Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
ACE2 serves as a receptor for...
Clinical and pathological...
Pathogenic roles of ACE2 in the...
Animal models for pathological...
Therapeutic approaches
Conclusion and Future Directions
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(16):7448-7464. doi:10.7150/thno.48076 This issue Cite
Review
SARS-CoV-2 pandemic and research gaps: Understanding SARS-CoV-2 interaction with the ACE2 receptor and implications for therapy
Prasun K. Datta1,2 ![]() , Fengming Liu1,2, Tracy Fischer1,2, Jay Rappaport1,2
, Fengming Liu1,2, Tracy Fischer1,2, Jay Rappaport1,2 ![]() , Xuebin Qin1,2
, Xuebin Qin1,2 ![]()
1. Division of Comparative Pathology, Tulane National Primate Research Center, Covington, LA 70433, USA.
2. Department of Immunology and Microbiology, Tulane University School of Medicine, New Orleans, LA 70112, USA.
Received 2020-5-11; Accepted 2020-5-28; Published 2020-6-12
Abstract

The COVID-19 pandemic is an emerging threat to global public health. While our current understanding of COVID-19 pathogenesis is limited, a better understanding will help us develop efficacious treatment and prevention strategies for COVID-19. One potential therapeutic target is angiotensin converting enzyme 2 (ACE2). ACE2 primarily catalyzes the conversion of angiotensin I (Ang I) to a nonapeptide angiotensin or the conversion of angiotensin II (Ang II) to angiotensin 1-7 (Ang 1-7) and has direct effects on cardiac function and multiple organs via counter-regulation of the renin-angiotensin system (RAS). Significant to COVID-19, ACE2 is postulated to serve as a major entry receptor for SARS-CoV-2 in human cells, as it does for SARS-CoV. Many infected individuals develop COVID-19 with fever, cough, and shortness of breath that can progress to pneumonia. Disease progression promotes the activation of immune cells, platelets, and coagulation pathways that can lead to multiple organ failure and death. ACE2 is expressed by epithelial cells of the lungs at high level, a major target of the disease, as seen in post-mortem lung tissue of patients who died with COVID-19, which reveals diffuse alveolar damage with cellular fibromyxoid exudates bilaterally. Comparatively, ACE2 is expressed at low level by vascular endothelial cells of the heart and kidney but may also be targeted by the virus in severe COVID-19 cases. Interestingly, SARS-CoV-2 infection downregulates ACE2 expression, which may also play a critical pathogenic role in COVID-19. Importantly, targeting ACE2/Ang 1-7 axis and blocking ACE2 interaction with the S protein of SARS-CoV-2 to curtail SARS-CoV-2 infection are becoming very attractive therapeutics potential for treatment and prevention of COVID-19. Here, we will discuss the following subtopics: 1) ACE2 as a receptor of SARS-CoV-2; 2) clinical and pathological features of COVID-19; 3) role of ACE2 in the infection and pathogenesis of SARS; 4) potential pathogenic role of ACE2 in COVID-19; 5) animal models for pathological studies and therapeutics; and 6) therapeutics development for COVID-19.
Keywords: COVID-19, ACE2, spike protein, pathogenesis, and animal model
Introduction
A recently identified novel betacoronavirus, severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), is the etiological agent of coronavirus disease-2019 (COVID-19). COVID-19 emerged in December 2019 in Wuhan City, China and rapidly spread to the rest of world. With a 4-12% mortality rate, COVID-19 is an emerging threat to global public health. According to the World Health Organization (WHO), China reported a cluster of pneumonia cases in Wuhan, Hubei province on December 31, 2019. It was reported that no deaths were associated with this pneumonia outbreak, which involved 44 patients as of January 4, 2020. On January 5, 2020, WHO announced that the pneumonia outbreak was due to a new virus that originated in the Huanan Seafood market in Wuhan, China, on January 12, 2020, the virus was confirmed as a novel coronavirus (nCoV). The first case of nCoV outside of China was reported in Thailand on January 13, 2020.
On the following day, the WHO announced that there was limited human-to-human transmission of nCoV, mainly through family members, and suggested the possibility of a wider outbreak. On January 30, 2020, the WHO declared that 2019-nCoV was a Public Health Emergency of International Concern (PHEIC). On February 11, 2020, 2019‐nCoV was named severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) by the coronavirus study group of the International Committee on Taxonomy of Viruses and the WHO officially named the disease caused by the 2019‐nCoV as coronavirus disease (COVID‐19). A month later, March 11, 2020, the WHO declared COVID-19 a pandemic, and SARS-CoV-2 infection has since reached pandemic status worldwide. The worldwide spread of this virus from China is associated with international travel and subsequent communal transmission.
Currently, the virus is aggressively and rapidly spreading, infecting millions of people globally, and causing high mortality and morbidity. Indeed, compared to SARS-CoV and MERS-CoV, SARS-CoV-2 is more virulent, infecting a greater number of individuals world-wide than these earlier viruses. The available public health tools to control transmission are passive approaches, such as isolation and quarantine, social distancing, and community containment measures. Moreover, after the current crisis abates, there remains a high probability of a reemergence of SRAS-CoV-2 and COVID-19 in the fall or upcoming years, as there are currently no vaccines or specific treatment available for COVID-19.
While previous recognition of SARS pathogenesis greatly facilitates our understanding of the pathogenesis of COVID-19, our current understanding remains limited and is based largely on clinical observations and a few autopsy findings. A better understanding of the pathogenesis will help us develop better, more specific treatment and prevention strategies for COVID-19. One potential target for therapeutic intervention is angiotensin converting enzyme 2 (ACE2), a peptidase in the renin-angiotensin system (RAS) (Figure 1). ACE2 plays a crucial role in SARS infection and is believed to serve as a major entry receptor for SARS-CoV-2 in humans (Figure 1).
Function of ACE2 in renin-angiotensin system (RAS) and SARS-CoV2 downregulation of ACE2 level in membrane. Angiotensinogen is converted by renin to angiotensin I (Ang I). Ang I is subsequently converted to angiotensin II (Ang II) by ACE, which is expressed on the surface of endothelial cells in lung and kidney. Angiotensin-converting enzyme inhibitors (ACEIs) inhibit the production of Ang II and angiotensin receptor blockers (ARBs) inhibits the binding of Ang II to angiotensin receptors. ACE2 negatively regulates the function of ACE by converting Ang I to Ang 1-9 and Ang II to Ang 1-7. SARS-CoV-2 interacts with ACE2 and infects ACE2-expressing epithelial and endothelial cells in lung and other organs, leading to the down-regulation of ACE2 on endothelium of lung and presumably, other organs, such as kidney. The downregulation of ACE2 leads to unopposed Ang II accumulation, which may accelerate the progress of COVID-19 via increased activity of RAS.
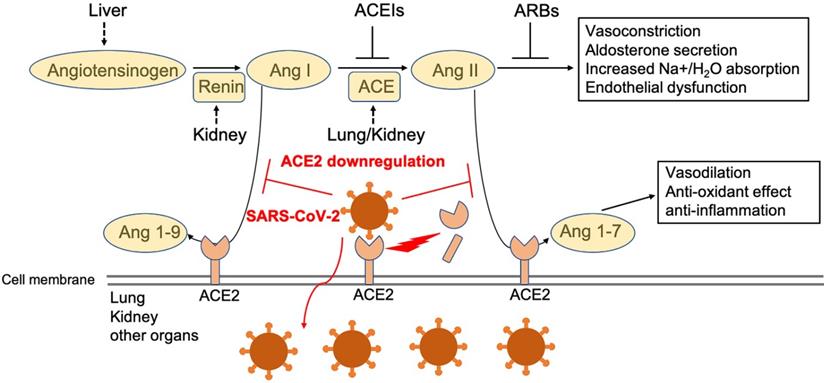
In infected patients, SARS-CoV-2 mainly infects the type II pneumocytes in lungs, and but also infects proximal tubule epithelial cells in kidney in severe cases [1]. These cells express its receptor ACE2, which facilitates viral entry [2, 3]. The endothelial cells in a variety of tissues may be also infected by the virus, although they appear to express little to no ACE2. Infection leads to the downregulation of ACE2 (Figure 1), which impacts the function of Ang II and RAS in a variety of tissues, including lung, heart, vasculature, and kidney, and may facilitate the progression of COVID-19 from mild and moderate to more severe disease (Figure 1). Indeed, direct infection of endothelial cells may explain some of the clinical features of advanced disease, including massive activation of coagulation pathways and platelets, as well as immune cells, such as monocytes and neutrophils in the lung, heart, and kidney. These observations suggest that ACE2 may play a critical pathogenic role in COVID-19, which warrants further extensive investigation. Here, we review the current view of ACE2 in the pathogenesis of SARS-CoV-2 infection.
ACE2 serves as a receptor for SARS-CoV-2
ACE2 as a receptor for SARS-CoV and SARS-CoV-2
ACE2 serves as an entry receptor for SARS-CoV-2 and SARS-CoV in humans by binding to the viral envelope protein spike (S) protein, which may contribute to pathogenesis of SARS [4-7]. The prompt identification of ACE2 as the receptor for SARS-CoV-2 is largely due to its identification as the receptor for SARS-CoV, which emerged approximately 17 years ago. At that time, ACE2 was identified as the functional receptor for SARS-CoV after the fusion protein gene of SARS-CoV was characterized [8]. Through in vitro studies, Li et al [9] found that: 1) ACE2 efficiently binds the S1 domain of the SARS-CoV S protein; 2) a soluble form of ACE2, but not ACE1, blocked association of the S1 domain with ACE2; 3) SARS-CoV replicated efficiently in ACE2-transfected but not mock-transfected 293T cells; and 4) anti-ACE2 but not anti-ACE1 antibody blocked viral replication on Vero E6 cells from African green monkey kidney, a cell sensitive to SARS-CoV [10], Middle East respiratory syndrome (MERS)-CoV [11], and SARS-Cov-2 infection [12]. In addition, exogenous ACE2 expression allows refractory cell lines to support SARS-CoV replication [13]. These results convincingly demonstrate that ACE2 is a functional receptor for SARS-CoV [9].
In vivo studies also consistently demonstrate that ACE2 is a crucial SARS-CoV receptor [14]. A deficiency of ACE2 in mice results in a dramatic decrease in viral replication and much less severe pathologic alterations in lungs as compared to wild-type mice [14, 15]. Transgenic overexpression of human ACE2 (hACE2) in mice renders them more likely to develop severe SARS phenotypes, similar to those seen in human patients [16-18]. The injection of SARS-CoV spike (S) protein into mice worsens acute lung failure in vivo [14]. Recombinant ACE2 can protect mice from severe acute lung injury by blocking SARS-CoV from binding to the membrane-bound form of ACE2 in pneumocytes, which suggests that soluble ACE2 may have a therapeutic potential for treatment of SARS [15], as well as COVID-19. Together, these extensive in vitro and in vivo results demonstrate that ACE2 serves as a major receptor for SARS-CoV infection.
Structural analysis of ACE2 interaction with spike (S) protein of SARS-CoV-2
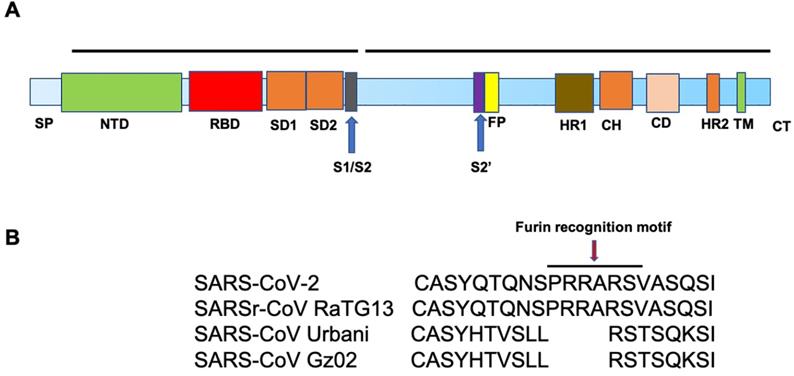
SARS-CoV-2 and SARS-CoV S proteins share 76.5% identity in amino acid sequences [19]. The S protein of SARS-CoV-2 is a 1273 amino acid (aa) protein, consisting of two very important regions referred to as S1 and S2, in addition to the N-terminal 19 aa signal peptide and, in the carboxy terminal, a short transmembrane domain and a short cytoplasmic domain. The S1 region harbors the N-terminal domain (NTD) and a C-terminal domain (CTD), both of which function as the receptor-binding domain (RBD) (Figure. 2A). ACE2 serves as an entry receptor for both SARS-CoV-2 and SARS-CoV in humans via binding to their S proteins [5, 6, 20], which have almost identical 3-D structures. Recent studies from three independent groups, using Biolayer interferometry binding analysis, reported KD values between ACE2 and SARS-CoV RBD of 31, 15.2, and 5.0 nM, and 15.2, 4.7, and 1.2 nM between ACE2 and SARS-CoV-2 RBD [21-23]. 2.5-Å crystal structure of SARS-CoV-2 CTD complexed with hACE2 revealed the SARS-CoV-2 RBD is similar to that of SARS-CoV RBD [24]. Through a separate study, using surface plasmon resonance, ACE2 was reported to bind the SARS-CoV2 S ectodomain with ~15 nM affinity, which is ~10- to 20-fold higher than ACE2 binding to SARS-CoV S [25].
