Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(15):7491-7506. doi:10.7150/thno.59196 This issue Cite
Research Paper
Microbial and genetic-based framework identifies drug targets in inflammatory bowel disease
Zhihan Wang1,2*, Kai Guo3*, Pan Gao2,4, Qinqin Pu2, Ping Lin2,5, Shugang Qin2,6, Na Xie1, Junguk Hur2, Changlong Li1 ![]() , Canhua Huang1,6
, Canhua Huang1,6 ![]() , Min Wu2
, Min Wu2 ![]()
1. West China School of Basic Medical Sciences & Forensic Medicine, Sichuan University, Chengdu, Sichuan 610041, China.
2. Department of Biomedical Sciences, School of Medicine and Health Sciences, University of North Dakota, Grand Forks, ND 58202, USA.
3. Department of Neurology, University of Michigan, Ann Arbor, MI 48109, USA.
4. Medical Research Institute, Wuhan University, Wuhan 430071, China.
5. State Key Laboratory of Trauma, Burns and Combined Injury, Institute of Surgery Research, Daping Hospital, Army Medical University, Chongqing 400038, China.
6. State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University, and Collaborative Innovation Center for Biotherapy, Chengdu, Sichuan 610041, China.
*These authors contributed equally to this work.
Received 2021-2-8; Accepted 2021-5-14; Published 2021-6-1
Abstract

Rationale: With increasing incidence and prevalence of inflammatory bowel disease (IBD), it has become one of the major public health threats, and there is an urgent need to develop new therapeutic agents. Although the pathogenesis of IBD is still unclear, previous research has provided evidence for complex interplays between genetic, immune, microbial, and environmental factors. Here, we constructed a gene-microbiota interaction-based framework to discover IBD biomarkers and therapeutics.
Methods: We identified candidate biomarkers for IBD by analyzing the publicly available transcriptomic and microbiome data from IBD cohorts. Animal models of IBD and diarrhea were established. The inflammation-correlated microbial and genetic variants in gene knockout mice were identified by 16S rRNA sequences and PCR array. We performed bioinformatic analysis of microbiome functional prediction and drug repurposing. Our validation experiments with cells and animals confirmed anti-inflammatory properties of a drug candidate.
Results: We identified the DNA-sensing enzyme cyclic GMP-AMP synthase (cGAS) as a potential biomarker for IBD in both patients and murine models. cGAS knockout mice were less susceptible to DSS-induced colitis. cGAS-associated gut microbiota and host genetic factors relating to IBD pathogenesis were also identified. Using a computational drug repurposing approach, we predicted 43 candidate drugs with high potency to reverse colitis-associated gene expression and validated that brefeldin-a mitigates inflammatory response in colitis mouse model and colon cancer cell lines.
Conclusions: By integrating computational screening, microbiota interference, gene knockout techniques, and in vitro and in vivo validation, we built a framework for predicting biomarkers and host-microbe interaction targets and identifying repurposing drugs for IBD, which may be tested further for clinical application. This approach may also be a tool for repurposing drugs for treating other diseases.
Keywords: Inflammatory bowel disease, cyclic GMP-AMP synthase (cGAS), host transcriptome-microbiome interaction, drug repurposing, brefeldin-a
Introduction
Inflammatory bowel disease (IBD), including ulcerative colitis (UC) and Crohn's disease (CD), is a complex chronic inflammatory disease, typically manifested with diarrhea and weight loss. Mucosal inflammation in CD can affect any portion of the gastrointestinal tract, while UC is restricted to the colon. IBD has become an urgent public health threat, affecting 1.6 million Americans with 70,000 new cases each year and more than 3.5 million people worldwide [1]. To date, the etiology of IBD remains incompletely understood though it is linked to genetic susceptibility, immunoregulatory factors, microbiome imbalance, and environmental factors [2].
Transcriptome analysis and microbiota profiling have emerged as promising approaches to assess host gene expression and survey microbiota composition quantitatively [3]. Understanding of host-microbiome interactions in IBD pathogenesis is a critical avenue for therapeutic development. Genome-wide association studies (GWAS) have identified approximately 240 driver genes of IBD through analyzing a large number of patients [4]. Many of these genes are associated with key immunological pathways, such as host-microbial interactions, innate immunity, adaptive immunity and autophagy [5]. For example, NOD2-associated dysregulation of microbial sensing contributes to microflora dysbiosis [6] and CARD9 impacts colitis by altering gut microbiota metabolism [7]. ATG16L1 affects autophagy in Paneth cells and goblet cells and regulates intestinal inflammation in an IL-22-dependent manner [8]. Moreover, NOD2 and ATG16L1 are required for human commensal Bacteroides fragilis-mediated protection from colitis [9]. Intestinal inflammation in Card9-/- mice is attenuated after inoculation of Lactobacillus strains [7]. As part of the Integrative Human Microbiome Project (iHMP), the IBD Multi'omics database (IBDMDB) is the first integrated study supporting a systems-level understanding of gut microbiome in IBD dynamics [10]. Altogether, the interactions between host and enteric microbiota may guide rapid development of treatments to improve intestinal health.
Innate immunity plays a vital role in the homeostasis of the intestine during the pathogenesis of IBD. Cyclic GMP-AMP synthase (cGAS) is a double-stranded DNA sensor and is essential for eliciting immune responses [11]. cGAS recognizes the DNA from various invading microbes to activate the adaptor, stimulator of the interferon genes (STING), and induce type I interferon (IFN) secretion following phosphorylation of interferon regulatory transcription factor 3 (IRF3) by TANK binding kinase 1 (TBK1), and other pro-inflammatory cytokine production through NF-κB nuclear translocation [11]. Intense research in recent years has implicated that cGAS signaling plays a role in the initiation and pathogenesis of autoimmune diseases, cancers, Aicardi-Gourtières syndrome (AGS), skin cancer, acute pancreatitis, and sepsis [12]. Although the role of STING in regulating intestinal inflammation [13] and tumorigenesiss [14] in mice has been reported, the effect of cGAS on commensal microbiota in intestinal homeostasis remains to be determined.
Application of current IBD drugs is severely limited by low efficacy, side effects or intolerance, and particularly a drastic loss of response after long-term use [15]. Since the discovery and development of new medications is time-consuming and labor-intensive, drug repurposing or drug repositioning becomes an attractive strategy for identifying new uses of 'old' drugs to potentially treat many debilitating diseases [16]. For instance, denosumab is an antibody to TNFSF11 and has been used in osteoporosis of postmenopausal women [16]. Since the TNFSF11 genetic variant was found to be associated with CD by GWAS, denosumab has been repurposed for treating CD and completed phase 1/2 trials in 2019 [17]. A large-scale computational method [18] demonstrates the potency to reverse disease signatures via drug discovery based on the Library of Integrated Network-Based Cellular Signatures (LINCS), a drug perturbation database maintained by the Broad Institute [19]. The Connectivity Map Linked User Environment (CLUE) platform of LINCS is used to indicate treatment possibility of IBD by analyzing relationships between drugs and differentially expressed genes (DEGs) [20].
Here, we established a novel microbiota and genetic-based framework using a system bioinformatics approach based on host-microbiome interactions and laboratory studies. We identified cGAS as a potential biomarker in IBD, and knockout of cGAS in mice attenuated the inflammatory response. Next, we predicted IBD candidate therapeutics and validated the efficacy of the top candidate, brefeldin-a (BFA, a membrane protein transporter inhibitor), using colon cancer cell lines and mouse models. Our strategy may be applied to potentially reposition candidate drug targets and drugs, promoting drug discovery and medical improvement for other diseases.
Methods
Integrated Microbiome and Host Transcriptome Analysis
Publicly available RNA-seq and microbiome data on human IBD were collected from IBDMDB (http://ibdmdb.org) [10] (Table S1) and imported into R software version 4.0.2 [21]. The RNA-seq data were then normalized using a regularized log (rlog) transformation implemented in the DESeq2 package [22]. Principal component analysis (PCA) of the host RNA-seq profile based on variance stabilizing transformation data was used to examine the overall similarity among all groups.
Only data from patients diagnosed as UC and non-IBD were used for integrated analyses, and a total of 4,811 DEGs were identified from the RNA-seq data with the adjusted p-value < 0.01 cutoff. Relative abundances of bacteria were calculated for microbiome data. Bacteria were filtered out with a relative abundance value < 0 in only three samples or less, and 169 bacteria were found for the following analysis.
We used two datasets from 43 common patients for the integrative analysis. Two-way orthogonal partial least squares (O2PLS), a method for pair-wise integration of multi-omics data, was used to identify novel key pathways and biomarkers that strongly correlated with the disease progression among different data matrix projects [23]. The loading values for the joint covariance part from the O2PLS analysis results were extracted to find highly inter-associated genes and bacteria. Reactome [24] functional enrichment analysis for the top 200 genes (around 5% of the total DEGs) with the highest loading values was performed with the richR (https://github.com/hurlab/richR) package.
Mice
Wild-type (WT) C57BL/6NHsd mice were obtained from ENVIGO (Indianapolis, IN). Cgastm1a(EUCOMM)Hmgu mice (Cgas-/- or cGAS KO mice) were kindly provided by Dr. Charlie Rice (Rockefeller University, NY) [25]. All mice were housed in the animal facility at the University of North Dakota (UND) and treated according to guidelines approved by the Institutional Animal Care and Use Committee (IACUC) of UND. The age- and gender-matched mice (8-12 weeks old) were randomly allocated to different experimental groups based on their genotypes.
Mice Treatments
The experimental model of acute colitis induced by dextran sulfate sodium (DSS, 3% wt/vol, 36,000-50.000 kD; MP Biomedicals, LLC, Aurora, OH) was administered in drinking water ad libitum for seven days, following four days of normal distilled water. Control mice were fed with distilled water for 11 days. Daily monitoring of mouse body weight, survival rate, and disease activity index (DAI), with a score of 1-4, including three parameters: weight loss, fecal blood, and stool consistency (0: no weight loss, no blood, normal stool; 1: 1-5% weight loss; 2: 5-10% weight loss, visible blood in the stool, loose stool; 3: 10-20% weight loss; 4: >20% weight loss, gross bleeding, diarrhea). Mice were sacrificed to collect intestine tissue and feces for further analysis at day 9. Colon lengths were also measured at autopsy. Fecal samples were collected for microbiome analysis.
