Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Overview of the novel degrader...
Intracellular proteins...
Extracellular protein degradation
Design and synthesis of LYTAC...
Future Perspectives
Conclusion
Abbreviations
Acknowledgements
References
Author Biographies
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(17):8337-8349. doi:10.7150/thno.62686 This issue Cite
Review
Emerging protein degradation strategies: expanding the scope to extracellular and membrane proteins
Jiayi Lin1*, Jinmei Jin1*, Yiwen Shen1*, Lijun Zhang1, Gang Gong1, Huiting Bian1, Hongzhuan Chen1, Dale G. Nagle3, Ye Wu1 ![]() , Weidong Zhang1,2
, Weidong Zhang1,2 ![]() , Xin Luan1
, Xin Luan1 ![]()
1. Institute of Interdisciplinary Integrative Medicine Research and Shuguang Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai, 201203, China
2. School of Pharmacy, Second Military Medical University, Shanghai, 201203, China
3. Department of Biomolecular Sciences and Research of Institute of Pharmaceutical Sciences, School of Pharmacy, University of Mississippi, University, MS, 38677-1848, USA
*These authors contributed equally to this work.
Received 2021-5-12; Accepted 2021-7-3; Published 2021-7-13
Abstract

Classic small molecule inhibitors that directly target pathogenic proteins typically rely on the accessible binding sites to achieve prolonged occupancy and influence protein functions. The emerging targeted protein degradation (TPD) strategies exemplified by PROteolysis TArgeting Chimeras (PROTACs) are revolutionizing conventional drug discovery modality to target proteins of interest (POIs) that were categorized as “undruggable” before, however, these strategies are limited within intracellular POIs. The novel new degrader technologies such as LYsosome-TArgeting Chimaeras (LYTACs) and Antibody-based PROTACs (AbTACs) have been successfully developed to expand the scope of TPD to extracellular and membrane proteins, fulfilling huge unmet medical needs. Here, we systematically review the currently viable protein degradation strategies, emphasize that LYTACs and AbTACs turn a new avenue for the development of TPD, and highlight the potential challenges and directions in this vibrant field.
Keywords: targeted protein degradation (TPD), extracellular and membrane proteins, LYTAC, AbTAC
Introduction
Remarkable advances in human genetic studies have revealed a broad spectrum of protein targets that associate with disease progression [1, 2]. The classical small molecule inhibitor paradigm has been pursued worldwide through large-scale screening and structure-based optimization by academic and industry groups. Nevertheless, traditional drug discovery approaches usually require accessible binding sites and measurable biochemical index of target proteins, thus, only a small portion of human proteome is pharmaceutically druggable [3]. The majority of disease-causing proteins, including scaffolding proteins, transcription factors, and other non-enzymatic proteins are rendered as undruggable targets for a long time [4, 5].
Targeted protein degradation (TPD) has emerged to be a powerful tool to handle these undruggable targets and exhibits significant therapeutic benefit over standard small-molecule inhibition strategy [6]. A full degradation of disease-causing proteins provides the chance for ablation of target protein as well as all of its associated biological functions. It has been demonstrated that small molecules can induce protein degradation at the post-translational level, which occur with estrogen receptor alpha (ERα), inhibitor of apoptosis protein (IAP), androgen receptor (AR) and others [7-9]. However, this strategy has limited applications because there is rare ligand possessing degradation potency and it is wrought with great uncertainties. Recently, TPD strategies have expanded to include new modalities: PROteolysis Targeting Chimeras (PROTACs) [10], autophagy-targeting chimeras (AUTACs) [11], autophagosome-tethering compounds (ATTECs) [12], molecular glue degraders [13, 14], the degradation tag (dTAG) system [15, 16], and Trim-Away [17, 18], etc. Among them, PROTAC is the most advanced degradation strategy with two oral PROTACs (ARV-110 and ARV-471) exhibiting encouraging outcomes in Phase Ⅰ clinical trials [19]. However, these novel degrader technologies have only been applied to intracellular protein targets, the extracellular and membrane proteins that consist of more than 40% of human proteome still lack of effective therapeutic degraders [20].
To broaden the spectrum of target proteins, researchers are turning their attentions on the lysosome degradation system. Two marvelous approaches, termed LYTACs [21] and AbTACs [22], were reported to degrade secreted and cell-surface proteins by hijacking cell-surface lysosomal targeting proteins. Bertozzi's group established the first-generation LYTAC, M6Pn-LYTAC, combining the ligand of the cation-independent mannose-6-phosphate receptor (CI-M6PR) with an extracellular protein binder [21]. Pioneering work from research groups of Bertozzi, Spiegel and Tang developed the second-generation LYTAC, GalNAc-LYTAC, with the advantage of cell-specific degradation by engaging the liver-specific asialoglycoprotein receptor (ASGPR) [23-25]. Wells and colleagues used the AbTAC, a bispecific antibody, to expand the scope of targeted degradation via transmembrane E3 ligases [22]. Up to now, the degradation role has been established in secreted protein apolipoprotein E4 (ApoE4) and membrane-bound proteins, including epidermal growth factor receptor (EGFR), CD71, programmed death-ligand 1 (PD-L1) [21, 22], etc. LYTACs are able to induce target protein degradation with a high degree of selectivity to POIs and tissues, while AbTACs exploit the new bispecific antibody's potential for protein degradation. Although substantial progress has been made in the development of TPD, the emerging degradation approaches demand high technical requirements and each faces its own set of challenges towards its way to the clinic. Hence, this review compares the emerging degradation platforms, summarizes the design, synthesis, and mechanism of LYTACs and AbTACs, and highlights their future directions and challenges.
Overview of the novel degrader technologies
Over the last two decades, the breakthrough progress in TPD technologies not only provides useful tools for biological discovery, but also offers viable therapeutic candidates in clinic. We herein summarize the novel technologies with different mechanisms, as shown in Figure 1, and discuss their potential applications and limitations.
An overview of the novel protein degradation technologies. LYTAC and AbTAC utilizes lysosome system to degrade extracellular and membrane POIs. Intracellular POIs are targeted and degraded by PROTAC through the UPS. AUTAC and ATTEC technologies take advantages of autophagy-lysosome system to selectively degrade intracellular proteins and even organelles. Figure created with BioRender.com.
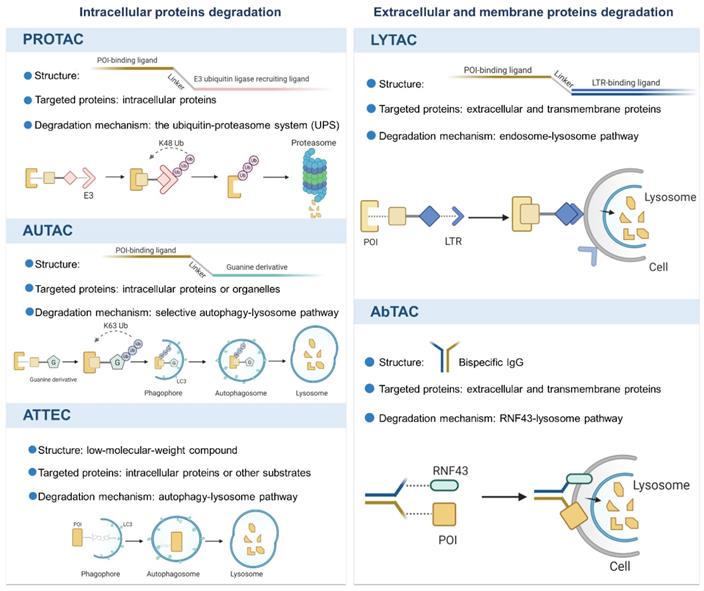
Intracellular proteins degradation
PROTAC
A PROTAC molecule consists of a targeting warhead for intracellular POI and an E3 ligase ligand for recruitment of E3, which are connected by a flexible linker [26]. The formation of a POI-PROTAC-E3 ternary complex induces ubiquitination of the POI and subsequent its degradation cascade by the 26S proteasome [27]. The PROTAC concept provides a powerful strategy for targeting those “undruggable” proteins that lack an active site for an inhibitor to bind or is not addressable by an inhibitor [28]. Of note, event-driven degradation induced by PROTAC is catalytic and sub-stoichiometric due to its multiple cycles of degradation mechanism [29,30]. Currently, a variety of PROTAC molecules have been designed and validated in preclinical settings and two oral PROTACs ARV-110 targeting AR (NCT03888612) and ARV-471 for ER (NCT04072952) have shown promising prospects in phase I clinical trials with good efficacy and safety profile [19,31,32]. Despite the promising clinical data, PROTACs have certain limitations to target those “undruggable” proteins with large shallow surfaces as well as extracellular proteins [10,33-35]. Small molecules and peptides are currently the main targeting warheads used by PROTAC design. However, those ligands have corresponding limitations. Among them, small molecules heavily rely on the binding pockets of POI [36], while peptide ligand application is limited to poor cellular membrane penetration and stability in vivo [37]. Moreover, PROTAC has faced challenges such as further expansion of human E3 ligases, reducing off-target toxicity, and the optimal linker-length determination, which have hindered its development. For example, because only a handful of E3 ubiquitin ligases are available, the current application of PROTAC is restricted. Similar to PROTAC, other UPS-based modalities using limited E3, like molecular glues, dTAG, and Trim-Away, are under restrictions.
