Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(6):2788-2805. doi:10.7150/thno.53330 This issue Cite
Research Paper
Lipin 1 deficiency causes adult-onset myasthenia with motor neuron dysfunction in humans and neuromuscular junction defects in zebrafish
Shuxian Lu1#, Zhaojie Lyu1#, Zhihao Wang1, Yao Kou1, Cong Liu1, Shengyue Li1, Mengyan Hu1, Hongjie Zhu1, Wenxing Wang1, Ce Zhang3, Yung-Shu Kuan4, Yi-Wen Liu5, Jianming Chen2,6 ![]() , Jing Tian1
, Jing Tian1 ![]()
1. Key laboratory of Resource Biology and Biotechnology in Western China, Ministry of Education, School of Medicine, Northwest University, Xi'an 710069, China.
2. Institute of Oceanography, Minjiang University, Fuzhou 350108, China.
3. State Key Laboratory of Photon-Technology in Western China Energy, Institute of Photonics and Photon-Technology, Northwest University, Xi'an 710069, China.
4. Institute of Biochemical Sciences, College of Life Science, National Taiwan University, Taipei, Taiwan.
5. Department of Life Science, Tunghai University, Taichung, Taiwan.
6. Sino-French Hoffmann Institute, School of Basic Medical Sciences, Guangzhou Medical University, Guangzhou 511436, China.
#These authors contributed equally to this work.
Received 2020-9-16; Accepted 2020-12-12; Published 2021-1-1
Abstract

Lipin 1 is an intracellular protein acting as a phosphatidic acid phosphohydrolase enzyme controlling lipid metabolism. Human recessive mutations in LPIN1 cause recurrent, early-onset myoglobinuria, a condition normally associated with muscle pain and weakness. Whether and how lipin 1 deficiency in humans leads to peripheral neuropathy is yet unclear. Herein, two novel compound heterozygous mutations in LPIN1 with neurological disorders, but no myoglobinuria were identified in an adult-onset syndromic myasthenia family. The present study sought to explore the pathogenic mechanism of LPIN1 in muscular and neural development.
Methods: The clinical diagnosis of the proband was compared to the known 48 cases of LPIN1 recessive homozygous mutations. Whole-exome sequencing was carried out on the syndromic myasthenia family to identify the causative gene. The pathogenesis of lipin 1 deficiency during somitogenesis and neurogenesis was investigated using the zebrafish model. Whole-mount in situ hybridization, immunohistochemistry, birefringence analysis, touch-evoke escape response and locomotion assays were performed to observe in vivo the changes in muscles and neurons. The conservatism of the molecular pathways regulated by lipin 1 was evaluated in human primary glioblastoma and mouse myoblast cells by siRNA knockdown, drug treatment, qRT-PCR and Western blotting analysis.
Results: The patient exhibited adult-onset myasthenia accompanied by muscle fiber atrophy and nerve demyelination without myoglobinuria. Two novel heterozygous mutations, c.2047A>C (p.I683L) and c.2201G>A (p.R734Q) in LPIN1, were identified in the family and predicted to alter the tertiary structure of LPIN1 protein. Lipin 1 deficiency in zebrafish embryos generated by lpin1 morpholino knockdown or human LPIN1 mutant mRNA injections reproduced the myotomes defects, a reduction both in primary motor neurons and secondary motor neurons projections, morphological changes of post-synaptic clusters of acetylcholine receptors, and myelination defects, which led to reduced touch-evoked response and abnormalities of swimming behaviors. Loss of lipin 1 function in zebrafish and mammalian cells also exhibited altered expression levels of muscle and neuron markers, as well as abnormally enhanced Notch signaling, which was partially rescued by the specific Notch pathway inhibitor DAPT.
Conclusions: These findings pointed out that the compound heterozygous mutations in human LPIN1 caused adult-onset syndromic myasthenia with peripheral neuropathy. Moreover, zebrafish could be used to model the neuromuscular phenotypes due to the lipin 1 deficiency, where a novel pathological role of over-activated Notch signaling was discovered and further confirmed in mammalian cell lines.
Keywords: LPIN1, syndromic myasthenia, zebrafish morphants, neuromuscular development, Notch signaling
Introduction
Muscle weakness, also known as myasthenia, is a symptom of muscular, neurological and metabolic disorders with a wide variety of mild to severe conditions that arise due to aging, infection, trauma, heart failure, diabetes, malignancy, autoimmune diseases and other abnormal processes. Muscle weakness can occur in any age group and various ethnic backgrounds [1-4]. Muscular dystrophy, inflammatory myopathy, neuromuscular disorders, motor neuron diseases and myasthenia gravis are the common genetic causes of muscle weakness [1-3, 5, 6]. The underlying molecular processes and genetic disorders that cause muscle weakness are still under intense study.
Lipin 1 (phosphatidic acid phosphatase 1, PAP1, EC 3.1.3.4) [7], encoded by LPIN1 gene, is a Mg2+-dependent phosphatidic acid phosphatase (PAP1) enzyme that controls lipid synthesis and the expression of genes involved in energy metabolism pathways [8, 9]. In the cytoplasm, lipin 1 protein acts as PAP1 enzyme converting phosphatidic acid (PA) to diacylglycerol (DAG) for the synthesis of triglycerides (TAG) and phospholipids; while translocated to the nucleus, lipin 1 functions as a transcriptional co-activator through direct interaction with transcription factors to regulate gene expressions [10, 11]. Lipin 1 is highly expressed in adipose tissue, skeletal muscle and is also present in other tissues including heart, liver, kidney, brain and peripheral nerve [12, 13], which indicates a complicated role for lipin 1 in development and homeostasis.
Autosomal recessive LPIN1 mutations in human have been identified as a major cause of recurrent, early-onset myoglobinuria and rhabdomyolysis (MIM#268200), a condition characterized by recurrent attacks of rhabdomyolysis due to damaged skeletal muscle, leading to the excretion of myocellular proteins (including creatine kinase and myoglobin) into the circulatory system. The clinical features are also accompanied by muscle weakness, myalgia, and occasional renal failure [4, 14]. The heterozygous mutations of LPIN1 might cause statin-induced myopathy [15]. Association studies between the metabolic syndrome and genetic variants of the LPIN1 gene revealed that LPIN1 polymorphisms contribute to several metabolic traits and obesity-related phenotypes which may differ among populations [16, 17]. Additional, functions of lipin 1 have also been revealed by a series of studies in mouse models. The whole-body deficiency of lipin 1 caused by spontaneous mutation in the fatty-liver dystrophic mouse (Lpin1fld/fld) was identified by progressive lipodystrophy [18], peripheral neuropathy and skeletal muscle myocyte damage [19, 20]. Furthermore, by a conditional knockout of Lpin1 in Schwann cells (SCs) in mouse (MPZcre+/LpfE2-3/fE2-3), Nadra et. al. [9] demonstrated that ablation of Lpin1 is sufficient to cause peripheral neuropathy; as well as endoneurial accumulation of phosphatidate in adipose tissues and peripheral nerves, which in turn lead to activation of MEK-Erk pathway and demyelination in SCs. Schweitzer et. al. [21] generated two muscle-specific LPIN1-deficienct mice models (MCK-Lpin1△115 and MCK-Lpin1-/-), and demonstrated that either selective loss of PAP activity or complete deletion of LPIN1 protein in skeletal muscles leads to chronic myopathy, myofibrillar degeneration, increased phosphatidic acid levels, mitochondrial dysfunction and impaired autophagy. These studies supported a critical role of lipin1, in addition to its biochemical functions in lipid synthesis and energy metabolism, in peripheral nerve and skeletal muscle development. However, whether and how lipin 1 mediates the formation, maintenance and function of neuromuscular junction (NMJ) remains unclear. Moreover, it remains elusive whether lipin 1 deficiency in human adults would cause peripheral neuropathy as in the mouse model.
In this study we identified two novel compound heterozygous variants, c.2047A>C (p.I683L) and c.2201G>A (p.R734Q) of LPIN1 from a syndromic myasthenia family. The two mutations were predicted to be damaging and disease-causing by in silico analysis. Unlike the reported human LPIN1 mutations which cause myoglobinuria and rhabdomyolysis mostly in early-onset pediatric form; the proband in this study presented adult-onset severe mixed peripheral neuropathy, neurogenic damage of skeletal muscle, and progressive muscle weakness, albeit without myoglobinuria or rhabdomyolysis. In order to explore the mechanisms underlying LPIN1 functions in muscle and neuron development and the potential signaling pathways involved, a zebrafish model of lipin 1 deficiency was established. By lpin1 morpholino knockdown, or overexpression of human point mutation LPIN1 mRNA, Lpin1-deficient zebrafish embryos exhibited myopathic phenotype with myotomes defects caused by myofibrillar disorganization, reduction of primary motor neurons (PMNs) and secondary motor neurons (SMNs) projections, morphological changes in the post-synapse, and myelination defects, leading to reduced touch-evoked response and mobility, which partially phenocopied human LPIN1 mutations and Lpin1 mouse models. Notably, we found that the Notch pathway was activated upon lipin 1 knockdown in zebrafish and mammalian cells, and the abnormally increased Notch signaling could be partially rescued by gamma-secretase inhibitor DAPT. These results indicated an essential and evolutionarily conserved role of lipin 1 in the neuromuscular system, and suggested a novel mechanism underlying the pathology of LPIN1-related neuromuscular disorders.
Materials and Methods
Clinical assessment
The study was carried out in accordance to the Declaration of Helsinki [22]. Written informed consent was obtained from all participants before their participation in this study. Individual II-5 of the family was initially diagnosed at the Peking University First Hospital in China. All human studies were approved by the Review Board of Northwest University.
Whole-exome sequencing
Genomic DNA was extracted from peripheral blood (II2, II3, II4, II5) and saliva samples (II1, II6, III1, III2, III3) by standard procedures. Whole-exome capture of subjects II2, II3, II4, II5 was performed using Agilent SureSelect XT Human All Exon v.4 kit, sequenced by 100nt paired-end reads on Illumina HiSeq2000 platform. The reads provided in FASTQ files were mapped to the human genome (hg19) using Burrows-Wheeler Aligner (BWA) [23]. Variants were called using SAMtools program and duplicated reads were marked by Picard [24]. All variants were screened with the SNP database version 142 (dbSNP142), 1000 Genomes Project (version 2014 October), and NHLBI Exome Sequencing Project (ESP) 6500. Functional prediction was carried out by Sorting Intolerant from Tolerant (SIFT), Polymorphism Phenotyping version 2 (PolyPhen-2), MutationTaster and MutationAssessor, and the remaining rare variants were annotated using ANNOVAR to separate non-synonymous substitutions, indel variants and splicing mutations. Further analysis was carried out by ModelR and Phenolyzer to screen the candidate variants in the transcribed regions.
