Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
High fat diet promotes...
HFD alters the composition of...
Intestinal microbial imbalance...
High fat diet promotes the...
Prospect
Others
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(12):5889-5910. doi:10.7150/thno.56157 This issue Cite
Review
High fat diet, gut microbiome and gastrointestinal cancer
Yao Tong1#, Huiru Gao1#, Qiuchen Qi1#, Xiaoyan Liu1, Juan Li1, Jie Gao1, Peilong Li1, Yunshan Wang1, Lutao Du1 ![]() , Chuanxin Wang1,2,3
, Chuanxin Wang1,2,3 ![]()
1. Department of Clinical Laboratory, The Second Hospital, Cheeloo College of Medicine, Shandong University, Jinan, Shandong, China.
2. Shandong Engineering & Technology Research Center for Tumor Marker Detection, Jinan, Shandong, China.
3. Shandong Provincial Clinical Medicine Research Center for Clinical Laboratory, Jinan, Shandong, China.
#These authors contributed equally to this article.
Received 2020-11-19; Accepted 2021-3-9; Published 2021-4-3
Abstract

Gastrointestinal cancer is currently one of the main causes of cancer death, with a large number of cases and a wide range of lesioned sites. A high fat diet, as a public health problem, has been shown to be correlated with various digestive system diseases and tumors, and can accelerate the occurrence of cancer due to inflammation and altered metabolism. The gut microbiome has been the focus of research in recent years, and associated with cell damage or tumor immune microenvironment changes via direct or extra-intestinal effects; this may facilitate the occurrence and development of gastrointestinal tumors. Based on research showing that both a high fat diet and gut microbes can promote the occurrence of gastrointestinal tumors, and that a high fat diet imbalances intestinal microbes, we propose that a high fat diet drives gastrointestinal tumors by changing the composition of intestinal microbes.
Keywords: high fat diet, gastrointestinal cancer, gut microbiome, inflammation, metabolic reprogramming
Introduction
According to 2020 statistics, 1,806,590 new cancer cases and 606,520 cancer deaths occurred in the United States, with gastrointestinal cancer being one of the leading causes of death [1]. Colorectal cancer (CRC) is the most diagnosed and fatal gastrointestinal cancer. Pancreatic cancer is the second, with a five-year survival rate of only 9% in America [1]. These are followed by liver, stomach and esophageal cancers. Obesity is a risk factor for gastrointestinal cancer [2], and the main cause of obesity is a high fat diet (HFD). It contains high amounts of fatty acids, but low amounts of fiber, vitamins and minerals. In recent years, with the economic development and speed-up of life, obesity has become a global health problem, leading to the prevalence of a variety of chronic diseases [3]. It is estimated that, by 2025, the global obesity rate will rise to 18% for men and 21% for women [4]. The increased prevalence of obesity may increase the global burden of gastrointestinal cancer.
With the development of the metagenome and macrotranscriptome in recent years, research into intestinal microorganisms has reached a new peak [5, 6]. These organisms are in a dynamic state, influenced by drugs, diet, lifestyle, genetics and environmental factors [7]. Researchers have found that gut microbes not only affect intestinal diseases like inflammatory bowel disease [8] and CRC [9], but also extra-intestinal disorders such as hepatic disease [10] and pancreatic disease [11]. In addition, researchers have demonstrated that HFD can significantly alter the composition of gut microbes [12]. We hypothesized that intestinal microorganisms may play an important role in the pathogenesis of gastrointestinal tumors induced by HFD. This review will provide a brief overview of recent studies on the relationship between HFD, gut microbes and gastrointestinal tumors, and discuss possible mechanisms by which HFD alters the composition of intestinal microorganisms to promote the development of gastrointestinal tumors.
High fat diet promotes gastrointestinal cancers
HFD can significantly promote the occurrence and development of gastrointestinal tumors, mainly involving metabolic reprogramming and the change of various carcinogenic molecules (Figure 1).
Association of HFD with gastrointestinal cancers. High fat diet can cause metabolic reprogramming in multiple organs and tissues of the human body with alterations in the content of various regulatory factors. It mainly acts on tumor cells themselves, nearby tissues and tumor microenvironment. The molecules marked in red are upregulated, while green downregulated. GGPPS: Geranylgeranyl diphosphate synthase.
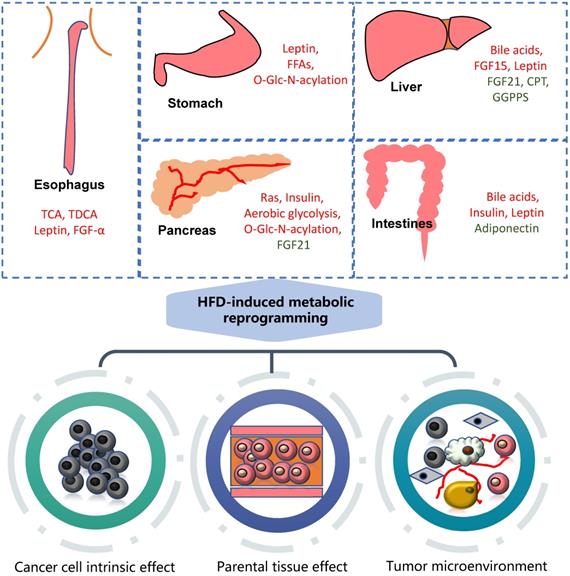
Esophagus cancer
As early as in 1994, it was found that mice fed HFD had a higher incidence of esophageal cancer, suggesting a relationship between HFD and esophageal cancer [13]. HFD can lead to changes in the composition of bile acids of mice, especially taurocholic acid and tauroursodeoxycholic acid, leading to an increased incidence of Barrett's esophagus and esophageal cancer in mice [14]. Fowler and colleagues found that esophageal adenocarcinoma in OE33 mice fed HFD had higher growth and metabolic activity and increased expression of pro-inflammatory and tumorigenic factors such as leptin, IGFBP, FGF-α in their adipose tissue, in contrast to decreased anti-inflammatory and growth inhibition molecules [15].
In clinical epidemiological studies, Ibiebele et al. found that “meat and fat” were well correlated with esophageal adenocarcinoma, gastric esophagus adenocarcinoma and esophageal squamous cell carcinoma [16]. Lagergren et al. also found that a higher proportion of fat exacerbated the occurrence of esophageal carcinoma and esophageal gastric adenocarcinoma, while carbohydrates lowered the occurrence of esophageal adenocarcinoma and esophageal gastric adenocarcinoma [17].
Gastric cancer
Many epidemiological studies have reported that dietary fat may be a risk factor for gastric cancer [18]. Leptin is thought to play an important role in obesity-related gastrointestinal malignancies because of its role in angiogenesis, apoptosis, cell proliferation and cell migration [19]. It has also been shown to promote mucin production and gastrointestinal tumor formation by regulating mTOR, STAT3 and ERK-dependent pathways [20], PI3K dependent pathways and MAPK dependent pathways [21]. Excessive leptin and leptin signaling activation leads to gastric tumors by inhibiting suppressors of cytokine signaling 3 in gastrointestinal epithelial cells [22] as well as increased expression of ectopic molecules related to intestinal epithelium, such as intestinal mucin 2 and Paneth cell marker PLA2, as well as decreased expression of the transcription factor SRY-box transcription factor 2 and H+/K+ ATPase [23].
Arita and colleagues found that lipotoxicity associated with HFD induced precancerous lesions due to the disruption of gastric epithelial organelle homeostasis, tissue integrity, and stemness gene expression mediated by ObR signaling [24]. In brief, through the up-regulation of the PI3K-Akt pathway in epithelial cells, HFD promotes β-catenin and disrupts organelle homeostasis, and can moreover up-regulate the characteristics of cancer stem cells. Various signaling molecules are expressed, such as LAMP2A, cytochrome c oxidase, LGR5 etc. Autophagy inhibition can be caused by an increase in LAMP2A, which is involved in poorly differentiated human gastric adenocarcinoma (GAC) [25].
A study found that, during 8-20 weeks of HFD feeding, mitochondrial damage was found in gastric wall cells accompanied by increased mucosal thickness. The addition of free fatty acids (FFAs) can replicate this expression and promote metagenetic changes, indicating that the lipid toxicity of FFAs induces the death of parietal cells and the occurrence of precancerous lesions. Jiang et al. found that HFD could provide sufficient energy for metastasis and increase the level of O-Glc-N-acylation, which promoted the transcriptional activation of CD36. Upregulation of CD36 leads to increased fat uptake in gastric cancer cells, forming a vicious cycle that promotes gastric cancer metastasis [26].
Hepatic carcinoma
Metabolic reprogramming, as a key intermediate, promotes the development of tumors by connecting many pro-oncogenic molecules. Studies have confirmed that HFD can cause significantly increased hepatic retention of hydrophobic bile acids, which are significantly associated with changes in intestinal microorganisms [27]. At the same time, the synthesis and transport of bile acids in the liver are disordered, leading to the release of a variety of inflammatory cytokines and the serious deposition of bile acids, and promoting the occurrence of liver cancer. In addition, a variety of molecules regulating metabolism are changed. For example, FGF21 and CPT2 were reduced in the livers of diet-induced obese mice, while FGF15 (19), IRE1α and leptin were upregulated, which were then connected with other pathological changes to promote carcinogenesis.
Reduced FGF21 is closely related to excessive proliferation, abnormal p53 and TGF-β/Smad signals [28], as well as the aberrant expression of epithelial-mesenchymal transition (EMT) and Wnt/β-catenin signals in the liver [29]. CPT2, a fatty acid oxidase, was significantly down-regulated in HFD-fed mice, leading to the accumulation of acylcarnitine in hepatocellular carcinoma (HCC) tissues and serum, which synergistically inhibited the oxidation of fatty acids and activated STAT3, jointly promoting the occurrence of HCC [30]. Liu and colleagues found that long-term HFD can decrease the expression of geranylgeranyl diphosphate synthase in mice [31]. Liver geranylgeranyl diphosphate synthase knockout enhanced liver kinase B1 hyper-farnesylation, damaged mitochondrial function through regulating AMPK activity and promoted glycolysis. These metabolic changes led to liver inflammation, infiltration of macrophages and proinflammatory cytokines, which then promoted the liver pathological progress.
IRE1α is linked to endoplasmic reticulum stress in liver cancer and drives pathogenesis [32]. On the one hand, IRE1α promotes the activation of an obesity-associated inhibitor of the NFκB pathway, leading to the production of typical pro-inflammatory cytokines such as TNF and IL-6 in the liver. On the other hand, it maintains STAT3 activation and thereby promotes hepatocyte proliferation [33]. The leptin signaling pathway can activate mTOR [34] through downstream PI3K/Akt signaling, while mTOR indirectly activates eukaryotic initiation factor 4E, thus stimulating the translation of mRNA encoding proliferation and anti-apoptotic factors [35]. Meanwhile, HFD can significantly increase the serum DPP4 level, promoting the cascade reaction of DPP4/CCL2/angiogenesis and the inflammatory response mediated by DPP4-regulated macrophage infiltration, all of which play a key role in HFD-related HCC progression [36].
Pancreatic cancer
HFD can promote proliferation and inhibit abnormal cell clearance. A western diet induced excessive proliferation of pancreatic epithelial cells in mice and led to an increase in the frequency and possibility of mutations [37]. HFD feeding significantly reduced the ability to remove RasV12 transformed cells, which impairs the epithelial defense against cancer [38].
HFD can create both inflammatory and immunosuppressive tumor microenvironments. Philip et al. found that the pancreatic tissues of LSL-KRas/ela-CreERT mice fed with HFD had higher KRAS activity, fibrotic matrix, shorter survival time and higher degree of pancreatic intraepithelial neoplasms (PanIN) and pancreatic ductal adenocarcinoma (PDAC) [39]. The possible mechanism is that the KRAS point mutation limits the ability of guanosine triphosphate hydrolysis, meaning Ras is continuously activated. High Ras activity can activate a positive feedback inflammatory cycle through the activation of COX-2 [40], increasing the expression of inflammatory mediators, and activating and extending KRAS activity. In another study, Incio et al. found that obese mice had increased tumor weight and disseminated mesenteric peritoneal metastases as well as a fibrotic tumor microenvironment which was characterized by a lack of oxygen and hypertrophy of fat cells. The recruitment of detrimental cytokines and immune cells resulted in an increase in tumor-associated neutrophils (TANs), which mediated the activation of pancreatic stellate cells (PSCs) and the growth of tumors [41]. Recently, Luo et al. found that pancreatic tissue also expressed FGF21 and its receptor proteins in addition to liver. KRAS mutation reduced the level of pancreatic FGF21, leading to increased abdominal fat accumulation, extensive inflammation, pancreatic cysts and high-grade PanIN in KC mice fed HFD [42].
Metabolic reprogramming is also involved in PDAC. For example, HFD can promote the development of pancreatic tumors by over-activating oncogenic KRAS to induce aerobic glycolysis [43]. According to Chang et al., HFD can also lead to hyperinsulinemia and accelerate the formation and progression of PanIN in KRASG12D-expressing mice [44]. Zhang et al. found that increased endogenous insulin promotes precancerous PanIN and pancreatic cancer induced by HFD, suggesting a possible carcinogenic mechanism [45].
Hu et al. found that the mechanism of PDAC development may be related to DNA damage [46]. A high sugar, high fat diet fed to C57BL/6 mice and normal pancreatic cell lines treated with high glucose in vitro showed significant DNA damage and increased KRAS mutation, and they also found that KRAS mutant cells had growth advantages in both normal and high glucose environments.
Colorectal cancer
Epidemiological studies on CRC and western diets has confirmed the link between them [47]. HFD promotes the occurrence and metastasis of CRC [48]. Several pathways play key roles in HFD promoting CRC. The JNK pathway plays a crucial role in obesity and insulin resistance [49] and promotes carcinogenic transformation and cell proliferation. Niku et al. found that a western diet accelerated the formation of colorectal adenoma, accompanied by the heterozygous loss of the APC gene and the downregulation of the ERK1/2, AKT and mTOR signaling pathways [50]. The STRA6 pathway serves as a bridge between HFD and CRC, contributing to the maintenance of CRC stem cells. HFD promotes the increase in STRA6 in tumor tissues, and STRA6 activation transduces JAK2-STAT3 signaling cascade [51]. Lysine homocysteinylation, which can be increased by HFD, modifies ATR and inhibits ATR activity, and thereby disconnects the normal ATR-interacting protein connection; it moreover inhibits the downstream checkpoint kinase-1 and p53 activity of ATR, reduces the repair of DNA damage and promotes cell proliferation [52]. Intestinal bile acids such as tauro-β-muricholic acid and deoxycholic acid can act as FXR antagonists, inhibit the FXR receptor, and promote the proliferation of lgr5+ tumor stem cells and DNA damage [53]. HFD not only inhibits the FXR receptor, but also reduces the reabsorption of bile acid in the colonic mucosa, leading to the aggravation of toxic and side effects of bile acid on colon epithelial cells and the promotion of CRC [54]. In addition, HFD can also activate the MAPK/ERK and PI3K/Akt/mTOR signaling pathways [55]. In a study by Park et al., obesity caused by HFD can promote the occurrence of inflammation-related CRC, driven by the PI3K/Akt pathway and an increase in IL-12, MCP-1, IL-6 and TNF-α in the tumor microenvironment [56].
There have also been many studies on the effects of HFD on cytokines or obesity factors. Zhu and colleagues found that elevated serum levels of insulin, leptin, TNF-α, IGF1, as well as increased levels of proliferating cell nuclear antigen, COX-2, cyclinD1, β-catenin and NFκB proteins in adenoma tissues indicate that HFD promotes the formation of colonic adenomas through inflammation and metabolic abnormalities, and influences cell cycle [57]. Zhang and colleagues reported that HFD can affect adipokines and cytokines in the serum and increase levels of TNF-α, IL-6 and CXCL10 [58].
HFD alters the composition of gut microbes
Many studies have demonstrated the key role of intestinal microorganisms in diseases induced by HFD, such as metabolic disorders [59] and endotoxemia [60]. Therefore, we hypothesized that HFD affects the development of gastrointestinal cancer by altering the composition of gut microbes.
High fat diet alters microbial composition regardless of duration
David et al. have shown that short-term HFD can change the composition of gut microbes [61]. HFD can increase the abundance of bile-tolerant microorganisms (Alistipes, Bilophila and Bacteroides) and reduce the level of Firmicutes that metabolize plant polysaccharides (Roseburia, Eubacterium rectale and Ruminococcus bromii). Some metabolites, such as bile acid and metabolic enzymes (bisulfite reductase), are increased in the intestinal tract of whole-meat eaters. [61]. On the other hand, Wu et al. have shown that a long-term HFD can alter the composition of gut microbes [62]. They investigated 98 healthy volunteers with long-term dietary habits and performed a cross-sectional analysis between 16S rDNA sequence information and metabolites, confirming the theory of Arumugam et al. about intestinal types, i.e. that Bacteroidetes is highly correlated with animal proteins, various amino acids and saturated fats [63]. Another study similarly showed that European children on a typical western diet, high in animal protein and fat, were dominated by the typical taxa of Bacteroides enterotypes, while children in Burkina Faso on a diet high in carbohydrates and low in animal protein were dominated by Privotella enterotypes [64].
High fat diet leads to an increase in the F: B ratio
Recently, Bisanz et al. conducted a meta-analysis on the sequencing data of 27 animal and human studies on diet and flora and summarized the similarities between studies on the impact of HFD on intestinal flora composition. In most studies, the HFD raised the ratio of Firmicutes to Bacteroidetes [12]. Moreover, the HFD group and the low fat diet group showed significant differences in the composition of intestinal flora, indicating that HFD could indeed cause changes to the intestinal flora. We have summarized the effects of HFD on intestinal microbes and the body (Table 1).
