Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(17):7635-7644. doi:10.7150/thno.44229 This issue Cite
Research Paper
The prognostic value of a Methylome-based Malignancy Density Scoring System to predict recurrence risk in early-stage Lung Adenocarcinoma
Lu Yang1#, Jing Zhang2#, Guangjian Yang1, Haiyan Xu3, Jing Lin4, Lin Shao4, Junling Li1, Changyuan Guo2, Yanru Du2, Lei Guo2, Xin Li2, Han Han-Zhang4, Chenyang Wang4, Shannon Chuai4, Junyi Ye4, Qiaolin Kang4, Hao Liu4, Jianming Ying2 ![]() , Yan Wang1
, Yan Wang1 ![]()
1. Department of Medical Oncology, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100021, China.
2. Department of Pathology, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100021, China.
3. Department of Comprehensive Oncology, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100021, China.
4. Burning Rock Biotech, Guangzhou, 510300, China.
#L. Yang and J. Zhang contributed equally to this article.
Received 2020-1-23; Accepted 2020-6-1; Published 2020-6-18
Abstract

Current NCCN guidelines do not recommend the use of adjuvant chemotherapy for stage IA lung adenocarcinoma patients with R0 surgery. However, 25% to 40% of patients with stage IA disease experience recurrence. Stratifying patients according to the recurrence risk may tailor adjuvant therapy and surveillance imaging for those with a higher risk. However, prognostic markers are often identified by comparing high-risk and low-risk cases which might introduce bias due to the widespread interpatient heterogeneity. Here, we developed a scoring system quantifying the degree of field cancerization in adjacent normal tissues and revealed its association with disease-free survival (DFS).
Methods: We recruited a cohort of 44 patients with resected stage IA lung adenocarcinoma who did not receive adjuvant therapy. Both tumor and adjacent normal tissues were obtained from each patient and subjected to capture-based targeted genomic and epigenomic profiling. A novel methylome-based scoring system namely malignancy density ratio (MD ratio) was developed based on 39 patients by comparing tumor and corresponding adjacent normal tissues of each patient. A MD score was then obtained by Wald statistics. The correlations of MD ratio, MD score, and genomic features with clinical outcome were investigated.
Results: Patients with a high-risk MD ratio showed a significantly shorter postsurgical DFS compared with those with a low-risk MD ratio (HR=4.47, P=0.01). The MD ratio was not associated with T stage (P=1), tumor cell fraction (P=0.748) nor inflammatory status (p=0.548). Patients with a high-risk MD score also demonstrated an inferior DFS (HR=4.69, P=0.039). In addition, multivariate analysis revealed EGFR 19 del (HR=5.39, P=0.012) and MD score (HR= 7.90, P=0.01) were independent prognostic markers.
Conclusion: The novel methylome-based scoring system, developed by comparing the signatures between tumor and corresponding adjacent normal tissues of individual patients, largely minimizes the bias of interpatient heterogeneity and reveals a robust prognostic value in patients with resected lung adenocarcinoma.
Keywords: stage IA lung adenocarcinoma, disease-free survival, methylome-based malignancy density, genomic and epigenetic signatures, recurrence
Introduction
Curative resection is the standard of care for patients with stage I non-small-cell lung cancer (NSCLC). Current NCCN guidelines do not recommend the use of adjuvant chemotherapy for stage IA lung adenocarcinoma patients with R0 surgery. However, approximately 20% to 40% of patients with stage IA NSCLC experience recurrence within 5 years after surgery [1-3]. Effective recurrence prediction in this group of patients is needed. In addition, although surveillance imaging is also recommended for all resected patients, the level of supportive evidences is low and adherence rates are limited [4, 5]. Hence, stratifying patients according to the predicted recurrence risk may tailor adjuvant therapy and surveillance imaging to patients with a higher risk [6]. Tremendous efforts have been invested to identify clinical and molecular characteristics that might help predict recurrence risk in addition to TNM stage in resected lung cancers [7-11]. Clinical parameters including but not limited to vascular invasion, visceral pleural involvement (VPI), poorly differentiated tumors, and wedge resections, as well as molecular signatures including Ki-67 expression, MACC1 gene amplification, microRNA expression were found to be indicative of prognosis; however, the prediction performance was still not satisfactory [12, 13]. In clinical practice, the prognostic features commonly used in clinical decision-making remain to be the tumor stage and the patient's performance status [14].
DNA methylation, a primary epigenetic modification in mammalian genome often occurring at CpG islands, is an important mechanism in gene and microRNA expression regulation [15] as well as in alternative gene splicing [16]. It plays an essential role in the development as well as the progression and metastasis of lung cancer [17]. Compared with mutation, copy number variation (CNV) and gene/microRNA expression [18-20], DNA methylation was utilized as the most promising marker for the early detection of cancer due to its stability and being easily detected qualitatively and quantitatively [21, 22]. In the past decade, there has been a blossoming of studies on early detection and risk of recurrence of lung cancer by analyzing methylation signatures [23-26]. However, limitations also exist such as small sample size, limited number of selected genes and qualitative instead of quantitative measurement of DNA methylation, which might explain the low reproducibility of these assays [25].
