Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(17):8218-8233. doi:10.7150/thno.59280 This issue Cite
Research Paper
GATA3 functions downstream of BRCA1 to suppress EMT in breast cancer
Feng Bai1,2,3, Li-Han Zhang3,4,5, Xiong Liu1,9, Chuying Wang3,6, Chenglong Zheng1,9, Jianping Sun7, Min Li4, Wei-Guo Zhu8, Xin-Hai Pei1,3,9 ![]()
1. Guangdong Provincial Key Laboratory of Regional Immunity and Diseases, International Cancer Center, Marshall Laboratory of Biomedical Engineering, Shenzhen University Health Science Center, Shenzhen 518060, China.
2. Department of Pathology, Shenzhen University Health Science Center, Shenzhen 518060, China.
3. Dewitt Daughtry Family Department of Surgery, University of Miami, Miami, FL 33136, USA.
4. School of Basic Medical Sciences, Lanzhou University, Lanzhou, Gansu, 730000, China.
5. The Affiliated Cancer Hospital of Zhengzhou University, Zhengzhou, Henan 450008, China.
6. The Second Affiliated Hospital of Xi'an Jiaotong University, Xi'an, Shaanxi, 710061, China.
7. Department of Mathematics and Statistics, University of North Carolina at Greensboro, Greensboro, NC 27402, USA
8. Department of Biochemistry and Molecular Biology, International Cancer Center, Shenzhen University Health Science Center, Shenzhen 518060, China.
9. Department of Anatomy and Histology, Shenzhen University Health Science Center, Shenzhen 518060, China.
Received 2021-2-10; Accepted 2021-7-5; Published 2021-7-13
Abstract

Purpose: Functional loss of BRCA1 is associated with poorly differentiated and metastatic breast cancers that are enriched with cancer stem cells (CSCs). CSCs can be generated from carcinoma cells through an epithelial-mesenchymal transition (EMT) program. We and others have previously demonstrated that BRCA1 suppresses EMT and regulates the expression of multiple EMT-related transcription factors. However, the downstream mediators of BRCA1 function in EMT suppression remain elusive.
Methods: Depletion of BRCA1 or GATA3 activates p18INK4C, a cell cycle inhibitor which inhibits mammary epithelial cell proliferation. We have therefore created genetically engineered mice with Brca1 or Gata3 loss in addition to deletion of p18INK4C, to rescue proliferative defects caused by deficiency of Brca1 or Gata3. By using these mutant mice along with human BRCA1 deficient as well as proficient breast cancer tissues and cells, we investigated and compared the role of Brca1 and Gata3 loss in the activation of EMT in breast cancers.
Results: We discovered that BRCA1 and GATA3 expressions were positively correlated in human breast cancer. Depletion of BRCA1 stimulated methylation of GATA3 promoter thereby repressing GATA3 transcription. We developed Brca1 and Gata3 deficient mouse system. We found that Gata3 deficiency in mice induced poorly-differentiated mammary tumors with the activation of EMT and promoted tumor initiating and metastatic potential. Gata3 deficient mammary tumors phenocopied Brca1 deficient tumors in the induction of EMT under the same genetic background. Reconstitution of Gata3 in Brca1-deficient tumor cells activated mesenchymal-epithelial transition, suppressing tumor initiation and metastasis.
Conclusions: Our finding, for the first time, demonstrates that GATA3 functions downstream of BRCA1 to suppress EMT in controlling mammary tumorigenesis and metastasis.
Keywords: BRCA1, GATA3, EMT, tumorigenesis, metastasis
Introduction
Breast cancer is mainly divided into estrogen receptor (ER) positive luminal and ER-negative basal-like tumors [1]. Basal-like breast cancers (BLBCs) are poorly differentiated and contain a number of distinct cell types which include cells that express luminal, basal, and mesenchymal biomarkers [2, 3]. Consistently, we and others have demonstrated that at least some of the BLBCs originate from luminal epithelial cells [4-7]. BLBCs are the most lethal, partly due to their enrichment of cancer stem cells (CSCs) that are thought to drive clinical relapse and metastasis [8]. The CSCs can be generated from luminal tumor cells by an epithelial-mesenchymal transition (EMT) program, a process in which epithelial cells lose many of their epithelial characteristics and acquire mesenchymal features [4-9]. The molecular mechanisms controlling EMT in breast cancers are poorly understood.
Mammary epithelia are mainly composed of luminal and basal cells that are maintained by luminal and basal progenitors, respectively [10]. The maintenance of luminal cell fate is orchestrated by networks of transcription factors (TFs), including BRCA1 and GATA3 [11]. Interestingly, functional loss of BRCA1 by germline or somatic mutation, or promoter methylation is associated with more than half of BLBCs [12-14]. Loss of GATA3 expression is also associated with BLBCs and tumor metastasis [13, 15-19]. Loss of function of BRCA1 in breast cancer cells enhances the expression of several EMT inducing transcription factors (EMT-TFs) including SLUG [5], TWIST [20], FOXC1/C2 [21], and inversely reduces the expression of a few of the EMT suppressing TFs, such as FOXA1 [22, 23], FOXO3 [24, 25]. However, no downstream mediator of BRCA1 in regulation of EMT in breast cancers has been identified. In cell line models, GATA3 functions as a tumor suppressor by inducing epithelial fates while suppressing mesenchymal fates [26-28]. We previously demonstrated that BRCA1 suppresses EMT in breast cancer development [20]. However, whether and how BRCA1 interacts with GATA3 to control EMT in mammary tumorigenesis remains elusive.
Due to growth and differentiation defects induced by Brca1 and Gata3 deficiency [15, 19, 29-32], mice deficient for either Brca1 or Gata3 rarely develop tumors, making it difficult to identify the role of Brca1 and Gata3 loss in regulating EMT in tumor development and metastasis. Targeted deletion of GATA3 in tumor cells leads to apoptosis preventing the analysis of the functional loss of GATA3 in tumor cell differentiation [33]. Most, if not all, genetic studies have utilized co-deletion of one of the genes in the p53 pathway to overcome the growth defects induced by Brca1 deficiency in mice [5, 6, 29-31, 34]. However, deletion of p53 induces EMT and BLBCs [35-38], masking the contribution of Brca1 depletion alone in these processes. Hence it is imperative that the role of Brca1 in controlling EMT and tumorigenesis be determined under a genetically intact p53 background.
The RB protein which is phosphorylated and inactivated by CDK4 and CDK6 (CDK4/6), controls the G1-to-S transition of the cell cycle. CDK4/6 are inhibited by inhibitors of CDK4/6 (INK4) such as p16INK4A (p16) and p18INK4C (p18). Inactivation of the INK4-CDK4/6-RB pathway (i.e. loss of INK4 or RB and amplification of cyclin D or CDK4/6) is a common event in breast cancers [13, 39]. Most BRCA1-deficient breast cancers carry a dysfunctional INK4-CDK4/6-RB pathway [13, 40, 41], All widely used BRCA1 mutant breast cancer cell lines have deletions in either RB or p16 [42, 43]. We previously demonstrated that depletion of Brca1 or Gata3 activates p16/p18-CDK4/6-RB pathway leading to cell cycle arrest [7, 15, 44, 45] which was later confirmed by two independent groups [32, 46]. Since inactivation of INK4-CDK4/6-RB pathway in mice induces mammary luminal type tumors with little effect on EMT activation [7, 15, 20, 47], p18;Brca1 and p18;Gata3 mutant mice provide us a unique opportunity to investigate and compare the role of Brca1 and Gata3 loss in activation of EMT in breast cancers under a genetically intact p53 background.
In this report, we used p18, Brca1, and Gata3 singly and doubly deficient mouse models as well as human BRCA1 proficient and deficient cancer cells to determine the mechanisms underlying the role of BRCA1 in the regulation of GATA3 in mammary tumorigenesis and metastasis. We demonstrated that the depletion of BRCA1 stimulated methylation of GATA3 promoter thereby repressing GATA3 transcription. We found that Gata3 deficient mammary tumors phenocopied Brca1 deficient tumors in the induction of EMT and promotion of tumorigenesis and progression. We discovered that GATA3 functions downstream of BRCA1 to suppress EMT in controlling mammary tumor initiation and metastasis.
Methods
Mice, histopathology, and immunostaining
The generation of p18-/-, p18+/-, p18-/-;Brca1MGKO (p18-/-;Brca1f/f;MMTV-Cre or p18-/-;Brca1f/-;MMTV-Cre), p18-/-;Brca1+/-, p18+/-;Brca1+/-, Gata3f/f, Gata3+/-, p18+/-;Gata3+/-, and p18-/-;Gata3+/- mice has been previously described [7, 15, 20, 48]. The Institutional Animal Care and Use Committee at the University of Miami and Shenzhen University approved all animal procedures. Histopathology and immunohistochemistry (IHC) were performed as previously described [7, 15, 20]. The primary antibodies used were: BRCA1, E-cadherin (E-Cad), Vimentin (Vim), FRA1 (Cell Signaling), Fibronectin (Fn), TWIST, DNA Methyltransferase 1 (DNMT1) (Abcam), CD29, GATA3 (Santa Cruz). Immunocomplexes were detected using the Vectastain ABC DAB kit according to the manufacturer's instructions (Vector Laboratories). The positive results of IHC were quantified by H-score, as previously described [49, 50].
Mammary tumor cell preparation, transplantation, and analysis of metastasis
Mammary tumors were dissected from female mice and tumor cell suspensions were prepared as previously described [7, 15, 20]. For the transplantation of primary p18-/- and p18-/-;Gata3+/- mammary tumor cells, cells derived from mutant mice were inoculated into the left inguinal mammary fat pads (MFPs) of 6-week-old female NSG mice (Jackson Laboratory) with subcutaneous implantation of estrogen pellets. Four weeks after transplantation, animals were euthanized for analysis of histopathology and immunohistochemistry. For transplantation of p18-/-;Brca1MGKO tumor cells, cells that were infected with pBabe-puro-empty or pBabe-puro-Gata3 and selected in puromycin were then inoculated into the left and/or right inguinal MFPs of 6-week-old female NSG mice, respectively. Four weeks after transplantation, animals were euthanized and mammary tumors were dissected for histopathological, immunohistochemical, and biochemical analyses. For analysis of lung metastasis from mammary tumors, p18-/-; Brca1MGKO tumor cells that were infected with pBabe-puro-empty or pBabe-puro-Gata3 and selected in puromycin were inoculated into the MFPs of NSG mice. When newly generated tumors either reached the IACUC designated endpoint size (1.3 cm3; in 4-10 weeks) or the mice became moribund, the lungs were examined for detection of metastasis. For quantification of the number of metastatic nodules in the lungs, fixed lung tissues of all five lobes were sagittally sectioned at 200-μm intervals. At least three sections for each lobe were prepared and stained with H.E. The metastatic nodules in each lobe of lung tissue were confirmed by H.E. staining, counted under a microscope, and averaged. The number of nodules in all lobes was then calculated.
