Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(2):893-905. doi:10.7150/thno.48080 This issue Cite
Research Paper
Human umbilical cord-derived mesenchymal stem cell therapy ameliorates lupus through increasing CD4+ T cell senescence via MiR-199a-5p/Sirt1/p53 axis
Tao Cheng1*, Shuai Ding1*, Shanshan Liu1, Yan Li2 ![]() , Lingyun Sun1
, Lingyun Sun1 ![]()
1. Department of Rheumatology and Immunology, The Affiliated Drum Tower Hospital, Medical School of Nanjing University, Nanjing, 210008 China
2. The State Key Laboratory of Pharmaceutical Biotechnology, Chemistry and Biomedicine Innovation Center, Model Animal Research Center of Nanjing University, Nanjing, 210061 China
*Contributed equally to this work.
Received 2020-5-11; Accepted 2020-10-26; Published 2021-1-1
Abstract

Rationale: Although human umbilical cord-derived mesenchymal stem cells (hUC-MSCs) transplantation has been proved to be an effective therapeutic approach to treat systemic lupus erythematosus (SLE), the detailed underlying mechanisms are not fully understood. Transferring miRNAs is one mean by which MSCs communicate with surrounding cells. Sirt1 is a NAD-dependent deacetylase that protects against cell senescence by deacetylating p53. Here we aimed to explore whether hUC-MSCs affected senescence of splenic CD4+ T cells through regulating Sirt1/p53 via miRNA in the MRL/lpr lupus mouse model.
Methods: The effects of hUC-MSCs on lupus syndrome and senescence pathways in MRL/lpr mice in vivo and in vitro were determined. The functional roles of miR-199a-5p in splenic CD4+ T cell senescence were studied by miRNA mimic or inhibitor in vitro. MRL/lpr mice were injected with miR-199a-5p agomir to evaluate the effects of miR-199a-5p on splenic CD4+ T cell senescence and disease in vivo.
Results: We showed that hUC-MSCs transplantation ameliorated lupus symptoms and increased senescence of splenic CD4+ T cells through Sirt1/p53 signaling via miR-199a-5p in MRL/lpr mice. Moreover, systemic delivery of miR-199a-5p in MRL/lpr mice increased splenic CD4+ T-cell senescence, mimicking the therapeutic effects of transplanted hUC-MSCs.
Conclusions: We have identified miR-199a-5p as one of the mechanisms employed by hUC-MSCs to alleviate lupus disease associated pathologies in MRL/lpr mice, which is attributable for promoting splenic CD4+ T cell senescence through Sirt1/p53 pathway.
Keywords: Mesenchymal stem cells, Lupus, Senescence, Sirt1/p53, miR-199a-5p
Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune and potentially fatal disease characterized by activation and proliferation of auto-reactive T cells and B cells with production of autoantibodies against nuclear and endogenous antigens [1]. Current treatments for lupus are limited to anti-inflammatory and steroids synthetic cortisone medications with side effects, and often do not lead to improved outcomes for severe patients. Our previous studies demonstrated that human umbilical cord-derived mesenchymal stem cells (hUC-MSCs) alleviate the SLE disease both in humans and experimental animal models by immunomodulation, including regulating the Treg/Th17 balance, inhibiting T follicular helper cell expansion and so on [2-4]. MSCs are widely known to suppress T cell responses through cell-cell contact and secretion of anti-inflammatory factors. As part of the mechanisms involved in this process, MSCs also inhibit T-cell proliferation by inducing T-cell unresponsiveness, arresting the cell cycle in the G1/G0 phase and promoting premature senescence [5, 6]. However, the molecular pathways whereby MSCs induce T-cell-cycle arrest and suppress proliferation still await investigation.
Recently, cellular senescence, a form of cell cycle arrest, has emerged as an important mechanism for orchestrating immune cells during homeostasis and active immunity [7]. It can be triggered by telomere shortening as well as a diversity of cellular stresses and signaling imbalances, also known as telomere-dependent senescence and telomere-independent senescence, respectively [8]. The expansion of CD28null senescent T cells has been associated with disease severity in lupus patients [9, 10]. Inconsistent with the definition of classical cellular senescence, previous researches focused on CD28null T cells, which has been described as a heterogeneous population and proliferate upon certain type of stimulation [11, 12]. To date, the relationship between CD28null T cells and SLE disease activity appears controversial. [13]. Furthermore, the function of CD28null senescent T cells has positive or adverse effect depend on senescence inducer and surrounding microenvironment [13-15]. All these intrigue us to investigate the role of hUC-MSCs induced T cell premature senescence in lupus and the molecular mechanism of senescence induction.
Cellular senescence usually relies on the activation of the cyclin-dependent kinase inhibitors p21 and p16, which are components of the tumor-suppressor pathways governed by the p53 [8, 16]. While p53 acts as a threshold factor of cell homeostasis and senescence [17], its key upstream regulator NAD-dependent protein deacetylase sirtuin-1 (Sirt1) is essentially responsible for preventing cell fate towards senescence [18, 19]. Consequently, silencing or inhibiting Sirt1 leads to p53 activation, and subsequently accelerate or drive senescence [20]. Moreover, microRNAs (miRNAs) have been shown to regulate the levels of p53-target genes and regulators in senescence induction [21].
Employing the above-mentioned studies and considering the fact that MSCs communicate with surrounding immune cells by releasing miRNAs [22], we hypothesized that hUC-MSCs regulate target genes in signaling pathways of senescence via miRNAs may account for the therapeutic effect of hUC-MSCs in lupus. To test this hypothesis, we utilized the MRL/lpr mice to model SLE in human. MRL/lpr mice can reproduce key clinical pathologies of SLE and more importantly, the hyperactivation and pathogenicity of CD4+ T cell make this mouse model suitable for investigation of senescence-inducing therapy [23-25].
On the above basis, we postulated that improved understanding of the molecular mechanisms used in the generation of T cell telomere-independent senescence, including their functional alterations in SLE, could present novel approaches to normalizing T-cell function and consequently down regulating disease processes. In this study, we present evidence that hUC-MSCs increase senescence of splenic CD4+ T cells through regulating Sirt1/p53 via miR-199a-5p.
Materials and methods
Mice
Female MRL/lpr and ICR mice were purchased from Shanghai SLAC Laboratory Animal Co. Ltd. (Shanghai, China). Mice were housed under specific-pathogen-free conditions in the animal center of the Affiliated Drum Tower Hospital, Medical School of Nanjing University. All experimental animal protocols were approved by the Committee of Experimental Animal Administration of the Affiliated Drum Tower Hospital, Medical School of Nanjing University.
Isolation, culture and identification of hUC-MSCs
The study protocol was approved by the Ethics Committee of the Affiliated Drum Tower Hospital, Medical School of Nanjing University. Umbilical cord specimen was collected from a 29-years-old parturient after taking signed informed consent forms. hUC-MSCs were prepared as previously described [26, 27], and cultured in DMEM/F12 supplemented with 10% fetal bovine serum and 100 U/ml penicillin/streptomycin (Gibco, Life Technologies, Grand Island, NY, USA) until confluent. The adherent cells were detached by 0.25% Trypsin-EDTA (Gibco) and reseeded into new flasks for further expansion. All the cell cultures were maintained at 37 °C in a 5% CO2 humidified atmosphere. Flow cytometry analysis showed hUC-MSCs used in this study were positive for the surface staining of CD73, CD90 and CD105, but lacked CD34, CD45, CD14, CD19 and HLA-DR expression.
hUC-MSCs transplantation
Female MRL/lpr mice were randomly divided into two groups (hUC-MSCs and PBS treatment, n = 5 per group) and transfused with 5 × 105 hUC-MSCs or PBS, respectively, via the tail vein at 17-weeks old. After 4 weeks, all treated mice were sacrificed for further analysis. Wild type (WT) ICR female mice were used as controls (n = 5) [28, 29].