The concept of field cancerization (also unknown as field effect or field defect) was first introduced by Slaughter et al. in 1953 to describe a field of normal-appearing tissue that has been preconditioned by unknown processes so as to predispose it towards development of cancer [27]. Even though initially based on histological observations, field cancerization now has been demonstrated to occur at the molecular level. Precancerous cells adjacent to the tumor cells acquiring tumor-primed genetic alterations have been identified in various organs [28-30]. In addition, epigenetic field cancerization has also been discovered in various types of cancers, including lung [31], stomach [32], liver [33], colon [34], bladder [35] and so on. Field cancerization is now recognized to underlie the development of many types of cancer, including lung carcinomas [36, 37] and might have an etiologic role in a substantial number of recurrences [38]. Therefore, sensitive detection of cancerized fields at high risk of developing malignancy by molecular profiling is highly desirable. However, the whole cancerization idea can't pinpoint the risk. Instead, biomarker based on quantifying the underlying evolutionary process within the cancerized field might have a more robust prognostic value [39]. Genetic diversity, genomic instability and size of clonal expansion have been identified as such evolutionary markers showing prognostic value in blood, Barrett's esophagus and ulcerative colitis [40-43]. Under the hypothesis that the field cancerization of adjacent tissues from the surgery site might be associated with patient's outcome, we developed a novel methylome-based scoring system namely malignancy density ratio (MD ratio) to characterize the degree of field cancerization of the adjacent normal tissues. The aim of the present study is to investigate the association of this MD ratio with the risk of disease recurrence in resected stage IA lung adenocarcinomas.
Methods
Patient information
We retrospectively recruited a total of 44 stage IA (T1a/T1b, N0) lung adenocarcinoma patients who underwent curative resection (without adjuvant therapies) from Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College between October 2011 and November 2015 (follow-up through April 2018). Curative resection was defined as the removal of all malignant (cancerous) tissue to cure the disease. The histopathological and clinical characteristics of patients as well as disease-free survival (DFS) were collected. The study was approved by the institutional review board of Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College. All patients provided written informed consent, in accordance with the Declaration of Helsinki.
DNA isolation from tissues
Paired tumor tissues and adjacent normal tissues were obtained during surgery. Adjacent normal tissues were biopsied >5cm distant from resection margins for lobectomy and segmentectomy, and > 3cm from margins for wedge resection. The absence of tumor cells in the normal tissue samples was confirmed by histopathological assessment. Both samples were subjected to DNA isolation using the QIAamp DNA FFPE Tissue Kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. DNA was quantified with the Qubit 2.0 fluorimeter (ThermoFisher Scientific, Waltham, MA, USA).
Bisulfite sequencing
Forty-four paired tumor and adjacent normal tissues were sequenced using a capture-based bisulfite sequencing panel as described previously [44]. The bisulfite sequencing (BS-seq) library was prepared using the brELSATM method (Burning Rock Biotech, Guangzhou, China). Briefly, purified DNA was treated with sodium bisulfite (D5046, EZ-96 DNA Methylation-Lightning™ MagPrep, Zymo Research, Orange, CA, USA). Subsequently, the converted single-strand DNA molecules were ligated to a splinted adapter, and amplified by a uracil-tolerating DNA polymerase to generate whole-genome BS-seq libraries. Custom-designed methylation profiling RNA baits were used for target enrichment which covers 80,672 CpG sites and spans 1.05 mega base of human genome. The target libraries were subsequently quantified by real-time PCR (Kapa Biosciences Wilmington, MA, USA) and sequenced on NovaSeq 6000 (Illumina, San Diego, CA, USA) with an average sequencing depth of 1,000X.
Methylation data analysis
Trimmomatic (v.0.32) was used to remove custom adaptor sequences and low-quality bases. Paired-end reads were aligned to C to T- and G to A-transformed hg19 genome by BWA-meth (v.0.2.2) [45]. After alignment, duplicate reads were marked by samblaster (v.0.1.20) [46], and low mapping quality (MAPQ<20) or improper pairing reads were removed by sambamba (v.0.4.7) [47] from further downstream analyses. Paired reads were merged by clipping overlapping reads to avoid double-counting of methylation calls. Methylation blocks (MBs) were defined as the genomic region consisting of the neighboring CpG sites which were not only close on distance but also correlated on methylation level. A total of 8,312 MBs were generated from 80,672 CpG sites using a proposed region-defined algorithm. Within all MBs, 84% were annotated in genes with 59% in promoter regions, 7% in exons and 18% in introns. We defined M and U as the methylated and unmethylated reads aligned on a CpG site, respectively. Mij and Uij sum the M and the U in the jth MB for the ith patient, respectively (Table S1). We defined βji as the methylation signature in the jth MB of the ith patient following the formula: βij = Mij/(Mij+Uij).
Generation of the MD ratio and MD score
MD ratio and MD score were generated as illustrated in Figure S1.The cancer-specific blocks namely differential methylated blocks (DMBs) were selected by testing the signature difference between tumor and normal tissues, and MBs with significant difference (P<0.05) were chosen. The personalized DMBs were selected by comparing the differential signatures in tumor and paired normal tissues of a given patient. We estimated the baseline methylation signature βj(0) based on the methylation data of normal lung tissues from an internal database via maximum likelihood estimation (MLE). We defined βij(a) and βij(t) as the methylation signature for adjacent normal tissue and tumor tissue of the ith patient respectively, which follow the equation:
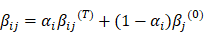
Assuming that the methylation signature follows a mixed beta-binomial model, the αi (MD ratio) of each patient was estimated by MLE which reflects the proportion of methylation signature in the adjacent normal tissue shared by its corresponding malignant tumor tissue. Hessian Matrix was used to estimate the variance of the MD ratio estimator. MD score was obtained by Wald statistics under the null hypothesis of MD ratio equals to 0, representing the density of malignant signature present in the adjacent normal tissue of an individual patient.
Targeted DNA sequencing for genomic characterization
Capture-based targeted sequencing for somatic mutation profiling was performed on 44 tumor samples using a panel consisting of 520 cancer-related genes (Table S2). Ten randomly selected adjacent normal tissues were also subjected to capture-based targeted sequencing for mutation profiling. The NGS library was prepared as previously described [48] and sequenced on a NextSeq 500 (Illumina, Inc., San Diego, CA, USA) with pair-end reads with an average depth of 1,000X. The sequencing data in a FASTQ format were mapped to the human genome (hg19) using BWA aligner 0.7[49]. Local alignment optimization, mark duplication, and variant calling were performed using the Genome Analysis ToolKit (GATK) 3.2[50], Picard (http://picard.sourceforge. net/) and VarScan[51]. Gene translocations were identified with FACTERA [52] and the CNV was called with an in-house algorithm based on sequencing depth [53].
