Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(12):6044-6057. doi:10.7150/thno.55092 This issue Cite
Research Paper
Repurposing 18F-FMISO as a PET tracer for translational imaging of nitroreductase-based gene directed enzyme prodrug therapy
Gorka Ruiz de Garibay1,2*, Elvira García de Jalón1,3*, Endre Stigen1, Kjetil B Lund1, Mihaela Popa1,4, Ben Davidson5, Mireia Mayoral Safont4, Cecilie B. Rygh6,7, Heidi Espedal6, Torill M Barrett8, Bengt Erik Haug3, Emmet McCormack1,9,10 ![]()
1. Centre for Cancer Biomarkers CCBIO, Department of Clinical Science, University of Bergen, Jonas Lies vei 65, N-5021, Bergen, Norway.
2. University Hospital 12 de Octubre, Madrid, Spain; Lung Cancer Unit H12O-CNIO, Spain.
3. Department of Chemistry and Centre for Pharmacy, University of Bergen, Allégaten 41, N-5007, Bergen, Norway.
4. KinN Therapeutics AS, Jonas Lies vei 65, 5021 Bergen, Norway.
5. Department of Pathology, Oslo University Hospital, Norwegian Radium Hospital, and Faculty of Medicine, Institute of Clinical Medicine, University of Oslo, Oslo, Norway.
6. Molecular imaging Center, Department of Biomedicine, University of Bergen, Jonas Lies vei 91, N-5009, Bergen, Norway.
7. Western Norway University College, Inndalsveien 28, N-5063, Bergen, Norway.
8. Department of Pathology, Haukeland University Hospital, Jonas Lies vei 65, N-5021, Bergen, Norway.
9. Centre for Pharmacy, Department of Clinical Science, University of Bergen, Jonas Lies vei 65, Bergen 5021, Norway.
10. Vivarium, Department of Clinical Medicine, University of Bergen, Jonas Lies vei 65, 5021 Bergen, Norway.
*These authors contributed equally to this work.
Received 2020-10-27; Accepted 2021-2-25; Published 2021-4-7
Abstract

Nitroreductases (NTR) are a family of bacterial enzymes used in gene directed enzyme prodrug therapy (GDEPT) that selectively activate prodrugs containing aromatic nitro groups to exert cytotoxic effects following gene transduction in tumours. The clinical development of NTR-based GDEPT has, in part, been hampered by the lack of translational imaging modalities to assess gene transduction and drug cytotoxicity, non-invasively. This study presents translational preclinical PET imaging to validate and report NTR activity using the clinically approved radiotracer, 18F-FMISO, as substrate for the NTR enzyme.
Methods: The efficacy with which 18F-FMISO could be used to report NfsB NTR activity in vivo was investigated using the MDA-MB-231 mammary carcinoma xenograft model. For validation, subcutaneous xenografts of cells constitutively expressing NTR were imaged using 18F-FMISO PET/CT and fluorescence imaging with CytoCy5S, a validated fluorescent NTR substrate. Further, examination of the non-invasive functionality of 18F-FMISO PET/CT in reporting NfsB NTR activity in vivo was assessed in metastatic orthotopic NfsB NTR expressing xenografts and metastasis confirmed by bioluminescence imaging. 18F-FMISO biodistribution was acquired ex vivo by an automatic gamma counter measuring radiotracer retention to confirm in vivo results. To assess the functional imaging of NTR-based GDEPT with 18F-FMISO, PET/CT was performed to assess both gene transduction and cytotoxicity effects of prodrug therapy (CB1954) in subcutaneous models.
Results: 18F-FMISO retention was detected in NTR+ subcutaneous xenografts, displaying significantly higher PET contrast than NTR- xenografts (p < 0.0001). Substantial 18F-FMISO retention was evident in metastases of orthotopic xenografts (p < 0.05). Accordingly, higher 18F-FMISO biodistribution was prevalent ex vivo in NTR+ xenografts. 18F-FMISO NfsB NTR PET/CT imaging proved useful for monitoring in vivo NTR transduction and the cytotoxic effect of prodrug therapy.
Conclusions: 18F-FMISO NfsB NTR PET/CT imaging offered significant contrast between NTR+ and NTR- tumours and effective resolution of metastatic progression. Furthermore, 18F-FMISO NfsB NTR PET/CT imaging proved efficient in monitoring the two steps of GDEPT, in vivo NfsB NTR transduction and response to CB1954 prodrug therapy. These results support the repurposing of 18F-FMISO as a readily implementable PET imaging probe to be employed as companion diagnostic test for NTR-based GDEPT systems.
Keywords: 18F-FMISO, Gene-directed enzymatic prodrug therapy, GDEPT, nitroreductase, NTR, cancer, xenograft, preclinical, mouse, PET/CT, imaging
Introduction
Gene directed enzyme prodrug therapy (GDEPT) is a promising anti-cancer strategy that aims to reduce off-target toxicity and limit severe side-effects by the combination of a prodrug and its activating enzyme [1]. In a first step, tumours are transduced to express a gene encoding for the prodrug-activating enzyme. In a second step, a non-toxic prodrug is delivered systemically and subsequently converted in situ to a cytotoxic derivative by the expressed enzyme [2]. In this way, GDEPT achieves high concentration of cytotoxic compounds locally, minimising systemic side effects associated with conventional cancer chemotherapy [3].
The most extensively studied prodrug/enzyme pairs for GDEPT are ganciclovir (GCV)/herpes simplex virus-thymidine kinase (HSV-TK) [4] and 5-(aziridin-1-yl)-2,4-dinitrobenzamide (CB1954, Figure 1B)/E. Coli nitroreductase NfsB (onwards referred as NTR) [5]. HSV-TK phosphorylates GCV, allowing its incorporation into DNA, resulting in toxicity due to interference with the DNA synthesis [6]. NTR reduces the nitro groups (R-NO2) present in CB1954, into hydroxylamines (R-NHOH), resulting in the formation of an alkylating agent with cytotoxic activity [7, 8]. Moreover, the diffusion of the activated compound into neighbouring non-targeted cells amplifies this cytotoxic effect (bystander effect) [9, 10]. Several trials have evaluated the clinical application of HSV-TK and NTR-based GDEPT strategies with contrasting results [5, 11, 12]. The largest clinical trial, including 124 patients receiving HSV-TK-based GDEPT, reported no difference in comparison to standard therapy. Modest HSV-TK expression was suggested as a potential limiting factor [11]. Indeed, a phase II HSV-TK-based GDEPT trial using a more efficient gene transduction strategy reported a significant increase in survival [12]. These studies illustrate the need for monitoring the gene transduction efficiency. One of the NTR-based GDEPT clinical trials assessed the transduction efficiency using immunohistochemistry in tumour biopsies [5]. However, immunohistochemistry is time consuming and may be affected by inadequate sampling of representative tumoral areas. For large clinical trials, non-invasive techniques such as imaging, which allows for easier clinical implementation, are required. In this respect, positron emission tomography (PET), which is routinely employed in clinical settings, enables detection of the activity for some of the enzymes used in the GDEPT strategies and may be suitable for interrogation of transduction efficiency in clinical trials [13, 14].
Nitroreductases and nitroaromatic substrates for multiple applications. (A) Nitroreductase subtrates employed in imaging or GDEPT. (B) Comparison of the catalytic mechanisms of oxygen-sensitive and oxygen-insensitive nitroreductases in normoxic and hypoxic conditions.
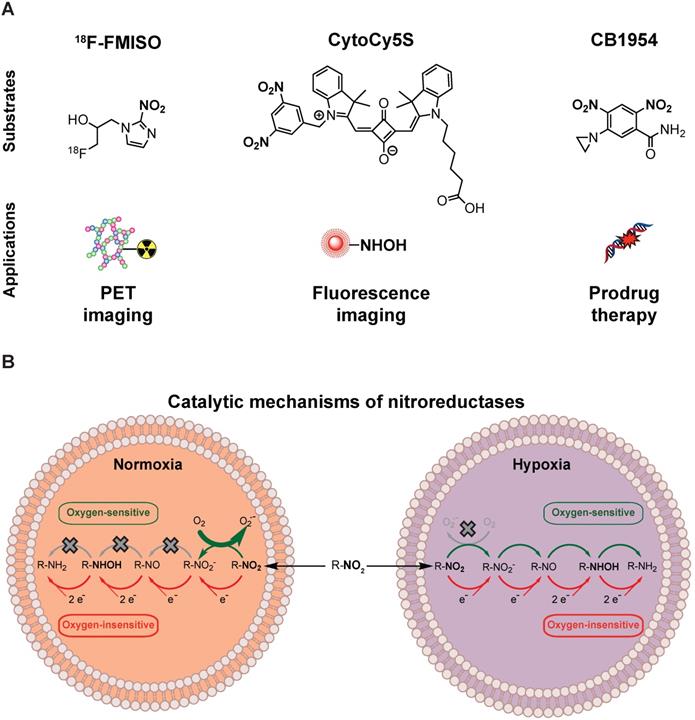
The activity of HSV-TK can be evaluated using radiolabeled probes, such as 18F-FIAU (1-(2- deoxy-2-[18F]-fluoro-1-D-arabinofuranoside)-5-iodouracil), 18F-FHBG (9-[4-[18F]fluoro-3-(hydroxymethyl) butyl]guanine) and GCV analogues [15, 16]. HSV-TK PET/CT imaging has indeed been successfully applied for monitoring transduction efficiency, tissue specificity and therapeutic effect preclinically [15, 17]. Subsequently, HSV-TK PET/CT imaging was incorporated into phase I clinical trials to monitor transduction efficiency in healthy individuals and in cancer patients [18, 19]. In an ongoing phase I clinical trial investigating the use of HSV-TK-based GDEPT in combination with immunotherapy (NCT04313868), HSV-TK PET/CT imaging with 18F-FHBG has been implemented to explore the effect of viral administration routes in transduction efficacy.
