Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviation
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(5):2410-2423. doi:10.7150/thno.47182 This issue Cite
Research Paper
Platelet-derived microvesicles induce calcium oscillations and promote VSMC migration via TRPV4
Shan-Shan Li1,2#, Shuang Gao1#, Yi Chen1, Han Bao1, Zi-Tong Li1, Qing-Ping Yao1, Ji-Ting Liu1, Yingxiao Wang3 ![]() , Ying-Xin Qi1,4,5
, Ying-Xin Qi1,4,5 ![]()
1. Institute of Mechanobiology& Medical Engineering, School of Life Sciences &Biotechnology, Shanghai Jiao Tong University, Shanghai, China
2. School of Perfume and Aroma Technology, Shanghai Institute of Technology, Shanghai, China
3. Department of Bioengineering, Institute of Engineering in Medicine, University of California, San Diego, San Diego, United States
4. Key Laboratory for Biomechanics and Mechanobiology of Ministry of Education, School of Biological Science and Medical Engineering, Beihang University, Beijing, 100083, China
5. Beijing Advanced Innovation Center for Biomedical Engineering, Beihang University, Beijing, 100083, China
# These authors contributed equally to this manuscript.
Received 2020-4-19; Accepted 2020-11-18; Published 2021-1-1
Abstract

Rationale: Abnormal migration of vascular smooth muscle cells (VSMCs) from the media to the interior is a critical process during the intimal restenosis caused by vascular injury. Here, we determined the role of platelet-derived microvesicles (PMVs) released by activated platelets in VSMC migration.
Methods: A percutaneous transluminal angioplasty balloon dilatation catheter was used to establish vascular intimal injury. Collagen I was used to activate PMVs, mimicking collagen exposure during intimal injury. To determine the effects of PMVs on VSMC migration in vitro, scratch wound healing assays were performed. Fluorescence resonance energy transfer was used to detect variations of calcium dynamics in VSMCs.
Results: Morphological results showed that neointimal hyperplasia was markedly increased after balloon injury of the carotid artery in rats, and the main component was VSMCs. PMVs significantly promoted single cell migration and wound closure in vitro. Fluorescence resonance energy transfer revealed that PMVs induced temporal and dynamic calcium oscillations in the cytoplasms of VSMCs. The influx of extracellular calcium, but not calcium from intracellular stores, was involved in the process described above. The channel antagonist GSK219 and specific siRNA revealed that a membrane calcium channel, transient receptor potential vanilloid 4 (TRPV4), participated in the calcium oscillations and VSMC migration induced by PMVs.
Conclusions: TRPV4 participated in the calcium oscillations and VSMC migration induced by PMVs. PMVs and the related molecules might be novel therapeutic targets for vascular remodeling during vascular injury.
Introduction
Vascular intimal injury occurs following cardiovascular disease treatments, such as bypass surgery, coronary vein graft, angioplasty and stent treatment [1]. As a consequence, intimal hyperplasia, even postangioplasty restenosis, may contribute to treatment failure [2,3]. Vascular smooth muscle cells (VSMCs), the dominant cellular component of arteries, are primarily present within the media layer of vessels under physiological conditions. However, it has been well established that abnormal VSMC media-to-intima migration is an important cellular process during intimal hyperplasia [4-6]. Therefore, studying the mechanism of abnormal VSMC migration during intimal hyperplasia is of great significance for improving the cure rate of cardiovascular disease.
Adult VSMCs retain the potential to alter their migrative properties, and this process can be regulated by extracellular matrix components, peptide growth factors, cytokines, RNA molecules, mechanical factors, ion signaling and other environmental cues [7-9]. For example, metalloproteinase-2 (MMP2) and metalloproteinase-9 (MMP9) participate in VSMC migration from the media to the intima following arterial injury and alter postinjury vascular remodeling [10,11]. Wu et al. revealed that kindlin-2 plays a critical role in VSMC proliferation, migration and intimal hyperplasia via Wnt signaling, and blocking the activity of kindlin-2 is an attractive therapeutic approach for vascular injury [12]. Blood flow and shear stress have also been found to abrogate the proliferative and migratory response of VSMCs in the early stages after injury [13,14]. In addition to these regulatory responses, numerous studies have revealed that the regulation of cell migration is critically dependent on calcium. Chemoattractants stimulate neutrophil migration by inducing repeated transient increases in intracellular calcium level [15]. Excitatory neurotransmitters initiate cell contraction, which is the key process in cell migration, by interacting with cell surface receptors to generate inositol 1,4,5-trisphosphate (IP3) [16]. IP3 binds to inositol 1,4,5-trisphosphate receptors (IP3Rs) on the sarcoplasmic reticulum to trigger calcium release, which sustains elevated levels of cytoplasmic calcium to regulate colonic smooth muscle contraction.
Although the studies described above have revealed many important factors that affect VSMC migration, the underlying mechanisms during intimal hyperplasia are still not fully understood. During vascular intimal injury, the adherence and accumulation of circulating platelets to the injured intima is an important pathological process [17,18]. Circulating platelets can be activated by exposure to collagen (mainly collagen I), which is caused by the injury of monolayer endothelial cells (ECs). Activated platelets can release a variety of heterogeneous platelet-derived microvesicles (PMVs) [17], which are mainly 100-1000 nm in diameter [19-21]. PMVs are capable of selectively carrying different types of biomolecules, such as membrane and cytoplasm proteins, lipids, RNAs, and other bioactive molecules, and transferring these biomolecules to recipient cells, thus participating in the regulation of recipient cell functions [17,18,22]. It has been reported that thrombin/collagen-induced PMVs can enhance the potential of early outgrowth cells (EOCs) to restore endothelial integrity by transferring chemokine receptor-4 (CXCR4) to EOCs after vascular injury [23].
PMVs have been reported to play critical roles in various cardiovascular diseases, including hypertension, atherosclerosis and thrombin formation [18,24]. Under conditions of intimal injury, the internal elastic lamina can be broken, and PMVs can thus directly contact the middle layer [25]. Even under conditions of mild intimal injury, the EC layer can be broken, and the fenestrae on the internal elastic lamina (1~3 µm in width) [26] can allow PMVs (10~1000 nm in diameter) to diffuse into the middle layer. However, the relationship between PMVs and VSMC migration during intimal hyperplasia remains poorly characterized. In this study, collagen I was used to activate PMVs, mimicking collagen exposure during intimal injury, and the roles of PMVs in VSMC migration were demonstrated. Our bioinformatic analysis, which was based on previously published proteomic data of collagen-induced PMVs [27], suggested that calcium may be the key node in the regulatory network of PMV-induced cell migration. We investigated the calcium dynamics induced by PMVs in live VMSCs at the single-cell and subcellular levels by using fluorescence resonance energy transfer (FRET) combined with a calcium biosensor. We then further identified the potential calcium channels that participate in PMV-induced calcium oscillations in VSMCs.
Materials and Methods
Animal model
The animal care and experimental protocols were conducted in accordance with the Animal Management Rules of China (55, 2001, Ministry of Health, China), and the study was approved by the Animal Research Committee of Shanghai Jiao Tong University.
Male Sprague Dawley rats were housed in a temperature-controlled room with a 12-h light/dark cycle and were given access to standard chow and tap water ad libitum. Vascular injury was established in 8-week-old rats, as previously described [3]. Briefly, a percutaneous transluminal angioplasty balloon dilatation catheter (Boston Scientific Corporation, Galway, Ireland) was used to establish vascular intimal injury. All the animals were anesthetized by isoflurane inhalation and treated under sterile conditions. The balloon was placed into the left carotid artery and repeatedly stretched to damage the blood vessels. The undamaged right carotid artery served as the self-control [3].
HE and immunofluorescence staining
Four weeks after balloon injury, the arteries were removed, fixed in 4% paraformaldehyde solution, embedded, and finally cut into 8-μm sections. HE staining [28] or immunofluorescence staining [29] was performed as previously described. For immunofluorescence staining, the paraffin-embedded sections were permeabilized with 0.1% Triton X-100 for 10 min. After treatment with phosphate-buffered saline (PBS) containing 10% goat serum for 1 h, the sections were coincubated with primary rabbit anti-von Willebrand Factor antibody (1:200, Dako, Copenhagen, Denmark A/S) and mouse monoclonal anti-α-SMA antibody (1:200, Dako, Copenhagen, Denmark A/S) for 24 h at 4°C. Secondary anti-mouse/rabbit IgG antibodies (1:1000, Cell Signaling Technology, Boston, MA) were used. The nuclei were stained with DAPI after immunofluorescence staining. The fluorescence images were acquired by using a fluorescence microscope (Olympus, Tokyo, Japan).