Statistical analysis
Statistical analysis was performed using R version 3.3.3 software. Differences in groups were calculated and presented either by Fisher's exact test or paired two-tailed Student's t-tests, as appropriate. Wilcox test was used to study the correlation of MD ratio with tumor cell fraction in tumor tissue. A receiver operating characteristic (ROC) curve was generated to identify the cut-off of MD ratio. Kaplan-Meier analysis was used to estimate survival functions, and a log-rank test was used to determine the difference in the survival curves between groups. p < 0.05 was considered statistically significant. Possible predictors of DFS were investigated using Cox univariate or multivariate proportional-hazards analysis.
Results
Characteristics of patients
We retrospectively recruited 44 patients with stage IA lung adenocarcinoma who underwent curative resection without adjuvant therapy. The demographic and clinical characteristics of patients were summarized in Table 1. The median age of this cohort was 61 years, ranged from 40 to 81 years. Among them, 28 (63.6%) were males. Twenty-five (56.8%) patients had a history of smoking. The median tumor diameter was 1.5cm. The perineural invasion (PNI) and spread through air spaces (STAS) were present in 6 (13.6%) and 18 (40.9%) patients, respectively. Visceral pleural invasion (VPI) was not found in any of the patients. Eleven (25%) patients underwent pulmonary lobectomy and 31 (70.5%) had thoracoscopic lobectomy. Only 2 (4.5%) were treated with thoracoscopic wedge resection. Twenty-nine (65.9%) patients had right lung resected. After a median follow-up time of 41.5 months, the median DFS of this cohort was 33 months, ranged from 3.3 - 69.3 months. None of the characteristics significantly correlated with DFS (Table 1).
Clinicopathological characteristics of patients
| Characteristics | All (n=44) | Correlation with DFS |
|---|---|---|
| Age, years | P=0.134 | |
| Median (Min, Max) | 61 (40,82) | |
| Gender, n (%) | P=0.051 | |
| Female | 16 (36.4%) | |
| Male | 28 (63.6%) | |
| Smoking history, n (%) | P=0.151 | |
| No | 18 (40.9%) | |
| Yes | 25 (56.8%) | |
| Unknown | 1 (2.3%) | |
| Tumor diameter, cm | P=0.068 | |
| Median (Min, Max) | 1.5 (0.7,2.0) | |
| PNI, n (%) | P=0.512 | |
| No | 38 (86.4%) | |
| Yes | 6 (13.6%) | |
| STAS, n (%) | P=0.141 | |
| No | 24 (54.6%) | |
| Yes | 18 (40.9%) | |
| Unknown | 2 (4.5%) | |
| VPI, n (%) | - | |
| No | 44 (100%) | |
| Surgical procedure, n (%) | P=0.085 | |
| Pulmonary lobectomy | 11 (25%) | |
| Thoracoscopic lobectomy | 31 (70.5%) | |
| Thoracoscopic wedge resection | 2 (4.5%) | |
| Surgery Location, n (%) | P=0.06 | |
| Right lung | 29 (65.9%) | |
| Left lung | 15 (34.1%) | |
| Follow-up time, months | - | |
| Median (Min, Max) | 41.5 (8.1, 74.9) | |
| DFS, months | - | |
| Median (Min, Max) | 33 (3.3, 69.3) |
PNI: Perineural invasion; STAS: Spread through air spaces; VPI: Visceral pleural invasion; DFS: Disease-free survival. P-value was calculated by Cox univariate proportional-hazards analysis.
The prognostic values of MD ratio and MD score
Paired tumor and adjacent normal tissue samples from 44 patients were subjected to bisulfite sequencing and 39 of them generated data with sufficient quality for both paired samples, therefore, underwent further methylation analysis and MD ratio calculation. The cancer-specific blocks for each patient were selected by comparing the methylome-based signatures between the patient's tumor and adjacent normal tissues. MD ratio of each patient was estimated by MLE, which reflects the proportion of malignant methylation signal in the adjacent normal tissue shared by its corresponding malignant tumor tissue. MD ratio ranged from 0 to 0.2 with a majority of samples close to 0 (Figure 1A). Next, we performed a ROC analysis to derive a cut-off for MD ratio to discriminate patients who relapsed during the follow-up from those who did not (Figure 1B). MD ratio of 0.00979 was identified, with an area under curve of 76.3%. We observed a significantly shorter postsurgical DFS in patients with a MD ratio greater than 0.00979 (high-risk) compared with those with a low-risk MD ratio (33 months vs. NR, HR=4.47, P=0.01, Figure 1C).
The association of malignancy density (MD) ratio and prognosis in resected stage IA adenocarcinoma patients (n=39) A. The distribution of MD ratio; B. The ROC curve of MD ratio to discriminate patients relapsed from those did not during the follow-up; C. The disease-free survival (DFS) in patients with different MD ratios, cut-off=0.00979.