NTR activity has been effectively imaged in a preclinical context using the near-infrared fluorescent probe CytoCy5S [20] and a caged bioluminescent substrate “NTR caged luciferin” (NCL) [21]. However, the non-invasive clinical application of optical imaging approaches is challenging due to limited tissue penetrance of light and associated autofluorescence. This could potentially be overcome by employing PET as the imaging modality, provided that an appropriate NTR specific tracer is available. For this purpose, we have focused on radiolabelled 2-nitroimidazoles (2-NI) that have been extensively utilised as hypoxia imaging agents (e.g. FMISO or FETNIM) [22, 23]. Hypoxia induces expression of oxygen-sensitive nitroreductases, able to convert 2-NI (e.g. FMISO, Figure 1A) into the corresponding hydroxylamines, which covalently bind to macromolecules in the tumour, allowing PET contrast [24]. The mechanistic similarity between oxygen-sensitive and E.coli nitroreductases, oxygen-insensitive, (Figure 1B) and their ability to convert aromatic nitro groups of various substrates (Figure 1A) into aromatic hydroxylamines, render 2-NI as plausible probes for clinical imaging of NTR activity [25].
Recently, Mowday and co-workers [26] have demonstrated effective NTR PET imaging employing NfsA from E. Coli in combination with the 2-NI PET tracer 18F-HX4. Similarly, in this work we have investigated whether 18F-FMISO, a PET tracer approved by FDA as an investigational new drug for human use, could be repurposed as a PET probe for non-invasive imaging of NTR for application in the context of GDEPT. 18F-FMISO was validated as an NTR specific PET imaging tracer using mammary carcinoma xenograft models. Further, the applicability of 18F-FMISO imaging was demonstrated in the two steps of GDEPT i.e. gene transduction and cytotoxicity effects of prodrug therapy. These results support 18F-FMISO as a readily implementable NTR specific PET imaging probe to be employed as a companion diagnostic test for NTR-based GDEPT.
Materials and methods
In vitro experiments
Cell culture
Cells were maintained in a humidified atmosphere at 37 °C in 5% CO2 in complete medium, consisting of DMEM (Sigma-Aldrich, MO, USA) or RPMI-1640 supplemented with 10% FBS, 1% penicillin/streptomycin (Sigma-Aldrich), and 1% L-glutamine (Sigma-Aldrich). 293T cells were purchased from DSMZ (Braunschweig, Germany). MDA-MB-231Luc+ and NCI-H460 Luc+ cells, stably expressing luciferase, (onwards referred to as NTR-) were kindly provided by Prof. James Lorens (University of Bergen). Generation of MDA-MB-231Luc+GFP+NTR+ and NCI-H460Luc+GFP+NTR+ cells, stably expressing luciferase, GFP and NfsB (onwards referred to as NTR+) has been previously described by McCormack et al. [20]. Characterisation of the cell lines by flow cytometry can be found in Figures S1 and S3. To avoid possible expression drift due to cell culturing, NTR+ cells were sorted prior to the experiments employing the flow cytometric methods described previously.
NTR lentivirus production
A custom-made lentiviral expression vector, pCDH-EF1α-NTR, containing the coding sequence of E. coli NfsB gene under the control of the EF1α promoter was produced by System Bioscience (CA, USA). Packaging into VSV-G pseudotyped viral particles was performed by co-transfection of pCDH-EF1α-NTR, pMD2.G and psPAX2 (Figure S1) into 293T cells. Viral titers were reported as transforming units (TU) per mL. TU were measured by transduction of 293T cells with serial dilutions of conditioned medium from virus packaging cells and analysis of NTR expression using a flow cytometric method described previously by McCormack et al. [20]. For validation purposes, it was confirmed that NTR expression levels in MDA-MB-231 cells transduced with pCDH-EF1α-NTR and in the NTR+ cell line were within the same range (Figure S1).
Flow cytometry
For the analysis of NTR expression 0.1 x 106 cells were incubated at 37 °C with DMEM supplemented with 1 µM of CytoCy5S for 1 hour. All samples were washed twice with PBS + 2% BSA before acquisition with the BD Accuri C6 flow cytometer (BD Biosciences, NJ, USA) with a laser excitation at 640 nm and emission filter at 675 ± 12.5 nm.
In vivo experiments
General animal care
All experiments were approved by The Norwegian Animal Research Authority (FOTS approval ID 9059) and conducted according to The European Convention for the Protection of Vertebrates Used for Scientific Purposes. NOD-scid IL2Rgnull mice (referred to as NSG) were bred at Vivarium (University of Bergen). Mice were housed in groups of ≤ 5 in individually ventilated cages (Techniplast, Italy). General condition and body weight were recorded twice per week.
MDA-MB-231 and NCI-H460 subcutaneous xenografts
Tumours were engrafted subcutaneously mono- or bi-laterally in the scapular area. 5 × 106 cells were prepared in 50 μL DMEM with 25% BD Matrigel™ (BD Science, CA, USA) and injected at each site to form a single tumour. Tumour volumes were measured weekly with a digital calliper and calculated using the ellipsoid volume formula: Volume = π (length × width × height)/6.
MDA-MB-231 orthotopic xenografts
1 × 106 cells were prepared in 50 μL of DMEM with 25% BD Matrigel™ (BD Science) and engrafted in the right inguinal mammary fat pad, as previously described [20, 27]. Prior to cell implantation, mice were injected with 0.1 mg/kg buprenorphine (Temgesic, Indivior, UK) for analgesia.
CB1954 treatment
CB1954 (Sigma-Aldrich) was resuspended in DMSO at 26.6 mg/mL. CB1954 treatment was initiated when mean tumour volumes reached 172 ± 65 mm3 in NTR+ and NTR- xenografts or four weeks after in vivo transduction in NTR transduced xenografts. Animals received two intraperitoneal (i.p.) treatments in a six-day interval with a dose of 40 mg/kg diluted in saline.
In vivo NTR transduction with lentiviral particles
NTR lentiviral particles were concentrated using Lenti-Pac Lentivirus Concentration solution (Genecopoeia, MD, USA). Subcutaneous xenografts were transduced in vivo by intratumoral injection of 8 × 107 TU resuspended in 75 μL of the complete medium supplemented with 5 μg/mL Polybrene (Sigma-Aldrich) when mean tumour volume was 70 ± 25 mm3. A single injection of viral particles per tumour was performed administering the viral suspension throughout the diameter of the tumour.
18F-FMISO biodistribution
Tumours and organs were harvested from four mice, two hours post i.v. administration of 18F-FMISO (150 µL, 8-12 MBq, 228-342 MBq/kg) and radiation was measured on a Wizard2® Automatic Gamma Counter (Perkin Elmer, MA, USA). The results are expressed as a percentage of the injected dose per gram of tissue (% ID/g).
Histopathology examinations
Following euthanasia, the tumour and the organs were fixed in neutral buffered formalin solution 10% (Sigma-Aldrich) at room temperature for 24 hours and later kept in PBS and stored in darkness at 4 °C. 5 μm thick sections were stained with haematoxylin and eosin (HE) and were examined by a pathologist for verification of malignancy.