Cell culture, treatment, transfection, and viral infection
MCF-7, T47D, MDA-MB-231, BT20, SUM149, and HCC1937 cells were cultured per ATCC recommendations. Primary murine mammary tumor cells were cultured in 10% FBS (Gibco). For drug treatment, cells were cultured in the presence of 5-aza-2'-deoxycytidine (DAC) or DMSO for 72 hours, and then were lysed for further analysis. For ectopic expression of BRCA1, HCC1937 and SUM149 cells were transfected with pBabe-empty or pBabe-HA-BRCA1, and for knockdown (KD) of BRCA1, MCF7 cells were infected with pGIPZ-empty or pGIPZ-shBRCA1, as previously described [20]. For ectopic expression of Gata3, murine mammary tumor cells were infected with retroviruses expressing wild-type (WT) GATA3, pBabe-GATA3, or control pBabe vector, pBabe-empty, as previously described [15].
Western blot, qRT-PCR, and flow cytometry
Western blot analysis was carried out as previously described [7, 15, 20, 51]. Primary antibodies used are as follows: BRCA1, HSP90, GAPDH (Ambion), E-Cad, Vim, GATA3 (D13C9), (Cell Signaling), TWIST (Abcam). For qRT-PCR, total RNA was extracted using the RNeasy kit (Qiagen) according to the manufacturer's protocol and cDNA was generated using the Omniscript RT Kit (Qiagen). qRT-PCR was performed as previously reported [7]. For surface marker analysis, tumor cells were isolated and stained. After exclusion of lymphocytes, dead cells, and Lin+ cells, the expression profile for CD24 and CD29 was determined by flow cytometry as previously described [15, 20].
Methylation analysis
For methylation specific PCR (MS-PCR) analysis, genomic DNA extracted from cells were treated with bisulfite and analyzed for GATA3 promoter methylation with specific primers amplifying the unmethylated or methylated allele as described [44, 52]. Primer sequences were as below: FW: 5'-ACGATTTTCGATTTTTCGACGGTAGGAGTTTTTC-3' and RV: 5'-GACTATACTCGCGCCCTCTCGCCGA-3' for methylated GATA3. FW: 5'-ATGATTTTTGATTTTTTGATGGTAGGAGTTT-3' and RV: 5'-TCAACTATACTCACACCCTCTCA-3' for unmethylated GATA3.
Human tumor samples and meta-analysis of data sets
Formalin fixed paraffin-embedded (FFPE) human breast cancer samples lacking patient-identifying information were obtained from the Tissue Bank Core Facility at the University of Miami as previously reported [20]. Regions from tumor samples were microdissected and only samples with a consistent tumor cell content >75% of tissues were used for RNA extraction. The expression of genes was determined by qRT-PCR as previously reported [20]. In addition, FFPE samples derived from BRCA1 WT and BRCA1 mutant patient-derived xenograft (PDX) tumors were prepared as previously reported [50].
For analysis of the correlation of GATA3 mRNA with BRCA1 mutation, the TCGA dataset with 974 cancer samples including 14 samples harboring BRCA1 mutations was analyzed [13]. For analysis of the correlation of GATA3 methylation with BRCA1 mutation, the TCGA dataset with 664 cancer samples including 13 samples harboring BRCA1 mutations was analyzed.
Statistical analysis
All data are presented as the mean ± SD for at least three repeated individual experiments for each group. Quantitative results were analyzed by two-tailed Student's t-test. P < 0.05 was considered statistically significant.
Results
Loss of BRCA1 abrogates GATA3 expression and activates EMT
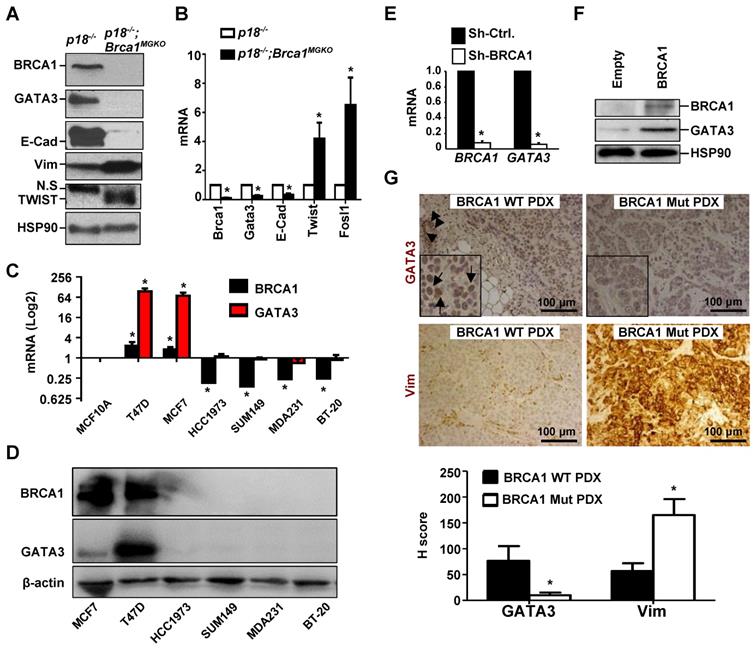
We previously found that heterozygous germline deletion of Brca1 (Brca1+/-) or mammary epithelia specific deletion of Brca1 (Brca1MGKO) in p18-deficient mice eliminated the expression of Gata3 in tumor-free mammary tissue and mammary epithelial cells (MECs) [7, 20]. We then followed mammary tumors and found that p18-/- tumors were well-differentiated tumors most of which were positive for GATA3 and E-Cad, a downstream target of GATA3, but p18-/- tumors were negative or weakly expressing Vim and EMT-TFs which include TWIST and FRA1. Whereas, p18-/-;Brca1+/- and p18-/-;Brca1MGKO tumors were poorly differentiated tumors most of which were negative for GATA3 and E-Cad, but strongly positive for Vim and EMT-TFs (Figure 1A, Table 1, and in reference [15, 20]). We isolated cells from primary mammary tumors and generated tumor cell lines, and consistently found that the expression of Gata3 and E-Cad was abrogated and the expression of Twist and Fosl1 (encoding Fra1) was enhanced in p18-/-;Brca1MGKO (Brca1 deficient) cells relative to the expression in p18-/- (Brca1 proficient) counterparts (Figure 1B, Figure S1A).
BRCA1 positively regulates GATA3 expression in breast cancer cells and tissues. (A, B) Mammary tumor tissues (A) or primary tumor cells (B) from p18-/- and p18-/-;Brca1MGKO mice were analyzed by western blot (A) or qRT-PCR (B). N.S., non-specific band. Data in (B) represent the mean ± SD. from triplicates of two independent primary cell lines of each genotype. The asterisk (*) in (B) denotes a statistical significance from p18-/- and p18-/-;Brca1MGKO samples determined by the T-test. (C, D) The mRNA and protein levels of BRCA1 and GATA3 in human breast cell lines were detected by qRT-PCR (C) and Western blot (D). BRCA1 and GATA3 mRNA levels in breast cancer cell lines were normalized to that of MCF-10A cell line. Data in (C) represent the mean ± SD from triplicates of each of the two independent experiments. The asterisk (*) denotes a statistical significance from MCF-10 and breast cancer cell lines determined by the T-test. (E) T47D cells were infected with either pGIPZ-empty (sh-Ctrl) or pGIPZ-sh-BRCA1 (sh-BRCA1). Cells stably expressing sh-Ctrl or sh-BRCA1 were analyzed by qRT-PCR. Data represent the mean ± SD from triplicate of each of the two independent experiments. The asterisk (*) denotes a statistical significance from sh-Ctrl and sh-BRCA1 samples determined by the T-test. (F) HCC1937 cells were transfected with pBabe-empty (Empty) or pBabe-HA-BRCA1 (BRCA1). Expression of genes indicated were determined by western blot 48 hours after transfection. (G) PDX tumors generated by BRCA1 WT and BRCA1 mutant (Mut) breast cancers were stained with antibodies against GATA3 and Vim. GATA3 positive tumor cells (arrows in inset) and luminal epithelial cells (arrowheads) in mouse endogenous mammary glands are indicated. The H-scores for GATA3 and Vim in IHC were calculated. The results represent the mean ± SD of four individual tumors per group. The asterisk (*) denotes a statistical significance from BRCA1 WT PDX and BRCA1 Mut PDX samples determined by the T-test.
Spontaneous mammary tumor development in Brca1 mutant mice.
| Genotype a | ||||
|---|---|---|---|---|
| Tumor | Wt | p18mt b | Brca1+/- | p18mt;Brca1+/- c |
| Mammary Tumor | 1/10 | 23/34 (68%) | 1/11 | 22/31 (71%) f |
| Metastasis d | 2/23 (9%) | 0/1 | 8/22 (36%) g | |
| EMT+ tumor No.e | 5/23 (22%) | 1/1 | 16/22 (73%) h | |
a All mice were in Balb/c background and were at 8-22 months of age.
b This group contains eleven p18+/- and twenty three p18-/- mice.
c This group contains ten p18+/-;Brca1+/- and twenty one p18-/-;Brca1+/- mice.
d Mammary tumors metastasized mostly to the lung except one to a blood vessel, and one to liver.
e At least two EMT markers (decreased E-Cad, increased Vim, Fn1, SMA or CD29) or two EMT-TFs, which include TWIST, SLUG, SNAIL, FRA1, FOXC1, and FOXC2, were detected in > 2% tumor cells by IHC, as we previously reported (Bai, Cancer Res., 2014).
f No significance from p18mt;Brca1+/- and p18mt tumors by a two-tailed Fisher's exact test (p = 0.7948); but p = 0.0008 from p18mt;Brca1+/- and Brca1+/- tumors by a two-tailed Fisher's exact test.
g A significance from p18mt;Brca1+/- and p18mt tumors by a two-tailed Fisher's exact test (p = 0.0351).
h A significance from p18mt;Brca1+/- and p18mt tumors by a two-tailed Fisher's exact test (p = 0.0009).
