Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Conclusion
Materials and Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(15):6854-6874. doi:10.7150/thno.44165 This issue Cite
Research Paper
Therapeutic potential of a TrkB agonistic antibody for Alzheimer's disease
Shudan Wang1,2,3, Hongyang Yao1*, Yihua Xu1,3*, Rui Hao4, Wen Zhang1, Hang Liu1,3, Ying Huang4, Wei Guo1,2,3 ![]() , Bai Lu1,2,3
, Bai Lu1,2,3 ![]()
1. School of Pharmaceutical Sciences, IDG/McGovern Institute for Brain Research, Tsinghua University, Beijing, China, 100084.
2. Beijing Tiantan Hospital, Advanced Innovation Center for Human Brain Protection, Capital Medical University, Beijing, China, 100070.
3. R&D Center for the Diagnosis and Treatment of Major Brain Diseases, Research Institute of Tsinghua University in Shenzhen, Shenzhen, Guangdong, China, 518057.
4. Center of Translational Medicine, Tongji Hospital, Tongji University School of Medicine, Shanghai, China, 200065.
*These authors have contributed equally to this work.
Received 2020-1-21; Accepted 2020-5-11; Published 2020-5-23
Abstract

Repeated failures of “Aβ-lowering” therapies call for new targets and therapeutic approaches for Alzheimer's disease (AD). We propose to treat AD by halting neuronal death and repairing synapses using a BDNF-based therapy. To overcome the poor druggability of BDNF, we have developed an agonistic antibody AS86 to mimic the function of BDNF, and evaluate its therapeutic potential for AD.
Method: Biochemical, electrophysiological and behavioral techniques were used to investigate the effects of AS86 in vitro and in vivo.
Results: AS86 specifically activated the BDNF receptor TrkB and its downstream signaling, without affecting its other receptor p75NTR. It promoted neurite outgrowth, enhanced spine growth and prevented Aβ-induced cell death in cultured neurons, and facilitated Long-Term Potentiation (LTP) in hippocampal slices. A single-dose tail-vein injection of AS86 activated TrkB signaling in the brain, with a half-life of 6 days in the blood and brain. Bi-weekly peripheral administration of AS86 rescued the deficits in object-recognition memory in the APP/PS1 mouse model. AS86 also reversed spatial memory deficits in the 11-month, but not 14-month old AD mouse model.
Conclusion: These results demonstrate the potential of AS86 in AD therapy, suggesting that neuronal and/or synaptic repair as an alternative therapeutic strategy for AD.
Keywords: Neurodegeneration, Therapy, Cognition, Antibody drug, Synaptic plasticity
Introduction
Great progress has been made in elucidating pathogenic mechanisms of neurodegenerative diseases such as Alzheimer's disease (AD). There is a general understanding that the accumulation of toxic protein aggregates such as Amyloid β (Aβ) and tau in the brain is the major pathogenic factor for AD and perhaps even the cause of the disease [1-3]. Molecular pathways for the pathogenesis of AD have begun to emerge, thanks to the advances in Genome-wide association (GWA) analyses and studies using animal models [4,5]. However, the progress in understanding AD biology has not been translated into effective therapy. Hundreds of clinical trials have been conducted, but until now, there are no disease-modifying agents that could halt or slow down AD progression [6,7]. Several recent clinical studies have shown that reducing the pathogenic toxins such as Aβ, either by immunological approaches (vaccine, antibodies) or by inhibiting its production (inhibitors for β- or γ-secretase), reduced Aβ pathology but failed to improve cognitive deficits [8]. These findings cast doubt on the “toxin-reducing” approach and call for a “paradigm shift” in developing therapies for this dreadful disease.
“Disease-modifying” is defined as a “pharmacologic treatment that retards the underlying process of AD by intervening in the neurobiological processes that constitute the pathology and pathophysiology of the disease and lead to cell death or dysfunction.” [9]. One emerging concept is to focus on pathophysiology rather than pathogenesis in developing therapeutic treatment for AD [10]. Major pathophysiological hallmarks for AD include neuronal death and synapse degeneration. The progression in AD is reflected by early synaptic deficiency [11-13], followed by progressive neuronal loss [14-17]. Increasing evidence suggests that synapse loss or neuronal death, but not toxin accumulation, is highly correlated with the progression of AD [2,7]. Synaptic integrity and functions are critical for the maintenance of neural circuits and behavioral functions. It is suggested that there is a ~10% neuronal loss, but perhaps more meaningful is ~20% synapse loss in Mild Cognitive Impairment (MCI). Similarly, synaptic loss (~60%) probably contributed more significantly than neuronal loss (~45%) to the disease manifestation in mild AD [12]. Unlike neuronal loss, dysfunctional synapses can be repaired or regenerated because synapses are highly dynamic and plastic [18-20]. Therefore, targeting mechanisms that can either stabilize/protect or repair/regenerate synapses would allow treatment of the diseases at a later stage (e.g. in prodromal AD or even mild AD).
BDNF, a member of the neurotrophin family, is by far the best known “synaptogenic” molecule and perhaps the only one proven to facilitate synaptic function in humans [21-23]. The biological functions of BDNF are mediated primarily by its high-affinity receptor TrkB (Tropomyosin Receptor Kinases B), which activates three major downstream signaling pathways: phosphatidylinositol-3 kinase (PI3 kinase)/Akt; Ras/extracellular regulated kinase (Erk); and PLCγ/PKC [24]. Numerous studies have demonstrated that BDNF potentiates synaptic transmission, modulates synaptic plasticity, and induces synapse formation [25-28], both during development and in the adult [23]. Behavioral experiments have revealed a role of BDNF in the formation and retention of hippocampal-dependent memory, fear memory extinction, motor learning, as well as mood control [29,30].
Two lines of evidence suggest that the BDNF-TrkB signaling pathway may be a therapeutic target for AD treatment. First, in human subjects, alteration of BDNF expression has been correlated with AD pathophysiology. BDNF mRNA and proteins are decreased in hippocampus, temporal cortex and parietal cortex in the post-mortem brains of human AD subjects [31-33]. Moreover, the BDNF Val66Met polymorphism, implicated in a slight reduction in activity-dependent BDNF release [34, 5], has been associated with impairments in episodic memory and hippocampal activity as measured by FDG-PET (Flurodeoxyglucose-Positron emission tomography) in both sporadic and autosomal dominant AD [36-39]. Longitudinal studies have revealed an accelerated decline of hippocampal volume and episodic memory, two key endophenotypes, in MCI patients [40-43]. Second, extensive studies have documented that BDNF enhances synaptic growth and functions in normal animals [26-28]. Moreover, viral expression of BDNF gene in the brain of APP (Amyloid precursor protein) or Tau transgenic AD mice attenuated synapse loss and neuronal abnormality, and rescued the deficits in hippocampal-dependent memory, but did not alleviate Aβ and Tau pathology [44-46]. Infusion of BDNF protein into the hippocampus of rat AD model (ventricular injection of Aβ), or daily i.p. injection of a BDNF-mimicking peptide was able to halt neuronal death and ameliorated cognitive impairment [47-49]. Therefore, the BDNF-TrkB signaling pathway may be used as a drug target for AD treatment.
Despite its promise, development of drugs targeting the BDNF-TrkB signaling pathway has been challenging. Previous studies have demonstrated that BDNF protein itself may not be used as a therapeutic agent due to its intrinsic physicochemical properties. A number of issues have been raised. First, the half-life of BDNF is very short in both blood (1-10 min) and CSF (1 hour) [50-52]. Second, BDNF protein is highly charged and difficult to diffuse in brain or spinal cord parenchyma [53-59]. Finally, in addition to activating TrkB, BDNF may also activate its low affinity receptor p75NTR, leading to cell death and synaptic depression [60, 61]. Efforts have been made to develop a small molecule activator for TrkB [62-66]. Unfortunately, more rigorous studies recently demonstrated that these molecules neither bind nor activate TrkB [67,68]. To overcome the aforementioned problems, we have attempted to use TrkB agonistic antibodies to activate BDNF signaling. A new TrkB agonist antibody AS86 has been developed, using hybridoma technologies. We evaluated its pharmacological and biological properties, examined its treatment efficacy in both in vitro cell model and in an AD mouse model, and outlined the mechanism involved in pathology and synaptic function in AD treatment. Our findings suggest that agonistic antibodies may be a new modality to activate the BDNF-TrkB signaling pathway, paving the way for a new avenue for AD treatment.
Results
Characterization of TrkB agonistic antibody AS86
Using human TrkB extracellular domain (TrkB-ECD) protein as the antigen, TrkB agonist antibodies were developed by immunization-hybridoma technologies. We have identified a new TrkB monocloncal antibody, AS86, which exhibited a dose-dependent activation of TrkB similar to BDNF, as measured by NFAT assay in TrkB-NFAT-bla CHO-K1 cells (Figure 1A). As a tyrosine receptor kinase (RTK), TrkB activation is mediated by ligand-induced receptor dimerization and endocytosis [24]. To determine whether AS86 could induce TrkB endocytosis, we incubated hippocampal neurons with AS86 or BDNF at 37 °C for different amounts of time to allow ligand-induced receptor endocytosis. A biotinylation experiment was performed to detect cell surface TrkB levels. We found that treatment with AS86 elicited a significant decrease in cell surface TrkB as well as total and phosphorylated TrkB, suggesting TrkB endocytosis and degradation upon AS86 binding (Figure S1).
Potency and signaling of TrkB agonistic antibody AS86. (A) Dose response of TrkB activation by AS86. hTrkB-CHO cells were treated with different doses of TrkB antibodies or BDNF for 4 h, and TrkB activation was analyzed using NFAT assay. (B) Dose response of TrkB activation and its downstream signaling in cultured hippocampal neurons. Primary hippocampal neurons (DIV10) were treated with different concentrations of AS86 or BDNF for 30 min, and then the cell lysates were analyzed using Western blotting (N = 3 independent culture experiments, n = 3 repeats for each experiment). Three different tyrosine-phosphorylated sites and downstream signaling pathways were examined. Representative Western blots are presented. (C) Time course of AS86 downstream signaling in cultured hippocampal neurons. Primary hippocampal neurons (DIV10) were stimulated with AS86 or BDNF for 0, 5 min, 15 min, 30 min, 60 min, 180 min, 360 min, 720 min and 1440 min, and then the cell lysates were examined for the activation of Akt, Erk and PLCγ with Western blots (N = 3 independent culture experiments, n = 3 repeats for each experiment). Representative Western blots are presented.
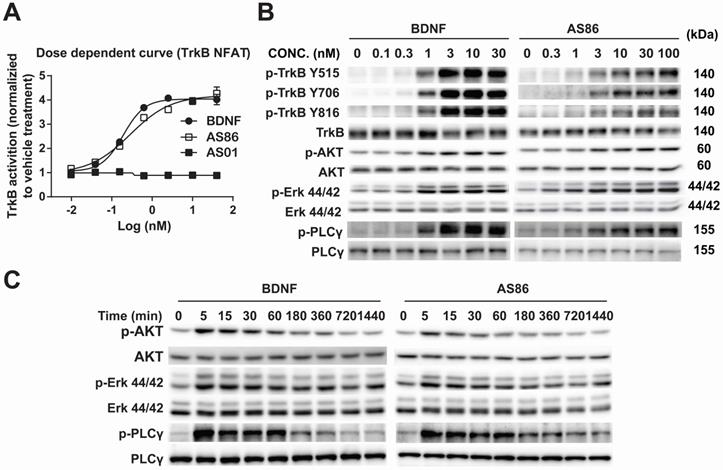
Next, we examined whether AS86 could activate TrkB and its downstream signaling pathways. In cultured hippocampal neurons, AS86 induced TrkB phosphorylation as well as the three major downstream signaling pathways (Akt, Erk and PLCγ) at a concentration as low as 3 nM (Figures 1B and S2A-B). The kinetics of Akt, Erk, and PLCγ signaling by AS86 (10 nM) and BDNF (3 nM) were similar, with the maximal activation at 5 min (Figures 1C and S2C). The antibody binds specifically to TrkB, but not to other neurotrophin receptors such as TrkA, TrkC or p75NTR (Figure 2A), and its ability to induce TrkB tyrosine phosphorylation (Y515 or Y816) was completely blocked by the Trk inhibitors K252a and AZD-1332 (Figure 2B-C). To further demonstrate the specificity of AS86, we performed immunostaining under non-permeable conditions. We found that in cells incubated with AS86, staining with a FITC labeled anti-mouse IgG antibody detected bright TrkB staining in TrkB-CHO or TrkB-PC12 cells, but no signal at all in control TrkA-CHO or normal PC12 cells (Figure S3A), suggesting that AS86 does not bind any other membrane proteins.
