Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(17):8185-8196. doi:10.7150/thno.59121 This issue Cite
Research Paper
Designer exosomes for targeted and efficient ferroptosis induction in cancer via chemo-photodynamic therapy
Jianbing Du1,2#, Zhuo Wan3#, Cong Wang4#, Fan Lu5, Mengying Wei5, Desheng Wang1 ![]() , Qiang Hao2
, Qiang Hao2 ![]()
1. Department of Hepatobiliary Surgery, Xijing Hospital, Fourth Military Medical University, Xi'an, 710032, China.
2. State Key Laboratory of Cancer Biology, Biotechnology Center, School of Pharmacy, Fourth Military Medical University, Xi'an, 710032, China.
3. Department of Hematology, Tangdu Hospital, Fourth Military Medical University, Xi'an, 710038, People's Republic of China.
4. Department of Clinical Laboratory, The Second People's Hospital of Hefei, Hefei, China.
5. State Key Laboratory of Cancer Biology, Department of Biochemistry and Molecular Biology, Fourth Military Medical University, Xi'an, 710032, People's Republic of China.
# These authors contributed equally to this article.
Received 2021-2-6; Accepted 2021-6-30; Published 2021-7-13
Abstract

Background: Efficient and specific induction of cell death in liver cancer is urgently needed. In this study, we aimed to design an exosome-based platform to deliver ferroptosis inducer (Erastin, Er) and photosensitizer (Rose Bengal, RB) into tumor tissues with high specificity.
Methods: Exosome donor cells (HEK293T) were transfected with control or CD47-overexpressing plasmid. Exosomes were isolated and loaded with Er and RB via sonication method. Hepa1-6 cell xenograft C57BL/6 model was injected with control and engineered exosomes via tail vein. In vivo distribution of the injected exosomes was analyzed via tracking the fluorescence labeled exosomes. Photodynamic therapy was conducted by 532 nm laser irradiation. The therapeutic effects on hepatocellular carcinoma and toxic side-effects were systemically analyzed.
Results: CD47 was efficiently loaded on the exosomes from the donor cells when CD47 was forced expressed by transfection. CD47 surface functionalization (ExosCD47) made the exosomes effectively escape the phagocytosis of mononuclear phagocyte system (MPS), and thus increased the distribution in tumor tissues. Erastin and RB could be effectively encapsulated into exosomes after sonication, and the drug-loaded exosomes (Er/RB@ExosCD47) strongly induced ferroptosis both in vitro and in vivo in tumor cells after irradiation of 532 nm laser. Moreover, compared with the control exosomes (Er/RB@ExosCtrl), Er/RB@ExosCD47 displayed much lower toxicity in liver.
Conclusion: The engineered exosomes composed of CD47, Erastin, and Rose Bengal, induce obvious ferroptosis in hepatocellular carcinoma (HCC) with minimized toxicity in liver and kidney. The proposed exosomes would provide a promising strategy to treat types of malignant tumors.
Keywords: Hepatocellular carcinoma, exosomes, ferroptosis, photodynamic therapy, synergistic effects
Introduction
Despite considerable progress in diagnosis and therapy, primary liver cancer ranks the fifth leading cause of cancer death [1]. Currently, beyond surgical resection, the treatments are mainly chemotherapy, targeted drug therapy and immunotherapy [2-4]. However, the effectiveness of these therapies in cancer is limited [5, 6]. Therefore, it is particularly urgent to explore other strategies to induce cell death specific in cancer.
Recent years, ferroptosis has attracted the attention of many researchers in the field of tumor research [7]. Ferroptosis is a new form of cell death first proposed in 2012, which has been accepted as a novel type of feature to eliminate tumor cells [8, 9]. Ferroptosis depends on iron-dependent Fenton reaction and the accumulation of lipid peroxides, is totally different from necrosis and apoptosis [10]. The characteristic of ferroptosis is the exhaustion of the intracellular pool of glutathione (reduced) (GSH), passivation of the glutathione peroxidase 4 (GPX4), eventually accumulation of lipid reactive oxygen species (ROS) [11]. The cystine-glutamate antiporter (system xc-), a critical transporter regulates glutamate homeostasis, plays a great role in ferroptosis [10, 12]. Existing evidence have showed ferroptosis played a therapeutic role in many apoptosis-resistant tumors, such as gastric cancer, pancreatic cancer, colorectal cancer and HCC [13-16]. Therefore, inducing tumor cell ferroptosis will open a new therapeutic avenue for hepatocellular carcinoma (HCC).
In 2012, researchers defined the form of cell death caused by Erastin as ferroptosis [10]. Erastin, as a classic ferroptosis inducer, has been explored in the treatment of tumors which can directly inhibit the activity of the system xc- [10, 17]. However, free Erastin has low water solubility and high nephrotoxicity, limiting its application [18]. In addition, locally induced ROS leads to the accumulation of lipid ROS in cells, which will promote ferroptosis and thus reduced the need of Erastin. Photodynamic therapy (PDT) is a technology that irradiating a specific wavelength light to excite the photosensitizer (PS) in the target site to result in a significant accumulation of ROS [19]. Rose Bengal (RB) is a famous photosensitizer which has a high ROS yield in PDT [20]. Therefore, efficiently delivering Erastin and RB to tumor tissues might synergistically induce efficient ferroptosis with low side effects is a promising method to treat cancer.
In the past decade, the research of drug delivery nanocarriers have been rapidly developed [21]. However, their application has been limited due to several reasons, such as, low load capacity, biodistribution problem and cationic ligand toxicity [22, 23]. Exosome, a small nanovesicle, is a new type of communication and drug delivery medium that can transport biologically active molecules between cells through a variety of biological molecules (such as proteins and nucleic acid) and regulates cells microenvironment and immune system [24-27]. Unlike other nanoparticle carriers, exosomes contain transmembrane proteins that may enhance endocytosis, thus promoting the delivery of their internal content [28]. As a delivery carrier, exosomes have the advantages of low immunogenicity, high biocompatibility and enhanced permeability and retention effect in tumor issues [29]. However, most injected nanomaterials are phagocytized by the mononuclear phagocyte system (MPS, e.g., liver and spleen), preventing their delivery to the desired area [30-33]. CD47, which is a widely expressed integrin-related transmembrane protein, is the ligand for signal regulatory protein alpha (SIRPα), and CD47-SIRPα binding initiates the “don't eat me” signal that inhibits phagocytosis [28, 34-36]. Engineering of CD47 on the exosomes could protect the exosomes from phagocytosis by macrophages.
In this study, we have engineered an exosome armed with three moieties, surface functionalization with CD47, membrane loading with ferroptosis inducer Erastin, and core with photosensitizer RB. The exosomes displayed high delivery efficiency to tumors. Upon irradiation of 532 nm laser in the tumor region, Er and RB synergistically induced cell death. By using the HCC xenograft model, we here showed that the designed exosomes display potent anti-tumor therapeutic effects, and amazing low toxicity, which would provide a promising strategy to treat malignant tumors.