Isolation and culture of CD4+ T cells with hUC-MSCs
Mononuclear spleen suspensions of 17- to -20 weeks-old female MRL/lpr mice were prepared by gentle disruption with cell strainers (Corning, 352360) and lysed erythrocytes by RBC Lysis Buffer. The CD4+ T cells were purified using immunomagnetic positive selection (EasySep™ mouse CD4+ positive selection Kit, 18952, Stemcell technologies, Canada). They were then co-cultured with hUC-MSCs for 48 h at ratios of 1:1, 10:1, and 50:1 (T cells: MSCs), in the presence of soluble anti-mouse CD3 (2 μg/ml) and anti-mouse CD28 (2 μg/ml) antibodies. In some experiments, a transwell system (0.4 μm pore size, Millipore) was used to block cell-cell contact.
CD4+ T cell culture and Sirt1 treatment
The CD4+ T cells were treated with either 50 μM EX527, a specific Sirt1 inhibitor, or 5 μM SRT1720, a Sirt1 activator (both from Selleck, Houston, TX, USA) for 12 h prior to coculture with hUC-MSCs for 24 h or 48 h in the presence of soluble anti-mouse CD3 (2 μg/ml) and anti-mouse CD28 (2 μg/ml) antibodies. Cells treated with DMSO alone were used as controls.
Histopathology examination
Kidneys obtained at sacrifice were divided along their longitudinal axis. One half was fixed in 10% formaldehyde and embedded in paraffin. Three-micrometer-thick sections were stained with hematoxylin-eosin (HE) for histopathology. The other half of the kidney was embedded in Tissue-Tec OCT medium, frozen in liquid nitrogen, and stored at -70 °C until section processed. Five-micrometer-thick frozen sections were fixed with 4% paraformaldehyde and blocked with 2% BSA prior to staining with goat anti-mouse IgG (1:150 dilutions; Abcam, Cambridge, UK), followed by FITC-labeled anti-goat antibodies. Sections were photographed using a fluorescence microscope fitted with a digital camera (Cannon Power shot G10, Cannon, Inc.). Histological scores of renal lesions were calculated. Mainly, the severity of glomerulonephritis was graded on a 0-4 scale as follows: 0, normal; 1, mild increase in mesangial cellularity and matrix; 2, moderate increase in mesangial cellularity and matrix, with thickening of the glomerular basement membrane (GBM); 3, focal endocapillary hyper cellularity with obliteration of capillary lumina and a substantial increase in the thickness and irregularity of the GBM; and 4, diffuse endocapillary hyper cellularity, segmental necrosis, crescents and hyalinized end-stage glomeruli [30]. The immunofluorescence (IF) intensity within the peripheral glomerular capillary walls and mesangial region were scored on a scale from 0 to 3 (0 = none; 1 = weak; 2 = moderate; 3 = strong) [27]. At least 10 glomeruli/section were analyzed.
Enzyme-linked Immunosorbent Assay (ELISA)
Serum levels of ANA, anti-dsDNA antibody, and total IgG were measured using a Mouse ANA ELISA Kit (Alpha Diagnostic, USA), a Mouse anti-dsDNA ELISA Kit (Shibayagi, Japan), and a Mouse IgG total ELISA Kit (Fcmacs, China) respectively, according to the manufacturers' instructions.
Capillary WES analyses
After immunomagnetic positive selection, CD4+ T cells were lysed and protein concentration was measured by BCA (Thermo Fisher). Equal amounts of proteins were taken and diluted with 0.1× sample buffer. Four parts of diluted samples and 1 part 5× Fluorescent Master mix were mixed and transferred to stacking gel. After blocking, anti-Sirt1 (1:25, Cell Signaling Technology, 9475) and anti-Beta-Tubulin (1:25, proteintech, 10094-1-AP) were probed. HRP-conjugated secondary antibodies and chemiluminescent substrate were dispensed into designated wells in an assay plate. A biotinylated ladder provided molecular weight standards for each assay. After plate loading, the separation electrophoresis and immunodetection steps take place in the fully automated capillary system (WES, Protein Simple, San Jose, CA).
Western blotting
Rabbit antibodies to Sirt1 (9475), acetyl-p53 (Lys379) (Cell Signaling Technology), p53 (10442-1-AP) (Proteintech), p16 (ab51243), p21 (ab109199) (Abcam), glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Cell Signaling Technology) and Goat anti-Rabbit IgG (H+L) (SA00001-2) (Proteintech) were used for western blotting. CD4+ T cells were washed twice with PBS and lysed on ice for 30 min with 1× RIPA buffer (Cell Signaling Technology) containing 1% 100× protease/phosphatase inhibitor Cocktail (Cell Signaling Technology). Lysates were centrifuged at 12,000 g at 4 °C for 20 min, and the supernatants were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Proteins were then transferred to polyvinylidene fluoride membranes (Millipore), blocked for 1 h in 5% nonfat milk (in 10 mM Tris pH 7, 150 mM NaCl, 0.1% Tween 20, TBST), and then immunoblotted with indicated primary antibodies and appropriate horseradish peroxidase-conjugated secondary antibodies. The bands were visualized in a luminol-based detection system with piodophenol enhancement.
Quantitative real-time reverse transcription-polymerase chain reaction (RT-PCR) analysis
Total cellular RNA was extracted using Trizol reagent (Invitrogen) and 1 ug RNA was used in RT reactions. cDNA was synthesized by Primer Script RT reagent Kit (TaKaRa). Sequences of the forward and reverse primers were: p16 5'-CCCAACGCCCCGAACT-3' and 5'-GCAGAAGAGCTGCTACGTGAA-3'; p21 5'-GAGCAGTTGCGTGACCGTTG-3' and 5'-GGGGAATCTTCAGGCCGCTC-3'; Sirt1 5'-TCTATGCTCGCCTTGCGGTTG-3' and 5'-CCTGCAACCTGCTCCAAGGT-3'; GAPDH 5'-TGGCCTTCCGTGTTCCTAC-3' and 5'-GAGTTGCTGTTGAAGTCGCA-3'. MiR-199a-5p was performed by poly(A)-tailed RT-PCR as previously described [31]. MiR-199a-5p primers and the 3'primer were: 5'-CCCAGUGUUCAGACUACCUGUUC-3'; 5'-ATTCTAGAGGCCGAGGCGGCCGACATGT-3'. Sequences of the U6 forward and reverse primers were: 5'-CGCTTCGGCAGCACATATACTA-3'; 5'-CGCTTCACGAATTTGCGTGTC3'. A HiPure miRNA Kit (Magen) was used to isolate miRNAs from cultured media samples; a total of 1000 μl of cultured media was used for each isolation. A cel-miR-39-3p standard RNA was added to samples as an external reference control. For real-time PCR experiments, reactions containing SYBR Premix EX Taq (Takara), ROX Reference Dye (50; Takara), cDNA, and gene primers were run on a Step One Plus real-time PCR system and analyzed using Step One Software, version 2.1 (Applied Biosystems). Relative gene quantification was calculated by the 2ΔCt method and then normalized to the level of GAPDH, U6 or miR-39-3p.
Transfection of CD4+ T cells with miR-199a-5p mimic or inhibitor
The CD4+ T cells were seeded at density of 6 × 105 cells/well in a 12-well plate. The cells were then transfected with either miR-199a-5p mimic or inhibitor obtained from GenePharma (Shanghai, China) using EntransterTM-R4000 Transfection reagent from Engreen (Beijing, China). The transfected cells were cultured for 48-72 h in the presence of soluble anti-mouse CD3 (2 μg/ml) and anti-mouse CD28 (2 μg/ml) antibodies. The transfected cells were then collected and used for total RNA and protein isolation.