The specificity of TrkB agonistic antibody AS86. (A) AS86 at different concentrations (0.1 nM, 1 nM and 10 nM) was added to the plates coated with different proteins (0.1 μg TrkA, TrkB, TrkC, or p75 respectively), and ELISA was used to examine the binding capacity of AS86. (B, C) Cultured hippocampal neurons (DIV10) were pretreated with the Trk inhibitors k252a (300 nM) or AZD-1332 (100 nM) for 60 min before incubation with mIgG (3 nM), BDNF (1 nM), or AS86 (3 nM) for 15 min (N = 2, n = 3). The Western blots of TrkB Y515 and Y816 sites activation (B) and the quantitative plots (C) are presented. Unless specifically indicated otherwise, statistical analyses in this and all other figures were carried out using one-way ANOVA followed by post hoc test. Symbols for P values (for both ANOVA and Student's t-test): *: p < 0.05; **: p < 0.01; ***: p < 0.001; ****: P < 0.0001. The quantification data in this and all other figures were presented as mean ± SEM.
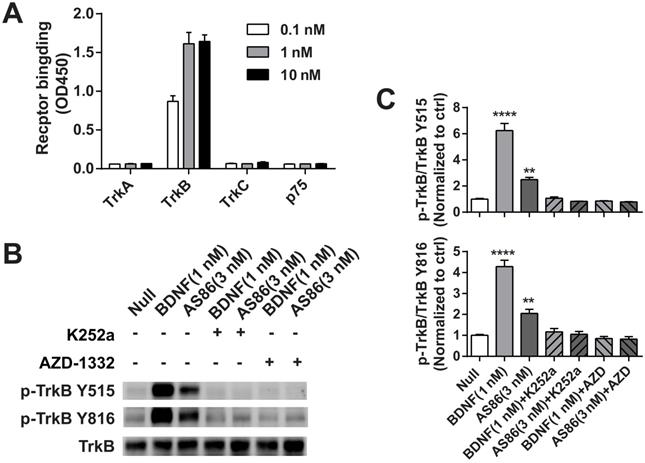
To determine which sub-domain of TrkB that AS86 binds to, we constructed a series of truncated TrkB plasmids and transfected them into 293-T cell line. Immunoprecipitation experiments revealed that only the deletion of D5, but not other sub-domains completely eliminated the AS86 binding to TrkB (data not shown). Competition experiments with BDNF at the saturated concentration for TrkB activation (4 nM) and increasing concentrations of AS86 revealed no significant difference between “BDNF alone” and “BDNF plus AS86” (Figure S3B-C), suggesting that AS86 is a non-competitive TrkB agonist.
Attenuation of Aβ-induced cell death by AS86
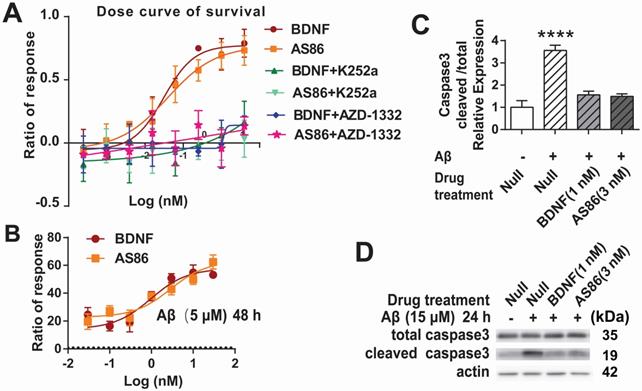
To determine whether the TrkB agonistic antibody could counter cell death and Aβ toxicity, we used PC12 cells expressing human TrkB. Apoptotic cell death, measured by fluorescent substrate cleaved by caspase-3, was induced by serum deprivation in hTrkB-PC12 cells. The hTrkB-PC12 cells were treated with BDNF or AS86, and the protective effects were examined after the cells were serum-deprived for 16 hours. Both AS86 and BDNF elicited dose-dependent rescue effects, with EC50 at 0.039 nM and 0.031 nM, respectively (Figures 3A and S4A). These effects were blocked by the TrkB inhibitors (K252a or AZD-1332) (Figure 3A). Next, we used an Aβ induced cell death assay to assess the potential of AS86 in Alzheimer's disease (AD) therapy. Hippocampal neurons (DIV10) were treated with Aβ (25-35), the most toxic fragment of Aβ (1-42) peptide, to induce cell death [69, 70]. Based on the dose-response curve (Figure S4B), 5 μM of Aβ (25-35) was used to induce moderate cell death. Under these conditions, pretreatment with AS86 or BDNF effectively prevented Aβ (25-35) induced neuronal death and elicited a dose-dependent increase in survival rate, when normalized to the “Aβ (25-35) treated alone” group (Figure 3B). Cleaved caspase 3, an apoptosis marker, was significantly increased after treatment with Aβ (25-35) for 24 hours (3.6 fold, p < 0.0001). This increase was almost completely attenuated if the neurons were pretreated with AS86 (3 nM) or BDNF (1 nM) (Figure 3C-D). Therefore, similar to BDNF, the TrkB agonistic antibody AS86 attenuates apoptotic cell death induced by serum deprivation or Aβ (25-35) through activation of TrkB.
Cell survival function of AS86. (A) Attenuation of serum-deprivation induced cell death by AS86. Different doses of BDNF or AS86 were applied to serum-deprived cultures of hTrkB-PC12 cells in the absence or presence of Trk inhibitors (300 nM K252a or 50 nM AZD-1332) for 16 hours, and the levels of apoptosis were determined by the ratio of the number of caspase 3 positive cells to total number of cells, using a caspase 3-substrate kit. Survival rates were measured by the decreased apoptotic levels normalized to that of vehicle treatment (n = 3). (B) Attenuation of Aβ induced cell death by AS86. Hippocampal neurons were pretreated with AS86 or BDNF for 30 minutes, followed by treatment with Aβ (25-35) (5 µM). Cell viability was analyzed with ATP level quantification assay 48 hours later (N = 2 independent experiments, n = 6 samples). (C, D) Inhibition of Aβ induced apoptotic signals by AS86. Hippocampal neurons were pretreated with AS86 or BDNF for 30 minutes, followed by treatment with Aβ (25-35) (15 µM) for 24 hours. Total and cleaved caspase3 levels were measured using Western blotting (N = 3 independent experiments, n = 3 replicates). Representative Western blots (D) and quantitative plots (C) are presented.
Facilitation of dendritic growth by AS86
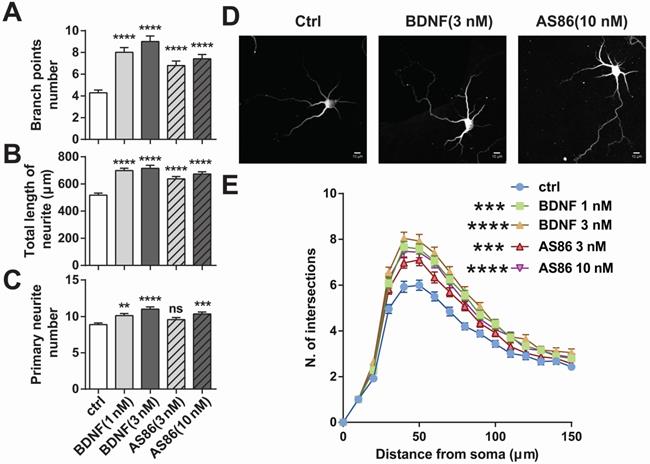
BDNF could elicit a variety of cellular functions including promoting neurite growth [27,71]. We examined whether AS86 has similar capacities using cultured hippocampal neurons derived from E18 rats. Treatment of hippocampal neurons (DIV1) with AS86 (3 nM, 10 nM) or BDNF (1 nM, 3 nM) for 3 days resulted in a significant increase in both neurite branch points and neurite total length (Figures 4A, B and D). Primary neurite number was also increased by AS86 at 10 nM (Figure 4C-D). Sholl analysis indicated that both AS86 and BDNF increased neurite complexity (Figure 4E).
Differential effects of AS86 and BDNF on neurite growth in hippocampal neurons. Cultured hippocampal neurons (DIV1) were treated with BDNF or AS86 for 3 days, and then fixed and stained with MAP2 antibody. Quantifications of branching points (A), total length of neurites (B) and primary neurite numbers (C), neuronal images with MAP2 staining (D) and sholl analysis (E) are presented (N = 3 independent experiments, n = 120-140 neurons). Scale bar represents 10 µm.
Enhancement of synaptic functions by AS86
Numerous studies have demonstrated that BDNF promotes dendritic spine growth and facilitates synaptic transmission and plasticity [25,27,28]. Thus, BDNF-mediated synaptic repair has been proposed as a new therapeutic strategy for neurodegenerative diseases including AD [10]. We examined whether AS86 could mimic BDNF on synaptic plasticity. Hippocampal neurons expressing fluorescent protein mCherry (red) were grown for 15 days in culture and treated with AS86 (3 nM or 10 nM) or BDNF (1 nM or 3 nM) for 24h, and neuronal images were examined by the confocal microscope. Similar to BDNF, AS86 significantly increased dendritic spine density (control = 4.6 ± 0.16, AS86 3 nM = 7.4 ± 0.23, AS86 10 nM = 7.6 ± 0.25). In particular, the density of mature, mushroom-shaped spines was markedly increased (control = 2.7 ± 0.12, AS86 3 nM=4.2 ± 0.14, AS86 10 nM= 4.7 ± 0.17) (Figure 5A-B; all p < 0.0001).
Facilitation of synaptic functions by AS86. (A-B) Effects of AS86 on dendritic spine growth. Cultured hippocampal neurons (DIV7) were transfected with mCherry, followed by treatment with AS86 or BDNF at DIV 15 for 24 h before fixation (N = 3 independent experiments, n = 60 dendrites). Images of dendritic spine (A) and the quantifications of dendritic protrusion densities (B) are presented. Ctrl in this and all other figures: Control, no treatment. (C) Enhancement of HFS-induced LTP in hippocampal slices treated with AS86. AS86 or mIgG was added into perfusate at least 30 min before LTP induction and maintained in perfusate during entire course of recording (65 min). Field EPSPs (fEPSPs) were recorded and the slopes of fEPSPs were plotted over time. The right panels show the representative fEPSPs before HFS (gray) or after stimulation (blue or black). (D) Quantification of slope of fEPSPs 1 hour after LTP induction (n = 7 slices) (Student's t-test). (E) Activation of TrkB by AS86 in hippocampal slices. P12 hippocampal slices were treated with AS86 (15 nM) for 0.5, 1 and 2 hours, and the lysates were analyzed by Western blotting using anti-pTrkB antibody Y515.
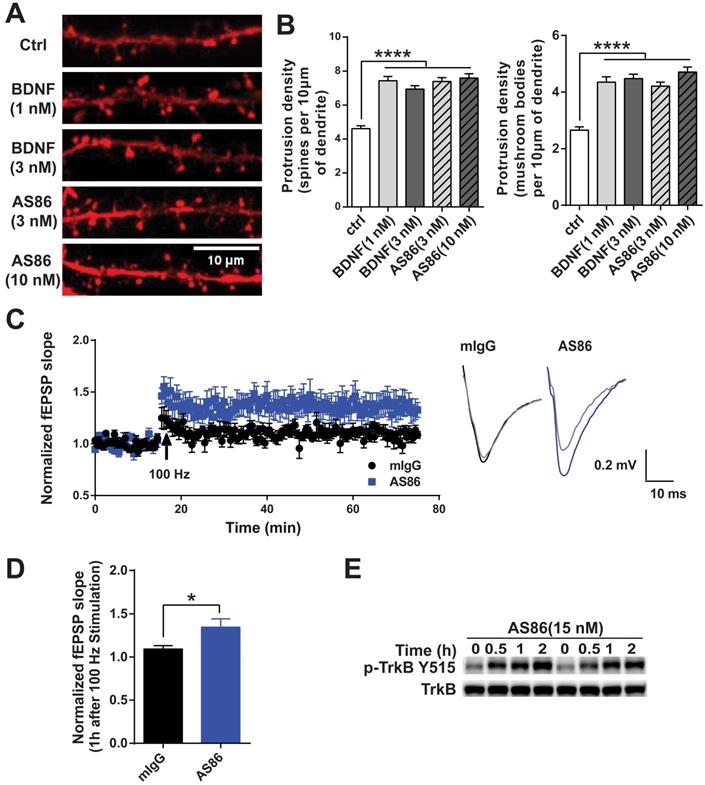
We have previously shown that exogenous BDNF facilitates LTP (long-term potentiation) in slices derived from young postnatal day 12-13 (P12-13) hippocampus in which endogenous BDNF is low [25]. Here we performed a series of experiments to determine whether AS86 could mimic the effects of BDNF on LTP using rat hippocampal slices. Time course and dose-response experiments indicated that treatment of hippocampal slices derived from P12 rats with 15 nM AS86 for 2 hours elicited a robust activation of TrkB (Figures 5E and S5A). Among all slices derived from rats of different ages (P10-P28) treated with AS86 under these conditions (2 hours, 15 nM), P12 slices exhibited the most robust activation of TrkB and its downstream signals such as Akt and Erk (Figure S5B). We therefore used hippocampal slices from P12-P13 rat for the LTP experiments.