Host cell proteases are required for SARS-CoV-2 entry
Similar to SARS-CoV, SARS-CoV-2 utilizes cellular proteases, such as transmembrane serine protease 2 (TMPRSS2) [26-29] and the endosomal cysteine proteases, cathepsin B and L [30], for S protein priming to enhance virus entry [6, 28]. Proteolysis of the S protein into S1 and S2 is essential for S protein-mediated CoV infection [31]. Interestingly, sequence analysis of SARS-CoV-2 demonstrates the presence of four novel amino acid (PRRA) insertions between S1 and S2 [21], in comparison to SARS-CoV (Figure. 2B), which results in the introduction of a cleavage site by furin, a member of the kexin-like subfamily of proprotein convertases [32]. This sequence is conserved among all SARS-CoV-2 isolates sequenced to date, but is not found in the sequence of the S protein of its closest relative, RaTG13 [33]. In addition, the S1/S2 cleavage site of SARS-CoV-2 S harbors several arginine residues that are not present in RaTG13. The functional significance of this multibasic cleavage site remains to be ascertained. Hoffmann and co-workers demonstrated that the endosomal pH modulator, ammonium chloride, which blocks cathepsin B and L activity, strongly inhibited SARS-CoV-2 entry in TMPRSS2- 293T cells [6]. Furthermore, the TMPRSS2 inhibitor, camostat mesylate, partially blocked SARS-CoV-2-S entry into cells, while E-64d, an inhibitor of cathepsin B and L, in combination with camostat mesylate, fully inhibited SARS-CoV-2 S-mediated entry into cells [6].
TMPRSS2 is required for activation of a viral fusion protein but not for the S protein synthesized in and transported to the surface of cells [28]. TMPRSS2 proteolytically cleaves and activates the S protein in subunit S1, which facilitates viral attachment to the surface of target cells [27, 34]. TMPRSS2 is expressed in lung tissue and subsegmental bronchial branches; in the subsegmental bronchial branches, ACE2 is predominantly expressed in a transient secretory cell type [35]. Colocalization of TMPRSS2 with ACE2 enhances cell entry, which correlates with TMPRSS2-mediated proteolysis of adsorbed S and ACE2 complexes [29]. The significance of TMPRSS2 in mediating efficient virus infection is demonstrated in mice deficient of TMPRSS2, which have reduced body weight loss and virus in lungs following challenge with a mouse-adapted SARS-CoV [36]. ADAM17, a metalloprotease can also cleave ACE2 [37]. TMPRSS2 was found to compete with the metalloprotease ADAM17 for ACE2 processing [37], suggesting that ADAM17-cleaved ACE2 may confer organ protection [38]. Conversely, with the help of TMPRSS2, ADAM17-regulated ectodomain shedding of ACE2 may also support SARS-CoV cell entry through endocytosis, thereby contributing to infection [38]. Thus, the exact role of ADAM17-mediated ACE2 shedding in SARS-CoV-2 infection of kidney and lungs requires further investigation. These findings suggest that a cell surface complex, comprising a primary receptor, ACE2, and TMPRSS2, operates as a major portal for activation of SARS-CoV [29] and SARS-CoV-2 cell entry [6].
Together, these observations demonstrate that priming of the viral S protein by host cell proteases is essential for SARS-CoV-2 entry into cells. This involves cleavage at the S1/S2 and S2′ sites by cellular protease, allowing fusion of the viral and cellular membranes and subsequent viral entry. These processes also contribute to the pathogenesis of SARS and COVID-19 [39].
Clinical and pathological features of COVID-19
Consistent with widespread expression of ACE2, several organ systems appear to be impacted by infection. Indeed, reports of new disease symptoms are disclosed almost daily; however, these may also reflect existing comorbidities or vulnerabilities of the infected individual. Typically, patients initially present with fever, chills, dry cough, and fatigue. Although less common, gastrointestinal complications, including abdominal pain, nausea, vomiting, and/or diarrhea have also been reported [40, 41]. Viral RNA levels from upper respiratory specimens appear to be higher soon after symptom onset, as compared with later times [42-44]. Thus, SARS-CoV-2 can be transmitted prior to the development of symptoms and throughout disease, particularly early in the course [42-44]. A large number of patients first show the infection in the lower respiratory tract (possibly requiring hospitalization) with mild or no symptoms [45]. SARS-CoV-2 has a mean incubation period of 5.2 days and a median duration of the onset of symptoms to death of 14 days. Patients ≥70 years of age have a shorter median duration (from the onset of initial symptoms to death) of 11.5 days, highlighting the vulnerability of this particular patient cohort [45]. Among the more severe cases requiring hospitalization, leukopenia (reduced white blood cells), lymphocytopenia (reduced lymphocytes), and shortness of breath that may advance to acute respiratory distress syndrome (ARDS) and requiring mechanical ventilation are frequently observed [41, 46, 47]. Bilateral lung ground-glass opacity is seen on computed tomography (CT) imaging in the majority of patients with COVID-19, consistent with pulmonary involvement [41, 48, 49, 50].
Studies focused on the clinical characteristics of infection report that patients with underlying comorbidities are more likely to progress to severe respiratory disease and even death [40, 41, 48]. Among these, aging, hypertension, diabetes, and obesity appear to confer the greatest risk for the development of severe disease [41, 47, 51, 52]. The majority of individuals eventually recover from infection, even among those requiring prolonged hospitalization. Concerns regarding whether patients attain full recovery prior to discharge have surfaced following anecdotal reports of a number of “cured” patients testing positive for the virus at a later date. Although it has been posited that this may reflect an absence of protective immunity among these individuals, a recent report demonstrated SARS-CoV-2 in postmortem lung acquired from a patient who had been slated for discharge prior to succumbing to sudden cardiac arrest [53]. Interestingly, this individual had three consecutive SARS-CoV-2 PCR negative nasopharyngeal swabs and improved significantly, indicating she had recovered from the infection. Immunohistochemistry and electron microscopy, however, revealed residual virus in the lung, and histopathological investigation showed diffuse alveolar damage consistent with lung injury previously observed in patients with SARS and MERS [53]. The findings from this postmortem investigation yield important insight into SARS-CoV-2/COVID-19 pathogenesis and argue against the idea that previously infected individuals remain at risk for a second SARS-CoV-2 infection. This remains a significant clinical and public health concern that requires further investigation.
Although lung appears to be the primary site of infection and injury, other tissues are also impacted by COVID-19, particularly the vascular system. A comprehensive autopsy study [54] revealed deep vein thrombosis in 7 of 12 patients (58%) studied, in whom venous thromboembolism was not suspected before death. In the same investigation, pulmonary embolism had been the direct cause of death in 4 patients. Consistently, postmortem examinations reveal diffuse alveolar damage with severe capillary congestion and variegated findings in lung and other organs, suggesting COVID-19-related endothelial cell infection, endotheliitis, and vascular dysfunction [55-58]. Postmortem investigation of tissues that may harbor virus demonstrated that high levels of SARS-CoV-2 RNA in lung of all 12 patients examined, while 5 of the 12 patients demonstrated high viral titers in liver, kidney, or heart [54]. These autopsy observations indicate that the pathogenesis of severe COVID-19 involves significant coagulation and platelet activation, endothelial cell and severe ongoing lung damage, and other organ failure. Although clinical observations and autopsy studies greatly help us to understand the pathogenesis of COVID-19, the mechanism underlying COVID-19 pathogenesis still require further experimental studies.
Pathogenic roles of ACE2 in the infection and pathogenesis of COVID-19
Previous recognition of the role of ACE2 in SARS pathogenesis greatly facilitates our understanding of the pathogenesis of COVID-19. In this section, we will review the function of ACE2 and the RAS and the potential pathogenic role of ACE2 in SARS.
Function of ACE2 and the renin-angiotensin system (RAS)
RAS consists of renin, angiotensin, and aldosterone. RAS plays an important role in regulating blood volume and systemic vascular resistance, which together influence cardiac output and arterial pressure [59-61]. ACE2 is a peptidase of the RAS, a primary cardiovascular regulatory system [59-61]. ACE, a dipeptidyl carboxypeptidase in the RAS, converts the inactive decapeptide Ang I into the active octapeptide and potent vasoconstrictor Ang II (Figure 1) and inactivates the vasodilator bradykinin [62]. ACE2 largely counteracts the activity of ACE by degrading Ang I into the nonapeptide, Ang 1-9, an inactive form of angiotensin. It can also degrade and hydrolyze the vasoconstrictor, Ang II, into a heptapeptide, Ang (1-7), which acts as a potent vasodilator [59-61]. Ang II, a peptide with multiple actions, promotes cardiovascular diseases (CVD), which can be antagonized by the effects of Ang (1-7) [63, 64]. It is important to note that the conversion of Ang II to Ang (1-7) is the primary function of ACE2 and kinetic studies reveal that Ang II is a much better substrate for ACE2 than Ang I [65] (Figure 1). The effects of Ang II are largely mediated through angiotensin II receptor type I (AT1), which is opposed by Ang (1-7) [63] (Figure 1). ACE breaks down Ang (1-7) into Ang 1-5, the major degrading pathway for Ang (1-7) [63].
Ang II is responsible for most of the physiological and pathophysiological effects of the RAS (Figure 1). Inhibitors of ACE that reduce the formation of Ang II have been successfully applied as an effective hypertension treatment (Figure 1). ACE inhibitors are also a standard therapy following myocardial infarction to delay the development of heart failure and reduce the progression of renal disease (Figure 1). ACE2 influences ACE by altering the ratio of Ang II and Ang (1-7), effectively modulating the balance between vasoconstrictors and vasodilators within the heart and kidney [66-68] (Figure 1). Human ACE2 has some homology (40% identity and 61% similarity at the amino acid level) to human ACE [69]. Despite sharing significant homology and many biochemical properties with ACE, ACE2 activity cannot be blocked by classical ACE inhibitors [70].
ACE2 expression pattern and factors influencing ACE2 expression level
The ACE2 gene contains 18 exons that map to chromosome Xp22. Thus, gender differences in the RAS, CVD, and renal diseases may be linked to the ACE2 gene [60, 69]. Both human and mouse ACE2 contain 805 amino acids that include an N-terminal signal sequence, a single active-site catalytic region, and a C-terminal hydrophobic membrane-anchor region [70]. ACE2 is expressed predominantly by epithelial cells of the lungs and kidneys, and cardio myocytes [66], primary targets of SARS-CoV and SARS-CoV-2 [64]. scRNA-seq data analyses recently revealed that ACE2 is highly expressed in organs at risk for injury in the context of COVID-19 and SARS, including lung, heart, esophagus, kidney, bladder, ileum, oral mucosa, and specific cell types (e.g., type II alveolar cells (AT2) in lung, myocardial cells, proximal tubule cells of the kidney, ileum and esophagus epithelial cells, and bladder urothelial cells) [42, 71]. ACE2 protein expression is high in renal and cardiovascular tissues and much of the gastrointestinal tract, particularly ileum, duodenum, jejunum, caecum, and colon [2]. Interestingly, ACE2 expression is regulated by aging and genetic alteration, which may explain the varying susceptibility among different ages and individuals. One study showed no gender-related differences in ACE2 expression between young and middle-aged adults, however, greater ACE2 expression is observed between young adults those of advanced age, which may contribute to the predominance in SARS-CoV-2 infection and development of more severe disease among older individuals [72]. A more recent study, however, does not support this finding [73]. Car, et al analyzed four large-scale datasets of normal lung tissue and found that race, age, or gender do not appear to influence ACE2 gene expression [73]. This study did reveal significantly higher ACE2 gene expression in lung of smokers compared to non-smokers, suggesting that smokers may be more susceptible to COVID-19 [73]. Genetic alterations in the expression of ACE2 develop a diverse pattern of phenotypes, ranging from hypertension and metabolic and behavioral dysfunctions to impairments in serotonin synthesis and neurogenesis [74, 75].