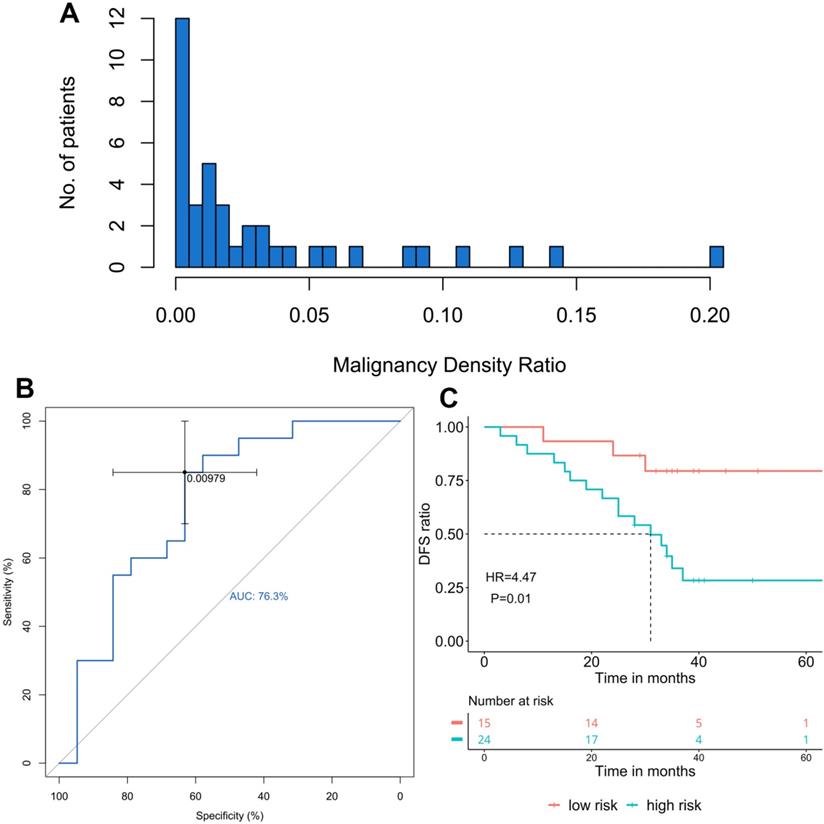
We also investigated the correlation between the MD ratio and some histological characteristics, including T stage, tumor cell fraction and inflammatory status (Table 2). No significant correlation between MD ratio and T1a/T1b was observed (P=1). Furthermore, neither tumor cell fraction (P=0.548) nor inflammatory status (P=0.748) of adjacent normal tissue correlated with MD ratio, suggesting that the MD ratio derived from this panel is an independent predictive factor for prognosis. Furthermore, we investigated the associations of tumor cell fraction and inflammatory status with DFS by univariate analysis, and neither of them significantly correlated with DFS (HR=0.125, P=0.097; HR=0.747, P=0.53).
The association of MD ratio with histopathological features
| Characteristics | MD ratio | P-value | ||
|---|---|---|---|---|
| All (n=39) | Low-risk (n=15) | High-risk (n=24) | ||
| Stage, no. (%) | Pa=1 | |||
| T1a | 4 (10.3%) | 1(6.7%) | 3 (12.5%) | |
| T1b | 35 (89.7%) | 14(93.3%) | 21 (87.5%) | |
| Adjacent normal tissue status, no. (%) | Pa=0.748 | |||
| Normal | 19(48.7%) | 8(53.3%) | 11 (45.8%) | |
| Inflammatory * | 20(51.3%) | 7(46.7%) | 13 (54.2%) | |
| % tumor cells in tumor, median (range) | 0.5(0.1-0.9) | 0.5(0.2-0.9) | 0.5 (0.1-0.8) | Pb=0.548 |
*Inflammatory status includes congestion, emphysema, pulmonary bullae with interstitial fibrosis, obstructive pneumonia; a. P-value was calculated by Fisher's exact test; b. P-value was calculated by Wilcox test.
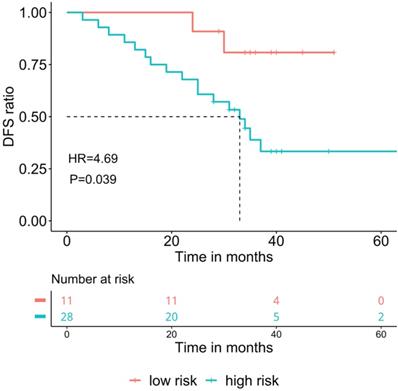
Since MD ratio is a point-estimation on the proportion of tumor-shared methylation signature in the adjacent normal tissue, a raw statistic which lacks the statistical significance. We further developed an MD score, which was obtained by Wald statistics under the null hypothesis that MD ratio equals to 0. Statistically, the MD score is positively correlated with the probability (p-value) of the condition that the malignant signature presents in the adjacent normal tissue of a given patient. As a Wald statistic, MD score follows a chi-square distribution with 1 degree of freedom and takes the variance of MD ratio into consideration, so it is a more robust marker to stratify patients. We used 1.96 (p-value<0.05) as the calling threshold of the high-risk score. Patients with a high-risk MD score also showed significantly poorer prognosis (DFS: 33 months vs. NR, HR=4.69, P=0.039, Figure 2).
The association of malignancy density (MD) score and prognosis in resected stage IA adenocarcinoma patients (n=39). The cut-off for high-risk group was 1.96, DFS: Disease-free survival.
The genomic profile and correlation with prognosis
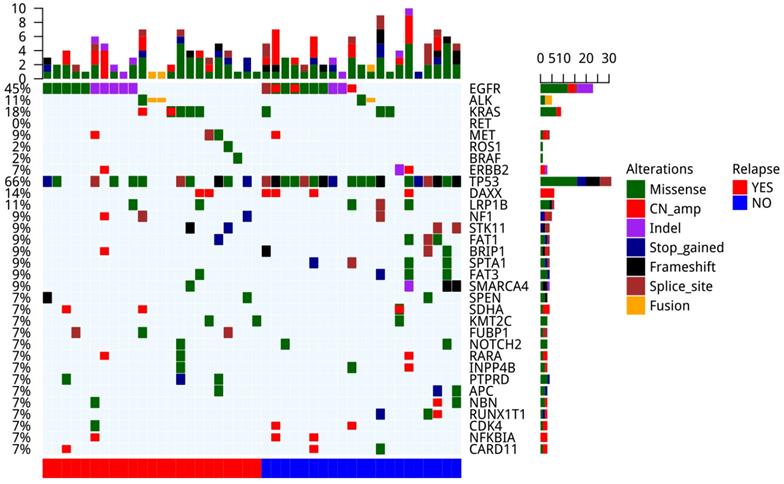
We performed a comprehensive analysis on the genomic alternations in 44 tumor tissue samples (Figure 3). Driver mutations were detected in 33 out of the 44 samples (70%), including EGFR driver mutations (n=18, 41%), ALK fusion (n=3, 7%), KRAS G12V (n=7, 16%), MET amp (n=2, 5%), MET 14 splicing (n=1, 2%), ERBB2 amp (n=2, 5%) and ERBB2 20ins (n=1, 2%). The most common concomitant alternations occurred in TP53 gene (66%), followed by DAXX (14%) and LRP1B (11%). TP53 (P=0.011) and SPTA1 (P=0.044) mutated less frequently in patients with recurrence compared with those without (Figure S2).