To consolidate the regulation of Gata3 by Brca1 in the control of EMT, we examined the expression of these genes in mouse embryos in which EMT plays a critical role during developmental stage [53]. We found that the expression of Gata3 along with E-Cad was significantly decreased in p18-/-; Brca1MGKO embryos at both E9.5 and E12.5 of age relative to their p18-/- counterparts (Figure S1B-C). We then knocked down Brca1 in murine mammary epithelial cell line, HC11, and found that depletion of Brca1 reduced the expression of Gata3 and E-cad, but enhanced that of Vim (Figure S1D). Together, these data indicate deletion of Brca1 in mice abrogates Gata3 expression and activates EMT.
We then determined the expression of GATA3 in human breast cancer cell lines and found that GATA3 mRNA and protein levels were high in BRCA1 proficient cells (T47D and MCF7) and low or undetectable in BRCA1 deficient (BT20, MDA-MB231, SUM149, and HCC1937) cells (Figure 1C-D). Knockdown of BRCA1 in T47D and MCF7 cells significantly reduced GATA3 expression (Figure 1E and Figure 3F-G). Consistently, reconstitution of WT BRCA1 in BRCA1 mutant HCC1937 and SUM149 cells drastically induced GATA3 expression (Figure 1F, Figure S2). We then performed IHC analysis and found that GATA3 expression was barely detectable in BRCA1 mutant PDX tumors, but readily detected in BRCA1 WT PDX tumors, whereas Vim was highly expressed in BRCA1 mutant PDX tumors (Figure 1G). When the number and intensity of positive cells were quantified, we found that the H scores for GATA3 in BRCA1 mutant tumor cells were significantly less than the H scores in BRCA1 WT counterparts, whereas, the H scores for Vim in BRCA1 mutant tumor cells were significantly more than the H scores in BRCA1 WT counterparts (Figure 1G, bottom panel). These results confirm that BRCA1 is required to maintain GATA3 expression in human breast cancer cells.
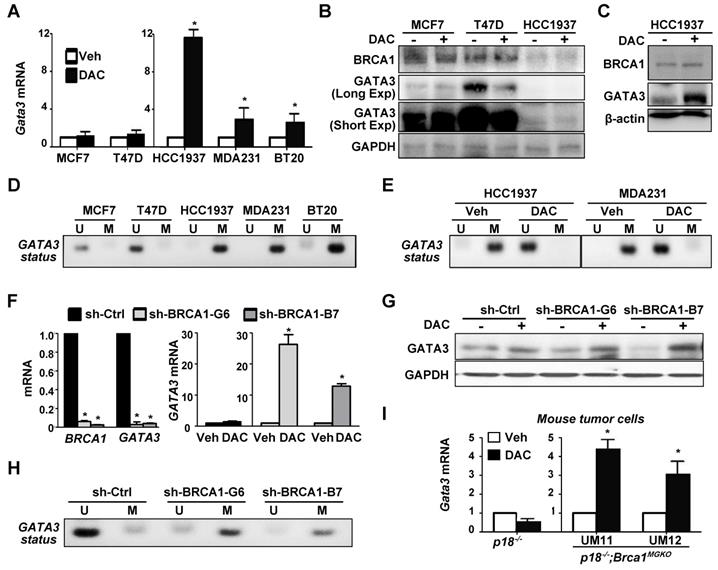
GATA3 gene is hypermethylated in BRCA1-deficient cells, and depletion of BRCA1 in BRCA1 proficient cells stimulates methylation of GATA3 gene. (A, B, C) Human breast cancer cells were treated with either DMSO (Veh) or DAC (5 µM) for 72 hours, and then analyzed by qRT-PCR (A) and western blot (B, C). Triple the amount of protein lysates from HCC1937 in (B) were analyzed in (C). MDA231, MDA-MB-231. Note the drastic increase of GATA3 mRNA and protein in DAC-treated HCC1937 cells. (D, E) MS-PCR analysis of GATA3 promoter methylation for a panel of cell lines (D), as well as HCC1937 and MDA231 cells treated with Veh or DAC for 72 hours (E). U, unmethylated; M, methylated. (F, G, H) MCF7 cells infected with either pGIPZ-empty (sh-Ctrl) or pGIPZ-shBRCA1 targeting different sequences of human BRCA1 (sh-BRCA1-G6, and sh-BRCA1-B7) were treated with or without DAC for 72 hours and then analyzed by qRT-PCR (F), western blot (G), or MS-PCR analysis of GATA3 promoter methylation (H). (I) p18-/- and p18-/-;Brca1MGKO mammary tumor cells were treated with either Veh or DAC (5 µM) for 72 hours, and then analyzed by qRT-PCR. (UM11) and (UM12) represent two independent primary cell lines derived from two individual mice (p18-/-;Brca1MGKO). Data in (A), (F), and (I) represent the mean ± SD from triplicates of each of the two independent experiments. The asterisk (*) denotes a statistical significance from DAC and Veh treated samples, or from sh-Ctrl and sh-BRCA1-G6 or sh-BRCA1-B7 samples determined via student T-test.
Expression of BRCA1 is positively correlated with that of GATA3 in human breast cancers
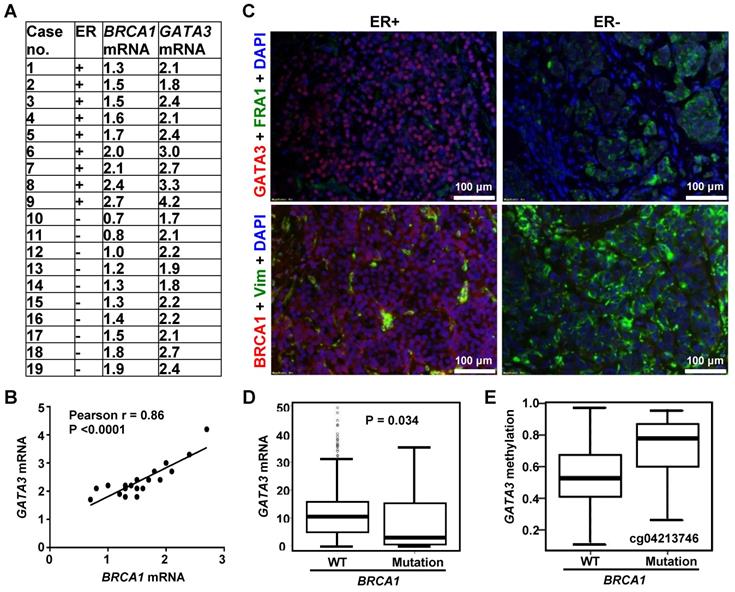
Prompted by the findings derived from mouse models, we then employed human breast cancer samples to determine the correlation of BRCA1 and GATA3. By using our previously published resource of 43 invasive human breast cancers and the RNA prepared from microdissected FFPE sections of tumors [20], we assessed BRCA1 and GATA3 mRNA levels in 9 ER-positive and 10 ER-negative samples. BRCA1 and GATA3 mRNA levels were significantly higher in ER-positive tumors when compared to ER-negative tumors (2.67 ± 0.74 vs. 2.13 ± 0.29, p = 0.049 for GATA3; 1.87 ± 0.47 vs. 1.29 ± 0.39, p = 0.009 for BRCA1). Importantly, across both ER-negative and ER-positive tumors, we observed a significant relationship between GATA3 mRNA and BRCA1 mRNA levels (Figure 2A-B). We then performed immunostaining for these human breast cancer samples and found that in the ER-positive group, BRCA1-positive tumors whose BRCA1 mRNA levels were equal to or higher than 1.5 expressed moderate to strong GATA3 protein. Conversely the human breast samples in ER-negative group, BRCA1-negative tumors whose BRCA1 mRNA levels were less than 1.5 expressed weak or undetectable GATA3 protein (Figure 2C, Figure S3, and data not shown). Together, these clinical findings are consistent with the results from our mice cohorts; thereby suggesting an opportunity to use murine systems to further explore how BRCA1 interacts with GATA3 to control EMT in breast cancers.
Correlation analysis of BRCA1 with GATA3 in human breast cancers. (A) Summary of expression of BRCA1 and GATA3 by qRT-PCR. The levels of BRCA1 and GATA3 mRNA are expressed relative to the corresponding values for T47D luminal tumor cell, as we previously reported [20]. (B) Correlation analysis of BRCA1 and GATA3 mRNA levels for breast cancer sample. (C) Representative immunostaining analysis for human breast cancer samples. Case# 8 (ER+) and case#10 (ER-) in (A) were selected for analysis. (D, E) Correlation analysis of GATA3 mRNA (D) and GATA3 promoter methylation (E) levels between breast cancers with BRCA1 WT and mutations in the TCGA dataset. Note, methylation levels in all 7 CpGs tested in GATA3 promoter are higher in BRCA1 mutants than in WTs. Representative methylation level in a CpG is shown.
BRCA1 mutation is associated with GATA3 promoter hypermethylation and reduced GATA3 expression in human breast cancers
We performed data mining in the TCGA breast cancer dataset. A one-sided Wilcoxon rank sum test was conducted to test the null hypothesis (GATA3 mRNA level is greater in BRCA1 mutant tumors than in WT tumors) versus the alternative hypothesis (GATA3 mRNA level is less in BRCA1 mutant tumors than in WT tumors). The resulted p-value (0.034 < 0.05) suggested that we should reject the null hypotheses and conclude that GATA3 mRNA level is significantly less in BRCA1 mutant tumors than in BRCA1 WT tumors (Figure 2D). Notably, median methylation levels in all 7 CpGs tested with the GATA3 promoter are higher in BRCA1 mutants than in WTs (Figure 2E, Figure S4). For each CpG site, Wilcoxon rank sum test was conducted to test the difference of methylation levels between BRCA1 mutants and WTs. The resulted p-values were then adjusted to correct for multiple testing by controlling overall false discover rate at 0.05. However, no CpGs were significant after adjustment, likely due to the limited number of BRCA1 mutant samples (13 only available). These data indicate that BRCA1 mutation is associated with GATA3 promoter hypermethylation and reduced GATA3 expression in human breast cancer samples.
