Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Conclusion
Materials and Methods
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(19):8528-8540. doi:10.7150/thno.46002 This issue Cite
Research Paper
Increased intracellular Cl- concentration improves airway epithelial migration by activating the RhoA/ROCK Pathway
Wenjie Huang1,3*, Meiling Tan1*, Yue Wang2,4, Lei Liu2, Yan Pan2, Jingjing Li2, Mingxing Ouyang2, Chunjiao Long1, Xiangping Qu1, Huijun Liu1, Chi Liu1, Jia Wang1, Linhong Deng2 ![]() , Yang Xiang1
, Yang Xiang1 ![]() , Xiaoqun Qin1
, Xiaoqun Qin1 ![]()
1. School of Basic Medicine, Central South University, Changsha, Hunan 410078, China.
2. Changzhou Key Laboratory of Respiratory Medical Engineering, Institute of Biomedical Engineering and Health Sciences, Changzhou, Jiangsu 213164, China.
3. Affiliated Liutie Central Hospital of Guangxi medical university, Liuzhou, Guangxi 545007, China.
4. School of Nursing, Changzhou University, Changzhou, Jiangsu 213164, China.
*These authors contributed equally to this work.
Received 2020-3-14; Accepted 2020-6-24; Published 2020-7-9
Abstract

In the airway, Cl- is the most abundant anion and is critically involved in transepithelial transport. The correlation of the abnormal expression and activation of chloride channels (CLCs), such as cystic fibrosis transmembrane conductance regulators (CFTRs), anoctamin-1, and CLC-2, with cell migration capability suggests a relationship between defective Cl- transport and epithelial wound repair. However, whether a correlation exists between intracellular Cl- and airway wound repair capability has not been explored thus far, and the underlying mechanisms involved in this relationship are not fully defined.
Methods: In this work, the alteration of intracellular chloride concentration ([Cl-]i) was measured by using a chloride-sensitive fluorescent probe (N-[ethoxycarbonylmethyl]-6-methoxyquinolium bromide).
Results: We found that clamping with high [Cl-]i and 1 h of treatment with the CLC inhibitor CFTR blocker CFTRinh-172 and chloride intracellular channel inhibitor IAA94 increased intracellular Cl- concentration ([Cl-]i) in airway epithelial cells. This effect improved epithelial cell migration. In addition, increased [Cl-]i in cells promoted F-actin reorganization, decreased cell stiffness, and improved RhoA activation and LIMK1/2 phosphorylation. Treatment with the ROCK inhibitor of Y-27632 and ROCK1 siRNA significantly attenuated the effects of increased [Cl-]i on LIMK1/2 activation and cell migration. In addition, intracellular Ca2+ concentration was unaffected by [Cl-]i clamping buffers and CFTRinh-172 and IAA94.
Conclusion: Taken together, these results suggested that Cl- accumulation in airway epithelial cells could activate the RhoA/ROCK/LIMK cascade to induce F-actin reorganization, down-regulate cell stiffness, and improve epithelial migration.
Keywords: Airway epithelial cells, Intracellular chloride, MQAE, Epithelial migration, Cell stiffness
Introduction
Airway epithelia, the first physical barrier, are vulnerable to injuries inflicted by bacterial virulence factors, allergens and environmental hazards. Defective capability epithelial wound repair due to the dysfunction of airway epithelial cells is involved in numerous respiratory illnesses, including chronic obstructive pulmonary disease, asthma, and cystic fibrosis [1, 2]. Interest in the overlooked role of intracellular Cl- concentration ([Cl-]i) in airway epithelial cells has recently increased when a number of studies highlighted the correlation between the above-mentioned diseases and the dysfunction of Cl- channels, such as Ca2+-activated chloride channels (CLCs), cystic fibrosis transmembrane conductance regulators (CFTRs), and voltage-gated CLCs in airway epithelial cells [3-8].
Previous researches have shown that the change in [Cl-]i is involved in cell proliferation [9], synaptic transmission [10-12], gene expression [13], and NF-κB-activation-related signaling pathways [14, 15]. The [Cl-]i of liver cancer cells markedly increases in response to acids and alkalis [16]. Lipopolysaccharide (LPS) stimulation increases [Cl-]i in airway epithelial cells [15, 17]. We also found that the [Cl-]i of 16HBE14o- cells markedly increases in response to various airway stimuli, including acids, alkalis, coldness, heat, LPS, and H2O2 (Figure S1). Interestingly, in a starfish oocyte wound resealing model, the plasma membrane begins to exhibit chloride permeability by as early as 1 s following wounding [18]. This observation prompted us to speculate that intracellular Cl- might be a driving signal of cells in response to injury. The increase in the [Cl-]i of injured cells fuels epithelial repair events by launching a signaling cascade that is associated with wound repair.
Cell migration plays an important role in the wound repair of airway epithelial cells. The dynamic polymerization and depolymerization of the filamentous actin (F-actin) cytoskeleton, as well as the remodeling of the mechanical performances of epithelial cells, are involved in multiple stages of cell migration [19, 20]. Among several possible control elements of cytophysical properties, the Rho family of GTPases and downstream signals appear to play the most prominent role in modulating the biochemical pathways of cell migration [21-23]. Although [Ca2+]i is known to be a prominent regulator that can exert multiple effects on the structure and dynamics of the actin cytoskeleton [24, 25], whether [Cl-]i is involved this response remains poorly defined.
In this work, we found the increased [Cl-]i of 16HBE14o- cells improved epithelial migration, promoted F-actin reorganization, and decreased cell stiffness. High [Cl-]i activated RhoA and increased LIMK1/2 phosphorylation without changing intracellular Ca2+ concentration ([Ca2+]i) in 16HBE14o- cells, indicating that [Cl-]i could play a vital role in initiating wound repair events and thus promote the biological and mechanical function renewal of 16HBE14o- cells.
Results
Double ionophore strategy artificially increased the [Cl-]i of 16HBE14o- cells
We used the double ionophore strategy described by Verkman et al. to artificially increase the [Cl-]i of 16HBE14o- cells [13, 26, 27] to study the role of Cl- concentration in wound repair. This method enables changes in [Cl-]i concentration by changing extracellular concentrations. We clamped the [Cl-]i of 16HBE14o- cells at 0 mM to 130 mM. Meanwhile, we used the fluorescence probe MQAE, a low-toxicity indicator of dynamic Cl- concentration in cells, in combination with laser scanning confocal fluorescence microscopy to measure changes in [Cl-]i. As shown in Figure S2, when [Cl-]i was increased from 0 mM to 130 mM, the MQAE fluorescence signal in 16HBE14o- cells was quenched by Cl-, and a linear correlation existed between [Cl-]i and the changes in MQAE fluorescence. Then, the data were fitted to the Stern-Volmer equation (F0/F - 1 = KSV[Cl-]) with the intracellular quenching constant KSV of 22.71 ± 1.53 M-1. In this equation, F0 indicates the mean fluorescence level in the high-KNO3 clamping buffer (without chloride), and the mean fluorescence at different clamping chloride concentrations is defined as F.
