Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Structure of MLKL
Necroptosis signaling
MLKL functions beyond necroptosis
MLKL inhibitors
Conclusions
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(10):4759-4769. doi:10.7150/thno.54072 This issue Cite
Review
MLKL: Functions beyond serving as the Executioner of Necroptosis
Chaoning Zhan*, Minchun Huang*, Xiaojun Yang ![]() , Jin Hou
, Jin Hou ![]()
Department of Stomatology, Nanfang Hospital, Southern Medical University, Guangzhou, P.R. China.
*These authors contribute equally to this work.
Received 2020-10-4; Accepted 2021-2-7; Published 2021-3-4
Abstract

Recently, necroptosis, as a programmed cell death pathway, has drawn much attention as it has been implicated in multiple pathologies, especially in the field of inflammatory diseases. Pseudokinase mixed lineage kinase domain-like protein (MLKL) serves as a terminal-known obligate effector in the process of necroptosis. To date, the majority of research on MLKL has focused on its role in necroptosis, and the prevailing view has been that the sole function of MLKL is to mediate necroptosis. However, increasing evidence indicates that MLKL can serve as a regulator of many diseases via its non-necroptotic functions. These functions of MLKL shed light on its functional complexity and diversity. In this review, we briefly introduce the current state of knowledge regarding the structure of MLKL, necroptosis signaling, as well as cross-linkages among necroptosis and other regulated cell death pathways, and we particularly highlight recent progress related to newly identified functions and inhibitors of MLKL. These discussions promote a better understanding of the role of MLKL in diseases, which will foster efforts to pharmacologically target this molecule in clinical treatments.
Keywords: MLKL, necroptosis, structure, inhibitors, therapeutic target.
Introduction
Cell death is an irreversible degeneration process that results in the loss of cell integrity and is crucial for organ development, tissue homeostasis, and host defense [1, 2]. There are two types of cell death, namely accidental cell death (ACD) and regulated cell death (RCD). While the former is unpreventable, the latter can be modulated by pharmacologic and/or genetic interventions [3]. In 1972, the caspase-dependent apoptosis pathway was the first type of RCD morphotype to be discovered [4], and our understanding of its molecular mechanism has grown considerably. However, only in the past decade has caspase-independent programmed necrosis or 'necroptosis' started to be a major focus in efforts to understand cell death and the pathology of inflammatory diseases. A key finding has been that cell fate can be determined by a switch from apoptosis to necroptosis via genetic ablation of caspase 8 (CASP8) or by using caspase inhibitors. In the natural state, if pathogens inhibit CASP8 to evade host defense, they will encounter the second guard pathway, namely necroptosis [5]. Although necroptosis may be a backup mechanism to clear pathogens, it can also trigger a strong inflammatory response, which substantiates the detrimental side-effect of necroptosis [6].
Because apoptosis is indispensable for mammalian development [7, 8], apoptosis is not an ideal target for controlling inflammation. Thus, its counterpart defense mechanism, necroptosis, may represent an attractive target goal. Studies have shown that mixed lineage kinase domain-like protein (MLKL)-deficient mice are viable, healthy, and fertile and do not display any physical or behavioral abnormalities [9, 10]. Thus, MLKL, acting as the most terminal obligate effector in necroptosis seems to be a desirable target for drug development [10]. Moreover, the traditional view suggests that the only function of MLKL is compromising cellular integrity, thus resulting in necroptosis, which would limit the side effects of MLKL inhibitors. However, more recent studies have found that in addition to its well-known role in necroptosis, MLKL has been found to play regulatory roles in a number of other physiological and pathophysiological events. These beneficial non-necroptotic functions of MLKL, such as limiting intracellular bacteria replication and promoting nerve regeneration [11, 12], would likely be affected by MLKL-targeting drugs aimed at controlling inflammation. Besides, a clear understanding of the cellular and molecular mechanisms of the newly discovered functions of MLKL is also needed to aid the development of new treatment strategies for relevant diseases, such as multiple sclerosis and cancer [13, 14]. Based on the existing studies, the non-necroptotic functions of MLKL are summarized for the purpose of sharing a contemporary understanding of MLKL-based biology.
Structure of MLKL
More than 500 protein kinases have been identified in the human genome, with approximately 10% of them appearing to be enzymatically inactive and having been classified as pseudokinases [15]. The primary function of protein kinases is to phosphorylate protein substrates, which is important in cellular homeostasis. In general terms, protein kinases rely on the ATP interactor 'VAIK' motif, the catalytic loop 'HRD' motif, and the metal co-factor binding 'DFG' motif [10]. All of these are crucial for phosphoryl transfer activity. However, the 'HRD' motif and 'DFG' motif are defective or absent in MLKL orthologs [16]. Thus, MLKL is classified as a pseudokinase based on its lack of two of the three conserved catalytic residues in its kinase-like domain [15]. Generally, divalent metals, most typically of Mg2+, can neutralize the net negative charge of the nucleotide and enable the kinase active site to interact with nucleotides. Intriguingly, MLKL can bind nucleotides in the absence of cations [10, 17].
MLKL, as a pseudokinase, consists of a C-terminal pseudokinase domain, a two-helix brace or linker, and an N-terminal four-helix bundle (4HB) (Figure 1) [10, 18]. The phosphorylation sites of RIPK3 exist on the MLKL activation loop residues, mouse serine 345 [16, 19] or human threonine 357/serine 358 [20], in the pseudokinase domain. Upon phosphorylation of MLKL, the conformation of the pseudokinase domain will change, which results in 4HB domain exposure [16, 21, 22]. Additionally, there are other phosphorylation events that modulate MLKL function. Mouse serine 158 is a MLKL phosphorylation site that is subject to as-yet-unknown protein-mediated phosphorylation, and its phosphorylation helps to suppress cell death [23].
Structure of MLKL and its regulation in necroptosis. MLKL contains a C-terminal pseudokinase domain, a two-helix brace and an N-terminal four-helix bundle (4HB). After MLKL is phosphorylated by RIPK3, TAM (Tyro3, Axl, and Mer) kinase phosphorylates MLKL at the pseudokinase domain to initiate oligomerization of MLKL. Then MLKL is conjugated to heat shock protein 90 (Hsp90) and Hsp70 to achieve membrane translocation. Furthermore, inositol phosphate metabolites are required for the IP6 production. And IP4, IP5 and IP6 serve as critical binders of MLKL in necroptosis. During the course of MLKL activation, Beclin 1 and B-cell lymphoma 2 (Bcl-2) play negative regulatory roles in the conformation and oligomerization of MLKL. Additionally, flotillin-mediated endocytosis and ALIX-syntenin-1-mediated exocytosis can preclude contact between MLKL and the plasma membrane to suppresses necroptosis.