Field excitatory postsynaptic potentials (fEPSPs) in the CA1 region of hippocampal slices were recorded in response to stimulation of the Schaffer collateral pathway. Similar to our previous observation [25], in P12-13 hippocampal slices of rats, high frequency stimulation (HFS, 100Hz, 1 sec) can only induce a subthreshold, small LTP (109 ± 3.9%, n = 7 slices) in control slices which were pretreated with 15 nM mouse IgG (mIgG). In contrast, in slices pretreated with AS86 (15 nM, 30 min), a HFS-induced stable LTP was observed (134 ± 9.6%, n = 7 slices) (Figure 5C-D). The slope of fEPSPs in AS86 treated slices 1 hour after HFS was significantly enhanced compared with that of mIgG treated slices (Figure 5D; p = 0.0328). However, AS86 administration did not change the input-output curve (Figure S5D). Similarly, in paired-pulse facilitation (PPF) test, the bath application of AS86 (15 nM) did not change PPF at different time points (Figure S5C). These results suggest that AS86 had no effect on basal synaptic transmission. The ability of AS86 to facilitate dendritic spine growth and LTP, together with its role in promoting neurite outgrowth and countering Aβ-induced cell death, forms the basis for further investigation of the therapeutic potential of AS86 for AD.
Target engagement of AS86 in mouse brains
To investigate the therapeutic effects of AS86 on AD in vivo, it is important to determine whether AS86 could engage its target TrkB in the mouse brain. We determined the pharmacokinetics of AS86 in blood and brain, and whether it can penetrate blood-brain barrier (BBB) when administered peripherally. We administered AS86 (1.5 mg/kg body weight) to mice through tail vein injection and collected the plasma and brain tissues (after perfusion) at different time points. AS86 concentrations were examined and quantified by ELISA. We found that AS86 had a half-life of approximately 6 days in both mouse plasma and brain. AS86 was almost eliminated completely in blood by 30 days but remained at approximately 20% levels in the brain (Figure 6A-B).
Target engagement of AS86 in mouse brains. (A-B) The pharmacokinetic curves of AS86 in plasma and brain tissues of mice. Mice were injected with AS86 (1.5 mg/kg) by tail vein injection, and the AS86 concentration at different time points were analyzed with ELISA, and plotted in time-curve graphs for plasma (A) and brain tissue (B) respectively (n = 3-4 mice). (C-E) Activation of TrkB downstream signals and gene expression by AS86. The hippocampal tissues were lysed after AS86 (1.5 mg/kg) was administrated through tail vein injection, and p-Akt, p-Erk, and EGR1 expression were analyzed at 1 day after injection (n = 3 mice). Representative Western blots (C, D) and quantitative plots (E) are presented (Student's t-test).
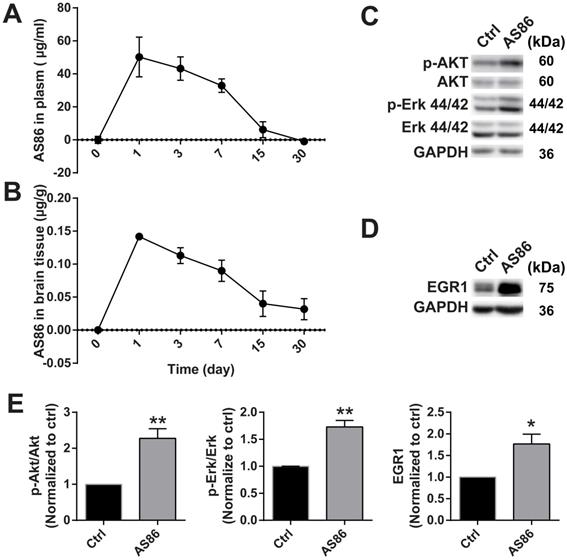
Body weight reduction cannot be a direct pharmacodynamic (PD) marker for the main effects we examined in this paper - the amelioration of cognitive impairments, which are largely mediated by cortical and hippocampal synapses. However, substantial evidence supports the view that BDNF-mediated body weight reduction is achieved by enhancement of synaptic function in the hypothalamus. Given the difficulties in finding a direct PD marker for brain diseases, a common strategy in CNS drug development is to find a surrogate marker linked to the key mechanism of the drug. Previous studies showed that treatment with TrkB agonistic antibodies resulted in weight loss [72], which is mainly caused by inhibition of food intake through enhancement of synaptic function by BDNF-TrkB signaling in the hypothalamus. Therefore, body weight could be used as a surrogate indicator for the PD measurement. We found that body weight reduction began from day-1 after a single-dose administration of AS86 and continued to a plateau level by day-7, and began to recover after day-14 (Figure S6A). The body weight always remained at a reduced level after repeated doses of AS86 (Figure S7C), revealing a PD profile for single dose AS86. We also examined the downstream signaling and the expression of EGR1, an early response gene downstream of TrkB activation. AS86 increased p-Akt, p-Erk and EGR1 expression (Figure 6C-E). These results suggest that AS86 can pass the BBB and penetrate into mouse brain.
Effect of AS86 on novel object recognition
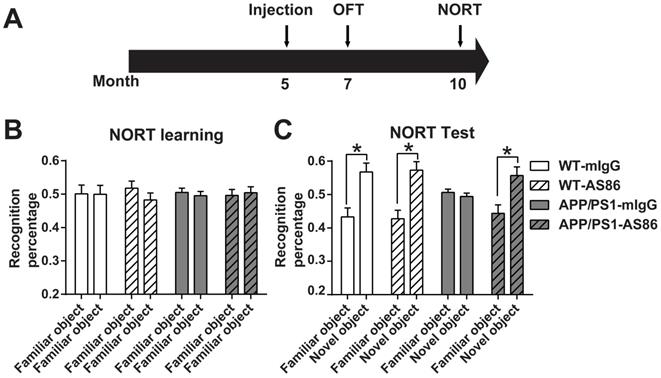
Previous studies have shown that BDNF injected to the brains of an AD mouse model could improve cognitive functions [44, 48]. Here we tested whether the TrkB agonistic antibody AS86, with significant improvements in PK and BBB penetration over BDNF shown above, could induce behavioral changes when administered peripherally to APP/PS1, a commonly used AD mouse model. The littermates APP/PS1 were divided into four groups: Wild-type (WT)-mIgG, WT-AS86, APP/PS1-mIgG and APP/PS1-AS86. AS86 or mIgG was administered into 5-month-old mice through tail vein injection every two weeks (Figure 7A). The peak value of antibody concentration in homogenate of brain tissue is about 1 nM (0.15 μg AS86/g brain weight) according to the PK analysis with the single dose of 1.5 mg/kg. In fact, the antibody cannot penetrate through cell membranes. Therefore, the actual concentration in brain was estimated to be about 3-5 nM, which could elicit more than 80% TrkB activation and promote cell survival in vitro. Based on the PK analysis (Figure 6B), there should still be approximately 30% AS86 left at 15-30 days, after a single dose of AS86. Considering the cumulative effect induced by repeated dosing with an interval of 2 weeks, we chose the dosage of 1 mg/kg body weight for our behavioral experiments.
Effects of AS86 on novel object recognition test (NORT) in APP/PS1 mice. (A) Schematic diagram showing the timeline of drug treatments and behavior tests were performed. (B-C) Performance of NORT behavior after AS86 treatment. Mice were subjected to NORT after AS86 (1 mg/kg) treatment for 5 months. WT-mIgG (n = 7), WT-AS86 (n = 10), APP/PS1-mIgG (n = 10) and APP/PS1-AS86 (n = 15) mice were subjected to learning paradigm (B), followed by the test trail (C) 3 hours later. Note that administration of AS86 significantly improved the ability to recognize novel objects by APP/PS1 mice. Paired student's t-test.
Consistent with previous reports [72], administration of AS86 resulted in a moderate loss of body weight (20%, Figure S7C). To determine whether the weight loss altered spontaneous motor activities, we performed the open-field task using 7-month old animals (AS86 treatment for 2 months). There were no significant differences in velocity, moving distance, time spent in center and time spent in corner among these four groups (Figure S7A; all p > 0.05). It has been demonstrated that BDNF may induce hyperalgesia in intact animals [73]. Therefore, we performed experiments of von Frey and Plantar test to examine the cutaneous sensitivity 1-day after AS86 treatment. Treatment with AS86 elicited no effect on sensory functions as determined by these tests. (Figure S6B-C).
We first examined the effect of AS86 treatment on novel object recognition test (NORT) using 10-month old APP/PS1 mice with 5-month treatment of AS86. We found that AS86 treatment had no effect on NORT learning of two familiar objects both in WT and APP/PS1 mice (Figure 7B). Moreover, long-term treatment with AS86 did not further improve NORT memory of control animals (Figure 7C). WT animals spent approximately 20% more time on novel object than familiar object, regardless whether they were treated with AS86 or not (Figure 7C; the first 4 columns). However, in the test phase, the APP/PS1 mice exhibited a severe impairment in NORT scores. APP/PS1 mice treated with mIgG spent equal amount time on familiar and novel objects (Figure 7C; Compare the 5th and 6th columns, p = 0.9124, paired student's t-test). In marked contrast, APP/PS1 mice treated with AS86 for 5 months exhibited huge preferences towards novel object, almost the same level as the WT animals (Figure 7C; Compare the 7th and 8th columns, p = 0.0113, paired student's t-test). These results indicate that long-term (5 months) treatment of APP/PS1 mice from 5 months to 10 months old is efficacious in rescuing the deficits in novel object recognition.
Effects of AS86 on spatial learning and memory
The NORT results suggested that long-term treatment of AS86 by tail vein injection could improve cognition in APP/PS1 mice. Next, the same mouse model was used to investigate the effect of AS86 on spatial memory deficits, a form of impairment most relevant to AD. Morris water maze (WM), which could be tested repeatedly when the platform location changed, was used. AS86 was administered twice monthly to APP/PS1 mice beginning at 5 months of age, and WM tests were performed 3 times (WM1, WM2 and WM3) when the animals were at the age of 8, 11, and 14 months (with drug treatments for 3, 6, and 9 months), respectively (Figure 8A).
Effects of long-term treatment of AS86 on spatial learning and memory in APP/PS1 mice. (A) Schematic diagram showing the time point when AS86 was first dosed as well as those when Morris water maze (WM) tests were performed. Mice were subjected to tail-vein injection with AS86 or mIgG every two weeks for 10 months (5 to 15 months old). Three tests of WM were performed during this period. (B-J) Morris water maze (WM) tests were performed to evaluate the effects of AS86 (1 mg/kg) treatments for different durations. (B-D) First evaluation: AS86 (1 mg/kg) treatment for 3 months (WM1). WT-mIgG (n = 10), WT-AS86 (n = 11), APP/PS1-mIgG (n = 14) and APP/PS1-AS86 (n = 18) mice were subjected to learning paradigm of water maze for 5 days, followed by a probe trial at day 6. (E-G) Second evaluation: AS86 treatment for 6 months (WM2). WT-mIgG (n = 9), WT-AS86 (n = 11), APP/PS1-mIgG (n = 12) and APP/PS1- AS86 (n = 18) mice were examined in this round (4 days of training trails, and followed by a probe trial at day 5). (H-J) Third evaluation: AS86 treatment for 9 months (WM3). WT-mIgG (n = 9), WT-AS86 (n = 9), APP/PS1-mIgG (n = 10) and APP/PS1- AS86 (n = 15) mice were subjected to WM (4 days of training trails, and followed by a probe trial at day 5). Statistics for the escape latency of learning trails (B, E, H): Two-way ANOVA with repeated measurement was used for the learning curves. Significant differences in WM2 (WT-mIgG and APP/PS1-mIgG) and WM3 (WT-mIgG and WT-AS86) were indicated by bracket plus “#” (p < 0.05). ns: not significant. Student's t-test was for individual day comparison. For data points significantly different from the corresponding point in WT-mIgG. *: p < 0.05; **: p < 0.01. Statistics for the primary latency (C, F, I) and platform crossover times (D, G, J) of probe trails: the results of two-way ANOVA (mIgG. vs. AS86, and WT. vs. APP/PS1) were shown in Table 1, 2. For the comparison between specific groups, the independent-samples t-test or non-parametric (Mann-Whitney) test was selected depending on whether the data followed normal distribution. *: p < 0.05; **: p < 0.01. ns: not significant.