Sanger sequencing and in silico analyses
Segregation validation was performed by direct Sanger sequencing in all available family members using specific oligonucleotide primers flanking the exons with ABI3500 sequencer (Applied Biosystems, USA). New LPIN1 variants were further screened in at least 100 unrelated healthy controls. The primer sequences were listed in Table S1. Sequence analysis was carried out using BigDye Terminator Cycle Sequencing Kit and PCR products were run on ABI PRISM 3730 Analyzer (Applied Biosystems, USA). In silico algorithms for predicting deleteriousness of nonsynonymous mutations were applied using dbNSFP. MultAlin was used for multiple sequence alignment. Crystal structural models of wild-type (wt) and mutant LPIN1 were constructed using the SWISS-MODEL online server. The predicted structures were displayed by PyMol software (version 1.5).
Plasmids construction and mRNA synthesis
Human LPIN1 cDNA (NM_145693.4) was amplified and cloned into vectors pCDNA3.1+ and pCS2+. Two missense mutations, p.I683L and p.R734Q, were introduced into hLPIN1 plasmid by site-direct mutagenesis PCR. Full length of zebrafish lpin1 cDNA (NM_001044353.1) was amplified and cloned into pCS2+ vector. All generated plasmids were validated by Sanger sequencing. Capped mRNA of human LPIN1wt, LPIN1 I683L, LPIN1 R734Q, LPIN1 I683L& R734Q and zebrafish lpin1 was generated using a mMESSAGE mMACHINE SP6 Transcription kit (Thermo Fisher Scientific, USA).
Zebrafish maintenance
All adult zebrafish, including AB strain and transgenic line Tg(mbp:eGFP) [25], were raised according to standard protocols [26]. Embryos were obtained by natural crosses and staged as described previously [27]. To inhibit pigmentation embryos were treated with 0.03% 1-phenyl-2-thiourea (Sigma). All experimental procedures for zebrafish were carried out in accordance with the approved guidelines by the Experimental Animal Management and Ethics Committees of Northwest University.
Microinjection of morpholinos (MOs) and mRNA
Antisense MO oligomers of zebrafish lpin1, a splicing-donor MO (lpin1 MO) and standard control MO (STD-MO) were obtained from Genetools, LLC (USA). The nucleotide sequences of the MOs were given in Table S1. MOs were resuspended in 1 X Danieau solution to a concentration of 2 mM stock solution before further dilutions into the required concentrations. 0.2 pmol lpin1 MO, 1.2 pmol STD-MO, 200 pg human wt or mutant LPIN1 mRNA was injected into each embryo at the 1-cell stage, respectively, using PLI-100A Plus Pico Injector (Harvard, USA).
Analysis of skeletal muscle structure by birefringence
For scoring the skeletal muscle lesions, 4 day-post-fertilization (dpf) embryos were anaesthetized with tricaine (0.04%), embedded in 5% methyl cellulose, and viewed under SMZ25 stereomicroscope equipped with a DS-Ri2 digital camera (Nikon, Japan). Birefringence was imaged as previously described [28]. For quantification analysis, 10 somites between the levels of somite 5 to 15 were imaged per embryo.
Whole-mount in situ hybridization (ISH) and immunohistochemistry (IHC)
Digoxigenin (DIG)-labeled probes of lpin1, deltaC, actn3a, neurog1 and isl1 were synthesized using SP6/T7 RNA Labeling Kit (Roche Diagnostics, USA). Zebrafish embryos at different stages were collected and fixed in 4% paraformaldehyde, dehydrated in methanol and stored at -20 °C until use. Whole-mount ISH assays were performed as described previously [29, 30]. Stained embryos from the ISH assay were cleared in 50% glycerol in PBS and photographed under Nikon SMZ25 microscope system.
In vivo IHC was performed on whole embryos according to the described protocol [29, 31]. To detect Lpin1 protein, fixed embryos were permeabilized with PBST, incubated with rabbit anti-LPIN1 antibody at 1:200 and mouse anti-acetylated tubulin antibody at 1:400 (Sigma, USA), followed by incubation with Alexa568-conjugated goat anti-rabbit IgG (Invitrogen) and Alexa488-conjugated goat anti-mouse IgG (Invitrogen, USA) as the secondary antibody at 1:200 dilution. To label acetylcholine receptors (AChR) clusters, embryos were incubated in Alexa fluor 488-conjugated α-Bungarotoxin (α-BGT) at 1:100 (Invitrogen, USA). Primary motoneurons (PMN) were labeled with mouse anti-Znp1 antibody (DSHB, USA) and secondary motoneurons (SMNs) were labeled with mouse anti-Zn8 antibody (DSHB, USA), followed by incubation with Dylight 594-conjugated goat anti-mouse IgG (Abcam, UK) at 1:200 dilution. The images were captured under a Leica TCS SP5 confocal microscope with Leica LAS AF Lite 4.0 software.
Touch-evoked escape response assays and tracking of swimming behavior
Touch-evoked escape response was tested in 4 dpf embryos and recorded under Nikon SMZ25 stereomicroscope equipped with a DS-Ri2 digital camera system, the images were processed using NIS Elements software [28].
Swimming behavior was monitored and locomotion was tracked using a DanioVision system and EthoVision XT 11.5 locomotion tracking software (Noldus Information, Netherlands) [32]. Zebrafish larvae (injected with morpholino or mRNA) developed to 5 dpf were placed in individual wells of 24-well cell culture plate containing embryo medium and monitored at room temperature (22 °C). For startle response analysis, one cycle included a 20-second (s) spontaneous movement tracking followed by cycle stimulation with light on for 5 s, interval 1 s, trigger tap 1 s, and interval 48 s. The distance, movement, and velocity parameters for individual locomotion plots were analyzed using the EthoVision XT 11.5 software.
RNA interference
Human glioblastoma U87 and mouse myoblast C2C12 cells were maintained in DMEM (Gibco, USA) supplemented with 10% FBS (Gibco, USA) and 50 units/mL streptomycin and penicillin (Gibco, USA) at 37 °C. Small interfering RNA (siRNA) duplexes targeting human and mouse LPIN1/Lpin1 genes were synthesized by GenePharma (China). Cells were transfected with two LPIN1 siRNAs for each cell line or standard siRNA as a negative control by Lipofectamine 3000 (Invitrogen, USA) according to the manufacturer's instructions. siRNA sequences were listed in Table S1.
Luciferase assays and Western blot analysis
HEK293T cells were seeded at 800,000 cells/well in 6-well plates. To evaluate the Notch signaling pathway activity, transfections were performed in triplicates with pGL4.10-HES1 reporter system and the indicated expression plasmids with the following amounts in each well: 2500 ng of wt or mutant LPIN1, 2500 ng of pGL4.10-HES1 vector, and 20 ng of pRL-TK (Renilla). At 2 days after transfection, cells were lysed and luciferase activity was measured with the use of the Dual-Luciferase Reporter Assay Kit (Promega, USA) and luminometer (Molecular Devices, USA). Each transfection was also measured in triplicates. For in vivo luciferase assay in zebrafish, 0.2 nM lpin1 MO or 200 ng pCS2-lpin1 with 60 ng pGL4.10-her1 and 5 ng pRL-TK were co-injected into one-cell stage embryos. Each experiment was performed in triplicates.
Western blot analysis was carried out according to the standard protocol described previously [33]. The following antibodies were used: rabbit anti-LPIN1 (1:1000, Abcam, UK), mouse anti-Actin (1:5000, TransGen Biotech, China).
Drug treatment
DAPT (MedChem Express, USA), an effective inhibitor of the Notch pathway, was dissolved at a concentration of 10 mM in DMSO as stock. STD-MO or lpin1 MO was injected into zebrafish embryos at the one-cell stage. 50 μM DAPT was added at the sphere stage and an equal amount of DMSO was used as the control.
Quantitative Real-Time PCR (qRT-PCR) analysis
Total RNA was isolated from U87 cells, C2C12 cells and zebrafish embryos using TRIzolTM reagent (Ambion, USA). 1 μg of RNA was reverse transcribed using SuperScriptTM system (Invitrogen). qRT-PCR was performed as described previously with SYBR Green PCR Master Mix (Kapa Biosystems, USA) [34]. Primer sequences are listed in Table S1. Image J software was used for densitometric measurement.
Statistical analysis
Each experiment was repeated at least three times and all data were expressed as means ± SEM. Student's t-test was applied for comparisons among different groups. Differences with the p value < 0.05 were considered significant.
Results
Two novel heterozygous mutations in LPIN1 were identified from a familial adult-onset muscle weakness proband
The proband (II5) in the family was diagnosed in the hospital for muscle weakness and convulsions lasting 17 years since he was 27-year-old. Laboratory evaluation revealed high blood creatine phosphokinase (CPK) (22013 UI/L; normal<150 UI/L), as well as elevated levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST). Skeletal muscle biopsy revealed muscle fibers with an abnormal lipid overload, the predominance of type I muscle fibers, type II muscle fibers atrophy, and mitochondria of muscle fibers exhibited morphological abnormality. Furthermore, electromyography disclosed a slowed gastrocnemius sensory nerve conduction velocity, and histology analysis of the upper limb muscles exhibited neurogenic damage with severely nerve demyelination (Table 1). The proband's eldest brother (II2) had similar symptoms.
Main clinical features of the investigated patient in current study compared to patients with LPIN1 mutations in the literature
| Features | Present patient | Reported patients | Percentage |
|---|---|---|---|
| Asian origin | + | 15/48 | 31.3% |
| Consanguinity | - | 17/48 | 35.4% |
| High Creatine kinase value | + | 48/48 | 100% |
| Rhabdomyolysis | - | 48/48 | 100% |
| Myohemoglobinuria | - | 40/48 | 83.3% |
| Infantile onset (5y) | - | 39/48 | 81.3% |
| Dyslipidemia | + | 2/48 | 4.2% |
| Increasd Alanine aminotransferase (ALT) and Aspartate aminotransferase (AST) | + | 12/48 | 25.0% |
| Increased lipid droplets in muscle fibers | + | 23/48 | 47.9% |
| Myasthenia | + | 20/48 | 41.7% |
| Myalgia | - | 16/48 | 33.3% |
| Amyotrophy | + | 2/48 | 4.2% |
| Predominance of type I muscle fibers | + | 20/48 | 41.7% |
| Nerve demyelination | + | n.m. | n.m. |
| Reduced myelinated nerves to the gastrocnemius muscle | + | n.m. | n.m. |
| Abnormal Electromyography | + | n.m. | n.m. |
| Neurogenic injury of skeletal muscle | + | 1/48 | 2.1% |
| Abnormal pyruvate/lactic acid ratio | + | n.m. | n.m. |
| Abnormal mitochondrial morphology of muscle fibers | + | n.m. | n.m. |
| Lysosomal polymerization of muscle fibers | + | n.m. | n.m. |
| History of kidney ailments | + | 5/48 | 10.4% |
| History of heart ailments | + | 2/48 | 4.2% |
+ affirmative; - negative; +/- variable; n.m., not mentioned.