Materials and methods
Cell culture
HEK293T, RAW264.7 cells (obtained from ATCC) and Hepa1-6 cells (carried luciferase reporter gene) (donated by Pro. Dou from the Fourth Military Medical University) were maintained in Dulbeccoo's modified Eagle's medium (DMEM) (Hyclone, USA) supplemented with 10% fetal bovine serum (HyClone, USA), 1% penicillin/streptomycin (HyClone, USA). All cells were cultured at 37 °C in 5% CO2.
Tumor-bearing models
Male C57BL/6 mice (6-8 weeks) were purchased from the animal laboratory center of the Fourth Military Medical University. All animal experiments were approved by the Animal Experiment Administration Committee of the Fourth Military Medical University. Hepa1-6 cells (3×106 cells in 100 μL phosphate buffered saline, PBS) were subcutaneously injected on the left back of mice for subcutaneous HCC model. The long diameter and short diameter of tumor were measured by vernier caliper. Tumor volume was estimated as (L × S2) × 0.5 (L, long diameter; S, short diameter).
Plasmid construction
CD47 was amplified with cDNA in Hepa1-6 cells by PCR using primers flanking with corresponding enzyme sites. Then the amplicons were inserted between BamH I and Hind III into a pcDNA3.1(-) vector, with the resultant correct plasmids designated as pcDNA 3.1(-)-CD47. All the clones were confirmed by sequencing and the right clones were stored at -80 °C for following application.
Transfection
To produce exosomes engineered with CD47, HEK293T cells were transfected with the corresponding plasmid with Lipofectamine 2000 (Invitrogen) as instructed. Briefly, when the HEK293T cells density grew to 60%-70% in 100-mm culture dish, culture medium was replaced with antibiotic-free medium. Then, 5 μg plasmid and 10 μL Lipofectamine 2000 were added to two centrifuge tubes containing 1 mL of DMEM medium respectively and incubated at room temperature for 5 min. Then, the contents of the two centrifuge tubes were mixed together and incubated at room temperature for 5 min. The above mixed culture medium was added to the 293T cell culture dish.
Exosome preparation
Exosomes were extracted from the culture supernatant of HEK293T. HEK293T cells were washed twice with PBS and replaced with serum-free medium. After the HEK293T cells were cultured in a serum-free medium at 37 °C in 5% CO2 for 48 h, the supernatant was collected. Exosomes were isolated by ultracentrifugation. Briefly, the culture medium was centrifuged at 3000 g for 15 min to remove cell debris, and the resulting supernatant was filtered using a 0.22 μm filter. Then the supernatant was ultracentrifuged at 100,000 g for 60 min and then the pellet was resuspended and repeated again. The resulting precipitate, namely exosomes, was diluted in PBS and stored at -80 ℃ until use.
Exosome characterization
The concentration of exosomes was measured by total proteins using the Pierce BCA Protein Assay Kit (Thermo Scientific, USA). The exosomes were diluted to 1 mg/mL, and the size distribution was analyzed by NanoPlus (Otsuka Electronics, Japan). The morphology of exosomes was observed by transmission electron microscopy (JEM-2000EX TEM, Japan). The expression of exosome proteins of TSG101, CD9 and CD47 were determined by western blot.
Exosome pulldown assay
For analysis of the expression of CD47 on exosomes, we performed exosome pull down assay using anti-CD47 (Abcam, ab175388) antibodies as described before [37]. Briefly, about 50 μL of Protein-A Sepharose beads in 0.2% BSA in 1 mL of PBS was first incubated with 5 μg of anti-CD47 or control IgG at 4 °C overnight. The unbound anti-CD47 was removed by centrifugation. Then, the antibody bound Protein-A Sepharose beads were incubated with the exosomes, followed by precipitation. The immunoprecipitated exosomes were then subjected to lysis buffer, followed by Western blotting assay.
Western blotting
Protein was extracted from exosomes, HEK293T cells, Hepa1-6 cells or tumor tissues, lysed with RIPA lysis buffer at 4 °C for 30 min, and then the protein concentration was measured using Pierce BCA protein kit (Thermo, USA). Protein samples were concentrated on 5% SDS-PAGE gel and separated by 12% SDS-PAGE gel. After that, the gel was transferred to a nitrocellulose filter membrane. After blocking with 5% BSA, NC membrane was incubated with primary antibody overnight at 4 ℃. The corresponding HRP-conjugated secondary antibodies were incubated for 1 h at room temperature and then the band was visualized by ECL tool (GE Healthcare, UK). Antibodies used were mouse anti-TSG101 (1:200, Santa, sc-7964), rabbit anti-CD9 (1:2,000, Abcam, ab92726), rabbit anti-GM130 (1:1,000, Abcam, ab30637), rabbit anti-CD47 (1:1,000, Abcam, ab175388), mouse anti-GPX4 (1:200, Santa, sc-166570), mouse anti-GAPDH (1:20,000, Proteintech, 60004-1-1g). The secondary antibodies used were horse anti-mouse (1:2,000, Cell Signaling Technology, 7076P2), goat anti-rabbit (1:2,000, Cell Signaling Technology, 7074P2).
Cel-miR-54 mimics loading into exosomes
Exosomes at a protein concentration of 0.5 mg/mL (0.8 mL) (BCA Protein Assay Kit, Thermo Scientific) were electroporated with 0.5 OD cel-miR-54 mimics (GenePharma, Shanghai, China) in 4 mm cuvette at 700 V and 50 μF for 3 pulses. Following electroporation, exosomes were treated with RNase to remove miRNAs that had not been loaded into the exosomes. Then, exosomes were washed with PBS and ultracentrifuged at 100,000 g for 60 min.
Quantitative real-time PCR for exosome phagocytosis in vitro
The exosomes (100 μg) loaded with cel-miR-54 mimics were added to Hepa1-6 cells or RAW264.7 cells cultured in 100-mm culture dish respectively. Then, the cells and exosomes were incubated for 6 h. Total RNA from exosomes or cells was extracted by Trizol (Invitrogen) following the manufacturer's instructions. The concentration of RNA was measured with a Nanodrop-2000 spectrophotometer (Thermo Scientific, USA). For analysis of miRNA expression, 2 μg total RNA was reverse transcribed using miRcute Plus miRNA First-Strand cDNA Kit (KR211-02, TIANGEN, China) and qPCR was performed using FastStart Essential DNA Green Master Kit (06924204001, Roche). U6 was used as an internal control. All kits were used following the manufacturers' instructions. The primer sequences are listed in Table S1.
Quantitative real-time PCR for exosome phagocytosis in the circulation
The exosomes (about 200 μg) loaded with cel-miR-54 mimics were injected into the mice via the tail vein. 2 h and 24 h after injection, blood was collected by removing eyeball and the serum was separated by 3000 g, 5 min centrifuge. Total RNA from the serum was extracted by Trizol. Then the expression of cel-miR-54 was analyzed as previous described.
