Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Tyrosine kinases
Small molecule inhibitors of TKs...
Nanomedicine
Nano-based combination therapy...
Theranostics
Future outlooks
Conclusions
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(4):1546-1567. doi:10.7150/thno.48662 This issue Cite
Review
Nanomedicine of tyrosine kinase inhibitors
Veronika Smidova1, Petr Michalek1,2, Zita Goliasova1, Tomas Eckschlager3, Petr Hodek4, Vojtech Adam1,2, Zbynek Heger1,2 ![]()
1. Department of Chemistry and Biochemistry Mendel University in Brno, Zemedelska 1, 613 00 Brno, Czech Republic.
2. Central European Institute of Technology, Brno University of Technology, Purkynova 656/123, 612 00 Brno, Czech Republic.
3. Department of Paediatric Haematology and Oncology, 2nd Faculty of Medicine, Charles University, and University Hospital Motol, V Uvalu 84, Prague 5 CZ-15006, Czech Republic.
4. Department of Biochemistry, Faculty of Science, Charles University, Albertov 2030, 128 40 Prague 2, Czech Republic.
Received 2020-5-25; Accepted 2020-9-21; Published 2021-1-1
Abstract

Recent progress in nanomedicine and targeted therapy brings new breeze into the field of therapeutic applications of tyrosine kinase inhibitors (TKIs). These drugs are known for many side effects due to non-targeted mechanism of action that negatively impact quality of patients' lives or that are responsible for failure of the drugs in clinical trials. Some nanocarrier properties provide improvement of drug efficacy, reduce the incidence of adverse events, enhance drug bioavailability, helps to overcome the blood-brain barrier, increase drug stability or allow for specific delivery of TKIs to the diseased cells. Moreover, nanotechnology can bring new perspectives into combination therapy, which can be highly efficient in connection with TKIs. Lastly, nanotechnology in combination with TKIs can be utilized in the field of theranostics, i.e. for simultaneous therapeutic and diagnostic purposes. The review provides a comprehensive overview of advantages and future prospects of conjunction of nanotransporters with TKIs as a highly promising approach to anticancer therapy.
Keywords: Bioavailability, Drug delivery, Nanotechnology, Targeted therapy
Introduction
With the incidence of 18.1 million new cases worldwide and death count reaching 9.6 million people in the year of 2018, cancer is a serious player on the field of human health [1]. Tyrosine kinases (TKs) are important targets for cancer treatment due to their role in signal transduction and subsequent cell proliferation that can turn into neoplastic cell transformation and tumor growth [2]. Inhibitors of these kinases are commonly used in the treatment of non-small cell lung cancer (NSCLC), chronic myeloid leukemia (CML) and some cases of breast and colorectal cancer, advanced gastrointestinal stromal tumor, renal cell, hepatocellular or thyroid carcinoma and soft tissue sarcoma [3].
Tyrosine kinase inhibitor (TKI) therapy, however beneficial, has its limitations and disadvantages. Off-target activity causes multitude of adverse events, negatively affecting the quality of patient's life and continuation of therapy [2]. Moreover, cancer tends to develop variety of mechanisms of acquired drug resistance, resulting in inefficacy of TKI medications. However, in recent years, more and more publications focus on the incorporation of these small molecule drugs into nanomedicinal applications in order to facilitate progress in cancer therapy [4].
This review is focused on two particular themes, concerning TKIs and nanomedicine. In its first part, we indulge in the matters of TKs and their small molecule inhibitors. Second part is dedicated to nanoformulations of TKIs and to the potential nanomedicine has to help overcome the limitations of TKI therapy and to improve it.
Tyrosine kinases
TKs are transmembrane (receptor) or unattached (non-receptor) enzymes that transfer γ‑phosphate from adenosine triphosphate (ATP) to tyrosine residue of a substrate molecule. This transfer causes allosteric or functional change of the protein and represents an essential mechanism for activation and direction of cell signaling [5]. There are ~90 members of protein TK family, 58 of which are receptor TKs (RTKs), and 32 are non-receptor TKs. Moreover, these two groups can be divided - according to Robinson et al. (2000) - into 20 and 10 subfamilies, respectively, based on kinase domain sequence [6], although designation and number of subfamilies can be variable. The 20 subfamilies of RTKs can be grouped according to the typology of the receptor. There are two important oncotargeted groups based on receptors of type I (EGFR/HER, alternate Cys and Leu rich domains) and type IV (VEGFR, Ig-like domains). In addition, there are two groups with influence on neoplastic growth, although not yet oncotargeted - type II Insulin receptor family (Leu-Cys-Leu domains attached with fibronectin) and type IX Ephrins (Ig-like + Cys-rich + two fibronectin domains). Lastly, there is a group of miscellaneous kinases with different (e.g. ALK, DDR, MET, RON, RYK) or truncated extracellular domains (LMR, STYK1, etc.) [5].
RTKs structure
RTKs are membrane glycoproteins that transfer signals from extracellular into intracellular environment. Therefore, their structure consists of an extracellular hydrophilic domain, a hydrophobic transmembrane segment and an intracellular domain. Extracellular part of RTKs can be very variable according to their corresponding ligand. Binding of the ligand needs to be specific and reversible. A number of low-energy bonds like hydrogen, ionic, hydrophobic or Van der Waals interactions are involved [7]. Intracellular part, however, is quite the opposite, consisting of a juxtamembrane and a TK domain with a flexible C-terminal tail. In some cases, TK domain can be divided in two parts by a kinase insertion [5]. ATP binding site, situated on the N-terminal lobe, has a conserved sequence, in which a loop with Gly residue serves to localize phosphate on the ATP molecule [8].
RTKs activation and activity
RTKs are mostly monomeric, undergoing homo- or heteromerization even before or upon ligand binding [9]. In the absence of ligand, RTKs show a basal kinase activity, possibly due to self- or exophosphorylation [5]. Mayer proposes a dynamic phospho-turnover model where constant autophosphorylation and phosphatase activity maintains a stable background “noise” signal [10]. Binding of the ligand causes the intracellular part of juxtaposed kinase domains to transphosphorylate juxtamembrane and kinase domains with C-terminal tail. This fully activates the RTK [5]. Another Mayer's suggestion envisions a dynamic ligand binding in a form of “hopping” from domain to domain, consequently activating up to 20 RTKs in the process [10]. Inhibition of kinase activity is executed by various mechanisms, such as dephosphorylation by protein phosphatases, endocytosis or ubiquitination [5]. Gelens et al. further extend the dynamic theory, showing that even when the signal seems to be stably switched on, there are rapid phosphorylation-dephosphorylation cycles happening [11].
After the RTK is activated, Src homology-2 (SH2) and phosphotyrosine-binding (PTB) domains of cytoplasmic proteins bind to the phosphorylated tyrosine to initiate the signaling cascade. These adaptive (SH2 containing) and anchoring (PTB containing) proteins have either intrinsic enzymatic activity themselves (Src, PLCγ, GAP, PTPN11, etc.) or recruit other enzymes, which are involved in various signaling pathways (Nck, Crk, Shc, Grb2, etc.) that lead to growth stimulation, survival, migration, and actin reorganization [12].
Roles of TKs in cancer
TKs and their phosphorylation mechanism are in control of four essential signaling networks mediating cancerogenesis, cancer progression and maintenance. These networks affect proliferation, viability, motility and cytostasis or differentiation. In detail, this entails upregulation or constitutive activation of proliferative signals, downregulation of cell death signals and maintenance of microenvironment conditions to favor tumor progression and diffusion [13].
Fleuren et al. created a consensus of 91 PKs out of 1,100 cancer driver genes, compiled from multiple pan-cancer studies. TKs stand for 40 % of these PKs; 36 of all 58 known RTKs being represented among them [14]. Both, the kinase catalytic domain and the “gatekeeper” residue in C‑terminal part, which controls access to ATP-binding pocket, are frequently involved in mutations. Genome-wide amino acid substitutions including Arg, Cys, Trp, Tyr and substitutions in the catalytic domain involving Gly and Asp make PKs more prone to developing a disease. In addition, virtually all organs are affected by mutations in PKs [13].
In the elaborate network of TK signaling (Figure 1), consisting mainly of MAPK, PI3K/AKT, Src, PLCγ and JAK/STAT pathways [7], not only genetic mutations are responsible for abnormal level of kinase activity; chromosomal translocations, overexpression or autocrine activation may also play this role [15]. On the other hand, for TKs to be considered an oncogenic agent, it is not necessary for their function to be altered. Cancer tissue can benefit even from a perfectly normal kinase activity; therefore, oncogenic kinase is a term encompassing more than just kinases derived from oncogenic genes [13]. However, the streamlining function of other kinases, phosphorylation-dephosphorylation cycle, internal molecular control by regulatory domains or subunits and local subcellular segregation of single kinases may be disrupted during oncogenesis and may cause overactivity [5]. Lahiry et al. scanned 67 germline disorders related to kinase function, identifying 50 causative kinases, approximately half of them being TKs [16]. More than 80% of the 915 detected mutations were located in the catalytic kinase domain gene or in its close proximity. In addition, a study of 3,000 cancer tissue samples from 12 tumor types revealed that most mutations were present in common oncogenic pathways RTK/RAS/RAF, PI3K/AKT/mTOR, cell cycle and p53-DNA repair, with the first route being affected in about half of the tumor samples [17].
Schematic representation of RTK downstream signaling - modified from KEGG: Kyoto Encyclopedia of Genes and Genomes Database.
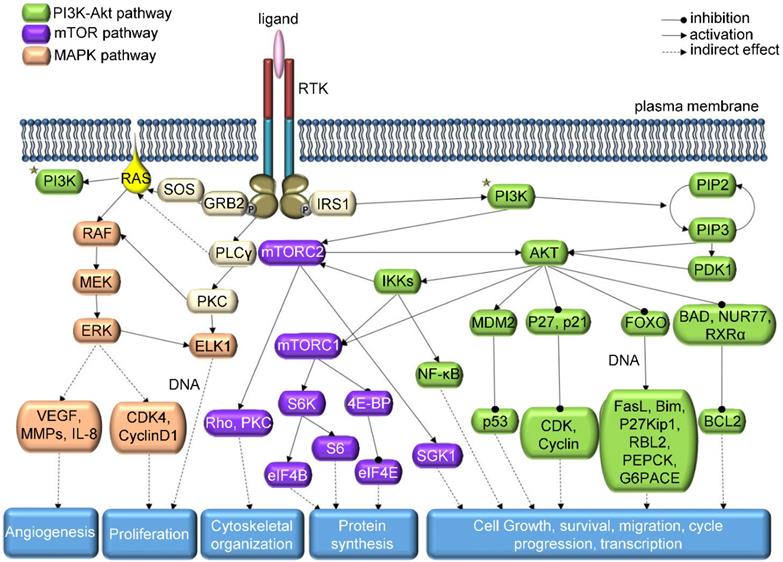
Each TK has its own specific function that can become a factor in the emergence of neoplastic growth and progression. For instance, EGFR is especially important in NSCLC. Many EGFR mutations co-occur with other genetic alterations (e.g. mutations in TP53, PIK3CA, BRAF, MET, etc.), causing tumor progression and therapy resistance [18]. EGFR overexpression is also common in colorectal cancer [19, 20] and phosphorylation of EGFR is considered a good prognostic biomarker in squamous cell carcinoma [21]. In genomic characterization of 91 aberrant out of 206 glioblastoma samples, EGFR alterations were found in 45% of cases, with 24.0% amplifications, 17.5% amplifications with point mutations and 3.3% point mutations [22].
Next generation sequencing of 4,853 tumors revealed involvement of FGFR aberrations in 7.1% of samples, mostly in urothelial, breast, endometrial, squamous lung and ovarian cancers - gene amplifications being the most frequent aberration involved [23]. In urothelial bladder cancer RNA sequencing study, RTKs were amplified, mutated or fused in 45% of samples, 15% of which had altered FGFR3 [24]. In brain metastatic breast cancer, FGFR1 was more frequently altered than other FGFR family members, and patients with this cancer type had more FGFR aberrations than non-brain metastatic breast cancer patients [25].
VEGFR has complex signaling affairs with both immune and cancer cells [26]. VEGFR-1 supports tumor angiogenesis, progression and invasiveness [27]. In obese mouse model, abrogation of VEGFR-1 expression normalized tumor immune environment phenotype and inhibited obesity-induced tumor progression [28]. Recently, it was found to be highly expressed in glioblastoma tissue, showing ~30% higher expression than in surrounding parenchyma tissue [29]. VEGFR-2 is expressed in breast cancer [30], NSCLC and other cancer types [31]; it promotes tumor angiogenesis and cancer cell survival through autocrine/paracrine VEGF/VEGFR-2 loop.
PDGFR impacts tumor development by affecting fibroblasts, vascular and cancer cells [32]. Alterations in PDGFR were found in approx. 13% of glioblastoma patients [22]. In gastrointestinal stromal tumor sequencing, 12% of samples harbored PDGFR mutations, and 78% of samples contained KIT gene mutations [33]. In prostate cancer, PDGFR-β was discovered to have higher expression in tumor stroma rather than in normal stroma and this expression was associated with disease recurrence [34].
In CML, fusion of chromosome 22 and 9 gives rise to chimeric Bcr-Abl protein, where Abl kinase domain is constitutively activated and drives the disease progression [35]. Src kinase was found to be overexpressed in liver [36] and breast cancer [37], which had mitigating effect on mitochondrial activity, and consequently supported tumor progression and metastasis. MET and HER2 gene amplifications are considered important driver oncogene alterations in lung adenocarcinomas [38]. HER2 amplification is also significantly present in breast cancer [39], with the rate of about 15-30%, and is also found in 10-30% of gastric or gastroesophageal cancers [40]. These were but a few examples of TKs involved in various types of cancers. Researchers have discovered many more over the decades of vigorous investigation, and thus are still markedly extending the number of druggable targets suitable for development of novel therapeutic modalities.
Small molecule inhibitors of TKs (TKIs)
From a drug discovery point of view, inhibition of PK activity can be achieved by a molecule that binds either the ligand or the PK itself to prevent ligand binding, dimerization or catalytic activity [41]. Alternatively, some drugs have been developed to cause kinase degradation [42]. Antibodies can bind either PK's ligand or extracellular domain [41]. However, they are only weak antagonists, as activating mutations in kinase domains are not inhibited by them [15]. Small-molecule inhibitors target ATP‑binding site, depriving kinases of phosphate, or induce allosteric changes in low sequence homology portions of PK molecules [43].
Classification of TKIs has gone through an evolution [44-49]. Now, according to Roskoski, there are six classes of protein kinase inhibitors (PKIs) with different binding properties [50] (summarized in Table 1).