Whole-exome sequencing was subsequently performed on II2, II3, II4, and II5 to find disease-causative mutation in the family. A total of 817 small nucleotide variants (SNPs) and 414 indels were identified (Table S2-S4). Since the deceased parents of the proband did not present related symptoms, autosomal recessive inheritance was considered. Only 7 protein-changing variants including 6 genes were remained after further bioinformatics filtering (Table S5). Using a combination of segregation validation by direct Sanger sequencing and in silico analysis, two heterozygous mutations in LPIN1 were identified in II2 and II5: (GRCh37 [hg19] chr2: g.11944690 A>C, GeneBank: NM_145693:c.2047A>C:p.I683L in exon 15 and GRCh37 [hg19] chr2: g. 11955273 G>A, GeneBank: NM_145693:c.2201G>A:p.R734Q in exon 17 (Figure 1A-C). Both of the two heterozygous mutations were predicted to be damaging and related to disease-causing by in silico prediction algorithms queried in dbNSFP (Table 2). The frequencies of two variants were extraordinarily low, only 2/121402 for c.2047A>C and 0/121220 for c.2201G>A in Exome Aggregation Consortium database, and were not found in the Chinese Millionome DataBase (Table 2). It was further confirmed by screening at least 100 unrelated healthy individuals.
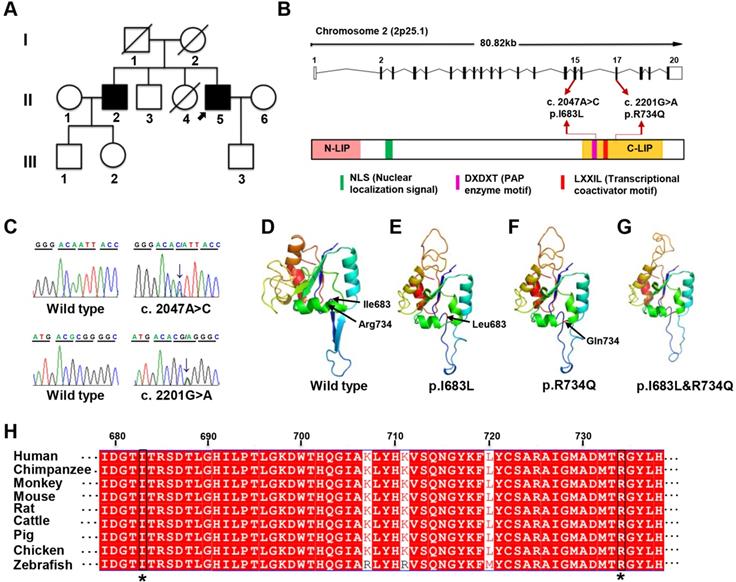
Identification of LPIN1 mutations from a family presenting with adult-onset muscle weakness. (A) Pedigree of the investigated family. The proband (II:5) was indicated by black arrow. (B) Schematic representation of the localization of identified LPIN1 mutations c.2047A>C (p.I683L) and c.2201G>A (p.R734Q) in the genome structure (top) and protein structure (bottom). (C) Sanger sequencing chromatographs showing two compound heterozygous mutations of c.2047A>C and c.2201G>A presenting in patients. (D-G) Crystal structures of human LPIN1 wild type and mutants carrying p.I683L or p.R734Q, or p.I683L/p.R734Q. (H) LPIN1was highly conserved across different vertebrate species; the conserved I683L and R734Q are highlighted with a star (*).
Mutation information
| Genome-level consequence | Gene | Gene-level consequence | |||
|---|---|---|---|---|---|
| NC_000002.12:g.11944690 A>C | LPIN1 | NM_145693:c.A2047C:p.I683L | |||
| NC_000002.12:g.11955273 G>A | NM_145693:c.G2201A:p.R734Q | ||||
| In silico nonsynonymous mutations deleteriousness prediction algorithms and corresponding predictions | |||||
| Mutation Variety | Polyphen2 HDIV | Polyphen2 HVAR | SIFT | MutationTaster | MutationAssessor |
| A>C | Damaging | Damaging | Damaging | Disease causing | Medium impact |
| G>A | Damaging | Damaging | Damaging | Disease causing | Medium impact |
| Frequency of variants in large-scale population studies (mutant allele count/total alleles) | |||||
| Mutation Variety | Exome Aggregation Consortium | TopMed | gnomAD-Exomes | gnomAD-Genomes | Chinese Millionome DataBase |
| A>C | 0.001647% (2/121402) | 0.00001 (1/125568) | 0 (0/246214) | 0 (0/30946) | 0 (0/141431) |
| G>A | 0 (0/121220) | 0.00001 (1/125568) | 0 (1/246214) | 0 (1/30946) | 0 (0/141431) |
The two missense mutations p.I683L and R734Q are located in C-terminal (C-LIP) domain of lipin 1 protein, which is crucial for PAP enzyme activity and transcriptional regulation (Figure 1B), and highly conserved across various species (Figure 1H). To predict the possible pathogenic effect of two missense mutations, LPIN1 wt, p.I683L, p.R734Q and p.I683L & p.R734Q were subjected to crystal structure modeling using SWISS-MODEL and PyMOL. In LPIN1 proteins with the mutations of p.I683L or p.R734Q, the β-sheets between Thr684 and Thr688, and between Gly697 and Thr701, of the wt protein were abolished; and the α-helices between Ile727 and Tyr736 were broken into two parts, rendering less compact protein structures (Figure 1D-G). Thus, it was likely that p.I683L and p.R734Q mutations affected the appropriate folding and biological functions of lipin 1 protein.
Mutations of LPIN1 in humans have been identified as a major cause of recurrent, early-onset myoglobinuria and rhabdomyolysis (MIM#268200). Hitherto, 46 homozygous or compound heterozygous mutations in LPIN1 gene from 48 cases have been identified in humans [14, 15, 35-46]; among which 83.3% cause myoglobinuria, and 100% lead to rhabdomyolysis symptoms, mostly in childhood. About 41.7% and 33.3% of the LPIN1 mutations result in myasthenia and myalgia respectively (Table 1). In addition to muscle phenotypes, the proband in this study manifested severe mixed peripheral neuropathy and nerve demyelination, which had not been reported in other LPIN1 mutations where only neurogenic damage of skeletal muscle was found in one case [44] (Table 1). It suggested a critical role for LPIN1, not only in skeletal muscle, but also in the peripheral nervous system.
Expression of zebrafish lpin1 was specifically enriched in somites and neural tissues
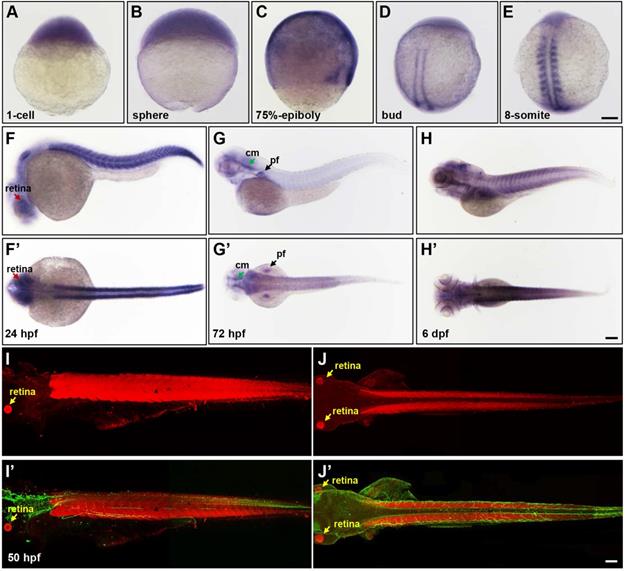
To gain insight into the pathogenesis of LPIN1 mutations in humans and LPIN1 functions in vivo, the zebrafish model was used in this study. The spatio-temporal expression patterns analyzed by whole-mount ISH analysis displayed a ubiquitous expression of lpin1 mRNA from 1-cell to the blastula and gastrula stages (Figure 2A-C). At the end of gastrulation, lpin1 mRNA was restricted to the bilateral stripes of presomitic mesoderm (Figure 2D). During somitogenesis, lpin1 was expressed in the elongated adaxial cells and the striped paraxial mesoderm, and continued to be highly expressed in somites till a total of about 30 somite pairs were formed at 24 hour-post-fertilization (hpf) (Figure 2E-F'). The lpin1 mRNA was also broadly expressed at the head region at 24 hpf, with a notable distribution in the retina. At 72 hpf, the lpin1 mRNA was only detected at the neural crest, cephalic musculature and pectoral fins (Figure 2G, G'), but the myotome-specific expression was restored at 6 dpf (Figure 2H, H'). The levels of lpin1 transcripts were confirmed by qRT-PCR (Figure S1). Consistent with the mRNA expression pattern of lpin1, IHC staining showed that the Lpin1 protein was specifically localized in myotomes as well as in the retina at 50 hpf (Figure 2I-J'). The enriched and specific expression of lpin1 in developing somites and neural tissues of zebrafish implicated an essential role of lpin1 in the formation and function of muscular and neural tissues.
The spatial and temporal expression patterns of lpin1 during zebrafish early development. The expression of lpin1 mRNA was ubiquitous during cleavage and gastrula stages (A-C), restricted to paraxial mesoderm at the bud stage (D), and abundant at somites from the 8-somite stage onwards (E-H'). The dynamic lpin1 mRNA expression was detected in the retina (red arrows in F and F'), pectoral fins (pf, black arrows in G and G') and the cephalic musculature (cm, green arrows in G and G'). (I-J') IHC staining showed the Lpin1 protein expression in myotomes and the retina (yellow arrows), acetylated α-tubulin was stained in green color. (F-J) Lateral views of embryos, (F'- J') Dorsal views of embryos. Scale bar: 100 µm.
Lpin1 deficiency caused defects of skeletal muscle development in zebrafish
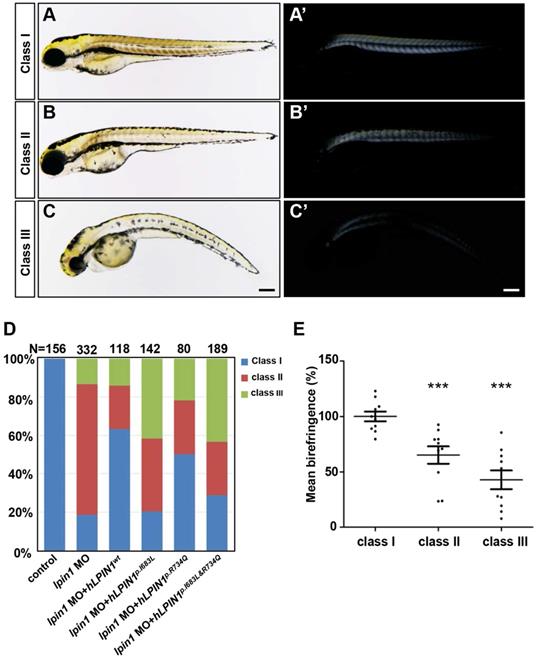
Previous studies reported that LPIN1 deficiency resulted in peripheral neuropathy and skeletal muscle myocyte damage in mice [21, 47, 48]. To further determine the biological function of lipin 1, we generated Lpin1-deficient zebrafish by lpin1 MO injections or by overexpression of mutated human LPIN1 mRNA. IHC analysis revealed that the Lpin1 protein expression in somites was dramatically decreased in lpin1 MO injected embryos at 24 hpf, as compared with that in control embryos, indicating a successful knockdown effect of lpin1 MO (Figure S2). lpin1 morphant embryos were classified into three categories according to the severity of skeletal muscle phenotypes as revealed by birefringence imaging (Figure 3). In control embryos, the skeletal muscle was highly birefringent under polarized light due to the organization of myofibrils (Figure 3A, A'); but lpin1 MO injected embryos displayed overall reduction in birefringence due to muscular lesions caused by myofibrillar disorganization in the major axial skeletal muscles (Figure 3B-C'). The quantification of the birefringence further revealed a significant reduction in the brightness in class II (35% reduction) and class III (58% reduction) of lpin1 MO injected embryos (Figure 3E). Overexpression of mutant human LPIN1p.I683L, LPIN1p.R734Q or LPIN1p.I683L& R734Q, but not LPIN1wt mRNA caused a similar overall reduction in the birefringence of the skeletal muscle (Figure S3). Moreover, the lpin1 MO induced skeletal muscle defects could be alleviated by the co-injected mRNA of human LPIN1wt but not LPIN1p.I683L, LPIN1p.R734Q or LPIN1p.I683L& R734Q (Figure 3D).
