Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(19):8606-8618. doi:10.7150/thno.46861 This issue Cite
Research Paper
Fructose-1, 6-bisphosphatase 1 interacts with NF-κB p65 to regulate breast tumorigenesis via PIM2 induced phosphorylation
Chao Lu1*, Chune Ren1*, Tingting Yang1*, Yonghong Sun2*, Pengyun Qiao1, Xue Han1, Zhenhai Yu1 ![]()
1. Department of Reproductive Medicine, Affiliated Hospital of Weifang Medical University, Weifang, Shandong Province, P.R. China.
2. Department of Pathology, Affiliated Hospital of Weifang Medical University, Weifang, Shandong Province, P.R. China.
*These authors contributed equally to this work.
Received 2020-4-9; Accepted 2020-6-24; Published 2020-7-9
Abstract

Rationale: Fructose-1, 6-bisphosphatase 1 (FBP1), a rate-limiting enzyme in gluconeogenesis, was recently shown to be a tumor suppressor and could mediate the activities of multiple transcriptional factors via its non-canonical functions. However, the underlying mechanism of posttranscriptional modification on the non-canonical functions of FBP1 remains elusive.
Methods: We employed immunoaffinity purification to identify binding partner(s) and used co-immunoprecipitation to verify their interactions. Kinase reaction was used to confirm PIM2 could phosphorylate FBP1. Overexpression or knockdown proteins were used to assess the role in modulating p65 protein stability. Mechanistic analysis was involved in protein degradation and polyubiquitination assays. Nude mice and PIM2-knockout mice was used to study protein functions in vitro and in vivo.
Results: Here, we identified Proviral Insertion in Murine Lymphomas 2 (PIM2) as a new binding partner of FBP1, which could phosphorylate FBP1 on Ser144. Surprisingly, phosphorylated FBP1 Ser144 abrogated its interaction with NF-κB p65, promoting its protein stability through the CHIP-mediated proteasome pathway. Furthermore, phosphorylation of FBP1 on Ser144 increased p65 regulated PD-L1 expression. As a result, phosphorylation of FBP1 on Ser144 promoted breast tumor growth in vitro and in vivo. Moreover, the levels of PIM2 and pSer144-FBP1 proteins were positively correlated with each other in human breast cancer and PIM2 knockout mice.
Conclusions: Our findings revealed that phosphorylation noncanonical FBP1 by PIM2 was a novel regulator of NF-κB pathway, and highlights PIM2 inhibitors as breast cancer therapeutics.
Keywords: PIM2, FBP1, phosphorylation, protein stability, tumor growth
Introduction
Fructose-1, 6-biphosphatase 1 (FBP1) is a rate-limiting enzyme in gluconeogenesis, and functions as a negative regulator of the Warburg effect [1]. As a tumor suppressor, FBP1 plays a crucial role in tumor progression in multiple cancers [2]. Low expression of FBP1 is associated with tumorigenesis and poor prognosis in patients with breast, kidney, colon, pancreas, lung, stomach and liver cancers [3-8]. In addition to its function as a metabolic enzyme, it also acts as a co-suppressor for multiple transcription factors to reduce downstream gene expression. For example, FBP1 can directly bind to hypoxia inducible factor 1α (HIF-1α), and regulates its transcriptional activity to oppose renal carcinoma progression [5]. FBP1 high-regulation inhibits the activity of the WNT/β-catenin pathway and reduces the level of its downstream target genes [9]. Moreover, FBP1 can directly destabilize c-MYC by disrupting the ERK-c-MYC axis, an action that has been shown to increase the sensitivity of pancreatic cancer cells to JQ1 [10]. Our previous study showed that FBP1 binds to NOTCH1 and enhances its ubiquitination, further leading to proteasomal degradation via the FBXW7 pathway [11]. However, the underlying mechanisms of post-transcriptional modification for the non-canonical function of FBP1 remain elusive in breast cancer.
The PIM2 kinase belongs to a serine/threonine kinase family of three members (PIM1, PIM2 and PIM3), and was first identified as a proviral integration site for Moloney murine leukemia virus 2-induced T-cell lymphoma [12]. These three kinases participate in several tumor progression factors, including cell cycle, survival, proliferation, migration, apoptosis, metabolism, and drug resistance [12]. Unlike PIM1 and PIM3, PIM2 can be constitutively activated because it lacks a regulatory domain, and thus could be used to design drug targets [13]. The function of PIM2 in cancer depends on its serine/threonine kinase activity, which can phosphorylate multiple substrates including p21, p27, NOTCH1, p65, BAD, AMPKα1, TSC2, PKM2, c-MYC, HK2 and HSF1 [12, 14-18]. Moreover, various special inhibitors, including JP11646, SMI-4a and SGI-1776, have been developed for PIM kinase activity and have been used for clinical treatment [19-21]. Recently, we found that PIM2 acts as an oncogene in breast cancer [15, 17, 22], but the underlying mechanism of its oncogene function remains largely unknown.
FBP1 has recently emerged as a broad co-repressor for multiple transcriptional factors via its non-canonical functions. In particular, we found that FBP1 inhibits NOTCH1 function via FBXW7-induced protein degradation [11]. In this study, we uncovered that PIM2 can phosphorylate FBP1 at the Ser144 site, and decrease FBP1 binding to p65 independent of its enzyme activity. FBP1 phosphorylation by PIM2 promoted breast tumor growth and p65-induced PD-L1 expression, highlighting the role of PIM2-dependent FBP1 phosphorylation in breast tumor progression.
Materials and Methods
Cell culture
HEK293T, MCF-7 and MB231 cells were cultured in DMEM supplemented with 10% FBS. All cells were cultured at 37℃ supplied with 5% CO2.
RNA interference
shRNAs were constructed into pLVX-shRNA1 vector. Viral packaging plasmids (pMD2.G and psPAX2) and shRNA plasmid were transfected to 293T cells by using lipofectamine 2000. After 24hr, virus culture medium was replaced with new DMEM containing 10% FBS. 48hr post transfection, the medium was collected and added to breast cancer cells added with polybrene. Breast cancer cells were harvested 48hr after puromycin selection. shRNA sequence information was provided in Table S2.
Immunoprecipitation (IP) and GST pull-down assays
Cells were harvested and lysed with IP buffer (50mM Tris-HCl, pH 7.5, 150mM NaCl, and 0.5% NP40) with multiple protease inhibitors (Sigma-Aldrich). On ice for more than 30min and the lysate was centrifuged at 12000 rpm at 4℃ for 10 min. The supernatant was rocked overnight with protein A/G agarose beads and indicated antibodies under 4℃. The beads were washed at least 5 times using IP buffer, and then used for subsequent experiments. The indicated proteins were expressed in E.coli BL21 (DE3), and GST-pull down assay was performed as described previously [17].
Phosphorylation assay
The kinase reaction buffer was performed as described previously [15, 16, 18]. The reactions were subjected to Western blotting analysis.
Putting back stable cell lines
To generate rescue stable cell pools, HA-tagged FBP1 (WT, S144A or S144D) was cloned into the lentiviral pLVX-IRES-Neo vectors and co-transfected with pMD2.G and psPAX2 package vectors in HEK293T cells to produce lentiviruses, The breast cancer cells with stable FBP1 knockdown were then infected, following a selection with G418 for 2 weeks. The single stable cells were selected by reseeded into 96-well plates.