Effects of high fat diet on intestinal microorganisms and body
| No. | Specimen type | Specimen Source | Technology | Microbial composition alteration | Metabolites and other alterations | First author, year |
|---|---|---|---|---|---|---|
| 1 | Mucosal and luminal contents | C57BL/6J mice | Whole-genome shotgun sequencing, 16S rRNA gene amplification | Order level: ↑Caudovirales order of temperate phages; Class level: ↑Bacteroidia, Bacilli and ↑Negativicutes | — | Kim M.S., 2016 [201] |
| 2 | Fecal samples | Rat | Pyrosequencing technology, NMR | ↑Firmicutes/Bacteroidetes ratio | ↑Fecal tyrosine and phenylalanine. ↓Fecal amino acids, SCFAs, purines, pyrimidines, niacin, bile acids, ethanol, hexoses, N-acetyl-D-glucosamine, TCA cycle intermediates, gut microbiota related metabolites including 4-hydroxyphenylacetate, trimethylamine and dimethylamine. | Lin H., 2016 [235] |
| 3 | Fecal samples | C57BL/6 mice | 16S rRNA sequencing | Phylum level: ↑Firmicutes; Family level: Lachnospiraceae, Streptococcaceae (phylum Firmicutes) | ↑Plasma: leptin, TNFα, IL-6. Colon and ileum: inducible nitric oxide synthase (iNOS) and Ki67. | Zeng H., 2016 [236] |
| 4 | Fecal samples | C57BL/6J mice, 129S6/Sv mice | HiSeq-based whole genome sequencing | Genus level: ↑Clostridium, Pseudoflavonifractor, Spirochaetes, Fusobacteria, Dorea, Synergistetes, Faecalibacterium, Eubacterium, Oscillibacter, Ruminococcus, Subdoligranulum, Anaerotruncu, Blautia, Euryarchaeotadramatic, Firmicutes/Bacteroidetes ratio; ↓Tannerella, Parabacteroides, Prevotella | ↑Genes expression involved in pathways and modules related to fatty acid metabolism, cell mobility, transport, methane metabolism, and xenobiotic degradation; capacity for glycerol utilization of the gut microbiota. ↓Genes expression involved in translation and vitamin biosynthesis. | Xiao L., 2017 [187] |
| 5 | Intestinal contents | C57BL/6 mice | Real-time PCR | Genus level: ↑Firmicutes, Lactobacillus; ↓Turicibacter, Prevotella, Bacteroides, Bifidobacteria. | ↑Intestinal inflammatory cytokines including TNF-α, IL-1β, and IL-6. ↑Serum IFNγ and TNF-α. | Guo X, 2017 [237] |
| 6 | Cecal contents | C57BL/6J, 129S1/SvImJ and 129S6/SvEvTac mice | 16S rRNA sequencing | Phylum level: ↑Bacteroidetes, Verrucomicrobia | ↑Cecum: bile acids, AMP, cAMP, ADP, and CMP and nucleosides; plasma: proinflammatory fatty acids, such as adrenic and stearic acid. ↓Plasma: anti-inflammatory fatty acids, such as eicosopentaenoic and docosohexanoic acids. | Fujisaka S., 2018 [238] |
| 7 | Fecal samples | Mice | 16S rRNA sequencing | Order level: ↑Lactobacillales; Bacteroidale, Erysipelotrichales, Burkholderiales (all are subject to Bacteroidete); Genus levels: Lactobacillus Firmicutes/Bacteroidetes ratio. | ↑Serum: triglyceride, cholesterol, and high density lipoprotein; membrane transport and carbohydrate metabolism. ↓Adipose tissue: genes related to lipid metabolism expression such as PPARɑ/γ, LXRɑ/β; Liver: genes related to lipid metabolism expression such as and PPARγ and LXRɑ; metabolism of amino acid, energy, and cofactors and vitamins. | Yin J., 2018 [239] |
| 8 | Cecum contents | Hens | 16S rRNA gene amplification, pyrosequencing | Family level: ↑Erysipelotrichaceae, Alcaligenaceae, Enterococcaceae, Lactobacillaceae, ↓Firmicutes/Bacteroidetes ratio; Rikenellaceae. | ↑TC, TG, low-density lipoprotein cholesterol Lactobacillaceae, Erysipelotrichaceae are positively linked with LDL-C, TC, and TG. Ruminococcaceae had a significantly positive association with glucose. Bacteroidaceae and Porphyromonadaceae had significantly negative relationships with glucose and TG. | Liu C., 2018 [240] |
| 9 | Faecal samples | Young adults | 16S rRNA sequencing | Genus level: ↑Alistipes, Bacteroides; Blautia, Faecalibacterium Firmicutes/Bacteroidetes ratio | ↑Four pathways: steroid hormone biosynthesis, lysosome pathway, arachidonic acid metabolism and lipopolysaccharide biosynthesis. Faecal metabolites: indole, palmitic acid, stearic acid, arachidonic acid and indoleacetic acid; plasma:hypersensitive-c-reactive-protein and thromboxane B2; ↓Faecal metabolites: butyric acid, valeric acid and ethylmethylacetic acid. | Wan Y., 2019 [241] |
↑: Increased; ↓: Decreased.
Intestinal microbial imbalance promotes gastrointestinal neoplasms
Previous studies have shown that intestinal microorganisms play an important pathogenic role in some gastrointestinal tumors. We hypothesized that intestinal microorganisms may have a causal effect on gastrointestinal tumors (Figure 2).
Association of intestinal microbiomes with gastrointestinal cancers. Intestinal microbes mainly through their bacteria or secreted metabolite components that lead to the development of gastrointestinal tumors.
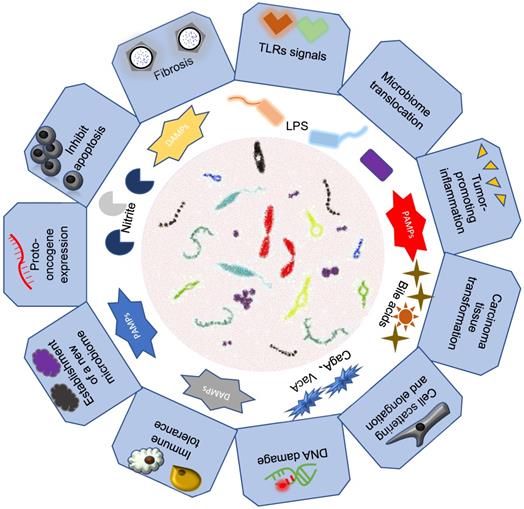
Esophagus cancer
Microorganisms in the esophagus are affected by oral and stomach microbes [65]. Helicobacter pylori infection has been shown to be negatively correlated with the occurrence of esophageal cancer [66]. The possible mechanism is that gastric atrophy caused by H. pylori infection reduces the occurrence of gastroesophageal reflux and thus reduces the occurrence of Barrett's esophagus and esophageal cancer [67]. Fusobacterium nucleatum may lead to aggressive tumor behavior through the activation of chemokines such as CCL20 in esophageal tissues [68]. Researchers have linked HFD and gut microbes to cancerous growth in Barrett's esophagus. They found that HFD accelerated esophageal dysplasia and tumor formation in L2-IL1β mice, accompanied by increased neutrophils, myeloid cells, IL-8 expression and changes in the intestinal microbiome. Under the same conditions, HFD did not accelerate the formation of esophageal tumors in germ-free mice, demonstrating that intestinal microorganisms play a key role in promoting the development of esophageal tumors in HFD mice [69]. Quante and colleagues found that mice stimulated by bile acid, which is an important intestinal microbial metabolite, were more likely to accelerate the transformation of Barrett's esophagus into esophageal cancer with the expansion of lgr5+ gastric and cardiac progenitor cells [70].
Gastric cancer
H. pylori induces complex inflammatory and immune responses that lead to a series of morphological events, beginning with chronic gastritis in almost all infected individuals, from atrophy to intestinal metaplasia to dysplasia and eventually to gastric cancer [71]. A number of toxin proteins including CagA, VacA and outer membrane proteins are directly or indirectly involved in carcinogenesis by Helicobacter [72]. CagA phosphorylates tyrosine by Src and Abl kinase; phospho-CagA then activates a number of host cell proteins, including Src homologous domain 2 (SH2) containing non-transmembrane phosphatases and SH2 domain-containing protein-tyrosine phosphatase, causing morphological changes such as cell scattering and elongation [73]. The CagA protein of some H. pylori strains can stimulate the expression of interleukin by activating the transcription factor NFκB [74]. It also causes DNA damage [75]. Wang et al. found that hydrogen metabolism plays an important role in H. pylori-induced gastric cancer, and the transmembrane potential mediated by H2 oxidation catalyzed by H. pylori hydrogenase is involved in CagA transport and also affects DNA conversion efficiency [76]. VacA results in a variety of changes in gastric epithelial cells, including vacuolation, plasma and mitochondrial membrane permeability, autophagy and apoptotic cell death [77]. In addition, studies have shown that VacA and CagA can also regulate each other, affecting host cell response [78].
In addition to toxin proteins, increased secretion of pro-inflammatory cytokines, such as TNF and IL-1β, has also been shown to be closely related to tumors [79]. IL-1β levels were elevated in gastric mucosa in patients with H. pylori infection. Recently, Koch and Muller proposed that H. pylori can activate the dendritic cell (DC) inflammasomes to secrete IL-1β and IL-18 [80]. IL-1β promotes Th1 differentiation while IL-18 promotes Treg differentiation, immune tolerance and persistent infection.
In addition, recent studies in humans have shown that the colonization of some non-H. pylori bacteria in the stomach affects the development of GAC [81]. Gastrointestinal intraepithelial neoplasia occurred earlier in insulin-gastrin (INS-GAS) mice without H. pylori but colonized with intestinal flora than in sterile mice [82]. Similar results were obtained in the K19-WNT1/C2mE GAC model [83]. In the other study, Gantuya et al. tested H. pylori negative gastritis, H. pylori positive gastritis and H. pylori negative non-gastritis groups. Streptococcus spp., Haemophilus parainfluenzae and Treponema spp. were possible pathogens [84].
H. pylori can also affect other microbiomes with an impact on gastric cancer. For example, a study showed that compared with the uninfected stomach, H. pylori infection increased Proteobacteria, Spirochetes and Acidobacteria, while it decreased the relative abundance of Actinobacteria, Bacteroidetes and Firmicutes [85]. Lertpiriyapong et al. proposed that restricted altered Schaedler's flora (including Clostridium and Lactobacillus) alone was sufficient to promote the gastric pathogenesis of INS-GAS mice, while H. pylori accelerated the occurrence and progression of gastrointestinal intraepithelial neoplasia in INS-GAS mice, and showed severe inflammation in serum and local gastric tissues [86]. High expression of TNF-α, IL-17, CCL2 (MCP-1) and IL-11 were found to be positively correlated with H. pylori infection and gastric atrophy [83]. The changes in the stomach caused by chronic H. pylori infection that reduce the secretion of gastric acid enable the successful establishment of a new microbiome that promotes malignant transformation [87].
Recently, Inasco et al. performed studies on gastric cancer and proposed that lactic acid bacteria (LAB) can affect gastric cancer through various mechanisms [88]. LAB increases thorough the development of GAC, rather than specimen contamination and dead bacteria. Lactic acid is a fuel source for cancer cells and promotes inflammation, angiogenesis, metastasis and EMT [89]. LAB is a inducer of reactive oxygen species (ROS) [90], which has been shown to cause DNA damage in cells [91]. In addition, LAB has been shown to reduce nitrate to nitrite, thus forming a large number of N-nitroso compounds, which promote mutation, angiogenesis, proto-oncogene expression and inhibit apoptosis [92]. When LAB were grown on atrophic gastric mucosa, it had the ability to induce immune tolerance [93], which was conducive to the colonization of other important carcinogenic pathological organisms including Veillonella, Prevotella, Fusobacterium and Leptotrichia [94].
Hepatic carcinoma
There is increasing evidence that the pathophysiology and treatment of various liver diseases may be strongly influenced by the gut microbiota [95]. Intestinal microflora imbalance is associated with liver diseases such as lipid accumulation, stellate cell activation, immune cell recruitment and cancer development [96]. In addition, increased bacterial translocations and disorders have been observed in the early stages of HCC and contribute to the progression of inflammation, fibrosis and cirrhosis [97]. The evidence that intestinal clearance with antibiotics can significantly reduce the incidence of HCC further confirms the close relationship between microorganisms and HCC [98].
Animal model studies have shown that liver inflammation is correlated with intestinal mucosal barrier dysfunction, suggesting that bacterial translocation and imbalance caused by intestinal mucosal barrier dysfunction affect the occurrence of non-alcoholic fatty liver disease, non-alcoholic steatohepatitis (NASH) and progression to HCC [99]. Firstly, bacteria components such as lipopolysaccharide (LPS) can promote the activation of TLR signals, and the release of inflammatory cytokines can promote the progress of NASH [100]. IL-6 activates JAK-STAT3 pathway [101] and IL-1β activates PI3K-MDM2 to negatively regulate the p53 pathway, thereby increasing DNA-damaged cell survival [102]. Secondly, the bacteria can produce ethanol and increase it in patients with NASH [103]. Ethanol is not only a hepatotoxin, but also a known carcinogen that affects the development of HCC [104]. Thirdly, gut bacteria can affect bile acid metabolism [105]. Long-term exposure to excessive bile acids in the liver leads to increased oxidative stress, resulting in DNA damage, mitochondrial damage and damage to the cell membrane of liver cells, finally activating Ras and NFκB to induce liver cancer [106]. Another mechanism controlling liver tumor growth in bile acid metabolism is the regulation of liver CXCL16 expression and CXCL16-mediated natural killer T cell recruitment [10].
Pancreatic cancer
In recent years, many studies have found that the occurrence and development of pancreatic cancer are correlated with intestinal microorganisms, and the microorganisms in the pancreas are affected by intestinal microorganisms. It was found that the Firmicutes/Bacteroidetes ratio was significantly reduced in the feces of pancreatic cancer patients [107]. Mendez et al. found that, compared with one-month KPC mice, the abundance of Firmicutes, Deferribacteres, Actinobacteria and Proteobacteria decreased significantly. Moreover, the metabolism of the microbiota changed from dominant energy metabolism to enhanced polyamine and lipid metabolism [108]. Ren and colleagues found that the α diversity of pancreatic cancer patients reduced. In the intestinal microorganisms of pancreatic cancer patients, there is an increase in some potential pathogens and lipid-producing bacteria, as well as a decrease in some probiotics and butyrate producing bacteria [109]. In addition, there are significant differences in the microbial composition of the duodenal mucosa [110], intraductal papillary mucinous tumor fluid [111] and bile [112] between patients and normal controls. In recent years, some researchers have focused on the correlation between pancreatic microbes and pancreatic cancer. Mitsuhashi and colleagues found that, compared with the Fusobacterium negative group, the cancer-specific mortality observed in the positive group was significantly higher, and the presence of Clostridium was significantly correlated with a poor prognosis of pancreatic cancer [113]. In mice, the intestinal microorganisms of PDAC patients affected the microorganisms and volume of pancreatic tumors, as the tumors of mice transplanted with fecal bacteria from patients with a short survival time were larger, indicating that intestinal microorganisms obviously influence the microorganisms in the pancreas and the development of pancreatic cancer [114].
Traditionally, the pancreas has been considered a sterile environment [115]. However, in recent years, research has shown that the pancreas contains microbes that can be affected by gut microbes [114]. How microbes get into the pancreas remains a matter of debate. Several possible mechanisms may be involved [116]. One is the oral route. The microorganisms in the upper digestive tract such as the oral cavity, esophagus, stomach, duodenum or biliary tract can enter the pancreatic parenchyma through the pancreatic duct [117]. Secondly, microorganisms may be transferred to mesenteric lymph nodes through mesenteric lymphatic drainage, and then enter the pancreatic parenthesis through immune cell trafficking. Thirdly, anatomically, because mesenteric vein drainage enters the liver through the pancreas, microorganisms can also transfer through the wall of the colon. As a result of environmental factors, the intestinal flora is imbalanced, increasing intestinal permeability and causing microorganisms to enter the blood and reach the pancreas through the blood circulation [118].
Regarding the mechanism of the influence of microorganisms on the occurrence of pancreatic cancer, Thomas and colleagues found that there is an extra-intestinal, long-distance interaction between intestinal microorganisms and the pancreas that can promote the development of pancreatic cancer and influence transcriptional changes in pancreatic cancer xenografts and the infiltration of innate immune cells [119]. Pushalkar and colleagues orally fed fluorescently labeled Enterococcus faecalis and GFP-labeled Escherichia coli into wild-type mice. They found that the bacteria could migrate to the pancreas [120]. They also found that pancreatic cancer microbes inhibit innate and adaptive immunity within the tumor. In the presence of pancreatic cancer microorganisms, immunosuppressive CD206+ M2-like tumor-associated macrophages (TAMs) increased, M1-like TAMs decreased, CD4+ T cell differentiation to Th1 cells decreased and cytotoxic CD8+ T cells decreased. Activation of pattern recognition receptors in the tumor microenvironment, especially TLR2, TLR4 and TLR5, can accelerate the growth of pancreatic cancer and enhance innate and adaptive immunosuppression [120]. Aykut et al. found that orally fed microbes could be transferred from the intestinal lumen to the pancreas within 30 minutes [11]. Malassezia can promote the occurrence of pancreatic cancer. Meanwhile, Sethi et al. found that the removal of microorganisms significantly inhibited pancreatic tumor volume and reduced the risk of pancreatic cancer liver metastasis. However, in Rag1 knockout mice lacking mature T and B lymphocytes, the tumor inhibitory effect was significantly eliminated after the removal of microorganisms, suggesting that the tumor inhibition effect may be mediated by the immune system [121].