Zebrafish lpin1 morphant embryos showed defects in the skeletal muscle at 4 dpf. (A-C') Classification of the lpin1 morphants was based on the trunk phenotype (A-C) and the corresponding reduction in birefringence observed in skeletal muscles (A'-C'). Class I (A, A') represents the wild-type-like phenotype with neither defect in embryonic trunk nor reduced birefringence in the skeletal muscle. Scale bar: 200 μm. (D) The phenotype of lpin1 morphants was rescued by the co-injected mRNA of human LPIN1wt but not LPIN1p.I683L, LPIN1p.R734Q or LPIN1p.I683L& R734Q. (E) Quantification data are calculated by the mean intensity of 10 somites (from somite 5 to 15) for each embryo using Image J, which is normalized to that of class I (control) to obtain percentage values. n = 10 embryos for each class. *** indicates p < 0.001 by Student's t-test.
Lpin1 deficiency led to defects of neuromuscular synapse formation in zebrafish
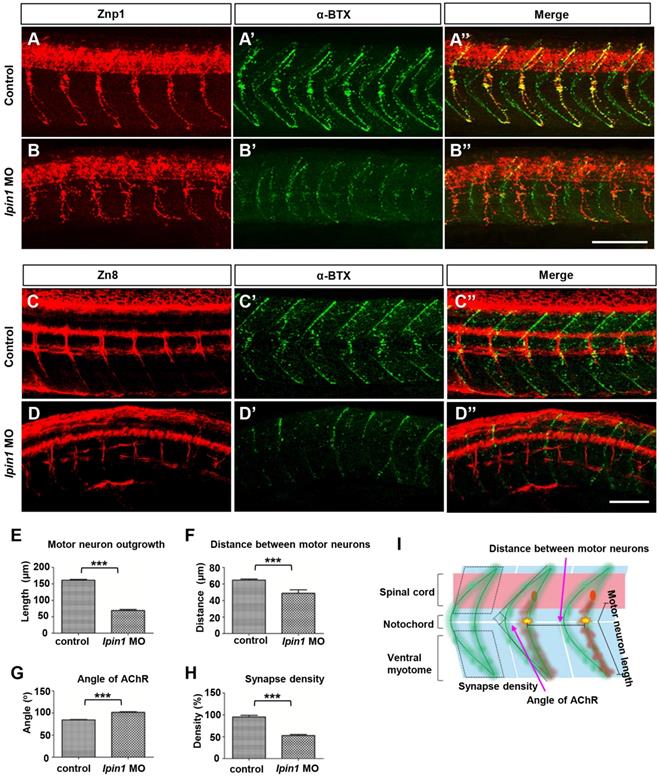
To address whether the loss of lpin1 function in zebrafish affects the connection between nerve and muscle, we analyzed the projections of motor axons and the formation of neuromuscular synapses in lpin1 MO injected embryos. Zebrafish have two populations of spinal motoneurons, PMNs and SMNs projections. In the control embryos at 26 hpf, PMNs were labeled by Znp1 antibody and exhibited a common axonal path, where motor neuron growth cones traversed the muscle territory into ventral and dorsal somitic muscle blocks [49] (Figure 4A). Meanwhile, AChR clusters (post-synapses) visualized by α-bungrotoxin (α-BTX) [49, 50] were formed properly and correlated well with motor axonal projections (Figure 4A', A''). However, lpin1 MO injected embryos displayed a significant reduction in both motor neuron axonal projections and AChR clusters (Figure 4B-B''). The length of the PMNs was significantly shorter in the lpin1 morphant (69.2 ± 19.3 μm, n = 9) than in the control embryo (161.0 ± 16.6 μm, n = 9) with a ~69% reduction (Figure 4E, I). The distance between motor axons in control embryos (64.6 ± 7.6 μm) was also reduced in the lpin1 morphant (49.1 ± 25 μm) (Figure 4F, I). The morphological changes of AChR clusters were measured by calculating the angle of AChR stripes with a ~20% increase in the lpin1 morphant (102 ± 8.8 o) compared to the control embryo (84.4 ± 6.6 o) (Figure 4G, I). Quantification of the post-synaptic densities showed that lpin1 morphants had lower density with a ~44% decrease compared to controls (Figure 4H, I). In addition, the co-localization of PMNs and post-synapses was disorganized in lpin1 morphants, indicating that the motor axons cannot innervate the muscle fiber properly.
Knockdown of lpin1 in zebrafish embryos affected neuron development and AChR clustering. Znp1 antibody staining (A-B) and α-BTX labeling (A'-B') of control embryos (A-A'') and lpin1 morphants (B-B'') at 26 hpf. Zn8 antibody staining (C-D) and α-BTX labeling (C'-D') of control embryos (C-C'') and lpin1 morphants (D-D'') at 60 hpf. Scale bar: 100 µm. Measurements of the length of motor neurons (E, n = 6 motor neurons/larvae), the distance between two adjacent motor neurons (F, n = 6 segments/larvae), the angle of AChR clusters (G, n = 6 AChR clusters/larvae), and the density of post-synapses (H, n = 6 areas of myotome/larvae), were carried out in 9 larva under each treatment. (I) Schematic representations of the parameters in E, F, G and H. *** indicates p < 0.001 by Student's t-test.
After the localization of PMNs was completed, at ~25 hpf, SMNs extend their axons along the paths pioneered by primary axons. Axons of both PMNs and SMNs eventually form the dorsal and ventral motor nerves [51]. Once the development of PMNs is ablated, the extension of SMNs to their targets will be delayed [52]. In the control embryos at 60 hpf, it was observed that SMNs labeled by Zn8 completed their axonal migration along the common paths and the axons of the ventral nerve extended to the ventral myotome (Figure 4C), and AChR clusters also exhibited a universally and regular distribution (Figure 4C', C''). In lpin1 morphants, axons of SMNs had very weak signal intensity, showed an additional decrease in the length of motor neurons and an additional increase in premature branching, failing to reach the ventral myotome (Figure 4D). The density of AChR clusters was greatly reduced in lpin1 morphants (Figure 4D', D''). At 5 dpf, the α-BTX signals in lpin1 morphants were reduced significantly in both fast muscles and slow muscles compared to that in control embryos (Figure S6A, B) [53]. qRT-PCR results also confirmed the expression levels of almost all nicotinic acetylcholine receptors were decreased in lpin1 morphants (Figure S6G). The malformation of PMNs and SMNs axonal projections, and AChR clusters were also observed in those embryos injected with the mRNA of human LPIN1p.I683L, LPIN1p.R734Q or LPIN1p.I683L& R734Q; but not in LPIN1wt injected embryos (Figure S4, S5, S6). Therefore, the common axonal paths as well as the neuromuscular junction were both affected in Lpin1-deficent zebrafish embryos.
Lpin1 deficiency resulted in impaired myelination in zebrafish
Myelin sheath surrounds nerve axons, allowing saltatory nerve conduction and maintenance of the axon at a long distance from the cell body [54]. Myelin basic protein (MBP) is produced by differentiated oligodendrocyte lineage cells, including oligodendrocytes in the central nervous system and Schwann cells in the peripheral nervous system [55]. In zebrafish, Tg(mbp:eGFP) transgenic strain facilitates the visualization of myelination [25]. At 5 dpf, the GFP signals expressed in oligodendrocytes and SCs were dramatically decreased in lpin1 morphants compared to control larvae (Figure 5A-B').The quantification of mbp fluorescence signals was also reduced by 70% in lpin1 morphants compared with control larvae (Figure 5C). It suggested that the loss of lpin1 function in zebrafish larvae caused impaired myelination, consistent with our patient phenotype, which neurohistological examination of left sural showed a significant decrease in myelinated nerve fibers and severe nerve demyelination. Tg(mbp:eGFP) embryos injected with the mRNA of human LPIN1 p.I683L , LPIN1 p.R734Q or LPIN1 p.I683L& R734Q, but not LPIN wt were also observed impaired myelination (Figure S7).
Lpin 1 deficiency impaired the maturation of schwann cells and myelin synthesis. (A-B') GFP signals of 5 dpf Tg(mbp:eGFP) larvae was visualized by fluorescent microscope, yellow arrows indicated GFP signal in the spinal cord, and red arrows indicated GFP signal motor neurons, lateral views, scale bar: 100 μm. (C) Mbp fluorescence signals of control embryos and lpin1 morphants were analyzed by Image J software. Ten independent samples were evaluated. (D) qRT-PCR measurements of immature SC markers, mature SC markers, and myelin gene markers in lpin1 morphants and STD-MO injected embryos at 3dpf. (E-G) qRT-PCR measurements of pou3f1, egr2b, and mpz dynamic expression at 1 dpf, 3 dpf, and 5 dpf respectively. Values in (D-G) represented means ± SE of data from three independent experiments, **p< 0.01, and ***p < 0.001 (Student's t-test).
