Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Recent developments
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
Author Biographies
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(16):7034-7052. doi:10.7150/thno.47406 This issue Cite
Review
Bioengineering tools to speed up the discovery and preclinical testing of vaccines for SARS-CoV-2 and therapeutic agents for COVID-19
Manuela Teresa Raimondi1 ![]() , Francesca Donnaloja1, Bianca Barzaghini1, Alberto Bocconi1, Claudio Conci1, Valentina Parodi1, Emanuela Jacchetti1*, Stephana Carelli2*
, Francesca Donnaloja1, Bianca Barzaghini1, Alberto Bocconi1, Claudio Conci1, Valentina Parodi1, Emanuela Jacchetti1*, Stephana Carelli2*
1. Department of Chemistry, Materials and Chemical Engineering G. Natta, Politecnico di Milano, Milano, Italy.
2. Pediatric Clinical Research Center “Fondazione Romeo ed Enrica Invernizzi”, Department of Biomedical and Clinical Sciences L. Sacco, University of Milano, Italy.
*Equally-contributing authors.
Received 2020-4-24; Accepted 2020-5-13; Published 2020-5-27
Abstract

This review provides an update for the international research community on the cell modeling tools that could accelerate the understanding of SARS-CoV-2 infection mechanisms and could thus speed up the development of vaccines and therapeutic agents against COVID-19.
Many bioengineering groups are actively developing frontier tools that are capable of providing realistic three-dimensional (3D) models for biological research, including cell culture scaffolds, microfluidic chambers for the culture of tissue equivalents and organoids, and implantable windows for intravital imaging.
Here, we review the most innovative study models based on these bioengineering tools in the context of virology and vaccinology. To make it easier for scientists working on SARS-CoV-2 to identify and apply specific tools, we discuss how they could accelerate the discovery and preclinical development of antiviral drugs and vaccines, compared to conventional models.
Keywords: Coronavirus, bioengineering, target, antiviral, vaccine, preclinical testing
Introduction
As of May 8, 2020, the 2019 novel coronavirus SARS-CoV-2 was responsible in 215 countries for 3,767,744 infections and 259,593 deaths from the related disease, defined as COronaVIrus Disease 2019 (COVID-19) [1]. The human population has no immunity to SARS-CoV-2 because this virus had never previously infected humans before its initial outbreak in December 2019. Until now, no drugs or biologics have been proven to be effective for the prevention or treatment of COVID-19. To deal with this pandemic, apart from non-pharmaceutical containment measures including quarantine and maintenance of social distance, possible interventions consist of the repurposing of existing pharmaceutical products, along with the development of new products and vaccines [2-4].
The development of a radically new pharmaceutical product, either traditional, or biological such as an antibody targeting a SARS-CoV-2 receptor, is subject to regulatory approval for use in humans based on three steps: 1) lab discovery in vitro on cell cultures; 2) preclinical testing in vivo in animals to confirm the mechanism of action and to measure the range of effective dosages and tolerability; and 3) clinical testing in patients to define the safe dosage and to confirm the efficacy and possible interactions. This process involves serious challenges both related to the efficiency, for example the development of a human vaccine from concept to licensing takes at least 15 years [5], and also to the effectiveness. Drug development currently involves a 99.9% overall failure rate, of which 96.4% is due to failure in the preclinical testing phase, meaning that the drug efficacy measured in vitro is almost never confirmed in animals [6].
The main issue is that the most widespread technology currently used to test therapeutic agents in vitro is obsolete: often it consists of a two-dimensional (2D) polystyrene culture dish, in which a single cell population is cultured on the bottom, the drug to be tested is added to the culture medium, and the expected modulation in specific processes or targets is measured.
However, in these simplified in vitro culture conditions, the drug elicits a cell response that is not representative of the in vivo response, which is based on the cell interactions that occur a) in three-dimensional (3D) non-flat environments, and b) within an heterogeneous cell population which is never limited to the cell population primarily addressed by the drug.
Another very serious issue is the unethical slaughter of lab animals, which more effective in vitro methods would replace, at least in part. Moreover, the methods adopted for monitoring the drug effects in animals are also obsolete, whereas new miniaturized intravital imaging techniques are now available that could greatly refine in vivo observations, even in terms of the temporal evolution in the same animal, thus reducing the number of animals sacrificed by 80-90%.
There are many on-going clinical trials evaluating potential vaccines and treatments for COVID-19, which cannot be accelerated without putting the safety of patients at risk. Instead, both the in vitro and in vivo preclinical phases of drug development could be accelerated by replacing some of the current unrepresentative and obsolete study models, with new ones based on improved modeling tools. For example, many groups have developed 3D static, microfluidic and intravital imaging models of viral infection and relevant therapeutics using new high-performance experimental devices that are user-friendly for operators and enable live organoids to be cultured and examined in 3D with high-resolution imaging.
Here, we review the published work based on these modeling tools in the specific fields of virology and pharmacology.
Recent developments
In silico discovery of drug targets for SARS-CoV-2 and prediction of its pathogenic mechanisms
SARS-CoV-2 infection can lead to COVID-19 infection, which causes massive damage to the pulmonary cells [7-11] due to a cytokine storm, i.e. an acute secretion of inflammatory signals by innate monocytes in response to the infection, which causes hyper inflammation. The WHO [12] and Clinicaltrials.gov database currently report many active trials on COVID-19 patients of drugs that have already been licensed for other infectious and inflammatory diseases. These include anti-malarial drugs, such as chloroquine and its derivative hydroxychloroquine, large spectrum antivirals (inhibitors of endonucleases, nucleosides, protease inhibitors, and other antiretrovirals), interferons, corticosteroids, immune suppressants, and anticoagulants.
In parallel, many groups are repurposing known compounds and are developing new vaccines [3] and therapeutic solutions [4] to specifically target the SARS-CoV-2 virus. The drug targets are primarily viral proteins, most of which have been resolved and are available on the protein data bank (PDB). Much progress has been made in the structural biology of SARS-CoV-2. Many structural and non-structural proteins have been determined experimentally by cryo-electron microscopy (cryo-EM) and X-ray crystallography. The proteins specific to SARS-CoV-2 that have already been determined experimentally are shown in Figure 1.
Drug targets specific to the SARS-CoV-2 virus. A) The SARS-CoV-2 whole genome consists of three main open reading frames (ORF): ORF1a, ORF1b and ORF2-10. The most frequent targets for therapies are reported in gray with their PDB codes. B) Some proteins encoded in ORF1a and ORF1b represent the primary I potential drug target. Their inhibition blocks the viral RNA synthesis and replication. Pp1ab encodes for 16 non-structural proteins (NSPs) including the papain-like protease (PLpro) in the Nsp3region, main protease or C-like protease (Mpro or 3CLpro), ADP ribose phosphatase (ADRP), RNA-dependent RNA polymerase (RdRp) and helicase. ORF2-10 encodes for accessory proteins (e.g. 7a) and the structural and accessory proteins, spike protein (S), envelope protein (E), membrane protein (M), and nucleocapsid protein (N). C) The spike protein is the most studied secondary II potential drug target. The virus cannot bind the cell receptors if the spike protein is inhibited [14]. The spike protein has been studied the most, has been solved by electron microscopy (EM) and consists of: N-ter domain (NTD); receptor binding domain (RBD) consisting of receptor binding motif (RBM), subdomain 1 (SD1) and subdomain 2 (SD2); fusion peptide (FP); heptad repeat 1 (HR1); heptad repeat 2 (HR2); transmembrane region (TM) and intracellular domain (IC). The most studied RBD domain is RBM (in red), the domain mainly involved in host-cell interaction [13]. RBM is believed to bind mainly with the ACE2 human cell receptor and, therefore, most of the relevant PDB codes, of which 6M0J, includes the RBD+ACE2 complex. The complete amino acid sequence for RBD is shown at the bottom of the figure. Amino acids corresponding to RBM are in red, the beta-sheet and alpha-helix structures are inside the dotted boxes. The contact residues at the RBD/ACE2 interface [14] are shown in bold squares. *https://www.ncbi.nlm.nih.gov/genbank/sars-cov-2-seqs/.
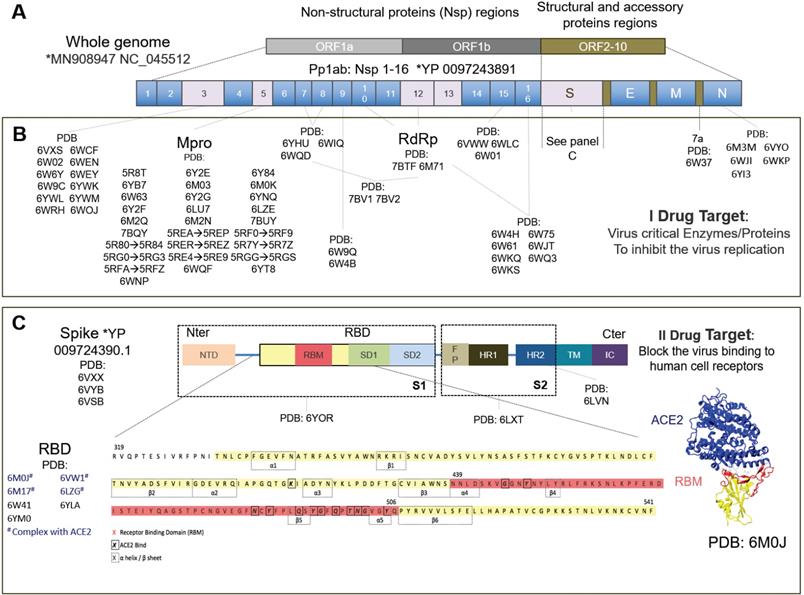
Drug therapies may act either a) on the host cells, affecting the specific receptor or restoring the innate immunity of the cells, or b) on the virus, by blocking the virus functions (I target) or acting on the virus structural proteins (II target). The primary (I) drug target is represented by several non-structural viral proteins (NSPs) whose inhibition blocks the viral RNA synthesis and replication [13]. These include the papain-like protease (PLpro) in the Nsp3 region, the main protease (Mpro) also known as C-like protease (3CLpro), the RNA-dependent RNA polymerase (RdRp) and the helicase.
The structural spike protein on the viral envelope is the most studied protein of the secondary (II) drug targets [13]. The virus cannot bind to the host cell receptors, fuse with cell membranes and enter cells, if the spike protein is inhibited, and thus it has also already been solved by electron microscopy [14]. This comprises a receptor binding domain (RBD), mainly involved in the host-cell interaction, incorporating a sub-domain called a receptor binding motif (RBM) believed to bind, similarly to SARS-CoV, mainly with the human angiotensin-converting enzyme 2 (ACE2) cell receptor, to gain entry inside cells [15]. The ACE2 receptor was discovered in 2000 and is expressed above all in the lungs, heart, kidneys, gastrointestinal tract, testis and other tissues or organs [16]. Most drugs against SARS-CoV-2 investigated today therefore target the complex RBD+ACE2 [17-19].
Solved SARS-CoV-2 molecular structures have been used to set-up docking analyses and molecular dynamics simulations that predict the affinity of chemical binding of these structures with known compounds. This in silico modeling technique has been used intensely for drug screening in virtual libraries. Table 1 shows the latest published results in terms of drugs predicted to bind with SARS-CoV-2 targets. It is worth noting that most of the in silico drug docking analyses listed in Table 1 use the homology modelling strategy to predict the structures of the target proteins that have not yet been experimentally resolved [20-25]. The homology modelling approach predicts the atomic-resolution model of a target protein based on the structure of the protein chosen as a template according to the genome sequence identity [26]. The genome of SARS-CoV-2 showed a high homology with the SARS virus, which makes it a reliable template [13].