Immunohistochemical (IHC) staining was done on tumour tissue sectioned at 5 µm thickness. Deparaffinisation and antigen retrieval was performed with EnVision FLEX Target Retrieval Solution, Low pH (DAKO, Copenhagen, Denmark) on a Dako PT Link instrument (Dako). After antigen retrieval, tissues were incubated for 8 min with peroxidase blocker (Peroxidase Blocking Reagent, cat #S2001, Agilent Technologies) and thereafter for 10 min with protein blocker (Protein Block Serum-free, cat # X090930-2, DAKO) at room temperature. The blocking solution was then removed and the slide was wiped dry around the tissue section before application of the primary antibody. The sections were then incubated overnight at 4 ºC degrees with the HIF1α primary antibody (ab51608, Abcam, Cambridge, UK) dilution 1:300. The staining was performed on a DAKO Autostainer using the EnVision+ System-HRP Labelled Polymer Anti-Rabbit K4002 as secondary antibody for 30 min (Agilent Technologies, Norway). 3,3`-Diaminobenzine (DAB+) Substrate-Chromogen was used as chromogen for 10 min. Sections were counterstained with haematoxylin (cat # S3301, Agilent Technologies) for 10 min, dehydrated, and mounted with a coverslip (Agilent Technologies) using Pertexx mounting medium (Histolab Products AB, Askim, Sweden). Human placenta with known reactivity to the selected marker was used as positive control.
Whole tissues were scanned employing an Olympus VS120 S6 Slide scanner (Olympus Corporation, Tokio, Japan). Quantification of the normalised HIF1α positive area was performed employing FIJI [28], following the protocol described by Crowe et al. [29].
PCR for monitoring in vivo NTR transduction
Following euthanasia, part of the tumour was snap-frozen on dry ice and stored at -80 ºC. DNA was purified using QIAamp DNA Mini Kit (QIAGEN, Germany). A 348 bp fragment of the coding sequence of E. coli NfsB gene was PCR amplified in a thermocycler under standard conditions using Taq DNA Polymerase (Invitrogen, CA, USA) and the following primers: NTR-F 5' GCGTCATTCCACTAAGGCAT 3' and NTR-R 5' GCGAAGAACTTGCGACCTTT 3'. DNA amplification was visualised by gel electrophoresis followed by imaging on a Gel Doc EZ system (Bio-Rad, CA, USA). Nancy-520 (Sigma-Aldrich) was used as an intercalating agent and 1 kb DNA Ladder (Sigma-Aldrich) to estimate DNA size.
Imaging techniques
During bioluminescence and fluorescence imaging mice were anaesthetised with 1.5% isoflurane (Abbot Labor Ltd, IL, USA), 3-4% sevoflurane (Abbot Labor Ltd) during PET/CT imaging.
Bioluminescence imaging
D-Luciferin (Biosynth, Switzerland) was administered i.p. at a dose of 150 mg/kg 10 minutes before imaging. Images were acquired using an In-Vivo FX Pro molecular imaging system (Carestream Health Inc., NY, USA). Analysis was performed with the Carestream molecular imaging software v5.0.6.20.
CytoCy5S-Fluorescence imaging
CytoCy5S was synthesised following a published method [20]. 100 µL of a 1 mM CytoCy5S solution were injected intravenously. Imaging was performed after a 1-hour washout using an IVIS Spectrum imaging system (PerkinElmer, MA, USA.). Analysis was performed with the Living Imaging® software v4.5 (PerkinElmer).
18F-FMISO PET/CT imaging
18F-FMISO synthesis
18F-fluoride was produced by the 18O(p,n)18F-reaction in a niobium target on a GE PETtrace6 cyclotron (GE Healthcare,) with EOB-activities in the range of 80 - 100 GBq. The activity was delivered to the synthesis hot-cell by a Safe Transfer System (Skistad Elektroautomasjon AS, Norway). 18F-FMISO was produced on a FASTlab2 (GE Healthcare, IL, USA) with the synthesis sequence provided by the supplier. The radiosynthesis has been described in detail previously [30, 31]. Reagents and synthesis cassette were purchased from GE Healthcare Norway AS (Norway). Purification of 18F-FMISO was performed through a series of SPE-cartridges, and thus did not require any semi-preparative HPLC. The final 18F-FMISO formulation contained approx. 7% ethanol. Total synthesis time was approximately 50 minutes and yields in the order of 20-30% (not decay corrected).
PET/CT imaging
PET/CT scans were acquired using the integrated nanoScan PC PET/CT (Mediso Ltd, Hungary) featuring spatial resolution of 800 μm and 300 μm of the PET and CT detector systems, respectively. The field of view (FOV) was 9.6 × 10 cm in axial direction and transaxial direction allowing whole-body imaging of the mice. The PET detectors consist of LYSO crystals, and acquisition was performed in a 1:5 coincidence and normal count mode. Mice were scanned using a dual mouse bed with integrated heating (37 °C). Each PET scan was conducted over 30 minutes, 1.5 hours post i.v. administration of 18F-FMISO (8-12 MBq, 228-342 MBq/kg). Prior to PET acquisition, a whole-body CT scan (helical projections with tube energy of 50kVp, exposure time 300 ms, 720 projections, max FOV, binning 1:4) was performed providing anatomical information, as well as attenuation correction PET image reconstruction.
PET/CT reconstruction and processing
PET images were reconstructed using the Nucline software by employing the Tera-Tomo 3D (OSEM) algorithm (four iterations and six subsets, 1-5 coincidence mode) and the following corrections: depth-of-interaction (DOI), randoms, crystal dead time, normalisation. Prior to PET, a whole-body CT (low energy) was acquired for anatomic reference and attenuation correction. CT images were reconstructed using a RamLak filter. The PET and CT images were co-registered automatically. Images were reconstructed with a voxel size of 0.25×0.25×0.25 mm3 for CT, and 0.4×0.4×0.4 mm3 for PET. Data analyses were performed using InterView Fusion version 3.03.078.0000 (Mediso Ldt.). Standard uptake value (SUV) was calculated using the equation: SUV = CPET(T)/(ID/BW), where CPET(T) was the measured activity in tissue, ID the injected dose measured in kBq, and BW the mouse's body weight in kg. For each scan a spherical volume of interest (VOI) with radius 2 mm was drawn manually over the muscle in the neck and SUVmean was calculated to serve as reference. This value enabled segmentation of putative tumour tissue having SUV ratios of twice higher than reference for 18F-FMISO. VOIs of primary tumours and of likely metastases were drawn semi-automatically in the PET images for estimation and calculation of SUVmax and SUVpeak values. SUVmax is the maximum SUV value of all voxels included in the VOI. For SUVpeak analysis a specific volume was set (5 mm3 for SUVp5 and 10 mm3 for SUVp10) and all the possible spheres with that volume fitting inside the VOI were identified. The average SUV of all voxels within the spheres was calculated for all possible spheres and the highest average SUV values from this analysis were reported as SUVp5 and SUVp10. The SUVpeak analyses were performed using PMOD software (Version 3.8).
Statistics
Results are given as mean ± standard deviation (SD). All statistical tests were performed using GraphPad Prism v 6.0h (GraphPad Software Inc, CA, USA) and p < 0.05 was considered significant. After randomisation, a one-way ANOVA was applied to ensure unbiased assignment of tumour volumes among the experimental groups. Comparison of means was performed using Student's t-tests. Test for equality of variances was performed using an F test. When variances were not equal Welch's correction was applied to Student's t-tests. Correlations were analysed by computing Pearson's correlation coefficients.
Results
18F-FMISO permits imaging of NTR expression in subcutaneous xenograft models
Although fluorescent [20] and chemiluminescent [21] substrates have been successfully applied to report NTR expression in preclinical models of cancers, their clinical potential is limited. We hypothesised that 18F-FMISO, a clinically approved PET tracer known to be sequestered and reduced in hypoxic cancer environments, may provide a PET-based imaging modality for the visualisation of NTR activity in vivo. To examine the potential of 18F-FMISO as a PET tracer of NTR expression in vivo, 18F-FMISO NTR PET/CT imaging and CytoCy5S-FLI were compared in a subcutaneous xenograft model.
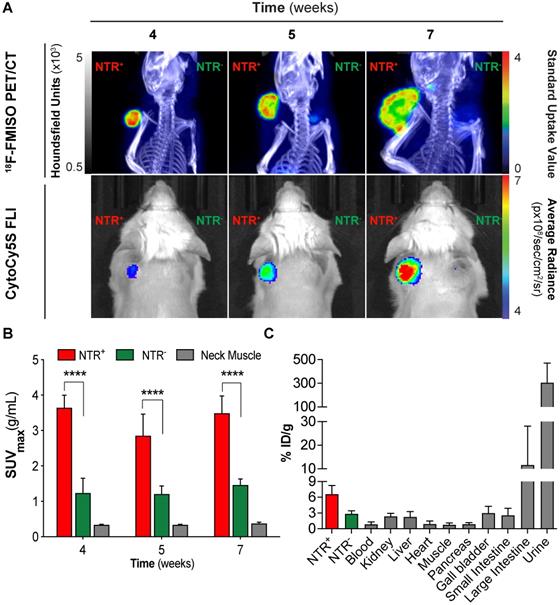
Briefly, mice were xenografted bi-laterally in the scapular region with either NTR+ or NTR- cells (n = 3 mice per group) in addition to one mouse implanted with both NTR+ and NTR- on adjacent scapulae. Mice were imaged at three timepoints post-engraftment (weeks four, five and seven) and imaging results compared (Figure 2A). CytoCy5S-FLI confirmed significantly higher contrast between NTR+ and NTR- xenografts from week five (p < 0.05, Figure 2A), when mean tumour volumes reached 243.6 ± 94.6 mm3. 18F-FMISO NTR PET/CT resulted in significantly higher contrast between NTR+ and NTR- xenografts earlier, at week four, when mean tumour volumes were 154.0 ± 75.8 mm3. At week four, NTR+ and NTR- xenograft SUVmax values were 3.64 ± 0.36 g/mL and 1.24 ± 0.42 g/mL respectively (p < 0.0001). Contrast remained significantly higher at weeks five and seven (Figure 2A and 2B). To confirm these results, 18F-FMISO tracer biodistribution was analysed ex vivo. The highest tracer retention was detected in the bladder, consistent with high renal clearance, and in the large intestine (Figure 2C), which is expected as a consequence of the expression of nitroreductases by the local microbiota. 18F-FMISO retention was higher in NTR+ compared to NTR- xenografts and remaining organs. Thus, our results suggest that 18F-FMISO can act as an NTR PET tracer.