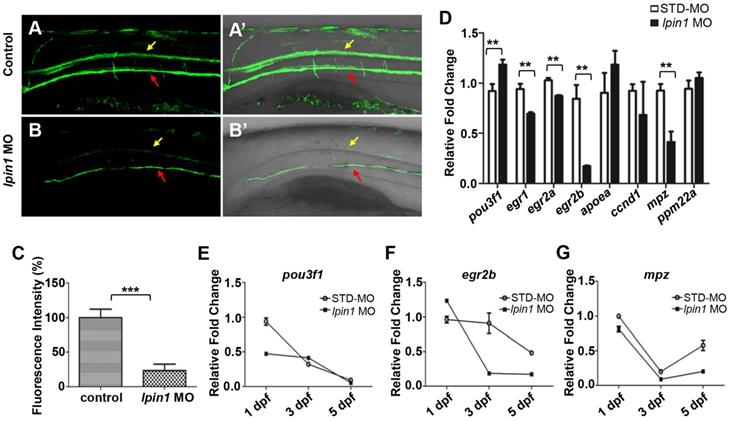
In order to quantitate the myelination defects in lpin1 morphants at the molecular level, we examined the expression of genes involved in myelination. qRT-PCR showed that the expression of immature SCs marker pou3f1 [57] was abnormally increased, while the expressions of mature SCs markers, egr1 [58], egr2a [59], egr2b [60] and myelin marker, mpz [61] were abornarmally decreased in lpin1 morphants at 3 dpf (Figure 5D). The dynamic expressions of these genes further revealed that the expression of pou3f1 was higher in lpin1 morphants at 3 dpf and was followed by decreased expression of egr2b and mpz at 3dpf and 5dpf. This indicated that SCs were kept in an immature state, resulting in a decrease in myelin synthesis in Lpin1-deficient zebrafish embryos (Figure 5E-G). Altogether, these findings demonstrated that lpin1 might mediate oligodendrocytes and Schwann cells differentiation, which was crucial for the normal progression of myelination in zebrafish.
Lpin1-deficient zebrafish embryos exhibited abnormalities of swimming behaviors
The severe defects in the skeletal muscle, neuromuscular synapses and myelination of Lpin1-deficient embryos directly affected the swimming behaviors of zebrafish larva. In contrast to the 4 dpf control larva that swam away rapidly after 5 milliseconds (ms) in response to touch (Figure 6A), lpin1 morphants showed an impaired response to touch and failed to swim away from the field of view even after 20 ms (Figure 6B). This touch-evoke failure could be partially rescued by human LPIN1wt mRNA (Figure 6C).
Locomotor deficits in lpin1 morphants. (A-C) lpin1 morphant embryos exhibited reduced touch-evoked response at 4 dpf (B) while control embryos swam away rapidly after mechanosensory stimulation (A). The reduced touch-evoked response of lpin1 morphant could be rescued by co-injected hLPIN1wt mRNA (C). Scale bar: 200 µm. (D) Cumulative plots of the position and velocity of control larva, lpin1 morphants, and lpin1 morphants co-injected with hLPIN1wt mRNA; during 5 min of behavioral recording. 8 representative individuals were recorded for each treatment group. (E) Representative heat map plots. Red dots indicate the long rest time without any movement. (F) Cumulative duration records showed that the lpin1 morphants were less active. (G) Velocity was dramatically decreased in lpin1 morphants, but can be rescued by the hLPIN1wt mRNA. (H) Total movement distance during a 5 min startle response analysis as follows: 20 s spontaneous movement tracking followed by cycle stimulation with light on 5 s, interval 1 s, trigger tap 1 s, and interval 48 s; repeated 5 cycles. Data shown are means of distance moved in 5 s ± SE (n = 24 for each group at 5 dpf).
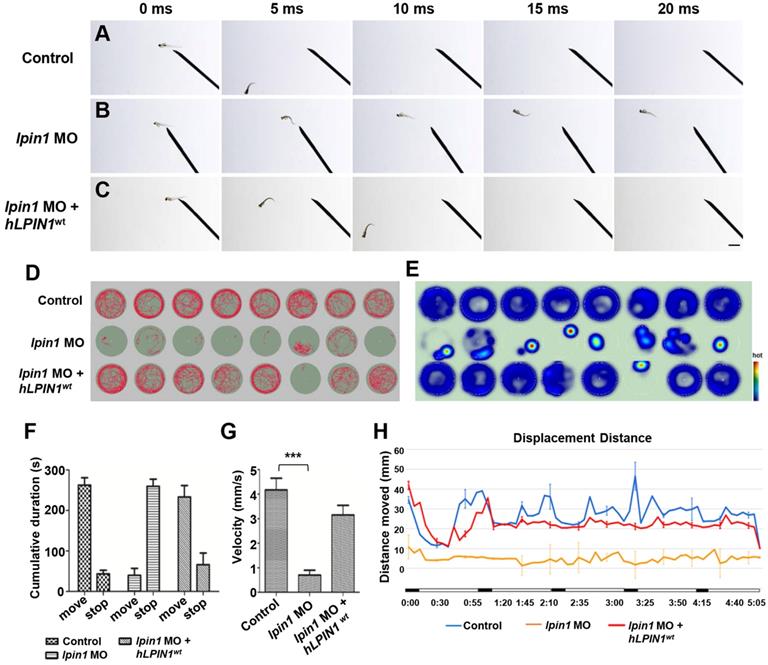
To further examine the effect of Lpin1 deficiency on swimming activity, we quantified the velocity and the movement distance of individual zebrafish larvae at 5 dpf by locomotion assay. The movement trajectory and the distance of zebrafish larva were generated over a 5 minutes (min) period. Individual control larvae showed high activity and mobility by measuring the travel trajectory and movement heat map at an interval of 0.5 s (Figure 6D, E). On the other hand, lpin1 MO morphants displayed severely reduced activity and mobility over the test period. The co-injection of lpin1 MO and human LPIN1wt mRNA rescued the defective locomotion phenotype (Figure 6D, E). The cumulative duration records during a period of 300 s indicated that lpin1 morphants had a dramatically reduced movement time (39.9 ± 49.2 s) and increased rest time (259.9 ± 49.1 s) compared to those of control larvae (movement time: 262.8 ± 50.3 s; rest time: 43.5 ± 24.8 s) and hLPIN1wt mRNA co-injected larvae (movement time: 233.5 ± 24.8 s; rest time: 66.3 ± 30.5 s) (Figure 6F, n = 24 for each group). Correspondingly, the velocity of lpin1 morphants was also significantly reduced (0.71 ± 0.56 mm/s) compared to the control larvae (4.17 ± 1.34 mm/s) and hLPIN1wt mRNA co-injected larvae (3.15 ± 1.13 mm/s) (Figure 6G, n = 24 for each group). The total distance moved was quantified over a 5-min period for individual zebrafish larvae. lpin1 morphants displayed a significant decrease in the mean distance swum (309.9 ± 4.9 mm) compared to the control larvae (1686.5 ± 26.8 mm) and rescued larvae (1402.9 ± 22.3 mm) (Figure 6H, n = 24 for each group). The locomotion assay was further applied to the 5 dpf zebrafish larvae overexpressing human LPIN1wt, LPIN1p.I683L, LPIN1p.R734Q or LPIN1p.I683L& R734Q respectively. Similar to the swimming behavior of lpin1 morphants, the zebrafish larvae injected with the mRNA of LPIN1p.I683L, LPIN1p.R734Q or LPIN1p.I683L& R734Q exhibited dramatically decreased movement velocities and distances compared to either control or LPIN1wt overexpressing larva (Figure S8). The reduced mobility caused by the lpin1 MO or the overexpressed mutant LPIN1 mRNA in larval zebrafish was consistent with the defective structures of myotome and NMJs; and partially phenocopied the abnormalities of human patients with mutations in LPIN1. Together, these results suggested a conserved and important role of LPIN1 in the function of neuromuscular tissues.
Lpin1 knockdown in zebrafish and mammalian cells caused abnormal expression of muscle and neuron markers
According to the dramatically decreased locomotion ability observed in lpin1 morphants, we further assessed several markers of muscle and neuron development including deltaC [61], deltaD [61], segmental plate and somite markers; actn3a [62], musk [63], expressed in muscle fibers; neurog1 [64], lrp4 [29], expressed in the central nervous system; and isl1 [65], a spinal cord motor neuron marker. At the 8-somite stage, the expression of deltaC and deltaD in lpin1 morphants was expanded in the presomitic mesoderm and abnormally distributed in newly formed somites, as compared to control embryos (Figure 7A, A', Figure S9A, A'). The expression of actn3a, a muscle-specific actin highly and specifically expressed in myotome at 25 hpf, indicated a morphological change in the myotome shape and arrangement in lpin1 morphants (Figure 7B, B'), similar change was observed using musk marker (Figure S9B, B'). The expression of neurog1 in midbrain, hindbrain, diencephalon and spinal cord of the lpin1 morphants at 50 hpf was obviously decreased compared to control embryo (Figure 7C, C'), suggesting a defective neurogenesis. The high expression in rhombic lip and pectoral fin of lrp4 was also reduced in lpin1 morphants at 50 hpf (Figure S9C, C'). The shape and location of PMNs in the spinal cord, as labeled by isl1 expression, were altered in 25 hpf lpin1 morphants compared to control embryos (Figure 7D, D'). In addition, the expression levels of muscle and neuron markers were significantly altered in lpin1 morphants as detected by qRT-PCR analysis (Figure 7E, F). Thus, both qualitative and quantitative analysis of gene expressions in zebrafish support that lpin1 was required for muscular and neuronal development.
lpin1 knockdown affects gene expressions in the muscle and neuron development of zebrafish larva. The expression of somite marker deltaC in control embryos (A) and lpin1 morphant (A') embryos at 8-somite stage. Red arrows indicate newly formed somites and red bars indicate the length of presomitic mesoderm. Control embryos (B) and lpin1 morphant (B') embryos at 25 hpf were stained for actn3a. Note the shape of myotome, indicated by red angle, was changed in lpin1 morphants. The expression of neuron markers neurog1 (C, C') at 50 hpf and isl1 (D, D') at 25 hpf in control embryos (C, D) and lpin1 morphants (C', D'). In lpin1 morphants, the expression of neurog1 was reduced in the midbrain (mb), hindbrain (hb), and spinal cord (sp) (pink arrows); and that of islet1 was reduced in PMNs (yellow arrows). Scale bar: 100 μm. The expressions of muscle (E) and neuron (F) markers, as determined by qRT-PCR, were significantly changed in lpin1 morphants at 4 dpf and 1 dpf respectively. Lpin1 siRNA knockdown in C2C12 (G) and U87 cells (H) resulted in abnormal expressions of muscle and neuron markers. Values in (E, F, G and H) represent means ± SE of data from three independent experiments, *p< 0.05, **p< 0.01, and ***p< 0.001 (Student's t-test).