Culture of CD4+ T cells with RNase
MRL/lpr splenic CD4+ T cells and hUC-MSCs were cultured alone or together in the presence or absence of RNase (Qiagen, final concentration: 5 µg/mL) using a transwell system. The cells were processed as described above after 48 h addition of RNase.
Dual luciferase reporter assay
To confirm miR-199a-5p targeted 3'-UTR of Sirt1, we constructed the fragment of sirt1 3'-UTR, which contains the predicted miR-199a-5p binding sites into pmirGLO Luciferase reporter vector (Promega, Madison, WI, USA). For the control, we introduced 507 A > T and 511 U > A point mutations located within the seed region of miR-199a-5p binding sites in Sirt1 3'UTR. Site-directed mutagenesis in the seed sequence of miR-199a binding site was introduced using In-Fusion cloning kit (Clontech, Mountain View, CA, USA) as per manufacturer's protocol. Dual-Luciferase Reporter Assay System was applied for luciferase reporter assay (Promega). HEK 293T cells were simultaneously transfected with WT or mutant (mut) type 3'-UTR Sirt1 and miR-199a-5p mimic or negative control (NC) mimic. Luciferase activity was detected using CytoFLEX Flow Cytomerter (Beckman Coulter). Firefly luciferase activity was normalized to renilla luciferase activity. The ratio of Sirt1 3'-UTR to NC mimic was set as 1; therefore, the results were presented as the fold changes relative to control.
MiR-199a-5p agomir treatment in MRL/lpr mice
The miR-199a-5p agomir and agomir negative control (nc) were synthesized by RiboBio (Guangzhou, China) (n = 6 per group). Each mouse received a tail vein injection of either miR-199a-5p agomir or agomir nc, at a dose of 20 nmol in 0.2 ml saline every 4 days for 4 weeks and sacrificed 48 h after the last injection. Serum, spleens, lymph nodes and kidneys were harvested. The splenic CD4+ T cells were purified using immunomagnetic positive selection (EasySep™ mouse CD4+ positive selection Kit, 18952, Stemcell technologies, Canada).
Flow cytometry
Single cell suspensions were prepared from the spleen by standard procedures. Fixable viability dye eFlour506 (ebioscience) were used to exclude dead cells. Flurochrome-conjugated antibodies from Biolegend were used in dilutions according to manufactures' instructions, which includes APC/Cyanine7 anti-mouse CD45, FITC anti-mouse CD3e, BV 711 anti-mouse CD4, PE anti-mouse CD27, BV421 anti-mouse CD28, APC/Cyanine7 anti-mouse CD45, FITC anti-mouse CD3e, BV510 anti-mouse CD8, BV711 anti-mouse CD44, BV605 anti-mouse CD62L, PE anti-mouse Foxp3, APC anti-mouse CD25. Foxp3 staining used Foxp3/Transcription Factor Staining Buffer (ebioscience) as recommended. Fortessa (BD biosciences) were used for acquisition and Flowjo (10.4) for analysis. Gating strategies for flow cytometry analysis are detailed in Supplementary information, Figure S1.
Statistical analysis
The data were expressed as mean ± standard error of mean and analyzed with Prism 5 (GraphPad Software). Student's t test was used to analyze significance between two groups and comparisons among more than two groups were analyzed using one-way ANOVA. The value of p < 0.05 was considered statistically significant.
Results
hUC-MSCs ameliorate disease progression of MRL/lpr mice
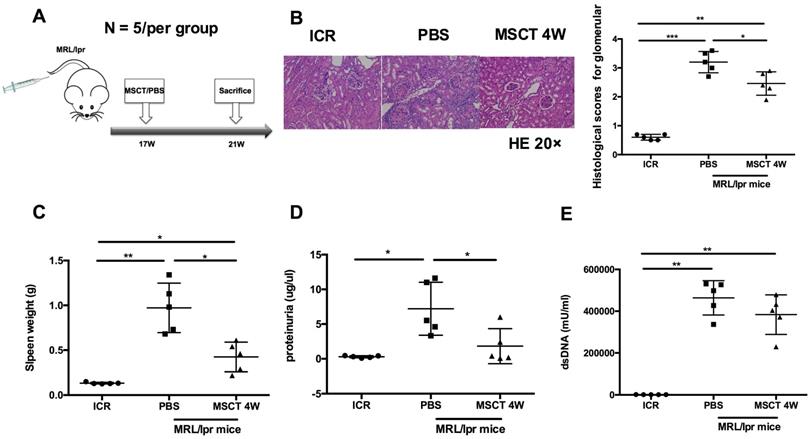
To assess the therapeutic effect on the lupus syndrome of MRL/lpr mice, we treated MRL/lpr mice with 5 × 105 hUC-MSCs or PBS starting at 17 weeks of age and sacrificed all mice at 21 weeks of age (Figure 1A). Compared to PBS-treated mice, the kidney lesions of hUC-MSC-treated mice were significantly reduced (Figure 1B). In addition, the spleen weight and proteinuria were much lower after hUC-MSCs treatment compared to the PBS-treated MRL/lpr mice (Figure 1C-D). Serum anti-dsDNA antibody levels were lower in the hUC-MSCs-treated group, but did not reach statistical significance (Figure 1E). The data indicate that hUC-MSCs ameliorate lupus disease in MRL/lpr mice.
hUC-MSCs ameliorate disease of MRL/lpr mice. (A) Experimental outline describing the use of hUC-MSCs to treat MRL/lpr mice (n = 5 per group). Kidney lesion (B), spleen weight (C), proteinuria (D) and serum anti-dsDNA antibody (E) levels in WT mice, PBS-treated MRL/lpr mice and hUC-MSCs-treated MRL/lpr mice. *p < 0.05; **p < 0.01; ***p < 0.001.
hUC-MSCs increase senescence of splenic CD4+ T cells through Sirt1/p53 signaling in MRL/lpr mice in vivo
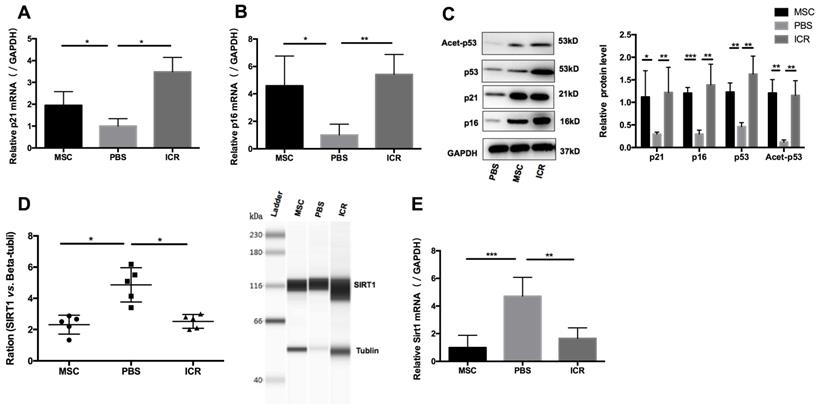
We postulated that a potential mechanism of the therapeutic efficacy of hUC-MSCs in MRL/lpr mice could be through suppression of the accumulation of CD4+ T cells by enhancing their senescence. To test this, we isolated mononuclear cells from splenic cell suspension and then purified CD4+ T cells for RNA and protein preparation. We measured levels of p21 and p16 as markers of premature senescence. Our analysis showed that the mRNA and protein levels of p21 and p16 were significantly reduced in MRL/lpr mice compared to the WT control group, meanwhile, an increase in p21 and p16 mRNA and protein expression were seen in hUC-MSCs treatment group compared to PBS-treated MRL/lpr mice (Figure 2A-C). We also found that MRL/lpr splenic CD4+ T cells had increased Sirt1 levels and decreased acetylation of p53 at Lys-379 compared to the WT mice, while treatment with hUC-MSCs was able to reverse the changes by decreasing Sirt1 and increasing p53 and acetylation of p53 (Figure 2C-E). With the immunostaining CD45/CD3e/CD4/CD27/CD28, the senescent T cells can be identified as intermediate differentiation CD27-CD28+ population and highly differentiated CD27-CD28- population [32]. There was a significant difference between two groups to the proportion of CD27-CD28+ cells, and an increasing trend toward CD27-CD28- cells in hUC-MSCs group (Supplementary information, Figure S2). Taken together, these results suggest that the senescence of splenic CD4+ T cells in MRL/lpr mice could be defective because of altered Sirt1/p53 signaling, which might result in CD4+ T-cell hyper proliferation. Thus, the clinical effects of hUC-MSCs could be associated with the restoration of senescence pathways in splenic CD4+ T cells.