The level of ACE2 is also influenced by some CVD and antihypertensive drugs targeted to RAS, such as Ang II receptor blockers (ARBs), but not ACE inhibitors (Figure 1) [76]. Plasma ACE2 activity is low in healthy subjects but elevated in patients with CVD [77]. ARBS, such as olmesartan, but not ACE inhibitors, increase urinary ACE2 [76]. This potentially offers additional renoprotective effects in patients who use olmesartan [76]. ACE inhibitors increase Ang (1-7) through Ang I conversion by neprilysin so altered ACE2 is not required [63]. Increased Ang II levels are suggested to upregulate ACE2 activity [60]. In ACE2-deficient mice, Ang II levels are approximately double that of wild-type mice, while Ang 1-7 levels are almost undetectable [60]. Loss of ACE2 accelerates diabetic kidney injury in these mice and contributes to the late development of Ang II-dependent glomerulosclerosis [78, 79]. Using adeno-associated virus (AAV) or adenoviral-mediated gene delivery of ACE2 or Ang (1-7) protects against diabetes-induced retinopathy and glomerular injury in rodent models [80-82]. Further, targeting the degradation of Ang II with recombinant ACE2 prevents angiotensin II-dependent hypertension [83] and human recombinant ACE2 reduces the progression of diabetic nephropathy [84]. Together, these findings suggest that ACE2 plays a crucial role in the RAS because it opposes the actions of Ang II [60, 69] (Figure 1). Moreover, ACE2 has a beneficial role and therapeutic potential in many diseases such as hypertension, diabetes, and cardiovascular disease. Because ACE2 performs these vital functions, as well as serving as a receptor for SARS-CoV-2 infection that result in its downregulation, it is conceivable that ACE2 plays a key role in the pathogenesis of SARS and COVID-19.
ACE2 in pathogenesis of SARS
SARS-CoV S protein downregulates ACE2 expression and thereby promotes lung injury [85]. This downregulation effect may result from enhanced shedding/internalizing processes and other unknown mechanisms [85, 86]. Consistently, SARS-CoV replicated efficiently in ACE2-positive Vero cells, which also reduced ACE2 expression, indicating robust receptor interference in the context of SARS-CoV [14]. Since a virus must dock and enter the cells before it can replicate, surface localization of ACE2 and the state of cell differentiation may have a strong impact on SARS-CoV disease and other pathologies triggered by acute lung injury [75]. As discussed above, the maintenance of normal ACE2 levels in lung is beneficial for combatting inflammatory lung disease [66-68]. The RAS acts both systemically and locally in a variety of tissues, including the lung [75]. These results suggest a molecular mechanism for SARS-CoV-associated severe and often lethal lung failure and support a potential therapeutic strategy for preventing or attenuating SARS and COVID-19 [14, 75].
Potential pathogenic role of ACE2 in COVID-19
Although ACE2 is widely recognized as the primary receptor for SARS-CoV-2, its role in the pathogenesis of COVID-19 remains elusive. ACE2 plays an essential role in virus infection and dissemination; however, whether ACE2 contributes to the progression of severe COVID-19 through additional means remains unclear. SARS-CoV-2 infects ACE2-expressing cells in the lung, leading to shedding of ACE2 from tissue, effectively reducing the level of ACE2 expression by infected cells. On the other hand, if ACE2 is shed into the bronchial space by SARS-Cov-2, the peptidase is still active and can cleave Ang II to Ang (1-7). Within the bronchial space, ACE2 can still bind SARS-CoV-2 but doesn't internalize, since it is no longer attached to the apical membrane of the epithelial cells (Figure 1). Whether this shedding prevents other cells from SARS-CoV-2 infection remains elusive and warrants further investigation.
In a mouse model, membrane-bound ACE2 was demonstrated to play a critical role in anti-inflammation through RAS signaling and conversion of Ang II to Ang (1-7), which protected animals against acute lung injury [15]. It is conceivable that the reduction of ACE2 expression may promote inflammation in the lung and the subsequent cytokine storm that presents in many severe COVID-19 patients (Figure 1). ACE2 maintains proper function of heart and kidney and downregulation of ACE2 by SARS-CoV-2 may compromise this protective feature and contribute to the damage caused by infection of these organs (Figure 1). Since ACE2 also serves as a binding site for SARS-CoV-2, enriched expression in lung and other tissues may play a dual role in facilitating virus entry (detrimental) and protection from additional viral attack (protective) (Figure 1), depending on the stage of infection [87, 88]. Of note, ACE2 has collectrin domains that facilitate amino acid transport. There is 47.8% identity between ACE2 collectrin domains and Tmem27 (collectrin) [89]. Since Tmem27 is expressed in multiple tissues including guts and lacks the extracellular peptide domain of ACE2, it is not expected to directly bind and enable internalization of SARS-CoV-2[64, 90]. Animal studies indicate that angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs) may increase ACE2 expression. Patients who take ACEIs and ARBs were assumed to be at increased risk for SARS-CoV-2 infections due to elevated ACE2 expression [91, 92]. Use of ACEIs were thought to cause increased morbidity and mortality of COVID-19 [93], however, this hypothesis is not supported by other clinical findings, where recent studies show that ACEIs/ARBs are unrelated to the severity or mortality of COVID-19 in such patients [94, 95] and may even improve the outcome of COVID-19 as discussed extensively below [96]. These clinical studies suggest that the downregulation of ACE2 caused by SARS-CoV-2 infections may accelerate the progression of COVID-19 from mild to severe disease via increased activity of the RAS (Figure 1).
Many patients with COVID-19 present with dysfunction of multiple organs including heart, kidney, and intestine, which are also rich in ACE2 expression. It is still unclear whether the damage is from direct infection of ACE2-expressed endothelial/epithelial cells at those tissues, or an indirect effect from the whole-body cytokine storm in severe COVID-19 patients. Enriched ACE2 expression in endothelial/epithelial cells might make these tissues vulnerable to the virus entering the circulation system from the lung. SARS-CoV-2 infection of kidney tubules and the digestive system has been documented through biopsy studies [97, 98]. This may result in renal injury/failure and digestive disorders, such as loss of appetite and diarrhea experienced by some infected individuals [97, 98]. Cardiovascular complications are rapidly emerging as a key threat in COVID-19, in addition to respiratory, renal and digestive diseases [99]. Recent findings show the presence of viral elements within endothelial cells and an accumulation of inflammatory cells, in addition to endothelial and inflammatory cell death [99]. These findings suggest that SARS-CoV-2 infection facilitates the induction of endotheliitis in several organs as a direct consequence of viral involvement (as noted by the presence of viral bodies) and of the host inflammatory response [99]. COVID‐19 patients also exhibit some neurological manifestations, including loss of smell, ataxia, and convulsions, indicative of a potential direct or indirect attack on neurons and/or glia [51, 100]. It is likely that disease exaggeration up to the late stage may be attributed not only to direct viral damage, but also generalized hypoxia and/or immune-mediated injury induced by SARS-CoV-2 [101]. Two pathological features occur in severe COVID-19 patients: progressive increase of immune activation and cytokine storm, and an unusual trend of hypercoagulation and platelet activation [101]. However, whether ACE2 expression on cells of multiple organs contribute to multi-organ damage in patients is unclear, as does the role of ACE2 in immune activation and cytokine storm. Whether downregulation of ACE2 in the infected organs facilitates multiple organ failure also remains unclear.
A recent study using an engineered human tissue system demonstrated that soluble human ACE2 can reduce viral growth in vitro and inhibit SARS-CoV-2 infection in blood vessels and human kidney organoids on the early stages of infection [102], suggesting exogenous ACE2 could block early entry of SARS-CoV-2. Further, because of the protective effects of ACE2 on chronic underlying diseases and ARDS, the development of therapeutic enhancers of ACE2 activity, such as diminazene aceturate (DIZE) [103], which are currently used as antiparasitic drugs, has been proposed for the treatment of COVID-19 patients in the late stages of disease [104]. However, due to lung, gut, liver and kidney toxicity, it is very unlikely to enter clinical trial [105, 106]. Further, whether or not administration of soluble ACE2 or ACE2 activator is beneficial in later stages of the disease requires further study. ACE2 gene polymorphisms do not affect the outcome of severe ARDS [107] and results obtained from human studies of the role of ACE2 gene polymorphisms in human hypertension are inconclusive. Some studies suggest that the gene polymorphism is association with human hypertension [108-115], which is not supported in other studies [116]. The role of ACE2 in the development of COVID-19 is complex. It may serve as a binding target, but it may also directly participate in the pathogenesis of disease and contribute to immune activation and dysfunction. Further investigation in animal models and clinical trials is much needed to clarify these mechanisms.
Animal models for pathological and preclinical studies of COVID-19
An ideal animal model for pathogenesis and preclinical studies should reflect the clinical signs, viral replication, and pathology seen in humans [117]. Pathologically, the model should show severe COVID-19 symptoms such as severe pneumonia, multiple organ failure, and even death, all of which are essential for pathogenesis studies. The animal models for COVID-19 will also facilitate the development of a vaccine and therapeutics for prevention and treatment of COVID-19, although they do not need to replicate all aspects of disease to provide useful information for preclinical studies [118]. For these reasons, many labs are rushing to develop COVID-19 models in various species and transgenic animals [119]. Considering SARS-CoV and SARS-CoV-2 utilize the same hACE2 receptor for entry into the host, leading to SARS and COVID-19 in humans, we will review animal models for both SARS and COVID-19.
Animal models for SARS
No single model offers a direct reproduction of what is seen in humans with SARS [118, 120, 121]. In the spontaneous condition without molecular engineering, monkeys, cats, ferrets, mice, swine, chickens, hamsters, guinea pigs, and rats have been experimentally infected [117, 122-124]. Most display mild signs with severe acute pneumonia-like symptoms after infection, and show SARS-CoV replication in their serum followed by spontaneously clearing the virus [117, 122, 123]. Some studies show that ferrets exhibit the strongest clinical symptoms and pathology [120, 123, 125]; however, these findings were not reproduced in later studies [121, 123, 126]. Studies conducted in various species of NHPs such as rhesus macaques, cynomolgus macaques, and African green monkeys support SARS-CoV replication, and the animals develop pneumonitis with variable mild clinical symptoms, depending upon the species employed [120, 127]. No single NHP species is highly preferred at this time [120].
Although mice are the best model for pathogenesis studies of SARS, inbred mouse strains such as B6, BALB/C, and 129 mice infected with SARS-CoV do not show associated signs of clinical illness or overt pathology that are reproducible and equivalent in severity to those observed in SARS patients [118]. In addition, SARS-CoV infects old BALB/C wild type mice (12-14 months old) and shows some severe phenotypes [128], while SARS-CoV-infected aged BALB/c mice demonstrate signs of clinical illness characterized by significant weight loss, hunching, ruffled fur, and slight dehydration measured by skin turgor [128]. Although this age-related increase in morbidity in BALB/c mice represents the clinical characteristics seen in humans in the 2003 SARS outbreak, as well as the current SARS-CoV-2 crisis, in which age is a significant risk factor for severe disease and poor outcome, old mice are very difficult to procure for experimental studies and their immune senescence also complicates pathogenesis studies [118, 129].