Depletion of BRCA1 enhances methylation of GATA3 promoter
Prompted by the finding that BRCA1 mutation is associated with enhanced methylation of the GATA3 promoter and reduced expression of GATA3, we hypothesized that loss of function of BRCA1 stimulates methylation of GATA3 promoter thereby suppressing its transcription. To test this hypothesis, we treated breast cancer cells with 5-aza-2'-deoxycytidine (DAC), a DNMT inhibitor, and found that GATA3 mRNA levels in all BRCA1-deficient cells, HCC1937, MDA-MB-231, and BT20, were enhanced more than 2.5 fold, whereas GATA3 mRNA levels in BRCA1-proficient cells, MCF7 and T47D, were enhanced less than 1.5 fold (Figure 3A). Consistently, western blot analysis revealed that DAC treatment did not increase GATA3 protein levels in MCF7 and T47D cells, whereas GATA3 protein levels in BRCA1 mutant HCC1937 cells were clearly enhanced, though the basal levels of GATA3 were low (Figure 3B-C). Interestingly, BRCA1 levels showed insignificant change in response to DAC treatment. We performed MS-PCR and confirmed that GATA3 promoter in HCC1937, MDA-MB-231, and BT20 cells was hypermethylated, whereas GATA3 promoter in MCF7 and T47D cells was unmethylated (Figure 3D). We also treated HCC1937 and MDA-MB-231 cells with DMSO and DAC and found that DAC, not DMSO, converted hypermethylated GATA3 promoter into its un-methylated form (Figure 3E). These results not only confirm the specificity of the MS-PCR for detection of hypermethylated GATA3 promoter, but also demonstrate the activity of DAC in the de-methylation of GATA3 promoter. Further, these data indicate that BRCA1 deficiency is indeed associated with methylation of GATA3 promoter. We knocked down BRCA1 in MCF7 cells using BRCA1 shRNA to target two different sequences. We found that treatment of BRCA1-KD, but not control-KD, MCF7 cells with DAC led to drastic increase of GATA3 mRNA and protein levels relative to Veh treatment (Figure 3F-G). MS-PCR analysis revealed that the GATA3 promoter in BRCA1-KD MCF7 cells was hypermethylated relative to the GATA3 Promotor in control MCF7 cells (Figure 3H). These results indicate that depletion of BRCA1 in breast cancer cell lines enhances methylation of the GATA3 promoter thereby repressing its transcription.
To genetically confirm the role of Brca1 in regulating the methylation of Gata3 gene we treated p18-/- (Brca1 proficient) and p18-/-;Brca1MGKO (Brca1 deficient) tumor cells with DAC. We found that DAC drastically enhanced Gata3 expression in p18-/-;Brca1MGKO, but not in p18-/-, tumor cells (Figure 3I). These results suggest that Gata3 promoter is hypermethylated in Brca1 deficient cells relative to that in Brca1 proficient cells, which confirms our findings derived from human breast cancer samples and cells. Since loss of p18 is required for Brca1 deficient mammary tumorigenesis [7, 20] and inactivation of the INK4-CDK4/6-RB pathway stimulates transcription of DNMT1 in a few cell lines [54], we performed IHC and consistently detected increased DNMT1 expression in p18-null MECs and stromal cells relative to DNMT1 expression in their WT counterparts (Figure S5). These data suggest that p18 loss stimulates DNMT1 expression in mammary gland, which promotes methylation of the Gata3 promoter when Brca1 is deficient. Together with the finding that mammary tumors developed in p18 deficient mice are Brca1- and Gata3-positive well-differentiated tumors (Figure 1, and Reference [7, 15, 20]), these results indicate that Brca1 protects the Gata3 promoter from methylation to maintain Gata3 transcription in the suppression of mammary tumors.
Heterozygous germline deletion of Gata3 in mice leads to poorly-differentiated mammary tumors with activation of EMT
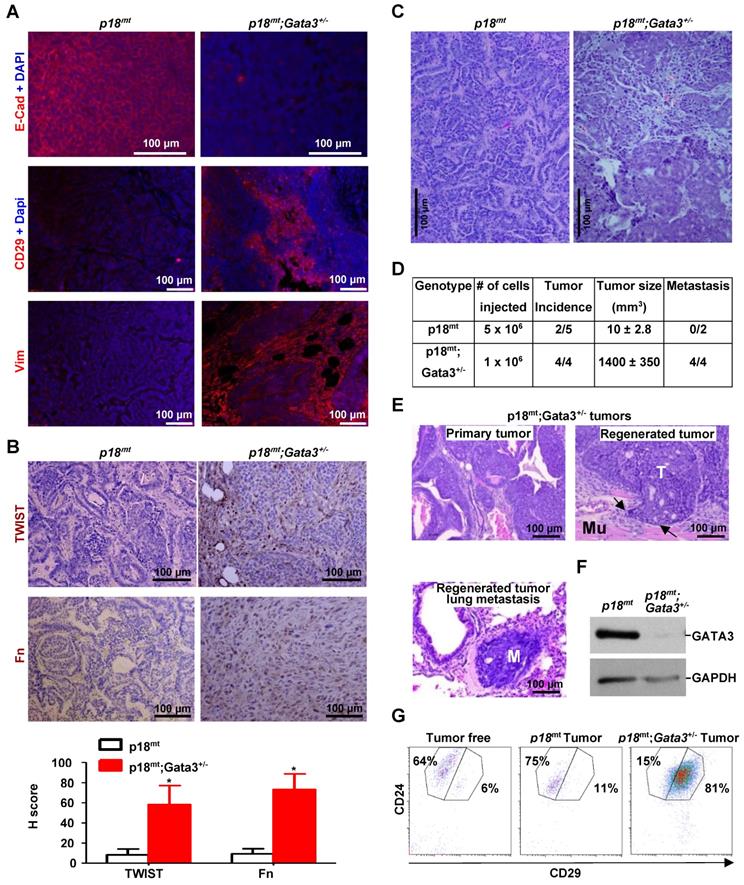
Given the direct regulation of GATA3 by BRCA1 shown above, and EMT-suppressive function of ectopic GATA3 in cell lines [26-28], we hypothesized that depletion of Gata3 in mice resulted in poorly differentiated mammary tumors with the induction of EMT which phenocopied Brca1-deficient tumors. To test this hypothesis, we generated p18-/-, p18+/-, Gata3+/-, p18-/-;Gata3+/-, and p18+/-;Gata3+/- mice in Balb/c-B6 mixed background. Due to the haploinsufficient role of p18 in mammary tumor development [7, 55] and the indistinguishable mammary tumor phenotype between p18-/- and p18+/-, as well as p18-/-;Gata3+/- and p18+/-;Gata3+/- mice, we combined p18-/- and p18+/- mice as p18mt group, p18-/-;Gata3+/- and p18+/-;Gata3+/- mice as p18mt;Gata3+/- group. We followed tumor development in these mice and found that 50% (n = 34) of p18mt;Gata3+/- and 30% (n = 27) of p18mt mice developed mammary tumors between 8-20 months, indicating that haploid loss of Gata3 in p18 mutant mice accelerates mammary tumorigenesis (Table 2). Though the mammary tumor incidence of p18mt mice in the Balb/c-B6 mixed background was lower when compared to the Balb/c background, as we previously reported [15], p18mt mammary tumors in a Balb/c-B6 mixed background were also E-cad positive, well-differentiated mammary tumors (Table 2, Figure 1, Figure 4, and data not shown). p18mt;Gata3+/- mammary tumors displayed typical pathological characteristics of poorly-differentiated tumors--highly heterogeneous cell types with increased necrosis, squamous metaplasia, spindle cells, nuclear-cytoplasm ratio, and mitotic indices (Table 2, Figure 4C, E, Figure 5A). 77% (13 out of 17) of p18mt;Gata3+/- mammary tumors were positive for EMT markers including fibronectin (Fn), vimentin (Vim), CD29, and EMT-TFs in 2-60% of the tumor cells. Whereas only a quarter (2 out of 8) of p18mt tumor was positively stained with mesenchymal markers in ~2% of the tumor cells (Table 2, Figure 4A-B, Figure 5B, Figure S6). Although 29% (5 out of 17) of p18mt;Gata3+/- mammary tumors and none (0 out of 8) of p18mt mammary tumors metastasized to lung, no significant increase of metastasis was observed in p18mt;Gata3+/- tumors relative to p18mt tumors when statistical analysis was conducted. This was partially caused by the development of kidney cysts and consequential renal failure, as well as various types of tumors in other organs including lymphoma and sarcoma in p18mt;Gata3+/- mice (Reference [48], and data will be published elsewhere), which prevented a thorough analysis of mammary tumor related metastasis. Together, these results indicate that haploid loss of Gata3 induces poorly-differentiated mammary tumors with the activation of EMT.
Spontaneous mammary tumor development in Gata3 mutant mice.
| Genotype a | ||||
|---|---|---|---|---|
| Tumor | Wt | p18mt b | Gata3+/- | p18mt;Gata3+/- c |
| Mammary Tumor | 0/9 | 8/27 (30%) | 0/8 | 17/34 (50%) f |
| Metastasis d | 0/8 | 5/17 (29%) g | ||
| EMT+ tumor No.e | 2/8 (25%) | 13/17 (77%) h | ||
a All mice were in Balb/c-B6 mixed background and were at 8-22 months of age.
b This group contains eight p18+/- and nineteen p18-/- mice.
c This group contains ten p18+/-;Gata3+/- and twenty four p18-/-;Gata3+/- mice.
d Three mammary tumors metastasized to lung, and the other two mammary tumors metastasized to lung and liver.
e At least two EMT markers (decreased E-Cad, increased Vim, Fn1, SMA or CD29) or two EMT-TFs, which include TWIST, SLUG, SNAIL, FRA1, FOXC1, and FOXC2, were detected in > 2% tumor cells by IHC, as we previously reported (Bai, Cancer Res., 2014).
f No significance from p18mt;Gata3+/- and p18mt tumors by a two-tailed Fisher's exact test (p = 0.1246). but p = 0.0135 from p18mt;Gata3+/- and Gata3+/- tumors by a two-tailed Fisher's exact test.
g No significance from p18mt;Gata3+/- and p18mt tumors by a two-tailed Fisher's exact test (p = 0.1399). Due to the development of lymphoma and kidney cyst in old p18mt;Gata3+/- mice, we were unable to follow the mammary tumor formation and metastasis in aged mice (mice older than 20 months).
h A significance from p18mt;Gata3+/- and p18mt tumors by a two-tailed Fisher's exact test (p = 0.028).