AUTAC and ATTEC
In addition to the extensively utilized UPS in current TPD approaches, novel techniques such as AUTAC and ATTEC have been developed to modulate and control protein levels by harnessing the autophagy/lysosome pathway (reviewed in [38]), which offer a glimpse into future possibilities [11,12,39].
An AUTAC molecule consists of a small molecular binder of target protein and a guanine derivative as a degradation tag to trigger K63 polyubiquitination (different from the K48 polyubiquitination triggered by PROTACs) [11]. Ubiquitinated POIs are recognized by autophagy receptors such as p62/SQSTM1 and are linked to phagophores through the LC3-interacting region [40-42]. Despite the unique advantages of AUTAC for its specific and broad degradation scope, the underlying mechanisms of selective autophagy and its effects on the overall cellular proteins remain unclear and require further investigation.
Similar to AUTAC based on autophagy-lysosome system, ATTEC is a linker compound that tethers the POI to the autophagosome by interacting with both POI and LC3 proteins [12]. Owing to the advantages of its small size, ATTEC manipulates the protein levels more effectively. Meanwhile, it also reminds us that a largely unexplored area of compounds regulating therapeutically relevant proteins or other cytoplasmic substrates needs to be further exploited and clarified. These degraders all provide orthogonality and optimization for TPD platforms.
Extracellular protein degradation
Despite the promising prospect of TPD strategy, non-cytosolic proteins lied beyond the scope of TPD for a long time, which limited their further application. Encouragingly, the novel technologies, LYTAC and AbTAC, have emerged to broaden the spectrum of protein targets.
LYTAC
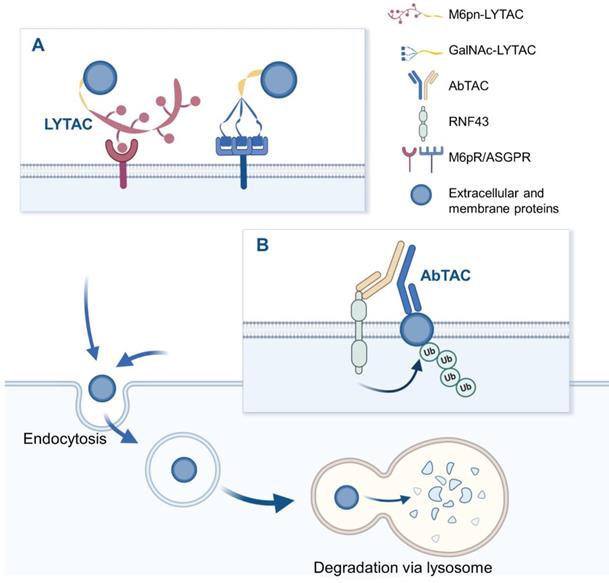
LYTAC is a novel technology that targets extracellular and/or membrane protein to induce degradation by harnessing the endosome/lysosome pathway. It is a bifunctional conjugate that simultaneously binds the extracellular domain of a target and a cell-surface lysosome-targeting receptor (LTR) to form a ternary complex, leading to protein internalization via clathrin-mediated endocytosis [43]. After being engulfed, the complex successively passes through early endosome (EE) and late endosome (LE) where a low pH enables the complex to be dissociated [44]. Subsequently, POI proceeds to lysosome to be degraded, while LTR is recycled into cell membrane via recycling endosome (RE). Degradation mechanism of LYTAC is shown in Figure 2. Notably, compared to POI inhibition, LYTAC directly exerts degradation effect on protein, and therefore avoids the potential activation of other downstream pathways that may be caused by inhibitors [21]. Moreover, this degradation strategy prevents molecular compensation and cellular adaptation due to their higher depletion efficiency compared with genetic techniques like CRISPR-Cas9 [45].
The schematic diagram of LYTAC and AbTAC. (A) M6Pn-LYTAC targets extracellular or membrane protein and is recognized by lysosome shuttling receptor CI-M6PR at the cell surface, to form ternary complex, while GalNAc-LYTAC binds target protein and liver cell-surface ASGPR simultaneously. The resulting complex is engulfed by the cell membrane, endocytosed into endosomes, and degraded in lysosomes. (B) AbTAC binds to RNF43 and cell-surface proteins simultaneously, inducing RNF43-AbTAC-protein complexes internalization and lysosomal degradation. Figure created with BioRender.com.
AbTAC
Bispecific antibodies (bsAbs) refer to a large family of molecules that recognize two different epitopes or antigens [46]. AbTAC is a fully recombinant bispecific immunoglobulins G (IgG) that can recruit transmembrane E3 ligases ring finger 43 (RNF43) [47] and cell-surface proteins simultaneously, inducing RNF43-AbTAC-protein complexes internalization and subsequent lysosomal degradation of POI [22], as shown in Figure 2. However, its mechanism of action is mainly remained elusive. Particularly, it remains unknown whether RNF43 ubiquitinates the intracellular regions of POI to induce endocytosis. Meanwhile, it needs to clarify what proteins are required for the AbTAC system and whether the RNF43-dependent degradation manner leads to other changes in cellular functions. Although there is no large cellular perturbation in whole-cell proteomics, the cell safety of AbTAC requires further proof [48]. In addition, when screening AbTAC for optimal degradation efficiency, we should also take the RNF43 cell specificity and endocytosis kinetics into account.
To fully understand the discussed techniques and choose the appropriate one for the problem at hand, we compare the advantages and disadvantages of intracellular protein degradation strategies and the two extracellular protein degradation approaches (LYTAC and AbTAC), as shown in Table 1.
Advantages and disadvantages of five TPD technologies
| POIs | Techs | Advantages | Disadvantages |
|---|---|---|---|
| Intracellular proteins | PROTAC | Clear mechanisms of action [49]; event-driven action [50]; catalytic degradation activity [51]; | Limitation to cytosolic domain of proteins [36]; In a proteasome-dependent manner [45]; Poor cellular penetration needs to be solved [36] |
| AUTAC | Targeting organelles and intracellular proteins; selective autophagy [11,39] | Degradation mechanisms need further investigation [11,39] | |
| ATTEC | Low molecular weight; selective degradation; Drug-like property [12] | Hard to design [12] | |
| Extracellular and membrane-bound proteins | LYTAC | Targeting extracellular and membrane-bound proteins [21]; high selectivity in POIs and cell types [23,24] | Poor tissue permeability [21]; Complex synthesis of LTR ligands M6Pn [21]; Possible immune response in vivo [52]; Lack of studies |
| AbTAC | Targeting membrane proteins; high bispecificity [22] | High cost; Potential immunogenicity [53]; Unclear endocytosis mechanism [22] |
Design and synthesis of LYTAC and AbTAC
To date, LYTAC and AbTAC have been the main two TPD technologies for extracellular and membrane protein degradation. Although they are recognized as tremendous milestones in the development of TPD, targeting extracellular protein degradation is still in its infancy. In this part, we mainly summarize the design principles, recent progress, and current synthesis strategies of LYTAC and AbTAC, providing theoretical basis for the future application. By focusing on the successive conceptual and technical innovations in this field, we hope to encourage and inspire further efforts to optimize current techniques and create new TPD methods.