18F-FMISO permits imaging of NTR expression in subcutaneous xenograft models. (A) Representative 18F-FMISO PET/CT MIP images and CytoCy5S-FLI 2D surface weighted images over four weeks. NTR- tumour (right flank) and NTR+ tumour (left flank). (B) SUVmax values, were significantly higher in NTR+ xenografts for all the time points (****, p < 0.0001, n=7). (C) Ex vivo biodistribution (n = 3) of 18F-FMISO obtained at week seven. Biodistribution observed mainly in large intestines and bladder. Biodistribution was higher in NTR+ tumours than in NTR- tumours and remaining organs. The p-values are represented as indicated: * p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001.
To confirm that the higher retention of 18F-FMISO observed in NTR+ is specific for NTR expression, we wanted to compare the hypoxic status of the NTR- and NTR+ xenografts. The hypoxic status of the subcutaneous MDA-MB-231 NTR- and NTR+ xenografts was assessed employing IHC to detect HIF1α accumulation (Figure S2A). Quantification of the normalised area positive for HIF1α shows no statistical significance between MDA-MB-231 NTR- and NTR+ xenografts (Figure S2B). This result suggests that 18F-FMISO retention in NTR+ xenografts is not related to increased levels of hypoxia, confirming 18F-FMISO as an NTR PET tracer.
Next, we wanted to analyse the performance of 18F-FMISO as an NTR PET tracer in the presence of other nitroreductases, both oxygen-insensitive and oxygen-sensitive. For this purpose we selected the non-small cell lung carcinoma NCI-H460 cell line, with high endogenous levels of the nitroreductase DT-diaphorase [32] and increased levels of hypoxia in comparison to MDA-MB-231 (hypoxic fractions of 13.6% and 4.7% respectively [33, 34]). First we confirmed in vitro that NTR+/NTR- ratio was notably lower for NCI-H460 than for MDA-MB-231 cell line, being respectively 5.4 and 50 (Figure S3A). Then, mice were xenografted bi-laterally in the scapular region with either NTR+ or NTR- NCI-H460 cells (n = 4 tumours per group). Mice were imaged two weeks after engraftment and imaging results compared (Figure S3B). 18F-FMISO NTR PET/CT resulted in significantly higher contrast between NCI-H460 NTR+ and NTR- xenografts with SUVmax values of 2.07 ± 0.36 g/mL and 1.51 ± 0.30 g/mL respectively (p < 0.05; Figure S3C). Interestingly, no significant differences were observed between SUVmax values from MDA-MB-231 NTR- xenografts (1.24 ± 0.42 g/mL) and from NCI-H460 NTR- xenografts. These results suggest that 18F-FMISO can act as an NTR PET tracer, even in models with notable presence of oxygen-insensitive and oxygen-sensitive nitroreductases.
18F-FMISO imaging of NTR+ metastatic lesions in orthotopic xenograft models
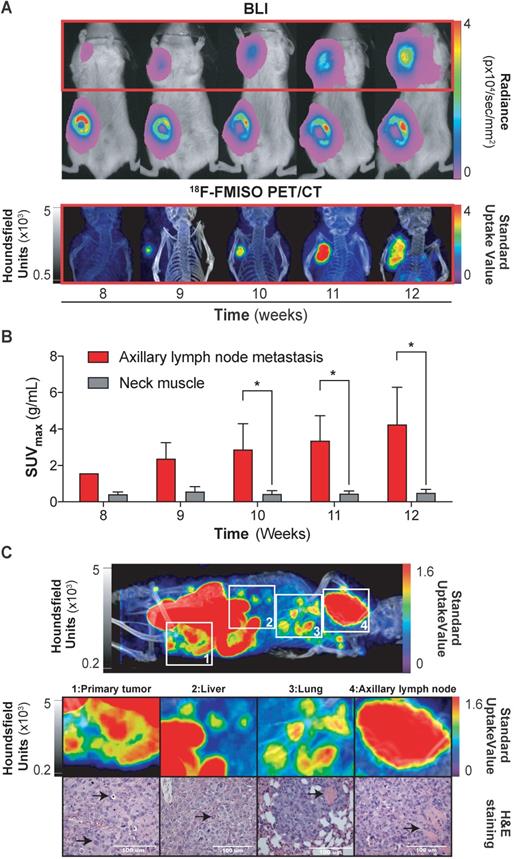
Having demonstrated the ability of 18F-FMISO to act as an NTR PET tracer in subcutaneous tumours, we next examined the suitability of the tracer for the detection of small metastatic lesions. We have previously visualised the axillary lymph node metastatic capacity of MDA-MB-231 cells when implanted orthotopically in immunodeficient mice via BLI [27]. Thus, 1 × 106 NTR+ cells were xenografted orthotopically in the mammary fat pad (n = 3) and imaged weekly with BLI and, from the initiation of metastasis, also with 18F-FMISO NTR PET/CT for five weeks (Figure 3A). Axillary lymph node metastases were clearly visible from week 8, with progressive increase in BLI up to week 12 (Figure 3A). Comparative 18F-FMISO NTR PET/CT imaging of the same mice demonstrated contrast at the axillary lymph node metastatic site with SUVmax values of 2.37 ± 0.88 g/mL from week 9 (Figure 3B). Metastatic contrast increased in weeks 10, 11 and 12 with SUVmax values of 2.88 ± 1.41, 3.36 ± 1.37 and 4.25 ± 2.04 g/mL, which were significant from background (SUVmax of reference tissue range: 0.4-0.56 g/mL; p < 0.05). Further analysis of 18F-FMISO NTR PET/CT images of NTR+ xenografts revealed additional contrast in the thoracic and lumbar regions of metastatic mice suggestive of hepatic and pulmonary metastasis (Figure 3C, upper). Subsequently, mice were euthanised, and suspected metastatic organs excised. Histological examination of HE stained sections of the lungs, liver and axillary lymph node demonstrated the presence of metastases in organs where accumulation of 18F-FMISO had been observed by 18F-FMISO NTR PET/CT imaging (Figure 3C, lower).
18F-FMISO imaging of NTR+ metastatic lesions in orthotopic xenograft models. (A) Representative images from week eight to 12. Presence of primary tumour (inguinal area) and axillary metastases (red square) by BLI. 18F-FMISO NTR PET/CT imaging in the axillary area. (B) Evolution of lymph node metastasis SUVmax compared to the reference tissue. Significant difference is observed at weeks ≥ 10 (*, p < 0.05), n = 3. (C) Whole body MIP images of 18F-FMISO NTR PET/CT show contrast in the primary tumour, liver, lung and axillary lymph node. HE staining shows widespread metastatic neoplasia in all the analysed organs. The histological features associated with neoplasia (marked with black arrows) include atypical mitosis, pyknosis, hyperchromatism, desmoplasia, irregular shape and size. Excessive extranodal tumour extension invading the adjacent muscle tissue can be seen in the axillary lymph (marked with black arrow). The lungs show signs of scattered micrometastases and venous infiltration (marked with black arrow). The p-values are represented as indicated: * p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001.
18F-FMISO allows assessment of NTR expression after in vivo transduction
While the aforementioned 18F-FMISO imaging results demonstrate the useful application of 18F-FMISO NTR PET/CT imaging for detection of constitutive NTR expression, the imaging demands of GDEPT are far greater. Initially, imaging of GDEPT should permit visual confirmation of in vivo viral transduction, which will occur in a discrete fraction of the tumoral cells [11, 12]. To examine the potential of 18F-FMISO NTR PET/CT imaging to assess in vivo transduction of tumour cells by NTR, an NTR lentiviral construct was designed (Figure S1). Viral particles were collected, concentrated and 8 x 107 TU injected intratumorally into NTR- subcutaneous xenografts (Figure 4A).