Gata3 deficiency in mice induces poorly differentiated mammary tumors with activation of EMT, and Gata3 deficient tumor cells harbor the enhanced tumor initiating and metastatic potential. (A, B, C) Representative IF staining (A), IHC (B), and H & E (C) analysis of primary mammary tumors developed in mice with the indicated genotypes. The H-scores for TWIST and Fn in (B) were calculated. The results represent the mean ± SD of four individual tumors per group. The asterisk (*) denotes a statistical significance from p18mt and p18mt;Gata3+/- samples determined by the T-test. (D) Primary tumor cells were transplanted into MFPs of NSG mice with estradiol supplement. Four weeks later, recipient mice were dissected, regenerated mammary tumors were counted, and metastasis in lungs was determined. Two mice that received p18mt;Gata3+/- tumor cell transplants died of lung metastasis in 4 weeks. (E) H & E. staining of p18mt;Gata3+/- primary and regenerated mammary tumors, as well as lung metastasis from regenerated mammary tumors. Note the regenerated mammary tumor (T) invasion into surrounding muscle (Mu) and metastasis (M) in the lung. (F, G) Representative tumors generated by transplantation of p18mt;Gata3+/- or p18mt tumor cells into mammary fat pad were analyzed by western blot (F) and FACS (G).
Gata3 deficient mammary tumors phenocopy Brca1 deficient tumors in induction of EMT. (A, B) Representative mammary tumors and their lung metastasis were analyzed and compared by H.E (A) and IHC (B). Two independent mammary tumors (Mammary Tumor 1 and Mammary Tumor 2) and one lung metastasis derived from p18mt;Gata3+/- and p18mt;Brca1+/- mice individually are shown in (A). The H-scores for Vim in (B) were calculated. The results represent the mean ± SD of four individual tumors per group. The number sign (#) denotes a statistical insignificance from p18mt;Gata3+/- and p18mt;Brca1+/- samples determined by the T-test. (C, D, E) The expression of genes and the morphology of the tumor cell lines derived from primary mammary tumors of different genotype were determined by qRT-PCR (C), western blot (D), and phase contrast microscopy (E). Data in (C) represent the mean ± SD. from triplicates of two independent primary cell lines of each genotype. The asterisk (*) in (C) denotes a statistical significance from p18mt and p18mt;Gata3+/- or p18mt;Brca1+/- samples determined by the T-test.
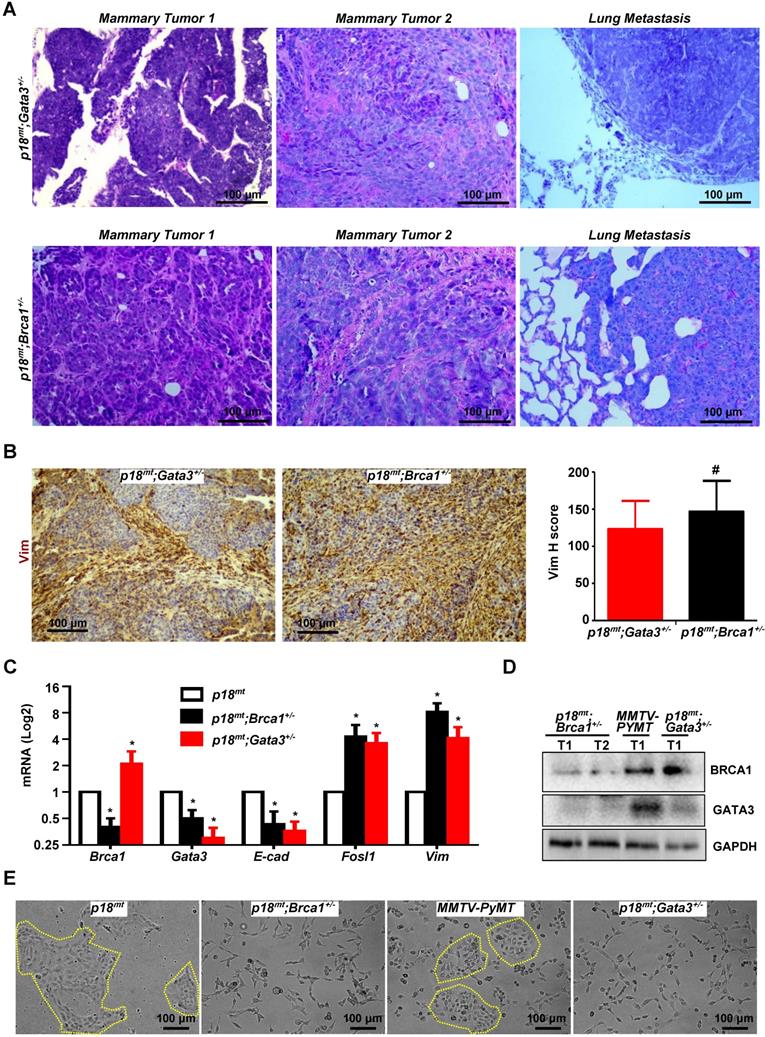
Gata3 deficiency promotes tumor initiating and metastatic potential
We transplanted primary tumor cells into mammary fat pads (MFPs) of NSG mice and found that 2 out of 5 mice received 5 x 106 p18mt tumor cell transplants produced tiny tumors (10 ± 2.8 mm3 in size) in 4 weeks. Regenerated p18mt mammary tumors, like primary p18mt tumors, were well differentiated tumors with typical epithelial features and markers, but with no metastasis (Figure 4A-D, Table 1, Table 2, and Ref [50]). Surprisingly, in the same time period all mice (4 out of 4) that received 1 x 106 p18mt;Gata3+/- tumor cell transplants developed huge mammary tumors (1440 ± 350 mm3 in size) and exhibited clinical sign of dyspnea (Figure 4D, E). Two mice that received p18mt;Gata3+/- tumor cell transplants died of lung metastasis in 4 weeks. Pathological analysis revealed that, like primary p18mt;Gata3+/- mammary tumors, regenerated p18mt;Gata3+/- mammary tumors were poorly differentiated, highly aggressive, invaded into the surrounding muscles, and metastasized to the lungs (Figure 4D-E). FACS analysis revealed that regenerated p18mt;Gata3+/- mammary tumors were composed of predominantly (81%) CD24+CD29high cells, previously demonstrated as CSC-enriched cell populations [56, 57], while regenerated p18mt tumors were composed of predominantly (75%) CD24+CD29low luminal/epithelial cells (Figure 4G), which was also detected in primary p18mt tumors [20]. The regenerated p18mt;Gata3+/- tumors expressed almost no detectable Gata3 relative to regenerated p18mt tumors (Figure 4F), suggesting that the regenerated tumors were enriched for cells with complete loss of Gata3. Notably, both primary and regenerated Brca1 deficient mammary tumors were also composed of predominantly CD24+CD29high cells, and Brca1 deficient mammary tumor cells harbored drastically enhanced potential for tumor initiation and metastasis [20, 50]. Together, these results indicate that Gata3 deficiency in mammary tumor cells promotes the potential for tumor initiation and metastasis.
Gata3 deficient mammary tumors phenocopy Brca1 deficient tumors in induction of EMT
Haploid loss of Gata3 activates EMT and promotes mammary tumorigenesis and metastasis, suggesting that Gata3 deficient tumors phenocopy Brca1 deficient tumors. To test this hypothesis, we compared the mammary tumors developed in Gata3+/- and Brca1+/- mice under the same p18mt background. Pathological analysis revealed that p18mt;Gata3+/- mammary tumors were highly heterogeneous and with various degrees of whorls and clusters of spindle-shaped cells, which are typical morphological characteristics of mesenchymal cells and also frequently observed in p18mt;Brca1+/- mammary tumors (Figure 4C, 5A, and Ref [7]). Importantly, when compared with p18mt tumors, both p18mt;Gata3+/- and p18mt;Brca1+/- mammary tumors more frequently metastasized to the lungs, and the lung metastasis were also enriched with mesenchymal-like cells. We examined EMT markers including Vim, Fn, E-cad, and EMT-TFs and found that all EMT markers positively stained in p18mt;Brca1+/- primary mammary tumors and its related metastasis were also detected in p18mt;Gata3+/- counterparts. Interestingly, very weak Brca1 and Gata3 expression were observed in less than 10% of p18mt;Brca1+/- lung metastatic tumor cells, while Gata3 was barely detectable in p18mt;Brca1+/- lung metastasis. (Table 1, Table 2, Figure 4A-B, Figure 5B, Fig. S6, S7, and Ref [50]). When the number and intensity of positive cells were quantified, we found that the H scores for Vim in p18mt;Gata3+/- tumor cells were comparable with the H scores in p18mt;Brca1+/- counterparts (Figure 5B, right panel).
To determine the similarity of cellular phenotype of Brca1 and Gata3 deficient tumor cells, we generated five p18mt;Brca1+/-, five p18mt;Gata3+/-, and three p18mt tumor cell lines, each of which was derived from a primary mammary tumor developed in an individual mouse. In addition, due to the slow proliferation rate of p18mt tumor cells in vitro, we isolated and characterized three cell lines from mammary tumors developed in MMTV-PYMT mice, which have been well characterized as luminal type mammary tumor model with genetically intact Brca1 and Gata3 [33, 58]. We confirmed the reduction of Brca1 and Gata3 expressions in 18mt;Brca1+/- and p18mt;Gata3+/- cells, respectively, when compared with those in p18mt and MMTV-PYMT cells (Figure 5C-D). Importantly, we noticed that Brca1 mRNA and protein levels were not decreased, rather slightly increased in p18mt;Gata3+/- cells relative to p18mt and MMTV-PYMT cells, whereas Gata3 mRNA and protein levels were, again, significantly reduced in p18mt;Brca1+/- and p18mt;Gata3+/- cells relative to p18mt and MMTV-PYMT cells (Figure 5C-D). To directly test if Gata3 loss impacts the expression of Brca1, we generated Gata3f/f MECs and transduced them with pMX-Cre and pMX-Empty. We found that deletion of Gata3 in MECs did not cause significant change of Brca1 mRNA level (Figure S8A). We demonstrated that knockdown of GATA3 in T47D human breast cancer cells or overexpression of WT Gata3 in Gata3 deficient mouse mammary tumor cells did not cause significant change of BRCA1 mRNA levels (Figure S8B-C). These data further support that Gata3 is downstream of Brca1, and that Brca1 is less likely a target of Gata3 in mammary epithelial and cancer cells. Consistent with the findings derived from tumor tissues, primary p18mt;Brca1+/- and p18mt;Gata3+/- tumor cells also exhibited a typical mesenchymal-like morphology with a high level of EMT markers, whereas, epithelial-like morphology were observed in p18mt and MMTV-PYMT tumor cells (Figure 5C, E). Taking into consideration of the similar potential of p18mt;Brca1+/- and p18mt;Gata3+/- tumor cells in promoting tumor initiation and metastasis, these results indicate that in breast cancer cells Gata3 deficiency functions similarly with Brca1 deficiency in activation of EMT and promotion of tumorigenesis and metastasis.