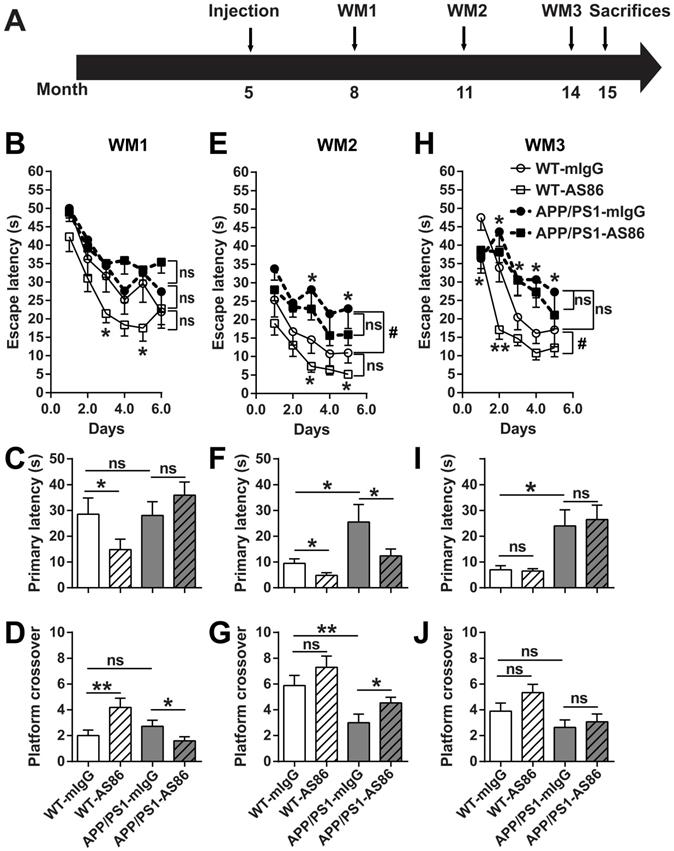
In WM1, the mice were trained for 6 days. The escape latency for the WT-mIgG group in last three training trials on the 6th day reached the test standard (20s). We found that there was no difference in spatial learning and memory between the WT and AD mouse model (WT vs. APP/PS1 groups, all p > 0.05, Figure 8B-D, Table 1, 2), indicating that APP/PS1 had not developed spatial cognition deficiency at the age of 8 months. Treatment of the WT animals with AS86 for 3 months slightly enhanced the learning performance (Figure 8B, compare WT-mIgG with WT-AS86 groups; F (1, 19) = 4.356, p = 0.0506), and this enhancement reached statistical significance on day 3 (p = 0.0310) and day 5 (p = 0.0366). In the probe test on day 7, treatment with AS86 elicited no effect in primary latency and platform crossover (Table 1, 2), compared with mIgG group. However, WT-AS86 group also exhibited better scores than WT-mIgG group in the primary latency (Figure 8C; p = 0.0436) and the platform crossover number (Figure 8D; p = 0.0093). These results suggest that AS86 could enhance cognitive functions of WT mice. Treatment of APP/PS1 with AS86 for the same duration (3 months) generally did not improve spatial learning and memory (Figure 8B-D). Paradoxically, the APP/PS1-AS86 group performed worse compared with the APP/PS1-mIgG group in the platform crossover (Figure 8D; p = 0.0262).
Two-way ANOVA analysis for Primary latency in the probe test of water maze
| WM1 | WM2 | WM3 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Source of Variation | DF | F | P value | DF | F | P value | DF | F | P value |
| Interaction | 1 | 3.834 | 0.0559 | 1 | 1.066 | 0.3077 | 1 | 0.06883 | 0.7944 |
| Factor A (WT. vs. APP/PS1) | 1 | 3.515 | 0.0668 | 1 | 8.276 | 0.0062 ** | 1 | 10.66 | 0.0022 ** |
| Factor B (mIgG. vs. AS86) | 1 | 0.2888 | 0.5934 | 1 | 4.68 | 0.0361 * | 1 | 0.03029 | 0.8627 |
| Residual (Error) | 49 | 43 | 40 | ||||||
In WM2, AS86 appeared to be most effective in improving cognitive functions for both WT and APP/PS1 mice. First, APP/PS1-mIgG group showed a dramatic deficit in spatial learning compared with WT-mIgG (Figure 8E; compare WT-mIgG with APP/PS1-mIgG groups; F (1, 19) = 4.905, p = 0.0392; day 3, p = 0.0166, day 5, p = 0.0450). The probe test also revealed a marked impairment of APP/PS1-mIgG group, compared with WT-mIgG group, in spatial memory, as reflected by the increases in primary latency (Figure 8F; p = 0.0270) and platform crossover number (Figure 8G; p = 0.0063). Second, treatment with AS86 for 6 months improved the spatial learning for WT (Figure 8E, compare WT-mIgG and WT-AS86 groups; F (1, 18) = 3.936, p = 0.0627; day 3: p = 0.0360, day 5: p = 0.0180) and exhibited a non-significant trend of improvement in ASP/PS1 mice (Figure 8E, compare APP/PS1-mIgG and APP/PS1-AS86 groups: F (1, 28) = 1.514, p = 0.2287). Most importantly, AS86 rescued the spatial memory deficiency of APP/PS1 mice in the primary latency (Figure 8F; p = 0.0464) and platform crossover (p = 0.0274). For WT mice, AS86 treatment improved the primary latency (Figure 8F, p = 0.0164) but not platform crossover (Figure 8G, p = 0.1268). Similar conclusions could be reached using two-way ANOVA analysis of the probe test data (Table 1, 2).
Two-way ANOVA analysis for Platform crossover in the probe test of water maze
| WM1 | WM2 | WM3 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Source of Variation | DF | F | P value | DF | F | P value | DF | F | P value |
| Interaction | 1 | 11.63 | 0.0013 ** | 1 | 0.00598 | 0.9387 | 1 | 0.6443 | 0.427 |
| Factor A (WT. vs. APP/PS1) | 1 | 3.754 | 0.0586 | 1 | 17.51 | 0.0001 *** | 1 | 7.811 | 0.008 ** |
| Factor B (mIgG. vs. AS86) | 1 | 1.185 | 0.2818 | 1 | 4.795 | 0.034 * | 1 | 2.234 | 0.1431 |
| Residual (Error) | 48 | 43 | 39 | ||||||
In WM3, 3 months after WM2, regardless whether the animals were treated with AS86 or not, the APP/PS1 exhibited poorer performances in spatial learning and memory than WT mice (Figure 8H-J, Table 1, 2). In animals of this age (14 months old), long-term treatment with AS86 (9 months) did not show any effect on spatial memory compared with mIgG group (Table 1, 2). AS86 failed to rescue the impairments in spatial learning (Figure 8H) or memory (Figure 8I-J) in APP/PS1 mice (all p > 0.05). For comparison, treatment of WT mice with AS86 also showed an improvement in spatial learning (Figure 8H, compare WT-mIgG with WT-AS86 groups; F (1, 16) = 8.282, p = 0.0109; day 1 p = 0.0373, day 2 p = 0.0011) and a small but non-significant effect on platform crossover (Figure 8J, p = 0.0649). However, treatment with AS86 seemed to enhance the lifespan of both WT and APP/PS1 mice (Figure S7B).
Given that AS86 treatment resulted in a moderate decrease in body weight, we examined whether body weight differences affected the performance of WM test. We analyzed the data from all groups and found there is no correlation between body weight and primary latency or platform crossover number (Figure S7D; all r square < 0.95, all p > 0.05, Linear Regression). Moreover, no difference in swimming velocity was found compared with WT-mIgG group (Figure S7E; all p > 0.05).
The WM experiments together suggested that activation of TrkB by AS86 administration seemed to improve spatial learning and memory slightly for WT but more so for the APP/PS1 mutant mice. In the AD model, treatment with AS86 elicited no effect before spatial memory deficiency (8-month old mice, treated for 3 months), significant effects at the early stage of spatial memory deficiency (11-month old mice, treated for 6 months), and no effects at the middle stage of memory deficiency (14-month old mice, treated for 9 months). Thus, the age of animals (stage of disease manifestation) and duration of AS86 are both important. Treatment of younger animals (8-month old, presumably at the early stage of AD) with AS86 for 3 months seems to be marginally effective. In older animals (14 months or older) in which the Aβ pathology becomes overwhelming, activation of TrkB would no longer be effective regardless of how long the TrkB agonist is administered.
Effects of AS86 on pathology and pathophysiology of AD mice
A hallmark of AD is the aggregation of Aβ42 peptide, leading to accumulation of amyloid plaques in the brain [7,74]. In APP/PS1 mice, the Aβ pathology occurs at around 6 months of age, and in 12-month old animals, amyloid plaques are seen throughout the brain [75,76]. We examined whether long-term treatment with AS86 could alter Aβ pathology in APP/PS1 mice. AS86 was administered twice monthly through tail vein injection to the APP/PS1 mice at 5 months of age, and Aβ42 level in forebrain was examined with ELISA after 10 months of treatment with AS86. As shown in Figure 9A, long-term treatment with AS86 had no effect on the Aβ42 level in APP/PS1 mice (p > 0.05). Next, we examined the amyloid plaques in the brain using Congo red staining method. The percentage of amyloid load in hippocampus and cortex were quantified. Again, a 10-month treatment with AS86 did not change amyloid load either in hippocampus or in cortex (Figure 9B-D; all p > 0.05). These results suggest that improvement of cognitive functions by AS86 was not mediated by a reduction of Aβ pathology.
Effects of AS86 on pathology and pathophysiology of AD mice. (A-D) Effect of AS86 treatment on Aβ pathology in APP/PS1 mice. (A) Detection of Aβ42 for each sub-group. Human Aβ42 in forebrain (15 months) was measured with Aβ42 Elisa (n = 6 for each group), followed by quantification. (B, C, D) Amyloid plaque load in hippocampus and cortex. Amyloid plaques were detected by Congo red staining in hippocampus and cortex (12 slices from 6 mice for each group). The quantification of amyloid plaque load percentage in cortex (B) and hippocampus (C) and the Congo red staining images (D) are presented. Scale bar represents 200 µm. The black and red arrows indicated the amyloid plaques in cortex and hippocampus respectively. The images in black and red squares were amplified at the upper right. (E, F) Rescue of synaptophysin loss in APP/PS1 mice by AS86. Synaptophysin puncta in hippocampal CA1 (15 slices from 6 mice of 15 months old for each group) were detected with immunohistochemistry. The density of synaptophysin puncta in each group was analyzed blindly by Imaris software with the same standard. The immunostaining images (upper) and calculated spot images (lower) with Imaris software (F), and the histogram of quantification for puncta numbers are presented (E). Scale bar represents 5 µm. Student's t-test.
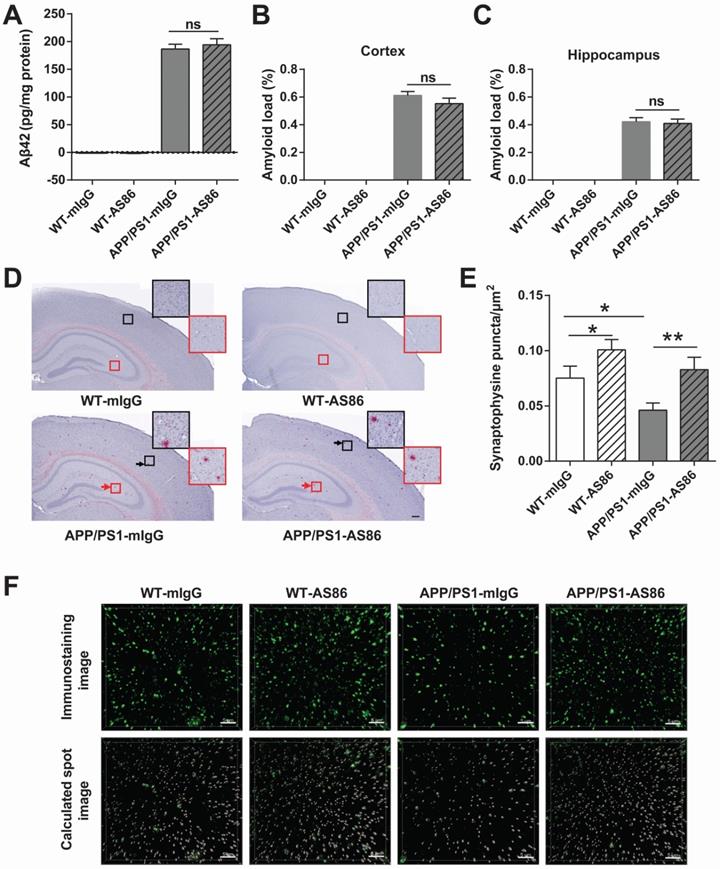
A key pathophysiological feature of AD brain is synaptic loss [11-13]. To determine whether AS86 attenuated cognitive impairment in APP/PS1 mice by synaptic repair, we examined the presynaptic marker protein synaptophysin in CA1 area of hippocampus. After 10-month treatment with AS86 by tail vein injection, the density of synaptophysin puncta was analyzed. Among the four groups of mice, the synaptophysin puncta density of APP/PS1-mIgG mice was significantly decreased compared with WT-mIgG mice (p = 0.0139). In contrast, the APP/PS1-AS86 group exhibited a significant increase in synaptophysin puncta density compared with APP/PS1-mIgG group (p = 0.0054) (Figure 9E-F). There was also a significant increase in WT-AS86 group (p = 0.0398) compared with WT-mIgG group. Therefore, AS86 treatment ameliorated synaptic loss as reflected by the synaptophysin puncta density in the 15-month-old APP/PS1 mice.