Development of mouse-sensitive SARS-CoV strains
To compensate for the disadvantage of using old mice, a mouse-sensitive SARS-CoV derived from the Urbani strain by serial passage in the respiratory tract of young BALB/c mice has been developed [129]. Fifteen passages resulted in a virus (MA15) that is lethal for mice following intranasal inoculation [129]. Lethality is preceded by rapid and high titer viral replication in lungs, viremia, and dissemination of virus to extrapulmonary sites accompanied by lymphopenia, neutrophilia, and pathological changes in the lungs [129]. Abundant viral antigen is extensively distributed in bronchial epithelial cells and alveolar pneumocytes, and necrotic cellular debris is present in airways and alveoli, with only mild and focal pneumonitis [129]. These observations suggest that mice infected with MA15 die from an overwhelming viral infection with extensive, virally mediated destruction of pneumocytes and ciliated epithelial cells [129]. The MA15 virus has six coding mutations associated with adaptation and increased virulence; when introduced into a recombinant SARS-CoV, these mutations result in a highly virulent and lethal virus (rMA15), duplicating the phenotype of the biologically derived MA15 virus [129]. Intranasal inoculation with MA15 reproduces many aspects of disease seen in severe human cases of SARS. Another new strain of SARS-CoV was further derived from serially passaged 25 times in BALB/c mice at 3-day intervals and resulted in a highly lethal strain designated as v2163 [130]. This v2163 strain was more severe, with higher lethality even in 5- to 6-week-old BALB/c mice than MA15 strains. It had nine mutations affecting 10 amino acid residues. Strain v2163 increased interleukin (IL)-1α, IL-6, macrophage inflammatory protein (MIP)-1α, monocyte chemoattractant protein (MCP)-1, and regulated on activation, normal T cell expressed and secreted (RANTES) in mice, with high IL-6 expression correlating with increased mortality [130]. The infection largely mimicked human disease, but lung pathology lacked hyaline membrane formation [130]. Nonetheless, these strains may be very helpful for pathogenesis studies [130]. However, since the strains are mutated to be sensitive to mice but not humans, there are some concerns regarding the clinical relevance obtained by using these strains for preclinical studies in mice.
hACE2 transgenic lines
To compensate for this drawback, transgenic overexpression of hACE2 in mice may serve as a viable alternative for SARS and COVID-19 models [16-18]. Introduction of hACE2 to mice renders them highly sensitive to the development of SARS, as seen in human subjects [16-18]. Yang and co-workers developed a mouse model of human SARS-CoV infection by introducing hACE2, driven by the mouse ACE2 promoter, into the mouse genome [16]. Mice infected with a human SARS strain show pulmonary lesions, including interstitial hyperemia and hemorrhage, monocytic and lymphocytic infiltration, protein exudation, alveolar epithelial cell proliferation and desquamation, and even death [16]. Additionally, SARS-CoV replicated more efficiently in the lungs of transgenic mice than in those of wild-type mice. Other pathologic changes, including vasculitis, degeneration, and necrosis, were found in the extrapulmonary organs of transgenic mice, and viral antigen was found in brain [16]. Two other transgenic mouse models that express hACE2 specifically in the airway and other epithelia, or systemically in multiple tissues and cells, develop a rapidly lethal infection after intranasal inoculation with SARS-CoV [17, 18]. Infection begins in airway epithelia, with subsequent alveolar involvement and extrapulmonary virus spread to the brain [17]. Infection results in macrophage and lymphocyte infiltration in the lungs and upregulation of proinflammatory cytokines and chemokines in both the lung and the brain [17]. Further, another rapid model for SARS and COVID-19 can be achieved by over-expressing hACE2 in lungs of wild type B6 mice by intranasally administering adenovirus 5-expressing hACE2 (Ad5-hACE2). An advantage of this model is that it can be easily adapted for performing pathogenesis studies in any given molecular-engineered mouse lines without multiple mouse crosses. This approach has been successfully used to generate a rapid mouse model for MERS via instilling an Ad5 vector encoding MERS receptor, hDPP4, and prior to MERS-CoV infection [131].
Animal models for COVID-19 for pathogenesis and preclinical studies
Some studies indicate that mice, cats, hamsters, ferrets, and nonhuman primates can be infected by SARS-CoV-2 [132]. After infection, however, only mild phenotypes in lung are seen that do not progress to severe disease. A recent study demonstrates that ferrets and cats are highly susceptible to SARS-CoV-2, while dogs have low susceptibility and livestock, including pigs, chickens, and ducks are not susceptible to the virus [132]. However, SARS-CoV-2 only replicates in the nasal turbinate, soft palate, and tonsils of ferrets, which is not comparable with SARS-CoV, which replicates in both the upper and lower respiratory tract of ferrets [132]. Another study reported that Syrian hamsters infected by SARS-CoV-2 developed mild to moderate clinical syndrome and mild lung pathology changes [133]. The maximal clinical signs included rapid breathing, weight loss, histopathological changes from the initial exudative phase of diffuse alveolar damage with extensive apoptosis to the later proliferative phase of tissue repair, airway and intestinal involvement with virus nucleocapsid protein expression, high lung viral load, and spleen and lymphoid atrophy associated with marked cytokine activation, all of which were observed within the first week of virus challenge [133]. Challenged index hamsters consistently infected naïve contact hamsters housed within the same cage, resulting in similar pathology but not weight loss [133]. All infected hamsters recovered and developed mean serum neutralizing antibody titer ≥1:427 fourteen days post-challenge. Immunoprophylaxis with early convalescent serum achieved a significant decrease in lung viral load but not in lung pathology [133]. Several laboratories across the world have shown with high reproducibility that rhesus macaques are infectable with SARS-CoV-2, showing evidence of virus replication and shedding in nasal swabs with mild clinical signs [119]. A very recent study by SARS-CoV-2-infected rhesus macaques show SARS-CoV-2 infection protects against rechallenge [134], indicating that immunologic approaches to the prevention and treatment of SARS-CoV-2 infection may be possible [134]. Other nonhuman primate models for COVID-19 are currently being developed by several labs, including those at the Tulane National Primate Research Center.
Using hACE2 transgenic mice, Bao et al documented that mice overexpressing hACE2 were infected with SARS-CoV-2. The infected mice showed weight loss and virus replication in lung with mild to moderate changes in lung pathology [135]. The typical histopathology was interstitial pneumonia with infiltration of significant lymphocytes and monocytes in alveolar interstitium, and accumulation of macrophages in alveolar cavities. Viral antigens were observed in bronchial epithelial cells, alveolar macrophages, and alveolar epithelia. This phenomenon was not found in wild type mice with SARS-CoV-2 infection [135]. The pathogenicity of SARS-CoV-2 in hACE2 mice was clarified and the Koch's postulates were fulfilled [135]. Although it does not develop severe clinical manifestations seen in severe COVID-19, this mouse model will facilitate our understanding the pathogenesis of COVID-19. Together with the SARS-CoV-2-infected nonhuman primate, ferret, cat, and hamster models, mouse models will be very useful for the development of therapeutics and vaccines against SARS-CoV-2. However, they require further optimization, including conducting experiments in old mice or mice with induced pre-existing conditions such as hypertension, obesity, diabetes, and respiratory deficits, as seen in human patients, or with immune compromised or conditionally induced-immune cell depleted conditions [136-140].
In summary, there are no perfect animal models that develop severe COVID-19 phenotypes as seen in severe human COVID-19. There is a pressing need to develop animal models that show reproducible and severe COVID-19 symptoms for pathogenesis studies of the role of ACE2 in COVID-19 and preclinical studies of potential therapeutics and vaccines.
Therapeutic approaches
Currently there is no effective antiviral therapy, therefore prevention or treatment of COVID-19 can be an alternative approach to mitigate rapid disease progression and high mortality. Clinical trial studies targeting ACE2/Ang 1-7 axis and ACE2 interaction with the S protein is drawing a great attention for treating COVID-19 patients with lung and cardiovascular damage and preventing the spreading of the disease [141]. Also, these clinical trial studies will greatly facilitate the pathogenesis of COVID-19. Therefore, we will review the current therapeutic and vaccine developments related to blocking ACE2 and S protein interactions and inhibiting RAS, anti-SARS-CoV-2 drugs, and immune-based therapy.
Spike and ACE2 interaction inhibitors
The spike (S) protein mediates viral receptor-binding and membrane fusion. As such, inhibitors that prevent either receptor binding or membrane fusion may serve as viable strategies for preventing further virus dissemination in infected patients. Specifically, disruption of the interaction between ACE2 and the critical motifs within the S2 subunit, consisting of a fusion peptide (FP) region and two heptad repeat regions: HR1 and HR2 (Figure 2) may be particularly effective. In this regard, a pan-coronavirus fusion inhibitor peptide, EK1, targeting the HR1 domains of HCoV S proteins, has been proven to be effective against SARS-CoV-2 S protein-mediated membrane fusion in vitro [142]. The same group has also shown that a lipopeptide version of EK1 known as EK1C4 prevented SARS-CoV-2 S protein-mediated membrane fusion and pseudovirus infection with IC50s of 1.3 and 15.8 nM. This inhibitory effect is 241- and 149-fold more potent than the original EK1 peptide, respectively [143]. With regards to spike protein and ACE2 receptor interaction, an alternative approach is to mop up the virus using high concentrations of APN01, a recombinant soluble form of human ACE2 (rhACE2) to not only block viral entry but also protect lungs from injury. It has been reported that S protein from SARS-CoV does not influence the hydrolysis of Ang II to Ang-(1-7) by soluble ACE2 [144]. Therefore, spike inhibitors may not bind and downregulate ACE2. Ang II or other peptide substrates would directly interfere with SAR-CoV-2 binding and internalization. A clinical trial assessing effects of rhACE2 in COVID19 patients was proposed in China but was later withdrawn (NCT04287686) [64]. However, APN01 (rhACE2), developed by APEIRON, is slated for a multicenter trial in Europe (NCT04335136). It is important to note, that giving ACE2 in the circulation would not necessarily prevent SARS-2 infection. Instead, an intranasal delivery of rhACE2, would likely increase the efficacy for prevention and treatment of COVID-19, which warrants further clinical trial studies. Some other potent recombinant ACE2 drugs such as ACE2-Ig that has a better pharmacological property and a potent neutralizing effect in SARS-CoV-2 pseudotyped virus is under the development for the countermeasure of COVID-19 [145].
A. Schematic representation of the structure of SARS-CoV-2 Spike protein. The different regions on the spike are follows. SP-Signal peptide; NTD-N-terminal domain; RBD-receptor binding domain; SD1-subdomain 1; SD2-subdomain 2; S1/S2- S1/S2 protease cleavage site; S2'-S2' protease cleavage site; FP- fusion peptide; HR1-heptad repeat 1; CH- central helix; CD- connector domain; HR2-heptad repeat 2; TM-transmembrane domain and CT-cytoplasmic tail. B. Illustration of the location of the Furin cleavage site (PRRARS) in SARS-CoV-2 and SARSr-CoV RaTG13 (bat) and the absence of such sequence in SARS-CoV strains, Urbani and GZ02.
Neutralizing antibodies
In contrast to vaccines, since an antibody offers immediate protection, development of an antibody that binds to a conserved epitope on the spike protein of SARS-CoV-2 can neutralize the virus in patients even before they produce their own antibodies. Following studies thus far have been successful in generating neutralizing antibodies targeting the S protein that are effective in vitro. Single domain antibodies (VHHs) from a llama immunized with prefusion-stabilized coronavirus S protein neutralizes SARS-CoV-2 S pseudotyped viruses [146]. A human monoclonal antibody mAb 47D11 that most likely targets the conserved core structure of the S1B RBD, neutralizes SARS-CoV-2 in cell culture [147]. A cross-neutralization antibody S309, potently neutralizes SARS-CoV-2 and SARS-CoV pseudoviruses as well as authentic SARS-CoV-2 by engaging the S RBD [148]. Spike-specifc monoclonal antibodies derived from single B cells of SARS-CoV-2 infected individuals have been isolated and characterized [149]. These neutralizing antibodies will be tested for preventing the spread of COVID-19 and treating COVID-19 severe patients [148]. Further, convalescent plasma therapy for COVID-19 is viable approach to reduce COVID-19 mortality since it appears safe, and clinically effective. In this regard, several studies showed that convalescent plasma obtained from recently recovered COVID-19 patients improved the clinical outcomes by neutralizing viremia in severe COVID-19 cases if started early but not in critically ill patients [150-153].
Immune-Based Therapy
The amplified immune response and cytokine release or “cytokine storm” seen in SARS-CoV-2 infection could result in multiorgan failure. Therefore, monoclonal antibodies directed against inflammatory cytokines or its receptors can be adjunctive therapies for COVID-19. In this regard, clinical studies show increased IL-6 to be a key driver of inflammation [7]. Several randomized clinical trials with Tocilizumab (monoclonal antibody against IL-6), alone or in combination (NCT04310228, ChiCTR20000297), and Sarilumab, another IL-6 receptor antagonist (NCT04315298) are underway in patients with COVID-19 worldwide.