Current classification of TKIs
| Class | Binding | ATP-competitive | Reversible binding | Examples of FDA approved TKIs |
|---|---|---|---|---|
| Type I | In the ATP-binding pocket of the active enzyme conformation | Yes | Yes | Cabozantinib, Dasatinib, Erlotinib, Gefitinib, Vandetanib, |
| Type I ½ A | In the ATP-binding pocket of a conformation with DFG-Asp in and αC-helix out with extension to back cleft of the ATP site | Yes | Yes | Lenvatinib, Lapatinib |
| Type I ½ B | In the ATP-binding pocket of a conformation with DFG-Asp in and αC-helix out without extension to back cleft of the ATP site | Yes | Yes | Alectinib, Ceritinib, Crizotinib, Erlotinib |
| Type II A | In the ATP-binding pocket of a conformation with DFG-Asp out with extension to back cleft of the ATP site | Yes | Yes | Axitinib, Imatinib, Nilotinib, Ponatinib, Sorafenib |
| Type II B | In the ATP-binding pocket of a conformation with DFG-Asp out without extension to back cleft of the ATP site | Yes | Yes | Bosutinib, Sunitinib, Nintedanib |
| Type III | Allosterically next to but outside of ATP-binding site with the extension to back cleft of the ATP site | No | No | - |
| Type IV | Allosterically not next to the ATP-binding site | No | No | - |
| Type V | Bivalently to two regions of PK domain | Variable | Yes | - |
| Type VI | Covalently to Cys residues | No | Usually no | Afatinib, Ibrutinib |
Up to this date, there are 55 small molecule PKIs approved by FDA out of which 39 are TKIs (Table 2). Most FDA-approved TKIs fall within Types I to II. An online PKI database, managed by SB&C Team from Institute of Organic and Analytical Chemistry of the University of Orleans, France, compiles all PKIs, even the ones investigated in clinical trials. At present, there are 117 TKIs listed. One in phase 0, seven in phase I, twenty-nine in phase II, thirty-six in phase III and lastly forty-three in phase IV, out of which five were not approved by FDA [51].
Summary of FDA approved small molecule TKIs according to the type of their target as of 19 March 2020; *- non-receptor TKs
| Type of primary target | TKIs |
|---|---|
| ALK | Alectinib, Brigatinib, Ceritinib, Crizotinib, Entrectinib, Lorlatinib |
| AXL | Sunitinib |
| BCR-Abl* | Bosutinib, Dasatinib, Imatinib, Nilotinib, Ponatinib, Regorafenib |
| BTK* | Acalabrutinib, Ibrutinib |
| c-Met | Cabozantinib, Crizotinib |
| DDR family | Nilotinib |
| EGFR family | Afatinib, Brigatinib, Dacomitinib, Dasatinib, Erlotinib, Gefitinib, Lapatinib, Neratinib, Osimertinib, Vandetanib |
| Ephrin-like | Cabozantinib, Dasatinib, Vandetanib |
| FRK family | Vandetanib |
| Insulin/Insulin-like GF | Brigatinib, Ceritinib, Crizotinib, Entrectinib |
| JAK family* | Baricitinib, Fedratinib, Ruxolitinib, Tofacitinib, Upadactinib |
| PDGFR α/β | Axitinib, Gefitinib, Imatinib, Lenvatinib, Nintedanib, Pazopanib, Regorafenib, Sorafenib, Sunitinib |
| RET | Cabozantinib, Lenvatinib, Regorafenib, Sorafenib, Sunitinib, Vandetanib |
| Src family* | Bosutinib, Dasatinib, Ponatinib, Vandetanib |
| SYK | Fostamatinib |
| TRK family | Cabozantinib, Entrectinib, Larotrectinib |
| VEGFR family | Axitinib, Brigatinib, Cabozantinib, Dasatinib, Erdafitinib, Fedratinib, Gilteritinib, Imatinib, Lenvatinib, Midostaurin, Nilotinib, Nintedanib, Regorafenib, Pazopanib, Pexidartinib, Ponatinib, Regorafenib, Sorafenib, Sunitinib, Vandetanib. |
The principle of TKI function is based on a phenomenon called “oncogene addiction” [52]. In other words, cancer cell's prosperity is dependent on a single pathway. Members of this pathway become drug targets, and their inhibition ideally suppresses neoplastic growth. However, kinase signaling is rather an intricate spider web than a linear pathway, and disruptions are eventually bypassed, causing tumor to regain its proliferation capacity. The higher the selectivity of therapy, the higher is the risk of the tumor to develop an escape scenario. Drug resistance against TKIs usually emerges within one year of therapy [13] and can be caused by various intrinsic and extrinsic factors, like secondary target gene amplification, mutation or alternative splicing, activation of compensatory pathways or loops, structural (phenotype/histotype/epithelial-mesenchymal transition) changes, increased production of growth factors, changes in glucose metabolism or paracrine-autocrine overactivation [53]. Nearly all TKI-resistant tumors show oversignaling activity by receptor overexpression or overactivation [54]. This leads to development of new drugs targeting resistant variants. However, even to these mutant-specific inhibitors, resistance will inevitably occur [42]. Therefore, more systemic approach might lead to better results in cancer therapy. Combination of different types of inhibitors or combination of TKIs with classic chemotherapy or immunotherapy might prove beneficial [13]. Another highly promising approach is a conjunction of TKIs with nanoscaled transporters, which could lead to elimination of adverse effects of treatment on healthy tissue, known for TKI-based therapy (disorders concerning nerves, eyes, heart, lungs, liver, gastrointestinal tract, kidneys, muscles, bones, circulatory and lymphatic system, skin and reproductive system) [54]. Beyond the adverse effects, nanomedicine has much more to offer in order to improve the outcomes of patients treated with TKIs. These phenomena are discussed in following chapters.
Nanomedicine
The main objective for nanomedicine is to secure the distribution of the drug to the desired target - tumor cells - in an adequate local concentration with minimal loss of their volume or activity in the blood circulation. Moreover, it is able to reduce the potential toxicity towards healthy cells and tissues, as well as to avoid acquired drug resistance [55]. Wide range of nanomaterials based on metals, non-metals, polymers and liposomes is available, and each of the delivery systems has their specific advantages and disadvantages stemming from their biocompatibility, stability, capacity of loading and release and costs [56, 57].
Composition of nanoparticles (NPs), as well as size, morphology and surface charge can have influence on their biological effect and distribution in the organism. NPs larger than 150 nm accumulate mostly in lungs, liver and spleen, but avoid bioaccumulation and clearance by kidneys. Small NPs (under 5 nm) behave in the opposite manner. NPs in between are less likely to be caught in lungs than in liver and spleen [58]. Spherically-shaped NPs are mostly captured in liver, whereas rod-like NPs stay in liver and spleen [59]. Extravasation of NPs out of the blood vessels is easier for discoidal-shaped NPs because they rarely exhibit laminar flow, and therefore have more chances to get into proximity with receptors on vascular endothelial cells or to move into adjacent capillaries. On the other hand, internalization of NPs into the cell is easier with smaller cell-NP contact points; therefore, spherical and ovoidal NPs will internalize more easily than elongated NPs that lie flat on the cell surface [60]. NPs with positive surface charge are more likely to get filtered out of bloodstream by lungs, liver and spleen, and are therefore least suitable for maintaining prolonged blood circulation [61]. Importantly, properties of NPs can be improved by various modifications affecting surface charge, providing functional groups or coating for attachment of ligands, such as antibodies, proteins, and peptides, to enhance drug bioavailability, prolong blood circulation or facilitate active targeting [62-67].
In the fashion of poorly soluble small molecule anticancer drugs [68, 69], TKIs are recently in the focus of nanomedicine research. Recent review written by Moradpour and Barghi discusses different types of targeted delivery of TKIs [70]. In contrast to this paper, we focus on the particulars of nanoconstruct applications in combination with TKIs. As a brief overview of the applicability of nanoscaled materials in conjunction with several types of TKIs, Table 3 summarizes recent reports concerning this topic.
List of TKI-nanoconstruct applications published recently
| Nanomaterial | TKI | Effect | Targeting | Targeting ligand | Ref. |
|---|---|---|---|---|---|
| Gold NPs | Afatinib | Improvement of efficacy and biocompatibility | Passive | - | [86] |
| Immunoliposomes | Afatinib Cetuximab | Protection from binding to hemoglobin, strongly enhanced drug delivery and anti-tumor efficacy, selectivity and potentially fewer side effects. | Active | Anti-EGFR antibody | [80] |
| Liposomes | Afatinib | Improved anti-tumor activity | Passive | - | [166] |
| Colloidal polyethylene glycolated (PEG) gold NPs | Afatinib | Higher cellular uptake, 5 and 20 times more potent than Afatinib alone | Passive | - | [167] |
| Cyclic arginylglycylaspartic acid (cRGD) and PEG-modified liposomes | Apatinib | Significant tumor treatment targeting ability, better inhibition of tumor growth, and less toxicity. | Active | cRGD | [91] |
| Enzyme responsive size-changeable gold NPs | Cediranib | Enhanced tumor vascular permeability, significant therapeutic effect | Passive | - | [168] |
| Poly(lactic-co-glycolic acid) (PLGA)-PEG NPs | Cediranib Verteporfin | Combination drug therapy with phototherapy resulted in significant in vitro cytotoxicity. | Passive | - | [169] |
| Poly(styrene-co-maleic acid) micelles | Crizotinib Dasatinib | Enhanced drug activity of drugs in combination, same anti-proliferative effect in vitro as free drug, potent anti-proliferative effect in vivo. | Passive | - | [170] |
| Human serum albumin (HSA) NPs | Dasatinib | As effective as free drug, reduced endothelial hyperpermeability | Passive | - | [171] |
| Poly-L-lactic acid(PLA) NPs modified with polyethyleneimine | Dasatinib Trastuzumab | Better in vitro efficacy and sustained release of dasatinib | Active | Anti-HER2 antibody | [172] |
| Poly(Cyclohexene Phthalate) NPs | Dasatinib | Superior in vitro efficacy | Passive | - | [173] |
| PLGA NPs | Dasatinib | Compared to free drug enhanced inhibition of proliferative vitreoretinopathy related cellular contraction. | Passive | - | [174] |
| PLGA-conjugated gold NPs | Dasatinib | Enhanced growth inhibition in vitro and bioavailability in vivo | Passive | - | [175] |
| Magnetic micelles | Dasatinib | Increased in vitro cytotoxicity and decreased cellular migration | Active | Lactoferrin | [176] |
| CdSe/ZnS quantum dots | Desmethyl Erlotinib | Cytotoxic enhancement | Passive | - | [177] |
| Nanoparticular platform utilizing fat and supercritical fluid | Erlotinib | Improved water solubility | Passive | - | [178] |
| Magnetic iron oxide NPs | Erlotinib | Enhancement of therapeutic efficacy, MRI visualization | Passive | - | [142] |
| Nanocrystals formulation | Erlotinib | Solubility and drug efficacy enhancement | Passive | - | [179] |
| Folate-conjugated thermosensitive O‑maleoyl modified chitosan micellar NPs | Erlotinib | Significantly enhanced cytotoxicity | Active | Folate | [180] |
| Solid lipid NPs | Erlotinib | Higher anticancer activity than free drug | Passive | - | [181] |
| Nanoparticulation platform utilizing fat and supercritical fluid | Erlotinib | More potent in inhibiting EGF signaling and in suppressing tumor cell proliferation. | Passive | - | [182] |
| Cyclodextrin nanosponge | Erlotinib | Increase of solubility, dissolution and oral bioavailability, higher cellular uptake and in vitro cytotoxicity. | Passive | - | [183] |
| Polyamidoamine dendrimers | Erlotinib Survivin shRNA Chloroquine | Promoted drug delivery and enhanced drug efficacy | Active | Anti-EGFR aptamer | [114] |
| Anti-EGFR aptamer-modified liposomal complexes | Erlotinib O2 | Superior anti-tumor activity, significant inhibition of cell proliferation and improved apoptosis induction. | Active | Anti-EGFR aptamer | [105] |
| Eudragit® RL100 | Gefitinib | Enhanced oral bioavailability | Passive | - | [184] |
| Human heavy chain apoferritin | Gefitinib | Enhanced anti-tumor activity against HER2 overexpressing SKBR3 breast cancer cell line; decreased uptake in cell line, which does not express HER2. | Passive | - | [185] |
| Gold colloidal NPs | Gefitinib | Greater cytotoxicity | Passive | - | [186] |
| Gelatin tri-block NPs | Gefitinib Cetuximab siRNA | Effective targeting and high bioavailability, very specific for KRAS G12C | Active | Anti-EGFR antibody | [117] |
| PEG-PLA NPs | Gefitinib Cyclosporin A | Improvement of drug efficacy, sensitization of gefitinib resistant cells | Passive | - | [187] |
| Chitosan NPs | Gefitinib Chloroquine | Potential to overcome acquired resistance and improve cancer treatment efficacy. | Passive | - | [106] |
| Anti‐PD‐L1‐modified liposomal system | Gefitinib Simvastatin | Remodeling the tumor microenvironment, reversing gefitinib resistance and enhancing EGFR T790M‐mutated NSCLC treatment outcomes. | Active | Anti-PD-L1 nanobody | [121] |
| Sialic acid-stearic acid conjugate modified on the surface of nanocomplexes | Ibrutinib | Suppressed tumor progression | Active | Sialic acid | [188] |
| HSA NPs | Imatinib base | 35% greater cytotoxicity | Passive | - | [189] |
| Galactoxyloglucan NPs | Imatinib mesylate | Enhancement of cytotoxic potential and reversal of multidrug resistance | Passive | - | [190] |
| PLGA NPs | Imatinib mesylate | Improved cytotoxic compared to free drug, 28 day‑long oral administration showed no significant cardiotoxicity or associated changes. | Passive | - | [87] |
| Poly(ε-caprolactone) NPs with chitosan | Imatinib mesylate | Improved drug's kinetics and efficacy, long-lasting inactivation of BCR-ABL autokinase activity. | Passive | - | [191] |
| Polycaprolactone nanocapsules | Lapatinib | Improvement of anti-tumor effects | Passive | - | [192] |
| Hyaluronic acid-D-α-tocopherol succinate-(4-carboxybutyl)triphenyl phosphonium bromide-based NPs (HA-TS-TPP) | Lapatinib | Better tumor growth suppression, triple negative breast cancer targeting | Active | HA; TS; TPP | [193] |
| HSA NPs | Lapatinib | Enhanced cell cytotoxicity and induction of apoptosis, inhibition of HER2 phosphorylation and superior anti-tumor efficacy in vivo, no subchronic toxicity within 60 days of treatment. | Passive | - | [88] |
| HSA NPs | Lapatinib | Inhibition of adhesion, migration and invasion ability of cells more effectively; extension of median survival time in mice. | Passive | - | [89] |
| HSA NPs | Lapatinib | Increased accumulation of Lapatinib in tumor tissue, better suppression effects both on primary breast cancer and lung metastasis in vivo. | Passive | - | [194] |
| PTX NPs and LAPA microparticles in a thermosensitive hydrogel | Lapatinib Paclitaxel | Synergistic effect of LAPA and PTX on cell line overexpressing HER2 and P-gp; significantly less nonspecific toxicity. | Passive | - | [115] |
| Liposomes | Ponatinib | Significant tumor growth inhibition (by 60.4%) and markedly reduced side effects. | Passive | - | [195] |
| Liposomes | Nintedanib | Significant tumor growth inhibition (by 60.4%) and markedly reduced side effects. | Passive | - | [195] |
| Linear-dendritic self-assembling polymeric drug carrier release-triggered by enzyme Cathepsin B | Saracatinib | Better suppression of metastasis | Passive | - | [196] |
| Reduced graphene oxide nanosheets | Sorafenib | Improved cytotoxicity | Passive | - | [197] |
| Lipid nanocapsules | Sorafenib | Early tumor vascular normalization, decreased proliferation | Passive | - | [198] |
| Focused ultrasound-triggered thermosensitive liposomes | Sorafenib | Significantly lower cell viability | Passive | - | [90] |
| Self-assembling PEG-vitamin E succinate derivative NPs | Sorafenib Curcumin | Enhanced in vitro cytotoxicity and anti-angiogenesis, greater drug concentration in organs in vivo and inhibition of tumor growth. | Passive | - | [132] |
| HSA encapsulated gold nanorods paired with photothermal ablation | Sorafenib | 100% tumor cell kill rate | Passive | - | [199] |
| Irradiated HSA gold nanorods | Sorafenib | Significantly induced hyperthermia, enhanced cytotoxicity | Passive | - | [200] |
| PLA-PEG-poly(L)-lysine-diethylenetriamine pentaacetic acid NPs with gadolinium and poly(L-histidine)-PEG-biotin modification | Sorafenib | Improved diagnostic abilities, higher anti-tumor effect in vitro and in vivo. | Active | Anti-VEGFR antibody | [141] |
| Styrene-co-maleic acid micelles | Sorafenib Nilotinib | Greater cytotoxicity, decreased cell proliferation, increased apoptosis relative to the free TKIs. | Passive | - | [110] |
| Lactobionic acid modified and pH-sensitive chitosan-conjugated mesoporous silica nanocomplex | Sorafenib Ursolic acid | Enhanced bioavailability, synergistic cytotoxicity, significant increase of cellular apoptosis and down-regulation of EGFR and VEGFR2 proteins expression, significant reduction of tumor burden in hepatocellular carcinoma. | Active | Lactobionic acid | [122] |
| Integrin-targeted cAmpRGD liposomes | Sunitinib | Inhibition of growth and adhesion, anti-angiogenic effect | Active | cAmpRGD | [201] |
| Self-nanoemulsifying drug delivery system | Sunitinib | Bioavailability and cytotoxicity increase | Passive | - | [79] |
| PLGA-PEG-MBA polymeric micelles combined with mannose-modified lipid calcium phosphate NPs-based Trp2 vaccine | Sunitinib | Abrogation of tumor-associated immune suppression, enhanced therapeutic efficacy. | Passive | - | [119] |
| Self-nanoemulsifying drug delivery system | Sunitinib malate | Two-fold increase in efficacy | Passive | - | [202] |
| PEG-NLG919-based immunostimulatory nanocarrier | Sunitinib Paclitaxel NLG919 | More active tumor immune microenvironment and further improved anti-tumor activity. | Passive | - | [203] |
| BSA-coated superparamagnetic iron oxide NPs | Sunitinib Curcumin | Significant tumor inhibition yet least drug-induced toxicity both in vitro and in vivo when compared with free drug formulations. | Passive | - | [104] |
| iRGD-PEG-PLA NPs | Vandetanib | More effective cytotoxic activity in vitro and tumor inhibition in vivo | Active | iRGD | [204] |
| Micellar gold NPs | Vandetanib | Inhibition of tumor growth | Passive | - | [205] |
Nanomaterials-based improvement of pharmacokinetics of TKIs
TKIs are characterized by poor solubility, and thus highly variable bioavailability manifesting through diverse characteristics regarding absorption from the gastrointestinal tract (Figure 2). Major determinant in TKI absorption is the pH-dependent solubility. TKIs exhibit weakly basic properties; therefore, the luminal pH of the gastrointestinal tract and the pKa of the drug determine whether they take the ionized or non‑ionized form. In the highly acidic environment of the stomach, the equilibrium is inclined to the ionized form, which normally dissolves more easily than a non‑ionized form. Bioavailability can be impaired by co‑administration of TKIs with an acid-suppressive medication [e.g. antacids, proton pump inhibitors (PPI)], which increases the stomach pH. The balance of both forms of the drug would shift to the less soluble, non-ionized one and may result in a significant subtherapeutic exposure and treatment failure [71-73]. TKIs that are lipophilic and non-polarized at physiological pH dissolve freely in lipids, and therefore easily diffuse passively through the lipid bilayer membrane. Polarized TKIs, on the other hand, are commonly transported by proteins that form transmembrane channels [74, 75]. These properties of TKIs pose a major challenge for their use in clinical practice, and it has to be noted that nanomedicine is highly promising to solve these issues. Through encapsulation or complexation of TKIs, nanomedicines are capable of: i) protecting TKIs from the harsh conditions of gastrointestinal tract, ii) improving the intestinal absorption of TKIs and iii) provide TKIs with a controlled release and sustained ionization rates. Interestingly, despite the fact that most of the TKIs get manufactured for the oral administration, nearly all of the FDA-approved nanomedicines are developed for i.v. applications. This implicates an essential need for more advanced nanovehicles able to cope with the obstacles for orally-administered nanomedicines.