Xenograft mouse model
The female 4 week old BALB/c nude mice were injected subcutaneously with 5×106/100μL PBS FBP1 (WT, S144A or S144D) stable expression MCF-7 cells. Tumor volume was measured during the tumor growth for 3 weeks. Tumor volume was calculated according to the following formula: Tumor volume = (length×width2)/2. After three weeks, the mice were killed, and tumors were weighed. Finally, the tumor tissues were harvested, embedded, fixed, and prepared for H&E and IHC staining. Animal experiments were performed in strict accordance with the protocols approved by the Institutional Animal Care and Use Committee of Weifang Medical University.
Breast cancer patient samples
The details of patient tissues samples were shown in Table S4. All experiments involving human participants were approved by the Review Board of the Affiliated Hospital of Weifang Medical University. The slides of tissues were prepared by Affiliated Hospital of Weifang Medical University.
Statistical analysis
All statistical analyses were determined using the SPSS version 17.0 (SPSS Inc., Chicago, IL, USA). Quantitative data were presented as means ± SD. Statistical significance of Student's t-test was used for two-group comparisons. Statistical significance was displayed as *P < 0.05, and n.s. was not significant.
Other materials and methods were shown in Supplementary Data.
Results
PIM2 interacts with FBP1 and phosphorylates it at Ser144
We recently used PIM2 as bait to identify FBP1 as a new binding partner [15]. PIM2 as an oncogene played an important role in breast tumorigenesis, but the underlying mechanism of its oncogene function remains elusive. The interaction between PIM2 and FBP1 was determined by co-immunoprecipitation (Co-IP) assay (Figure 1A-1D). In addition, a GST-pull down assay suggested that PIM2 directly interacted with FBP1 (Figure 1E). Furthermore, we used immunofluorescent staining to determine endogenous PIM2 was colocalized with endogenous FBP1 primarily in the nucleus but also slightly in the cytoplasm (Figure 1F). To determine which domains of PIM2 and FBP1 are responsible for regulating this interaction, truncated constructs of the functional domains of PIM2 and FBP1 were made for further analysis (Figure 1G and 1I) [5, 22]. Co-IP analyses suggested that the kinase domain of PIM2 (33-286aa) was associated with FBP1 (Figure 1H). Moreover, PIM2 demonstrated strong binding to FBP1 domains (E1, E3, E4, E5, and E6), whereas other mutants did not bind to PIM2 (Figure 1J). Taken together, these data indicate that PIM2 physically interacts with FBP1.
PIM2 interacts with FBP1 and phosphorylates it at Ser144. Immunoprecipitation and immunoblotting analyses were performed with the indicated antibodies. (A, B) 293T cells were transfected with indicated plasmids, followed by IP with anti-HA (a) or Flag (b) and IB with indicated antibodies. (C, D) MCF-7 cells were collected, followed by IP with anti-FBP1 (c) or PIM2 (d) and IB with indicated antibodies. (E) Purified GST-tagged FBP1 or GST was mixed with His-PIM2 for GST pull-down assay. (F) Confocal immunofluorescence microscopy was performed to analyze localization of PIM2 and FBP1 in MCF-7 cells. (G) The PIM2 truncation mutants used in this study. (H) 293T cells were overexpressed the indicated HA-tagged FBP1 and GFP-tagged PIM2 fragments proteins. Immunoprecipitation with an anti-HA antibody was performed. (I) The FBP1 truncation mutants used in this study. (J) 293T cells were overexpressed the indicated HA-tagged PIM2 and GFP-tagged FBP1 fragments proteins. Immunoprecipitation with an anti-HA antibody was performed. (K) 293T cells were overexpressed the indicated HA-tagged FBP1 and Flag-tagged PIM2 (WT or K61A) proteins. Immunoprecipitation with an anti-HA antibody was performed. (L, M) 293T cells were overexpressed the indicated HA-tagged FBP1 (S144A or WT) and Flag-tagged PIM2 (WT or K61A) proteins. Immunoprecipitation with an anti-HA antibody was performed. (N, O) Purified GST-tagged FBP1 (WT or S144A) was mixed with the indicated bacterially purified His-tagged PIM2 proteins. An in vitro kinase assay was performed.
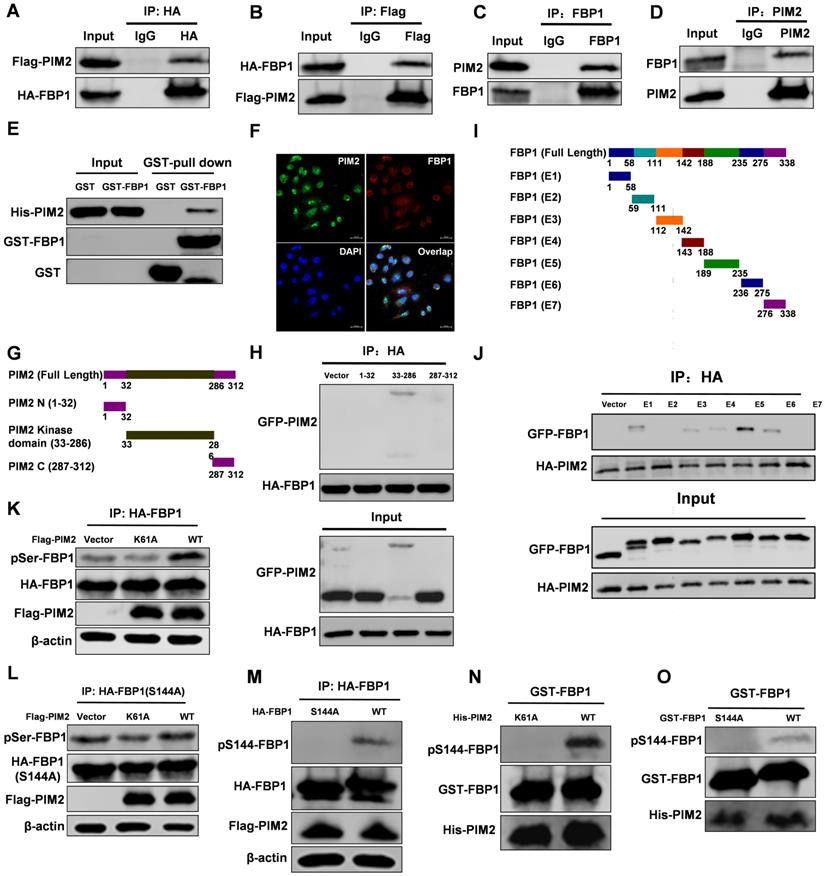
As a serine/threonine kinase, PIM2 mediates tumor progression via the phosphorylation and activation of a variety of its substrate proteins [12]. Thus, we evaluated whether PIM2 could phosphorylate FBP1. Wild-type PIM2 increased FBP1 serine phosphorylation levels compared with the control vector or kinase-inactive (Figure 1K) but failed to promote threonine phosphorylation levels (Figure S1A). Interestingly, we found potential PIM substrate motifs in FBP1 (Figure S1B). Moreover, phosphorylation of FBP1 at Ser144 was identified by proteomic analyses according to the protein post-translational modifications database (https://www.phosphosite.org/) [23], which was consistent with our speculation. As we expected, mutant FBP1 S144A abrogated the effect on PIM2-induced serine phosphorylation (Figure 1L). Furthermore, we produced an antibody that specifically recognizes FBP1 Ser144 phosphorylation and verified its validity via immunohistochemistry assays in breast cells and tissue samples (Figure S1C and S1D). We then used this FBP1-phosphorylation antibody and determined that PIM2 has no effect on the FBP1 S144A mutant (Figure 1M). Moreover, an in vitro kinase assay demonstrated that PIM2 directly phosphorylated FBP1 at Ser144 (Figure 1N and 1O). However, PIM1 and PIM3 had no effect on FBP1 Ser144 phosphorylation (Figure S1E). To further test whether this phosphorylation also happens in breast cancer cells, we used PIM inhibitor-SMI-4a. SMI-4a abrogated the effects on FBP1 Ser144 phosphorylation (Figure S1F). Taken together, our results provide convincing evidence that PIM2 is a direct kinase for FBP1.