PMV isolation, collection and size analysis
PMVs were isolated according to the previous literatures [30,31]. As shown in Supplementary Figure 1A, whole blood was collected from the abdominal aorta of anesthetized rats into a syringe containing 100 μL/mL anticoagulant in 0.5% sodium chloride solution. The blood was centrifuged at 1500 rpm for 10 min to obtain platelet-rich plasma and then centrifuged at 2800 rpm for 15 min to obtain platelets. The platelets were resuspended using Tyrode solution and activated with collagen I at 37°C. After activating for 90 min, the solution was centrifuged at 2800 rpm for 15 min to remove the platelets, and the PMVs were collected at 20500 g/min for 90 min. The PMVs were analyzed with a flow cytometry column (FACSCalibur TM; BD Biosciences) combined with a platelet-specific marker (anti-rat CD41) to demonstrate the platelet origin[30,31], and with a NanoSight3000 high-sensitivity detection system (Malvern Panalytical, Malvern, England) to analyze the particle size.
PMV treatment
After the carotid artery balloon injury model was established, PMVs (6×109 per ml) or normal saline (as a control group) were injected into the tail veins of the rats on alternate days for 2 weeks after the surgery. On day 14 after the surgery, the injured carotid artery was collected.
Cell culture and transfection
VSMCs were isolated from the thoracic aortas of male Sprague Dawley rats via an explant method, as previously described [32]. The VSMCs were cultured in Dulbecco's modified Eagle's medium (DMEM) (GIBCO, NY, USA) with 10% calf serum (FCS, GIBCO, NY, USA), 2 mM glutamine (GIBCO, NY, USA), 100 U/mL penicillin and 100 μg/mL streptomycin (BBI Company, Shanghai, China) at 37°C in a 5% CO2 atmosphere. The VSMCs were characterized by antibody specific for α-smooth muscle actin (1:1000, Dako, Copenhagen, Denmark A/S). In all the experiments, the purity of the cell populations was universally higher than 95%, and only VSMCs between passages 4 and 7 were used.
For the transfection process, a FRET-based calcium biosensor, which includes a pair of fluorescent proteins (ECFP and YPet) and calmodulin-linked light chain protein kinase M13, was utilized. The structure of the biosensor is altered after binding to calcium ions [33]. Adenovirus vectors (Genepharma, Shanghai, China) were used to introduce the reconstructed plasmid containing the FRET-based calcium biosensor into VSMCs.
Microscopy, image acquisition and analysis
Cells were starved in DMEM without FCS for 6 h before PMV stimulation. Then, PMVs were gently added to the culture dish. During the imaging experiments, the cells were maintained in streptomycin-free medium to prevent possible effects on calcium ion channels. All the images were obtained by using a Leica inverted microscope (DMi8, Wetzlar, Germany) equipped with a charge-coupled device (CCD) camera (Andor iXon 897, Belfast, UK) and two emission filters controlled by a filter changer (480DF20 for ECFP and 535DF15 for YPet). During the image capturing process, a temperature-control system with CO2 supplement was used to maintain cellular viability. Time-lapsed fluorescence images were acquired at 30-s intervals by Leica LASX software (Leica Biosystems GmbH). The emission ratio of FRET/ECFP was directly computed and generated by the Leica LASX software before being subjected to quantification and analysis. The max ratio and calcium oscillation frequency were further analyzed.
Scratch wound healing assay
To assess VSMC migration in vitro, scratch wound healing assays were performed. A wound healing cell migration assay was performed using 95% confluent cells, as described in previous studies [34,35]. A line was scratched across a monolayer of cells using a sterile 10-μL pipette tip. Images of the scratched line were immediately captured, and the cells were imaged again after 12 h and 24 h (4X objective, IX-71, Olympus, Japan). The wound area was measured using ImageJ software (NIH, USA). The cell migration abilities were calculated as (S0 - St)/ S0, where S0 is the wound area at the initial time point, and St is the wound area at the observation time point t (12 h or 24 h).
Solutions and chemicals
For the experiments that required calcium-free conditions, calcium-free DMEM (GIBCO, NY, USA) was used. The chemical reagents 2-aminoethoxydiphenyl borate (2-APB) (0.1 mM) [36] and nifedipine (10 μM) [37] were purchased from Sigma-Aldrich (St. Louis, MO). GSK219 (0.1 mM) [38] was obtained from Merck (NY, USA). All the drugs were preincubated with VSMCs for 20 min before the addition of the PMVs. The amount of drug administered was based on previous publications.
RNA interference
For RNA interference, VSMCs were transfected with 50 nM small interfering RNA (siRNA) fragments (si-1/2/3) or control nonsilencing siRNA (si-NC) (Genepharma, Shanghai, China) for 24 h using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions [39]. The sequences of the small interfering RNA fragments targeting TRPV4 (sequence accession number NM_023970.1) and si-NC are listed in Table S1.
Western blot
Lysates were separated by 10% SDS-PAGE. The proteins were detected using primary antibodies against TRPV4 (1:500, Alomone Labs, Jerusalem, Israel) and GAPDH (1:1000, Protein tech, Beijing, China). An HRP-labeled IgG was used as the secondary antibody at a 1:1000 dilution, and the bands were visualized using an ECL kit (Beyotime, Shanghai, China) and quantified with Quantity One software (Bio-Rad, Hercules, CA, USA).
Statistical analysis
All the values are expressed as the mean ± standard error (S.E.M.) of the mean, and all the data are consistent with variance after R studio software. The statistical analysis of the data was performed by unpaired Student's t-test to identify significant differences between two mean values, and a value of P < 0.05 was considered statistically significant. The statistical analysis was performed using Excel (Microsoft Corporation, Washington, USA) and GraphPad 9.0 (GraphPad Software, San Diego, CA).
Results
Vascular intimal injury promotes VSMC migration and neointimal hyperplasia
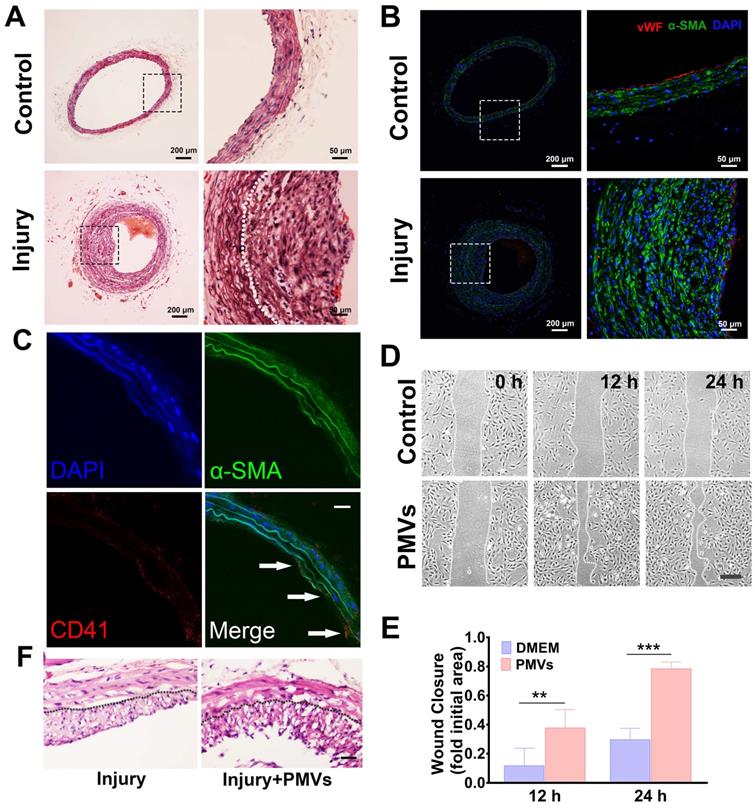
Four weeks after intimal injury, HE staining revealed that compared with that of the self-contralateral noninjured artery, the neointimal layer of the intimal injured artery was markedly thickened, and the area of the vascular cavity was markedly reduced (Figure 1A). In addition, immunofluorescence staining revealed the expression of α-smooth muscle actin (α-SMA) in neointimal hyperplasia, which suggested that the main component of the vascular neointima was VSMCs (Figure 1B). During vascular intimal injury, collagen exposure activates platelets, which accumulate and release a variety of heterogeneous vesicles, including PMVs. In vivo immunofluorescence staining for CD41 (a marker of PMVs [40]) and α-SMA (a marker of VSMCs [41]) showed that CD41-positive particles were closely adjacent to α-SMA-positive cells, indicating that PMVs accumulate at the injured artery and contact VSMCs in vivo (Figure 1C). We then demonstrated the role of PMVs in VSMC migration and the potential mechanisms.