For the diarrheal model induced by antibiotic treatment, a broad-spectrum antibiotic cocktail or individual antibiotic (5 mg/mL streptomycin, 0.5 mg/mL vancomycin, 1 mg/mlL colistin, and 1 mg/mL Ampicillin) (Sigma-Aldrich, St. Louis, MO) was added to drinking water for five days until analysis, as described previously [26]. Streptomycin (STR) is a broad-spectrum antibiotic inhibiting both gram-positive and gram-negative bacteria, vancomycin (VAN) targets gram-positive bacteria, colistin (COL) targets Enterobacteriaceae, and ampicillin (AAM) targets gram-positive and some gram-negative bacteria [27].
Myeloperoxidase (MPO) Assay
Colonic MPO levels were measured to determine the degree of neutrophil infiltration. The colon was weighed and homogenized in 50 mM hexadecyl trimethyl ammonium bromide (HTAB) - 50 mM potassium phosphate (pH 6.0) buffer. The homogenate was centrifuged at 13,400 × g for 6 min at 4 °C. The supernatant was collected or restored at -80 °C until use. 7 μL of tissue homogenate was mixed with 200 μL of reaction buffer (16.7 mg/mL o-dianisidine dihydrochloride, 50 mM potassium phosphate buffer [pH 6.0], 0.0006% H2O2) and was read at 450 nm using a spectrophotometer at 30 s intervals as previously performed [28].
Enzyme-linked Immunosorbent Assay (ELISA)
Colonic intestinal tissue lysed with radiation immunoprecipitation (RIPA) buffer (Thermo Scientific, Waltham, MA) for cytokine analysis. The lysate was centrifuged at 15000 × g for 10 min, and then the supernatant was collected. Paired (capture and detection) antibodies and standard recombinant mouse IL-1β (eBioscience, San Diego, CA), TNF-α (eBioscience), and IFN-β (Invitrogen, Waltham, MA) were used to determine cytokine concentrations following the manufacturer's instructions [29].
Immunoblotting
Mouse colon biopsies were freeze-thawed in RIPA buffer with protease and phosphatase inhibitor cocktail (ThermoFisher Scientific). The supernatant's protein concentration was quantified using the Pierce BCA protein assay kit (ThermoFisher Scientific). We resuspended the samples in 5X loading buffer (ThermoFisher Scientific) and heated to 100 ℃ for 10 min. An equal amount of samples was resolved by SDS-PAGE gel electrophoresis and transferred to the nitrocellulose membranes (Cytiva Amersham, LLC, Marlborough, MA). After blocked in TBS-T buffer supplemented with 5% blotting-grade non-fat milk at room temperature for 1.5 h, the membrane was subsequently incubated with antibodies against cGAS (D3O8O, Cell Signaling Technology [CST], Danvers, MA), STING (D2P2F, CST), p-TBK1 (D52C2, CST), TBK1 (D1B4, CST), TNF-α (D2D4, CST), NF-κB (D14E12, CST), Casp-1 (pro, sc-56036, Santa Cruz Biotechnology, Dallas, TE; cleaved, D57A2, CST), IL-1β (pro, D3H1Z, CST; cleaved, 52718S, CST), and β-actin (sc-517582, Santa Cruz), followed with the appropriate HRP-conjugated secondary antibody (Invitrogen) incubation, and detected by enhanced chemiluminescence (Cytiva Amersham) [30].
Quantitative Polymerase Chain Reaction (qPCR)
Total RNA was extracted from mouse colon tissue homogenates with TRIzol reagent (Invitrogen), reverse transcribed into cDNAs by the Superscript III kit (11752, Invitrogen), and real-time PCR was performed using SYBR Green Master Mix (A25742, Applied Biosystems, LLC, Marlborough, MA) on the CFX Connect real-time PCR detection system (Bio-Rad, Hercules, California) as previously described [31]. The primer sequences are as follows: Cgas F: 5'- CCACTGAGCTCACCAAAGAT-3'; Cgas R: 5'- CAGGCGTTCCACAACTTTATTC-3'; Sting F: 5'- GGTCACCGCTCCAAATATGTAG-3'; Sting R: 5'- CAGTAGTCCAAGTTCGTGCGA-3'; Ifn-β F: 5'- TCCGAGCAGAGATCTTCAGGAA-3'; Ifn-β R: 5'- TGCAACCACCACTCATTCTGAG-3'; Ifit-1 F: 5'- CGTAGCCTATCGCCAAGATTTA-3'; Ifit-1 R: 5'- AGCTTTAGGGCAAGGAGAAC-3'; Gapdh F: 5'- TTGCTGTTGAAGTCGCAGGAG-3'; Gapdh R: 5'- TGTGTCCGTCGTGGATCTGA -3'. These genes were normalized to the housekeeping gene Gapdh using 2- ΔΔCT method.
Histology and Immunofluorescence
Colon samples were extracted and washed with cold PBS, then immediately fixed with 10% formalin for 24 h and embedded in paraffin. 5-μm histological sections were deparaffinized and stained with hematoxylin and eosin (H&E) or Periodic acid-Schiff (PAS) and analyzed by light microscopy (Nikon Eclipse 80i). Histological assessment of colitis was performed with blinded genotypes as previously described [32, 33]. The score criteria (0-3) included monocyte infiltration, epithelial injury, polymorphonuclear cell filtration, and crypt hyperplasia.
For immunofluorescence analysis, the mouse colon was immersed in the optimal cutting temperature (OCT) medium mold, snap-frozen in liquid nitrogen, and stored at -80 °C. 4 μm frozen sections were fixed in pre-chilled acetone for 5 min, stained with primary antibody MUC5AC (sc-21701, Santa Cruz), and then incubated with fluorescent-labeled secondary antibody (R&D Systems, Minneapolis, MN). DNA was stained with 20 µg/mL 4',6'-diamidino-2-phenylindole (DAPI, Invitrogen) [34]. Zeiss LSM-510 Meta Confocal Microscope was used to obtain fluorescent images.
Fecal DNA Extraction and 16S rRNA Library Preparation
According to the manufacturer, bacterial genomic DNA was extracted from approximately 0.2 g of frozen fecal samples using the QIAamp Fast DNA Stool Mini Kit (51604, Qiagen, Hilden, Germany). As previously described [35], the V3 and V4 hypervariable regions of the 16S rRNA gene were amplified using the following primers: F 5'-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG-3', R 5'-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC-3', and incorporated to Illumina (San Diego, CA) adapters and indexing barcodes. The amplified, barcoded products were purified using AMPure XP beads (Beckman Coulter, Indianapolis, IN) and pooled and sequenced on the Illumina MiSeq platform.
16S rRNA Sequencing Analysis
The raw pair-end sequencing data in FASTQ files were first assessed for quality by FastQC (version 0.11.3) [36] and filtered with Trimmomatic (version 0.36) [37] to remove low-quality reads (threshold quality < 30). Trimmed reads were assessed again for quality by FastQC. Taxonomy classification for trimmed reads was performed by Kraken 2 (version 2.0.8-beta) [38] based on the MiniKraken2_v1 database (version 201904), and sample report files were generated. Alpha diversity metrics based on bacterial counts at the genus level were performed using the estimate richness function within the phyloseq package (version 1.32.0) [39] and Shannon and Simpson indexes to measure the richness of the communities within samples. Principal Coordinates Analysis (PCoA) plot using Bray-Curtis dissimilarity was implemented based on bacterial counts at the genus level to reveal the differences between various groups. The 'Adonis' feature from vegan (version 2.5-6) was used to assess whether sample grouping by metadata factor accounted for inter-sample differences (https://CRAN.R-project.org/package=vegan). A significance P-value was generated by comparing that obtained by R2 to that obtained from 1000 random data permutations. Treemaps were generated from the genus percentage in report files by the treemapify package (version 2.5.3) (https://CRAN.R-project.org/package=treemapify) to show the relative abundance between different groups. Differential abundance analysis was performed using the DESeq2 package (version 1.28.1) with p < 0.05 and |log2Fold Change| > 1 based on bacterial counts at the genus level, and adjusted p < 0.05 at the species level [22]. The taxon sets for TSEA were downloaded from MicrobiomeAnalyst, designed to predict biologically or ecologically meaningful patterns from public datasets and known microbial signatures [40]. The TSEA of biologically meaningful taxon sets were performed with the richR package for predictive potential microbial targets to develop effective therapies.
Relative Gene Expression Analysis by PCR Arrays
The real-time PCR primer sets of 85 genes (Table S7) were coated on 96-well plates (Eurofins Genomics) following the manufacturer's instructions, optimized according to the mouse RT2 Profiler PCR Array (PAMM-169Z, Qiagen) and relevant literature [41]. After normalizing the three housekeeping genes (Eef2, Gapdh, and Tbp), the qPCR results were expressed as relative fold change over the WT control. The detection limit was 40 Ct. All experiments were performed in three biological replicates. The mouse transcriptomes were clustered according to genotype. Volcano plots and the heatmap were generated in the R package ggplot2 with a cut-off of fold change > 2 and p-value < 0.05.
Drug Repurposing Using the LINCS Drug-perturbation Data
DEGs from PCR arrays were first sorted by log2FC values. The up-regulated and down-regulated genes were then chosen to identify drugs and compounds against the LINCS database using the Connectivity Map Linked User Environment (CLUE) platform [42]. The drug connectivity score (CS) with a negative value smaller than -80 was used to identify candidates and compounds.
Cell Culture
Murine macrophage RAW264.7 cells and Human colon epithelial carcinoma cell lines, SW480 (ATCC, CCL-228) and DLD-1 (ATCC, CCL-221) cells were maintained in RPMI 1640 (Gibco, #A1049101) and DMEM/F12 (Gibco, #11330032) medium, supplemented with 10% fetal bovine serum, 1X penicillin-streptomycin solution (Gibco, #15140163) at 37 °C with 5% CO2.