FBP1 Ser144 phosphorylation regulates binding to p65
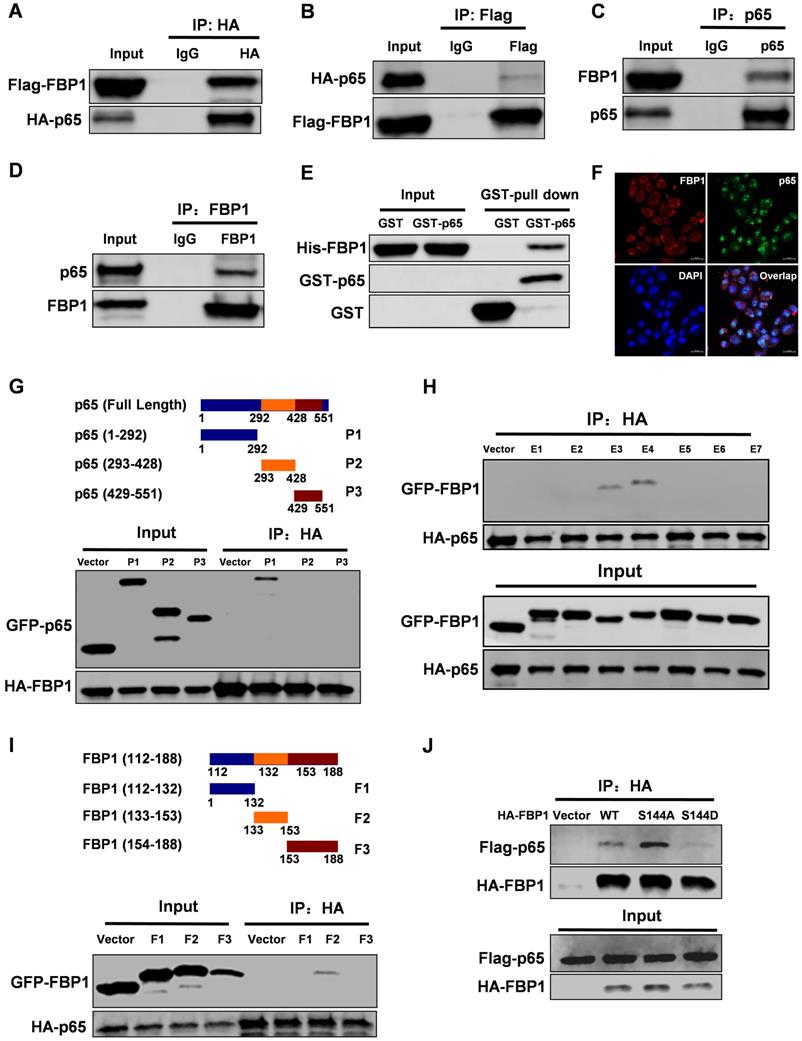
Interestingly, PIM2 failed to affect the enzymatic activity of FBP1 (Figure S2A). To further investigate the FBP1 non-canonical functions that are regulated by Ser144 phosphorylation, we used FBP1 as bait to screen for interaction partners. Interestingly, we found that NF-κB p65 was a new binding partner of FBP1. To further confirm their interaction, we performed a co-IP assay. The data showed that FBP1 could bind to p65 in breast cancer (Figure 2A-2D). Moreover, the enzymatic activity of FBP1 was dispensable for their interaction (Figure S2B). We next evaluated the binding with other NF-κB family proteins and found that FBP1 could interact with p50, but not RELB (Figure S2C and S2D). Furthermore, in vitro experiments demonstrated that FBP1 direct binding to p65 was independent of other proteins (Figure 2E). Immunofluorescence assays showed the co-localization of FBP1 and p65 in MCF-7 cells (Figure 2F). To determine which domains of FBP1 and p65 are responsible for regulating their interaction, truncated constructs of FBP1 and p65 were constructed according to their functional domains (Figure 1G and 2G). Co-IP analyses suggested that the DNA-binding domain of p65 (1-292aa) was associated with FBP1 (Figure 2G). Moreover, p65 exhibited strong binding to the domains of FBP1 (E3 and E4), whereas other mutants could not bind to p65 (Figure 2H). Furthermore, we found that the FBP1 (F2) domain containing the PIM2 phosphorylation site interacted with p65 (Figure 2I). Next, we tested whether this phosphorylation contributed to their interaction. As we expected, phosphorylation of the PIM2 site abrogated FBP1 interaction with p65 (Figure 2J). Thus, these data demonstrate that PIM2 phosphorylation of FBP1 inhibits its interaction with p65.
FBP1 Ser144 phosphorylation regulates binding to p65. Immunoprecipitation and immunoblotting analyses were performed with the indicated antibodies. (A, B) 293T cells were transfected with indicated plasmids, followed by IP with anti-HA (a) or Flag (b) and IB with indicated antibodies. (C, D) MCF-7 cells were collected, followed by IP with anti-p65 (c) or FBP1 (d) and IB with indicated antibodies. (E) Purified GST-tagged p65 or GST was mixed with His-FBP1 for GST pull-down assay. (F) Confocal immunofluorescence microscopy was performed to analyze localization of FBP1 and p65 in MCF-7 cells. (G) The P65 truncation mutants used in this study. 293T cells were overexpressed the indicated HA-tagged FBP1 and GFP-tagged p65 fragments proteins. Immunoprecipitation with an anti-HA antibody was performed. (H, I) 293T cells were overexpressed the indicated HA-tagged p65 and GFP-tagged FBP1 fragments proteins. Immunoprecipitation with an anti-HA antibody was performed. (J) 293T cells were overexpressed the indicated HA-tagged FBP1 (WT, S144A or S144D) and Flag-tagged p65 proteins. Immunoprecipitation with an anti-HA antibody was performed.
FBP1 Ser144 phosphorylation regulates p65 stability via the CHIP-mediated ubiquitin proteasome pathway
Previous studies found that FBP1 regulated the protein stability of some transcriptional factors [10, 11]. To determine whether FBP1 regulates p65 protein stability, we examined the effect of FBP1 manipulation on p65 protein levels. We found that FBP1 decreases p65 protein levels in a dose-dependent manner (Figure 3A). Moreover, mutant FBP1 G260R had an inhibitory effect on p65 protein levels similar to that of wild-type FBP1, suggesting that its enzymatic activity was not required for this regulation (Figure 3B). Because our previous study demonstrated that FBP1 protein expression was higher in MCF-7 cells than in MB231 cells, MCF-7 cells were used for knockdown, and MB231 cells were used for overexpression (Figure S3A) [11]. FBP1 knockdown efficiency was validated in MCF-7 cells (Figure S3B), and the data showed that FBP1 negatively mediated endogenous p65 protein levels in breast cancer cells (Figure 3C and 3D). To further determine whether FBP1 regulates p65 stability, we next examined the protein half-life of p65 in response to the manipulation of FBP1 levels in the presence of cycloheximide to inhibit new protein synthesis. FBP1 knockdown significantly increased the protein half-live of p65 (Figure 3E), whereas the opposite effect on its half-life was seen with FBP1 overexpression (Figure 3F).
