Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(21):9528-9543. doi:10.7150/thno.42971 This issue Cite
Research Paper
m6A RNA modification modulates PI3K/Akt/mTOR signal pathway in Gastrointestinal Cancer
Qijie Zhao1,2,3*, Yueshui Zhao1,2*, Wei Hu4,5*, Yan Zhang6*, Xu Wu1,2, Jianwei Lu6, Mingxing Li1,2, Wei Li7,8, Weiqing Wu8, Jianhong Wang7,8, Fukuan Du1,2, Huijiao Ji1,2, Xiao Yang1, Zhenyu Xu9, Lin Wan10, Qinglian Wen11, Xiang Li1, Chi Hin Cho1,2, Chang Zou7,8 ![]() , Jing Shen1,2
, Jing Shen1,2 ![]() , Zhangang Xiao1,2
, Zhangang Xiao1,2 ![]()
1. Laboratory of Molecular Pharmacology, Department of Pharmacology, School of Pharmacy, Southwest Medical University, Luzhou, 646000, Sichuan, PR China.
2. South Sichuan Institute of Translational Medicine, Luzhou, 646000, Sichuan, PR China.
3. Department of Pathophysiology, College of Basic Medical Science, Southwest Medical University, Luzhou, 646000, Sichuan, PR China.
4. Department of Gastroenterology, Shenzhen Hospital, Southern Medical University, Shenzhen, Guangdong, PR China.
5. Department of Anaesthesia and Intensive Care, The Chinese University of Hong Kong, Hong Kong SAR, PR China.
6. Department of Oncology, Jiangsu Cancer Hospital and Jiangsu Institute of Cancer Research and The Affiliated Cancer Hospital of Nanjing Medical University, Nanjing 210000, PR China.
7. Clinical Medical Research Center, the Second Clinical Medical College of Jinan University, The First Affiliated Hospital of Southern University, Shenzhen People's Hospital, Shenzhen, Guangdong 518020, PR China.
8. Department of Breast and Thyroid Surgery, the Second Clinical Medical College of Jinan University, The First Affiliated Hospital of Southern University, Shenzhen People's Hospital, Shenzhen, Guangdong 518020, PR China.
9. Department of Pharmacy, Yijishan Affiliated Hospital of Wannan Medical College, Wuhu, Anhui, PR China.
10. Department of Hematology and Oncology, The Children's Hospital of Soochow, Jiangsu, PR China.
11. Department of Oncology, The Affiliated Hospital of Southwest Medical University, Luzhou, Sichuan 646000, PR China.
*These authors contributed equally to this work.
Received 2019-12-11; Accepted 2020-7-10; Published 2020-7-25
Abstract

Rationale: Methylation at the N6 position of adenosine (m6A) is the most prevalent RNA modification within protein-coding mRNAs in mammals, and it is a reversible modification with various important biological functions. The formation and function of m6A are regulated by methyltransferases (writers), demethylases (erasers), and special binding proteins (readers) as key factors. However, the underlying modification mechanisms of m6A in gastrointestinal (GI) cancer remain unclear. Here, we performed comprehensive molecular profiling of the nine known m6A writer, eraser, and reader proteins in GI cancer.
Methods: Data from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) were used. Gene alteration and pathway analysis were done in cBioportal. The protein network of m6A regulators and its related pathway members was analyzed in STRING online platform. Phylogenetic tree was constructed in MEGA7. m6A modification sites were predicted by SRAMP. m6A related SNPs were analyzed by m6ASNP. The modulation of m6A on its related pathway members was validated by m6A-seq, real-time PCR and phosphor-MAPK array.
Results: We found that m6A regulators were mostly upregulated in GI cancer and their differential expression significantly influenced the overall survival of patients with GI cancer. The phosphatidylinositol-3-kinase (PI3K)/Akt and mammalian target of rapamycin (mTOR) signaling pathways were found to be potentially affected by m6A modification in most human cancers, including GI cancer, which was further verified by m6A-Seq and phospho-MAPK array.
Conclusions: Our findings suggest that m6A RNA modification has a fundamental role in the regulation of PI3K/Akt and mTOR signaling pathway function in cancer.
Keywords: m6A RNA methylation, gastrointestinal cancer, bioinformatics, PI3K/Akt signal pathway, mTOR signaling pathway
Introduction
N6 methylation of adenosine (m6A) is the most common reversible mRNA modification in eukaryotes since its emergence a decade ago [1, 2]. It was recently recognized that reversible m6A modification influences mRNA fate in cells [3-6]. Unlike DNA methylation and histone modification, RNA modifications have recently been emphasized as a new layer of epigenetic regulation at the post-transcriptional RNA level [7-9]. All m6A have strict sequence specificity and no requirement for extended sequences or secondary structures, and only occur in a fraction of transcripts [2-5, 7-10]. The cellular pathway of m6A modification of RNA is shown in (Figure 1A). m6A is catalyzed by a methytransferase complex consisting of the “writer” proteins methyltransferase-like 3 (METTL3), methyltransferase-like 14 (METTL14), and Wilms tumor 1 associated protein (WTAP). Its demethylation is catalyzed by two “eraser” demethylases, fat mass and obesity-associated protein (FTO) and AlkB homolog 5 (ALKBH5) [11-13]. YTHDF1, YTHDF2, YTDHF3, YTHDC1, and NHRNPA2B1, members of the YTH domain family of proteins; are m6A “readers” that recognize its modification and affect pre-mRNA splicing as well as mRNA transport, stability, and translation [14, 15]. The METTL3-METTL14 hetero methyltransferase complex (stoichiometric ratio=1:1) [16] interacted with WTAP to affect nuclear RNA m6A load (Figure 1A) [12]. The writer proteins can colocalize the mammalian RNA methylation through m6A deposition [17]. The methyl donor S-adenosylmethionine is catalyzed by writer proteins and is present during RNA methylation (Fig. 1a) [18]. As a reversible internal modification, FTO and ALKBH5 catalyze the demethylation of m6A [5, 7-10, 19]. The YTHDF1/2/3 reader proteins selectively bind m6A-containing mRNA in the cytoplasm of eukaryotes [20], and NHRNPA2B1 is widely distributed in the nucleus where it functions as a special m6A-binding protein [14]. To date, more than 12,000 m6A sites in the transcripts of more than 7,000 mammal genes have been characterized by methylated RNA immunoprecipitation sequencing (MeRIP-Seq) [15, 21]. RNA modification plays a crucial role in important biological processes in mammals. Recently, some key studies demonstrated that the alteration of core genes during m6A modification affects tumorigenesis, cancer cell proliferation, tumor microenvironment, and prognosis in cancers such as glioblastoma, lung cancer, liver cancer, and breast cancer [22-26]. However, the biological function and critical target genes of these m6A regulators remain unknown for the majority of human cancers.
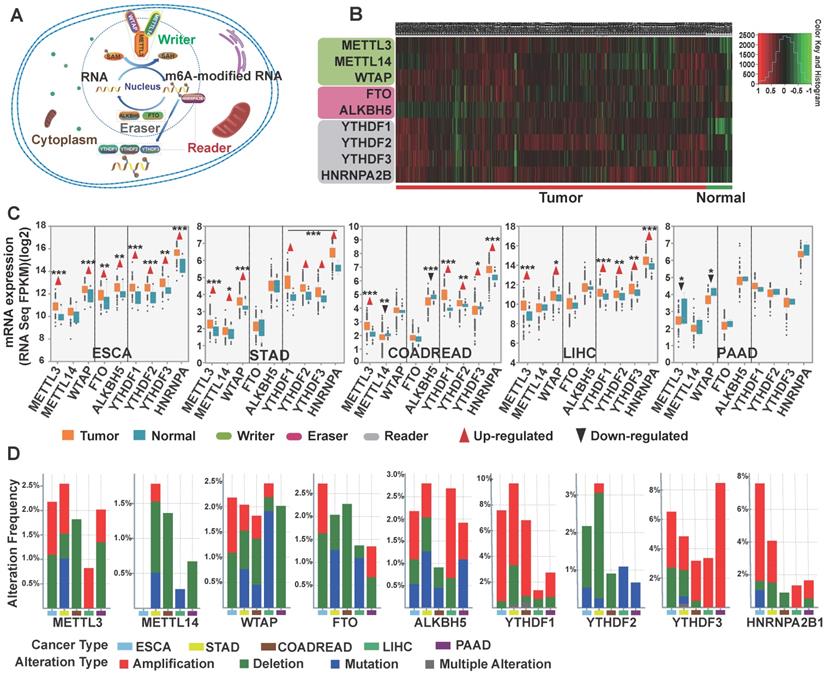
Comprehensive molecular profiling of m6A writers, erasers, and readers in GI cancer. (A) Schematic of eukaryotic cellular m6A methylatransferases (writers: METTL3, METTL14, and/or WTAP), demethylases (erasers: FTO and ALKBH5), and binding proteins (readers: YTHDF1-3, HNRNPA2B1). These proteins led to dynamic and reversible m6A modification of RNA. (B) Heatmap depicting the RNA expression profiles of the nine m6A regulators in GI cancer based on RNA sequencing data from TCGA database. Data were obtained from cancer patients (n=1727) and healthy patients (n=160), and each sample was normalized by counting the number of reads. The red and green regions represent higher and lower expression levels, respectively. (C) The expression level of the nine m6A regulators in different GI cancer types was analyzed using the RNA sequencing data from TCGA database consisting of 1727 cancer patients and 160 healthy patients. (D) The frequency of genetic alterations of m6A regulators in GI cancer. Cases with mutations, copy number alterations (amplification and deletion), and multiple alterations were selected from TCGA database and analyzed in cBioPortal. About 24.7% (46/186) of esophageal cancer, 26.8% (99/396) of stomach cancer, 13.7 (88/640) of colorectal cancer, 18.3 (67/366) of liver cancer, and 13.4(20/149) of pancreatic cancer clinical samples had genomic alterations of m6A regulators. (*p < 0.05, **p < 0.01, ***p < 0.001 between tumor and normal group).