Colorectal cancer
In clinical studies, gut microbes have been shown to be directly associated with the development of CRC. Yu et al. found that F. nucleatum, Peptostreptococcus stomatis, Parvimonas micra and Solobacterium moorei were significantly correlated with the occurrence of CRC [122]. In a case-control study, the researchers found that antibiotics play a promoting role in pancreatic cancer by comparing 28,980 cases of CRC patients and 137,077 healthy controls [123]. Cao et al. found that exposure to antibiotics early in life was significantly positively associated with the risk of colorectal adenoma after age 60, suggesting a potential mediating role of the intestinal microbiota in carcinogenesis [124]. In addition, adenoma is an important precancerous lesion in CRC and the intestinal microbial diversity of adenoma patients is significantly reduced [125]. In the classification analysis, Ruminococcaceae, Clostridiaceae and Lachnospiracea in the feces of patients significantly decreased, while Bacillus and Gammaproteobacteria significantly increased. In addition to bacteria, Temperate bacteriophages have also been shown to be associated with the occurrence and progression of CRC, as they interact with the host bacterial community and accelerate the occurrence of CRC by changing its composition [126]. In terms of intestinal microbial functions, the production of LPS, polyamine synthesis, butyrate metabolism and oxidative phosphorylation are related to the occurrence and development of CRC. For example, Mucispirillum schaedleri can promote the production of LPS and aggravate the inflammation. Lachnospiraceae bacterium A4 reduces butyrate generation and promotes the formation of cancers [127].
The role of some specific microbial communities in the development of CRC has been clarified, such as F. nucleatum, E. coli and Bacteroides fragilis. F. nucleatum is one of the common bacterial species in CRC [128]. In clinical practice, it has been shown to be related to the occurrence of CRC, as well as metastasis and prognosis [129]. It can bind with human TIGIT via fibroblast activation protein 2, which is an inhibitory receptor on all human NK cells and various T cells; this inhibits the cytotoxicity and T cell activity of NK cells against tumor cells and protects tumor cells from attack by immune cells [130]. Fibroblast activation protein 2 can also bind to D-galactose-B (1-3)-N-acetyl-D-galactosamine on the surface of CRC cells, promoting the development and metastasis of CRC [131]. In CRC cells, F. nucleatum infections can activate the TLR4 and MyD88 signaling pathways, leading to the activation of NFκB and increased miR21 expression. It can also increase the expression of anoctamin-1 in CRC cells and prevent apoptosis [132]. The specific Fusobacterium adhesin A (FadA) of F. nucleatum can bind to E-cadherin, promoting intracellular annexin A1 expression, forming a FadA/E-cadherin/annexin A1/β-catenin tetramer, thereby activating the β-catenin pathway and promoting CRC cell proliferation [133]. In addition, F. nucleatum was shown to up-regulate CARD3, activate the autophagy pathway in cells, and then promote the metastasis of CRC [134]. For E. coli, researchers have known for years that it promotes CRC [135]. Colibactin-producing E. coli infection can induce the phosphorylation of H2AX (a marker of DNA double-strand break), transient DNA damage, incomplete DNA repair, chromosomal aberrations and increased cell division [136]. E. coli infection can also promote the occurrence and development of CRC by activating the P38 MAPK pathway in macrophages to up-regulate COX-2 expression [137]. In addition, B2 E. coli isolated from CRC tissues was found to increase the invasion of multinucleated cells and the formation of crypt abscess and promote the expression of proliferative nuclear antigen in epithelial cells [138]. It has been shown that genotoxic pks+ E. coli resulted in a distinct mutational signature to promote CRC [139]. Enterotoxigenic Bacteroides fragilis (ETBF)can activate the Stat3 pathway in colonic epithelial cells through Bacteroides fragilis toxin (BFT), combine with extracellular IL-17 to activate the NFκB signaling pathway via IL17R, and trigger the expression of chemokines such as CXCL1 which can lead to the accumulation of myeloid cells in the distal colon and lead to cancer [140]. Thiele Orberg et al. found that ETBF tumorigenesis required the synergistic effect of BFT and IL-17 driven inflammatory response to coordinate the recruitment of myeloid cells to tumor microenvironment and activate the immunosuppressive myeloid-derived suppressor cells (MDSCs), especially iNOShiMO-MDSCs [141]. BFT can also up-regulate the production of spermine oxidase in colon epithelial cells, resulting in increased ROS and DNA damage. Spermine oxidase is a source of ROS induced by bacteria, which is directly related to tumorigenesis [142]. ETBF can lead to colonic mucus degradation and thereby increase the adhesion of E. coli, promoting colibactin-induced DNA damage to colonic epithelial cells [143]. In addition to these three bacteria, it was found that Campylobacter jejuni 81-176 clinically isolated from humans could activate the cytolethal distending toxin (CDT) B gene, promote the production of the CDT subunit CDTB, and thereby play the role of gene toxin and promote the occurrence of CRC [144]. Infection of C. jejuni 81-176 also changes the expression of host genes, up-regulates the PPAR signaling and calcium signaling pathways and alters the intestinal microbial composition and gene transcription of the host. Lysinibacillus sphaericus in the gut degrades aspirin and reduces its chemopreventive effects regarding CRC in mice [145].
Intestinal microbial imbalance can promote the occurrence of colorectal inflammation and then lead to CRC. For example, under normal circumstances, symbiotic fungi in the intestine can be recognized by C-type lectin receptors, activating downstream SYK and CARD9, which in turn mediates the activation of inflammasomes and promotes the maturation of IL-18 and the anti-tumor T cell response, thereby limiting the development of CRC. Imbalanced changes in intestinal microorganisms may inhibit the anti-tumor effect of the SYK-CARD9 axis [146]. In addition, fungal dysregulation increases and accumulates the MDSCs in the lamina propria of the colon, thereby promoting the development of CRC, while CARD9 can limit the accumulation of MDSCs and inhibit cancer development [147]. In CRC, several flagellar microorganisms such as Proteobacteria increase in intestinal microorganisms. Flagellin is an important component of microorganisms, which can increase the secretion of IL-6 and CCL2/MCP-1 mRNA expression by C26 CRC cells, reduce caspase-1 activity and active oxygen production, thereby increasing cytotoxicity, leading to an increase in the inflammatory response, which plays a role in promoting cancer [148].
In addition, intestinal microbial metabolites can also promote CRC. For example, short-chain fatty acids (SCFAs) are products of microbial catabolism of dietary fiber in the colon and cecum, including acetate, propionate and butyrate [149], which can be combined with Ffar2 expressed on ILC3s [150]. Through binding with Ffar2, acetate increases the number of ILC3s in the colon, and propionate promotes the number of IL-22+ ILC3s, reducing inflammation in the colorectum [151]. The down-regulation of Ffar2 leads to increased tumor bacterial load, promotes the failure of CD8+ T cells, excessively activates DCs and promotes the occurrence of colorectal tumors [152] (Table 2).
Intestinal microbial action on cancer mechanism
| No. | Types of cancer | Intestinal microbes | Molecules | Tumor promoter or suppressor | Mechanism | First author, year |
|---|---|---|---|---|---|---|
| 1 | Colon cancer | Fusobacterium nucleatum | Promoter | Fusobacterium nucleatum increases tumor multiplicity and selectively recruits tumor-infiltrating myeloid cells, which can promote tumor progression. | Kostic A.D., 2013 [242] | |
| 2 | Colon cancer | Fusobacterium nucleatum | Promoter | Fap2 protein of F. nucleatum directly interacted with TIGIT, leading to the inhibition of NK cell cytotoxicity. | Gur C., 2015 [130] | |
| 3 | Colon cancer | Fusobacterium nucleatum | Promoter | Fusobacterium nucleatum is inversely associated with CD3+ T-cell density in colorectal carcinoma tissue. | Mima K., 2015 [188] | |
| 4 | Colon cancer | Fusobacterium nucleatum | Fap2 (fusobacterial lectin) | Promoter | Fap2 mediates attachment of F. nucleatum to Gal-GalNAc which is highly expressed in human CRC, metastases, and a preclinical CRC model. | Abed J., 2016 [131] |
| 5 | Colon cancer | Fusobacterium nucleatum | Promoter | F. nucleatum activates TLR4 signaling to MYD88, leading to activation of NFκB and increased expression of miR21; this miRNA reduces levels of the RAS GTPase RASA1. | Yang Y., 2017 [243] | |
| 6 | Colon cancer | Fusobacterium nucleatum | Promoter | Fusobacterium nucleatum induces Annexin A1 expression in cancerous cells through FadA and E-cadherin, and FadA, E-cadherin, Annexin A1, and β-catenin form a complex. | Rubinstein M.R., 2019 [133] | |
| 7 | Colon cancer | Escherichia coli | Promoter | Colibactin-producing E. coli contribute to the emergence of senescent cells, which enhance tumour promotion via growth factor secretion. | Cougnoux A., 2014 [244] | |
| 8 | Colon cancer | Escherichia coli | Promoter | Colon cancer-associated E. coli bacteria induce COX-2 expression in human macrophages by p38 MAPK. | Raisch J.,2015 [137] | |
| 9 | Colon cancer | Bacteroides fragilis | Promoter | ETBF-triggered colon tumorigenesis is associated with an IL-17-driven myeloid signature characterized by subversion of steady-state myelopoiesis in favor of the generation of protumoral monocytic-MDSCs. | Thiele Orberg E., 2017 [141] | |
| 10 | Colon cancer | Bacteroides fragilis | Promoter | BFT triggers a pro-carcinogenic, multi-step inflflammatory cascade requiring IL-17R, NF-κB, and Stat3 signaling in colonic epithelial cells. | Chung L., 2018 [140] | |
| 11 | Colon cancer | (1)Lachnospiraceae bacterium A4 (2)Helicobacter hepaticus (3) Mucispirillum schaedleri | Suppressor; promoter; promoter | (1)Lachnospiraceae bacterium A4 is related to the production of Butyrate by promoting butyrate kinase synthes. (2)Helicobacter hepaticus has increased RNA counts of genes involved in oxidative phosphorylation,which suggests it exerts an oncogenic effect through oxidative damage. (3) Mucispirillum schaedleri is increasing inflammation through increased LPS production. | Daniel S.G., 2017 [127] | |
| 12 | Colon cancer | Commensal gut fungi | Suppressor | Commensal gut fungi mediate inflammasome activation by SYK-CARD9 Signaling Axis to restrict colon cancer. | Malik A., 2018 [146] | |
| 13 | Colon cancer | Sirtuin-3 (Sirt3) | Suppressor | Gut microbiota (mainly Escherichia/Shigella, Lactobacillus reuteri and Lactobacillus taiwanensis) and Sirtuin-3 can interact with another and exert an anti-inflammatory and tumor-suppressing impact. | Zhang Y., 2018 [245] | |
| 14 | Colon cancer | Campylobacter jejuni | Cytolethal distending toxin (microbial metabolites) | Promoter | Campylobacter jejuni promotes colorectal cancer through the genotoxic action of cytolethal distending toxin, which has DNAse activity and causes DNA double-strand breaks. | He Z., 2019 [144] |
| 15 | Colon cancer | P-cresol (microbial metabolites) | Promoter | Exogenous p-cresol further increased DNA damage, and independently p-cresol induced DNA damage in a dose-dependent manner against HT29 and Caco-2 cells and influenced cell cycle kinetics. | Al Hinai E.A., 2019 [246] | |
| 16 | Colon cancer | Flagellin (microbial componments) | Promoter | Flagellin increase IL6 and CCL2/MCP-1 mRNA and IL6 excretion and cytotoxicity, decrease caspase-1 activity and the production of reactive oxygen species of CRC cells. | Pekkala S., 2019 [148] | |
| 17 | Colon cancer liver metastasis | Lipopolysaccharide (microbial componments) | promoter | LPS promote CRC metastasis by stimulating TLR4 signaling and increasing β1 integrin-mediated cell adhesion. | Hsu R.Y., 2011 [247] | |
| 18 | Colon cancer liver metastasis | Lipopolysaccharide (microbial componments) | Promoter | Trapping LPS reduced liver metastasis of primary CRC and attenuated metastasized tumor growth in the liver. | Song W., 2018 [248] | |
| 19 | Gastric cancer | Helicobacter, intestinal commensals | Promoter | The gastric carcinoma microbiota is dysbiotic and characterised by reduced microbial diversity, reduced Helicobacter abundance and over-representation of bacterial genera that include intestinal commensals.The microbial community found in gastric carcinoma has increased nitrosating functions consistent with increased genotoxic potential. | Ferreira RM, 2018 [249] | |
| 20 | Gastric cancer | Helicobacter pylori | P-cresol (microbial metabolites) | Promoter | H. pylori increase proliferation in a strain-specific manner in a novel gastroid system. H. pylori also alter expression and localisation of claudin-7 in gastroids and human epithelial cells, which is mediated by β-catenin and snail activation. | Wroblewski LE, 2015 [250] |
| 21 | Gastric cancer | Helicobacter pylori | CagPAI | Promoter | EMT-like morphological changes, specifically induced by cagPAI+ H. pylori in gastric epithelial cells, are associated to enhanced expression of mesenchymal genes and are regulated by a tripartite NF-κB/ZEB1 signaling pathway | Jessica Baud, 2013 [251] |
| 22 | Gastric cancer | Helicobacter pylori | CagA | Promoter | Degradation of p53 induced by bacterial CagA protein is mediated by host HDM2 and ARF-BP1 E3 ubiquitin ligases, while the p14ARF protein counteracts H. pylori-induced signalling. | Jinxiong Wei, 2015 [252] |
| 23 | Gastric cancer | Bacterial overgrowth and diversification | Promoter | Lactobacillus and Lachnospiraceae uncultured are enriched in GAC. The gastric microbiota is altered in patients with GAC and is correlated with bacterial overgrowth and diversification. Enrichment of microbiota potentially associated with cancerpromoting activities. | Wang, 2016 [253] | |
| 24 | Gastric cancer | LAB, oral bacterial species | SCFA, lactic | Promoter | 16S rRNA transcript sequencing Helicobacter pylori infection status affects overall constitution of the gastric microbiota. Increased bacterial diversity in GAC. Enrichment of proinflammatory oral bacterial species in GAC. Increased abundance of LAB and upregulated SCFAs production metabolism. | Castaño-Rodriguez, 2017 [254] |
| 25 | Liver cancer | Bile acids | Promoter | The altered gut microbiota causes sustained retention of high concentrations of hepatic bile acids, and then promote liver carcinogenesis. | Xie G., 2016 [27] | |
| 26 | Liver cancer | Lipoteichoic acid (microbial componments) deoxycholic acid (microbial metabolites) | Promoter | Deoxycholic acid and lipoteichoic acid derived from the gram-positive gut microbiota cooperated to upregulate the expression of SASP factors and COX2 in DCA-induced senescent hepatic stellate cells through TLR2. | Loo T.M., 2017 [172] | |
| 27 | Liver cancer | Bile acid | Suppressor/promoter | Primary bile acids increases CXCL16 expression, which recruits CXCR6+ natural killer T cells to the liver, and mediate liver tumor inhibition, whereas secondary bile acids showed the opposite effect. | Ma C., 2018 [10] | |
| 28 | Liver cancer | SCFA-producing bacteria | SCFA (microbial metabolites) | promoter | Dietary soluble fibers are fermented by gut bacteria into SCFAs, which promotes hepatocyte proliferation, liver fibrosis and induces cholestatic liver cancer. | Singh V., 2018 [255] |
| 29 | Liver cancer | Interleukin-25 | promoter | Dysbiosis of gut microbiota results in secretion of IL-25, which promotes the progression of HCC through inducing alternative activation and CXCL10 secretion of macrophages in tumor microenvironment. | Li Q., 2019 [256] | |
| 30 | Breast cancer | Lithocholic acid (microbial metabolites) | Suppressor | Lithocholic acid can limit the proliferation of breast cancer cells in vitro and in vivo through activating TGR5 receptor. | Mikó E., 2018 [257] | |
| 31 | Breast cancer | Gut microbiome | Promoter | Commensal dysbiosis promoted early inflammation within the mammary gland, enhanced fibrosis and collagen deposition both systemically and locally within the tumor microenvironment and induced significant myeloid infiltration into the mammary gland and breast tumor. | Buchta Rosean C., 2019 [258] | |
| 32 | Breast cancer | Lithocholic acid (microbial metabolites) | Suppressor | Lithocholic acid decreases nuclear factor E2-related factor 2 expression, increases KEAP1 expression via activation of TGR5 and constitutive androstane receptor, elicits oxidative stress that slows down the proliferation of breast cancer cells. | Kovács P., 2019 [259] | |
| 33 | Breast cancer | Cadaverine (microbial metabolites) | Suppressor | Cadaverine exerts fuctions through trace amino acid receptors to reduce breast cancer metastasis and induce a mesenchymal-to-epithelial transition and invasion. | Kovács T., 2019 [260] | |
| 34 | Pancreatic cancer | Gut microbiome | Promoter | Gut microbiome interacts with immune system and affects cancer progression, gut microbiome depletion causes a significant anti-tumor influence in TME, such as increase in Th1 and Tc 1 cells. | Sethi V., 2018 [121] | |
| 35 | Pancreatic cancer | Bifidobacterium pseudolongum | Promoter | A distinct gut microbiome was associated with immunogenic reprogramming of the PDAC tumor microenvironment. Bifidobacterium pseudolongum promoted mitigating M1 differentiation of macrophages. | Pushalkar S., 2018 [120] | |
| 36 | Pancreatic cancer | Malassezia | Promoter | Malassezia acting as pathogenic fungi promote PDAC by driving the C3 complement cascade through the activation of MBL. | Aykut B., 2019 [11] | |
| 37 | Esophagus cancer | Bile acids | Promoter | Bile acids exposed mice were easier to developed EAC and Barrett esophagus, with acute and chronic immune response, activate differential gene expression and expansion of gastric cardia progenitor cells. | Quante M., 2012 [70] | |
| 40 | Esophagus cancer | Gut microbiome | Promoter | HFD promoted dysplasia by altering the esophageal micro-environment and gut microbiome, thereby inducing inflammation and stem cell expansion. | Münch N.S., 2019 [69] | |
| 41 | Lung cancer | Propionate (microbial metabolites) | Suppressor | Propionate inhibited lung cancer cell proliferation by inducing cell cycle arrest, especially in the G2/M phase. It increased cleaved PARP-1 and caspase 3 expression by down- and upregulating survivin and p21. | Kim K., 2019 [261] | |
| 42 | Melanoma | Bifidobacterium | Suppressor | Bifidobacterium showed a positive association with antitumor T cell responses within the tumor, and it promoted expression of genes associated with antitumor immunity of dendritic cells. | Sivan A., 2015 [262] |
High fat diet promotes the development of gastrointestinal tumors by changing intestinal microbes
Esophageal cancer
Changes in intestinal microbes due to HFD will cause an increase in immature myeloid cells and neutrophils of esophagus tissues. The progenitor cells, i.e. lgr5+ cells, expand into the esophagus accompanied by chronic inflammation, thereby causing metaplasia and dysplasia Meanwhile, the intestinal microbiota promotes the development of esophageal cancer by activating IL-8 via TLR signaling, which has an impact on the immune system and activates granulocytic myeloid cells [69]. Hydrogen sulfide (H2S) has been shown to promote the occurrence of various cancers, which is a metabolite of intestinal microorganisms [153]. Studies have found that HFD can induce gut microorganisms such as Desulfovibrio spp. and Clostridium lavalense to produce carcinogenic H2S [154], while H2S in the blood circulation is mainly microbial origin, and the amount of H2S in the blood of germ-free mice is very low [155]. After the increased H2S reaches the esophageal tissue through the blood circulation, by up-regulating HSP90, it promotes the proliferation, anti-apoptosis, angiogenesis and migration of esophageal cancer cells [156].