Major sites of absorption, pharmacokinetic drug-drug interactions and elimination in TKI therapy. Abbreviations: TKI, tyrosine kinase inhibitor; MRPs, multidrug resistance protein drug transporters; P-gp, P-glycoprotein; CYPs, cytochrome P450 enzymes. *With increasing stomach pH the bioavailability of TKIs decreases. †Protein-bound molecules are not available to exert pharmacological effects. ‡Shows the association between CYPs and drug transporters in TKI absorption or metabolism. Adapted from Van Leeuwen et al. [71] with publisher´s permission (license no. 4802950894525).
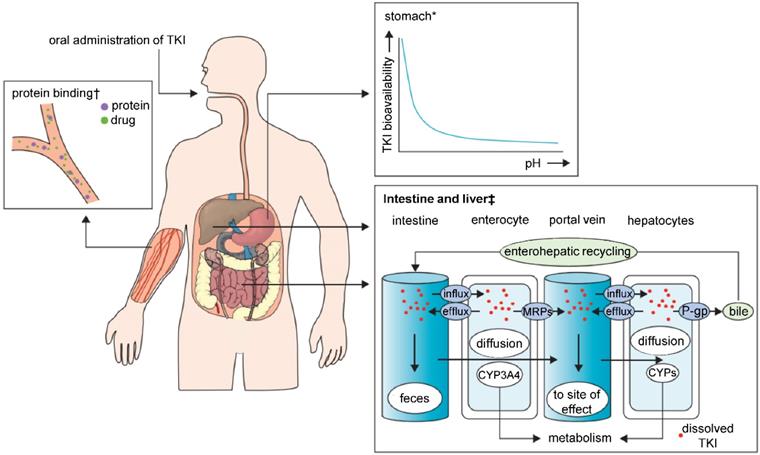
When administered i.v., all TKIs are highly bound (most of them over 90%) to plasma proteins (most often to α(1)-acid glycoprotein and/or albumin), which makes these inhibitors ineffective [76]. TKIs are also characterized by relatively long plasma half‑life, ranging from shorter time spans, like dasatinib (3-5 h), to long ones, like sunitinib (40-60 h). In this unilateral point of view, the drug stability in the bloodstream should be sufficient to reach desired target without the help of nanoformulation. However, short term maximum tolerable dose of TKIs can be transformed into intolerable toxic dose with chronic long-term drug exposure. Another reason to consider nanoformulation could be the low bioavailability of TKIs that is affected by their hydrophobicity, even though there are exceptions (e.g. imatinib, erlotinib) [54].
Nowadays, mostly rat and mouse models are used for in vivo studies of TKI nanoformulations. Therefore, we cannot compare actual data from clinical usage of TKIs with pharmacokinetic abilities of nanoformulated drugs tested on animals. However, recent studies showed that nanoformulations can markedly improve TKIs pharmacokinetic properties in in vivo settings.
For example, lipid-based nanoformulations, being amphipathic, are able to enhance solubility and bioavailability of TKIs. This phenomenon was demonstrated by Dora et al., who constructed a phospholipid complex with erlotinib. Pharmacokinetic assessment showed ∼1.7-fold (p < 0.05) higher bioavailability of the nanoformulated complex compared to free drug, with ∼1.3-fold (p < 0.01) higher maximum plasma concentration and longer half-life. [77]. The profound pharmacokinetic improvement of TKIs can be explained by the enhancement of their hydrophilicity achieved through complexation with phospholipid complexes together with prolonged exposure to the molecular complex due to higher mean-residence time in a proximity of tumor tissue. Similarly, Qiu et al. prepared a phospholipid complex with ibrutinib, which exhibited 9.14-fold increase in bioavailability compared to free drug. Maximum plasma concentration was also higher and was achieved in shorter time period, while tumor volume was significantly decreased [78].
Nazari-Vanani et al. used sunitinib maleate in combination with self-nanoemulsifying delivery system (SNEDDS) that is characterized by a high hydrophilic-lipophilic balance. In an in vivo experiment, oral administration of nanoformulation had higher plasma concentration over time, with 1.45 times higher maximum concentration achieved in less than half the time [79]. In another recently published study, Lu et al. constructed liposomal afatinib with cetuximab (anti-EGFR antibody) coating. These immuno-NPs showed tumor growth inhibition and higher afatinib concentration in tumor tissue, as well as in liver and lungs. Due to the prolonged blood circulation and inhibited binding rate to endogenous proteins, afatinib nanoformulation exhibited higher area under curve (AUC) and plasma terminal half-life, when it could be detected even after 72 h in the bloodstream [80].
Gao et al. utilized human serum albumin (HSA), which, as the main protein for plasma transport in physiological conditions, can escape systemic clearance naturally and has higher uptake in cancer cells, because they are in need of nutrients for their uncontrolled growth [81]. Aware of these advantages, they prepared NPs coated with sorafenib and with or without grafted folic acid (FA), and studied their effect on hepatocellular carcinoma. Nanoformulation increased AUC, mean residence time and plasma half-life and decreased total plasma clearance, which is a positive result. NPs without grafted FA showed better values than the ones with FA. However, the latter ones markedly increased sorafenib accumulation in tumor tissue and reduced the tumor volume [82].
Li et al. produced biomimetic galactose-modified polymeric NPs loaded with imatinib that exhibited enhanced oral bioavailability, higher maximum concentration and AUC with increased intestinal permeability and affinity to intestinal epithelial cells. Fast entrance into the bloodstream resulted in fast clearance in liver, and NPs were not able to extend plasma residence time. Authors suggest NP modification with functional biomaterials to maintain imatinib plasma concentration for longer period. However, benefit of this nanoformulation lies in fast bloodstream penetration, as it might result in adverse events reduction [83].
Taken together, inherent physico-chemical properties of TKIs are substantially complicating their treatment efficiency. Abovementioned studies clearly demonstrate that distinct types of nanomedicines can markedly improve the pharmacokinetic behavior of TKIs. This in particular is due to improved solubility of TKIs incorporated into delivery vehicles, followed by a subsequent enhancement of TKIs hydrophilicity and improved stability in the blood stream that results in a prolonged blood circulation of nanoformulations and their higher residence time in tissues adjacent to tumor mass. Even passive, non-targeted nanoformulations can be then accumulated in the tumor mass through “enhanced permeability and retention” (EPR) effect [84]. Additional fine-tuning of nanovehicles (e.g. involvement of ligands mediating active targeting of specific subsets of cancer cells or utilization of tumor microenvironment stimuli-responsive domains) could further improve the pharmacokinetics of TKI nanoformulations.
Reduction of adverse effects of TKIs-based therapy
Although targeted therapy is, in general, regarded as more moderate than classic chemotherapy, it has more intricate mechanisms of action concerning their structure and targets. Even in targeted therapy, severe adverse events occur and resulting dose adjustments or treatment discontinuation impair its better treatment efficacy [85].
Eckstein et al. summarized toxicity characteristics of eleven TKI drugs available on the market. They pooled the side effects classification scale into two groups, the first including rare and uncommon events, the second involving common and very common events. All eleven inhibitors caused nerve, lung and airways disorders with the higher abundance. Also, in the same abundance category, ten caused skin disorders, nine caused gastrointestinal disorders, carcinogenic, mutagenic and toxic effects on reproductive system, and eight of them caused eye, blood and lymphatic disorders. Other adverse effects were related to liver, bile, vascular system, kidneys or musculoskeletal system. Pazopanib, as the most severe inhibitor from the list, had 12 out of 14 indications with the frequency of common, very common. On the other hand, sorafenib had only 5 common or very common effects out of 14 [54].
As demonstrated in the previous paragraph, pooling of adverse events grading quite commonly occurs in studies and articles. This bad habit may distort the actual graveness of some events. For example, in Common terminology criteria for adverse events (CTCAE v5.0), grade 1 plus grade 2 and grade 3 plus grade 4 events are regularly pooled, where main focus goes to the grade 3/4 group. However, in the case of long-term drug administration, group 1/2 should not be overlooked, if we want to improve patient's quality of life. It is important to keep in mind that the milder but more common side effects put a significant impairment on patient's everyday psyche [85].
In this sense, well-thought-out design in drug development, substantial drug toxicity profiling and proper administration are needed to minimize adverse events. Nanoformulations have good potential to reduce these events even more by specific biodistribution of anti-cancer drugs. In preclinical in vitro and in vivo models, number of papers provide evidence that drug-loaded NPs show enhanced properties compared to free drugs. For example, Cryer et al. developed gold NPs conjugated with an afatinib analog that mediated cancer cell-specific growth inhibition and attenuated inflammatory cytokine release. The selectivity to cancer cells is plausibly due to highly efficient uptake of gold NPs concomitantly inhibiting the efflux of afatinib to the extracellular space [86]. Marslin et al. evaluated performance of imatinib mesylate-loaded poly(lactide-co-glycolide) NPs that exhibited increased cytotoxicity in vitro and significantly reduced cardiotoxicity in vivo compared to free drug. [87]. Despite the fact that the authors did not investigate phenomenon responsible for inhibition of imatinib side effects, it is likely that loading of imatinib into NPs results in improved blood circulation and preferential bioaccumulation of imatinib in tumor mass due to the EPR effect. Wan et al. worked with lapatinib-loaded HSA NPs on HER2 positive [88] and triple negative metastatic breast cancer [89]. The former, apart from showing enhanced anti-tumor activity, manifested no subchronic toxicity throughout the duration of the experiment. The latter one improved inhibition of adhesion, invasion and migration of brain-metastatic breast cancer cells. In vivo, the nanoformulation increased delivery to brain tissue and significantly prolonged median survival period of tested mice.
The relative selectivity of nanodrugs in cancer is based on the so-called passive targeting by the abovementioned EPR effect. This mechanism is based on substantial extravasation of the nanodrugs into the interstitial fluid at the tumor site. Nanodrugs can better penetrate the tumor due to poor quality vessels in the tumor and are retained for a longer time at the tumor site because of the defective lymphatic vessel system in tumor microenvironment. EPR affects nanodrugs that are 20-100 nm in size [90]. Improved therapeutic effects of drug nanocarriers can be achieved by targeting molecules specific to a particular tissue or cell type. Song et al. prepared cyclic arginylglycylaspartic acid (cRGD) and polyethylene glycol (PEG) modified liposomes with apatinib that specifically recognize integrin αvβ3 [91]. In comparison with free drug and untargeted liposomes, the cRGD-Lipo-PEG/Apa showed enhanced cytotoxicity and cellular uptake in vitro, plus it performed better targeting, faster clearance from murine organism, significantly reduced tumor weight and no visible damage to internal organs. Lu et al. intended to overcome adverse effects of combination therapy with afatinib and cetuximab by encapsulating the TKI into liposomes and using cetuximab as targeting and therapeutic agents bound to the surface of liposomes [80]. This solution resulted in better cellular uptake and in vivo prolonged half-life, better drug delivery, prevention of afatinib binding to hemoglobin, enhanced tumor selectivity and growth inhibition. This study suggested the drug dosage could be significantly decreased in clinical application, therefore potentially reducing adverse events incidence.
