Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(11):5232-5247. doi:10.7150/thno.53417 This issue Cite
Research Paper
c-MYC-directed NRF2 drives malignant progression of head and neck cancer via glucose-6-phosphate dehydrogenase and transketolase activation
Ya-Chu Tang1,2, Jenn-Ren Hsiao3,4, Shih-Sheng Jiang5, Jang-Yang Chang2,5,6, Pei-Yi Chu5,7, Ko-Jiunn Liu5, Hsun-Lang Fang8, Li-Mei Lin2, Huang-Hui Chen2, Yen-Wen Huang5, Yu-Tsen Chen5, Fang-Yu Tsai5, Su-Fang Lin5, Yung-Jen Chuang9,10 ![]() , Ching-Chuan Kuo2,11
, Ching-Chuan Kuo2,11 ![]()
1. Graduate Program of Medical Biotechnology, National Tsing Hua University, Hsinchu, Taiwan.
2. Institute of Biotechnology and Pharmaceutical Research, National Health Research Institutes, Miaoli, Taiwan.
3. Department of Otolaryngology, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, Tainan, Taiwan.
4. Institute of Clinical Medicine, College of Medicine, National Cheng Kung University, Tainan, Taiwan.
5. National Institute of Cancer Research, National Health Research Institutes, Miaoli, Taiwan.
6. Department of Internal Medicine, College of Medicine, National Cheng Kung University, Tainan, Taiwan.
7. Department of Pathology, Show Chwan Memorial Hospital, Changhua, Taiwan.
8. Department of Cosmetology and Health Care, Min-Hwei College of Health Care Management, Tainan, Taiwan.
9. Institute of Bioinformatics and Structural Biology, National Tsing Hua University, Hsinchu, Taiwan.
10. Department of Medical Science, National Tsing Hua University, Hsinchu, Taiwan.
11. Graduate Institute of Biomedical Sciences, China Medical University, Taichung, Taiwan.
Received 2020-9-18; Accepted 2021-2-17; Published 2021-3-11
Abstract

Rationale: NRF2, a redox sensitive transcription factor, is up-regulated in head and neck squamous cell carcinoma (HNSCC), however, the associated impact and regulatory mechanisms remain unclear.
Methods: The protein expression of NRF2 in HNSCC specimens was examined by IHC. The regulatory effect of c-MYC on NRF2 was validated by ChIP-qPCR, RT-qPCR and western blot. The impacts of NRF2 on malignant progression of HNSCC were determined through genetic manipulation and pharmacological inhibition in vitro and in vivo. The gene-set enrichment analysis (GSEA) on expression data of cDNA microarray combined with ChIP-qPCR, RT-qPCR, western blot, transwell migration/ invasion, cell proliferation and soft agar colony formation assays were used to investigate the regulatory mechanisms of NRF2.
Results: NRF2 expression is positively correlated with malignant features of HNSCC. In addition, carcinogens, such as nicotine and arecoline, trigger c-MYC-directed NRF2 activation in HNSCC cells. NRF2 reprograms a wide range of cancer metabolic pathways and the most notable is the pentose phosphate pathway (PPP). Furthermore, glucose-6-phosphate dehydrogenase (G6PD) and transketolase (TKT) are critical downstream effectors of NRF2 that drive malignant progression of HNSCC; the coherently expressed signature NRF2/G6PD/TKT gene set is a potential prognostic biomarker for prediction of patient overall survival. Notably, G6PD- and TKT-regulated nucleotide biosynthesis is more important than redox regulation in determining malignant progression of HNSCC.
Conclusions: Carcinogens trigger c-MYC-directed NRF2 activation. Over-activation of NRF2 promotes malignant progression of HNSCC through reprogramming G6PD- and TKT-mediated nucleotide biosynthesis. Targeting NRF2-directed cellular metabolism is an effective strategy for development of novel treatments for head and neck cancer.
Keywords: Nuclear factor erythroid 2-related factor 2 (NRF2), head and neck squamous cell carcinoma (HNSCC), c-MYC, pentose phosphate pathway (PPP), glucose-6-phosphate dehydrogenase (G6PD), transketolase (TKT)
Introduction
Nuclear factor erythroid 2-related factor 2 (NRF2) is a member of Cap'n'Collar family of transcription factors that share a highly conserved basic region‐leucine zipper structure. NRF2 is tightly regulated by Kelch‐like ECH associating protein 1 (KEAP1), a substrate adaptor protein for a Cullin3‐based E3 ubiquitin ligase, resulting in expression of NRF2 at low transactivating levels in all human organs [1, 2]. When cells are exposed to electrophiles or oxidative stress, NRF2 is released from KEAP1 and transported into the cell nucleus to activate the transcription of specific cytoprotective genes by binding to the antioxidant response element (ARE) as a heterodimer with the small Maf protein [1, 2]. Abundant evidence has shown that activation of NRF2 can suppress carcinogenesis [3, 4]. However, recent evidence has revealed that aberrant activation of NRF2 is associated with cancer hallmarks [4-10], including promoted tumorigenesis [11, 12], sustained proliferative signaling and survival advantage [7, 13, 14], resistance to treatment [15-18], deregulated redox homeostasis [19-21], enhanced tissue invasion and metastasis [14, 22, 23], and altered cellular metabolism [13, 24-26].
Head and neck squamous cell carcinoma (HNSCC) is the fifth most common malignancy worldwide and comprises a diverse set of cancers arising from the squamous epithelium of the oral cavity, larynx, and pharynx, including the nasopharynx, oropharynx, hypopharynx, etc. [27]. The primary risk factors for HNSCC include alcohol consumption, betel nut chewing, cigarette smoking, human papillomavirus (HPV) infection (for oropharyngeal cancer), and Epstein-Barr virus (EBV) infection (for nasopharyngeal cancer) [28]. Even though the curability of early-stage HNSCC is high, the survival rate of patients with recurrent or metastatic HNSCC diminishes drastically [29]. Exploring robust biomarkers that contribute to the malignant progression of HNSCC may aid the development of useful therapeutic strategies. For example, identification of aberrant signaling pathways and targeting these molecules (e.g., EGFR) has been an effective approach to HNSCC treatment [30].
Genomic studies using The Cancer Genome Atlas Network revealed that the activation of the NRF2/KEAP1/CUL3 pathway is a common feature of HPV-negative HNSCC [29]. In addition, several groups demonstrated that increased NRF2 expression was observed in HNSCC and positively correlated with poor clinical outcome [19, 31-33]. There is limited research on the effects of NRF2 on the malignant progression of HNSCC, which includes NRF2-mediated radiation tolerance and drug resistance [16, 34, 35], activated oncogenic Notch [14] or mTOR signaling [36], and NRF2 activity of as a central node in the maintenance of low ROS levels and stemness properties [19]. Thus, the biological consequences of NRF2 in head and neck cancer remain unclear, and the critical NRF2 downstream effectors that participate in NRF2-promoted malignant progression have yet to be conclusively identified in HNSCC. In this study, we provide evidence that activation of NRF2 induces tumorigenesis and promotes tumor growth and metastasis. We also report our discovery of potential upstream modulators and downstream effectors of NRF2 in head and neck cancer. Among them, we identified c-MYC, which is associated with chemical induced carcinogenesis and a modulator of NRF2 activation. In addition, glucose-6-phosphate dehydrogenase (G6PD) and transketolase (TKT) exhibit exclusive roles in determining how NRF2 drives malignant progression of HNSCC.
Materials and Methods
HNSCC patient samples
Human HNSCC clinical specimens at different clinicopathological stages were obtained from National Cheng Kung University Hospital, Tainan, Taiwan in accordance with the associated Institutional Review Board (A-ER-107-147) and National Health Research Institutes, Zhunan, Miaoli, Taiwan (EC1070105).
Cell lines
Normal human oral keratinocytes (NHOK), human dysplastic oral keratinocytes (DOK), and human HNSCC cells (Ca9-22, HSC-3, OC3, HONE-1 and OEC-M1) were maintained as described previously [37-40]. The Ca9-22-D1 cell line and the carcinogen-transformed DOK cells were established in Dr. Ching-Chuan Kuo's laboratory (Figure S1, Figure S2). See Supplementary Materials and Methods for details. All cell lines were authenticated using Short-tandem-repeat (STR) profiling (DNA fingerprinting).
Genetic manipulation
For transient gene silencing, the siRNAs specific to NRF2, G6PD, and TKT and siRNA negative controls were purchased from Invitrogen and delivered into cells using Lipofectamine RNAiMAX reagent (Thermo Fisher Scientific, Waltham, MA., USA). For establishment of stable NRF2-knockdown HNSCC cells, a set of pGFP-C-shLenti vectors containing four independent 29-mer shRNAs against human NRF2 and a non-targeting shRNA were purchased from OriGene Technologies Inc. (Rockville, MD., USA). After transfection, the cells were selected by puromycin treatment. For generation of stable NRF2-overexpressing DOK cells, human NRF2 was amplified from Gene PoolTM normal human prostate cDNA (Life Technologies, Carlsbad, CA., USA) and cloned into the pIRES2-EGFP vector between the Nhe1 and Xho1 cloning sites. The specific primers used for plasmid cloning are listed in Table S1. After transfection, the cells were selected by geneticin (G418) treatment.
Evaluation of mRNA and protein levels
The real-time polymerase chain reaction (PCR) primer pairs used for amplification of target genes are listed in Table S2. Gene expression was determined by quantitative real-time PCR. Western blot analysis and immunohistochemistry (IHC) staining were used to analyze protein levels of cell lysates and tissue sections, respectively. The antibodies used for recognizing specific proteins are listed in Table S3.
Assessment of malignant features
(1) In vitro: Cell proliferation was assessed by methylene blue staining. Anchorage-independent growth was examined using the soft agar colony formation assay. Trans-well migration and invasion assays were performed to evaluate cell motility and invasiveness, respectively. (2) In vivo: Animals used in this study were purchased from BioLASCO Taiwan Co., Ltd (Taipei, Taiwan) and maintained at the Laboratory Animal Center of National Health Research Institutes (NHRI), Taiwan. The in vivo tumorigenic assay and the experimental lung metastasis assay were performed. See Supplementary Materials and Methods for details.