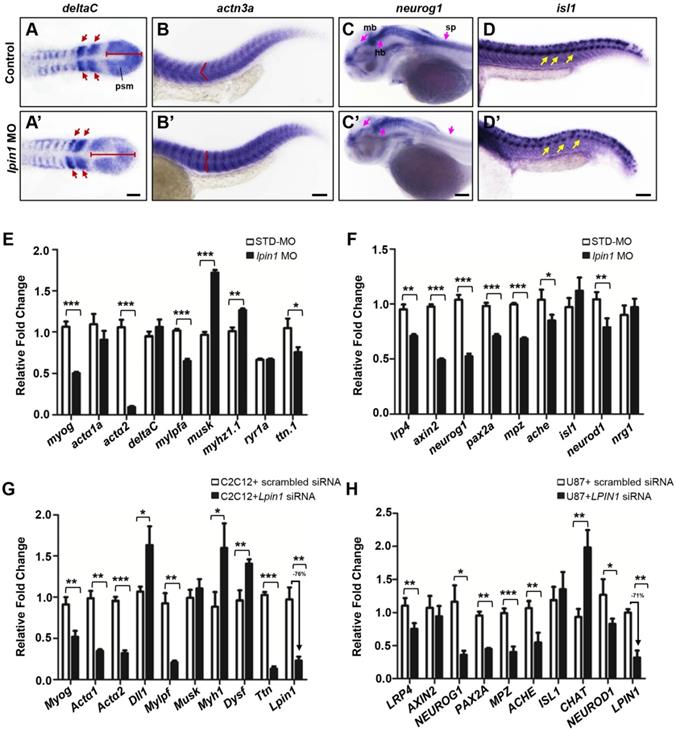
In order to address the muscular and neuronal anomalies due to lipin 1 deficiency, we further modeled the loss of LPIN1 by siRNA knockdown in mammalian cells. In mouse myoblast C2C12 and human primary glioblastoma U87 cells, LPIN1 siRNA achieved > 70% knockdown efficiency, resulting in abnormal expression levels of muscular and neuronal markers (Figure 7G, H). Therefore, the results of quantitative analysis of gene expressions in LPIN1-deficient mammalian cells were consistent with the Lpin1-deficient phenotypes in zebrafish, suggesting a conserved and essential role for LPIN1 in the regulation of muscular and neuronal gene expressions.
Loss of lipin 1 function led to increased Notch signaling in zebrafish and mammalian cells
In zebrafish, the highly specific expression of lpin1 mRNA and protein at somites, and the dysfunction of myogenesis and neurogenesis due to the loss of Lpin1 function, are reminiscent of the gene expressions and phenotypes regulated by Notch signaling [66, 67]. Therefore, we assessed whether the Notch pathway was affected by the loss of lipin 1 function in zebrafish. Dual-luciferase Notch reporter assay performed in zebrafish indicated that the Notch pathway activity was upregulated in Lpin1-deficient embryos (Figure 8A). qRT-PCR analysis further confirmed that the relative transcript abundance of jag1b, a Notch ligand; notch2, a Notch receptor; and her1, an effector of Notch; were significantly upregulated in the lpin1 morphant. In contrast, the expression of lim1 known to be repressed by Notch signaling was decreased markedly. The addition of DAPT, a Notch signaling inhibitor, could partially rescue the over-activation of Notch pathway in Lpin1-deficient embryos (Figure 8B). These results suggested that the Notch activity was negatively regulated by Lpin1 in zebrafish.
Loss of lipin 1 function results in activation of Notch signaling in zebrafish and mammalian cells. (A) Dual luciferase reporter assay to quantify the Notch signaling activity as her1 was coinjected with lpin1 morpholino into zebrafish embryos. (B) Lpin1-deficient zebrafish embryos showed upregulated gene expressions of Notch signaling components, which can be partially rescued by Notch inhibitor DAPT. (C) Dual luciferase reporter assay to quantify the Notch signaling activity as HES1 was coexpressed with the indicated LPIN1 cDNA in HEK293T cells. (D) Western blot and quantification analysis of LPIN1 wild type and mutant proteins. (E) LPIN1 siRNA in HEK293T cells achieved 65% knockdown with upregulated gene expressions of NOTCH signaling components. The abnormal upregulation of NOTCH signaling can be rescued by NOTCH inhibitor DAPT. Values in (A-E) represent means ± SE of data from three independent experiments, *p< 0.05, **p< 0.01, and ***p< 0.001 (Student's t-test).
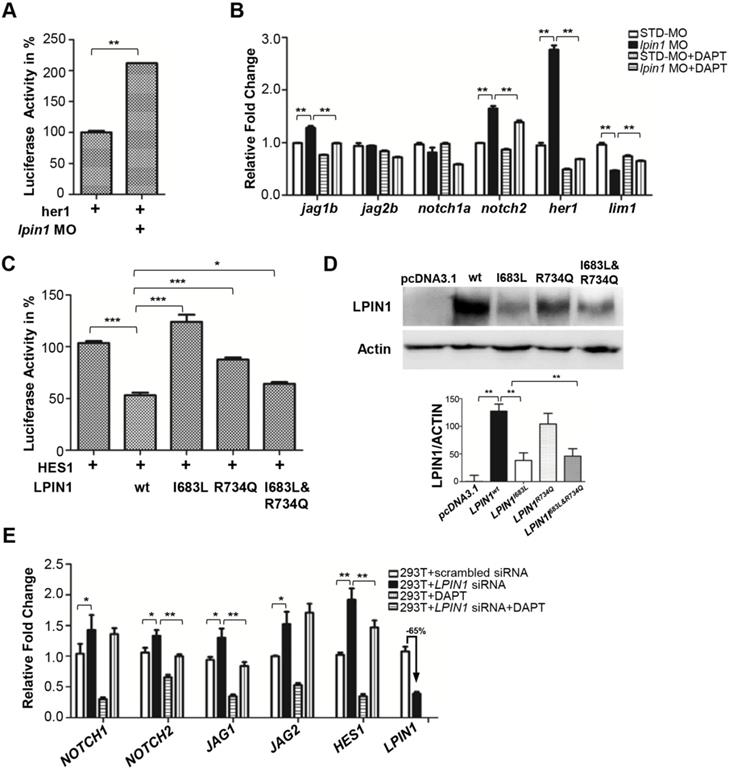
To investigate whether the two identified mutations in patients lead to a loss of LPIN1 function in Notch pathway, we analyzed the effect of I683L and R734Q on the activation of NOTCH signaling by Dual-luciferase reporter assay in transiently transfected HEK293T cells. Consistent with the findings in zebrafish, LPIN1 was able to significantly inhibit NOTCH signaling as measured by HES1 reporter. In contrast, the mutated LPIN1 harboring I683L, or R734Q, or both I683L and R734Q abolished the antagonistic effect of LPIN1 on the NOTCH signaling activity (Figure 8C). Western blot analysis further revealed that the protein abundance of LPIN1p.I683L, LPIN1p.R734Q and LPIN1p.I683L& R734Q were lower than that of LPIN1wt protein, implicating compromised stability of mutant LPIN1 proteins (Figure 8D).
To further substantiate the influence of LPIN1 on NOTCH signaling, we carried out siRNA knockdowns of LPIN1 in HEK293T, human primary glioblastoma U87, mouse myoblast C2C12, and human osteosarcoma U2OS cells, respectively. In HEK293T cells where LPIN1 siRNA achieved > 65% knockdown efficiency, the levels of NOTCH1, NOTCH2, JAG1, JAG2 and HES1were increased significantly. The abnormal augmentation of NOTCH pathway components could be rescued by the addition of DAPT (Figure 8E). Similarly, the siRNA knockdown of LPIN1 in U87, C2C12, and U2OS cells also led to an abnormal increase of NOTCH signaling at different levels (Figure S10). Taken together, these results from zebrafish and mammalian cell lines showed that LPIN1 possessed widespread repressive activity on NOTCH signaling, which might be a possible pathomechanism for the muscle and neuron phenotypes induced by lipin 1 deficiency in human and zebrafish.
Discussion
Since Zeharia et.al [15] identified six mutations of the LPIN1 gene in 25 patients aged 2-7 years with recurrent myoglobinuria for the first time, up to now there are 48 LPIN1 mutation sites reported in human. More than 90% recessive LPIN1 mutations, including homozygous or compound heterozygous genotypes, cause myoglobinuria and rhabdomyolysis of which more than 80% of patients developed in early childhood (<5 years old). Although the majority of human LPIN1 mutations manifested myoglobinuria, no obvious myoglobinuria phenotype was found in the Lpin1 mutant mice [19, 20, 68]. LPIN1-deficient mice exhibited defects not only in lipid metabolism, but also in muscle and neuron development. For example, the first reported Lpin1 mutant mice Lpin1fld/fld was identified by neonatal fatty liver, lipodystrophy, insulin resistance, and peripheral neuropathy [18, 19]. Nadra et. al. [9] verified the function of Lpin1 for myelin formation and maintenance through the analysis of Lpin1fld/fld mice. Sellers et. al. [20] also reported the phenotypic changes in skeletal muscle fibers of Lpin1fld/fld mice. Recently, Stepien et. al. [44] reported a case of 25-year-old Irish woman who carried compound heterozygous mutations of LPIN1 (c.2295-866_2410-30del1763; c.942C>A). The patient was reported to have persistent myalgia and bilateral common peroneal neuropathies. This is the first description of a human patient with nerve and muscle phenotypes caused by LPIN1 mutation. However, the patient with LPIN1 deficiency also had an episode of rhabdomyolysis requiring intensive care admission. Here, we were first to report two novel compound heterozygous mutations, c. 2047A>C (p.I683L) and c. 2201G>A (p.R734Q) in LPIN1, which caused adult-onset syndromic myasthenia. The clinical diagnosis of the proband revealed the abnormality of skeletal muscle fibers and neurogenic damage with severe nerve demyelination. The progressive muscle weakness appeared in adulthood without any episode of myoglobinuria. Previously, Jama et. al. [48] generated conditional knockout mice Lpin1Myf5cKO and found that LPIN1 deficiency induced reduced muscle mass and myopathy. In addition, HSA-Cre;Lipin1floxed/floxed mice, where a skeletal muscle-specific lipin 1 deficiency was achieved, exhibited a severe sarcoplasmic reticulum stress, necrosis of skeletal muscle fibers, stress disorder of gastrocnemius muscle, and triggered myopathy [68]. The phenotypes of skeletal muscle-specific lipin 1 deficiency in mice were consistent with the clinical features of our patient, which included predominance of type I muscle fibers, atrophy of type II muscle fibers, morphological abnormality of muscle fiber mitochondria, and progressive myopathy. On the other hand, by inactivating of Lpin1 gene in Schwann cells, MPZCre/+/Lp fE2-3 / fE2-3 mice showed demyelination of nerve cells, dedifferentiation of Schwann cells, and reduced nerve conduction velocity [9]. Consistently, the neurohistological examination of left sural in our patient showed a significant decrease in myelinated nerve fibers and severe nerve demyelination, which was similar to the neural phenotypes observed in LPIN1-deficient mice. Furthermore, Lpin1-deficiency in the zebrafish larvae induced by either lpin1 MO knockdown or overexpression of human point mutations caused myofibrillar disorganization in the axial skeletal muscles, abnormal formation of PMNs, SMNs and post-synaptic clusters, and myelination defects; which in turn led to serve defects in locomotion ability and swimming behaviors (Figure 3,4,5,6,S4, S5, S6, S7 and S8). The phenotypic analysis of lipin 1 deficiency in previous studies using mice, and in our study of human mutations and the zebrafish model, together supported a highly conserved role for lipin 1 in the development and function of neuromuscular tissues.