Reconstitution of Gata3 in Brca1-deficient tumor cells activates mesenchymal-epithelial transition (MET) suppressing tumor initiation and metastasis
Prompted by the finding that Gata3 functions downstream of Brca1 in suppressing EMT in breast cancers, we then examined if ectopic Gata3 in Brca1-deficinet tumor cells suppresses their potential for tumor initiation and metastasis. We transduced p18-/-;Brca1MGKO tumor cells with pBabe-Empty and pBabe-Gata3 respectively, and found that ectopic Gata3 restored expression of Cdh1, a target of Gata3, but inhibited expression of Vim and EMT-TFs including Slug, Snail, and Twist1 (Figure 6A, B), which was stimulated by loss of Brca1 in mammary tumors (Figure 1A, Figure S1). Morphology analysis revealed that some of the Gata3-expressing p18-/-;Brca1MGKO tumor cells were cuboidal shaped epithelial-like cells whereas all Empty-expressing cells were spindle-shaped, mesenchymal-like cells (Figure 6C). These results suggest that reconstitution of Gata3 in p18-/-;Brca1MGKO tumor cells activates MET. When transplanted, Gata3-expressing p18-/-;Brca1MGKO tumor cells produced significantly smaller tumors with more E-cad but less Vim than Empty-expressing counterparts (Figure 6D, 6F, and Figure S9). Pathological analysis showed that relative to tumors generated by Empty-expressing cells, tumors generated by Gata3-expressing cells were well differentiated with glandular structures, and less aggressive (decreased necrosis, squamous metaplasia, spindle cells, and mitotic indices) (Figure 6G). FACS and IHC analysis revealed that tumors generated by Gata3-expressing p18-/-;Brca1MGKO cells exhibited drastically enhanced CD24-positive and significantly reduced Vim-positive cells relative to those generated by Empty-expressing cells (Figure 6E, G-H, and Figure S9).
Ectopic Gata3 in Brca1-deficient tumor cells activates MET suppressing tumorigenesis and metastasis. (A-C) p18-/-;Brca1MGKO tumor cells were infected with pBabe-Empty (Empty) or pBabe-Gata3 (Gata3) and selected in puromycin. The expression of genes (A, B) and cell morphology (C) were determined. Data in (B) represent the mean ± SD. from triplicates of two independent experiments. The asterisk (*) denotes a statistical significance from Empty and Gata3 samples determined by the T-test. (D) 2×106 Empty and Gata3-expressing p18-/-;Brca1MGKO cells were transplanted into left and right inguinal MFP of NSG mice, respectively, in a pairwise manner. Four weeks later, regenerated tumor volumes were determined. The relative volumes of tumors generated by Gata3-expressing cells to tumors produced by Empty-expressing cells in each mouse were calculated and averaged. Data in the right represented as mean ± SD of five tumors in each group. Representative gross pictures of tumors were shown in the left. (E-G) Tumors generated from (D) were analyzed by flow cytometry (E), western blot (F), as well as H.E. and IHC staining (G). Note drastically decreased expression of Vim and increased that of E-cad in Gata3-expressing tumors relative to Empty-expressing tumors (F, G). (H) The H-scores for Vim in (G) were calculated. The results represent the mean ± SD of four individual tumors per group. The asterisk (*) denotes a statistical significance from Empty and Gata3 samples determined by the T-test. (I, J) 2×106 Empty and Gata3-expressing p18-/-;Brca1MGKO cells were transplanted into the MFPs of NSG mice. When newly generated tumors reached the maximum size allowed by IACUC in 4-10 weeks, or the mice became moribund, lungs were examined for H.E. staining (I) and IHC analysis (insets in I), and quantification of the number of metastatic nodules (J). M, metastatic nodules. Insets in (I) show representative IHC analysis of Vim in lung metastasis. Data in (J) represented as mean ± SD for the numbers of metastatic nodules detected in all lobes of the lungs in each group (n = 4).
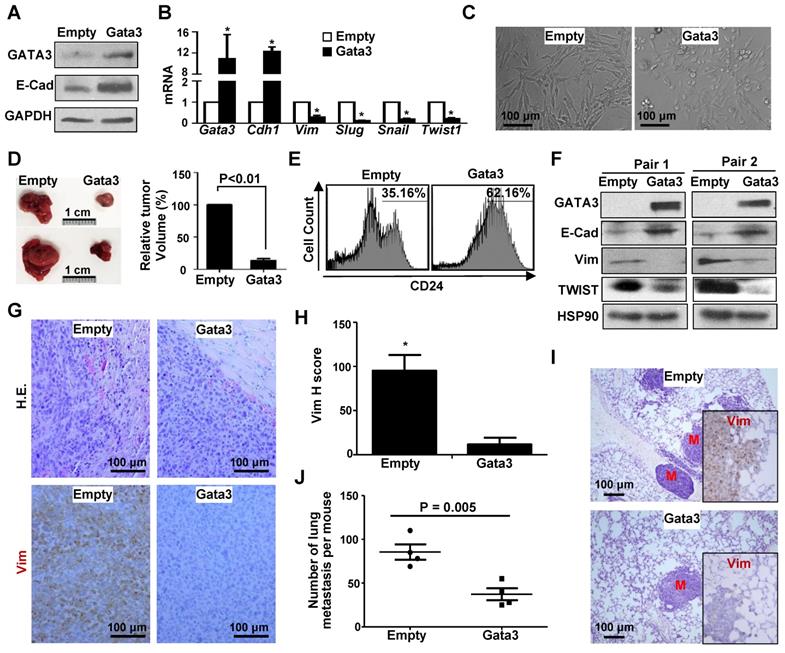
To determine if Gata3 affects Brca1 deficient mammary tumor metastasis, we transplanted Empty- and Gata3-expressing p18-/-; Brca1MGKO tumor cells into MFPs of NSG mice. We discovered that mammary tumors generated by Gata3-expressing cells produced significantly less metastatic nodules in the lung when compared with the mammary tumors initiated by Empty-expressing cells (Figure 6I-J), suggesting that ectopic Gata3 inhibits the metastatic potential of Brca1 deficient mammary tumor cells. In line with these findings, we also observed a drastic reduction of Vim expression in lung metastasis caused by Gata3-expressing cells relative to that in the metastasis done by Empty-expressing cells (Figure 6I, insets). Together, these data suggest that reconstitution of Gata3 activates MET and suppresses Brca1-deficient tumor development and metastasis.
Discussion
In this paper, we found that BRCA1 and GATA3 expressions are positively correlated and that BRCA1 mutation is associated with the enhanced methylation of the GATA3 promoter and reduced expression of the GATA3 gene in human breast cancers. We demonstrated that deletion of BRCA1 promotes methylation of the GATA3 promoter, therefore repressing GATA3 transcription. We discovered that Gata3 deficiency induces poorly-differentiated mammary tumors with the activation of EMT and promotes the potential for tumor initiation and metastasis. We demonstrated that Gata3 deficient mammary tumors phenocopy Brca1 deficient tumors in induction of EMT, and that reconstitution of Gata3 in Brca1-deficient tumor cells activates MET suppressing tumor initiation and metastasis. Our finding demonstrates that GATA3 functions downstream of BRCA1 to suppress EMT in controlling mammary tumor initiation and metastasis.
Functional loss of BRCA1 is associated with more than half of BLBCs with EMT features [8, 12-14]. Though it has been reported that loss of function of BRCA1 in breast cancers or cell lines enhances the expression of several EMT inducing transcription factors including SLUG [5], TWIST [20], FOXC1/C2 [21], and reduces the expression of a few EMT suppressing transcription factors, such as FOXA1 [22, 23], FOXO3 [24, 25], none of these transcription factors has been identified as downstream mediators of BRCA1 in the regulation of EMT in mammary tumor development and progression. Among the transcription factors upregulated by BRCA1 deficiency, overexpression of these EMT-TF induces EMT, and knockdown of SLUG, FOXC1, or FOXC2 in either BRCA1 mutant (SUM149 or SUM1315) or BRCA1 deficient (MDA-MB-231) breast cancer cell lines promotes expression of luminal and epithelial markers in vitro [5, 21]. For FOXA1 and FOXO3, though overexpression of either of these two transcription factors in cancer cell lines suppresses EMT, loss of either one in mice does not induce EMT in mammary tumor development [23, 25, 59, 60]. If a transcription factor primarily functions downstream of Brca1 controlling EMT in breast cancers, genetically loss of or gain of function of this transcription factor in mice with the same genetic background should produce similar EMT phenotype with Brca1 deficient mice, and restoration or removal of the function of the transcription factor should eliminate Brca1 deficient EMT phenotype in mammary tumors, in addition to the regulation of the transcription factor by BRCA1 in vitro. In the present study, we determined and compared the EMT phenotype in mammary tumors developed in mice deficient for Brca1 or Gata3 under the same p18 deficient background. We discovered that Gata3 deficiency activates EMT in the induction of mammary tumors and promotes tumor initiation as well as the metastatic potential of cancer cells, which phenocopy Brca1 deficient tumors and tumor cells. We demonstrated that reconstitution of Gata3 in Brca1-deficient tumor cells activates MET and eliminates potential for tumor initiation and metastasis. Our finding identifies GATA3 as the first transcription factor that functions downstream of BRCA1 to suppress EMT in breast cancers.
GATA3, a lineage specifier, is critical in controlling the fate of mammary epithelial [10, 11, 61] and lymphoid cells [62]. The function of GATA3 in suppressing EMT and metastasis in breast cancers has been well studied in cell line models [19, 26-28]. In oncogene transgenic mouse models, loss of Gata3 marks the loss of tumor differentiation and the onset of tumor dissemination, and deletion of Gata3 stimulates tumor progression [33, 63]. However, due to growth defects induced by long-term loss of Gata3 and apoptosis caused by acute loss of Gata3 in differentiated tumor cells [15, 32, 33], it remains elusive if Gata3 loss regulates EMT in breast cancer development and progression. We and others have previously identified p18 as a downstream target of Gata3 in the control of cell proliferation, and that loss of p18 rescues the proliferative defects induced by Gata3 deficiency [15, 32, 48]. p18;Gata3 double mutant mice provide us a genetic model and a unique opportunity to dissect the role of Gata3 loss in the regulation of tumor cell differentiation in vivo. Taking this advantage in our study, we demonstrated that haploid loss of Gata3 in p18 deficient background converts well-differentiated mammary tumors into poorly-differentiated mammary tumors with the activation of EMT and promotes tumor initiating and metastatic potential. Our findings suggest a critical role of Gata3 loss in driving CSC function and metastasis, and support the development of therapeutic drugs enhancing GATA3 function or targeting its downstream pathway to treat metastatic breast cancers.