Discussion
Despite plenty of evidence for the role of Aβ in the pathogenesis of Alzheimer's disease, therapies aimed at reducing Aβ accumulation have so far been unsuccessful. A paradigm shift is needed to treat the devastating illness. A large body of literature now supports the view that synapse loss, but not toxin accumulation, correlates with disease progression [2,7]. Thus, an emerging new approach is to address AD using neuronal and/or synaptic repair mechanisms. BDNF, the best known “synaptogenic” molecule demonstrated by animal studies and human genetics, may pave the way for this paradigm shift. However, decades of R&D efforts by many biopharmaceutical companies have suggested that BDNF protein itself may not be a good drug candidate due to its intrinsic properties. Here we have identified a TrkB agonistic antibody AS86 that could effectively penetrate into the brain and activate the BDNF-TrkB signaling pathways in the central nervous system (CNS). We provide a set of coherent evidence that AS86 could inhibit Aβ-induced neuronal death, promote spine formation, and facilitate LTP at hippocampal CA1 synapses. The antibody has also been shown to have a long blood half-life, good brain penetration, and sufficient target engagement. Most importantly, long-term treatment with AS86 attenuated the deficits of recognition and spatial memories at the early stage of spatial memory deficiency in a mouse model of AD. Taken together, the present study has laid a foundation for a new therapy for AD, based on the use of TrkB agonistic antibody.
The idea of targeting the BDNF-TrkB is not new. Several groups have attempted to infuse the BDNF-expressing viruses or BDNF itself into the brains of AD animal models [44,46,48,77,78]. While useful in proof of concept (PoC), the methods of delivering viruses or protein directly into the brain would be difficult to implement in the clinic, and acceptance by patients and doctors would be low. Daily administration of a BDNF-mimicking peptide has also been shown to improve memory deficits in rodent models of AD [49]. However, the pharmacokinetics of the peptide was not known and cost of goods (CoG) for daily injection of the peptide could be extremely high. Several papers have claimed that small molecule TrkB activators could mimic BDNF in cellular assays and therefore be potentially useful in treating neurodegenerative diseases including AD [62-66]. However, two recent studies using more systematic approaches and rigorous methods have reported contrary results: these small molecules neither bind nor activate TrkB [67,68]. Based on the structural model of TrkB [79], these compounds are too small to hold the two TrkB monomers together for TrkB dimerization, which is required for the activation of receptor tyrosine kinase of TrkB. To this end, bivalent macromolecules such as monoclonal antibody would be more appropriate for dimerization and activation of TrkB. Several TrkB agonistic antibodies have been developed in the past [67,80,81]. However, treatment of chronic illnesses such as AD would require long-term drug administration that demands a high bar for safety, and none of the previously reported TrkB antibodies have been tested in AD animal models.
The present study is the first to systemically examine whether long-term administration of TrkB agonistic antibody could be a feasible approach for AD therapy. We show that AS86, a monoclonal TrkB agonistic antibody, exhibited several features superior to BDNF: it binds TrkB but not p75NTR (Figure 2A), is more likely to be diffusible in brain tissues [82], and has a much longer half-life (about 6 days) in blood and brain (Figure 6A-B). In a TrkB activation assay with saturated concentration of BDNF, AS86 exhibited non-competition with BDNF suggesting that AS86 may elicit additional benefits in the presence of endogenous BDNF in vivo (Figure S2A-B). In addition to its non-competitiveness with BDNF, we selected AS86 among many other TrkB-agonistic antibodies as a drug candidate for AD for a number of reasons. First, although TrkB activation induced by AS86 is lower than by BDNF (Figure 1B), the two TrkB activators elicited similar levels of downstream signaling, especially ERK and Akt (Figure 1B-C, Figures S2A, S2C), the two signaling pathways thought to be more important for cell survival and synaptic growth [24]. Second, AS86 performed better than other TrkB agonistic antibodies that induced higher TrkB activation (data not shown), but similarly to BDNF in regulating neuronal survival (Figure 3), neurite growth (Figure 4) and synaptic functions (Figure 5). Third, TrkB over-activation may induce potential side-effects during long-term treatment for AD.
Behaviorally, bi-weekly administration of AS86 through tail vein injection for more than 5 months attenuated deficits in spatial as well as recognition memory in a mouse model of AD (Figure 8F-G; Figure 7C), without any impact on motor behavior (Figure S7A). It appears that the drug-induced improvements in spatial memory were not as robust as those on object recognition memory, raising the possibility that AS86 may preferentially alter synapses in various cortical areas over hippocampus. However, it should be noted that hippocampus is also a key area involved in recognition memory. Although we only examined the changes in synaptophysin puncta in the hippocampus, it is our belief that AS86 ameliorated synaptic deficits in hippocampus as well as various cortical regions. In this study, we focused on AS86-induced changes in the hippocampus because we performed spines and LTP analyses in this area. Many previous studies using APP/PS1 mice have also used hippocampus to examine synaptic changes [83]. Future studies should be designed to address why the TrkB agonistic antibody affects water maze test and object recognition test differentially, and whether AS86 could also rescue synaptic impairment and improve synapses in the cortex. Interestingly, long-term treatment with AS86 even elicited a slight increase in lifespan (Figure S7B). We further observed that bi-weekly administration of AS86 for as long as 9 months did not induce any visible side-effects. Taken together, we have developed a TrkB-specific agonistic antibody as a drug candidate potentially useful for the treatment of Alzheimer's disease in humans.
It is well established that BDNF in hypothalamus regulates energy metabolism including feeding. Several studies have also shown that treatment with TrkB agonistic antibody resulted in a gradual weight loss in mice [72]. Therefore, TrkB agonistic antibody had been considered as a potential drug for obesity. We found that long-term treatment with AS86 resulted in a 20% reduction of body weight in the first 20 days, but the body weight maintained unchanged in the next 9 months (Figure S7C). Interestingly, both WT and AD mice exhibited the same magnitude and time course of weight reduction. In general, this reduction in body weight is considered not harmful to health. More importantly, our own data indicate that this body weight loss did not influence the velocity, moving distance, or the time spent in center and corner in the open field task (Figure S7A). On the contrary, mice may benefit from the weight loss according to the preliminary data on the survival curve shown in Figure S7B. Thus, the mild reduction in body weight may be tolerable even for aged animals.
A major challenge for the treatment of CNS diseases with antibody drugs is whether they can effectively penetrate into the brain and engage their targets, although several techniques have been established to enhance brain delivery of antibody drugs [84,85]. It is believed that the BBB is slightly leaky in the AD brain. Based on data from rat studies, it is estimated that approximately 0.1% - 0.2% of the antibody could pass through the BBB [86]. Clinical studies using anti-Aβ antibodies also demonstrate the penetration through BBB and functions of antibodies in the brain through peripheral treatment. In the present study, we show that administration of AS86 at 1.5 mg/kg (body weight) through tail vein injection could effectively activate TrkB and its downstream signaling in the brain (Figure 6C-E). The pharmacokinetic curve of AS86 in brain is matched to that in plasma, and the half-life of AS86 in brain is a little longer (Figure 6A-B).
An interesting observation from the water maze experiments was that bi-weekly administration of AS86 to APP/PS1 mice beginning at 5-months old was ineffective at 3-month treatment (WM1), but effective at 6-month treatment (WM2), and ineffective again at 9-month treatment (WM3). While the mechanisms underlying this biphasic curve remain to be investigated, a number of points could be discussed. First, in WM1, the lack of efficacy in APP/PS1 mice was not due to the inability of AS86 to reach and engage its target TrkB, because the weight reduction occurred shortly after AS86 administration was initiated (Figure S7C), and AS86 treatment improved learning and memory for WT animals (Figure 8B-D). One may speculate that while AS86 was effective in repairing synapses in the APP/PS1 brains, re-building of the lost connectivity may take longer time to accomplish. Second, in WM2, AS86 treatment enhanced learning for both WT and APP/PS1 mice, and even improved memory for both WT and the mutant mice (Figure 8E-G). One may again speculate that learning and memory may have different sensitivity to AS86. Regardless, a critical window is identified in which the therapeutic effects of AS86 could be clearly demonstrated. These results further suggest the length of clinical trial should not be too short for BDNF-based therapy. Finally, in WM3, continuous treatment with AS86 for 9 months was no longer beneficial for learning or memory in either WT or APP/PS1 mice (Figure 8H-J). However, such treatment was effective in increasing synaptophysin-positive puncta and repairing synapses (Figure 9E-F). Thus, the TrkB agonistic antibody, like many other therapeutic approaches, may not work when the disease is progressed to a very severe stage.
Does activation of BDNF-TrkB signaling inhibit Aβ production? In normal cells or animals, there are several conflicting results on the role of BDNF in APP or Aβ expression [87-90]. In one study, Wu et al reported that treatment of APPswe mice with a BDNF-mimetic peptide reduced the levels of Aβ42, Aβ40, the Aβ42/Aβ40 ratio, as well as the protein levels of APP, APP fragments, BACE1 and PS1 [91]. However, the specificity and the PK of this peptide were not examined, and it is unclear these effects were truly mediated by the BDNF-TrkB signaling pathway. In another study [78], BDNF infusion into lateral ventricles for 6 weeks (0.5 μg, twice a week) decreased Thioflavin-positive Aβ plaques in APP/PS1 mice, although the number of total plaques was unaffected. Given that the reduction in Aβ plaques level by the BDNF was mild (less than 20%), it is less likely that such a reduction would help improve cognition when compared with those by Aβ antibodies [92-94]. In marked contrast, Nagahara et al. reported that the expression of BDNF by virus in the brain of J20 transgenic mice did not affect the amyloid plaque density in the hippocampus [44]. In addition, some drugs that increase BDNF indirectly also elicit cognitive improvement without reducing Aβ pathology [95,96]. Further, transcriptional analyses have revealed that treatment with BDNF could alter the expression of hundreds of genes, many of which are transcription factors [97]. Thus, the beneficial effects of BDNF to AD may be achieved through many downstream genes, irrespective of whether it alters Aβ metabolism or not.
In the present study, we found that treatment with AS86 for a prolonged period did not elicit any change in Aβ42 levels or amyloid load, but significantly repair the synaptic deficits and slow down the progression of cognitive impairments in the APP/PS1 model of AD. This is an important finding, because it suggests that disease-modifying therapy of AD could be achieved even in the presence of (or without reducing) Aβ pathology. Further, it opens up the potential of greater benefits of combinational therapies. Future efforts should be directed towards whether a combination of AS86 and the anti-Aβ antibody (such as aducanumab) could achieve a greater therapeutic effect.
A number of important issues remain to be addressed in future research. First, an important feature of Alzheimer's disease is progressive neuronal loss, leading to brain atrophy [14-17]. We show that AS86 could rescue Aβ-induced neuronal death in an in vitro cell model (Figure 3B-D). Given the lack of obvious neuronal loss in the APP/PS1 model, we could not address whether AS86 is capable of inhibiting neuronal death in vivo [98]. As far as we know, most if not all Aβ-perturbing animal models published so far exhibit no obvious neuronal death. The neuroprotective function of AS86 should be rigorously tested when such animal models become available. Second, although BDNF could not alleviate the Aβ pathology in APP/PS1 mouse model (Figure 9A-D), an APP-overexpression model, it would be interesting to test whether AS86 will have an effect on Aβ pathology in an APP knock-in model. Finally, synaptic loss and neuronal degeneration are general features that occur in many neurodegenerative diseases, including Huntington's disease [99] and Parkinson's disease [100]. It will be interesting to test whether AS86 could be useful in treating other neurodegenerative diseases.