18F-FMISO allows assessment of NTR expression after in vivo transduction. (A) Experimental set-up for in vivo NTR lentiviral transduction, CB1954 treatment and 18F-FMISO PET/CT imaging. (B) Representative 18F-FMISO PET/CT MIP images suggesting the expression of NTR in the in vivo transduced tumours both at week two and four after transduction. (C) Four weeks after transduction, the SUVmax in the in vivo transduced tumours was significantly higher (*, p < 0.05 and ***, p < 0.001), n = 10). (D) Representative 18F-FMISO PET/CT MIP images suggesting a decrease in 18F-FMISO PET contrast in NTR transduced xenografts treated with CB1954 (E) SUVmax of the in vivo transduced tumours treated with vehicle showed a non-significant increase. SUVmax of the in vivo transduced tumours treated with CB1954 decreased significantly (*, p < 0.05 n = 6) (F) Confirmation of NTR transduction by PCR amplification of NTR coding sequences only in NTR+ xenografts and NTR transduced xenografts treated with vehicle. The p-values are represented as indicated: * p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001.
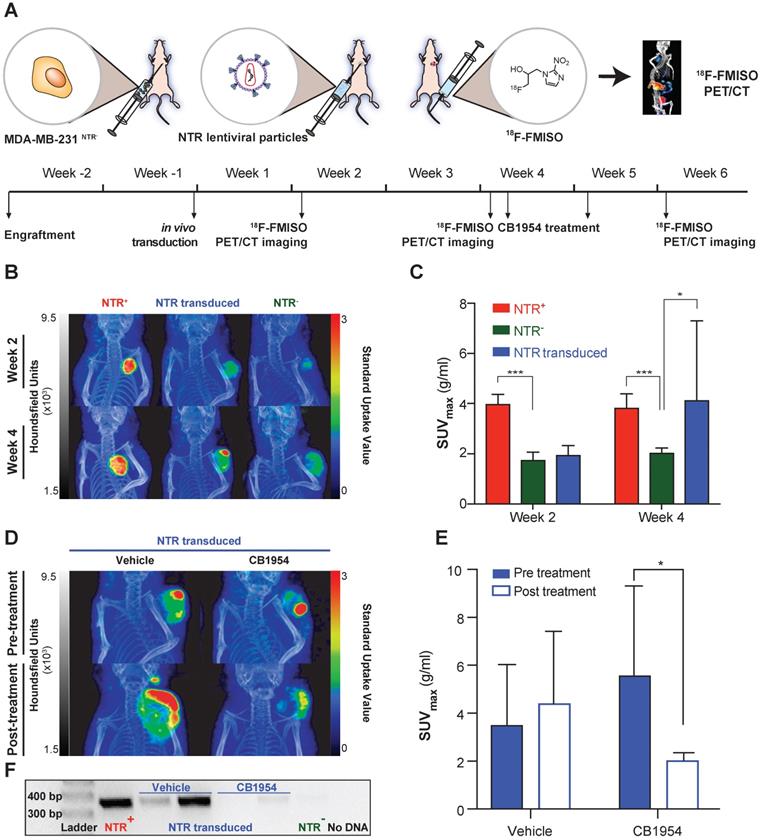
NTR transduced xenografts (n = 10), in addition to NTR+ and NTR- controls (n = 4 per group) were 18F-FMISO NTR PET/CT imaged, two and four weeks post-transduction (Figure 4A). Clear contrast of NTR+ controls could be demonstrated over NTR- controls both at week 2 and 4 (SUVmax of 3.97 ± 0.40 and 3.82 ± 0.57 g/mL respectively; p <0.001 Figure 4B & C). At week 2, minimal contrast was visualised in NTR transduced xenografts, albeit non-significant compared to NTR- controls (SUVmax of 1.94 ± 0.38 and 1.74 ± 0.32 g/mL respectively; Figure 4C). Imaging at week 4 demonstrated significant contrast compared with NTR- controls (SUVmax of 4.12 ± 3.18 and 2.03 ± 0.20 g/mL respectively; p < 0.05). Similar results were obtained when SUVpeak was analysed (Figure S4). Interestingly, contrast in lentiviral transduced tumours was observed at discrete foci (Figure 4B).
At week 4, NTR transduced xenografts were randomised to receive CB1954 (two 40 mg/kg i.p. injections in a six-day interval) or vehicle (Figure 4A). Post-treatment 18F-FMISO NTR PET/CT imaging, at week 6, showed a further increase in contrast in xenografts treated with vehicle, although non-significant (pre-treatment SUVmax of 3.5 ± 2.5 and post-treatment SUVmax of 4.4 ± 3.0 g/mL Figure 4D & E). On the other hand, a significant decrease in contrast was observed in xenografts treated with CB1954 (pre-treatment SUVmax of 5.57 ± 3.73 and post-treatment SUVmax of 2.02 ± 0.32 g/mL; p < 0.05; Figure 4D & E).
To confirm these imaging results, tumours were isolated post necropsy, DNA isolated and NTR transduction was corroborated by PCR amplification of an NTR coding sequence in the NTR transduced xenografts treated with vehicle and NTR+ controls (Figure 4F). These results confirm the utility of 18F-FMISO NTR PET/CT imaging for detecting NTR expression after in vivo viral transduction.
To exclude any possible confounding effect on 18F-FMISO NTR PET/CT imaging arising from hypoxia induced by intratumoral injection, at the end of the experiment the accumulation of HIF1α was measured in NTR- transduced xenografts and compared with NTR- tumours not receiving an intratumoral injection. The spatial pattern of HIF1α accumulation was similar in both tumour groups (Figure S5A) and the mean normalised HIF1α positive area was indeed significantly lower in intratumorally injected tumours (Figure S5B). All the above-mentioned data confirms 18F-FMISO NTR PET/CT imaging as a technique able to detect in vivo NTR viral transduction.
18F-FMISO NTR PET/CT imaging reports CB1954 treatment efficacy in NTR expressing xenografts
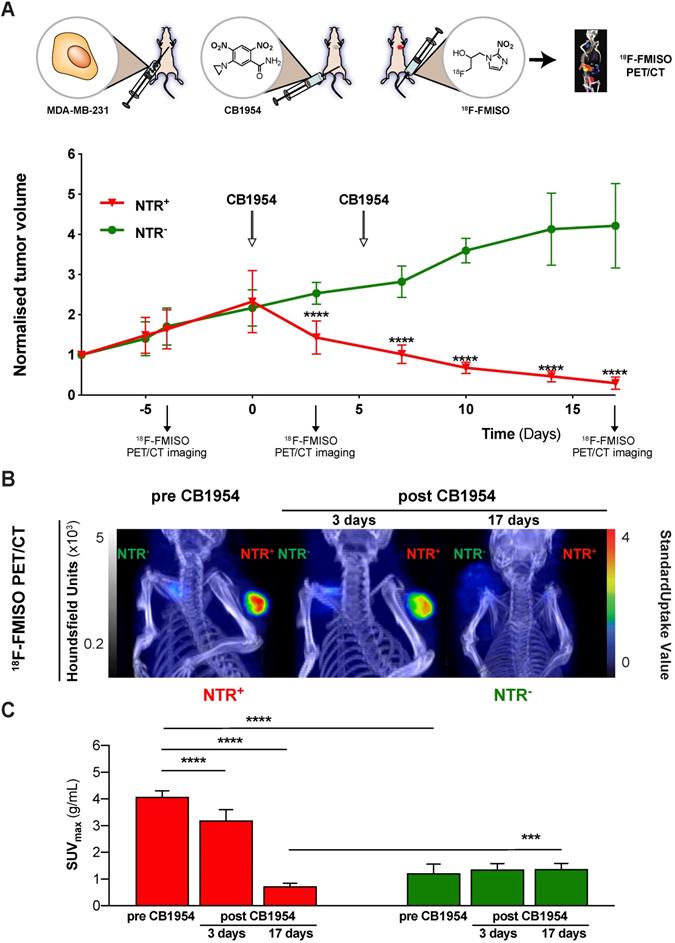
Having demonstrated the potential of 18F-FMISO NTR PET/CT imaging to successfully visualise in vivo NTR transduction, the capacity of 18F-FMISO NTR PET/CT imaging to report on CB1954 prodrug treatment efficacy was examined. NTR- and NTR+ subcutaneous xenografts (n = 7 per group) were treated with CB1954 (two 40 mg/kg i.p. injections in a six-day interval) (Figure 5). 18F-FMISO NTR PET/CT imaging was performed pre-treatment (day -4), during treatment (day 3) and following completion of treatment (day 17), in addition to calliper measurements of tumour volumes (Figure 5A). As expected, pre-treatment 18F-FMISO NTR PET/CT imaging of NTR+ xenografts resulted in significant contrast in comparison to NTR- xenografts (SUVmax of 4.07 ± 0.23 and 1.22 ± 0.35 g/mL respectively; p < 0.001 (Figure 5B and C). At day 3, a significant decrease in NTR+ tumour volumes was observed versus corresponding NTR- xenografts (normalised tumour volumes 2.53 ± 0.27 and 1.43 ± 0.41 mm3; p < 0.0001). Similarly, significant decrease in NTR+ tumour SUVmax values was also noted in 18F-FMISO PET contrast performed at day 3 in comparison to pre-treatment SUVmax values (SUVmax 4.07 ± 0.23 and 3.19 ± 0.41 g/mL; p < 0.001). Progressive reductions in NTR+ xenograft tumour volume continued following the completion of CB1954 treatment at days 7, 10 and 14 (p < 0.001), with normalised volumes of 0.1 ± 0.02 at termination of the experiment (Figure 5A). Final PET imaging of xenografts on day 17 (Figure 5B) confirmed calliper measurement results, with observed SUVmax values reduced to 0.72 ± 0.12 g/mL (p < 0.0001) (Figure 5C). These preclinical results validate translational development of 18F-FMISO PET/CT for prodrug treatment efficacy monitoring in the context of NTR-based GDEPT.