FBP1 Ser144 phosphorylation regulates p65 stability via the CHIP-mediated ubiquitin proteasome pathway. Immunoprecipitation and immunoblotting analyses were performed with the indicated antibodies. (A, B) 293T cells were overexpressed the indicated HA-tagged p65 and Flag-tagged FBP1 (WT or G260R) proteins. Total cell lysates were prepared. (C) MCF-7 cells were knocked down FBP1 with shRNA. Total cell lysates were prepared. (D) MB231 cells were overexpressed the indicated Flag-tagged FBP1 proteins. Total cell lysates were prepared. (E) MCF-7 cells with stable knockdown FBP1 proteins were treated with CHX for indicated time. Total cell lysates were prepared. (All data represent mean ± SEM n = 3), *p<0.05. (F) MB231 cells with overexpressed Flag-tagged FBP1 proteins were treated with CHX for indicated time. Total cell lysates were prepared. (All data represent mean ± SEM n = 3), *p<0.05. (G) 293T cells with overexpression of the indicated both Flag-tagged FBP1 and HA-tagged p65 proteins were treated with CHX or CHX+MG132 for 12hr. Total cell lysates were prepared. (H) MCF-7 cells were knocked down FBP1 by shRNA. Total cell lysates were prepared. Immunoprecipitation with an anti-p65 antibody was performed. (I) MB-231 cells were over-expressed Flag-tagged FBP1. Total cell lysates were prepared. Immunoprecipitation with an anti-p65 antibody was performed. (J) MCF-7 cells were co-overexpressed the indicated Flag-tagged p65 and HA-tagged FBP1 (WT, S144A or S144D) proteins. Total cell lysates were prepared. Immunoprecipitation with an anti-Flag antibody was performed. (K) MCF-7 cells were overexpressed HA-tagged FBP1 (WT, S144A or S144D) proteins. Total cell lysates were prepared. (L) MCF-7 cells with CHIP knocked down were overexpressed the indicated Flag-tagged FBP1 proteins. Total cell lysates were prepared. (M) MCF-7 cell lysates were prepared. (N) MCF-7 cells with overexpression of the indicated both Flag-tagged PIM2 or CHIP and HA-tagged p65 proteins. Total cell lysates were prepared.
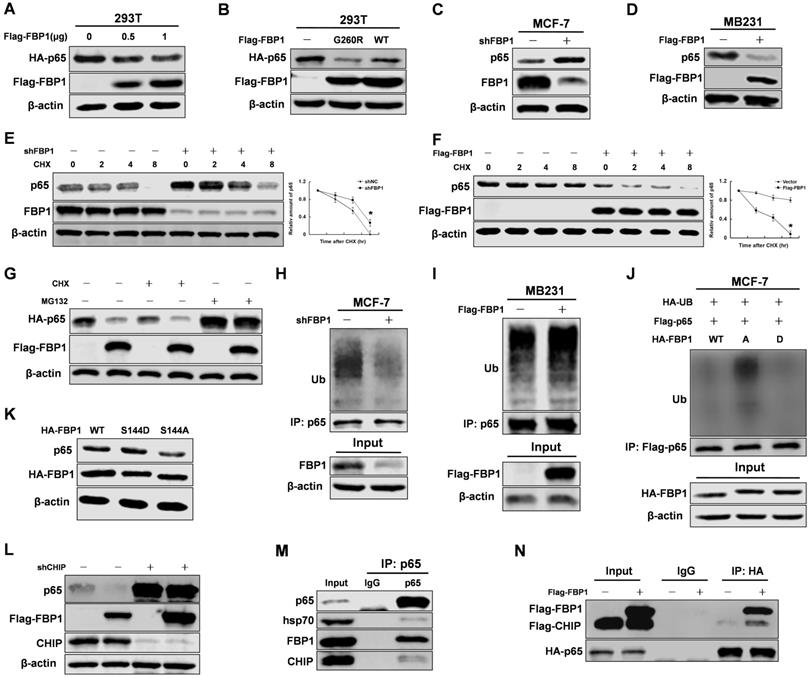
We next investigated whether the proteasome pathway was involved in regulating of p65 protein stability. Our data suggested that MG132 blocked the FBP1-mediated degradation of p65 protein (Figure 3G). The proteasome pathway often increases protein ubiquitination levels, so we tested whether FBP1 regulated p65 ubiquitination levels. Consistent with previous results, FBP1 significantly enhanced p65 ubiquitination (Figure 3H and 3I). To evaluate if FBP1 Ser144 phosphorylation is required for regulation p65 protein stability, we over-expressed HA-tagged FBP1 (WT, S144A or S144D) in MCF-7 cells. Consistently, FBP1 Ser144 phosphorylation increased p65 protein levels via reducing p65 ubiquitination (Figure 3J and 3K). According to previous studies, CHIP binds to p65 and promotes its ubiquitination and degradation through the proteasome pathway [24, 25]. To investigate whether CHIP is responsible for FBP1-mediated p65 degradation, we first determined whether this degradation was affected by CHIP. Indeed, the FBP1-mediated p65 degradation was abrogated by CHIP knockdown (Figure 3L). Finally, p65 interacted with the CHIP cooperative protein-Hsp70 (Figure 3M), and FBP1 overexpression enhanced CHIP interaction with p65 (Figure 3N). Again, FBP1 Ser144 phosphorylation reduced its affinity with CHIP (Figure S3C). However, FBP1 Ser144 phosphorylation enhanced p65 protein stability, and PIM2 had no effect on FBP1 protein level (Figure S3D). Taken together, these data demonstrate that FBP1 Ser144 phosphorylation regulates p65 stability via the CHIP-mediated ubiquitin proteasome pathway.
FBP1 Ser144 phosphorylation contributes to p65 transcriptional activity
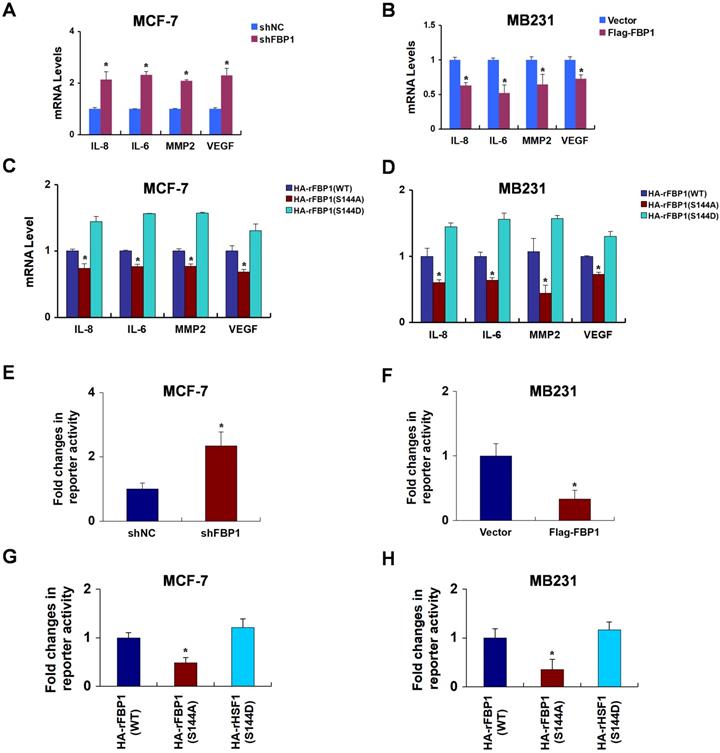
Our study demonstrated that FBP1 binds to the DNA binding domain of p65. Thus, we predicted that the manipulation of FBP1 would affect p65 transcriptional progression. To examine this notion, we rescued FBP1 expression in FBP1-knockdown breast cancer cells (Figure S4A and S4B). Previously reported p65 transcriptional target genes, such as IL-8, IL-6, MMP2, and VEGF, emerged as responsive sensitive to FBP1 manipulation (Figure 4A and 4B). Compare with the mutant FBP1 S144A, wide type FBP1 and mimic FBP1 S144D enhanced the expression of p65 transcriptional target genes, suggesting FBP1 phosphorylation by PIM2 contributed to p65 transcriptional activity (Figure 4C and 4D). Moreover, luciferase reporter assay confirmed that FBP1 knockdown or overexpression affected p65 transcriptional activity (Figure 4E and 4F). Similarly, p65 transactivation activity increased upon FBP1 Ser144 phosphorylation (Figure 4G and 4H). Taken together, we conclude that FBP1 Ser144 phosphorylation leads to enhanced p65 transcriptional activity in breast cancer cells.