LYTAC
LYTAC is designed with one end binding to the POI and the other end recognized by LTR. The incorporation of a ligand for efficient drug delivery via a LTR has been widely explored [54]. Based on structure of two substrates and the certain degradation mechanism, LYTAC has been rationally designed and synthesized, as shown in Figure 3.
A toolbox of functional LYTAC. (A) The design of LTR-binding ligands. (B) The currently targeted extracellular and transmembrane POIs and their ligands. Figure created with BioRender.com.
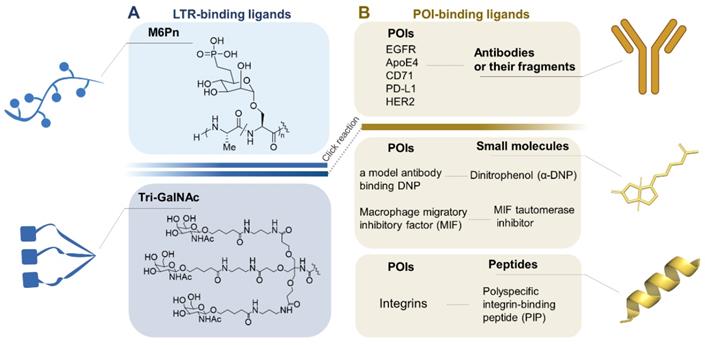
LTR-binding ligands
PolyM6Pn
Cation-independent mannose-6-phosphate receptor (CI-M6PR), one member of cell-surface LTR families, can enclose hydrolases or other cargoes in endosomes, and subsequently deliver those proteins into lysosomes for depletion [55]. Based on the structure of CI-M6PR, studies have demonstrated that CI-M6PR can bind with mannose-6-phosphate (M6P) glycoproteins [56]. So far, M6P derivatives have been utilized in enzyme replacement therapies for lysosomal diseases or in neoplastic drug targeting usage [55]. In order to increase affinity, stability, and avoid cytotoxicity, multiple M6P analogues were designed and tested, and an appropriate ligand M6Pn glycopolypeptide was obtained. Additionally, factors like length, chemical modification or side-chains should be further studied to achieve optimal CI-M6PR agonism [57].
M6Pn glycopolypeptides synthesis strategy starts with the conversion of mannose pentaacetate to M6Pn-NCA in 13 steps and ends with copolymerization of M6Pn-NCA to acquire poly(M6Pn) polypeptides [21]. However, as a ubiquitous receptor, the expression levels of CI-M6PR in various cell types may impact LYTAC-mediated degradation efficiency [21], or result in potential off-target effects. Moreover, some studies indicated that CI-M6PRs are overexpressed in specific cancer types (such as breast cancer) [58], and play a role in regulating cancer cell growth and motility [59]. However, whether this regulation effect is disrupted under the action of LYTAC is still unknown.
Tri-GalNAc
Unlike CI-M6PR, Asialoglycoprotein receptor (ASGPR) is a LTR which is highly expressed in hepatocytes [60]. Due to the ligand specificity and the ability of supporting multiple rounds of uptake [61], ASGPR has enormous potentials for liver cell specific degradation of proteins. On the basis of structure studies, it is clear that glycoproteins bearing N-acetylgalactosamine (GalNAc) or galactose (Gal) ligands can be recognized by ASGPR and internalized via clathrin-mediated endocytosis [62]. Indeed, GalNAc has already been applied as therapeutic nucleic acid agents in preclinical or clinical settings [63,64]. Studies also revealed that triantenerrary GalNAc (tri-GalNAc) ligands engage ASGPR with higher affinity compared to GalNAc [65,66]. Tri-GalNAc ligands are synthesized from peracetylated GalNAc to tri-GalNAc-DBCO in 8 steps, and then are conjugated to azides on non-specifically labeled antibodies [23]. Tang's group directly used the commercially available Tri-GalNAc-COOH. After being converted to Tri-GalNAc-NHS (N-hydroxysuccinimide) ester, Tri-GalNAc ligands are conjugated with the lysine residues on the antibody [24]. Compared to M6Pn, selectively delivering undesired proteins to liver by tri-GalNAc ligands is much safer than ubiquitously delivery of POIs to various types of cells [24]. We compare the advantages and disadvantages of polyM6Pn and tri-GalNAc in Table 2.
Advantages and disadvantages of polyM6Pn and tri-GalNAc
| Advantages | Disadvantages | |
|---|---|---|
| PolyM6Pn | High affinity [59]; overexpression in specific cancer cells [58] | Complex inhomogeneity of structure [68]; complicated synthesis process [21] |
| Tri-GalNAc | Homogeneous structure [24]; cell type-specific degradation [23,24] | Limitation on liver cells [23,24]; |
POI-binding ligands
The degradation ability of LYTAC also relies on the affinity between POI and its ligand. For targeting the significant disease-associative proteins, there are three choices for POI ligands, including antibodies, small molecules, and peptides [21,23-25]. Although having specific affinity with POI, antibodies with high molecular weight have lower tissue permeability than small molecules in several cases [68]. Take it further, the large molecular LYTAC labeled with an antibody needs to be miniaturized [52]. With the development of structure biology and virtual screening, small molecular targeting warheads possessing high affinity are selected [69]. Considering their inability to target “undruggable” proteins with large shallow surface, researchers have drawn their attentions towards specific binding peptides, which have advantages of lower production cost and amenability to chemical synthesis [70]. Compared to small molecules, peptides possess greater potential in structural modification by point mutation or truncation [34,71,72]. Generally, based on the crystal structure of endogenous complex of POI and binding protein, the key interacting residues can be analyzed to design the peptide targeting warheads [73]. To fully understand those POI ligands, we compared the advantages and disadvantages between them as shown in Table 3.
Advantages and disadvantages of POI ligands
| Advantages | Disadvantages | |
|---|---|---|
| Antibodies | Specific affinity with POIs [74] | High cost; low stability and tissue permeability [21]; potential immunogenicity [52] |
| Small molecules | High stability [75]; good penetrability [25] | Inability to target “undruggable proteins” [25] |
| Peptides | Low cost; easy to synthesise [61] | Low stability; limited penetrability [36] |
Construction strategy: Click reaction chemistry
Chemically, a LYTAC molecule can be assembled via azide-alkyne cycloaddition (click chemistry) between a TLR ligand and a POI ligand. For example, following modular design and synthesis, a POI ligand labeling with azide group is conjugated with a TLR ligand with alkynylation via copper-free strain-promoted click reaction, in turn, it also works that a POI ligand labeled with DBCO (dibenzocyclooctyne) or BCN can conjugate to an azide-labeled TLR ligand. After the conjugation reaction, the product is interrogated by native gel electrophoresis [21,23].