In vitro and in vivo fluorescence tracing of exosomes
For in vitro tracing of exosomes in RAW264.7 cells and Hepa1-6 cells, exosomes (1 μg/μL, 300 μL) were labeled with DiI (Invitrogen, USA) by incubating with the dye (1 mM) at the ratio of 400:1 in volume for 20 min, followed by exosomes isolation as described above. RAW264.7 cells or Hepa1-6 cells cultured in glass bottom cell culture dish were incubated with DiI-labeled exosomes (about 50 μg) for 6 h. The cells were washed with PBS three times and fixed with 4% paraformaldehyde for 10 min and washed with PBS twice. The cells nuclei were counter-stained with 1 μg/mL Hoechst 33342 (C1022, Beyotime, China) for 10 min in dark room at 37 °C. In the end, the cells were washed with PBS for 3 times and observed using a Nikon A1 Spectral Confocal Microscope (Nikon, Japan).
For in vivo tracing of exosomes, the exosomes (about 200 μg) were labeled with the fluorescent dye DiR (Invitrogen, USA) by incubating with the dye (1 mM) at the ratio of 400:1 in volume and injected into the tumor-bearing mice via the tail vein to analyze the distribution of exosomes. 6 h later, the mice were sacrificed by cervical dislocation, then the organs and tumors were taken out, and the distribution of fluorescently labeled exosomes in various organs was observed using in vivo imaging system (PerkinElmer, USA). To further examine the location of exosomes in different organs and tumors. The exosomes were labeled with the fluorescent dye DiI and injected into the tumor-bearing mice via the tail vein. 6 h later, the mice were sacrificed by cervical dislocation, then, the organs and tumors were taken out. The organs and tumors were stored in the optimal cutting temperature (O.C.T) specimen matrix for cryosections at -20 °C by cryostat, fixed with 4% paraformaldehyde at room temperature for 10 min, washed with PBS three times, and then stained with 1 μg/mL Hoechst 33342 for 10 min. Next, the tissue sections were imaged by Nikon A1 Spectral Confocal Microscope.
Erastin (Er) and Rose Bengal (RB) loading into exosomes
Erastin (MedChemExpress, USA) and Rose Bengal (Sigma-Aldrich, USA) were added to the exosomes (1 mL) and the mixture was sonicated [38] (20% amplitude, 6 cycles, 10 s on/off, 3 min duration, 2 min cooling period between each cycle) by Ultrasonic Homogenizer (Cole-Parmer, USA). The solution was then incubated at 37 °C for 4 h to restore the exosome membrane to form Er/RB@Exos. Er/RB@Exos was obtained by centrifugation and excess Er and RB were eliminated in the supernatant.
Determination of loading capacity
Er and RB were respectively dissolved in DMSO and PBS at a concentration of 10 mM and stored at -20 ℃. To determine the maximum drug loading dose of RB in exosomes, different dose of RB was added to the exosomes (600 μg/mL, 1 mL, total protein) (the group of exosomes) and the same dose RB was added to PBS (the group of RB). Then, the UV-vis absorption spectrum of RB was collected by the Nanodrop-2000 spectrophotometer (Thermo Scientific, USA) and the absorption intensity at the maximum absorption peak was used to calculate the drug concentration. For the group of exosomes, after sonication, different mixtures were centrifuged at 100,000 g for 60 min and then the absorption intensity (A1) of the supernatant at the peak was measured. For the group of RB, the absorption intensity (A2) of the aqueous solution at the peak was also measured. The loading rate of RB in exosomes were calculated by the following equation: loading rate = (A2 - A1) / A2 × 100%. The same method was applied to measure the loading rate of Er in exosomes. In the meantime, the UV-vis absorption spectrum of exosomes was collected by the Nanodrop-2000 spectrophotometer.
Cells experiments
Hepa1-6 cells were cultured in 6-well plates, then different groups of exosomes (about 100 μg) were added to per well and incubated for 6 h and cells without any treatment were used as the control groups. 6 h later, the medium was replaced with fresh medium to remove the rest exosomes that had not been phagocytosed by cells. 6 h later, the light groups were given laser light (532 nm, 10 mW/cm2, 5 min).
MTT assay
Hepa1-6 cells were cultured in 96-well plates, then different groups of exosomes (about 10 μg) were added to per well. 24 h after different treatments, the cell viability was detected by MTT assay according to protocol of MTT kit (ST316, Beyotime, China).
Flow cytometry analysis for lipid ROS
Hepa1-6 cells were cultured in 6-well plates. Cells were treated and then 50 μM C11-BODIPY 581/591 (MX5211-1MG, MKBio, China) was added and incubated for 1 h. Lipid ROS generation was measured by a flow cytometer according to the method of Gao M, et al [39].
Animal experiments
Mice were injected with control or engineered exosomes through the tail vein for two consecutive weeks and the light groups were given laser light (532 nm, 100 mW/cm2, 15 min), twice a day. At the end of the experiment, the mice were sacrificed and the inoculated tumors were removed, and then the tumor volume was measured and histologically analyzed. From the treatment to the end of the experiment, the size of the subcutaneous tumor was measured every two days using a vernier caliper, and the weight change of the mice was recorded.
Total ROS detection
Hepa1-6 cells with density of 104 cells per mL were seeded on glass bottom cell culture dish. Treated cells were incubated with 1μM dihydroethidium (DHE) (S0063, Beyotime) for 30 min at 37 °C, fixed with 4% paraformaldehyde at room temperature for 10 min, then washed with PBS three times and subsequently followed by nuclei staining with 1 μg/mL Hoechst 33342 for 10 min. Images were then captured by Nikon A1 Spectral Confocal Microscope (Nikon, Japan). In order to evaluate the production of superoxide histologically, the tumors cut from the mice were stored in the optimal cutting temperature (O.C.T) specimen matrix (SAKURA, USA) for cryosections at 20 °C by cryostat (Leica, USA). Frozen tumor tissue sections (about 8 μm thick) were stained with 1μM dihydroethidium (DHE) at 37 °C for 30 min, washed with PBS three times, fixed with 10% paraformaldehyde at room temperature for 10 min, washed with PBS three times. Then tissue sections were stained with 1 μg/mL Hoechst 33342 for 10 min. The images were observed under Nikon A1 Spectral Confocal Microscope (Nikon, Japan).
Tumor lipid ROS measurement
In order to evaluate the production of lipid ROS histologically, the tumors cut from the mice were stored in the optimal cutting temperature (O.C.T) specimen matrix (SAKURA, USA) for cryosections at -20 °C by cryostat (Leica, USA). Frozen tumor tissue sections (about 8 μm thick) were stained with 2 μM C11 BODIPY 581/591 at 37 °C for 20 min, washed with PBS three times, fixed with 10% paraformaldehyde at room temperature for 10 min, washed with PBS three times. Then tissue sections were stained with 1 μg/mL Hoechst 33342 for 10 min. The oxidized C11 BODIPY 581/591 were observed under Nikon A1 Spectral Confocal Microscope (Nikon, Japan).