FBP1 Ser144 phosphorylation contributes to p65 transcriptional activity. (A) MCF-7 cells were stably knocked down FBP1 by shRNA. mRNA levels were quantitated by RT-PCR. (B) MB-231 cells were stably over-expressed HA-tagged FBP1. mRNA levels were quantitated by RT-PCR. (C, D) MCF-7 or MB231 cells expressing FBP1 shRNA were reconstituted expression of HA-rFBP1 (WT, S144A or S144D). mRNA levels were quantitated by RT-PCR. (E) Stable knockdown FBP1 MCF-7 cells were transfected with dual p65 reporter plasmids, and detected by luciferase reporter assay. (F) Stable overexpression HA-tagged FBP1 MB231 cells were transfected with dual p65 reporter plasmids, and detected by luciferase reporter assay. (G) FBP1-depleted MCF-7 cells with reconstituted expression of HA-FBP1 (WT, S144A or S144D) were transfected with dual p65 reporter plasmids, and detected by luciferase reporter assay. (H) FBP1-depleted MB231 cells with reconstituted expression of HA-FBP1 (WT, S144A or S144D) were transfected with dual p65 reporter plasmids, and detected by luciferase reporter assay. (All data represent mean ± SEM n = 3), *p<0.05.
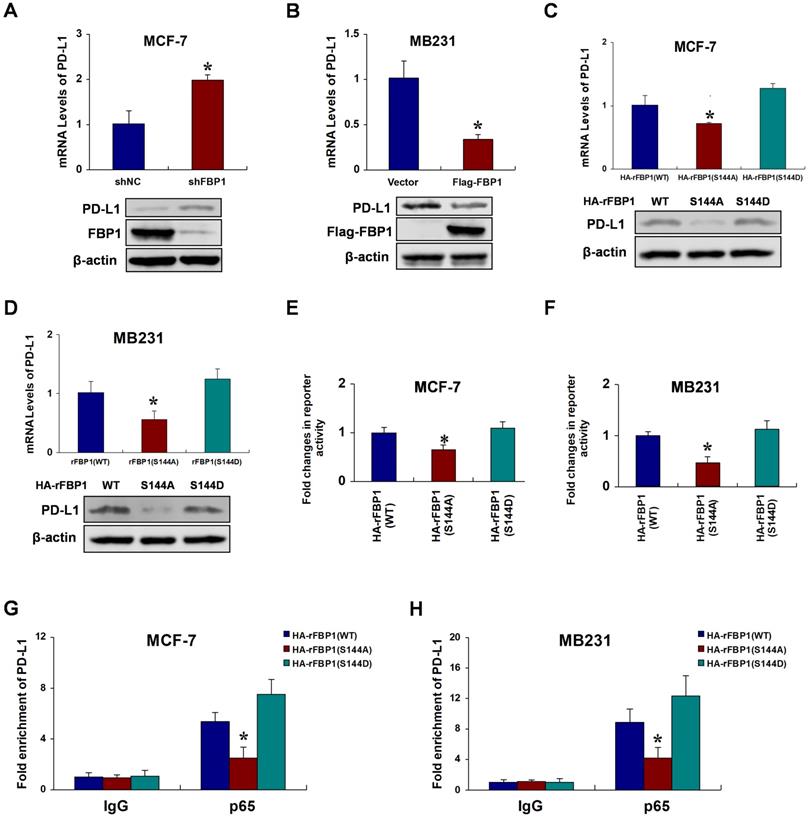
FBP1 Ser144 phosphorylation promotes p65-induced PD-L1 expression
Previous studies have identified programmed death ligand 1 (PD-L1) as a target gene of p65 [26, 27]. Thus, we speculated that FBP1 Ser144 phosphorylation regulates PD-L1 expression via p65. To investigate the potential relationships between FBP1 and PD-L1, we first silenced or overexpressed FBP1 to test PD-L1 expression in breast cancer cells. The results showed that FBP1 repressed PD-L1 expression (Figure 5A and 5B). We further examined whether PD-L1 expression was regulated by FBP1 Ser144 phosphorylation. Compare with that of the mutant FBP1 S144A, the ectopic expression of wild type FBP1 substantially increased PD-L1 expression, and this effect was largely enhanced in cells expressing the mutant FBP1 S144D (Figure 5C and 5D). Consistently, luciferase reporter assays demonstrated that FBP1 Ser144 phosphorylation highlights p65 transactivation activity (Figure 5E and 5F). Moreover, we performed chromatin immunoprecipitation assays, and the results suggested that FBP1 Ser144 phosphorylation enhanced p65 binding to the PD-L1 promoter (Figure 5G and 5H). To further validate whether FBP1 Ser144 phosphorylation regulates PD-L1 expression depending on p65, we knocked out p65 using a special single guide RNA. We found that p65 was involved in FBP1 Ser144 phosphorylation regulating PD-L1 expression (Figure S5A and S5B). These data indicate that FBP1 Ser144 phosphorylation augments p65-induced PD-L1 expression.
FBP1 Ser144 phosphorylation promotes p65-induced PD-L1 expression. (A) MCF-7 cells were stably transfected with FBP1 shRNA. PD-L1 mRNA levels were quantitated by RT-PCR. (B) MB231 cells were transfected with Flag-tagged FBP1. PD-L1 mRNA levels were quantitated by RT-PCR. (C, D) FBP1-depleted MCF-7 or MB231 cells were reconstituted expression of HA-rFBP1 (WT, S144A or S144D). PD-L1 mRNA levels were quantitated by RT-PCR. (E, F) Reconstituted expression of HA-rFBP1 (WT, S144A or S144D) MCF-7 or MB231 cells were transfected with dual PD-L1 reporter plasmids, and detected by luciferase reporter assay. (G, H) CHIP assays were performed using FBP1-depleted MCF-7 or MB231 cells with reconstituted expression of HA-rFBP1 (WT, S144A or S144D). The results were normalized against the values of IgG controls. (All data represent mean ± SEM n = 3), *p<0.05.