In silico molecular mechanics modeling of drug interaction with SARS-CoV-2
| Refs. | Therapeutic agent | Target (Viral subunit) | Suggested drug based on the predicted binding energy |
|---|---|---|---|
| [101] | Commercially available antiviral drugs. | Mpro, RdRp, helicase, 3'-to-5' exonuclease, endoRNAse 2'-O-ribose methyltransferase. | Atazanavir against all the virus endonucleases. |
| [20] | Sofosbuvin and ribavirin, remdesvir, IDX-184 guanosine triphosphate, uracil triphoshate, cinnamaldehyde, thymoquinone. | RdRp. | Sofosbuvir and ribavirin, remdesivir and IDX-184 were shown to tightly bind to the target. |
| [21] | 7173 purchasable drugs. | Mpro. | Ledipasvir or velpatasvir (minimal side effects) + Epclusa® (velpatasvir/sofosbuvir) and Harvoni® (ledipasvir/sofosbuvir) with inhibitory actions on two viral proteases. |
| [13] | Drug repurposing (ZINC database + database of traditional Chinese medicine and natural products). | All the main proteins encoded by SARS-CoV-2: 3-chymotrypsin-like protease (3CLpro), Spike, RdRp and PLpro. | Remdesivir (GS-5734) inhibitor of RdRp. Its efficacy was verified in vitro. It is also hypothesized to inhibit Nsp3b (score=-36.5), RdRp (mfScores=-112.8), E-channel (mfScore=-125.1), and TMPRSS2 (score=-36.23, mfScores=-109.4). |
| [102] | FDA-approved drugs against viral protease + in-house database of natural and drug-like compounds of synthetic origin. | Mpro | FDA-approved drugs: remdesivir, saquinavir, darunavir + two natural compounds: flavone and coumarin derivatives. |
| [103] | Designed inhibitor. | ACE2-Spike interface. | Designed inhibitors consisting of α1+α2 helixes. |
| [104] | Designed inhibitor. | Mpro. | Designed drug that needs to be optimized and tested in vitro and vivo. |
| [22] | Zinc15 - Approved drugs in major jurisdictions, including the FDA, i.e. DrugBank approved. | Spike protein + Mpro. | Modeled the action of many approved drugs: zanamivir, indinavir, saquinavir, remdesivir (against Mpro)+flavin adenine dinucleotide (FAD), adeflavin (against both spike and Mpro), and coenzyme A (against spike protein). |
| [23] | Specific drug. | Spike via homology modeling (template PDB: 2GHV)+spike-ACE2 interaction (template: PDB:2AjF). | Cobicistat, carfilzomib and ombitasvir. |
| [24] | From a specific drug to its possible target. | 20 virus protein + 22 human proteins. | Development of molecular docking based webserver, namely D3Targets-2019-nCoV for target prediction (in vitro/in vivo studies), to identify: (1) the potential target proteins and their different conformations were included; (2) all the potential ligand-binding sites with volume larger than 200 A°^3; (3) correlations among some conformations or binding sites was annotated; (4) easily to be updated, and freely accessible. |
| [25] | Medicinal plant library (32, 297 potential anti-viral phytochemicals/traditional Chinese medicinal compounds). | Mpro. | 5,7,3',4'-tetrahydroxy-2'-(3,3-dimethylallyl) isoflavone. |
The sequence alignment with the previous SARS-like viruses have also been applied to perform the virus evolutionary analysis [27], or to highlight particular features of the SARS-CoV-2 virus (Table 2). Several studies have compared SARS-CoV-2 with SARS in terms of interaction of the spike protein with the host cell receptor. Structural- and free-binding energy-based analyses have revealed accurate details of SARS-CoV-2. These observations may explain the aggressiveness of SARS-CoV-2, at least in part [17-19,28].
In silico prediction of the pathogenic mechanisms of SARS-CoV-2
| Refs. | Viral/cellular subunit | Modelling method | Primary results |
|---|---|---|---|
| [18] | Receptor ACE2 + SARS-CoV-2 spike. | 1) Structure alignment between SARS-CoV-2 and SARS-COV; 2) SARS-CoV-2 + ACE2 complex modeling via single point mutation from solved SARS-CoV +ACE2; 3) SARS-CoV-2 and SARS-CoV interaction with ACE2 hot spots (residues mainly involved in RBD binding) comparison; 4) comparison with animals ACE2. | The two spike proteins are quite similar and therefore the authors analyzed in detail the hot spots revealed for SARS-CoV; 2) two virus-binding hot spots identified: ACE2 Hot spot 31(salt bridge Lys31-Glu35) and Hot spot 353 (salt bridge Lys353-Asp38); 3) (a) SARS-CoV-2 Q493 (vs SARS-CoV N479) was compatible but had less of an affinity with hotspot 3, (b) SARS-CoV-2 Q501 (vs SARS-CoV T487) had less affinity with hotspot 353, but was still compatible, (c) SARS-CoV-2 L455 (vs SARS-CoV Y442) preferred interaction with Hotspot 31, (d) SARS-CoV-2 F586 (vs SARS-CoV L472) enhances interaction with Hotspot 31 (e) SARS-CoV-2 S494 (vs SARS-CoV D480) made the interaction weaker but compatible with the hotspot 353; 4) mouse or rat ACE2 contained a hotspot at the 353 position, which did not fit into the RBD of SARS-CoV-2 which instead recognized ACE2 from pigs, ferrets, cats, orangutans, monkeys, and humans. |
| [19] | Receptor ACE2 + SARS-CoV-2 spike. | 1) Genomic analysis; 2) homology modeling for SARS-CoV-2 spike; 3) structural superimposition in SARS-CoV+ACE2 model; 4) binding free energy evaluation. | 1) Low homology in RBD domain between SARS-CoV-2 and SARS-CoV, but high similarity (similar polarity); 2) From free energy binding the authors observed that even though it was weaker than SARS-CoV (-78.6 kcal mol-1), SARS-CoV-2 spike still showed an high affinity with ACE2 (-50.6 kcal mol-1); 3) Differences in aa sequences did not alter the protein conformation and maintained similar Van der Waals and electrostatic properties. |
| [17] | receptor CD26 + SARS-CoV-2 Spike. | 1) Homology modeling for SARS-CoV-2 Spike (S1+S2) using the SARS-CoV as template (PDB: 6ACD) to obtain inactive configuration and using SARS-CoV+ACE2 complex structure as a template (PDB:6ACG) to obtain the active structure; 2) analysis of glycosylation sites via the server; 3) comparison (SARS-CoV-2 vs SARS-CoV) of the glycosylation shield structure obtained by server; 4) docking between the modeled trimeric spike and human CD26 (PDB: 4QZV). | 1) SARS-CoV-2 Spike tetramer configuration in both active and inactive state; 2) Based on the Spike+ACE2 model the authors identified the 3C-like proteinase cleavage site + glycosylation sites; 3) from SARS-CoV and SARS-CoV-2 comparison: SARS-CoV-2 showed several unique glycolysation sites in addition to the conserved from SARS-CoV. This suggested a different shielding or camouflage pattern; 4) SARS-CoV-2 spike showed a large interaction surface with CD26 with many unique residues (different in SARS-CoV) involved in the interaction. SARS-CoV-2, compared to previous SARS-CoV viruses, interacts in a different way with the human receptors. |
| [28] | ORF1ab (Nsp2 and Nsp3). | Provides information on how quickly the virus could potentially increase its genetic variability, with implications for contagiousness and drugs via comparison of ORF1ab SARS structure: 1) The ORF1ab of 15 SARS-CoV-2 sequence alignment with five sequences of SARS virus and five sequences from Bat SARS‐like virus; 2) selective pressure analysis; 3) homology modeling for NSp2 Nsp3; 4) trans-membrane analysis. | Potential sites under positive selective pressure were found (501); transmembrane helices were predicted. At the residue 501, Bat SARS‐like coronavirus showed an apolar amino acid (threonine), while SARS and SARS-CoV-2 showed a polar amino acid, glycine and glutamine, respectively. Hypothesis: polarity, and potential to form H‐bonds may confer higher stability to the protein. 723 aa. SARS-CoV-2 sequence showed a Serine replacing for Glycine in Bat SARSlike and SARS coronaviruses. Hypothesis: substitution increases the local stiffness of the polypeptide chain due to a steric effect and to the ability of Serine side chain to form H‐bonds. At 1010 aa, SARS-CoV-2 has Prolin, the Bat SARS‐like coronavirus and SARS virus showed a polar and an apolar a. Hypothesis: steric bulge and stiffness of the Proline may cause local conformation perturbation compared with the proteins of the other two viruses. |
| [27] | Spike-ACE2. | Prediction of the interaction ACE2 RBD-spike. Evolutionary analysis and search for the possible virus reservoirs. Comparisons of the spike sequences between SARS‐CoV‐2 with SARS‐CoV, Bat SARS‐like CoV, and other coronaviruses. Analysis of the ACE2 structures and binding motif alignment:1) Alignment of spike protein sequences from different sources. Full-length and RBD sequences of spike protein from SARS‐CoV, bat, or pangolin SARS-like CoV and SARS‐CoV‐2 were aligned; 2) comparison of ACE2 enzyme among different species; 4) prediction of spike protein model and spike‐ACE2 binding model. | Bat SARS‐like CoV RaTG13, is an inner joint neighbor of SARS‐CoV‐2 (6.2% overall genome sequence identity); the full‐length sequence of S pangolin SARS‐like CoV and SARS‐CoV‐2 seem to be a little different but SARS‐CoV‐2 RBD sequence (329 to 521) like CoV, had a higher than 89% similarity with bat SARS‐like CoV; pangolin SARS‐like CoV has a higher probability to cross host barriers and infect humans; from infection-involved aa comparison, they identified pangolins, turtles as possible intermediate hosts. |
| [26] | ORF1ab. | 1) The authors showed a typical workflow for homology modeling applied to the SARS-CoV-2 ORF1ab proteins; 2) aa sequence comparison. They performed sequence alignment investigations on 10 primary sequences of SARS-CoV-2 via BLAST, SWISS‐MODEL, and Clustal Omega. | 1) According to BLAST analysis, the sequence identity of ORF1ab protein between SARS-CoV-2 and SARS‐CoV was over 90% with the query cover of about 100%; 2) the authors showed a typical workflow for homology modeling applied to the SARS-CoV-2 ORF1ab proteins. |
| [29] | Spike + ACE2 receptor. | This study explores the binding of the proteins encoded by different human ACE2 allelic variants with SARS‐CoV‐2 spike protein. 1) They selected the coding variants of ACE2 and predicted in silico the possible allelic variants; 2) they modelled the 17 obtained coding variants of ACE2 via homology modeling with native structure and superimposed the structures for comparison; 3) They superimposed the obtained models over the native ACE2‐spike protein complex; 4) inter‐residual interaction maps. | 1) The authors selected the allelic structures involved in the ACE2-spike interaction (17 variants); 2) The protein architecture of ACE2 allelic variants was similar to the wild type but the spatial orientation of substituting residues varied notably; 3) ACE2 variants bind with spike protein identical to the native complex + intermolecular contacts between SARS‐CoV‐2 spike protein and ACE2 variants are comparable. Among the differences, rs73635825 (S19P) and rs143936283 (E329G) showed a low binding affinity, which may confer resistance against the virus infection. |
A similar approach has also been taken to explore the efficiency of the binding of the proteins encoded by different human ACE2 allelic variants with the SARS‐CoV‐2 spike protein. From the binding analysis, two variants showed variations in the orientation of the amino acid side chain, altering the intramolecular interactions and the complex formation. This aspect may represent partial intrinsic susceptibility or resistance to the SARS‐CoV‐2 infection [29].
3D cell models to improve pre-clinical in vitro studies
Once discovered in silico, the drug has to be tested in vitro and in vivo in experimental models of viral infection.
Our aim was to include any advanced study model developed for virology and vaccinology since the SARS coronavirus outbreak of 2003 and we analyzed the literature pertaining to the last two decades. We found 40 papers, 75% of which were published in the last five years. 3D cell models are generally based on cell aggregates or organoids, cultured within a matrix or scaffold that provide a 3D environment for cell growth (Figure 2). Organoids are usually obtained starting with pluripotent stem cells (most frequently iPS), while engineered tissues are mainly derived from multipotent or unipotent cells.
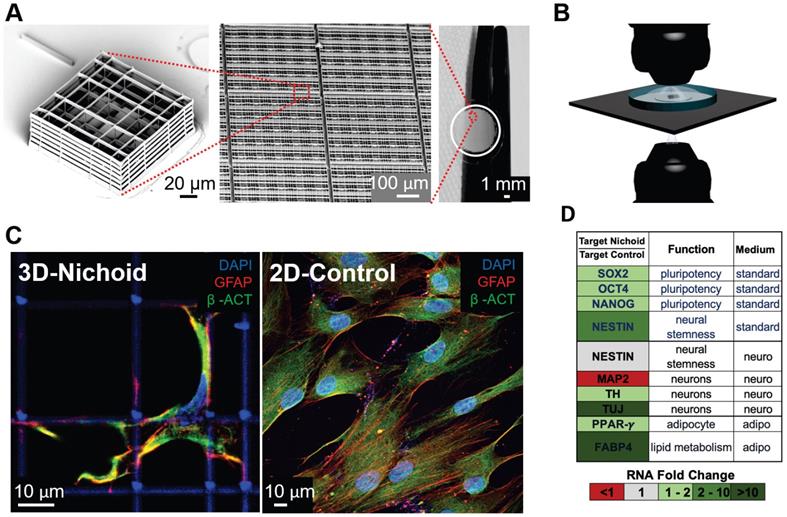
Micro-fabricated “Nichoid” scaffold for 3D cell culture. A) Nichoid architecture. From left to right: scanning electron microscopy (SEM) image of a Nichoid single-module; SEM image of up-scaled repetitive matrixes of Nichoids; picture of 50 mm2 micro patterned glass coverslip with Nichoids. Nichoid culture 3D-substrates are produced via two-photon laser polymerization of SZ2080 negative photoresist with near-infrared exposure following a CAD geometry. B) Possible microscopy configurations to use on Nichoid culture substrates. Thanks to the device's versatility, this scaffold enables optical accessibility both in transmission and in reflection modality. C) Immunofluorescence images obtained via confocal fluorescence microscopy of GFAP (red), β-ACTIN (green) and DNA (blue), in human Adipose derived Stem Cells (hADSCs) after a 7-day expansion inside the Nichoid and in standard conditions (2D-Control). The images demonstrate that nuclear morphology and protein organization and localization (both β-ACTIN and GFAP) differed between the two culture systems. Cytoskeletal markers merge (yellow signal) into cellular protrusions inside Nichoids, while a poor signal appears in flat conditions. D) Real-time PCR analysis of specific gene targets shows a significant gene expression difference (up-regulated in green, and down-regulated in red) between Nichoid and Control conditions, both using a standard culture medium and also using a culture medium that induces adipogenic or neural differentiation.