Vaccines
There is an unprecedented need to develop effective COVID-19 vaccine. To meet this challenge there is a world-wide multipronged strategic approach for vaccine development around the globe. In this effort, just released clinical data from phase I study of mRNA-1273, vaccine candidate encoding for a perfusion-stabilized form of the SARS-CoV-2 spike protein, report that patients dosed at 25 µg or 100 µg had seroconverted by day 15 after a single dose. mRNA-1273 vaccination resulted in high neutralizing antibody titers in eight patients that exceeded levels seen in convalescent sera (National Institute of Allergy and Infectious Diseases (NIAID) and Moderna).
In a recent study, DNA vaccine candidates expressing different forms of the SARS-CoV-2 Spike (S) protein was evaluated in rhesus macaques. Vaccinated animals developed humoral and cellular immune responses and neutralizing antibody titers comparable to those found in convalescent humans and macaques infected with SARS-CoV- 2 [154].
The other strategy involves using Ad5-nCoV, a recombinant vaccine incorporating the adenovirus type 5 vector by CanSino, which has received approval from the Chinese authorities to begin human trials. Inovio Pharmaceuticals in collaboration with Beijing Advaccine Biotechnology is developing a DNA vaccine against COVID-19 called INO-4800. Development of a heterologous SARS-CoV RBD recombinant protein has been proposed to be a vaccine approach potential against COVID-19 [155].
Angiotensin converting enzyme inhibitors (ACEI) and angiotensin receptor blockers (ARBs)
Hypertension is a common risk factor for mortality among individuals with COVID-19. It is postulated that the angiotensin system modulation by ACEI, such as enalapril, lisinopril, captopril, and ramipril, or ARBs, such as losartan, candesartan, and valsartan, could be used in prevention and treatment of SARS-CoV-2. The use of these two angiotensin system modulators in clinical management of COVID-19 has taken center stage due to controversial findings in animal studies that show increase in ACE2 expression with use of the ACEI, lisinopril, or ARBs, losartan, at doses that are much higher than that prescribed to patients [156]. Inpatient use of ACEI/ARBs is associated with a lower risk of all-cause mortality and improvement of clinical outcomes compared with ACEI/ARBs non-users [157, 158]. Treatment with ACEIs or ARBs continues to provide cardiovascular and renal protection in patients diagnosed with COVID-19 [157, 158], so discontinuing these medications may be harmful in this patient population [159]. Therefore, several scientific societies highly recommend that patients continue their current hypertensive medication regimen as we await the results of randomized controlled trials addressing the impact of renin-angiotensin system inhibitors (RASIs) on COVID-19 morbidity and mortality [160]. For example, the current Centers for Disease (CDC) Control and Prevention guidance for clinical care of patients with COVID-19 (April 3, 2020) highlights that “…there are no data to suggest a link between ACEIs or ARBs with worse COVID-19 outcomes”. The American Heart Association (AHA), the Heart Failure Society of America (HFSA), and the American College of Cardiology (ACC) recommends continuation of these drugs for patients already receiving them for heart failure, hypertension, or ischemic heart disease (HFSA/ACC/AHA Statement Addresses Concerns Re: Using RAAS Antagonists in COVID-19).
It is important to note that data from human studies show no increase ACE2 expression by ACEI or ARBs [161-163]. A caveat of ARB or ACEi effects on ACE2 expression in human is that only the shed form of ACE2 is studied in plasma or urine where ACE2 content is quite low and local tissue concentration of ACE2 are unknown and require further investigation. Very recent study shows that ACEI offers greater protection than ARBs in COVID-19 patients [164].
Since hyper-inflammation state seen in COVID-19 cases, can be a result of angiotensin peptide (1-7) deficiency (Figure 1), a non-randomized interventional clinical trial to evaluate the possible effect of plasma derived angiotensin peptide (1-7) supplementation on treatment of COVID-19 cases (NCT04375124) is in progress. To examine the therapeutic values of ACEI and ARBs in COVID-19 patients, several clinical trial studies have recently launched. Two randomized clinical trials examining the role of losartan in COVID-19 in USA (NCT04312009, NCT04311177) is underway. A double-blind, placebo-controlled 1:1 randomized clinical trial to assess if ARB valsartan may prevent the development of ARDS and avert morbidity (admission to intensive care unit (ICU) and mechanical ventilation is initiated in Netherland (NCT04335786).
Antiviral drugs
Existing antiviral drugs are effective in treating related, lopinavir-ritonavir, and hydroxychloroquine and chloroquine used in patients during SARS or MERS outbreak, to assess their effectiveness in inhibiting SARS-CoV-2 replication.
Remdesivir
Remdesivir is an inhibitor of the RNA-dependent RNA polymerase. To date, studies on remdesivir show that is highly effective in inhibiting 2019-nCoV infection in Vero E6 cells [165, 166]. In a recent study compassionate-use of remdesivir in patients hospitalized for severe COVID-19 showed clinical improvement in 36 of 53 patients [167]. In a randomized, double-blind, placebo-controlled, multicenter trial in severe COVID-19 patient Hubei, China showed no statistically significant clinical benefit [168]. Preliminary results from a randomized, controlled clinical trial with remdesivir indicate that critically ill COVID-19 patients who were given intravenous infusion of remdesivir had a 31% faster time to recovery than those who received placebo. The median time to recovery was 11 days for patients treated with remdesivir compared with 15 days who were in placebo group (NCT04280705).
Lopinavir-Ritonavir
Lopinavir was shown to have antiviral effect against SARS-CoV-2 virus in Vero E6 cells [166]. A study with lopinavir/ritonavir showed that it does not shorten the duration of SARS CoV-2 shedding [169], however, in a study from Hong Kong, compared with ribavirin alone, patients treated with lopinavir/ritonavir plus ribavirin had a lower risk of ARDS or death caused by SARS-CoV after the onset of symptoms [170].
Hydroxychloroquine and Chloroquine
Chloroquine (CQ) and hydroxychloroquine (HCQ) are aminoquinolines, which have been used to treat malaria and autoimmune diseases. Recent studies show that CQ and HCQ is effective against SARS-CoV-2 infection in Vero E6 cells [165, 171]. However, a prospective randomized trial of 30 adults with COVID-19 in China revealed that HCQ with conventional treatment were compared with 15 patients treated with conventional treatment only showed no effectiveness of HCQ, and one patient in the HCQ group progressed to severe disease [172]. An open-label non-randomized clinical trial showed that azithromycin added to HCQ was significantly more efficient for virus elimination [173]. The outcome of a randomized, double-blind, phase IIb clinical trial (CloroCovid-19 Study) showed that at two different CQ doses as adjunctive therapy for SARS-CoV-2 patients, patients on high dose CQ arm presented more QTc > 500 ms (25%), and higher fatality (17%) than the lower dosage in the first recruited 81 patients [174]. In a recent observational study assessing the effects of chloroquine or hydroxychloroquine, with or without a macrolide, in 96032 hospitalized COVID-19 patients indicate any benefit of 4-aminoquinoline-based treatments in this population and suggest that they could even be increased risk of de-novo ventricular arrythmia [175] These data prove that HCQ or CQ has no therapeutic role either in the treatment or in the prophylaxis of COVID-19.
Advantages and Limitations of therapeutics
SARS-CoV-2 continues to spread globally without any approved therapies and researchers are urgently searching for effective prophylactic and therapeutic interventions. Therefore, repurposing existing drugs approved for other uses could greatly benefit COVID-19 patients around the world despite benefits and limitations. As an example, in vitro studies using drugs such as lopinavir/ritonavir, HCQ and CQ, and remdesivir have shown effectiveness against SARS-CoV-2 however, small non-randomized trials using these drugs alone or in combination have shown mixed results. In the context of convalescent plasma therapy and vaccines, earlier studies have shown that high concentrations of anti-sera against SARS-CoV neutralized SARS-CoV infection, while highly diluted anti-sera significantly increased SARS-CoV infection (Wang et al, 2014). In another study certain epitopes against the S protein enhanced infection in rhesus macaques, and was attributed to antibody-dependent disease enhancement (ADE) [176, 177]. Therefore, even with the selection of perfect adjuvant and appropriate antigen or antigens for SARS-CoV-2 vaccine development, ADE remains a concern when neutralizing antibodies are elicited at sub-neutralizing doses. Randomized, placebo-controlled clinical trials with existing drugs approved for other diseases is an attractive strategy since it may reduce the costs and time. Furthermore, with the existing pharmaceutical supply chain for formulation and distribution there is the possibility of rapid repositioning of drugs for COVID-19 treatment.
In summation, the COVID-19 Treatment Guidelines Panel recommends against the use of the following drugs for the treatment of COVID-19, combination of HCQ and azithromycin because of potential toxicities. The use of lopinavir/ritonavir or other HIV protease inhibitors because of unfavorable pharmacodynamics and negative clinical trial data. We are waiting for the results regarding the therapeutic and vaccine developments related to blocking ACE2 and S protein interactions and inhibiting RAS.
Conclusion and Future Directions
SARS-Cov2 appears to have gained increased capacity for transmission over time, infecting millions of people globally and causing significant morbidity and mortality. This is seen particularly among the elderly and those with comorbid conditions, including advanced age, hypertension, diabetes, and obesity. The severity of the pandemic is far beyond expectation and much worse than previous coronavirus epidemics, including SARS and MERS, which had a comparatively faster but limited spread. Efforts to minimize transmission, including closure of businesses and limitation of many activities, have had significant and life-changing impacts, including personal economic hardships and damage to global economies. There is significant debate regarding the best way to restore economies and bring people back to work, without enabling a second wave of infection. It is critical that we identify a means to end the current pandemic and prevent the emergence of a “second” wave of infection. While the clinical and pathological features of COVID-19 are similar to those of SARS, there are several gaps on our current understanding of COVID-19 pathogenesis. First, although ACE2 serves as a receptor for SARS-CoV2, the pathogenic role of ACE2 may be more complex but remains unclear. Second, the SARS-CoV-2 infection also downregulates ACE2 expression in ACE2-expressing cells via shedding ACE2 from tissue and reducing the level of ACE2 in the infected cells. Whether these changes in tissue or blood have any prognostic value for predicting the disease progress remains unclear [178]. Third, although clinical trial studies with Ang II inhibitors and angiotensin receptor blockers indicates the critical roles of ACE2 COVID-19 pathogenesis, there remains a pressing need to develop effective therapeutic strategies through clinical trials and studies in animal models. Animal models for dissecting the pathogenic role of ACE2 in COVID-19, and for the evaluation of therapeutic strategies targeting to block ACE2 interaction with the S protein and inhibit the immune activation/cytokine storm for modulating severe COVID-19 are also sorely needed. Finally, current and future preclinical and clinical trials of passive antibodies and vaccine strategies focusing on the interaction of ACE2 with the S protein are urgently needed to inform the development of safe and effective vaccines to mitigate morbidity and mortality of COVID-19 infection. Filling these gaps with multiple approaches and effort will add in our understanding of COVID-19 pathogenesis and allow for the identification of targets for therapeutic intervention and the development of safe and effective neutralizing vaccine.
Acknowledgements
This work was supported by NIH 5 P51OD011104-58 (JR) and FAST COVID-19 Grant (TF) and by Tulane start-up funds (XQ, TF, and JP).