Blood-brain barrier
A number of targets have been identified for potential treatment of brain malignancies by TKIs (e. g. EGFR, VEGFR, AKT, ALK, c-Met) [92]. At least 50% of brain metastases are caused by lung cancer. Second most common (15-25%) metastasis originates in breast cancer tissue [93]. Both of their non-metastatic precursors are commonly treated with TKIs. However, major problem in brain cancer therapy is connected to the blood-brain barrier (BBB) - the specific lining of brain blood vessels characterized by intercellular adherens and paracellular tight junctions, specific efflux transporters for nutrients (solute carriers) and metabolites (ATP binding cassette) and absence of intracellular vesicular transport. Nature of the BBB helps protect the brain tissue from passive diffusion of molecules, mainly to avoid toxins and harmful substances [94]. Although the BBB gets disrupted by tumor to better supply nutrients, this only happens in the center of the tumor mass. In the peripheral areas, blood-brain tumor barrier exists, and it is similar to BBB [95]. All of this, unfortunately, works against brain disease therapy, and many small-molecule inhibitors did not find success in reaching their target behind the BBB [92].
Nanomedicine gives drugs the advantage of prolonged blood circulation time, which increases their chance to cross the BBB (Figure 3) [96]. Alternatively, NPs can increase drug concentration in the proximity of BBB cells, which could cause a local disruption of the BBB and allow the drug to penetrate inside the brain [97]. More importantly, NPs can be designed to possess the ability to cross the BBB themselves and successfully deliver the drug. For example, surface charge of NPs has an effect on the BBB - neutral or lower concentrations of anionic NPs are suitable for brain penetration, as opposed to cationic NPs, which exhibit toxicity to brain endothelium [98]. Targeting ligand, such as transferrin, insulin, glutathione, HIV-1 trans-activating transcriptor or peptide derived from apolipoprotein-E, can be attached to the surface of NPs to ensure crossing the BBB by receptor-mediated transcytosis [99]. However, after overcoming the BBB, NPs have to be able to internalize into tumor cells as well [96].
Pathways across the BBB. BBB consists of endothelial cells linked by tight junctions, encircled by pericytes and astrocytes, together with basal lamina and neurons. In between adjacent cells (paracellular pathway - A), some ions and solutes cross freely by passive diffusion. Other molecules utilize various transcellular pathways (B-F), like specific transporter proteins or receptor-mediated transcytosis and diffusion. Efflux transporters, such as P-glycoprotein (P-gp), prevent some substances from crossing the BBB. Adapted from Wang and Wu [206] with publisher´s permission (license no. 4892530338842).
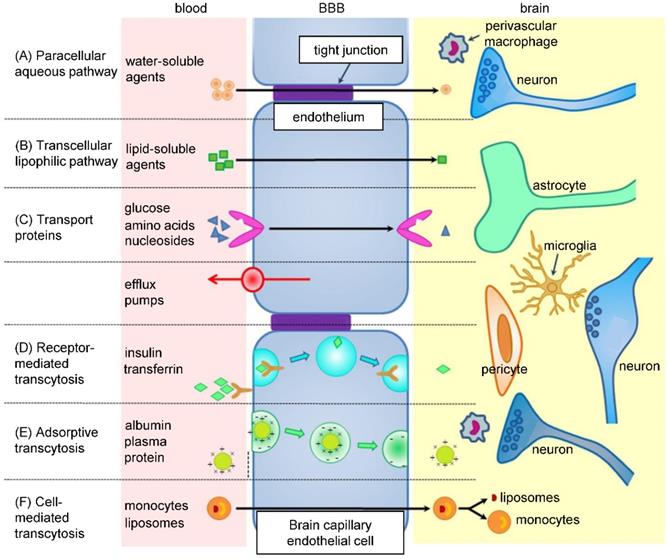
Wan et al. prepared HSA NPs for treatment of triple negative breast cancer brain metastases with lapatinib [89]. In vitro analysis showed that, compared to free drug, the nanoconstruct suppressed the adhesion, migration and invasion ability of 4T1 cell line with better efficacy. In vivo, lapatinib concentration in the brain of 4T1 injected mice was the highest in NPs-treated group at all time points after administration. High brain tissue accumulation of lapatinib was plausibly due to capability of HSA NPs to inhibit the efflux of the drug by P-gp and BCRP transporters, restricting the retention of free lapatinib in the brain. In a survival experiment, brain metastatic nude mice were treated with three different concentrations of nanoformulated lapatinib, as well as free drug solution and commercially available drug Tykerb. Each nanoformulation prolonged mice survival compared to non-nano formulations, while the highest concentration (30 mg/kg) resulted in the longest survival time.
To improve the transcytosis of lapatinib across the BBB, Wan et al. relied on enhanced EPR effect plus attraction of HSA NPs by SPARC (secreted protein acidic and rich in cysteine), as well as on beneficial properties of HSA that include preferential tumor uptake and binding to 60-kDa glycoprotein receptor on vascular endothelial cells [89]. Lakkadwala et al. took up different approach - liposomes modified with transferrin and penetratin loaded with erlotinib and doxorubicin for treatment of glioblastoma [100]. Surface functionalization mediated intracellular uptake, which was demonstrated in this study. Erlotinib and doxorubicin concentration in brain tissue was significantly higher using dual functionalized nanocarriers than free drugs or single peptide-modified liposomes, suggesting utmost importance of receptor-mediated endocytosis (transferrin) and enhanced cell penetration (penetratin) for efficient translocation of erlotinib-loaded liposomes across the BBB. Importantly, survival of glioblastoma bearing nude mice was significantly prolonged and tumor area was minimized.
Although research in brain-targeted nanomedicine is in full bloom, not many researchers focus on using nanoformulations for TKI treatment of brain cancer or metastases. However, these few promising examples show that TKIs could be used in this manner. We postulate that future investigations might focus on development of highly biocompatible nanovehicles of endogenous origin, able to utilize the inherent properties of BBB to mediate transcytosis of nanoformulations. In addition, nanovehicles for TKIs brain delivery might also be capable of inhibiting frequent brain-associated efflux mechanisms causing undesired transport of the payload to the extracellular region.
Nano-based combination therapy with TKIs
Therapy that combines two or more anticancer agents is applied for two main reasons - to bypass/delay drug resistance or to enhance drug efficacy. Moreover, combination of drugs usually allows reducing the doses of individual drugs, and thus alleviates side effects. Main strategy would be to co-target compensatory signaling pathways with either TKIs or monoclonal antibodies. Some strategies are focusing on Heat shock protein 90 that is necessary for protein stabilization and plays a role in the folding of many RTKs [101-103]. Another possibility is to combine TKIs with immune therapy for better tumor recognition by the immune system [53]. TKIs can be also used in combination with standard chemotherapy or radiotherapy [2]. However, recent research brought to attention novel combinations of TKIs with diverse spectrum of other agents, for example curcumin [104], oxygen [105], chloroquine [106] or even miRNA for regulation of expression of cancer-associated genes [107]. In addition, a number of clinical trials studying TKI combination therapy are currently underway or recruiting participants. Studies frequently focus on TKI usage with immunotherapy (NCT04055792, NCT04044378, NCT04042116, and NCT03580928) and chemotherapy (NCT04035486, NCT01931098, NCT00390793, NCT00557193). Combination with different TKIs (NCT04028778, NCT01531361, NCT01982955, NCT02824458 and NCT02767804) is also very common. Fewer trials investigate applications of TKIs with hormonal therapy (NCT04033172) or other agents (PPI - NCT02800330, dendritic cell vaccines - NCT01876212, hydroxychloroquine - NCT00813423, various inhibitors - NCT00335764, NCT00655655, eicosapentaenoic acid - NCT04006847, etc.). Several trials explore combination therapy of TKIs and nanoformulation of other drugs - most frequently it is nanoformulated paclitaxel (e.g. NCT01455389, NCT00733408, NCT00331630, NCT00709761, NCT03942068 and NCT00313599). Importantly, no downright nano-TKI applications reached the stage of clinical research yet.
Multidrug approach has been previously found beneficial in the control of HIV, showing that specific drug combinations were able to overcome treatment difficulties [85]. However, the approach to TKI combination therapy should not be global. Tumor/patient specific approach is a prerequisite for making a diagnostic decision whether to use one multi-targeted TKI or several single-targeted inhibitors. Multidrug therapy is generally more suitable for tumors with higher number of overexpressed kinases responsible for resistance and for tumors with resistance induced by a mutation in the TK. Combination TKI therapy requires knowledge of toxicity profiles and metabolism routes for each drug. Tumor microenvironment is also quite important, because gene expression of its stromal cells may be different from the neoplastic cells. Therefore the effect of each TKI on tumor stroma might also be different [108].
Use of drug combination in nanoformulation should be well devised, as there are some drugs that properly function only in specific order and timeframe. Their simultaneous release would cause severe side effects [109].
TKIs in combination with other small molecule drugs
Combination therapy of two different TKIs has its benefits in targeting multiple TK signaling pathways, generating more potent anti-tumor effects and limiting possibilities of bypassing the inhibited pathway, thus avoiding or postponing the occurrence of resistance. Archibald et al. showed this on castration-resistant prostate cancer cell lines with the combination of sorafenib and nilotinib [110]. Greish et al. proved that crizotinib and dasatinib combination in micellar formulation shows enhanced anti-tumor efficacy against glioblastoma multiforme, possibly due to increased blood circulation and improved bioaccumulation of TKIs at the tumor site [111].
There are records of using chloroquine in combination with nanoformulated TKIs based on its multiple mechanisms of action affecting tumor microenvironment. Chloroquine can inhibit late stage autophagy, activate tumor suppressor protein p53, normalize tumor vasculature, which decreases hypoxia, etc. [112]. In this way, Zhao et al. delivered chloroquine/gefitinib cocktail encapsulated in chitosan NPs to resistant hepatocellular carcinoma cells and managed to overcome said resistance through inhibition of the autophagic effects, evidenced by down-regulation of the LC3 II and LC3 I ratio [113]. Similarly, Lv et al. made polyamidoamine NPs functionalized with anti-EGFR aptamers for targeted co-delivery of erlotinib and survivin-shRNA with simultaneous systemic administration of chloroquine. Purpose of chloroquine in this case was mainly vascular normalization for smooth NPs delivery and endosomal escape, while therapeutic effects were mediated by erlotinib and shRNA down-regulation of survivin that inhibits drug-induced apoptosis [114].
Interestingly, Hu et al. developed a thermosensitive hydrogel for peritumoral use, containing a nanoformulated Pluronic F127-based chemotherapeutic agent paclitaxel with short release time and lapatinib microparticles with long-term release kinetics [115]. Initial high concentration of paclitaxel strongly suppressed tumor growth, and the stable lapatinib release maintained its efficiency to enhance the therapeutic impact. In vivo experiment showed that in comparison with oral administration of lapatinib, the gel administration performed comparably, although with much smaller amount of drug needed. In addition, by using the gel, steadier bioaccumulation of lapatinib in tumors together with lower undesired toxicity was achieved, and nanoformulated lapatinib exhibited good synergistic effects with paclitaxel.
TKIs in combination with immunotherapy
In some cases, antibodies are used in combination with TKIs in nanoconstructs to target TK domains for precise delivery and for their anti-tumor activity in the form of competitive ligand binding [116, 117]. Other times, TKIs like sunitinib are used in combination with immunotherapy for their ability to modulate tumor microenvironment [118]. For example, Huo et al. used sunitinib encapsulated in polymeric (anisamide-modified poly-lactic-glycolic-acid-PEG) micellar nanovehicle to support their tyrosinase-related protein 2 (Trp2) nanovaccine in melanoma therapy, with results showing attenuated tumor-associated immune suppression [119]. Polymeric nanovehicles enabled an efficient delivery of a low dose of sunitinib and decreased the number of myeloid-derived suppressor and regulatory T cells with enhanced infiltration of cytotoxic T cells, thereby abrogating tumor-associated immune suppression. Kim et al. combined their Toll-like receptor 7/8 agonist nanovaccine with free sunitinib and programmed death-ligand 1 (PD-L1) antibody, which, again, abrogated tumor-associated immune suppression and significantly decreased tumor weight in an in vivo experiment [120]. Yin et al. also used PD-L1; however, they produced a nanobody acting as a targeting ligand of liposomal delivery system of gefitinib and simvastatin. Their goal, to overcome gefitinib resistance in T790M EGFR-mutated NSCLC, was achieved in an in vivo experiment [121]. The studies have clearly demonstrated that nanoformulations of TKIs could be useful as synergistic modalities to improve the outcomes of existing immunotherapies.
TKIs in combination with other biologically active agents
There have been studies combining TKIs with natural substances that have multiple effects on human metabolism. For example, Zhao et al. combined sorafenib with ursolic acid in mesoporous silica NPs coated with pH sensitive chitosan and lactobionic acid - targeting agent for asialoglycoprotein receptor overexpressed in hepatocellular carcinoma. They used ursolic acid for its reported ability to down-regulate VEGFR and EGFR [122]. Ursolic acid is said to have profound anticancer properties [123]. Zhao's data hint a synergy of both free drugs combined; however, NPs showed the best results in anti-tumor activity, particularly due to their ability to trigger the release of payloads in response to acidic pH of tumor microenvironment.
Chen et al. combined sunitinib with curcumin in superparamagnetic iron oxide NPs coated with BSA [104]. Curcumin was found to have an anti-invasion and anti-metastatic effect [124, 125] and is quite popular in nanomedicine nowadays [126-131]. In Chen's study, nanoformulation showed markedly better results than free drugs alone or in combination. This was achieved through the capability of the vehicle to efficiently deliver the payload and to strictly maintain the optimal ratio of sunitinib and curcumin at the tumor mass toward the most optimal synergistic cytotoxic effect [104]. Cao et al. used sorafenib/curcumin cocktail encapsulated in directed self-assembled nanoparticles composed of PEG derivative of vitamin E succinate with similar results. Upon oral administration, significantly increased biodistribution of sorafenib and curcumin in gastrointestinal tract was found. The authors stated that this phenomenon is most likely due to the improvement of the affinity and accessibility of nanoformulations to the intestinal membrane that results in their enhanced absorption in the intestine [132].
Hypoxia occurs in the center mass of solid tumors. It stimulates overexpression of HIF-1α that mediates signaling pathways leading to epithelial-mesenchymal transition, neovascularization, cell survival, metastasis and therapy resistance [133]. In NSCLC with mutated and wild-type EGFR subjected to hypoxia, significantly upregulated HIF-1α and TGFα were found, and resistance to gefitinib treatment occurred [134]. Li et al. co-delivered oxygen-releasing perfluorooctylbromide with erlotinib encapsulated in anti-EGFR aptamer-modified liposomal NPs to overcome hypoxia-induced resistance to chemotherapy. NPs with combination of drugs had significant reducing effect on tumor volume compared to NPs with erlotinib alone, which suggests good therapeutic synergy. The study highlights the importance of active targeting moiety on the surface of nanoformulation. Anti-EGFR aptamer enables specific ligand-receptor mediated accumulation of the payload enhancing efficiency of encapsulated drugs with concomitant inhibition of undesired side-effects in vivo [105].
Theranostics
Theranostic approach unites within itself the therapeutic and diagnostic aspects of medicine. In another words, it is the combination of therapy and pharmacokinetic imaging in situ that enables long-term monitoring, and therefore adaptation to modality of disease prognosis. This approach addresses pharmacokinetic variability of drug responses in the population of patients, hopefully resulting in a reduction in adverse event occurrence and in an improvement of treatment planning [135]. Nanotechnology became a suitable theranostic tool, bringing in its benefits, like the ability to carry a single drug or drug combination, controlled and sustained release of its load, ability to bind targeting moiety for specific delivery, etc. [136].