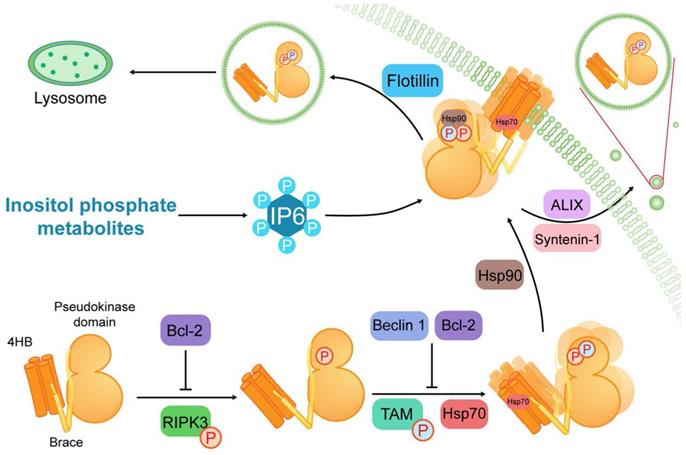
The N-terminal region of MLKL consists of a 4HB domain interacting with a two-helix brace [10]. Generally, MLKL disulfide bond-dependent oligomerization and membrane translocation are essential for the formation of membrane pores [21, 22, 24-27]. The oligomerization is initiated by TAM (Tyro3, Axl, and Mer) kinase, which can phosphorylate MLKL at tyrosine 376 [28]. The membrane translocation is based on patches of positively charged amino acids that are localized over the surface of the 4HB, which can interact with phosphatidylinositol phosphates (PIPs) [29, 30]. This process requires several inositol phosphate (IP) (e.g., IP4, IP5 and IP6) and inositol pentakisphosphate 2-kinase, which transforms IP5 into IP6 [31]. Conversely, flotillin-mediated endocytosis and ALIX-syntenin-1-mediated exocytosis can prevent contact between phosphorylated MLKL (p-MLKL) and the plasma membrane to inhibit necroptosis [32]. As a consequence, oligomerization and membrane translocation are subject to complex multilevel regulation to prevent these processes from getting out of control.
The late formation of small pores around 4 nm in diameter is a core event in necroptosis [33]. Furthermore, MLKL-induced cell death is positively correlated with the activity of cation channels formed by MLKL [34]. In addition to phosphatidylinositol phosphates (e.g., PI(5)P and PI(4,5)P2) within plasma membranes [29, 35], cardiolipin can also bind MLKL oligomer [36]. Cardiolipin is mainly distributed in the mitochondrial inner membrane. Although several studies have implicated mitochondria and mitochondrial reactive oxygen species (ROS) production as being involved in necroptosis [37-39], specific removal of mitochondria does not compromise necroptosis [40].
It is significant to note that MLKL can act as a client protein in interacting with different molecular chaperone proteins. Heat shock protein 90 (Hsp90) binds the pseudokinase domain of MLKL via its cochaperone CDC37. This weak or transient interaction is required for MLKL function and may be involved in the phosphorylation of MLKL [41]. Another molecular chaperone, heat shock protein 70 (Hsp70), can stabilize MLKL and promote MLKL polymerization via binding to the N-terminal region of MLKL [42]. Such evidence demonstrates that the molecular chaperone proteins can promote necroptosis via interacting with different sites in the MLKL structure. In contrast, some proteins function as negative modulators of MLKL. For example, Beclin 1 and B-cell lymphoma 2 (Bcl-2) can bind to the coiled-coil domain and Bcl-2 homology (BH)-3 domain of MLKL, respectively, to inhibit necroptosis [43, 44].
Necroptosis signaling
Phenotypically, apoptosis is characterized by nuclear shrinkage, plasma membrane blebbing, chromatin condensation, and regular DNA fragmentation [45], whereas, necroptotic cells show cell swelling and plasma membrane damage, resulting in cell lysis and a release of damage-associated molecular patterns (DAMPs) [46]. As RCD pathways, both extrinsic apoptosis and necroptosis can be initiated by common death receptor ligation, among which the tumor necrosis factor receptor (TNFR) is the most widely studied (Figure 2). Ligation of the TNFR promotes the assembly of a dynamic multi-protein complex at the cytoplasmic side of the receptor, which is referred to as “TNFR complex I” and contains TNF-associated death domain (TRADD), TNFR-associated factor 2 (TRAF2), receptor-interacting protein kinases 1 (RIPK1), cellular inhibitor of apoptosis 1 /2 (cIAP1/2), and linear ubiquitin chain assembly complex (LUBAC) [47]. Within complex I, cIAP1/2-induced RIPK1 ubiquitination is upstream of nuclear factor-κB (NF-κB) nuclear translocation and is followed by increased expression of a series of pro-inflammatory cytokines and products of anti-apoptotic endogenous proteins, such as cellular FLICE-like inhibitory protein (cFLIP) and cIAP1/2 [48-51].
Overview of the molecular pathways involved in necroptosis. Ligation of tumor necrosis factor receptor 1 (TNFR) by TNF-α rapidly leads to the formation of complex I, which is made up of TNFR, TNF-associated death domain (TRADD), TNFR-associated factor 2 (TRAF2), receptor interacting protein kinases 1 (RIPK1), cellular inhibitor of apoptosis 1/2 (cIAP1/2), and linear ubiquitin chain assembly complex (LUBAC). Ubiquitin linkages on RIPK1 are decorated by cIAP1/2, followed by NF-κB activation and upregulation of anti-apoptotic proteins (a). Once NF-κB is blocked by Smac mimetic, complex I shifts into cytoplasmic complex II, which is comprised of TRADD, TRAF2, RIPK1, Fas associated via death domain (FADD), caspase 8 (CASP8), and cellular FLICE-like inhibitory protein (cFLIP). The cell then moves towards apoptosis (b). If CASP8 is compromised by z-VAD-fmk, then RIPK1 and receptor interacting protein kinases 3 (RIPK3) are activated and form a necrosome, facilitating necroptosis (c). In some contexts, the necroptosis executioner mixed lineage kinase domain-like protein (MLKL) activates peptidylarginine deiminase 4 (PAD4)-dependent NETosis (d). Although there is no direct association between necroptosis and pyroptosis, MLKL can activate pyrin domain-containing 3 (NLRP3)-CASP1 inflammasomes. MLKL causes K+ efflux, and then NLRP3 inflammasomes are activated by high-low intracellular K+ levels (e). Active MLKL inhibits autophagy flux through mammalian target of rapamycin (mTOR)-dependent signaling or by directly suppressing the formation of autophagosomes (f). Abbreviations: CASP3, caspase 3; CASP1, caspase 1.
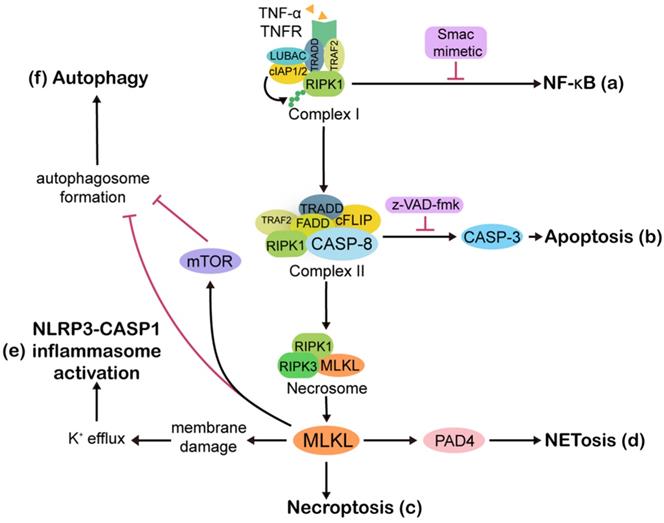
When the complex I-driven NF-κB signaling is blocked by Smac mimetic, cytoplasmic complex II, which consists of TRADD, TRAF2, RIPK1, Fas associated via death domain (FADD), CASP8, and cFLIP, exerts pro-apoptotic functions through CASP8 dimerization and activation [52]. If CASP8 or its adaptor protein FADD is compromised, RIPK1 and receptor interacting protein kinases 3 (RIPK3) activation will lead to amyloid necrosomes formation, followed by MLKL oligomerization and translocation to the cell membrane, thus steering cell death from apoptosis towards necroptosis [9, 53, 54]. Treating cells with a mixture composed of TNF-α, a Smac mimetic that degrades cIAPs, and the caspase inhibitor z-VAD-fmk is one of the most common methods to induce necroptosis in in vitro experiments [55, 56].