Increased [Cl-]i in 16HBE14o- cells enhanced cell repair activity
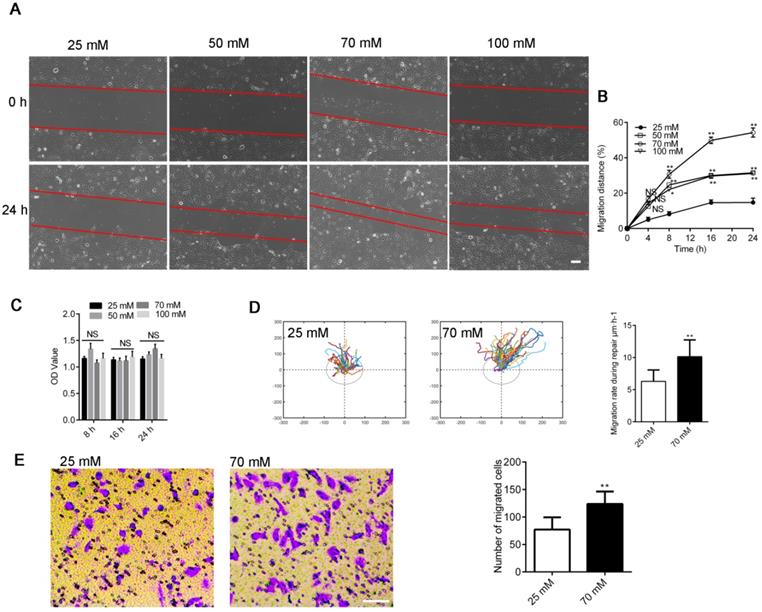
We assayed wound healing after mechanical injury [28] to investigate repair activities in 16HBE14o- cells to investigate the influence of increased [Cl-]i on airway wound repair. [Cl-]i was clamped at the concentrations of 25 (resting [Cl-]i), 50, 70, and 100 mM. As shown in Figure 1A-B, the artificially clamped high levels of [Cl-]i markedly promoted wound repair in 16HBE14o- cells in a concentration-dependent manner. The maximum repair rate was shown by the 70 mM [Cl-]i group. Furthermore, we tracked and evaluated the migratory capability of different cells in each group by randomly selecting one cell at the wound margin and recording its migration through video-microscopy [6]. The mean migration rate of cells in the 70 mM group was significantly faster than that of cells in the group of 25 mM (Figure 1D), and the number of migrating cells was markedly increased in the 70 mM group than in the 25 mM group (Figure 1E). We tested the proliferation of 16HBE14o- cells via the MTT assay to determine if cell proliferation was involved in this process [29]. As shown by Figure 1C, the proliferation of 16HBE14o- cells was slightly enhanced although proliferation affected wound repair in response to [Cl-]i.
Effects of clamping [Cl-]i at high levels on 16HBE14o- cell wound healing. A) Cell images taken at 0 and 24 h after injury (Scale bars: 200 µm). B) Standardized migration distances at 0, 4, 8, 16 and 24 h after injury. (n = 3 independent experiments, *P < 0.05; **P < 0.01; nonsignificant [NS]). C) Proliferation of 16HBE14o- cells was estimated through MTT over different time points (8, 16 and 24 h; n = 3 independent experiments; nonsignificant [NS]). D) Mean cell migration rates were calculated from single-cell tracking at the wound edge (45 cells at the wound edge) over a 24 h period after injury (n = 3 independent experiments; **P < 0.01). E) Transwell assay following clamping [Cl-]i of 16HBE14o- cells at 25 or 70 mM for 1 h. The number of migrated cells was compared between groups (n = 3 independent experiments, **P < 0.01 versus 25 mM, scale bar, 100 µm). Data are presented as mean ± SD.
Accumulation of Cl- in 16HBE14o- cells induced by CFTR channels and chloride intracellular channel inhibitors promoted wound repair
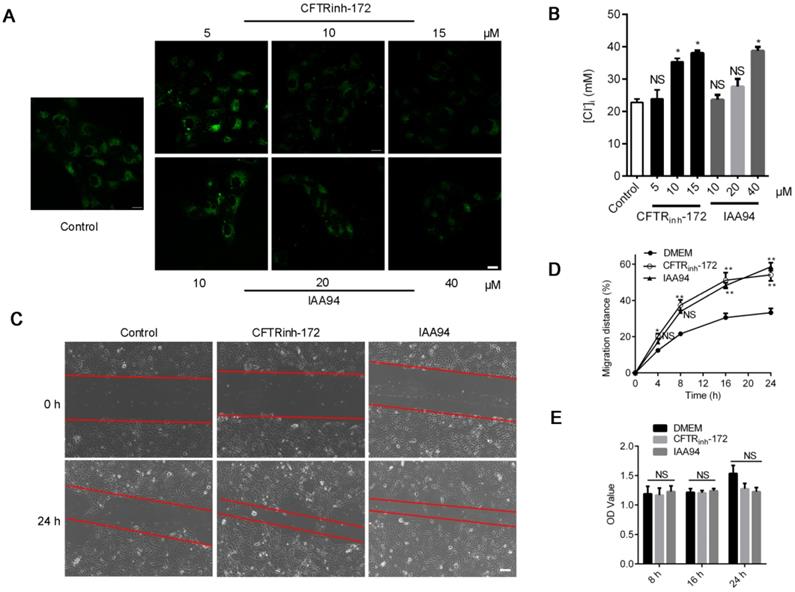
We also induced Cl- accumulation in 16HBE14o- cells through treatment with the CFTR blocker CFTRinh-172 (1, 10, and 15 μM) [26] and chloride intracellular channel (CLIC) inhibitor IAA94 (10, 20, and 40 μM) for 1 h [30]. As shown in Figure 2A, Cl- accumulated in accordance with the increase in the concentrations of both inhibitors. The Stern-Volmer equation (Figure S2) showed that the [Cl-]i of 16HBE14o- cells drastically increased from the baseline value of 22.74 ± 0.83 mM to 35.36 ± 1.29 (CFTRinh-172, 10 μM) and 38.74 ± 1.41 mM (IAA94, 40 μM) (Figure 2B). Moreover, as depicted in Figure 2C and D, we found that the high level of [Cl-]i induced by CFTRinh-172 or IAA94 in 16HBE14o- cells elicited a significant increase in migration rate but only slightly affected cell proliferation (Figure 2E).
Effects of CLIC-inhibitor-induced high [Cl-]i on the wound-healing capacity of 16HBE14o- cells. A) Confocal fluorescent images of living 16HBE14o- cells stained with MQAE followed by treatment with CFTRinh-172 (5, 10, and 15 µM) and IAA94 (10, 20, and 40 µM) for 1 h (n = 100-180 cells for each group; scale bars: 20 µm). B) [Cl-]i of 16HBE14o- cells was calculated in accordance with the Stern-Volmer plot. C, D) Standardized migration distances were measured at 0, 4, 8, 16 and 24 h after cells were treated with the CFTR blocker CFTRinh-172 (10 µM) or the CLIC inhibitor IAA94 (40 µM) for 1 h (n = 3 independent experiments, *P < 0.05; **P < 0.01; nonsignificant [NS]). E) Proliferation of 16HBE14o- cells at 8, 16 and 24 h of repair was evaluated via MTT assay (n = 3 independent experiments; nonsignificant [NS]). Data are presented as mean ± SD.
Increased [Cl-]i of 16HBE14o- cells promoted cytoskeletal reorganization
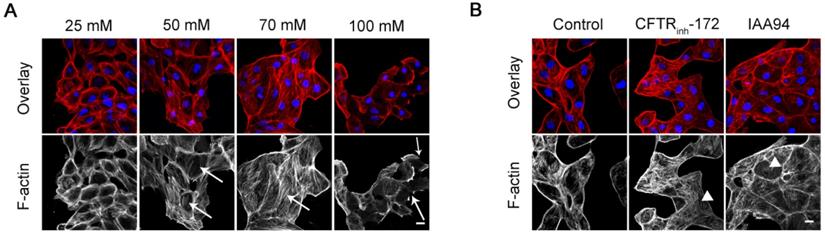
Cell migration requires dramatic changes in cell shape. To a large degree, the dynamic remodeling of F-actin is linked to the events of morphological changes and physical forces that occur during migration [31]. Typically, 16HBE14o- cells showed highly concentrated F-actin structures around cell peripheries as depicted by Figure 3A and B. The high levels of [Cl-]i induced by the double ionophore technique and treatment with CFTRinh-172 (10 μM) and IAA94 (40 μM) promoted F-actin reorganization in 16HBE14o- cells. Compared with those of the control cells, the peripheral F-actin fibers of treated epithelial cells were disassembled, and the amounts of threadlike stress fibers were markedly increased throughout the cell body (Figure 3A, arrows; 3B, triangles). The development of perinuclear thick stress fibers is closely associated with elongation capability and contractility [32]. The reorganization of F-actin fibers owing to the high level of [Cl-]i in epithelial cells suggested an increased potential for migration. However, we also observed numerous discontinuous punctuate peripheral F-actin structures in 16HBE14o- cells treated with 100 mM [Cl-]i (Figure 3A, arrows). This result might be in agreement with the finding in Figure 2A, which shows that the migration rate of cells in this group was lower than that of cells in the 70 mM [Cl-]i group.