Anti-inflammatory Assay
The cytotoxicity of brefeldin-a (BFA, MedChemExpress, HY-16592) and flubendazole (FLU, MedChemExpress, HY-B0294) on SW480 and DLD-1 cells was assessed by cell counting kit-8 (CCK-8) assay (APExBIO, #K1018). Cells were cultured overnight in 96-well (1 × 104 cells/well) plate. Drugs were diluted in distilled water to reach final concentrations of 0.01, 0.1, 1, 10, and 25 µM and 0.05% DMSO as the negative control. Plates were cultured with the fresh drug-containing medium at 37 °C for 24 h. The absorbance of each well was determined by using a 96-well multiscanner. After subtracting background absorption, the results were expressed as a percentage of viability relative to control cultures. Drug concentrations that inhibited cell viability by 50% (IC50) were determined using GraphPad Prism 8 software.
RAW264.7 and SW480 cells were plated in 6-well (5 × 105 cells/well) plates for ELISA or RNA/protein isolation, respectively. To evaluate the anti-inflammation efficacy of BFA and FLU, RAW264.7 cells were stimulated with Escherichia coli-derived lipopolysaccharides (LPS, O111:B4; SIGMA, L3024) at 50 ng/ml concentration for different times alone or with BFA at indicated concentrations in triplicates. The supernatant was collected for ELISA detection. SW480 cells were stimulated with LPS (50 ng/mL) alone or with BFA (25 µM) for 24 h. Total RNA was isolated from cells using TRIzol reagent and reverse transcribed to cDNA. Changes in gene transcripts of CGASs, STING, IFN-β, and IFIT-1 were analyzed by RT-qPCR. The total protein lysates were collected from SW480 cells using RIPA buffer. Changes in protein levels of cGAS, STING, TBK1, NFκB, pro-IL-1β and pro-Casp-1, mature IL-1β (p17), cleaved Casp-1 (p20), and NLRP3 (from CST, Invitrogen, and Santa Cruz) were detected by western blot. Three technical replicates were used for each sample.
Mouse Model Validation of Drug Efficacy
Acute colitis was induced in mice by 3% DSS in drinking water ad libitum for seven days and followed with four days of normal distilled water. Control mice were fed with distilled water for 11 days. The colitis group mice were randomly grouped and intraperitoneally injected with 100 µL vehicle (0.01% DMSO) or BFA (2.5, 5, and 10 mg/kg) at day 4 and 6 post-DSS. Mice were evaluated daily for changes of body weight. At day 9 and 11, mice were euthanized to harvest colons and blood for further analysis. Colon lengths were measured at autopsy. Mouse colon biopsies were lysed in HTAB buffer to determine the MPO activity and in RIPA buffer for IL-1β secretion (ELISA) and cGAS expression (immunoblotting). Serum was separated from clotted blood by centrifugation. Serum AST and ALT were measured by ELISA (IT5530 and IT5508, G-Biosciences) according to the manufacturer's instructions.
Statistical Analysis
All data are mean ± s.d. For animal experiments, GraphPad Prism software version 8 (GraphPad Software, CA) was used for the preparation of graphs and comparison by using unpaired t-test, one-way ANOVA (Tukey's post hoc), or two-way ANOVA between groups, Kaplan-Meier curves (log-rank test) for survival analysis. Statistical analysis was also carried out in R (version 4.0.2) and plotted using ggplot (version 3.3.2) [21] and pheatmap (version 1.0.12) (https://CRAN.R-project.org/package=pheatmap). For all experiments, P < 0.05 was considered to be statistically significant.
Results
cGAS is Positively Associated with Human IBD
To identify the potential pathways in the pathogenesis of IBD, we built a novel potential strategy based on host-microbiome interaction for biomarker discovery and drug repurposing for IBD (Figure 1A). We analyzed the host transcriptome data from the publicly available dataset (IBDMDB), including 90 participants (43 CD, 25 UC, and 22 non-IBDs (controls), with no statistically significant differences in age and BMI (Figure S1A, Table S1). A total of 243 biopsies of transcriptional profiling were selected, including 98 rectal, 53 colonic, and 92 ileal subjects. We observed a clearer separation between ileum with colon or rectum samples (Figure S1A), consistent with the previous analysis that tissue location was a driver of intestinal gene expression [10].
cGAS is Highly Associated with Human IBD. (A) Workflow for the exploration of drugs for IBD including three steps: I) target discovery, identify highly inter-correlated microbiome-transcriptome interactions by using O2PLS based on the human cohort to reveal potential biomarkers in IBD; II) experimental validation, studies on intestinal bacteria for functional prediction based on TSEA and transcriptomes for drug screening based on LINCS, respectively; III) drug validation, showing the working model of brefeldin-a for treatment of IBD. (B) Functional enrichment network of Reactome based on the top 200 highly inter-correlated genes. cGAS signaling pathway and downstream effectors are highlighted in orange. Each node corresponds to a Reactome term; the node's size is the number of genes annotated to the term. Edges between nodes indicate genes shared between Reactome terms. IL, interleukin. (C) cGAS expression levels in the rectum. n = 49, 26, 23 independent samples for CD, UC, non-IBD. One-way ANOVA with Tukey's post hoc test determined significance. See also Figure S1.
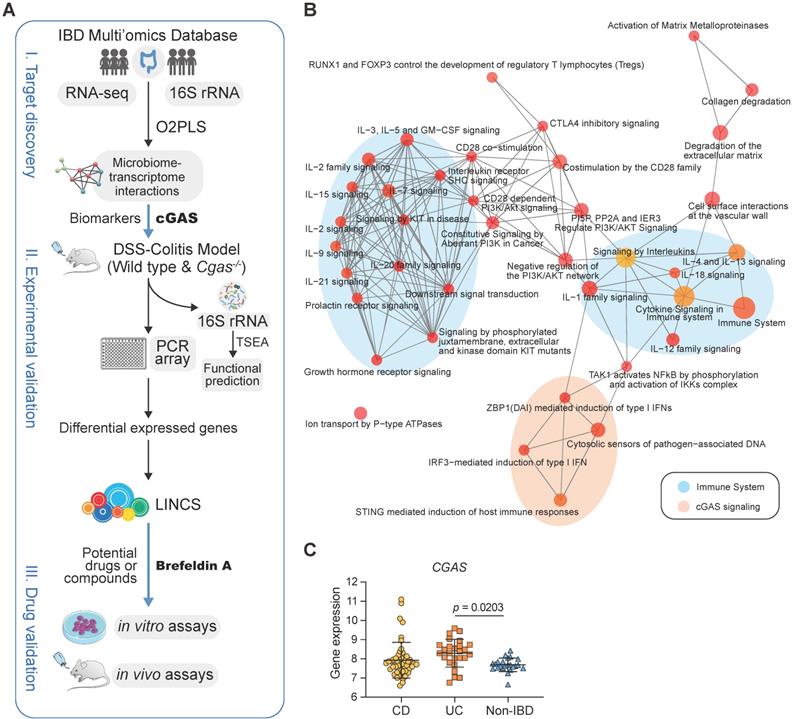
To identify genes that are highly related to gut microflora in IBD, a total of 4,811 differentially expressed genes and 169 bacteria were selected to identify the joint covariation between the transcriptomic and microbiome datasets by using the O2PLS approach and the loading value were shown in Table S2. The joint variation between UC and non-IBD subjects revealed a strong positive linear correlation (R2 = 0.711; Figure S1B). Reactome pathways [24] enrichment analysis confirmed that the immune-related pathways were significantly over-represented (Figure 1B, Table S3). In particular, cGAS signaling and its downstream effectors were significantly enriched, including cytosolic sensors of pathogen-associated DNA, STING-mediated induction of host immune responses, and IRF3- or ZBP1(DAI)-mediated induction of type I IFN pathways.
The cGAS-STING pathway was enriched in up-regulated genes in ileum biopsies with CD and in rectum with both CD and UC, which had the same trends as IL-17 signaling pathway [43] and the complement cascade [10] that have been confirmed by overrepresentation enrichment and implicated in the inflamed intestines of IBD (Figure S1C). Compared to subjects without IBD, cGAS, STING, and TNF involved in the cGAS-STING signaling and IL1B and MUC5AC involved in IL-17 signaling pathways were significantly differentially expressed in inflamed rectum biopsies (Figure 1C, S1D). Overall, these data supported that cGAS may play a causal role in colitis.
Colitis and Dysbacteriosis Activate cGAS and Induce Type I IFN in Mice
We next used a standard model of 3% DSS (epithelial irritant) in drinking water ad libitum to induce IBD (Figure S2A). This colitis model was appropriately established by DSS treatment. It significantly aggravated colitis's clinical signs, including weight loss, DAI, colon length, colonic MPO activity, pathological changes, and goblet cell secretion (Figure S2B-E, S2J-K). On the 9th-day post DSS challenge, where the severity of colitis nearly reached its peak, we observed a dramatic up-regulation of cGAS and downstream effectors at protein and mRNA levels in the inflamed colon following DSS treatment compared to the drinking water groups as controls (Figure S2F-G). Higher levels of type I IFN induction IL-1β and TNFα were observed in the colon of DSS-exposed mice (Figure S2H-I). Taken together, these findings indicate that cGAS may play a vital role in promoting innate immune responses in IBD.