Furthermore, necroptosis can be triggered by intricate pathways. For example, viral RNAs can be sensed by DNA-dependent activator of interferon regulatory factors (DAI), and this elicits necroptosis through an interaction between DAI and RIPK3 [57]. Similarly, Toll-like receptor 3 (TLR3) and Toll-like receptor 4 (TLR4) detect viral RNAs and lipopolysaccharide (LPS) from bacterium, respectively, and this is followed by activation of RIPK3 via the TIR domain-containing adapter-inducing interferon-β (TRIF) [58]. Whatever the upstream signaling may be, it is clear that activation of RIPK3 and phosphorylation of MLKL are central events in necroptosis [59-61].
MLKL functions beyond necroptosis
In the past decade, MLKL has been considered perhaps the only substrate of RIPK3 that can effectively execute necroptosis. Hence, it may represent a molecular target for anti-inflammation and cancer therapies. More recently, its functions other than necroptosis have been gradually recognized. Neglecting the novel functions of MLKL could potentially lead to serious side-effects if MLKL inhibitors are used for the treatment of inflammation or cancer in humans. In addition, because MLKL is involved in many types of diseases (Table 1), a better of understanding of all the functions of MLKL will yield insights for developing control strategies in numerous other physiological and pathological processes.
Reported non-necroptosis functions of MLKL and its role in several specific diseases
| Disease | Cell type | RIPK3 activation | MLKL phosphorylation | MLKL oligomerization | Phenotype | Function | Reference |
|---|---|---|---|---|---|---|---|
| Type 2 diabetes | Hepatocyte | + | + | + | Insulin resistance | Inhibiting PIP3 production | [86] |
| Non-alcoholic fatty liver disease | Hepatocyte | - | + | + | Hepatocellular injury | Suppressing autophagic flux | [87] |
| Listeria monocytogenes infection | Intestine epithelial cell | + | + | - | Slow-multiplying bacterial states of Listeria in epithelial cells | Impinging on bacterium to disrupt their plasma membranes | [11] |
| Multiple sclerosis | Oligodendrocyte | - | + | Unknown | Increased demyelination | Unknown | [13] |
| Nerve regeneration | Schwann cell | - | + | Unknown | Increased demyelination | Loss of the myelin structure maintenance function of sulfatide | [12] |
| Nasopharyngeal carcinoma | Radioresistant nasopharyngeal cell | - | - | Unknown | Irradiation-induced invasion | Promoting epithelial-mesenchymal transition | [14] |
| Loss of the pregnancy | Syncytiotrophoblast | - | - | Unknown | Release of dangerous proteins | Facilitating endosomal trafficking and generation of extracellular vesicles | [94] |
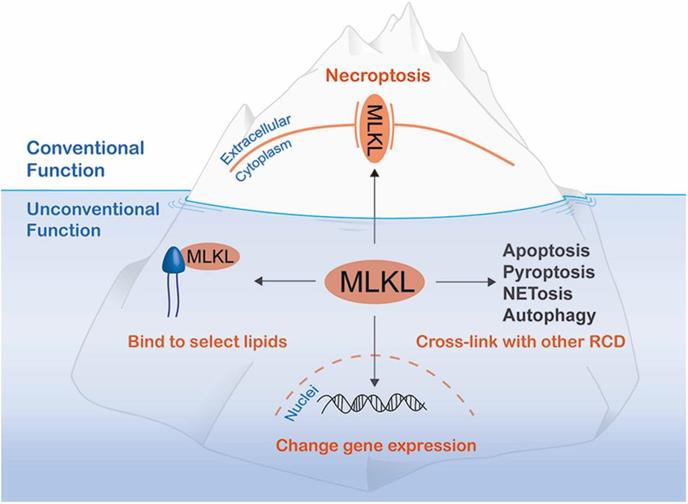
To our knowledge, functions of MLKL beyond serving as the executioner of necroptosis can be attributed to the following three aspects: interaction with other RCD pathways; regulation of gene expression; and combining with lipids to block specific processes (Figure 3). Of note, interaction with other RCD pathways often requires necroptosis. However, the other functions of MLKL can be realized without the risk of necrotic death.
Additional MLKL functions below sea level. In addition to its well-known function as a molecule that disrupts plasma membrane integrity to exert necroptosis, MLKL has non-canonical functions that can be divided into the three categories: cross-linking with other regulated cell death (RCD) mediators, translocating to the nucleus to alter gene expression and binding to select lipids to damage bacteria or inhibit a specific metabolic process.
Non-necroptosis functions of MLKL accompanying necroptosis
Necroptosis does not operate in isolation as is often described in schematic models. In fact, it might affect other RCD or cell death-related pathways, and other RCD pathways also regulate necroptosis [62]. This cross-linkages among cell death pathways not only indicates the complexity of RCD but also shows that MLKL participates in many pathways in addition to necroptosis activation (Figure 2).
Cross-linkages with other RCD
With respect to the cross-linkages among RCD, the linkage between the above-mentioned apoptosis and necroptosis has been most broadly studied, but many other linkages are implicated. In contrast to the opposing relationship between apoptosis and necroptosis, the relationship between necroptosis and other RCD prefers to be synergistic. For example, necroptosis is the basis of NETosis in some situations. NETosis is a type of cell death characterized by neutrophil extracellular trap (NET) formation. Phorbol-12-myristate-13-acetate (PMA) can stimulate NET generation independently of RIPK3 and MLKL signaling [63, 64]. In contrast, upon antineutrophil cytoplasmic antibody stimulation or respiratory syncytial virus infection, NET formation occurs via necroptosis [65, 66]. Necroptotic NET formation requires the activation of peptidylarginine deiminase 4 (PAD4) [64, 66], and necroptotic NETs located on the membrane are surrounded by MLKL [64]. Similar to necroptosis, pyroptosis is also a form of lytic cell death that is triggered by nucleotide-binding oligomerization domain (NOD)-like receptor protein 3 (NLRP3) inflammasome activation and gasdermin D (GSDMD) cleavage. No direct connection has been reported between necroptosis and pyroptosis. However, MLKL can activate NLRP3-caspase 1 (CASP1) inflammasome and interleukin (IL)-1β secretion as a consequence of necroptosis signaling, but before cell lysis. In this case, GSDMD, the pore-forming CASP1 substrate, is dispensable for IL-1β release [67]. However, another study found that IL-1β is secreted simultaneously from the dying cells. Further, a MLKL deficiency can reduce the LPS-induced release of IL-1β from LPS-stimulated A20-deficient macrophages. An in vivo study reported that necroptosis contributed to inflammasome-associated arthritis and this effect could be inhibited by zinc finger 7 of A20 [68]. These findings hinted at the possibility that different RCD can co-occur in some cases. And it is difficult to clearly define which type of cell death is predominant in these cases. Regardless, the expression of MLKL and the subsequent cell lysis are key determinants in the co-occurrence of the RCD. Therefore, targeting MLKL might constitute a new approach for therapeutic intervention of relevant diseases such as arthritis.