Increased VSMC migration after intimal injury in vivo and PMV treatment in vitro and in vivo. (A) HE staining revealed that the neointima was markedly thickened 4 weeks after vascular intimal injury compared with the self-artery control (control). The white dotted line represents the inner elastic fibers. Scale bar: 200 μm (left panel); 50 μm (right panel). (B) Immunofluorescence staining revealed that neointimal hyperplasia mainly consists of VSMCs. VSMCs were identified by α-SMA (green), and ECs were identified by von Willebrand factor (vWF, red). vWF, a marker of ECs, was discontinuously expressed on the lumen of damaged blood vessels in the intimal injury models (bottom panel). Cell nuclei were stained with DAPI (blue). Scale bar: 200 μm (left panel); 50 μm (right panel). (C) PMVs adhered to VSMCs in neointimal hyperplasia in vivo. Twenty-four hours after injury, carotid arteries were collected and then examined by immunofluorescence staining. PMVs were identified by CD41 (red), and VSMCs were identified by α-SMA (green). Cell nuclei were stained with DAPI (blue). The white arrows indicate typical locations where PMVs adhere to VSMCs. Scale bar: 50 μm. (D) The migration of VSMCs stimulated with DMEM control or PMVs for 12 h/24 h, as detected by the wound healing assay. Scale bar: 200 μm. (E) The histogram shows the fold change in the level of VSMC migration stimulated by PMVs (n = 8) relative to that stimulated by the DMEM control (n = 7). The values are shown as the mean ± S.E.M. for each condition. ** P < 0.01, *** P < 0.001. (F) PMVs promote neointimal hyperplasia in injured carotid arteries on Day 14 after surgery. Scale bar: 100 μm.
After using collagen I to simulate the pathological exposure of extracellular matrix components during intimal injury, PMVs were extracted, and the diameter of the PMVs was further examined by NanoSight3000. The results illustrated that the sizes of the PMVs were distributed within a range of 100~600 nm, and there were 3 peaks at 121.6 nm, 183.0 nm and 291.5 nm (Figure S1).
PMVs can adhere to VSMCs in vitro (Figure S2A). Our scratch wound data showed that PMVs induced a marked increase in the wound repair capabilities of VSMCs at 12 h and 24 h (Figure 1D). The averaged wound closure areas at 24 h are 78.42% and 29.64% smaller than the initial area at 0 h for the PMVs group and the control, respectively (Figure 1E). Moreover, the track results of the free migration of single cells revealed that compared with the control (Figure S2B, Video S1A), PMV treatment increased the migration distance of single VSMCs in a time-dependent manner, and a significant increase was detected at approximately 10.5 h (Figure S2B, Video S1B).
These results suggested that the PMVs released by collagen I-activated platelets promote the migration of VSMCs, which may participate in the intimal hyperplasia caused by intimal injury in vivo. We further conducted in vivo experiments to investigate PMV-induced neointimal hyperplasia in arteries. First, a carotid artery balloon injury model was established. Then, PMVs (6×109 per 1 mL) or normal saline (as a control group) were injected through the tail veins of the rats on alternate days for 2 weeks after the surgery. Fourteen days after the surgery, the injured carotid artery was collected. Neointimal hyperplasia was examined with HE staining. As shown in Figure 1F, the thickness of the neointimal layer of the rats was considerably increased by PMVs.
PMVs induce calcium oscillations in VSMCs
Based on previously published proteomic data [27], IPA software was used to analyze the potential mechanism by which PMVs participate in VSMC migration. The results revealed that 31 proteins expressed in PMVs were correlated with calcium signaling (Figure 2A, Table S2), and cellular movement was one of the critical identified functions that are regulated by PMVs (Figure 2B). Then, the spatiotemporal characteristics of the calcium dynamics in VSMCs treated with PMVs were characterized with the aid of FRET real-time microscopy.
Intercellular calcium oscillations in VSMCs in response to PMVs. (A) The proteomic data were analyzed by IPA, which revealed that calcium signaling may be the key node in regulating cell migration. (B) Bar graphs demonstrate the top cell and disease functions regulated by PMV-expressed proteins. (C) Time-lapse FRET images of changes in cytoplasmic calcium in VSMCs treated with PMVs and the DMEM control. The hot and cold colors represent high and low FRET ratios, indicating high and low levels of cytoplasmic calcium change, respectively. Scale bar: 30 μm. (D) The time courses represent the normalized FRET/ECFP ratio averaged over the cell body in VSMCs treated with PMVs (n = 23) and DMEM (n = 10), and all shadowed areas indicate the S.E.M. Comparison of the max normalized FRET/ECFP ratio (E) and frequency of cytoplasmic calcium oscillations (F) between VSMCs treated with PMVs (n = 23) and those treated with DMEM (n = 10). The data are expressed as the mean ± S.E.M. ** P < 0.01.
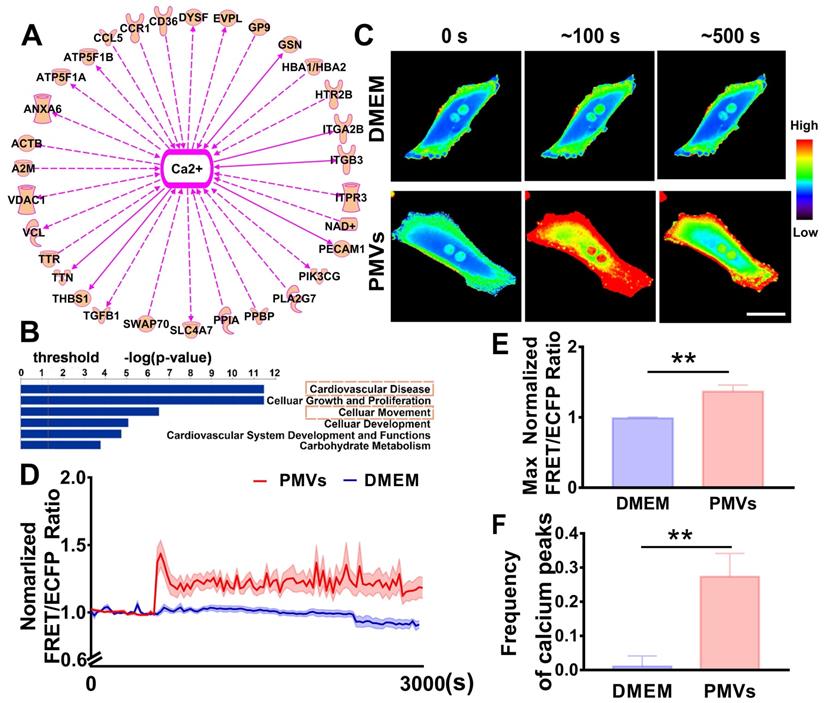
The representative heat map of the FRET/ECFP ratio showed that compared with the DMEM control, PMVs triggered marked increases in the calcium levels in live VSMCs (Figure 2C, Video S2A-B). The average FRET/ECFP ratio increased by ~1.5-fold within 60 s and fluctuated at high levels for a period of 3000 s in VSMCs treated with PMVs (Figure 2D). Furthermore, the maximum ratio of FRET/ECFP in response to PMV treatment also significantly increased (1.3646 ± 0.0934 vs. 0.9869 ± 0.0149) (Figure 2E), and the frequency of calcium peaks upon PMV stimulation was significantly increased with those induced by the DMEM control (0.2733 ± 0.0681 vs. 0.0105 ± 0.0307) (Figure 2F). Emission spectral analysis showed that after PMV treatment, the ECFP emission peak at 475 nm decreased, and the YPet emission peak at 525 nm increased. The results revealed that PMVs enhanced YPet emission at the expense of ECFP emission, indicating a calcium-induced gain of FRET (Figure S3).
Calcium peaks that increase in amplitude modulation and frequency modulation are defined as calcium oscillations [42], and these results suggested that PMVs induced calcium oscillations in target VSMCs. There are two main sources of cytosolic calcium, the influx of calcium from the extracellular matrix and the release of calcium from intracellular stores; thus, the role of these two sources in PMV-induced calcium oscillation were explored in further studies.
Extracellular calcium participates in calcium oscillations and VSMC migration
To identify the calcium source, calcium-free medium was used to remove the extracellular calcium. The results showed that the calcium oscillations induced by PMV treatment were immediately abrogated in the calcium-free medium (Figure 3A, Video S3A), and the time-lapsed FRET/ECFP ratio line was horizonal and steady throughout the time course (Figure 3B). Compared with the DMSO control, PMV treatment led to significantly decreased frequencies of calcium peaks (0.0733 ± 0.0281 vs. 0.2733 ± 0.0681) and max FRET/ECFPs ratios (1.265 ± 0.0690 vs 1.8810 ± 0.1580) in the calcium-free medium (Figure 3C-D). Moreover, the wound healing assay revealed that the calcium-free medium also markedly reversed the migration of VSMCs induced by PMVs at 12 h and 24 h (Figure 3E-F).