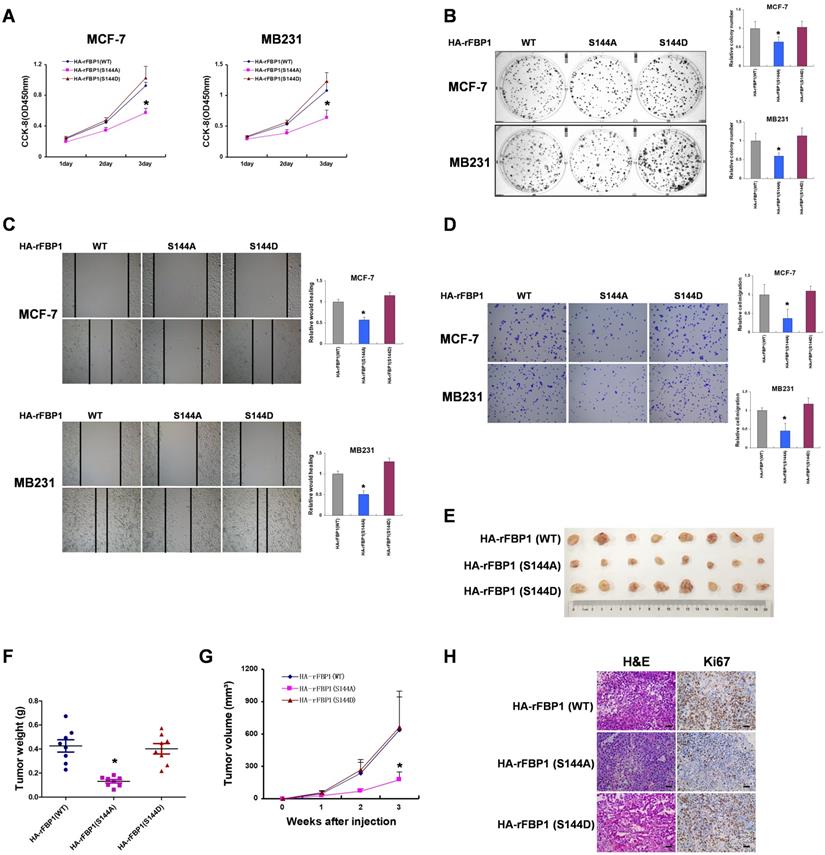
FBP1 Ser144 phosphorylation promotes breast tumorigenesis
To investigate the biological significance of FBP1 Ser144 phosphorylation, we measured the effect of FBP1 Ser144 phosphorylation on breast tumorigenesis in vitro. These results showed that FBP1 Ser144 phosphorylation promoted cell proliferation in breast cancer cells (Figure 6A and 6B). Consistently, FBP1 Ser144 phosphorylation increased cell migration and invasion (Figure 6C and 6D). Furthermore, FBP1 Ser144 phosphorylation stimulated breast tumor growth in vivo, as detected in athymic nude mice (Figure 6E-6G). Again, we used immunohistochemical analysis to demonstrate low Ki67 expression in mutant FBP1 S144A, which could reflect the proliferative ability of cells. Collectively, these data suggest that FBP1 Ser144 phosphorylation contributes to breast tumorigenesis.
FBP1 Ser144 phosphorylation promotes breast tumorigenesis. (A) MCF-7 or MB231 cells with stable expression of HA-rFBP1 (WT, S144A or S144D) were seeded in a new plate. CCK-8 assay was performed to determine cell proliferation. (B-D) MCF-7 or MB231 cells with stable expression of HA-rFBP1 (WT, S144A or S144D) were seeded in a new plate. Clone formation, wound healing assay and cell invasion assays were performed. (E-G) MCF-7 cells with stable expression of HA-rFBP1 (WT, S144A or S144D) were subcutaneously injected into nude mice. After 3 weeks, the mice were sacrificed and dissected at the endpoint. Tumor growth and weight were examined. (H) Representative images of H/E staining and Ki67 staining of tumor samples (Scale bar, 20μm). (All data represent mean ± SEM n = 3), *p<0.05.
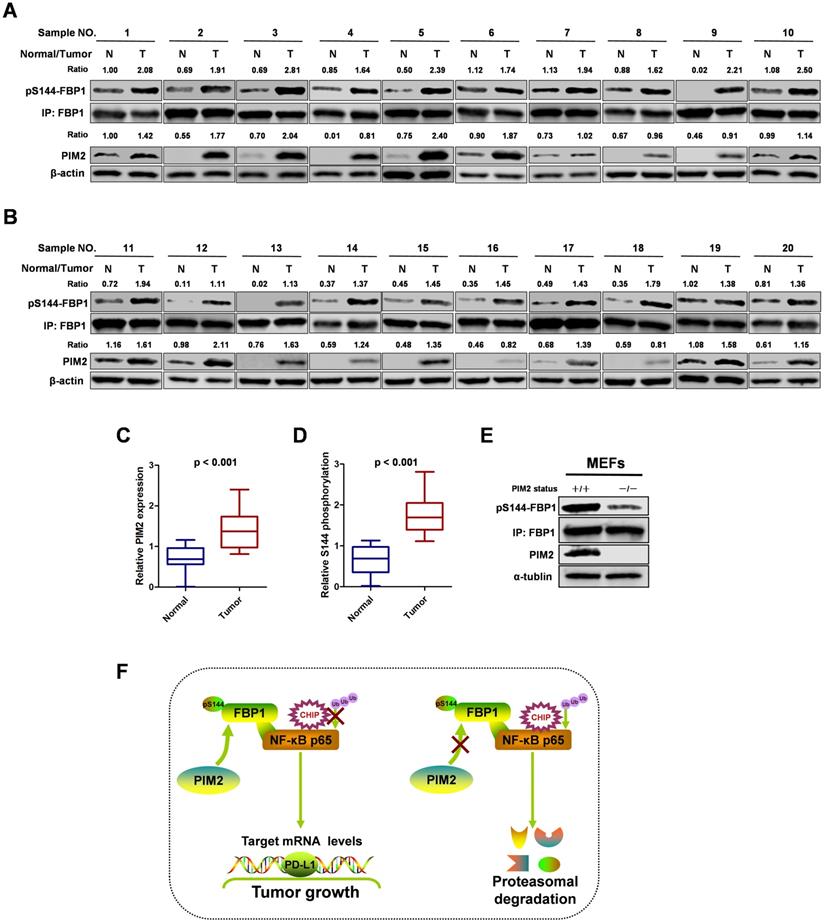
FBP1 Ser144 phosphorylation is upregulated in breast cancer
To study the clinical relevance of FBP1 Ser144 phosphorylation, we collected 20 breast cancer samples with paired surrounding normal breast tissues. We analyzed PIM2 and FBP1 Ser144 phosphorylation expression levels by western blot in breast tumors (T) and their adjacent normal tissues (N) (Figure 7A and 7B). Consistently, PIM2 and FBP1 Ser144 phosphorylation were expressed at higher levels in the tumor samples than in the normal control samples (Figure 7C and 7D). Finally, we generated a PIM2 knockout mouse model and used mouse embryonic fibroblasts (MEFs) derived from the embryos for further analysis. Indeed, PIM2 depletion caused a reduction in FBP1 Ser144 phosphorylation levels (Figure 7E). These data further support the crucial role of FBP1 Ser144 phosphorylation in breast cancer development.
FBP1 Ser144 phosphorylation is upregulated in breast cancer. (A, B) The expression of PIM2 and FBP1 Ser144 phosphorylation in 20 breast cancer samples (T) with paired adjacent normal tissues (N) was analyzed by IB. PIM2 was quantified and normalized to β-actin. Ser144 phosphorylation of immunopurified FBP1 was determined and normalized to FBP1 protein (Ratio). (C, D) PIM2 and FBP1 Ser144 phosphorylation protein levels in normal and tumor tissues were statistically analyzed. (E) PIM2 depletion causes decrease of FBP1 Ser144 phosphorylation protein level. Primary MEFs generated from E12.5-13.5 embryos with PIM2 knockout were followed by IB with the indicated antibodies. (F) Working model for PIM2-induced phosphorylation of FBP1. (All data represent mean ± SEM n = 3), *p<0.05.