Cross-linkages with cell death-related pathways
Moreover, necroptosis can inhibit pro-survival signaling pathways to promote cell death. Autophagy is a lysosomal digestion and recycling process for cellular contents that promotes cell survival [69, 70]. Macroautophagy, the best-characterized form of autophagy, requires the fusion of an autophagosome and lysosome to form an autolysosome. However, necroptosis might inhibit autolysosomal function by compromising lysosomal membrane integrity, subsequently blocking autophagy flux [71]. In this context, neither AMP-activated protein kinase (AMPK; autophagy activator) nor the mammalian target of rapamycin (mTOR; autophagy inhibitor) activity is affected by necroptosis. However, when cells are exposed to oxidized low-density lipoprotein, overexpressed MLKL can inhibit autophagy mainly via activation of the mTOR-dependent signaling pathway [72]. Thus, necroptotic-mediated regulation of autophagy may vary, depending on the stimulus applied.
Necroptosis-independent functions of MLKL
Regulation of gene expression
Cytoplasmic membrane translocation of cytoplasmic MLKL is necessary for the induction of necroptosis. In addition to cytoplasmic membrane translocation, Yoon et al. found that MLKL also translocates to the nucleus before cell death in necroptosis. Three-dimensional analysis of immunocytochemistry showed that the translocated MLKL exists within the nuclei instead of associating with the nuclear membrane. Of note, the deadly effector function of MLKL is independent of the translocation, and the pharmacological blocking of necroptosis by necrosulfonamide (NSA) has no effect on the nuclear translocation of MLKL [73]. Therefore, the nuclear translocation of MLKL might serve some novel functions instead of inducing necroptosis. Given that MLKL does not have a direct DNA or RNA binding domain, therefore MLKL might not directly regulate gene expression. It needs to cooperate with other molecules to exert its regulatory functions. A recent study revealed that MLKL can interact with RNA-binding motif protein 6 (RBM6) to promote expression of adhesion molecules intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and E-selectin in endothelial cells. Additionally, nuclear translocation of the MLKL-RBM6 complex promotes expression of these molecules by increasing mRNA stability [74]. This study confirmed the hypothesis that MLKL can influence gene expression in the nucleus.
Inducing apoptosis is considered to be a promising approach to cancer treatment. Chelerythrine (CHE), a natural benzo[c]phenanthridine alkaloid, can induce apoptosis in tumor cells by promoting ROS production [75, 76]. With ROS generation, MLKL translocates from the cytoplasm to the nuclei, facilitating CHE-promoted apoptosis [76]. In this case, neither oligomerization nor membrane translocation of MLKL occurs after treatment with CHE. These findings show that MLKL, besides mediating necroptosis, may also contribute to apoptosis via nuclear translocation. Further studies are needed to reveal the nuclear function of MLKL and confirm the necessity of MLKL nuclear translocation in apoptosis.
Additionally, another study reported that depletion of MLKL severely compromises invasion of the nasopharyngeal carcinoma cells accompanied by the reverse of epithelial-mesenchymal transition (EMT). Although the specific mechanism is not clear, it should be noted that MLKL depletion leads to downregulation of 8 hub genes (VIM, EGFR, JUN, CD44, SPP1, FGF13, PLAU, and MMP1) and upregulation of 2 hub genes (CHD1 and MMP9) [14]. However, another study found no difference in lung metastasis when wild type or MLKL knockout breast cancer cells (MVT-1) were injected intravenously [77]. This means that MLKL might function in tumor metastasis via regulation of gene expression only in specific tumor cell types. Such cell specificity is not surprising given that tumor metastasis is regulated by numerous proteins [78-80]. In particular, the determination of kinases or proteins that activate this non-necroptotic aids in elucidating the cell type-specific manner.
Many cancer cells generally downregulate the expression of necroptotic factors to evade necroptosis and survive [81-83]. This suggests that low expression of MLKL may contribute to both tumor survival and regulate gene expression, thus promoting metastasis. Whereas, if MLKL expression is upregulated dramatically, necroptosis may also be induced in cancer cells, which results in cell death. For example, shikonin, a Chinese herbal medicine extract, can trigger necroptosis in nasopharyngeal carcinoma cells via upregulation of MLKL expression [84]. Also of note, the inflammatory response can be irritated by necroptosis in the body, which may provide a microenvironment suitable for cancer progression [85]. Thus, MLKL is a potential target for anti-tumor treatments but upregulation of MLKL is not a one-size-fits-all approach to cancer.
Blocking specific processes via the affinity of MLKL for select phospholipids and cardiolipin
Similar to perforation in necroptosis, MLKL may exert its necroptosis-independent functions via affinity for select molecules, such as phosphatidylinositol, cardiolipin, sulfatide, and so on. For example, p-MLKL can bind to PI(4,5)P2 to inhibit insulin-stimulated PI(3,4,5)P3 production at the plasma membrane in hepatocytes [86]. This process leads to the pathophysiology of hepatic insulin resistance and type 2 diabetes. Intriguingly, MLKL does not affect hepatic inflammation and liver cell death under obese conditions. Murine models of non-alcoholic fatty liver disease provide another example that MLKL translocates to autophagosomes, a spherical structure with double-layer membranes, prior to reaching the plasma membrane and blocks the autophagic flux resulting in liver injury [87]. The necroptosis-independent functions of MLKL seem to rely on the interactions between 4HB and other molecules.
Necroptosis plays a positive role in at least two ways. First, it limits pathogen spread by preventing the host cell from becoming a pathogen factory, and second, it galvanizes the adaptive immune response via the release of debris into the extracellular space [88]. Both of these pathways involve cell destruction, leading to cell death. However, a recently study found that infection by Listeria monocytogenes, a gram-positive enteropathogenic bacteria, can activate the RIPK3-MLKL pathway in the infected cell without causing necroptosis [11]. Additionally, the monomer p-MLKL can bind to cardiolipin of Listeria, which is immediately followed by decreased Listeria replication. Of note, cardiolipin is more abundant on the Listeria cell membrane compared with the outer membranes of gram-negative bacteria, which perhaps explains why p-MLKL does not associate with Salmonella typhimurium [11]. Intriguingly, although MLKL can be activated by RIPK3 in Listeria-infected intestine epithelial cells, no MLKL oligomer formation and membranal translocation occurs in these cells [11]. Considering the fact that pharmacologic or genetic targeting of TAM kinases has no impact on the membranal translocation of MLKL, other mechanisms, apart from inhibition of TAM kinase, must be responsible for this phenomenon [28].