To date, only five very recent studies have addressed SARS-CoV-2 with such an innovative approach: specifically they replicate the SARS-CoV-2 infection in models of the human airway epithelium [30-32], human liver organoids [33] and in engineered human kidney and vessel organoids [34]. In addition, three of the above studies include therapeutic agents to inhibit SARS-CoV-2 infection: clinical-grade human recombinant soluble angiotensin converting enzyme 2 (hrsACE2) [34], remdesivir also in association with diltiazdem [31] and β-D-N4-hydroxycytidine [32]. Table 3 reports the findings of the published 3D cell models used to study viral infections, and Table 4 reports those that also include the effects of an antiviral therapy.
In vitro 3D cell models of viral infection
| Refs. | Pathogen | Cell type/s | Cell culture configuration |
|---|---|---|---|
| [30] | SARS-CoV-2. | Primary human epithelial cells from airway epithelium. | Epithelial cells were isolated and expanded in transwells, then an air-liquid-interface (ALI) model was provided with a pseudo-stratified and fully-differentiated system, showing basal, ciliated and goblet cells which secreted mucus and proteins. |
| [33] | SARS-CoV-2. | Primary human cholangiocytes from primary bile ducts. | Liver ductal organoids (LDO) embedded in Matrigel. |
| [46] | MERS-CoV. | Primary human intestinal epithelial cells, LGR5+ intestinal adult stem cells and Caco2 cells. | A) 2D epithelial intestinal cell (2D/EICs) differentiation for 7 days. B) Human intestinal tissue (HTI) excised and maintained in transwells coated with Matrigel. C) Intestinal Organoid from LGR5+ cells embedded in Matrigel. D) Caco-2 cells seeded on transwells and polarized (2 weeks). |
| [105] | Zika virus. | Primary human testicular cells: spermatogonial stem cells (SSC), Sertoli cells (SC), Leydig cells (LC) and peritubular cells. | Human testicular organoid (HTO): SSC, SC, LC and peritubular cells centrifugated and seeded in ultra-low attachment plate maintained with medium + testis ECM after 48 h self-assembled in spheroids. |
| [35] | Respiratory syncytial virus (RSV). | Human iPSCs from fetal tissue. | Lung bud organoid (LBO) from hPSCs embedded in Matrigel and induced to anterior foregut endoderm differentiation (CD184+ +CXCR4/c-KiT) and airway branch formation. |
| [36] | Respiratory syncytial virus (RSV). | Primary epithelial cells isolated from non-small cell lung cancer. | Cells were embedded in basement membrane extract (BME) and grown as adult human epithelial airway organoids (pseudostratified airway epithelium with basal, secretory, multi-ciliated cells). |
| [37] | Avian influenza (IAV) H1N1/H3N2. | Primary human small airway epithelial cells (SAEpCs). | HSAEpCs were loaded onto a 3D chitosan-collagen scaffold and exposed to air-liquid interface (ALI) on the upper part of transwell, alternatively the cells were totally covered with media. 2D culture conditions were performed by seeding cells directly on the upper part of a transwell plate replicating the same ALI and immerged conditions as the 3D one. |
| [38] | Avian influenza (IAV) (H1N1 - H7N9/Ah - H5N1 - H7N2 -H7N9/ah). | Human lung resident adult stem cells (ASC). | Airway organoids and proximally differentiated airway organoids (AO) made by ASCs embedded in Matrigel and grown in transwells for 16 days. |
| [39] | Parainfluenza virus-3 (HPIV3) + measles virus (MeV). | A) Human airway epithelium (HAE) system: normal human derived trachea/bronchial epithelial cells; B) Organoid system: embryonic stem cell line 2 (RUES2) + feeder layer mouse embryonic fibroblasts. | A) HAE system: epithelial cells cultured to form a highly differentiated pseudostratified mucociliary epithelium; B) lung bud organoids grown to the last-second trimester of lung development (LBO): endoderm (c-KiT, CXCR4 positive)+anterior endoderm foregut formation + ventralization inductions followed by branching airways formation after Matrigel embedding. |
| [45] | Measles virus (MeV). | Human primary fibroblast (FBCs)+H358 lung adenocarcinoma epithelial cell line+dendritic cells (DCs) human primary monocytes. | Decellularized porcine small intestinal submucosa (SIS) seeded on the apical side with FBCs and H358-eFLUOR670 cells, DCs-GFP+ and DCs-GFP- added in suspension on the basolateral side. |
| [48] | HIV-1 virus. | Primary human CD4+ T-lymphocytes. | Cells were loaded in custom-made collagen type I gels derived from rat tail and bovine. |
| [40] | Andes-hantavirus (ANDV). | Human lung fibroblast MRC-5+h-bronchial epithelial cells 16HBE14o-. | Layered construct in transwell system: bottom bovine collagen-I, MRC-5 + b-coll-I, top 16HBE14o- guarantee air-liquid interface (ALI). ANDV inoculated from the apical side of the construct. |
| [41] | Human rhinovirus C (HRV-C) and human bocavirus (HBoV). | Human primary epithelial cells (HAE). | 2D culture systems provided via HAE seeding in monolayers. 3D model obtained seeding HAEs in 4 different ways on transwells coated with Matrigel. 3D possible configurations of HAEs were: ON, WITHIN, ON and WITHIN and UNDER Matrigel. |
In vitro 3D cell models of viral infection and evaluation of antiviral therapy effects
| Refs. | Pathogen | Therapeutic agent | Cell type/s | Cell culture configuration |
|---|---|---|---|---|
| [31] | SARS-CoV-2. | Remdesivir, remdesivir + diltiazem. | A) Vero E6 cell line for monolayer culture, B) nasal and bronchial human primary epithelial cells for pseudostratified epithelial model. | A) Vero E6 cultured in standard static condition, B) cells after isolation cultured in transwell inserts to reach a full differentiation until pseudostratified mucociliary epithelium (human airway epithelium HAE). |
| [32] | SARS-CoV-2, SARS-CoV, MERS-CoV. | β-D-N4-hydroxycytidine (NHC, EIDD-1931). | A) 2D model with Calu-3 human lung epithelial cell line; B) human primary tracheobronchial epithelial cells (HAE). | Human airway epithelium cultures (HAEs) were generated by an air-liquid interface for 6 to 8 weeks to form well- differentiated, polarized cultures that resembled in vivo mucociliary epithelium. Primary cells were expanded and plated at a density of 250,000 cells per well on transwell-COL (12mm diameter) supports. |
| [34] | SARS-CoV-2. | Human recombinant soluble angiotensin converting enzyme 2 (hrsACE2). | iPSCs and human embryonic stem cells (hESCs). For bidimensional culture: Vero-E6 cells. | Human capillary organoids from iPSCs and kidney organoids form hESCs. |
| [42] | H7N9 influenza A virus. | Recombinant human interferons α2b (rhIFN-α2b) and λ1 (rhIFN-λ1). | Well-differentiated pseudostratified human airway epithelium (HAE) cells; controls: A549 adenocarcinomic human alveolar basalepithelial cell line. | All cell types were grown at the air-liquid interface: 2D HAE in monolayer on collagen, 3D HAE in pseudostratified layers on transwell; A549 on transwell. |
| [43] | Rhinovirus species (RV-A16, RV-B14 and RV-C15); enteroviruses EV-D68 and influenza A (H1N1 and H3N2). | Rupintrivir, an irreversible inhibitor of human rhinovirus protease, was chosen as a reference drug. Oseltamivir, a neuraminidase inhibitor, approved to treat and prevent influenza infections. | Airway cells were obtained from 14 different patients undergoing surgical polypectomy. | Cells were cultured at the air-liquid interface in MucilAir culture medium in 24-well plates with 6.5-mm transwell inserts. |
| [44] | Syncytial virus (RSV). | Entry/fusion inhibitors (GS-580619 and TMC353121); nucleoside viral polymerase inhibitor; two non-nucleoside replication inhibitors (AZ-2721 and PC786). | Fully-differentiated human airway epithelium cells (HuAECs) of bronchial origin from healthy donors. | HuAECs of bronchial origin were seeded in an air-liquid interface cell culture system with MucilAir medium. |
| [49] | Rotavirus. | Interferon-alpha (IFN-α) and ribavirin. | For mouse and human organoids: resection on small intestine of mouse and intestinal biopsies or surgically resected intestinal tissues for patients; for 2D culture: Caco2 cell line. | The crypts obtained from the biopsies were suspended in growth factors reduced phenol-red free Matrigel. This was placed in the center of each well of a 24-well plate and was subsequently incubated with a specific culture medium. |
| [47] | Hepatitis B (HBV). | Entecavir. | HepG2-NTCP cell line (transfected cell line for HBV infection) + primary human hepatocytes (PHHs). | Human decellularized liver scaffold from healthy and cirrhotic donors (hDCS-hDLCS) seeded with PHHs and with HepG2-NTCP. |
| [50] | Herpes Simplex virus 1 (HSV-1). | Antivirals 5BVdU and interferon-alpha (IFN-α). | Human fibroblast derived iPSCs and HFF1S cell lines. | 2D neural culture obtained by hiPSCs seeded and cultured with neurobasal medium and brain neurothrophic factor to become neural progenitor cells (NPCs) and mature neurons. 3D brain organoids obtained via hiPSCs differentiation towards NPCs self-assembled neurospheroids in transwell and maturing in a toroidal shape while cultured in low-attachment plates. |
| [51] | Herpes simplex virus-1 (HSV-1). | Acyclovir (ACV). | 3T3 fibroblasts; for viral stock: Vero cell lines. | In vitro 3D acrylated hyaluronic acid (AHA) hydrogel model encapsulating fibroblasts. |
| [99] | Herpes simplex virus type 1 and type 2 (HSV) and varicella-zoster virus (VZV). | Acyclovir, penciclovir, brivudin, foscarnet, and cidofovir. | Primary human keratinocytes isolated from neonatal foreskins. | Organotypic epithelial raft cultures that permit full differentiation of keratinocyte monolayers. The cells were cultures on collagen gels at the air-liquid interface. |
| [100] | Cowpox virus. | Gefitinib. | Pooled primary normal human epidermal keratinocytes (NHEK); for viral stock: HEp-2 cells and Vero E6 cells. | For 3D culture, decellularized equine pericardium used as a biological ECM for cell culture of 1×105 NHEK cells. For monolayer control, NHEK were seeded in 24-well cell culture plates. |
Most studies on viruses other than SARS-CoV-2 consist in modeling respiratory infections diffused in an engineered human pseudostratified airway epithelium [35-44]. In these models, the cellular constructs are based on primary epithelial cells embedded in a matrix of biological origin replicating the basal membrane, and are grown to full differentiation, preferably at the air-liquid interface, into an organoid with basal, secretory, and multi-ciliated cells. Most studies also use specific control cell lines cultured in monolayer, for example derived from lung and intestinal adenocarcinoma epithelial cells [32, 45-46], or from lung primary fibroblasts and epithelial cells [40]. A comparison of the results from 3D models with monolayer cultures highlights the following significant advantages of 3D cell models in replicating conditions observed in vivo.
1) Cell morphology, virus penetration and cell damage
These parameters can be imaged and are replicated similarly to in-vivo in 3D cell and organoid models, unlike in cell monolayers. For example, cells in 3D engineered airway epithelium infected with SARS-CoV-2 showed apical-apical polarity, which is known to restrict the infection to the airway lumen in vivo [30], and reproduced infection peaks and significant morphological alterations earlier in nasal cells compared to bronchial cells [32]. Cells in liver organoids infected by SARS-CoV-2 [33] and cells in intestinal organoids infected by MERS-CoV [46] had syncytia and membrane fusions as observed in mice in vivo models. When infected by the respiratory syncytial virus (RSV), lung organoids had a morphological structure and infection features matching human fetal lung tissue [35], and airway epithelium organoids had massive epithelial modifications as in mature tissue infection in vivo [36]. In a 3D engineered airway epithelium, cell morphology was close to in-vivo characteristics, with a reorganization of cytoskeletal components, an abundance of ciliated cells [38], and a mucociliary layer [41], with surfactant and mucus often produced [37]. Here, the H7N9 influenza virus infected both ciliated and non-ciliated cells, causing cilia and tight junction damage seen in vivo [42]. A 3D engineered liver model reproduced and demonstrated greater physiological hepatocyte morphology, polarity, and apical/basal adhesion to ECM compared to a classical cell monolayer [47].
2) Susceptibility to viral infection
3D cell and organoid models are significantly more permissive to viral infection, compared to cell monolayers. For example, when infected by SARS-CoV-2, the peak infection in 3D-engineered human airway epithelium was reached 30% faster [31]; also, when infected by avian influenza virus H7N9, viral proliferation was faster [42]. In addition, propagation of human rhinovirus C and bocavirus was two and three orders of magnitude greater, respectively [41]. In airway epithelium organoids infected with avian influenza virus such as H1N1, the viral proliferation was faster and with a time-order equivalent to the in vivo proliferation time [38]. MERS-CoV was shown to infect intestinal organoids more than epithelial monolayers [46]. In a model of HIV-1 infection of primary human CD4+ T-lymphocytes, 3D collagen culture, unlike suspension conditions, promoted cell-associated HIV-1 transmission, consistent with spread mechanisms described in vivo [48]. Human intestinal organoids infected with rotavirus displayed a 10,000-fold increase in genomic viral RNA than cells infected in monolayer [49]. Hepatitis B replicated one order of magnitude faster in a 3D-engineered liver model, compared to a cell monolayer [47].