AbTAC
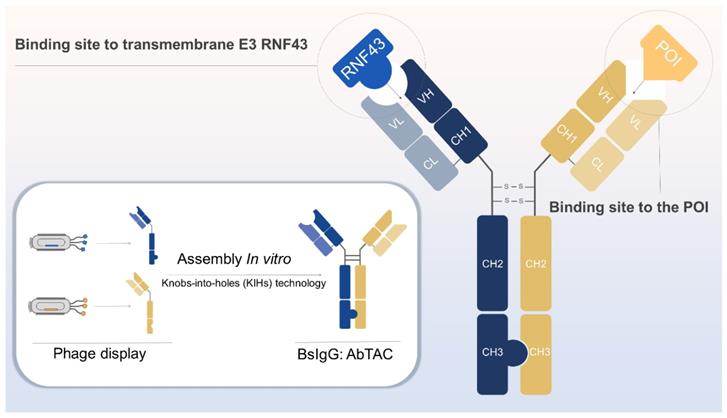
A single-pass E3 ligase: RNF43
As previously described, the AbTAC acts by assembling separately expressed half IgGs to form a bispecific IgG for degrading cell-surface proteins via RNF43 [22], which comprises a structured ectodomain, a transmembrane region, and an intracellular RING domain [46]. It was documented that RNF43 is widely expressed in a variety of cancer cells, such as MDA-MB-231, HCC827, and T24, etc., which provides generalizability for the degradation of RNF43-driven AbTAC [22]. In addition to that, RNF43 can negatively regulate the Wnt signaling pathway by ubiquitinating the Frizzled receptor, causing its endocytosis and degradation [76], which may open up possibilities to degrade the cell-surface POIs via endocytosis.
Construction strategy: Knobs-into-holes (KIHs) technology
The production of antibodies binding to RNF43 and POI begins with the phage display technique [77]. After multi-round selection strategies, clones with high affinities were identified by sequencing and assessing the on-cell binding ability [22]. Knobs-into-holes (KIHs) technology can prevent light chain mismatch pairing of half IgGs of anti-RNF43 and anti-POI, by creating either a “knob” or a “hole” in each heavy chain to promote heterodimerization [78]. The composition of AbTAC and its generation strategy are shown in Figure 4.
Generation of bispecific IgG AbTAC. The recombinant antibody for RNF43 and POI was generated using phage display. These two IgGs are assembled to form an AbTAC utilizing the knobs-into-holes technology. Figure created with BioRender.com.
Future Perspectives
Optimization of LYTAC and AbTAC
LYTAC
CI-M6PR is highly expressed in fetal and neonatal tissues but it decreases postnatally; however, it is overexpressed in some pathological conditions, such as fibrosis and some cancerous diseases [79-81]. For example, CI-M6PR is induced in a number of human carcinoma cells, such as breast cancer, pancreatic cancer, gastric cancer, melanoma, and prostate cancer [80]. Therefore, M6Pn-LYTAC is regarded as a more promising strategy for tumor therapy compared to GalNAc-LYTAC. Nevertheless, the construction of CI-M6PR-binding ligand, M6Pn glycopolypeptides, lacks precise manipulation, leading to the heterogeneity of products [21,57]. It is adverse for the subsequent study on structure optimization and structure-activity relationship. For instance, LYTACs with various glycopolypeptide length was observed only minor differences in uptake efficiency, suggesting that there should be a shortest M6Pn length to achieve high degradation efficiency. More important, smaller molecular size means lower immunogenicity [21]. Further, it was demonstrated that the projection topology of the saccharide units is of crucial importance for an efficient cell penetration mediated by CI-M6PR [59]. The spatial arrangement of glycopolypeptide needs to be further investigated.
AbTAC
AbTAC represents a new archetype to LYTAC with the ability to deplete cell-surface proteins. Despite its great promise in protein degradation, it is unclear how AbTAC recruit membrane-bound E3 ligases to induce cell-surface POI degradation via lysosome pathway. Only when the detailed regulation process is elucidated, could we make good use of this degradation pathway better. Moreover, a potential problem is whether there is appropriate distance between half IgGs for binding of membrane-bound E3 ligase and POIs at the same time. In this case, we envision that a bifunctional peptide with two warhead and a flexible linker can be used to target transmembrane E3 ligase and POIs simultaneously. We can also utilize phage display to rapidly generate targeting peptide for membrane-bound E3 ligases with high affinity and high specificity [82]. Meanwhile, there are so many existing tool peptides for disease related membrane proteins like PD-L1 [83], CD47 [84] and VEGFR [85]. Intermediate linker between two peptides is chosen according to actual situation, resulting in a “peptide version” AbTAC. We hope that accessible molecule will inspire the advancement of extracellular proteins degradation as a novel class of drug candidates.
Potential cell-surface lysosome targeting proteins
Currently, the number of E3 ubiquitin ligase ligands used in PROTAC technology is limited, which restricts their subsequent application and development. Also, it was verified that the loss of core components of E3 ligases leads to resistance to PROTACs [86]. Therefore, rather than overusing the same lysosome shuttling receptors, we do need to exploit new lysosome targeting proteins. For example, CD22 recycling receptor specifically expressing on B-cells [87] or mannose receptors (MR, CD206) presenting on tumor-associated macrophages (TAMs) surface [88,89] may be may be good choices for cell-specific degradation. Similarly, ZNRF3, another cell surface E3 ligase, provides a potential opportunity for the development of AbTACs [47]. In addition to cell surface receptors, intracellular lysosomal receptors, such as the lysosomal integral membrane protein (LIMP-2) and sortilin [90], might provide us with some enlightenments for targeting cytosolic proteins via lysosome system.
New possibilities for clinic
LYTAC gives failed drugs another chance
LYTAC provides possibilities to change protein ligands into degraders. TPD strategy possesses kinetic advantages by binding to any “nook” or “cranny” on POIs with low dose [91,92]. Generally, ligands that bind to the desired targets but were set aside because they could not adequately block protein function may be possible to be permitted as warheads of LYTAC. On the other hand, many preclinical or clinical 'occupancy-driven' molecules that have clear affinity with POI but not have gained FDA-approval due to their side effects can also be the warheads of LYTACs.
Multi-headed LYTAC and AbTAC-drug conjugates
Drug resistance and relapse are two major obstacles in cancer treatment [93], which can be avoided by rational polytherapy to target distinct pathogenetic mechanisms [94]. Recently, a series of PROTACs (MT-802, SJF620, and L18I) have been reported that they can overcome the resistance to ibrutinib induced by BTK mutations [95]. Based on this fact, we hypothesized that the “star molecule” multi-headed LYTACs might be able to overcome resistance with high efficiency and low toxicity [96,97].
Antibody-drug conjugates (ADC) are monoclonal antibodies conjugated to cytotoxic agents by environment-responsive linkers, which enable traditional drugs to have high tumor specificity and potency [98]. Five ADCs have since achieved FDA approval and more than 40 are now in or nearing clinical trials [99,100]. Here, we speculate the possibility of forming an AbTAC-drug conjugate (ATDC) as a multi-target agent. Ideally, after rational design of AbTAC linked to a drug, ATDC is able to degrade membrane protein to block the upstream signal, and further be internalized rapidly to deliver the linked drug intracellularly. Additionally, it requires that the linker is stable in blood circulation yet is readily cleavable at the target site. Future studies in MOA of AbTAC will possibly help to further develop AbTAC as a mature technology and discover effective multi-target agent.
Delivery nanosystem
Because LYTACs are macromolecules different from those of traditional protein-targeting small molecules, their aqueous solubility, drug delivery, and pharmacokinetics, and adverse reaction remain elusive. We estimate that the universal expression of cell-surface LTR and the instability of LYTACs may cause off-target effect and short drug half-life. Nanoparticles (NPs) have emerged as a powerful drug-delivery tool to enhance the specificity of drug actions, while reducing the systemic side effects [101]. For instance, The ARV-loaded NPs successfully improved the solubility, permeability, pharmacokinetics, and delivery of ARV, showing promising anticancer activity. Moreover, surface modification of PLGA NPs with PEG can impart high serum stability and prolonged half-life to PROTAC [102,103]. To ensure PROTAC delivery at the desired site in the required proportions, Ocaña and colleagues established PROTAC-loading NPs conjugated with antibody trastuzumab (selectively binds to the extracellular domain of HER2) to improve target selectivity, and cytotoxic efficiency for the treatment of breast cancer [104]. According to these cases of PROTAC-loading NPs, we propose that LYTAC-loading NPs might enable prolonged circulation and targeted delivery through elaborate design. Besides, “passively” targeted NPs are the most extensively explored strategy targeting cancer, by utilizing the enhanced permeability and retention (EPR) effect [105]. Moreover, stimuli-responsive NPs based on extrinsic stimuli have reached clinical trials and received approval [106]. We believe that it is promising to explore the feasibility and application of LYTACs delivery nanosystem.