In the United States, there were about 310,000 new cases diagnosed of gastrointestinal (GI) cancer in 2017 and 2018, including cancers of the esophagus, stomach, rectum, and colon, as well as cancers of the liver, pancreas, and gallbladder, and more than 150,000 deaths occurred [27]. GI cancer shares several characteristics such as similar endodermal development, high mortality, influenced by multiple factors, and chromosomal instability [28, 29]. In recent years, genetic alterations, single nucleotide polymorphisms (SNPs), and epigenetic modification have been increasingly recognized as important regulators of GI cancer [30]. However, there has been little progress towards understanding the biological functions and mechanisms of m6A in malignant tumors [31]. To gain further insight into the role of m6A in tumors, we studied the correlation between five GI subtypes (esophageal, stomach, colorectal, liver, and pancreatic) and m6A writer, reader, and eraser proteins. To this end, bioinformatics analysis of tumor profiles obtained from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) databases and comprehensive molecular profiling of the m6A regulators in GI cancer were performed. The results showed that genomic alterations in m6A caused significant changes in the overall survival (OS) of patients with GI cancer. The identification of risk factors for GI cancer may help with the development of targets and early detection and therapeutic strategies for the treatment of cancer.
Methods
Data Processing
We started with a set of 10,037 samples that were included in the final whitelist for the TCGA (http://cancergenome.nih.gov). There were 1920/10037 (19.1%) GI cancer samples. After filtering with eligibility formats, a total of 9,628 samples were used for pathway alteration analysis. Gene expression (generated by RNA-sequencing) was downloaded using the bioinformatics tool. Protein enrichment data (generated by Reverse phase protein array) were analyzed using cBioportal pipeline (http://www.cbioportal.org/). RNA-Sequence data was processed using Fragments per Kilobase of transcript per Million fragments mapped (FPKM). The American, East Asian, and European Genomic SNPs were collected from 1,000 Genome Project Data.
Pathway Analysis
Proteomic data were collected by Reverse Phase Protein Array (RPPA) based on TCGA data and analyzed by cBioportal in 27 kinds of human cancers. RPPA quality control and methodology stander were previously explained [32]. For the enriched proteins, significant change in expression was determined by the standard of a log ratio of Log2 based ratio (μ mean altered/ σ mean unaltered) (log>0 for overexpression and log<0 for underexpression) and queried event results p value<0.05. The selected proteins from this criterion were used to predict pathways by two conditions: (a) the sum of altered protein in each pathway, and (b) the statistical P-value score of significant pathway. DAVID function annotation tool (https://david.ncifcrf.gov) is a method that infers conditional dependencies between target genes and KEGG pathway. The significance of pathway level association was empirically estimated by P-value score and enriched counts, and the estimating score threshold was restricted with FDR (False Discovery Rate) of Benjamini-Hochberg method. We still considered the corrected P value smaller than 0.05 based on:
Q-value(i)=P(i)*length(P)/rank(P)
Correlation Analysis
The gene co-expression and DNA methylation correlation in GI cancer from TCGA data were analyzed in cBioportal and Prism6 with Pearson and Spearman correlation significances. In human, pathway gene co-expression was analyzed utilizing NCBI GEO microarray expression experiments deposit. The score from single- and dual-channel arrays (thresholds of 0.75 and 0.95) of gene-to-gene Pearson correlation were combined to get final scores, and calibration was depended on KEGG benchmark improvement in STRING [33]. The PPI network among the final cluster of genes was derived from the STRING online platform (http://string-db.org). STRING is a widely used database and online resource devoted to investigating PPI, including physical and function interaction [34]. All of the associations available in STRING were denoted with a probability confidence score. Interaction with a confidence score greater than 0.4 was selected to construct the PPI network. To define the phylogenetic characterization of target genes, a total of 66 whole genome sequences were recruited from GenBank for the phylogenetic tree construction. Neighbour-Joining (NJ) algorithm in MEGA7 was employed, respectively, with random addition of taxa and tree-bisection reconnection branch swapping.
m6A Methylation Site Prediction
Genetic mRNA sequence was used in m6A prediction tool SRAMP. Results were based on three type random forest classifiers, namely, Binary (Positional binary encoding of nucleotide sequence), KNN (K-nearest neighbor) and Spectrum (Nucleotide pair spectrum). Positive samples (with m6A sites) were retained by DRACH (D=A, G or U; R=A or G; H=A, C or U) consensus motif and 10-fold likelihood to correct proof negative sample (non-m6A sites) to avoid prominent bias, keeping the 1:10 positive-to-negative calculate ratio. For multiple transcripts with same locus, only the longest transcript with the largest number of m6A sit was retained. The different prediction scores of random forest classifiers were combined using the weighted summing formula:
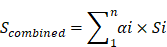
RNA Sequencing Data Analysis
In order to confirm the m6A function in human cancer, we applied the MeT-DB v2.0 bioinformatics tool with different Input FASTQ file from immunoprecipitation experiment. High-throughput sequencing data of expression profile after METTL3 knockdown for U2SO cells (GSE48037), Hela cells (GSE46705), HEK293T cells (GSE55572), and A549 cells (GSE76367) was obtained from Gene Expression Omnibus (GEO). Pathway member gene symbols with its chromosomal start/end position were used to query based on NCBI GEO high-throughput Input data. For each peak/site, the genomic location, gene ID, and read counts were displayed in the column of the newly lightweight JBrowse. Interesting target genes were screened by JBrowse track filtering panel. The target genetic exon peak region output were reconstructed and calculated within Origin 9.
m6A SNP Analysis
Genetic variant files variant call format (VCF) from the 1,000 Genome Project database was used in m6ASNP, a new m6A modification site predictor. There were 55,548 human m6A sites according to the literature [35, 36]. The sequences of 30 nucleotides upstream and downstream of m6A residue in the flanking regions were extracted for random forest prediction. Genetic variants were first mapped to known transcript structures. Based on PA-m6A-seq, MeRIP-seq, and miCLIP-seq data in m6ASNP, a total of 13,703 high-confidence, 54,222 medium-confidence, and 243,880 low-confidence m6A-associated variants were found for human. To avoid any bias, the deleteriousness of each variant score was aggregated from 0 to 5 using the five kinds of software. LRT [37], PolyPhen2 HVAR [38], PolyPhen2 HDIV [38], SIFT [39], and FATHMM [40], with a higher score indicating higher possibility of deleteriousness.
m6A-seq coverage analysis
The m6A-seq was performed as previously described [41]. Fragmented RNA (5μg mRNA) was incubated for 2 h at 4°C with 10 μg of affinity purified anti-m6A polyclonal antibody (Synaptic Systems) in 450 μL of immunoprecipitation (IP) buffer (150 mM NaCl, 0.1% NP-40 (v/v), 10mM Tris-HCl, pH 7.4) and 300 U mL-1 RNase inhibitor (Promega). The mixture was then immunoprecipitated by incubation with protein-Abeads (Repligen) at 4°C for an additional 2h. Then, the eluted mRNAs were re-covered with phenol-chloroform extraction and ethanol precipitation in IP buffer. 50 ng of immunoprecipitated mRNAs and pre-immunoprecipitated mRNAs (input control) was used for library construction with NEBNext ultra RNA library prepare kit for Illumina (NEB). High-throughput sequencing was performed on the Illumina HiSeq X sequencer with a paired-end read length of 150 bp according to the standard protocols. From the smoothed mountain plots for both the control and IPed data, we compute the potential m6A situs for any given site on a gene (e.g., an DRACH position).
Mammalian Cell Culture, siRNA Knockdown
Human gastric cancer cell TMK1, pancreatic cancer cell PANC-1, liver cancer cell Huh-7 were grown in DMEM media (Gibco) with 10% FBS (Gibco) and 1% 100X Pen/Strep (Gibco). Human colorectal cancer cell line HCT116, and esophageal cancer cell TE-1 were grown in IMEM and RPMI1640 media, respectively, with 10% FBS and 1% 100X Pen/Strep. Control siRNA and METTL3 siRNA were purchased from GenePharma (Shanghai, China) with reported sequences[12]. Transfection was achieved by using Lipofectamine Lipofectamine 3000 (Invitrogen) with 20 nM of siRNAs. At 48 h post-transfection, the mRNA levels of METTL3, AKT1, mTOR, PIK3CA, and PTEN were checked by Q-PCR.
m6A RNA Methylation Quantification
Cancer cells were treated with SAH or MA for 48 h, then total RNAs were extracted by TRIZOL, concentrations of RNAs were analyzed by Nanodrop 2000. Then, 200 ng of RNA for each sample were used for m6A RNA methylation analyses. m6A levels were analyzed by relative quantification method according to the manufacturer's instruction (Abcam, ab185912).