Discussion
PIM2, a serine/threonine kinase, has been shown to be highly expressed in many cancers, including breast cancer [22], liver cancer [28], stomach cancer [29], lymph cancer [30], ovarian cancer [31], endometrial cancer [16], prostate cancer [32], and lung cancer [33]. In addition, PIM2 plays an important role in tumor progression by phosphorylating its downstream substrate proteins. Our data demonstrate that PIM2 interacts with FBP1 and phosphorylates it at Ser144, inhibiting FBP1 binding to p65 and enhancing its protein stability. Moreover, the effects of FBP1 Ser144 phosphorylation on downstream signaling cascades result in phenotypic changes related to breast tumorigenesis and progression. Lastly, breast cancer tissues exhibited higher PIM2 and FBP1 Ser144 phosphorylation expression than normal, tumor-adjacent breast tissues, suggesting its importance for breast cancer progression.
FBP1 functions as a tumor suppressor via the promotion of glycogen synthesis and inhibition of glycolysis in many types of cancer [34]. However, the non-canonical functions of FBP1 are independent of its enzymatic activity. For example, noncanonical FBP1 acts as a co-suppressor for many transcriptional factors, including HIF-1α [5], β-catenin [35], NOTCH1 [11], STAT3 [36] and c-MYC [10], and it also regulates their functions through direct interactions, possibly leading to protein degradation. In addition, FBP1 binds to the WW domain of IQGAP1, and impedes IQGAP1-dependent ERK1/2 phosphorylation independent of FBP1 enzymatic activity [37]. However, the posttranscriptional modification of noncanonical FBP1 has never been elucidated in breast cancer. Our findings indicate that PIM2 phosphorylates FBP1 at the Ser144 site to perform its non-canonical function of regulating p65 and PD-L1 expression.
A recent study revealed a new tumor suppressor function of FBP1 to inhibit PD-L1 expression and enhance cancer immunity [36]. They found that FBP1 inhibits STAT3-dependent PD-L1 transcription, a finding that is consistent with our discovery in breast cancer. Thus, we do not rule out that other regulatory pathways are involved in FBP1-mediated PD-L1 expression. Moreover, phosphorylation is one of the most important post-translational modifications for tumor progression [38]. However, to the best of our knowledge, the phosphorylation of FBP1 has yet to be reported in cancer. Therefore, this study is the first to report PIM2 phosphorylates FBP1 at Ser144, uncovering a new pathway to regulate the non-canonical functions of FBP1.
NF-κB p65 is a highly expressed transcription factor in cancer [39-41]. NF-κB p65 is activated by many cytokines, including IL-8, IL-6, TNFα and many others [42, 43]. Besides regulating some canonical pro-growth genes, NF-κB p65 is also known to mediate PD-L1 mRNA level in multiple types of cancers [26, 44]. However, although PIM2 can directly phosphorylate p65 and enhance its transcriptional activity, there is no indirect pathway to regulate p65. In the present study, we identified an indirect pathway wherein phosphorylated FBP1 acts as a major promoter of p65 activity and PD-L1 transcription.
In summary, our results demonstrate that the phosphorylation of non-canonical FBP1 by PIM2 promotes breast tumorigenesis via augmenting NF-κB transcriptional activity and PD-L1 expression (Figure 7F). Our findings further suggest that PIM2-mediated non-canonical FBP1 phosphorylation may be targeted in breast cancer therapies.
Abbreviations
Co-IP: co-immunoprecipitation; FBP1: fructose-1,6-bisphosphatase 1; HIF-1α: hypoxia inducible factor 1α; PD-L1: programmed death ligand 1.
Supplementary Material
Supplementary figures, tables, methods.
Acknowledgements
The study was supported by research grants from National Natural Science Foundation of China (Grant no. 81972489), Shandong Province College Science and Technology Plan Project (Grant no. J17KA254), Projects of medical and health technology development program in Shandong province (Grant no. 2017WS398 and 2018WS057).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chen R, Li J, Zhou X, Liu J, Huang G. Fructose-1,6-Bisphosphatase 1 Reduces (18)F FDG Uptake in Hepatocellular Carcinoma. Radiology. 2017;284:844-53
2. Liao K, Deng S, Xu L, Pan W, Yang S, Zheng F. et al. A Feedback Circuitry between Polycomb Signaling and Fructose-1, 6-Bisphosphatase Enables Hepatic and Renal Tumorigenesis. Cancer Res. 2020;80:675-88
3. Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S. et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell. 2013;23:316-31
4. Hirata H, Sugimachi K, Komatsu H, Ueda M, Masuda T, Uchi R. et al. Decreased Expression of Fructose-1,6-bisphosphatase Associates with Glucose Metabolism and Tumor Progression in Hepatocellular Carcinoma. Cancer Res. 2016;76:3265-76
5. Li B, Qiu B, Lee DS, Walton ZE, Ochocki JD, Mathew LK. et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature. 2014;513:251-5
6. Cong J, Wang X, Zheng X, Wang D, Fu B, Sun R. et al. Dysfunction of Natural Killer Cells by FBP1-Induced Inhibition of Glycolysis during Lung Cancer Progression. Cell Metab. 2018;28:243-55 e5
7. Liu X, Wang X, Zhang J, Lam EK, Shin VY, Cheng AS. et al. Warburg effect revisited: an epigenetic link between glycolysis and gastric carcinogenesis. Oncogene. 2010;29:442-50
8. Zhang Y, Xie Z, Zhou L, Li L, Zhang H, Zhou G. et al. The zinc finger protein ZBTB20 regulates transcription of fructose-1,6-bisphosphatase 1 and beta cell function in mice. Gastroenterology. 2012;142:1571-80 e6
9. Li K, Ying M, Feng D, Du J, Chen S, Dan B. et al. Fructose-1,6-bisphosphatase is a novel regulator of Wnt/beta-Catenin pathway in breast cancer. Biomed Pharmacother. 2016;84:1144-9
10. Wang B, Fan P, Zhao J, Wu H, Jin X. FBP1 loss contributes to BET inhibitors resistance by undermining c-Myc expression in pancreatic ductal adenocarcinoma. J Exp Clin Cancer Res. 2018;37:224
11. Lu C, Ren C, Yang T, Sun Y, Qiao P, Wang D. et al. A Noncanonical Role of Fructose-1, 6-Bisphosphatase 1 Is Essential for Inhibition of Notch1 in Breast Cancer. Mol Cancer Res. 2020;18:787-96
12. Narlik-Grassow M, Blanco-Aparicio C, Carnero A. The PIM family of serine/threonine kinases in cancer. Med Res Rev. 2014;34:136-59
13. Bullock AN, Russo S, Amos A, Pagano N, Bregman H, Debreczeni JE. et al. Crystal structure of the PIM2 kinase in complex with an organoruthenium inhibitor. PLoS One. 2009;4:e7112