3) Cell expression of virus receptors, antiviral genes, antigens to viruses and inflammatory markers
The expression of all these markers is more similar in time and space to in-vivo values, in 3D cell and organoid models, compared to cell monolayers. For example, a 3D-engineered airway epithelium infected by SARS-CoV-2 showed different expression levels of immune-response genes (type I and type III interferons and a subset of genes associated with the NF-kB and TNFα pathways) between nasal cells (with strong upregulation at 24 hours post infection) and bronchial cells (with low expression) [31]. Cells in liver organoids expressed the ACE2 receptor, thus enabling SARS-CoV-2 infection, unlike in the mouse model [33]. When infected by the respiratory syncytial virus (RSV), lung organoids had gene and marker expression (primarily the mesenchymal markers PDGFR-α, PDGFR-β, α-SMA, vimentin and CD90) that matched infected human fetal lung tissue [35], while airway epithelium organoids expressed levels of genes involved in migration (KRT16, KRT6b), interferon signaling (IL1RN, IFI44L) and viral response (MX2, OAS1), comparable to in vivo values [36].
In 3D-engineered airway epithelium, expression of inflammatory cytokines (aquaporin-5 and cytokeratin-14) was two-fold higher than monolayers when infected with avian influenza viruses H1N1 and H3N2 [37], and two orders of magnitude higher (for IL-6 and IL-8) when infected with human rhinovirus C and human bocavirus [41], closely recapitulating the immune response seen in vivo. In a model of stratified airway epithelium infected by the andes-hantavirus, long-term culture at seven days showed a switch in immune response from anti-viral (transient interferon) to antinflammatory (cytokines and VEGF-A), as occurs in-vivo [40]. Hepatitis B induced a two-fold higher expression of specific antigens (HBsAg) in a 3D-engineered liver model, compared to cell monolayers [47].
4) Sensitivity to antiviral drugs
In 3D models, the sensitivity to antiviral drugs is significantly lower and more similar to in vivo than cell monolayer observations. For example, the efficacy of clinical-grade soluble human ACE2 in reducing SARS-CoV-2 infection in both human capillary organoids and kidney organoids was one order of magnitude lower than in monolayers [34]. In a 3D-engineered airway epithelium infected with the H7N9 influenza virus and treated with recombinant human interferons α2b and λ1, the expression of interferon-stimulated antiviral genes was significantly reduced, and the level of gene expression induced by the two agents was reversed, consistently with data in vivo, compared to monolayers [42]. Human intestinal organoids infected by human rotavirus, compared to the simple cell line model in monolayers, were twice less sensitive to interferon α and ribavirin, with a range of effective doses comparable to that administered in in vivo models [49].
The response to the antiviral drug entecavir in a 3D human engineered liver model infected by hepatitis B was more similar to in vivo, compared to the cell line model [47]. In conventional cell monolayers, the response of herpes simplex infection to antivirals is unrealistically fast with 100% reactivation. For herpes simplex 1, virus reactivation after exposure to interferon α was reduced five-fold in infected 3D brain organoids compared to cell monolayers, as reported in animal models [50]. In addition, in hydrogels made of acrylated hyaluronic acid and loaded with fibroblasts, the time for cell recovery and reaggregation after infection and exposure to the antiviral drug, acyclovir was three-fold greater than in cell monolayers, similarly to in vivo [51].
The evidence outlined above shows that 3D cells and organoid models consistently develop properties that are able to determine the in vivo-like dynamics of infection by pathogenic viruses. They thus may represent a valuable tool towards a more rapid assessment of infectivity to humans of emerging respiratory viruses such as the SARS-CoV-2, and of relevant drug responses.
From a technological point of view, there are two principal limitations of the previously analyzed 3D models compared to conventional monolayer models, which are essentially related to the 3D scaffolds. The current scaffolds are mainly represented by either hydrogels [52-53] or solid ECM analogues [54], which are placed on the bottom of culture plates or in trans-well inserts, resulting in constructs which are a few millimeters thick and cannot be examined under optical microscopy. The second problem in current 3D scaffolds is the very limited control over the culture time of some physical and chemical properties known to profoundly affect cell behavior, such as biodegradation with the release of various solutes, and mechanical properties such as stiffness, which influence cell adhesion and migration [54]. To overcome all these limitations, many groups of researchers are developing miniaturized 3D scaffolds by a cutting-edge fabrication technique called two-photon laser polymerization [55].
For example, our group has developed an innovative 3D micro-scaffold called the Nichoid (Figure 2), which is optically and physically accessible to the cells cultured inside, over time, and to all the existing biological assays. The Nichoid is a miniaturized rigid, transparent, ultra-precise 3D grid fabricated in a biocompatible resin [56-58]. This scaffold is up to 100 microns thick, thus truly 3D for cells but 100% optically accessible to high-resolution fluorescence diagnostics. By mimicking the 3D stiffness of a physiological cell niche [59], the Nichoid can guide the self-organization of several types of stem and progenitor cells [60-61], including embryonic stem cells [62] and mesenchymal stem cells (MSC) [63]. It also maintains their phenotype in expansion culture, by regulating hundreds of genes involved in mechanotransduction processes such as cell migration, cytoskeletal organization, and membrane plasticity [64]. The Nichoid could thus provide an effective culture environment to test mechanobiological effects such as the overexpression of interferon-induced transmembrane proteins that can rigidify the cell membrane, thus markedly inhibiting viral membrane fusion and entry. It could thus be used for screening candidate antiviral drugs and vaccines based on the targeting coronavirus spike protein. We also observed that the Nichoid potentiates the therapeutic efficacy of stem cells expanded inside [65]. This substrate is a powerful candidate to maintain the safety and function of expanded MSC [63], which is a new therapy already being tested in clinical trials to treat post-COVID-19 pulmonary fibrosis [66].
Bioreactor-perfused cell models
In bioreactor culture systems, the cellular constructs or organoids are hosted in culture chambers, which are connected to a hydraulic circuit that perfuses the culture medium with a pump (Figure 3). Microfluidic systems are the miniaturized version of bioreactors, generally consisting in thin transparent devices, similar to microscopy slides, with integrated micro-channels that can host cell-loaded hydrogels or suspensions. Table 5 shows the viral infection models that have been developed using bioreactor devices. The literature reports a pioneering model of SARS-CoV infection in a 3D bioreactor-engineered respiratory epithelium [67-68] and several other more recent models of infection based on different types of viruses. These studies demonstrate the following significant advantages of perfused culture in replicating conditions seen in vivo, compared to non-perfused culture.
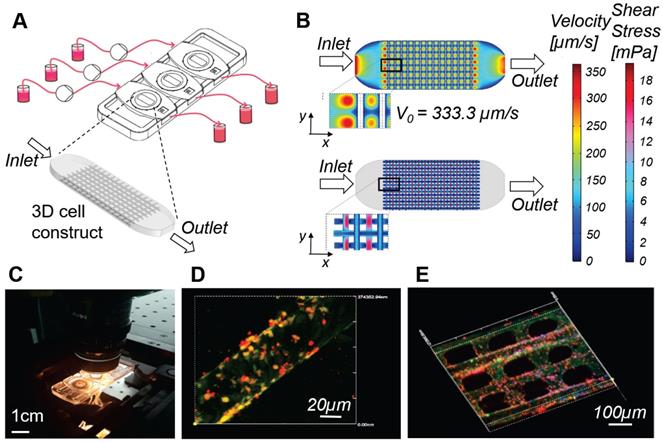
Millifluidic optically accessible bioreactor (MOAB) for perfused culture of 3D cell constructs. A) The system is composed of, from left to right: culture medium reservoirs, microfluidic pumps, bioreactor chamber and reservoirs for medium collection. CAD model of the 3D cell construct or organoid, cultured in the culture chambers, used for numerical simulations. B) Results of the computational fluid dynamics (CFD) simulations for a single culture chamber of the system. Top: fluid velocity (modulus of the velocity vector) is mapped on the whole culture chamber. Bottom: wall shear stress (WSS) is computed and mapped at the cell-culture medium interface. C) Photo of the MOAB showing the three independent chambers connected to the perfusion chamber by oxygenator tubes; the device is placed on a confocal microscope connected to a CPU allowing real time imaging of a perfused lymph-node-on-a-chip model, for the development and testing of vaccines and agents for cancer immune-therapy. D) Fibroblast reticular cells (yellow) are seeded on the 3D fiber scaffold. Dendritic cells introduced in suspension during culture medium flow adhere to the fibroblast reticular cells, migrate to the scaffold, and are activated to express the adhesion receptor ICAM-1 (red). E) Antigen-specific T cells (cyan) introduced with culture medium flow tend to adhere to dendritic cells expressing ICAM-1 (red) while crawling on the scaffold.
In vitro cell models of viral infection based on bioreactor-perfused cultures
| Refs. | Pathogen | Cell type/s | Cell culture configuration |
|---|---|---|---|
| [67-68] | SARS-CoV. | Primary human bronchial-tracheal cells (HBTCs) used to establish a human lung tissue-like assembly (TLA) with an overlay of human bronchial epithelial (BEAS-2B) cells. 2D control established with BEAS-2B monoculture. | 3D tissue dynamic culture system established using a Rotating Wall Vessel (RWV) bioreactor. Static 2D controls: tissue culture flasks of near confluent BEAS-2B monocultures. |
| [70] | Hepatitis B virus (HBV). | Primary Human Hepatocytes (PHH) and co-culture with primary Kupffer cells (KC). | Perfused 3D bioreactor. Medium was recirculated via a pneumatically driven micro-pump, and collagen-coated scaffolds were used for cell adherence. Each plate consisted of 12 individual bioreactors, in which the flow rate was regulated. 3D perfused: PHH seeded in 3D scaffolds. Non-perfused controls: 3D spheroid, static 2D PHH, and self-assembling co-cultures (SACC) of PHH. |
| [68-69] | Varicella zoster virus (VZV). | Normal human neural progenitor (NHNP) cells used to establish human neuronal tissue- like assembly (TLA). Gene mutations, infection study and VZV genome maintenance were performed on the TLA and validated with a 2D control of human melanoma (MeWo) cells. | Optimized 3D tissue culture systems created using a Rotating Wall Vessel (RWV) bioreactor. The 3D model was compared to a 2D control of NHNP cells. |
| [71] | Adenovirus carrying enhanced green fluorescent protein (AdEGFP). | Mouse embryonic fibroblasts (MEFs) and human foreskin fibroblasts (HFFs) both for microfluidic study and static control. | Microfluidic device composed of a supporting glass slide with a PDMS slab carved to accommodate the cell culture coverslip, a membrane-based vacuum system for the reversible sealing, and microfluidic channels delivering fluids to the cultured cells. The control was performed in static conditions. |
| [106] | Enhanced green fluorescent protein (EGFP) plasmid. | 3T3 fibroblasts used for 2D control, static and dynamic conditions. | Microfluidic perfused device for dynamic cell culture and virus infection in a micro channel, control was performed in 2D conventional culture, and in the microfluidic device was evaluated in both static and dynamic conditions. |
1) Perfusion increases the degree of cellular differentiation
For example, a 3D bioreactor-engineered respiratory epithelium infected with SARS-CoV was found to more closely resemble normal human tissue in terms of cell apical polarity, essential to viral cell-cell transport, compared to non-perfused culture [67-68]. A bioreactor-engineered liver model of hepatitis B infection recapitulated the functional hepatic microarchitecture and complete cell polarization, which is critical for the cells' susceptibility to infection, compared to non-perfused organoids which show de-differentiation over 10-13 days [70]. A bioreactor-engineered model of neuronal tissue based on neural progenitors showed some features of the human trigeminal ganglia, the site of latency of the varicella-zoster virus, not seen in non-perfused cultures [68-69].
2) Perfusion extends cell viability, cell metabolism and functional stability
A 3D bioreactor-engineered respiratory epithelium infected with SARS-CoV remained viable three-times longer than non-perfused controls [67-68]. A bioreactor-engineered liver model of hepatitis B infection was metabolically and functionally stable for at least 40 days, i.e. four times longer than conventional non-perfused cultures [70]. A bioreactor-engineered model of neuronal tissue infected by the varicella-zoster virus was successfully maintained in culture for 180 days, compared to the limit of a few days for primary human ganglia in non-perfused culture [68-69].
3) Perfusion increases the infection efficiency for longer incubation times
In a microfluidic model of fibroblast infection by the adenovirus, a 50% higher infection efficiency was maintained at a lower multiplicity of infection (the minimum virus/cell ratio able to cause infection) for longer incubation times, with respect to non-perfused controls [71]. A bioreactor-engineered liver model of hepatitis B infection led to a 10,000-fold lower multiplicity of infection, compared to non-perfused culture [70]. A bioreactor-engineered model of neuronal tissue infected by the varicella-zoster virus maintained persistent viral infection for 90 days, which is not possible with non-perfused culture [68-69].
4) Perfusion increases the level and stability of cell marker expression
In a 3D bioreactor-engineered respiratory epithelium infected with SARS-CoV, the antibody response to SARS-CoV-specific spike and nucleocapsid glycoproteins was two-fold, compared to non-perfused culture [67-68]. A bioreactor-engineered liver model of hepatitis B infection maintained stable expression levels of receptors of innate immune cells and downstream effectors, compared to non-perfused cultures [70].