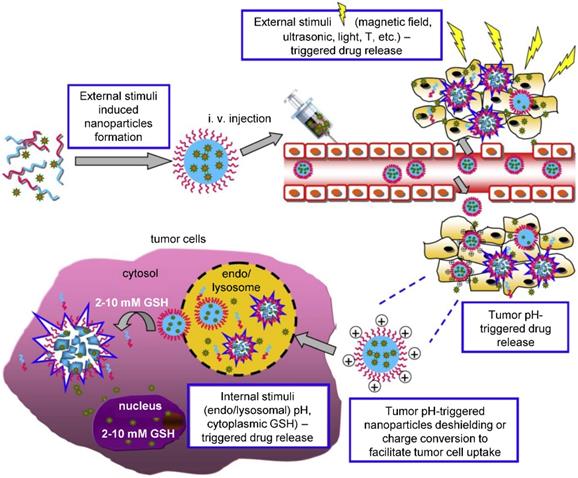
Theranostic agents typically consist of a therapeutic molecule and a traceable label (Figure 4). In case of using nanocarriers, targeting agent can be added, or solely the EPR effect of vehicles in the tumor tissue can be utilized. Targeting agents are typically small molecules, peptides, proteins or antibodies. The variety of labels used in theranostics corresponds to the selection of imaging techniques. For instance, gamma scintigraphy (positron emission or single-photon emission computed tomography) is suitable for visualization of radionuclides (e.g. 18F, 111In, 64Cu). Optical methods commonly detect fluorescent labels, quantum dots, carbon nanotubes or gold nanoshells. Magnetic resonance imaging (MRI) is a good method for para- or superparamagnetic metals, like gadolinium, iron and manganese or NPs from respective materials. Heavy elements, such as iodine, barium and sulphate are visualized by computed tomography, and last but not least, ultrasound can be used to view liposomes and perfluorocarbon nanodroplets [137, 138]. Drug release can be controlled by one or more stimuli (Figure 5). Some approaches rely on internal cellular stimuli, like pH, redox potential or enzymes. Others trigger drug release with external stimuli, temperature, light, ultrasound and magnetic field. The stimuli combination concept boosts tumor cellular uptake and intracellular drug release while decreasing nonspecific cytotoxicity thanks to improved nanocarrier stability [139].
Schematic representation of the complexity of theranostic agents'. Basic approach employs a therapeutic molecule conjugated with a label. Nanoformulation allows attaching variety of different ligands and functional surface modifications to achieve prolonged blood circulation (e.g. PEGylation), controlled release of therapeutic agent (e.g. chitosan) or targeting for particular tissue or cell type (e.g. antibodies, peptides).
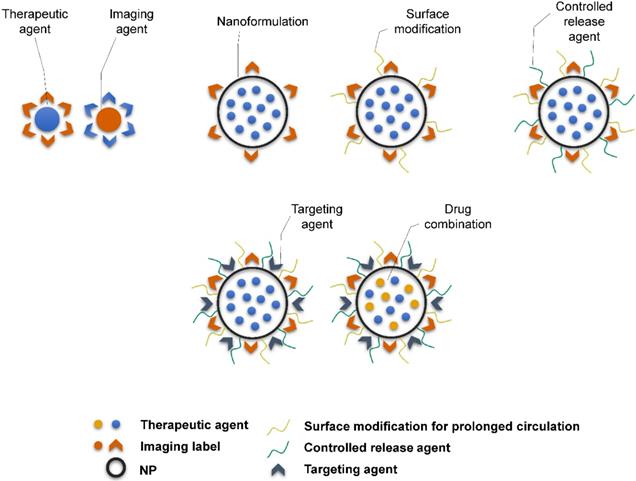
External and internal stimuli for controlled drug release. External stimuli allow for time and site-specific drug release triggered by the application of physical forces, as is magnetic field, temperature, ultrasound, electromagnetic field, light etc. On the other hand, internal stimuli stem from the nature of the tumor and its microenvironment, where specific pH, redox potential or enzyme activity act as a trigger in the tumor tissue or intracellularly. Adapted from Cheng et al. [139] with publisher´s permission (license no. 4892530661903).
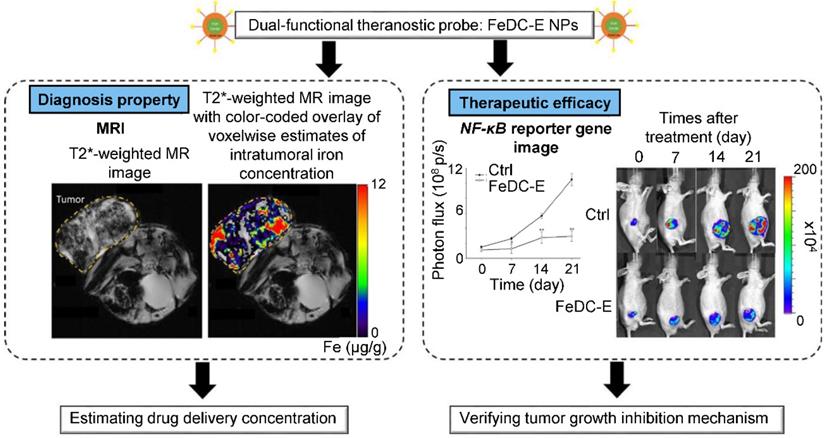
One of the earliest mentions of nano-TKI application in theranostics was made by Noh et al., who developed cross-linked iron oxide NPs conjugated with sunitinib for estimation of expression status of VEGFR and PDGFR by MRI. It has been shown that the iron oxide NPs serve as highly biocompatible nanoplatform with exceptional inherent MRI contrast capabilities, offering facile conjugation of sunitinib [140]. Liu et al. constructed sorafenib-loaded pH-sensitive multi-block polymer [poly(lactic acid)-poly-(ethylene glycol)-poly(l-lysine)-diethylenetriamine glycol)-biotin] NPs with VEGFR targeting by anti-VEFGR antibody and gadolinium ions as MRI contrast agent. In vitro, NPs with anti-VEGFR antibody showed higher cellular uptake than NPs without the antibody. In vivo, NPs showed better anti-tumor efficacy than sorafenib alone with prolonged imaging time and signal enhancement compared to commercial contrast agent Magnevist®. In addition, NPs exhibited exceptional biocompatibility and did not alter organ histology. Overall, the devised NPs served as highly promising biocompatible, actively-targeted, multifunctional platform capable of facile dual loading with TKI and imaging agent [141]. Hsu et al. developed erlotinib delivery monitoring system via iron oxide NPs with MRI imaging and demonstrated the ability to estimate intake concentration of NPs into the tumor (Figure 6) [142]. In vivo experiments revealed that the erlotinib-conjugated NPs effectively inhibited progression of lung cancer. Ex vivo protein expression analysis revealed efficient suppression of antiapoptotic and activation of apoptotic proteins mediated by NF‑κB signaling.
Representation of diagnostic and therapeutic properties of erlotinib-conjugated iron oxide NPs (FeDC-E NPs). Additionally to FeDC-E NPs capability of tumor growth inhibition, demonstrated by decrease of ectopic lung tumor mass and suppression of NF-κB expression captured (right), assessment of delivery efficacy and drug concentration estimation can be made, using MRI (left). Abbreviations: CTRL, control; FeDC-E NPs, erlotinib-conjugated iron oxide NPs; NF-κB, nuclear factor kappa light-chain enhancer of activated B cells; T2* decay of transverse magnetization caused by a combination of spin-spin relaxation and magnetic field inhomogeneity. Adapted from Hsu et al. [207] with publisher´s permission (license no. 4802960989730).
Zhang et al. harnessed the full potential of theranostic approach by fabricating indocyanine green-loaded mesoporous silica NPs lidded with zinc oxide quantum dots and coated with erlotinib-modified chitosan [143]. This redox/pH dual-responsive system targeted EGFR-mutated NSCLC and worked synergistically with NIR irradiation, which provided targeted therapeutic effects in the form of reactive oxygen species generation and regulation of EGFR and apoptosis-related protein expression. As a last example of possible conjunction of TKIs and nanomaterials for theranostics, Sang et al. constructed a complex nanodelivery system with multiple functions. Chitosan oligosaccharide was conjugated with IR780 cyanine fluorescent label and Black Hole Quencher for visualization of fluorescence triggered by NIR irradiation. This complex then self-assembled, encapsulating sorafenib and superparamagnetic iron oxide NPs that were intended for induction of ferroptosis characterized by a high rate of lipid peroxidation [144]. This elaborate structure was capable of causing explosive lipid peroxidation upon irradiation with NIR (> 18-fold compared to non-irradiated structure) and exhibited > 26-fold increase in half-life of sorafenib.
The few examples discussed above clearly demonstrate that TKIs-based chemotherapy might substantially benefit from involvement of nanoparticles with theranostic properties. Such structures could markedly improve the efficiency of anticancer therapy by real-time tracing of TKIs accumulation and continuous visualization of chemotherapy outcomes. Theranostic nanoformulations could also offer a reduction of the dose requirements of administered TKIs, which are frequently characterized by narrow therapeutic indices [145].
Future outlooks
Nanotechnology offers unique properties in drug development. There has already been several nanomedicines approved by FDA [146], and close to 200 clinical studies with “recruiting”, “enrolling”, “active” or “completed” status, are listed on Clinicaltrials.gov under key words “nanoparticle” and “cancer”. However, nanomedicine development still has a lot of room for improvement. Wu et al. summarize challenges in this field in their review [147]. If we follow the path of the first FDA approved nanomedicine - Doxil [148], we see an eight-year-long path from the initial idea to clinical trials. There are plenty of studies that reached in vivo phase, but we do not see these nanodrugs continue to clinical trials yet. For the advancement from in vivo to clinical trials, scale-up in nanodrug manufacturing must be achieved. That requires reproducibility and uniformity in production, drug stability and the possibility to involve automation in the production process, if the trials are successful. We see these requirements and hefty financial investment as the bottleneck of clinical transfer of today's nanomedicine. Especially TKI-nanoapplications are in the beginning phases of development and, although they have great potential, they still have a long way ahead of them.
Harnessing the potential of bioinformatics, artificial intelligence (AI) and deep learning could marginally increase the advancement in nanomedicine [149]. Shamay et al. demonstrated the strengths of computational methods when they showed precise prediction of NP formulation and properties based on molecular structures of precursor molecules, producing NPs with selective targeting ability and impressive therapeutic impact [150]. Nanoinformatics could eliminate or at least alleviate the labor-intensive trial and error-based NP development.
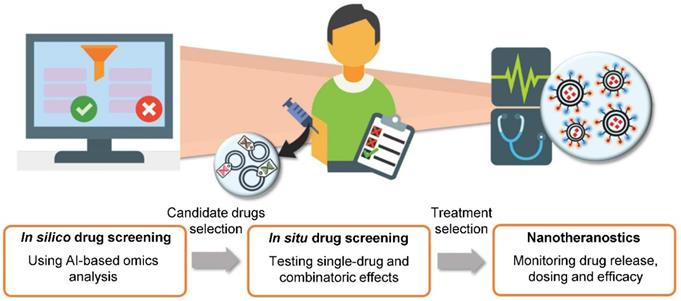
With growing number of public databases [151] and high-throughput methods, mining for information in large datasets becomes more and more important. The application of machine learning and AI technology introduces broad possibilities, such as drug discovery [152], target discovery [153, 154], predictions of NP properties [150, 155], prediction of phenotypic responses [156, 157], image analysis for clinical diagnosis [158, 159], determination of suitable drug combinations for individualized therapy [160, 161] with subsequent efficacy evaluation and dose adjustments [162] and so on [163]. Overall, computational methods and AI could be an integral part of processes vital to human medicine (Figure 7). Tendencies to apply these methods are apparent today, even in clinical trials [164].
The idea of future personalized medicine. AI-based technologies could be utilized in de novo target and drug discovery, prediction of drug efficacy, dosage or combination and response to therapy. Suitable drug candidates and strategies would then be tested and resulting final therapeutic approaches would be monitored by nanotheranostics. Obtained data could be fed back to AI to generate prediction of treatment success and adjustments of treatment plan. Adapted from Adir et al. [149] with publisher's permission (license no. 4892531028481).
RNA interference is a potent therapeutic path especially suitable for undruggable targets. However, it faces multiple challenges, like systemic toxicity and obstacles with delivery to tissues other than kidney or liver [165]. It has been shown that encapsulation of RNAi molecules with TKIs is able to sensitize resistant cells against TKI therapy [114, 117]. Therefore, it possesses a promising future perspective for combination cancer therapy.
Conclusions
Current focus on nanomedicines in cancer research stems from their unparalleled characteristics, starting with increased bioavailability that is especially important for poorly soluble drugs, prolonged blood circulation, passage through biological barriers, tumor targeting, improvement of drug efficacy, ability to effectively deliver multiple drugs at once and exploitation of their own physical attributes - optical, electrical or magnetic - to convey visualization, localization, additional therapeutic effect or controlled drug release. These features will be the foundation of future cutting-edge applications ranging from specific tissue targeting, real-time imaging and theranostic approach, hopefully leading to personalized cancer therapy.
All in all, even though TKIs are fundamentally imperfect as any other type of cancer therapy, and there is a long way to reach clinical trials with currently researched TKIs-NPs combinations, there is a great potential in this union - potential in improving patients' quality of life and efficacy of TKI therapy. Attention should be paid to developing NPs with sound safety profiles that avoid damage caused by systemic exposure of the nanocarrier itself. NPs that will stand their ground in the clinical settings and that will be easy to handle by patients themselves, stable and prone to slight deviations from timely medication adherence.
Abbreviations
AUC: area under curve; ATP: adenosine triphosphate; BBB: blood-brain barrier; CTCAE: common terminology criteria for adverse events; EPR: enhanced permeability and retention; FA: folic acid; i.v.: intravenous; MRI: magnetic resonance imaging; NP: nanoparticle; PK: protein kinase; PPI: proton pump inhibitor; PTB: phosphotyrosine-binding; RTK: receptor tyrosine kinase; SH2: Src homology-2; SNEDDS: self-nanoemulsifying delivery system; TK: tyrosine kinase; TKI: tyrosine kinase inhibitor.
Acknowledgements
This manuscript is dedicated in Memory of Professor Marie Stiborova.