The landscape of genomic mutations in tumor lesions of patients (n=44).
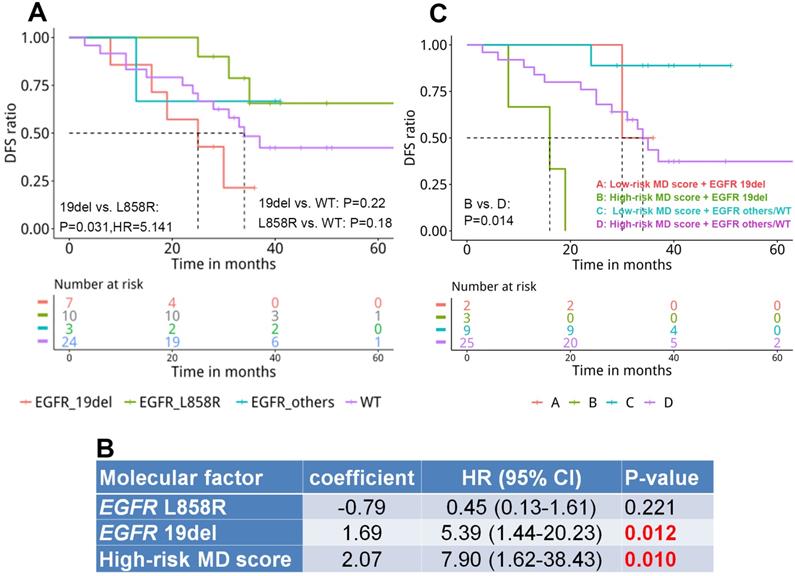
By investigating the correlation between mutation landscape and DFS, we revealed that patients harboring EGFR 19del displayed significantly shorter DFS compared with those carrying EGFR L858R (25 months vs. NR, HR=5.141, P=0.031, Figure 4A). A median DFS of 34 months was observed in WT patients without significant difference compared with EGFR-mutant groups.
The prognostic value of genomic and epigenetic signatures in resected stage IA adenocarcinoma patients. A. The correlation of EGFR driver mutation subtype with disease-free survival (DFS) (n=44); B. Cox multivariate proportional-hazards analysis of the correlation of molecular factors with disease-free survival (n=39); C. The correlation of EGFR driver mutation subtype and MD score with DFS (n=39).
We randomly selected 10 adjacent normal tissues for somatic mutation profiling. All sample had no mutation identified except for one, which was detected with an EGFR G719A with an allele frequency (AF) of 1.43%. This patient's paired tumor tissue also harbored EGFR G719A with a higher AF of 40.30%. Concordantly, both MD ratio and MD score classified the patient into high-risk group. On the other hand, 7 out of the 9 patients, whose adjacent normal tissues were free of mutation, were stratified into to high-risk by both MD ratio and score, suggesting a superior sensitivity of MD ratio/score to predict recurrence over somatic mutation.
Stratifying patients with both genomic signature and MD score
MD score and EGFR driver mutation, that were significantly associated with DFS in univariate analysis, were included in Cox multivariate proportional-hazards analysis. The multivariate analysis revealed that EGFR 19 del (HR=5.39, P=0.012) and MD score (HR= 7.90, P=0.01) remained as predictors for the risk of developing postsurgical recurrence (Figure 4B). Next, we stratified patients by integrating both prognostic factors and assessed the difference in DFS among subgroups. Our results displayed a significantly shorter DFS in patients with a high-risk MD score and an EGFR 19 del compared with those with a high-risk MD score but without the EGFR 19 del (EGFR others or WT)(P=0.014, Figure 4C). No significant difference between other subgroups was observed due to the small number of patients.
Discussion
Since the global change of DNA methylation occurs very early in the process of carcinogenesis, DNA methylation has been considered as one of the most powerful biomarkers for early detection and screening in cancer. Extensive efforts have been invested in the discovery of methylation biomarkers for lung cancer screening and early detection [25, 54-56]; however, fewer studies have assessed the utility of aberrant methylation profiles to predict recurrence risk after resecting NSCLC. Brock et al. demonstrated that methylation of the promoter region of p16, CDH13, RASSF1A, and APC was associated with early recurrence in surgically-treated patients with stage I (T1-2N0) NSCLC [23]. Belinsky et al. reported the methylation detection of 8 selected genes (CDKN2, MGMT, DAPK1, RASSF1, GATA4, GATA5, PAX5α and PAX5β) in sputum and blood had prognostic value for recurrence in stage IA (pT1N0) or stage IB (pT2N0) NSCLC [24]. More recently, Wang and colleagues derived a classifier based on 16-CpG sites to predict the overall survival of lung adenocarcinoma patients [26]. Sandoval et al. discovered a methylation signature based on 10 sites (HOXA9, C1orf114, TRH, HIST1H4F, SP9, PCDHGB6, OTX2, NPBWR1, TRIM58, and ALX1) that effectively distinguished stage I NSCLC patients with high recurrence risk and low risk [57].Conventionally, molecular or epigenetic biomarkers are often identified by comparing the genomic or epigenomic landscapes of two groups with diverse outcomes. This identification method might be influenced by selection bias present in the groups due to the widespread interpatient and intra-patient heterogeneity [6]. Notably, our panel covers 7 of the 10 sites identified in Sandoval et al. 2013 [57] (Table S3). However, neither any single nor combination of the 7 sites correlated significantly with DFS in our cohort (Table S3, Figure S3).