Next, we used mouse models of antibiotic-associated diarrhea to investigate the effects of the dysbiosis of the gut microbiota on cGAS signaling. Mice were treated with antibiotics cocktail (MIX group), including streptomycin (STR), vancomycin (VAN), colistin (COL), and ampicillin (AAM), or treated individual antibiotic in the drinking water ad libitum (Figure S3A). The MIX mice with acquired depleted intestinal microbiota were usually considered as pseudo-germ-free. STR, VAN, and AAM treatment regimens caused a higher degree of colonic shortening, while MIX mice showed no colonic changes (Figure S3B). Histopathological assays showed no structural changes of the colon in the MIX mice, while the STR, VAN, COL, and AAM groups displayed a varying degree of pathological changes including inflammatory infiltrates, destroyed crypts, and goblet cell loss (Figure S3E-F). cGAS pathway activation was shown by overexpression in STR, VAN, and COL mice, although the degree was different (Figure S3C-D). Our data showed that dysbacteriosis-induced intestinal inflammation resulted in cGAS activation. The differences observed between MIX and single antibiotic-treated mice may be explained by the absence of the normal microbiota or the presence of a remnant microbiota [44].
Loss of Cgas Attenuated Inflammatory Responses
To explore the effect of cGAS loss on the colon, we performed DSS-colitis experiments in cGAS germline knockout (KO) mice (Figure 2A). The Cgas-/- mice have no apparent gross phenotype and no aberrant colonic inflammation compared with WT control mice (Figure 2B-D). No significant difference was found in intestinal shortening, MPO activity, and injury or proliferation between WT and cGAS KO mice (Figure 2E-F, S4A-C). MUC5AC is a major gel-forming mucin secreted by goblet cells, which has shown a protective role against helminth infection in mice [45] and controls injury in the acutely inflamed colon [46]. The number of goblet cells and the fluorescence intensity of Muc5ac in intestines were not significantly altered in KO mice than in the controls (Figure S4D-E). Immunoblotting of colon extracts confirmed the absence of cGAS (Figure 2H).
cGAS Regulates Acute Intestinal Inflammation. (A) Experimental design. WT and Cgas-/- mice treated with water or 3% DSS in drinking water for 7 days and followed for normal water for 4 days. (B) Weight loss in DSS-exposed mice. (C) Disease activity index (DAI) of DSS-exposed mice. (D) Kaplan-Meier survival curve for 11 days after DSS treatment. (E) Length of the colons of WT or Cgas-/- mice treated with DSS or control at day 9. (F) Myeloperoxidase (MPO) activity in colon homogenates at day 9. (G) Colonic Cgas, Sting, Ifit-1, and Ifn-β transcripts determined in WT or Cgas-/- mice undergoing DSS-colitis at day 9. (H) Western blot analyses of colons at day 9. Lysates were probed against cGAS, STING, p65, p-TBK1, TBK1, and β-actin. (I) Inflammatory cytokine expression was quantified in whole colon tissues from WT or Cgas-/- mice at day 9 by ELISA. Data are presented as the mean ± s.d. n = 5-7 mice/group from three independent experiments; statistical significance determined by one-way ANOVA (Turkey's post hoc) or two-way ANOVA. ns, no significant. See also Figure S4.
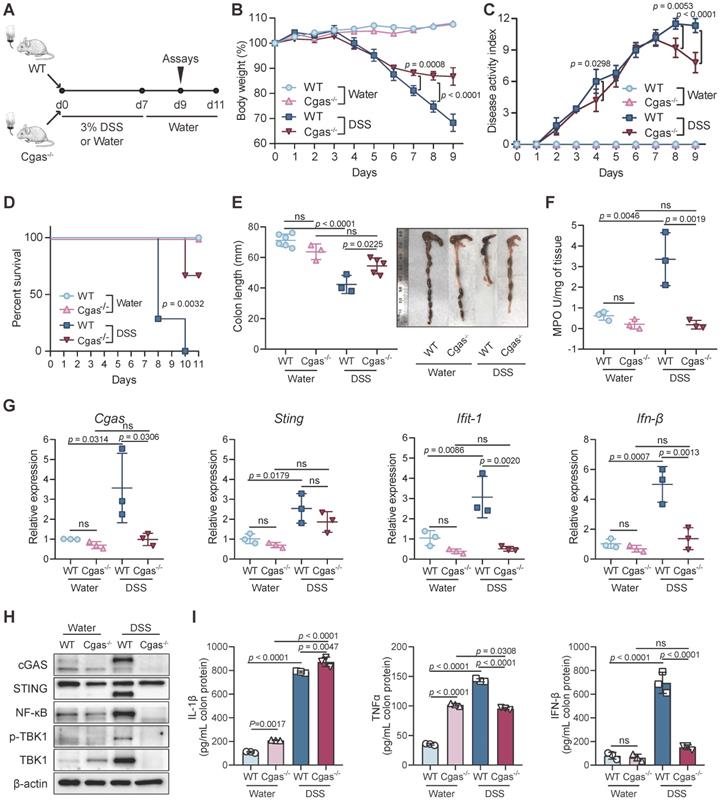
After 7 days of DSS treatment following 4 days of recovery, Cgas-/- mice exhibited dramatically reduced disease severity as measured by less weight loss, DAI scores, colonic shortening, and MPO expression (Figure 2B-C, 2E-F). WT mice began to die after 8 days of DSS induction. No Cgas-/- mice died within 9 days, which indicates a higher survival rate compared to WT mice (Figure 2D). Furthermore, Cgas-/- mice presented significantly decreased intestinal injury and inflammation to those in WT mice, displaying only mild histological inflammation features, less inflammatory infiltration, and no gross ulceration or overall tissue damage in colons and small intestines at day 9 (Figure S4A-C). Consistent with the reduced disease symptoms and inflammation, Cgas-/- mice showed no decrease in goblet cell numbers and fluorescence intensity of Muc5ac in intestines by day 9 (Figure S4D-E). Importantly, inhibition of NF-κB, p-TBK1, and TBK1 at protein levels and transcription of Ifit-1, and Ifn-β were observed in the colon of cGAS deficient DSS-colitis mice (Figure 2G-H). The colonic pro-inflammatory cytokines TNF-α and IFN-β were also decreased, while IL-1β increased in Cgas-/- mice compared to WT mice (Figure 2I). Taken together, our results demonstrated that cGAS plays an essential role in promoting the inflammatory response in colitis.
cGAS Deficiency Alters the Composition of Gut Microbiota
To probe the impact of cGAS signaling on microflora, we explored the effect of cGAS deficiency on fecal microbiota with or without induction of acute colitis with DSS. No difference was found in observed microbial richness in WT mice between both phenotypes (Figure 3A). Moreover, Cgas-/- mice showed no significant microbial richness difference from WT mice. On the other hand, DSS-treated Cgas-/- mice showed significantly lower taxa richness than the Cgas-/- control group and DSS-treated WT group. We discovered a distinct clustering of global microbiota composition between groups at the genus level driven by both phenotype (water vs. DSS, Pr(>F)=0.001) and genotype (WT vs. Cgas-/-, Pr(>F)=0.001) evaluated by PCoA analysis using Bray-Curtis distances metrics (Figure 3B). DSS treatment and Cgas deficiency strongly influenced the microbiota composition at the phylum level (Figure 3C, S5A). The most abundant phyla in WT vs. Cgas-/- mice included Bacteroidetes, Firmicutes, and Proteobacteria. DSS-treated mice showed increased relative abundance of Firmicutes, Proteobacteria, and Cyanobacteria, and decreased relative abundances of Bacteroidetes vs. control mice.
Difference of Microbiota in Stool Samples between WT and Cgas-/- Group. (A) Alpha diversity metrics to measure the richness of the communities in the feces. Dissecting filtered bacteria with Shannon and Simpson indexes (significance analyzed by t-test). Colors stand for different treatments. (B) Principal Coordinates Analysis (PCoA) based on bacterial 16S rRNA gene sequence abundance in fecal content. All samples were subjected to the beta diversity analysis of bacteria abundances generated using the Bray-Curtis distance metric. The shape of the dot represents different groups. Colors stand for different treatments. (C) Bacterial-taxon-based analysis at the phylum level in the feces. Relative abundance between different groups was expressed as a percentage (%) represented as treemap layout. Colors indicate the phylum information. Ten most abundant phyla of each group are shown. (D-E) Comparison of the significant differential microbiome at the genus level. Only bacteria with significant differences (p-value < 0.05 & |log2Fold Change|>1) between the WT and Cgas-/- controls (D) and subjected to DSS (E) are depicted. (F) Taxon set enrichment analysis (TSEA) of potential biological taxon sets in inflamed Cgas-/- versus control and WT. 1, GOS plus FOS, inulin and anthocyanins; 2, Bifidobacterium lactis HN019 and Lactobacillus acidophilus NCFM. Throughout, n = 6 in WT mice and n = 5 in cGAS KO mice for control, and n = 6 in WT mice and n = 3 in cGAS KO mice for DSS treatment. See also Figure S5-6.
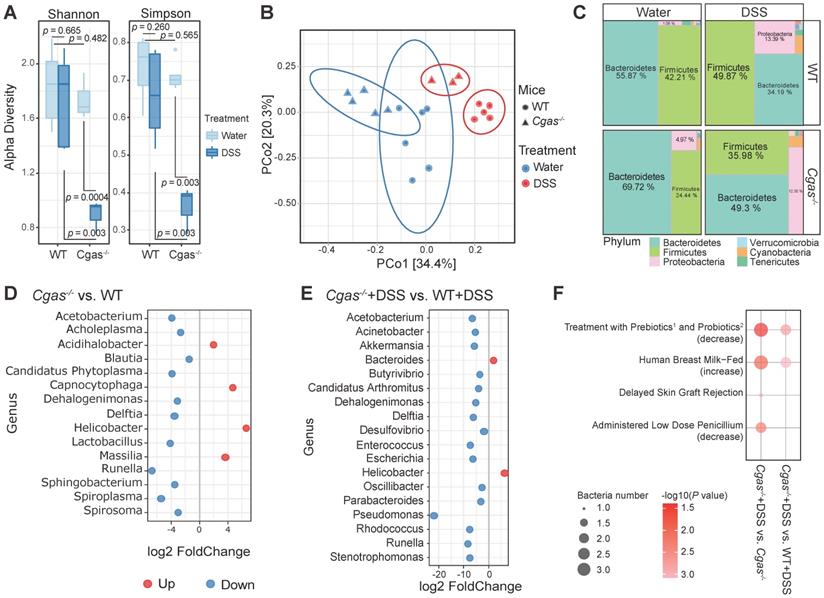
To understand the phylogenic detail, we examined differential bacterial levels down to the genera and species classification (Figure 3D-E, S5B-C and S6, Table S4-5). Consistent with studies of microbiome in IBD patients, Desulfovibrio, Enterococcus, and Escherichia at the genus level, and Desulfovibrio fairfieldensis, Escherichia coli, and Salmonella enterica at the species level were more abundant, while genus Prevotella was significantly less prevalent in DSS-treated WT mice (Figure S5B, S6B). In the fecal pellets of Cgas-/- control mice, most bacteria were significantly decreased vs. the WT control group, with genus Helicobacter and species Bacteroides vulgatus increased and genera Blautia and Lactobacillus decreased (Figure 3D, S6A). Significantly, after disease onset, the genera Bacteroides and Helicobacter were increased, and Parabacteroides and Pseudomonas were decreased in Cgas-/- control and WT+DSS groups (Figure 3E, S5C and S6C-D). Strikingly, the genera Desulfovibrio, Enterococcus, and Escherichia were diminished in Cgas-/- mice compared to WT mice under DSS treatment (Figure 3E). Meanwhile, Bacteroides vulgatus increased, and Clostridium perfringens, Enterococcus faecalis, Escherichia sp., and Salmonella sp. decreased at the species level (Figure S6D).