18F-FMISO NTR PET/CT imaging reports CB1954 treatment efficacy in NTR expressing xenografts. (A) CB1954 treatment response and experimental set-up for treatment monitoring employing 18F-FMISO PET/CT imaging (B) Representative 18F-FMISO PET/CT MIP images from NTR- tumour (left flank) and NTR+ tumour (right flank) before treatment and three and 17 days after CB1954 dosing. (C) Before treatment, the SUVmax values were significantly higher in NTR+ than in NTR- tumours (****, p < 0.0001, n = 7). Three and 17 days after, the SUVmax became significantly lower in NTR+ xenografts in comparison to before treatment (****, p < 0.0001, n = 7). After 17 days, SUVmax of NTR+ xenografts became significantly lower than the corresponding SUVmax of NTR- xenografts (***, p < 0.0001, n = 7). The p-values are represented as indicated: * p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001.
Discussion
GDEPT has been suggested as an attractive novel therapeutic strategy aiming to address both drug resistance and off-target cytotoxicity [35], with promising preclinical results [36-39] and a number of HSV-TK-based GDEPT phase II/III clinical trials opened in the last five years (NCT02831933; NCT02768363; NCT03004183; NCT03541928; NCT03603405; NCT03596086; NCT02446093). Recent phase I studies (NCT03281382; NCT04313868) are evaluating the use of HSV-TK-based PET imaging as a companion diagnostic tool, facilitating the analysis of HSV-TK transduction efficacy and prodrug therapy response.
In the current study, we have investigated the repurposing of 18F-FMISO, an FDA approved PET tracer as investigational new drug for human use, as a companion diagnostic tool for NTR-based GDEPT. Employing a mammary carcinoma xenograft model constitutively expressing NTR, we demonstrated that 18F-FMISO PET/CT imaging is sensitive for detection of NTR, even in small metastases. 18F-FMISO NTR PET/CT imaging was found to be effective for detection of in vivo NTR transduction and for monitoring response to CB1954 treatment, demonstrating the feasibility of 18F-FMISO NTR PET/CT imaging in the context of GDEPT.
Whilst this work has utilised breast and lung cancer models, 18F-FMISO NTR PET/CT imaging is expected to be applicable to any solid tumour type. As suggested by our results, the interaction between 18F-FMISO NTR and 18F-FMISO hypoxia PET/CT imaging does not appear to compromise this NTR imaging strategy. Indeed, preclinical 18F-FMISO hypoxia PET/CT imaging in many different tumour types reported SUVmax values in the range of 0.19-0.70 mg/mL [40-45], notably lower than the SUVmax values values for 18F-FMISO NTR PET/CT imaging reported in this study (1.5-9.95 mg/mL).
The general applicability of 18F-FMISO NTR PET/CT imaging might be limited by the distribution of the tracer to the bladder and large intestine [46], which might interfere with the imaging of tumours in the abdominal cavity. One strategy to reduce PET signal in the abdominal cavity is to increase the bowel motility using laxatives prior to imaging, this has been explored in preclinical models [47]. Another possible solution would be to employ NfsA, which is an NTR orthologue with higher affinity towards 18F-FMISO [26]. NfsA may permit the use of lower 18F-FMISO doses, resulting in a decrease in the retention in the abdominal cavity.
We acknowledge the limitations and sub-optimal PK properties of 18F-FMISO. However, the extensive clinical use of this tracer for PET imaging of hypoxia makes the process of approval for a repurposed application easier. In parallel, novel tracers with optimised pharmacokinetic properties, in order to achieve high tumour accumulation and rapid healthy tissue clearance, are under development. A second generation 2-NI, 18F-DiFA, has displayed slightly improved hypoxia sensitivity in clinical studies, accompanied by minimal distribution into the gastrointestinal tract [46, 48] and is an interesting candidate for further studies in the context of NTR imaging.
In comparison to HSV-TK-based GDEPT, NTR-based GDEPT possesses higher therapeutic potential, as NTR-based GDEPT targets cells independently of the cell cycle phase or proliferation status [49] and it results in a notable bystander effect caused by the diffusion of activated derivatives through the cell membrane [9, 10]. Despite the superior therapeutic potential of NTR-based GDEPT, clinical development is still in the early phases, as several of the steps for the NTR-based GDEPT strategy still need optimisation.
Efforts for optimisation of NTR-based GDEPT have in recent years focused on the key aspects of the technology, namely the delivery system, the NTR-enzyme and the prodrug. In an NTR GDEPT clinical trial, immune responses to the viral particles were detected [5]. Although it is not known if immune mediated responses will compromise the therapeutic efficacy, new gene delivery systems amenable for clinical translation, such as extracellular vesicles [50] and ultrasound and microbubble mediated sonoporation have been investigated [51]. While NfsB from E. coli has been the most commonly employed gene for NTR-based GDEPT, different NTR orthologs and homologs, with a much higher catalytic activity, such as NfsA from E. coli and Nme from N. meningitidis have been evaluated [26, 52, 53]. Finally, development of improved prodrug candidates is ongoing [54], since CB1954 has been associated with dose-limiting hepatotoxicity in humans [55]. Indeed, the last two aspects, NTR-enzyme and prodrug, have been optimised simultaneously through the engineering of a modified NTR enzyme that confers higher catalytic activity for an improved prodrug. Remarkable efforts by Gungor and co-workers employing an NTR from S. saprophyticus, Ssap-NtrB, and a library of different nitro-containing scaffolds has led to promising in vitro prodrug candidates, such as benzamide derivatives N-(2,4-dinitrophenyl)-4-nitrobenzamide, A5 or A20, and piperazine derivatives NHN12 or NHN14 [54, 56-58]. Other examples of such a strategy include HChrR6 with CNOB [59, 60] and NfsA variant no. 22 with PR-104A [61]. Interestingly, NfsA variant no. 22 retained reductive activity towards the hypoxia PET tracers, 18F-EF5 and 18F-HX4, allowing the possibility of NTR PET/CT imaging as companion diagnostic tool, as shown in the recently published work of Mowday et al. [26, 61].
Increasing preclinical knowledge will contribute significantly to the successful clinical translation of NTR-based GDEPT. The combination of a clinically amenable gene delivery system, optimised NTR enzymes and improved prodrugs is expected to boost the clinical application of NTR-based GDEPT. 18F-FMISO NTR PET/CT imaging should be integrated into the clinical testing of future NTR-based GDEPT strategies, providing a robust and sensitive technology to monitor forthcoming clinical trials. 18F-FMISO NTR PET/CT is an FDA methodology readily available for clinical application. Our preclinical studies further establish this strategy as a strong candidate for companion diagnostic testing of NTR-based GEDPT.
Supplementary Material
Supplementary figures.
Acknowledgements
The authors wish to thank Marianne Enger and André Sulen for cell sorting analysis performed in the Flow Cytometry Core Facility (Department of Clinical Science, UoB), Endy Spriet, Linda Sandven and Anne Karin Nyhaug (Molecular Imaging Center, Dept. of Biomedicine, UoB) for embedment and staining of histology samples, Molecular Imaging Center (MIC, Department of Biomedicine, UoB) for provision of optical imaging facility. Tom Christian Holm Adamsen and Anne Vaag Eikeland, from Center for Nuclear Medicine/PET, Haukeland University Hospital, are thanked for facilitating production of radiotracer and Hans Olav Rolfsnes (MIC, Department of Biomedicine, UoB) for assistance with the small animal PET/CT. All preclinical work was performed at the Vivarium, University of Bergen. This work was supported by The Research Council of Norway (SonoCURE grant no. 250317), the Norwegian Cancer Society (grant numbers 6833652, 421828 and 732200), the Western Norway Regional Health Authority (grant numbers 911779, 911182 and 912162), the University of Bergen and the Trond Mohn Foundation.