Effect of increased [Cl-]i on the F-actin filaments of 16HBE14o- cells. A) Confocal images of F-actin filaments in 16HBE14o- cells after incubation with buffers with various chloride concentrations for 1 h. B) Confocal images of F-actin filaments in 16HBE14o- cells after treatment with CFTRinh-172 (10 µM) and IAA94 (40 µM) for 1 h (n = 3 independent experiments, F-actin, red; nucleus, blue). Scale bars: 20 µm.
Increased [Cl-]i of 16HBE14o- cells decreased cell stiffness
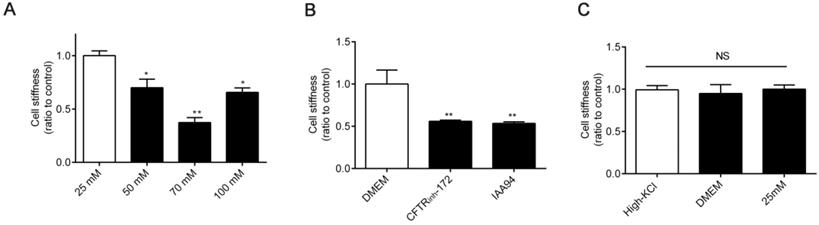
The mechanical rigidity of cells is conferred by the cytoskeleton, which forms an internal polymer network that extends throughout the cell body. Therefore, dynamic cytoskeleton remodeling modulates cell stiffness [33, 34]. Thus, we next detected cell stiffness by using OMTC [35]. As shown in Figure 4A-B, the stiffness of 16HBE14o- cells decreased under treatment with high [Cl-]i and 1 h of treatment with CFTRinh-172 (10 μM) and IAA94 (40 μM). Cell stiffness did not differ among cells under treatment with DMEM, high-KCl buffer (without ionophores), and 25 mM [Cl-]i (resting [Cl-]i) (Figure 4C).
Effect of increased [Cl-]i on cellular mechanical properties. A-B) Normalized stiffness (G'/G0') of 16HBE14o- cells after treatment with buffers with various chloride concentrations and 1 h of treatment with CFTRinh-172 (10 µM) and IAA94 (40 µM). C) Stiffness of 16HBE14o- cells treated with high-KCl buffer, DMEM, and buffer with 25 mM [Cl-]i (resting [Cl-]i), for 1 h (n = 3 independent experiments, *P < 0.05; **P < 0.01 versus 25 mM). Data are presented as mean ± SD.
Increased [Cl-]i in 16HBE14o- cells activated RhoA signaling in 16HBE14o- cells
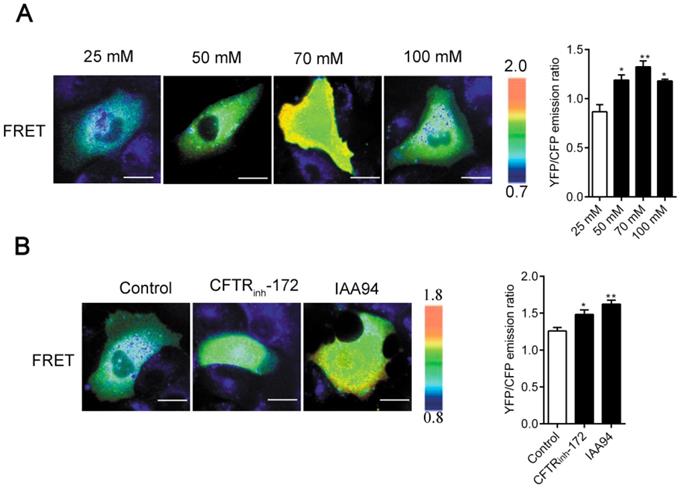
The regulation of cell physical structures is driven by a variety of signaling molecules, among which the most prominent are the Rho family of GTPases. Here, by using the real-time FRET reporters of RhoA activity, we investigated whether increased [Cl-]i could activate RhoA signaling in 16HBE14o- cells. As shown in Figure 5A-B, a remarkable increase in RhoA activity was observed in the cells under nigericin and tributyltin treatment (peak induction was observed at 70 mM [Cl-]i) and those under 1 h of treatment with CFTRinh-172 (10 μM) and IAA94 (40 μM). In the control cells, the constitutive activity of RhoA mainly localized at the perinuclear region toward one end of the nuclei. Upon elevated [Cl-]i stimulation, a substantial increase in RhoA activity was observed in the whole cytoplasm. These findings suggested that elevated [Cl-]i induced the activation of RhoA in airway epithelial cells.
Increased [Cl-]i activated RhoA in 16HBE14o- cells. A) Images of the emission ratios of YFP/CFP-based RhoA biosensor in 16HBE14o- cells after incubation with buffers with various chloride concentrations. B) Images of the emission ratios of the YFP/CFP-based RhoA biosensor in 16HBE14o- cells after 1 h of treatment with CFTRinh-172 (10 µM) and IAA94 (40 µM). Cells are shown on the left. The right panels represent the emission ratios of the YFP/CFP-based RhoA biosensors. Emission ratios were measured by taking the mean intensity of a ROI of multiple cells under the same conditions (n = 3 independent experiments; *P < 0.05; **P < 0.01. Scale bars: 20 µm). Data are presented as mean ± SD.
Elevated [Cl-]i of 16HBE14o- cells enhanced the phosphorylation of LIMK1/2 by ROCK1 without changing the [Ca2+]i of 16HBE14o- cells
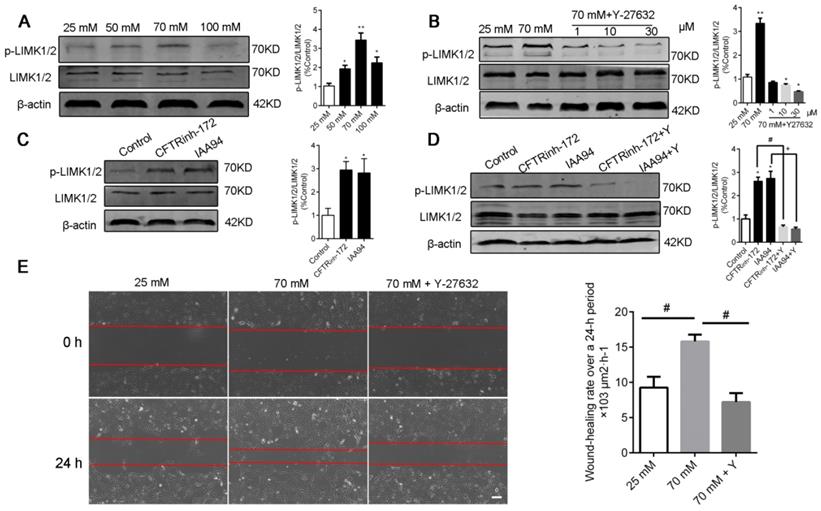
LIM kinases (LIMK1/2) are serine/threonine kinases that are involved in actin cytoskeletal regulation downstream of the RhoA-ROCK signaling pathway [36]. As shown in Figure 6A and C, a high level of [Cl-]i increased the phosphorylation of LIMK1/2 in the groups under nigericin and tributyltin treatment and those under 1 h of treatment with CFTRinh-172 (10 μM) and IAA94 (40 μM). Under treatment with Y-27632, a specific ROCK inhibitor (Figure 6B and D), or ROCK1 siRNA (Figure 7A-B), the dysregulation of the ROCK pathway significantly abolished the phosphorylation of LIMK1/2 that was induced by a high level of [Cl-]i. We also observed that ROCK inhibition with Y-27632 (10 μM) decreased the acceleration of wound healing caused by high [Cl-]i in 16HBE14o- cells (Figure 6E).