hUC-MSCs treatment normalizes markers of senescence in MRL/lpr mice splenic CD4+ T cells in vivo. (A, B) qPCR analysis showing the expression levels of p21, p16 in WT mice, PBS-treated MRL/lpr mice and hUC-MSCs-treated MRL/lpr mice. (C) Representative western blot showing the expression levels of p21, p16, p53 and acetylation of p53 in WT mice, PBS-treated MRL/lpr mice and hUC-MSCs-treated MRL/lpr mice. (D, E) Capillary WES and qPCR analysis showing the expression of Sirt1 in WT mice, PBS-treated MRL/lpr mice and hUC-MSCs-treated MRL/lpr mice. GAPDH was used as a protein loading control. *p < 0.05; **p < 0.01; ***p < 0.001.
hUC-MSCs increase senescence of MRL/lpr splenic CD4+ T cells through Sirt1 in vitro
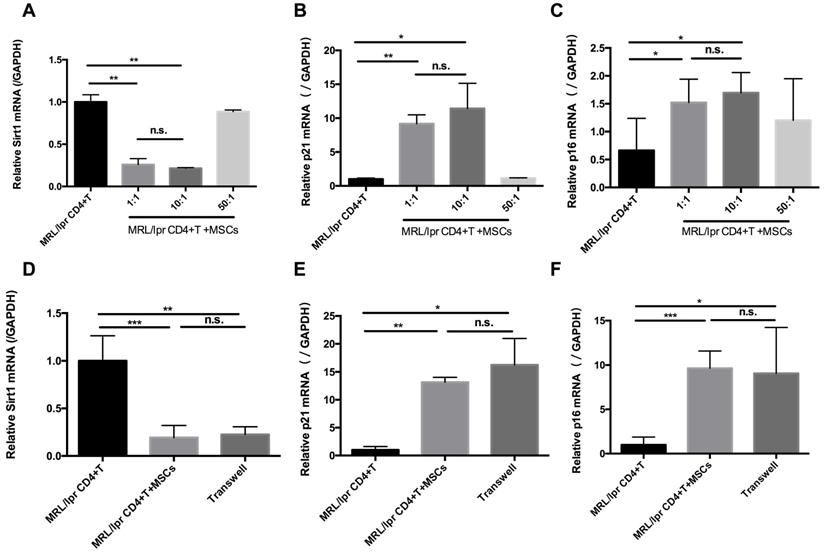
In parallel with the in vivo experiments, we determined the effects of hUC-MSCs on T cell senescence in vitro. Splenic CD4+ T cells from MRL/lpr mice were co-cultured with hUC-MSCs at different ratios (CD4+ T cells: MSCs: 1:1, 10:1, 50:1) for 48 h. We found that hUC-MSCs significantly up-regulated the levels of p21 and p16, but down-regulated Sirt1 levels in a dose-dependent manner (Figure 3A-C). Moreover, the modulation of cell senescence was not dependent on cell-cell contact, as similar results were found when the cultured CD4+ T cells and MSCs were separated by a transwell system (Figure 3D-F). These data suggest that soluble factors mediate the regulatory effects of hUC-MSCs on splenic CD4+ T cell senescence in MRL/lpr mice.
hUC-MSCs increase markers of senescence in splenic CD4+ T cells in vitro. (A-C) Splenic CD4+ T cells from MRL/lpr mice were cultured alone or with hUC-MSCs at ratios of 1:1, 10:1, or 50:1 for 48 h. (D-F) In further experiments, MRL/lpr splenic CD4+ T cells and hUC-MSCs (10:1) were cultured separated by transwells. Sirt1 (A, D), p21 (B, E) and p16 (C, F) RNA levels of CD4+ T cells were quantitated by qPCR. All experimental data were verified in at least two independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., not significant.
Sirt1 is required for hUC-MSCs-mediated senescence of CD4+ T cells in MRL/lpr mice
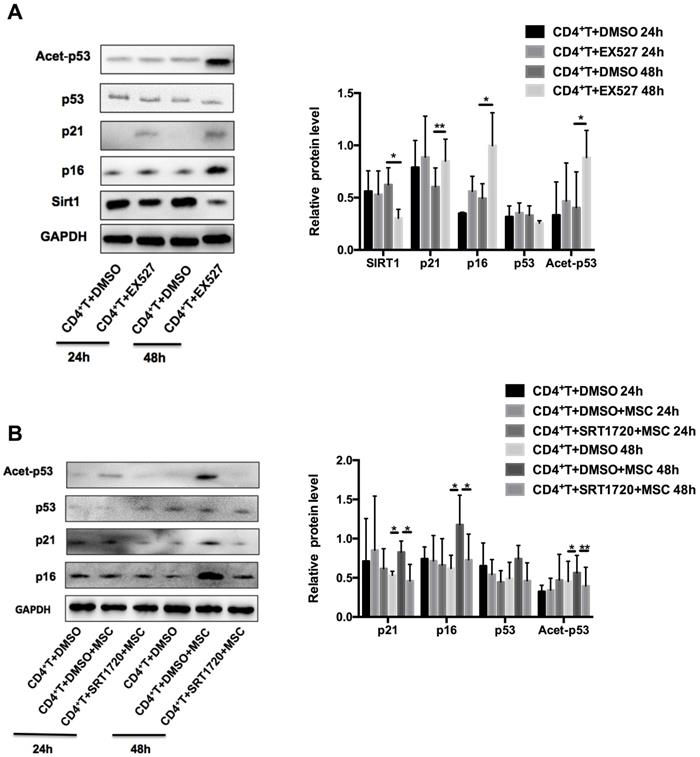
Next, we examined whether Sirt1 is sufficient and necessary for hUC-MSCs to enhance the senescence of MRL/lpr splenic CD4+ T cells. We found that pretreatment of MRL/lpr CD4+ T cells with the Sirt1 inhibitor-EX527 attenuated expression of Sirt1 and increased expression p21 and p16, as well as the acetylation of p53 (Figure 4A). Conversely, pretreatment of T cells with a selective activator of Sirt1, SRT1720, inhibited the senescence induced by hUC-MSCs (Figure 4B). Collectively, these data reveal that Sirt1 is a key mediator whereby hUC-MSCs increase senescence of splenic CD4+ T cells in MRL/lpr mice.