Conclusion
Nowadays, TPD has been recognized as a game-changing strategy by inducing the degradation of the 'undruggable' targets and PROTACs have broken the classical Lipinski's rule of five [107]. However, the substrates of PROTACs are limited to intracellular proteins through proteasomal clearance. LYTACs and AbTACs have emerged as potential therapeutics by taking advantage of the lysosome system to degrade disease-relevant extracellular and membrane proteins. Notably, there are also challenges in this nascent field, such as limited studies on length of linker, structure optimization, ternary complex equilibria, and pharmacokinetics. The critical next step is to develop them into mature technologies through interdisciplinary collaboration, which can provide more solutions. For example, synthetic procedure of M6Pn glycoproteins is interminable and inefficient. To increase efficiency, we can develop CI-M6PR-binding oligopeptides to replace the long M6P chain or simplify synthetic steps in chemistry, or search for other TLRs from a biological standpoint. Furthermore, the screening of LYTACs generally depends on measuring protein levels through western blot assay or mass spectroscopy, which is time-consuming. Development of computational tools that provide high-throughput methods to design and screen LYTACs may be a trend [108]. Moreover, the concept of degradation strategy offers possibilities to degrade RNA genome, further establishing more therapeutic tools at the level of genetic degradation [109,110].
In conclusion, by focusing on TPD strategies towards extracellular and membrane proteins, we hope to provide readers with a resource to help navigate in this booming field, and inspire further efforts to create new degradation modalities.
Abbreviations
TPD: targeted protein degradation; PROTAC: PROteolysis TArgeting Chimera; POI: protein of interest; LYTAC: LYsosome-TArgeting Chimaera; AbTAC: Antibody-based PROTAC; Erα: estrogen receptor alpha; AR: androgen receptor; AUTAC: autophagy-targeting chimera; ATTEC: autophagosome-tethering compounds; dTAG: degradation tag; CI-M6PR: cation-independent mannose-6-phosphate receptor; ASGPR: asialoglycoprotein receptor; ApoE4: apolipoprotein E4; EGFR: epidermal growth factor receptor; PD-L1: programmed death-ligand 1; UPS: ubiquitin-proteasome system; LTR: lysosome-targeting receptor; EE: early endosome; LE: late endosome; RE: recycling endosome; bsAbs: Bispecific antibodies; IgG: immunoglobulins G; RNF43: cell-surface transmembrane E3 ubiquitin ligase ring finger 43; ZNRF3: cell-surface transmembrane E3 ubiquitin ligase zinc and ring finger 3; VEGFR: vascular endothelial growth factor receptor; M6P: mannose-6-phosphate; GalNAc: glycoproteins bearing N-acetylgalactosamine; Gal: galactose; tri-GalNAc: triantenerrary glycoproteins bearing N-acetylgalactosamine; DBCO: dibenzocyclooctyne; BCN: cyclopropane cyclooctyne; KIHs: Knobs-into-holes; MR: mannose receptor; TAM: tumor-associated macrophage; LIMP: lysosomal integral membrane protein; ADC: Antibody-drug conjugate; ATDC: AbTAC-drug conjugate; MOA: mode of action; NPs: Nanoparticles.
Acknowledgements
Funding
This work was supported by funds from the National Natural Science Foundation of China (No.81903654), Shanghai Science and Technology Innovation Action Plan (21S11902800), Program for Professor of Special Appointment (Young Eastern Scholar) at Shanghai Institutions of Higher Learning (QD2018035), Shanghai Sailing Program (19YF1449400), State Key Laboratory of Innovative Natural Medicine and TCM Injections (No. QFSKL2020017 and QFSKL2020019), National Key Research and Development Program of China (2017YFC1700200), Graduate Innovation Training research project of the Shanghai University of Traditional Chinese Medicine (No. Y2021003).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ. et al. Identification and characterization of essential genes in the human genome. Science. 2015;350:1096-101
2. Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS. et al. Defining a cancer dependency map. Cell. 2017;170:564-76
3. Dang CV, Reddy EP, Shokat KM, Soucek L. Drugging the 'undruggable' cancer targets. Nat Rev Cancer. 2017;17:502-8
4. Ferguson FM, Gray NS. Kinase inhibitors: the road ahead. Nat Rev Drug Discov. 2018;17:353-77
5. Roskoski RJ. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol Res. 2019;144:19-50
6. Schapira M, Calabrese MF, Bullock AN, Crews CM. Targeted protein degradation: expanding the toolbox. Nat Rev Drug Discov. 2019;18:949-63
7. Fukui S, Nakai Y, Matsumoto Y, Kagebayashi Y, Samma S. Investigation of incidence and risk factors of subcutaneous granulomas induced by injection of leuprorelin acetate. Hinyokika Kiyo. 2015;61:55-9
8. Feltham R, Bettjeman B, Budhidarmo R, Mace PD, Shirley S, Condon SM. et al. Smac mimetics activate the E3 ligase activity of cIAP1 protein by promoting RING domain dimerization. J Biol Chem. 2011;286:17015-28
9. Preisler-Mashek MT, Solodin N, Stark BL, Tyriver MK, Alarid ET. Ligand-specific regulation of proteasome-mediated proteolysis of estrogen receptor-alpha. Am J Physiol Endocrinol Metab. 2002;282:E891-8
10. Jin J, Wu Y, Chen J, Shen Y, Zhang L, Zhang H. et al. The peptide PROTAC modality: a novel strategy for targeted protein ubiquitination. Theranostics. 2020;10:10141-53
11. Takahashi D, Moriyama J, Nakamura T, Miki E, Takahashi E, Sato A. et al. AUTACs: Cargo-specific degraders using selective autophagy. Mol Cell. 2019;76:797-810
12. Li Z, Zhu C, Ding Y, Fei Y, Lu B. ATTEC: a potential new approach to target proteinopathies. Autophagy. 2020;16:185-7
13. Słabicki M, Kozicka Z, Petzold G, Li Y, Manojkumar M, Bunker RD. et al. The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature. 2020;585:293-7
14. Mogaki R, Okuro K, Ueki R, Sando S, Aida T. Molecular glue that spatiotemporally turns on protein-protein interactions. J Am Chem Soc. 2019;141:8035-40
15. Nabet B, Roberts JM, Buckley DL, Paulk J, Dastjerdi S, Yang A. et al. The dTAG system for immediate and target-specific protein degradation. Nat Chem Biol. 2018;14:431-41
16. Nabet B, Ferguson FM, Seong B, Kuljanin M, Leggett AL, Mohardt ML. et al. Rapid and direct control of target protein levels with VHL-recruiting dTAG molecules. Nat Commun. 2020;11:4687
17. Clift D, So C, McEwan WA, James LC, Schuh M. Acute and rapid degradation of endogenous proteins by Trim-Away. Nat Protoc. 2018;13:2149-75
18. Clift D, McEwan WA, Labzin LI, Konieczny V, Mogessie B, James LC. et al. A method for the acute and rapid degradation of endogenous proteins. Cell. 2017;171:1692-706