Inhibition of ROCK abolished the phosphorylation of LIMK1/2 and the improvement in wound healing caused by high [Cl-]i in 16HBE14o- cells. A) Western blot analysis images of LIMK1/2 phosphorylation in 16HBE14o- cells after 1 h of incubation with buffers with various chloride concentrations. B) Phosphorylation of LIMK1/2 was measured in 16HBE14o- cells after 1 h of treatment with high [Cl-]i in the presence of Y-27632 (1, 10, and 30 µM). C-D) Phosphorylation of LIMK1/2 was measured in 16HBE14o- cells after 1 h of treatment with CLICs (CFTRinh-172 and IAA94) in the presence of Y-27632 (10 µM). The signal in each lane was quantified by using ImageJ software, and the ratios of p-LIMK1/2/LIMK1/2 to β-actin were determined (n = 3 independent experiments, *P < 0.05; **P < 0.01). E) 16HBE14o- cells were wounded, and wound-healing rates were monitored over a 24 h period under high [Cl-]i conditions in the presence of Y-27632 (10 µM) for 1 h (n = 3 independent experiments; #P < 0.05). Data are presented as mean ± SD.
LIMK1/2 phosphorylation caused by high [Cl-]i was regulated by ROCK1 without changing the [Ca2+]i of 16HBE14o- cells. A) ROCK1 RNA and protein levels in 16HBE14o- cells after 48 h of transfection with ROCK1 siRNA were detected through real-time PCR and Western blot analysis accordingly. B) LIMK1/2 phosphorylation caused by high [Cl-]i in 16HBE14o- cells treated with ROCK1 siRNA was monitored through Western blot analysis. C-D) Emission ratio images of the YFP/CFP-based Ca2+ biosensor in 16HBE14o- cells after treatment with buffers with various chloride concentrations and 1 h of treatment with CFTRinh-172 (10 µM) and IAA94 (40 µM). Cells are shown in the left panel. The right panels represent the emission ratios of the YFP/CFP-based Ca2+ biosensor. Emission ratios were measured by taking the mean intensity of a ROI of multiple cells under the same conditions (n = 3 independent experiments; nonsignificant [NS]). Data are presented as mean ± SD.
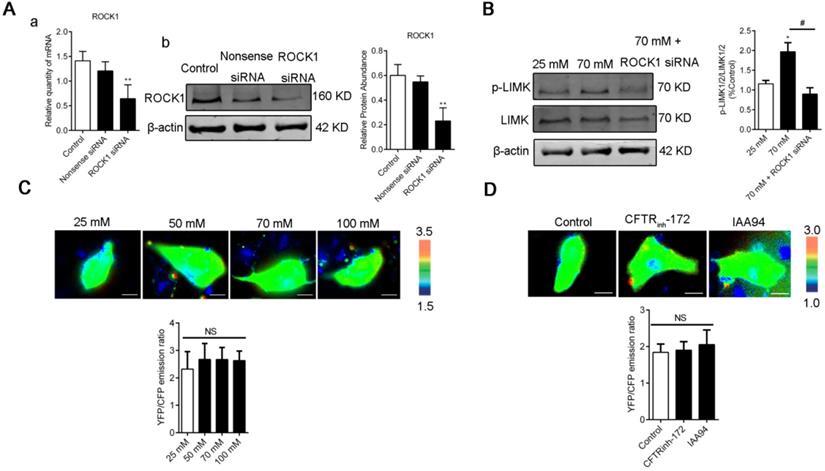
We next examined whether [Ca2+]i was affected by [Cl-]i clamping buffers or the CLIC inhibitors CFTRinh-172 and IAA94. As shown in Figure. 7C-D, [Cl-]i increased under treatment with [Cl-]i clamping buffers and CLICs without changing the [Ca2+]i of 16HBE14o- cells. These data suggested that increased [Cl-]i may activate the RhoA/ROCK/LIMK cascade pathway to improve the migration of airway epithelial cells.
Discussion
Cl- is the most abundant anion in the human body. Previous studies have mainly focused on its functional cellular role in regulating intracellular pH, cell volume, electrical charge balance, salt secretion, and cell reabsorption [1, 37-40]. Cl- is also the most important anion that is critically involved in transepithelial transport in the airway. The abnormal expression and activation of CLCs, such as CFTR, ANO1, and CLC-2, are correlated with cell migration capability, suggesting a relationship between defective Cl- transport and epithelial wound repair [6, 8, 41, 42]. We previously demonstrated that the expression and function of the CFTR of 16HBE14o- cells (human bronchial epithelial cells) were inhibited under ozone exposure and respiratory syncytial virus infection [43]. CFTR failure may delay the wound healing of airway epithelial cells [6]. However, whether [Cl-]i is implicated in the wound repair of airway epithelial cells remains unclear.
Given that prolonged treatment with nigericin-tributyltin might result in severe cell toxicity, an optimal time for treatment with clamping buffers was used to explore the physical functions of Cl- [13, 15, 27]. Here, the [Cl-]i of 16HBE14o- cells (25, 50, 70, and 100 mM) was artificially elevated by equalizing [Cl-]i and external chloride concentrations for 1 h via the double ionophore strategy and then validated them through 36Cl- and 3-O-[3H] methylglucose double-label experiments [44]. Figure S2 shows the fluorescence changes and the corresponding [Cl-]i of 16HBE14o- cells. The KSV was 22.71 ± 1.53 M-1, and the mean resting [Cl-]i of 16HBE14o- cells in our study was 22.74 ± 0.83 mM. In other tissues, KSV values varied between 3 and 26 M-1 [45-48]. Kondo and Froemter previously reported cell chloride concentrations between 25 and 30 mM [27]. Calculations with the Stern-Volmer equation showed that CFTR and CLIC blockers induced Cl- accumulation inside 16HBE14o- cells by hampering Cl- efflux [49, 50].
Surprisingly, our results demonstrated that elevated [Cl-]i caused by CFTR and CLIC blockers improved the migration capability of 16HBE14o- cells (Figure 2). In contrast to our findings, previous results indicate that CFTR mutation or inhibition in cells significantly suppresses Cl- secretion and delays wound healing [6, 7, 15, 51]. This discrepancy is likely due to the fact that we only performed 1 h of CLIC treatment to increase [Cl-]i instead of stimulating cells for a long time to affect the physiological function of CLCs. Furthermore, we found that cell migration showed the maximal acceleration when [Cl-]i was increased to 70 mM and then decreased when [Cl-]i was further increased from 70 mM to 100 mM. Interestingly, these results were similar to previous results for the expression of chloride-dependent genes, such as the multifunctional ribosomal protein RPS27, which exhibits biphasic regulation in response to different [Cl-]i (25, 75, and 125 mM) [13, 26]. During the repair process, cell proliferation in the groups subjected to elevated [Cl-]i was not evident in our study. Therefore, the effect of elevated [Cl-]i on repair capability observed in this study suggested an intimate relationship between [Cl-]i and cell migration.
Cell migration plays a crucial role in the wound repair of airway epithelial cells. The multiple stages of cell migration are closely associated with the dynamic assembly and disassembly of the F-actin cytoskeleton and with the remodeling of the mechanical properties of epithelial cells [52-55]. Similar to other ions (for example Ca2+ and K+), elevated [Cl-]i might serve as an intracellular signal and is involved in cell migration [56-58]. Our data showed that elevated [Cl-]i markedly increased the amount of threadlike stress fibers in the central area of the cells and decreased cytoskeletal stiffness and thus improved cell migration [55]. A Transwell assay showed that the number of migrating cells increased when [Cl-]i was clamped at a high level, indicating that, in agreement with the reduction in cell stiffness, deformation capacity increased. Notably, the well-organized peripheral F-actin bundles of 16HBE14o- cells showed some disruption when [Cl-]i was clamped at 100 mM (Figure 3); this effect was accompanied by an increase in cell stiffness (Figure 4). These results could account for the decreased migration rate of the 100 mM [Cl-]i group compared with that of the 70 mM [Cl-]i group. The significant inhibition of wound closure in response to the silencing of CFTR expression or the inhibition of CFTR in airway epithelial cells demonstrated in previous studies may be partially ascribed to the function of CFTR as a cytoskeletal complex component that contains ezrin and actin [3, 59].