Real-Time PCR
Total RNA was isolated from cultured cells using TRIzol Reagent (Thermo Fisher), and cDNA was made with the FastKing RT Kit (With gDNase, KR116). The RNA concentration was measured by NanoDrop. Total RNA isolation for Quantitative Real-time PCR (qPCR) was performed using the SuperReal PreMix Plus (SYBR Green, FP205) according to the manufacturer's instruction.
mRNA Lifetime Measurements
The half-life time of mRNA was measured using reported method [12, 42]. In brief, HCT116 and TMK1 cells were treated with SAH or MA for 48 h, then actinomycin D (ActD, 5 μg/ml, Millipore) was used to treat cells for 6 h, 3 h, and 0 h. Total RNAs were isolated, and reverse-transcripted into cDNA, the mRNA levels of AKT1, mTOR, PIK3CA, and PTEN were detected by qPCR, and the mRNA half-life time of these genes were calculated using reported method [12].
Human Phosphor-MAPK Array
Phosphorylation of PI3K/Akt and mTOR signaling effectors were detected using human phosphor-MAPK array kit (R&D, USA) following protocol provided by manufacturer. The signals were detected by the ChemiDocTM Imaging System and quantified using the software Image-Pro plus 6.0.
Statistical Analysis
Students' t-test was used in comparison of two groups and one-way ANOVA was applied to compare multiple groups. Overall survival analysis was performed with the Kaplan-Meier curve with P value calculated using the log-rank test.
Results
Deregulation of m6A Writers, Erasers, and Readers in GI Cancer
First, we used TCGA database to investigate the profile of m6A writers, erasers, and readers, including METTL3, METTL14, WTAP, FTO, ALKBH5, YTHDF1, YTHDF2, YTHDF3, in GI cancer. After comparing the expression pattern of m6A-related genes between 1,782 cancer samples and 148 normal control samples, heatmap analysis showed that the expression of m6A regulators was remarkably higher in GI cancer (Figure 1B). Further comparison between cancer and normal samples in the five subtypes of GI cancer (esophageal, stomach, colorectal, liver, and pancreatic) (Figure 1C) indicated that readers and writers were mostly upregulated in GI cancer compared to the normal control, except in pancreatic cancer. It is worth noting that nearly all m6A-related genes were overexpressed in esophageal cancer while such overexpression was not seen in pancreatic cancer. On the contrary, two of the writers were significantly downregulated in pancreatic cancer (Figure 1C). We further analyzed genetic alterations of m6A regulators using the cBioPortal and applied Significant Targets in Cancer algorithm (GISTIC 2.0). The frequency of copy number amplification, deletion and genetic mutation of m6A regulators is shown in Figure 1D. Overall, about 11.9% of the clinical samples (204/1710) had genetic amplification of m6A regulators, and nearly 4.6% of the samples (79/1710) had genomic mutations. Of note, some m6A-binding proteins (readers), YTHDF1, YTHDF3, and HNRNPA2B1, showed relatively higher frequency of copy number amplification.
Impact of m6A Regulator Alterations on the Survival Rate of GI Cancer Patients
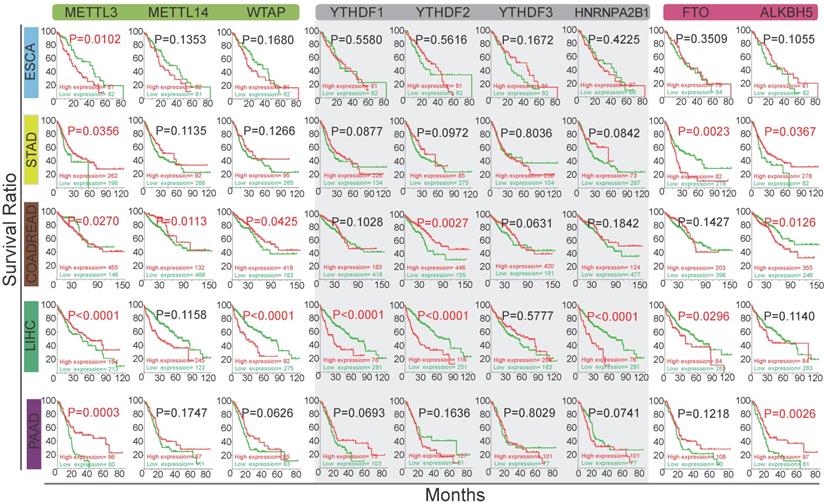
Next, we performed OS analysis on m6A regulators in five subtypes of GI cancer using TCGA database (Figure 2). Kaplan-Meier analysis revealed that the differential expression of m6A regulators was significantly associated with OS. Specifically, high expression of METTL3 was significantly associated with poor survival in esophageal and colorectal cancers (p<0.05). In contrast, low expression of METTL3 was significantly associated with poor survival in gastric cancer, liver cancer and pancreatic cancer. High expression of ALKBH5 was significantly associated with low OS in colorectal cancer while it was associated with better survival in stomach and pancreatic cancer. In liver cancer, high expression of writer, reader, and eraser proteins was associated with poor survival except METTL3, YTHDF3 and ALKBH5. Together, these results indicate that m6A regulators may have important influence in GI cancer survival.
The relationship between the alterations of m6A methylation regulators and the overall survival rate of GI cancer patients. Kaplan-Meier analysis of overall survival of GI patients based on the expression level of m6A regulators. The red and green curves indicate the survival curves of patients in the high expression and low expression groups, respectively.
Identification of Signaling Pathways Associated with the Deregulation of m6A in GI Cancer
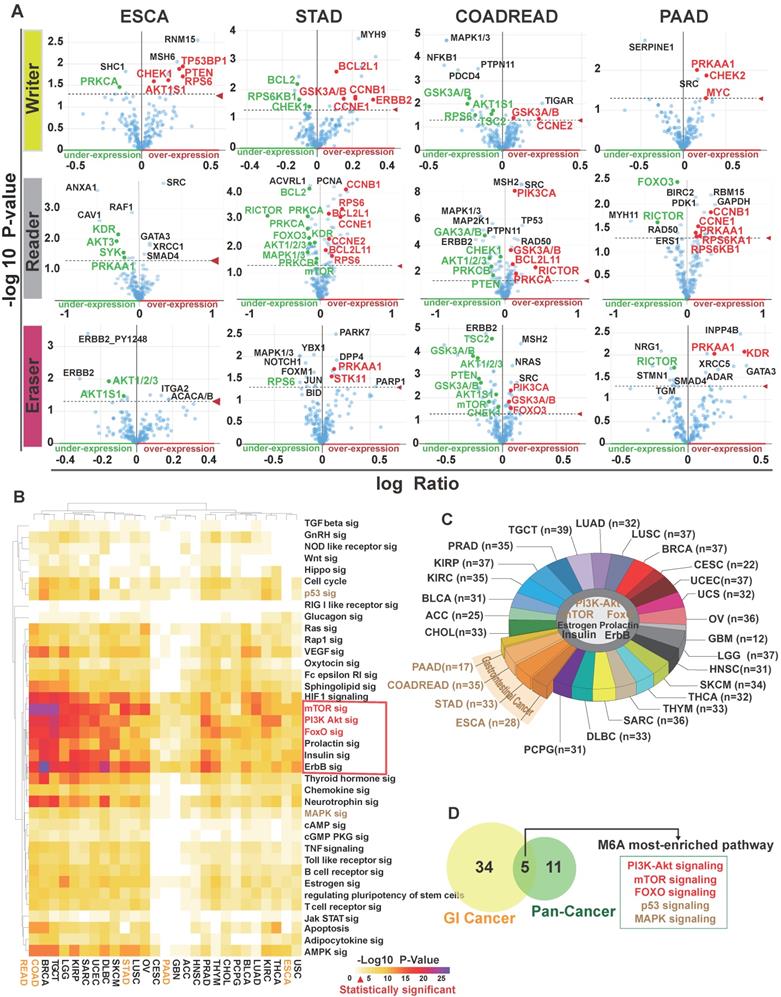
As m6A modification is frequently involved in both mRNA metabolism and translation to influence protein expression levels [43], we tried to find out proteins associated with m6A regulators. We applied correlational analysis of protein expression (both parent and phosphorylated proteins) changes with m6A regulatory gene alterations (mutations, copy number alterations, mRNA expression changes) in GI and other cancers in cBioportal. The main proteins with significant changes (p<0.05) are shown in Figure 3A and Table S1. Then, we used DAVID to analyze these proteins in 27 individual human cancers relying on the KEGG pathway database (Table S2). 41 different pathways were shown with P-value scores in heatmap (Figure 3B). The enriched proteins were related to several key pathways, including the phosphatidylinositol-3-kinase (PI3K)/Akt, mammalian target of rapamycin (mTOR), class O of forkhead box transcription factors (FoxO), prolactin, estrogen, insulin, and ErbB signaling pathways, which typically exist in 27 major types of human cancers (Figure 3C). Importantly, five pathways (PI3K/Akt, mTOR, FoxO, mitogen-activated protein kinase [MAPK], and p53) that were positively associated with m6A modification in GI cancer, overlapped with KEGG cancer pathways (Figure 3D).