Chromatin immunoprecipitation-quantitative polymerase chain reaction (ChIP-qPCR)
Chromatin immunoprecipitation (ChIP) was performed using the Magna ChIP™ A/G Chromatin Immunoprecipitation Kit (Merck Millipore, Burlington, MA, USA) with an antibody specific for NRF2 (Abcam, Cambridge, MA., USA), c-MYC (Abcam, Cambridge, MA., USA) or normal rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, CA., USA). Following ChIP, quantitative PCR was utilized to amplify and quantify the immunoprecipitated DNA using primers specific for the NRF2-targeted antioxidant response element (ARE) within G6PD or TKT, as well as primers for a c-MYC binding site within the NRF2 promoter region (Table S4). The c-MYC binding site within the NRF2 promoter region was obtained from the ENCODE Consortium. The ChIP-qPCR values were normalized to that of input control and represented as fold enrichment relative to the anti-normal rabbit IgG control.
Statistical analysis
The experimental results were analyzed and expressed as the mean ± standard deviation (S.D.) or mean ± standard error (S.E.) for the in vitro and in vivo experiments, respectively. One-way analysis of variance (ANOVA) and Student's t-test were used to determine statistical significance.
Results
Expression of NRF2 is positively correlated with the malignant characteristics of HNSCC
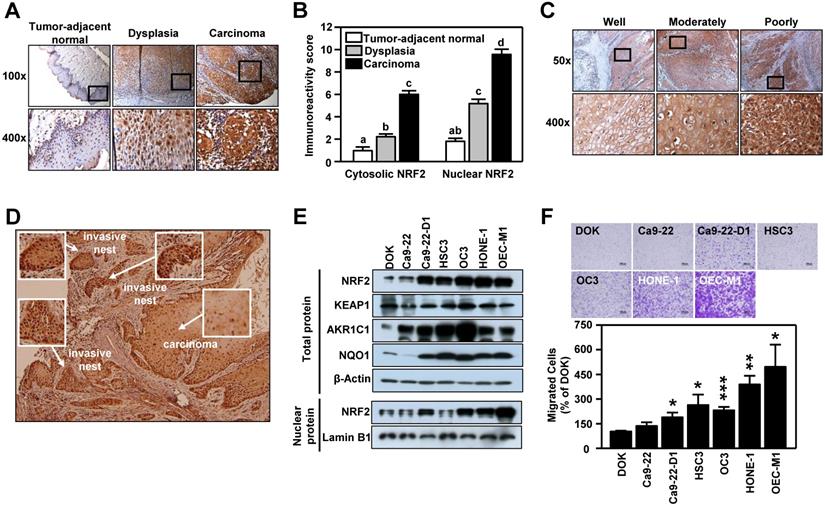
The clinicopathological significance of NRF2 was validated in an in-house cohort of clinical specimens of HNSCC (n=77) by IHC staining. In tumor-adjacent normal epithelium, few cells with positive staining for NRF2 were detected and most of the NRF2-positive cells were found in the active para-basal and basal layers. Conversely, in the oral epithelial dysplastic lesions and oral squamous cell carcinoma, NRF2-positive cells spread into the intermediate layer and the staining intensity was dramatically higher than in cancer-adjacent normal epithelia (Figure 1A). Based on immuno-reactive score (IRS) analysis, we noted that NRF2 was localized predominantly in cell nuclei, and both nuclear and cytosolic NRF2 levels increased significantly with malignant progression, i.e., progression from tumor-adjacent normal epithelia to epithelial dysplasia to squamous cell carcinoma (Figure 1B). We also compared differential expression of NFE2L2, the gene encoding NRF2, in HNSCC and normal tissue from clinical cohorts using the online cancer microarray database Oncomine [41]. As shown in Table S5, increased NRF2 expression in tumors compared to normal tissue appeared in many HNSCC datasets. While analyzing NRF2 expression with histological differentiation, we demonstrated that NRF2 level was positively correlated with the degree of differentiation (Figure 1C). Intriguingly, IHC analysis demonstrated higher staining intensity of NRF2 in the invasive oral squamous cell carcinoma and invasive nests than in primary carcinoma (Figure 1D). Similar to clinical findings, the expression of NRF2 protein was markedly higher in the HNSCC cell lines than in dysplasia oral keratinocytes (DOK; Figure 1E), and lower levels of NRF2 protein were observed in normal human oral keratinocytes (NHOK) compared to DOK and HNSCC cells (Figure S3). Increased nuclear localization of NRF2 led to upregulation of NRF2 downstream targets, such as AKR1C1, NQO1 (Figure 1E), and TrxR (Figure S3). Notably, overexpression of NRF2 was positively related to motility of HNSCC cells (Figure 1F). These results indicate that expression of NRF2 is positively correlated with malignant features of HNSCC.
NRF2 expression is positively associated with malignancy in HNSCC. (A, C, D) Representative IHC staining of NRF2 in the specimens obtained from HNSCC patients. Brown staining indicates NRF2 positivity. The cell nuclei and background were counterstained with hematoxylin (blue coloration). (A) NRF2 protein expression in HNSCC specimens with different clinicopathological stages. Magnification, 100× (upper panel) and 400× (lower panel). (B) The correlation between the level of nuclear and cytosolic expression of NRF2 and tumor stage was estimated using the immunoreactivity scoring system (IRS). The IRS was calculated by multiplying the intensity of NRF2 staining (scale, 0-3) by the percentage of positive cells (4, > 80%; 3, 51-80%; 2, 10-50%; 1, < 10%; 0, 0%), which resulted in values ranging from 0 to 12. Groups with different letters are significant different (p < 0.05) from each other. (C) NRF2 protein expression was positively correlated with the degree of tumor cell differentiation in human head and neck squamous cell carcinoma. Magnification, 50× (upper panel) and 400× (lower panel). (D) NRF2 expression was highest in the invasive carcinoma nests. Magnification 50×. (E) Levels of NRF2 and NRF2 downstream targets in dysplasia oral keratinocyte (DOK) and HNSCC cell lines. (F) Motility of DOK and various HNSCC cell lines via trans-well migration assay. * p < 0.05; ** p < 0.01; *** p < 0.001.
NRF2 is upregulated by c-MYC and acts as an important determinant for malignant progression of HNSCC
To confirm that NRF2 drives the malignant progression of HNSCC, we knocked down the NRF2 gene using RNA interference in three HNSCC cell lines (HONE-1, Ca9-22-D1, and OEC-M1; Figure S4) and found that both transient (Figure 2A) and stable (Figure S5A-B) knockdown of NRF2 markedly reduced cell migratory, invasive (Figure 2B), and growth (Figure 2C, Figure S5C) capacity compared to control. In contrast, stably overexpressed NRF2 facilitates cellular motility and growth; however, when NRF2 is re-suppressed, the migration and growth abilities were decreased in NRF2 overexpressing DOK cells (Figure 2D-E). Furthermore, by performing the soft agar colony formation assay, we found that knockdown of NRF2 significantly suppressed the ability of anchorage-independent growth in HNSCC cells (Figure S5D) and vice versa in NRF2 overexpressing cells (Figure S5E). In addition, the commercially available NRF2 inhibitor ML-385 produced results similar to genetic manipulation (Figure S6). These in vitro findings suggest that NRF2 is important for maintaining the malignant characteristics of HNSCC.
NRF2 promotes malignant features of HNSCC in vitro. Three human HNSCC cell lines, HONE-1, Ca9-22-D1 and OECM-1, were transiently transfected with NRF2 siRNA (siNRF2 #1 and siNRF2 #2) or scramble control (siControl). The motility and invasiveness of the transfected cells were evaluated by trans-well migration (A) and invasion (B) assays. The cells that had migrated or invaded were quantified and expressed as a percentage relative to siControl group. (C) The effects of NRF2-knockdown on cell growth of HNSCC cells. Cell number was determined using the methylene blue assay. (D) Migration ability was determined in DOK cells stably overexpressing NRF2 that were treated with NRF2 siRNA or a non-targeted control. To present relative cell migration, the value from the vector-only control DOK cells (blue bar) was set at 100%. (E) The growth rates of DOK cells stably overexpressing NRF2 and treated with NRF2 siRNA or a non-targeted control under low serum conditions were measured. The data were expressed as the percentage relative to the vector-only control DOK cell (MOCK) which was treated with negative control siRNA (blue bar). (F) Alteration of cellular properties with long-term exposure to nicotine or arecoline in dysplastic DOK cells. The nicotine- and arecoline-transformed DOK cells were established from DOK cells treated with non-toxic concentrations of carcinogens (nicotine: 500 µM; arecoline: 50 µM) for 1 year. The levels of NRF2, KEAP1, phospho-EGFR (Tyr1068), EGFR, B-RAF, and c-MYC in total lysates were assessed using Western blot analysis. The cell proliferation rates were measured by methylene blue assay after 72 h incubation and calculated as the fold increase compared to proliferation of the parental DOK cells (black bar). Top: representative Western blots. Bottom: the cell proliferation index. The effects of knockdown of MYC on NRF2 protein level in HNSCC cells (G) and carcinogen-transformed DOK cells (H). (I) RT-qPCR analysis of NRF2 mRNA levels in MYC-knockdown OEC-M1 cells. (J) ChIP-qPCR analysis of c-MYC binding at NRF2 promoter in MYC-knockdown OEC-M1 cells. All data are expressed as the mean ± S.D. from three individual experiments. * p < 0.05; ** p < 0.01; *** p < 0.001. vs. control.