Cytotoxicity evaluation
Mice were injected with PBS (as control), Er/RB@ExosCtrl or Er/RB@ExosCD47 through the tail vein for two consecutive weeks. Blood was collected by removing eyeball, and then the serum was separated by 3000 g, 5 min centrifuge, which was used to detect alanine transaminase (ALT), aspartate transaminase (AST), blood urea nitrogen (BUN), creatinine (Cr), creatine kinase (CK) and creatine kinase-MB (CK-MB). After the mice were sacrificed by cervical dislocation, the organs of livers and kidneys were taken out for H&E staining.
Statistical analysis
The data were expressed as the mean ± SEM. Student t-test was used for two group comparison and one-way ANOVA was used for comparison among more than two groups. The differences were considered statistically significant when the P value was < 0.05.
Results
Engineering and characterization of engineered exosomes
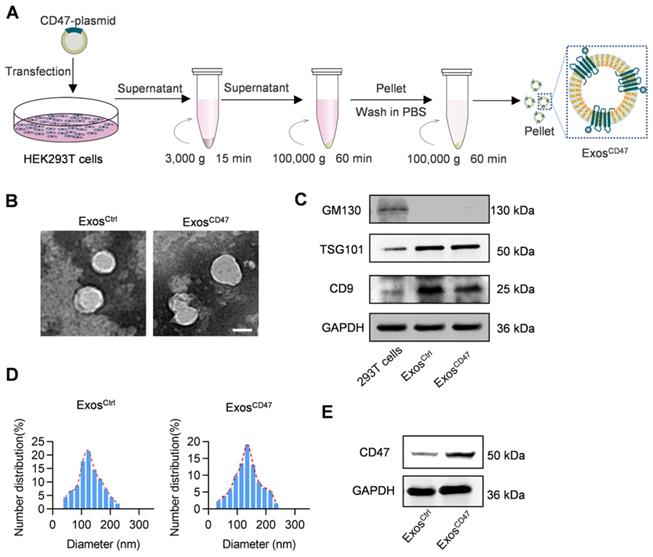
To functionalize the exosomes with CD47 on the surface, the donor cells (HEK293T) were transfected with CD47 overexpressing plasmid (Figure 1A). Transmission electron microscope analysis revealed that the isolated vesicles showed similar morphology as the typical exosomes (Figure 1B). Western blot analysis showed that there were abundant TSG101 and CD9 in both ExosCtrl and ExosCD47. In contrast, the Golgi complex marker GM130 was not found in the isolated exosomes (Figure 1C). Moreover, the expression of CD47 from ExosCD47 increased significantly in comparison with the ExosCtrl (Figure 1E). To further confirm whether CD47 was expressed on the exosomal surface, the exosomes were captured by anti-CD47, followed by western blot analysis of CD9 exosomal marker. As expected, anti-CD47 could capture the exosomes, suggesting that CD47 was mainly expressed on the exosome membrane (Figure S1). As shown in Figure 1D, the diameter of ExosCD47 ranged from 30 to 150 nm, which was indistinguishable from typical exosomes. These data confirmed the exosome feature of ExosCD47.
Preparation and characterization of ExosCD47. (A) Schematic illustration of exosomes preparation. (B) Representative images of transmission electron micrographs of ExosCtrl and ExosCD47. Scale bar = 100 nm, n = 3 samples per group. (C) Western blot analysis of exosome markers TSG101, CD9 and exclusive exosomal marker GM130 on 293T cells, ExosCtrl and ExosCD47. GAPDH served as internal control. (D) Size distribution of exosomes ExosCtrl and ExosCD47. n = 3 samples per group. (E) Western blot analysis of CD47 on exosomes (ExosCtrl and ExosCD47). GAPDH served as internal control.
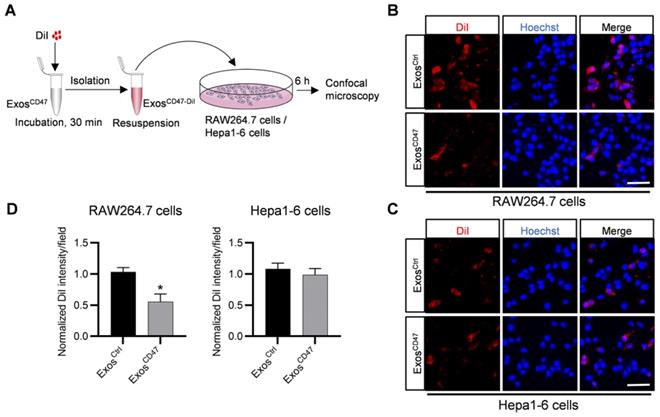
Enhanced tumor targeting of ExosCD47 via mononuclear phagocytic system escape
In order to explore whether ExosCD47 can escape the phagocytosis of the MPS and increase tumor targeting specificity, DiI-labeled ExosCtrl and ExosCD47 were incubated with RAW264.7 cells and Hepa1-6 cells respectively (Figure 2A). As expected, in vitro fluorescence imaging showed that the amount of ExosCD47 phagocytosed by RAW264.7 cells was much less than the ExosCtrl (Figure 2B-D). However, there were no significant differences of the phagocytosis efficiency by tumor cells between ExosCtrl and ExosCD47 (Figure 2C-D). In addition, qPCR tracing of the cel-miR-54-5p encapsulated in the exosomes was also applied to verify these results (Figure S2A). ExosCtrl and ExosCD47 were loaded with same amount of cel-miR-54-5p mimics by electroporation, followed by incubation with RAW264.7 and Hepa1-6 cells (Figure S2A-B). As expected, the expression of cel-miR-54-5p in RAW264.7 in ExosCD47 group was notably less than that in ExosCtrl group (Figure S2C). Moreover, there was no difference of cel-miR-54-5p in Hepa1-6 cells (Figure S2D).
ExosCD47 can escape the phagocytosis from macrophages. (A) Schematic illustration of the experiment. (B-C) Representative confocal fluorescence images of the DiI-labeled exosomes (red) in RAW264.7 cells and Hepa1-6 cells. The nuclei were counter-stained with Hoechst (blue). Scale bar = 20 μm, n = 3 samples per group. (D) Quantification of fluorescence intensity of the DiI-labeled exosomes in RAW264.7 cells and Hepa1-6 cells. n = 3, *, P < 0.05.