Cell migration is associated with several signaling cascades and their downstream effectors, including the MAPK cascade, scaffold proteins, and lipid kinase signaling pathways. Nevertheless, the Rho family of GTPases appears to play a prominent role in modulating cell migration [21-23]. Moreover, high extracellular NaCl concentration activates GEF-H1, a guanine nucleotide exchange factor (GEF) that directly induces RhoA activation [60]. This effect suggests that GEF-H1 may be regulated by [Cl-]i and may be implicated in airway epithelial cell migration. However, further experimental verifications are needed. Here, we assessed the activation of Rho GTPases in airway epithelial cells by using a FRET-based method (Figure 5), which accurately reflects cellular RhoA activity. Given that the existence of chloride-dependent genes and multiple Cl- sensing proteins have been documented [13, 15], we hypothesized that the substantial increase in RhoA activity and dispersed distribution in the whole cytoplasm with increased amounts of threadlike stress fibers throughout the cell upon stimulation by [Cl-]i elevation might be responsible for cytoskeleton reorganization (Figure 3, indicated by arrows and triangles). Moreover, previous reports have indicated that the phosphorylation of LIM-kinase by ROCK contributes to the Rho-induced reorganization of cellular physical properties in cytoskeletal formation and stiffness [61-63].
Ca2+ contributes to RhoA activation and wound repair [64-66]. The Cl- conductance stimulated by increased [Ca2+]i in epithelial cells exhibits a time-dependent relaxation of the Cl- current [67, 68], suggesting that intracellular Cl- associates with Rho GTPases. Our study showed that elevated [Cl-]i activated Rho GTPase and caused LIMK1/2 phosphorylation, which is sensitive to Y-27632 (the Rho-associated kinase ROCK inhibitor) and ROCK1 siRNA, without changing [Ca2+]i (Figure 6 and 7). Similar to other studies, our study showed that elevated [Cl-]i augmented glucocorticoid inducible protein kinase 1 activity [15] and up-regulated glutaredoxin-related protein 5 [13] in a concentration-dependent fashion. These effects plateaued at approximately 70 mM [Cl-]i. Moreover, in CF cells, the RPS27 gene is positively regulated by 5-75 mM [Cl-]i and negatively regulated by 75-125 mM [Cl-]i [13]. Our study is the first to show that the phospholylation of LIMK1/2 was promoted through biphasic modulation in response to increased [Cl-]i and showed maximal promotion under treatment with 70 mM [Cl-]i. Given that LIM kinases are involved in the reorganization of the actin cytoskeleton [69], we postulated that Cl- may interact with some cytoskeletal regulators. This interaction results in changes in cell stiffness. However, further investigations are needed. Our results were acquired by using cells that were cultured on glass supports. These cells cannot fully reflect the physiological characteristics of polarized epithelial cells. Nevertheless, our results will provide some basic principles related to the relationship among [Cl-]i, the actin cytoskeleton, and cell movement.
Conclusion
We investigated the role of intracellular Cl- in the wound repair of bronchial epithelial cells. The RhoA/ROCK/LIMK pathway was activated by elevated [Cl-]i to promote the migration of airway epithelial cells. This effect was evidenced by increased RhoA activity, augmented LIMK1/2 phosphorylation, elicited F-actin stress-fiber reorganization, and decreased cellular stiffness. In addition, the ROCK inhibitor Y-27632 and ROCK1 siRNA abolished downstream effects without changing [Ca2+]i. Our results demonstrated that intracellular Cl- might exert a modulatory effect on the cellular physical properties and functions of airway epithelial cells, thus providing new insight into the pathophysiological function of Cl-.
Materials and Methods
Cell culture
16HBE14o- cells (an in vitro cultured engineered human bronchial epithelial cell line) were obtained from Professor Dieter Gruenert, University of California San Francisco [70, 71]. The cells were cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS), 100 U/ml streptomycin, and 100 U/ml penicillin in a humidified air atmosphere containing 5% CO2 at 37 °C. Cell culture reagents were purchased from Gibco (Invitrogen, Grand Island, NY, USA). Before the assays, 16HBE14o- cells were starved (no serum) for 12 h. Inhibitors, such as R(+)-IAA94 (40 μM [30]; Sigma-Aldrich, St Louis, USA), CFTRinh-172 (10 μM [15]; Sigma-Aldrich), and Y-27632 (10 μM, Sigma-Aldrich, St Louis, USA) were added to the cells for 1 h.
Measurement of the Stern-Volmer constant for N-(ethoxycarbonylmethyl)-6-methoxyquinolium bromide quenching by Cl-
The double ionophore strategy was used with some modifications as previously described [13, 27] to define the relationship between the fluoescence intensity of the quinolinium-salt-based halide-sensitive fluorescence probe N-(ethoxycarbonylmethyl)-6-methoxyquinolium bromide (MQAE; ab145418, Abcam) and [Cl-]i [72]. Briefly, 16HBE14o- cells were cultured on glass-bottomed cell culture dishes (801002, NEST, China). The cells were subsequently loaded with 5 mM MQAE in the dark for 1 h at 37 °C, washed twice to remove the unbound probe, and then clamped with different [Cl-]i for 1 h. Solutions with different [Cl-]i were prepared by mixing two high-K+ buffers (high KO3 buffer [in mM]: KNO3 [140], CaCl2 [1.3], NaH2PO4 [3.7], KH2PO4 [0.4], NaHCO3 [4.2], MgSO4 [0.7], HEPES [10], D-glucose [5.5] [pH = 7.4]; High-KCl buffer [in mM]: KCl [140], CaCl2 [1.3], NaH2PO4 [3.7], KH2PO4 [0.4], NaHCO3 [4.2], MgSO4 [0.7], HEPES [10], and D-glucose [5.5] [pH = 7.4]) containing the Cl-/OH- exchanger tributyltin (10 μM) and K+/H+ exchanger nigericin (5 μM) to equilibrate extracellular and intracellular pH and Cl-. As previously described, 15 min of tributyltin/nigericin treatment was sufficient to equilibrate [Cl-]e and [Cl-]i, and osmolarity was in the normal range (293-302 mOsm/Kg) [27]. Fluorescence intensity was recorded by using a laser scanning confocal microscope (Zeiss LSM710, Carl Zeiss, Jena, Germany). Fluorescence images of MQAE were acquired at an excitation wavelength of λ = 350 nm. Then, in accordance with the Stern-Volmer plot (F0/F = 1 + KCl [Cl-]), the calibration curve was acquired by fitting fluorescence intensity to the corresponding [Cl-]i. In this equation, F0 represents the mean fluorescence level of the Cl--free clamping solution (high-KNO3 buffer), and the mean fluorescence intensity at different clamping [Cl-]i is defined as F. The Stern-Volmer constant KCl (M-1) is the reciprocal of a linear regression fit [73].
Cell stiffness measurement
The stiffness of 16HBE14o- cells was measured through optical magnetic twisting cytometry (OMTC). The details of this method have been previously described [74]. Briefly, cells were incubated with RGD-coated magnetic ferrimagnetic beads (4.5 μm in diameter) at 37 °C for 20 min. The beads were fabricated in Dr. Jeffery Fredberg's lab at the Harvard School of Public Health. Subsequently, the cells were washed with PBS for three times to remove unbound beads. The cells were placed on a microscope stage with magnetization and twisting coils (OMTC-1D, EOL, Switzerland). The beads were rotated and displaced by a homogeneous magnetic twisting field. Then, images were acquired during a twisting cycle by using a Cell Observer System (Zeiss, Göttingen, Germany). The stiffness of F-actin (G′) was calculated by using the ratio of the applied magnetic torque to the measured lateral bead displacement. The measurement of G′ was repeated 12 times under each experimental condition by using Matbin. We defined G′0 as the baseline cellular stiffness, and G′ was normalized to G′0 in each experiment to compare stiffness values among different experimental batches and groups.
Immunofluorescence assay
Cells were washed three times with PBS and then fixed with 4% paraformaldehyde for 15 min at room temperature. Cells were then rinsed three times with PBS (2 min each time) and subsequently permeabilized with 0.3% Triton X-100 in PBS for 5-6 min. Cells on slides were blocked with 10% BSA in PBS for 30 min at 37 °C. F-actin was stained with phalloidin (teramethylrhodamine isothiocyanate-phalloidin, 1:200 dilution, Yesdrn Biotechnology Co. Ltd, China) at room temperature for 30 min. Nuclei were stained with 4′,6-diamidino-2-phenylindole (Sigma-Aldrich, USA) for 2-3 min. Fluorescence images were taken by using a laser scanning confocal microscope (Zeiss LSM710, Carl Zeiss).