Sirt1 is a mediator of hUC-MSCs increasing senescence of splenic CD4+ T cells in MRL/lpr mice. (A) Western blotting showing the expression of Sirt1, p21, p16, p53 and acetylation of p53 in EX527 and DMSO treated MRL/lpr splenic CD4+ T cells. (B) MRL/lpr splenic CD4+ T cells were pretreated with SRT1720 12 h before exposed to hUC-MSCs for 24-48 h, western blotting showing the expression of p21, p16, p53 and acetylation of p53. GAPDH was used as a protein loading control. All experimental data were verified in at least three independent experiments. *p < 0.05; **p < 0.01.
hUC-MSCs regulate Sirt1/p53 signaling in MRL/lpr splenic CD4+ T cells through transferring miR-199a-5p
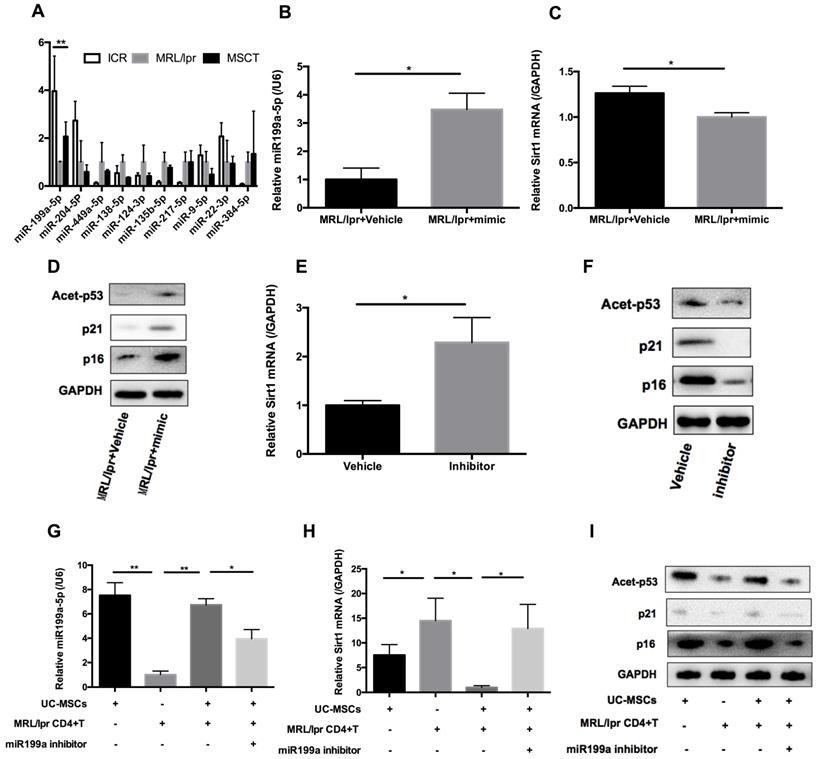
hUC-MSCs are able to transfer miRNAs, which are small non-coding RNAs (~22 nucleotides) capable of silencing gene expression at a post-transcriptional level, to surrounding cells in cancer and autoimmune disease models [33]. We therefore considered whether miRNAs could be involved in hUC-MSCs-mediated regulation of Sirt1 expression in splenic CD4+ T cells. We used software available online, Target scan, to identify 10 miRNAs that would be predicted computationally to target Sirt1 gene expression (Supplementary information, Table S1-2). qPCR analysis showed that of these 10 miRNAs, only miR-199a-5p was significantly decreased in MRL/lpr splenic CD4+ T cells (Figure 5A). Furthermore, miR-199a-5p was the only tested miRNA to show increased expression after hUC-MSCs treatment (Figure 5A). According to bioinformatic prediction, we also designed luciferase experiment to provide evidence of the direct interaction between the miR-199a-5p and Sirt1 mRNA (Supplementary information, Figure S3). In addition, we demonstrated a functional role of miR-199a-5p by showing that a miR-199a-5p mimic downregulated and a miR-199a-5p inhibitor upregulated Sirt1 expression in splenic CD4+ T cells in vitro respectively (Supplementary information, Figure S4). In fact, the miR-199a-5p mimic could not only increased the levels of miR-199a-5p and decreased Sirt1 expression in MRL/lpr splenic CD4+ T cells (Figure 5B-C), but also elevated expression of the senescence markers p21, p16 and acetyl-p53 (Figure 5D). Conversely, miR-199a-5p inhibitor treatment elevated the expression of Sirt1 in WT splenic CD4+ T cells (Figure 5E), and decreased the senescence markers p21, p16 and acetyl-p53 (Figure 5F). Finally, hUC-MSCs showed up to an increase in the production of miR-199a-5p as compared to MRL/lpr splenic CD4+ T cells, indicating that hUC-MSCs is the source of the key regulator miR-199a-5p (Figure 5G). Co-culture of hUC-MSCs with MRL/lpr splenic CD4+ T cells in a transwell system for 48 h showed increased intracellular miR-199a-5p along with decreased Sirt1 gene expression and restored expression levels of senescence markers in the CD4+ T cells, while miR-199a-5p inhibitor treatment remained depressed status (Figure 5G-I). The transwell cultures had also been undertaken with addition of RNase to the medium (Supplementary information, Figure S5). In addition, presence of miR-199a-5p in the supernatant from hUC-MSCs cultures was verified (Supplementary information, Figure S6). Collectively, the data suggest that hUC-MSCs increase splenic CD4+ T cell senescence through miR-199a-5p-mediated downregulation of Sirt1 and subsequent reduction of p53 deacetylation.
hUC-MSCs induced miR-199a-5p can increase MRL/lpr splenic CD4+ T cell senescence. (A) qPCR analysis of the levels of ten potential miRNAs in WT mice, PBS-treated MRL/lpr mice and hUC-MSCs-treated MRL/lpr mice splenic CD4+ T cells. (B-D) qPCR and western blotting analysis of the levels of miR-199a-5p, Sirt1, p21, p16 and acetyl-p53 in vehicle and miR-199a-5p mimic-treated MRL/lpr splenic CD4+ T cells. (E-F) qPCR and western blotting analysis of the levels of Sirt1, p21, p16 and acetyl-p53 in vehicle and miR-199a-5p inhibitor-treated WT splenic CD4+ T cells. (G-I) MRL/lpr splenic CD4+ T cells and hUC-MSCs were cultured alone or together in the presence or absence of miR-199a inhibitor using a transwell system. MiR-199a-5p, Sirt1, p21, p16 and acetyl-p53 were quantified in hUC-MSCs (the first bar) or splenic CD4+ T cells (the last three bars). GAPDH was used as a protein loading control. All experimental data were verified in at least two independent experiments. *p < 0.05; **p < 0.01.
MiR-199a-5p ameliorates disease in MRL/lpr mice
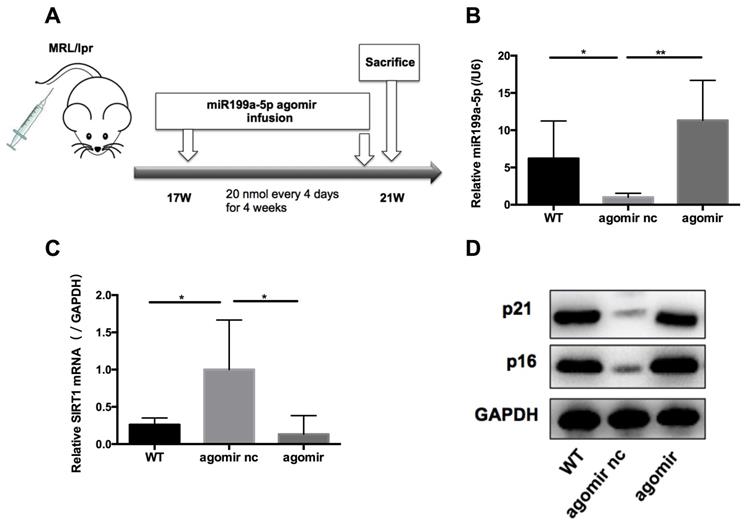
We next assessed the effects of miR-199a-5p on MRL/lpr mice in vivo. 17-weeks old MRL/lpr mice were injected with 20 nmol miR-199a-5p agomir or control (agomir nc) every 4 days for 4 weeks (Figure 6A). 48 h after the last injection, mice were sacrificed and splenic CD4+ T cells were isolated for examination. The miR-199a-5p agomir-treated group showed elevated levels of miR-199a-5p (Figure 6B), reduced expression of Sirt1 (Figure 6C), and marked up regulation of the senescence markers p21 and p16 compared to control treated cells (Figure 6D). Moreover, miR-199a-5p agomir-treated group showed increased CD27-CD28+ cells and CD27-CD28- cells compared to control group (Supplementary information, Figure S7). The in vivo data suggest that hUC-MSCs increase MRL/lpr splenic CD4+ T cell senescence through miR-199a-5p.