To determine intrinsic and extrinsic factors associated with the disruption on microbial communities, we performed taxon set enrichment analysis (TSEA) among the microbiome differentially between inflamed Cgas-/- versus control and WT+DSS groups (Figure 3F, Table S6). Interestingly, the cGAS-associated microbiome was predicted affecting pathways including treatment with prebiotics (GOS plus FOS, inulin and anthocyanins) [47] and probiotics (Bifidobacterium lactis HN019 and Lactobacillus acidophilus NCFM) [48], human breast milk-fed [49], and administered low dose penicillium [50], which have been implicated to be beneficial to IBD or irritable bowel syndrome. In summary, these observations indicate that cGAS has a functional role in shaping the bacterial gut microbiota and is required to adjust microbiota expansion in colitis, which would be a niche of host-microbiome based potential therapeutic strategy.
Loss of Cgas Affects IBD-Associated Genes Expression
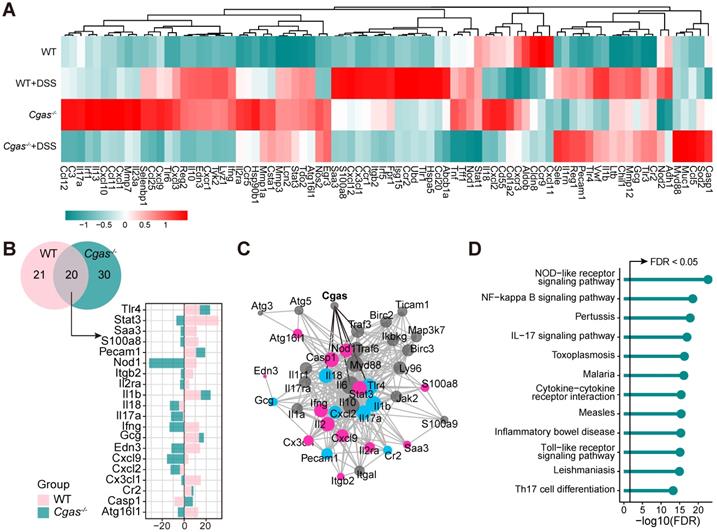
To explore the effect of cGAS on the gene expression relevant to mouse IBD, we compared the expression of 85 genes in colons, including inflammation, immunity, apoptosis, cell adhesion, and known IBD-related genes, in WT and Cgas-/- mice before and after DSS-colitis on day 9 (Figure 4A, Tables S7 and S8). A total of 20 differentially expressed genes were detected both in WT and Cgas-/- mice, including Tlr4 and Il18 undergoing the same change, and Stat3, Ifng, Cxcl9, Atg16l1, and Casp1 changing in the opposite direction (Figure 4B). Furthermore, we mapped these shared DEGs between WT and Cgas-/- mice based on the STRING database to obtain potentially linked genes (Figure 4C), and the Mb21d1(CGAS) gene showed association with Tlr4, Il18, and Casp1. Enriched pathways from STRING showed that the induced transcripts may be involved in inflammatory bowel disease and dominance of immune-related pathways, including NF-kappa B, IL-17, Toll-like receptor signaling pathways, and Th17 cell differentiation, confirming the specific role of cGAS and its potential interactions in response to bacterial infections under the colitis (Figure 4D).
Real-time PCR-array Analysis of Dysregulated Genes Implicated by cGAS Gene Depletion. PCR-array analysis of colon tissue from WT and Cgas-/- mice subjected to DSS-induced colitis at day 9 (n = 5 per genotype for control and DSS treatment groups). (A) Heatmap represents up- and down-regulated genes profiling among groups. Genes with higher expression are depicted in red, with lower expression in green. (B) Venn diagram represents the number of genes significant DEGs between WT and Cgas-/- groups subjected to DSS-induced colitis (p < 0.05). The barplot shows the log2 fold change of the shared significant DEGs. Colors stand for different groups. (C) Protein-protein interaction network built using STRING. The nodes represent the proteins (genes); the edges represent the interaction of proteins (genes); blue and pink represent the same or opposite direction of gene changes in WT and Cgas-/- groups, respectively. (D) Lollipop chart represents KEGG pathways associated genes in Figure 4C displayed as the -log10 (FDR). The line is drawn at FDR = 0.05. FDR, false discovery rate.
Prediction of Candidate Compounds for IBD
To identify potential drugs and compounds that can be used for IBD treatment, we first selected the over- and under-expressed genes between control and DSS treatment groups in WT and Cgas-/- mice as a disease signature, and then used these signatures to query in CLUE platform, respectively. The closer the CS is to -100, the better the response showed by drugs to reverse the expression of DEGs. The top 200 up- and down-scored medications were picked out with the lowest CS score, in which 43 common candidate drugs were in WT and Cgas-/- groups under DSS and water treatment at day 9 (Figure 5A, Table S9). 7 of the 43 candidates were published relevant to IBD. Azacitidine (DNA methyltransferase inhibitor) [51], ISOX (HDAC inhibitor) [52], ascorbic-acid (antioxidant) [53], cosmosiin (cytochrome P450 inhibitor) [54], IB-MECA (adenosine receptor agonist) [55], fostamatinib (SYK inhibitor) [56], and iloprost (platelet aggregation inhibitor) [57] have been used as a treatment or providing protective effects for IBD.
Drug Repurposing on the LINCS Database for IBD. (A) The Venn diagram represents the top 200 drugs using the DEGs from WT and Cgas-/- groups under DSS and water treatment searching against LINCS, individually. Heatmap shows the common 43 candidate drugs with the score values. (B) Experimental design. B6 WT mice were treated with water or 3% DSS in drinking water for 7 days and followed with drinking water for 4 days. Brefeldin-a (BFA, 2.5, 5, and 10 mg/kg) or vehicle (0.01% DMSO) was injected intraperitoneally (i.p.) in mice at day 4 and day 6 post-DSS. (C) Weight change. a, p < 0.0001, 0.0001 and 0.0004 (2.5, 5, and 10 mg/kg BFA groups) for comparison with vehicle group at day 6 post-DSS; b, p < 0.0001 for comparison with vehicle group. (D) Lengths and gross morphological changes of colons. (E) Colonic MPO activity. (F) Colonic IL-1β levels. (G) Serum AST and ALT levels were quantified by ELISA. (H) Western blot measured cGAS expression in the colons of these mice. (I) Schematic diagram of BFA exerting anti-inflammatory activity through inhibiting cGAS-STING pathway. In addition to blocking the protein trafficking from ER to the Golgi and the translocation of STING to the ER-Golgi intermediate compartment, BFA inhibits cGAS to block type I interferon production. BFA also effectively impairs NLRP3 inflammasome activation. Data are presented as the mean ± s.d. by two-way ANOVA or one-way ANOVA (Turkey's post hoc); n = 5 mice per group from three independent experiments. See also Figure S7.
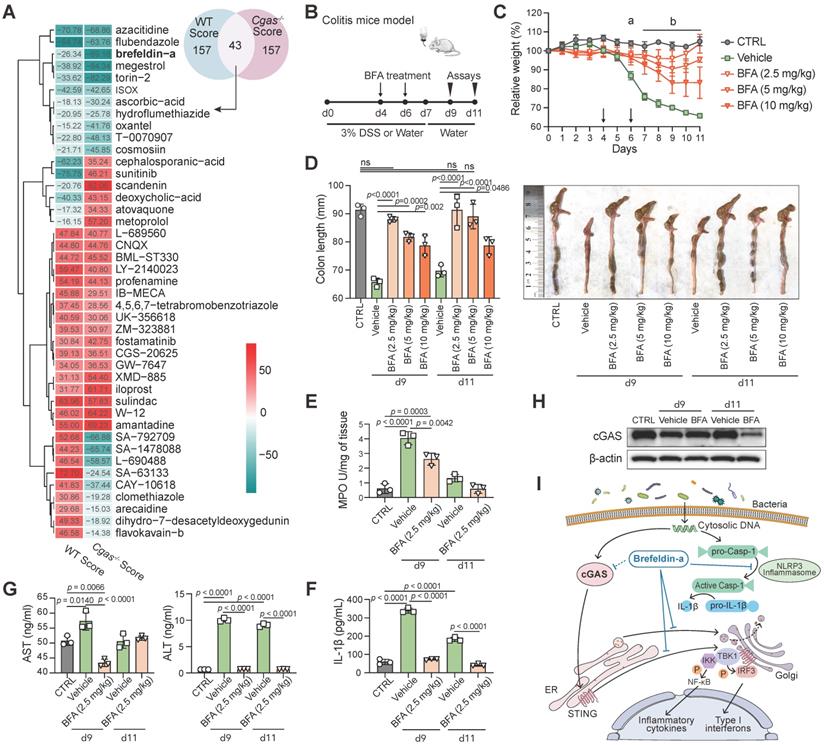
Among the top compounds, we investigated the anti-inflammatory activity of the protein synthesis inhibitor brefeldin-a (BFA) and the tubulin inhibitor flubendazole (FLU), which have not been previously studied in IBD. The anti-inflammatory activities of these drugs were examined in vitro in LPS-induced murine macrophage RAW264.7 cells. BFA decreased LPS-induced IL-6 and TNFα in the supernatant of RAW264.7 in a dose-dependent manner; however, FLU increased IL-6 at high concentrations (Figure S7A). Therefore, we next focused on the role of BFA, whose treatment also inhibited IL-1β, IL-6, TNFα, and IFN-β in a time-dependent manner compared to vehicle controls (Figure S7D). BFA showed less cytotoxicity in SW480 human colon cancer cell line (IC50=4.652 µM) than that in DLD-1 cells (IC50=0.167 µM) (Figure S7B-C). Thus, SW480 cells were selected for subsequent experiments. To validate the mechanism of drug therapy, we observed decreased transcript and protein levels of the cGAS-STING pathway and NLRP3 inflammasome in LPS-induced SW480 by qPCR and western blot (Figure S7E-F).