Author contribution
PD, FL, TF, JR and XQ developed concept and wrote the manuscript and all authors participated in the review and critique of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Farkash EA, Wilson AM, Jentzen JM. Ultrastructural Evidence for Direct Renal Infection with SARS-CoV-2. J Am Soc Nephrol. 2020 doi: 10.1681/ASN.2020040432
2. Harmer D, Gilbert M, Borman R, Clark KL. Quantitative mRNA expression profiling of ACE 2, a novel homologue of angiotensin converting enzyme. FEBS letters. 2002;532:107-10
3. Soler MJ, Wysocki J, Batlle D. ACE2 alterations in kidney disease. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2013;28:2687-97
4. Li Y, Zhou W, Yang L, You R. Physiological and pathological regulation of ACE2, the SARS-CoV-2 receptor. Pharmacological research. 2020;157:104833
5. Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor recognition by novel coronavirus from Wuhan: An analysis based on decade-long structural studies of SARS. Journal of virology. 2020 doi: 10.1128/JVI.00127-20
6. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S. et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:271-80.e8
7. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z. et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054-62
8. Kuhn JH, Li W, Choe H, Farzan M. Angiotensin-converting enzyme 2: a functional receptor for SARS coronavirus. Cellular and molecular life sciences: CMLS. 2004;61:2738-43
9. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA. et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450-4
10. Ng ML, Tan SH, See EE, Ooi EE, Ling AE. Proliferative growth of SARS coronavirus in Vero E6 cells. The Journal of general virology. 2003;84:3291-303
11. de Wilde AH, Raj VS, Oudshoorn D, Bestebroer TM, van Nieuwkoop S, Limpens R. et al. MERS-coronavirus replication induces severe in vitro cytopathology and is strongly inhibited by cyclosporin A or interferon-alpha treatment. The Journal of general virology. 2013;94:1749-60
12. Matsuyama S, Nao N, Shirato K, Kawase M, Saito S, Takayama I. et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc Natl Acad Sci U S A. 2020;117:7001-3
13. Mossel EC, Huang C, Narayanan K, Makino S, Tesh RB, Peters CJ. Exogenous ACE2 expression allows refractory cell lines to support severe acute respiratory syndrome coronavirus replication. Journal of virology. 2005;79:3846-50
14. Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B. et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11:875-9
15. Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B. et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112-6
16. Yang XH, Deng W, Tong Z, Liu YX, Zhang LF, Zhu H. et al. Mice transgenic for human angiotensin-converting enzyme 2 provide a model for SARS coronavirus infection. Comparative medicine. 2007;57:450-9
17. McCray PB Jr, Pewe L, Wohlford-Lenane C, Hickey M, Manzel L, Shi L. et al. Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus. Journal of virology. 2007;81:813-21
18. Tseng CT, Huang C, Newman P, Wang N, Narayanan K, Watts DM. et al. Severe acute respiratory syndrome coronavirus infection of mice transgenic for the human Angiotensin-converting enzyme 2 virus receptor. Journal of virology. 2007;81:1162-73
19. Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive care medicine. 2020;46:586-90
20. Li G, Clercq ED. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nature Reviews Drug Discovery. 2020;19:149-50
21. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020;181:281-92 e6
22. Tian X, Li C, Huang A, Xia S, Lu S, Shi Z. et al. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg Microbes Infect. 2020;9:382-5
23. Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S. et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215-20
24. Wang X, Xu W, Hu G, Xia S, Sun Z, Liu Z. et al. SARS-CoV-2 infects T lymphocytes through its spike protein-mediated membrane fusion. Cell Mol Immunol. 2020 doi: 10.1038/s41423-020-0424-9
25. Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O. et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260-3
26. Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S. et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:271-80 e8
27. Glowacka I, Bertram S, Muller MA, Allen P, Soilleux E, Pfefferle S. et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. Journal of virology. 2011;85:4122-34
28. Matsuyama S, Nagata N, Shirato K, Kawase M, Takeda M, Taguchi F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. Journal of virology. 2010;84:12658-64
29. Shulla A, Heald-Sargent T, Subramanya G, Zhao J, Perlman S, Gallagher T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. Journal of virology. 2011;85:873-82
30. Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci U S A. 2005;102:11876-81
31. Lu G, Wang Q, Gao GF. Bat-to-human: spike features determining 'host jump' of coronaviruses SARS-CoV, MERS-CoV, and beyond. Trends Microbiol. 2015;23:468-78
32. Izaguirre G. The Proteolytic Regulation of Virus Cell Entry by Furin and Other Proprotein Convertases. Viruses. 2019 11; doi: 10.1128/JVI.00127-20
33. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270-3
34. Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S. et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020; https://doi.org/10.1016/j.cell. 2020 02.052
35. Lukassen S, Chua RL, Trefzer T, Kahn NC, Schneider MA, Muley T. et al. SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. The EMBO journal. 2020: e105114.
36. Iwata-Yoshikawa N, Okamura T, Shimizu Y, Hasegawa H, Takeda M, Nagata N. TMPRSS2 Contributes to Virus Spread and Immunopathology in the Airways of Murine Models after Coronavirus Infection. Journal of virology. 2019 93; doi: 10.1128/JVI.01815-18
37. Heurich A, Hofmann-Winkler H, Gierer S, Liepold T, Jahn O, Pohlmann S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. Journal of virology. 2014;88:1293-307
38. Xiao L, Sakagami H, Miwa N. ACE2: The key Molecule for Understanding the Pathophysiology of Severe and Critical Conditions of COVID-19: Demon or Angel? Viruses. 2020 12; doi: 10.3390/v12050491
39. Bosch BJ, Smits SL, Haagmans BL. Membrane ectopeptidases targeted by human coronaviruses. Curr Opin Virol. 2014;6:55-60
40. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J. et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA. 2020 doi: 10.1001/jama.2020.1585
41. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497-506
42. Zou L, Ruan F, Huang M, Liang L, Huang H, Hong Z. et al. SARS-CoV-2 Viral Load in Upper Respiratory Specimens of Infected Patients. N Engl J Med. 2020;382:1177-9
43. Wölfel R, Corman VM, Guggemos W, Seilmaier M, Zange S, Müller MA. et al. Virological assessment of hospitalized patients with COVID-2019. Nature. 2020;581:465-9
44. He X, Lau EHY, Wu P, Deng X, Wang J, Hao X. et al. Temporal dynamics in viral shedding and transmissibility of COVID-19. Nat Med. 2020;26:672-5
45. Sohrabi C, Alsafi Z, O'Neill N, Khan M, Kerwan A, Al-Jabir A. et al. World Health Organization declares global emergency: A review of the 2019 novel coronavirus (COVID-19). Int J Surg. 2020;76:71-6
46. Zhou S, Wang Y, Zhu T, Xia L. CT Features of Coronavirus Disease 2019 (COVID-19) Pneumonia in 62 Patients in Wuhan, China. AJR Am J Roentgenol. 2020:1-8
47. Grasselli G, Zangrillo A, Zanella A, Antonelli M, Cabrini L, Castelli A. et al. Baseline Characteristics and Outcomes of 1591 Patients Infected With SARS-CoV-2 Admitted to ICUs of the Lombardy Region, Italy. JAMA. 2020 doi: 10.1001/jama.2020.5394
48. Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y. et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507-13
49. Cheng Z, Lu Y, Cao Q, Qin L, Pan Z, Yan F. et al. Clinical Features and Chest CT Manifestations of Coronavirus Disease 2019 (COVID-19) in a Single-Center Study in Shanghai, China. AJR Am J Roentgenol. 2020:1-6
50. Li L, Yang L, Gui S, Pan F, Ye T, Liang B. et al. Association of clinical and radiographic findings with the outcomes of 93 patients with COVID-19 in Wuhan, China. Theranostics. 2020;10:6113-21
51. Zhou L, Zhang M, Wang J, Gao J. Sars-Cov-2: Underestimated damage to nervous system. Travel Med Infect Dis. 2020: 101642.
52. Xu PP, Tian RH, Luo S, Zu ZY, Fan B, Wang XM. et al. Risk factors for adverse clinical outcomes with COVID-19 in China: a multicenter, retrospective, observational study. Theranostics. 2020;10:6372-83
53. Yao XH, He ZC, Li TY, Zhang HR, Wang Y, Mou H. et al. Pathological evidence for residual SARS-CoV-2 in pulmonary tissues of a ready-for-discharge patient. Cell Res. 2020
54. Wichmann D, Sperhake JP, Lutgehetmann M, Steurer S, Edler C, Heinemann A. et al. Autopsy Findings and Venous Thromboembolism in Patients With COVID-19: A Prospective Cohort Study. Annals of internal medicine. 2020 doi: 10.7326/M20-2003
55. Menter T, Haslbauer JD, Nienhold R, Savic S, Hopfer H, Deigendesch N. et al. Post-mortem examination of COVID19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings of lungs and other organs suggesting vascular dysfunction. Histopathology. 2020 doi: 10.1111/his.14134
56. Konopka KE, Wilson A, Myers JL. Postmortem Lung Findings in an Asthmatic Patient With Coronavirus Disease 2019. Chest. 2020 doi: 10.1016/j.chest.2020.04.032
57. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS. et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395:1417-8
58. Liu Q, Wang RS, Qu GQ, Wang YY, Liu P, Zhu YZ. et al. Gross examination report of a COVID-19 death autopsy. Fa Yi Xue Za Zhi. 2020;36:21-3
59. Tipnis UR, He GY, Li S, Campbell G, Boor PJ. Attenuation of isoproterenol-mediated myocardial injury in rat by an inhibitor of polyamine synthesis. Cardiovasc Pathol. 2000;9:273-80
60. Tikellis C, Thomas MC. Angiotensin-Converting Enzyme 2 (ACE2) Is a Key Modulator of the Renin Angiotensin System in Health and Disease. Int J Pept. 2012;2012:256294
61. Patel VB, Zhong JC, Grant MB, Oudit GY. Role of the ACE2/Angiotensin 1-7 Axis of the Renin-Angiotensin System in Heart Failure. Circulation research. 2016;118:1313-26
62. Erdos EG. Conversion of angiotensin I to angiotensin II. The American journal of medicine. 1976;60:749-59
63. Chappell MC. Biochemical evaluation of the renin-angiotensin system: the good, bad, and absolute? American journal of physiology. 2016;310:H137-52
64. South AM, Diz DI, Chappell MC. COVID-19, ACE2, and the cardiovascular consequences. American journal of physiology. 2020;318:H1084-H90
65. Chappell MC, Marshall AC, Alzayadneh EM, Shaltout HA, Diz DI. Update on the Angiotensin converting enzyme 2-Angiotensin (1-7)-MAS receptor axis: fetal programing, sex differences, and intracellular pathways. Front Endocrinol (Lausanne). 2014;4:201
66. Burrell LM, Johnston CI, Tikellis C, Cooper ME. ACE2, a new regulator of the renin-angiotensin system. Trends in endocrinology and metabolism: TEM. 2004;15:166-9
67. Johnston CI. Tissue angiotensin converting enzyme in cardiac and vascular hypertrophy, repair, and remodeling. Hypertension. 1994;23:258-68
68. Dzau VJ. Theodore Cooper Lecture: Tissue angiotensin and pathobiology of vascular disease: a unifying hypothesis. Hypertension. 2001;37:1047-52
69. Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. The Journal of biological chemistry. 2000;275:33238-43
70. Oudit GY, Imai Y, Kuba K, Scholey JW, Penninger JM. The role of ACE2 in pulmonary diseases-relevance for the nephrologist. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2009;24:1362-5