MiR-199a-5p agomir treatment increases senescence of MRL/lpr splenic CD4+ T cells. (A) Experimental outline. CD4+ T cells were harvested to measures miR-199a-5p (B), Sirt1 (C), p21 and p16 (D). GAPDH was used as a protein loading control. (n = 6 per group). *p < 0.05; **p < 0.01.
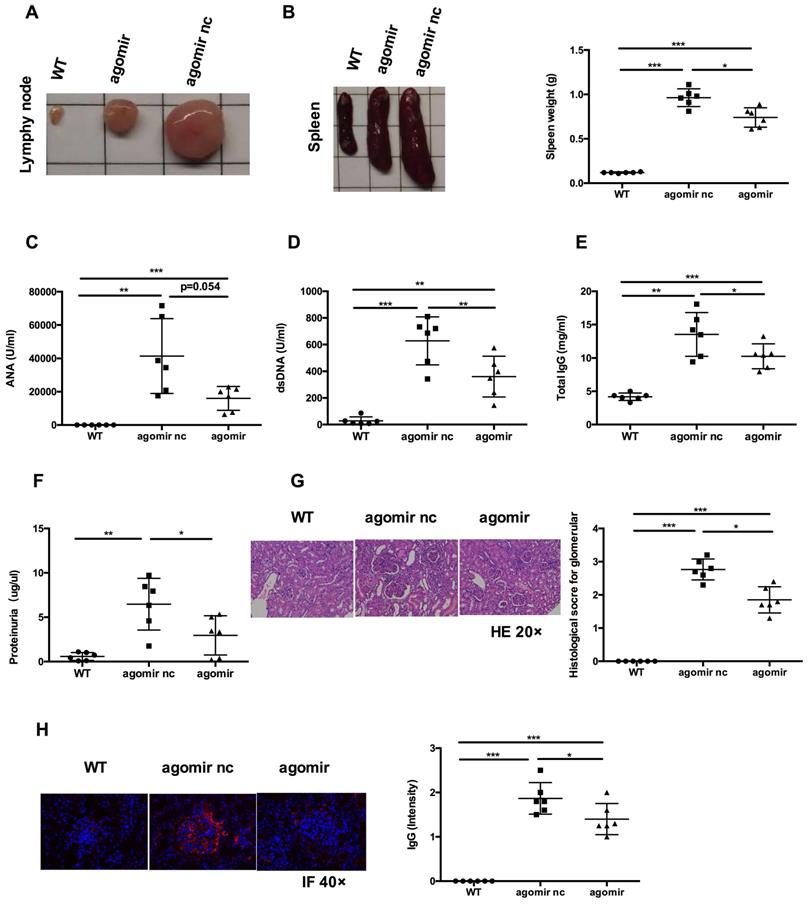
Next, we examined whether systemic administration of miR-199a-5p agomir rescued the lupus phenotype in MRL/lpr mice. When compared to agomir nc treatment group, miR-199a-5p agomir treatment significantly reduced the size of spleens and lymph nodes (Figures 7A-B) and decreased serum levels of anti-dsDNA antibody, IgG (Figure 7D-E). Serum ANA levels showed a tendency to decrease (Figure 7C). Renal impairments were also ameliorated in the miR-199a-5p agomir treated group, as shown by lower proteinuria (Figure 7F), reduced glomerular enlargement and hyper cellularity (Figure 7G) and less IgG deposition in the peripheral capillary loops (Figure 7H). Collectively, these data suggest that hUC-MSCs transplantation ameliorate disease phenotype in MRL/lpr mice through miR-199a-5p-mediated downregulation of Sirt1 signaling, and consequently increase senescence in splenic CD4+ T cells (see schematic diagram, Supplementary information, Figure S8).
MiR-199a-5p agomir treatment ameliorates disease phenotype and immune disorder in MRL/lpr mice. MiR-199a-5p treatment reduced lymph node size (A), spleen index (B), serum levels of ANA (C), anti-dsDNA antibody (D), total IgG (E) and proteinuria (F) in MRL/lpr mice. (G, H) Representative images of renal pathology and immunohistochemical analysis after treatment with miR-199a-5p agomir. *p < 0.05; **p < 0.01; ***p < 0.001.
Discussion
Excessive T-cell proliferation with the subsequent clonal expansion is common immune imbalance phenomenon during the progression of SLE. As such, senescence of hyperactive T cells, accompanied with cell cycle arrest, could potentially reduce systemic inflammation and ameliorate SLE symptoms. In fact, researchers have already established the protective role of cellular senescence in tumor suppression [8, 34], wound healing [35], embryogenesis [36] and protection against tissue fibrosis [37]. On the other hand, the negative effects of T cell senescence have been shown in tissue remodeling, aging and age-related disease [38]. In brief, the effects of T cell senescence are context- and senescence inducer-dependent, and moreover, vary upon which subset that is being targeted. Given that MSCs transplantations inhibit T cells proliferation in SLE patients and mouse models [5, 39], we seek to establish connections between T cell premature senescence and beneficial effect of hUC-MSCs treatment. We prove the significant changes of senescence markers in splenic CD4+ T cells of MRL/lpr mice and then demonstrate that hUC-MSCs rescue lupus phenotype with induction of CD4+ T cell senescence. Our findings were in agreement with previous evidences of reduced p21 levels in lupus patient T cells and enhanced p21 expression for therapeutic potential in lupus-prone mice [40, 41]. Taken together, these results suggest that the machinery of senescence induction in splenic CD4+ T cells of MRL/lpr mice is compromised and this might be associated with consequent hyper proliferation of CD4+ T cells.
Epigenetic modifications are characteristic of senescent changes. Taking into consideration the importance of histone modifying enzymes in fundamental processes such as cell proliferation and senescence, Sirt1 is one of the most important histone deacetylase catalyzing both histone and non-histone targets including p53 and FoxO proteins [19, 42]. Sirt1 has been implicated in abnormal T-cell activation in the development of autoimmune syndrome, since Sirt1 deacetylates a variety of transcriptional factors and cofactors involved in apoptosis [43, 44]. However, the impact of Sirt1 on T cell senescence is still unknown. In this study, we have demonstrated that MRL/lpr CD4+ T cells show increased Sirt1 expression and decreased acetylation of p53 in vivo. However, treatment with hUC-MSCs was able to decrease Sirt1 levels both in vivo and in vitro, and thus increase markers of senescence in splenic CD4+ T cells. Moreover, Sirt1 inhibitor could attenuate expression of Sirt1 and increase expression of acetylated p53, p21 and p16 in MRL/lpr splenic CD4+ T cells, while hUC-MSCs could not promote senescence when MRL/lpr splenic CD4+ T cells were pretreated with Sirt1 activator. These data suggest that Sirt1 is a mediator for hUC-MSCs induced senescence of splenic CD4+ T cells in MRL/lpr mice.