Gastric cancer
A recent study found that gastric leptin signal regulates gastric flora and intestinal metaplasia of gastric mucosa. Stomach flora transplantation in HFD group induces intestinal metaplasia in recipient mice, indicating that microbial changes promote intestinal metaplasia [157]. In this study, Lactobacillales increased in the stomach of mice fed HFD, while Bifidobacteriales decreased. Lactobacillus converts lactose to lactic acid, acidifying the surface of the gastric mucosa [158]. Lactic acid can serve as a fuel source for cancer cells and promote inflammation, angiogenesis, metastasis and EMT [159]. LAB is also an inducer of ROS [90], causing DNA damage to cells. Some LAB can reduce nitrate to nitrite [160], which promote mutation, angiogenesis, proto-oncogene expression, and inhibit apoptosis [92]. It has the ability to induce immune tolerance [93], which is beneficial to colonize other important carcinogenic pathological organisms including Veillonella, Prevotella, Fusobacterium, Leptotrichia and promote tumorigenesis in various ways [161], but this still needs further study.
He et al. found that, after 12 weeks of HFD, the gastric flora was abnormal and the diversity of the flora decreased [162]. Besides, HFD-induced overgrowth of Enterobacteriaceae can increase endotoxin production, thereby further triggering chronic inflammation and accelerating obesity [163]. The research of Xiao et al. showed that Desulfovibrionaceae may be an important group of endotoxin producers that can produce LPS and induce chronic inflammation and metabolic endotoxemia [164], which can be observed in the stomach of mice fed with HFD. LPS stimulation induces the expression of CXCR7 in gastric cancer and promotes the proliferation and migration of gastric cancer cells through the TLR4/MD-2 signaling pathway [165]. In addition, TLR4/CD74/MIF can also promote cell proliferation [166]. TLR4 signal activation can also promote gastric cancer cell proliferation by generating mROS [167]; LPS-NFκB-PD-L1 axis can also affect immune escape [168]. Meanwhile, Desulfovibrionaceae reduces sulfate to H2S and destroys the intestinal barrier [169]. H2S upregulates the expression of the fatty acid receptor CD36 in gastric cancer cells and directly activates CD36, triggering lipid metabolism reprogramming and thereby promoting gastric cancer metastasis [170].
Hepatic carcinoma
There exist some scholars who have done research on HFD promoting HCC through intestinal microorganisms. Xie et al. found in a mouse model of NASH-HCC induced by streptozotocin and HFD that hydrophobic bile acids significantly increased intrahepatic retention, and these are significantly associated with changes in intestinal microbes [27]. At the same time, the dysregulation of bile acids synthesis and transport in the liver leads to the release of various inflammatory cytokines and the severe accumulation of bile acids, which promotes the occurrence of liver cancer. Long-term mechanical exposure to excessive liver bile acids leads to increased oxidative stress, which can lead to DNA damage, mitochondrial damage and destruction of liver cell membranes, and activate Ras and NFκB [106]. The activated NFκB is transferred to the nucleus, and promotes the expression of genes encode proinflammatory cytokines, such as TNF-α, IL-1β and IL-6. IL-6 can activate the JAK-STAT3 pathway, leading to reduced apoptosis and HCC progression [101]. IL-1β activates PI3K-MDM2 and negatively regulates the p53 pathway, thus increasing the survival of DNA-damaged cells, which may lead to the development of liver cancer [102]. Disturbance of bile acids on cell membranes can also activate intracellular PLA2, allowing cell membranes to release arachidonic acid, which is metabolized by lipoxygenase and COX in liver cells and goes on to generate ROS. Reactive oxygen can directly activate NFκB or induce direct DNA damage to cells, which may lead to HCC [171]. Another mechanism for controlling liver tumor growth in bile acid metabolism is that secondary bile acids inhibit liver CXCL16 expression and CXCL16-mediated natural killer T cell recruitment to suppress antitumor immunity [10].
Similarly, a study found that HFD can cause excessive growth of Gram-positive bacteria in the intestine and increase the level of deoxycholic acid in the liver. Moreover, the aging-related hepatic stellate cell secretory phenotype produces various inflammatory and tumor-promoting factors, thereby promoting the development of HCC in mice exposed to the chemical carcinogen DMBA [98]. Deoxycholic acid and lipoteichoic acid in the liver are excessively displaced, and recognized by the TLR2 receptor of hepatic atellate cells to upregulate the senescence associated secretory phenotype (SASP) factors and COX-2, and then produce PGE2 to suppress anti-tumor immunity through EP4 [172]. In addition, Xie et al. used the NASH-HCC C57BL/6J mouse model induced by streptozotocin and HFD to observe the changes in intestinal microbes. They found that Atopobium spp., Bacteroides spp., Bacteroides vulgatus, Bacteroides acidifaciens, Bacteroides uniformis, Clostridium cocleatum, Clostridium xylanolyticum and Desulfovibrio spp. were significantly increased in model mice, positively correlated with LPS levels and the liver disease pathological progress [173]; the interaction between LPS and TLR is a possible mechanism.
In addition to the pathways mentioned above, we have also investigated other pathways. For example, while HFD can cause abnormal accumulation of bile acids under the action of microorganisms, and Cui et al. found that the liver FGF15 (19)/FGFR4 signal is significantly enhanced by HFD, we infer that bile acids enhance FGF15 (19)/FGFR4 and then activate EMT and the Wnt/β-catenin pathway, leading to the cancerous conversion of liver cancer [174]. In addition, HFD can reduce the proportion of Akkermansia muciniphila [175] and increase lipotoxicity [176]; we assume that HFD reduces the content of this flora, increases the damage due to lipotoxicity and activates the UPR signal conducting molecule, IRE1α [32, 177]. In addition, intestinal microbes may be related to the level of Dpp4 [178], so the Dpp4 pathway we mentioned above is likely to involve intestinal microbes.
Pancreatic cancer
We propose several mechanisms to support the hypothesis that HFD promotes the development of pancreatic cancer by affecting the composition of intestinal microbes. A number of studies have shown that HFD can affect human blood glucose levels through the intestinal flora. For example, HFD can cause the decline of Lactobacillus in the upper small intestine. This flora is related to the expression of sodium glucose cotransporter-1 in the small intestine and the secretion of GLP 1 [179]. Therefore, we speculate that HFD results in a reduced ability to regulate blood sugar and insulin resistance, leading to hyperglycemia. High glucose can increase fructose-6-phosphate/glucose-6-phosphate in pancreatic cells, which upregulates the level of glycosylation modification of O-GlcNAc, destroys functional formation of the ribonucleotide reductase complex, depletes the dNTP pool and triggers DNA replication pressure. Collectively, this leads to genome instability, increased gene mutations and a higher risk of pancreatic cancer [46].
HFD proliferates pro-inflammatory flora such as Bilophila wadsworthia, promotes the translocation of LPS to pancreatic tissues and accelerates the occurrence of pancreatic cancer through inflammation. We speculate that HFD exacerbates intestinal barrier dysfunction, resulting in the accumulation of flora and LPS, which may translocate into pancreatic tissue through the destroyed intestinal barrier and stimulate an inflammatory positive feedback loop to synergistically enhance Ras to promote pancreatic cancer. Since NFκB is a common targeting molecule downstream of Ras, we surmise that translocated LPS stimulates TLRs of pancreatic tissue and stimulates the downstream NFκB pathway, enables downstream COX-2 and promotes a positive inflammatory feedback loop through the production of PGE2 [180], thereby sustaining inflammatory response and fibrosis activation to promote tumorigenesis. COX-2 signaling can also activate glucose-regulated protein 78 through the cAMP/PKA pathway via the EP receptor of epithelial cells [181], then activate the PI3K/AKT pathway, and play an important role in tumor development in various ways [182].
It has been pointed out that complement activation in the tumor microenvironment can promote tumor immune escape, proliferation, and metastasis by maintaining T cell immunosuppression and chronic inflammation [183]. Meanwhile, a variety of complement-derived effectors and downstream signaling molecules participate in these processes, including the anchoring and proliferation of tumor cells, as well as tumor-related angiogenesis, matrix remodeling, migration, tissue invasion and metastasis [184]. Based on a study by Aykut et al. [11], we speculate that HFD can promote the translocation or migration of fungi like Malassezia spp. to pancreatic tissue, which combines with mannose binding lectin (MBL) and stimulates the complement cascade by the lectin pathway, leading to the release of C3a, C5a and other substances. C3aR-mediated signal transduction triggers the release of neutrophil extracellular traps and the polarization of neutrophils to the tumorigenic phenotype [185], while C5a can recruit MDSCs into solid tumors. MDSCs suppress effector T cells by exerting a powerful immunosuppressive effect by depriving amino acids, producing nitric oxide and ROS, up-regulating the expression of PD-L1 and secreting angiogenic factors [186], forming an immunosuppressive inflammatory microenvironment. HFD increases the abundance of F. nucleatum in the intestine, which may transfer from the intestine to the pancreas through mesenteric vein drainage, and promote the occurrence of pancreatic cancer. In HFD-fed mice, the abundance of Fusobacterium increased significantly [187]. As mentioned above, intestinal microorganisms may enter the pancreas through mesenteric vein drainage due to the disordered intestinal barrier. The fibroblast activation protein 2 protein of F. nucleatum combines with TIGIT in the pancreas [130], regulates the tumor microenvironment, reduces CD3+ T cells and CD4+ T cells, increases M2-like TAMs, suppresses the attack of the immune system on tumor cells, and promotes the development of pancreatic cancer [188]. Since HFD can promote pancreatic fat tissue accumulation and this pancreatic fat infiltration is significantly associated with the occurrence of pancreatic cancer [189], an excessive diet in healthy men has been shown to increase plasma LPS levels. LPS activates pro-caspase-11, corresponding to human caspase-4/5 in mice through an atypical reaction independent of TLR4, and induces an immune response [190]. Therefore, we also speculate that the activated caspase-11 (caspase-4/5) controls the assembly of apoptotic speck-associate protein by NLRP3 [191], and then activates capase-1 to release IL-1β and IL-18 [192]. IL-1β plays an important joint amplification effect between cancer-associated adipocytes, TANs and PSCs [41].
Colorectal cancer
Regarding the pathogenesis of CRC, some scholars have found that intestinal microbes can serve as a bridge in the process of HFD promoting CRC development, connecting the causal relationship between the two.
Schulz and colleagues found that HFD feeding promoted the formation of tumors in the small intestine of mice, along with changes in the composition of intestinal microbes. This transformation of intestinal microbes is related to the reduction of host antibacterial defenses mediated by Paneth cells, which reduces the recruitment of DCs and the expression of MHC class II molecules in intestinal associated lymphoid tissues. In terms of microbial metabolites, HFD can cause a significant reduction of SCFAs in the small intestine and feces [193]. Gaines et al. found that the formation of colorectal tumors is relevant with collagenase-producing microbes in the intestine such as Enterococcus faecalis, Proteus mirabilis and Candida parapsilosis. The colonization of these bacteria at the anastomosis destroys the healing intestine, resulting in increased permeability and transmigration of cancer cells, which promotes the recurrence of CRC. HFD increased collagenase-producing microbes [194], accelerating this effect. MCP-1 is a cell factor that acts through its receptor CCR2 to recruit the circulating leukocytes to the site of inflammation [195]. The intestinal microbial imbalance caused by HFD mediates the activation of the MCP-1/CCR2 axis, recruits monocytes to the tumor microenvironment and promotes polarization to TAMs, thereby altering the tumor immune microenvironment [196].
Based on previous related studies, we propose other possible mechanisms. First, bile acids are one of the important metabolites of intestinal microbes [197]. HFD can accelerate the increase of bile secretion induced by saturated fatty acids into the intestine [198]. Gram-positive bacteria such as Clostridium in the intestine can convert primary bile acids synthesized by liver into secondary bile acids [199], and excessive exposure to bile acids can promote CRC [199]. Second, considering the effect of HFD on intestinal microbes and the promotion of carcinogenic microorganisms on CRC, we found that, under the effect of HFD, the abundance of Parabacteroides in the intestine decreased [187]. Some studies found that Parabacteroides distasonis can reduce the activation of TLR4 signaling pathway and Akt, and inhibit the promoting role of HFD on CRC. Therefore, we speculate that HFD reduces the abundance of Parabacteroides distasonis and indirectly promotes the development of CRC [200]. Similarly, we have also found that HFD can lead to an increase in Temperate phage and Fusobacterium [201]. As mentioned above, both types of microorganisms have a cancer-promoting effect. For F. nucleatum, it was found that the western diet has a strong correlation with F. nucleatum-positive CRC in an epidemiological survey, and it plays an important role in diet-mediated CRC, which further demonstrates the accuracy of our hypothesis [202]. Figure 3 describes the possible mechanisms of the abovementioned cancers.
High fat diet changes intestinal microbes and then promotes gastrointestinal cancer. (A) Esophageal Cancer: HFD induced intestinal microbiota dybiosis (1) increases the number of Lgr5+ cells in squamocolumnar junction; (2) activates TLR signal, and promotes the activation of IL8, (3) promotes the production of H2S by increasing the abundance of Desulfovibrio spp. and Clostridium Lavalense. H2S upregulates HSP90 to promote the esophageal cancer. (B) Gastric Cancer: Lactic acid bacteria can produce lactic acid, reactive oxygen species, nitrite and other harmful products to promote gastric cancer in a variety of ways like heterogenesis, angiogenesis and etc. Microbiomes like Enterobacterium and Desulfovibrionaceae can stimulate TLRs and induce carcinogenic changes by producing LPS. LPS can also bind to PD-L1 to produce immune escape. (C) Liver Cancer: HFD changes intestinal microbial composition, leading to accumulation of certain bacterial components and metabolites. FGF and bile acids(BAs)can activate corresponding receptors on hepatocytes, trigger key oncogenic pathways then promote proliferation of cancer cells, release of inflammatory factors or DNA damage. bile acids can also activate TLR on hepatic atellate cells leading to immunosuppression. Increased LPS can activate TLR receptors on Kupffer cells to produce IL-1β and promote fibrosis (D) Pancreatic Cancer: HFD induced gut dysbiosis leads to the hyperglycemia, the increased LPS as well as pathogenic microorganisms such as Malassezia and F. nucleatum. These changes cause chronic inflammation, immune suppression, KRAS mutation fibrosis of pancreas through various mechanisms, which promotes the initiation and progression of cancer. (E) Colorectal Cancer: HFD induced gut microbiome dysbiosis including some specific microbes changes such as Collagenolytic microbes, Fusobacterium nucleatum, Parabacteroides distasoni and microbial metabolites for instance bile acids, resulting in activation of carcinogenic pathways, changes of tumor immune microenvironment, DNA damage. There also exist reduction in DCs, MHC class II molecule and SCFAs. The above changes urge the development in colorectal cancer. SCJ: Squamocolumnar junction.