PMV-induced calcium oscillations in VSMCs and wound closure in calcium-free medium or 2-APB-containing medium. (A) FRET images of calcium upon PMV treatment of VSMCs pretreated with DMSO control (top), 2-APB (middle) and calcium-free medium (bottom). The hot and cold colors represent high and low FRET ratios, indicating high and low levels of cytoplasmic calcium change, respectively. Scale bar: 30 μm. (B) The time courses represent the normalized FRET/ECFP ratio averaged over the cell body in VSMCs pretreated with 2-APB (n = 14, blue), calcium-free medium (n = 19, red) and DMSO control (n = 10, orange) after PMV treatment, and all shadowed areas indicate the S.E.M. Comparation of the max normalized FRET/ECFP ratio (C) and frequency of cytoplasmic calcium oscillations (D) among VSMCs pretreated with 2-APB (n = 14, blue), calcium-free medium (n = 19, red) and DMSO control (n = 10, orange) after PMV treatment. (E) The migration of VSMCs pretreated with DMSO control (top), 2-APB (middle) and calcium-free medium (bottom) after PMV treatment. Scale bar: 200 μm. (F) The histogram shows the fold change in the level of 2-APB- and calcium-free medium-pretreated VSMC migration relative to the DMSO control-pretreated VSMC migration. The values are shown as the mean ± S.E.M. for each condition (n = 6). * P < 0.05, ** P < 0.01, *** P < 0.001.
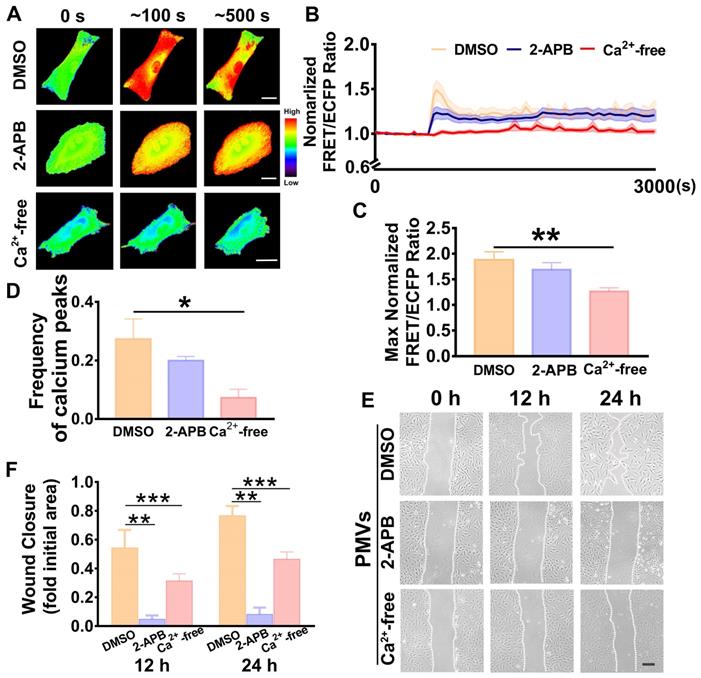
Binding of IP3 to IP3Rs on the sarcoplasmic reticulum triggers the release of intracellular calcium [16]. Hence, 2-APB, an antagonist of IP3Rs [36], was used to block the release of calcium from intracellular stores. The representative FRET/ECFP ratio maps are shown in Figure 3A (Video S3B-C). There was no significant difference in the average FRET/ECFP ratio (Figure 3B), the frequency of calcium peaks (0.2000 ± 0.014) (Figure 3C) or the max ratio (1.691 ± 0.1347) (Figure 3D) of the calcium dynamics between the 2-APB and DMSO control groups (Figure 3C-D).
These results suggested that oscillations were diminished by calcium-free medium, indicating that the influx of extracellular calcium was an important source of the calcium oscillations in the VSMCs.
TRPV4 channel, but not L-type voltage-gated calcium channel, mediates PMV-induced calcium oscillations
Since transient receptor potential vanilloid 4 (TRPV4) and L-type voltage-dependent calcium channel (L-VDCC) are widely reported to be abundantly expressed in VSMCs and related to important functions [42-44], the possible roles of these two channels that regulate the influx of extracellular calcium were further demonstrated.
GSK219 (GSK2193874, 0.1 mM), the specific antagonist of TRPV4, significantly reduced the calcium oscillations in response to PMV treatment (Figure 4A-D, Video S4B). Compared with the DMSO solvent control, GSK219 significantly decreased the max ratio of FRET/ECFP and the frequency of VSMC calcium peaks, and these values were 1.1770 ± 0.0635 vs. 1.8420 ± 0.1642 and 0.02 ± 0.0013 vs. 0.36 ± 0.098, respectively (Figure 4C-D, Video S4A-B). VSMC wound closure was suppressed at 12 h and 24 h by preincubation with GSK219 (Figure 4F). Nifedipine (Nife), the specific chemical inhibitor of L-VDCC, was used. Compared with the DMSO solvent control (Video S4A), Nife (10 μM) had no significant effect on the calcium oscillations (Figure 4A-B, Video S4C), the max ratio of FRET/ECFP (1.6341 ± 0.0633 vs. 1.8420 ± 0.1642) or the frequency (0.2901 ± 0.1056 vs. 0.36 ± 0.098) of the VSMC calcium peaks induced by PMVs. The wound closure rates induced by Nife and the solvent DMSO were also similar (Figure 4E-F).
The PMV-induced calcium oscillations in VSMCs and wound closure are abolished by the TRPV4 antagonist GSK219 but not by Nife. (A) The color images represent the time-lapse FRET images of the changes in cytoplasmic calcium upon PMV treatment of VSMCs pretreated with GSK219 (middle), Nife (bottom) and the DMSO control (top). The hot and cold colors represent high and low FRET ratios, indicating high and low levels of cytoplasmic calcium change, respectively. Scale bar: 30 μm. (B) The time courses represent the normalized FRET/ECFP ratio averaged over the cell body in VSMCs pretreated with GSK219 (n = 19, blue), Nife (n = 8, red) and DMSO (n = 12, orange) after PMV treatment, and all shadowed areas indicate the S.E.M. Comparation of the max normalized FRET/ECFP ratio (C) and frequency of cytoplasmic calcium oscillations (D) among VSMCs pretreated with GSK219 (n = 19, blue), Nife (n = 8, red) and DMSO control (n = 12, orange) after PMV treatment. (E) The migration of VSMCs pretreated with GSK219 (middle), Nife (bottom) and DMSO control (top) after PMV treatment. Scale bar: 200 μm. (F) The histogram shows the fold change in the level of GSK219- and Nife-pretreated VSMC migration relative to the that of DMSO control-pretreated VSMC migration. The values are shown as the mean ± S.E.M. for each condition (n = 6). * P < 0.05, ** P < 0.01. (G) The protein level of TRPV4 was increased after PMV treatment for 24 h. *** P < 0.001 vs. DMEM control (n = 5). (H) Immunofluorescence staining of TRPV4 (red) and nuclei (blue) of VSMCs after treatment with PMVs (top panel) or the DMEM control (bottom panel) for 5 min. Arrows indicate typical regions of dense and punctate TRPV4 staining. Scale bar: 40 μm. (I) TRPV4 inhibitor GSK219 attenuated neointimal hyperplasia in injured carotid arteries on Day 28 after the surgery. Scale bar: 50 μm.
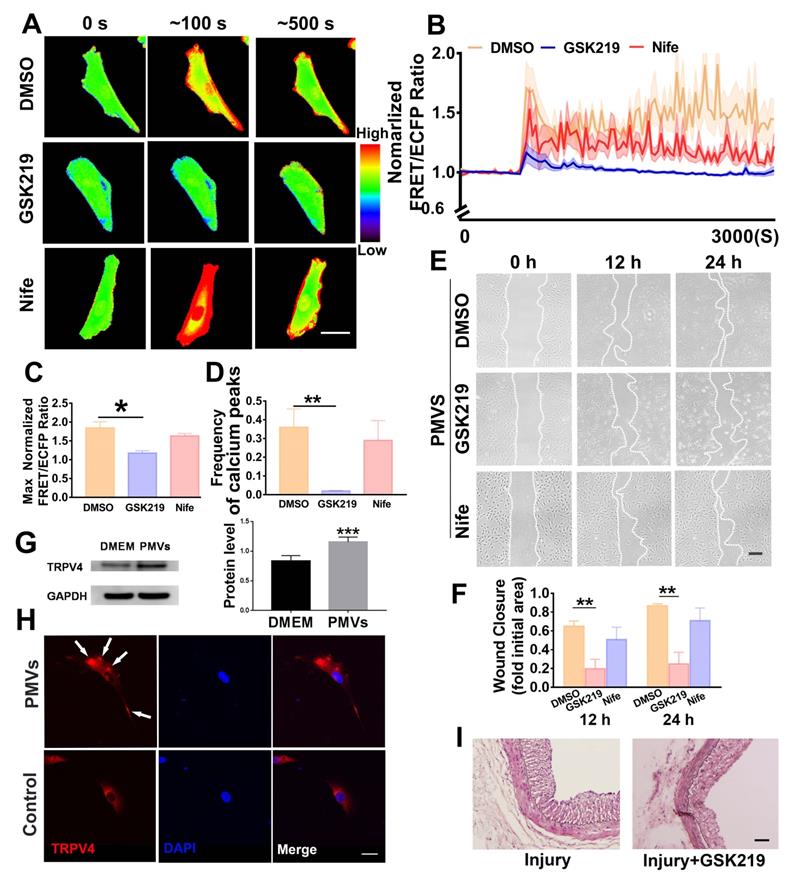
The change in TRPV4 protein expression in VSMCs before and after PMV treatment was examined with Western blot assays. The Western blot results indicated that the protein level of TRPV4 was increased after PMV treatment for 24 h (Figure 4G). Immunofluorescence staining demonstrated that the TRPV4 ion channels were distributed more densely in punctate regions on the cell membrane after PMV treatment for 5 min (Figure 4H).