As for the MLKL-sulfatide interaction, it plays an important role in demyelination, not only in the central nervous system (CNS), but also in the peripheral nervous system (PNS). Sulfatide is crucial to the maintenance of an intact myelin structure [89]. The myelin sheath is a lipid-rich substance formed by oligodendrocytes in the CNS and by Schwann cells in the PNS [90]. Multi-layered specialized plasma membranes protect the axons and speed the transmission of electrical impulses along myelinated axons. In the CNS, multiple sclerosis (MS), as a demyelination disease, is coordinated by necroptosis-independent MLKL [13]. Lack of MLKL protein delays chemical-induced (cuprizone) demyelination of MS, whereas, intriguingly, a recent study in two siblings reported that a variant in MLKL led to a deficiency of MLKL resulting in a disorder that shares clinical features with primary progressive MS and other neurodegenerative diseases [91]. In the PNS, successful regeneration of myelinated nerves requires the breakdown and clearance of myelin. A study showed that injury-induced phosphorylation of serine 441 in mouse MLKL targets the myelin sheath membrane of Schwann cells to promote demyelination and regeneration of axons [12]. Because the necroptosis- and nerve injury-associated phosphorylation pathways represent non-overlapping pathways, it is clear that different functions of MLKL result from different phosphorylation sites via different upstream kinases.
Existing evidence shows that MLKL and/or p-MLKL can be released within the extracellular vesicles (EVs) [92, 93], suggesting that the above-mentioned functions of MLKL may be regulated by endosomal trafficking and generation of EVs. This release appears to inhibit cell death. For example, syncytiotrophoblasts release EVs that contain Hsp 70, misfolded proteins, and MLKL, to eliminate intracellular increased toxins [94]. However, these dangerous/toxic EVs can lead to maternal endothelial cell activation, causing preeclampsia. Interestingly, MLKL is involved in endosomal trafficking and the generation of small EVs by interaction with endosomal sorting complexes required for transport (ESCRT) proteins and flotillins [92]. MLKL facilitates the generation of EVs, and this function is independent of phosphorylation but can be enhanced by it. MLKL also contributes to direct budding of the damaged plasma membranes, which results in the generation of phosphatidylserine positive and p-MLKL containing EVs [93]. This process also depends on the function of ESCRT-III activity. ESCRT-III components can sustain the survival of cells when MLKL activation is subsequently halted.
The current evidence from these studies suggests both the C-terminal pseudokinase domain and the N-terminal 4HB domain are the bases of non-necroptosis functions of MLKL. On the one hand, the nuclear localization signal motif-containing pseudokinase domain of MLKL is required for nuclear shuttling [73], but on the other hand, clear evidence indicates that 4HB is a required region of MLKL in necroptosis [95]. Currently, most studies focusing on necroptosis-independent functions of MLKL suggest that these functions are also dependent on MLKL to phosphatidylinositol, cardiolipin, sulfatide, and so on. However, the reasons for MLKL translocation to specific structures instead of the plasma membrane have not yet been clarified. Furthermore, these studies also raise other fascinating questions as do all other interesting findings [11, 12, 87]. For example, why does p-MLKL not oligomerize and damage the membrane structure in some contexts? Apart from RIPK3, what other proteins can phosphorylate MLKL? Apart from the known phosphorylation sites, are there several other phosphorylation sites for which the functional role is unclear? Furthermore, at present, no study has confirmed the regulation mode of MLKL in the same way as has been achieved in other pseudokinase situations. More experiments are therefore needed to investigate whether MLKL can serve as a mediator through the regulation of specific kinase function [96].
MLKL inhibitors
Increasing evidence shows that necroptosis contributes to the pathology of many severe diseases [97], especially inflammation and cancer, and numerous researchers have attempted to develop drugs to block MLKL function. A full understanding of the mechanism of action of MLKL inhibitors may be helpful in devising more potent drug therapies. And more importantly, it might also be speculated whether these currently available agents can inhibit non-necroptotic functions of MLKL. The first well-defined MLKL inhibitor is compound 1. Compound 1 (an ATP mimetic also named GW806742X) interacts with the nucleotide-binding site in the mouse MLKL pseudokinase domain, to exposure the MLKL activation loop. This interaction inhibits necroptosis by suppressing MLKL conformational change [95]. Of note, compound 1 can affect cell viability at high concentrations and may have off-target activity against RIPK1, RIPK3, and vascular endothelial growth factor receptor 2 [95, 98, 99]. A later study showed that the binding of compound 1 to the ATP-pocket of MLKL could not rescue cells from necroptosis, and the effect of compound 1 on necroptosis might be due to its non-specific binding to RIPK1 and other kinases [100]. Thus, considering the fact that different kinases activate different functions of MLKL, compound 1, as a potent pharmacological inhibitor of necroptosis, might be unable to completely inhibit the non-necroptotic function of MLKL. Unlike compound 1 described above, necroptosis-blocking compound 1 (NBC1) is another small molecule that conjugates with Hsp70 to inhibit MLKL polymerization, instead of directly acting on MLKL [42]. Up to date, no study has yet reported the effects of Hsp (e.g., Hsp70 and Hsp90) on non-necroptotic functions of MLKL. Therefore, it is difficult to speculate that whether NBC1 can act on the non-necroptotic functions.
To date, NSA is the most widely used MLKL inhibitor in experimental research. NSA can directly target the Cys86 residue in the N-terminal domain and block necroptosis in human cells [20, 25, 101]. Furthermore, NSA can cross-link Cys86 of human MLKL to Cys32 of thioredoxin-1, which then suppresses necroptosis by inhibiting disulfide bond formation between monomeric MLKL [102]. In addition to NSA, compound TC13172 is another MLKL inhibitor that induces covalent binding at Cys86 of MLKL [99]. Nevertheless, NSA and TC13172 do not inhibit necroptosis of mouse cells, because position 86 in mouse MLKL is a tryptophan residue, instead of a cysteine residue [20, 99]. This precludes the use of these inhibitors in pre-clinical mouse models. Recently, Petrie et al. developed inhibitory monobodies, synthetic binding proteins, that potently block necroptosis [103]. Both NSA and inhibitory monobodies only block MLKL translocation to membranes, but do not prevent human MLKL phosphorylation or oligomerization [36, 103, 104]. As we stated earlier, MLKL is to elicit non-necroptotic functions by employing a cytomembrane translocation-independent mechanism in most cases. The increase in intracellular MLKL may facilitate the regulation of gene expression and the affinity of MLKL for select phospholipids and cardiolipin to some extent. Therefore, rather than contributing to inhibit non-necroptotic functions, MLKL inhibitors, which aim to halt necroptosis, may instead promote these functions.
Even though existing MLKL inhibitors can effectively inhibit necroptosis, their inhibitory abilities for most of the non-necroptotic functions remain undetermined. The investigation of currently available MLKL inhibitors will be crucial to determining their effectiveness in regulating non-necroptotic functions. Besides, the functional complexity of MLKL requires further development of novel inhibitors. There are several approaches that could be adopted to inhibit non-necroptotic functions of MLKL: (a) blocking MLKL activation; (b) occupying MLKL-binding sites on phosphatidylinositol and cardiolipin; or (c) inhibiting intracellular movement of activated MLKL. Considering the beneficial role of MLKL in certain cases, future experiments are also needed to develop an inhibitor of necroptosis that does not compromise the beneficial functions of MLKL.