The evidence above therefore shows that perfused models enable infection studies to be extended even up to several months, on stable cell cultures of highly differentiated cells, with a higher infection efficiency and lower multiplicity of infection, compared to non-perfused models. This makes it potentially feasible to extend the study of the life cycle and genetic stability of new viruses such as SARS-CoV-2 to in vitro systems, despite being derived from minimal doses from patients, and to test sequential antiviral drug treatments [70]. Most importantly, perfused cell models make it feasible to reproduce dynamic interactions between tissue-resident cells and circulating cells, including the response to a pathogen of innate and adaptive immune cells and thus, to extend the testing of vaccines to in vitro models.
A first step forward in this regard is a recent microfluidic-based model of a lymph node [72]. Here, the stroma was modeled with adherent murine tumor dendritic cells, and CD4+ and CD8+ T lymphocytes were put in suspension in the culture medium and recirculated interstitially within the engineered organ. This system selectively promoted the adhesion of antigen-specific T cells through affinity isolation by serial cell contacts, with fluid-induced shear stresses maintained in the range 0.1-1 dyn/cm2. In contrast to non-perfused cultures, this model was able to compare in parallel and in real time, the effect on T cell adhesion to dendritic cells of different treatments, inhibitors, activators and immunogens.
With regard to vaccine testing, bioreactors that host cm-sized 3D scaffolds such as the rotating wall vessel are able to provide tens of millions cells for confirmation assays such as PCR or FACS, as required by regulatory agencies. Their technological limitation, however, is that they are too thick to be accessed thoroughly for real time optical inspection of cell interactions [54,73]. Microfluidic bioreactors, instead, are 2D and have micron-sized channels that are also fully inspectable in real time [74-75]. However, they can host only a few thousand cells which are difficult to harvest and in any case would not be sufficient for robust confirmation assays [76].
To overcome these limitations, our group developed a millifluidic optically-accessible bioreactor (MOAB), which is a compromise between the two categories mentioned above. The MOAB (Figure 3) is a chambered microscope slide allowing the culture of fully 3D cell models with dimensions of up to a few millimeters, containing several million cells, under interstitial perfusion of the culture medium, with the infusion of cells in suspension and the therapeutic agents to be tested. The culture chambers are only 400 microns thick and enable full-thickness high-resolution fluorescence diagnostics, both in real time and post-cultivation [77-81]. In in vitro tests, the MOAB led to a comparable cell response to biodrugs to the one seen in experimental animals, in specific fields characterized by slowly-developing diseases and slowly-developing healing induced by the therapeutic agent: stem cell therapy for neurodegeneration [82], gene therapy for muscular dystrophy [83], and chemotherapy for bone metastases [84].
By adding lymphocytes in suspension in the perfused medium (Figure 3), the MOAB easily extended to 3D a microfluidic model of lymphocytes instruction within a lymph node [72]. Millifluidic bioreactors in fact have a high potential to speed up the discovery and testing of drugs and new vaccines for SARS-CoV-2 and are not limited to the interactions with the immune system. For example, they can successfully maintain human primary neurons infected by a virus in long-term culture for up to 180 days, compared to the limit of a few days in non-perfused culture [68-69]. Millifluidic bioreactors can even be hydraulically connected in multiple units, to reproduce transport of neurotoxic agents through multiple body barriers pertaining to distant body compartments, for example from the gut endothelial barrier to the blood-brain-barrier [85]. This method can be exploited to assess neurotoxicity, which is a primary concern for new antiviral drugs and vaccines for SARS-CoV-2.
In addition, the SARS-CoV-2 virus itself could hold a neurotropic potential. New observations on the loss of smell and taste by patients later manifesting symptoms of COVID-19, together with evidence showing that coronaviruses such as SARS-CoV and MERS-CoV could target the central nervous system, open up a new window in this regard [86]. In fact, a long-term bioreactor co-culture of olfactory neurons and ensheathing glial cells in 3D, could not only confirm the expression of ACE2 in human olfactory neurons, thus their role as a potential entry site for the SARS-CoV-2 virus, but also could help to clarify the potential mechanism of virus propagation to the brain through neural cell populations, i.e. from the olfactory neurons to their ensheathing glial cells. Such a model could verify whether, in SARS-CoV-2 infection, the defense mechanism provided by olfactory ensheathing cells is present, and whether it is maintained during aging. Such an experiment could add crucial insights into unveiling the possible contribution of neurological tissue damage to the morbidity and mortality caused by COVID-19 [11], also suggesting new prevention measures, such as the vaporization of nasal disinfectants in the elderly population.
Intravital imaging models in vivo for drug testing
Immunization addresses the response of innate and adaptive immune cells to a pathogen, which is a very complex and slowly-developing phenomenon commonly studied in vivo. In advanced study models of the immune response to influenza viruses, the experimental animals were directly accessed with a microscope for intravital real time optical inspection of organs including the nasal cavity [87], trachea [88], and lungs [89-91].
Table 6 shows some examples of the intravital imaging models published in the field of immunization. The most elegant procedures generally consist in surgically creating a direct percutaneous access for the microscope objective, often using metallic holders, in transgenic mice with fluorescent CD4+ T cells and/or T cell receptors, and in visualizing their interactions with inoculated recombinant vaccine viruses also expressing fluorescent proteins [87-90,92-96]. Compared to conventional post-mortem histopathology, several key advantages of intravital imaging techniques can be highlighted in speeding-up the preclinical evaluation of vaccines and novel antiviral therapies.
In vivo models of viral infection based on intravital imaging
| Refs. | Pathogen | Function monitored | Imaged Cell type/s | Imaging window configuration | Imaged Organ/District |
|---|---|---|---|---|---|
| [91] | Influenza viruses: PR8-H1N1, PR8-H5N1. | Correlated pathological changes in the lungs of live animals at the cellular level (labeled immune-related cells) in response to influenza viruses. | Madin-Darby canine kidney cells for virus propagation.Mice immune-related cells. | Thoracotomy incision and left lung immobilization by vacuum. | Left lung. |
| [89-90] | Influenza A virus (IAV). | CD4+ T cells responding to IAV in the mice lung. | Mouse transgenic CD4+ T cells. | Thoracotomy incision and left lung immobilization by vacuum. | Lung. |
| [88] | Influenza virus (PR8- H1N1). | Neutrophil and dendritic cell interactions with influenza virus at the tracheal site. | Neutrophils, dendritic cells. | Tracheal exposure by removing the tissue and muscles. | Trachea. |
| [87] | Respiratory Syncytial Virus (RSV) A2-Line19F. | Kinetics of RSV infection in the nasal cavity and in the lungs of live BALB/c mice infected with a recombinant virus strain expressing firefly luciferase gene (RSV A2-Line19FFL). Immunization versus therapy by either vaccination with 5 μg of DS-Cav1, a glycoprotein of the syncytial virus, or treatment with the monoclonal antibody Palivizumab. | Virus expansion: A549, Hep-2. | Whole body live imaging without specific localization of window. | Whole body imaging detected strong bioluminescencesignal in the nasal cavity and in the lungs. |
| [92-93] | HIV. | HIV infection on T cell migration. | HEK293T cell line (virus expansion), Human CD4+ central memory-like T cells. | Abdominal exposure with permanent imaging device implanted. | Popliteal lymph nodes (popLN). |
| [94-95] | Oncolytic Viruses (OV): vesicular stomatitis virus (VSV); maraba virus; reovirus. | Interaction between OV and tumor in vivo. | Virus expansion: Vero cells (Vesicular Virus); L929 (Reovirus). Tumor model: CT-26 (murine colon adenocarcinoma). | Dorsal/Abdominal exposure with no permanent imaging device implanted. | Skin/endothelium. |
| [96] | Vaccinia virus (VACV). | VACV infection of mice to study both the movement of infected cells and the response of innate and adaptive immune cells into skin and mucosa. | Infected cells. | Craniocaudal exposure: (1) Ear sealing with no external device or wound generation; (2) Lower labial mucosa exposed into a steel stage. | (1) Ear blood vessels; (2) Labial mucosa. |
1) Dynamics of infection progression and the effects of vaccines and drugs
These can be quantified in real time with a resolution at the cellular scale. For example, intravital imaging studies in lungs quantified in time the recruitment of CD4+ T cells from blood vessels in response to the influenza A virus [89-90] and, most recently, the recruitment of neutrophils in response to the PR8-H1N1 and H5N5 influenza viruses [91]. Multiphoton intravital microscopy in abdominal lymph nodes was used to quantify a partial reduction in the motility of HIV-infected T cells as well as their tethering to and fusion with CD4+ immune cells [92-93]. Intravital imaging within vessels of colon adenocarcinoma tumors was used to quantify the timely evolution of several oncolytic viruses binding to intravascular leukocytes and endothelial cells [94-95].
2) Sensitivity and robustness of intravital imaging to assess immunization
These parameters are greater than in in vitro assays. For example, in an immunization study, using a glycoprotein of the respiratory syncytial virus, associated with an antiviral treatment with the specific monoclonal antibody, live imaging was used to visualize and capture virus-infected cells in the nasal cavity and in the lungs of experimental mice with a higher sensitivity compared to the in vitro plaque assay [87]. Intravital imaging of live mice was sufficiently sensitive to quantify vaccinia viruses in ear blood vessels and labial mucosa [96], and single oncolytic virus particles interacting with individual leukocytes and endothelial cells within blood, tumors, and visceral organs [94-95].
Intravital imaging is thus emerging as a key tool to observe the spatial-temporal dynamics of very complex phenomena, even as complex as embryonic development, at a cellular resolution [97].
In the field of immunology, however, intravital imaging models have significant limitations. Temporary “suction” window chambers are highly invasive and always require animal euthanization at the end of each observation, preventing repeated observations at subsequent time points. In contrast, implantable windows allow for long-term repeated observations in the same animal. However, they are currently too invasive and affected by chronic inflammation at the surgical site, thus interfering with the desired measurements [88]. In addition, implanted imaging windows in general do not retain lymphatic circulation, thus preventing the recruitment of cells from the bone marrow or lymphatic system [89-90].
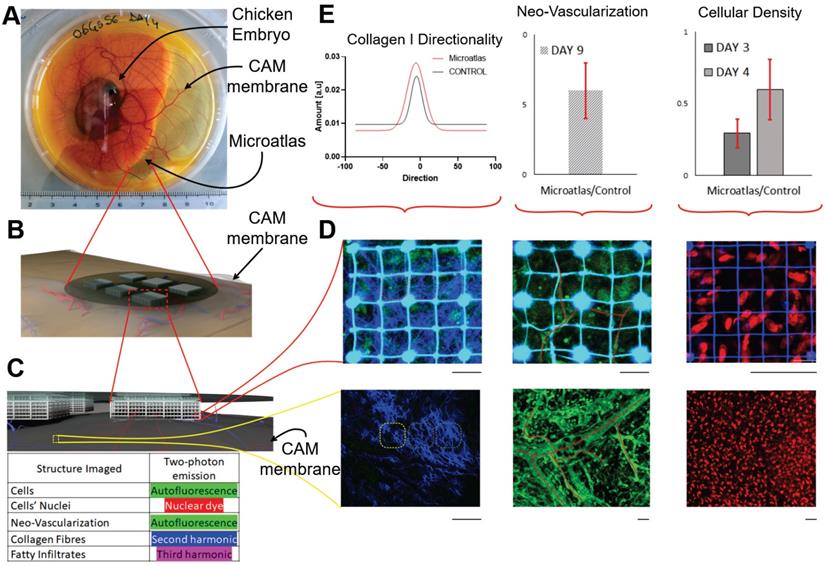
To overcome these limitations, we developed an implantable miniaturized intravital imaging device called Microatlas (Figure 4). This minimally invasive device, which is 5 mm in diameter and 1 mm in thickness, is implantable in chicken embryos or subcutaneously in mice to quantify in vivo aspects of the immune response to biomaterials and drugs [98]. Microatlas integrates a micro-fabricated fluorescent 3D scaffold that guides and hosts the intravital regeneration of vascularized tissue. In addition, this scaffold enables the repositioning of the observation field of view of a two-photon microscope, which is necessary for repeated and quantitative intravital measurements of immune cell recruitment, neo-angiogenesis and fibrotic reaction, on the same animal at different time-points. The fluorescent grid is currently designed to provide beacons to correct aberrations derived from deep optical sectioning of the 3D micro-scaffold repopulated by cells during ex ovo experiments.
Miniaturized “Microatlas” imaging window for intravital microscopy. A frontier imaging window device used for mini invasive quantitative analyses of living organisms, without any persistent percutaneous access. The device incorporates micro-scaffolds micro-fabricated by two-photon polymerization. The device was developed for animal examinations with optical fluorescence microscopy techniques. A) The device was validated by studying the reaction of the foreign body of the implant, and the animal model used was the chicken embryo. The widely employed Chorioallantoic Membrane (CAM) assay was used to quantify and characterize the amount of reaction occurring inside the imaging device. Here an image of a living embryo on the 8th day of incubation: the device was implanted in the membrane (red dot-circle). B) Rendering representation of the implant set-up. The device lies on the membrane. No conditioning factors were administered. C) Zoomed rendered detail of the tissue infiltration inside the micro-scaffold device. The mechanical conditioning, alone, guides the tissue regeneration in situ, allowing a fast neovascularization in the structure porosity. D) Example of possible fluorescence acquisitions that quantify the foreign reaction. In the upper part is the window imaging, with respect to the control, which is reported into the lower part. Second Harmonic Generation (left), tissue autofluorescence (middle) and nuclear dyes (right) were used for the examination in real-time. E) Graphical quantitative representation of the reaction occurring in terms of collagen fibers orientation, differential neo-vascularization rate and differential cellular density in subsequent incremental time-points. Scale bars: 50 µm.