Wound healing assay
The wound healing assay was conducted with a modified method as previously described [28, 75]. Briefly, human airway epithelial cells were grown to 100% confluence and then starved for 12 h. The cells were then clamped with different [Cl-]i and treated with the CLC inhibitors CFTRinh-172 and IAA94 for 1 h after mechanical injury with a p200 pipette tip. The cells were subsequently washed twice, and culture media were substituted with starvation medium (DMEM without FBS and inhibitors). Wound areas were observed and recorded at 0, 4, 8, 16, and 24 h by using a Cell Observer System (Zeiss, Germany) equipped with a CO2 control chamber and temperature. The rate of wound closure after repair was measured by applying ImageJ software (Image-Pro Plus, Version 7.0) and compared with the initial wound area, which was calculated from three representative wound areas and presented in μm2·h-1. ImageJ software and Matlab (The MathWorks) were used to analyze migration rate and trajectories.
Transwell assay
Cell migration was measured by using Transwell chambers (Corning, New York, USA) in accordance with previously described [76]. Briefly, the cells were clamped with different [Cl-]i for 1 h and then plated in Transwell chambers at a density of 2 × 104 in 200 μl of serum-free medium. Next, 800 μl of medium with 10% FBS was added into the lower chambers. After 24 h of incubation at 37 °C, the cells that had migrated to the lower surface were fixed with 4% paraformaldehyde for 15 min and stained with 0.5% crystal violet (Ameresco, LLC, USA) for 6 min. Then, the migrating cells were photographed with an inverted microscope (×200 magnification) and counted by using Image J software.
Cell proliferation assay
The proliferation of 16HBE14o- cells was evaluated through MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide) assay as previously described [28]. Briefly, cells were inoculated into 96-well assay plates at a density of 104 cells per well and then starved for 12 h before stimulation with different [Cl-]i, CFTRinh-172, and IAA94. Then, the cells were incubated with 100 μl of 0.5% MTT solution in each well at 37 °C for 4 h. Subsequently, the supernatants were removed, and dimethyl sulfoxide (AR, Yonghua chemical Technology, China) was added to each well. The mixture was shaken for 10 min to dissolve crystals. Absorbance was acquired by using an automatic microplate reader at 570 nm (Elx100, Thermo Fisher Scientific, Inc, Waltham, MA, USA).
Fluorescence resonance energy transfer microscopy of 16HBE14o- cells
Biosensors with intramolecular fluorescence resonance energy transfer (FRET) that responded to RhoA activation were used to study RhoA activation in living epithelial cells. The RhoA FRET biosensor used in this work was gifted by Professor Klaus Hahn at University of North Carolina and designed as previously described [77]. This biosensor contains a Rho-binding domain (RBD), which specifically binds to GTP·RhoA. This domain is followed by a cyan fluorescent protein (CFP), an unstructured linker of optimized length, a yellow fluorescent protein (YFP), and full-length RhoA. Upon activation by GTP-loading, RBD binds to Rho, changing the relative orientation of the two fluorophores and thus increasing FRET. RhoA activation is approximately proportional to the FRET/CFP emission ratio due to the attachment of fluorescent proteins to one another. Similarly, intracellular Ca2+ was detected by using a fluorescent Ca2+ biosensor that is based on green fluorescent proteins and calmodulin [78, 79]. The binding of Ca2+ causes calmodulin wrapping, thus increasing the FRET between the flanking GFPs.
After transfection with RhoA and Ca2+ biosensors for 36-48 h, 16HBE14o- cells were seeded on fibronectin-coated 15 mm glass-bottomed cell culture dishes (801002, NEST, China) and then starved in 0.5% FBS for 12 h before stimulation. Cell images were collected with a Cell Observer System (Zeiss), which was equipped with a CO2 control chamber and temperature. The following filters were used to acquire images (excitation; dichroic; emission) CFP (424/24 nm; 455; 460/40 nm), and YFP (426/20 nm; 455; 520/30 nm). The emission ratios of YFP/CFP were computed and generated by using Excel (Microsoft) and MetaFluor software to reflect RhoA activation and Ca2+ signals throughout the live cell.
Western blot analysis
Western blot analysis was conducted with a modified method as previously described [76, 80]. Briefly, the total protein of the cells was collected with RIPA Lysis Buffer (Thermo Scientific, USA) containing 1% phenylmethanesulfonyl fluoride on ice. After the detection of protein concentration by using a BCA Kit (Takara, Tokyo, Japan, T9300A), the cell lysates (50 μg of protein) were subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and subsequently transferred onto a PVDF membrane that was blocked with 5% bovine serum albumin (BSA) and then incubated overnight at 4 °C with the following primary antibodies: β-actin (Sigma-Aldrich, St Louis, MO, USA, A5441), LIMK1/2, and p-LIMK1/2 (1:1000, Affinity, USA). Then, the membrane was washed with TBST (TBS 0.1% Tween-20) three times and subsequently incubated with the following secondary antibodies at room temperature for 1 h: IRDye800CW antimouse IgG and goat antirabbit IgG diluted 1/5000 from LO-COR, Biosciences (Lincoln, NE, USA). Immunoblots were evaluated by using an Odyssey Imaging System, and β-actin was used as the loading control.
Silencing of gene expression via small interfering RNA technique
Small interfering RNAs (siRNAs) were transfected into the indicated cells by using Lipofectamine 3000 (Thermo Fisher Scientific) in accordance with standard procedures. The human ROCK1-gene-specific siRNA sequences (designed and synthesized by Ribobio lnc.) were as follows: 5′-GTT CTA TAA TGA CGA ACA A-3′ for siRNA-1, 5′-GCT GCA AGC TAT ATT AGA A-3′ for siRNA-2, and 5′-GTA GAA GAA TGT GCA CAT A-3′ for siRNA-3. The nonsense siRNA sequence 5′-TTC TCC GAA CGT GTC ACG T-3′ was used as the negative control. The efficiency of gene silencing after siRNA transfection was detected through Western blot and real-time PCR analyses.
Statistical analysis
Statistical analysis was conducted by using GraphPad Prism 5.01 software (Graph-Pad Software, San Diego California, USA). Data were presented as mean values ± SD (1 standard deviation) from a representative experiment. Each experiment was independently carried out at least three times. Student's t-test (two-tailed) was used to evaluate the statistical difference between groups. Data for three or more groups were analyzed with one-way analysis of variance followed by Bonferroni for multiple comparisons. P < 0.05 was assumed to denote statistical significance.
Supplementary Material
Supplementary figures.
Acknowledgements
The RhoA FRET Biosensor is a gift from Professor Klaus Hahn at University of North Carolina. This study is supported by the Natural Science Foundation of China (31671188, 81670002, 11532003, 31670950), Youth Science Fund Project of Guangxi medical university (Grant no. GXMUYSF201939).