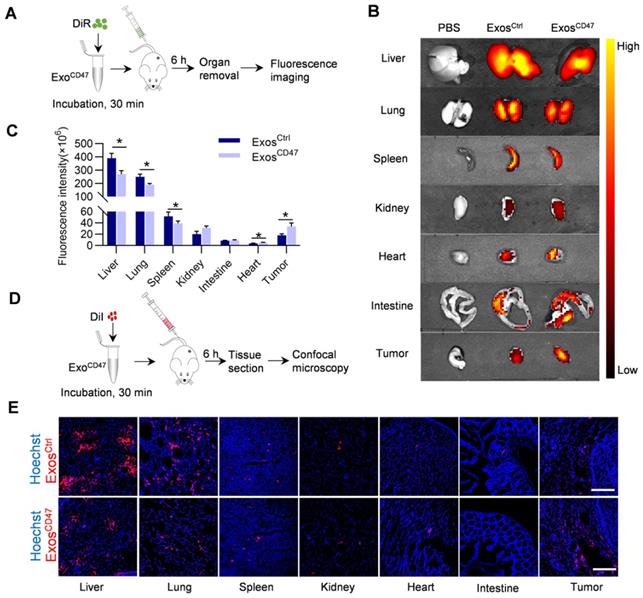
In order to detect the targeted delivery of ExosCD47 in target organs, DiI-labeled or DiR-labeled Exos were injected into the mice via the tail vein. Compared with ExosCtrl, the distribution of ExosCD47 in the livers, lungs and spleens was significantly reduced, and the distribution in the intestines and kidneys was slightly increased. Most importantly, the distribution in tumors was increased, as revealed by ex vivo imaging (Figure 3A-C). Consistent with the ex vivo fluorescent imaging data, confocal imaging analysis of organ tissue sections showed similar results (Figure 3D-E). In addition, the results of Quantitative real-time PCR showed that ExosCD47 had longer circulation time than ExosCtrl (Figure S2A, Figure S2E).
ExosCD47 escape from MPS in vivo. (A) Schematic illustration of the experimental procedure. (B) Representative ex vivo fluorescent images of the DiR-labeled exosomes in tumors and main organs. n = 5 mice. (C) Quantification of fluorescence intensity of different organs and tumors. Data are presented as the mean ± SEM, n = 5, *, P < 0.05. (D) Schematic illustration of the experimental procedure. (E) Representative fluorescence images of the DiI-labeled exosomes (red) in tumors and main organs. The nuclei were counter-stained with Hoechst (blue). Scale bar = 100 μm, n = 5 mice.
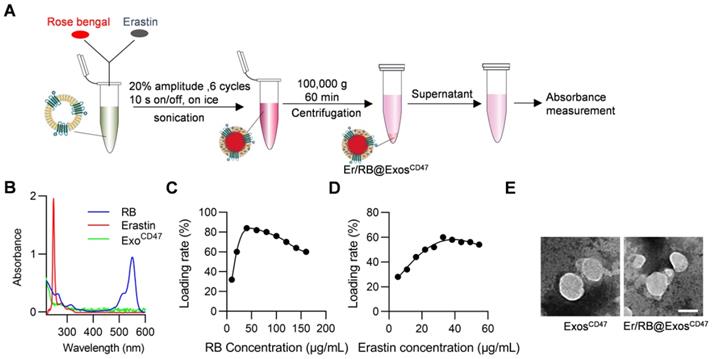
Encapsulation of Er and RB into exosomes via sonication
We next encapsulate Er/RB into the exosomes via sonication (Figure 4A). Er and RB concentrations were determined by the absorption measurement. The maximum absorption peaks of Er and RB in PBS are 249 nm and 550 nm, respectively (Figure 4B). Within a certain concentration range, the results (Figure S3) showed that the intensity and concentration of the maximum absorption peak were linearly related, so we used the intensity of the maximum absorption peak to calculate the concentration of the drugs. When the exosomes at the concentration of 600 μg/mL, the maximum encapsulation rates of RB and Er reached 84% and 60%, respectively. In other words, 32.8 μg Er was loaded per mg exosomes, while the 56 μg RB was loaded per mg exosomes. Importantly, the morphology of the exosomes had no significant change after sonication, as observed by transmission electron microscope (Figure 4E).
Efficient loading of Erastin and RB into exosomes. (A) Schematic illustration of the procedure how Erastin and RB are loaded into exosomes. (B) UV spectroscopy of RB and Erastin in PBS, the maximum UV absorption peak of RB is at 550 nm and Erastin at 249 nm. (C-D) Different amount of RB/Erastin was respectively added to ExosCD47 (600 μg/mL), the curves show the relationship between the loading rate of RB or Erastin in ExosCD47 and its concentration. (E) Representative images of transmission electron micrographs of ExoCD47, Er/RB@ExoCD47. Scale bar = 100 nm, n = 3 samples per group.
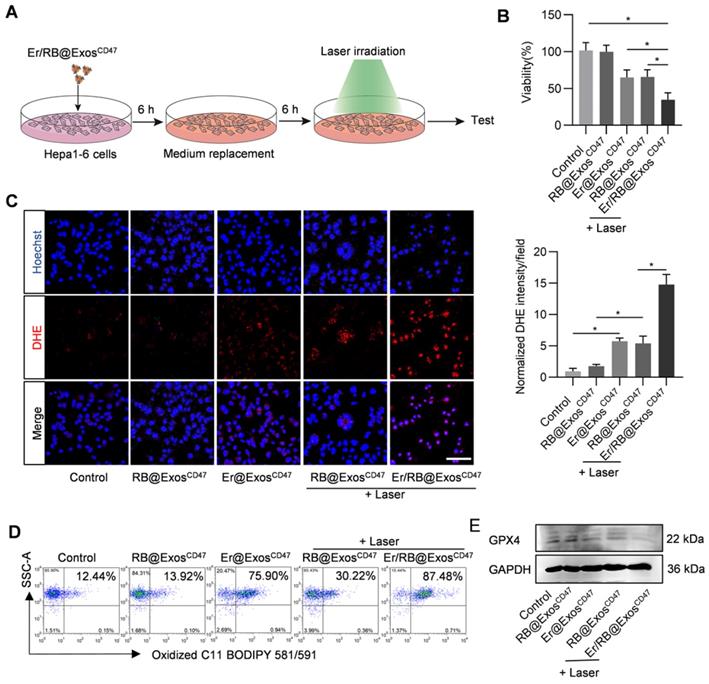
Er/RB@ExosCD47 together with laser irradiation induce ferroptosis in Hepa1-6 cells
In order to verify the therapeutic effect of Er/RB@ExosCD47, Hepa1-6 cells were treated with Control, RB@ExosCD47, Er@ExosCD47, RB@ExosCD47(L) and Er/RB@ExosCD47 (L) (Figure 5A). Er/RB@ExosCD47 (L) significantly induce cell death (Figure 5B). To further confirm whether Er/RB@ExosCD47 (L) induce cell death in the form of ferroptosis, lipid ROS and GPX4 expression were analyzed. As expected, Er@ExosCD47 and RB@ExosCD47(L) treatment moderately increased the ROS (Figure 5C), while the level of ROS in Hepa1-6 cells treated with Er/RB@ExosCD47(L) was significantly higher than that in all other groups. Notably, photosensitizer RB alone had no effect on ROS level of Hepa1-6 cells. Similar results were observed by flow cytometry analysis of C11 BODIPY 581/591 (Figure 5D, Figure S4). Consistent with the increased ROS, GPX4 expression was obviously downregulated by Er@ExosCD47and Er/RB@ExosCD47(L) (Figure 5E, Figure S4). These data indicated that Er/RB@ExosCD47(L) could significantly induce tumor cells ferroptosis and RB@ExosCD47(L) could induce ferroptosis by accumulation of lipid ROS not downregulating the GPX4 expression.