The signaling pathways associated with the deregulation of m6A in GI cancer. (A) Protein enrichment in esophageal, stomach, colorectal, and pancreatic cancer upon alteration of writer, eraser, or reader proteins. p<0.05 was considered significantly changed. The fold change in expression level was based on the log ratio (mean of altered expression/mean of unaltered expression). (B) Heatmap of pathways related to m6A gene alterations in 27 human cancers was analyzed by KEGG pathway analysis via DAVID. The degree of correlation was denoted by P values in different cancer types. (C) Pathway enrichment in different cancer types. Seven pathways were universally altered by m6A in human cancers. (n=number of pathways altered in each type of cancer.) (D) Identification of five m6A regulator-related pathways in cancer. Overlap was seen among 39 m6A regulator-related pathways and 16 cancer pathways from KEGG. The overlap resulted in five pathways that was represented in the Venn diagram.
Deregulation of PI3K/Akt, FoxO, mTOR, MAPK and p53 Signaling Pathways in Cancer and Their Relationship with m6A Members
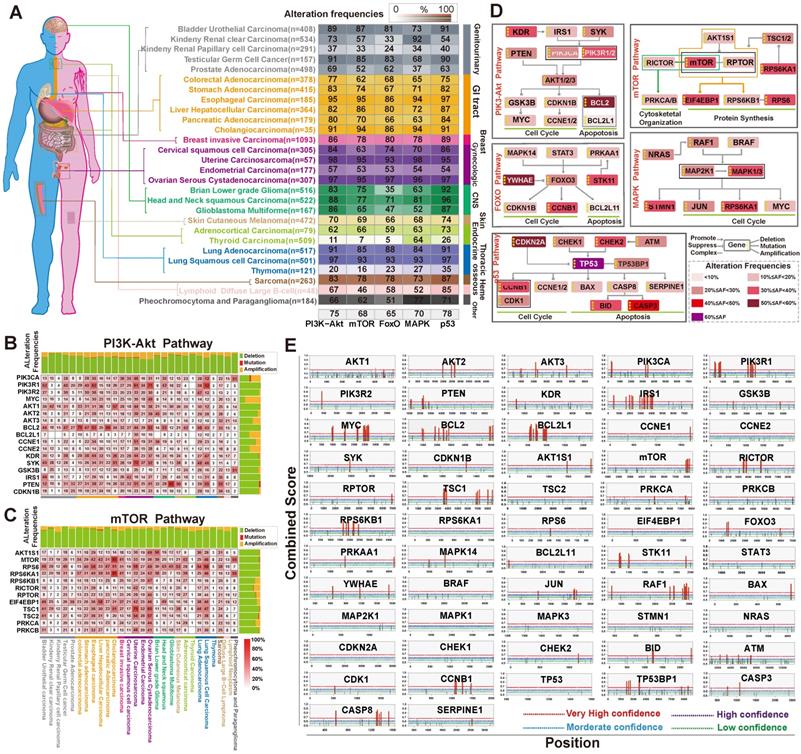
Next, to assess alterations of PI3K/Akt, FoxO, mTOR, MAPK, and p53 signaling in cancer, we collected data of somatic mutations and DNA copy number variations (amplification, shallow deletion, and deep deletion) from tumor patients (n=9,628) from 27 different cancer types in TCGA through cBioPortal for Cancer Genomics [44]. For each cancer type, target genes with at least one alteration were regard as being altered in a given pathway if one or more genes were involved in the pathway. As shown in Figure 4A and Table S3, the PI3K/Akt pathway showed the highest frequency of alterations (75%) in 27 human cancer types. The alteration frequency of GI cancer subtypes in this pathway were esophageal cancer (95% of patients), stomach cancer (83% of patients), colorectal cancer (77% of patients), liver cancer (82% of patients), and pancreatic cancer (80% of patients). The mTOR pathway also had a relatively high rate of alterations in GI cancer (75%), with an alteration frequency of 95%, 74%, 62%, 86% and 70% among the five cancer subtypes, respectively (Figure 4A). The FoxO, MAPK, and p53 signaling pathways also displayed high alteration frequency (Figure 4A). Further heatmap analysis indicated that the main genes in these five pathways had high frequency alterations across 27 cancer types (Figure 4B-C, Figure S1A-C). PIK3CA, PIK3R1, AKT1, PTEN, BCL2, and SYK in the PI3K/AKT pathway were among the most frequently altered genes (>20% of all patients; Figure 4B). In the mTOR pathway, the alteration of crucial members was the highest in GI and development tract cancer (43.4% across patients) than in other tumor types (Figure 4C). A simplified pathway diagram in Figure 4D shows the most frequently altered genes in GI cancer that were affected by m6A directly or indirectly in five pathways including the frequency and type of alteration, cellular function, and type of interaction with other genes in the pathway.
The relationship between the selected signaling pathways and m6A regulators. (A) The alteration of PI3K/Akt, mTOR, FoxO, MAPK and p53 pathways in different types of human cancer. Analysis was carried out in cBioPortal for Cancer Genomics using TCGA data. (B,C) Detailed heatmap of the alteration frequency in the PI3K/Akt and mTOR pathways. (D) Pathway diagram showing the frequency and type of alteration, cellular function, and interaction with other genes of the key genes in the five pathways. Different alteration frequencies were indicated by the color intensity of each gene from the entire GI cancer database. (E) m6A modification sites in the main pathway member mRNA sequences were predicted by SRAMP. The three random forest classifiers prediction scores were combined as combined score with weighted summing formula. The combined scores were divided into four degrees (very high confidence, high confidence, moderate confidence, low confidence).
To get an overview of whether the five pathways are regulated by DNA methylation in tumor and normal samples, we used DiseaseMeth version 2.0 for further analysis based on dataset in TCGA HumanMethylation450. The expression of TP53, AKT3, PIK3R1, CHEK1, PRKAA1, and RAF1 were affected by DNA methylation in GI cancer (Figure S3). Interestingly, STRING analysis [33] of the protein-protein interaction (PPI) networks among the five pathway members (n=57 genes) and m6A regulators (writers: METTL3, METTL14 and WTAP; readers: YTHDF1, YTHDF2, YTHDF3, HNRNPA2B1 and erasers: FTO and ALKBH5) revealed that the node in Figure S4A could be classified into two large groups: one group consisted of m6A regulators and the other group was connected by the interaction of the five pathway proteins. AKT1 functionally interlinked the pathway proteins with the FTO (eraser) and HNRNPA2B1 (reader) proteins. The proxy of co-expression is a strong indicator of functional correlation [45]. We found that there was strong similarity in expression between METTL14 (writer) and PIK3CA (score=0.195), suggesting functional correlation between the two proteins (Figure S4B).
To further explore the structural interlinks within the m6A regulators, we constructed a Phylogram tree of all genes (n=66) based on the nucleotide sequences (Figure S4C). In the tree, the length of branches was calculated from the likelihood ratio mapping the evolutionary messages among nuclear factors. The Phylogram tree showed a strong relationship between AKT1 and YTHDF1 (reader), indicating that they may have been a common ancestry or the same point of evolutionary origin. Moreover, the PI3K/Akt/mTOR pathway members (PIK3CA, AKTIS1, AKT2, AKT3, RPS6KB1, GSK3B) and m6A regulators (METTL3, METTL14, WTAP, FTO, YTHDF3, HNRNPA2B1) composed a monophyletic clade indicating a common ancestor. Moreover, we performed m6A site prediction. The special DRACH (D = A, G or U; R = A or G; H = A, C or U) and GAC consensus motif may be recognized as m6A sites and calculated by two random forest prediction modes of SRAMP to improve identity thresholds [46]. As shown in Figure 4E and Table S4, very high confidence m6A sites universally existed in members of the PI3K/Akt (AKT1/2/3, PI3KCA, PTEN, PI3KR1/2, BCL2, BCL2L1, IRS1, GSK3B, MYC) and mTOR pathways (mTOR, AKT1S1, TSC1, RICTOR, RPTOR, RPS6KB1, PRKCA), as well as members from the FoxO, MAPK, and p53 pathways.
Taken together, these results indicated that m6A plays a fundamental role in the regulation cancer pathway such as the PI3K/Akt/mTOR pathway.