Expression of the human ACE2 receptor can be engineered in transgenic mice. In contrast with histopathology techniques, imaging through a miniaturized intravital window can be used at incremental time points within the same mouse, in vital conditions to study the localization and distribution, pharmacokinetics, and the pharmacodynamics of antiviral therapies and vaccines. While seeking solutions for COVID-19, it could significantly speed-up the preclinical testing of experimental antiviral drugs and vaccines, while eliminating the need for animal sacrifice at the various time points, thus reducing by up to 90% the number of animals required by the regulation for ex vivo assessments.
Intravital imaging could be very effective in this model also to test therapeutic agents based on an engineered messenger RNA (mRNA) that directs cells to make a protein which could work as a drug or vaccine for SARS-CoV-2. Human safety trials have already begun for mRNA-based vaccines against two flu strains and the zika virus. In each case, the mRNA encodes for viral proteins that infected cells would physiologically present to activate the immune system against infections. To succeed, the engineered mRNA must hide from dendritic cells, enter the cells, avoid degradation, and be translated efficiently. The efficiency off all these stages could be monitored and measured by the intravital imaging of fluorescently-labeled mRNA.
Conclusions
In this review, we have discussed various cutting-edge bioengineering tools already available in the fields of virology and pharmacology, which are based on i) 3D co-cultures that are highly predictive of in vivo mechanisms and, ii) high-resolution intravital microscopy. We have also discussed the potential strategies aimed at repurposing these tools in order to significantly accelerate the development of therapeutics and vaccines against COVID-19.
Although several academic institutions have already adopted these improved tools for research into basic immune biology and drug discovery, pharmaceutical companies still use conventional cell lines and standardized animal models for confirmatory studies, as requested by the standards for the passage of new drugs through the preclinical steps of regulatory approval, but despite the limited predictive capability of these models.
The particular urgency to find clinical solutions for the SARS-CoV-2 pandemic makes it even more evident that an adequate validation of these new tools could lead to an update of the current preclinical testing standards, with the new ones based on the concepts of 3D culture, perfused culture and intravital imaging presented here.
In virology and vaccinology, as well as in many other pharmaceutical sectors, the new standards suggested in our review would improve the efficiency and effectiveness of the whole drug development process, with the added benefit of drastically reducing, refining and widely replacing animal testing.
Abbreviations
3CLpro: 3-chymotrypsin-like protease; ACE2: angiotensin-converting enzyme 2; ACV: acyclovir; ADRP: ADP ribose phosphatase; AHA: acrylated hyaluronic acid; ALI: air-liquid-interface; ANDV: andes-hantavirus; AO: airway organoids; ASC: adult stem cells; BME: basement membrane extract; CAD: computer aided design; CAM: chorioallantoic membrane; CD: cluster differentiation; CFD: computational fluid dynamics; CoV: coronavirus; COVID-19: coronavirus disease 2019; CPU: central processing unit; cryo-EM: cryo-electron microscopy; DC: dendritic cell; E: envelope protein; ECM: extracellular matrix; EGFP: enhanced green fluorescent protein; EIC: epithelial intestinal cell; EM: electron microscopy; EV: enterovirus; FACS: fluorescent-activated cell sorter; FAD: flavin adenine dinucleotide; FBC: human primary fibroblast; FP: fusion peptide; GFAP: glial fibrillary acidic protein; hADSC: human adipose derived stem cell; HAE: human airway epithelium; HBoV: human bocavirus; HBsAg: hepatitis B surface antigen; HBTC: human bronchial-tracheal cell; HBV: hepatitis b virus; hESC: human embryonic stem cell; HFF: human foreskin fibroblasts; HIV: human immunodeficiency virus; HPIV3: parainfluenza virus-3; HR1: heptad repeat 1; HR2: heptad repeat 2; hrsACE2: human recombinant soluble ACE2; HRV-C: human rhinovirus c; HSAEpCs: human primary small airway epithelial cells; HSV: herpes simplex virus; HTI: human intestinal tissue; HTO: human testicular organoid; HuAEC: human airway epithelium cell; IAV: avian Influenza; IC: intracellular domain; ICAM-1: intercellular adhesion molecule 1; IFI44L: interferon-induced protein 44-like; IFN-α: interferon-α; IL-6: interleukin 6; IL-8: Interleukin 8; IL1RN: interleukin 1 receptor antagonist; iPS: Induced pluripotent stem cells; KC: kupffer cells; KRT: keratin; LBO: lung bud organoid; LC: leydig cells; LDO: liver ductal organoids; M: membrane protein; MEF: mouse embryonic fibroblast; MERS: middle east respiratory syndrome; MeV: measles virus; MOAB: miniaturised optically accessible bioreactor; Mpro: main protease; MSC: mesenchymal stem cell; N: nucleocapsid protein; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; NHEK: normal human epidermal keratinocytes; NHNP: normal human neural progenitor; NPC: neural progenitor cell; NSP: non-structural proteins; NTD: N-ter domain; OAS1: 2'-5'-Oligoadenylate Synthetase 1; ORF: open reading frame; OV: oncolytic viruses; PCR: polymerase chain reaction; PDB: protein data bank; PDGFR-α: platelet-derived growth factor receptor α; PDGFR-β: platelet-derived growth factor receptor β; PDMS: polydimethylsiloxane; PHH: primary human hepatocytes; Plpro: papain-like protease; popLN: popliteal lymph nodes; RBD: receptor binding domain; RBM: receptor binding motif; RdRp: RNA-dependent RNA polymerase; rhIFN: recombinant human interferon; RSV: respiratory syncytial virus; RV: rhinovirus; RWV: rotating wall vessel; S: spike protein; SACC: self-assembling co-culture; SARS: severe acute respiratory syndrome; SC: sertoli cells; SD: subdomain; SEM: scanning electron microscopy; SIS: small intestinal submucosa; SSC: spermatogonial stem cell; TLA: tissue-like assembly; TM: transmembrane region; TMPRSS2: transmembrane serine protease 2; TNFα: tumor necrosis factor alpha; VACV: vaccinia virus; VEGF-A: vascular endothelial growth factor A; VSV: vesicular stomatitis virus; VZV: varicella-zoster virus; WSS: wall shear stress; α-SMA: alpha-smooth muscle actin.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This research has received funding from: the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (G.A. No. 646990 - NICHOID, No. 754467 - NICHOIDS and No. 825159 - MOAB); the Italian Ministry of University and Research (MIUR) under the grant program MIUR-FARE-2016 (G.A. No. R16ZNN2R9K - BEYOND); the National Centre for the Replacement, Refinement and Reduction of Animals in Research (NC3Rs) under the CleanCut 2020 Challenge (G.A. No. NC/C01903/1 - MOAB). We thank Dr. Matteo Laganà (Gemma Prototipi Studio), Profs. Giulio Cerullo (Politecnico di Milano, Italy), Giberto Chirico and Laura Sironi (Bicocca University, Milan, Italy), Oyvind Halaas (NTNU, Trondheim, Norwey), Roberto Osellame (IFN-CNR, Milan, Italy) and Andrea Remuzzi (University of Bergamo, Italy) for their contribution to the miniaturized platforms shown in the figures. Special thanks to Dr. Adrian Wallwork for his highly professional English editing. SC is deeply grateful to Professor Gian Vincenzo Zuccotti (University of Milan, Italy) for support and unswerving encouragement to the work and would like to acknowledge the “Romeo ed Enrica Invernizzi” Foundation, Milan, Italy.
Competing Interests
MTR is a co-founder of a university spin-off company, MOAB S.r.l., and holds shares.
References
1. [Internet 1] WHO, 8 May 2020: https://experience.arcgis.com/experience/685d0ace521648f8a5beeeee1b9125cd
2. Zumla A, Chan JF, Azhar EI, Hui DS, Yuen KY. Coronaviruses - drug discovery and therapeutic options. Nat Rev Drug Discov. 2016;15(5):327-47
3. Thanh Le T, Andreadakis Z, Kumar A, Gómez RR, Tollefsen S, Saville M. et al. The COVID-19 vaccine development landscape. Nat Rev Drug Discov. 2020;19:305-306
4. Li G, De Clercq E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat Rev Drug Discov. 2020;19(3):149-150
5. Bregu M, Draper SJ, Hill AV, Greenwood BM. Accelerating vaccine development and deployment: report of a Royal Society satellite meeting. Philos Trans R Soc & Lond B Biol Sci. 2011;366(1579):2841-2849
6. Saeidnia S, Manayi A, Abdollahi M. From in vitro experiments to in vivo and clinical studies; pros and cons. Curr Drug Discov Technol. 2015;12(4):218-24
7. Li L, Yang L, Gui S, Pan F, Ye T, Liang B. et al. Association of clinical and radiographic findings with the outcomes of 93 patients with COVID-19 in Wuhan, China. Theranostics. 2020 doi:10.7150/thno.46569; in press
8. Liu F, Zhang Q, Huang C, Shi C, Wang L, Shi N. et al. CT quantification of pneumonia lesions in early days predicts progression to severe illness in a cohort of COVID-19 patients. Theranostics. 2020Apr27;10(12):5613-5622
9. Yu Q, Wang Y, Huang S, Liu S, Zhou Z, Zhang S. et al. Multicenter cohort study demonstrates more consolidation in upper lungs on initial CT increases the risk of adverse clinical outcome in COVID-19 patients. Theranostics. 2020Apr27;10(12):5641-5648
10. Zhao W, Zhong Z, Xie X, Yu Q, Liu J. CT Scans of Patients with 2019 Novel Coronavirus (COVID-19) Pneumonia. Theranostics. 2020Mar15;10(10):4606-4613
11. Xu PP, Tian RH, Luo S, Zu ZY, Fan B, Wang XM. et al. Risk factors for adverse clinical outcomes with COVID-19 in China: a multicenter, retrospective, observational study. Theranostics. 2020 doi:10.7150/thno.46833; in press
12. [Internet 2]: WHO, Landscape analysis of therapeutics as 21 March 2020: https://www.who.int/blueprint/priority-diseases/key-action/Table_of_therapeutics_Appendix_17022020.pdf?ua=1
13. Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y. et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B. 2020:2211-3835
14. Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S. et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020:1-9
15. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020;181(2):281-292.e6
16. [Internet 3]: The Human Protein Atlas, 20 April 2020: www.proteinatlas.org/ENSG00000130234-ACE2/tissue
17. Vankadari N, Wilce JA. Emerging WuHan (COVID-19) coronavirus: glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26. Emerg Microbes Infect. 2020;9(1):601-604
18. Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor Recognition by the Novel Coronavirus from Wuhan: an Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J Virol. 2020;94(7):e00127-20
19. Xu X, Chen P, Wang J, Feng J, Zhou H, Li X. et al. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci China Life Sci. 2020;63:457-460
20. Elfiky AA. Anti-HCV, nucleotide inhibitors, repurposing against COVID-19. Life Sci. 2020;248:117477
21. Chen YW, Yiu CB, Wong K. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CL) structure: virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Res. 2020;9:129
22. Hall DC Jr, Ji HF. A search for medications to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2 spike glycoprotein and 3CL protease. Travel Med Infect Dis. 2020Apr;12:101646 doi: 10.1016/j.tmaid.2020.101646; in press
23. Ekins S, Mottin M, Ramos PRPS, Sousa BKP, Neves BJ, Foil DH. et al. Déjà vu: Stimulating open drug discovery for SARS-CoV-2. Drug Discov Today. 2020 S1359-6446(20):30145-8
24. Shi Y, Zhang X, Mu K, Peng C, Zhu Z, Wang X. et al. D3Targets-2019-nCoV: a webserver for predicting drug targets and for multi-target and multi-site based virtual screening against COVID-19. Acta Pharm Sin B. 2020 Apr 20; doi: 10.1016/j.apsb. 2020 04.006; in press
25. Ul Qamar MT, Alqahtani SM, Alamri MA, Chen LL. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J Pharm Anal. 2020 Mar 26; doi: 10.1016/j.jpha. 2020 03.009; in press
26. Dong S, Sun J, Mao Z, Wang L, Lu YL, Li J. A guideline for homology modeling of the proteins from newly discovered betacoronavirus, 2019 novel coronavirus (2019-nCoV). J Med Virol. 2020 Mar 17; doi: 10.1002/jmv.25768; in press
27. Liu Z, Xiao X, Wei X, Li J, Yang J, Tan H. et al. Composition and divergence of coronavirus spike proteins and host ACE2 receptors predict potential intermediate hosts of SARS-CoV-2. J Med Virol. 2020 Feb 26; doi: 10.1002/jmv.25726; in press
28. Angeletti S, Benvenuto D, Bianchi M, Giovanetti M, Pascarella S, Ciccozzi M. COVID-2019: The role of the nsp2 and nsp3 in its pathogenesis. J Med Virol. 2020 Feb 21. doi: 10.1002/jmv.25719; in press
29. Hussain M, Jabeen N, Raza F, Shabbir S, Baig AA, Amanullah A. et al. Structural variations in human ACE2 may influence its binding with SARS-CoV-2 spike protein. J Med Virol. 2020 Apr 6; doi: 10.1002/jmv.25832; in press
30. Milewska A, Kula-Pacurar A, Wadas J, Suder A, Szczepanski A, Dabrowska A. et al. Replication of SARS-CoV-2 in human respiratory epithelium. doi: 10.1101/2020.03.20.999029.