Ferroptosis induction by Er/RB@ExosCD47 combined with PDT. (A) Schematic illustration of the experimental procedure. (B) Cell viability detection by MTT assay. Data are presented as the mean ± SEM, n = 3, *, P < 0.05. (C) Total ROS generation in Hepa1-6 cells after different treatments was detected by fluorescence microscopy, Scale bar = 50 μm. Data are presented as the mean ± SEM, n = 3, *, P < 0.05. (D) Lipid ROS of Hepa1-6 cells detected by flow cytometry. Representative data of three different experiments. (E) Western blot analysis of GPX4 in Hepa1-6 cells after treated with different formulations. GAPDH served as internal control.
Therapeutic effects of Er/RB@ExosCD47 together with PDT in HCC xenograft model
In the following experiments, we explored the therapeutic effect of Er/RB@ExosCD47 combined with laser irradiation in the HCC xenograft model, in which Hepa1-6-luc cells were used. When the tumors reached 80 mm3, the tumor-bearing mice were divided into 5 groups: Control, Control (L), Er@ExosCD47, RB@ExosCD47 (L) and Er/RB@ExosCD47 (L). Exosomes were injected via tail vein every three days, and green laser light irradiation was administered for 15 min in the laser groups twice a day (Figure 6A). After 14 days treatment, the tumor fluorescence signal of the Er/RB@ExosCD47 (L) was the weakest, while the Er@ExoCD47 and RB@ExosCD47(L) was moderately lowered in comparison with Control and Control (L) group (Figure 6B). Accordingly, the tumor volume of in the Er/RB@ExoCD47 (L) group was the smallest (Figure 6C-D).
Therapeutic efficacy of Er/RB@ExosCD47 combined with PDT in vivo. (A) Schematic illustration of the experimental procedure. (B) In vivo representative fluorescence imaging of subcutaneous Hepa1-6-luc tumor-bearing C57BL/6 mice with different treatments. Data are presented as the mean ± SEM, n = 5, *, P < 0.05. (C) Tumor images of mice with different treatment for 14 days. (D) Tumor growth curves of tumor volume after different treatment for 14 days. (E) Representative images of DHE and C11 BODIPY 581/591 staining of tumor sections. Scale bar = 25 μm. n = 5 mice. (F) Western blot analysis of GPX4 in tumors after treated with different formulations. GAPDH served as internal control.
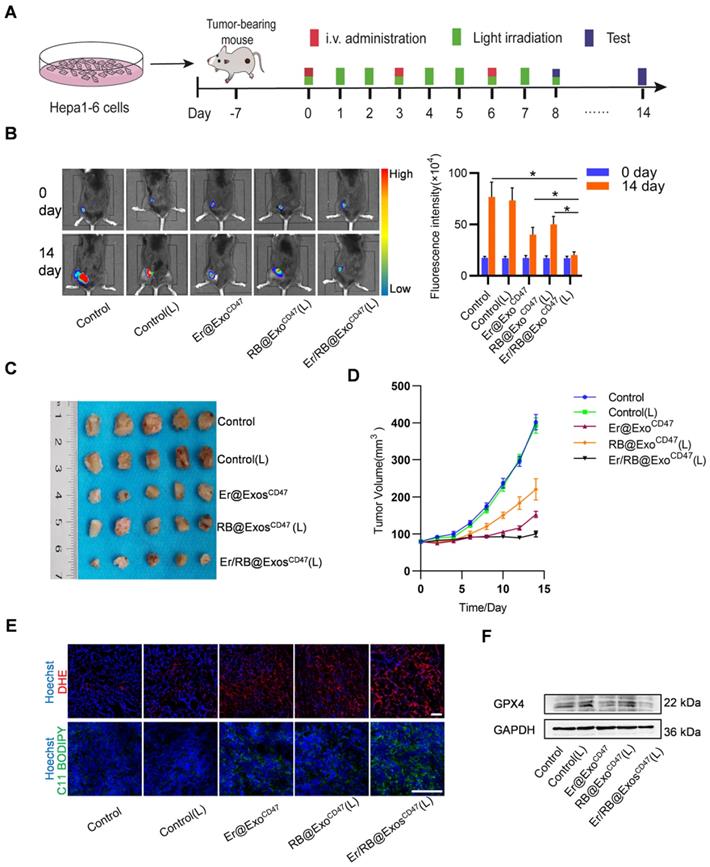
Consistent with the tumor growth inhibition in Er/RB@ExoCD47 (L) group, the total ROS and lipid ROS in tumors of Er/RB@ExoCD47 (L) group was the highest (Figure 6E), suggesting the most potent ferroptosis induction in the group. In addition, GPX4 expression of tumors was evaluated by western blotting. GPX4 expression was obviously downregulated in Er/RB@ExosCD47(L) group, moderately Er/RB@ExosCD47 and slightly RB@ExosCD47(L) compared to Control group (Figure 6F). The results were consistent with that in vitro. The cytotoxicity of Er/RB@ExosCD47 was also evaluated. As shown in Figure S5A. there was much obvious side-effects in liver Er/RB@ExosCtrl treated group, as seen from HE staining (Figure S5A) and biochemistry analysis (Figure S5B-D).
Discussion
In this study, we successfully constructed CD47-functionalized exosomes that could evade MPS and enhance tumor targeting. In addition, we used sonication to efficiently load Er and RB into exosomes, and in vivo and in vitro experiments have verified that it could promote the ferroptosis process of tumors and play anti-tumor effect.
In our research, we used the strategy of inducing ferroptosis to kill tumors. Existing evidence has proved that the method of inducing ferroptosis is to effectively kill cancer cells through the accumulation of ROS in the cells and is iron dependent [10]. It has been reported that gene inactivation or drug inhibition of GPX4 can promote cells ferroptosis [40, 41]. This strategy enhanced ferroptosis by reducing the degradation of ROS and increasing its accumulation in cells. However, GPX4 gene ablation resulted in increased accumulation of oxidized phospholipid products in the kidney, leading to AKI and early death in mice [42]. Furthermore, there have been many recent studies [43-45] that used iron-based nanomaterials as ferroptosis-inducing agents to increase iron supply and enhance ferroptosis to achieve the effect of treating tumors. Unfortunately, these methods could not solve the problem of the harmful toxicity of the vector and the low ROS conversion efficiency.