Impact of m6A Deregulation on the PI3K/Akt/mTOR Pathway in GI Cancer
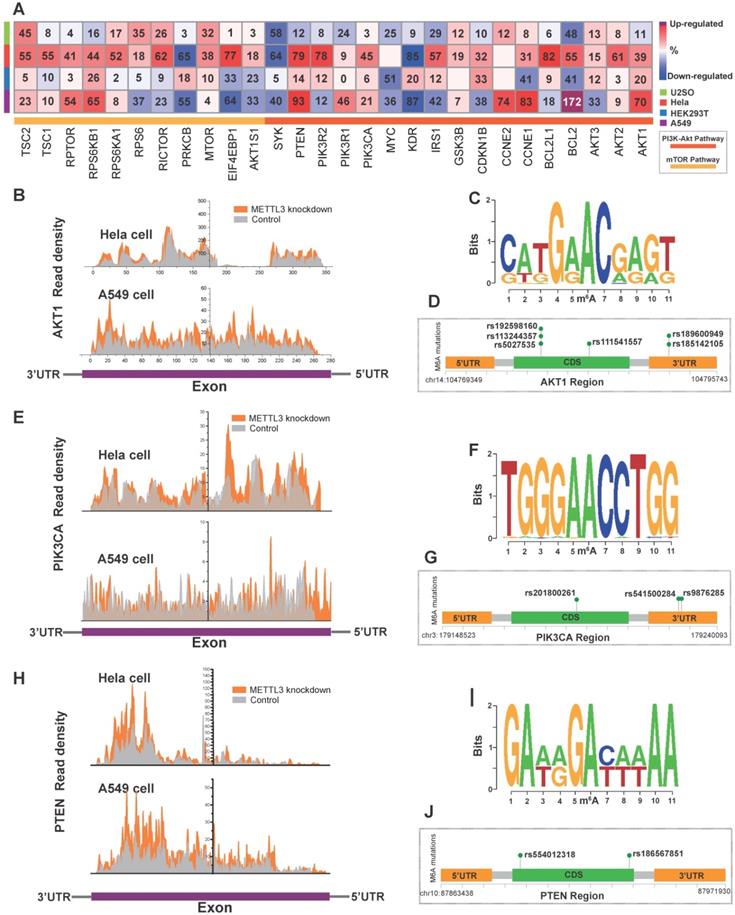
We found co-occurrence of PI3K/Akt and mTOR pathway alteration in cancer (Figure S2A) and the PI3K/Akt and mTOR pathway were interconnected (Figure S2B). To investigate the direct effects of m6A on the PI3K/Akt/mTOR pathway, we first employed MeT-DB V2.0 [47] to detect genomic expression profiles based on the GEO database. Since the presence of m6A can induce mRNA degradation in eukaryotes [48], we focused on upregulated genes with m6A writer downregulation based on the GEO database. Knockdown of the “writer” METTL3 in U2SO cells (GSE48037), Hela cells (GSE46705), HEK293T cells (GSE55572), and A549 cells (GSE76367) resulted in a relatively elevated expression of PI3K/Akt/mTOR signaling pathway proteins such as AKT1, AKT2, PTEN, PIK3CA and BCL2 as shown in the heatmap (Figure 5A, Figure S4D). The gene read density of AKT1, PTEN, and PIK3CA in Hela and A549 cells was shown in Figure 5B, E and H, A recent study indicated that post-transcription is influenced by genomic variants and is closely related to multiple disease such as cancer and genetic diseases [49]. To investigate the dysregulation of PI3K/Akt pathway in relation to the abnormity of variants in critical gene site, we analyzed SNPs from the 1000 Genomes Project database (among AMR, EAS, and EUR). Then we applied m6A SNP to detect the m6A site that is altered by variants (nonsynonymous and synonymous) around these sites [50]. The DRACH motif of AKT1 mutation sequence is shown in Figure 5C. We identified SNPs (rs185142105, rs189600949, rs5027535, rs113244357, rs192598160 and rs111541557 with dbSNP annotation) variants in AKT1 that disrupted m6A modification in the same or diverse sites (Figure 5D). The same phenomenon was found in PIK3CA (Figure 5F-G) with rs201800261, rs541500284, and rs9876285 and PTEN with rs554012318 and rs186567851 (Figure 5I-J) and AKT1S1 with rs546549861 (Figure S4E-G).
The modulation of m6A regulators on PI3K/Akt/mTOR pathway members. (A) Alteration in expression of PI3K/Akt and mTOR pathway genes in U2SO, Hela, HEK293T and A549 cells upon METTL3 knockdown based on the MeT-DB V2.0 and GEO databases (GSE48037, GSE46705, GSE55572, and GSE76367). (B, E, H) The transcript reads coverage peaks visualized exon region expression of AKT1, PIK3CA, and PTEN upon METTL3 knockdown in Hela and A549 cells. Figure was reconstructed by Origin 9. (C, F, I) To present the SNP alteration of m6A motif, the sequence logo was generated automatically based on DRA(m6A)CH structure with the 1000 Genome Project database SNP. (D, G, J) Recurrent known SNP mutation relevant to m6A modification in AKT1, PIK3CA, and PTEN. Recurrent known SNP mutations were color coded in green, and each mutation was annotated with dbSNP label. Distribution of m6A-related SNP variants within the gene across their 3'-UTR, 5'-UTR and exon regions was shown.
In vitro Study of m6A Deregulation on the PI3K/Akt/mTOR Pathway in GI Cancer
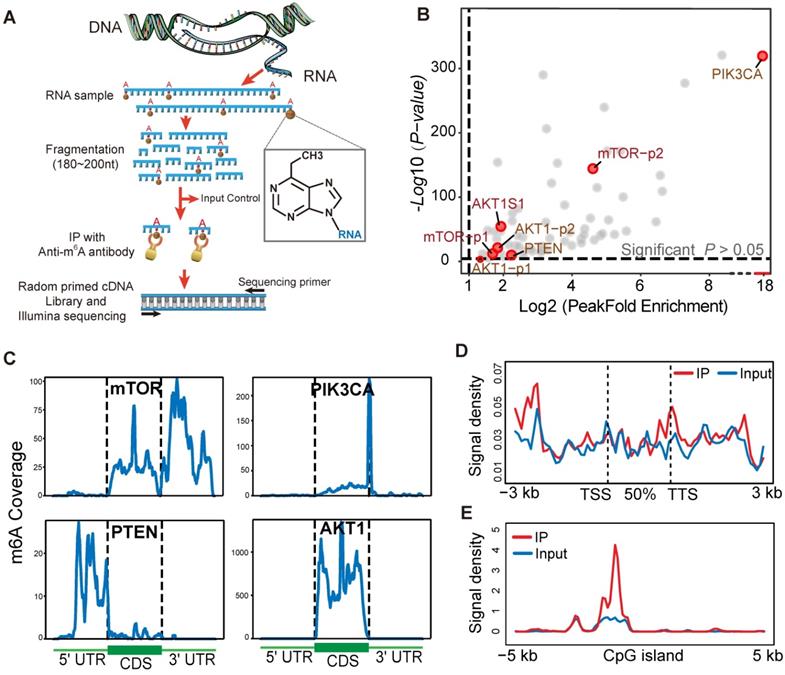
To identify and localize m6A sites at a transcriptome-wide level we applied m6A-seq to RNA purified from a human gastric cancer cell line (TMK1). Libraries were prepared from immunoprecipitated as well as input control fragments, and subjected to massively parallel sequencing (Figure 6A). This analysis yielded ~20,000 putative m6A sites peak (Figure S5A) and the overlapping transcript can be divided into five segments, TSS, 5' UTR; coding sequence (CDS); stop codon; and 3'UTR (Figure S5B-C). mTOR, AKT1, PIK3CA and PTEN were identified to possess a sequence element for specifying m6A RNA-methylation (Figure 6B). In mTOR and PIK3CA, the 3' UTR and contiguous segment stood out as most enriched in m6A peaks (Figure 6C). Moreover, PTEN showed m6A peaks enrichment near the 5' UTR region, and a corresponding decrease of m6A levels after the start codon (Figure 6C). AKT1 m6A modification predominantly target codon region. Furthermore, in these four genes, the average binding location analysis shows that m6A-binding activity a little bit higher in transcription termination sites (TTS) (Figure 6D). As shown in Figure 6E, mTOR, AKT1, PIK3CA and PTEN m6A modification peak was obviously enriched at the centre of CpGI (CpG islands). m6A/MeRIP-seq of the polyA+ RNA from human TMK-1 cell identified multiple peaks of varying intensities and one m6A-consensus sequences consisting of DRACH was present in the largest peak of mTOR and AKT1 (Figure S5G). This result may explain the structural context of the PI3K/AKT/mTOR pathway alteration by m6A methylation sites. The TMK1 cell genome m6A modification distribution landscape was showed in supplementary (Figure S5D-F).
m6A-seq capture of modified RNA in PI3K/Akt/mTOR pathway members. (A) Schematic diagram of the immunoprecipitation with high-throughput RNA sequencing. (B) Scatter plot of m6A Peak enrichment fold change in TMK1 cell. Within part of PI3K/AKT/ mTOR pathway genes, AKT1 mTOR, PTEN and PIK3CA show a significant m6A peak enrichment. (C) Representative gene transcripts harbouring m6A peaks in TMK1 cell. Black dashed lines signify CDS borders and flanking thin parts corresponding to UTRs. Uniquely, peak signal summarizing were divided into 100 bin for each regions. (D) Distribution of AKT1 mTOR, PTEN and PIK3CA-binding peaks across gene bodies. The signal density of m6A signaling was estimated on these gene bodies (between transcription start site, TSS and transcription termination site, TTS), as well as 3-kb upstream of TSS and 3-kb downstream of TTS regions. (E) Enrichment of m6A-binding peaks on CpG islands (CpGI). The signal density of m6A-binding peaks were estimated on CpG islands and 5-kb side regions.