31. Pizzorno A, Padey B, Julien T, Trouillet-Assant S, Traversier A, Errazuriz-Cerda E, et al. Characterization and treatment of SARS-CoV-2 in nasal and bronchial human airway epithelia; doi: 10.1101/2020.03.31.017889
32. Sheahan TP, Sims AC Zhou S, Graham RL Pruijssers AJ, Agostini ML et al. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci Transl Med. 2020 12(541)
33. Zhao B, Ni C, Gao R, Wang Y, Yang L, Wei J. et al. Recapitulation of SARS-CoV-2 Infection and Cholangiocyte Damage with Human Liver Organoids. bioRxivorg. 2020
34. Monteil V, Kwon H, Prado P, Hagelkrüys A, Wimmer R, Stahl M. et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 2020 pr 17. pii: S0092-8674(20)30399-8; doi: 10.1016/j.cell. 2020 04.004; in press
35. Chen YW, Huang SX, De Carvalho ALRT, Ho SH, Islam MN, Volpi S. et al. A three-dimensional model of human lung development and disease from pluripotent stem cells. Nat Cell Biol. 2017;19(5):542-54
36. Sachs N, Papaspyropoulos A, Zomer-van Ommen DD, Heo I, Böttinger L, Klay D. et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 2019 38(4)
37. Bhowmick R, Derakhshan T, Liang Y, Ritchey J, Liu L, Gappa-Fahlenkamp H. A Three-Dimensional Human Tissue-Engineered Lung Model to Study Influenza A Infection. Tissue Engineering Part A. 2018;24(19-20):1468-1480
38. Zhou J, Li C, Sachs N, Chiu MC, Wong BHY, Chu H, Song W. Differentiated human airway organoids to assess infectivity of emerging influenza virus. PNAS. 2018;115(26):6822-6827
39. Porotto M, Ferren M, Chen YW, Siu Y, Makhsous N, Rima B. et al. Authentic modeling of human respiratory virus infection in human pluripotent stem cell-derived lung organoids. mBio. 2019;10(3):e00723-19
40. Sundström KB, Hoang ATN, Gupta S, Ahlm C, Svensson M, Klingström J. Andes hantavirus-infection of a 3D human lung tissue model reveals a late peak in progeny virus production followed by increased levels of proinflammatory cytokines and VEGF-A. PloS one. 2016;11(2):e0149354
41. Chen YX, Xie GC, Dong PAN, Du YR, Pang LL, Song JD. et al. Three-dimensional culture of human airway epithelium in matrigel for evaluation of human rhinovirus C and bocavirus infections. Biomed Environ Sci. 2018;31(2):136-145
42. Chen AJ, Dong J, Yuan XH, Bo H, Li SZ, Wang C. et al. Anti-H7N9 avian influenza A virus activity of interferon in pseudostratified human airway epithelium cell cultures. Virol J. 2019;16(1):44
43. Boda B, Benaoudia S, Huang S, Bonfante R, Wiszniewski L, D. Tseligka E, et al. Antiviral drug screening by assessing epithelial functions and innate immune responses in human 3D airway epithelium model. Antiviral Res. 2018;156:72-79
44. Mirabelli C, Jaspers M, Boon M, Jorissen M, Koukni M, Bardiot D. et al. Differential antiviral activities of respiratory syncytial virus (RSV) inhibitors in human airway epithelium. J Antimicrob Chemother. 2018;73(7):1823-1829
45. Derakhshani S, Kurz A, Japtok L, Schumacher F, Pilgram L, Steinke M. et al. Measles virus infection fosters dendritic cell motility in a 3D environment to enhance transmission to target cells in the respiratory epithelium. Front Immunol. 2019;10:1294
46. Zhou J, Li C, Zhao G, Chu H, Wang D, Yan HHN, Chiu MC. Human intestinal tract serves as an alternative infection route for Middle East respiratory syndrome coronavirus. Sci Adv. 2017;3(11):eaao4966
47. Zhang Z, Xu H, Mazza G, Zhang M, Frenguelli L, Liu Q, Chen X. Decellularized human liver scaffold-based three-dimensional culture system facilitate hepatitis B virus infection. J Biomed Mater Res A. 2019 107(8), 1744-1753
48. Imle A, Kumberger P, Schnellbächer ND, Fehr J, Carrillo-Bustamante P, Ales J. et al. Experimental and computational analyses reveal that environmental restrictions shape HIV-1 spread in 3D cultures. Nat Commun. 2019;10(1):2144
49. Yin Y, Bijvelds M, Dang W, Xu L, van der Eijk A, Knipping K. et al. Modeling rotavirus infection and antiviral therapy using primary intestinal organoids. Antiviral Res. 2015;123:120-131
50. D'Aiuto L, Bloom DC, Naciri JN, Smith A, Edwards TG, McClain L. et al. Modeling herpes simplex virus 1 infections in human central nervous system neuronal cells using two-and three-dimensional cultures derived from induced pluripotent stem cells. J Virol. 2019 93(9)
51. Zhu X, Ding X. Study on a 3D hydrogel-based culture model for characterizing growth of fibroblasts under viral infection and drug treatment. SLAS Discov. 2017 22(5) 626-634
52. Maclean FL, Ims GM, Horne MK, Williams RJ, Nisbet DR. A Programmed Anti-Inflammatory Nanoscaffold (PAIN) as a 3D Tool to Understand the Brain Injury Response. Adv Mater. 2018Dec;30(50):e1805209
53. Weiden J, Voerman D, Dölen Y, Das RK, van Duffelen A, Hammink R. et al. Injectable Biomimetic Hydrogels as Tools for Efficient T Cell Expansion and Delivery. Front Immunol. 2018Nov28;9:2798
54. He B, Chen G, Zeng Y. Three-dimensional cell culture models for investigating human viruses. Virol Sin. 2016;31(5):363-379
55. Song J, Michas C, Chen CS, White AE, Grinstaff MW. From Simple to Architecturally Complex Hydrogel Scaffolds for Cell and Tissue Engineering Applications: Opportunities Presented by Two-Photon Polymerization. Adv Healthc Mater. 2020Jan;9(1):e1901217
56. Raimondi MT, Eaton SM, Nava MM, Laganà M, Cerullo G, Osellame R. Two-photon laser polymerization: from fundamentals to biomedical application in tissue engineering and regenerative medicine. J Appl Biomater Funct Mater. 2012;10(1):55-65
57. Ricci D, Nava MM, Zandrini T, Cerullo G, Raimondi MT, Osellame R. Scaling-up techniques for the nanofabrication of cell culture substrates via two-photon polymerization for industrial-scale expansion of stem cells. Materials (Basel). 2017;10(1):66
58. Zandrini T, Shan O, Parodi V, Cerullo G, Raimondi MT, Osellame R. Multi-foci laser microfabrication of 3D polymeric scaffolds for stem cell expansion in regenerative medicine. Sci Rep. 2019;9(1):11761
59. Nava MM, Raimondi MT, Credi C, De Marco C, Turri S, Cerullo G. et al. Interactions between structural and chemical biomimetism in synthetic stem cell niches. Biomed Mater. 2015;10(1):015012
60. Raimondi MT, Eaton SM, Laganà M, Aprile V, Nava MM, Cerullo G. et al. Three-dimensional structural niches engineered via two-photon laser polymerization promote stem cell homing. Acta Biomater. 2013;9(1):4579-84
61. Raimondi MT, Nava MM, Eaton SM, Bernasconi S, Vishnubhatla KC, Cerullo G. et al. Optimization of femtosecond laser polymerized structural niches to control mesenchymal stromal cell fate in culture. Micromachines. 2014;5(2):341-358
62. Nava MM, Piuma A, Figliuzzi M, Cattaneo I, Bonandrini B, Zandrini T. et al. Two-photon polymerized "nichoid" substrates maintain function of pluripotent stem cells when expanded under feeder-free conditions. Stem Cell Res Ther. 2016;7(1):132
63. Nava MM, Di Maggio N, Zandrini T, Cerullo G, Osellame R, Martin I. et al. Synthetic niche substrates engineered via two-photon laser polymerization for the expansion of human mesenchymal stromal cells. J Tissue Eng Reg Med. 2017;11(10):2836-2845
64. Carelli S, Colli M, Vinci V, Caviggioli F, Klinger M, Gorio A. Mechanical Activation of Adipose Tissue and Derived Mesenchymal Stem Cells: Novel Anti-Inflammatory Properties. Int J Mol Sci. 2018 Jan 16;19(1). pii: E267
65. Carelli S, Giallongo T, Rey F, Colli M, Tosi D, Bulfamante G. et al. Neuroprotection, Recovery of Function and Endogenous Neurogenesis in Traumatic Spinal Cord Injury Following Transplantation of Activated Adipose Tissue. Cells. 2019 Apr 8;8(4). pii: E329
66. Golchin A, Seyedjafari E, Ardeshirylajimi A. Mesenchymal Stem Cell Therapy for COVID-19: Present or Future. Stem Cell Rev Rep. 2020 Apr 13; doi: 10.1007/s12015-020-09973-w; in press
67. Suderman MT, McCarthy M, Mossell M, Watts DM, Peters CJ, Shope R. et al. Three-Dimensional Human Bronchial-Tracheal Epithelial Tissue- Like Assemblies (TLAs) as Hosts For Severe Acute Respiratory Syndrome (SARS)-CoV Infection SARS-CoV Infection in a 3-D HBTE Tissue-Like Assembly. 2006. NASA/TP-2006-213723.
68. Goodwin TJ, McCarthy M, Cohrs RJ, Kaufer BB. 3D tissue-like assemblies: A novel approach to investigate virus-cell interactions. Methods. 2015;90:76-84
69. Goodwin TJ, McCarthy M, Osterrieder N, Cohrs RJ, Kaufer BB. Three-dimensional normal human neural progenitor tissue-like assemblies: a model of persistent varicella-zoster virus infection. PLoS Pathog. 2013;9(8):e1003512
70. Ortega-Prieto AM, Skelton JK, Wai SN, Large E, Lussignol M, Vizcay-Barrena G. et al. 3D microfluidic liver cultures as a physiological preclinical tool for hepatitis B virus infection. Nat Commun. 2018;9(1):682
71. Cimetta E, Franzoso M, Trevisan M, Serena E, Zambon A, Giulitti S. et al. Microfluidic-driven viral infection on cell cultures: Theoretical and experimental study. Biomicrofluidics. 2012;6(2):24127-2412712
72. Moura Rosa P, Gopalakrishnan N, Ibrahim H, Haug M, Halaas O. The intercell dynamics of T cells and dendritic cells in a lymph node-on-a-chip flow device. Lab Chip. 2016;16(19):3728-3740
73. Hou X, Zhang YS, Santiago GTD, Alvarez MM, Ribas J, Jonas SJ. et al. Interplay between materials and microfluidics. Nat Rev Mater. 2017;2(5):17016
74. Park SE, Georgescu A, Huh D. Organoids-on-a-chip. Science. 2019Jun7;364(6444):960-965
75. Esch EW, Bahinski A, Huh D. Organs-on-chips at the frontiers of drug discovery. Nat Rev Drug Discov. 2015Apr;14(4):248-60
76. Parrish J, Lim K, Zhang B, Radisic M, Woodfield TBF. New Frontiers for Biofabrication and Bioreactor Design in Microphysiological System Development. Trends Biotechnol. 2019Dec;37(12):1327-1343
77. Laganà M, Raimondi MT. A miniaturized, optically accessible bioreactor for systematic 3D tissue engineering research. Biomed Microdevices. 2012;14(1):225-234
78. Nava MM, Raimondi MT, Pietrabissa R. A multiphysics 3D model of tissue growth under interstitial perfusion in a tissue-engineering bioreactor. Biomech Model Mechanobiol. 2013;12(6):1169-1179
79. Raimondi MT, Giordano C, Pietrabissa R. Oxygen measurement in interstitially-perfused cellularised constructs cultured in a miniaturized bioreactor. J Appl Biomater Funct Mater. 2015;13(4):e313-e319
80. Credi C, De Marco C, Molena E, Nava MM, Raimondi MT, Levi M. et al. Direct photo-patterning of hyaluronic acid baits onto a fouling-release perfluoropolyether surface for selective cancer cells capture and immobilization. Mater Sci Eng C Mater Biol Appl. 2016;62:414-22
81. Izzo L, Tunesi M, Boeri L, Laganà M, Giordano C, Raimondi MT. Influence of the static magnetic field on cell response in a miniaturized optically accessible bioreactor for 3D cell culture. Biomed Microdevices. 2019;21:29
82. Tunesi M, Fusco F, Fiordaliso F, Corbelli A, Biella G, Raimondi MT. Optimization of a 3D dynamic culturing system for in vitro modeling of Frontotemporal Neurodegeneration-relevant pathologic features. Front Aging Neurosci. 2016;8:146
83. Frattini P, Villa C, De Santis F, Meregalli M, Belicchi M, Erratico S. et al. Autologous intramuscular transplantation of engineered satellite cells induces exosome-mediated systemic expression of Fukutin-related protein and rescues disease phenotype in a murine model of limb-girdle muscular dystrophy type 2I. Hum Mol Genet. 2017;26(19):3682-3698
84. Marturano-Kruik A, Nava MM, Yaeger K, Chramiec A, Hao L, Robinson ST. et al. Human bone perivascular niche-on-a-chip for studying metastatic colonization. Proc Natl Acad Sci USA. 2018;115(6):1256-1261
85. Raimondi MT, Albani D, Giordano C. An organ-on-a-chip engineered platform to study the microbiota-gut-brain axis in neurodegeneration. Trends Mol Med. 2019;25(9):737-740
86. Baig AM, Khaleeq A, Ali U, Syeda H. Evidence of the COVID-19 virus targeting the CNS: tissue distribution, host-virus interaction, and proposed neurotropic mechanisms. ACS Chem Neurosci. 2020;11(7):995-998
87. Fuentes S, Arenas D, Moore MM, Golding H, Khurana S. Development of bioluminescence imaging of respiratory syncytial virus (RSV) in virus-infected live mice and its use for evaluation of therapeutics and vaccines. Vaccine. 2017 35(4), 694-702
88. Palomino-Segura M, Virgilio T, Morone D, Pizzagalli DU, Gonzalez SF. Imaging cell interaction in tracheal mucosa during influenza virus infection using two-photon intravital microscopy. J Vis Exp. 2018 138
89. Benson RA, Lawton JC, MacLeod MKL. T Cell Response in the Lung Following Influenza Virus Infection. T-Cell Trafficking. 2017;1591:235-248
90. Looney MR, Thornton EE, Sen D, Lamm WJ, Glenny RW, Krummel MF. Stabilized imaging of immune surveillance in the mouse lung. Nat Methods. 2011;8(1):91-96