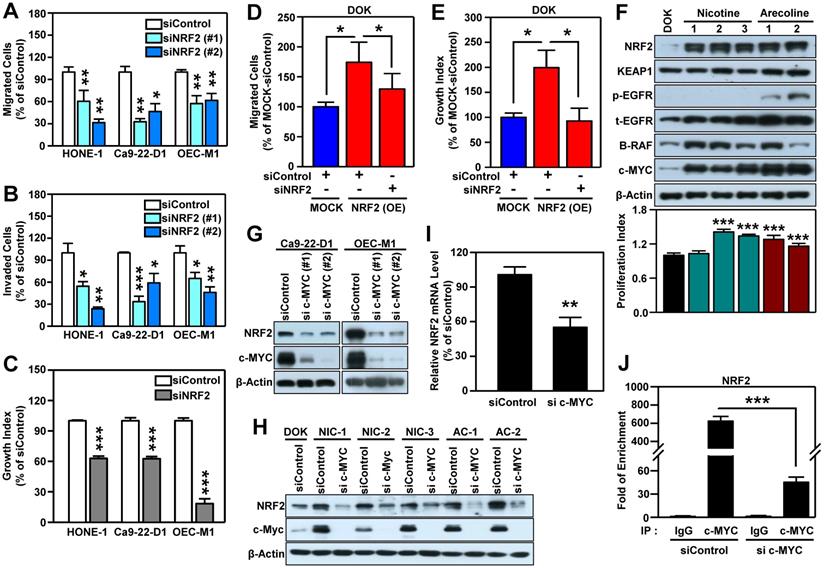
Next, we questioned whether carcinogenic substances modulate NRF2 during head and neck carcinogenesis. Cigarette smoking, alcohol consumption, and betel quid chewing are the leading risk factors for HNSCC [42-44]. IHC staining showed that HNSCC patients who smoke, drink, and chew betel nut have higher nuclear NRF2 levels than those who do not have those habits (Table S6). Therefore, we used nicotine and arecoline to further investigate the role of NRF2 in HNSCC, as long-term nicotine (e.g., cigarette smoking) and arecoline (e.g., betel nut chewing) exposure have been reported to promote head and neck carcinogenesis and progression [28, 45, 46]. We found that long-term exposure to nicotine or arecoline increased NRF2 expression, which was accompanied by an increased proliferation index in carcinogen-transformed DOK cells (Figure 2F). We previously established a 4-NQO (simulation of smoking) plus arecoline (simulation of betel nut chewing) induced mouse model of oral carcinogenesis [47], which mimics the pathogenesis of squamous cell carcinoma (SCC) in patients from Southeast Asia and Taiwan [48-50]. For in vivo validation, we analyzed normal and SCC samples from tongue tissue from the 4NQO/arecoline co-induction oral SCC mouse model. The results demonstrated that expression and nuclear localization of NRF2 protein was increased in SCC compared to normal tongue tissue (Figure S7).
It is known that NRF2 is modulated by several oncogenes [20]. By performing micro-western arrays, which enable quantitative, sensitive, and high-throughput assessment of protein abundance and modifications, we identified that BRAF, phospho-EGFR and c-MYC are significantly increased in both nicotine (NIC)- and arecoline (AC)-transformed DOK cells (Figure S8). Further validation revealed that increased expression levels of BRAF and c-MYC were observed in most carcinogen-transformed DOK cells (Figure 2F). Thus, we investigated whether BRAF or c-MYC were upstream modulators to upregulate NRF2 in HNSCC cells. As shown in Figure S9, we found silencing BRAF did not suppress NRF2 level in HNSCC cells. On the other hand, knockdown of MYC gene significantly inhibits NRF2 expression (Figure 2G). Similar results were observed in both nicotine (NIC)- and arecoline (AC)-transformed DOK cells (Figure 2H).
It is noteworthy that increased and differential MYC expression was observed in tumors of relevant clinical cancer datasets (Table S5). Therefore, we further explored the relationship between NRF2 and c-MYC. In addition to DOK cells, we confirmed the positive correlation between the expression of NFE2L2 (NRF2) and MYC in long-term arecoline-transformed normal human oral keratinocytes (NHOK) (Figure S10). Next, we determined whether NRF2 could regulate the expression of c-MYC and found that knockdown of NRF2 does not affect the expression of c-MYC protein in HNSCC cells (Figure S11). To investigate how c-MYC regulates NRF2 expression, we performed RT-qPCR analysis, and found that knockdown of c-MYC significantly decreased NRF2 mRNA levels in HNSCC cells (Figure 2I). Furthermore, we confirmed that c-MYC binds to the NRF2 promoter, and knockdown of c-MYC significantly reduced the binding strength of c-MYC at the NRF2-promoter in HNSCC cells (Figure 2J). These results indicate that c-MYC is involved in directing NRF2 expression in head and neck cancer.
NRF2 drives tumor progression of HNSCC in vivo
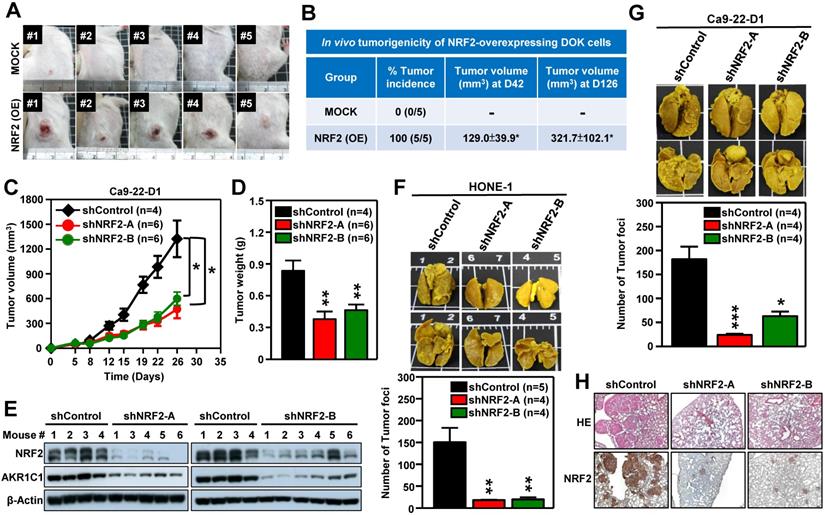
We next performed in vivo experiments to substantiate whether aberrant activation of NRF2 enhanced malignant progression of HNSCC. Firstly, we generated DOK cells with stable overexpression of NRF2 to examine the role of NRF2 in promoting malignant transformation in vivo. The pre-malignant epithelial cell line DOK is derived from human dorsal tongue tissue and is non-tumorigenic in mice [40]. There was no tumor formation after implantation of mock control DOK cells in vivo (Figure 3A, upper panel). In contrast, implantation of DOK cells with stable overexpression of NRF2 resulted in tumor development in vivo (Figure 3A, lower panel). The tumor incidences of the NRF2-overexpressing and mock control groups were 100% and 0%, respectively (Figure 3B). In our next experiment, we investigated how NRF2 expression could affect tumor growth using the Ca9-22-D1 (human origin) xenograft tumor model. As shown in Figure 3C-D, tumor growth was inhibited in mice bearing stable NRF2-knockdown Ca9-22-D1 xenograft tumors. In addition, we found that the expression levels of NRF2 and AKR1C1 (the classical NRF2 downstream target) proteins were significantly reduced in NRF2-knockdown tumors compared to controls (Figure 3E). To evaluate whether the depletion of NRF2 suppresses metastasis of HNSCC in vivo, we used an experimental metastasis model by injecting mice with HONE-1 or Ca9-22-D1 cells, either with stably expressing NRF2-shRNA or empty expression vector. Compared with the mock control group, a decrease in lung-specific metastatic foci formation was observed in NRF2-knockdown groups (Figure 3F-G), suggesting that genetic ablation of NRF2 remarkably hindered pulmonary metastasis. The hematoxylin and eosin (H&E) and IHC staining of lung tissue sections demonstrated that mice bearing NRF2-knockdown Ca9-22-D1 cells significantly inhibited lung-specific metastatic tumor foci formation compared with mice bearing mock control cells, and NRF2 expression level is positively correlated with the number of tumor foci (Figure 3H). These results suggest that augmentation of NRF2 expression drastically enhances the potential for malignant transformation, tumor growth, and metastasis of HNSCC in vivo.
NRF2 promotes tumorigenesis and malignant progression of HNSCC in vivo. (A, B) In vivo tumorigenicity of NRF2 overexpressing dysplastic oral keratinocytes. DOK cells with stably overexpressed NRF2 (NRF2-OE) or vector-only control (MOCK) were subcutaneously injected into NOD/SCID mice (5×106 cells per mouse), and tumor volume and body weight were measured weekly. (A) Representative photographs of mice with control (upper) and NRF2-overexpressing (lower) xenograft tumors. (B) The tumor incidence was defined as the percentage of mice in each group with a tumor volume that exceeded 50 mm3 in each group. Mean tumor volumes (mm3) of mice at days 42 and 126 were represented as mean ± S.E. (C, D) In vivo tumor growth assay of stable NRF2-knockdown Ca9-22-D1 cells. Tumor growth rate (C) and tumor weight (D) were compared between shControl (black) and two different shNRF2 groups (red and green). Mice were injected subcutaneously with Ca9-22-D1 cells. The tumor growth curves represent tumor volume data measured twice per week throughout the experiment. Mice were sacrificed and tumors were isolated and weighed at Day 26 after cell injection. (E) Expression of NRF2 and AKR1C1 was measured by Western blot assays of excised tumors from Ca9-22-D1 xenograft tumor-bearing mice. (F, G) In vivo experimental pulmonary metastasis assay of stable NRF2-knockdown HNSCC cells. HONE-1 (F) or Ca9-22-D1 (G) cells stably expressing NRF2-shRNA or control vector were delivered via tail vein injection into NOD/SCID mice. After 50 (HONE-1) or 25 (Ca9-22-D1) days, lungs were removed from all mice and fixed in Bouin's solution. The lung nodules were counted, and lung sections were prepared. Upper panel: Representative images of pulmonary metastatic foci produced after intravenous injection. Lower panel: Numbers of detectable tumor foci on the surface of whole lungs were quantified and indicated as mean ± S. E. * p < 0.05; ** p < 0.01; and *** p < 0.001 compared with the control group (shControl). (H) Representative histological photographs showing H&E staining and IHC analyses of lung sections taken under bright-field at 50× magnification using an upright microscope.