Conclusions
Most studies of MLKL to date have been restricted to necroptosis, and its non-necroptotic functions have received scant attention. The current review shows that MLKL is integrated into a network of pleiotropic functions. As a consequence, how exactly MLKL exerts its non-necroptotic functions is not yet clear. However, based on the existing literature, we can speculate that some of the non-necroptotic functions of MLKL, are also dependent upon its affinity for specific membrane structures. Likewise, MLKL can regulate other RCD pathways and even the expression of some genes. We are optimistic that further studies will provide insight avoiding the side effects of MLKL inhibitors in future applications and for controlling diseases through regulation of non-necroptotic functions of MLKL.
Abbreviations
MLKL: mixed lineage kinase domain-like protein; ACD: accidental cell death; RCD: regulated cell death; CASP 8: caspase 8; 4HB: four-helix bundle; TAM kinase: Tyro3, Axl, and Mer kinase; PIPs: phosphatidylinositol phosphates; Hsp90: heat shock protein 90; Hsp70: heat shock protein 70; Bcl-2: B-cell lymphoma 2; BH: Bcl-2 homology; DAMPs: damage-associated molecular patterns; TNFR: tumor necrosis factor receptor; TRADD: TNF-associated death domain; TRAF2: TNFR-associated factor 2; RIPK1: receptor-interacting protein kinases 1; cIAP1/2: cellular inhibitor of apoptosis 1 /2; LUBAC: linear ubiquitin chain assembly complex; NF-κB: nuclear factor-κB; cFLIP: cellular FLICE-like inhibitory protein; FADD: Fas associated via death domain; RIPK3: receptor interacting protein kinases 3; DAI: DNA-dependent activator of interferon regulatory factors; TLR3: Toll-like receptor 3; TLR4: Toll-like receptor 4; TRIF: TIR domain-containing adapter-inducing interferon-β; NET: neutrophil extracellular trap; PMA: phorbol-12-myristate-13-acetate; PAD4: peptidylarginine deiminase 4; NOD: nucleotide-binding oligomerization domain; NLRP3: NOD-like receptor protein 3; GSDMD: gasdermin D; CASP1: caspase 1; AMPK: AMP-activated protein kinase; mTOR: mammalian target of rapamycin; NSA: necrosulfonamide; RBM6: RNA-binding motif protein 6; CHE: chelerythrine; ROS: reactive oxygen species; EMT: epithelial-mesenchymal transition; p-MLKL: phosphorylated MLKL; CNS: central nervous system; PNS: peripheral nervous system; MS: multiple sclerosis; EVs: extracellular vesicles; ESCRT: endosomal sorting complexes required for transport; NBC1: necroptosis-blocking compound 1.
Acknowledgements
This work was supported by the Natural Science Foundation of China (Grant No. 81500870, 81971902).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361:1570-83
2. Lockshin RA, Zakeri Z. Cell death in health and disease. J Cell Mol Med. 2007;11:1214-24
3. Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D. et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. 2015;22:58-73
4. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239-57
5. Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nat Rev Immunol. 2017;17:151-64
6. Choi ME, Price DR, Ryter SW, Choi AMK. Necroptosis: a crucial pathogenic mediator of human disease. JCI Insight. 2019;4:e128834
7. Dillon CP, Oberst A, Weinlich R, Janke LJ, Kang T-B, Ben-Moshe T. et al. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep. 2012;1:401-7
8. Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C. et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363-7
9. Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y. et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 2013;23:994-1006
10. Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang J-G, Alvarez-Diaz S. et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39:443-53
11. Sai K, Parsons C, House JS, Kathariou S, Ninomiya-Tsuji J. Necroptosis mediators RIPK3 and MLKL suppress intracellular replication independently of host cell killing. J Cell Biol. 2019;218:1994-2005
12. Ying Z, Pan C, Shao T, Liu L, Li L, Guo D. et al. Mixed Lineage Kinase Domain-like Protein MLKL Breaks Down Myelin following Nerve Injury. Mol Cell. 2018;72:457-68.e5
13. Zhang S, Su Y, Ying Z, Guo D, Pan C, Guo J. et al. RIP1 kinase inhibitor halts the progression of an immune-induced demyelination disease at the stage of monocyte elevation. Proc Natl Acad Sci USA. 2019;116:5675-80
14. Dong Y, Sun Y, Huang Y, Fang X, Sun P, Dwarakanath B. et al. Depletion of MLKL inhibits invasion of radioresistant nasopharyngeal carcinoma cells by suppressing epithelial-mesenchymal transition. Ann Transl Med. 2019;7:741
15. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912-34
16. Czabotar PE, Murphy JM. A tale of two domains - a structural perspective of the pseudokinase, MLKL. FEBS J. 2015;282:4268-78
17. Kung JE, Jura N. Prospects for pharmacological targeting of pseudokinases. Nat Rev Drug Discov. 2019;18:501-26
18. Su L, Quade B, Wang H, Sun L, Wang X, Rizo J. A plug release mechanism for membrane permeation by MLKL. Structure. 2014;22:1489-500
19. Rodriguez DA, Weinlich R, Brown S, Guy C, Fitzgerald P, Dillon CP. et al. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 2016;23:76-88
20. Sun L, Wang H, Wang Z, He S, Chen S, Liao D. et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213-27
21. Petrie EJ, Sandow JJ, Jacobsen AV, Smith BJ, Griffin MDW, Lucet IS. et al. Conformational switching of the pseudokinase domain promotes human MLKL tetramerization and cell death by necroptosis. Nat Commun. 2018;9:2422
22. Tanzer MC, Matti I, Hildebrand JM, Young SN, Wardak A, Tripaydonis A. et al. Evolutionary divergence of the necroptosis effector MLKL. Cell Death Differ. 2016;23:1185-97
23. Tanzer MC, Tripaydonis A, Webb AI, Young SN, Varghese LN, Hall C. et al. Necroptosis signalling is tuned by phosphorylation of MLKL residues outside the pseudokinase domain activation loop. Biochem J. 2015;471:255-65
24. Davies KA, Tanzer MC, Griffin MDW, Mok YF, Young SN, Qin R. et al. The brace helices of MLKL mediate interdomain communication and oligomerisation to regulate cell death by necroptosis. Cell Death Differ. 2018;25:1567-80
25. Liu S, Liu H, Johnston A, Hanna-Addams S, Reynoso E, Xiang Y. et al. MLKL forms disulfide bond-dependent amyloid-like polymers to induce necroptosis. Proc Natl Acad Sci USA. 2017;114:E7450-E9
26. Chen X, Li W, Ren J, Huang D, He WT, Song Y. et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014;24:105-21
27. Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J. et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16:55-65
28. Najafov A, Mookhtiar AK, Luu HS, Ordureau A, Pan H, Amin PP. et al. TAM Kinases Promote Necroptosis by Regulating Oligomerization of MLKL. Mol Cell. 2019;75:457-68.e4
29. Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I. et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014;7:971-81
30. Dovey CM, Diep J, Clarke BP, Hale AT, McNamara DE, Guo H. et al. MLKL Requires the Inositol Phosphate Code to Execute Necroptosis. Mol Cell. 2018;70:936-48.e7
31. McNamara DE, Dovey CM, Hale AT, Quarato G, Grace CR, Guibao CD. et al. Direct Activation of Human MLKL by a Select Repertoire of Inositol Phosphate Metabolites. Cell Chem Biol. 2019;26:863-77.e7
32. Fan W, Guo J, Gao B, Zhang W, Ling L, Xu T. et al. Flotillin-mediated endocytosis and ALIX-syntenin-1-mediated exocytosis protect the cell membrane from damage caused by necroptosis. Sci Signal. 2019;12:eaaw3423
33. Ros U, Peña-Blanco A, Hänggi K, Kunzendorf U, Krautwald S, Wong WW-L. et al. Necroptosis Execution Is Mediated by Plasma Membrane Nanopores Independent of Calcium. Cell Rep. 2017;19:175-87
34. Xia B, Fang S, Chen X, Hu H, Chen P, Wang H. et al. MLKL forms cation channels. Cell Res. 2016;26:517-28
35. Quarato G, Guy CS, Grace CR, Llambi F, Nourse A, Rodriguez DA. et al. Sequential Engagement of Distinct MLKL Phosphatidylinositol-Binding Sites Executes Necroptosis. Mol Cell. 2016;61:589-601
36. Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF. et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54:133-46
37. Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148:228-43
38. Vanlangenakker N, Vanden Berghe T, Bogaert P, Laukens B, Zobel K, Deshayes K. et al. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ. 2011;18:656-65
39. Vanden Berghe T, Vanlangenakker N, Parthoens E, Deckers W, Devos M, Festjens N. et al. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010;17:922-30
40. Tait SWG, Oberst A, Quarato G, Milasta S, Haller M, Wang R. et al. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep. 2013;5:878-85
41. Jacobsen AV, Lowes KN, Tanzer MC, Lucet IS, Hildebrand JM, Petrie EJ. et al. HSP90 activity is required for MLKL oligomerisation and membrane translocation and the induction of necroptotic cell death. Cell Death Dis. 2016;7:e2051
42. Johnston AN, Ma Y, Liu H, Liu S, Hanna-Addams S, Chen S. et al. Necroptosis-blocking compound NBC1 targets heat shock protein 70 to inhibit MLKL polymerization and necroptosis. Proc Natl Acad Sci USA. 2020;117:6521-30
43. Shi CS, Kehrl JH. Bcl-2 regulates pyroptosis and necroptosis by targeting BH3-like domains in GSDMD and MLKL. Cell Death Discov. 2019;5:151
44. Seo J, Seong D, Nam YW, Hwang CH, Lee SR, Lee CS. et al. Beclin 1 functions as a negative modulator of MLKL oligomerisation by integrating into the necrosome complex. Cell Death Differ. 2020;27:3065-3081
45. Kroemer G, Dallaporta B, Resche-Rigon M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol. 1998;60:619-42
46. Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209-23
47. Newton K, Manning G. Necroptosis and Inflammation. Annu Rev Biochem. 2016;85:743-63
48. Bertrand MJM, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J. et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. 2008;30:689-700
49. Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181-90
50. Horie R, Watanabe M, Okamura T, Taira M, Shoda M, Motoji T. et al. DHMEQ, a new NF-kappaB inhibitor, induces apoptosis and enhances fludarabine effects on chronic lymphocytic leukemia cells. Leukemia. 2006;20:800-6
51. Nakano H, Piao X, Shindo R, Komazawa-Sakon S. Cellular FLICE-Inhibitory Protein Regulates Tissue Homeostasis. Curr Top Microbiol Immunol. 2017;403:119-41
52. Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008;133:693-703
53. Zhang D-W, Shao J, Lin J, Zhang N, Lu B-J, Lin S-C. et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332-6
54. Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao Y-S. et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150:339-50
55. Li L, Thomas RM, Suzuki H, De Brabander JK, Wang X, Harran PG. A small molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death. Science. 2004;305:1471-4
56. Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148:228-43
57. Upton JW, Kaiser WJ. DAI Another Way: Necroptotic Control of Viral Infection. Cell Host Microbe. 2017;21:290-3
58. Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ. et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288:31268-79
59. Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J. et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci U S A. 2012;109:5322-7
60. He S, Wang L, Miao L, Wang T, Du F, Zhao L. et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100-11
61. Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M. et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112-23
62. Schwarzer R, Laurien L, Pasparakis M. New insights into the regulation of apoptosis, necroptosis, and pyroptosis by receptor interacting protein kinase 1 and caspase-8. Curr Opin Cell Biol. 2020;63:186-93
63. Amini P, Stojkov D, Wang X, Wicki S, Kaufmann T, Wong WW-L. et al. NET formation can occur independently of RIPK3 and MLKL signaling. Eur J Immunol. 2016;46:178-84
64. D'Cruz AA, Speir M, Bliss-Moreau M, Dietrich S, Wang S, Chen AA. et al. The pseudokinase MLKL activates PAD4-dependent NET formation in necroptotic neutrophils. Sci Signal. 2018 11
65. Schreiber A, Rousselle A, Becker JU, von Mässenhausen A, Linkermann A, Kettritz R. Necroptosis controls NET generation and mediates complement activation, endothelial damage, and autoimmune vasculitis. Proc Natl Acad Sci USA. 2017;114:E9618-E25
66. Muraro SP, De Souza GF, Gallo SW, Da Silva BK, De Oliveira SD, Vinolo MAR. et al. Respiratory Syncytial Virus induces the classical ROS-dependent NETosis through PAD-4 and necroptosis pathways activation. Sci Rep. 2018;8:14166
67. Conos SA, Chen KW, De Nardo D, Hara H, Whitehead L, Núñez G. et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc Natl Acad Sci USA. 2017;114:E961-E9
68. Polykratis A, Martens A, Eren RO, Shirasaki Y, Yamagishi M, Yamaguchi Y. et al. A20 prevents inflammasome-dependent arthritis by inhibiting macrophage necroptosis through its ZnF7 ubiquitin-binding domain. Nat Cell Biol. 2019;21:731-42
69. Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F. et al. Molecular definitions of autophagy and related processes. EMBO J. 2017;36:1811-36
70. Radonjic-Hoesli S, Wang X, de Graauw E, Stoeckle C, Styp-Rekowska B, Hlushchuk R. et al. Adhesion-induced eosinophil cytolysis requires the receptor-interacting protein kinase 3 (RIPK3)-mixed lineage kinase-like (MLKL) signaling pathway, which is counterregulated by autophagy. J Allergy Clin Immunol. 2017;140:1632-42