14. Warfel NA, Kraft AS. PIM kinase (and Akt) biology and signaling in tumors. Pharmacol Ther. 2015;151:41-9
15. Yang T, Ren C, Qiao P, Han X, Wang L, Lv S. et al. PIM2-mediated phosphorylation of hexokinase 2 is critical for tumor growth and paclitaxel resistance in breast cancer. Oncogene. 2018;37:5997-6009
16. Han X, Ren C, Yang T, Qiao P, Wang L, Jiang A. et al. Negative regulation of AMPKalpha1 by PIM2 promotes aerobic glycolysis and tumorigenesis in endometrial cancer. Oncogene. 2019;38:6537-49
17. Yang T, Ren C, Lu C, Qiao P, Han X, Wang L. et al. Phosphorylation of HSF1 by PIM2 Induces PD-L1 Expression and Promotes Tumor Growth in Breast Cancer. Cancer Res. 2019;79:5233-44
18. Yu Z, Zhao X, Huang L, Zhang T, Yang F, Xie L. et al. Proviral insertion in murine lymphomas 2 (PIM2) oncogene phosphorylates pyruvate kinase M2 (PKM2) and promotes glycolysis in cancer cells. J Biol Chem. 2013;288:35406-16
19. Nair JR, Caserta J, Belko K, Howell T, Fetterly G, Baldino C. et al. Novel inhibition of PIM2 kinase has significant anti-tumor efficacy in multiple myeloma. Leukemia. 2017;31:1715-26
20. Beharry Z, Mahajan S, Zemskova M, Lin YW, Tholanikunnel BG, Xia Z. et al. The Pim protein kinases regulate energy metabolism and cell growth. Proc Natl Acad Sci U S A. 2011;108:528-33
21. Chen LS, Redkar S, Bearss D, Wierda WG, Gandhi V. Pim kinase inhibitor, SGI-1776, induces apoptosis in chronic lymphocytic leukemia cells. Blood. 2009;114:4150-7
22. Ren C, Yang T, Qiao P, Wang L, Han X, Lv S. et al. PIM2 interacts with tristetraprolin and promotes breast cancer tumorigenesis. Mol Oncol. 2018;12:690-704
23. Mertins P, Yang F, Liu T, Mani DR, Petyuk VA, Gillette MA. et al. Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol Cell Proteomics. 2014;13:1690-704
24. Wang S, Wu X, Zhang J, Chen Y, Xu J, Xia X. et al. CHIP functions as a novel suppressor of tumour angiogenesis with prognostic significance in human gastric cancer. Gut. 2013;62:496-508
25. Wang Y, Ren F, Feng Y, Wang D, Jia B, Qiu Y. et al. CHIP/Stub1 functions as a tumor suppressor and represses NF-kappaB-mediated signaling in colorectal cancer. Carcinogenesis. 2014;35:983-91
26. Bouillez A, Rajabi H, Jin C, Samur M, Tagde A, Alam M. et al. MUC1-C integrates PD-L1 induction with repression of immune effectors in non-small-cell lung cancer. Oncogene. 2017;36:4037-46
27. Jin X, Ding D, Yan Y, Li H, Wang B, Ma L. et al. Phosphorylated RB Promotes Cancer Immunity by Inhibiting NF-kappaB Activation and PD-L1 Expression. Mol Cell. 2019;73:22-35 e6
28. Kronschnabl P, Grunweller A, Hartmann RK, Aigner A, Weirauch U. Inhibition of PIM2 in liver cancer decreases tumor cell proliferation in vitro and in vivo primarily through the modulation of cell cycle progression. Int J Oncol. 2020;56:448-59
29. Xin H, Deng Y, Cao J. Proviral insertion in murine lymphomas 2 promotes stomach cancer progression by regulating apoptosis via reactive oxygen species-triggered endoplasmic reticulum stress. Biochem Biophys Res Commun. 2018;506:145-52
30. Gomez-Abad C, Pisonero H, Blanco-Aparicio C, Roncador G, Gonzalez-Menchen A, Martinez-Climent JA. et al. PIM2 inhibition as a rational therapeutic approach in B-cell lymphoma. Blood. 2011;118:5517-27
31. Musiani D, Hammond DE, Cirillo L, Erriquez J, Olivero M, Clague MJ. et al. PIM2 kinase is induced by cisplatin in ovarian cancer cells and limits drug efficacy. J Proteome Res. 2014;13:4970-82
32. Dai H, Li R, Wheeler T, Diaz de Vivar A, Frolov A, Tahir S. et al. Pim-2 upregulation: biological implications associated with disease progression and perinueral invasion in prostate cancer. Prostate. 2005;65:276-86
33. Yu Z, Huang L, Qiao P, Jiang A, Wang L, Yang T. et al. PKM2 Thr454 phosphorylation increases its nuclear translocation and promotes xenograft tumor growth in A549 human lung cancer cells. Biochem Biophys Res Commun. 2016;473:953-8
34. Snaebjornsson MT, Schulze A. Non-canonical functions of enzymes facilitate cross-talk between cell metabolic and regulatory pathways. Exp Mol Med. 2018;50:34
35. Zhao W, Yang S, Chen J, Zhao J, Dong J. Forced overexpression of FBP1 inhibits proliferation and metastasis in cholangiocarcinoma cells via Wnt/beta-catenin pathway. Life Sci. 2018;210:224-34
36. Wang B, Zhou Y, Zhang J, Jin X, Wu H, Huang H. Fructose-1,6-bisphosphatase loss modulates STAT3-dependent expression of PD-L1 and cancer immunity. Theranostics. 2020;10:1033-45
37. Jin X, Pan Y, Wang L, Ma T, Zhang L, Tang AH. et al. Fructose-1,6-bisphosphatase Inhibits ERK Activation and Bypasses Gemcitabine Resistance in Pancreatic Cancer by Blocking IQGAP1-MAPK Interaction. Cancer Res. 2017;77:4328-41
38. Brautigan DL, Shenolikar S. Protein Serine/Threonine Phosphatases: Keys to Unlocking Regulators and Substrates. Annu Rev Biochem. 2018;87:921-64
39. Taniguchi K, Karin M. NF-kappaB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. 2018;18:309-24
40. Yu C, Chen S, Guo Y, Sun C. Oncogenic TRIM31 confers gemcitabine resistance in pancreatic cancer via activating the NF-kappaB signaling pathway. Theranostics. 2018;8:3224-36
41. Meng Q, Liang C, Hua J, Zhang B, Liu J, Zhang Y. et al. A miR-146a-5p/TRAF6/NF-kB p65 axis regulates pancreatic cancer chemoresistance: functional validation and clinical significance. Theranostics. 2020;10:3967-79
42. Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005;30:43-52
43. Liu C, Zhou Y, Li M, Wang Y, Yang L, Yang S. et al. Absence of GdX/UBL4A Protects against Inflammatory Diseases by Regulating NF-small ka, CyrillicB Signaling in Macrophages and Dendritic Cells. Theranostics. 2019;9:1369-84
44. Peng J, Hamanishi J, Matsumura N, Abiko K, Murat K, Baba T. et al. Chemotherapy Induces Programmed Cell Death-Ligand 1 Overexpression via the Nuclear Factor-kappaB to Foster an Immunosuppressive Tumor Microenvironment in Ovarian Cancer. Cancer Res. 2015;75:5034-45
Author contact
![]() Corresponding author: Zhenhai Yu, Department of Reproductive Medicine, Affiliated Hospital of Weifang Medical University, Weifang, Shandong Province, P.R. China. Fax: +865363083802, Tel: +865363081391, E-mail: tomsyucom.
Corresponding author: Zhenhai Yu, Department of Reproductive Medicine, Affiliated Hospital of Weifang Medical University, Weifang, Shandong Province, P.R. China. Fax: +865363083802, Tel: +865363081391, E-mail: tomsyucom.