Funding
This work was supported by The Research Council of Norway (SonoCURE grant no. 250317), the Norwegian Cancer Society (grant numbers 6833652 and 182735), the Western Norway Regional Health Authority (grant numbers 911779, 911182, 912146 and 912035), Research Council of Norway through its Centers of excellence funding scheme, the University of Bergen and the Trond Mohn Foundation.
Author contributions
E.G.J. and G.R.G. contributed equally to this work. E.G.J., E.S., G.R.G. and K.B.L. participated in the in vitro and in vivo work. E.G.J., E.S., G.R.G., K.B.L. and E.M.C. conceived and designed the study. E.G.J provided optical probes for NTR verification supervised by B.E.H. M.P. and M.M.S assisted in animal handling and in vivo work. B.D. performed immunohistochemical analyses. C.B.R and H.E. ran the PET/CT acquisition and reconstructed the images. T.B. performed histopathology on obtained tissue. E.G.J. and G.R.G wrote the manuscript, E.M.C. and B.E.H. contributed to writing and editing the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zhang J, Kale V, Chen M. Gene-directed enzyme prodrug therapy. AAPS J. 2015;17:102-10
2. Mishra AP, Chandra S, Tiwari R, Srivastava A, Tiwari G. Therapeutic Potential of Prodrugs Towards Targeted Drug Delivery. Open Med Chem J. 2018;12:111-23
3. Bashraheel SS, Domling A, Goda SK. Update on targeted cancer therapies, single or in combination, and their fine tuning for precision medicine. Biomed Pharmacother. 2020;125:110009
4. Li N, Zhou J, Weng D, Zhang C, Li L, Wang B. et al. Adjuvant adenovirus-mediated delivery of herpes simplex virus thymidine kinase administration improves outcome of liver transplantation in patients with advanced hepatocellular carcinoma. Clin Cancer Res. 2007;13:5847-54
5. Patel P, Young JG, Mautner V, Ashdown D, Bonney S, Pineda RG. et al. A phase I/II clinical trial in localized prostate cancer of an adenovirus expressing nitroreductase with CB1954 [correction of CB1984]. Mol Ther. 2009;17:1292-9
6. Ladd B, O'Konek JJ, Ostruszka LJ, Shewach DS. Unrepairable DNA double-strand breaks initiate cytotoxicity with HSV-TK/ganciclovir. Cancer Gene Ther. 2011;18:751-9
7. Knox RJ, Boland MP, Friedlos F, Coles B, Southan C, Roberts JJ. The nitroreductase enzyme in Walker cells that activates 5-(aziridin-1-yl)-2,4-dinitrobenzamide (CB 1954) to 5-(aziridin-1-yl)-4-hydroxylamino-2-nitrobenzamide is a form of NAD(P)H dehydrogenase (quinone) (EC 1.6.99.2). Biochem Pharmacol. 1988;37:4671-7
8. Pitsawong W, Hoben JP, Miller A-F. Understanding the broad substrate repertoire of nitroreductase based on its kinetic mechanism. J Biol Chem. 2014;289:15203-14
9. Denny WA, Wilson WR. Bioreducible mustards: a paradigm for hypoxia-selective prodrugs of diffusible cytotoxins (HPDCs). Cancer Metastasis Rev. 1993;12:135-51
10. William RW, Kevin OH, Susan MP, Dianne MF, Nuala AH, Adam VP. Bystander Effects of Bioreductive Drugs: Potential for Exploiting Pathological Tumor Hypoxia with Dinitrobenzamide Mustards. Radiat Res. 2007;167:625-36
11. Rainov NG. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum Gene Ther. 2000;11:2389-401
12. Immonen A, Vapalahti M, Tyynela K, Hurskainen H, Sandmair A, Vanninen R. et al. AdvHSV-tk gene therapy with intravenous ganciclovir improves survival in human malignant glioma: a randomised, controlled study. Mol Ther. 2004;10:967-72
13. Shah K, Jacobs A, Breakefield XO, Weissleder R. Molecular imaging of gene therapy for cancer. Gene Ther. 2004;11:1175-87
14. Sekar TV, Foygel K, Ilovich O, Paulmurugan R. Noninvasive theranostic imaging of HSV1-sr39TK-NTR/GCV-CB1954 dual-prodrug therapy in metastatic lung lesions of MDA-MB-231 triple negative breast cancer in mice. Theranostics. 2014;4:460-74
15. Tjuvajev JG, Doubrovin M, Akhurst T, Cai S, Balatoni J, Alauddin MM. et al. Comparison of radiolabeled nucleoside probes (FIAU, FHBG, and FHPG) for PET imaging of HSV1-tk gene expression. J Nucl Med. 2002;43:1072-83
16. Gambhir SS, Barrio JR, Wu L, Iyer M, Namavari M, Satyamurthy N. et al. Imaging of adenoviral-directed herpes simplex virus type 1 thymidine kinase reporter gene expression in mice with radiolabeled ganciclovir. J Nucl Med. 1998;39:2003-11
17. Deng W-P, Yang WK, Lai W-F, Liu R-S, Hwang J-J, Yang D-M. et al. Non-invasive in vivo imaging with radiolabelled FIAU for monitoring cancer gene therapy using herpes simplex virus type 1 thymidine kinase and ganciclovir. Eur J Nucl Med Mol Imaging. 2004;31:99-109
18. Yaghoubi SS, Gambhir SS. PET imaging of herpes simplex virus type 1 thymidine kinase (HSV1-tk) or mutant HSV1-sr39tk reporter gene expression in mice and humans using [18F]FHBG. Nat Protoc. 2006;1:3069-75
19. Penuelas I, Mazzolini G, Boan JF, Sangro B, Marti-Climent J, Ruiz M. et al. Positron emission tomography imaging of adenoviral-mediated transgene expression in liver cancer patients. Gastroenterology. 2005;128:1787-95
20. McCormack E, Silden E, West RM, Pavlin T, Micklem DR, Lorens JB. et al. Nitroreductase, a near-infrared reporter platform for in vivo time-domain optical imaging of metastatic cancer. Cancer Res. 2013;73:1276-86
21. Vorobyeva AG, Stanton M, Godinat A, Lund KB, Karateev GG, Francis KP. et al. Development of a Bioluminescent Nitroreductase Probe for Preclinical Imaging. PLoS One. 2015;10:e0131037
22. Koch CJ, Evans SM. Optimizing hypoxia detection and treatment strategies. Semin Nucl Med. 2015;45:163-76
23. Horsman MR, Mortensen LS, Petersen JB, Busk M, Overgaard J. Imaging hypoxia to improve radiotherapy outcome. Nat Rev Clin Oncol. 2012;9:674-87
24. Shimizu Y, Zhao S, Yasui H, Nishijima KI, Matsumoto H, Shiga T. et al. A Novel PET Probe "[(18)F]DiFA" Accumulates in Hypoxic Region via Glutathione Conjugation Following Reductive Metabolism. Mol Imaging Biol. 2019;21:122-9
25. Williams EM, Little RF, Mowday AM, Rich MH, Chan-Hyams JVE, Copp JN. et al. Nitroreductase gene-directed enzyme prodrug therapy: insights and advances toward clinical utility. Biochem J. 2015;471:131-53
26. Mowday AM, Copp JN, Syddall SP, Dubois LJ, Wang J, Lieuwes NG. et al. E. coli nitroreductase NfsA is a reporter gene for non-invasive PET imaging in cancer gene therapy applications. Theranostics. 2020;10:10548-62
27. Gjerdrum C, Tiron C, Høiby T, Stefansson I, Haugen H, Sandal T. et al. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc Natl Acad Sci U S A. 2010;107:1124-9
28. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T. et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676-82
29. Crowe AR, Yue W. Semi-quantitative Determination of Protein Expression using Immunohistochemistry Staining and Analysis: An Integrated Protocol. Bio Protoc. 2019;9:e3465
30. Lim JL, Berridge MS. An efficient radiosynthesis of [18F]fluoromisonidazole. Appl Radiat Isot. 1993;44:1085-91
31. Adamsen TCH, Grierson JR, Krohn KA. A new synthesis of the labeling precursor for [18F]-fluoromisonidazole. J Labelled Comp Radiopharm. 2005;48:923-7
32. Fitzsimmons SA, Workman P, Grever M, Paull K, Camalier R, Lewis AD. Reductase enzyme expression across the National Cancer Institute Tumor cell line panel: correlation with sensitivity to mitomycin C and EO9. J Natl Cancer Inst. 1996;88:259-69