71. Frank D, Vaux DL, Murphy JM, Vince JE, Lindqvist LM. Activated MLKL attenuates autophagy following its translocation to intracellular membranes. J Cell Sci. 2019;132:jcs220996
72. Guo F-X, Wu Q, Li P, Zheng L, Ye S, Dai X-Y. et al. The role of the LncRNA-FA2H-2-MLKL pathway in atherosclerosis by regulation of autophagy flux and inflammation through mTOR-dependent signaling. Cell Death Differ. 2019;26:1670-87
73. Yoon S, Bogdanov K, Kovalenko A, Wallach D. Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ. 2016;23:253-60
74. Dai J, Zhang C, Guo L, He H, Jiang K, Huang Y. et al. A necroptotic-independent function of MLKL in regulating endothelial cell adhesion molecule expression. Cell Death Dis. 2020;11:282
75. Tang Z-H, Cao W-X, Wang Z-Y, Lu J-H, Liu B, Chen X. et al. Induction of reactive oxygen species-stimulated distinctive autophagy by chelerythrine in non-small cell lung cancer cells. Redox Biol. 2017;12:367-76
76. Cao W-X, Li T, Tang Z-H, Zhang L-L, Wang Z-Y, Guo X. et al. MLKL mediates apoptosis via a mutual regulation with PERK/eIF2α pathway in response to reactive oxygen species generation. Apoptosis. 2018;23:521-31
77. Jiao D, Cai Z, Choksi S, Ma D, Choe M, Kwon H-J. et al. Necroptosis of tumor cells leads to tumor necrosis and promotes tumor metastasis. Cell Res. 2018;28:868-70
78. Alaseem A, Alhazzani K, Dondapati P, Alobid S, Bishayee A, Rathinavelu A. Matrix Metalloproteinases: A challenging paradigm of cancer management. Semin Cancer Biol. 2019;56:100-15
79. Liu J, Zhang Y, Li Q, Wang Y. Transgelins: Cytoskeletal Associated Proteins Implicated in the Metastasis of Colorectal Cancer. Front Cell Dev Biology. 2020;8:573859
80. Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119:1429-37
81. Raden Y, Shlomovitz I, Gerlic M. Necroptotic extracellular vesicles - present and future. Semin Cell Dev Biol. 2020 [Epub ahead of print]
82. Li S, Zhang T, Xu W, Ding J, Yin F, Xu J. et al. Sarcoma-Targeting Peptide-Decorated Polypeptide Nanogel Intracellularly Delivers Shikonin for Upregulated Osteosarcoma Necroptosis and Diminished Pulmonary Metastasis. Theranostics. 2018;8:1361-75
83. Shi F, Zhou M, Shang L, Du Q, Li Y, Xie L. et al. EBV(LMP1)-induced metabolic reprogramming inhibits necroptosis through the hypermethylation of the RIP3 promoter. Theranostics. 2019;9:2424-38
84. Liu T, Sun X, Cao Z. Shikonin-induced necroptosis in nasopharyngeal carcinoma cells via ROS overproduction and upregulation of RIPK1/RIPK3/MLKL expression. Onco Targets Ther. 2019;12:2605-14
85. Gong Y, Fan Z, Luo G, Yang C, Huang Q, Fan K. et al. The role of necroptosis in cancer biology and therapy. Mol Cancer. 2019;18:100
86. Xu H, Du X, Liu G, Huang S, Du W, Zou S. et al. The pseudokinase MLKL regulates hepatic insulin sensitivity independently of inflammation. Mol Metab. 2019;23:14-23
87. Wu X, Poulsen KL, Sanz-Garcia C, Huang E, McMullen MR, Roychowdhury S. et al. MLKL-dependent signaling regulates autophagic flux in a murine model of non-alcoholic fatty liver and steatohepatitis. J Hepatol. 2020;73:616-27
88. Upton JW, Shubina M, Balachandran S. RIPK3-driven cell death during virus infections. Immunol Rev. 2017;277:90-101
89. Marcus J, Honigbaum S, Shroff S, Honke K, Rosenbluth J, Dupree JL. Sulfatide is essential for the maintenance of CNS myelin and axon structure. Glia. 2006;53:372-81
90. Baumann N, Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev. 2001;81:871-927
91. Faergeman SL, Evans H, Attfield KE, Desel C, Kuttikkatte SB, Sommerlund M. et al. A novel neurodegenerative spectrum disorder in patients with MLKL deficiency. Cell Death Dis. 2020;11:303
92. Yoon S, Kovalenko A, Bogdanov K, Wallach D. MLKL, the Protein that Mediates Necroptosis, Also Regulates Endosomal Trafficking and Extracellular Vesicle Generation. Immunity. 2017;47:51-65
93. Gong Y-N, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P. et al. ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell. 2017;169:286-300
94. Tang Y, Chen Y, Nursalim Y, Groom K, Hickey A, Chamley L. et al. Endoplasmic reticulum stress occurs in association with the extrusion of toxic extracellular vesicles from human placentae treated with antiphospholipid antibodies. Clin Sci. 2020;134:459-72
95. Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P. et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci USA. 2014;111:15072-7
96. Reiterer V, Eyers PA, Farhan H. Day of the dead: pseudokinases and pseudophosphatases in physiology and disease. Trends Cell Biol. 2014;24:489-505
97. Liu ZY, Zheng M, Li YM, Fan XY, Wang JC, Li ZC. et al. RIP3 promotes colitis-associated colorectal cancer by controlling tumor cell proliferation and CXCL1-induced immune suppression. Theranostics. 2019;9:3659-73
98. Sammond DM, Nailor KE, Veal JM, Nolte RT, Wang L, Knick VB. et al. Discovery of a novel and potent series of dianilinopyrimidineurea and urea isostere inhibitors of VEGFR2 tyrosine kinase. Bioorg Med Chem Lett. 2005;15:3519-23
99. Yan B, Liu L, Huang S, Ren Y, Wang H, Yao Z. et al. Discovery of a new class of highly potent necroptosis inhibitors targeting the mixed lineage kinase domain-like protein. Chem Commun (Camb). 2017;53:3637-40
100. Ma B, Marcotte D, Paramasivam M, Michelsen K, Wang T, Bertolotti-Ciarlet A. et al. ATP-Competitive MLKL Binders Have No Functional Impact on Necroptosis. PLoS ONE. 2016;11:e0165983
101. Liao D, Sun L, Liu W, He S, Wang X, Lei X. Necrosulfonamide inhibits necroptosis by selectively targeting the mixed lineage kinase domain-like protein. Med Chem Commun. 2014;5:333-7
102. Reynoso E, Liu H, Li L, Yuan AL, Chen S, Wang Z. Thioredoxin-1 actively maintains the pseudokinase MLKL in a reduced state to suppress disulfide bond-dependent MLKL polymer formation and necroptosis. J Biol Chem. 2017;292:17514-24
103. Petrie EJ, Birkinshaw RW, Koide A, Denbaum E, Hildebrand JM, Garnish SE. et al. Identification of MLKL membrane translocation as a checkpoint in necroptotic cell death using Monobodies. Proc Natl Acad Sci U S A. 2020;117:8468-75
104. Murai S, Yamaguchi Y, Shirasaki Y, Yamagishi M, Shindo R, Hildebrand JM. et al. A FRET biosensor for necroptosis uncovers two different modes of the release of DAMPs. Nat Commun. 2018;9:4457
Author contact
![]() Corresponding authors: Jin Hou and Xiaojun Yang. Department of Stomatology, Nanfang Hospital, Southern Medical University, Guangzhou Da Dao Bei 1838, 510515, Guangzhou, P.R. China. E-mail address: houjinedu.cn (Jin Hou) or yxj_dencom (Xiaojun Yang)
Corresponding authors: Jin Hou and Xiaojun Yang. Department of Stomatology, Nanfang Hospital, Southern Medical University, Guangzhou Da Dao Bei 1838, 510515, Guangzhou, P.R. China. E-mail address: houjinedu.cn (Jin Hou) or yxj_dencom (Xiaojun Yang)