19. Proof-of-concept with PROTACs in prostate cancer. Cancer Discov. 2020; 10: 1084.
20. Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A. et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419
21. Banik SM, Pedram K, Wisnovsky S, Ahn G, Riley NM, Bertozzi CR. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature. 2020;584:291-7
22. Cotton AD, Nguyen DP, Gramespacher JA, Seiple IB, Wells JA. Development of antibody-based PROTACs for the degradation of the cell-surface immune checkpoint protein PD-L1. J Am Chem Soc. 2021;143:593-8
23. Ahn G, Banik SM, Miller CL, Riley NM, Cochran JR, Bertozzi CR. LYTACs that engage the asialoglycoprotein receptor for targeted protein degradation. Nat Chem Biol. 2021 DOI: 10.1038/s41589-021-00770-1
24. Zhou Y, Teng P, Montgomery NT, Li X, Tang W. Development of triantennary N-acetylgalactosamine conjugates as degraders for extracellular proteins. ACS Cent Sci. 2021;7:499-506
25. Caianiello D, Zhang M, Ray J, Swartzel J, Branham E, Chirkin E. et al. Bifunctional small molecules that mediate the degradation of extracellular proteins. ChemRxiv. Preprint. https://doi.org/10.26434/chemrxiv.12732689.v2. 2020
26. Dale B, Cheng M, Park KS, Kaniskan HÜ, Xiong Y, Jin J. Advancing targeted protein degradation for cancer therapy. Nat Rev Cancer. 2021 DOI: 10.1038/s41568-021-00365-x
27. Moreau K, Coen M, Zhang AX, Pachl F, Castaldi MP, Dahl G. et al. Proteolysis-targeting chimeras in drug development: A safety perspective. Br J Pharmacol. 2020;177:1709-18
28. An S, Fu L. Small-molecule PROTACs: An emerging and promising approach for the development of targeted therapy drugs. Ebiomedicine. 2018;36:553-62
29. Olson CM, Jiang B, Erb MA, Liang Y, Doctor ZM, Zhang Z. et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat Chem Biol. 2018;14:163-70
30. Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K. et al. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem Biol. 2015;22:755-63
31. Neklesa T, Snyder LB, Willard RR, Vitale N, Pizzano J, Gordon DA. et al. ARV-110: An oral androgen receptor PROTAC degrader for prostate cancer. J Clin Oncol. 2019;37:259
32. Flanagan JJ, Qian Y, Gough SM, Andreoli M, Bookbinder M, Cadelina G. et al. Abstract P5-04-18: ARV-471, an oral estrogen receptor PROTAC degrader for breast cancer. Cancer Res. 2019;79:P4-5
33. Mullard A. First targeted protein degrader hits the clinic. Nat Rev Drug Discov. 2019 DOI: 10.1038/d41573-019-00043-6
34. Dai Y, Yue N, Gong J, Liu C, Li Q, Zhou J. et al. Development of cell-permeable peptide-based PROTACs targeting estrogen receptor α. Eur J Med Chem. 2020;187:111967
35. Lazo JS, Sharlow ER. Drugging undruggable molecular cancer targets. Annu Rev Pharmacol Toxicol. 2016;56:23-40
36. Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001;98:8554-9
37. Burslem GM, Crews CM. Small-molecule modulation of protein homeostasis. Chem Rev. 2017;117:11269-301
38. Pei J, Wang G, Feng L, Zhang J, Jiang T, Sun Q. et al. Targeting lysosomal degradation pathways: new strategies and techniques for drug discovery. J Med Chem. 2021;64:3493-507
39. Takahashi D, Arimoto H. Targeting selective autophagy by AUTAC degraders. Autophagy. 2020;16:765-6
40. Lamark T, Svenning S, Johansen T. Regulation of selective autophagy: the p62/SQSTM1 paradigm. Essays Biochem. 2017;61:609-24
41. Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014;16:495-501
42. Schaaf MB, Keulers TG, Vooijs MA, Rouschop KM. LC3/GABARAP family proteins: autophagy-(un)related functions. Faseb J. 2016;30:3961-78
43. Kaksonen M, Roux A. Mechanisms of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol. 2018;19:313-26
44. Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema K. et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14:1435-55
45. Wu T, Yoon H, Xiong Y, Dixon-Clarke SE, Nowak RP, Fischer ES. Targeted protein degradation as a powerful research tool in basic biology and drug target discovery. Nat Struct Mol Biol. 2020;27:605-14
46. Labrijn AF, Janmaat ML, Reichert JM, Parren P. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov. 2019;18:585-608
47. Serra S, Chetty R. Rnf43. J Clin Pathol. 2018;71:1-6
48. Zebisch M, Jones EY. ZNRF3/RNF43-A direct linkage of extracellular recognition and E3 ligase activity to modulate cell surface signalling. Prog Biophys Mol Biol. 2015;118:112-8
49. Lu M, Liu T, Jiao Q, Ji J, Tao M, Liu Y. et al. Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway. Eur J Med Chem. 2018;146:251-9
50. Burslem GM, Crews CM. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell. 2020;181:102-14
51. Bondeson DP, Mares A, Smith IE, Ko E, Campos S, Miah AH. et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol. 2015;11:611-7
52. Ding Y, Fei Y, Lu B. Emerging new concepts of degrader technologies. Trends Pharmacol Sci. 2020;41:464-74
53. Li H, Er SP, Song E. Challenges and strategies for next-generation bispecific antibody-based antitumor therapeutics. Cell Mol Immunol. 2020;17:451-61
54. Zimmermann TS, Karsten V, Chan A, Chiesa J, Boyce M, Bettencourt BR. et al. Clinical proof of concept for a novel hepatocyte-targeting GalNAc-siRNA conjugate. Mol Ther. 2017;25:71-8
55. Gary-Bobo M, Nirdé P, Jeanjean A, Morère A, Garcia M. Mannose 6-phosphate receptor targeting and its applications in human diseases. Curr Med Chem. 2007;14:2945-53
56. Ghosh P, Dahms NM, Kornfeld S. Mannose 6-phosphate receptors: new twists in the tale. Nat Rev Mol Cell Biol. 2003;4:202-12
57. Jeanjean A, Gary-Bobo M, Nirdé P, Leiris S, Garcia M, Morère A. Synthesis of new sulfonate and phosphonate derivatives for cation-independent mannose 6-phosphate receptor targeting. Bioorg Med Chem Lett. 2008;18:6240-3
58. Hébert E. Mannose-6-phosphate/insulin-like growth factor II receptor expression and tumor development. Biosci Rep. 2006;26:7-17
59. Ali L, Simon M, El CK, Aguesseau-Kondrotas J, Godefroy A, Nguyen C. et al. Topological requirements for CI-M6PR-mediated cell uptake. Bioconjug Chem. 2019;30:2533-8
60. Spiess M. The asialoglycoprotein receptor: a model for endocytic transport receptors. Biochemistry-Us. 1990;29:10009-18
61. Wu J, Nantz MH, Zern MA. Targeting hepatocytes for drug and gene delivery: emerging novel approaches and applications. Front Biosci. 2002;7:d717-25
62. Schwartz AL, Fridovich SE, Lodish HF. Kinetics of internalization and recycling of the asialoglycoprotein receptor in a hepatoma cell line. J Biol Chem. 1982;257:4230-7
63. Glazier DA, Liao J, Roberts BL, Li X, Yang K, Stevens CM. et al. Chemical synthesis and biological application of modified oligonucleotides. Bioconjug Chem. 2020;31:1213-33
64. Huang Y. Preclinical and clinical advances of GalNAc-decorated nucleic acid therapeutics. Mol Ther Nucleic Acids. 2017;6:116-32
65. Huang X, Leroux JC, Castagner B. Well-defined multivalent ligands for hepatocytes targeting via asialoglycoprotein receptor. Bioconjug Chem. 2017;28:283-95
66. Lee YC, Townsend RR, Hardy MR, Lönngren J, Arnarp J, Haraldsson M. et al. Binding of synthetic oligosaccharides to the hepatic Gal/GalNAc lectin. Dependence on fine structural features. J Biol Chem. 1983;258:199-202
67. Whitworth C, Ciulli A. New class of molecule targets proteins outside cells for degradation. Nature. 2020;584:193-4
68. Rensen PC, Sliedregt LA, Ferns M, Kieviet E, van Rossenberg SM, van Leeuwen SH. et al. Determination of the upper size limit for uptake and processing of ligands by the asialoglycoprotein receptor on hepatocytes in vitro and in vivo. J Biol Chem. 2001;276:37577-84
69. Zengerle M, Chan KH, Ciulli A. Selective small molecule induced degradation of the bet bromodomain protein BRD4. Acs Chem Biol. 2015;10:1770-7