We established a scoring system by comparing the methylation signatures in a tumor and its adjacent normal tissue of the same individual to reflect the malignant progression of a cancerized field. This classification is independent of patients' DFS and largely attenuates the effect of interpatient heterogeneity. MD ratio demonstrated a significant association with DFS, which was independent of clinicopathological factors including T stage and tumor cell fraction, suggesting its robustness in a heterogeneous population. In addition, epigenetic modifications arising from exposure to the environment, such as the disturbance of DNA methylation in the context of transcriptional level, as well as the signal-transduction and cellular pathways of the inflammatory cascade, has been closely linked with the pathophysiology of inflammatory diseases [58, 59]. Conceivably, the inflammatory status might affect the methylation signatures in the tissue. In our study, the methylome-based MD ratio was independent of the inflammatory status (congestion, emphysema, pulmonary bullae with interstitial fibrosis or obstructive pneumonia) in the adjacent normal tissue because our scoring system was based on the degree of field cancerization of the adjacent normal tissues. In other words, it is a comparison between the tumor tissue and adjacent normal tissue of the same individual and minimizes the selection bias that could be introduced during the identification of conventional markers. Our method indicates the merit of evolutionary markers over selective markers.
The prognostic role of EGFR mutations also remains controversial in patients with resectable lung adenocarcinoma [60, 61]. A recent study [62] conducted in 835 patients, who underwent complete surgical resection for lung adenocarcinoma without EGFR TKIs as a neoadjuvant or adjuvant therapy, showed that patients with 19del had a significantly higher incidence of extrathoracic recurrence than patients with L858R (p =0.004), and the L858R group had a significantly longer recurrence-free survival than the WT group (p < 0.001) and the 19del group (p = 0.016). Concordantly, our results also showed that 19del was an independent genetic predictor significantly associated with a worse prognosis.
Our study was limited by the number of patients enrolled and its retrospective and non-randomized nature. To extend the interesting findings from our work, prospective studies with larger cohorts are required to validate the prognostic value of the MD ratio/score. Nonetheless, we established a methylome-base scoring system to quantitatively assess the malignant progression of adjacent normal tissue based on personalized cancer-specific methylation signatures of individual. The scoring system revealed robust prognostic value in patients with resected stage IA lung adenocarcinoma and was independent of clinicopathological factors and genetic signatures. Using it to characterize the risk of lung cancer and recurrence will facilitate a personalized utility of adjuvant therapy and surveillance imaging in completely resected NSCLCs.
Abbreviations
AF: allele frequency; CNV: copy number variation; DFS: disease-free survival; MBs: methylation blocks; MD: methylome-based malignancy density; NGS: next-generation sequencing; NR: not reached; NSCLC: non-small-cell lung cancer; PNI: perineural invasion; STAS: spread through air spaces; ROC: receiver operating characteristic; VPI: visceral pleural involvement.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
Funding
The study was supported by the CAMS Innovation Fund for Medical Sciences (2017-I2M-2-003).
Competing Interests
Jin Lin, Lin Shao, Han Han-Zhang, Chenyang Wang, Shannon Chuai, Junyi Ye, Qiaolin Kang, and Hao Liu are employees of Burning Rock Biotech.
References
1. Port JL, Kent MS, Korst RJ, Libby D, Pasmantier M, Altorki NK. Tumor size predicts survival within stage IA non-small cell lung cancer. Chest. 2003;124:1828-33
2. Gajra A, Newman N, Gamble GP, Abraham NZ, Kohman LJ, Graziano SL. Impact of tumor size on survival in stage IA non-small cell lung cancer: a case for subdividing stage IA disease. Lung Cancer. 2003;42:51-7
3. Birim O, Kappetein AP, Takkenberg JJ, van Klaveren RJ, Bogers AJ. Survival after pathological stage IA nonsmall cell lung cancer: tumor size matters. Ann Thorac Surg. 2005;79:1137-41
4. Srikantharajah D, Ghuman A, Nagendran M, Maruthappu M. Is computed tomography follow-up of patients after lobectomy for non-small cell lung cancer of benefit in terms of survival? Interact Cardiovasc Thorac Surg. 2012;15:893-8
5. Backhus LM, Farjah F, Zeliadt SB, Varghese TK, Cheng A, Kessler L. et al. Predictors of imaging surveillance for surgically treated early-stage lung cancer. Ann Thorac Surg. 2014;98:1944-51 discussion 51-2
6. Thornblade LW, Mulligan MS, Odem-Davis K, Hwang B, Waworuntu RL, Wolff EM. et al. Challenges in Predicting Recurrence After Resection of Node-Negative Non-Small Cell Lung Cancer. Ann Thorac Surg. 2018;106:1460-7
7. Zhang Y, Sun Y, Xiang J, Zhang Y, Hu H, Chen H. A clinicopathologic prediction model for postoperative recurrence in stage Ia non-small cell lung cancer. J Thorac Cardiovasc Surg. 2014;148:1193-9