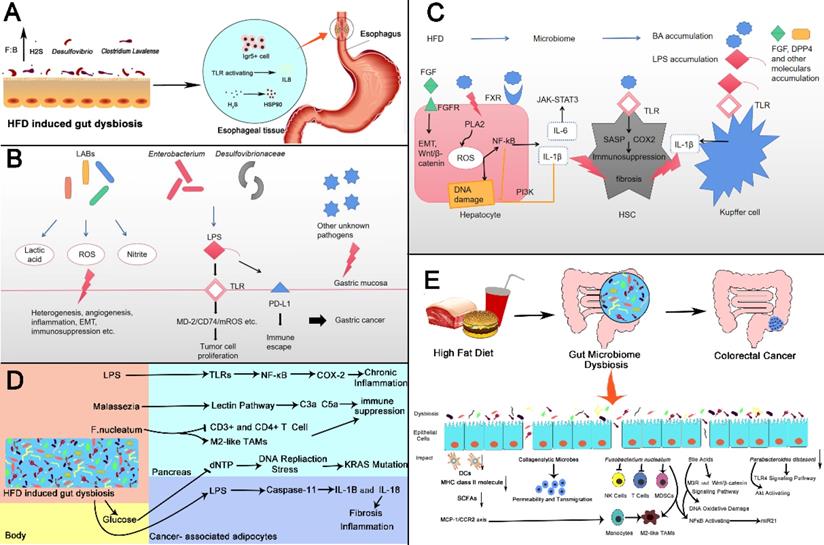
Prospect
In the preceding narrative, we demonstrated that HFD, intestinal microbes and gastrointestinal tumors are related to each other. There is a solid theoretical basis for the suggestion that the HFD can promote the occurrence of gastrointestinal cancer by changing intestinal microbes. It can be used as a direction for future research and experiments to verify whether this proposal is correct, but in the research process, we need to pay attention to distinguish whether HFD promotes tumors directly or requires microbial mediation. In addition, because the intestinal tract and various organs are linked in many ways, even if HFD unbalances intestinal microorganisms, its cancer-promoting effect is mediated through a long-distance method or when the pathogenic microorganisms directly enter the organs. These all need further research and elaboration.
In the treatment of tumors, the relationship between HFD, intestinal microbes and gastrointestinal tumors also provides us with new ideas. First, we suggest changing the lifestyle of HFD and reduce the amount of fat in the diet to prevent the occurrence of gastrointestinal tumors, and effectively control the incidence of tumors from food-borne pathways. Secondly, for patients already suffering from disease, it is also extremely important to take effective treatment measures to prevent or even interrupt the development of tumors, and intestinal microorganisms may be an effective target. Geller and colleagues found that removing cytidine deaminase or using antibiotics such as ciprofloxacin to remove Gammaproteobacteria may make the tumor sensitive to gemcitabine [117]. Antibiotics may be a strong candidate for future clinical trials. However, excessive exposure to antibiotics can cause dysbiosis and promote tumorigenesis [203]. Therefore, there is still a need for more research on antibiotic administration methods and the feasibility of treating gastrointestinal tumors. Given that gastrointestinal tumors may have an immunosuppressive tumor microenvironment, we reason that targeted immunotherapy may also be a valuable therapeutic direction. Targeted therapy can be specifically designed based on these immune cells, their action molecules and cytokines to improve the survival rate of gastrointestinal cancer. For example, the removal of Treg can reduce the tumor burden of mouse models [204], inhibit indoleamine 2,3-dioxygenase-1 activity and enhance the tumor-specific T cell response, reduce the conversion to Treg-like cells [205]. However, there is a need for a deeper understanding of tumor microenvironment components, which may make immunotherapy a more effective treatment.
Others
Among many lifestyle factors, the diet is one of the most significant factors affecting the intestinal microbiome [206]. In addition to HFD, many other lifestyle factors also have important influences on the occurrence and development of the microbiome and cancer. Human dietary habits are complex and diverse, and when studied individually, each major micronutrient and many micronutrients have been shown to alter the gut microbiome [207]. In a mouse study, the chronic high-protein diet fed for 24 weeks was shown to increase intestinal permeability, cause intestinal leakage and result in changes to the β-diversity of intestinal microbes and the microbial community structure in mice [208]. In another experiment in mice, the researchers reduced the dietary carbohydrate content and increased the protein content, and found that this dietary pattern can not only limit weight gain in mice, but it can also limit the occurrence and progress of breast cancer [209]. In addition, high protein diets have also been shown to significantly increase the overall survival of patients with cancer [210]. A high salt diet may affect gut microbial components, such as lower Lactobacillus spp. and change the fecal contents of short chain fatty acids such as butyrate. This can induce proinflammatory genes such as Rac1, Map2k1, Map2k6 and Atf2, as well as many cytokines and chemokines (Ccl3, Ccl4, Cxcl2, Cxcr4 and Ccr7), thereby affecting intestinal immunity and promoting inflammation [211, 212]. In terms of cancer, a high-salt diet has been shown to inhibit tumor growth in mice by regulating the activity of MDSCs, activating anti-tumor immune surveillance [213]. A high-sugar diet can also reduce the cecal microbial diversity [214, 215] and can be a factor that promotes the occurrence of cancer. For a high-fiber diet, researchers such as Marques found that a large amount of fiber intake can increase acetate-producing bacteria. The presence of fiber and acetate can improve intestinal imbalance [216]. A high-fiber diet can extend the survival time of patients with CRC and improve the prognosis of patients with CRC. For the immune system, it regulated the activation of B and T cells to exert a positive impact on immune disorders [217, 218]. Meanwhile, some micronutrients such as vitamins and zinc can also affect the composition of intestinal microbes [219, 220].
To a certain extent, the composition of the gut microbiota may depend on the individual's genetics, although studies by scholars have shown that genes play a minor influence on the formation of gut microbes [221]. Khan and colleagues found immune genes regulated the selected bacterial lineages in the mouse intestine. For example, the abundance of segmented filamentous bacteria and Mucispirillum is related with MHC-allele-dependent IgA response [222]. Some scholars have also explored the impact of host genetics on gut microbes in patients with IBD by combining whole exome sequencing and metagenomic shotgun sequencing, and found that the reduction in the levels of the microbial acetyl-CoA and glyoxylate metabolic pathways correlated with the minor allele (C) of a variant located in the gene MYRF, while the immune-related gene CABIN1 provided a more conducive environment to the growth of the E. coli [223]. In NOD2 knock-out mice, gut microbes changed significantly. Compared with WT mice, Alistipes (Rikenellaceae) and Bacteroides (Bacteroidaceae) genera were over-represented in Nod2 knock-out mice. Conversely, the Prevotella (Prevotellaceae) genus was reduced [224]. Researchers such as Bonder have systematically studied the impact of host genotype on gut microbes. They found multiple associations between genetic variation and the composition and function of the human gut microbiome, such as the recessive effect of an lactase functional variant on the abundance of Bifidobacterium [225]. In a study of twins, researchers also found that the host gene ALDH1L1 was potentially related to the bacterium SHA-98, which was a component of the Christensenellaceae consortium [226]. In general, host gene-related mutations, including single nucleotide polymorphism, gene copy number variation, non-coding RNA regulation, etc., will cause changes in host microorganisms [227]. Gene-microbe interactions can also affect the occurrence of immune diseases. In the research center of Chu, H. et al., it was found that microorganisms, as an environmental factor, interact with ATG16L1/NOD2 genes to promote anti-inflammatory immune responses [228].
Molecular pathology epidemiology is also an important direction of microbial research in recent years. The tumorigenesis promoted by HFD has obvious molecular pathological characteristics. For example, we mentioned above that mice fed with HFD have higher activity of KRAS [229]. Under HFD conditions, mutations in the KRAS will also promote the occurrence and development of pancreatic cancer by down-regulating the expression of the key molecule FGF21 [230]. In liver cancer, HFD can significantly increase the serum level of DPP4. The cascade of DPP4/CCL2/angiogenesis and the inflammatory response formed by DPP4-regulated macrophage infiltration play key roles in the progression of HFD-related HCC [231]. Niku and colleagues found that Western diet accelerates the formation of colorectal adenomas, which was accompanied by the loss of APC gene heterozygosity [232]. When specific pathogenic microorganisms change in the intestine, the tumors catalyzed by these pathogenic microorganisms may also have obvious molecular pathological characteristics. Hale found that in the two subtypes of deficient and proficient mismatch repair CRC, intestinal microbes are significantly different, and microbes have different roles in the two subtypes of CRC [233]. C. jejuni 81-176 infection will up-regulate two cancer-promoting signal pathways including Peroxisome proliferator-activated receptors signaling pathway and calcium signaling pathway, and promote the occurrence of CRC [234]. In addition, there are some other molecular pathological epidemiology related to HFD and microbes and intestinal tumors, which was involved in the former paper.
Abbreviations
HFD: high fat diet; BFT: Bacteroides fragilis toxin; CDT: cytolethal distending toxin; CRC: colorectal cancer; DC: dendritic cell; EMT: epithelial-mesenchymal transition; ETBF: Enterotoxigenic Bacteroides fragilis; FadA: Fusobacterium adhesin A; FFA: free fatty acid; GAC: gastric adenocarcinoma; H2S: Hydrogen sulfide; HCC: hepatocellular carcinoma; INS-GAS: insulin-gastrin; LAB: lactic acid bacteria; LPS: lipopolysaccharide; MBL: mannose binding lectin; MDSC: myeloid-derived suppressor cell; NASH: Non-alcoholic steatohepatitis; PanIN: pancreatic intraepithelial neoplasms; PDAC: pancreatic ductal adenocarcinoma; PSC: pancreatic stellate cell; ROS: reactive oxygen species; SASP: senescence associated secretory phenotype; SCFA: short-chain fatty acid; SH2: Src homologous domain 2; TAM: tumor-associated macrophage; TAN: tumor-associated neutrophil.
Acknowledgements
Authors' contributions
YT, HG and QQ wrote the first draft of the manuscript. Conceptualization was carried out by YW. CW and LD assisted in reviewing literature, guided the analysis, and provided intellectual input in the manuscript. JG, XL, PL and JL helped in reviewing the first draft of the manuscript. YT, HG, QQ and CW reviewed and edited the final manuscript. All authors read and approved the final manuscript.
Funding
The authors were supported by grant from the National Natural Science Foundation of China (81972007, 81873977, 81874040 and 81902882), the National Key Research and Development Program of China (2018YFC0114700), the Natural Science Foundation of Shandong Province (ZR2019PH074 and ZR2019BH066), the Key Research and Development Program of Shandong Province (2019GHZ003, 2018YFJH0505, 2019GSF108091 and 2019GSF108218), Taishan Scholar Program of Shandong Province.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7-30
2. Murphy N, Jenab M, Gunter MJ. Adiposity and gastrointestinal cancers: epidemiology, mechanisms and future directions. Nat Rev Gastroenterol Hepatol. 2018;15:659-70
3. Imamura F, Micha R, Khatibzadeh S, Fahimi S, Shi P, Powles J. et al. Dietary quality among men and women in 187 countries in 1990 and 2010: a systematic assessment. Lancet Glob Health. 2015;3:e132-42
4. Collaboration NCDRF. Trends in adult body-mass index in 200 countries from 1975 to 2014: a pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet. 2016;387:1377-96
5. Adolph TE, Mayr L, Grabherr F, Schwärzler J, Tilg H. Pancreas-Microbiota Cross Talk in Health and Disease. Annu Rev Nutr. 2019;39:249-66
6. Akshintala VS, Talukdar R, Singh VK, Goggins M. The Gut Microbiome in Pancreatic Disease. Clin Gastroenterol Hepatol. 2019;17:290-5
7. Schmidt TSB, Raes J, Bork P. The Human Gut Microbiome: From Association to Modulation. Cell. 2018;172:1198-215
8. Cohen LJ, Cho JH, Gevers D, Chu H. Genetic Factors and the Intestinal Microbiome Guide Development of Microbe-Based Therapies for Inflammatory Bowel Diseases. Gastroenterology. 2019;156:2174-89
9. Wong SH, Yu J. Gut microbiota in colorectal cancer: mechanisms of action and clinical applications. Nat Rev Gastroenterol Hepatol. 2019;16:690-704
10. Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M. et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science. 2018 360
11. Aykut B, Pushalkar S, Chen R, Li Q, Abengozar R, Kim JI. et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature. 2019;574:264-7
12. Bisanz JE, Upadhyay V, Turnbaugh JA, Ly K, Turnbaugh PJ. Meta-Analysis Reveals Reproducible Gut Microbiome Alterations in Response to a High-Fat Diet. Cell Host Microbe. 2019 26
13. Clark GW, Smyrk TC, Mirvish SS, Anselmino M, Yamashita Y, Hinder RA. et al. Effect of gastroduodenal juice and dietary fat on the development of Barrett's esophagus and esophageal neoplasia: an experimental rat model. Ann Surg Oncol. 1994;1:252-61
14. Chen K-H, Mukaisho K-I, Sugihara H, Araki Y, Yamamoto G, Hattori T. High animal-fat intake changes the bile-acid composition of bile juice and enhances the development of Barrett's esophagus and esophageal adenocarcinoma in a rat duodenal-contents reflux model. Cancer Sci. 2007;98:1683-8
15. Fowler AJ, Richer AL, Bremner RM, Inge LJ. A high-fat diet is associated with altered adipokine production and a more aggressive esophageal adenocarcinoma phenotype in vivo. J Thorac Cardiovasc Surg. 2015;149:1185-91
16. Ibiebele TI, Hughes MC, Whiteman DC, Webb PM. Dietary patterns and risk of oesophageal cancers: a population-based case-control study. Br J Nutr. 2012;107:1207-16
17. Lagergren K, Lindam A, Lagergren J. Dietary proportions of carbohydrates, fat, and protein and risk of oesophageal cancer by histological type. PLoS ONE. 2013;8:e54913
18. Han J, Jiang Y, Liu X, Meng Q, Xi Q, Zhuang Q. et al. Dietary Fat Intake and Risk of Gastric Cancer: A Meta-Analysis of Observational Studies. PLoS ONE. 2015;10:e0138580
19. Howard JM, Pidgeon GP, Reynolds JV. Leptin and gastro-intestinal malignancies. Obes Rev. 2010;11:863-74
20. Chang MS, Byeon S-j, Yoon SO, Kim B-h, Lee HS, Kang GH. et al. Leptin, MUC2 and mTOR in appendiceal mucinous neoplasms. Pathobiology. 2012;79:45-53
21. El Homsi M, Ducroc R, Claustre J, Jourdan G, Gertler A, Estienne M. et al. Leptin modulates the expression of secreted and membrane-associated mucins in colonic epithelial cells by targeting PKC, PI3K, and MAPK pathways. Am J Physiol Gastrointest Liver Physiol. 2007;293:G365-G73
22. Inagaki-Ohara K, Mayuzumi H, Kato S, Minokoshi Y, Otsubo T, Kawamura YI. et al. Enhancement of leptin receptor signaling by SOCS3 deficiency induces development of gastric tumors in mice. Oncogene. 2014;33:74-84
23. Inagaki-Ohara K, Okamoto S, Takagi K, Saito K, Arita S, Tang L. et al. Leptin receptor signaling is required for high-fat diet-induced atrophic gastritis in mice. Nutr Metab (Lond). 2016;13:7
24. Arita S, Kinoshita Y, Ushida K, Enomoto A, Inagaki-Ohara K. High-fat diet feeding promotes stemness and precancerous changes in murine gastric mucosa mediated by leptin receptor signaling pathway. Arch Biochem Biophys. 2016;610:16-24
25. Wei S-H, Li W, Liu Y, Gao DEK, Pan J, Gu C-W. et al. Disturbance of autophagy-lysosome signaling molecule expression in human gastric adenocarcinoma. Oncol Lett. 2014;7:635-40
26. Jiang M, Wu N, Xu B, Chu Y, Li X, Su S. et al. Fatty acid-induced CD36 expression via O-GlcNAcylation drives gastric cancer metastasis. Theranostics. 2019;9:5359-73
27. Xie G, Wang X, Huang F, Zhao A, Chen W, Yan J. et al. Dysregulated hepatic bile acids collaboratively promote liver carcinogenesis. Int J Cancer. 2016;139:1764-75
28. Zhang Q, Li Y, Liang T, Lu X, Liu X, Zhang C. et al. Loss of FGF21 in diabetic mouse during hepatocellular carcinogenetic transformation. Am J Cancer Res. 2015;5:1762-74
29. Liu X, Zhang P, Martin RC, Cui G, Wang G, Tan Y. et al. Lack of fibroblast growth factor 21 accelerates metabolic liver injury characterized by steatohepatities in mice. Am J Cancer Res. 2016;6:1011-25
30. Fujiwara N, Nakagawa H, Enooku K, Kudo Y, Hayata Y, Nakatsuka T. et al. CPT2 downregulation adapts HCC to lipid-rich environment and promotes carcinogenesis via acylcarnitine accumulation in obesity. Gut. 2018;67:1493-504
31. Liu J, Jiang S, Zhao Y, Sun Q, Zhang J, Shen D. et al. Geranylgeranyl diphosphate synthase (GGPPS) regulates non-alcoholic fatty liver disease (NAFLD)-fibrosis progression by determining hepatic glucose/fatty acid preference under high-fat diet conditions. J Pathol. 2018;246:277-88