70. Li C, Zhang N, Zhou J, Ding C, Jin Y, Cui X. et al. Peptide blocking of PD-1/PD-L1 interaction for cancer immunotherapy. Cancer Immunol Res. 2018;6:178-88
71. Jiang Y, Deng Q, Zhao H, Xie M, Chen L, Yin F. et al. Development of stabilized peptide-based PROTACs against estrogen receptor α. Acs Chem Biol. 2018;13:628-35
72. Au YZ, Wang T, Sigua LH, Qi J. Peptide-based PROTAC: The predator of pathological proteins. Cell Chem Biol. 2020;27:637-9
73. Eskandari S, Guerin T, Toth I, Stephenson RJ. Recent advances in self-assembled peptides: Implications for targeted drug delivery and vaccine engineering. Adv Drug Deliv Rev. 2017;110-111:169-87
74. Tas JM, Mesin L, Pasqual G, Targ S, Jacobsen JT, Mano YM. et al. Visualizing antibody affinity maturation in germinal centers. Science. 2016;351:1048-54
75. Pelay-Gimeno M, Glas A, Koch O, Grossmann TN. Structure-based design of inhibitors of protein-protein interactions: Mimicking peptide binding epitopes. Angew Chem Int Ed. 2015;54:8896-927
76. Zebisch M, Xu Y, Krastev C, MacDonald BT, Chen M, Gilbert RJ. et al. Structural and molecular basis of ZNRF3/RNF43 transmembrane ubiquitin ligase inhibition by the Wnt agonist R-spondin. Nat Commun. 2013;4:2787
77. Barderas R, Benito-Peña E. The 2018 Nobel Prize in Chemistry: phage display of peptides and antibodies. Anal Bioanal Chem. 2019;411:2475-9
78. Xu Y, Lee J, Tran C, Heibeck TH, Wang WD, Yang J. et al. Production of bispecific antibodies in "knobs-into-holes" using a cell-free expression system. Mabs-Austin. 2015;7:231-42
79. Ye Z, Cheng K, Guntaka RV, Mahato RI. Targeted delivery of a triplex-forming oligonucleotide to hepatic stellate cells. Biochemistry-Us. 2005;44:4466-76
80. Vaillant O, El CK, Warther D, Brevet D, Maynadier M, Bouffard E. et al. Mannose-6-phosphate receptor: a target for theranostics of prostate cancer. Angew Chem Int Ed Engl. 2015;54:5952-6
81. Prakash J, Beljaars L, Harapanahalli AK, Zeinstra-Smith M, de Jager-Krikken A, Hessing M. et al. Tumor-targeted intracellular delivery of anticancer drugs through the mannose-6-phosphate/insulin-like growth factor II receptor. Int J Cancer. 2010;126:1966-81
82. Saw PE, Song EW. Phage display screening of therapeutic peptide for cancer targeting and therapy. Protein Cell. 2019;10:787-807
83. Chang HN, Liu BY, Qi YK, Zhou Y, Chen YP, Pan KM. et al. Blocking of the PD-1/PD-L1 interaction by a D-peptide antagonist for cancer immunotherapy. Angew Chem Int Ed. 2015;54:11760-4
84. Wang H, Sun Y, Zhou X, Chen C, Jiao L, Li W. et al. CD47/SIRPα blocking peptide identification and synergistic effect with irradiation for cancer immunotherapy. J Immunother Cancer. 2020;8:e000905
85. Feng J, Zhao C, Wang L, Qu L, Zhu H, Yang Z. et al. Development of a novel albumin-based and maleimidopropionic acid-conjugated peptide with prolonged half-life and increased in vivo anti-tumor efficacy. Theranostics. 2018;8:2094-106
86. Zhang L, Riley-Gillis B, Vijay P, Shen Y. Acquired resistance to BET-PROTACs (Proteolysis-Targeting Chimeras) caused by genomic alterations in core components of E3 ligase complexes. Mol Cancer Ther. 2019;18:1302-11
87. O'Reilly MK, Tian H, Paulson JC. CD22 is a recycling receptor that can shuttle cargo between the cell surface and endosomal compartments of B cells. J Immunol. 2011;186:1554-63
88. Jaynes JM, Sable R, Ronzetti M, Bautista W, Knotts Z, Abisoye-Ogunniyan A. et al. Mannose receptor (CD206) activation in tumor-associated macrophages enhances adaptive and innate antitumor immune responses. Sci Transl Med. 2020;12:eaax6337
89. Stahl PD. The macrophage mannose receptor: current status. Am J Respir Cell Mol Biol. 1990;2:317-8
90. Coutinho MF, Prata MJ, Alves S. A shortcut to the lysosome: the mannose-6-phosphate-independent pathway. Mol Genet Metab. 2012;107:257-66
91. Lai AC, Crews CM. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov. 2017;16:101-14
92. Coll-Martínez B, Delgado A, Crosas B. The potential of Proteolytic Chimeras as pharmacological tools and therapeutic agents. Molecules. 2020;25:5956
93. Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y. et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N Engl J Med. 2020;382:41-50
94. Chatterjee N, Bivona TG. Polytherapy and targeted cancer drug resistance. Trends Cancer. 2019;5:170-82
95. Buhimschi AD, Armstrong HA, Toure M, Jaime-Figueroa S, Chen TL, Lehman AM. et al. Targeting the C481S ibrutinib-resistance mutation in Bruton's tyrosine kinase using PROTAC-mediated degradation. Biochemistry-Us. 2018;57:3564-75
96. Gao J, Wang F, Wang S, Liu L, Liu K, Ye Y. et al. Hyperthermia-triggered on-demand biomimetic nanocarriers for synergetic photothermal and chemotherapy. Adv Sci. 2020;7:1903642
97. Wang Y, Han L, Liu F, Yang F, Jiang X, Sun H. et al. Targeted degradation of anaplastic lymphoma kinase by gold nanoparticle-based multi-headed proteolysis targeting chimeras. Colloids Surf B Biointerfaces. 2020;188:110795
98. Chau CH, Steeg PS, Figg WD. Antibody-drug conjugates for cancer. Lancet. 2019;394:793-804
99. Thomas A, Teicher BA, Hassan R. Antibody-drug conjugates for cancer therapy. Lancet Oncol. 2016;17:e254-62
100. Panowski S, Bhakta S, Raab H, Polakis P, Junutula JR. Site-specific antibody drug conjugates for cancer therapy. Mabs-Austin. 2014;6:34-45
101. Fam SY, Chee CF, Yong CY, Ho KL, Mariatulqabtiah AR, Tan WS. Stealth coating of nanoparticles in drug-delivery systems. Nanomaterials. 2020;10:787
102. Minko T. Nanoformulation of BRD4-Degrading PROTAC: Improving druggability to target the 'undruggable' MYC in pancreatic cancer. Trends Pharmacol Sci. 2020;41:684-6
103. Saraswat A, Patki M, Fu Y, Barot S, Dukhande VV, Patel K. Nanoformulation of PROteolysis TArgeting Chimera targeting 'undruggable' c-Myc for the treatment of pancreatic cancer. Nanomedicine (Lond). 2020;15:1761-77
104. Cimas FJ, Niza E, Juan A, Noblejas-López M, Bravo I, Lara-Sanchez A. et al. Controlled delivery of BET-PROTACs: In Vitro evaluation of MZ1-Loaded polymeric antibody conjugated nanoparticles in breast cancer. Pharmaceutics. 2020;12:986
105. Danhier F. To exploit the tumor microenvironment: Since the EPR effect fails in the clinic, what is the future of nanomedicine? J Control Release. 2016;244:108-21
106. Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751-60
107. Chagas CM, Moss S, Alisaraie L. Drug metabolites and their effects on the development of adverse reactions: Revisiting Lipinski's Rule of Five. Int J Pharm. 2018;549:133-49
108. Douglass EJ, Miller CJ, Sparer G, Shapiro H, Spiegel DA. A comprehensive mathematical model for three-body binding equilibria. J Am Chem Soc. 2013;135:6092-9
109. Liu X, Haniff HS, Childs-Disney JL, Shuster A, Aikawa H, Adibekian A. et al. Targeted degradation of the oncogenic microRNA 17-92 cluster by structure-targeting ligands. J Am Chem Soc. 2020;142:6970-82
110. Haniff HS, Tong Y, Liu X, Chen JL, Suresh BM, Andrews RJ. et al. Targeting the SARS-CoV-2 RNA genome with small molecule binders and ribonuclease targeting chimera (RIBOTAC) degraders. ACS Cent Sci. 2020;6:1713-21
Author Biographies
 Xin Luan, Ph.D., Professor, earned his Bachelor's degree (2011) in Pharmaceutics from China Pharmaceutical University. He received his PhD degree (2016) in Pharmacology at Shanghai Jiao Tong University, under the supervision of Prof. Hongzhuan Chen and Prof. Chao Fang. Then he joined the research group of Prof. Duxin Sun at the University of Michigan as a postdoctoral fellow. In late 2018, he started his independent career at Shanghai University of Traditional Chinese Medicine. His current research focus on the discovery, modification, and targeted delivery of anti-cancer compounds from Traditional Chinese Medicine.