71. Xu H, Zhong L, Deng J, Peng J, Dan H, Zeng X. et al. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int J Oral Sci. 2020;12:8 doi: 10.1038/s41368-020-0074-x
72. Xie X, Chen J, Wang X, Zhang F, Liu Y. Age- and gender-related difference of ACE2 expression in rat lung. Life sciences. 2006;78:2166-71
73. Cai G. Tobacco-Use Disparity in Gene Expression of ACE2. Preprints. 2020 2020020051
74. Alenina N, Bader M. ACE2 in Brain Physiology and Pathophysiology: Evidence from Transgenic Animal Models. Neurochemical research. 2019;44:1323-9
75. Jia H. Pulmonary Angiotensin-Converting Enzyme 2 (ACE2) and Inflammatory Lung Disease. Shock (Augusta, Ga. 2016;46:239-48
76. Furuhashi M, Moniwa N, Mita T, Fuseya T, Ishimura S, Ohno K. et al. Urinary angiotensin-converting enzyme 2 in hypertensive patients may be increased by olmesartan, an angiotensin II receptor blocker. American journal of hypertension. 2015;28:15-21
77. Patel SK, Velkoska E, Freeman M, Wai B, Lancefield TF, Burrell LM. From gene to protein-experimental and clinical studies of ACE2 in blood pressure control and arterial hypertension. Front Physiol. 2014;5:227
78. Oudit GY, Herzenberg AM, Kassiri Z, Wong D, Reich H, Khokha R. et al. Loss of angiotensin-converting enzyme-2 leads to the late development of angiotensin II-dependent glomerulosclerosis. The American journal of pathology. 2006;168:1808-20
79. Wong DW, Oudit GY, Reich H, Kassiri Z, Zhou J, Liu QC. et al. Loss of angiotensin-converting enzyme-2 (Ace2) accelerates diabetic kidney injury. The American journal of pathology. 2007;171:438-51
80. Verma A, Shan Z, Lei B, Yuan L, Liu X, Nakagawa T. et al. ACE2 and Ang-(1-7) confer protection against development of diabetic retinopathy. Mol Ther. 2012;20:28-36
81. Liu CX, Hu Q, Wang Y, Zhang W, Ma ZY, Feng JB. et al. Angiotensin-converting enzyme (ACE) 2 overexpression ameliorates glomerular injury in a rat model of diabetic nephropathy: a comparison with ACE inhibition. Mol Med. 2011;17:59-69
82. Nadarajah R, Milagres R, Dilauro M, Gutsol A, Xiao F, Zimpelmann J. et al. Podocyte-specific overexpression of human angiotensin-converting enzyme 2 attenuates diabetic nephropathy in mice. Kidney international. 2012;82:292-303
83. Wysocki J, Ye M, Rodriguez E, Gonzalez-Pacheco FR, Barrios C, Evora K. et al. Targeting the degradation of angiotensin II with recombinant angiotensin-converting enzyme 2: prevention of angiotensin II-dependent hypertension. Hypertension. 2010;55:90-8
84. Oudit GY, Liu GC, Zhong J, Basu R, Chow FL, Zhou J. et al. Human recombinant ACE2 reduces the progression of diabetic nephropathy. Diabetes. 2010;59:529-38
85. Glowacka I, Bertram S, Herzog P, Pfefferle S, Steffen I, Muench MO. et al. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. Journal of virology. 2010;84:1198-205
86. Dijkman R, Jebbink MF, Deijs M, Milewska A, Pyrc K, Buelow E. et al. Replication-dependent downregulation of cellular angiotensin-converting enzyme 2 protein expression by human coronavirus NL63. The Journal of general virology. 2012;93:1924-9
87. Imai Y, Kuba K, Penninger JM. Angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Cellular and molecular life sciences: CMLS. 2007;64:2006-12
88. Imai Y, Kuba K, Penninger JM. [Lessons from SARS: a new potential therapy for acute respiratory distress syndrome (ARDS) with angiotensin converting enzyme 2 (ACE2)]. Masui. 2008;57:302-10
89. Zhang H, Wada J, Hida K, Tsuchiyama Y, Hiragushi K, Shikata K. et al. Collectrin, a collecting duct-specific transmembrane glycoprotein, is a novel homolog of ACE2 and is developmentally regulated in embryonic kidneys. The Journal of biological chemistry. 2001;276:17132-9
90. Danilczyk U, Sarao R, Remy C, Benabbas C, Stange G, Richter A. et al. Essential role for collectrin in renal amino acid transport. Nature. 2006;444:1088-91
91. Diaz JH. Hypothesis: angiotensin-converting enzyme inhibitors and angiotensin receptor blockers may increase the risk of severe COVID-19. J Travel Med. 2020
92. Kuster GM, Pfister O, Burkard T, Zhou Q, Twerenbold R, Haaf P. et al. SARS-CoV2: should inhibitors of the renin-angiotensin system be withdrawn in patients with COVID-19? European heart journal. 2020 DOI: 10.1093/eurheartj/ehaa235
93. Cure E, Cumhur Cure M. Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers may be harmful in patients with diabetes during COVID-19 pandemic. Diabetes Metab Syndr. 2020;14:349-50
94. Li J, Wang X, Chen J, Zhang H, Deng A. Association of Renin-Angiotensin System Inhibitors With Severity or Risk of Death in Patients With Hypertension Hospitalized for Coronavirus Disease 2019 (COVID-19) Infection in Wuhan, China. JAMA cardiology. 2020 doi:10.1001/jamacardio.2020.1624
95. Sunden-Cullberg J. Chronic Use of Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers Is High Among Intensive Care Unit Patients With Non-COVID-19 Sepsis but Carry a Moderately Increased Risk of Death. Hypertension. 2020: DOI: 10.1161/HYPERTENSIONAHA.120.15178.
96. Vaduganathan M, Vardeny O, Michel T, McMurray JJV, Pfeffer MA, Solomon SD. Renin-Angiotensin-Aldosterone System Inhibitors in Patients with Covid-19. The New England journal of medicine. 2020;382:1653-9
97. Bo Diao CW, Rongshuai Wang, Zeqing Feng, Yingjun Tan, Huiming Wang, Changsong Wang, Liang Liu, Ying Liu, Yueping Liu, Gang Wang, Zilin Yuan, Liang Ren, Yuzhang Wu, Yongwen Chen. Human Kidney is a Target for Novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection. MedRxiv. 2020; April 10. 2020 doi: https://doi.org/10.1101/2020.03.04.20031120
98. Xiao F, Tang M, Zheng X, Liu Y, Li X, Shan H. Evidence for Gastrointestinal Infection of SARS-CoV-2. Gastroenterology. 2020;158:1831-3 e3
99. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS. et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020 DOI:https://doi.org/10.1016/S0140-6736(20)30937-5
100. Baig AM. Neurological manifestations in COVID-19 caused by SARS-CoV-2. CNS Neurosci Ther. 2020;26:499-501
101. Cao W, Li T. COVID-19: towards understanding of pathogenesis. 2020; 30, 367-369.
102. Monteil V, Kwon H, Prado P, Hagelkruys A, Wimmer RA, Stahl M. et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell. 2020; https://doi.org/10.1016/j.cell. 2020 04.004
103. Cure E, Cumhur Cure M. Comment on "Organ-protective Effect of Angiotensin-converting Enzyme 2 and its Effect on the Prognosis of COVID-19". Journal of medical virology. 2020 DOI: 10.1002/jmv.25848
104. Cheng H, Wang Y, Wang GQ. Organ-protective effect of angiotensin-converting enzyme 2 and its effect on the prognosis of COVID-19. Journal of medical virology. 2020 DOI: 10.1002/jmv.25785
105. Peregrine AS, Mamman M. Pharmacology of diminazene: a review. Acta Trop. 1993;54:185-203
106. Baldissera MD, Goncalves RA, Sagrillo MR, Grando TH, Ritter CS, Grotto FS. et al. Effects of treatment with the anti-parasitic drug diminazene aceturate on antioxidant enzymes in rat liver and kidney. Naunyn Schmiedebergs Arch Pharmacol. 2016;389:429-38
107. Chiu RW, Tang NL, Hui DS, Chung GT, Chim SS, Chan KC. et al. ACE2 gene polymorphisms do not affect outcome of severe acute respiratory syndrome. Clin Chem. 2004;50:1683-6
108. Yang W, Huang W, Su S, Li B, Zhao W, Chen S. et al. Association study of ACE2 (angiotensin I-converting enzyme 2) gene polymorphisms with coronary heart disease and myocardial infarction in a Chinese Han population. Clinical science. 2006;111:333-40
109. Vasku A, Bienertova-Vasku J, Parenica J, Goldbergova MP, Lipkova J, Zlamal F. et al. ACE2 gene polymorphisms and invasively measured central pulse pressure in cardiac patients indicated for coronarography. J Renin Angiotensin Aldosterone Syst. 2013;14:220-6
110. Liu D, Chen Y, Zhang P, Zhong J, Jin L, Zhang C. et al. Association between circulating levels of ACE2-Ang-(1-7)-MAS axis and ACE2 gene polymorphisms in hypertensive patients. Medicine. 2016;95:e3876
111. Lieb W, Graf J, Gotz A, Konig IR, Mayer B, Fischer M. et al. Association of angiotensin-converting enzyme 2 (ACE2) gene polymorphisms with parameters of left ventricular hypertrophy in men. Results of the MONICA Augsburg echocardiographic substudy. Journal of molecular medicine. 2006;84:88-96
112. Huang W, Yang W, Wang Y, Zhao Q, Gu D, Chen R. Association study of angiotensin-converting enzyme 2 gene (ACE2) polymorphisms and essential hypertension in northern Han Chinese. J Hum Hypertens. 2006;20:968-71
113. Feng W, Sun L, Qu XF. Association of AGTR1 and ACE2 gene polymorphisms with structural atrial fibrillation in a Chinese Han population. Die Pharmazie. 2017;72:17-21
114. Fan X, Wang Y, Sun K, Zhang W, Yang X, Wang S. et al. Polymorphisms of ACE2 gene are associated with essential hypertension and antihypertensive effects of Captopril in women. Clinical pharmacology and therapeutics. 2007;82:187-96
115. Dhangadamajhi G, Mohapatra BN, Kar SK, Ranjit M. Gene polymorphisms in angiotensin I converting enzyme (ACE I/D) and angiotensin II converting enzyme (ACE2 C->T) protect against cerebral malaria in Indian adults. Infection, genetics and evolution: journal of molecular epidemiology and evolutionary genetics in infectious diseases. 2010;10:337-41
116. Benjafield AV, Wang WY, Morris BJ. No association of angiotensin-converting enzyme 2 gene (ACE2) polymorphisms with essential hypertension. American journal of hypertension. 2004;17:624-8
117. Gretebeck LM, Subbarao K. Animal models for SARS and MERS coronaviruses. Curr Opin Virol. 2015;13:123-9
118. Subbarao K, Roberts A. Is there an ideal animal model for SARS? Trends Microbiol. 2006;14:299-303
119. Callaway E. Labs rush to study coronavirus in transgenic animals - some are in short supply. Nature. 2020;579:183
120. Roberts A, Subbarao K. Animal models for SARS. Advances in experimental medicine and biology. 2006;581:463-71
121. Roberts A, Wood J, Subbarao K, Ferguson M, Wood D, Cherian T. Animal models and antibody assays for evaluating candidate SARS vaccines: summary of a technical meeting 25-26 August 2005, London, UK. Vaccine. 2006;24:7056-65
122. Sutton TC, Subbarao K. Development of animal models against emerging coronaviruses: From SARS to MERS coronavirus. Virology. 2015;479-480:247-58
123. Gong SR, Bao LL. The battle against SARS and MERS coronaviruses: Reservoirs and Animal Models. Animal Model Exp Med. 2018;1:125-33
124. van den Brand JM, Haagmans BL, Leijten L, van Riel D, Martina BE, Osterhaus AD. et al. Pathology of experimental SARS coronavirus infection in cats and ferrets. Vet Pathol. 2008;45:551-62
125. Martina BE, Haagmans BL, Kuiken T, Fouchier RA, Rimmelzwaan GF, Van Amerongen G. et al. Virology: SARS virus infection of cats and ferrets. Nature. 2003;425:915
126. Weingartl H, Czub M, Czub S, Neufeld J, Marszal P, Gren J. et al. Immunization with modified vaccinia virus Ankara-based recombinant vaccine against severe acute respiratory syndrome is associated with enhanced hepatitis in ferrets. Journal of virology. 2004;78:12672-6
127. McAuliffe J, Vogel L, Roberts A, Fahle G, Fischer S, Shieh WJ. et al. Replication of SARS coronavirus administered into the respiratory tract of African Green, rhesus and cynomolgus monkeys. Virology. 2004;330:8-15
128. Roberts A, Paddock C, Vogel L, Butler E, Zaki S, Subbarao K. Aged BALB/c mice as a model for increased severity of severe acute respiratory syndrome in elderly humans. Journal of virology. 2005;79:5833-8