32. Wu Y, Shan B, Dai J, Xia Z, Cai J, Chen T. et al. Dual role for inositol-requiring enzyme 1α in promoting the development of hepatocellular carcinoma during diet-induced obesity in mice. Hepatology. 2018;68:533-46
33. Garbers C, Rose-John S. Dissecting Interleukin-6 Classic- and Trans-Signaling in Inflammation and Cancer. Methods Mol Biol. 2018;1725:127-40
34. Mittenbühler MJ, Sprenger H-G, Gruber S, Wunderlich CM, Kern L, Brüning JC. et al. Hepatic leptin receptor expression can partially compensate for IL-6Rα deficiency in DEN-induced hepatocellular carcinoma. Mol Metab. 2018;17:122-33
35. Maya-Monteiro CM, Bozza PT. Leptin and mTOR: partners in metabolism and inflammation. Cell Cycle. 2008;7:1713-7
36. Qin C-J, Zhao L-H, Zhou X, Zhang H-L, Wen W, Tang L. et al. Inhibition of dipeptidyl peptidase IV prevents high fat diet-induced liver cancer angiogenesis by downregulating chemokine ligand 2. Cancer Lett. 2018;420:26-37
37. Xue L, Yang K, Newmark H, Leung D, Lipkin M. Epithelial cell hyperproliferation induced in the exocrine pancreas of mice by a western-style diet. J Natl Cancer Inst. 1996;88:1586-90
38. Kon S, Ishibashi K, Katoh H, Kitamoto S, Shirai T, Tanaka S. et al. Cell competition with normal epithelial cells promotes apical extrusion of transformed cells through metabolic changes. Nat Cell Biol. 2017;19:530-41
39. Philip B, Roland CL, Daniluk J, Liu Y, Chatterjee D, Gomez SB. et al. A high-fat diet activates oncogenic Kras and COX2 to induce development of pancreatic ductal adenocarcinoma in mice. Gastroenterology. 2013;145:1449-58
40. Huang H, Daniluk J, Liu Y, Chu J, Li Z, Ji B. et al. Oncogenic K-Ras requires activation for enhanced activity. Oncogene. 2014;33:532-5
41. Incio J, Liu H, Suboj P, Chin SM, Chen IX, Pinter M. et al. Obesity-Induced Inflammation and Desmoplasia Promote Pancreatic Cancer Progression and Resistance to Chemotherapy. Cancer Discov. 2016;6:852-69
42. Luo Y, Yang Y, Liu M, Wang D, Wang F, Bi Y. et al. Oncogenic KRAS Reduces Expression of FGF21 in Acinar Cells to Promote Pancreatic Tumorigenesis in Mice on a High-Fat Diet. Gastroenterology. 2019 157
43. Wang D, Bi Y, Hu L, Luo Y, Ji J, Mao AZ. et al. Obesogenic high-fat diet heightens aerobic glycolysis through hyperactivation of oncogenic KRAS. Cell Commun Signal. 2019;17:19
44. Chan MT, Lim GE, Skovsø S, Yang YHC, Albrecht T, Alejandro EU. et al. Effects of insulin on human pancreatic cancer progression modeled in vitro. BMC Cancer. 2014;14:814
45. Zhang AMY, Magrill J, de Winter TJJ, Hu X, Skovsø S, Schaeffer DF. et al. Endogenous Hyperinsulinemia Contributes to Pancreatic Cancer Development. Cell Metab. 2019;30:403-4
46. Hu C-M, Tien S-C, Hsieh P-K, Jeng Y-M, Chang M-C, Chang Y-T. et al. High Glucose Triggers Nucleotide Imbalance through O-GlcNAcylation of Key Enzymes and Induces KRAS Mutation in Pancreatic Cells. Cell Metab. 2019 29
47. Meyerhardt JA, Niedzwiecki D, Hollis D, Saltz LB, Hu FB, Mayer RJ. et al. Association of dietary patterns with cancer recurrence and survival in patients with stage III colon cancer. JAMA. 2007;298:754-64
48. Park H, Kim M, Kwon GT, Lim DY, Yu R, Sung M-K. et al. A high-fat diet increases angiogenesis, solid tumor growth, and lung metastasis of CT26 colon cancer cells in obesity-resistant BALB/c mice. Mol Carcinog. 2012;51:869-80
49. Hirosumi J, Tuncman G, Chang L, Görgün CZ, Uysal KT, Maeda K. et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333-6
50. Niku M, Pajari A-M, Sarantaus L, Päivärinta E, Storvik M, Heiman-Lindh A. et al. Western diet enhances intestinal tumorigenesis in Min/+ mice, associating with mucosal metabolic and inflammatory stress and loss of Apc heterozygosity. J Nutr Biochem. 2017;39:126-33
51. Karunanithi S, Levi L, DeVecchio J, Karagkounis G, Reizes O, Lathia JD. et al. RBP4-STRA6 Pathway Drives Cancer Stem Cell Maintenance and Mediates High-Fat Diet-Induced Colon Carcinogenesis. Stem Cell Reports. 2017;9:438-50
52. Wang D, Zhao R, Qu Y-Y, Mei X-Y, Zhang X, Zhou Q. et al. Colonic Lysine Homocysteinylation Induced by High-Fat Diet Suppresses DNA Damage Repair. Cell Rep. 2018 25
53. Fu T, Coulter S, Yoshihara E, Oh TG, Fang S, Cayabyab F. et al. FXR Regulates Intestinal Cancer Stem Cell Proliferation. Cell. 2019 176
54. Dermadi D, Valo S, Ollila S, Soliymani R, Sipari N, Pussila M. et al. Western Diet Deregulates Bile Acid Homeostasis, Cell Proliferation, and Tumorigenesis in Colon. Cancer Res. 2017;77:3352-63
55. Tang F-Y, Pai M-H, Chiang E-PI. Consumption of high-fat diet induces tumor progression and epithelial-mesenchymal transition of colorectal cancer in a mouse xenograft model. J Nutr Biochem. 2012;23:1302-13
56. Day SD, Enos RT, McClellan JL, Steiner JL, Velázquez KT, Murphy EA. Linking inflammation to tumorigenesis in a mouse model of high-fat-diet-enhanced colon cancer. Cytokine. 2013;64:454-62
57. Zhu Q-C, Gao R-Y, Wu W, Guo B-M, Peng J-Y, Qin H-L. Effect of a high-fat diet in development of colonic adenoma in an animal model. World J Gastroenterol. 2014;20:8119-29
58. Zhang J, Guo S, Li J, Bao W, Zhang P, Huang Y. et al. Effects of high-fat diet-induced adipokines and cytokines on colorectal cancer development. FEBS Open Bio. 2019;9:2117-25
59. Luck H, Khan S, Kim JH, Copeland JK, Revelo XS, Tsai S. et al. Gut-associated IgA immune cells regulate obesity-related insulin resistance. Nat Commun. 2019;10:3650
60. Pendyala S, Walker JM, Holt PR. A high-fat diet is associated with endotoxemia that originates from the gut. Gastroenterology. 2012 142
61. David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE. et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559-63
62. Wu GD, Chen J, Hoffmann C, Bittinger K, Chen Y-Y, Keilbaugh SA. et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105-8
63. Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR. et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174-80
64. De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S. et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci USA. 2010;107:14691-6
65. Nardone G, Compare D, Rocco A. A microbiota-centric view of diseases of the upper gastrointestinal tract. Lancet Gastroenterol Hepatol. 2017;2:298-312
66. Polyzos SA, Zeglinas C, Artemaki F, Doulberis M, Kazakos E, Katsinelos P. et al. Helicobacter pylori infection and esophageal adenocarcinoma: a review and a personal view. Ann Gastroenterol. 2018 31
67. Maret-Ouda J, Wahlin K, Artama M, Brusselaers N, Färkkilä M, Lynge E. et al. Risk of Esophageal Adenocarcinoma After Antireflux Surgery in Patients With Gastroesophageal Reflux Disease in the Nordic Countries. JAMA Oncol. 2018;4:1576-82
68. Baba Y, Iwatsuki M, Yoshida N, Watanabe M, Baba H. Review of the gut microbiome and esophageal cancer: Pathogenesis and potential clinical implications. Ann Gastroenterol Surg. 2017 1
69. Münch NS, Fang H-Y, Ingermann J, Maurer HC, Anand A, Kellner V. et al. High-Fat Diet Accelerates Carcinogenesis in a Mouse Model of Barrett's Esophagus via Interleukin 8 and Alterations to the Gut Microbiome. Gastroenterology. 2019 157
70. Quante M, Bhagat G, Abrams JA, Marache F, Good P, Lee MD. et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasia. Cancer Cell. 2012;21:36-51
71. Correa P, Houghton J. Carcinogenesis of Helicobacter pylori. Gastroenterology. 2007;133:659-72
72. Chmiela M, Gonciarz W. Molecular mimicry in infections. World J Gastroenterol. 2017;23:3964-77
73. Mueller D, Tegtmeyer N, Brandt S, Yamaoka Y, De Poire E, Sgouras D. et al. c-Src and c-Abl kinases control hierarchic phosphorylation and function of the CagA effector protein in Western and East Asian Helicobacter pylori strains. J Clin Invest. 2012;122:1553-66
74. Brandt S, Kwok T, Hartig R, König W, Backert S. NF-kappaB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc Natl Acad Sci USA. 2005;102:9300-5
75. Chaturvedi R, Asim M, Romero-Gallo J, Barry DP, Hoge S, de Sablet T. et al. Spermine oxidase mediates the gastric cancer risk associated with Helicobacter pylori CagA. Gastroenterology. 2011 141
76. Wang G, Romero-Gallo J, Benoit SL, Piazuelo MB, Dominguez RL, Morgan DR. et al. Hydrogen Metabolism in Helicobacter pylori Plays a Role in Gastric Carcinogenesis through Facilitating CagA Translocation. mBio. 2016 7
77. Cover TL, Blanke SR. Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat Rev Microbiol. 2005;3:320-32
78. Backert S, Tegtmeyer N. the versatility of the Helicobacter pylori vacuolating cytotoxin vacA in signal transduction and molecular crosstalk. Toxins (Basel). 2010;2:69-92
79. Kamangar F, Cheng C, Abnet CC, Rabkin CS. Interleukin-1B polymorphisms and gastric cancer risk-a meta-analysis. Cancer Epidemiol Biomarkers Prev. 2006;15:1920-8
80. Koch KN, Müller A. Helicobacter pylori activates the TLR2/NLRP3/caspase-1/IL-18 axis to induce regulatory T-cells, establish persistent infection and promote tolerance to allergens. Gut Microbes. 2015;6:382-7
81. Stearns JC, Lynch MDJ, Senadheera DB, Tenenbaum HC, Goldberg MB, Cvitkovitch DG. et al. Bacterial biogeography of the human digestive tract. Sci Rep. 2011;1:170
82. Lofgren JL, Whary MT, Ge Z, Muthupalani S, Taylor NS, Mobley M. et al. Lack of commensal flora in Helicobacter pylori-infected INS-GAS mice reduces gastritis and delays intraepithelial neoplasia. Gastroenterology. 2011;140:210-20
83. Oshima H, Hioki K, Popivanova BK, Oguma K, Van Rooijen N, Ishikawa T-O. et al. Prostaglandin E₂ signaling and bacterial infection recruit tumor-promoting macrophages to mouse gastric tumors. Gastroenterology. 2011 140
84. Gantuya B, El-Serag HB, Matsumoto T, Ajami NJ, Oyuntsetseg K, Azzaya D. et al. Gastric Microbiota in -Negative and -Positive Gastritis Among High Incidence of Gastric Cancer Area. Cancers (Basel). 2019 11
85. Maldonado-Contreras A, Goldfarb KC, Godoy-Vitorino F, Karaoz U, Contreras M, Blaser MJ. et al. Structure of the human gastric bacterial community in relation to Helicobacter pylori status. ISME J. 2011;5:574-9
86. Lertpiriyapong K, Whary MT, Muthupalani S, Lofgren JL, Gamazon ER, Feng Y. et al. Gastric colonisation with a restricted commensal microbiota replicates the promotion of neoplastic lesions by diverse intestinal microbiota in the Helicobacter pylori INS-GAS mouse model of gastric carcinogenesis. Gut. 2014;63:54-63
87. Plottel CS, Blaser MJ. Microbiome and malignancy. Cell Host Microbe. 2011;10:324-35
88. Vinasco K, Mitchell HM, Kaakoush NO, Castaño-Rodríguez N. Microbial carcinogenesis: Lactic acid bacteria in gastric cancer. Biochim Biophys Acta Rev Cancer. 2019;1872:188309
89. San-Millán I, Brooks GA. Reexamining cancer metabolism: lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis. 2017;38:119-33
90. Koller VJ, Marian B, Stidl R, Nersesyan A, Winter H, Simić T. et al. Impact of lactic acid bacteria on oxidative DNA damage in human derived colon cells. Food Chem Toxicol. 2008;46:1221-9
91. Forsythe SJ, Cole JA. Nitrite accumulation during anaerobic nitrate reduction by binary suspensions of bacteria isolated from the achlorhydric stomach. J Gen Microbiol. 1987;133:1845-9
92. Feng CW, Wang LD, Jiao LH, Liu B, Zheng S, Xie XJ. Expression of p53, inducible nitric oxide synthase and vascular endothelial growth factor in gastric precancerous and cancerous lesions: correlation with clinical features. BMC Cancer. 2002;2:8
93. van Baarlen P, Troost FJ, van Hemert S, van der Meer C, de Vos WM, de Groot PJ. et al. Differential NF-kappaB pathways induction by Lactobacillus plantarum in the duodenum of healthy humans correlating with immune tolerance. Proc Natl Acad Sci USA. 2009;106:2371-6
94. Flemer B, Lynch DB, Brown JMR, Jeffery IB, Ryan FJ, Claesson MJ. et al. Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut. 2017;66:633-43
95. Chassaing B, Etienne-Mesmin L, Gewirtz AT. Microbiota-liver axis in hepatic disease. Hepatology. 2014;59:328-39
96. Son G, Kremer M, Hines IN. Contribution of gut bacteria to liver pathobiology. Gastroenterol Res Pract. 2010. 2010
97. Tripathi A, Debelius J, Brenner DA, Karin M, Loomba R, Schnabl B. et al. The gut-liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol. 2018;15:397-411
98. Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S. et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013 499
99. Gäbele E, Dostert K, Hofmann C, Wiest R, Schölmerich J, Hellerbrand C. et al. DSS induced colitis increases portal LPS levels and enhances hepatic inflammation and fibrogenesis in experimental NASH. J Hepatol. 2011;55:1391-9
100. Orci LA, Lacotte S, Delaune V, Slits F, Oldani G, Lazarevic V. et al. Effects of the gut-liver axis on ischaemia-mediated hepatocellular carcinoma recurrence in the mouse liver. J Hepatol. 2018;68:978-85
101. Jung IH, Choi JH-K, Chung Y-Y, Lim G-L, Park Y-N, Park SW. Predominant Activation of JAK/STAT3 Pathway by Interleukin-6 Is Implicated in Hepatocarcinogenesis. Neoplasia. 2015;17:586-97
102. Nag S, Qin J, Srivenugopal KS, Wang M, Zhang R. The MDM2-p53 pathway revisited. J Biomed Res. 2013;27:254-71
103. Engstler AJ, Aumiller T, Degen C, Dürr M, Weiss E, Maier IB. et al. Insulin resistance alters hepatic ethanol metabolism: studies in mice and children with non-alcoholic fatty liver disease. Gut. 2016;65:1564-71
104. Seitz HK, Stickel F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat Rev Cancer. 2007;7:599-612
105. Ridlon JM, Kang D-J, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47:241-59
106. Péan N, Doignon I, Tordjmann T. Bile acids and liver carcinogenesis: TGR5 as a novel piece in the puzzle? Clin Res Hepatol Gastroenterol. 2013;37:226-9
107. Half E, Keren N, Reshef L, Dorfman T, Lachter I, Kluger Y. et al. Fecal microbiome signatures of pancreatic cancer patients. Sci Rep. 2019;9:16801
108. Mendez R, Kesh K, Arora N, Di Martino L, McAllister F, Merchant N. et al. Microbial dysbiosis and polyamine metabolism as predictive markers for early detection of pancreatic cancer. Carcinogenesis. 2020;41:561-70
109. Ren Z, Jiang J, Xie H, Li A, Lu H, Xu S. et al. Gut microbial profile analysis by MiSeq sequencing of pancreatic carcinoma patients in China. Oncotarget. 2017;8:95176-91
110. Mei Q-X, Huang C-L, Luo S-Z, Zhang X-M, Zeng Y, Lu Y-Y. Characterization of the duodenal bacterial microbiota in patients with pancreatic head cancer vs. healthy controls. Pancreatology. 2018;18:438-45
111. Gaiser RA, Halimi A, Alkharaan H, Lu L, Davanian H, Healy K. et al. Enrichment of oral microbiota in early cystic precursors to invasive pancreatic cancer. Gut. 2019;68:2186-94
112. Di Carlo P, Serra N, D'Arpa F, Agrusa A, Gulotta G, Fasciana T. et al. The microbiota of the bilio-pancreatic system: a cohort, STROBE-compliant study. Infect Drug Resist. 2019;12:1513-27
113. Mitsuhashi K, Nosho K, Sukawa Y, Matsunaga Y, Ito M, Kurihara H. et al. Association of Fusobacterium species in pancreatic cancer tissues with molecular features and prognosis. Oncotarget. 2015;6:7209-20
114. Riquelme E, Zhang Y, Zhang L, Montiel M, Zoltan M, Dong W. et al. Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell. 2019 178
115. Pagliari D, Saviano A, Newton EE, Serricchio ML, Dal Lago AA, Gasbarrini A. et al. Gut Microbiota-Immune System Crosstalk and Pancreatic Disorders. Mediators Inflamm. 2018;2018:7946431
116. Thomas RM, Jobin C. Microbiota in pancreatic health and disease: the next frontier in microbiome research. Nat Rev Gastroenterol Hepatol. 2020;17:53-64
117. Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D. et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science. 2017;357:1156-60
118. Zambirinis CP, Pushalkar S, Saxena D, Miller G. Pancreatic cancer, inflammation, and microbiome. Cancer J. 2014;20:195-202
119. Thomas RM, Gharaibeh RZ, Gauthier J, Beveridge M, Pope JL, Guijarro MV. et al. Intestinal microbiota enhances pancreatic carcinogenesis in preclinical models. Carcinogenesis. 2018;39:1068-78
120. Pushalkar S, Hundeyin M, Daley D, Zambirinis CP, Kurz E, Mishra A. et al. The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune Suppression. Cancer Discov. 2018;8:403-16
121. Sethi V, Kurtom S, Tarique M, Lavania S, Malchiodi Z, Hellmund L. et al. Gut Microbiota Promotes Tumor Growth in Mice by Modulating Immune Response. Gastroenterology. 2018 155