Xin Luan, Ph.D., Professor, earned his Bachelor's degree (2011) in Pharmaceutics from China Pharmaceutical University. He received his PhD degree (2016) in Pharmacology at Shanghai Jiao Tong University, under the supervision of Prof. Hongzhuan Chen and Prof. Chao Fang. Then he joined the research group of Prof. Duxin Sun at the University of Michigan as a postdoctoral fellow. In late 2018, he started his independent career at Shanghai University of Traditional Chinese Medicine. His current research focus on the discovery, modification, and targeted delivery of anti-cancer compounds from Traditional Chinese Medicine.
 Weidong Zhang, Ph.D., Professor, is currently a professor of the Shanghai University of Traditional Chinese Medicine and Second Military Medical University. He obtained his Bachelor's degree and Master's degree from Second Military Medical University in 1988 and 1991, respectively. He received his PhD in Natural Product Chemistry in 1998 from Shanghai Institute of Pharmaceutical Industry with Professor Hui-Ting Li. His research focuses on total synthesis, biological function, structural modification, structure-activity relationship of structurally complex and bioactive natural products.
Weidong Zhang, Ph.D., Professor, is currently a professor of the Shanghai University of Traditional Chinese Medicine and Second Military Medical University. He obtained his Bachelor's degree and Master's degree from Second Military Medical University in 1988 and 1991, respectively. He received his PhD in Natural Product Chemistry in 1998 from Shanghai Institute of Pharmaceutical Industry with Professor Hui-Ting Li. His research focuses on total synthesis, biological function, structural modification, structure-activity relationship of structurally complex and bioactive natural products.
 Ye Wu, Ph.D., obtained his Bachelor's degree (2013) and Master's degree (2017) in Medicinal Chemistry at Chengdu Medical College. He received his PhD in Shanghai University of Traditional Chinese Medicine in 2021 with the supervision of Prof. Wei-Dong Zhang and Prof. Xin Luan. In the same year, he became a Postdoctoral Research Fellow with Prof. Hong-Zhuan Chen and Prof. Xin Luan. His research focuses on the conformational constraint and targeted delivery of peptide drugs.
Ye Wu, Ph.D., obtained his Bachelor's degree (2013) and Master's degree (2017) in Medicinal Chemistry at Chengdu Medical College. He received his PhD in Shanghai University of Traditional Chinese Medicine in 2021 with the supervision of Prof. Wei-Dong Zhang and Prof. Xin Luan. In the same year, he became a Postdoctoral Research Fellow with Prof. Hong-Zhuan Chen and Prof. Xin Luan. His research focuses on the conformational constraint and targeted delivery of peptide drugs.
 Jiayi Lin obtained her Bachelor's degree from the School of Chinese Medicine, Southern Medical University in 2020. She is currently studying for a master's degree at Shanghai University of Traditional Chinese Medicine, under the supervision of Prof. Xin Luan. Her research is centered on development of LYTAC and related oncotherapy.
Jiayi Lin obtained her Bachelor's degree from the School of Chinese Medicine, Southern Medical University in 2020. She is currently studying for a master's degree at Shanghai University of Traditional Chinese Medicine, under the supervision of Prof. Xin Luan. Her research is centered on development of LYTAC and related oncotherapy.
 Jinmei Jin obtained her Master's degree (2019) in institute of Chinese Materia Medica at Shanghai University of Traditional Chinese Medicine. She is currently a Ph.D. student under the supervision of Prof. Hong-Zhuan Chen. Her current research is centered on the synthesis of peptide PROTAC and related oncotherapy.
Jinmei Jin obtained her Master's degree (2019) in institute of Chinese Materia Medica at Shanghai University of Traditional Chinese Medicine. She is currently a Ph.D. student under the supervision of Prof. Hong-Zhuan Chen. Her current research is centered on the synthesis of peptide PROTAC and related oncotherapy.
 Yiwen Shen received her Bachelor's degree in Zhejiang Agriculture and Forestry University in 2019 and now studies in Shanghai University of Traditional Chinese Medicine under the supervision of Prof. Wei-Dong Zhang. Her current research is focused on oncology drug discovery.
Yiwen Shen received her Bachelor's degree in Zhejiang Agriculture and Forestry University in 2019 and now studies in Shanghai University of Traditional Chinese Medicine under the supervision of Prof. Wei-Dong Zhang. Her current research is focused on oncology drug discovery.
![]() Corresponding authors: Dr. Ye Wu, Institute of Interdisciplinary Integrative Medicine Research, Shanghai University of Traditional Chinese Medicine, Shanghai, 201203, China; orcid.org/0000-0003-4974-0774; Email: wuyeedu.cn. Prof. Weidong Zhang, Institute of Interdisciplinary Integrative Medicine Research, Shanghai University of Traditional Chinese Medicine, Shanghai, 201203, China; School of Pharmacy, Second Military Medical University, Shanghai, 201203, China; orcid.org/0000-0002-7384-2490; Email: dzhangycom. Prof. Xin Luan, Institute of Interdisciplinary Integrative Medicine Research, Shanghai University of Traditional Chinese Medicine, Shanghai, 201203, China; orcid.org/0000-0003-3674-256X; Email: luanxinedu.cn.
Corresponding authors: Dr. Ye Wu, Institute of Interdisciplinary Integrative Medicine Research, Shanghai University of Traditional Chinese Medicine, Shanghai, 201203, China; orcid.org/0000-0003-4974-0774; Email: wuyeedu.cn. Prof. Weidong Zhang, Institute of Interdisciplinary Integrative Medicine Research, Shanghai University of Traditional Chinese Medicine, Shanghai, 201203, China; School of Pharmacy, Second Military Medical University, Shanghai, 201203, China; orcid.org/0000-0002-7384-2490; Email: dzhangycom. Prof. Xin Luan, Institute of Interdisciplinary Integrative Medicine Research, Shanghai University of Traditional Chinese Medicine, Shanghai, 201203, China; orcid.org/0000-0003-3674-256X; Email: luanxinedu.cn.