resistant Tumor Growth in Combination with CDK Inhibition by Dinaciclib. Cancers (Basel). 2020 12
Author contact
![]() Corresponding author: Pro. Taotao Ma, Inflammation and Immune Mediated Diseases Laboratory of Anhui Province, Anhui Institute of Innovative Drugs, School of Pharmacy, Anhui Medical University, Hefei, 230032, China. Email: mataotaoedu.cn
Corresponding author: Pro. Taotao Ma, Inflammation and Immune Mediated Diseases Laboratory of Anhui Province, Anhui Institute of Innovative Drugs, School of Pharmacy, Anhui Medical University, Hefei, 230032, China. Email: mataotaoedu.cn