NRF2-mediated antioxidant capacity is not the primary cause of malignant progression of HNSCC
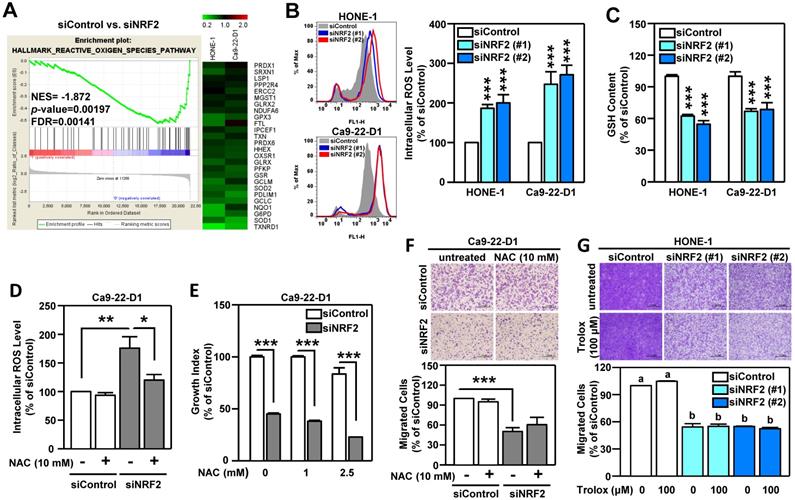
Oxidative stress is known to play a pivotal role in the development of HNSCC [51]. The primary function of NRF2 is to maintain cellular redox homeostasis through enhancing antioxidant gene expression. Therefore, we investigated whether the change in redox balance is the primary cause of NRF2-mediated malignant progression in HNSCC. Gene Set Enrichment Analysis (GSEA) revealed that the HALLMARK REACTIVE OXYGEN SPECIES pathway genes were highly enriched in NRF2-silenced HNSCC cells and that there was a concomitant decrease in the expression of a panel of NRF2-mediated antioxidant genes (Figure 4A). Profoundly increased intracellular ROS levels were also found in both Ca9-22-D1 and HONE-1 cells (Figure 4B). In addition, DOK cells with stable overexpression of NRF2 exhibited lower ROS levels than mock controls (Figure S12A). When cells were challenged with oxidative stress, the reduced form of glutathione (GSH) was oxidized to form glutathione-disulfide (GSSG), which brought on a decrease in intracellular GSH levels. Knockdown of NRF2 significantly reduced intracellular GSH levels in HNSCC cells (Figure 4C). Although treatment of cells with N-acetyl cysteine (NAC), a widely used pharmacological antioxidant, effectively reduced ROS production (Figure 4D, Figure S12B), addition of NAC did not restore cell growth (Figure 4E, Figure S12C) and migration ability (Figure 4F, Figure S12D) in NRF2-knockdown HNSCC cells. Similarly, treatment with another antioxidant, Trolox, had no effect in the rescue of cell motility in NRF2-knockdown HNSCC cells (Figure 4G). These results suggested that NRF2-mediated antioxidant capacity was not a primary cause of malignant progression of HNSCC.
NRF2-mediated antioxidant defense is not a primary cause of malignant progression of HNSCC cells. (A) GSEA enrichment plots of the HALLMARK REACTIVE OXYGEN SPECIES PATHWAY and the corresponding heat map for siControl versus siNRF2 groups in HONE-1 and Ca9-22-D1 cells. (B, C) NRF2 modulates ROS and glutathione. The intracellular ROS level (B) and reduced form of glutathione (GSH; C) levels were measured in NRF2 knockdown HNSCC cell lines by fluorescent staining of 2', 7'-dichlorodihydrofluorescein (DCF) and monochlorobimane (MBC)-GSH adducts, respectively. (D) NRF2-knockdown Ca9-22-D1 cells were assayed for intracellular ROS levels in the absence and presence of 10 mM N-acetylcysteine (NAC). The relative intracellular ROS levels were normalized to the siControl group without NAC treatment. The effects of elimination of ROS by NAC on cell survival (E) and migration (F) in NRF2-knockdown Ca9-22-D1 cells. (G) The effects of elimination of ROS by Trolox (100 μM) on cell migration in NRF2-knockdown HONE-1 cells. All data are expressed as the mean ± S.D. from three individual experiments. Groups with different letters are significant different (p < 0.05) from each other. * p < 0.05; ** p < 0.01; *** p < 0.001.
Pentose phosphate pathway (PPP) is the most highly enriched NRF2-mediated metabolic pathway in HNSCC cells
After a close examination of GSEA data, we noticed that 33% (10/30) of the top 30 most highly enriched down-regulated gene sets in NRF2-knockdown cells were related to cell metabolism (Figure S13A). Among them, the highest-ranking metabolic pathway is the KEGG PENTOSE PHOSPHATE PATHWAY (PPP; Figure S13B). The enrichment plot and heat map of differentially expressed genes displayed obvious suppression of PPP in NRF2-knockdown HONE-1 and Ca9-22-D1 cells (Figure 5A). Since the PPP is a major source of reduced nicotinamide adenine dinucleotide phosphate (NADPH), we checked and observed that NADPH was also significantly suppressed upon NRF2 inhibition (Figure S14).
The pentose phosphate pathway is altered in NRF2-knockdown HNSCC cells. (A) GSEA enrichment plots and heat maps of differentially expressed genes belonging to the KEGG PENTOSE PHOSPHATE PATHWAY (PPP) and associated with knockdown of NRF2 in HNSCC cells. (B) GSEA of the TCGA-HNSC dataset showed that NRF2 level was positively correlated with the PPP pathway. (C) GSEA demonstrated that upregulation of the PPP was associated with poor survival of HNSCC according to gene expression and overall survival data in the TCGA-HNSC dataset. (D) Heat-map of relative gene expression, assessed via real-time PCR, in NRF2-knockdown cells compared to the non-targeted negative control. Gene expression in the NRF2-knockdown cells was significantly different (p < 0.05) compared to scramble control and is represented as the average value of three individual experiments per heat map square (siControl = 100%). (E) Total G6PD and TKT protein levels were assessed in NRF2-knockdown HONE-1 and Ca9-22-D1 cells by Western blot. β-Actin was detected as a loading control. (F) G6PD and TKT protein levels were assessed in NRF2-overexpressing DOK cells treated with NRF2 siRNA or non-targeted control. Examination of the changes of G6PD (G) and TKT (H) enzyme activities in NRF2-overexpressing DOK cells treated with NRF2 siRNA or non-targeted control. G6PD (I) and TKT (J) enzyme activity assays were performed on the excised tumors from Ca9-22-D1 xenograft tumor-bearing mice. ChIP-qPCR analysis of G6PD (K) and TKT (L) were performed on HONE-1 and Ca9-22-D1 cells by using normal rabbit IgG or an anti-NRF2 antibody, and the result was normalized to input control values and represented as the fold enrichment relative to the anti-normal rabbit IgG control. All data are expressed as the mean ± S.D. from three individual experiments. * p < 0.05; ** p < 0.01; *** p < 0.001.
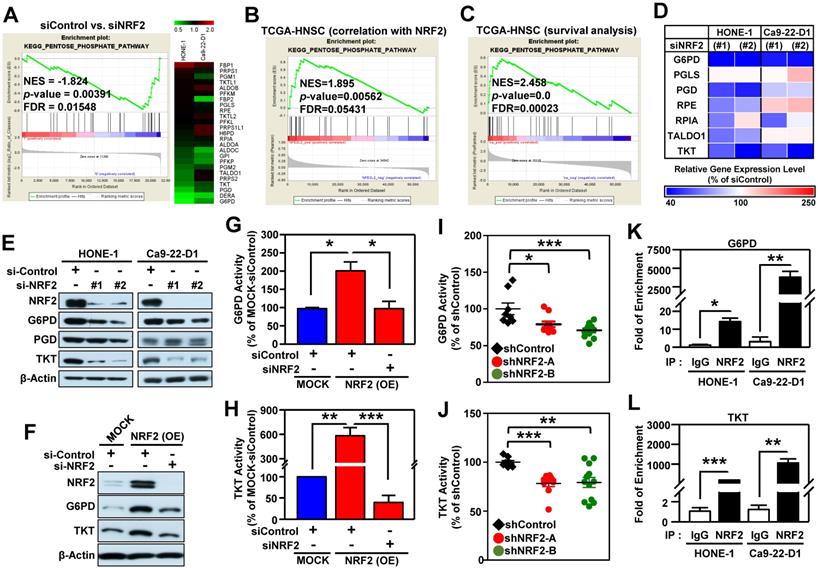
To explore which biological processes or molecular pathways could be regulated by NRF2 signaling in the milieu of tumors, we again performed GSEA analysis on head and neck tumor samples annotated in The Cancer Genome Atlas Head-Neck Squamous Cell Carcinoma (TCGA-HNSC) dataset. We queried significant enrichment of gene sets that were co-expressed with NRF2. At nominal (NOM) p-value < 0.05 and FDR < 0.25, we found 511 gene sets were highly co-expressed with NRF2. As expected, PPP pathway also showed significant positive enrichment (NES = 1.895) among these gene sets, indicating a positive correlation between PPP genes and NRF2 level (Figure 5B). We conducted another independent GSEA assay based on Cox regression model to query whether any enrichment of genes was associated with clinical outcomes of HNSCC. Notably, the PPP pathway was found to be significantly associated with patient survival (NES = 2.458; Figure 5C). These results indicate that increased PPP activity may predict poor overall survival of the patients with HNSCC.
We next identified the specific PPP genes enriched in HNSCC by performing qPCR analysis, and the results demonstrated that G6PD, 6-phosphogluconate dehydrogenase (PGD), and TKT were significantly suppressed in NRF2-knockdown HONE-1 and Ca9-22-D1 cells (Figure 5D). Remarkably, knockdown of NRF2 drastically suppressed the protein levels and enzymatic activity of G6PD and TKT in HNSCC cells (Figure 5E, Figure S15A-B). Consistently, the protein levels or enzyme activity of G6PD and TKT were increased in stable NRF2-overexpressing cells (Figure 5F-H, Figure S15C) and could be re-suppressed by NRF2-targeted siRNA treatment (Figure 5F-H). Furthermore, the in vivo study revealed that knockdown of NRF2 markedly decreased the protein levels (Figure S15D) and enzymatic activities (Figure 5I-J) of G6PD and TKT in the excised tumors from Ca9-22-D1 xenograft tumor-bearing mice.