Herein, we adopted the strategy of ferroptosis inducer-Er combined with PDT to amplify the effect of ferroptosis in tumor treatment. In PDT, molecular oxygen can be converted into reactive oxygen species (ROS) by photosensitizers (PSs) in their respective excited states [46]. We used the large amount of ROS produced in PDT to supplement the accumulation of lipid ROS required for ferroptosis, thereby enhancing the therapeutic effect of ferroptosis. In our research, in vitro and in vivo experiments have fully verified our research assumption. ROS has a short lifespan in biological systems, and the diffusion radius of its generation site (< 30 nm) is limited [47], so PDT selectively targets the position of PS during light irradiation. Therefore, our strategy of combined PDT and chemotherapy could induce efficient ferroptosis in tumors with low dose of drugs, which could not only reduce side effects, but also avoid the biological safety issues of knocking out and inactivating GPX4 in the previous research methods, as well as the low ROS conversion rate and carrier toxicity issues in the iron overloading research method.
Previous studies have shown that exosome was an effective biological carrier for drugs for therapeutic purposes [48]. However, a major obstacle to exosome-based treatment was its phagocytosis by MPS, which limited its clinical application [49]. Therefore, in our study, we designed ExosCD47 as a delivery vehicle for these two drugs. It has been widely reported that CD47 was mainly expressed on the surface of circulating hematopoietic stem cells [36], normal erythrocytes [50] and most types of tumor cells [51]. CD47 was a transmembrane molecule which bound to SIRPα on the surface of macrophages to produce “don't eat me” signal to prevent phagocytosis [52]. Several drug delivery nanocarriers have been used in cancer treatment, but they have certain limitations. For example, nanoparticles have a limited load capacity, which is less than 5%. Another major problem is that the payload will be released suddenly before reaching the predetermined position [22]. Another drug delivery carrier, gold nanoparticles have problems such as biodistribution and cationic ligand toxicity [23]. The ExosCD47 that we successfully constructed have been verified in vivo distribution experiments to evade the phagocytosis of MPS, and increase the distribution in the tumor. And through our experimental verification, the encapsulation rate of each drug in this bio-delivery carrier had reached 60%. In the experiment, we used CD47-overexpressed exosomes to load both Er and RB at the same time instead of simply loading one drug. On the one hand, this experimental design can better exert the superimposed effect of the combination of the two methods. On the other hand, the operability is more convenient and safer.
Although our treatment method had a good therapeutic effect on tumors in vitro and in vivo, we still need to conduct further experimental research in the future. For example, according to the current technology, it is not possible to prepare exosomes in large quantities. In view of this, exploring new technologies to generate exosomes in sufficient quantities will be a major research project in our future.
In conclusion, we have engineered an exosome armed with three moieties, surface functionalization with CD47, membrane loading with ferroptosis inducer Er, and core with photosensitizer RB. By using the HCC xenograft model, we here showed that the designed exosomes display potent anti-tumor therapeutic effects, and amazing low toxicity. Thus, the engineered exosomes would provide a promising strategy to treat malignant tumors.
Supplementary Material
Supplementary figures and table.
Acknowledgements
We are grateful to the technical help from Zhenzhen Hao from the Department of Biochemistry and Molecular Biology, Fourth Military Medical University.
Funding sources
This study was funded by grants from the National Natural Science Foundation of China (NO.81672341, 82072658) and Shaanxi Provincial Natural Science Foundation (2020SF-079).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71:7-33
2. Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391:1301-14
3. Inarrairaegui M, Melero I, Sangro B. Immunotherapy of Hepatocellular Carcinoma: Facts and Hopes. Clin Cancer Res. 2018;24:1518-24
4. Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell. 2019;35:830-49
5. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714-26
6. Su Z, Yang Z, Xie L, DeWitt JP, Chen Y. Cancer therapy in the necroptosis era. Cell Death Differ. 2016;23:748-56
7. Zhang W, Wang F, Hu C, Zhou Y, Gao H, Hu J. The progress and perspective of nanoparticle-enabled tumor metastasis treatment. Acta Pharm Sin B. 2020;10:2037-53
8. Mou Y, Wang J, Wu J, He D, Zhang C, Duan C. et al. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol. 2019;12:34
9. Dixon SJ. Ferroptosis: bug or feature? Immunol Rev. 2017;277:150-7
10. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060-72
11. Wang SF, Liao HW, Li FY, Ling DS. A mini-review and perspective on ferroptosis-inducing strategies in cancer therapy. Chinese Chemical Letters. 2019;30:847-52
12. Jiang Q, Wang K, Zhang XY, Ouyang BS, Liu HX, Pang ZQ. et al. Platelet Membrane-Camouflaged Magnetic Nanoparticles for Ferroptosis-Enhanced Cancer Immunotherapy. Small. 2020 16. doi: 10.1002/smll.202001704
13. Hao S, Yu J, He W, Huang Q, Zhao Y, Liang B. et al. Cysteine Dioxygenase 1 Mediates Erastin-Induced Ferroptosis in Human Gastric Cancer Cells. Neoplasia. 2017;19:1022-32
14. Yamaguchi Y, Kasukabe T, Kumakura S. Piperlongumine rapidly induces the death of human pancreatic cancer cells mainly through the induction of ferroptosis. Int J Oncol. 2018;52:1011-22
15. Guo J, Xu B, Han Q, Zhou H, Xia Y, Gong C. et al. Ferroptosis: A Novel Anti-tumor Action for Cisplatin. Cancer Res Treat. 2018;50:445-60
16. Sun X, Niu X, Chen R, He W, Chen D, Kang R. et al. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology. 2016;64:488-500
17. Timmerman LA, Holton T, Yuneva M, Louie RJ, Padro M, Daemen A. et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell. 2013;24:450-65
18. Yu M, Gai C, Li Z, Ding D, Zheng J, Zhang W. et al. Targeted exosome-encapsulated erastin induced ferroptosis in triple negative breast cancer cells. Cancer Sci. 2019;110:3173-82