Conclusion
We have developed a TrkB agonist antibody AS86 which specifically activates TrkB and its downstream signaling, enhances neurite outgrowth and cell survival in in vitro model. Based on its capacity of synaptic enhancement, AS86 could rescue the deficiency of spatial cognition and novel object recognition in APP/PS1 mouse model. Taken together, these results suggest that AS86 could be further developed as a potential therapeutic agent for AD.
Materials and Methods
Antibody and reagents
The antibodies used in this study were against: phospho-Akt (S473, Cell Signaling Technology, 9271), phospho-TrkA (Y490, Cell Signaling Technology, 9141), phospho-TrkA (Y706/707, Cell Signaling Technology, 4621), phospho-TrkA (Y816, Cell Signaling Technology, 4168), Erk (Cell Signaling Technology, 4695), phospho-Erk (T202/Y204, Cell Signaling Technology, 4370), phospho- PLCγ(Y738, Cell Signaling Technology, 2821), PLCγ(Cell Signaling Technology, 2822), TrkB (80E3, Cell Signaling Technology, 4603), Akt (Santa Cruz Biotechnology, H-136), MAP2 (EMD Millipore Corporation, MAB3418), GAPDH (Bioeasy (Beijing) Technology, BE0023), EGR1 (Cell Signaling Technology, 4153), Synaptophysin1 (Synaptic Systems, 101 002), β-actin (CWBIO, CW0096), and Caspase-3 (Cell Signaling Technology, 8610). The reagents included BDNF protein (Sino Biological Inc, 50240-MNAS), Mouse IgG (YEASEN, 3611ES10), and β-Amyloid protein (25-35) (Synpeptide), K252a (Bio Vision, 2013-500), AZD-1332 (Alomeone labs, A-495).
Production of TrkB agonistic antibodies by the hybridoma technology
The antigen (recombinant TrkB-ECD-hFc) in DPBS was used to immunize BALB/C mice of about 6~8 weeks old via subcutaneous route injections following a procedure of fast immunization. All experiments involving animals in this study were approved by Tsinghua University Committees on Animal Care. After immunization, animals were selected by tittering using ELISA. Immunized mice with the highest titer were sacrificed and lymph nodes were harvested. The lymphoid cells were suspended in DMEM before fusion with a myeloma cell line Sp2/0-Ag14 by PEG (P7306, sigma). The fused cells were cultured in HAT selecting medium (21060-017, Gibco). On day 7 or 10, one-half medium was changed. After 14 days of culturing, hybridoma supernatants were screened for TrkB-specific monoclonal antibodies. ELISA was used to analyze the affinity of the antibodies with both human TrkB-ECD-His and rat TrkB-ECD-His, and NFAT assay was used to select active TrkB agonists. After selection of positive cell pools, sub-cloning was done by limiting dilution. Hybridoma sequencing was performed with the 5'RACE kit (cat.634858, &634859, Clontech).
Cell culture and treatment
CHO-TrkB cells were grown in standard DMEM (Gibco, 11960-051) containing 10% FBS (Invitrogen, 26400-044) and GlutaMax supplement (Gibco, 35050061). Embryonic hippocampal neurons (embryonic day 18) were dissociated and plated on 100 ng/ml poly-D-lysine-coated 12-well plates at 200,000 cells per well as described previously [101]. Neurons were cultured in Neurobasal Medium supplemented with B27 and GlutaMax (Gibco) for 10 d and then processed for biochemical experiments. Dendritic morphology was studied with cultures grown for only 1 d on coverslips (18 mm) at 10,000 cells per coverslip. Spine density and morphology were studied with cultures grown at 100,000 cells per coverslip (18 mm). Cell survival test was performed with cultures grown at 20000 cells per well on black 96well plates (Corning, 3603). All cultures were maintained at 37 °C in 5% CO2 and 95% air.
NFAT
The procedure was described previously [82].
Analysis of neurite complexity
Cultured hippocampal neurons at day 1 in vitro (DIV1) were treated with TrkB agonist antibody or BDNF and grown for an additional 3 days. The neurons were fixed and stained with a mouse antibody against MAP2 (1:1000, Millipore). Images were acquired with a Zeiss confocal microscope (20 ×, 488-nm laser, LSM710) in a double-blind manner. Four parameters of neurite growth were analyzed using the Image J analysis software (Neuron J plugins and ShollAnalysis plugins). Typically, images of 100 neurons per group were captured, and three independent experiments were performed.
Western blot analysis
Cells were lysed in the lysis buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 1% CA-630, 1% sodium deoxycholate, 0.1% SDS, 2 mM EDTA and a phospho-protease inhibitor cocktail purchased from Roche) on ice for 30 min. After 10 min centrifugation at 10000 rpm, the supernatants of lysate were subjected to the loading buffer, incubated at 95 °C for 10 minutes and resolved by SDS-PAGE. Proteins were transferred to PVDF membrane for 1.5 hours at 100 V. The membrane was blocked with 5% bovine serum albumin (BSA) for 1 hour at room temperature and incubated with the primary antibodies in blocking solutions at 4 °C overnight before detection with HRP-conjugated secondary antibodies. Chemiluminescence was detected with ECL solution.
Survival Assay
The assay of serum deprivation model using hTrkB-PC12 cells was described by previous work [82]. For AD cell death model, 2 mM β-Amyloid protein (25-35) stock solution was prepared in sterile distilled water, stored at -80 °C, and incubated for 3 days at 37 °C to aggregate oligomers before use. Hippocampal neurons were cultured for 8 d (DIV8). After the neurons were pretreated with TrkB agonist antibodies or BDNF or control medium for 30 minutes, Aβ25-35 oligomer was added for an additional 48 h. Cell viability was determined by the CellTiter-Glo Luminescent Cell Viability Assay (Promega, G7570).
Analysis of spine density
Hippocampal neurons (7 days in vitro (DIV7)) were transfected with membrane-target mCherry by the calcium phosphate method (Takara, 631312) and examined at 16 d in vitro using a Nikon confocal microscope (60 × Oil objective at a 1.5 × zoom, 561 nm laser). Each image consisted of a z stack of pictures taken at a depth interval of 0.15 μm and then projected into one image (max intensity). Morphometric quantifications were performed by investigators blind to the experimental condition. Morphometric measurements were performed with Image J. The density of spine and mushroom bodies were calculated by dividing the spine number by the measured length (in 10 μm) of imaged secondary dendritic stretches (longer than 20 μm). In each experiment, 60 secondary dendrites from 30 neurons were analyzed. Three independent experiments were performed.
Aβ42 quantification
The brain tissues were homogenized in the lysis buffer described as Western blots analysis on ice, and the homogenates were treated with ultrasonic to be lysed thoroughly. Then the tissue lysates were centrifuged at 12000 rpm for 30 min at 4 °C and the supernatants were collected. Before the analysis with ELISA, the sample protein concentrations were determined by BCA protein assay kit (Pierce, 23225) according to the manufacturer's instruction, and normalized by dilution as the lowest one. Aβ42 ELISA kit (Invitrogen, KHB3441) was used to determine the Aβ42 concentration according to the manufacturer's instruction. Optical signals at 450 nm were read on a microplate spectrophotometer (BioTek, CYTATION 5).
ELISA Assay
Coating protein (TrkB-ECD , p75, TrkA or TrkC) was added into microplate in Coating Buffer (0.1 M Carbonate Buffer, pH 9.6, NaHCO3 8.4 g/L) overnight at 4 °C. The plate was washed with washing buffer (0.05% tween20 in PBS) for 3 times and then blocked with 0.5% BSA in PBS for 1h at RT, followed by 3-time washing. Standard proteins or samples (serum or protein lysates) diluted to suitable concentrations were added into the microplates and incubated for 2 hours at RT. After samples were washed with washing buffer for 3 times, the HRP-conjugated second antibody in 1% BSA was added into the wells and incubated for 0.5 hours at RT. After 3 times of washing, 100 μl chromogen (TMB) was added into each well and incubated for 15 min in dark, and then 50 μl 2 M HCl was added to stop the reaction. Optical signals at 450 nm were read on a microplate spectrophotometer (BioTek, CYTATION 5).
Animals and housing conditions
The APPswe/PS1dE9 (APP/PS1) mice were generated by Jackson Laboratories (Bar Harbor, ME, USA) [102]. Heterozygous male mice of APP/PS1 and their littermate wild type (WT) mice at 4 weeks old with a B6C3F1 background for behavioral tasks were obtained from the Model Animal Research Center of Nanjing University. These mice were randomly assigned into four groups: WT-mIgG (mouse normal IgG), WT-AS86, APP/PS1-mIgG and APP/PS1-AS86. All the mice in other experiments with a C57BL/6J background and Sprague-Dawley (SD) rats were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. The animals were maintained in a temperature-controlled room (21 ± 2 °C) with a 12 h light-dark cycle. The ventilation system of mice cages was independent, and food and water were freely available. All these operations were only conducted at the dark cycle. All procedures followed the Institutional Animal Care and Use Committee (IACUC) guidelines provided by Tsinghua University.
Slice Preparation
P12-13 male SD rats were anesthetized with isoflurane and sacrificed by cervical removal. The brains were rapidly dissected and placed in ice-cold artificial cerebrospinal fluid (ACSF, saturated with 95% O2 and 5% CO2), containing (in mM): NaCl (125.8), NaHCO3 (26), NaH2PO4 (1.2), KCl (3.1), CaCl2 (2.5), MgCl2 (1.5) and D-glucose (10). Subsequently, coronal hippocampal slices (400 μm) were prepared in oxygenated ACSF using a Leica VT1000S vibratome (Leica Instruments) at 4-6 °C and maintained in ACSF at 25°C for at least 1h before use.
Electrophysiological recording
The slices were submerged in the recording chamber and perfused with ACSF containing (in mM): NaCl (125.8), NaHCO3 (26), NaH2PO4 (1.2), KCl (3.1), CaCl2 (2.2), MgCl2 (1.4) and D-glucose (10) at a rate of 2-4 ml/min at 25 °C. AS86 or mIgG were perfused at a concentration of 15 nM. For LTP experiments, brain slices were perfused for at least 30 min prior to the induction of LTP.
All electrophysiological recordings were performed with an Axopatch-700B amplifier (Molecular Devices, USA) at the sampling rate of 10 kHz and filtered at 5 kHz using a Digidata 1550B analog-digital converter (Axon Instruments). Field EPSPs were recorded using 1.5-3.5 MΩ glass pipettes filled with ACSF and placed in the stratum radiatum of the CA1 region. Data were acquired and subsequently analyzed using ClampFit 10.4 software (Molecular Devices, USA). A bipolar stimulating electrode was positioned at the terminals of the Schaffer collateral (SC). Input/output relation of EPSP was assessed by electrical stimulation with intensities ranging from 100 μA to 1000 μA in 100 μA increments. Stimuli were delivered every 30 s and repeated for five times at each intensity. Paired-pulse responses were evoked at inter-stimulus intervals of 10, 20, 40, 60, 100, 150, 250 and 500 ms using a stimulation intensity of 0.5 mA. The paired-pulse ratio is defined as the ratio of second population spike amplitude to the first population spike amplitude. For LTP experiments, baseline responses were recorded for 15 min prior to stimulation. Electrical stimulation was delivered every 30 s in recordings, consisting of low-intensity, square-wave pulses (0.1 ms). LTP was induced by high frequency stimulation (1 s burst of equally spaced pulses at 100 Hz). Responses were subsequently recorded for an additional 60 min to monitor changes in synaptic transmission. The synaptic strength was determined by the slope from 10% to 90% of the rising phase of the field excitatory postsynaptic potential (fEPSP). The magnitude of LTP was quantified as the normalized average slope of the fEPSP taken from the last 10 min of recording.
Congo red staining
Congo red staining was performed according to standard protocols. Frozen brain sections were prepared at the thickness of 30 μm and immersed in congo red solution (160 ml methanol, 40 ml glycerol mixed with 1 g Congo Red (Sigma, C6767)) for 15 min, and then dissolved in 200 ml 80% ethanol mixed with 0.4 g KOH for 1 min twice. After rinsing with distilled water, the slices were counterstained in hematoxylin (Solarbio life sciences, G1142), rinsed again, and dehydrated in ethanol and xylene. The bright field images were captured using ZEISS Axio Scan.Z1 (10 ×), and analyzed with Image J.