Notch signaling is well-known to play critical roles in the regulation of somitogenesis and post-natal myogenesis. During early zebrafish embryogenesis, a number of Notch pathway components including deltaC, deltaD, notch1a, notch6, her1, her4 are expressed in the presomitic mesoderm, paraxial mesoderm and corresponding somites, which are important to the maintenance of segmental clock and establishment of the somite polarity [69-71]. ISH analysis showed that lpin1 mRNA was expressed in the presomitic mesoderm at the tailbud stage, and in the adaxial cells and segmental somites during somitogenesis, similar to the expression pattern of myoD, a member of the bHLH transcription factors acting sequentially in myogenic differentiation [72]. Antibody staining further revealed a highly specific expression of Lpin1 protein in the myotome (Figure 2). The expression patterns and deficiency phenotypes of lpin1 in zebrafish were reminiscent of those regulated by Notch signaling during somitogenesis and myogenesis. Consistently, deltaC and deltaD expressions in lpin1 morphants were expanded at the presomitic mesoderm and abnormally distributed in newly formed somites, as compared to control embryos (Figure 7, S9); thereby suggesting an alteration of Notch signaling during somite formation. The her1 reporter activity assay and the notch inhibitor treatment led to the discovery that Lpin1 acted as an antagonist of Notch signaling pathway during zebrafish embryogenesis. Further analysis in human and mouse cells confirmed the negative regulation of Notch pathway by LPIN1. HES1 reporter assay in HEK293T cells revealed that the repression of Notch activity by LPIN1 was partially abrogated by I683L and R734Q mutations. Strikingly, LPIN1 deficiency facilitated by siRNA knockdown in human and mouse cells caused abnormally upregulated Notch pathway, which can be partially rescued by the Notch inhibitor DAPT (Figure 8). Therefore, the experiments in both zebrafish and mammalian cells confirmed the conserved inhibitory functions of LPIN1 upon Notch signaling activity during development.
Another interesting phenotype observed in Lpin1-deficient zebrafish embryos was the defects in motor neuron outgrowth and synapse formation. Analysis of PMNs and SMNs in Lpin1-deficient embryos revealed impaired axonal outgrowth, as well as an increased arborization of the PMNs and SMNs that altered the normal ventral pathfinding. The morphological changes and mis-patterning of post-synapses were also observed in Lpin1-deficient embryos. Furthermore, the co-localization of primary motor axons and post-synapses was disorganized, indicating that the NMJ was deformed and dysfunctional in lpin1 morphants. NMJ is the synapse formed between motor neurons and skeletal muscle fibers, and is covered by SCs in vertebrates [73, 74]. NMJ-related diseases include congenital myasthenic syndromes, myasthenia gravis, and Lambert-Eaton myasthemic syndrome, etc. [75]. Other neuromuscular diseases also include amyotrophic lateral sclerosis, muscular dystrophy, and different kinds of muscle weakness. AGRIN-LRP4-MuSk is a well-known signaling complex during NMJ formation. In zebrafish mutant unplugged, the homologue of muscle specific tyrosine kinase (MuSK), AChR clustering could be initiated but the approaching of growth cones to the muscle center is restricted, which is similar to the phenotype observed in Lpin1-deficient zebrafish embryos [76, 77]. The expression patterns of lpin1 is also very similar to that of unplugged; both of genes are highly expressed in somites during somitegenesis and in myotomes at 24 hpf, subsequently the expressions are restricted to pectoral fins and cephalic musculature at later stages. In lpin1 morphants, the expression patterns and levels of musk and lrp4 were altered as well. These evidences implicate that LPIN1 may play a role in the NMJ formation and function, either directly or indirectly through the AGRIN-LRP4-MuSk signaling.
Abbreviations
Lipin 1: phosphatidic acid phosphatase 1, PAP1; LPIN1: human LPIN1 gene; Lpin1: mouse LPIN1 gene; lpin1: zebrafish LPIN1 gene; PAP: phosphatidic acid phosphohydrolase; AChR: acetylcholine receptor; PA: phosphatidic acid; DAG: diacylglycerol; Schwann cells (SCs); TAG: triglycerides; PMNs: primary motor neurons; SMNs: secondary motor neurons; NMJ: neuromuscular junction; qRT-PCR: quantitative real time polymerase chain reaction; MOs: morpholinos; ISH: in situ hybridization; IHC: immunohistochemistry; DIG: Digoxigenin; CPK: creatine phosphokinase; ALT: Alanine aminotransferase; AST: Aspartate aminotransferase; SNPs: small nucleotide variants; C-LIP: C-terminal; α-BTX: α-bungrotoxin; MBP: Myelin basic protein; MuSK: muscle specific tyrosine kinase; dpf: day-post-fertilization; hpf: hour-post-fertilization.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We are indebted to the family for kindly partaking in this study. We are grateful to Mr. Baijun Du for assistance on gene screening; Northwest University Zebrafish Core Facility for assistance with fish culture. This work was supported by Shaanxi key industry innovation chain (Group) Foundation in social development field, China (2019ZDLSF02-05); Opening Foundation of Key Laboratory of Resource Biology and Biotechnology in Western China, Ministry of Education, (ZSK2018010); Project 2018N2001 from Department of Fujian Science and Technology and Program for Innovative Research Team in Science and Technology in Fujian Province University; Ministry of Science and Technology, Taiwan (MOST) 106-2313-B-029-002-MY3.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Goronzy JJ, Weyand CM. Immune aging and autoimmunity. Cell Mol Life Sci. 2012;69:1615-23
2. Lorenzoni PJ, Scola RH, Kay CS, Werneck LC. Congenital myasthenic syndrome: a brief review. Pediatr Neurol. 2012;46:141-48
3. Suzuki S, Utsugisawa K, Yoshikawa H, Motomura M, Matsubara S, Yokoyama K. et al. Autoimmune targets of heart and skeletal muscles in myasthenia gravis. Arch Neurol. 2009;66:1334-38
4. Tonin P, Lewis P, Servidei S, DiMauro S. Metabolic causes of myoglobinuria. Ann Neurol. 1990;27:181-85
5. Hellmann MA, Mosberg-Galili R, Steiner I. Myasthenia gravis in the elderly. J Neurol Sci. 2013;325:1-5
6. Al-Chalabi A, Visscher PM. Motor neuron disease: Common genetic variants and the heritability of ALS. Nat Rev Neurol. 2014;10:549-50
7. Harris TE, Huffman TA, Chi A, Shabanowitz J, Hunt DF, Kumar A. et al. Insulin controls subcellular localization and multisite phosphorylation of the phosphatidic acid phosphatase, lipin 1. J Biol Chem. 2007;282:277-86
8. Donkor J, Zhang P, Wong S, O'Loughlin L, Dewald J, Bernard P C. et al. A conserved serine residue is required for the phosphatidate phosphatase activity but not the transcriptional coactivator functions of lipin-1 and lipin-2. J Biol Chem. 2009;284:29968-78
9. Nadra K, de Preux Charles AS, Médard JJ, Hendriks WT, Han GS, Grès S. et al. Phosphatidic acid mediates demyelination in Lpin1 mutant mice. Genes Dev. 2008;22:1647-61
10. Péterfy M, Harris TE, Fujita N, Reue K. Insulin-stimulated interaction with 14-3-3 promotes cytoplasmic localization of lipin-1 in adipocytes. J Biol Chem. 2010;285:3857-64
11. Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E. et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146:408-20
12. Han GS, Carman GM. Characterization of the human LPIN1-encoded phosphatidate phosphatase isoforms. J Biol Chem. 2010;285:14628-38
13. Bou Khalil M, Sundaram M, Zhang HY, Links PH, Raven JF, Manmontri B. et al. The level and compartmentalization of phosphatidate phosphatase-1 (lipin-1) control the assembly and secretion of hepatic VLDL. J Lipid Res. 2009;50:47-58
14. Jaradat SA, Amayreh W, Al-Qa'qa' K, Krayyem J. Molecular analysis of LPIN1 in Jordanian patients with rhabdomyolysis. Meta Gene. 2015;7:90-94
15. Zeharia A, Shaag A, Houtkooper RH, Hindi T, de Lonlay P, Erez G. et al. Mutations in LPIN1 cause recurrent acute myoglobinuria in childhood. Am J Hum Genet. 2008;83:489-94
16. Pelosi M, Testet E, Le Lay S, Dugail I, Tang XT, Mabilleau G. et al. Normal human adipose tissue functions and differentiation in patients with biallelic LPIN1 inactivating mutations. J Lipid Res. 2017;58:2348-64
17. Van Harmelen V, Rydén M, Sjölin E, Hoffstedt J. A role of lipin in human obesity and insulin resistance: relation to adipocyte glucose transport and GLUT4 expression. J Lipid Res. 2007;48:201-06
18. Langner CA, Birkenmeier EH, Ben-Zeev O, Schotz MC, Sweet HO, Davisson MT. et al. The fatty liver dystrophy (fld) mutation. A new mutant mouse with a developmental abnormality in triglyceride metabolism and associated tissue-specific defects in lipoprotein lipase and hepatic lipase activities. J Biol Chem. 1989;264:7994-03
19. Péterfy M, Phan J, Xu P, Reue K. Lipodystrophy in the fld mouse results from mutation of a new gene encoding a nuclear protein, lipin. Nat Genet. 2001;27:121-24
20. Sellers RS, Mahmood SR, Perumal GS, Macaluso FP, Kurland IJ. Phenotypic Modulation of Skeletal Muscle Fibers in LPIN1-Deficient Lipodystrophic (fld) Mice. Vet Pathol. 2019;56:322-31
21. Schweitzer GG, Collier SL, Chen Z, Eaton JM, Connolly AM, Bucelli RC. et al. Rhabdomyolysis-Associated Mutations in Human LPIN1 Lead to Loss of Phosphatidic Acid Phosphohydrolase Activity. JIMD Rep. 2015;23:113-22
22. Helsinki Declaration. World Medical Association (AMM). Ethical principles for medical research involving human subjects. Assist Inferm Ric. 2001;20:104-07
23. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754-60
24. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078-79
25. Wu PY, Chuang PY, Chang GD, Chan YY, Tsai TC, Wang BJ. et al. Novel Endogenous Ligands of Aryl Hydrocarbon Receptor Mediate Neural Development and Differentiation of Neuroblastoma. ACS Chem Neurosci. 2019;10(9):4031-42