We further dissected the mechanisms by which NRF2 regulates G6PD and TKT in HNSCC cells. ChIP-qPCR assays demonstrated direct binding of NRF2 to the ARE consensus sequence in the promoter region of G6PD and TKT in both HONE-1 and Ca9-22-D1 cells (Figure 5K-L). Analysis of data from the TCGA-HNSC cohort demonstrated that the level of G6PD or TKT gene expression was positively correlated with NRF2 (G6PD vs. NRF2: Pearson correlation = 0.49, p < 4.7e-33; TKT vs. NRF2: Pearson correlation = 0.36, p < 2.7e-17; Figure S16). These results indicated that NRF2 repurposes PPP metabolism by direct modulation of G6PD and TKT in head and neck cancer.
G6PD and TKT are key NRF2-downstream effectors to drive malignant progression of HNSCC
We next investigated whether G6PD and TKT were critical downstream effectors in driving NRF2-mediated malignant progression of HNSCC. First, we found that the levels of G6PD and TKT expression (Figure 6A) were positively correlated with hallmarks of cell malignancy (i.e., motility as shown in Figure 1F). As expected, knockdown of G6PD or TKT lead to suppression of cell migration, invasion, and anchorage-independent growth of HNSCC cells (Figure 6B-D). We also observed that increase of NRF2 drives cell motility, invasiveness, and proliferation in stably NRF2-overexpressing DOK cells, and these effects could be re-suppressed after blocking the function of G6PD and TKT (Figure 6E-G). These results indicated that G6PD and TKT are key NRF2-downstream effectors to drive malignant features of HNSCC.
G6PD and TKT are downstream effectors of NRF2-mediated malignant progression of HNSCC. (A) Expression patterns of G6PD and TKT were positively related to malignancy and NRF2 protein level in HNSCC cells. (B) Cell motility and (C) invasiveness were inhibited in G6PD- and TKT-knockdown HNSCC cells. Three different HNSCC cell lines, HONE1, Ca9-22-D1, and OEC-M1, were transiently transfected with siRNA against G6PD (siG6PD), TKT (siTKT), or with a non-targeted control siRNA (siControl) for 48 h, resulting in down-regulation of G6PD and TKT expression. The motility (B) and invasiveness (C) of G6PD- or TKT-knockdown HNSCC cells were evaluated by trans-well migration or invasion assays. (D) Anchorage-independent growth of G6PD- and TKT-knockdown HONE-1 cells was evaluated by soft agar colony formation. Silencing of the G6PD or TKT genes by siRNA diminished cell migration (E), invasion (F), and proliferation (G) of NRF2-overexpressing DOK cells. (H) Inhibition of G6PD induced synergistic cytotoxicity with cisplatin in HNSCC cells. Ca9-22-D1 cells were treated with the G6PD inhibitor DHEA, cisplatin, or a combination of the two agents for 72h. Cell viability was evaluated via the methylene blue assay and presented as the percentage of viable cells compared to the control group (white bar). Groups with different letters had significantly different percent viability (p < 0.05). (I) Kaplan-Meier curves demonstrated correlation between high NRF2/G6PD/TKT expression and poor overall survival of HNSCC patients via analysis of the TCGA dataset. The data are expressed as the mean ± S.D. from three individual experiments. * p < 0.05; ** p < 0.01; *** p < 0.001.
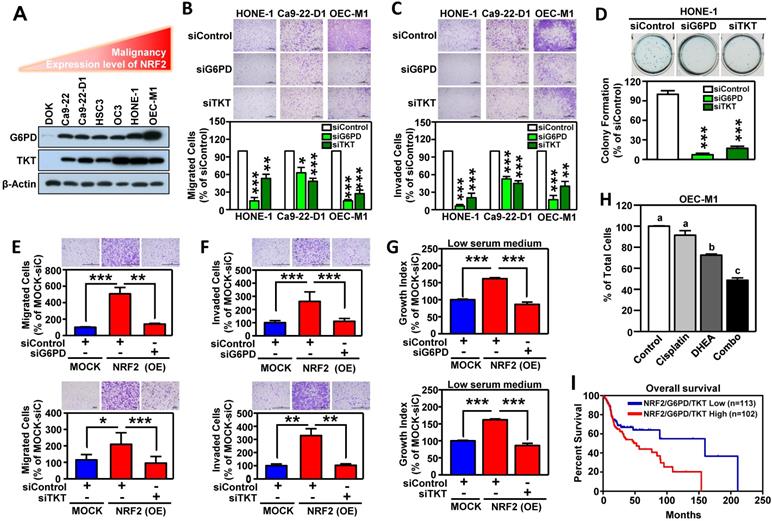
The combination of pathway targeted therapies with standard-of-care (S.O.C.) treatment is a promising therapeutic strategy that may generate synthetic lethality [52]. Cisplatin concurrent chemoradiotherapy (CCRT) is a S.O.C. therapy for treatment of HNSCC, therefore, we questioned whether targeted G6PD or TKT could elicit a synergistic interaction with cisplatin. We treated HNSCC cells with either low dose cisplatin alone, or in combination with G6PD or TKT inhibitors. Strikingly, three different G6PD inhibitors, including dehydroepiandrosterone (DHEA) [53], trans-polydatin [54], and 6-aminonicotinamide (6-AN) [55] were found to cause synthetic lethality in HNSCC cells when combined with cisplatin (Figure 6H, Figure S17), while co-treatment with the TKT inhibitor (oxythiamine chloride hydrochloride) and cisplatin showed no synergistic cytotoxicity (data not shown). These results indicate that combined treatment with cisplatin plus a G6PD inhibitor may be particularly effective as a synthetic lethal therapeutic regimen for HNSCC, and clinical application of this strategy merits further investigation. Although the combination of a TKT inhibitor and cisplatin did not show synergy in HNSCC cells, targeting TKT may have a synergistic interaction with other approved drugs; further research is needed to explore additional combinations.
To expand the level of evidence, the clinical outcome of NRF2/G6PD/TKT-gene signature in HNSCC patients was analyzed in TCGA-HNSC cohort. A total of 215 patient samples were divided into the NRF2/G6PD/TKT-high group (red; n = 102) and the NRF2/G6PD/TKT-low group (blue; n = 113) based on gene expression patterns. The survival probability estimates for the two groups were visualized as Kaplan-Meier plots. Notably, overall survival analysis revealed that the patients in the NRF2/G6PD/TKT-high (red) group had worse survival outcomes than the NRF2/G6PD/TKT-low (blue) group (p = 0.05; Figure 6I). These results suggested that the NRF2/G6PD/TKT gene set could be a potential prognostic biomarker to predict the clinical outcome of HNSCC.
Ribose-5-phosphate generation is more important than NADPH production in determining malignant features of head and neck cancer
NADPH and ribose 5-phosphate (R5P) are the main products or intermediates of G6PD and TKT. NADPH in cells maintains glutathione in the reduced state (GSH) and is important for reductive biosynthesis (e.g., fatty acid synthesis). R5P is required for de novo nucleotide biosynthesis [56]. The metabolite analysis showed that intracellular GSH, NADPH, NADP+ and nucleotide metabolism-related metabolites, especially R5P, were down-regulated in NRF2-knockdown HNSCC cells (Table S7). We found that replenishment of metabolites can effectively rescue the levels of NADPH, NADP+, GSH and R5P in NRF2-knockdown cells (Figure 7A-D). However, addition of NADPH and GSH does not significantly restore cell motility in NRF2-knockdown HNSCC cells (Figure 7E). Notably, unlike NADPH and GSH, R5P significantly restored cell motility and invasiveness in NRF2-knockdown HNSCC cells (Figure 7F-G; Figure S18).
The role of PPP-related metabolites in rescuing cell motility and invasiveness of NRF2-knockdown HNSCC cells. Effects of NADPH supply on intracellular NADPH (A) and NADP+ (B) levels in NRF2-knockdown Ca9-22-D1 cells. Intracellular NADPH and NADP+ levels were measured after incubation with or without 100 μM NADPH for 4 h. (C) Effects of GSH supply on intracellular GSH levels in NRF2-knockdown Ca9-22-D1 cells. Intracellular GSH was measured after incubation with or without 20 mM GSH for 2 h or 4 h. (D) Effects of R5P supply on intracellular R5P in NRF2-knockdown Ca9-22-D1 cells. Intracellular R5P was measured after incubation with or without 240 μM R5P for 1 h or 20 h. Intracellular metabolite levels were normalized to those of the respective siControl without treatment. (E) The effects of NADPH and GSH on cell migration ability in NRF2-knockdown Ca9-22-D1 cells. Cell motility (F) and invasiveness (G) were determined by trans-well migration or invasion assay in NRF2-knockdown Ca9-22-D1 cells in the presence or absence of R5P. The data are expressed as the mean ± S.D. from three individual experiments. * p < 0.05; ** p < 0.01; *** p < 0.001.
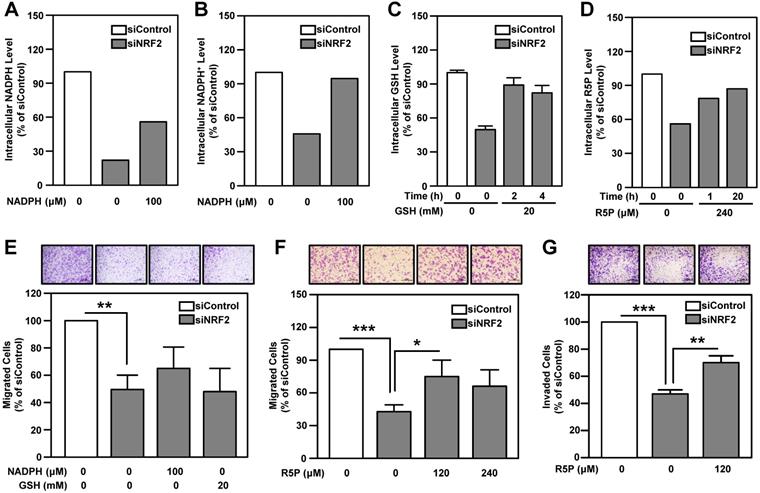
Nucleotide biosynthesis begins with the conversion of R5P to 5-phosphoribosyl-1-pyrophosphate (PRPP), which is then catalyzed to become phosphoribosylamine, a nucleotide precursor. Therefore, we performed a rescue experiment by adding PRPP to the growth media of NRF2-knockdown HNSCC cells. The result demonstrated that, similar to R5P, PRPP could rescue the cell migration ability under NRF2 knockdown condition (Figure S18). These results suggested that the regulatory function of G6PD/TKT on nucleotide biosynthesis appears to be more important than redox regulation for malignant progression of head and neck cancer.