We gratefully acknowledge the Czech Science Agency (project no. 18-10251S), Internal Grant Agency of Mendel University in Brno (project no. AF-IGA2019-IP010) and CEITEC 2020 (LQ1601) for financial support of this work.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA-Cancer J Clin. 2018;68:394-424
2. Bhullar KS, Lagarón NO, McGowan EM, Parmar I, Jha A, Hubbard BP. et al. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer. 2018;17:1-48
3. Jiao QL, Bi L, Ren YD, Song SL, Wang Q, Wang YS. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol Cancer. 2018;17:12
4. Yin YL, Yuan X, Gao HL, Yang Q. Nanoformulations of small molecule protein tyrosine kinases inhibitors potentiate targeted cancer therapy. Int J Pharm. 2020;573:17
5. Tridente G. Kinases. In: Tridente G, editor. Adverse Events and Oncotargeted Kinase Inhibitors: Academic Press. 2017 p: 9-56
6. Robinson DR, Wu Y-M, Lin S-F. The protein tyrosine kinase family of the human genome. Oncogene. 2000;19:5548-57
7. Segaliny AI, Tellez-Gabriel M, Heymann M-F, Heymann D. Receptor tyrosine kinases: Characterisation, mechanism of action and therapeutic interests for bone cancers. J Bone Oncol. 2015;4:1-12
8. Knape MJ, Ballez M, Burghardt NC, Zimmermann B, Bertinetti D, Kornev AP. et al. Divalent metal ions control activity and inhibition of protein kinases. Metallomics. 2017;9:1576-84
9. Moriki T, Maruyama H, Maruyama IN. Activation of preformed EGF receptor dimers by ligand-induced rotation of the transmembrane domain. J Mol Biol. 2001;311:1011-26
10. Mayer BJ. Perspective: Dynamics of receptor tyrosine kinase signaling complexes. FEBS Lett. 2012;586:2575-9
11. Gelens L, Saurin AT. Exploring the function of dynamic phosphorylation-dephosphorylation cycles. Dev Cell. 2018;44:659-63
12. Heldin C-H. Protein tyrosine kinase receptor signaling overview. In: Bradshaw RA, Dennis EA, editors. Handbook of Cell Signaling (Second Edition). San Diego: Academic Press. 2010 p: 419-26
13. Tridente G. Kinase Inhibitors. In: Tridente G, editor. Adverse Events and Oncotargeted Kinase Inhibitors: Academic Press. 2017 p: 57-80
14. Fleuren EDG, Zhang L, Wu J, Daly RJ. The kinome 'at large' in cancer. Nat Rev Cancer. 2016;16:83-98
15. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117-34
16. Lahiry P, Torkamani A, Schork NJ, Hegele RtA. Kinase mutations in human disease: interpreting genotype-phenotype relationships. Nat Rev Genet. 2010;11:60-74
17. Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45:1127-33
18. Blakely CM, Watkins TBK, Wu W, Gini B, Chabon JJ, McCoach CE. et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat Genet. 2017;49:1693-704
19. Garouniatis A, Zizi-Sermpetzoglou A, Rizos S, Kostakis A, Nikiteas N, Papavassiliou AG. FAK, CD44v6, c-Met and EGFR in colorectal cancer parameters: tumour progression, metastasis, patient survival and receptor crosstalk. Int J Colorectal Dis. 2013;28:9-18
20. Huang C-W, Tsai H-L, Chen Y-T, Huang C-M, Ma C-J, Lu C-Y. et al. The prognostic values of EGFR expression and KRAS mutation in patients with synchronous or metachronous metastatic colorectal cancer. BMC Cancer. 2013;13:1-12
21. Bossi P, Resteghini C, Paielli N, Licitra L, Pilotti S, Perrone F. Prognostic and predictive value of EGFR in head and neck squamous cell carcinoma. Oncotarget. 2016;7:74362-79
22. McLendon R, Friedman A, Bigner D. et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061-8
23. Helsten T, Elkin S, Arthur E, Tomson BN, Carter J, Kurzrock R. The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next-Generation Sequencing. Clin Cancer Res. 2016;22:259-67
24. Weinstein JN, Akbani R, Broom BM, Wang W, Verhaak RGW, McConkey D. et al. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315-22
25. Xie N, Tian C, Wu H, Yang X, liu L, Li J. et al. FGFR aberrations increase the risk of brain metastases and predict poor prognosis in metastatic breast cancer patients. Ther Adv Med Oncol. 2020;12:1-15
26. Li Y-L, Zhao H, Ren X-B. Relationship of VEGF/VEGFR with immune and cancer cells: staggering or forward? Cancer Biol Med. 2016;13:206-14
27. Lacal PM, Graziani G. Therapeutic implication of vascular endothelial growth factor receptor-1 (VEGFR-1) targeting in cancer cells and tumor microenvironment by competitive and non-competitive inhibitors. Pharm Res. 2018;136:97-107
28. Incio J, Tam J, Rahbari NN, Suboj P, McManus DT, Chin SM. et al. PlGF/VEGFR-1 signaling promotes macrophage polarization and accelerated tumor progression in obesity. Clin Cancer Res. 2016;22:2993-3004
29. Lisi L, Ciotti GMP, Chiavari M, Ruffini F, Lacal PM, Graziani G. et al. Vascular endothelial growth factor receptor 1 in glioblastoma-associated microglia/macrophages. Oncol Rep. 2020;43:2083-92
30. Guo S, Colbert LS, Fuller M, Zhang Y, Gonzalez-Perez RR. Vascular endothelial growth factor receptor-2 in breast cancer. Biochim Biophys Acta. 2010;1806:108-21
31. Chatterjee S, Heukamp LC, Siobal M, Schöttle J, Wieczorek C, Peifer M. et al. Tumor VEGF:VEGFR2 autocrine feed-forward loop triggers angiogenesis in lung cancer. J Clin Invest. 2013;123:1732-40
32. Paulsson J, Ehnman M, Östman A. PDGF receptors in tumor biology: prognostic and predictive potential. Future Oncol. 2014;10:1695-708
33. Kiwerska K, Wroblewska J, Kaluzna A, Marszalek A. Justification of direct Sanger sequencing application for detection of KIT and PDGFR gene mutations in formalin-fixed, paraffin-embedded samples from gastrointestinal stromal tumours. J Clin Pathol. 2020;73:1-7
34. Nordby Y, Richardsen E, Rakaee M, Ness N, Donnem T, Patel HRH. et al. High expression of PDGFR-β in prostate cancer stroma is independently associated with clinical and biochemical prostate cancer recurrence. Sci Rep. 2017;7:1-9
35. Quintás-Cardama A, Cortes J. Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood. 2009;113:1619-30
36. Hunter HA, Koc H, Koc EC. c-Src kinase impairs the expression of mitochondrial OXPHOS complexes in liver cancer. Cell Signal. 2020;72:1-11
37. Djeungoue-Petga M-A, Lurette O, Jean S, Hamel-Cote G, Martín-Jimenez R, Bou M. et al. Intramitochondrial Src kinase links mitochondrial dysfunctions and aggressiveness of breast cancer cells. Cell Death Dis. 2019;10:1-15
38. Collisson EA, Campbell JD, Brooks AN, Berger AH, Lee W, Chmielecki J. et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543-50
39. Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF. et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61-70
40. Iqbal N, Iqbal N. Human Epidermal Growth Factor Receptor 2 (HER2) in Cancers: Overexpression and Therapeutic Implications. Mol Biol Int. 2014;2014:1-9
41. Bellinger AM, Arteaga CL, Force T. et al. Cardio-oncology. Circulation. 2015;132:2248-58
42. Ferguson FM, Gray NS. Kinase inhibitors: the road ahead. Nat Rev Drug Discov. 2018;17:353
43. Wu P, Clausen MH, Nielsen TE. Allosteric small-molecule kinase inhibitors. Pharmacol Therapeut. 2015;156:59-68
44. Dar AC, Shokat KM. The Evolution of Protein Kinase Inhibitors from Antagonists to Agonists of Cellular Signaling. Anu Rev Biochem. 2011;80:769-95
45. Zuccotto F, Ardini E, Casale E, Angiolini M. Through the “Gatekeeper Door”: Exploiting the Active Kinase Conformation. J Med Chem. 2010;53:2681-94
46. Gavrin LK, Saiah E. Approaches to discover non-ATP site kinase inhibitors. MedChemComm. 2013;4:41-51
47. Lamba V, Ghosh I. New directions in targeting protein kinases: focusing upon true allosteric and bivalent inhibitors. Current pharmaceutical design. 2012;18:2936-45
48. Liao JJ. Molecular Recognition of Protein Kinase Binding Pockets for Design of Potent and Selective Kinase Inhibitors. J Med Chem. 2007;50:409-24
49. van Linden OPJ, Kooistra AJ, Leurs R, de Esch IJP, de Graaf C. KLIFS: A Knowledge-Based Structural Database To Navigate Kinase-Ligand Interaction Space. J Med Chem. 2014;57:249-77
50. Roskoski R. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharm Res. 2016;103:26-48
51. Carles F, Bourg S, Meyer C, Bonnet P. PKIDB: A curated, annotated and updated database of protein kinase inhibitors in clinical trials. Molecules. 2018;23:1-18
52. Weinstein IB, Joe A. Oncogene addiction. Cancer Res. 2008;68:3077-80
53. Lovly CM, Shaw AT. Molecular pathways: resistance to kinase inhibitors and implications for therapeutic strategies. Clin Cancer Res. 2014;20:2249-56
54. Eckstein N, Röper L, Haas B, Potthast H, Hermes U, Unkrig C. et al. Clinical pharmacology of tyrosine kinase inhibitors becoming generic drugs: the regulatory perspective. J Exp Clin Cancer Res. 2014;33:15 -
55. Du W, Elemento O. Cancer systems biology: embracing complexity to develop better anticancer therapeutic strategies. Oncogene. 2015;34:3215-25
56. Han HJ, Ekweremadu C, Patel N. Advanced drug delivery system with nanomaterials for personalised medicine to treat breast cancer. J Drug Deliv Sci Tec. 2019;52:1051-60
57. Jain K, Mehra NK, Jain NK. Potentials and emerging trends in nanopharmacology. Curr Opin Pharmacol. 2014;15:97-106
58. Longmire M, Choyke PL, Kobayashi H. Clearance properties of nano-sized particles and molecules as imaging agents: considerations and caveats. Nanomedicine. 2008;3:703-17
59. Black KCL, Wang Y, Luehmann HP, Cai X, Xing W, Pang B. et al. Radioactive 198Au-doped nanostructures with different shapes for in vivo analyses of their biodistribution, tumor uptake, and intratumoral distribution. ACS Nano. 2014;8:4385-94
60. Decuzzi P, Pasqualini R, Arap W, Ferrari M. Intravascular Delivery of Particulate Systems: Does Geometry Really Matter? Pharm Res. 2008;26:1-9
61. Xiao K, Li Y, Luo J, Lee JS, Xiao W, Gonik AM. et al. The effect of surface charge on in vivo biodistribution of PEG-oligocholic acid based micellar nanoparticles. Biomaterials. 2011;32:3435-46
62. Baetke SC, Lammers T, Kiessling F. Applications of nanoparticles for diagnosis and therapy of cancer. Br J Radiol. 2015;88:20150207 -
63. Din FU, Aman W, Ullah I, Qureshi OS, Mustapha O, Shafique S. et al. Effective use of nanocarriers as drug delivery systems for the treatment of selected tumors. Int J Nanomedicine. 2017;12:7291-309
64. Kou L, Sun J, Zhai Y, He Z. The endocytosis and intracellular fate of nanomedicines: Implication for rational design. Asian J Pharm Sci. 2013;8:1-10
65. Nichols JW, Bae YH. Odyssey of a cancer nanoparticle: from injection site to site of action. Nano Today. 2012;7:606-18
66. Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33:941-51
67. Cao Y. The toxicity of nanoparticles to human endothelial cells. In: Saquib Q, Faisal M, Al-Khedhairy AA, Alatar AA, editors. Cellular and Molecular Toxicology of Nanoparticles. Cham: Springer International Publishing. 2018 p: 59-69
68. Han W, Xie B, Li Y, Shi L, Wan J, Chen X. et al. Orally Deliverable Nanotherapeutics for the Synergistic Treatment of Colitis-Associated Colorectal Cancer. Theranostics. 2019;9:7458-73
69. Wan J, Qiao Y, Chen X, Wu J, Zhou L, Zhang J. et al. Structure-Guided Engineering of Cytotoxic Cabazitaxel for an Adaptive Nanoparticle Formulation: Enhancing the Drug Safety and Therapeutic Efficacy. Adv Funct Mater. 2018;28:1-12
70. Moradpour Z, Barghi L. Novel approaches for efficient delivery of tyrosine kinase inhibitors. Journal of pharmacy & pharmaceutical sciences: a publication of the Canadian Society for Pharmaceutical Sciences, Societe canadienne des sciences pharmaceutiques. 2019;22:37-48
71. van Leeuwen RWF, van Gelder T, Mathijssen RHJ, Jansman FGA. Drug-drug interactions with tyrosine-kinase inhibitors: a clinical perspective. Lancet Oncol. 2014;15:315-26
72. Puszkiel A, Noe G, Thomas-Schoemann A, Bellesoeur A, Boudou-Rouquette P, Vidal M. et al. Pharmacokinetics and pharmacodynamics of tyrosine kinase inhibitors in the treatment of metastatic renal cell carcinoma. Int J Pharmacokinet. 2017;2:257-83
73. Augustijns P, Wuyts B, Hens B, Annaert P, Butler J, Brouwers J. A review of drug solubility in human intestinal fluids: implications for the prediction of oral absorption. Eur J Pharm Sci. 2014;57:322-32
74. Managuli RS, Raut SY, Reddy MS, Mutalik S. Targeting the intestinal lymphatic system: a versatile path for enhanced oral bioavailability of drugs. Exp Opin Drug Deliv. 2018;15:787-804
75. Herbrink M, Nuijen B, Schellens JHM, Beijnen JH. Variability in bioavailability of small molecular tyrosine kinase inhibitors. Cancer Treatment Rev. 2015;41:412-22
76. Di Gion P, Kanefendt F, Lindauer A, Scheffler M, Doroshyenko O, Fuhr U. et al. Clinical pharmacokinetics of tyrosine kinase inhibitors. Clin Pharm. 2011;50:551-603
77. Dora CP, Kushwah V, Katiyar SS, Kumar P, Pillay V, Suresh S. et al. Improved oral bioavailability and therapeutic efficacy of erlotinib through molecular complexation with phospholipid. Int J Pharm. 2017;534:1-13
78. Qiu Q, Lu M, Li C, Luo X, Liu X, Hu L. et al. Novel self-assembled ibrutinib-phospholipid complex for potently peroral delivery of poorly soluble drugs with pH-dependent solubility. AAPS PharmSciTech. 2018;19:3571-83
79. Nazari-Vanani R, Azarpira N, Heli H, Karimian K, Sattarahmady N. A novel self-nanoemulsifying formulation for sunitinib: Evaluation of anticancer efficacy. Colloids Surf B Biointerfaces. 2017;160:65-72