Author Contributions
WH and MT designed the experiments, interpreted and analysed the results and wrote the manuscript. YW, LL and YP helped with data acquisition and analysis in immunofluorescence and OMTC. YW, JL performed the cell culture and western blot. OM assisted with FRET Microscopy. JW, HL, CL, XQ helped with manuscript review and data interpretation. YX, XQ, LD: conception and design, financial support, data analysis and interpretation, and reviewed the manuscript. All authors reviewed and approved of the submitted manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sacco O, Silvestri M, Sabatini F, Sale R, Defilippi AC, Rossi GA. Epithelial cells and fibroblasts: structural repair and remodelling in the airways. Paediatr Respir Rev. 2004;5:S35-S40
2. Kippelen P, Anderson SD. Airway injury during high-level exercise. Br J Sports Med. 2012;46:385-90
3. Monterisi S, Favia M, Guerra L, Cardone RA, Marzulli D, Reshkin SJ. et al. CFTR regulation in human airway epithelial cells requires integrity of the actin cytoskeleton and compartmentalized cAMP and PKA activity. J Cell Sci. 2012;125:1106-17
4. Favia M, Guerra L, Fanelli T, Cardone RA, Monterisi S, Di Sole F. et al. Na+/H+ exchanger regulatory factor 1 overexpression-dependent increase of cytoskeleton organization is fundamental in the rescue of F508del cystic fibrosis transmembrane conductance regulator in human airway CFBE41o- cells. Mol Biol Cell. 2010;21:73-86
5. Lasalvia M, Castellani S, D'Antonio P, Perna G, Carbone A, Colia AL. et al. Human airway epithelial cells investigated by atomic force microscopy: A hint to cystic fibrosis epithelial pathology. Exp Cell Res. 2016;348:46-55
6. Trinh NTN, Bardou O, Privé A, Maillé E, Adam D, Lingée S. et al. Improvement of defective cystic fibrosis airway epithelial wound repair after CFTR rescue. Eur Respir J. 2012;40:1390-400
7. Cheng SH, Rich DP, Marshall J, Gregory RJ, Welsh MJ, Smith AE. Phosphorylation of the R domain by cAMP-dependent protein kinase regulates the CFTR chloride channel. Cell. 1991;66:1027-36
8. Zhang JT, Jiang XH, Xie C, Cheng H, Da Dong J, Wang Y. et al. Downregulation of CFTR promotes epithelial-to-mesenchymal transition and is associated with poor prognosis of breast cancer. Biochim Biophys Acta. 2013;1833:2961-9
9. Shiozaki A, Otsuji E, Marunaka Y. Intracellular chloride regulates the G(1)/S cell cycle progression in gastric cancer cells. World J Gastrointest Oncol. 2011;3:119-22
10. Kuner T, Augustine GJ. A Genetically Encoded Ratiometric Indicator for Chloride: Capturing Chloride Transients in Cultured Hippocampal Neurons. Neuron. 2000;27:447-59
11. Berglund K, Schleich W, Krieger P, Loo LS, Wang D, Cant NB. et al. Imaging synaptic inhibition in transgenic mice expressing the chloride indicator, Clomeleon. Brain Cell Biol. 2006;35:207-28
12. Succol F, Fiumelli H, Benfenati F, Cancedda L, Barberis A. Intracellular chloride concentration influences the GABAA receptor subunit composition. Nat Commun. 2012;3:738
13. Valdivieso AG, Clauzure M, Massip-Copiz M, Santa-Coloma TA. The Chloride Anion Acts as a Second Messenger in Mammalian Cells - Modifying the Expression of Specific Genes. Cell Physiol Biochem. 2016;38:49-64
14. Yang H, Huang LY, Zeng DY, Huang EW, Liang SJ, Tang YB. et al. Decrease of intracellular chloride concentration promotes endothelial cell inflammation by activating nuclear factor-kappaB pathway. Hypertension. 2012;60:1287-93
15. Zhang Y-L, Chen P-X, Guan W-J, Guo H-M, Qiu Z-E, Xu J-W. et al. Increased intracellular Cl- concentration promotes ongoing inflammation in airway epithelium. Mucosal Immunol. 2018;11:1149
16. Li P, Zhang S, Fan N, Xiao H, Zhang W, Zhang W. et al. Quantitative fluorescence ratio imaging of intralysosomal chloride ions with single excitation/dual maximum emission. Chemistry. 2014;20:11760-7
17. Mariángeles C, Valdivieso AG, Copiz MM, Gustavo S, María Luz T, Santa-Coloma TA. Disruption of interleukin-1β autocrine signaling rescues complex I activity and improves ROS levels in immortalized epithelial cells with impaired cystic fibrosis transmembrane conductance regulator (CFTR) function. Plos One. 2014;9:e99257
18. Fein A, Terasaki M. Rapid increase in plasma membrane chloride permeability during wound resealing in starfish oocytes. J Gen Physiol. 2005;126:151-9
19. Lauffenburger DA, Horwitz AF. Cell Migration: A Physically Integrated Molecular Process. Cell. 1996;84:359-69
20. Gardel ML, Schneider IC, Aratyn-Schaus Y, Waterman CM. Mechanical integration of actin and adhesion dynamics in cell migration. Annu Rev Cell Dev Biol. 2010;26:315-33
21. Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol. 2004;265:23-32
22. Grunwald IC, Klein R. Axon guidance: receptor complexes and signaling mechanisms. Curr Opin Neurobiol. 2002;12:250-9
23. Meyer G, Feldman EL. Signaling mechanisms that regulate actin-based motility processes in the nervous system. J Neurochem. 2002;83:490-503
24. Ruth F, Andrew M, Thomson SAM, Lim RWL, Stokes JV, Marcus F. Calcium regulation of actin crosslinking is important for function of the actin cytoskeleton in Dictyostelium. Int Arch Allergy Immunol. 2003;124:172-5
25. Zhang Y, Yan J, Xu H, Yang Y, Li W, Wu H. et al. Extremely low frequency electromagnetic fields promote mesenchymal stem cell migration by increasing intracellular Ca2+ and activating the FAK/Rho GTPases signaling pathways in vitro. Stem Cell Res Ther. 2018;9:143
26. Valdivieso ÁG, Mori C, Clauzure M, Massip-Copiz M, Santa-Coloma TA. CFTR modulates RPS27 gene expression using chloride anion as signaling effector. Arch Biochem Biophys. 2017;633:103-9
27. Krapf R, Berry CA, Verkman AS. Estimation of intracellular chloride activity in isolated perfused rabbit proximal convoluted tubules using a fluorescent indicator. Biophys J. 1988;53:955-62
28. Tan M, Liu C, Huang W, Deng L, Qin X, Xiang Y. CTNNAL1 inhibits ozone-induced epithelial-mesenchymal transition in human bronchial epithelial cells. Exp Physiol. 2018;103:1157-69
29. Liu Y, Lv H, Ren L, Xue G, Wang Y. Improving the moisturizing properties of collagen film by surface grafting of chondroitin sulfate for corneal tissue engineering. J Biomater Sci Polym Ed. 2016;27:758-72
30. Ushio-Fukai M, Xu Y, Zhu J, Hu X, Wang C, Lu D. et al. CLIC1 Inhibition Attenuates Vascular Inflammation, Oxidative Stress, and Endothelial Injury. Plos One. 2016;11:e0166790
31. Gardel ML, Schneider IC, Yvonne AS, Waterman CM. Mechanical Integration of Actin and Adhesion Dynamics in Cell Migration. Annu Rev Cell Dev Biol. 2010;26:315-33
32. Beli P, Mascheroni D, Xu D, Innocenti M. WAVE and Arp2/3 jointly inhibit filopodium formation by entering into a complex with mDia2. Nat Cell Biol. 2008;10:849-57
33. Elson EL. Cellular mechanics as an indicator of cytoskeletal structure and function. Annu Rev Biophys. 1988;17:397
34. Guck J, Schinkinger S, Lincoln B, Wottawah F, Ebert S, Romeyke M. et al. Optical deformability as an inherent cell marker for testing malignant transformation and metastatic competence. Biophys J. 2005;88:3689-98
35. Madl CM, Lesavage BL, Dewi RE, Dinh CB, Stowers RS, Khariton M. et al. Maintenance of neural progenitor cell stemness in 3D hydrogels requires matrix remodelling. Nat Mater. 2017 16
36. Bernard O. Lim kinases, regulators of actin dynamics. Int J Biochem Cell Biol. 2007;39:1071-6
37. Hoffmann EK, Dunham PB. Membrane Mechanisms and Intracellular Signalling in Cell Volume Regulation. Int Rev Cytol. 1995;161:173-262