122. Yu J, Feng Q, Wong SH, Zhang D, Liang QY, Qin Y. et al. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut. 2017;66:70-8
123. Zhang J, Haines C, Watson AJM, Hart AR, Platt MJ, Pardoll DM. et al. Oral antibiotic use and risk of colorectal cancer in the United Kingdom, 1989-2012: a matched case-control study. Gut. 2019;68:1971-8
124. Cao Y, Wu K, Mehta R, Drew DA, Song M, Lochhead P. et al. Long-term use of antibiotics and risk of colorectal adenoma. Gut. 2018;67:672-8
125. Peters BA, Dominianni C, Shapiro JA, Church TR, Wu J, Miller G. et al. The gut microbiota in conventional and serrated precursors of colorectal cancer. Microbiome. 2016;4:69
126. Hannigan GD, Duhaime MB, Ruffin MT, Koumpouras CC, Schloss PD. Diagnostic Potential and Interactive Dynamics of the Colorectal Cancer Virome. mBio. 2018 9
127. Daniel SG, Ball CL, Besselsen DG, Doetschman T, Hurwitz BL. Functional Changes in the Gut Microbiome Contribute to Transforming Growth Factor β-Deficient Colon Cancer. mSystems. 2017 2
128. Serna G, Ruiz-Pace F, Hernando J, Alonso L, Fasani R, Landolfi S. et al. Fusobacterium nucleatum persistence and risk of recurrence after preoperative treatment in locally advanced rectal cancer. Ann Oncol. 2020
129. Bullman S, Pedamallu CS, Sicinska E, Clancy TE, Zhang X, Cai D. et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science. 2017;358:1443-8
130. Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M. et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity. 2015;42:344-55
131. Abed J, Emgård JEM, Zamir G, Faroja M, Almogy G, Grenov A. et al. Fap2 Mediates Fusobacterium nucleatum Colorectal Adenocarcinoma Enrichment by binding to Tumor-Expressed Gal-GalNAc. Cell Host Microbe. 2016;20:215-25
132. Lu P, Xu M, Xiong Z, Zhou F, Wang L. Fusobacterium nucleatum prevents apoptosis in colorectal cancer cells via the ANO1 pathway. Cancer Manag Res. 2019;11:9057-66
133. Rubinstein MR, Baik JE, Lagana SM, Han RP, Raab WJ, Sahoo D. et al. Fusobacterium nucleatum promotes colorectal cancer by inducing Wnt/β-catenin modulator Annexin A1. EMBO Rep. 2019 20
134. Chen Y, Chen Y, Zhang J, Cao P, Su W, Deng Y. et al. Fusobacterium nucleatum Promotes Metastasis in Colorectal Cancer by Activating Autophagy Signaling via the Upregulation of CARD3 Expression. Theranostics. 2020;10:323-39
135. Jin Y, Tang S, Li W, Ng SC, Chan MWY, Sung JJY. et al. Hemolytic E. coli Promotes Colonic Tumorigenesis in Females. Cancer Res. 2016;76:2891-900
136. Cuevas-Ramos G, Petit CR, Marcq I, Boury M, Oswald E, Nougayrède J-P. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci USA. 2010;107:11537-42
137. Raisch J, Rolhion N, Dubois A, Darfeuille-Michaud A, Bringer M-A. Intracellular colon cancer-associated Escherichia coli promote protumoral activities of human macrophages by inducing sustained COX-2 expression. Lab Invest. 2015;95:296-307
138. Raisch J, Buc E, Bonnet M, Sauvanet P, Vazeille E, de Vallée A. et al. Colon cancer-associated B2 Escherichia coli colonize gut mucosa and promote cell proliferation. World J Gastroenterol. 2014;20:6560-72
139. Pleguezuelos-Manzano C, Puschhof J, Rosendahl Huber A, van Hoeck A, Wood HM, Nomburg J. et al. Mutational signature in colorectal cancer caused by genotoxic pks(+) E. coli. Nature. 2020;580:269-73
140. Chung L, Thiele Orberg E, Geis AL, Chan JL, Fu K, DeStefano Shields CE. et al. Bacteroides fragilis Toxin Coordinates a Pro-carcinogenic Inflammatory Cascade via Targeting of Colonic Epithelial Cells. Cell Host Microbe. 2018 23
141. Thiele Orberg E, Fan H, Tam AJ, Dejea CM, Destefano Shields CE, Wu S. et al. The myeloid immune signature of enterotoxigenic Bacteroides fragilis-induced murine colon tumorigenesis. Mucosal Immunol. 2017;10:421-33
142. Goodwin AC, Destefano Shields CE, Wu S, Huso DL, Wu X, Murray-Stewart TR. et al. Polyamine catabolism contributes to enterotoxigenic Bacteroides fragilis-induced colon tumorigenesis. Proc Natl Acad Sci USA. 2011;108:15354-9
143. Dejea CM, Fathi P, Craig JM, Boleij A, Taddese R, Geis AL. et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science. 2018;359:592-7
144. He Z, Gharaibeh RZ, Newsome RC, Pope JL, Dougherty MW, Tomkovich S. et al. Campylobacter jejuni promotes colorectal tumorigenesis through the action of cytolethal distending toxin. Gut. 2019;68:289-300
145. Zhao R, Coker OO, Wu J, Zhou Y, Zhao L, Nakatsu G. et al. Aspirin Reduces Colorectal Tumor Development in Mice and Gut Microbes Reduce its Bioavailability and Chemopreventive Effects. Gastroenterology. 2020
146. Malik A, Sharma D, Malireddi RKS, Guy CS, Chang T-C, Olsen SR. et al. SYK-CARD9 Signaling Axis Promotes Gut Fungi-Mediated Inflammasome Activation to Restrict Colitis and Colon Cancer. Immunity. 2018 49
147. Wang T, Fan C, Yao A, Xu X, Zheng G, You Y. et al. The Adaptor Protein CARD9 Protects against Colon Cancer by Restricting Mycobiota-Mediated Expansion of Myeloid-Derived Suppressor Cells. Immunity. 2018 49
148. Pekkala S, Keskitalo A, Kettunen E, Lensu S, Nykänen N, Kuopio T. et al. Blocking Activin Receptor Ligands Is Not Sufficient to Rescue Cancer-Associated Gut Microbiota-A Role for Gut Microbial Flagellin in Colorectal Cancer and Cachexia? Cancers (Basel). 2019 11
149. Wang G, Yu Y, Wang Y-Z, Wang J-J, Guan R, Sun Y. et al. Role of SCFAs in gut microbiome and glycolysis for colorectal cancer therapy. J Cell Physiol. 2019;234:17023-49
150. Koh A, De Vadder F, Kovatcheva-Datchary P, Bäckhed F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell. 2016;165:1332-45
151. Chun E, Lavoie S, Fonseca-Pereira D, Bae S, Michaud M, Hoveyda HR. et al. Metabolite-Sensing Receptor Ffar2 Regulates Colonic Group 3 Innate Lymphoid Cells and Gut Immunity. Immunity. 2019 51
152. Lavoie S, Chun E, Bae S, Brennan CA, Gallini Comeau CA, Lang JK. et al. Expression of Free Fatty Acid Receptor 2 by Dendritic Cells Prevents Their Expression of Interleukin 27 and Is Required for Maintenance of Mucosal Barrier and Immune Response Against Colorectal Tumors in Mice. Gastroenterology. 2020 158
153. Zaiss MM, Jones RM, Schett G, Pacifici R. The gut-bone axis: how bacterial metabolites bridge the distance. J Clin Invest. 2019;129:3018-28
154. Lee SM, Kim N, Yoon H, Nam RH, Lee DH. Microbial Changes and Host Response in F344 Rat Colon Depending on Sex and Age Following a High-Fat Diet. Front Microbiol. 2018;9:2236
155. Shen X, Carlström M, Borniquel S, Jädert C, Kevil CG, Lundberg JO. Microbial regulation of host hydrogen sulfide bioavailability and metabolism. Free Radic Biol Med. 2013;60:195-200
156. Lei Y, Zhen Y, Zhang W, Sun X, Lin X, Feng J. et al. Exogenous hydrogen sulfide exerts proliferation, anti-apoptosis, angiopoiesis and migration effects via activating HSP90 pathway in EC109 cells. Oncol Rep. 2016;35:3714-20
157. Arita S, Inagaki-Ohara K. High-fat-diet-induced modulations of leptin signaling and gastric microbiota drive precancerous lesions in the stomach. Nutrition. 2019;67-68:110556
158. Carr FJ, Chill D, Maida N. The lactic acid bacteria: a literature survey. Crit Rev Microbiol. 2002;28:281-370
159. Colegio OR, Chu N-Q, Szabo AL, Chu T, Rhebergen AM, Jairam V. et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559-63
160. Yun SH, Hwang TS, Park DH. Metabolic characterization of lactic acid bacterium Lactococcus garvieae sk11, capable of reducing ferric iron, nitrate, and fumarate. J Microbiol Biotechnol. 2007;17:218-25
161. Li S, Konstantinov SR, Smits R, Peppelenbosch MP. Bacterial Biofilms in Colorectal Cancer Initiation and Progression. Trends Mol Med. 2017;23:18-30
162. He C, Cheng D, Peng C, Li Y, Zhu Y, Lu N. High-Fat Diet Induces Dysbiosis of Gastric Microbiota Prior to Gut Microbiota in Association With Metabolic Disorders in Mice. Front Microbiol. 2018;9:639
163. Kim K-A, Gu W, Lee I-A, Joh E-H, Kim D-H. High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS ONE. 2012;7:e47713
164. Xiao S, Fei N, Pang X, Shen J, Wang L, Zhang B. et al. A gut microbiota-targeted dietary intervention for amelioration of chronic inflammation underlying metabolic syndrome. FEMS Microbiol Ecol. 2014;87:357-67
165. Li N, Xu H, Ou Y, Feng Z, Zhang Q, Zhu Q. et al. LPS-induced CXCR7 expression promotes gastric Cancer proliferation and migration via the TLR4/MD-2 pathway. Diagn Pathol. 2019;14:3
166. Zheng Y-X, Yang M, Rong T-T, Yuan X-L, Ma Y-H, Wang Z-H. et al. CD74 and macrophage migration inhibitory factor as therapeutic targets in gastric cancer. World J Gastroenterol. 2012;18:2253-61
167. Yuan X, Zhou Y, Wang W, Li J, Xie G, Zhao Y. et al. Activation of TLR4 signaling promotes gastric cancer progression by inducing mitochondrial ROS production. Cell Death Dis. 2013;4:e794
168. Li H, Xia J-Q, Zhu F-S, Xi Z-H, Pan C-Y, Gu L-M. et al. LPS promotes the expression of PD-L1 in gastric cancer cells through NF-κB activation. J Cell Biochem. 2018 119
169. Zhang C, Zhang M, Wang S, Han R, Cao Y, Hua W. et al. Interactions between gut microbiota, host genetics and diet relevant to development of metabolic syndromes in mice. ISME J. 2010;4:232-41
170. Wang R, Tao B, Fan Q, Wang S, Chen L, Zhang J. et al. Fatty-acid receptor CD36 functions as a hydrogen sulfide-targeted receptor with its Cys333-Cys272 disulfide bond serving as a specific molecular switch to accelerate gastric cancer metastasis. EBioMedicine. 2019;45:108-23
171. Ouyang X, Ghani A, Mehal WZ. Inflammasome biology in fibrogenesis. Biochim Biophys Acta. 2013;1832:979-88
172. Loo TM, Kamachi F, Watanabe Y, Yoshimoto S, Kanda H, Arai Y. et al. Gut Microbiota Promotes Obesity-Associated Liver Cancer through PGE-Mediated Suppression of Antitumor Immunity. Cancer Discov. 2017;7:522-38
173. Xie G, Wang X, Liu P, Wei R, Chen W, Rajani C. et al. Distinctly altered gut microbiota in the progression of liver disease. Oncotarget. 2016;7:19355-66
174. Cui G, Martin RC, Jin H, Liu X, Pandit H, Zhao H. et al. Up-regulation of FGF15/19 signaling promotes hepatocellular carcinoma in the background of fatty liver. J Exp Clin Cancer Res. 2018;37:136
175. Yang Y, Zhong Z, Wang B, Xia X, Yao W, Huang L. et al. Early-life high-fat diet-induced obesity programs hippocampal development and cognitive functions via regulation of gut commensal Akkermansia muciniphila. Neuropsychopharmacology. 2019;44:2054-64
176. Zhang L, Qin Q, Liu M, Zhang X, He F, Wang G. Akkermansia muciniphila can reduce the damage of gluco/lipotoxicity, oxidative stress and inflammation, and normalize intestine microbiota in streptozotocin-induced diabetic rats. Pathog Dis. 2018 76
177. Singh R, Mishra MK, Aggarwal H. Inflammation, Immunity, and Cancer. Mediators Inflamm. 2017;2017:6027305
178. Olivares M, Schüppel V, Hassan AM, Beaumont M, Neyrinck AM, Bindels LB. et al. The Potential Role of the Dipeptidyl Peptidase-4-Like Activity From the Gut Microbiota on the Host Health. Front Microbiol. 2018;9:1900
179. Bauer PV, Duca FA, Waise TMZ, Rasmussen BA, Abraham MA, Dranse HJ. et al. Metformin Alters Upper Small Intestinal Microbiota that Impact a Glucose-SGLT1-Sensing Glucoregulatory Pathway. Cell Metab. 2018 27
180. Natividad JM, Lamas B, Pham HP, Michel M-L, Rainteau D, Bridonneau C. et al. Bilophila wadsworthia aggravates high fat diet induced metabolic dysfunctions in mice. Nat Commun. 2018;9:2802
181. Chang S-H, Ai Y, Breyer RM, Lane TF, Hla T. The prostaglandin E2 receptor EP2 is required for cyclooxygenase 2-mediated mammary hyperplasia. Cancer Res. 2005;65:4496-9
182. Hill R, Li Y, Tran LM, Dry S, Calvopina JH, Garcia A. et al. Cell intrinsic role of COX-2 in pancreatic cancer development. Mol Cancer Ther. 2012;11:2127-37
183. Vadrevu SK, Chintala NK, Sharma SK, Sharma P, Cleveland C, Riediger L. et al. Complement c5a receptor facilitates cancer metastasis by altering T-cell responses in the metastatic niche. Cancer Res. 2014;74:3454-65
184. Mamidi S, Höne S, Kirschfink M. The complement system in cancer: Ambivalence between tumour destruction and promotion. Immunobiology. 2017;222:45-54
185. Guglietta S, Chiavelli A, Zagato E, Krieg C, Gandini S, Ravenda PS. et al. Coagulation induced by C3aR-dependent NETosis drives protumorigenic neutrophils during small intestinal tumorigenesis. Nat Commun. 2016;7:11037
186. Umansky V, Blattner C, Gebhardt C, Utikal J. The Role of Myeloid-Derived Suppressor Cells (MDSC) in Cancer Progression. Vaccines (Basel). 2016 4
187. Xiao L, Sonne SB, Feng Q, Chen N, Xia Z, Li X. et al. High-fat feeding rather than obesity drives taxonomical and functional changes in the gut microbiota in mice. Microbiome. 2017;5:43
188. Mima K, Sukawa Y, Nishihara R, Qian ZR, Yamauchi M, Inamura K. et al. Fusobacterium nucleatum and T Cells in Colorectal Carcinoma. JAMA Oncol. 2015;1:653-61
189. Matsuda A, Makino N, Tozawa T, Shirahata N, Honda T, Ikeda Y. et al. Pancreatic fat accumulation, fibrosis, and acinar cell injury in the Zucker diabetic fatty rat fed a chronic high-fat diet. Pancreas. 2014;43:735-43
190. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S. et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246-9
191. Eldridge MJG, Shenoy AR. Antimicrobial inflammasomes: unified signalling against diverse bacterial pathogens. Curr Opin Microbiol. 2015;23:32-41
192. Bauernfeind F, Hornung V. Of inflammasomes and pathogens-sensing of microbes by the inflammasome. EMBO Mol Med. 2013;5:814-26
193. Schulz MD, Atay C, Heringer J, Romrig FK, Schwitalla S, Aydin B. et al. High-fat-diet-mediated dysbiosis promotes intestinal carcinogenesis independently of obesity. Nature. 2014;514:508-12
194. Gaines S, van Praagh JB, Williamson AJ, Jacobson RA, Hyoju S, Zaborin A. et al. Western Diet Promotes Intestinal Colonization by Collagenolytic Microbes and Promotes Tumor Formation After Colorectal Surgery. Gastroenterology. 2020 158
195. Xu L, Sharkey D, Cantley LG. Tubular GM-CSF Promotes Late MCP-1/CCR2-Mediated Fibrosis and Inflammation after Ischemia/Reperfusion Injury. J Am Soc Nephrol. 2019;30:1825-40
196. Liu T, Guo Z, Song X, Liu L, Dong W, Wang S. et al. High-fat diet-induced dysbiosis mediates MCP-1/CCR2 axis-dependent M2 macrophage polarization and promotes intestinal adenoma-adenocarcinoma sequence. J Cell Mol Med. 2020;24:2648-62
197. Sánchez B. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis: a role for bifidobacteria and lactobacilli? Nat Rev Gastroenterol Hepatol. 2018;15:205
198. Zeng H, Umar S, Rust B, Lazarova D, Bordonaro M. Secondary Bile Acids and Short Chain Fatty Acids in the Colon: A Focus on Colonic Microbiome, Cell Proliferation, Inflammation, and Cancer. Int J Mol Sci. 2019 20
199. Schramm C. Bile Acids, the Microbiome, Immunity, and Liver Tumors. N Engl J Med. 2018;379:888-90
200. Koh GY, Kane A, Lee K, Xu Q, Wu X, Roper J. et al. Parabacteroides distasonis attenuates toll-like receptor 4 signaling and Akt activation and blocks colon tumor formation in high-fat diet-fed azoxymethane-treated mice. Int J Cancer. 2018;143:1797-805
201. Kim M-S, Bae J-W. Spatial disturbances in altered mucosal and luminal gut viromes of diet-induced obese mice. Environ Microbiol. 2016;18:1498-510
202. Mehta RS, Nishihara R, Cao Y, Song M, Mima K, Qian ZR. et al. Association of Dietary Patterns With Risk of Colorectal Cancer Subtypes Classified by Fusobacterium nucleatum in Tumor Tissue. JAMA Oncol. 2017;3:921-7
203. Boursi B, Mamtani R, Haynes K, Yang Y-X. Recurrent antibiotic exposure may promote cancer formation-Another step in understanding the role of the human microbiota? Eur J Cancer. 2015;51:2655-64
204. Viehl CT, Moore TT, Liyanage UK, Frey DM, Ehlers JP, Eberlein TJ. et al. Depletion of CD4+CD25+ regulatory T cells promotes a tumor-specific immune response in pancreas cancer-bearing mice. Ann Surg Oncol. 2006;13:1252-8
205. Liu X, Shin N, Koblish HK, Yang G, Wang Q, Wang K. et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood. 2010;115:3520-30
206. Kolodziejczyk AA, Zheng D, Elinav E. Diet-microbiota interactions and personalized nutrition. Nat Rev Microbiol. 2019;17:742-53
207. Gentile CL, Weir TL. The gut microbiota at the intersection of diet and human health. Science. 2018;362:776-80
208. Snelson M, Clarke RE, Nguyen TV, Penfold SA, Forbes JM, Tan SM. et al. Long Term High Protein Diet Feeding Alters the Microbiome and Increases Intestinal Permeability, Systemic Inflammation and Kidney Injury in Mice. Mol Nutr Food Res. 2021: e2000851.