8. Zhang Y, Zheng D, Xie J, Li Y, Wang Y, Li C. et al. Development and Validation of Web-Based Nomograms to Precisely Predict Conditional Risk of Site-Specific Recurrence for Patients With Completely Resected Non-small Cell Lung Cancer: A Multiinstitutional Study. Chest. 2018;154:501-11
9. Lopez Guerra JL, Gomez DR, Lin SH, Levy LB, Zhuang Y, Komaki R. et al. Risk factors for local and regional recurrence in patients with resected N0-N1 non-small-cell lung cancer, with implications for patient selection for adjuvant radiation therapy. Ann Oncol. 2013;24:67-74
10. Wu CF, Fu JY, Yeh CJ, Liu YH, Hsieh MJ, Wu YC. et al. Recurrence Risk Factors Analysis for Stage I Non-small Cell Lung Cancer. Medicine (Baltimore). 2015;94:e1337
11. Wang X, Janowczyk A, Zhou Y, Thawani R, Fu P, Schalper K. et al. Prediction of recurrence in early stage non-small cell lung cancer using computer extracted nuclear features from digital H&E images. Sci Rep. 2017;7:13543
12. Uramoto H, Tanaka FJTLCR. Recurrence after surgery in patients with NSCLC. Transl Lung Cancer Res. 2014;3:242-9
13. Uramoto H, Tanaka F. Prediction of Recurrence after Complete Resection in Patients with NSCLC. Anticancer Res. 2012;32:3953-60
14. Kerr KM, Nicolson MC. Prognostic factors in resected lung carcinomas. EJC Suppl. 2013;11:137-49
15. He Y, Cui Y, Wang W, Gu J, Guo S, Ma K. et al. Hypomethylation of the hsa-miR-191 locus causes high expression of hsa-mir-191 and promotes the epithelial-to-mesenchymal transition in hepatocellular carcinoma. Neoplasia. 2011;13:841-53
16. Flores K, Wolschin F, Corneveaux JJ, Allen AN, Huentelman MJ, Amdam GV. Genome-wide association between DNA methylation and alternative splicing in an invertebrate. BMC Genomics. 2012;13:480
17. Kerr KM, Galler JS, Hagen JA, Laird PW, Laird-Offringa IA. The role of DNA methylation in the development and progression of lung adenocarcinoma. Dis Markers. 2007;23:5-30
18. Goncalves CS, Vieira de Castro J, Pojo M, Martins EP, Queiros S, Chautard E. et al. WNT6 is a novel oncogenic prognostic biomarker in human glioblastoma. Theranostics. 2018;8:4805-23
19. Shi J, Bao X, Liu Z, Zhang Z, Chen W, Xu Q. Serum miR-626 and miR-5100 are Promising Prognosis Predictors for Oral Squamous Cell Carcinoma. Theranostics. 2019;9:920-31
20. Wang H, Liang L, Dong Q, Huan L, He J, Li B. et al. Long noncoding RNA miR503HG, a prognostic indicator, inhibits tumor metastasis by regulating the HNRNPA2B1/NF-kappaB pathway in hepatocellular carcinoma. Theranostics. 2018;8:2814-29
21. Zhao Y, Sun J, Zhang H, Guo S, Gu J, Wang W. et al. High-frequency aberrantly methylated targets in pancreatic adenocarcinoma identified via global DNA methylation analysis using methylCap-seq. Clin Epigenetics. 2014;6:18
22. Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer. 2003;3:253-66
23. Brock MV, Hooker CM, Ota-Machida E, Han Y, Guo M, Ames S. et al. DNA methylation markers and early recurrence in stage I lung cancer. N Engl J Med. 2008;358:1118-28
24. Belinsky SA, Leng S, Wu G, Thomas CL, Picchi MA, Lee SJ. et al. Gene Methylation Biomarkers in Sputum and Plasma as Predictors for Lung Cancer Recurrence. Cancer Prev Res (Phila). 2017;10:635-40
25. Guo S, Yan F, Xu J, Bao Y, Zhu J, Wang X. et al. Identification and validation of the methylation biomarkers of non-small cell lung cancer (NSCLC). Clin Epigenetics. 2015;7:3
26. Wang Y, Deng H, Xin S, Zhang K, Shi R, Bao X. Prognostic and Predictive Value of Three DNA Methylation Signatures in Lung Adenocarcinoma. Front Genet. 2019;10:349
27. Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6:963-8
28. Franklin WA, Gazdar AF, Haney J, Wistuba II, La Rosa FG, Kennedy T. et al. Widely dispersed p53 mutation in respiratory epithelium. A novel mechanism for field carcinogenesis. J Clin Invest. 1997;100:2133-7
29. McDonald SA, Greaves LC, Gutierrez-Gonzalez L, Rodriguez-Justo M, Deheragoda M, Leedham SJ. et al. Mechanisms of field cancerization in the human stomach: the expansion and spread of mutated gastric stem cells. Gastroenterology. 2008;134:500-10
30. Yu YP, Landsittel D, Jing L, Nelson J, Ren B, Liu L. et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol. 2004;22:2790-9
31. Guo M, House MG, Hooker C, Han Y, Heath E, Gabrielson E. et al. Promoter hypermethylation of resected bronchial margins: a field defect of changes? Clin Cancer Res. 2004;10:5131-6
32. Nakajima T, Maekita T, Oda I, Gotoda T, Yamamoto S, Umemura S. et al. Higher methylation levels in gastric mucosae significantly correlate with higher risk of gastric cancers. Cancer Epidemiol Biomarkers Prev. 2006;15:2317-21
33. Kondo Y, Kanai Y, Sakamoto M, Mizokami M, Ueda R, Hirohashi S. Genetic instability and aberrant DNA methylation in chronic hepatitis and cirrhosis-A comprehensive study of loss of heterozygosity and microsatellite instability at 39 loci and DNA hypermethylation on 8 CpG islands in microdissected specimens from patients with hepatocellular carcinoma. Hepatology. 2000;32:970-9
34. Shen L, Kondo Y, Rosner GL, Xiao L, Hernandez NS, Vilaythong J. et al. MGMT promoter methylation and field defect in sporadic colorectal cancer. J Natl Cancer Inst. 2005;97:1330-8
35. Wolff EM, Chihara Y, Pan F, Weisenberger DJ, Siegmund KD, Sugano K. et al. Unique DNA methylation patterns distinguish noninvasive and invasive urothelial cancers and establish an epigenetic field defect in premalignant tissue. Cancer Res. 2010;70:8169-78