26. Westerfield M. The zebrafish book: A guide for the laboratory use of zebrafish (Danio rerio). Univ. of Oregon Press, Eugene. 2000
27. Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203:253-310
28. Smith LL, Beggs AH, Gupta VA. Analysis of skeletal muscle defects in larval zebrafish by birefringence and touch-evoke escape response assays. J Vis Exp. 2013;82:e50925
29. Tian J, Shao J, Liu C, Hou HY, Chou CW, Shboul M. et al. Deficiency of lrp4 in zebrafish and human LRP4 mutation induce aberrant activation of Jagged-Notch signaling in fin and limb development. Cell Mol Life Sci. 2019;76:163-78
30. Tian J, Yam C, Balasundaram G, Wang H, Gore A, Sampath K. A temperature-sensitive mutation in the nodal-related gene cyclops reveals that the floor plate is induced during gastrulation in zebrafish. Development. 2003;130:3331-42
31. Chou CW, Zhuo YL, Jiang ZY, Liu YW. The hemodynamically-regulated vascular microenvironment promotes migration of the steroidogenic tissue during its interaction with chromaffin cells in the zebrafish embryo. PLoS One. 2014;9:e107997
32. Grone BP, Marchese M, Hamling KR, Kumar MG, Krasniak CS, Sicca F. et al. Epilepsy, Behavioral Abnormalities, and Physiological Comorbidities in Syntaxin-Binding Protein 1 (STXBP1) Mutant Zebrafish. PLoS One. 2016;11:e0151148
33. Tian J, Ling L, Shboul M, Lee H, O'Connor B, Merriman B. et al. Loss of CHSY1, a secreted FRINGE enzyme, causes syndromic brachydactyly in humans via increased NOTCH signaling. Am J Hum Genet. 2010;87:768-78
34. Lu S, Hu M, Wang Z, Liu H, Kou Y, Lyu Z, Tian J. Generation and Application of the Zebrafish heg1 Mutant as a Cardiovascular Disease Model. Biomolecules. 2020;10(11):1542
35. Bergounioux J, Brassier A, Rambaud C, Bustarret O, Michot C, Hubert L. et al. Fatal rhabdomyolysis in 2 children with LPIN1 mutations. J Pediatr. 2012;160:1052-54
36. Burstal RJ. Volatile anesthesia for a child with LPIN1 gene mutation and recurrent rhabdomyolysis. Paediatr Anaesth. 2018;28:813-14
37. Legendre A, Khraiche D, Ou P, Mauvais FX, Madrange M, Guemann AS. et al. Cardiac function and exercise adaptation in 8 children with LPIN1 mutations. Mol Genet Metab. 2018;123:375-81
38. Meijer IA, Sasarman F, Maftei C, Rossignol E, Vanasse M, Major P. et al. LPIN1 deficiency with severe recurrent rhabdomyolysis and persistent elevation of creatine kinase levels due to chromosome 2 maternal isodisomy. Mol Genet Metab Rep. 2015;5:85-88
39. Michot C, Hubert L, Brivet M, Meirleir LD, Valayannopoulos V, Felber FM. et al. LPIN1 gene mutations: a major cause of severe rhabdomyolysis in early childhood. Hum Mutat. 2010;31:1564-73
40. Nunes D, Nogueira C, Lopes A, Chaves P, Rodrigues E, Cardoso T. et al. LPIN1 deficiency: A novel mutation associated with different phenotypes in the same family. Mol Genet Metab Rep. 2016;9:29-30
41. Pichler K, Scholl-Buergi S, Birnbacher R, Freilinger M, Straub S, Brunner J. et al. A novel therapeutic approach for LPIN1 mutation-associated rhabdomyolysis - The Austrian experience. Muscle Nerve. 2015;52:437-39
42. Spriet LL. Regulation of skeletal muscle fat oxidation during exercise in humans. Med Sci Sports Exerc. 2002;34:1477-84
43. Minton T, Forrester N, Baba SA, Urankar K, Brady S. A rare case of adult onset LPIN1 associated rhabdomyolysis. Neuromuscul Disord. 2020;30:241-45
44. Stepien KM, Schmidt WM, Bittner RE, O'Toole O, McNamara B, Treacy EP. Long-term outcomes in a 25-year-old female affected with lipin-1 deficiency. JIMD Rep. 2019;46:4-10
45. Yim SW, Chan TYC, Belaramani KM, Man SS, Wong FCK, Chen SPL. et al. Case Report: The first probable Hong Kong Chinese case of LPIN1-related acute recurrent rhabdomyolysis in a boy with two novel variants. F1000Res. 2019;8:1566
46. Zayegh A, Lee J, Peters H, Efron D. A critically ill child with dark urine. Arch Dis Child Educ Pract Ed. 2019;104:15
47. Mul JD, Nadra K, Jagalur NB, Nijman IJ, Toonen PW, Médard JJ. et al. A hypomorphic mutation in Lpin1 induces progressively improving neuropathy and lipodystrophy in the rat. J Biol Chem. 2011;286(30):26781-93
48. Jama A, Huang D, Alshudukhi AA, Chrast R, Ren H. Lipin1 is required for skeletal muscle development by regulating MEF2c and MyoD expression. J Physiol. 2019;597:889-01
49. Chen YH, Huang YH, Wen CC, Wang YH, Chen WL, Chen LC. et al. Movement disorder and neuromuscular change in zebrafish embryos after exposure to caffeine. Neurotoxicol Teratol. 2008;30:440-47
50. Remédio L, Gribble KD, Lee JK, Kim N, Hallock PT, Delestrée N. et al. Diverging roles for Lrp4 and Wnt signaling in neuromuscular synapse development during evolution. Genes Dev. 2016;30(9):1058-69
51. Buckley CE, Goldsmith P, Franklin RJ. Zebrafish myelination: a transparent model for remyelination? Dis Model Mech. 2008;1(4-5):221-8
52. Pike SH, Melancon EF, Eisen JS. Pathfinding by zebrafish motoneurons in the absence of normal pioneer axons. Development. 1992;114(4):825-31
53. Zempo B, Yamamoto Y, Williams T, Ono F. Synaptic silencing of fast muscle is compensated by rewired innervation of slow muscle. Sci Adv. 2020;6(15):eaax8382
54. Nave KA, Trapp BD. Axon-glial signaling and the glial support of axon function. Annu Rev Neurosci. 2008;31:535-61
55. Fang Y, Lei X, Li X, Chen Y, Xu F, Feng X. et al. A novel model of demyelination and remyelination in a GFP-transgenic zebrafish. Biol Open. 2014;4(1):62-8
56. Armant O, März M, Schmidt R, Ferg M, Diotel N, Ertzer R. et al. Genome-wide, whole mount in situ analysis of transcriptional regulators in zebrafish embryos. Dev Biol. 2013;380(2):351-62
57. Nadra K, de Preux Charles AS, Médard JJ, Hendriks WT, Han GS, Grès S. et al. Phosphatidic acid mediates demyelination in Lpin1 mutant mice. Genes Dev. 2008;22(12):1647-61
58. Nikolaou N, Watanabe-Asaka T, Gerety S, Distel M, Köster RW, Wilkinson DG. Lunatic fringe promotes the lateral inhibition of neurogenesis. Development. 2009;136(15):2523-33
59. Bisgrove BW, Su YC, Yost HJ. Maternal Gdf3 is an obligatory cofactor in Nodal signaling for embryonic axis formation in zebrafish. Elife. 2017;6:e28534
60. Antonellis A, Dennis MY, Burzynski G, Huynh J, Maduro V, Hodonsky CJ. et al. NISC Comparative Sequencing Program. A rare myelin protein zero (MPZ) variant alters enhancer activity in vitro and in vivo. PLoS One. 2010;5(12):e14346
61. Gallagher TL, Tietz KT, Morrow ZT. et al. Pnrc2 regulates 3'UTR-mediated decay of segmentation clock-associated transcripts during zebrafish segmentation. Dev Biol. 2017;429:225-39
62. Holterhoff CK, Saunders RH, Brito EE, Wagner DS. Sequence and expression of the zebrafish alpha-actinin gene family reveals conservation and diversification among vertebrates. Dev Dyn. 2009;238:2936-47
63. Zhang J, Lefebvre JL, Zhao S, Granato M. Zebrafish unplugged reveals a role for muscle-specific kinase homologs in axonal pathway choice. Nat Neurosci. 2004;7:1303-09
64. Gariano G, Guarienti M, Bresciani R, Borsani G, Carola G, Monti E. et al. Analysis of three μ1-AP1 subunits during zebrafish development. Dev Dyn. 2014;243:299-14
65. Ke Z, Kondrichin I, Gong Z, Korzh V. Combined activity of the two Gli2 genes of zebrafish play a major role in Hedgehog signaling during zebrafish neurodevelopment. Mol Cell Neurosci. 2008;37:388-01
66. Cornell RA, Eisen JS. Delta/Notch signaling promotes formation of zebrafish neural crest by repressing Neurogenin 1 function. Development. 2002;129:2639-48
67. Dias TB, Yang YJ, Ogai K, Becker T, Becker CG. Notch signaling controls generation of motor neurons in the lesioned spinal cord of adult zebrafish. J Neurosci. 2012;32:3245-52
68. Rashid T, Nemazanyy I, Paolini C, Tatsuta T, Crespin P, Villeneuve DD. et al. Lipin1 deficiency causes sarcoplasmic reticulum stress and chaperone-responsive myopathy. EMBO J. 2019;38:e99576
69. Holley SA, Jülich D, Rauch GJ, Geisler R, Nüsslein-Volhard C. her1 and the notch pathway function within the oscillator mechanism that regulates zebrafish somitogenesis. Development. 2002;129:1175-83
70. Wahi K, Bochter MS, Cole SE. The many roles of Notch signaling during vertebrate somitogenesis. Semin Cell Dev Biol. 2016;49:68-75
71. Buas MF, Kadesch T. Regulation of skeletal myogenesis by Notch. Exp Cell Res. 2010;316:3028-33
72. Weinberg ES, Allende ML, Kelly CS, Abdelhamid A, Murakami T, Andermann P. et al. Developmental regulation of zebrafish MyoD in wild-type, no tail and spadetail embryos. Development. 1996;122:271-80
73. Tintignac LA, Brenner HR, Rüegg MA. Mechanisms Regulating Neuromuscular Junction Development and Function and Causes of Muscle Wasting. Physiol Rev. 2015;95:809-52
74. Wu H, Lu Y, Shen C, Patel N, Gan L, Xiong WC, Mei L. Distinct roles of muscle and motoneuron LRP4 in neuromuscular junction formation. Neuron. 2012;75:94-07
75. Li L, Xiong WC, Mei L. Neuromuscular Junction Formation, Aging, and Disorders. Annu Rev Physiol. 2018;80:159-88
76. Choi HY, Liu Y, Tennert C, Sugiura Y, Karakatsani A, Kröger S. et al. APP interacts with LRP4 and agrin to coordinate the development of the neuromuscular junction in mice. Elife. 2013;2:e00220
77. Zong Y, Zhang B, Gu S. et al. Structural basis of agrin-LRP4-MuSK signaling. Genes Dev. 2012;26:247-58
Author contact
![]() Corresponding authors: Key laboratory of Resource Biology and Biotechnology in Western China, Ministry of Education, School of Medicine, Northwest University, 229 Taibai North Road, Xi'an 710069, China. Phone: 86-29-88302339; Fax: 86-29-88302411; E-mail: tianjingedu.cn (J.T.); Institute of Oceanography, Minjiang University, Fuzhou 350108, China. Phone: 86-13459201013; E-mail: chenjmedu.cn (J. C.).
Corresponding authors: Key laboratory of Resource Biology and Biotechnology in Western China, Ministry of Education, School of Medicine, Northwest University, 229 Taibai North Road, Xi'an 710069, China. Phone: 86-29-88302339; Fax: 86-29-88302411; E-mail: tianjingedu.cn (J.T.); Institute of Oceanography, Minjiang University, Fuzhou 350108, China. Phone: 86-13459201013; E-mail: chenjmedu.cn (J. C.).