Our results show that soluble factors released from hUC-MSCs are responsible for promoting senescence by cell-to-cell contact-independent manner. Transferring miRNAs is one mean by which MSCs communicate with surrounding cells. A growing number of miRNAs were reported as key modulators of senescence in both human and mouse models [45], but their roles in regulating senescence in immune cells, especially in the context of SLE, are unclear. In the present study, we identified a novel target, miR-199a-5p, delivering from hUC-MSCs to splenic CD4+ T cells and assisting in T cell senescence. Through enhancing the expression of miR-199a-5p, the senescence of MRL/lpr splenic CD4+ T cells was increased. MiR-199a has been found to regulate osteogenic and chondrogenic differentiation of MSCs [46]. In addition, regulatory role of miR-199a from adipose tissue-derived MSCs has also been reported to enhance the chemosensitivity in hepatocellular carcinoma through mTOR pathway [47]. Notably, miRNAs have different target genes in various diseases and exert diverse regulatory functions [48, 49]. In this study, we have shown that in vivo delivery of agomir produced mature miR-199a-5p in recipient cells and significantly reduced Sirt1 expression, resulting increased MRL/lpr splenic CD4+ T cell senescence. This result encouraged us to examine whether miR-199a-5p overexpression could rescue autoimmune disorders in MRL/lpr mice. Our data indeed indicated that miR-199a-5p agomir treatment successfully ameliorated disease phenotypes and restored immune homeostasis to some extent. However, it should be noted that its potential off-target effects needs to be explored in future study, in order to develop effective and safe miR-199a-5p-based gene therapy in clinical settings.
In the recent decade, therapies targeting cellular senescence pathways have been developed for a wide variety of diseases. Our study is based on the lupus-prone mouse model and support the therapeutic value of enhancing CD4+ T cell senescence in lupus mice via miR-199a-5p agomir. The clinical and epidemiological studies indicated that senescent CD4+ CD28null T cells were positively correlated with disease progression and renal damage in SLE patients [9, 10], but it remains to be clarified whether CD4+ CD28null T cells that expand as phenotypically and functionally heterogeneous population under chronic inflammation are truly senescent. In addition to CD28, terminally differentiated T cells in human can be better identified by expression of various other surface markers such as CD27, CD45R, CCR7 and CD57, resulting in a specific subset of “truly senescent” [32]. Herein we identified CD27-CD28+ cells and CD27-CD28- cells in the MRL/lpr mouse that referenced as intermediate/highly differentiation population, which frequencies were higher in hUC-MSCs treated group and miR-199a-5p agomir-treated group when compared to controls. Furthermore, as compared with chronic pro-inflammatory state, hUC-MSCs infusion probably trigger and produce different premature senescent T cell subsets in a short-term. Therefore, further clinical studies are required to compare the hUC-MSCs induced senescent T cells and CD4+ CD28null T cells generated under SLE chronic inflammatory milieu.
In conclusion, we have shown here that hUC-MSCs increase splenic CD4+ T cell senescence through the Sirt1/p53 pathway via miR-199a-5p in lupus-prone MRL/lpr mice. These findings not only extend our knowledge of the immunoregulatory function of hUC-MSCs in SLE, but also offer miR-199a-5p as a potential biomarker or therapeutic target to further explore in the future.
Abbreviations
SLE: Systemic lupus erythematosus; hUC-MSCs: human umbilical cord-derived mesenchymal stem cells; Sirt1: sirtuin-1; miRNAs: microRNAs; WT: wild type; GBM: glomerular basement membrane; nc: negative control; HE: hematoxylin-eosin; IF: immunofluorescence; RT-PCR: Quantitative real-time reverse transcription-polymerase chain reaction; GAPDH: glyceraldehyde 3-phosphate dehydrogenase.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
The authors thank Professor Wanjun Chen from Mucosal Immunology Section, NIDCR, NIH for critically reading and English editing the manuscript. We also thank Dr. Xiaojun Tang and Zutong Li for sorting cells and mice supports.
Funding
This work was supported by Key Program of National Natural Science Foundation of China (81930043); Major International (Regional) Joint Research Project of China (81720108020); National Key Research and Development Program of China (2019YFA0802900).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Tsokos GC. Systemic lupus erythematosus. New Engl J Med. 2011;365:2110-21
2. Dandan Wang, Saisai Huang, Xinran Yuan, Jun Liang, Renju Xu, Genhong Yao. et al. The regulation of the Treg/Th17 balance by mesenchymal stem cells in human systemic lupus erythematosus. Cell Mol Immunol. 2017;14:423-31
3. Zhang Z, Feng R, Niu L, Huang S, Deng W, Shi B. et al. Human umbilical cord mesenchymal stem cells inhibit T follicular helper cell expansion through the activation of iNOS in lupus-prone B6.MRL-Faslpr mice. Cell Transplant. 2017;26:1031-42
4. Wang D, Li J, Zhang Y, Zhang M, Chen J, Li X. et al. Umbilical cord mesenchymal stem cell transplantation in active and refractory systemic lupus erythematosus: a multicenter clinical study. Arthritis Res Ther. 2014;16:R79
5. Glennie S, Soeiro I, Dyson PJ, Lam EW, Dazzi F. Bone marrow mesenchymal stem cells induce division arrest anergy of activated T cells. Blood. 2005;105:2821-7
6. Giuliani M, Fleury M, Vernochet A, Ketroussi F, Clay D, Azzarone B. et al. Long-lasting inhibitory effects of fetal liver mesenchymal stem cells on T-lymphocyte proliferation. PLoS One. 2011;6:e19988
7. Vicente R, Mausset-Bonnefont AL, Jorgensen C, Louis-Plence P, Brondello JM. Cellular senescence impact on immune cell fate and function. Aging Cell. 2016;15:400-6
8. Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729-40
9. Patricia L ́opez, Javier Rodr ́ıguez-Carrio, Aleida Mart ́ınez-Zapico, Luis Caminal-Montero, Ana Suarez. Senescent profile of angiogenic T cells from systemic lupus erythematosus patients. J Leukoc Biol. 2016;99:405-12
10. Ugarte-Gil MF, Sanchez-Zuniga C, Gamboa-Cardenas RV, Aliaga-Zamudio M, Zevallos F, Tineo-Pozo G. et al. Circulating CD4+CD28null and extra-thymic CD4+CD8+ double positive T cells are independently associated with disease damage in systemic lupus erythematosus patients. Lupus. 2016;25:233-40
11. Warrington KJ, Vallejo AN, Weyand CM, Goronzy JJ. CD28 loss in senescent CD4+ T cells: reversal by interleukin-12 stimulation. Blood. 2003;101:3543-9
12. Chiu WK, Fann M, Weng NP. Generation and growth of CD28null CD8+ memory T cells mediated by IL-15 and its induced cytokines. J Immunol. 2006;177:7802-10
13. van den Hoogen LL, Sims GP, van Roon JA, Fritsch-Stork RD. Aging and Systemic Lupus Erythematosus-Immunosenescence and Beyond. Curr Aging Sci. 2015;8:158-77
14. Tulunay A, Yavuz S, Direskeneli H, Eksioglu-Demiralp E. CD8+CD28-, suppressive T cells in systemic lupus erythematosus. Lupus. 2008;17:630-7