Discussion
In HNSCC, activation of NRF2 occurs through gain-of-function mutations in NFE2L2 (gene encoding NRF2) [57-59], loss-of-function mutations in the KEAP1 gene [57], disruption of the KEAP1/CUL3/RBX1 E3-ubiquitin ligase complex [31], modulation by IL-6 [18] or GRP78/PERK signaling [19], and increased protein stability of NRF2 via O-GlcNAcylation [23]. In this study, we are the first to provide evidence that aberrant activation of NRF2 is modulated by c-MYC in chemical-induced head and neck tumorigenesis. Major chemical-associated risk factors for development of HNSCC, including alcohol consumption, betel nut chewing, and cigarette smoking, all generate oxidative stress [60]. NRF2 is a key determinant for cells coping with oxidative stress and a tobacco exposure-related signature in HNSCC [57, 61]. Long-term exposure of dysplastic DOK cells to nicotine (simulating smoking) or arecoline (simulating betel nut chewing) was used to mimic the malignant transformation in vitro, and the result demonstrated that increased cell proliferation was accompanied by increased NRF2, B-RAF, and c-MYC expression in carcinogen-transformed cells (Figure 2F, Figure S8). Through additional experiments we excluded B-RAF and confirmed that c-MYC positively modulates NRF2 through binding to the NRF2 promoter region in HNSCC cells (Figure 2G-2J). ChIP-seq data from the ENCODE consortium demonstrated that c-MYC binds directly to the NFE2L2 locus and increases NRF2 transcription [20]. The MYC gene has been shown to be amplified in HNSCC, particularly in HPV(-) HNSCC [57]. Our study demonstrated that long-term exposure of DOK and NHOK cells with carcinogens induces c-MYC expression (Figure 2F, Figure S8 and S10), suggesting that chemical factors may be more important to initiate dysregulation of c-MYC oncogene than viral factors during head and neck carcinogenesis.
Through systematic investigation by combining RNAi, microarrays, and GSEA approach, we noted that 33% (10/30) of the 30 most highly enriched down-regulated gene sets in NRF2-knockdown HNSCC cells were related to cellular metabolism (Figure S13). Metabolic reprogramming is firmly established as a hallmark of cancer [62]. During malignant development, cancer cells are obliged to rewire their metabolism to support aberrant cell growth, enhance their metastatic capacity, and adapt to the stress of survival in the austere tumor microenvironment [63]. A characteristic feature of head and neck cancer is enhanced glycolysis, providing the basis for clinical use of FDG-PET as a diagnostic imaging agent [64]. In addition, recent studies on NRF2-mediated cellular metabolism in HNSCC are focused on glycolysis (the Warburg effect). Chang et al. demonstrated that the Warburg effect and the stemness of cancer-initiating cells of the head and neck were mediated by GRP78/p-PERK/NRF2 signaling [19]. Fu et al. found that hyperactive NRF2 causes metabolic reprogramming and up-regulation of glycolysis genes, such as pyruvate kinase M2 (PKM2), in the mouse esophagus [26]. In this study, we observed that KEGG GLYCOLYSIS GLUCONEOGENESIS pathway is the fourth most highly enriched down-regulated gene set in NRF2-knockdown HNSCC cells (Figure S13), which mirrored previous reports [19, 26].
In addition to highlighting glycolysis, we have now identified the PPP pathway as the most highly enriched down-regulated metabolism gene set in NRF2-knockdown HNSCC cells (Figure S13). More importantly, as supported by clinical evidence, the expression of PPP genes is positively correlated with NRF2 (Figure 5B), and increased PPP predicted poor overall survival of patients with HNSCC (Figure 5C). PPP branches off from glycolysis and comprises irreversible oxidative and reversible non-oxidative phases. The enzymes involved in the oxidative phase are G6PD, 6-phosphogluconolactonase (PGLS), and PGD. Ribose-5-phosphate isomerase (RPI), ribulose 5-phosphate 3-epimerase (RPE), transaldolase (TALDO1), and TKT function in the non-oxidative phase of the PPP. To date, very little is known regarding these PPP enzymes in head and neck cancer. NRF2-regulated PPP genes, including G6PD, PGD, TKT, and TALDO1 were first identified in lung adenocarcinoma A549 cells [13]. In this study, we confirmed that G6PD and TKT mRNA level, protein level, and enzyme activity were consistently regulated by NRF2 in HNSCC in vitro and in vivo (Figure 5, Figure S15).
G6PD is overexpressed in a number of cancer types [65], including HNSCC (Table S5). Several oncogenic pathways associated with PPP flux regulation are linked with G6PD [65]. G6PD can be transcriptionally activated by TAp73 [66], YY1 [67], and NRF2 [13]. However, only one prior study revealed that G6PD was modulated by FOXM1 for participating radio-resistance in head and neck cancer [68]. TKT had been reported to be involved in the malignant progression of breast cancer [69] and liver cancer [70], however, there is very limited information on TKT in HNSCC. Based on Oncomine database analysis, increased TKT expression was observed in tumors in several clinical HNSCC datasets (Table S5). It has been shown that transketolase like 1 (TKTL1), a TKT isoform, is activated by promoter hypomethylation and contributes to HNSCC carcinogenesis [71]. TKT, TKTL1, and TKTL2 encoded enzymes all have transketolase activity. Our study showed that transketolase activity was significantly reduced in NRF2-knockdown HNSCC cells and xenograft tumors (Figure 5, Figure S15), and NRF2 did not affect the expression of TKTL1 or TKTL2 gene in HNSCC (data not shown). These results suggested that knockdown of NRF2-mediated TKT suppression did not cause compensatory effects by increasing TKTL1 and TKTL2. Knockdown of TKT has been reported to inhibit the proliferation of oral cancer cells and be accompanied by an increased uptake of glucose and glutamine, as well as increased production of lactate [72], which suggested that cancer cells may overcome single defective pathways through secondary metabolic network adaptations. Therefore, inhibition of multiple metabolic pathways or their common upstream regulator may be required to impede tumor progression. Accordingly, inhibition of NRF2 may be more advantageous compared to targeting a particular downstream metabolic pathway. Although there have yet to be any FDA-approved drugs targeting NRF2 activity in cancer, there have been increasing efforts toward the development of novel NRF2 inhibitors [73] and continued research in this area has great potential for clinical application.
NRF2 has been shown to activate PPP genes through direct binding to the ARE in the gene promoters [13, 21], producing effects such as attenuation of miR-1 and miR-206 [12] or driving telomerase reverse transcriptase [74]. We demonstrated that NRF2 bound directly to the ARE consensus sequence in the promoters of G6PD and TKT in HNSCC cells (Figure 5K-5L), and G6PD and TKT acted as the key NRF2-downstrem effectors of malignant progression of HNSCC (Figure 6). G6PD is the irreversible rate limiting enzyme in the oxidative phases of the PPP that ensures sufficient NADPH and R5P levels. The reversible enzyme TKT bridges the PPP with glycolysis, and is therefore a key enzyme controlling the direction of PPP and R5P levels [56]. Although the requirement for NADPH is higher than that for R5P in cell physiological functions, the addition of R5P can restore cell motility in NRF2-knockdown HNSCC cells, whereas the addition of NADPH does not rescue motility (Figure 7). These results suggest that G6PD- and TKT-regulated nucleotide biosynthesis may be more important than redox regulation in driving malignant progression of head and neck cancer. NADPH could be generated by both PPP and the folate-mediated one-carbon metabolism and malic enzymes in cancer cells and other proliferating cells [75, 76], therefore, we speculate that inhibition of the PPP in HNSCC cells may reprogram other NADPH homeostasis systems. Additional research is needed to explore this possibility.
In fact, the mechanism by which NRF2 regulates nucleotide biosynthesis varies among cancer types. Saigusa et al. found that TALDO1 may involve in NRF2-mediated de novo purine nucleotide synthesis in non-small-cell lung carcinoma [77]. However, we noted that in all test HNSCC cells, the expression level of the TALDO1 gene did not consistently change after genetic ablation of NRF2 (Figure 5D). In addition, Mitsuishi et al. has reported that MTHFD2, the metabolism enzyme for nucleotide biosynthesis, was decreased in lung cancer cell lines with NRF2 knockdown [13]. DeNicola et al. demonstrated that NRF2 controls the expression of PHGDH, PSAT1, PSPH, and SHMT2 via ATF4 to support nucleotide production in non-small cell lung cancer [25]. However, according to our real-time PCR validation, the expression levels of these genes (PHGDH, PSAT1, PSPH, SHMT1/2, MTHFD1/2) did not change upon NRF2 silencing in HNSCC cells (Figure S19), suggesting that NRF2-mediated regulatory mechanisms may vary substantially in head and neck versus lung cancer, further supporting the unique role of NRF2-mediated G6PD and TKT expression in nucleotide biosynthesis of head and neck cancer. More importantly, we have identified the NRF2/G6PD/TKT gene signature as a potential prognostic biomarker for prediction of HNSCC survival outcomes (Figure 6I). Even though it was known that NRF2 can regulate PPP [12, 13, 21], no study has demonstrated that NRF2 promotes cancer progression directly through PPP components, particularly in head and cancer. In this study, we provided the first evidence to confirm that G6PD and TKT are critical downstream effectors of NRF2-driven malignant progression in HNSCC. We believe this is an important breakthrough for clarifying the role of NRF2 in cancer development.