38. Deitmer JW, Rose CR. pH regulation and proton signalling by glial cells. Prog Neurobiol. 1996;48:73-103
39. Kidd JF, Thorn P. Intracellular Ca2+ and Cl- Channel Activation in Secretory Cells. Annu Rev Physiol. 2000;62:493-513
40. Bregestovski P, Waseem T, Mukhtarov M. Genetically encoded optical sensors for monitoring of intracellular chloride and chloride-selective channel activity. Front Mol Neurosci. 2009;2:15
41. Li Y, Zhang J, Hong S. ANO1 as a marker of oral squamous cell carcinoma and silencing ANO1 suppresses migration of human SCC-25 cells. Med Oral Patol Oral Cir Bucal. 2013;19:313-9
42. Nighot PK, Moeser AJ, Ryan KA, Ghashghaei T, Blikslager AT. ClC-2 is required for rapid restoration of epithelial tight junctions in ischemic-injured murine jejunum. Exp Cell Res. 2009;315:110-8
43. Qu F, Qin XQ, Cui YR, Xiang Y, Tan YR, Liu HJ. et al. Ozone stress down-regulates the expression of cystic fibrosis transmembrane conductance regulator in human bronchial epithelial cells. Chem Biol Interact. 2009;179:219-26
44. Chao AC, Dix JA, Sellers MC, Verkman AS. Fluorescence measurement of chloride transport in monolayer cultured cells. Mechanisms of chloride transport in fibroblasts. Biophys J. 1989;56:1071-81
45. Lau K, Evans R, Case R. Intracellular Cl- concentration in striated intralobular ducts from rabbit mandibular salivary glands. Pflugers Arch. 1994;427:24-32
46. Eberhardson M, Patterson S, Grapengiesser E. Microfluorometric analysis of Cl- permeability and its relation to oscillatory Ca2+ signalling in glucose-stimulated pancreatic β-cells. Cell Signal. 2000;12:781-6
47. Gilbert D, Franjic-Würtz C, Funk K, Gensch T, Frings S, Möhrlen F. Differential maturation of chloride homeostasis in primary afferent neurons of the somatosensory system. Int J Dev Neurosci. 2007;25:479-89
48. Hille C, Lahn M, Löhmannsröben H-G, Dosche C. Two-photon fluorescence lifetime imaging of intracellular chloride in cockroach salivary glands. Photochem Photobiol Sci. 2009;8:319-27
49. Berryman M, Bruno J, Price J, Edwards JC. CLIC-5A functions as a chloride channel in vitro and associates with the cortical actin cytoskeleton in vitro and in vivo. J Biol Chem. 2004;279:34794-801
50. Ma T, Thiagarajah JR, Yang H, Sonawane ND, Folli C, Galietta LJ. et al. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J Clin Invest. 2002;110:1651-8
51. Valdivieso AG, Marin MC, Clauzure M, Santa-Coloma TA. Measurement of cystic fibrosis transmembrane conductance regulator activity using fluorescence spectrophotometry. Anal Biochem. 2011;418:231-7
52. Wakatsuki T, Schwab B, Thompson NC, Elson EL. Effects of cytochalasin D and latrunculin B on mechanical properties of cells. J Cell Sci. 2001;114:1025-36
53. Wu HW, Kuhn T, Moy VT. Mechanical properties of L929 cells measured by atomic force microscopy: effects of anticytoskeletal drugs and membrane crosslinking. Scanning. 2010;20:389-97
54. Rotsch C, Radmacher M. Drug-induced changes of cytoskeletal structure and mechanics in fibroblasts: an atomic force microscopy study. Biophys J. 2000;78:520-35
55. Remmerbach TW, Wottawah F, Dietrich J, Lincoln B, Wittekind C, Guck J. Oral cancer diagnosis by mechanical phenotyping. Cancer Res. 2009;69:1728-32
56. Molitoris BA, Dahl R, Geerdes A. Cytoskeleton disruption and apical redistribution of proximal tubule Na(+)-K(+)-ATPase during ischemia. Am J Physiol. 1992;263:488-95
57. Ho WT, Chiang TH, Chang SW, Chen YH, Hu FR, Wang IJ. Enhanced corneal wound healing with hyaluronic acid and high-potassium artificial tears. Clin Exp Optom. 2013;96:536-41
58. Moe AM, Golding AE, Bement WM. Cell healing: Calcium, repair and regeneration. Semin Cell Dev Biol. 2015;45:18-23
59. Edelman A. Cytoskeleton and CFTR. Int J Biochem Cell Biol. 2014;52:68-72
60. Ly DL, Waheed F, Lodyga M, Speight P, Masszi A, Nakano H. et al. Hyperosmotic stress regulates the distribution and stability of myocardin-related transcription factor, a key modulator of the cytoskeleton. Am J Physiol Cell Physiol. 2013;304:C115-C27
61. Shu-Er C, Jong-Shyan W, Ming-Rung L, Chien Lin L. Downregulation of p57kip2 promotes cell invasion via LIMK/cofilin pathway in human nasopharyngeal carcinoma cells. J Cell Biochem. 2011;112:3459-68
62. Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A. et al. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 1999;285:895
63. Sharma S, Santiskulvong C, Rao J, Gimzewski JK, Dorigo O. The role of Rho GTPase in cell stiffness and cisplatin resistance in ovarian cancer cells. Integr Biol. 2014;6:611
64. Singh I, Knezevic N, Ahmmed GU, Kini V, Malik AB, Mehta D. Gαq-TRPC6-mediated Ca2+ entry induces RhoA activation and resultant endothelial cell shape change in response to thrombin. J Biol Chem. 2007;282:7833-43
65. Gong MC, Fujihara H, Somlyo AV, Somlyo AP. Translocation of rhoA associated with Ca2+ sensitization of smooth muscle. J Biol Chem. 1997;272:10704-9
66. Sank A, Chi M, Shima T, Reich R, Martin G. Increased calcium levels alter cellular and molecular events in wound healing. Surgery. 1989;106:1141-8
67. Cliff WH, Frizzell RA. Separate Cl-conductances activated by cAMP and Ca2+ in Cl (-)-secreting epithelial cells. Proc Natl Acad Sci. 1990;87:4956-60
68. Pusch M. Ca2+-activated chloride channels go molecular. J Gen Physiol. 2004;123:323-5
69. Fu Q, Wu C, Shen Y, Zheng S, Chen R. Effect of LIMK2 RNAi on reorganization of the actin cytoskeleton in osteoblasts induced by fluid shear stress. J Biomech. 2008;41:3225-8
70. Gruenert DC, Finkbeiner WE, Widdicombe JH. Culture and transformation of human airway epithelial cells. Am J Physiol. 1995;268:347-60
71. Cozens AL, Yezzi MJ, Kunzelmann K, Ohrui T, Chin L, Eng K. et al. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am J Respir Cell Mol Biol. 1994;10:38-47
72. Verkman AS. Development and biological applications of chloride-sensitive fluorescent indicators. Am J Physiol. 1990;259:375-88
73. Kovalchuk Y, Garaschuk O. Two-photon chloride imaging using MQAE in vitro and in vivo. Cold Spring Harb Protoc. 2012;2012:778-85
74. Deng L, Fairbank NJ, Fabry B, Smith PG, Maksym GN. Localized mechanical stress induces time-dependent actin cytoskeletal remodeling and stiffening in cultured airway smooth muscle cells. Am J Physiol Cell Physiol. 2004;287:C440
75. Wang X, Jiang B, Sun H, Zheng D, Xu R-H. Noninvasive application of mesenchymal stem cell spheres derived from hESC accelerates wound healing in a CXCL12-CXCR4 axis-dependent manner. Theranostics. 2019;9:6112-28
76. Hamyeh M, Bernex F, Larive RM, Naldi A, Urbach S, Simony-Lafontaine J. et al. PTPN13 induces cell junction stabilization and inhibits mammary tumor invasiveness. Theranostics. 2020;10:1016-32
77. Olivier P, Louis H, Klemke RL, Hahn KM. Spatiotemporal dynamics of RhoA activity in migrating cells. Nature. 2006;440:1069-72
78. Miyawaki A, Llopis J, Heim R, Mccaffery JM, Adams JA, Ikura M. et al. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 1997;388:882-7
79. Wallrabe H, Periasamy A. Imaging protein molecules using FRET and FLIM microscopy. Curr Opin Biotechnol. 2005;16:19-27
80. Su D, Guo X, Huang L, Ye H, Li Z, Lin L. et al. Tumor-neuroglia interaction promotes pancreatic cancer metastasis. Theranostics. 2020;10:5029-47
Author contact
![]() Corresponding authors: Dr. Yang Xiang (xiangyangedu.cn); Dr. Xiaoqun Qin (qinxiaoqunedu.cn); Dr. Linhong, Deng (dlhedu.cn).
Corresponding authors: Dr. Yang Xiang (xiangyangedu.cn); Dr. Xiaoqun Qin (qinxiaoqunedu.cn); Dr. Linhong, Deng (dlhedu.cn).