80. Lu X, Liu S, Han M, Yang X, Sun K, Wang H. et al. Afatinib-loaded immunoliposomes functionalized with cetuximab: A novel strategy targeting the epidermal growth factor receptor for treatment of non-small-cell lung cancer. Int J Pharm. 2019;560:126-35
81. Hoogenboezem EN, Duvall CL. Harnessing albumin as a carrier for cancer therapies. Adv Drug Deliv Rev. 2018;130:73-89
82. Gao W, Jia X, Wu J, Song Y, Yin J, Zhang M. et al. Preparation and evaluation of folate-decorated human serum albumin nanoparticles for the targeted delivery of sorafenib to enhance antihepatocarcinoma efficacy. J Drug Deliv Sci Tec. 2019;54:1-10
83. Li Y, Yang B, Zhang X. Oral delivery of imatinib through galactosylated polymeric nanoparticles to explore the contribution of a saccharide ligand to absorption. Int J Pharm. 2019;568:1-9
84. Shi Y, van der Meel R, Chen X, Lammers T. The EPR effect and beyond: Strategies to improve tumor targeting and cancer nanomedicine treatment efficacy. Theranostics. 2020;10:7921-4
85. Tridente G. Adverse Events. In: Tridente G, editor. Adverse Events and Oncotargeted Kinase Inhibitors: Academic Press. 2017 p: 81-93
86. Cryer AM, Chan C, Eftychidou A, Maksoudian C, Mahesh M, Tetley TD. et al. Tyrosine Kinase Inhibitor Gold Nanoconjugates for the Treatment of Non-Small Cell Lung Cancer. ACS Appl Mater Interfaces. 2019;11:16336-46
87. Marslin G, Revina AM, Khandelwal VKM, Balakumar K, Prakash J, Franklin G. et al. Delivery as nanoparticles reduces imatinib mesylate-induced cardiotoxicity and improves anticancer activity. Int J Nanomedicine. 2015;10:3163-70
88. Wan X, Zheng X, Pang X, Zhang Z, Zhang Q. Incorporation of lapatinib into human serum albumin nanoparticles with enhanced anti-tumor effects in HER2-positive breast cancer. Colloids Surf B Biointerfaces. 2015;136:817-27
89. Wan X, Zheng X, Pang X, Pang Z, Zhao J, Zhang Z. et al. Lapatinib-loaded human serum albumin nanoparticles for the prevention and treatment of triple-negative breast cancer metastasis to the brain. Oncotarget. 2016;7:34038-51
90. Abshire C, Murad HY, Arora JS, Liu J, Mandava SH, John VT. et al. Focused Ultrasound-Triggered Release of Tyrosine Kinase Inhibitor From Thermosensitive Liposomes for Treatment of Renal Cell Carcinoma. J Pharm Sci. 2017;106:1355-62
91. Song Z, Lin Y, Zhang X, Feng C, Lu Y, Gao Y. et al. Cyclic RGD peptide-modified liposomal drug delivery system for targeted oral apatinib administration: enhanced cellular uptake and improved therapeutic effects. Int J Nanomedicine. 2017;12:1941-58
92. Heffron TP. Small molecule kinase inhibitors for the treatment of brain cancer. J Med Chem. 2016;59:10030-66
93. Soffietti R, Cornu P, Delattre JY, Grant R, Graus F, Grisold W. et al. EFNS Guidelines on diagnosis and treatment of brain metastases: report of an EFNS Task Force. Eur J Neurol. 2006;13:674-81
94. Bhowmik A, Khan R, Ghosh MK. Blood brain barrier: a challenge for effectual therapy of brain tumors. Biomed Res Int. 2015;2015:320941
95. van Tellingen O, Yetkin-Arik B, de Gooijer MC, Wesseling P, Wurdinger T, de Vries HE. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist Update. 2015;19:1-12
96. Han W, Shi L, Ren L, Zhou L, Li T, Qiao Y. et al. A nanomedicine approach enables co-delivery of cyclosporin A and gefitinib to potentiate the therapeutic efficacy in drug-resistant lung cancer. Signal Transduct Target Ther. 2018;3:1-10
97. Saraiva C, Praca C, Ferreira R, Santos T, Ferreira L, Bernardino L. Nanoparticle-mediated brain drug delivery: Overcoming blood-brain barrier to treat neurodegenerative diseases. J Control Release. 2016;235:34-47
98. Lockman PR, Koziara JM, Mumper RlJ, Allen DD. Nanoparticle surface charges alter blood-brain barrier integrity and permeability. J Drug Target. 2004;12:635-41
99. Niu X, Chen J, Gao J. Nanocarriers as a powerful vehicle to overcome blood-brain barrier in treating neurodegenerative diseases: Focus on recent advances. Asian J Pharm Sci. 2019;14:480-96
100. Lakkadwala S, dos Santos Rodrigues B, Sun C, Singh J. Dual functionalized liposomes for efficient co-delivery of anti-cancer chemotherapeutics for the treatment of glioblastoma. J Control Release. 2019;307:247-60
101. Wagner AJ, Agulnik M, Heinrich MC, Mahadevan D, Riedel RF, von Mehren M. et al. Dose-escalation study of a second-generation non-ansamycin HSP90 inhibitor, onalespib (AT13387), in combination with imatinib in patients with metastatic gastrointestinal stromal tumour. Eur J Cancer. 2016;61:94-101
102. Bhatia S, Diedrich D, Frieg B, Ahlert H, Stein S, Bopp B. et al. Targeting HSP90 dimerization via the C terminus is effective in imatinib-resistant CML and lacks the heat shock response. Blood. 2018;132:307-20
103. Park SE, Kim DE, Kim MJ, Lee JS, Rho JK, Jeong S-Y. et al. Vorinostat enhances gefitinib-induced cell death through reactive oxygen species-dependent cleavage of HSP90 and its clients in non-small cell lung cancer with the EGFR mutation. Oncol Rep. 2019;41:525-33
104. Chen S, Liang Q, Liu E, Yu Z, Sun L, Ye J. et al. Curcumin/sunitinib co-loaded BSA-stabilized SPIOs for synergistic combination therapy for breast cancer. J Mater Chem B. 2017;5:4060-72
105. Li F, Mei H, Gao Y, Xie X, Nie H, Li T. et al. Co-delivery of oxygen and erlotinib by aptamer-modified liposomal complexes to reverse hypoxia-induced drug resistance in lung cancer. Biomaterials. 2017;145:56-71
106. Zhao L, Yang G, Shi Y, Su C, Chang J. Co-delivery of gefitinib and chloroquine by chitosan nanoparticles for overcoming the drug acquired resistance. J Nanobiotechnol. 2015;13:57
107. Joo LJS, Weiss J, Gill AJ, Clifton-Bligh R, Brahmbhatt H, MacDiarmid JA. et al. RET kinase-regulated microRNA-153-3p improves therapeutic efficacy in medullary thyroid carcinoma. Thyroid. 2019;29:830-44
108. Broekman F, Giovannetti E, Peters GJ. Tyrosine kinase inhibitors: Multi-targeted or single-targeted? World J Clin Oncol. 2011;2:80-93
109. Knight ZA, Lin H, Shokat KM. Targeting the cancer kinome through polypharmacology. Nat Rev Cancer. 2010;10:130-7
110. Archibald M, Pritchard T, Nehoff H, Rosengren RJ, Greish K, Taurin S. A combination of sorafenib and nilotinib reduces the growth of castrate-resistant prostate cancer. Int J Nanomedicine. 2016;11:179-200
111. Greish K, Jasim A, Parayath N, Abdelghany S, Alkhateeb A, Taurin S. et al. Micellar formulations of Crizotinib and Dasatinib in the management of glioblastoma multiforme. J Drug Target. 2018;26:692-708
112. Verbaanderd C, Maes H, Schaaf MB, Sukhatme VP, Pantziarka P, Sukhatme V. et al. Repurposing Drugs in Oncology (ReDO)-chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermedicalscience. 2017;11:1-35
113. Zhao L, Yang G, Shi Y, Su C, Chang J. Co-delivery of Gefitinib and chloroquine by chitosan nanoparticles for overcoming the drug acquired resistance. Journal of Nanobiotechnology. 2015;13:57
114. Lv T, Li Z, Xu L, Zhang Y, Chen H, Gao Y. Chloroquine in combination with aptamer-modified nanocomplexes for tumor vessel normalization and efficient erlotinib/Survivin shRNA co-delivery to overcome drug resistance in EGFR-mutated non-small cell lung cancer. Acta Biomater. 2018;76:257-74
115. Hu H, Lin Z, He B, Dai W, Wang X, Wang J. et al. A novel localized co-delivery system with lapatinib microparticles and paclitaxel nanoparticles in a peritumorally injectable in situ hydrogel. J Control Release. 2015;220:189-200
116. Lu X, Liu S, Han M, Yang X, Sun K, Wang H. et al. Afatinib-loaded immunoliposomes functionalized with cetuximab: A novel strategy targeting the epidermal growth factor receptor for treatment of non-small-cell lung cancer. International Journal of Pharmaceutics. 2019;560:126-35
117. Srikar R, Suresh D, Zambre A, Taylor K, Chapman S, Leevy M. et al. Targeted nanoconjugate co-delivering siRNA and tyrosine kinase inhibitor to KRAS mutant NSCLC dissociates GAB1-SHP2 post oncogene knockdown. Sci Rep. 2016;6:1-14
118. Tan H-Y, Wang N, Lam W, Guo W, Feng Y, Cheng Y-C. Targeting tumour microenvironment by tyrosine kinase inhibitor. Mol Cancer. 2018;17:1-15
119. Huo M, Zhao Y, Satterlee AB, Wang YS, Xu Y, Huang L. Tumor-targeted delivery of sunitinib base enhances vaccine therapy for advanced melanoma by remodeling the tumor microenvironment. J Control Release. 2017;245:81-94
120. Kim H, Khanna V, Kucaba TA, Zhang W, Ferguson DM, Griffith TS. et al. Combination of Sunitinib and PD-L1 Blockade Enhances Anticancer Efficacy of TLR7/8 Agonist-Based Nanovaccine. Mol Pharm. 2019;16:1200-10
121. Yin W, Yu X, Kang X, Zhao Y, Zhao P, Jin H. et al. Remodeling Tumor-Associated Macrophages and Neovascularization Overcomes EGFRT790M-Associated Drug Resistance by PD-L1 Nanobody-Mediated Codelivery. Small. 2018;14:1802372
122. Zhao R, Li T, Zheng G, Jiang K, Fan L, Shao J. Simultaneous inhibition of growth and metastasis of hepatocellular carcinoma by co-delivery of ursolic acid and sorafenib using lactobionic acid modified and pH-sensitive chitosan-conjugated mesoporous silica nanocomplex. Biomaterials. 2017;143:1-16
123. Iqbal J, Abbasi BA, Ahmad R, Mahmood T, Kanwal S, Ali B. et al. Ursolic acid a promising candidate in the therapeutics of breast cancer: Current status and future implications. Biomed Pharmacother. 2018;108:752-6
124. Naizhi W, Tao F, Xiaona L, Qin L. Curcumin inhibits migration and invasion of non-small cell lung cancer cells through up-regulation of miR-206 and suppression of PI3K/AKT/mTOR signaling pathway. Acta Pharm. 2020;70:399-409
125. Li W, Sun L, Lei J, Wu Z, Ma Q, Wang Z. Curcumin inhibits pancreatic cancer cell invasion and EMT by interfering with tumor-stromal crosstalk under hypoxic conditions via the IL-6/ERK/NF-κB axis. Oncol Rep. 2020;44:382-92
126. Almutairi FM, El Rabey HA, Tayel AA, Alalawy AI, Al-Duais MA, Sakran MI. et al. Augmented anticancer activity of curcumin loaded fungal chitosan nanoparticles. Int J Biol Macromolecules. 2020;155:861-7
127. de Freitas CF, Kimura E, Rubira AF, Muniz EC. Curcumin and silver nanoparticles carried out from polysaccharide-based hydrogels improved the photodynamic properties of curcumin through metal-enhanced singlet oxygen effect. Mater Sci Eng C. 2020;112:1-14
128. Mansourizadeh F, Alberti D, Bitonto V, Tripepi M, Sepehri H, Khoee S. et al. Efficient synergistic combination effect of Quercetin with Curcumin on breast cancer cell apoptosis through their loading into Apo ferritin cavity. Colloids Surf B Biointerfaces. 2020;191:1-9
129. Nosrati H, Danafar H, Rezaeejam H, Gholipour N, Rahimi-Nasrabadi M. Evaluation radioprotective effect of curcumin conjugated albumin nanoparticles. Bioorg Chem. 2020;100:1-7
130. Dong Y, Yang Y, Wei Y, Gao Y, Jiang W, Wang G. et al. Facile synthetic nano-curcumin encapsulated Bio-fabricated nanoparticles induces ROS-mediated apoptosis and migration blocking of human lung cancer cells. Process Biochem. 2020;95:91-8
131. Chen F-P, Liu L-L, Tang C-H. Spray-drying microencapsulation of curcumin nanocomplexes with soy protein isolate: Encapsulation, water dispersion, bioaccessibility and bioactivities of curcumin. Food Hydrocoll. 2020;105:105821
132. Cao H, Wang Y, He X, Zhang Z, Yin Q, Chen Y. et al. Codelivery of Sorafenib and Curcumin by Directed Self-Assembled Nanoparticles Enhances Therapeutic Effect on Hepatocellular Carcinoma. Mol Pharm. 2015;12:922-31
133. Muz B, de la Puente P, Azab F, Azab AK. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia. 2015;3:83-92
134. Minakata K, Takahashi F, Nara T, Hashimoto M, Tajima K, Murakami A. et al. Hypoxia induces gefitinib resistance in non-small-cell lung cancer with both mutant and wild-type epidermal growth factor receptors. Cancer Sci. 2012;103:1946-54
135. Kalash RS, Lakshmanan VK, Cho C-S, Park I-K. Theranostics. In: Ebara M, editor. Biomaterials Nanoarchitectonics: William Andrew Publishing. 2016 p: 197-215
136. Muthu MS, Leong DT, Mei L, Feng S-S. Nanotheranostics - application and further development of nanomedicine strategies for advanced theranostics. Theranostics. 2014;4:660-77
137. Pysz MA, Gambhir SS, Willmann JK. Molecular imaging: current status and emerging strategies. Clin Radiol. 2010;65:500-16
138. Janib SM, Moses AS, MacKay JA. Imaging and drug delivery using theranostic nanoparticles. Adv Drug Deliv Rev. 2010;62:1052-63
139. Cheng R, Meng F, Deng C, Klok H-A, Zhong Z. Dual and multi-stimuli responsive polymeric nanoparticles for programmed site-specific drug delivery. Biomaterials. 2013;34:3647-57
140. Noh GT, Kim M, Suh J-Y, Song Y, Lee CK, Baek JH. et al. Sunitinib-CLIO conjugate: A VEGFR/PDGFR-targeting active MR probe. Mol Imaging Biol. 2014;16:340-9
141. Liu Y, Feng L, Liu T, Zhang L, Yao Y, Yu D. et al. Multifunctional pH-sensitive polymeric nanoparticles for theranostics evaluated experimentally in cancer. Nanoscale. 2014;6:3231-42
142. Hsu F-T, Liu H-S, Ali AAA, Tsai PH, Kao Y-C, Lu C-F. et al. Assessing the selective therapeutic efficacy of superparamagnetic erlotinib nanoparticles in lung cancer by using quantitative magnetic resonance imaging and a nuclear factor kappa-B reporter gene system. Nanomed-Nanotechnol. 2018;14:1019-31
143. Zhang Y, Zhang L, Lin X, Ke L, Li B, Xu L. et al. Dual-responsive nanosystem for precise molecular subtyping and resistant reversal of EGFR targeted therapy. Chem Eng. 2019;372:483-95