We further conducted in vivo experiments to investigate if GSK219 ameliorates neointimal hyperplasia in rats. GSK219 (1 mg/kg) or normal saline (as a control group) was intraperitoneally injected 1 h before carotid artery balloon injury and on day 7, day 14 and day 21 after the surgery. On Day 28 after the surgery, the injured carotid artery was collected. Neointimal hyperplasia was examined by HE staining. As shown in Figure 4I, the thickness of the neointimal layer in the rats was considerably decreased by GSK219.
These results suggested that TRPV4, a member of the transient receptor potential (TRP) family, participates in the influx of extracellular calcium induced by PMVs, which may subsequently modulate VSMC migration.
TRPV4 knockdown abolishes calcium oscillations and cell migration
To further elucidate whether the effects of PMVs on VSMC migration and calcium oscillation were dependent on TRPV4, TRPV4 small interference RNA (siRNA) was transfected into VSMCs to knock down TRPV4. Then, FRET and wound healing were assessed. Three pairs of specific siRNAs were designed, and the most efficient siRNA, si-1, was identified (Figure 5A).
TRPV4 siRNA abolished calcium oscillations and VSMC wound closure. (A) Western blot results indicate the interference efficiencies of 3 TRPV4 siRNA sequences. (B) Time-lapse FRET images of the chances in cytoplasmic calcium in VSMCs transfected with TRPV4 si-1-Cy3 or si-NC during the PMV treatment process. The hot and cold colors represent high and low FRET ratios, indicating high and low levels of cytoplasmic calcium change, respectively. Scale bar: 30 μm. (C) The time courses represent the normalized FRET/ECFP ratio averaged over the cell body in VSMCs transfected with TRPV4 si-1-Cy3 (n = 16) or si-NC (n = 13) after PMV treatment, and all the shadowed areas indicate the S.E.M. Comparation of the max normalized FRET/ECFP ratio (D) and frequency of the cytoplasmic calcium oscillations (E) between the VSMCs transfected with TRPV4 si-1-Cy3 (n = 16) or si-NC control (n = 13) after PMV treatment. (F) The migration of VSMCs transfected with TRPV4 si-1 or si-NC control after 24 h of PMV treatment. Scale bar: 200 μm. (G) The histogram shows the fold change in the level of TRPV4 si-1-transfected VSMC migration relative to that of the si-NC control-transfected VSMC migration. The values are shown as the mean ± S.E.M. for each condition (n = 6). ** P < 0.01, **** P < 0.0001.
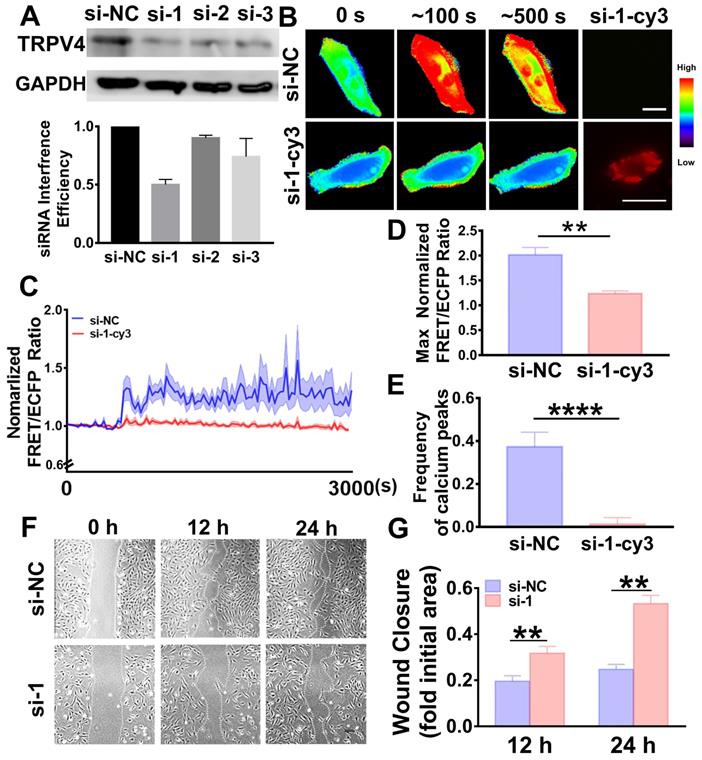
For further study, si-1 was labeled with the red fluorescent probe Cy3 (si-1-Cy3) and used to verify the effective silencing of TRPV4. Heat maps of the FRET/ECFP ratio revealed that in the si-1-Cy3 positive cells, the calcium oscillations were remarkably abolished and exhibited a steady state (Figure 5B, Video S5B); these results were not observed in the si-NC-positive cells (Figure 5B, Video S5A). After PMV treatment, the max ratio of FRET/ECFP was 1.2341 ± 0.0544 and the frequency of calcium peaks was 0.0125 ± 0.0307 in the si-1-Cy3-positive cells, indicating that these effects were all significantly abolished (Figure 5D-E); these results were not observed in the si-NC-positive cells (the max ratio was 2.0141 ± 0.1464 and the frequency was 0.3733 ± 0.06806). VSMC migration was also significantly suppressed by TRPV4 si-1 transfection (Figure 5F-G), and similar results were observed after incubation with the specific inhibitor GSK219.
Here, we validated that TRPV4 in VSMCs is essential for eliciting the calcium oscillations and VSMC migration triggered by PMVs.
Discussion
PMVs are submicroscopic (∼<1000 nm) membrane vesicles released by platelets during activation and selectively carry different molecules, including GP IIb/IIIa, GP Ib, P-selectin, and CXCR4 [45]. Low concentrations of PMVs are observed in normal circulation. Highly increased concentrations of PMVs may be an important indicator of thrombosis formation [18,24], atherosclerosis, hypertension and cardiopulmonary bypass [24]. PMVs have been studied in the pathogenesis of many diseases. Kim et al. reported that in vitro, PMVs promote human umbilical vein endothelial cell (HUVEC) survival and stimulate migration and elicit tube-like structure formation in angiogenesis [46]. PMVs stimulate p42/p44 MAP kinase phosphorylation, cellular oncogene Fos (c-Fos) induction and DNA synthesis in a concentration-dependent manner to promote the proliferation and migration of coronary artery smooth muscle cells under conditions of thrombosis formation [47]. However, the molecular mechanisms underlying these functions of PMVs are still largely unknown.
It has been reported that platelets, which immediately accumulate at the sites of injury once the intact endothelium is damaged during vascular injury, become activated and release PMVs [48]. Our present research revealed that PMVs promote the migration of VSMCs, which may participate in neointimal hyperplasia during vascular injury, and calcium is the crucial molecule underlying this process.
Calcium is the simplest and most versatile second messenger involved in regulating various cellular functions, both under physiological and pathological conditions [49-51]. The calcium levels in the cytoplasm are usually low and become significantly increased when cells respond to stimulation, such as stimulation by growth factors and mechanical factors. In the present study, the increased calcium levels have been demonstrated as calcium influx from the extracellular environment. Increased calcium levels lead to further calcium binding to calmodulin to form a complex that activates downstream pathways, such as phosphatidylinositol 3-kinase (PI3K), calcium-dependent protein kinases II (CaMKII), and myosin light chain kinase (MLCK) [38]. As a downstream molecule of calcium, CaMKII promotes VSMC migration during vascular injury through the posttranscriptional regulation of MMP9 [11,52]. Increased calcium activates PI3K/threonine-specific protein kinase (Akt) signaling via calmodulin in different cell lines [53,54]. In addition, MLCK causes changes in the fluorescence emission and the polarization excitation spectra of the enzyme by binding to calmodulin [55]. Due to its multifaceted roles, calcium signaling is the crucial coordinator of cell migration, this regulation occurs partly through local calcium pulses activating MLCK and modulating nascent focal adhesions [56]. In regulating the persistence of cell movement, calcium levels are highest at the rear and lowest at the leading edge of cells [57]. Previous literatures have demonstrated that extracellular calcium influx can active small GTPases Cdc42 [58], Rac1 [59] and RhoA [60], and thus promote cell migration. Here we found that the protein levels of all these three molecules were significantly increased by the PMVs (Figure S5), which suggested that extracellular calcium influx induced by PMVs may regulate VSMC migration via small GTPases.