Finally, we validated whether m6A deregulation causes alterations in the PI3K/Akt/mTOR signaling pathway in GI cancer by inhibiting the activity of the methyltransferase (METTL3-METTL14 complex) with S-adenosylhomocysteine (SAH) or by METTL3 siRNA, and by inhibiting the activity of demethylase (FTO) with meclofenamic acid (MA). m6A levels were deregulated in GI cancer cells under SAH or MA treatment (Figure S6A). The mRNA and protein levels of PTEN, AKT1, PIK3CA, and mTOR were significantly upregulated under SAH treatment in GI cancer cells, including gastric cancer cell TMK1, colorectal cancer cell line HCT116, esophageal cancer cell TE-1, pancreatic cancer cell PANC-1, liver cancer cell lines Huh-7 (Figure 7A-B, and S6B) or knocking down the expression levels of METTL3 in TMK1 and HCT116 cells (Figure 7C); Simultaneously, the expression levels were decreased under MA treatment in GI cancer cells (Figure 7A-B).
Validation of m6A regulation on PI3K/Akt/mTOR pathway. (A,B) mRNA expression level of m6A candidate targets AKT1, PTEN, PIK3CA, and mTOR after m6A manipulation in stomach cancer cell TMK1 and colon cancer cell HCT116. Expression mRNA levels and protein levels of target genes were measured by qPCR and by western blotting, respectively, after treatment with 1 μM of METTL3-METTL14 inhibitor S-adenosylhomocysteine (SAH) or m6A demethylase FTO inhibitor meclofenamic acid (MA) for 48 h. *P<0.05, **P<0.01, ***P<0.0001 compared with control. (C) m6A-related phosphorylation of PI3K/Akt/mTOR signaling effectors. Human phosphor-MAPK array was used to detect the phosphorylation of PI3K/Akt/mTOR signaling effectors in TMK1 cell. Cells were treated with culture media, 1µM SAH or 1µM MA for 48 h. (D) Quantification of protein phosphorylation for the phosphor-MAPK array. Integrated optical density was determined by densitometry analysis of the spots using Image-Pro Plus software.
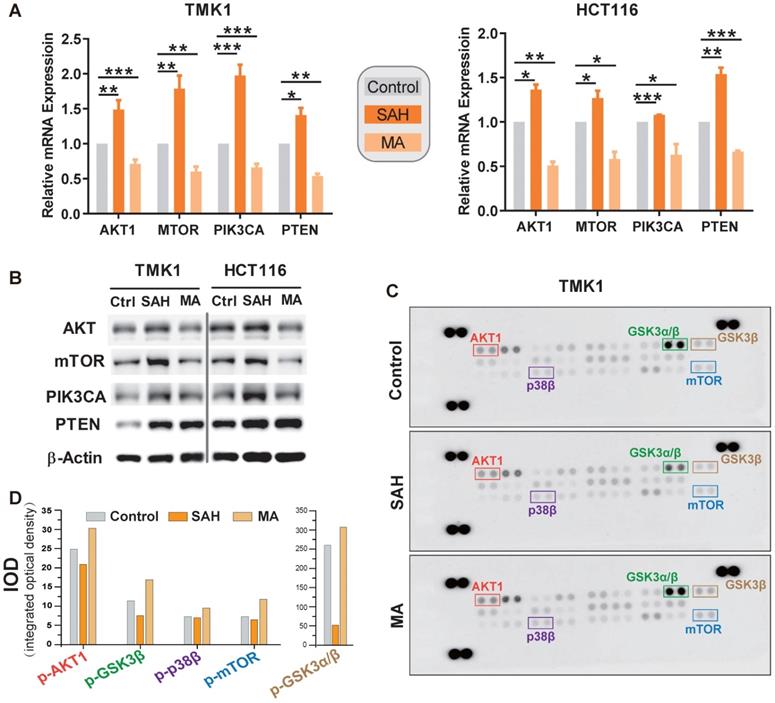
One possible explanation for the dynamic changes observed among the four genes could be compensatory feedback of m6A modification induced by methyltransferase or demethylase. Previous studies showed that the lifetime of mRNAs was elongated under reduced global m6A methylation condition [12, 42], to check whether m6A regulate the degradation of the components of PI3K/Akt/mTOR signaling pathway, after 48 h of SAH or MA treatment, TMK1 and HCT116 were treated with Act D, an inhibitor of gene transcription. We found that the lifetime of PTEN, AKT1, PIK3CA, and mTOR were significantly increased under SAH and ActD treatment, while they were significantly decreased under MA and ActD treatment (Figure S6D), indicating that m6A methylation regulates the expression levels of PTEN, AKT1, PIK3CA, and mTOR by inhibiting the degradation of these genes. To investigate the effect of m6A alteration on the aforementioned signaling pathway activation, we analyzed m6A-related phosphorylation of PI3K/Akt and mTOR signaling effectors using the phospho-MAPK array kit. In TMK1 cell, densitometry analysis of the signal demonstrated that phosphorylation of AKT1, GSK3α/β, p38β, and mTOR was clearly inhibited or activated after treatment with SAH and MA, respectively (Figure 7C-D).
Discussion
To date, several m6A modification mechanisms have been explored in cancer; however, the specific functions of the various regulators of m6A have yet to be fully identified and characterized in human cancer [22, 24, 25, 51]. In this study, we observed dysregulation of m6A methyltransferase, demethylase, and special binding protein expression in GI cancer (Figure 1B-C), indicating that m6A was potentially involved in GI cancer carcinogenesis. Due to the tumor heterogeneity and the limited clinical data, in our study, the METTL3 and ALKBH5 presented conflicting results in overall survival of stomach, liver and pancreatic cancer, further studies need to be performed to investigate the detailed mechanisms. We demonstrated that m6A regulators had high frequency copy-number amplification and other genetic alteration in GI cancer (Figure 1D). Moreover, we found significant connection between the abnormal expression of several m6A regulators and certain GI cancer survival (Figure 2). Up to now, there are only a few studies addressing the relationship of m6A regulator expression and survival. High expression of YTHDF1 and HNRNPA1 were associated with poor survival in liver cancer [52, 53]. High FTO expression was reported to be significantly associated with poor prognosis in gastric cancer [54], METTL3 downregulation acts as an adverse prognostic indicator in liver cancer similar to METTL14 [25]. These results were all consistent with our bioinformatics analysis result.
The m6A levels vary in different tissues and m6A regulators widely regulate cellular mRNAs and can contribute to pathway dysregulation, ultimately affecting human physiological processes [15]. A previous study demonstrated that m6A accelerates mRNA metabolism and translation to influence protein function [11]. In this study, protein enrichment in human cancers demonstrated that the alteration in m6A regulator expression influenced the expression of multiple proteins in GI cancer (Figure 3A, Table S1). Furthermore, we performed a comprehensive analysis of pathways influenced by m6A in 27 human cancers (Figure 3B). The results highlighted five crucial pathways (PI3K/Akt, mTOR, FoxO, MAPK, and p53) that may be affected by m6A and they were also cancer pathways in KEGG (Figure 3C-D). Of the enriched five crucial pathways (PI3K-Akt, mTOR, FoxO, MAPK, and p53 pathways), PI3K/Akt/mTOR closely influences the MAPK pathway function by ERK feedback activation [55, 56]. Activation or alteration of these pathways are closely involved in cell fate, tumorigenesis, cancer progress, and drug resistance in many cancer types, the therapies targeting these pathways are still investigational [57-63]. Of note, the PI3K/Akt and mTOR pathways were found universally altered in different cancers (Figure 3C). Our further analysis discovered that the above mentioned five pathways were frequently altered in cancers (Figure 4A-D). The crucial molecules PIK3CA, AKT1, PTEN, and mTOR in the PI3K/Akt and mTOR pathways were all frequently dysregulated in different types of cancer (Figure 4B-C).
We further carried out structural and functional investigation between m6A regulators and the five pathways by protein-protein interaction (PPI), genetic Phylogram tree, co-expression, and m6A sites prediction (Figure 4E, Figure S4A-C). Mechanistically, m6A regulators and PI3K-Akt pathway proteins possessed structural homology (Figure S4A). m6A was post-transcriptionally installed by “writer” (methyltransferase) within the DRA(m6A)CH (D = A, G or U; R = A or G; H = A, C or U) consensus motif and GA(m6A)C consensus motif [46, 64]. To gain mechanistic understanding of m6A function on the five pathways, we analyzed the m6A distribution in 57 genes mRNA and found that m6A modification sites were mostly enriched in the PI3K-Akt pathway and the mTOR pathway. AKT1/2/3, PI3KCA, PTEN, PI3KR1/2, BCL2, BCL2L1, IRS1, GSK3B, MYC, mTOR, AKT1S1, TSC1, RICTOR, RPTOR, and RPS6KB1 had very high confidence m6A modification sites (Figure 4E). Consistent with the result, the direct modification of m6A on MYC, BCL2 and PTEN has been reported in the literature [65, 66]. Taken together, our results demonstrated that the PI3K/Akt/mTOR pathway was most closely related to m6A in cancer and m6A modification sites were generally distributed in the PI3K/Akt and mTOR pathways. In accordance with our finding, several reports have revealed the influence of m6A regulators on the PI3K/Akt/mTOR pathway [51, 67-73], especially for FTO and HNRNPA2B1. However, our study demonstrates for the first time the comprehensive regulation of m6A regulators on PI3K/Akt/mTOR pathway members.