The therapeutic application of BFA was also examined in the acute 3% DSS-induced colitis model (Figure 5B). BFA-administered mice were protected from DSS-induced inflammatory disease with less weight loss (Figure 5C), DSS-induced colon shortening (Figure 5D), reduced MPO activity (Figure 5E), and decreased colonic inflammatory cytokine IL-1β (Figure 5F). We next evaluated the impact of BFA doses on the preventive efficacy. Remarkably, the low concentration of BFA (2.5 mg/kg) produced a more pronounced effect than higher concentrations (5 and 10 mg/kg). Furthermore, BFA had no effect on the serum AST and ALT levels in both DSS treatment and vehicle groups, indicating that it has not caused liver toxicity (Figure 5G). Consistent with the above cell experiment, BFA treatment can inhibit the expression of cGAS in the colon (Figure 5H). These results suggest that BFA can inhibit inflammatory response and mitigate colonic inflammation, through blocking cGAS (Figure 5I), which may be exerted by inhibiting NLRP3 inflammasome [58] and STING [59]. Taken together, we identified a top drug candidate BFA for potential IBD treatment consideration through the drug repurposing approach and validated the anti-inflammatory activity and potential mechanisms in vitro and in vivo.
Discussion
In this study, we designed a novel potential strategy based on host-microbiome interaction for biomarker discovery and drug repurposing for IBD (Figure 1A). By integrating public transcriptomic and microbiome data sets from IBD cohorts, we observed a significant correlation between cGAS signaling and IBD-associated genes and pathways. Hence, we focused on cGAS and revealed increased cGAS in IBD patient biopsies and experimental models, which subsequently activated immune reactions and inflammatory responses that may be associated with gut microbiota.
Previous studies have generally focused on the function of the cGAS-STING pathway in viral infection, cancer immunity, and autoimmune disorders [12]. Our results demonstrated that cGAS was significantly up-regulated in inflamed human samples with CD and UC, and also in two independent experimental models, DSS-colitis and antibiotic-associated diarrhea. Critically, we analyzed transcriptome and microbiome data of IBD patients and screened out cGAS as a potential biomarker and drug target. Further analysis using genetic knockout of cGAS in the inflammatory model strongly supported our notion that cGAS inhibition could rescue aspects of the inflammatory phenotype, consistent with the literature in other autoimmune disorders [60]. In IBD, ATG16L1 mutations promote IL-22 production through the STING pathway to excess epithelial cell death [13], while clear evidence for cGAS' role in IBD has not been presented. Recent research has implicated that STING agonists might have a potential therapeutic benefit in anticancer immune therapy [61] and antiviral responses [62], and STING inhibition may be promising for treating inflammatory diseases (such as AGS) through genetic approaches [63] or chemical inhibitors [64]. Accordingly, therapeutic avenue through targeting the cGAS-STING pathway may be realized one day for infections, autoimmunity, inflammatory diseases and cancers.
We subsequently investigated variations in the composition and relationship of host colonic transcriptomes and the intestinal microbiota in humans and mouse models. First, we found that cGAS was critically associated with the abundance of specific microbial taxa. cGAS-associated changes impacted multiple gut microbiota, such as Bacteroides, Lactobacillus, Prevotella, and Pseudomonas. Some of them are generally associated with gut health, including Bacteroides, Bifidobacterium, and Prevotella as key SCFA producers [65, 66], Coprococcus and Lachnospiraceae for producing butyrate [65, 67], Blautia relating to bile metabolism [65, 68], and Lactobacillus exhibiting probiotic ability [68]. On the other hand, Desulfovibrio [69], Enterococcus [70], Escherichia [71], and Pseudomonas [72] are frequently found at high ratios in IBD patients than healthy individuals and considered to be potential pathogens. Interestingly, Helicobacter has been recognized as a primary pathogenic factor for gastric and duodenal ulcers and gastric adenocarcinoma but prevents allergic disorders and chronic inflammatory conditions (i.e., IBD) in increasingly robust experimental and epidemiological evidence [73]. Additionally, we observed significantly increased strain diversity in beneficial species such as Anaerostipes hadrus [74], Bacteroides vulgatus [68], and Faecalibacterium prausnitzii [75] in cGAS deficient mice, which may play unknown roles in inflammation-associated immunosuppression.
Notably, our functional enrichment analysis predicted several potential targets to develop effective therapies. By using TSEA [40] analysis, our microbiome data were functionally profiled and compared to public datasets and known microbial signatures. Acinetobacter, Alistipes, and Parabacteroides (decreased abundance in Cgas-/-+DSS group) were enriched in prebiotics treatment (GOS plus FOS, inulin and anthocyanins), which significantly decreased the pro-inflammatory cytokines both in infected Caco-2 cells and post-infectious irritable bowel syndrome mouse models [47], and probiotics treatment (Bifidobacterium lactis HN019 and Lactobacillus acidophilus NCFM), which has positive effects on the treatment and maintenance of UC and other associated IBD pathologies (pouchitis and cholangitis) [48]. Acinetobacter, Klebsiella, and Pseudomonas (decreased abundance in Cgas-/-+DSS group) were enriched in human breast milk-fed, which is a protective factor against aberrant intestinal inflammation and necrotizing enterocolitis in preterm infants [49]. Candidatus Phytoplasma and Prevotella (decreased abundance in Cgas-/-+DSS group) were enriched in administered low dose penicillium, which reduces Th17 cells and subsequently suppresses the mouse colitis induced by DSS [50].
In addition to the functional prediction from the microbiome, we took a drug repurposing approach to identify drugs with potential to treat IBD using CLUE with the significant DEGs between cGAS KO and WT after DSS treatment. Forty-three common candidate drugs were identified in both WT and Cgas-/- groups, which can reverse the DEGs in inflamed colons Remarkably, seven candidate compounds in our screened results have already been reported for their potential protective or therapeutic effects in IBD [51-57]. The DNA methyltransferase inhibitor azacytidine has been indicated to induce complete remission in myelodysplastic syndrome associated with IBD by suppressing pro-inflammatory cytokine responses [51]. There were no reports on the direct association between ISOX and IBD, but some other HDAC inhibitors are already tested as an anticancer treatment, and one of them (SCFA butyrate) is used to treat IBD [52]. Ascorbic acid (AA), known as vitamin C, is important for patients suffering IBD since IBD patients are at risk of vitamin C deficiency. AA reduced oxidative stress and inflammatory response in DSS-induced colitis and could modify the gut microbiota composition related to the pathogenesis of IBD [53]. Since some critical CYP isoforms have been reportedly linked to metabolism and pathogenesis or etiology of UC, cosmosiin, as a cytochrome P450 (CYP) inhibitor, may play an effective role against IBD [54]. The adenosine receptor agonist IB-MECA was shown to alleviate inflammation in DSS-induced colitis and spontaneous colitis found in IL-10-deficient mice [55]. Fostamatinib, an SYK inhibitor, reduced the inflammatory damage in the acetic acid-induced colitis [56]. Platelet aggregation inhibitor iloprost has been demonstrated to be an antioxidant and protective against colitis induced oxidative stress in experimental colitis [57]. Hence our bioinformatics-based approach can effectively infer important repurposing drug candidates in both animal models and clinical trials.
To validate our drug repurposing findings, we chose to assess the anti-inflammatory properties of BFA and investigate a possible underlying mechanism of benefiting IBD patients. BFA has been widely studied for its antitumor, antifungal, and antiviral effects and apoptosis-inducing property [76]. BFA not only effectively inhibits protein trafficking from the endoplasmic reticulum (ER) to the Golgi apparatus [58], but also blocks type I IFN production by impeding the translocation of STING to the ER-Golgi intermediate compartment [59]. Apart from the above, we demonstrated that BFA inhibited inflammation through blocking cGAS expression and type I IFN response in colitis model and human colon cancer cells. Admittedly, acute DSS-induced colitis model may not accurately represent the IBD disease occurring in humans. Our validation assay presents a preliminary test for developing repurposing drugs, which is yet to be confirmed for its correlation with human IBD. Therefore, chronic colitis models and human specimens should be considered in future studies to clarify the significance of cGAS in the pathogenesis of human IBD as well as the efficacy and mechanisms of therapeutic candidates (i.e., BFA).
Altogether, we provide a new framework to identify biomarkers correlated with gut microbiota and immunity in the pathogenesis of IBD. We used these inflammation-correlated microbial and genetic variants as potential drug targets for IBD. Notably, our findings are twofold, including modified microbiota affecting the host's immune response and leading to intestinal inflammation, and in turn, the effect of the altered immune response in Cgas-/- mice on the microbiota. Furthermore, we combined multi-pronged approaches for drug repurposing and validation, including bioinformatic analysis of IBD cohorts and drugs, gene transcriptome and protein interactome, as well as cell culture and animal experimental studies. Collectively, our studies here offered a promising systemic framework to connect disease gene expression with microbiome for biomarker and therapeutics discovery applicable to IBD and successfully demonstrated the potential therapeutic effects and underlying mechanisms of repurposing drugs. This strategy may be broadly applied to other cases and have the potential to speed up drug discovery.