36. Curtius K, Wright NA, Graham TA. An evolutionary perspective on field cancerization. Nat Rev Cancer. 2018;18:19-32
37. Kadara H, Wistuba II. Field cancerization in non-small cell lung cancer: implications in disease pathogenesis. Proc Am Thorac Soc. 2012;9:38-42
38. Dakubo GD, Jakupciak JP, Birch-Machin MA, Parr RL. Clinical implications and utility of field cancerization. Cancer Cell Int. 2007;7:2
39. Dhawan A, Graham TA, Fletcher AG. A Computational Modeling Approach for Deriving Biomarkers to Predict Cancer Risk in Premalignant Disease. Cancer Prev Res (Phila). 2016;9:283-95
40. Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF. et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477-87
41. Maley CC, Galipeau PC, Finley JC, Wongsurawat VJ, Li X, Sanchez CA. et al. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet. 2006;38:468-73
42. Martinez P, Timmer MR, Lau CT, Calpe S, Sancho-Serra Mdel C, Straub D. et al. Dynamic clonal equilibrium and predetermined cancer risk in Barrett's oesophagus. Nat Commun. 2016;7:12158
43. Salk JJ, Salipante SJ, Risques RA, Crispin DA, Li L, Bronner MP. et al. Clonal expansions in ulcerative colitis identify patients with neoplasia. Proc Natl Acad Sci U S A. 2009;106:20871-6
44. Xia S, Ye J, Chen Y, Lizaso A, Huang L, Shi L. et al. Parallel serial assessment of somatic mutation and methylation profile from circulating tumor DNA predicts treatment response and impending disease progression in osimertinib-treated lung adenocarcinoma patients. Transl Lung Cancer Res. 2019;8:1016-1028
45. Pedersen BS, Eyring K, De S, Yang IV, Schwartz DA. Fast and accurate alignment of long bisulfite-seq reads. arXiv:1401.1129v2 [q-bioGN]. 2014
46. Faust GG, Hall IM. SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics. 2014;30:2503-5
47. Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics (Oxford, England). 2015;31:2032-4
48. Mao X, Zhang Z, Zheng X, Xie F, Duan F, Jiang L. et al. Capture-Based Targeted Ultradeep Sequencing in Paired Tissue and Plasma Samples Demonstrates Differential Subclonal ctDNA-Releasing Capability in Advanced Lung Cancer. J Thorac Oncol. 2017;12:663-72
49. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754-60
50. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297-303
51. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L. et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568-76
52. Newman AM, Bratman SV, Stehr H, Lee LJ, Liu CL, Diehn M. et al. FACTERA: a practical method for the discovery of genomic rearrangements at breakpoint resolution. Bioinformatics. 2014;30:3390-3
53. Yang L, Ye F, Bao L, Zhou X, Wang Z, Hu P. et al. Somatic alterations of TP53, ERBB2, PIK3CA and CCND1 are associated with chemosensitivity for breast cancers. Cancer Sci. 2019;110:1389-400
54. Leng S, Do K, Yingling CM, Picchi MA, Wolf HJ, Kennedy TC. et al. Defining a gene promoter methylation signature in sputum for lung cancer risk assessment. Clin Cancer Res. 2012;18:3387-95
55. Hong Y, Choi HM, Cheong HS, Shin HD, Choi CM, Kim WJ. Epigenome-Wide Association Analysis of Differentially Methylated Signals in Blood Samples of Patients with Non-Small-Cell Lung Cancer. J Clin Med. 2019;8:1307
56. Nikolaidis G, Raji OY, Markopoulou S, Gosney JR, Bryan J, Warburton C. et al. DNA methylation biomarkers offer improved diagnostic efficiency in lung cancer. Cancer Res. 2012;72:5692-701
57. Sandoval J, Mendez-Gonzalez J, Nadal E, Chen G, Carmona FJ, Sayols S. et al. A prognostic DNA methylation signature for stage I non-small-cell lung cancer. J Clin Oncol. 2013;31:4140-7
58. Surace AEA, Hedrich CM. The Role of Epigenetics in Autoimmune/Inflammatory Disease. Front Immunol. 2019;10:1525
59. Kominsky DJ, Keely S, MacManus CF, Glover LE, Scully M, Collins CB. et al. An endogenously anti-inflammatory role for methylation in mucosal inflammation identified through metabolite profiling. J Immunol. 2011;186:6505-14
60. D'Angelo SP, Janjigian YY, Ahye N, Riely GJ, Chaft JE, Sima CS. et al. Distinct clinical course of EGFR-mutant resected lung cancers: results of testing of 1118 surgical specimens and effects of adjuvant gefitinib and erlotinib. J Thorac Oncol. 2012;7:1815-22
61. Zhang Z, Wang T, Zhang J, Cai X, Pan C, Long Y. et al. Prognostic value of epidermal growth factor receptor mutations in resected non-small cell lung cancer: a systematic review with meta-analysis. PLoS One. 2014;9:e106053
62. Hayasaka K, Shiono S, Matsumura Y, Yanagawa N, Suzuki H, Abe J. et al. Epidermal Growth Factor Receptor Mutation as a Risk Factor for Recurrence in Lung Adenocarcinoma. Ann Thorac Surg. 2018;105:1648-54
Author contact
![]() Corresponding authors: Yan Wang, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100021, China. Tel: +86 13911793771, Fax: +86 1067734107, E-mail:wangyanyifucom; Jianming Ying, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100021, China. E-mail: jmyingac.cn.
Corresponding authors: Yan Wang, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100021, China. Tel: +86 13911793771, Fax: +86 1067734107, E-mail:wangyanyifucom; Jianming Ying, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100021, China. E-mail: jmyingac.cn.