15. Kaneko H, Saito K, Hashimoto H, Yagita H, Okumura K, Azuma M. Preferential elimination of CD28+ T cells in systemic lupus erythematosus (SLE) and the relation with activation-induced apoptosis. Clin Exp Immunol. 1996;106:218-29
16. Bringold F, Serrano M. Tumor suppressors and oncogenes in cellular senescence. Exp Gerontol. 2000;35:317-29
17. Reed SM, Quelle DE. p53 Acetylation: Regulation and Consequences. Cancers (Basel). 2014;7:30-69
18. Ong ALC, Ramasamy TS. Role of Sirtuin1-p53 regulatory axis in aging, cancer and cellular reprogramming. Ageing Res Rev. 2018;43:64-80
19. Yi J, Luo J. SIRT1 and p53, effect on cancer, senescence and beyond. Biochim Biophys Acta. 2010;1804:1684-9
20. Lee SH, Lee JH, Lee HY, Min KJ. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019;52:24-34
21. Faraonio R, Salerno P, Passaro F, Sedia C, Iaccio A, Bellelli R. et al. A set of miRNAs participates in the cellular senescence program in human diploid fibroblasts. Cell Death Differ. 2012;19:713-21
22. Chen JQ, Papp G, Szodoray P, Zeher M. The role of microRNAs in the pathogenesis of autoimmune diseases. Autoimmun Rev. 2016;15:1171-80
23. Singer GG, Carrera AC, Marshak-Rothstein A, Martínez C, Abbas AK. Apoptosis, Fas and systemic autoimmunity: the MRL-lpr/lpr model. Curr Opin Immunol. 1994;6:913-20
24. Cohen PL, Eisenberg RA. The lpr and gld genes in systemic autoimmunity: life and death in the Fas lane. Immunol Today. 1992;13:427-8
25. Koh DR, Ho A, Rahemtulla A, Fung-Leung WP, Griesser H, Mak TW. Murine lupus in MRL/lpr mice lacking CD4 or CD8 T cells. Eur J Immunol. 1995;25:2558-62
26. Zhao C, Zhang L, Kong W, Liang J, Xu X, Wu H. et al. Umbilical cord-derived mesenchymal stem cells inhibit cadherin-11 expression by fibroblast-like synoviocytes in rheumatoid arthritis. J Immunol Res. 2015;2015:137695
27. Geng L, Tang X, Zhou K, Wang D, Wang S, Yao G. et al. MicroRNA-663 induces immune dysregulation by inhibiting TGF-β1 production in bone marrow-derived mesenchymal stem cells in patients with systemic lupus erythematosus. Cell Mol Immunol. 2019;16:260-74
28. Ji L, Hou X, Liu W, Deng X, Jiang Z, Huang K. et al. Paeoniflorin inhibits activation of the IRAK1-NF-κB signaling pathway in peritoneal macrophages from lupus-prone MRL/lpr mice. Microb Pathog. 2018;124:223-9
29. Umeuchi H, Kawashima Y, Aoki CA, Kurokawa T, Nakao K, Itoh M. et al. Spontaneous scratching behavior in MRL/lpr mice, a possible model for pruritus in autoimmune diseases, and antipruritic activity of a novel nopioid receptor agonist nalfurafine hydrochloride. Eur J Pharmacol. 2005;518:133-9
30. Q-Z Li, J Zhou, R Yang, M Yan, Q Ye, K Liu. et al. The lupus-susceptibility gene kallikrein downmodulates antibody-mediated glomerulonephritis. Genes Immun. 2009;10:503-8
31. Fu HJ, Zhu J, Yang M, Zhang ZY, Tie Y, Jiang H. et al. A novel method to monitor the expression of microRNAs. Mol Biotechnol. 2006;32:197-204
32. Arne N Akbar, Sian M Henson. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Immunol. 2011;11:289-95
33. Rostami Z, Khorashadizadeh M, Naseri M. Immunoregulatory properties of mesenchymal stem cells: Micro-RNAs. Immunol Lett. 2020;219:34-45
34. Anna Lewinska, Jagoda Adamczyk-Grochala, Anna Deregowska, Maciej Wnuk. Sulforaphane-Induced Cell Cycle Arrest and Senescence are accompanied by DNA Hypomethylation and Changes in microRNA Profile in Breast Cancer Cells. Theranostics. 2017;7:3461-77
35. Marco Demaria, Naoko Ohtani, Sameh A Youssef, Francis Rodier, Wendy Toussaint, James R Mitchell. et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. 2014;31:722-33
36. Muñoz-Espín D, Cañamero M, Maraver A, Gómez-López G, Contreras J, Murillo-Cuesta S. et al. Programmed cell senescence during mammalian embryonic development. Cell. 2013;155:1104-18
37. Schafer MJ, Haak AJ, Tschumperlin DJ, LeBrasseur NK. Targeting Senescent Cells in Fibrosis: Pathology, Paradox, and Practical Considerations. Curr Rheumatol Rep. 2018;20:3
38. Chou JP, Effros RB. T cell replicative senescence in human aging. Curr Pharm Des. 2013;19:1680-98
39. Wang D, Feng X, Lu L, Konkel JE, Zhang H, Chen Z. et al. A CD8 T cell/indoleamine 2,3-dioxygenase axis is required for mesenchymal stem cell suppression of human systemic lupus erythematosus. Arthritis Rheumatol. 2014;66:2234-45
40. Tang H, Tan G, Guo Q, Pang R, Zeng F. Abnormal activation of the Akt-GSK3beta signaling pathway in peripheral blood T cells from patients with systemic lupus erythematosus. Cell Cycle. 2009;8:2789-93
41. Ji S, Guo Q, Han Y, Tan G, Luo Y, Zeng F. Mesenchymal stem cell transplantation inhibits abnormal activation of Akt/GSK3β signaling pathway in T cells from systemic lupus erythematosus mice. Cell Physiol Biochem. 2012;29:705-12
42. Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W. et al. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551-63
43. Kwon HS, Brent MM, Getachew R, Jayakumar P, Chen LF, Schnolzer M. et al. Human immunodeficiency virus type 1 Tat protein inhibits the SIRT1 deacetylase and induces T cell hyperactivation. Cell Host Microbe. 2008;3:158-67
44. Habib HM, Taher TE, Isenberg DA, Mageed RA. Enhanced propensity of T lymphocytes in patients with systemic lupus erythematosus to apoptosis in the presence of tumour necrosis factor alpha. Scand J Rheumatol. 2009;38:112-20
45. Liu FJ, Wen T, Liu L. MicroRNAs as a novel cellular senescence regulator. Ageing Res Rev. 2012;11:41-50
46. Salla K Laine, Jessica J Alm, Sanna P Virtanen, Hannu T Aro, Tiina K Laitala-Leinonen. MicroRNAs miR-96, miR-124 and miR-199a regulate gene expression in human bone marrow-derived mesenchymal stem cells. Cell Biochem. 2012;113:2687-95
47. Guohua Lou, Liang Chen, Caixia Xia, Weina Wang, Jinjin Qi, Aichun Li. et al. MiR-199a-modified exosomes from adipose tissue-derived mesenchymal stem cells improve hepatocellular carcinoma chemosensitivity through mTOR pathway. Exp Clin Cancer Res. 2020;39:4
48. Wang Q, Ye B, Wang P, Yao F, Zhang C, Yu G. Overview of microRNA-199a Regulation in Cancer. Cancer Manag Res. 2019;11:10327-35
49. Xu Chen, Tian Yang, Wei Wang, Wenjin Xi, Tianze Zhang, Qi Li. et al. Circular RNAs in immune responses and immune diseases. Theranostics. 2019;9:588-607
Author contact
![]() Corresponding authors: Lingyun Sun, MD, PhD, Department of Rheumatology and Immunology, The Affiliated Drum Tower Hospital, Medical School of Nanjing University, Nanjing, 210008 China. Email: lingyunsunedu.cn; Yan Li, PhD, The State Key Laboratory of Pharmaceutical Biotechnology, Chemistry and Biomedicine Innovation Center, Model Animal Research Center of Nanjing University, Nanjing, Jiangsu 210061, China. Email: yanliedu.cn.
Corresponding authors: Lingyun Sun, MD, PhD, Department of Rheumatology and Immunology, The Affiliated Drum Tower Hospital, Medical School of Nanjing University, Nanjing, 210008 China. Email: lingyunsunedu.cn; Yan Li, PhD, The State Key Laboratory of Pharmaceutical Biotechnology, Chemistry and Biomedicine Innovation Center, Model Animal Research Center of Nanjing University, Nanjing, Jiangsu 210061, China. Email: yanliedu.cn.