The concentration and real-time distribution of calcium can be visualized at the molecular level in single live cells. In our present study, a calcium biosensor based on FRET was used to detect the calcium dynamics induced by PMVs. The genetically encoded calcium biosensor based on FRET consists of a pair of fluorescent indicators [enhanced cyan fluorescent protein (ECFP) and a yield YFP for energy transfer (YPet)] and calmodulin-linked light chain protein kinase M13, and this biosensor has been shown to be a useful tool to characterize intracellular cytoplasmic calcium in live cells [63]. Upon increased binding of free calcium to calmodulin-linked light chain protein kinase M13, conformational changes lead to a decrease in the distance between the fluorescent protein pairs and an increase in FRET. The changes in the FRET/ECFP ratio are used to characterize the calcium dynamics in live cells.
VSMCs express a variety of ion channels in their cell membranes that mediate calcium influx in response to many environmental stimuli [64]. However, the identity of plasma membrane-associated calcium permeable pathways has not been reported in PMV-stimulated VSMCs. Among the calcium ion channels on VSMCs, the TRPV4 channels play crucial roles in regulating cellular functions [65,66]. TRPV4 channels are calcium-permeable nonselective cation channels on the cell membrane that are widely expressed in the cardiovascular system, including on endothelial cells, cardiac fibroblasts, and VSMCs [67]. In the present study, TRPV4 were indicated abundantly expressing in the neointimal hyperplasia (Figure S6). Hu et al. revealed that TRPV4 channels mediate the FSS-induced calcium influx and osteogenic differentiation of MSCs, and these effects were inhibited by the selective TRPV4 inhibitor HC-067047 and TRPV4-specific siRNA [32]. TRPV4 is required for the transforming growth factor-β (TGF-β)-induced differentiation of cardiac fibroblasts into myofibroblasts, which is critically involved in cardiac remodeling [68]. Activation of the TRPV4 channel at the plasma membrane appears to reflect the activation of existing channel structures with conformational changes within the homotetrameric structure that lead to channel opening [69]. Cao et al. found that protein kinase A-mediated Ser-824 phosphorylation is required for TRPV4 activation in endothelial cells and other systems [29]. Note that the 2-APB investigated in the present study (Figure 3) is a typical inhibitor of IP3R that blocks intracellular calcium release from the endoplasmic reticulum [36,63] and thus suppresses cell migration. The FRET results indicate that 2-APB does not significantly affect the PMV-induced calcium increase, which further demonstrates that the calcium oscillation in the present study mainly occurs due to the influx of extracellular calcium. To reveal the mechanism of the effect of PMVs on TRPV4, we first conducted bioinformatics analysis with IPA software (Supplement Figure S7), and the results suggested that AGTR1, P2RY1 and Pka are the most important regulatory proteins upstream of TRPV4. Based on the 457 proteins previously reported to be expressed in PMVs by Dean et al. [27], the bioinformatics analysis suggested that 31 potential molecules in PMVs may participate in calcium regulation (Supplement Table 1). We further analyzed the connections between the three regulatory proteins upstream of TRPV4 (AGTR1, P2RY1 and Pka) and molecules from PMVs. As shown in Supplement Figure S7, the results identified the following possible pathways: 1) TGFB1 in PMVs affected TRPV4 by regulating P2RY1 [70], and 2) CCL5 and CD36 in PMVs affected TRPV4 by regulating Pka [71,72]. The effects of microvesicles from endothelial cells and VSMCs on calcium oscillations and TRPV4 channel activation in VSMCs were examined with FRET, and the results are presented in Figure S8-S10. These results suggested that the regulatory mechanism of PMVs described above could not be the same in endothelial cell microvesicles or VSMC microvesicles. However, it still requires further studies to determine the specific stimulating composition in PMVs in order to fully understand the regulating mechanisms.
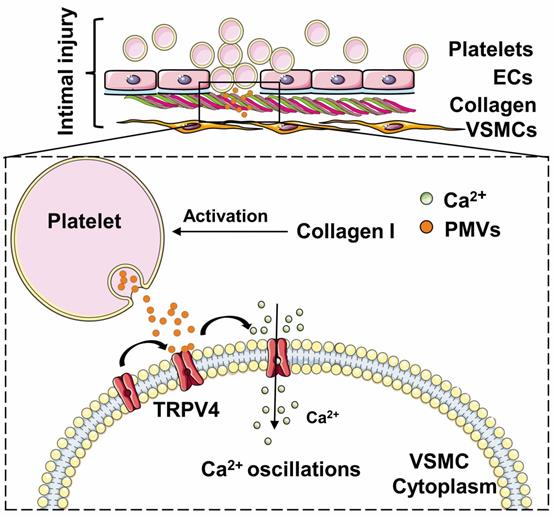
In conclusion, our findings identified the role of PMVs, specifically those induced by collagen exposure during vascular injury, in increasing VSMC migration (Figure 6). PMVs trigger the influx of extracellular calcium via the TRPV4 channel, which subsequently induces calcium oscillations and promotes VSMC migration. The study may provide new insights into the mechanism of abnormal VSMC migration after intimal injury and may have potential clinical applications for attenuating neointimal hyperplasia after intimal injury during vascular stent surgery. Further studies are encouraged to reveal the influences and mechanisms of antiplatelet drugs, such as aspirin [73-75] and clopidogrel [76], in PMV-induced calcium oscillations and in vivo regulatory functions.
Schematic drawing of the role of the calcium oscillations induced by PMVs in VSMC migration. Collagen-induced PMVs targeted TRPV4 and induced VSMC migration.
Abbreviation
2-APB, 2-aminoethoxydiphenyl borate; α-SMA, α-smooth muscle actin; CXCR4, chemokine receptor-4; DMEM, Dulbecco's modified Eagle's medium; ECs, endothelial cells; EOCs, early outgrowth cells; FRET, fluorescence resonance energy transfer; IP3, inositol 1,4,5-trisphosphate; IP3Rs, inositol 1,4,5-trisphosphate receptors; L-VDCC, L-type voltage-dependent calcium channel; MMP, metalloproteinase; siRNA, small interfering RNAs; PMVs, platelet-derived microvesicles; TRPV4, transient receptor potential vanilloid 4; VSMCs, vascular smooth muscle cells.
Supplementary Material
Supplementary figures and tables.
Supplementary video 1A.
Supplementary video 1B.
Supplementary video 2A.
Supplementary video 2B.
Supplementary video 3A.
Supplementary video 3B.
Supplementary video 3C.
Supplementary video 4A.
Supplementary video 4B.
Supplementary video 4C.
Supplementary video 5A.
Supplementary video 5B.