91. Ueki H, Wang IH, Fukuyama S, Katsura H, Silva Lopes TJ, Neumann G. et al. In vivo imaging of the pathophysiological changes and neutrophil dynamics in influenza virus-infected mouse lungs. PNAS. 2018;115(28):e6622-e6629
92. Murooka TT, Deruaz M, Marangoni F, Vrbanac VD, Seung E, von Andrian UH. et al. HIV-infected T cells are migratory vehicles for viral dissemination. Nature. 2012;490(7419):283-287
93. N TT, Mempel TR. Multiphoton intravital microscopy to study lymphocyte motility in lymph nodes. Methods Mol Biol. 2012;757:247-257
94. Naumenko V, Jenne C, Mahoney DJ. Intravital microscopy for imaging the tumor microenvironment in live mice. Methods Mol Biol. 2016;1458:217-230
95. Naumenko V, Van S, Dastidar H, Kim DS, Kim SJ, Zeng Z. et al. Visualizing oncolytic virus-host interactions in live mice using intravital microscopy. Mol Ther Oncolytics. 2018;10:14-27
96. Shannon JP, Kamenyeva O, Reynoso GV, Hickman HD. Intravital imaging of vaccinia virus-infected mice. Methods Mol Biol. 2019;2023:301-311
97. Huang Q, Cohen MA, Alsina FC, Devlin G, Garrett A, McKey J. et al. Intravital imaging of mouse embryos. Science. 2020;368(6487):181-186
98. Conci C, Jacchetti E, Zandrini T, Sironi L, Collini M, Chirico G. et al. Quantification of the foreign body reaction by means of a miniaturized imaging window for intravital nonlinear microscopy. Biomed Sci and Eng. 2019;3:106. doi:10.4081/bse.2019.106
99. Andrei G, Van Den Oord J, Fiten P, Opdenakker G, De Wolf-Peeters C, De Clercq E. et al. Organotypic epithelial raft cultures as a model for evaluating compounds against alphaherpesviruses. Antimicrob Agents Chemother. 2005;49(11):4671-4680
100. Koban R, Neumann M, Daugs A, Bloch O, Nitsche A, Langhammer S. et al. A novel three-dimensional cell culture method enhances antiviral drug screening in primary human cells. Antiviral Res. 2018;150:20-29
101. Beck BR, Shin B, Choi Y, Park S, Kang K. Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARSCoV-2) through a drug-target interaction deep learning model. Comput Struct Biotechnol J. 2020;18:784-790
102. Khan SA, Zia K, Ashraf S, Uddin R, Ul-Haq Z. Identification of Chymotrypsin-like Protease Inhibitors of SARS-CoV-2 Via Integrated Computational Approach. J Biomol Struct Dyn. 2020 [Epub ahead of print]
103. Han Y, Král P. Computational Design of ACE2-Based Peptide Inhibitors of SARS-CoV-2. ACS Nano. 2020;14(4):5143-5147
104. Macchiagodena M, Pagliai M, Procacci P. Identification of potential binders of the main protease 3CLpro of the COVID-19 via structure-based ligand design and molecular modeling. Chem Phys Lett. 2020;750:137489
105. Strange DP, Zarandi NP, Trivedi G, Atala A, Bishop CE, Sadri-Ardekani H. et al. Human testicular organoid system as a novel tool to study zika virus pathogenesis. Emerg Microbes Infect. 2018;7(1):1-7
106. Kim JA, Choi HJ, Kim CM, Jin HK, Bae JS, Kim GM. Enhancement of virus infection using dynamic cell culture in a microchannel. Micromachines (Basel). 2018;9(10):482
Author Biographies
 Manuela Teresa Raimondi is a full professor of bioengineering at the dept. of chemistry, materials and chemical engineering “Giulio Natta” at Politecnico di Milano, Italy. She holds a M.Sc. in mechanical engineering and a PhD in bioengineering. She has activated and directs the Mechanobiology group, based at the Mechanobiology Lab and at the Interdepartmental Live Cell Imaging lab. She develops frontier tools for cell modelling, including nanoengineered stem cell niches, microfluidic bioreactors and miniaturized windows for intravital imaging. She has coauthored over 200 publications and holds 10 patents in this field. She is the principal Investigator of three ERC-funded grants focused on frontier tools for cell modelling: NICHOID, NICHOIDS and MOAB (see www.nichoid.polimi.it).
Manuela Teresa Raimondi is a full professor of bioengineering at the dept. of chemistry, materials and chemical engineering “Giulio Natta” at Politecnico di Milano, Italy. She holds a M.Sc. in mechanical engineering and a PhD in bioengineering. She has activated and directs the Mechanobiology group, based at the Mechanobiology Lab and at the Interdepartmental Live Cell Imaging lab. She develops frontier tools for cell modelling, including nanoengineered stem cell niches, microfluidic bioreactors and miniaturized windows for intravital imaging. She has coauthored over 200 publications and holds 10 patents in this field. She is the principal Investigator of three ERC-funded grants focused on frontier tools for cell modelling: NICHOID, NICHOIDS and MOAB (see www.nichoid.polimi.it).
 Francesca Donnaloja obtained her M.Sc. in biomedical engineering in 2017 at Politecnico di Milano. Here, she attends a PhD program in bioengineering focused on the ERC-funded project NICHOID. She investigates the nuclear pore complex mechanics experimentally and computationally. She has been awarded a "R. Rocca" fellowship for a one-year stay at Prof. Thomas U. Schwartz group at MIT in Boston, USA.
Francesca Donnaloja obtained her M.Sc. in biomedical engineering in 2017 at Politecnico di Milano. Here, she attends a PhD program in bioengineering focused on the ERC-funded project NICHOID. She investigates the nuclear pore complex mechanics experimentally and computationally. She has been awarded a "R. Rocca" fellowship for a one-year stay at Prof. Thomas U. Schwartz group at MIT in Boston, USA.
 Bianca Barzaghini obtained her M.Sc. in biomedical engineering in 2019 at Politecnico di Milano, Italy. Here, she is currently attending a PhD program in bioengineering focused on the ERC-funded project NICHOID. Her research is focused on the microfabrication of micro engineered stem cell niches by two-photon laser polymerization and their biological validation.
Bianca Barzaghini obtained her M.Sc. in biomedical engineering in 2019 at Politecnico di Milano, Italy. Here, she is currently attending a PhD program in bioengineering focused on the ERC-funded project NICHOID. Her research is focused on the microfabrication of micro engineered stem cell niches by two-photon laser polymerization and their biological validation.
 Alberto Bocconi obtained his M.Sc. in biomedical engineering in 2019 at Politecnico di Milano, Italy. Here, he is currently attending a PhD program in bioengineering in collaboration with Prof. Alessandra Balduini's lab at Università degli Studi di Pavia. His research focuses on computational modeling and development of millifluidic bioreactors to mimic the human bone marrow niche for platelet production ex-vivo.
Alberto Bocconi obtained his M.Sc. in biomedical engineering in 2019 at Politecnico di Milano, Italy. Here, he is currently attending a PhD program in bioengineering in collaboration with Prof. Alessandra Balduini's lab at Università degli Studi di Pavia. His research focuses on computational modeling and development of millifluidic bioreactors to mimic the human bone marrow niche for platelet production ex-vivo.
 Claudio Conci obtained his M.Sc. in biomedical engineering in 2017 at Politecnico di Milano, Italy. Here, he is currently attending a PhD program in bioengineering focused on the MIUR-funded project BEYOND, in collaboration with the Prof. Roberto Osellame group at the IFN-CNR in Milano and the Prof. Giberto Chirico group at Università degli Studi di Milano Bicocca. His research focuses on nonlinear microscopy techniques applied in intravital conditions and on the development of implantable micro devices fabricated by two-photon laser polymerization.
Claudio Conci obtained his M.Sc. in biomedical engineering in 2017 at Politecnico di Milano, Italy. Here, he is currently attending a PhD program in bioengineering focused on the MIUR-funded project BEYOND, in collaboration with the Prof. Roberto Osellame group at the IFN-CNR in Milano and the Prof. Giberto Chirico group at Università degli Studi di Milano Bicocca. His research focuses on nonlinear microscopy techniques applied in intravital conditions and on the development of implantable micro devices fabricated by two-photon laser polymerization.
 Valentina Parodi obtained her M.Sc. in biomedical engineering in 2017 at Politecnico di Milano, Italy. Here, she attends an interdisciplinary PhD program in bioengineering and physics, focused on two ERC-funded projects MOAB and VIBRA, in collaboration with the Prof. Dario Polli group at the Dept. of Physics of Politecnico di Milano. She investigates cell function in 3D organoids with nonlinear microscopy techniques.
Valentina Parodi obtained her M.Sc. in biomedical engineering in 2017 at Politecnico di Milano, Italy. Here, she attends an interdisciplinary PhD program in bioengineering and physics, focused on two ERC-funded projects MOAB and VIBRA, in collaboration with the Prof. Dario Polli group at the Dept. of Physics of Politecnico di Milano. She investigates cell function in 3D organoids with nonlinear microscopy techniques.
 Emanuela Jacchetti is an assistant professor of bioengineering at Politecnico di Milano, Italy. She holds a M.Sc. in physics and a PhD in physics, astrophysics and applied physics. Her research is focalized on mechanobiological processes that govern cell morphology and functions like migration or stem cell differentiation, as well as on the diffusion and localization of proteins. She has a strong experience in the fields of innovative cell culture scaffolds and fluorescence microscopy.
Emanuela Jacchetti is an assistant professor of bioengineering at Politecnico di Milano, Italy. She holds a M.Sc. in physics and a PhD in physics, astrophysics and applied physics. Her research is focalized on mechanobiological processes that govern cell morphology and functions like migration or stem cell differentiation, as well as on the diffusion and localization of proteins. She has a strong experience in the fields of innovative cell culture scaffolds and fluorescence microscopy.
 Stephana Carelli is a group leader at the pediatric research center “Romeo ed Enrica Invernizzi” foundation, dept. of biomedical and clinical sciences, University of Milan, Italy, and an aggregate professor of pharmacology at the faculty of medicine, University of Milan. She holds a M.Sc. in biological sciences and a PhD in physiopathology and pharmacology of metabolic diseases. She has coauthored over 45 publications and holds one patent in this field. Her current research interests include: (1) role of RNAs, particularly lncRNAs in epigenetic mechanisms and functions in neurodegeneration/regeneration (Parkinson's Disease, ALS) and in the programming and re-programming of stem cells fate; (2) regenerative medicine for curing neurodegenerative diseases such as spinal cord injury and Parkinson's disease, focusing on both cell therapies based on the application of adult stem cells (adult mouse neural stem cells and human mesenchymal stem cells) and on the neuroprotective action of erythropoietin; (3) application of advanced cell manipulation technologies in regenerative medicine.
Stephana Carelli is a group leader at the pediatric research center “Romeo ed Enrica Invernizzi” foundation, dept. of biomedical and clinical sciences, University of Milan, Italy, and an aggregate professor of pharmacology at the faculty of medicine, University of Milan. She holds a M.Sc. in biological sciences and a PhD in physiopathology and pharmacology of metabolic diseases. She has coauthored over 45 publications and holds one patent in this field. Her current research interests include: (1) role of RNAs, particularly lncRNAs in epigenetic mechanisms and functions in neurodegeneration/regeneration (Parkinson's Disease, ALS) and in the programming and re-programming of stem cells fate; (2) regenerative medicine for curing neurodegenerative diseases such as spinal cord injury and Parkinson's disease, focusing on both cell therapies based on the application of adult stem cells (adult mouse neural stem cells and human mesenchymal stem cells) and on the neuroprotective action of erythropoietin; (3) application of advanced cell manipulation technologies in regenerative medicine.
![]() Corresponding author: Manuela Teresa Raimondi, Department of Chemistry, Materials and Chemical Engineering “Giulio Natta”, Politecnico di Milano, Milano, Italy; E-mail: manuela.raimondiit.
Corresponding author: Manuela Teresa Raimondi, Department of Chemistry, Materials and Chemical Engineering “Giulio Natta”, Politecnico di Milano, Milano, Italy; E-mail: manuela.raimondiit.