In addition to PPP and glycolysis, the ten most highly enriched down-regulated metabolism pathways in NRF2 knockdown HNSCC cells also included amino acid, nucleotide, xenobiotics, heme, and fatty acid metabolism as well as oxidative phosphorylation (Figure S13B), and many of them have been known to modulate head and neck oncogenesis [78]. Interestingly, we noted that the KEGG VALINE, LEUCINE AND ISOLUCINE DEGRADATION pathway was the second most highly enriched downregulated gene set in response to NRF2 silencing in HNSCC cells. Valine, leucine, and isoleucine are branched-chain amino acids (BCAAs). BCAA metabolism can influence diverse cellular processes, ranging from protein synthesis to epigenetic regulation, and dysregulation of BCAA metabolism contributes to cancer progression by diverse mechanisms [79]. To date, no literature has been published on the regulation between NRF2 and BCAA metabolism. In addition, the role of valine, leucine, and isoleucine degradation in the progression of head and neck cancer remains to be clarified. Further exploration of the link between BCAA metabolism and NRF2 in HNSCC is warranted.
This is the first study providing data from systematic analysis of the regulatory mechanisms and function of NRF2 in HNSCC. We discovered that aberrant activation of NRF2 occurred via c-MYC-mediated upregulation and is associated with chemical-induced carcinogenesis. NRF2 is involves in reprogramming the broad metabolic milieu in HNSCC, and PPP is the most highly enriched NRF2-mediated metabolic pathway. Notably, G6PD and TKT are critical NRF2-downstream effectors that drive malignant progression of HNSCC. Furthermore, G6PD- and TKT-regulated nucleotide biosynthesis is more important than redox regulation for maintaining the malignant features of head and neck cancer. Taken together, these data support the potential for use of the NRF2/G6PD/TKT gene signature as a prognostic biomarker and for the development of NRF2-targeted therapies that alter cellular metabolism as novel and promising treatment options for head and neck cancer.
Abbreviations
AC: arecoline; ARE: antioxidant response element; BCAAs: branched-chain amino acids; CCRT: cisplatin concurrent chemoradiotherapy; DHEA: dehydroepiandrosterone; DOK: human dysplastic oral keratinocytes; F6P: fructose 6-phosphate; G3P: glyceraldehyde 3-phosphate; G6P: glucose 6-phosphate; G6PD: glucose-6-phosphate dehydrogenase; GSH: glutathione; GSSG: glutathione-disulfide; HNSCC: head and neck squamous cell carcinoma; KEAP1: Kelch‐like ECH associating protein 1; NAC: N-acetyl cysteine; NHOK: normal human oral keratinocytes; NIC: nicotine; NRF2: nuclear factor erythroid 2-related factor 2; OT: oxythiamine chloride hydrochloride; PGD: 6-phosphogluconate dehydrogenase; PGLS: 6-phosphogluconolactonase; PKM2: pyruvate kinase M2; PPP: pentose phosphate pathway; PRPP: 5-phosphoribosyl-1-pyrophosphate; R5P: ribose 5-phosphate; ROS: reactive oxygen species; RPE: ribulose 5-phosphate 3-epimerase; RPI: ribose-5-phosphate isomerase; S.O.C.: standard-of-care; TALDO1: transaldolase; TKT: transketolase; TKTL1: transketolase like 1.
Acknowledgements
This study was supported by Research Grants MOST 109-2320-B-400-012, 109-2314-B-400-046, 108-2314-B-400-007, and MOST 107-2314-B-400-020 from Ministry of Science and Technology, Taiwan; and by NHRI-BP-109-PP-02 and NHRI-BP-SP-04 from National Health Research Institutes, Taiwan. The authors thank Dr. Loretta Collins (WriteScience, LLC, Hannawa Falls, NY, USA) for scientific editing. The authors thank the Core Facilities for Proteomics and Chemistry Research and Bioinformatics at NHRI (Miaoli, Taiwan).
Author Contributions
Y.-C. T. contributed to design and conduct of the main experiments, analysis of data, and preparation of manuscript drafts. J.-R. H. contributed to collection of clinical specimens and interpretation of clinical results. S.-S. J contributed to design and conduct of the data mining approach. J.-Y. C. and K.-J.L. provided study materials and suggestions for research design. P.-Y.C. and H.-L. F. contributed to analysis of results of the pathology-related experiments. L.-M.L. conducted in vivo animal studies. H.-H.C., Y.-W. H. and Y.-T. C. assisted with in vitro experiments. F.-Y. T. and S.-F. L assisted with web-based data-mining analysis. Y.-J.C. co-supervised the research project and edited the manuscript. C.-C.K. initiated the research idea, guided research directions, supervised the research project, coordinated teamwork, and edited the manuscript.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Baird L, Yamamoto M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol Cell Biol. 2020;40:e00099-20
2. Tonelli C, Chio IIC, Tuveson DA. Transcriptional regulation by Nrf2. Antioxid Redox Signal. 2018;29:1727-45
3. Kensler TW, Wakabayashi N. Nrf2: friend or foe for chemoprevention? Carcinogenesis. 2010;31:90-9
4. Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer. 2012;12:564-71
5. Cloer EW, Goldfarb D, Schrank TP, Weissman BE, Major MB. NRF2 activation in cancer: from DNA to protein. Cancer Res. 2019;79:889-98
6. Kansanen E, Kuosmanen SM, Leinonen H, Levonen AL. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013;1:45-9
7. Leinonen HM, Kansanen E, Pölönen P, Heinäniemi M, Levonen AL. Role of the Keap1-Nrf2 pathway in cancer. Adv Cancer Res. 2014;122:281-320
8. Leinonen HM, Kansanen E, Pölönen P, Heinäniemi M, Levonen AL. Dysregulation of the Keap1-Nrf2 pathway in cancer. Biochem Soc Trans. 2015;43:645-9
9. Rojo de la Vega M, Chapman E, Zhang DD. NRF2 and the hallmarks of cancer. Cancer Cell. 2018;34:21-43
10. Taguchi K, Yamamoto M. The KEAP1-NRF2 System in Cancer. Front Oncol. 2017;7:85
11. Feng L, Li J, Yang L, Zhu L, Huang X, Zhang S. et al. Tamoxifen activates Nrf2-dependent SQSTM1 transcription to promote endometrial hyperplasia. Theranostics. 2017;7:1890-900
12. Singh A, Happel C, Manna SK, Acquaah-Mensah G, Carrerero J, Kumar S. et al. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J Clin Invest. 2013;123:2921-34
13. Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H. et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012;22:66-79
14. Fan H, Paiboonrungruan C, Zhang X, Prigge JR, Schmidt EE, Sun Z. et al. Nrf2 regulates cellular behaviors and Notch signaling in oral squamous cell carcinoma cells. Biochem Biophys Res Commun. 2017;493:833-9
15. Xu P, Jiang L, Yang Y, Wu M, Liu B, Shi Y. et al. PAQR4 promotes chemoresistance in non-small cell lung cancer through inhibiting Nrf2 protein degradation. Theranostics. 2020;10:3767-78
16. Lu BC, Li J, Yu WF, Zhang GZ, Wang HM, Ma HM. Elevated expression of Nrf2 mediates multidrug resistance in CD133(+) head and neck squamous cell carcinoma stem cells. Oncology letters. 2016;12:4333-8
17. Roh JL, Kim EH, Jang H, Shin D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 2017;11:254-62
18. Matsuoka Y, Nakayama H, Yoshida R, Hirosue A, Nagata M, Tanaka T. et al. IL-6 controls resistance to radiation by suppressing oxidative stress via the Nrf2-antioxidant pathway in oral squamous cell carcinoma. British journal of cancer. 2016;115:1234-44
19. Chang CW, Chen YS, Tsay YG, Han CL, Chen YJ, Yang CC. et al. ROS-independent ER stress-mediated NRF2 activation promotes warburg effect to maintain stemness-associated properties of cancer-initiating cells. Cell Death Dis. 2018;9:194
20. DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K. et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106-9
21. Hirotsu Y, Katsuoka F, Funayama R, Nagashima T, Nishida Y, Nakayama K. et al. Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012;40:10228-39
22. Li H, Li J, Zhang G, Da Q, Chen L, Yu S. et al. HMGB1-Induced p62 Overexpression Promotes Snail-Mediated Epithelial-Mesenchymal Transition in Glioblastoma Cells via the Degradation of GSK-3β. Theranostics. 2019;9:1909-22
23. Dai W, Jin X, Jiang B, Chen W, Ji Z, Xu X. et al. Elevated O-GlcNAcylation Promotes Malignant Phenotypes of Hypopharyngeal Squamous Cell Carcinoma by Stabilizing Nrf2 through Regulation of the PI3K/Akt Pathway. Anticancer Agents Med Chem. 2020;20:1933-42
24. Bi Z, Zhang Q, Fu Y, Wadgaonkar P, Zhang W, Almutairy B. et al. Nrf2 and HIF1α converge to arsenic-induced metabolic reprogramming and the formation of the cancer stem-like cells. Theranostics. 2020;10:4134-49
25. DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D. et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet. 2015;47:1475-81
26. Fu J, Xiong Z, Huang C, Li J, Yang W, Han Y. et al. Hyperactivity of the transcription factor Nrf2 causes metabolic reprogramming in mouse esophagus. J Biol Chem. 2019;294:327-40
27. Vokes EE, Weichselbaum RR, Lippman SM, Hong WK. Head and neck cancer. N Engl J Med. 1993;328:184-94
28. Wenig BM. Atlas of head and neck pathology. Philadelphia: Elsevier Inc. 2016
29. Hammerman PS, Hayes DN, Grandis JR. Therapeutic insights from genomic studies of head and neck squamous cell carcinomas. Cancer Discov. 2015;5:239-44
30. Leemans CR, Snijders PJF, Brakenhoff RH. The molecular landscape of head and neck cancer. Nat Rev Cancer. 2018;18:269-82