Abbreviations
AAM: ampicillin; AGS: Aicardi-Gourtières syndrome; ALT: alanine aminotransferase; AST: asparate aminotransferase; BFA: brefeldin-a; Casp-1: caspase-1; CCK-8: cell counting kit-8; CD: Crohn's disease; cGAS: cyclic GMP-AMP synthase; CLUE: Connectivity Map Linked User Environment; COL: colistin; CS: connectivity score; CST: Cell Signaling Technology; DAI: disease activity index; DAPI: 4',6'-diamidino-2-phenylindole; DEGs: differential expressed genes; ELISA: enzyme-linked immunosorbent assay; ER: endoplasmic reticulum; FLU: flubendazole; GWAS: genome-wide association studies; H&E: hematoxylin and eosin; IACUC: Institutional Animal Care and Use Committee; IBD: inflammatory bowel disease; IBDMDB: IBD Multi's omics database; IC50: 50% inhibiting concentration; IFN: type I interferon; iHMP: Integrative Human Microbiome Project; IL: interleukins; i.p.: intraperitoneally; IRF3: interferon regulatory transcription factor 3; LINCS: Library of Integrated Network-Based Cellular Signatures; LPS: lipopolysaccharides; MPO: myeloperoxidase; O2PLS: orthogonal partial least squares; OCT: optimal cutting temperature; PAS: Periodic acid-Schiff; PCoA: Principal Coordinates Analysis; qPCR: Quantitative Polymerase Chain Reaction; RIPA: radiation immunoprecipitation; SLE: systemic lupus erythematosus; STING: stimulator of the interferon genes; STR: streptomycin; TBK1: TANK binding kinase 1; Tregs: regulatory T lymphocytes; TSEA: Taxon Set Enrichment Analysis; UC: ulcerative colitis; UND: university of North Dakota; VAN: vancomycin; WT: wild type.
Supplementary Material
Supplementary figures.
Supplementary tables.
Acknowledgements
The authors thank Charlie Rice (Rockefeller University) for kindly providing Cgastm1a(EUCOMM)Hmgu mice. This project was supported by the National Institutes of Health (NIH) grants R01 AI138203, AI109317 grants, P20 GM103442, and GM113123 to MW, the National Key Research and Development Project (2020YFA0509400, 2020YFC2002705), Guangdong Basic and Applied Basic Research Foundation (2019B030302012), and National Natural Science Foundation of China (81821002 and 81790251) to CH, and the Science and Technology Department of Sichuan Province (No. 2019YJ0050) to CL. The Human core, Histology core, and Imaging core at UND were acknowledged for the support of the work. Histological services were provided by the UND Histology Core Facility supported (NIH) / NIGMS award P20GM113123, DaCCoTA CTR NIH grant U54GM128729, and UND SMHS funds. Figures 1A and 5I were created by modifying illustrations provided by Servier Medical Art (SMART, smart.servier.com) licensed under a Creative Commons Attribution 3.0 Unported License and Vecteezy.com.
Data Availability
All relevant data supporting this study's key findings are available within the supplementary information files and available from the corresponding author upon reasonable request. The original 16S rDNA-seq data is deposited in the NCBI Bio-Project ID: PRJNA684767. Data underlying Figures 1 and S1 are provided in Tables S1-3 and IBD multi's omics database (http://ibdmdb.org). Data underlying Figure 3F is provided in Table S6 and the taxon sets were downloaded from MicrobiomeAnalyst (https://www.microbiomeanalyst.ca/MicrobiomeAnalyst/faces/docs/Resources.xhtml). Source data in the figures are provided in Table S10.
Code Availability
For this study, R software (v 4.0.2) was used. The code is available at https://github.com/guokai8/IBD.
Author Contributions
MW, CH, CL, ZW, and KG conceived of and designed the project. ZW performed most of the experiments, obtained profiled feces and prepared sequencing library. KG designed implemented bioinformatic methods and processed and aligned sequencing data. PG participated in bioinformatics analysis and interpreted results. PG, QP, SQ, and PL participated in experiments. MW, CH, CL, JH, and NX supervised and supported the work. ZW and KG wrote the manuscript and all authors reviewed and edited the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kaplan GG. The global burden of IBD: from 2015 to 2025. Nat Rev Gastroenterol Hepatol. 2015;12:720-7
2. de Souza HSP, Fiocchi C, Iliopoulos D. The IBD interactome: an integrated view of aetiology, pathogenesis and therapy. Nat Rev Gastroenterol Hepatol. 2017;14:739-49
3. Franzosa EA, Hsu T, Sirota-Madi A, Shafquat A, Abu-Ali G, Morgan XC. et al. Sequencing and beyond: integrating molecular 'omics' for microbial community profiling. Nature reviews Microbiology. 2015;13:360-72
4. de Lange KM, Moutsianas L, Lee JC, Lamb CA, Luo Y, Kennedy NA. et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet. 2017;49:256-61
5. Pu Q, Lin P, Wang Z, Gao P, Qin S, Cui L. et al. Interaction among inflammasome, autophagy and non-coding RNAs: new horizons for drug. Precis Clin Med. 2019;2:166-82
6. Ramanan D, Tang MS, Bowcutt R, Loke Pn, Cadwell K. Bacterial sensor Nod2 prevents inflammation of the small intestine by restricting the expansion of the commensal Bacteroides vulgatus. Immunity. 2014;41:311-24
7. Lamas B, Richard ML, Leducq V, Pham H-P, Michel M-L, Da Costa G. et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nature medicine. 2016;22:598-605
8. Kabat AM, Harrison OJ, Riffelmacher T, Moghaddam AE, Pearson CF, Laing A. et al. The autophagy gene Atg16l1 differentially regulates Treg and TH2 cells to control intestinal inflammation. eLife. 2016;5:e12444
9. Chu H, Khosravi A, Kusumawardhani IP, Kwon AHK, Vasconcelos AC, Cunha LD. et al. Gene-microbiota interactions contribute to the pathogenesis of inflammatory bowel disease. Science (New York, NY). 2016;352:1116-20
10. Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, Poon TW. et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019;569:655-62
11. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786-91
12. Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet. 2019;20:657-74
13. Aden K, Tran F, Ito G, Sheibani-Tezerji R, Lipinski S, Kuiper JW. et al. ATG16L1 orchestrates interleukin-22 signaling in the intestinal epithelium via cGAS-STING. J Exp Med. 2018;215:2868-86
14. Zhu Q, Man SM, Gurung P, Liu Z, Vogel P, Lamkanfi M. et al. Cutting edge: STING mediates protection against colorectal tumorigenesis by governing the magnitude of intestinal inflammation. Journal of immunology (Baltimore, Md: 1950). 2014;193:4779-82
15. Neurath MF. Current and emerging therapeutic targets for IBD. Nat Rev Gastroenterol Hepatol. 2017;14:269-78
16. Pushpakom S, Iorio F, Eyers PA, Escott KJ, Hopper S, Wells A. et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov. 2019;18:41-58
17. Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T. et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet. 2010;42:1118-25
18. Chen B, Ma L, Paik H, Sirota M, Wei W, Chua MS. et al. Reversal of cancer gene expression correlates with drug efficacy and reveals therapeutic targets. Nat Commun. 2017;8:16022
19. Koleti A, Terryn R, Stathias V, Chung C, Cooper DJ, Turner JP. et al. Data Portal for the Library of Integrated Network-based Cellular Signatures (LINCS) program: integrated access to diverse large-scale cellular perturbation response data. Nucleic acids research. 2018;46:D558-D66
20. Keenan AB, Jenkins SL, Jagodnik KM, Koplev S, He E, Torre D. et al. The Library of Integrated Network-Based Cellular Signatures NIH Program: System-Level Cataloging of Human Cells Response to Perturbations. Cell Syst. 2018;6:13-24