19. Dolmans DE, Fukumura D, Jain RK. Photodynamic therapy for cancer. Nat Rev Cancer. 2003;3:380-7
20. Wang B, Wang JH, Liu Q, Huang H, Chen M, Li K. et al. Rose-bengal-conjugated gold nanorods for in vivo photodynamic and photothermal oral cancer therapies. Biomaterials. 2014;35:1954-66
21. Peng Q, Zhang S, Yang Q, Zhang T, Wei XQ, Jiang L. et al. Preformed albumin corona, a protective coating for nanoparticles based drug delivery system. Biomaterials. 2013;34:8521-30
22. Couvreur P. Nanoparticles in drug delivery: past, present and future. Adv Drug Deliv Rev. 2013;65:21-3
23. Arvizo R, Bhattacharya R, Mukherjee P. Gold nanoparticles: opportunities and challenges in nanomedicine. Expert Opinion on Drug Delivery. 2010;7:753-63
24. Chen R, Xu X, Tao Y, Qian Z, Yu Y. Exosomes in hepatocellular carcinoma: a new horizon. Cell Commun Signal. 2019;17:1
25. Roma-Rodrigues C, Raposo LR, Cabral R, Paradinha F, Baptista PV, Fernandes AR. Tumor Microenvironment Modulation via Gold Nanoparticles Targeting Malicious Exosomes: Implications for Cancer Diagnostics and Therapy. Int J Mol Sci. 2017; 18. doi: 10.4143/crt. 2016 572
26. Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W. et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018;560:382-6
27. Pegtel DM, Gould SJ. Exosomes. Annu Rev Biochem. 2019;88:487-514
28. Kamerkar S, LeBleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA. et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017;546:498-503
29. Qiu X, Li Z, Han X, Zhen L, Luo C, Liu M. et al. Tumor-derived nanovesicles promote lung distribution of the therapeutic nanovector through repression of Kupffer cell-mediated phagocytosis. Theranostics. 2019;9:2618-36
30. Ni D, Wei H, Chen W, Bao Q, Rosenkrans ZT, Barnhart TE. et al. Ceria Nanoparticles Meet Hepatic Ischemia-Reperfusion Injury: The Perfect Imperfection. Adv Mater. 2019;31:e1902956
31. Ni D, Ferreira CA, Barnhart TE, Quach V, Yu B, Jiang D. et al. Magnetic Targeting of Nanotheranostics Enhances Cerenkov Radiation-Induced Photodynamic Therapy. J Am Chem Soc. 2018;140:14971-9
32. Ni D, Jiang D, Ehlerding EB, Huang P, Cai W. Radiolabeling Silica-Based Nanoparticles via Coordination Chemistry: Basic Principles, Strategies, and Applications. Acc Chem Res. 2018;51:778-88
33. Li Z, Zhou X, Gao X, Bai D, Dong Y, Sun W. et al. Fusion protein engineered exosomes for targeted degradation of specific RNAs in lysosomes: a proof-of-concept study. J Extracell Vesicles. 2020;9:1816710
34. Chao MP, Weissman IL, Majeti R. The CD47-SIRPalpha pathway in cancer immune evasion and potential therapeutic implications. Curr Opin Immunol. 2012;24:225-32
35. Kibria G, Ramos EK, Lee KE, Bedoyan S, Huang S, Samaeekia R. et al. A rapid, automated surface protein profiling of single circulating exosomes in human blood. Sci Rep. 2016;6:36502
36. Jaiswal S, Jamieson CH, Pang WW, Park CY, Chao MP, Majeti R. et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138:271-85
37. Li Z, Zhou X, Wei M, Gao X, Zhao L, Shi R. et al. In Vitro and in Vivo RNA Inhibition by CD9-HuR Functionalized Exosomes Encapsulated with miRNA or CRISPR/dCas9. Nano Lett. 2019;19:19-28
38. Kim MS, Haney MJ, Zhao Y, Mahajan V, Deygen I, Klyachko NL. et al. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomedicine. 2016;12:655-64
39. Gao MH, Monian P, Pan QH, Zhang W, Xiang J, Jiang XJ. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26:1021-32
40. Hu CL, Nydes M, Shanley KL, Morales Pantoja IE, Howard TA, Bizzozero OA. Reduced expression of the ferroptosis inhibitor glutathione peroxidase-4 in multiple sclerosis and experimental autoimmune encephalomyelitis. J Neurochem. 2019;148:426-39
41. Li C, Deng X, Xie X, Liu Y, Friedmann Angeli JP, Lai L. Activation of Glutathione Peroxidase 4 as a Novel Anti-inflammatory Strategy. Front Pharmacol. 2018;9:1120
42. Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ. et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180-91
43. Shen Z, Liu T, Li Y, Lau J, Yang Z, Fan W. et al. Fenton-Reaction-Acceleratable Magnetic Nanoparticles for Ferroptosis Therapy of Orthotopic Brain Tumors. ACS Nano. 2018;12:11355-65
44. Zhang C, Bu WB, Ni DL, Zhang SJ, Li Q, Yao ZW. et al. Synthesis of Iron Nanometallic Glasses and Their Application in Cancer Therapy by a Localized Fenton Reaction. Angew Chem Int Edit. 2016;55:2101-6
45. Zheng DW, Lei Q, Zhu JY, Fan JX, Li CX, Li C. et al. Switching Apoptosis to Ferroptosis: Metal-Organic Network for High-Efficiency Anticancer Therapy. Nano Lett. 2017;17:284-91
46. Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO. et al. Photodynamic therapy of cancer: an update. CA Cancer J Clin. 2011;61:250-81
47. Moan J, Berg K. The photodegradation of porphyrins in cells can be used to estimate the lifetime of singlet oxygen. Photochem Photobiol. 1991;53:549-53
48. Barile L, Vassalli G. Exosomes: Therapy delivery tools and biomarkers of diseases. Pharmacol Ther. 2017;174:63-78
49. Yong SB, Song Y, Kim HJ, Ain QU, Kim YH. Mononuclear phagocytes as a target, not a barrier, for drug delivery. J Control Release. 2017;259:53-61
50. Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science. 2000;288:2051-4
51. Matlung HL, Szilagyi K, Barclay NA, van den Berg TK. The CD47-SIRPα signaling axis as an innate immune checkpoint in cancer. Immunol Rev. 2017;276:145-64
52. Logtenberg MEW, Scheeren FA, Schumacher TN. The CD47-SIRPα Immune Checkpoint. Immunity. 2020;52:742-52
Author contact
![]() Corresponding authors: Desheng Wang, Department of Hepatobiliary Surgery, Xijing Hospital, Fourth Military Medical University, Xi'an, 710032, China, email: wangdeshcom; Tel: +860293375015. Qiang Hao, The State Key Laboratory of Cancer Biology, Biotechnology Center, School of Pharmacy, Fourth Military Medical University, Changlexi Road NO.169th, 710032, Xi'an, China, email: haosuqedu.cn; Tel: +862984774516, Fax: +862984774937.
Corresponding authors: Desheng Wang, Department of Hepatobiliary Surgery, Xijing Hospital, Fourth Military Medical University, Xi'an, 710032, China, email: wangdeshcom; Tel: +860293375015. Qiang Hao, The State Key Laboratory of Cancer Biology, Biotechnology Center, School of Pharmacy, Fourth Military Medical University, Changlexi Road NO.169th, 710032, Xi'an, China, email: haosuqedu.cn; Tel: +862984774516, Fax: +862984774937.