How GATA3 is regulated is largely unknown. BRCA1 regulates GATA3 expression likely through multiple mechanisms. We and others have demonstrated that BRCA1 binds to GATA3 binding sites on the promoter of a few genes repressing their transcription [20, 21, 64], and that GATA3 promoter harbors multiple GATA3 binding sites and thus GATA3 transcription is autoregulatorily activated [65, 66]. It is possible that BRCA1 binds to the promoter of GATA3 to directly activate the transcription of GATA3. In addition, since histone methyltransferase EZH2 is overexpressed in BRCA1-deficient breast tumor cells [67], and EZH2 binds to Gata3 promoter repressing its transcription [68, 69], it is also possible that BRCA1 prevents EZH2-mediated H3K27 trimethylation at the GATA3 locus to maintain GATA3 transcription. In the present study, we demonstrated that BRCA1 mutation is associated with GATA3 promoter hypermethylation and reduced GATA3 expression in human breast cancer samples. Although deletion of p18 stimulates expression of DNMT1 in MECs and stromal cells, mice lacking p18 develop Gata3-positive well-differentiated mammary tumors. Conversely mice deficient for both Brca1 and p18 generate Gata3-negative poorly-differentiated tumors in which Gata3 gene is hypermethylated. These data suggest that when Brca1 is present, methylation of Gata3 gene in tumor cells is significantly suppressed even if the high level of DNMT1 is available in the tumor and surrounding cells. When Brca1 is depleted, the Gata3 gene is hypermethylated; likely by DNMT. Our finding that suggests Brca1 protects the Gata3 promoter from methylation is, at least, one of the important mechanisms by which BRCA1 regulates GATA3 transcription, though it remains to be investigated that whether BRCA1 directly transactivates GATA3 and how BRCA1 interacts with DNA methyltransferases (DNMTs) and EZH2 to control transcription of GATA3. Our findings also suggest that DNMT inhibitors can be used to induce GATA3 and its mediated differentiation for treatment of BRCA1-deficient breast cancers.
Conclusions
Our finding, for the first time, demonstrates that GATA3 functions downstream of BRCA1 to suppress EMT in controlling mammary tumor initiation and metastasis.
Abbreviations
BLBC: basal-like breast cancer; Brca1MGKO: Brca1f/f, MMTV-Cre or Brca1f/-, MMTV-Cre; ChIP: chromatin-immunoprecipitation; CSC: cancer stem cell; DAC: 5-aza-2'-deoxycytidine; E-cad: E-cadherin; EMT: epithelial-mesenchymal transition; EMT-TFs: EMT-inducing transcription factors; ER: estrogen receptor; FFPE: formalin-fixed paraffin-embedded; Fn: fibronectin; IHC: immunohistochemistry; INK4: inhibitors of CDK4/6; KD: knockdown; MEC: mammary epithelial cell; MET: mesenchymal-epithelial transition; MFP: mammary fat pad; MS-PCR: methylation specific PCR; p16: p16INK4A; p18: p18INK4C; TFs: transcription factors; Vim: vimentin.
Supplementary Material
Supplementary figures.
Acknowledgements
This study was supported by the Guangdong Provincial Science and Technology Program (2019B030301009), National Natural Science Foundation of China (81972637), High-level university phase 2 construction funding from Shenzhen University (860-00000210), Natural Science Foundation of Shenzhen City (JCYJ20190808115603580 and JCYJ20190808165803558), Natural Science Foundation of Guangdong Province (2019A1515011343 and 2021A1515011145), DOD Idea Expansion Award (W81XWH-13-1-0282), the Bankhead-Coley Cancer Research grant (TBC07), and research funds from Shenzhen University. We thank Drs. Beverly Koller, Chuxia Deng, I-Cheng Ho, and Lothar Hennighausen for Brca1 mutant, Gata3 mutant, and MMTV-cre mice, Yue Xiong for discussion, Ho Lam Chan, Shiqin Liu, Emely Pimentel and Alexandria Scott for technical support, the FACS core facility at University of Miami and Shenzhen University for cell sorting, the DVR core facility at University of Miami for animal husbandry. Li-Han Zhang thanks the China Scholarship Council for financial support.
Author Contributions
FB, LHZ, and XHP designed the research studies. FB, LHZ, XL, CW, CZ and XHP conducted experiments and analyzed data. JS performed data mining in human breast cancer datasets; ML and WGZ provided administrative and material support; FB, LHZ and XHP wrote the manuscript. FB and XHP provided financial support, XHP supervised the project. All authors made comments on the manuscript.
Ethics approval and consent to participate
The Institutional Animal Care and Use Committee at the University of Miami and Shenzhen University approved all animal procedures.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Althuis MD, Fergenbaum JH, Garcia-Closas M, Brinton LA, Madigan MP, Sherman ME. Etiology of hormone receptor-defined breast cancer: a systematic review of the literature. Cancer Epidemiol Biomarkers Prev. 2004;13:1558-68
2. Kim MJ, Ro JY, Ahn SH, Kim HH, Kim SB, Gong G. Clinicopathologic significance of the basal-like subtype of breast cancer: a comparison with hormone receptor and Her2/neu-overexpressing phenotypes. Hum Pathol. 2006;37:1217-26
3. Livasy CA, Karaca G, Nanda R, Tretiakova MS, Olopade OI, Moore DT. et al. Phenotypic evaluation of the basal-like subtype of invasive breast carcinoma. Mod Pathol. 2006;19:264-71
4. Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH. et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009;15:907-13
5. Proia TA, Keller PJ, Gupta PB, Klebba I, Jones AD, Sedic M. et al. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell. 2011;8:149-63
6. Molyneux G, Geyer FC, Magnay FA, McCarthy A, Kendrick H, Natrajan R. et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell. 2010;7:403-17
7. Bai F, Smith MD, Chan HL, Pei XH. Germline mutation of Brca1 alters the fate of mammary luminal cells and causes luminal-to-basal mammary tumor transformation. Oncogene. 2013;32:2715-25
8. Fedele M, Cerchia L, Chiappetta G. The epithelial-to-mesenchymal transition in breast cancer: focus on basal-like carcinomas. Cancers. 2017;9:134. doi: 10.3390/cancers9100134
9. Ye X, Tam WL, Shibue T, Kaygusuz Y, Reinhardt F, Ng Eaton E. et al. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature. 2015;525:256-60
10. Visvader JE, Stingl J. Mammary stem cells and the differentiation hierarchy: current status and perspectives. Genes Dev. 2014;28:1143-58
11. Siegel PM, Muller WJ. Transcription factor regulatory networks in mammary epithelial development and tumorigenesis. Oncogene. 2010;29:2753-9
12. De Summa S, Pinto R, Sambiasi D, Petriella D, Paradiso V, Paradiso A. et al. BRCAness: a deeper insight into basal-like breast tumors. Ann Oncol. 2013;24(Suppl 8):viii13-viii21
13. Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF. et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;487:330-7
14. Zhu X, Shan L, Wang F, Wang J, Wang F, Shen G. et al. Hypermethylation of BRCA1 gene: implication for prognostic biomarker and therapeutic target in sporadic primary triple-negative breast cancer. Breast Cancer Res Treat. 2015;150:479-86
15. Pei XH, Bai F, Smith MD, Usary J, Fan C, Pai SY. et al. CDK inhibitor p18(INK4c) is a downstream target of GATA3 and restrains mammary luminal progenitor cell proliferation and tumorigenesis. Cancer Cell. 2009;15:389-401
16. Asch-Kendrick R, Cimino-Mathews A. The role of GATA3 in breast carcinomas: a review. Hum Pathol. 2016;48:37-47
17. Liu J, Prager-van der Smissen WJ, Look MP, Sieuwerts AM, Smid M, Meijer-van Gelder ME. et al. GATA3 mRNA expression, but not mutation, associates with longer progression-free survival in ER-positive breast cancer patients treated with first-line tamoxifen for recurrent disease. Cancer Lett. 2016;376:104-9
18. Mehra R, Varambally S, Ding L, Shen R, Sabel MS, Ghosh D. et al. Identification of GATA3 as a breast cancer prognostic marker by global gene expression meta-analysis. Cancer Res. 2005;65:11259-64
19. Chou J, Provot S, Werb Z. GATA3 in development and cancer differentiation: cells GATA have it!. J Cell Physiol. 2010;222:42-9
20. Bai F, Chan HL, Scott A, Smith MD, Fan C, Herschkowitz JI. et al. BRCA1 suppresses epithelial-to-mesenchymal transition and stem cell dedifferentiation during mammary and tumor development. Cancer Res. 2014;74:6161-72
21. Tkocz D, Crawford NT, Buckley NE, Berry FB, Kennedy RD, Gorski JJ. et al. BRCA1 and GATA3 corepress FOXC1 to inhibit the pathogenesis of basal-like breast cancers. Oncogene. 2012;31:3667-78
22. Gong C, Fujino K, Monteiro LJ, Gomes AR, Drost R, Davidson-Smith H. et al. FOXA1 repression is associated with loss of BRCA1 and increased promoter methylation and chromatin silencing in breast cancer. Oncogene. 2015;34:5012-24
23. Song Y, Washington MK, Crawford HC. Loss of FOXA1/2 is essential for the epithelial-to-mesenchymal transition in pancreatic cancer. Cancer Res. 2010;70:2115-25
24. Gong C, Yao S, Gomes AR, Man EP, Lee HJ, Gong G. et al. BRCA1 positively regulates FOXO3 expression by restricting FOXO3 gene methylation and epigenetic silencing through targeting EZH2 in breast cancer. Oncogenesis. 2016;5:e214. doi: 10.1038/oncsis.2016.23
25. Belguise K, Guo S, Sonenshein GE. Activation of FOXO3a by the green tea polyphenol epigallocatechin-3-gallate induces estrogen receptor alpha expression reversing invasive phenotype of breast cancer cells. Cancer Res. 2007;67:5763-70