129. Roberts A, Deming D, Paddock CD, Cheng A, Yount B, Vogel L. et al. A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice. PLoS pathogens. 2007;3:e5
130. Day CW, Baric R, Cai SX, Frieman M, Kumaki Y, Morrey JD. et al. A new mouse-adapted strain of SARS-CoV as a lethal model for evaluating antiviral agents in vitro and in vivo. Virology. 2009;395:210-22
131. Zhao J, Li K, Wohlford-Lenane C, Agnihothram SS, Fett C, Zhao J. et al. Rapid generation of a mouse model for Middle East respiratory syndrome. Proc Natl Acad Sci U S A. 2014;111:4970-5
132. Shi J, Wen Z, Zhong G, Yang H, Wang C, Huang B. et al. Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS-coronavirus 2. Science. 2020 DOI: 10.1126/science.abb7015
133. Chan JF, Zhang AJ, Yuan S, Poon VK, Chan CC, Lee AC. et al. Simulation of the clinical and pathological manifestations of Coronavirus Disease 2019 (COVID-19) in golden Syrian hamster model: implications for disease pathogenesis and transmissibility. Clin Infect Dis. 2020 https://doi.org/10.1093/cid/ciaa325
134. Chandrashekar A, Liu J, Martinot AJ, McMahan K, Mercado NB, Peter L. et al. SARS-CoV-2 infection protects against rechallenge in rhesus macaques. Science. 2020 DOI: 10.1126/science.abc4776
135. Bao L, Deng W, Huang B, Gao H, Liu J, Ren L. et al. The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature. 2020 https://doi.org/10.1038/ s41586-020-2312-y (2020)
136. Liu F, Dai S, Feng D, Qin Z, Peng X, Sakamuri S. et al. Distinct fate, dynamics and niches of renal macrophages of bone marrow or embryonic origins. Nat Commun. 2020;11:2280
137. Hu W, Ferris SP, Tweten RK, Wu G, Radaeva S, Gao B. et al. Rapid conditional targeted ablation of cells expressing human CD59 in transgenic mice by intermedilysin. Nat Med. 2008;14:98-103
138. Feng D, Dai S, Liu F, Ohtake Y, Zhou Z, Wang H. et al. Cre-inducible human CD59 mediates rapid cell ablation after intermedilysin administration. The Journal of clinical investigation. 2016;126:2321-33
139. Liu F, Dai S, Feng D, Peng X, Qin Z, Kearns AC. et al. Versatile cell ablation tools and their applications to study loss of cell functions. Cellular and molecular life sciences: CMLS. 2019;76:4725-43
140. Dai S, Liu F, Qin Z, Zhang J, Chen J, Ding WX, Feng D, Ji Y, Qin X. Kupffer cells promote T-cell hepatitis by producing CXCL10 and limiting liver sinusoidal endothelial cell permeability. Theranostics. 2020;10:7163-7177
141. Gheblawi M, Wang K, Viveiros A, Nguyen Q, Zhong JC, Turner AJ. et al. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of ACE2. Circulation research. 2020;126:1456-74
142. Xia S, Zhu Y, Liu M, Lan Q, Xu W, Wu Y. et al. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cellular & molecular immunology. 2020 https://doi.org/10.1038/s41423-020-0374-2
143. Xia S, Liu M, Wang C, Xu W, Lan Q, Feng S. et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020;30:343-55
144. Jia HP, Look DC, Tan P, Shi L, Hickey M, Gakhar L. et al. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am J Physiol Lung Cell Mol Physiol. 2009;297:L84-96
145. Lei C, Qian K, Li T, Zhang S, Fu W, Ding M. et al. Neutralization of SARS-CoV-2 spike pseudotyped virus by recombinant ACE2-Ig. Nat Commun. 2020;11:2070
146. Wrapp D, De Vlieger D, Corbett KS, Torres GM, Wang N, Van Breedam W. et al. Structural Basis for Potent Neutralization of Betacoronaviruses by Single-Domain Camelid Antibodies. Cell. 2020; https://doi.org/10.1016/j.cell. 2020 04.031
147. Wang C, Li W, Drabek D, Okba NMA, van Haperen R, Osterhaus A. et al. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat Commun. 2020;11:2251
148. Pinto D, Park YJ, Beltramello M, Walls AC, Tortorici MA, Bianchi S. et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature. 2020 https://doi.org/10.1038/s41586-0202349-y (2020)
149. Catalan-Dibene J. Human antibodies can neutralize SARS-CoV-2. Nature reviews Immunology. 2020 https://doi.org/10.1038/s41577-020-0313-6
150. Shen C, Wang Z, Zhao F, Yang Y, Li J, Yuan J. et al. Treatment of 5 Critically Ill Patients With COVID-19 With Convalescent Plasma. JAMA. 2020;323(16):1582-1589 doi:10.1001/jama.2020.4783
151. Ahn JY, Sohn Y, Lee SH, Cho Y, Hyun JH, Baek YJ. et al. Use of Convalescent Plasma Therapy in Two COVID-19 Patients with Acute Respiratory Distress Syndrome in Korea. J Korean Med Sci. 2020;35:e149
152. Duan K, Liu B, Li C, Zhang H, Yu T, Qu J. et al. Effectiveness of convalescent plasma therapy in severe COVID-19 patients. Proc Natl Acad Sci U S A. 2020;117:9490-6
153. Zeng QL, Yu ZJ, Gou JJ, Li GM, Ma SH, Zhang GF. et al. Effect of Convalescent Plasma Therapy on Viral Shedding and Survival in COVID-19 Patients. J Infect Dis. 2020 https://doi.org/10.1093/infdis/jiaa228
154. Yu J, Tostanoski LH, Peter L, Mercado NB, McMahan K, Mahrokhian SH. et al. DNA vaccine protection against SARS-CoV-2 in rhesus macaques. Science. 2020 DOI: 10.1126/science.abc6284
155. Chen WH, Hotez PJ, Bottazzi ME. Potential for developing a SARS-CoV receptor-binding domain (RBD) recombinant protein as a heterologous human vaccine against coronavirus infectious disease (COVID)-19. Hum Vaccin Immunother. 2020:1-4
156. Ferrario CM, Jessup J, Chappell MC, Averill DB, Brosnihan KB, Tallant EA. et al. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. 2005;111:2605-10
157. Meng J, Xiao G, Zhang J, He X, Ou M, Bi J. et al. Renin-angiotensin system inhibitors improve the clinical outcomes of COVID-19 patients with hypertension. Emerg Microbes Infect. 2020;9:757-60
158. Zhang P, Zhu L, Cai J, Lei F, Qin JJ, Xie J. et al. Association of Inpatient Use of Angiotensin Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers with Mortality Among Patients With Hypertension Hospitalized With COVID-19. Circulation research. 2020 https://doi.org/10.1161/CIRCRESAHA.120.317134
159. Rossi GP, Sanga V, Barton M. Potential harmful effects of discontinuing ACE-inhibitors and ARBs in COVID-19 patients. Elife. 2020 Apr 6;9:e57278. doi: 10.7554/eLife.57278
160. Tedeschi S, Giannella M, Bartoletti M, Trapani F, Tadolini M, Borghi C. et al. Clinical impact of renin-angiotensin system inhibitors on in-hospital mortality of patients with hypertension hospitalized for COVID-19. Clin Infect Dis. 2020 doi: 10.1093/cid/ciaa492
161. Epelman S, Tang WH, Chen SY, Van Lente F, Francis GS, Sen S. Detection of soluble angiotensin-converting enzyme 2 in heart failure: insights into the endogenous counter-regulatory pathway of the renin-angiotensin-aldosterone system. Journal of the American College of Cardiology. 2008;52:750-4
162. Mizuiri S, Aoki T, Hemmi H, Arita M, Sakai K, Aikawa A. Urinary angiotensin-converting enzyme 2 in patients with CKD. Nephrology (Carlton). 2011;16:567-72
163. Reynolds HR, Adhikari S, Pulgarin C, Troxel AB, Iturrate E, Johnson SB. et al. Renin-Angiotensin-Aldosterone System Inhibitors and Risk of Covid-19. N Engl J Med. 2020 DOI: 10.1056/NEJMoa2008975
164. Mehra MR, Desai SS, Kuy S, Henry TD, Patel AN. Cardiovascular Disease, Drug Therapy, and Mortality in Covid-19. N Engl J Med. 2020 DOI: 10.1056/NEJMoa2007621
165. Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M. et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269-71
166. Choy KT, Wong AY, Kaewpreedee P, Sia SF, Chen D, Hui KPY. et al. Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS-CoV-2 replication in vitro. Antiviral research. 2020;178:104786
167. Grein J, Ohmagari N, Shin D, Diaz G, Asperges E, Castagna A. et al. Compassionate Use of Remdesivir for Patients with Severe Covid-19. N Engl J Med. 2020 DOI: 10.1056/NEJMoa2007016
168. Wang Y, Zhang D, Du G, Du R, Zhao J, Jin Y. et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2020;395:1569-78
169. Cheng CY, Lee YL, Chen CP, Lin YC, Liu CE, Liao CH. et al. Lopinavir/ritonavir did not shorten the duration of SARS CoV-2 shedding in patients with mild pneumonia in Taiwan. J Microbiol Immunol Infect. 2020; https://doi.org/10.1016/j.jmii. 2020 03.032
170. Chu CM, Cheng VC, Hung IF, Wong MM, Chan KH, Chan KS. et al. Role of lopinavir/ritonavir in the treatment of SARS: initial virological and clinical findings. Thorax. 2004;59:252-6
171. Yao X, Ye F, Zhang M, Cui C, Huang B, Niu P. et al. In Vitro Antiviral Activity and Projection of Optimized Dosing Design of Hydroxychloroquine for the Treatment of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). Clin Infect Dis. 2020 https://doi.org/10.1093/cid/ciaa237
172. CHEN J LD, LIU L, LIU P, XU Q, XIA L, LING Y, HUANG D, SONG S, ZHANG D, QIAN Z, LI T, SHEN Y, LU H. A pilot study of hydroxychloroquine in treatment of patients with common coronavirus disease-19 (COVID-19). J Zhejiang Univ (Med Sci). 2020 49; DOI, zjujournals.com
173. Gautret P, Lagier JC, Parola P, Hoang VT, Meddeb L, Mailhe M. et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int J Antimicrob Agents. 2020: 105949; DOI: 10.1016/j.ijantimicag. 2020 105949
174. Borba MGS, Val FFA, Sampaio VS, Alexandre MAA, Melo GC, Brito M. et al. Effect of High vs Low Doses of Chloroquine Diphosphate as Adjunctive Therapy for Patients Hospitalized With Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection: A Randomized Clinical Trial. JAMA Netw Open. 2020;3:e208857 doi:10.1001/jamanetworkopen.2020.8857
175. Mehra MR, Desai SS, Ruschitzka F, Patel AN. Hydroxychloroquine or chloroquine with or without a macrolide for treatment of COVID-19: a multinational registry analysis. Lancet. 2020 DOI:https://doi.org/10.1016/S0140-6736(20)31180-6
176. Wang SF, Tseng SP, Yen CH, Yang JY, Tsao CH, Shen CW. et al. Antibody-dependent SARS coronavirus infection is mediated by antibodies against spike proteins. Biochemical and biophysical research communications. 2014;451:208-14
177. Wang Q, Zhang L, Kuwahara K, Li L, Liu Z, Li T. et al. Immunodominant SARS Coronavirus Epitopes in Humans Elicited both Enhancing and Neutralizing Effects on Infection in Non-human Primates. ACS Infect Dis. 2016;2:361-76
178. Liu F, Zhang Q, Huang C, Shi C, Wang L, Shi N. et al. CT quantification of pneumonia lesions in early days predicts progression to severe illness in a cohort of COVID-19 patients. Theranostics. 2020;10:5613-22
Author contact
![]() Corresponding author: Xuebin Qin, MD, PhD, Division of Comparative Pathology, Tulane National Primate Research Center, Health Sciences Campus, 18703 Three Rivers Road, Covington, LA 70433, USA. Tel: 985 871 6296, E-mail: xqin2edu; Prasun K. Datta, PhD, Division of Comparative Pathology, Tulane National Primate Research Center, 18703 Three Rivers Road, Covington, LA 70433, Tel: 985-871-6278, E-mail: pdattaedu; and Jay Rappaport, Ph.D., Division of Comparative Pathology, Tulane National Primate Research Center, Health Sciences Campus, 18703 Three Rivers Road, Covington, LA 70433, USA, Tel: 985-871-6201, E-mail: jrappaportedu.
Corresponding author: Xuebin Qin, MD, PhD, Division of Comparative Pathology, Tulane National Primate Research Center, Health Sciences Campus, 18703 Three Rivers Road, Covington, LA 70433, USA. Tel: 985 871 6296, E-mail: xqin2edu; Prasun K. Datta, PhD, Division of Comparative Pathology, Tulane National Primate Research Center, 18703 Three Rivers Road, Covington, LA 70433, Tel: 985-871-6278, E-mail: pdattaedu; and Jay Rappaport, Ph.D., Division of Comparative Pathology, Tulane National Primate Research Center, Health Sciences Campus, 18703 Three Rivers Road, Covington, LA 70433, USA, Tel: 985-871-6201, E-mail: jrappaportedu.