Acknowledgements
This research was supported by grants from the National Natural Science Foundation of China, nos. 11625209, 31700816 and 11222223.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wu B, Mottola G, Schaller M, Upchurch GR, Conte MS. Resolution of vascular injury: specialized lipid mediators and their evolving therapeutic implications. Mol Aspects Med. 2017;58:72-82
2. Cai X. Regulation of smooth muscle cells in development and vascular disease: current therapeutic strategies. Expert Rev Cardiovas. 2006;4(6):789-800
3. Tsaousi A, Williams H, Lyon CA, Taylor V, Swain A, Johnson JL. et al. Wnt4/β-catenin signaling induces VSMC proliferation and is associated with intimal thickening. Circ Res. 2011;108(4):427-436
4. Yao Y, Hu Z, Ye J, Hu C, Song Q, Da X. et al. Targeting AGGF1 (angiogenic factor with G patch and FHA domains 1) for blocking neointimal formation after vascular injury. J Am Heart Assoc. 2017;6(6):e005889
5. Hara T, Fukuda D, Tanaka K, Higashikuni Y, Hirata Y, Yagi S. et al. Inhibition of activated factor X by rivaroxaban attenuates neointima formation after wire-mediated vascular injury. Eur J Clin Pharmacol. 2018;820:222-228
6. Yan W, Li Y, Yin T, Hou Z, Qu K, Wang N. et al. M2 macrophage-derived exosomes promote the c- KIT phenotype of vascular smooth muscle cells during vascular tissue repair after intravascular stent implantation. Theranostics. 2020;10(23):10712-10728
7. Gerthoffer TW. Mechanisms of vascular smooth muscle cell migration. Circ Res. 2007;100(5):607-621
8. Kiyan Y, Kurselis K, Kiyan R, Haller H, Chichkov BN, Dumler L. Urokinase receptor counteracts vascular smooth muscle cell functional changes induced by surface topography. Theranostics. 2013;3(7):516-526
9. Yang GS, Zheng B, Qin Y, Zhou J, Yang Z, Zhang XH. et al. Salvia miltiorrhiza-derived miRNAs suppress vascular remodeling through regulating OTUD7B/KLF4/NMHC IIA axis. Theranostics. 2020;10(17):7787-7811
10. Furman C, Luo Z, Walsh K, Duverger N, Copin C, Fruchart JC. et al. Systemic tissue inhibitor of metalloproteinase-1 gene delivery reduces neointimal hyperplasia in balloon-injured rat carotid artery. FEBS Lett. 2002;531(2):122-126
11. Mason DP, Kenagy RD, Hasenstab D, Bowen-Pope DF, Seifert RA, Coats S. et al. Matrix metalloproteinase-9 overexpression enhances vascular smooth muscle cell migration and alters remodeling in the injured rat carotid artery. Circ Res. 1999;85(12):1179-1185
12. Wu XL, Liu WW, Jiang H, Chen J, Wang JC, Zhu R. et al. Kindlin-2 siRNA inhibits vascular smooth muscle cell proliferation, migration and intimal hyperplasia via Wnt signaling. Int J Mol Med. 2016;37(2):436-444
13. Song RH, Kocharyan HK, Fortunato JE, Glagov S, Bassiouny HS. Increased flow and shear stress enhance in vivo transforming growth factor-beta 1 after experimental arterial injury. Arterioscler Thromb Vasc Biol. 2000;20(4):923-930
14. Wang HQ, Huang LX, Qu MJ, Yan ZQ, Liu B, Shen BR. et al. Shear stress protects against endothelial regulation of vascular smooth muscle cell migration in a coculture system. Endothelium. 2006;13(3):171-180
15. Lawson MA, Maxfield FR. Ca2+- and calcineurin-dependent recycling of an integrin to the front of migrating neutrophils. Nature. 1995;377(6544):75-79
16. Kovac JR, Chrones T, Sims SM. Temporal and spatial dynamics underlying capacitative calcium entry in human colonic smooth muscle. Am J Physiol Gastrointest Liver Physiol. 2008;294(1):G88-98
17. Fingerle J, Johnson R, Clowes AW, Majesky MW, Reidy MA. Role of platelets in smooth muscle cell proliferation and migration after vascular injury in rat carotid artery. Proc Natl Acad Sci U S A. 1989;86(21):8412-8416
18. Zaldivia MTK, McFayden JD, Lim B, Wang X, Peter K. Platelet-derived microvesicles in cardiovascular diseases. Front Cardiovasc Med. 2017;4:74
19. Aatonen M, Gronholm M, Siljander P. Platelet-derived microvesicles: multitalented participants in intercellular communication. Semin Thromb Hemost. 2012;38(01):102-113
20. Tricarico C, Clancy J, D'Souza-Schorey C. Biology and biogenesis of shed microvesicles. Small GTPases. 2017;8(4):220-232
21. Melki I, Tessandier N, Zufferey A, Boilard E. Platelet microvesicles in health and disease. Platelets. 2017;28(3):214-221
22. Nunzio I, Tommaso L, Florian G, Beatriz V, Stefano P. Focus on extracellular vesicles: physiological role and signalling properties of extracellular membrane vesicles. Int J Mol Sci. 2016;17(2):171
23. Mause SF, Ritzel E, Liehn EA, Hristov M, Bidzhekov K, Müller-Newen G. et al. Platelet microparticles enhance the vasoregenerative potential of angiogenic early outgrowth cells after vascular injury. Circulation. 2010;122(5):495-506
24. Nieuwland R, Berckmans RJ, Rotteveel-Eijkman RC, Maquelin KN, Sturk A. Cell-derived microparticles generated in patients during cardiopulmonary bypass are highly procoagulant. Circulation. 1997;96(10):3534-3541
25. Zeng Z, Xia L, Fan X, Ostriker AC, Yarovinsky T, Su M, Zang Y. et al. Platelet-derived miR-223 promotes a phenotypic switch in arterial injury repair. J Clin Invest. 2019;129(3):1372-1386
26. Sandow SL, Gzik DJ, Lee RM. Arterial internal elastic lamina holes: relationship to function. J Anat. 2009;214(2):258-266
27. Dean WL, Lee MJ, Cummins TD, Schultz DJ, Powell DW. Proteomic and functional characterisation of platelet microparticle size classes. J Thromb Haemost. 2009;102(4):711-718
28. Mause SF, Ritzel E, Liehn EA, Hristov M, Bidzhekov K, Müller-Newen G. et al. Platelet microparticles enhance the vasoregenerative potential of angiogenic early outgrowth cells after vascular injury. Circulation. 2010;122(5):495-506
29. Brandtzaeg P, Rognum TO. Evaluation of tissue preparation methods and paired immunofluorescence staining for immunocytochemistry of lymphomas. Histochem J. 1983;15(7):655-689
30. Bao H, Chen YX, Huang K, Zhuang F, Bao M, Han Y. et al. Platelet-derived microparticles promote endothelial cell proliferation in hypertension via miR-142-3p. FASEB J. 2018;32(7):3912-3923
31. Yan J, Bao H, Fan YJ, Jiang ZL, Qi YX, Han Y. Platelet-derived microvesicles promote endothelial progenitor cell proliferation in intimal injury by delivering TGF-β1. FEBS J. 2020 febs.15293
32. Heath DE, Kang GC, Ye C, Yin FP, Chan-Park MB. Biomaterials patterned with discontinuous microwalls for vascular smooth muscle cell culture: biodegradable small diameter vascular grafts and stable cell culture substrates. J Biomater Sci Polym Ed. 2016;27(15):1-39
33. Liu B, Lu S, Zheng S, Jiang Z, Wang Y. Two distinct phases of calcium signalling under flow. Cardiovasc Res. 2011;91(1):124-133
34. Goueffic Y, Guilluy C, Guerin P, Patra P, Pacaud P, Loirand G. Hyaluronan induces vascular smooth muscle cell migration through RHAMM-mediated PI3K-dependent Rac activation. Cardiovas Res. 2006;72(2):339-348
35. Li P, Liu Y, Yi B, Wang G, You X, Zhao X, et al. MicroRNA-638 is highly expressed in human vascular smooth muscle cells and inhibits PDGF-BB-induced cell proliferation and migration through targeting orphan nuclear receptor NOR1.Cardiovasc Res. 2013; 99(1): 185-193
36. Bootman MD1, Collins TJ, Mackenzie L, Roderick HL, Berridge MJ, Peppiatt CM. 2-aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J. 2002;7(6):429-439
37. Reid K, Guo TZ, Davies MF, Maze M. Nifedipine, an L-type calcium channel blocker, restores the hypnotic response in rats made tolerant to the alpha-2 adrenergic agonist dexmedetomidine. J Pharmacol Exp Ther. 1997;83(3):993-999
38. Cheung M, Bao W, Behm DJ, Brooks CA, Bury MJ, Dowdell SE. et al. Discovery of GSK2193874: an orally active, potent, and selective blocker of transient receptor potential vanilloid 4. ACS Med Chem Lett. 2017;8(5):549-554
39. Dalby B, Cates S, Harris A, Ohki EC, Tilkins ML, Price PJ. et al. Advanced transfection with Lipofectamine 2000 reagent: primary neurons, siRNA, and high-throughput applications. Methods. 2004;33(2)0-103