To further support of this mechanism, we next applied two independent bioinformatics tools, MeT-DB V2.0 [47] and m6A SNP [50], to validate the genes m6A modulated in the PI3K/Akt/mTOR pathway in tumors. Since the function of m6A in eukaryotes involves mRNA degradation [48], we first investigated the effect of METTL3 knockdown in human cancer cells (U2SO, Hela, and A549) and HEK293T cells based on RNA Sequencing analysis (Figure 5A, Figure S4C). Results showed that gene expression in the PI3K/Akt/mTOR pathways was generally upregulated after METTL3 knockdown, suggesting METTL3 inhibited their expression. Furthermore, the expression upregulation after METTL3 knockdown was obviously higher in tumor cell lines than in HEK293T. Second, to further confirm the relationship between these genes and m6A modification, we employed m6A SNP analysis. More than 55,548 human m6A sites were referenced for prediction of genetic variants with m6A modification sites [35, 36]. In the PI3K-Akt pathway, the potential variants that may influence m6A modification on AKT1, PTEN, and PIK3CA were mapped on core AC motif (DRACH motif) (Figure 5C, F, I). Noteworthy, the C→T transition at reliable m6A position in PTEN was in accordance with previous m6A variants study [35]. The results of detailed m6ASNP within AKT1, PTEN, and PIK3CA in the PI3K-Akt pathway are shown in genomic regions (Figure 5D, G, J). Such variants may disrupt m6A modification and cause disease, including cancer.
With the epigenetic regulation is an important mechanism to m6A modification in mRNA [74]. It is vital to clarify whether it also plays dual roles in PI3K/AKT/mTOR pathway dysregulation. Our m6A sequencing data demonstrated that m6A modification are wildly present in TMK1 cells, like AKT1, mTOR, PIK3CA and PTEN m6A binding peak enrichment. Moreover, the distribution of m6A in these vital pathway genes provides hints as to its functions in expression regulation (Figure 6C). Broadly, the appearance of mRNA methylation in translational regulation might be interwoven with other function of m6A in RNA biology, such as RNA status and lifetime [48]. We further validated modulation of m6A on target genes by in vitro study using methyltransferase and demethylase selective inhibitors (Figure 7A-B). The m6A methyltransferase inhibitor SAH [75] elevated the expression of AKT1, PTEN, mTOR, and PIK3CA in stomach and colon cancer cell lines. At the same time, demethylase inhibitor MA decreased their expression [76]. In addition, we employed low-scale semiquantitative phosphoproteomic analysis of the PI3K-Akt and mTOR signal pathway critical members to analyze protein phosphorylation in the presence or absence of m6A modification. The intracellular kinases activity of AKT1, GSK3B, p38, and mTOR inhibited by the methyltransferase inhibitor SAH and promoted by demethylase inhibitor MA (Figure 7C-D). Our current finding demonstrated for the first time that m6A modification directly modulate PI3K/Akt and mTOR signal pathway activity by regulating critical kinases in GI cancer.
In summary, we present here a comprehensive study of the major target of m6A modification in GI cancer, especially its influence on signal pathways. The findings from the current study reveal the critical influence of m6A in GI cancer and contribute to the knowledge of this prevalently existed mRNA regulation mechanism in cancer. This pioneering work provides potential avenues to further investigate the underlying mechanism of m6A modification in human cancer.
Abbreviations
m6A: Methylation at the N6 position of Adenosine; GI: Gastrointestinal; TCGA: The Cancer Genome Atlas; GEO: Gene Expression Omnibus; SNPs: Single Nucleotide Polymorphisms; PI3K: Phosphatidylinositol-3-kinase; mTOR: Mammalian Target of Rapamycin; METTL: Methyltransferase Like; WTAP: Wilms Tumor 1 Associated Protein; YTHDF: YTH N6-methyladenosine RNA Binding Protein; ALKBH5: AlkB Homolog 5; FTO: Fat mass and Obesity-associated protein; MeRIP-Seq: Methylated RNA Immunoprecipitation Sequencing; OS: Overall Survival; FoxO: Forkhead Box Transcription Factors; MAPK: Mitogen-activated Protein Kinase; UTR: Untranslated Region; CDS: Coding Sequence; TSS: Transcription Start Sites; TTS: Transcription Termination Sites; SAH: S-adenosylhomocysteine; MA: Meclofenamic Acid; PPI: Protein-Protein Interaction; FPKM: Per Kilobase of Transcript Per Million Fragments mapped; RPPA: Reverse Phase Protein Array; FDR: False Discovery Rate; NJ: Neighbour-Joining; KNN: K-Nearest Neighbor; VCF: Variant Call Format; qPCR: Quantitative Real-time PCR; KEGG: Kyoto Encyclopedia of Genes and Genomes.
Supplementary Material
Supplementary materials and methods, figures, and tables.
Acknowledgements
We thank the supports of the South Sichuan Institute of Translational Medicine and Southwest Medical University.
Author Contributions
ZX, JS and CZ conceived and designed the study. QZ, YZ, WH, YZ carry out the experiments and methodology. XW, JL, QJZ, YSZ, WH, WL, WQW, JHW and FKD contributed to analysis and interpretation of data. HJJ, XY, ZYX and LW provide administrative, technical, or material support. QLW, XL and CHC supervised the research. All authors write, read, and approved the final manuscript.
Financial support
This work was supported by the National Natural Science Foundation of China (Grant Nos. 81503093, 81602166, and 81672444), the Joint Funds of the Southwest Medical University & Luzhou (2016LZXNYD-T01, 2017LZXNYD-Z05, 2017LZXNYD-J09, and 2019LZXNYD-J45).
Availability of data and materials
All data generated or analyzed during this study are available from the corresponding author upon reasonable request.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Molinie B, Giallourakis CC. Genome-Wide Location Analyses of N6-Methyladenosine Modifications (m(6)A-Seq). Methods Mol Biol. 2017;1562:45-53
2. Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A. 1974;71:3971-5
3. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511-5
4. Sanchez-Diaz P, Penalva LO. Post-transcription meets post-genomic: the saga of RNA binding proteins in a new era. RNA Biol. 2006;3:101-9
5. Tang C, Klukovich R, Peng H, Wang Z, Yu T, Zhang Y. et al. ALKBH5-dependent m6A demethylation controls splicing and stability of long 3'-UTR mRNAs in male germ cells. Proc Natl Acad Sci U S A. 2018;115:E325-e33
6. Chen Z, Wu L, Zhou J, Lin X, Peng Y, Ge L. et al. -methyladenosine-induced ERRγ triggers chemoresistance of cancer cells through upregulation of ABCB1 and metabolic reprogramming. Theranostics. 2020;10:3382-96
7. He C. Grand challenge commentary: RNA epigenetics? Nat Chem Biol. 2010;6:863-5
8. Nguyen S, Meletis K, Fu D, Jhaveri S, Jaenisch R. Ablation of de novo DNA methyltransferase Dnmt3a in the nervous system leads to neuromuscular defects and shortened lifespan. Dev Dyn. 2007;236:1663-76
9. Pereira JD, Sansom SN, Smith J, Dobenecker MW, Tarakhovsky A, Livesey FJ. Ezh2, the histone methyltransferase of PRC2, regulates the balance between self-renewal and differentiation in the cerebral cortex. Proc Natl Acad Sci U S A. 2010;107:15957-62
10. Harper JE, Miceli SM, Roberts RJ, Manley JL. Sequence specificity of the human mRNA N6-adenosine methylase in vitro. Nucleic Acids Res. 1990;18:5735-41
11. Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2017;18:31-42
12. Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L. et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93-5
13. Li X-C, Jin F, Wang B-Y, Yin X-J, Hong W, Tian F-J. The m6A demethylase ALKBH5 controls trophoblast invasion at the maternal-fetal interface by regulating the stability of mRNA. Theranostics. 2019;9:3853-65
14. Alarcon CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 Is a Mediator of m(6)A-Dependent Nuclear RNA Processing Events. Cell. 2015;162:1299-308
15. Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell. 2012;149:1635-46
16. Wang P, Doxtader KA, Nam Y. Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol Cell. 2016;63:306-17
17. Horiuchi K, Kawamura T, Iwanari H, Ohashi R, Naito M, Kodama T. et al. Identification of Wilms' tumor 1-associating protein complex and its role in alternative splicing and the cell cycle. J Biol Chem. 2013;288:33292-302
18. Gowda RM, Dogan OM, Tejani FH, Khan IA. Left main coronary artery aneurysm. Int J Cardiol. 2005;105:115-6
19. Ben-Haim MS, Moshitch-Moshkovitz S, Rechavi G. FTO: linking m6A demethylation to adipogenesis. Cell Res. 2015;25:3-4
20. Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518:560-4
21. Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S. et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201-6
22. Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G. et al. m(6)A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep. 2017;18:2622-34
23. Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z. et al. m(6)A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell. 2017;31:591-606.e6
24. Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m(6)A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol Cell. 2016;62:335-45
25. Ma JZ, Yang F, Zhou CC, Liu F, Yuan JH, Wang F. et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6) -methyladenosine-dependent primary MicroRNA processing. Hepatology. 2017;65:529-43
26. Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I. et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m(6)A-demethylation of NANOG mRNA. Proc Natl Acad Sci U S A. 2016;113:E2047-56
27. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7-30
28. Liu Y, Sethi NS, Hinoue T, Schneider BG, Cherniack AD, Sanchez-Vega F. et al. Comparative Molecular Analysis of Gastrointestinal Adenocarcinomas. Cancer Cell. 2018;33:721-35.e8
29. Xu H, Wang H, Zhao W, Fu S, Li Y, Ni W. et al. SUMO1 modification of methyltransferase-like 3 promotes tumor progression via regulating Snail mRNA homeostasis in hepatocellular carcinoma. Theranostics. 2020;10:5671-86
30. Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. Lancet. 2016;388:73-85
31. Wang S, Sun C, Li J, Zhang E, Ma Z, Xu W. et al. Roles of RNA methylation by means of N(6)-methyladenosine (m(6)A) in human cancers. Cancer Lett. 2017;408:112-20
32. Li J, Zhao W, Akbani R, Liu W, Ju Z, Ling S. et al. Characterization of Human Cancer Cell Lines by Reverse-phase Protein Arrays. Cancer Cell. 2017;31:225-39
33. Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J. et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447-52
34. Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J. et al. STRING 8-a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37:D412-6
35. Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12:767-72
36. Ke S, Alemu EA, Mertens C, Gantman EC, Fak JJ, Mele A. et al. A majority of m6A residues are in the last exons, allowing the potential for 3' UTR regulation. Genes Dev. 2015;29:2037-53
37. Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res. 2009;19:1553-61
38. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P. et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248-9
39. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073-81
40. Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ. et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013;34:57-65
41. Dominissini D, Moshitch-Moshkovitz S, Salmon-Divon M, Amariglio N, Rechavi G. Transcriptome-wide mapping of N(6)-methyladenosine by m(6)A-seq based on immunocapturing and massively parallel sequencing. Nat Protoc. 2013;8:176-89
42. Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D. et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117-20
43. Yue Y, Liu J, He C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 2015;29:1343-55
44. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401-4
45. Marcotte EM, Pellegrini M, Thompson MJ, Yeates TO, Eisenberg D. A combined algorithm for genome-wide prediction of protein function. Nature. 1999;402:83-6
46. Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H. et al. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell. 2015;161:1388-99
47. Liu H, Wang H, Wei Z, Zhang S, Hua G, Zhang SW. et al. MeT-DB V2.0: elucidating context-specific functions of N6-methyl-adenosine methyltranscriptome. Nucleic Acids Res. 2018;46:D281-d7
48. Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D. et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117-20
49. Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA Modifications in Gene Expression Regulation. Cell. 2017;169:1187-200
50. Jiang S, Xie Y, He Z, Zhang Y, Zhao Y, Chen L. et al. m6ASNP: a tool for annotating genetic variants by m6A function. Gigascience. 2018 7
51. Jiao Y, Zhang J, Lu L, Xu J, Qin L. The Fto Gene Regulates the Proliferation and Differentiation of Pre-Adipocytes in Vitro. Nutrients. 2016;8:102
52. Zhao X, Chen Y, Mao Q, Jiang X, Jiang W, Chen J. et al. Overexpression of YTHDF1 is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Biomark. 2018;21:859-68
53. Zhou ZJ, Dai Z, Zhou SL, Fu XT, Zhao YM, Shi YH. et al. Overexpression of HnRNP A1 promotes tumor invasion through regulating CD44v6 and indicates poor prognosis for hepatocellular carcinoma. Int J Cancer. 2013;132:1080-9
54. Xu D, Shao W, Jiang Y, Wang X, Liu Y, Liu X. FTO expression is associated with the occurrence of gastric cancer and prognosis. Oncol Rep. 2017;38:2285-92
55. Polivka J Jr, Janku F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol Ther. 2014;142:164-75
56. Briest F, Grabowski P. PI3K-AKT-mTOR-signaling and beyond: the complex network in gastroenteropancreatic neuroendocrine neoplasms. Theranostics. 2014;4:336-65
57. Mayer IA, Arteaga CL. The PI3K/AKT Pathway as a Target for Cancer Treatment. Annu Rev Med. 2016;67:11-28
58. Matter MS, Decaens T, Andersen JB, Thorgeirsson SS. Targeting the mTOR pathway in hepatocellular carcinoma: current state and future trends. J Hepatol. 2014;60:855-65
59. Boreddy SR, Pramanik KC, Srivastava SK. Pancreatic tumor suppression by benzyl isothiocyanate is associated with inhibition of PI3K/AKT/FOXO pathway. Clin Cancer Res. 2011;17:1784-95
60. Song G, Xu S, Zhang H, Wang Y, Xiao C, Jiang T. et al. TIMP1 is a prognostic marker for the progression and metastasis of colon cancer through FAK-PI3K/AKT and MAPK pathway. J Exp Clin Cancer Res. 2016;35:148
61. Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov. 2014;13:217-36
62. Mangé A, Coyaud E, Desmetz C, Laurent E, Béganton B, Coopman P. et al. FKBP4 connects mTORC2 and PI3K to activate the PDK1/Akt-dependent cell proliferation signaling in breast cancer. Theranostics. 2019;9:7003-15
63. Li X, Chen W, Li P, Wei J, Cheng Y, Liu P. et al. Follicular Stimulating Hormone Accelerates Atherogenesis by Increasing Endothelial VCAM-1 Expression. Theranostics. 2017;7:4671-88
64. Wei CM, Moss B. Nucleotide sequences at the N6-methyladenosine sites of HeLa cell messenger ribonucleic acid. Biochemistry. 1977;16:1672-6
65. Wang XS, He JR, Yu S, Yu J. [Methyltransferase-like 3 Promotes the Proliferation of Acute Myeloid Leukemia Cells by Regulating N6-methyladenosine Levels of MYC]. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2018;40:308-14
66. Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G. et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. 2017;23:1369-76
67. Gulati P, Cheung MK, Antrobus R, Church CD, Harding HP, Tung YC. et al. Role for the obesity-related FTO gene in the cellular sensing of amino acids. Proc Natl Acad Sci U S A. 2013;110:2557-62
68. Zhang Z, Zhou D, Lai Y, Liu Y, Tao X, Wang Q. et al. Estrogen induces endometrial cancer cell proliferation and invasion by regulating the fat mass and obesity-associated gene via PI3K/AKT and MAPK signaling pathways. Cancer Lett. 2012;319:89-97
69. Choi HS, Lee HM, Jang YJ, Kim CH, Ryu CJ. Heterogeneous nuclear ribonucleoprotein A2/B1 regulates the self-renewal and pluripotency of human embryonic stem cells via the control of the G1/S transition. Stem Cells. 2013;31:2647-58
70. Shi X, Ran L, Liu Y, Zhong SH, Zhou PP, Liao MX. et al. Knockdown of hnRNP A2/B1 inhibits cell proliferation, invasion and cell cycle triggering apoptosis in cervical cancer via PI3K/AKT signaling pathway. Oncol Rep. 2018;39:939-50
71. Wang X, Huang N, Yang M, Wei D, Tai H, Han X. et al. FTO is required for myogenesis by positively regulating mTOR-PGC-1alpha pathway-mediated mitochondria biogenesis. Cell Death Dis. 2017;8:e2702
72. Li X, Tang J, Huang W, Wang F, Li P, Qin C. et al. The M6A methyltransferase METTL3: acting as a tumor suppressor in renal cell carcinoma. Oncotarget. 2017;8:96103-16
73. Liu Y, Wang R, Zhang L, Li J, Lou K, Shi B. The lipid metabolism gene FTO influences breast cancer cell energy metabolism via the PI3K/AKT signaling pathway. Oncol Lett. 2017;13:4685-90
74. Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ. et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18-29
75. Li F, Kennedy S, Hajian T, Gibson E, Seitova A, Xu C. et al. A Radioactivity-Based Assay for Screening Human m6A-RNA Methyltransferase, METTL3-METTL14 Complex, and Demethylase ALKBH5. J Biomol Screen. 2016;21:290-7
76. Huang Y, Yan J, Li Q, Li J, Gong S, Zhou H. et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015;43:373-84
Author contact
![]() Corresponding authors: Zhangang Xiao, Laboratory of Molecular Pharmacology, Department of Pharmacology, School of Pharmacy, Southwest Medical University, Luzhou, 646000, Sichuan, PR China; E-mail: zhangangxiaoedu.cn; xzg555898com. Jing Shen, Laboratory of Molecular Pharmacology, Department of Pharmacology, School of Pharmacy, Southwest Medical University, Luzhou, 646000, Sichuan, PR China; E-mail: crystal_straycom. Chang Zou, Clinical Medical Research Center, the Second Clinical Medical College of Jinan University, The First Affiliated Hospital of Southern University, Shenzhen People's Hospital, Shenzhen, Guangdong 518020, P.R. China; E-mail: zouchang.cuhkcom; Fax: (86)755 22942850.
Corresponding authors: Zhangang Xiao, Laboratory of Molecular Pharmacology, Department of Pharmacology, School of Pharmacy, Southwest Medical University, Luzhou, 646000, Sichuan, PR China; E-mail: zhangangxiaoedu.cn; xzg555898com. Jing Shen, Laboratory of Molecular Pharmacology, Department of Pharmacology, School of Pharmacy, Southwest Medical University, Luzhou, 646000, Sichuan, PR China; E-mail: crystal_straycom. Chang Zou, Clinical Medical Research Center, the Second Clinical Medical College of Jinan University, The First Affiliated Hospital of Southern University, Shenzhen People's Hospital, Shenzhen, Guangdong 518020, P.R. China; E-mail: zouchang.cuhkcom; Fax: (86)755 22942850.