209. Ho VW, Leung K, Hsu A, Luk B, Lai J, Shen SY. et al. A low carbohydrate, high protein diet slows tumor growth and prevents cancer initiation. Cancer Res. 2011;71:4484-93
210. Pimentel GD, Pichard C, Laviano A, Fernandes RC. High protein diet improves the overall survival in older adults with advanced gastrointestinal cancer. Clin Nutr. 2020
211. Bier A, Braun T, Khasbab R, Di Segni A, Grossman E, Haberman Y. et al. A High Salt Diet Modulates the Gut Microbiota and Short Chain Fatty Acids Production in a Salt-Sensitive Hypertension Rat Model. Nutrients. 2018 10
212. Miranda PM, De Palma G, Serkis V, Lu J, Louis-Auguste MP, McCarville JL. et al. High salt diet exacerbates colitis in mice by decreasing Lactobacillus levels and butyrate production. Microbiome. 2018;6:57
213. He W, Xu J, Mu R, Li Q, Lv DL, Huang Z. et al. High-salt diet inhibits tumour growth in mice via regulating myeloid-derived suppressor cell differentiation. Nat Commun. 2020;11:1732
214. Do MH, Lee E, Oh MJ, Kim Y, Park HY. High-Glucose or -Fructose Diet Cause Changes of the Gut Microbiota and Metabolic Disorders in Mice without Body Weight Change. Nutrients. 2018 10
215. Yue S, Zhao D, Peng C, Tan C, Wang Q, Gong J. Effects of theabrownin on serum metabolites and gut microbiome in rats with a high-sugar diet. Food Funct. 2019;10:7063-80
216. Marques FZ, Nelson E, Chu PY, Horlock D, Fiedler A, Ziemann M. et al. High-Fiber Diet and Acetate Supplementation Change the Gut Microbiota and Prevent the Development of Hypertension and Heart Failure in Hypertensive Mice. Circulation. 2017;135:964-77
217. Folkerts J, Stadhouders R, Redegeld FA, Tam SY, Hendriks RW, Galli SJ. et al. Effect of Dietary Fiber and Metabolites on Mast Cell Activation and Mast Cell-Associated Diseases. Front Immunol. 2018;9:1067
218. Song M, Wu K, Meyerhardt JA, Ogino S, Wang M, Fuchs CS. et al. Fiber Intake and Survival After Colorectal Cancer Diagnosis. JAMA Oncol. 2018;4:71-9
219. Cantorna MT, Snyder L, Arora J. Vitamin A and vitamin D regulate the microbial complexity, barrier function, and the mucosal immune responses to ensure intestinal homeostasis. Crit Rev Biochem Mol Biol. 2019;54:184-92
220. Lopez CA, Skaar EP. The Impact of Dietary Transition Metals on Host-Bacterial Interactions. Cell Host Microbe. 2018;23:737-48
221. Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D. et al. Environment dominates over host genetics in shaping human gut microbiota. Nature. 2018;555:210-5
222. Khan AA, Yurkovetskiy L, O'Grady K, Pickard JM, de Pooter R, Antonopoulos DA. et al. Polymorphic Immune Mechanisms Regulate Commensal Repertoire. Cell Rep. 2019;29:541-50.e4
223. Hu S, Vich Vila A, Gacesa R, Collij V, Stevens C, Fu JM. et al. Whole exome sequencing analyses reveal gene-microbiota interactions in the context of IBD. Gut. 2020
224. Mondot S, Barreau F, Al Nabhani Z, Dussaillant M, Le Roux K, Doré J. et al. Altered gut microbiota composition in immune-impaired Nod2(-/-) mice. Gut. 2012;61:634-5
225. Bonder MJ, Kurilshikov A, Tigchelaar EF, Mujagic Z, Imhann F, Vila AV. et al. The effect of host genetics on the gut microbiome. Nat Genet. 2016;48:1407-12
226. Goodrich JK, Davenport ER, Beaumont M, Jackson MA, Knight R, Ober C. et al. Genetic Determinants of the Gut Microbiome in UK Twins. Cell Host Microbe. 2016;19:731-43
227. Tang J, Wu X, Mou M, Wang C, Wang L, Li F. et al. GIMICA: host genetic and immune factors shaping human microbiota. Nucleic Acids Res. 2021;49:D715-D22
228. Chu H, Khosravi A, Kusumawardhani IP, Kwon AH, Vasconcelos AC, Cunha LD. et al. Gene-microbiota interactions contribute to the pathogenesis of inflammatory bowel disease. Science. 2016;352:1116-20
229. Philip B, Roland CL, Daniluk J, Liu Y, Chatterjee D, Gomez SB. et al. A high-fat diet activates oncogenic Kras and COX2 to induce development of pancreatic ductal adenocarcinoma in mice. Gastroenterology. 2013;145:1449-58
230. Luo Y, Yang Y, Liu M, Wang D, Wang F, Bi Y. et al. Oncogenic KRAS Reduces Expression of FGF21 in Acinar Cells to Promote Pancreatic Tumorigenesis in Mice on a High-Fat Diet. Gastroenterology. 2019;157:1413-28.e11
231. Qin CJ, Zhao LH, Zhou X, Zhang HL, Wen W, Tang L. et al. Inhibition of dipeptidyl peptidase IV prevents high fat diet-induced liver cancer angiogenesis by downregulating chemokine ligand 2. Cancer Lett. 2018;420:26-37
232. Niku M, Pajari AM, Sarantaus L, Päivärinta E, Storvik M, Heiman-Lindh A. et al. Western diet enhances intestinal tumorigenesis in Min/+ mice, associating with mucosal metabolic and inflammatory stress and loss of Apc heterozygosity. J Nutr Biochem. 2017;39:126-33
233. Hale VL, Jeraldo P, Chen J, Mundy M, Yao J, Priya S. et al. Distinct microbes, metabolites, and ecologies define the microbiome in deficient and proficient mismatch repair colorectal cancers. Genome Med. 2018;10:78
234. He Z, Gharaibeh RZ, Newsome RC, Pope JL, Dougherty MW, Tomkovich S. et al. Campylobacter jejuni promotes colorectal tumorigenesis through the action of cytolethal distending toxin. Gut. 2019;68:289-300
235. Lin H, An Y, Hao F, Wang Y, Tang H. Correlations of Fecal Metabonomic and Microbiomic Changes Induced by High-fat Diet in the Pre-Obesity State. Sci Rep. 2016;6:21618
236. Zeng H, Ishaq SL, Zhao F-Q, Wright A-DG. Colonic inflammation accompanies an increase of β-catenin signaling and Lachnospiraceae/Streptococcaceae bacteria in the hind gut of high-fat diet-fed mice. J Nutr Biochem. 2016;35:30-6
237. Guo X, Li J, Tang R, Zhang G, Zeng H, Wood RJ. et al. High Fat Diet Alters Gut Microbiota and the Expression of Paneth Cell-Antimicrobial Peptides Preceding Changes of Circulating Inflammatory Cytokines. Mediators Inflamm. 2017;2017:9474896
238. Fujisaka S, Avila-Pacheco J, Soto M, Kostic A, Dreyfuss JM, Pan H. et al. Diet, Genetics, and the Gut Microbiome Drive Dynamic Changes in Plasma Metabolites. Cell Rep. 2018;22:3072-86
239. Yin J, Li Y, Han H, Chen S, Gao J, Liu G. et al. Melatonin reprogramming of gut microbiota improves lipid dysmetabolism in high-fat diet-fed mice. J Pineal Res. 2018;65:e12524
240. Liu C, Zhang J, Li M, Zhao L, Ji C, Ma Q. Alterations and structural resilience of the gut microbiota under dietary fat perturbations. J Nutr Biochem. 2018 61
241. Wan Y, Wang F, Yuan J, Li J, Jiang D, Zhang J. et al. Effects of dietary fat on gut microbiota and faecal metabolites, and their relationship with cardiometabolic risk factors: a 6-month randomised controlled-feeding trial. Gut. 2019;68:1417-29
242. Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M. et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe. 2013;14:207-15
243. Yang Y, Weng W, Peng J, Hong L, Yang L, Toiyama Y. et al. Fusobacterium nucleatum Increases Proliferation of Colorectal Cancer Cells and Tumor Development in Mice by Activating Toll-Like Receptor 4 Signaling to Nuclear Factor-κB, and Up-regulating Expression of MicroRNA-21. Gastroenterology. 2017 152
244. Cougnoux A, Dalmasso G, Martinez R, Buc E, Delmas J, Gibold L. et al. Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut. 2014;63:1932-42
245. Zhang Y, Wang X-L, Zhou M, Kang C, Lang H-D, Chen M-T. et al. Crosstalk between gut microbiota and Sirtuin-3 in colonic inflammation and tumorigenesis. Exp Mol Med. 2018;50:21
246. Al Hinai EA, Kullamethee P, Rowland IR, Swann J, Walton GE, Commane DM. Modelling the role of microbial p-cresol in colorectal genotoxicity. Gut Microbes. 2019;10:398-411
247. Hsu RYC, Chan CHF, Spicer JD, Rousseau MC, Giannias B, Rousseau S. et al. LPS-induced TLR4 signaling in human colorectal cancer cells increases beta1 integrin-mediated cell adhesion and liver metastasis. Cancer Res. 2011;71:1989-98
248. Song W, Tiruthani K, Wang Y, Shen L, Hu M, Dorosheva O. et al. Trapping of Lipopolysaccharide to Promote Immunotherapy against Colorectal Cancer and Attenuate Liver Metastasis. Adv Mater Weinheim. 2018;30:e1805007
249. Ferreira RM, Pereira-Marques J, Pinto-Ribeiro I, Costa JL, Carneiro F, Machado JC. et al. Gastric microbial community profiling reveals a dysbiotic cancer-associated microbiota. Gut. 2018;67:226-36
250. Wroblewski LE, Piazuelo MB, Chaturvedi R, Schumacher M, Aihara E, Feng R. et al. Helicobacter pylori targets cancer-associated apical-junctional constituents in gastroids and gastric epithelial cells. Gut. 2015;64:720-30
251. Baud J, Varon C, Chabas S, Chambonnier L, Darfeuille F, Staedel C. Helicobacter pylori initiates a mesenchymal transition through ZEB1 in gastric epithelial cells. PLoS ONE. 2013;8:e60315
252. Wei J, Noto JM, Zaika E, Romero-Gallo J, Piazuelo MB, Schneider B. et al. Bacterial CagA protein induces degradation of p53 protein in a p14ARF-dependent manner. Gut. 2015;64:1040-8
253. Wang L, Zhou J, Xin Y, Geng C, Tian Z, Yu X. et al. Bacterial overgrowth and diversification of microbiota in gastric cancer. Eur J Gastroenterol Hepatol. 2016;28:261-6
254. Castaño-Rodríguez N, Goh K-L, Fock KM, Mitchell HM, Kaakoush NO. Dysbiosis of the microbiome in gastric carcinogenesis. Sci Rep. 2017;7:15957
255. Singh V, Yeoh BS, Chassaing B, Xiao X, Saha P, Aguilera Olvera R. et al. Dysregulated Microbial Fermentation of Soluble Fiber Induces Cholestatic Liver Cancer. Cell. 2018 175
256. Li Q, Ma L, Shen S, Guo Y, Cao Q, Cai X. et al. Intestinal dysbacteriosis-induced IL-25 promotes development of HCC via alternative activation of macrophages in tumor microenvironment. J Exp Clin Cancer Res. 2019;38:303
257. Mikó E, Vida A, Kovács T, Ujlaki G, Trencsényi G, Márton J. et al. Lithocholic acid, a bacterial metabolite reduces breast cancer cell proliferation and aggressiveness. Biochim Biophys Acta Bioenerg. 2018;1859:958-74
258. Buchta Rosean C, Bostic RR, Ferey JCM, Feng T-Y, Azar FN, Tung KS. et al. Preexisting Commensal Dysbiosis Is a Host-Intrinsic Regulator of Tissue Inflammation and Tumor Cell Dissemination in Hormone Receptor-Positive Breast Cancer. Cancer Res. 2019;79:3662-75
259. Kovács P, Csonka T, Kovács T, Sári Z, Ujlaki G, Sipos A. et al. Lithocholic Acid, a Metabolite of the Microbiome, Increases Oxidative Stress in Breast Cancer. Cancers (Basel). 2019 11
260. Kovács T, Mikó E, Vida A, Sebő É, Toth J, Csonka T. et al. Cadaverine, a metabolite of the microbiome, reduces breast cancer aggressiveness through trace amino acid receptors. Sci Rep. 2019;9:1300
261. Kim K, Kwon O, Ryu TY, Jung C-R, Kim J, Min J-K. et al. Propionate of a microbiota metabolite induces cell apoptosis and cell cycle arrest in lung cancer. Mol Med Rep. 2019;20:1569-74
262. Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM. et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350:1084-9
Author contact
![]() Corresponding authors: Chuanxin Wang, Ph.D. Department of Clinical Laboratory, The Second Hospital, Cheeloo College of Medicine, Shandong University, 247 Beiyuan Street, Jinan 250033, Shandong, China. Tel.: +86-0531-85875002, Fax: +86-0531-88962544, E-mail: cxwangedu.cn; Lutao Du, Ph.D. Department of Clinical Laboratory, The Second Hospital, Cheeloo College of Medicine, Shandong University, 247 Beiyuan Street, Jinan 250033, Shandong, China. Tel.: +86-17660085177, Fax: +86-0531-88962544, E-mail: lutaoduedu.cn.
Corresponding authors: Chuanxin Wang, Ph.D. Department of Clinical Laboratory, The Second Hospital, Cheeloo College of Medicine, Shandong University, 247 Beiyuan Street, Jinan 250033, Shandong, China. Tel.: +86-0531-85875002, Fax: +86-0531-88962544, E-mail: cxwangedu.cn; Lutao Du, Ph.D. Department of Clinical Laboratory, The Second Hospital, Cheeloo College of Medicine, Shandong University, 247 Beiyuan Street, Jinan 250033, Shandong, China. Tel.: +86-17660085177, Fax: +86-0531-88962544, E-mail: lutaoduedu.cn.