31. Martinez VD, Vucic EA, Thu KL, Pikor LA, Lam S, Lam WL. Disruption of KEAP1/CUL3/RBX1 E3-ubiquitin ligase complex components by multiple genetic mechanisms: Association with poor prognosis in head and neck cancer. Head Neck. 2015;37:727-34
32. Namani A, Matiur Rahaman M, Chen M, Tang X. Gene-expression signature regulated by the KEAP1-NRF2-CUL3 axis is associated with a poor prognosis in head and neck squamous cell cancer. BMC Cancer. 2018;18:46
33. Stacy DR, Ely K, Massion PP, Yarbrough WG, Hallahan DE, Sekhar KR. et al. Increased expression of nuclear factor E2 p45-related factor 2 (NRF2) in head and neck squamous cell carcinomas. Head Neck. 2006;28:813-8
34. Wang T, Hu P, Li B, Zhang JP, Cheng YF, Liang YM. Role of Nrf2 signaling pathway in the radiation tolerance of patients with head and neck squamous cell carcinoma: an in vivo and in vitro study. Onco Targets Ther. 2017;10:1809-19
35. Hu J, Li Y, Li H, Shi F, Xie L, Zhao L. et al. Targeting Epstein-Barr virus oncoprotein LMP1-mediated high oxidative stress suppresses EBV lytic reactivation and sensitizes tumors to radiation therapy. Theranostics. 2020;10:11921-37
36. Shibata T, Saito S, Kokubu A, Suzuki T, Yamamoto M, Hirohashi S. Global downstream pathway analysis reveals a dependence of oncogenic NF-E2-related factor 2 mutation on the mTOR growth signaling pathway. Cancer Res. 2010;70:9095-105
37. Chen HH, Chen YT, Huang YW, Tsai HJ, Kuo CC. 4-Ketopinoresinol, a novel naturally occurring ARE activator, induces the Nrf2/HO-1 axis and protects against oxidative stress-induced cell injury via activation of PI3K/AKT signaling. Free radical biology & medicine. 2012;52:1054-66
38. Kuo CC, Liu TW, Chen LT, Shiah HS, Wu CM, Cheng YT. et al. Combination of arsenic trioxide and BCNU synergistically triggers redox-mediated autophagic cell death in human solid tumors. Free radical biology & medicine. 2011;51:2195-209
39. Yen YC, Shiah SG, Chu HC, Hsu YM, Hsiao JR, Chang JY. et al. Reciprocal regulation of microRNA-99a and insulin-like growth factor I receptor signaling in oral squamous cell carcinoma cells. Mol Cancer. 2014;13:6
40. Chang SE, Foster S, Betts D, Marnock WE. DOK, a cell line established from human dysplastic oral mucosa, shows a partially transformed non-malignant phenotype. Int J Cancer. 1992;52:896-902
41. Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D. et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1-6
42. Ko YC, Huang YL, Lee CH, Chen MJ, Lin LM, Tsai CC. Betel quid chewing, cigarette smoking and alcohol consumption related to oral cancer in Taiwan. J Oral Pathol Med. 1995;24:450-3
43. Ngan HL, Wang L, Lo KW, Lui VWY. Genomic Landscapes of EBV-Associated Nasopharyngeal Carcinoma vs. HPV-Associated Head and Neck Cancer. Cancers (Basel). 2018 10
44. Saleh K, Eid R, Haddad FG, Khalife-Saleh N, Kourie HR. New developments in the management of head and neck cancer - impact of pembrolizumab. Ther Clin Risk Manag. 2018;14:295-303
45. Kao SY, Lim E. An overview of detection and screening of oral cancer in Taiwan. Chin J Dent Res. 2015;18:7-12
46. Song H, Wan Y, Xu YY. Betel quid chewing without tobacco: a meta-analysis of carcinogenic and precarcinogenic effects. Asia Pac J Public Health. 2015;27:Np47-57
47. Lee CH, Chang JS, Syu SH, Wong TS, Chan JY, Tang YC. et al. IL-1β promotes malignant transformation and tumor aggressiveness in oral cancer. J Cell Physiol. 2015;230:875-84
48. Chang NW, Pei RJ, Tseng HC, Yeh KT, Chan HC, Lee MR. et al. Co-treating with arecoline and 4-nitroquinoline 1-oxide to establish a mouse model mimicking oral tumorigenesis. Chem Biol Interact. 2010;183:231-7
49. Lai CH, Chang NW, Lin CF, Lin CD, Lin YJ, Wan L. et al. Proteomics-based identification of haptoglobin as a novel plasma biomarker in oral squamous cell carcinoma. Clin Chim Acta. 2010;411:984-91
50. Sheu JJ, Hua CH, Wan L, Lin YJ, Lai MT, Tseng HC. et al. Functional genomic analysis identified epidermal growth factor receptor activation as the most common genetic event in oral squamous cell carcinoma. Cancer Res. 2009;69:2568-76
51. Thomas SJ, MacLennan R. Slaked lime and betel nut cancer in Papua New Guinea. Lancet. 1992;340:577-8
52. Burtness B, Golemis EA. Molecular determinants of head and neck cancer. 2 ed: Springer. 2018
53. Fang Z, Jiang C, Feng Y, Chen R, Lin X, Zhang Z. et al. Effects of G6PD activity inhibition on the viability, ROS generation and mechanical properties of cervical cancer cells. Biochim Biophys Acta. 2016;1863:2245-54
54. Mele L, Paino F, Papaccio F, Regad T, Boocock D, Stiuso P. et al. A new inhibitor of glucose-6-phosphate dehydrogenase blocks pentose phosphate pathway and suppresses malignant proliferation and metastasis in vivo. Cell Death Dis. 2018;9:572
55. Zhelev Z, Ivanova D, Bakalova R, Aoki I, Higashi T. Inhibition of the Pentose-phosphate Pathway Selectively Sensitizes Leukemia Lymphocytes to Chemotherapeutics by ROS-independent Mechanism. Anticancer Res. 2016;36:6011-20
56. Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer's Achilles' heel. Cancer cell. 2008;13:472-82
57. CancerGenomeAtlasNetwork. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576-82
58. Kim YR, Oh JE, Kim MS, Kang MR, Park SW, Han JY. et al. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J Pathol. 2010;220:446-51
59. Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K. et al. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci U S A. 2008;105:13568-73
60. Choudhari SK, Chaudhary M, Gadbail AR, Sharma A, Tekade S. Oxidative and antioxidative mechanisms in oral cancer and precancer: a review. Oral Oncol. 2014;50:10-8
61. Chung CH, Parker JS, Karaca G, Wu J, Funkhouser WK, Moore D. et al. Molecular classification of head and neck squamous cell carcinomas using patterns of gene expression. Cancer cell. 2004;5:489-500
62. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
63. Vander Heiden MG, DeBerardinis RJ. Understanding the Intersections between Metabolism and Cancer Biology. Cell. 2017;168:657-69
64. Creff G, Devillers A, Depeursinge A, Palard-Novello X, Acosta O, Jegoux F. et al. Evaluation of the prognostic value of FDG PET/CT parameters for patients with surgically treated head and neck cancer: a systematic review. JAMA Otolaryngol Head Neck Surg. 2020;146:471-9
65. Yang HC, Wu YH, Yen WC, Liu HY, Hwang TL, Stern A. et al. The redox role of G6PD in cell growth, cell death, and cancer. Cells. 2019;8:1055
66. Du W, Jiang P, Mancuso A, Stonestrom A, Brewer MD, Minn AJ. et al. TAp73 enhances the pentose phosphate pathway and supports cell proliferation. Nat Cell Biol. 2013;15:991-1000
67. Wu S, Wang H, Li Y, Xie Y, Huang C, Zhao H. et al. Transcription factor YY1 promotes cell proliferation by directly activating the pentose phosphate pathway. Cancer Res. 2018;78:4549-62
68. Eckers JC, Kalen AL, Sarsour EH, Tompkins VS, Janz S, Son JM. et al. Forkhead box M1 regulates quiescence-associated radioresistance of human head and neck squamous carcinoma cells. Radiat Res. 2014;182:420-9
69. Tseng CW, Kuo WH, Chan SH, Chan HL, Chang KJ, Wang LH. Transketolase Regulates the Metabolic Switch to Control Breast Cancer Cell Metastasis via the α-Ketoglutarate Signaling Pathway. Cancer Res. 2018;78:2799-812
70. Qin Z, Xiang C, Zhong F, Liu Y, Dong Q, Li K. et al. Transketolase (TKT) activity and nuclear localization promote hepatocellular carcinoma in a metabolic and a non-metabolic manner. J Exp Clin Cancer Res. 2019;38:154
71. Sun W, Liu Y, Glazer CA, Shao C, Bhan S, Demokan S. et al. TKTL1 is activated by promoter hypomethylation and contributes to head and neck squamous cell carcinoma carcinogenesis through increased aerobic glycolysis and HIF1alpha stabilization. Clin Cancer Res. 2010;16:857-66
72. Zhang M, Chai YD, Brumbaugh J, Liu X, Rabii R, Feng S. et al. Oral cancer cells may rewire alternative metabolic pathways to survive from siRNA silencing of metabolic enzymes. BMC Cancer. 2014;14:223
73. Jiang ZY, Lu MC, You QD. Nuclear factor erythroid 2-related factor 2 (Nrf2) inhibition: an emerging strategy in cancer therapy. J Med Chem. 2019;62:3840-56
74. Ahmad F, Dixit D, Sharma V, Kumar A, Joshi SD, Sarkar C. et al. Nrf2-driven TERT regulates pentose phosphate pathway in glioblastoma. Cell Death Dis. 2016;7:e2213
75. Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 2014;510:298-302
76. Liu L, Shah S, Fan J, Park JO, Wellen KE, Rabinowitz JD. Malic enzyme tracers reveal hypoxia-induced switch in adipocyte NADPH pathway usage. Nat Chem Biol. 2016;12:345-52
77. Saigusa D, Motoike IN, Saito S, Zorzi M, Aoki Y, Kitamura H. et al. Impacts of NRF2 activation in non-small-cell lung cancer cell lines on extracellular metabolites. Cancer Sci. 2020;111:667-78
78. Hsieh YT, Chen YF, Lin SC, Chang KW, Li WC. Targeting cellular metabolism modulates head and neck oncogenesis. Int J Mol Sci. 2019;20:3960
79. Sivanand S, Vander Heiden MG. Emerging roles for branched-chain amino acid metabolism in cancer. Cancer cell. 2020;37:147-56
Author contact
![]() Corresponding author: C.-C.K. (E-mail: cckuoorg.tw) or Y.-J.C. (E-mail: yjchuangnthu.edu.tw).
Corresponding author: C.-C.K. (E-mail: cckuoorg.tw) or Y.-J.C. (E-mail: yjchuangnthu.edu.tw).