33. Franco M, Man S, Chen L, Emmenegger U, Shaked Y, Cheung AM. et al. Targeted anti-vascular endothelial growth factor receptor-2 therapy leads to short-term and long-term impairment of vascular function and increase in tumor hypoxia. Cancer Res. 2006;66:3639-48
34. Sun JD, Liu Q, Wang J, Ahluwalia D, Ferraro D, Wang Y. et al. Selective tumor hypoxia targeting by hypoxia-activated prodrug TH-302 inhibits tumor growth in preclinical models of cancer. Clin Cancer Res. 2012;18:758-70
35. Moolten FL. Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: paradigm for a prospective cancer control strategy. Cancer Res. 1986;46:5276-81
36. McNeish IA, Green NK, Gilligan MG, Ford MJ, Mautner V, Young LS. et al. Virus directed enzyme prodrug therapy for ovarian and pancreatic cancer using retrovirally delivered E. coli nitroreductase and CB1954. Gene Ther. 1998;5:1061-9
37. Djeha AH, Hulme A, Dexter MT, Mountain A, Young LS, Searle PF. et al. Expression of Escherichia coli B nitroreductase in established human tumor xenografts in mice results in potent antitumoral and bystander effects upon systemic administration of the prodrug CB1954. Cancer Gene Ther. 2000;7:721-31
38. Caruso M, Panis Y, Gagandeep S, Houssin D, Salzmann JL, Klatzmann D. Regression of established macroscopic liver metastases after in situ transduction of a suicide gene. Proc Natl Acad Sci U S A. 1993;90:7024-8
39. Kuriyama S, Mitoro A, Yamazaki M, Tsujinoue H, Nakatani T, Akahane T. et al. Comparison of gene therapy with the herpes simplex virus thymidine kinase gene and the bacterial cytosine deaminase gene for the treatment of hepatocellular carcinoma. Scand J Gastroenterol. 1999;34:1033-41
40. Wobb J, Krueger SA, Kane JL, Galoforo S, Grills IS, Wilson GD. et al. The Effects of Pulsed Radiation Therapy on Tumor Oxygenation in 2 Murine Models of Head and Neck Squamous Cell Carcinoma. Int J Radiat Oncol Biol Phys. 2015;92:820-8
41. Mena-Romano P, Cheng C, Glowa C, Peschke P, Pan L, Haberkorn U. et al. Measurement of hypoxia-related parameters in three sublines of a rat prostate carcinoma using dynamic (18)F-FMISO-Pet-Ct and quantitative histology. Am J Nucl Med Mol Imaging. 2015;5:348-62
42. Arvold ND, Heidari P, Kunawudhi A, Sequist LV, Mahmood U. Tumor Hypoxia Response After Targeted Therapy in EGFR-Mutant Non-Small Cell Lung Cancer: Proof of Concept for FMISO-PET. Technol Cancer Res Treat. 2015;15:234-42
43. Zhao SJ, Kuge YJ, Zhao Y, Takeuchi S, Murakami M, Li H. et al. FMISO PET can early evaluate antiangiogenic response to anti-EGFR molecular-targeting therapy (cetuximab) in human lung cancer xenograft. J Nucl Med. 2011 52
44. Sorace AG, Syed AK, Barnes SL, Quarles CC, Sanchez V, Kang H. et al. Quantitative [(18)F]FMISO PET Imaging Shows Reduction of Hypoxia Following Trastuzumab in a Murine Model of HER2+ Breast Cancer. Mol Imaging Biol. 2017;19:130-7
45. Jiang H, Zhang R, Jiang H, Zhang M, Guo W, Feng G. et al. Evaluation of [18F]FDG/[18F]FLT/[18F]FMISO-based micro-positron emission tomography in detection of liver metastasis in human colorectal cancer. Nucl Med Biol. 2019;72-73:36-44
46. Nakata N, Kiriu M, Okumura Y, Zhao S, Nishijima K-I, Shiga T. et al. Comparative evaluation of [18F]DiFA and its analogs as novel hypoxia positron emission tomography and [18F]FMISO as the standard. Nucl Med Biol. 2019;70:39-45
47. Ruggiero A, Brader P, Serganova I, Zanzonico P, Cai S, Lipman NS. et al. Different strategies for reducing intestinal background radioactivity associated with imaging HSV1-tk expression using established radionucleoside probes. Mol Imaging. 2010;9:47-58
48. Watanabe S, Shiga T, Hirata K, Magota K, Okamoto S, Toyonaga T. et al. Biodistribution and radiation dosimetry of the novel hypoxia PET probe [(18)F]DiFA and comparison with [(18)F]FMISO. EJNMMI research. 2019;9:60
49. Denny WA. Nitroreductase-based GDEPT. Curr Pharm Des. 2002;8:1349-61
50. Forterre AV, Wang JH, Delcayre A, Kim K, Green C, Pegram MD. et al. Extracellular Vesicle-Mediated In vitro Transcribed mRNA Delivery for Treatment of HER2(+) Breast Cancer Xenografts in Mice by Prodrug CB1954 without General Toxicity. Mol Cancer Ther. 2020
51. Devulapally R, Lee T, Barghava-Shah A, Sekar TV, Foygel K, Bachawal SV. et al. Ultrasound-guided delivery of thymidine kinase-nitroreductase dual therapeutic genes by PEGylated-PLGA/PIE nanoparticles for enhanced triple negative breast cancer therapy. Nanomedicine (Lond). 2018;13:1051-66
52. Heap JT, Theys J, Ehsaan M, Kubiak AM, Dubois L, Paesmans K. et al. Spores of Clostridium engineered for clinical efficacy and safety cause regression and cure of tumors in vivo. Oncotarget. 2014;5:1761-9
53. Vass SO, Jarrom D, Wilson WR, Hyde EI, Searle PF. E. coli NfsA: an alternative nitroreductase for prodrug activation gene therapy in combination with CB1954. Br J Cancer. 2009;100:1903-11
54. Gungor T, Onder FC, Tokay E, Gulhan UG, Hacioglu N, Tok TT. et al. PRODRUGS FOR NITROREDUCTASE BASED CANCER THERAPY- 2: Novel amide/Ntr combinations targeting PC3 cancer cells. Eur J Med Chem. 2019;171:383-400
55. Chung-Faye G, Palmer D, Anderson D, Clark J, Downes M, Baddeley J. et al. Virus-directed, enzyme prodrug therapy with nitroimidazole reductase: a phase I and pharmacokinetic study of its prodrug, CB1954. Clin Cancer Res. 2001;7:2662-8
56. Gungor T, Yetis G, Onder FC, Tokay E, Tok TT, Celik A. et al. Prodrugs for Nitroreductase Based Cancer Therapy- 1: Metabolite Profile, Cell Cytotoxicity and Molecular Modeling Interactions of Nitro Benzamides with Ssap-NtrB. Med Chem. 2018;14:495-507
57. Tokay E, Güngör T, Hacıoğlu N, Önder FC, Gülhan ÜG, Tok TT. et al. Prodrugs for nitroreductase-based cancer therapy-3: Antitumor activity of the novel dinitroaniline prodrugs/Ssap-NtrB enzyme suicide gene system: Synthesis, in vitro and in silico evaluation in prostate cancer. Eur J Med Chem. 2020;187:111937
58. Güngör T, Tokay E, Güven Gülhan Ü, Hacıoğlu N, Çelik A, Köçkar F. et al. Prodrugs for nitroreductase based cancer therapy-4: Towards prostate cancer targeting: Synthesis of N-heterocyclic nitro prodrugs, Ssap-NtrB enzymatic activation and anticancer evaluation. Bioorg Chem. 2020;105:104450
59. Forterre AV, Wang J-H, Matin AC, Delcayre A, Kim K, Green C. et al. Extracellular Vesicle-Mediated In vitro Transcribed mRNA Delivery for Treatment of HER2(+) Breast Cancer Xenografts in Mice by Prodrug CB1954 without General Toxicity. Mol Cancer Ther. 2020;19:858-67
60. Wang J-H, Forterre AV, Zhao J, Frimannsson DO, Matin AC, Delcayre A. et al. Anti-HER2 scFv-Directed Extracellular Vesicle-Mediated mRNA-Based Gene Delivery Inhibits Growth of HER2-Positive Human Breast Tumor Xenografts by Prodrug Activation. Mol Cancer Ther. 2018;17:1133-42
61. Copp JN, Mowday AM, Williams EM, Guise CP, Ashoorzadeh A, Sharrock AV. et al. Engineering a Multifunctional Nitroreductase for Improved Activation of Prodrugs and PET Probes for Cancer Gene Therapy. Cell Chem Biol. 2017;24:391-403
Author contact
![]() Corresponding authors: Professor Emmet Mc Cormack, Tel: +47 55 97 3097; Fax: +47 55 97 29 50; E-mail: emmet.mc.cormackno.
Corresponding authors: Professor Emmet Mc Cormack, Tel: +47 55 97 3097; Fax: +47 55 97 29 50; E-mail: emmet.mc.cormackno.