144. Sang M, Luo R, Bai Y, Dou J, Zhang Z, Liu F. et al. BHQ-Cyanine-Based “Off-On” Long-Circulating Assembly as a Ferroptosis Amplifier for Cancer Treatment: A Lipid-Peroxidation Burst Device. ACS Apl Mater Interfaces. 2019;11:42873-84
145. Eckstein N, Roper L, Haas B, Potthast H, Hermes U, Unkrig C. et al. Clinical pharmacology of tyrosine kinase inhibitors becoming generic drugs: the regulatory perspective. J Exp Clin Cancer Res. 2014;33:10
146. Anselmo AC, Mitragotri S. Nanoparticles in the clinic: An update. Bioeng Transl Med. 2019;4:1-16
147. Wu L-P, Wang D, Li Z. Grand challenges in nanomedicine. Mater Sci Eng C. 2020;106:1-7
148. Barenholz YC. Doxil® - The first FDA-approved nano-drug: From an idea to a product. Handbook of Harnessing Biomaterials in Nanomedicine. 2012 p: 335-98
149. Adir O, Poley M, Chen G, Froim S, Krinsky N, Shklover J. et al. Integrating Artificial Intelligence and Nanotechnology for Precision Cancer Medicine. Adv Mater. 2020;32:1-15
150. Shamay Y, Shah J, Isık M, Mizrachi A, Leibold J, Tschaharganeh DF. et al. Quantitative self-assembly prediction yields targeted nanomedicines. Nat Mater. 2018;17:361-8
151. Tropsha A, Mills KC, Hickey AJ. Reproducibility, sharing and progress in nanomaterial databases. Nat Nanotechnol. 2017;12:1111-4
152. Kadurin A, Nikolenko S, Khrabrov K, Aliper A, Zhavoronkov A. druGAN: An Advanced Generative Adversarial Autoencoder Model for de novo Generation of New Molecules with Desired Molecular Properties in Silico. Mol Pharm. 2017;14:3098-104
153. Bello T, Gujral TS. KInhibition: A Kinase Inhibitor Selection Portal. iScience. 2018;8:49-53
154. Lampa S, Alvarsson J, Arvidsson Mc Shane S, Berg A, Ahlberg E, Spjuth O. Predicting Off-Target Binding Profiles With Confidence Using Conformal Prediction. Frontiers in Pharmacology. 2018;9:1-13
155. Wang W, Sedykh A, Sun H, Zhao L, Russo DP, Zhou H. et al. Predicting Nano-Bio Interactions by Integrating Nanoparticle Libraries and Quantitative Nanostructure Activity Relationship Modeling. ACS Nano. 2017;11:12641-9
156. Vijay S, Gujral TS. Non-linear Deep Neural Network for Rapid and Accurate Prediction of Phenotypic Responses to Kinase Inhibitors. iScience. 2020;23:1-49
157. Silva A, Silva MC, Sudalagunta P, Distler A, Jacobson T, Collins A. et al. An Ex vivo Platform for the Prediction of Clinical Response in Multiple Myeloma. Cancer Res. 2017;77:3336-51
158. Xie Q, Faust K, Van Ommeren R, Sheikh A, Djuric U, Diamandis P. Deep learning for image analysis: Personalizing medicine closer to the point of care. Crit Rev Cl Lab Sci. 2019;56:61-73
159. Min J, Im H, Allen M, McFarland PJ, Degani I, Yu H. et al. Computational Optics Enables Breast Cancer Profiling in Point-of-Care Settings. ACS Nano. 2018;12:9081-90
160. Rashid MBMA, Toh TB, Hooi L, Silva A, Zhang Y, Tan PF. et al. Optimizing drug combinations against multiple myeloma using a quadratic phenotypic optimization platform (QPOP). Sci Transl Med. 2018;10:1-18
161. Lim JJ, Goh J, Rashid MBMA, Chow EK-H. Maximizing Efficiency of Artificial Intelligence-Driven Drug Combination Optimization through Minimal Resolution Experimental Design. Adv Ther. 2020;3:1-7
162. Blasiak A, Khong J, Kee T. CURATE.AI: Optimizing Personalized Medicine with Artificial Intelligence. SLAS Technol. 2019;25:95-105
163. Cova TFGG, Bento DlJ, Nunes SCC. Computational Approaches in Theranostics: Mining and Predicting Cancer Data. Pharmaceutics. 2019;11:1-28
164. Ho D, Quake SR, McCabe ERB, Chng WJ, Chow EK, Ding X. et al. Enabling Technologies for Personalized and Precision Medicine. Trends Biotechnol. 2020;38:497-518
165. Setten RL, Rossi JJ, Han S-P. The current state and future directions of RNAi-based therapeutics. Nat Rev Drug Discov. 2019;18:421-46
166. Almurshedi AS, Radwan M, Omar S, Alaiya AA, Badran MM, Elsaghire H. et al. A novel pH-sensitive liposome to trigger delivery of afatinib to cancer cells: Impact on lung cancer therapy. J Mol Liq. 2018;259:154-66
167. Coelho SC, Almeida GM, Pereira MC, Santos-Silva F, Coelho MAN. Functionalized gold nanoparticles improve afatinib delivery into cancer cells. Exp Opin Drug Deliv. 2016;13:133-41
168. Xiao W, Ruan S, Yu W, Wang R, Hu C, Liu R. et al. Normalizing tumor vessels to increase the enzyme-induced retention and targeting of gold nanoparticle for breast cancer imaging and treatment. Mol Pharm. 2017;14:3489-98
169. Kydd J, Jadia R, Rai P. Co-Administered Polymeric Nano-Antidotes for Improved Photo-Triggered Response in Glioblastoma. Pharmaceutics. 2018;10:1-18
170. Greish K, Jasim A, Parayath N, Abdelghany S, Alkhateeb A, Taurin S. et al. Micellar formulations of Crizotinib and Dasatinib in the management of glioblastoma multiforme. J Drug Target. 2018;26:692-708
171. Dong C, Li B, Li Z, Shetty S, Fu J. Dasatinib-loaded albumin nanoparticles possess diminished endothelial cell barrier disruption and retain potent anti-leukemia cell activity. Oncotarget. 2016;7:49699-709
172. Niza E, Noblejas-López MD, Bravo I, Nieto-Jiménez C, Castro-Osma JA, Canales-Vázquez J. et al. Trastuzumab-Targeted Biodegradable Nanoparticles for Enhanced Delivery of Dasatinib in HER2+ Metastasic Breast Cancer. Nanomaterials. 2019;9:1-14
173. Niza E, Nieto-Jiménez C, Noblejas-López MD, Bravo I, Castro-Osma JA, de la Cruz-Martínez F. et al. Poly(Cyclohexene Phthalate) Nanoparticles for Controlled Dasatinib Delivery in Breast Cancer Therapy. Nanomaterials. 2019;9:1-14
174. Chauhan R, Balgemann R, Greb C, Nunn BM, Ueda S, Noma H. et al. Production of dasatinib encapsulated spray-dried poly (lactic-co-glycolic acid) particles. J Drug Deliv Sci Technol. 2019;53:1-8
175. Adena SKR, Upadhyay M, Vardhan H, Mishra B. Gold nanoparticles for sustained antileukemia drug release: development, optimization and evaluation by quality-by-design approach. Nanomedicine. 2019;14:851-70
176. Sabra SA, Sheweita SA, Haroun M, Ragab D, Eldemellawy MA, Xia Y. et al. Magnetically Guided Self-Assembled Protein Micelles for Enhanced Delivery of Dasatinib to Human Triple-Negative Breast Cancer Cells. J Pharm Sci. 2019;108:1713-25
177. Kulkarni NS, Parvathaneni V, Shukla SK, Barasa L, Perron JC, Yoganathan S. et al. Tyrosine kinase inhibitor conjugated quantum dots for non-small cell lung cancer (NSCLC) treatment. Eur J Pharm Sci. 2019;133:145-59
178. Kim DH, Choi YJ, Sung KJ, Yoo S-A, Sung YH, Kim JK. et al. Efficacy of nano-particulated, water-soluble erlotinib against intracranial metastases of EGFR-mutant lung cancer. Mol Oncol. 2018;12:2182-90
179. Thakkar S, Sharma D, Misra M. Comparative evaluation of electrospraying and lyophilization techniques on solid state properties of Erlotinib nanocrystals: Assessment of In-vitro cytotoxicity. Eur J Pharm Sci. 2018;111:257-69
180. Fathi M, Zangabad PS, Aghanejad A, Barar J, Erfan-Niya H, Omidi Y. Folate-conjugated thermosensitive O-maleoyl modified chitosan micellar nanoparticles for targeted delivery of erlotinib. Carbohydr Polym. 2017;172:130-41
181. Bakhtiary Z, Barar J, Aghanejad A, Saei AA, Nemati E, Ezzati Nazhad Dolatabadi J. et al. Microparticles containing erlotinib-loaded solid lipid nanoparticles for treatment of non-small cell lung cancer. Drug Dev Ind Pharm. 2017;43:1244-53
182. Yang KM, Shin IC, Park JW, Kim K-S, Kim DK, Park K. et al. Nanoparticulation improves bioavailability of Erlotinib. Drug Dev Ind Pharm. 2017;43:1557-65
183. Dora CP, Trotta F, Kushwah V, Devasari N, Singh C, Suresh S. et al. Potential of erlotinib cyclodextrin nanosponge complex to enhance solubility, dissolution rate, in vitro cytotoxicity and oral bioavailability. Carbohydr Polym. 2016;137:339-49
184. Kola Srinivas NS, Verma R, Pai Kulyadi G, Kumar L. A quality by design approach on polymeric nanocarrier delivery of gefitinib: formulation, in vitro, and in vivo characterization. Int J Nanomedicine. 2016;12:15-28
185. Kuruppu AI, Zhang L, Collins H, Turyanska L, Thomas NR, Bradshaw TD. An Apoferritin-based Drug Delivery System for the Tyrosine Kinase Inhibitor Gefitinib. Adv Healthc Mater. 2015;4:2816-21
186. Lam ATN, Yoon J, Ganbold E-O, Singh DK, Kim DH, Cho K-H. et al. Colloidal gold nanoparticle conjugates of gefitinib. Colloids Surf B Biointerfaces. 2014;123:61-7
187. Han W, Shi L, Ren L, Zhou L, Li T, Qiao Y. et al. A nanomedicine approach enables co-delivery of cyclosporin A and gefitinib to potentiate the therapeutic efficacy in drug-resistant lung cancer. Signal Tranduct Tar. 2018;3:1-16
188. Qiu Q, Li C, Song Y, Shi T, Luo X, Zhang H. et al. Targeted delivery of ibrutinib to tumor-associated macrophages by sialic acid-stearic acid conjugate modified nanocomplexes for cancer immunotherapy. Acta Biomater. 2019;92:184-95
189. Kamali M, Dinarvand R, Maleki H, Arzani H, Mahdaviani P, Nekounam H. et al. Preparation of imatinib base loaded human serum albumin for application in the treatment of glioblastoma. RSC Adv. 2015;5:62214-9
190. James AR, Unnikrishnan BS, Priya R, Joseph MM, Manojkumar TK, Raveendran PK. et al. Computational and mechanistic studies on the effect of galactoxyloglucan: Imatinib nanoconjugate in imatinib resistant K562 cells. Tumor Biol. 2017;39:1010428317695946
191. Cortese B, D'Amone S, Gigli G, Palama IE. Sustained anti-BCR-ABL activity with pH responsive imatinib mesylate loaded PCL nanoparticles in CML cells. MedChemComm. 2015;6:212-21
192. Buss JH, Begnini KR, Bruinsmann FA, Ceolin T, Sonego MS, Pohlmann AR. et al. Lapatinib-loaded nanocapsules enhances antitumoral effect in human bladder cancer cell. Front Oncol. 2019;9:203 -
193. Lee SY, Cho H-J. Mitochondria targeting and destabilizing hyaluronic acid derivative-based nanoparticles for the delivery of lapatinib to triple-negative breast cancer. Biomacromolecules. 2019;20:835-45
194. Wan X, Zheng X, Pang X, Zhang Z, Jing T, Xu W. et al. The potential use of lapatinib-loaded human serum albumin nanoparticles in the treatment of triple-negative breast cancer. Int J Pharm. 2015;484:16-28
195. Kallus S, Englinger B, Senkiv J, Laemmerer A, Heffeter P, Berger W. et al. Nanoformulations of anticancer FGFR inhibitors with improved therapeutic index. Nanomed-Nanotechnol. 2018;14:2632-43
196. Lang L, Shay C, Xiong Y, Thakkar P, Chemmalakuzhy R, Wang X. et al. Combating head and neck cancer metastases by targeting Src using multifunctional nanoparticle-based saracatinib. J Hematol Oncol. 2018;11:1-13
197. Xu X, Tang X, Wu X, Feng X. Biosynthesis of sorafenib coated graphene nanosheets for the treatment of gastric cancer in patients in nursing care. J Photoch Photobio B. 2019;191:1-5
198. Clavreul A, Roger E, Pourbaghi-Masouleh M, Lemaire L, Tetaud C, Menei P. Development and characterization of sorafenib-loaded lipid nanocapsules for the treatment of glioblastoma. Drug Deliv. 2018;25:1756-65
199. Liu J, Abshire C, Carry C, Sholl AB, Mandava SH, Datta A. et al. Nanotechnology combined therapy: tyrosine kinase-bound gold nanorod and laser thermal ablation produce a synergistic higher treatment response of renal cell carcinoma in a murine model. BJU Int. 2017;119:342-8
200. Callaghan C, Peralta D, Liu J, Mandava SH, Maddox M, Dash S. et al. Combined treatment of tyrosine kinase inhibitor-labeled gold nanorod encapsulated albumin with laser thermal ablation in a renal cell carcinoma model. J Pharm Sci. 2016;105:284-92
201. Bianchini F, De Santis A, Portioli E, Russo Krauss I, Battistini L, Curti C. et al. Integrin-targeted AmpRGD sunitinib liposomes as integrated antiangiogenic tools. Nanomed-Nanotechnol. 2019;18:135-45
202. Alshahrani SM, Alshetaili AS, Alalaiwe A, Alsulays BB, Anwer MK, Al-Shdefat R. et al. Anticancer efficacy of self-nanoemulsifying drug delivery system of sunitinib malate. AAPS PharmSciTech. 2018;19:123-33
203. Chen Y, Sun J, Huang Y, Lu B, Li S. Improved Cancer Immunochemotherapy via Optimal Co-delivery of Chemotherapeutic and Immunomodulatory Agents. Mol Pharm. 2018;15:5162-73
204. Wang J, Wang H, Li J, Liu Z, Xie H, Wei X. et al. iRGD-Decorated Polymeric Nanoparticles for the Efficient Delivery of Vandetanib to Hepatocellular Carcinoma: Preparation and in vitro and in vivo Evaluation. ACS Appl Mater Interfaces. 2016;8:19228-37
205. Sarkar S, Konar S, Prasad PN, Rajput S, Kumar BNP, Rao RR. et al. Micellear gold nanoparticles as delivery vehicles for dual tyrosine kinase inhibitor ZD6474 for metastatic breast cancer treatment. Langmuir. 2017;33:7649-59
206. Wang D, Wu L-P. Nanomaterials for delivery of nucleic acid to the central nervous system (CNS). Mater Sci Eng C. 2017;70:1039-46
207. Hsu F-T, Liu H-S, Ali AAA, Tsai P-H, Kao Y-C, Lu C-F. et al. Assessing the selective therapeutic efficacy of superparamagnetic erlotinib nanoparticles in lung cancer by using quantitative magnetic resonance imaging and a nuclear factor kappa-B reporter gene system. Nanomed-Nanotechnol. 2018;14:1019-31
Author contact
![]() Corresponding author: Zbynek Heger, Department of Chemistry and Biochemistry, Mendel University in Brno, Zemedelska 1, CZ-613 00 Brno, Czech Republic; E-mail: hegercz; phone: +420-5-4513-3350; fax: +420-5-4521-2044.
Corresponding author: Zbynek Heger, Department of Chemistry and Biochemistry, Mendel University in Brno, Zemedelska 1, CZ-613 00 Brno, Czech Republic; E-mail: hegercz; phone: +420-5-4513-3350; fax: +420-5-4521-2044.