21. Team RC. R: A language and environment for statistical computing. 2013.
22. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550
23. Bouhaddani SE, Houwing-Duistermaat J, Salo P, Perola M, Jongbloed G, Uh H-W. Evaluation of O2PLS in Omics data integration. BMC bioinformatics. 2016;17(Suppl 2):11
24. Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P. et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018;46:D649-d55
25. Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL. et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature. 2014;505:691-5
26. Ohnmacht C, Park JH, Cording S, Wing JB, Atarashi K, Obata Y. et al. MUCOSAL IMMUNOLOGY. The microbiota regulates type 2 immunity through RORgammat(+) T cells. Science. 2015;349:989-93
27. Lichtman JS, Ferreyra JA, Ng KM, Smits SA, Sonnenburg JL, Elias JE. Host-Microbiota Interactions in the Pathogenesis of Antibiotic-Associated Diseases. Cell Rep. 2016;14:1049-61
28. Kim JJ, Shajib MS, Manocha MM, Khan WI. Investigating intestinal inflammation in DSS-induced model of IBD. J Vis Exp. 2012
29. Guo Q, Shen N, Yuan K, Li J, Wu H, Zeng Y. et al. Caveolin-1 plays a critical role in host immunity against Klebsiella pneumoniae by regulating STAT5 and Akt activity. European journal of immunology. 2012;42:1500-11
30. Wu M, Stockley PG, Martin WJ 2nd. An improved western blotting technique effectively reduces background. Electrophoresis. 2002;23:2373-6
31. Lin P, Qin S, Pu Q, Wang Z, Wu Q, Gao P. et al. CRISPR-Cas13 Inhibitors Block RNA Editing in Bacteria and Mammalian Cells. Mol Cell. 2020;78:850-61 e5
32. Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, Bock J, Martinez-Naves E. et al. Paneth cells as a site of origin for intestinal inflammation. Nature. 2013;503:272-6
33. Li G, Fox J 3rd, Liu Z, Liu J, Gao GF, Jin Y. et al. Lyn mitigates mouse airway remodeling by downregulating the TGF-beta3 isoform in house dust mite models. Journal of immunology (Baltimore, Md: 1950). 2013;191:5359-70
34. Wu M, Pasula R, Smith PA, Martin WJ 2nd. Mapping alveolar binding sites in vivo using phage peptide libraries. Gene Ther. 2003;10:1429-36
35. Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013;41:e1
36. Andrews S. FastQC: a quality control tool for high throughput sequence data.; 2010
37. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics (Oxford, England). 2014;30:2114-20
38. Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2. Genome biology. 2019;20:257
39. McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PloS one. 2013;8:e61217
40. Chong J, Liu P, Zhou G, Xia J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat Protoc. 2020;15:799-821
41. Larabi A, Barnich N, Nguyen HTT. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy. 2020;16:38-51
42. Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X. et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell. 2017;171:1437-52.e17
43. Haberman Y, Tickle TL, Dexheimer PJ, Kim M-O, Tang D, Karns R. et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J Clin Invest. 2014;124:3617-33
44. Hernández-Chirlaque C, Aranda CJ, Ocón B, Capitán-Cañadas F, Ortega-González M, Carrero JJ. et al. Germ-free and Antibiotic-treated Mice are Highly Susceptible to Epithelial Injury in DSS Colitis. J Crohns Colitis. 2016;10:1324-35
45. Hasnain SZ, Evans CM, Roy M, Gallagher AL, Kindrachuk KN, Barron L. et al. Muc5ac: a critical component mediating the rejection of enteric nematodes. The Journal of experimental medicine. 2011;208:893-900
46. Olli KE, Rapp C, O'Connell L, Collins CB, McNamee EN, Jensen O. et al. Muc5ac Expression Protects the Colonic Barrier in Experimental Colitis. Inflamm Bowel Dis. 2020;26:1353-67
47. Chen Q, Ren Y, Lu J, Bartlett M, Chen L, Zhang Y. et al. A Novel Prebiotic Blend Product Prevents Irritable Bowel Syndrome in Mice by Improving Gut Microbiota and Modulating Immune Response. Nutrients. 2017 9
48. Saez-Lara MJ, Gomez-Llorente C, Plaza-Diaz J, Gil A. The role of probiotic lactic acid bacteria and bifidobacteria in the prevention and treatment of inflammatory bowel disease and other related diseases: a systematic review of randomized human clinical trials. Biomed Res Int. 2015;2015:505878
49. Thai JD, Gregory KE. Bioactive Factors in Human Breast Milk Attenuate Intestinal Inflammation during Early Life. Nutrients. 2020 12
50. Jin S, Zhao D, Cai C, Song D, Shen J, Xu A. et al. Low-dose penicillin exposure in early life decreases Th17 and the susceptibility to DSS colitis in mice through gut microbiota modification. Sci Rep. 2017;7:43662
51. Kono M, Komeda Y, Sakurai T, Okamoto A, Minaga K, Kamata K. et al. Induction of Complete Remission by Azacitidine in a Patient with Myelodysplastic Syndrome-Associated Inflammatory Bowel Disease. J Crohns Colitis. 2018;12:499-502
52. Meddens CA, van der List ACJ, Nieuwenhuis EES, Mokry M. Non-coding DNA in IBD: from sequence variation in DNA regulatory elements to novel therapeutic potential. Gut. 2019;68:928-41
53. Yan H, Wang H, Zhang X, Li X, Yu J. Ascorbic acid ameliorates oxidative stress and inflammation in dextran sulfate sodium-induced ulcerative colitis in mice. Int J Clin Exp Med. 2015;8:20245-53
54. Sen A, Stark H. Role of cytochrome P450 polymorphisms and functions in development of ulcerative colitis. World J Gastroenterol. 2019;25:2846-62
55. Mabley J, Soriano F, Pacher P, Haskó G, Marton A, Wallace R. et al. The adenosine A3 receptor agonist, N6-(3-iodobenzyl)-adenosine-5'-N-methyluronamide, is protective in two murine models of colitis. Eur J Pharmacol. 2003;466:323-9
56. Can G, Ayvaz S, Can H, Demirtas S, Aksit H, Yilmaz B. et al. The Syk Inhibitor Fostamatinib Decreases the Severity of Colonic Mucosal Damage in a Rodent Model of Colitis. J Crohns Colitis. 2015;9:907-17
57. Aytaç E, Teksöz S, Saygılı S, Tortum OB, Yavuz N, Sözer V. et al. Iloprost reduces colitis induced oxidative stress: An experimental study in rats. Turk J Gastroenterol. 2013;24:224-9
58. Hong S, Hwang I, Gim E, Yang J, Park S, Yoon SH. et al. Brefeldin A-sensitive ER-Golgi vesicle trafficking contributes to NLRP3-dependent caspase-1 activation. Faseb j. 2019;33:4547-58
59. Dobbs N, Burnaevskiy N, Chen D, Gonugunta VK, Alto NM, Yan N. STING Activation by Translocation from the ER Is Associated with Infection and Autoinflammatory Disease. Cell Host Microbe. 2015;18:157-68
60. Gao D, Li T, Li XD, Chen X, Li QZ, Wight-Carter M. et al. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc Natl Acad Sci U S A. 2015;112:E5699-705
61. An X, Zhu Y, Zheng T, Wang G, Zhang M, Li J. et al. An Analysis of the Expression and Association with Immune Cell Infiltration of the cGAS/STING Pathway in Pan-Cancer. Mol Ther Nucleic Acids. 2019;14:80-9
62. Yamamoto T, Kanuma T, Takahama S, Okamura T, Moriishi E, Ishii KJ. et al. STING agonists activate latently infected cells and enhance SIV-specific responses ex vivo in naturally SIV controlled cynomolgus macaques. Sci Rep. 2019;9:5917
63. Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA. et al. Mutations in the gene encoding the 3'-5' DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet. 2007;39:1065-7
64. Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A. et al. Targeting STING with covalent small-molecule inhibitors. Nature. 2018;559:269-73
65. Forbes JD, Chen CY, Knox NC, Marrie RA, El-Gabalawy H, de Kievit T. et al. A comparative study of the gut microbiota in immune-mediated inflammatory diseases-does a common dysbiosis exist? Microbiome. 2018;6:221
66. Imhann F, Vich Vila A, Bonder MJ, Fu J, Gevers D, Visschedijk MC. et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut. 2018;67:108-19
67. Schirmer M, Franzosa EA, Lloyd-Price J, McIver LJ, Schwager R, Poon TW. et al. Dynamics of metatranscription in the inflammatory bowel disease gut microbiome. Nat Microbiol. 2018;3:337-46
68. Gevers D, Kugathasan S, Denson LA, Vázquez-Baeza Y, Van Treuren W, Ren B. et al. The treatment-naive microbiome in new-onset Crohn's disease. Cell Host Microbe. 2014;15:382-92
69. Rowan F, Docherty NG, Murphy M, Murphy B, Calvin Coffey J, O'Connell PR. Desulfovibrio bacterial species are increased in ulcerative colitis. Dis Colon Rectum. 2010;53:1530-6
70. Seishima J, Iida N, Kitamura K, Yutani M, Wang Z, Seki A. et al. Gut-derived Enterococcus faecium from ulcerative colitis patients promotes colitis in a genetically susceptible mouse host. Genome Biol. 2019;20:252
71. Negroni A, Costanzo M, Vitali R, Superti F, Bertuccini L, Tinari A. et al. Characterization of adherent-invasive Escherichia coli isolated from pediatric patients with inflammatory bowel disease. Inflamm Bowel Dis. 2012;18:913-24
72. Wagner J, Short K, Catto-Smith AG, Cameron DJ, Bishop RF, Kirkwood CD. Identification and characterisation of Pseudomonas 16S ribosomal DNA from ileal biopsies of children with Crohn's disease. PLoS One. 2008;3:e3578
73. Yu Y, Zhu S, Li P, Min L, Zhang S. Helicobacter pylori infection and inflammatory bowel disease: a crosstalk between upper and lower digestive tract. Cell Death Dis. 2018;9:961
74. Zhang Q, Wu Y, Wang J, Wu G, Long W, Xue Z. et al. Accelerated dysbiosis of gut microbiota during aggravation of DSS-induced colitis by a butyrate-producing bacterium. Sci Rep. 2016;6:27572
75. Miquel S, Leclerc M, Martin R, Chain F, Lenoir M, Raguideau S. et al. Identification of metabolic signatures linked to anti-inflammatory effects of Faecalibacterium prausnitzii. mBio. 2015 6
76. Paek SM. Recent Synthesis and Discovery of Brefeldin A Analogs. Mar Drugs. 2018 16
Author contact
![]() Corresponding authors: Dr. Changlong Li, E-mail: changlongliedu.cn, West China School of Basic Medical Sciences & Forensic Medicine, Sichuan University, Chengdu, Sichuan 610041, China. Dr. Canhua Huang, E-mail: hcanhuacom, State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, and West China School of Basic Medical Sciences & Forensic Medicine, Sichuan University, and Collaborative Innovation Center for Biotherapy, Chengdu, Sichuan 610041, China. Dr. Min Wu, E-mail: min.wuedu, Department of Biomedical Sciences, School of Medicine and Health Sciences, University of North Dakota, Grand Forks, ND 58202, USA.
Corresponding authors: Dr. Changlong Li, E-mail: changlongliedu.cn, West China School of Basic Medical Sciences & Forensic Medicine, Sichuan University, Chengdu, Sichuan 610041, China. Dr. Canhua Huang, E-mail: hcanhuacom, State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, and West China School of Basic Medical Sciences & Forensic Medicine, Sichuan University, and Collaborative Innovation Center for Biotherapy, Chengdu, Sichuan 610041, China. Dr. Min Wu, E-mail: min.wuedu, Department of Biomedical Sciences, School of Medicine and Health Sciences, University of North Dakota, Grand Forks, ND 58202, USA.