26. Dydensborg AB, Rose AA, Wilson BJ, Grote D, Paquet M, Giguere V. et al. GATA3 inhibits breast cancer growth and pulmonary breast cancer metastasis. Oncogene. 2009;28:2634-42
27. Chou J, Lin JH, Brenot A, Kim JW, Provot S, Werb Z. GATA3 suppresses metastasis and modulates the tumour microenvironment by regulating microRNA-29b expression. Nat Cell Biol. 2013;15:201-13
28. Yan W, Cao QJ, Arenas RB, Bentley B, Shao R. GATA3 inhibits breast cancer metastasis through the reversal of epithelial-mesenchymal transition. J Biol Chem. 2010;285:14042-51
29. Drost RM, Jonkers J. Preclinical mouse models for BRCA1-associated breast cancer. Br J Cancer. 2009;101:1651-7
30. Cao L, Kim S, Xiao C, Wang RH, Coumoul X, Wang X. et al. ATM-Chk2-p53 activation prevents tumorigenesis at an expense of organ homeostasis upon Brca1 deficiency. EMBO J. 2006;25:2167-77
31. Cao L, Li W, Kim S, Brodie SG, Deng CX. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev. 2003;17:201-13
32. Hosokawa H, Tanaka T, Kato M, Shinoda K, Tohyama H, Hanazawa A. et al. Gata3/Ruvbl2 complex regulates T helper 2 cell proliferation via repression of Cdkn2c expression. Proc Natl Acad Sci U S A. 2013;110:18626-31
33. Kouros-Mehr H, Bechis SK, Slorach EM, Littlepage LE, Egeblad M, Ewald AJ. et al. GATA-3 links tumor differentiation and dissemination in a luminal breast cancer model. Cancer Cell. 2008;13:141-52
34. Kumar P, Mukherjee M, Johnson JP, Patel M, Huey B, Albertson DG. et al. Cooperativity of Rb, Brca1, and p53 in malignant breast cancer evolution. PLoS Genet. 2012;8:e1003027. doi: 10.1371/journal.pgen.1003027
35. Chang CJ, Chao CH, Xia W, Yang JY, Xiong Y, Li CW. et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011;13:317-23
36. Wang SP, Wang WL, Chang YL, Wu CT, Chao YC, Kao SH. et al. p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of Slug. Nat Cell Biol. 2009;11:694-704
37. Wu WS, Heinrichs S, Xu D, Garrison SP, Zambetti GP, Adams JM. et al. Slug antagonizes p53-mediated apoptosis of hematopoietic progenitors by repressing puma. Cell. 2005;123:641-53
38. Herschkowitz JI, Zhao W, Zhang M, Usary J, Murrow G, Edwards D. et al. Comparative oncogenomics identifies breast tumors enriched in functional tumor-initiating cells. Proc Natl Acad Sci U S A. 2012;109:2778-83
39. Pei XH, Xiong Y. Biochemical and cellular mechanisms of mammalian CDK inhibitors: a few unresolved issues. Oncogene. 2005;24:2787-95
40. Jonsson G, Staaf J, Vallon-Christersson J, Ringner M, Gruvberger-Saal SK, Saal LH. et al. The retinoblastoma gene undergoes rearrangements in BRCA1-deficient basal-like breast cancer. Cancer Res. 2012;72:4028-36
41. Stefansson OA, Jonasson JG, Olafsdottir K, Hilmarsdottir H, Olafsdottir G, Esteller M. et al. CpG island hypermethylation of BRCA1 and loss of pRb as co-occurring events in basal/triple-negative breast cancer. Epigenetics. 2011;6:638-49
42. Hollestelle A, Nagel JH, Smid M, Lam S, Elstrodt F, Wasielewski M. et al. Distinct gene mutation profiles among luminal-type and basal-type breast cancer cell lines. Breast Cancer Res Treat. 2010;121:53-64
43. Stephens PJ, McBride DJ, Lin ML, Varela I, Pleasance ED, Simpson JT. et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. 2009;462:1005-10
44. Scott A, Bai F, Chan HL, Liu S, Ma J, Slingerland JM. et al. p16INK4a suppresses BRCA1-deficient mammary tumorigenesis. Oncotarget. 2016;7:84496-507
45. Scott A, Bai F, Chan HL, Liu S, Slingerland JM, Robbins DJ. et al. p16 loss rescues functional decline of Brca1-deficient mammary stem cells. Cell Cycle. 2017;16:759-64
46. Sedic M, Skibinski A, Brown N, Gallardo M, Mulligan P, Martinez P. et al. Haploinsufficiency for BRCA1 leads to cell-type-specific genomic instability and premature senescence. Nature communications. 2015;6:7505. doi: 10.1038/ncomms8505
47. Jiang Z, Deng T, Jones R, Li H, Herschkowitz JI, Liu JC. et al. Rb deletion in mouse mammary progenitors induces luminal-B or basal-like/EMT tumor subtypes depending on p53 status. J Clin Invest. 2010;120:3296-309
48. Liu S, Lam Chan H, Bai F, Ma J, Scott A, Robbins DJ. et al. Gata3 restrains B cell proliferation and cooperates with p18INK4c to repress B cell lymphomagenesis. Oncotarget. 2016;7:64007-64020
49. Goulding H, Pinder S, Cannon P, Pearson D, Nicholson R, Snead D. et al. A new immunohistochemical antibody for the assessment of estrogen receptor status on routine formalin-fixed tissue samples. Hum Pathol. 1995;26:291-4
50. Wang C, Bai F, Zhang LH, Scott A, Li E, Pei XH. Estrogen promotes estrogen receptor negative BRCA1-deficient tumor initiation and progression. Breast Cancer Res. 2018;20:74. doi: 10.1186/s13058-018-0996-9
51. Zhang LH, Yang AJ, Wang M, Liu W, Wang CY, Xie XF. et al. Enhanced autophagy reveals vulnerability of P-gp mediated epirubicin resistance in triple negative breast cancer cells. Apoptosis. 2016;21:473-88
52. Cooper SJ, Zou H, Legrand SN, Marlow LA, von Roemeling CA, Radisky DC. et al. Loss of type III transforming growth factor-beta receptor expression is due to methylation silencing of the transcription factor GATA3 in renal cell carcinoma. Oncogene. 2010;29:2905-15
53. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420-8
54. Kimura H, Nakamura T, Ogawa T, Tanaka S, Shiota K. Transcription of mouse DNA methyltransferase 1 (Dnmt1) is regulated by both E2F-Rb-HDAC-dependent and -independent pathways. Nucleic Acids Res. 2003;31:3101-13
55. Bai F, Pei XH, Godfrey VL, Xiong Y. Haploinsufficiency of p18(INK4c) sensitizes mice to carcinogen-induced tumorigenesis. Mol Cell Biol. 2003;23:1269-77
56. Shackleton M, Vaillant F, Simpson KJ, Stingl J, Smyth GK, Asselin-Labat ML. et al. Generation of a functional mammary gland from a single stem cell. Nature. 2006;439:84-8
57. Shafee N, Smith CR, Wei S, Kim Y, Mills GB, Hortobagyi GN. et al. Cancer stem cells contribute to cisplatin resistance in Brca1/p53-mediated mouse mammary tumors. Cancer Res. 2008;68:3243-50
58. Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z. et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007;8:R76. doi: 10.1186/gb-2007-8-5-r76
59. Castrillon DH, Miao L, Kollipara R, Horner JW, DePinho RA. Suppression of ovarian follicle activation in mice by the transcription factor Foxo3a. Science. 2003;301:215-8
60. Bernardo GM, Lozada KL, Miedler JD, Harburg G, Hewitt SC, Mosley JD. et al. FOXA1 is an essential determinant of ERalpha expression and mammary ductal morphogenesis. Development. 2010;137:2045-54
61. Kouros-Mehr H, Kim JW, Bechis SK, Werb Z. GATA-3 and the regulation of the mammary luminal cell fate. Curr Opin Cell Biol. 2008;20:164-70
62. Wan YY. GATA3: a master of many trades in immune regulation. Trends Immunol. 2014;35:233-42
63. Asselin-Labat ML, Sutherland KD, Vaillant F, Gyorki DE, Wu D, Holroyd S. et al. Gata-3 negatively regulates the tumor-initiating capacity of mammary luminal progenitor cells and targets the putative tumor suppressor caspase-14. Mol Cell Biol. 2011;31:4609-22
64. Bai F, Liu S, Liu X, Hollern DP, Scott A, Wang C. et al. PDGFRbeta is an essential therapeutic target for BRCA1-deficient mammary tumors. Breast Cancer Res. 2021;23:10. doi: 10.1186/s13058-021-01387-x
65. Gunne-Braden A, Sullivan A, Gharibi B, Sheriff RSM, Maity A, Wang YF. et al. GATA3 mediates a fast, irreversible commitment to BMP4-driven differentiation in human embryonic stem cells. Cell Stem Cell. 2020;26:693-706 e9
66. Ouyang W, Lohning M, Gao Z, Assenmacher M, Ranganath S, Radbruch A. et al. Stat6-independent GATA-3 autoactivation directs IL-4-independent Th2 development and commitment. Immunity. 2000;12:27-37
67. Puppe J, Drost R, Liu X, Joosse SA, Evers B, Cornelissen-Steijger P. et al. BRCA1-deficient mammary tumor cells are dependent on EZH2 expression and sensitive to Polycomb Repressive Complex 2-inhibitor 3-deazaneplanocin A. Breast Cancer Res. 2009;11:R63. doi: 10.1186/bcr2354
68. Tumes DJ, Onodera A, Suzuki A, Shinoda K, Endo Y, Iwamura C. et al. The polycomb protein Ezh2 regulates differentiation and plasticity of CD4(+) T helper type 1 and type 2 cells. Immunity. 2013;39:819-32
69. Onodera A, Yamashita M, Endo Y, Kuwahara M, Tofukuji S, Hosokawa H. et al. STAT6-mediated displacement of polycomb by trithorax complex establishes long-term maintenance of GATA3 expression in T helper type 2 cells. J Exp Med. 2010;207:2493-506
Author contact
![]() Corresponding author: Xin-Hai Pei, Guangdong Provincial Key Laboratory of Regional Immunity and Diseases, International Cancer Center, Marshall Laboratory of Biomedical Engineering, Department of Anatomy and Histology, Shenzhen University Health Science Center, Shenzhen 518060, China. Phone: 86-755-26931350; Email: peixinhaiedu.cn. ORCID: 0000-0001-6441-1691
Corresponding author: Xin-Hai Pei, Guangdong Provincial Key Laboratory of Regional Immunity and Diseases, International Cancer Center, Marshall Laboratory of Biomedical Engineering, Department of Anatomy and Histology, Shenzhen University Health Science Center, Shenzhen 518060, China. Phone: 86-755-26931350; Email: peixinhaiedu.cn. ORCID: 0000-0001-6441-1691