40. Badimon L, Suades R, Fuentes E, Palomo I, Padró T. Role of platelet-derived microvesicles as crosstalk mediators in atherothrombosis and future pharmacology targets: a link between inflammation, atherosclerosis, and thrombosis. Front Pharmacol. 2016;7:293
41. Allahverdian S, Chaabane C, Boukais K, Francis GA. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc Res. 2018;114(4):540-550
42. Brundage R, Fogarty K, Tuft R, Fay F. Calcium gradients underlying polarization and chemotaxis of eosinophils. Science. 1991;254(5032):703-706
43. Cao S, Anishkin A, Zinkevich NS, Nishijima Y, Korishettar A, Wang Z. et al. Transient receptor potential vanilloid 4 (TRP4) activation by arachidonic acid requires protein kinase A-mediated phosphorylation. J Bio Chem. 2018 M117.811075
44. Hu K, Sun H, Gui B, Sui C. TRPV4 functions in flow shear stress induced early osteogenic differentiation of human bone marrow mesenchymal stem cells. Biomed Pharmacother. 2017;91:841-848
45. Plant TD, Strotmann R. TRPV4. Handb Exp Pharmacol. 2014;222:293-319
46. Horstman LL, Ahn YS. Platelet microparticles: a wide-angle perspective. Crit Rev Oncol Hematol. 1999;30(2):111-142
47. Kim HK, Song KS, Chung JH, Lee KR, Lee SN. Platelet microparticles induce angiogenesis in vitro. Br J Haematol. 2004;124(3):376-384
48. Weber AA, Köppen HO, Schrör K. Platelet-derived microparticles stimulate coronary artery smooth muscle cell mitogenesis by a PDGF-independent mechanism. Thromb Res. 2000;98(5):461-466
49. Wu B, Mottola G, Schaller M, Upchurch GR, Conte MS. Resolution of vascular injury: Specialized lipid mediators and their evolving therapeutic implications. Mol Aspects Med. 2017;58:72-82
50. Zhu M, Chen L, Zhao P. et al. Store-operated Ca2+ entry regulates glioma cell migration and invasion via modulation of Pyk2 phosphorylation. J Exp Clin Canc Res. 2014;33(1):98
51. Iamshanova O, Pla AF, Prevarskaya N. Molecular mechanisms of tumour invasion: regulation by calcium signals. J Physiol. 2017;595(10):3063-3075
52. Liou J, Kim ML, Heo WD, Jones JT, Myers JW, James EF. et al. STIM is a Ca2+ Sensor Essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15(13):1235-1241
53. Scott JA, Xie LT, Li H, Li WW, He JB, Sanders PN. et al. The multifunctional Ca2+/calmodulin-dependent kinase II regulates vascular smooth muscle migration through matrix metalloproteinase 9. Am J Physiol-Heart C. 2012;302(10):H1953-H1964
54. Pene F, Claessens Y-E, Muller O, Viguié F, Mayeux P, Dreyfus F. et al. Role of the phosphatidylinositol 3-kinase/Akt and mTOR/P70S6-kinase pathways in the proliferation and apoptosis in multiple myeloma. Oncogene. 2002;21(43):6587-6597
55. Xu J, Zhang QG, Li C, Zhang GY. Subtoxic N-methyl-D-aspartate delayed neuronal death in ischemic brain injury through TrkB receptor- and calmodulin-mediated PI-3K/Akt pathway activation. Hippocampus. 2007;17:525-537
56. Malencik DA, Anderson SR, Bohnert JL, Shalitin Y. Functional interactions between smooth muscle myosin light chain kinase and calmodulin. Biochemistry. 1982;21(17):4031-4039
57. Wei C, Wang X, Chen M, Ouyang K, Song LS, Cheng H. Calcium flickers steer cell migration. Nature. 2009;457(7231):901-905
58. Kholmanskikh SS, Koeller HB, Wynshaw-Boris A, Gomez T, Letourneau PC, Ross ME. Calcium-dependent interaction of Lis1 with IQGAP1and Cdc42 promotes neuronal motility. Nat Neurosci. 2006;9(1):50-57
59. Lam JGT, Vadia S, Pathak-Sharma S, McLaughlin E, Zhang X, Swanson J. et al. Host cell perforation by listeriolysin O (LLO) activates a Ca2+-dependent cPKC/Rac1/Arp2/3 signaling pathway that promotes Listeria monocytogenes internalization independently of membrane resealing. MBoC. 2018;29:270-284
60. Masiero L, Lapidos KA, Ambukar I, Kohn EC. Regulation of the RhoA pathway in human endothelial cell spreading on type IV collagen: role of calcium influx. J Cell Sci. 1999;112:3205-3213
61. Giannone G, Dubin-Thaler BJ, Rossier O, Cai Y, Chaga O, Jiang G. et al. Lamellipodial actin mechanically links myosin activity with adhesion-site formation. Cell. 2007;128(3):561-575
62. Zhu LJ, Klutho PJ, Scott JA, Xie L, Luczak ED, Dibbern ME. et al. Oxidative activation of the Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates vascular smooth muscle migration and apoptosis. Vasc Pharmacol . 2014;60(2):75-83
63. Kim TJ, Joo C, Seong J, Vafabakhsh R, Botvinick EL, Berns MW. et al. Distinct mechanisms regulating mechanical force-induced Ca2+ signals at the plasma membrane and the ER in human MSCs. Elife. 2015;4:e04876
64. Wu S, Jian MY, Xu YC, Zhou C, Almehdi AB, Liedtke W. et al. Ca2+ entry via alpha1G and TRPV4 channels differentially regulates surface expression of P-selectin and barrier integrity in pulmonary capillary endothelium. Am J Physiol. Lung C. 2009;297(4):L650-L657
65. Montell C. The TRP superfamily of cation channels. Sci. STKE. 2005(272):re3-re3
66. Pedersen SF, Owsianik G, Nilius B. TRP channels: an overview. Cell Calcium. 2005;38(3-4):233-252
67. Randhawa PK, Jaggi AS.TRPV4 channels. physiological and pathological role in cardiovascular system. Basic Res Cardiol. 2015;110(6):54
68. Adapala RK, Thoppil RJ, Luther DJ, Paruchuri S, Meszaros JG, Chilian WM. et al. TRPV4 channels mediate cardiac fibroblast differentiation by integrating mechanical and soluble signals. J Mol Cell Cardiol. 2013;54:45-52
69. Jin M, Wu Z, Chen L, Jaimes J, Collins D, Walters ET. et al. Determinants of TRPV4 activity following selective activation by small molecule agonist GSK1016790A. PLoS One. 2011;6(2):e16713
70. Rajasekhar P, Poole DP, Liedtke W, Bunnett NW, Veldhuis NA. P2Y1 receptor activation of the TRPV4 ion channel enhances purinergic signaling in satellite glial cells. J Bio Chem. 2015;290(48):29051-29062
71. Pais R, Zietek T, Hauner H, Daniel H, Skurk T. RANTES (CCL5) reduces glucose-dependent secretion of glucagon-like peptides 1 and 2 and impairs glucose-induced insulin secretion in mice. Am J Physiol-Gastr L. 2014;307(3):G330-G337
72. Zhou D, Samovski D, Okunade AL, Stahl PD, Su X. CD36 level and trafficking are determinants of lipolysis in adipocytes. FASEB J. 2012;26(11):4733-4742
73. Ho KJ, Spite M, Owens CD, Lancero H, Kroemer AH, Pande R. et al. Aspirin-triggered lipoxin and resolvin E1 modulate vascular smooth muscle phenotype and correlate with peripheral atherosclerosis. Am J pathol. 2010;177(4):2116-2123
74. Mottola G, Chatterje A, Wu B, Chen M, Conte MS. Aspirin-triggered resolvin D1 attenuates PDGF-induced vascular smooth muscle cell migration via the cyclic adenosine monophosphate/protein kinase A (cAMP/PKA) pathway. PLoS One. 2017 12(3), e0174936
75. Petri MH, Laguna-Fernandez A, Tseng CN, Hedin U, Perretti M, Bäck M. Aspirin-triggered 15-epi-lipoxin A4 signals through FPR2/ALX in vascular smooth muscle cells and protects against intimal hyperplasia after carotid ligation. Int J Cardiol. 2015;179:370-372
76. Niu X, Pi SL, Baral S, Xia YP, He QW, Li YN. et al. P2Y12 promotes migration of vascular smooth muscle cells through cofilin dephosphorylation during atherogenesis. Arterioscl Throm Vas. 2017;37(3):515-524
Author contact
![]() Corresponding author: Dr. Ying-Xin Qi, Institute of Mechanobiology & Medical Engineering, School of Life Sciences & Biotechnology, P.O.Box 888, Shanghai Jiao Tong University, 800 Dongchuan Road, Minhang, Shanghai 200240, China. Tel: +86-21-34204863; Fax: +86-21-34204118; E-mail: qiyxedu.cn. Dr. Yingxiao Wang, Department of Bioengineering, University of California, La Jolla, San Diego, California CA 92093, USA. Tel: +1 217 333 6727; Fax: +1 217 265 0246; E-mail: yiw015ucsd.edu
Corresponding author: Dr. Ying-Xin Qi, Institute of Mechanobiology & Medical Engineering, School of Life Sciences & Biotechnology, P.O.Box 888, Shanghai Jiao Tong University, 800 Dongchuan Road, Minhang, Shanghai 200240, China. Tel: +86-21-34204863; Fax: +86-21-34204118; E-mail: qiyxedu.cn. Dr. Yingxiao Wang, Department of Bioengineering, University of California, La Jolla, San Diego, California CA 92093, USA. Tel: +1 217 333 6727; Fax: +1 217 265 0246; E-mail: yiw015ucsd.edu