Synaptophysin immunostaining
Immunohistochemistry with 30 µm frozen brain sections was performed as previously described [103]. Images were captured with a Nikcon confocal microscope in 12 bit (1024 × 1024) using a 100 × Oil-immersion objective at a 3 × zoom. Each image was taken from CA1 of hippocampus and consisted of five steps of z stack pictures with a depth interval of 0.8 μm. The spine numbers were analyzed using Imaris software SPOT module with the same spot definition standard, and the spine numbers were calculated automatically.
Behavioral tasks
The mice were handled more than 5 days (5 min for each mouse per day) before each behavioral task.
Open field test (OFT)
The open field test was performed in a cubic open filed arena (made of white PVC). Mice were allowed to explore in this tank for 10 min in dark. The activity of mice was monitored by an infrared camera which is mounted above the arena and connected to an automated video tracking system and EthoVision XT software (Noldus, Netherlands). The arena was thoroughly cleaned with 75% alcohol after each trial.
Analysis of cutaneous sensitivity with von Frey and Plantar test
One day after the tail vein injection of the TrkB antibody AS86, analyses of cutaneous sensitivity were performed with von Frey and Plantar hot plate tests. For the von Frey test, the up and down method and threshold calculation, first described by Chaplan et al., (1994) was used. The mice adapted on the mesh surface for at least 1 hour. Then, von Frey filaments were used to stimulate the bottom (midplantar) surface of the mice hind foot. Nine filaments represented 0.02, 0.04, 0.07, 0.16, 0.40, 0.6, 1.0, 1.4 or 2.0 g respectively. The first filament applied is a medium force (0.40 g). The positive response was described as rapid claw extraction, licking or shaking during stimulation or after removal of filaments. Here, we followed a modified protocol [104]. For hot plate test, we used the instruments of Plantar Test (Ugo Basile, Comerio VA, Italy). The standard operation manual was followed. The strength of stimulation (IR value) was adjusted to produce baseline latencies of 8 to 10 seconds. Animals were first acclimatized in the apparatus for at least 1 hour before measurements. Each measurement was repeated at least 4 times with the intermission of 10 minutes.
Novel object recognition test (NORT)
The novel object recognition test was conducted in cubic arena (33 cm × 33 cm × 20 cm made of white PVC). The experiments were performed in the dark. The mice were allowed to accommodate the empty arena for three consequent days (5 min/day). At the fourth day, the mice were allowed to explore two same adjacent objects (40 ml flasks filled of yellow sand) situated at 10 cm away from the arena wall for 5 min. After a 3h interval, one of the flasks was replaced by a new object (60 ml glass reagent bottle filled with blue sand), and the mice were allowed to explore for 5 min. The arena and the objects were thoroughly cleaned with 75% alcohol, and the alcohol was blown away by a fan after each trial. The numbers of nose pokes of the mice to each object were scored by blinded observers.
Morris water maze (WM) task
The maze was made of a white inside circular tank (diameter = 120 cm) and was filled with white water at 21 ± 1 °C. The tank was equally divided into four quadrants (north, south, east, and west). An escape circular platform (diameter = 12 cm) covered with gauze for gripping was placed in the center of one quadrant and was hidden 1cm below water level. Four spatial cues were located on the inside walls of the circular tank, and the human tester always left the room in the same location during testing. For three rounds of testing, the platform was posited in a fixed position (WM1: SW, WM2: NE, WM3: SE) during the 4-5 days of learning trials. A camera located above the tank was interfaced with a computer, and EthoVision XT software (Noldus, Netherlands) was used to collect and analyze data. The mice were subjected to learning trials four times a day for 5-6 consecutive days, with a 25-30 min inter-trial interval. During each trial, the mice were released into water gently facing the tank wall from a principle start location as described previously [105], and were allowed to find the hidden platform for 60 s. They were to be put on the hidden platform manually for 10 s if the mice failed to find it within 60 s. On the probe day, a trial was carried out where the platform was removed, and the mice were allowed to swim for 60 s in the tank.
Abbreviations
BDNF: brain-derived neurotrophic factor; TrkB: tropomyosin-related kinase B; AD: alzheimer's disease; Aβ: amyloid β; LTP: long-term potentiation; PI3K: phosphatidylinositol-3 kinase; Erk: extracellular regulated kinase; PLCγ: phospholipase C-gamma; p75NTR: p75 neurotrophin receptor; RTK: tyrosine receptor kinase; hTrkB: human TrkB; IgG: immunoglobulin G; EGR1: early growth response 1; NORT: novel object recognition test; WM: water maze; CNS: central nervous system.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
The authors are grateful to Jingyu Shao and Yang Dou for contributing to antigen and antibody purification respectively, Yue Chen for technical assistance, Dr. Guan Wang and Dr. He You for advice on the manuscript, Yanhui Chen, Pengcheng Chu, Fang Han and Tingting Wang for help on the experiments for revision.
Funding
This work was supported by the National Key Research and Development Program of China (2017YFE0126500), the National Natural Science Foundation of China (81501105, 81861138013, 31730034), Beijing Advanced Innovation Center for Human Brain Protection, Shenzhen Science Technology and Innovation Commission (JCYJ20170411152419928 and JCYJ20180508152240368), and Beijing Municipal Science & Technology Commission (Z151100003915118).
Authors' contribution
SW, WG and BL initiated and designed the project. SW and WG managed all the experiments, and SW conducted all experiments except for the descriptions as following. SW and YX conducted the experiments of target engagement and tail vein injection. WG conducted the serum deprivation survival assay and analysis of cutaneous sensitivity. HY, WZ and WG conducted antibody screening. WG and HL conducted the experiment of specificity. RH and YH conducted the electrophysiology experiment, data analysis of this part and wrote the electrophysiology part of the manuscript. SW, WG and BL wrote the manuscript.
Competing Interests
BL, WG and HY are co-inventors of the filed patents related to the TrkB agonistic antibodies. The remaining authors declare no conflict of interest.
References
1. Polanco JC, Li C, Bodea L-G, Martinez-Marmol R, Meunier FA, Götz J. Amyloid-β and tau complexity — towards improved biomarkers and targeted therapies. Nat Rev Neurol. 2017;14:22
2. Chen X-Q, Mobley W. Alzheimer Disease Pathogenesis: Insights From Molecular and Cellular Biology Studies of Oligomeric Aβ and Tau Species. Front Neurosci. 2019 13
3. Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL. Alzheimer's disease. Nat Rev Dis Pri. 2015;1:15056
4. Beecham GW, Hamilton K, Naj AC, Martin ER, Huentelman M, Myers AJ. et al. Genome-wide association meta-analysis of neuropathologic features of Alzheimer's disease and related dementias. PLoS Genet. 2014;10(9):e1004606-e1004606
5. Hall AM, Roberson ED. Mouse models of Alzheimer's disease. Brain Res Bull. 2012;88(1):3-12
6. Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K. Alzheimer's disease drug development pipeline: 2019. Alzheime Dement: Transl Res Clin Interv. 2019;5:272-293
7. Long JM, Holtzman DM. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell. 2019;179(2):312-339
8. Panza F, Lozupone M, Logroscino G, Imbimbo BP. A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat Rev Neurol. 2019;15(2):73-88
9. Cummings JL. Challenges to demonstrating disease-modifying effects in Alzheimer's disease clinical trials. Alzheimers Dement. 2006;2(4):263-271
10. Lu B, Nagappan GH, Guan XM, Nathan PJ, Wren P. BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat Rev Neurosci. 2013;14(6):401-416
11. DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer's disease: Correlation with cognitive severity. Ann Neurol. 1990;27(5):457-464
12. Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007;68(18):1501
13. Scheff SW, Price DA. Synapse loss in the temporal lobe in Alzheimer's disease. Ann Neurol. 1993;33(2):190-199
14. Ball MJ. Neuronal loss, neurofibrillary tangles and granulovacuolar degeneration in the hippocampus with ageing and dementia. Acta Neuropathol. 1977;37(2):111-118
15. Whitehouse PJ, Price DL, Clark AW, Coyle JT, DeLong MR. Alzheimer disease: Evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann Neurol. 1981;10(2):122-126
16. Gómez-Isla T, Price JL, McKeel Jr DW, Morris JC, Growdon JH, Hyman BT. Profound Loss of Layer II Entorhinal Cortex Neurons Occurs in Very Mild Alzheimer's Disease. J Neurosci. 1996;16(14):4491
17. Andrade-Moraes CH, Oliveira-Pinto AV, Castro-Fonseca E, da Silva CG, Guimarães DM, Szczupak D. et al. Cell number changes in Alzheimer's disease relate to dementia, not to plaques and tangles. Brain. 2013;136(Pt 12):3738-3752
18. Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E. et al. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature. 2002;420(6917):788-794
19. Matsuzaki M, Honkura N, Ellis-Davies GCR, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429(6993):761-766
20. Bourne JN, Harris KM. Nanoscale analysis of structural synaptic plasticity. Curr Opin Neurobiol. 2012;22(3):372-382
21. Poo M-m. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2(1):24-32
22. Lu B. BDNF and activity-dependent synaptic modulation. Learn Mem. 2003;10(2):86-98
23. Lu B, Nagappan G, Lu Y. BDNF and synaptic plasticity, cognitive function, and dysfunction. Handb Exp Pharmacol. 2014;220:223-250
24. Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361(1473):1545-1564
25. Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381(6584):706-709
26. Ying S-W, Futter M, Rosenblum K, Webber MJ, Hunt SP, Bliss TVP. et al. Brain-Derived Neurotrophic Factor Induces Long-Term Potentiation in Intact Adult Hippocampus: Requirement for ERK Activation Coupled to CREB and Upregulation of Synthesis. J Neurosci. 2002;22(5):1532-1540
27. Ji Y, Lu Y, Yang F, Shen W, Tang TT-T, Feng L. et al. Acute and gradual increases in BDNF concentration elicit distinct signaling and functions in neurons. Nat Neurosci. 2010;13(3):302-309
28. Tyler WJ, Pozzo-Miller L. Miniature synaptic transmission and BDNF modulate dendritic spine growth and form in rat CA1 neurones. J Physiol. 2003;553(2):497-509
29. Lu Y, Christian K, Lu B. BDNF: a key regulator for protein synthesis-dependent LTP and long-term memory? Neurobiol Learn Mem. 2008;89(3):312-323
30. Bekinschtein P, Cammarota M, Izquierdo I, Medina JH. BDNF and memory formation and storage. Neuroscientist. 2008;14(2):147-156
31. Connor B, Young D, Yan Q, Faull RLM, Synek B, Dragunow M. Brain-derived neurotrophic factor is reduced in Alzheimer's disease. Mol Brain Res. 1997;49(1):71-81
32. Isidro Ferrer M, Conxita Marín, Ma Jesús Rey, Teresa Ribalta, Esther Goutan, Rosa Blanco, Tech Eduard Tolosa, Eulalia Martí. BDNF and Full-length and Truncated TrkB Expression in Alzheimer Disease. Implications in Therapeutic Strategies. J Neuropathol Exp Neurol. 1999;58(7):729-739
33. Holsinger RMD, Schnarr J, Henry P, Castelo VT, Fahnestock M. Quantitation of BDNF mRNA in human parietal cortex by competitive reverse transcription-polymerase chain reaction: decreased levels in Alzheimer's disease. Mol Brain Res. 2000;76(2):347-354
34. Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A. et al. The BDNF val66met Polymorphism Affects Activity-Dependent Secretion of BDNF and Human Memory and Hippocampal Function. Cell. 2003;112(2):257-269
35. Chen Z-Y, Jing D, Bath KG, Ieraci A, Khan T, Siao C-J. et al. Genetic Variant BDNF (Val66Met) Polymorphism Alters Anxiety-Related Behavior. Science. 2006;314(5796):140
36. Lim YY, Villemagne VL, Laws SM, Ames D, Pietrzak RH, Ellis KA. et al. BDNF Val66Met, Aβ amyloid, and cognitive decline in preclinical Alzheimer's disease. Neurobiol Aging. 2013;34(11):2457-2464
37. Lim YY, Villemagne VL, Laws SM, Pietrzak RH, Snyder PJ, Ames D. et al. APOE and BDNF polymorphisms moderate amyloid β-related cognitive decline in preclinical Alzheimer's disease. Mol Psychiatry. 2015;20(11):1322-1328
38. Lim YY, Hassenstab J, Cruchaga C. et al. BDNF Val66Met moderates memory impairment, hippocampal function and tau in preclinical autosomal dominant Alzheimer's disease. Brain. 2016;139(10):2766-2777
39. Lim YY, Hassenstab J, Goate A, Fagan AM, Benzinger TLS, Cruchaga C. et al. Effect of BDNFVal66Met on disease markers in dominantly inherited Alzheimer's disease. Ann Neurol. 2018;84(3):424-435
40. Jack CR Jr, Petersen RC, Xu YC, O'Brien PC, Smith GE, Ivnik RJ. et al. Prediction of AD with MRI-based hippocampal volume in mild cognitive impairment. Neurology. 1999;52(7):1397-1403
41. Henneman WJP, Sluimer JD, Barnes J, van der Flier WM, Sluimer IC, Fox NC. et al. Hippocampal atrophy rates in Alzheimer disease: added value over whole brain volume measures. Neurology. 2009;72(11):999-1007
42. Dudas RB, Clague F, Thompson SA, Graham KS, Hodges JR. Episodic and semantic memory in mild cognitive impairment. Neuropsychologia. 2005;43(9):1266-1276
43. Irish M, Lawlor BA, Coen RF, O'Mara SM. Everyday episodic memory in amnestic mild cognitive impairment: a preliminary investigation. BMC Neurosci. 2011;12:80-80
44. Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM. et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer's disease. Nat Med. 2009;15:331
45. Nagahara AH, Mateling M, Kovacs I, Wang L, Eggert S, Rockenstein E. et al. Early BDNF treatment ameliorates cell loss in the entorhinal cortex of APP transgenic mice. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33(39):15596-15602
46. Jiao SS, Shen LL, Zhu C, Bu XL, Liu YH, Liu CH. et al. Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer's disease. Transl Psychiatry. 2016;6:e907
47. Arancibia S, Silhol M, Moulière F, Meffre J, Höllinger I, Maurice T. et al. Protective effect of BDNF against beta-amyloid induced neurotoxicity in vitro and in vivo in rats. Neurobiol Dis. 2008;31(3):316-326
48. Zhang L, Fang Y, Lian Y, Chen Y, Wu T, Zheng Y. et al. Brain-Derived Neurotrophic Factor Ameliorates Learning Deficits in a Rat Model of Alzheimer's Disease Induced by Aβ1-42. PLoS One. 2015;10(4):e0122415
49. Wu Y, Luo X, Liu X, Liu D, Wang X, Guo Z. et al. Intraperitoneal Administration of a Novel TAT-BDNF Peptide Ameliorates Cognitive Impairments via Modulating Multiple Pathways in Two Alzheimer's Rodent Models. Sci Rep. 2015;5:15032-15032
50. Poduslo JF, Curran GL. Permeability at the blood-brain and blood-nerve barriers of the neurotrophic factors: NGF, CNTF, NT-3, BDNF. Mol Brain Res. 1996;36(2):280-286
51. Sakane T, Pardridge WM. Carboxyl-directed Pegylation of Brain-derived Neurotrophic Factor Markedly Reduces Systemic Clearance with Minimal Loss of Biologic Activity. Pharm Res. 1997;14(8):1085-1091
52. Soderquist RG, Milligan ED, Sloane EM, Harrison JA, Douvas KK, Potter JM. et al. PEGylation of brain-derived neurotrophic factor for preserved biological activity and enhanced spinal cord distribution. J Biomed Mater Res A. 2009;91(3):719-729
53. Morse JK, Wiegand SJ, Anderson K, You Y, Cai N, Carnahan J. et al. Brain-derived neurotrophic factor (BDNF) prevents the degeneration of medial septal cholinergic neurons following fimbria transection. J Neurosci. 1993;13(10):4146
54. Croll SD, Chesnutt CR, Rudge JS, Acheson A, Ryan TE, Siuciak JA. et al. Co-infusion with a TrkB-Fc Receptor Body Carrier Enhances BDNF Distribution in the Adult Rat Brain. Exp Neurol. 1998;152(1):20-33
55. Ankeny DP, McTigue DM, Guan Z, Yan Q, Kinstler O, Stokes BT. et al. Pegylated Brain-Derived Neurotrophic Factor Shows Improved Distribution into the Spinal Cord and Stimulates Locomotor Activity and Morphological Changes after Injury. Exp Neurol. 2001;170(1):85-100
56. Anderson KD, Alderson RF, Altar CA, DiStefano PS, Corcoran TL, Lindsay RM. et al. Differential distribution of exogenous BDNF, NGF, and NT-3 in the brain corresponds to the relative abundance and distribution of high-affinity and low-affinity neurotrophin receptors. J Comp Neurol. 1995;357(2):296-317
57. Burke MA, Mobley WC, Cho J, Wiegand SJ, Lindsay RM, Mufson EJ. et al. Loss of Developing Cholinergic Basal Forebrain Neurons Following Excitotoxic Lesions of the Hippocampus: Rescue by Neurotrophins. Exp Neurol. 1994;130(2):178-195
58. Dittrich F, Ochs G, Große-Wilde A, Berweiler U, Yan Q, Miller JA. et al. Pharmacokinetics of Intrathecally Applied BDNF and Effects on Spinal Motoneurons. Exp Neurol. 1996;141(2):225-239
59. Jakeman LB, Wei P, Guan Z, Stokes BT. Brain-Derived Neurotrophic Factor Stimulates Hindlimb Stepping and Sprouting of Cholinergic Fibers after Spinal Cord Injury. Exp Neurol. 1998;154(1):170-184
60. Lu B, Pang PT, Woo NH. The yin and yang of neurotrophin action. Nat Rev Neurosci. 2005;6(8):603-614
61. Friedman WJ. Neurotrophins Induce Death of Hippocampal Neurons via the p75 Receptor. J Neurosci. 2000;20(17):6340
62. Jang SW, Liu X, Yepes M, Shepherd KR, Miller GW, Liu Y. et al. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc Natl Acad Sci U S A. 2010;107(6):2687-2692
63. Jang SW, Liu X, Chan CB, France SA, Sayeed I, Tang W. et al. Deoxygedunin, a natural product with potent neurotrophic activity in mice. PLoS One. 2010;5(7):e11528
64. Massa SM, Yang T, Xie Y, Shi J, Bilgen M, Joyce JN. et al. Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J Clin Invest. 2010;120(5):1774-1785
65. Jang S-W, Liu X, Chan C-B, Weinshenker D, Hall RA, Xiao G. et al. Amitriptyline is a TrkA and TrkB Receptor Agonist that Promotes TrkA/TrkB Heterodimerization and Has Potent Neurotrophic Activity. Chem Biol. 2009;16(6):644-656
66. Wilkie N, Wingrove PB, Bilsland JG, Young L, Harper SJ, Hefti F. et al. The non-peptidyl fungal metabolite L-783,281 activates TRK neurotrophin receptors. J Neurochem. 2001;78(5):1135-1145
67. Todd D, Gowers I, Dowler SJ, Wall MD, McAllister G, Fischer DF. et al. A monoclonal antibody TrkB receptor agonist as a potential therapeutic for Huntington's disease. PLoS One. 2014;9(2):e87923
68. Boltaev U, Meyer Y, Tolibzoda F, Jacques T, Gassaway M, Xu Q. et al. Multiplex quantitative assays indicate a need for reevaluating reported small-molecule TrkB agonists. Sci Signal. 2017;10(493):eaal1670
69. Butterfield DA, Kanski J. Methionine residue 35 is critical for the oxidative stress and neurotoxic properties of Alzheimer's amyloid beta-peptide 1-42. Peptides. 2002;23(7):1299-1309
70. Butterfield DA, Bush AI. Alzheimer's amyloid beta-peptide (1-42): involvement of methionine residue 35 in the oxidative stress and neurotoxicity properties of this peptide. Neurobiol Aging. 2004;25(5):563-568
71. Guo W, Ji Y, Wang S, Sun Y, Lu B. Neuronal activity alters BDNF-TrkB signaling kinetics and downstream functions. J Cell Sci. 2014;127(10):2249-2260
72. Perreault M, Feng G, Will S, Gareski T, Kubasiak D, Marquette K. et al. Activation of TrkB with TAM-163 Results in Opposite Effects on Body Weight in Rodents and Non-Human Primates. PLoS One. 2013;8(5):e62616
73. Coull JAM, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K. et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438(7070):1017-1021
74. Gu L, Guo Z. Alzheimer's Aβ42 and Aβ40 peptides form interlaced amyloid fibrils. J Neurochem. 2013;126(3):305-311
75. Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA. et al. Mutant presenilins specifically elevate the levels of the 42 residue β-amyloid peptide in vivo: evidence for augmentation of a 42-specific γ secretase. Hum Mol Genet. 2003;13(2):159-170
76. Garcia-Alloza M, Robbins EM, Zhang-Nunes SX, Purcell SM, Betensky RA, Raju S. et al. Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol Dis. 2006;24(3):516-524
77. Nagahara AH, Tuszynski MH. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat Rev Drug Discov. 2011;10(3):209-219
78. Zhang W, Wang P-j, Li M-h, Wang G-l, Li P, Gao X-l. 1H-MRS Assessment of the Therapeutic Effect of Bilateral Intraventricular BDNF Infusion into APP/PS1 Double Transgenic Mice. J Mol Neurosci. 2013;50(3):434-442
79. Banfield MJ, Naylor RL, Robertson AG, Allen SJ, Dawbarn D, Brady RL. Specificity in Trk receptor:neurotrophin interactions: the crystal structure of TrkB-d5 in complex with neurotrophin-4/5. Structure. 2001;9(12):1191-1199
80. Bai Y, Xu J, Brahimi F, Zhuo Y, Sarunic MV, Saragovi HU. An Agonistic TrkB mAb Causes Sustained TrkB Activation, Delays RGC Death, and Protects the Retinal Structure in Optic Nerve Axotomy and in Glaucoma. Invest Ophthalmol Vis Sci. 2010;51(9):4722-4731
81. Vugmeyster Y, Rohde C, Perreault M, Gimeno RE, Singh P. Agonistic TAM-163 antibody targeting tyrosine kinase receptor-B: applying mechanistic modeling to enable preclinical to clinical translation and guide clinical trial design. MAbs. 2013;5(3):373-383
82. Guo W, Pang K, Chen Y, Wang S, Li H, Xu Y. et al. TrkB agonistic antibodies superior to BDNF: Utility in treating motoneuron degeneration. Neurobiol Dis. 2019;132:104590
83. Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S. et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352(6286):712-716
84. Hultqvist G, Syvänen S, Fang XT, Lannfelt L, Sehlin D. Bivalent Brain Shuttle Increases Antibody Uptake by Monovalent Binding to the Transferrin Receptor. Theranostics. 2017;7(2):308-318
85. Dong X. Current Strategies for Brain Drug Delivery. Theranostics. 2018;8(6):1481-1493
86. Poduslo JF, Curran GL, Berg CT. Macromolecular permeability across the blood-nerve and blood-brain barriers. Proc Natl Acad Sci U S A. 1994;91(12):5705-5709
87. Nature Reviews. NeurologyRuiz-León Y, Pascual A. Brain-derived neurotrophic factor stimulates β-amyloid gene promoter activity by a Ras-dependent/AP-1-independent mechanism in SH-SY5Y neuroblastoma cells. J Neurochem. 2001;79(2):278-285
88. Ruiz-León Y, Pascual A. Induction of tyrosine kinase receptor b by retinoic acid allows brain-derived neurotrophic factor-induced amyloid precursor protein gene expression in human sh-sy5y neuroblastoma cells. Neuroscience. 2003;120(4):1019-1026
89. Ruiz-León Y, Pascual A. Regulation of β-amyloid precursor protein expression by brain-derived neurotrophic factor involves activation of both the Ras and phosphatidylinositide 3-kinase signalling pathways. J Neurochem. 2004;88(4):1010-1018
90. Rohe M, Synowitz M, Glass R, Paul SM, Nykjaer A, Willnow TE. Brain-Derived Neurotrophic Factor Reduces Amyloidogenic Processing through Control of SORLA Gene Expression. J Neurosci. 2009;29(49):15472
91. Wu Y, Luo X, Liu X, Liu D, Wang X, Guo Z. et al. Intraperitoneal Administration of a Novel TAT-BDNF Peptide Ameliorates Cognitive Impairments via Modulating Multiple Pathways in Two Alzheimer's Rodent Models. Sci Rep. 2015;5(1):15032
92. Hartman RE, Izumi Y, Bales KR, Paul SM, Wozniak DF, Holtzman DM. Treatment with an Amyloid-β Antibody Ameliorates Plaque Load, Learning Deficits, and Hippocampal Long-Term Potentiation in a Mouse Model of Alzheimer's Disease. J Neurosci. 2005;25(26):6213
93. Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M. et al. The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature. 2016;537(7618):50-56
94. Xiao S, Song L-L, Li J-T, Wang H, Yu N, Wang Z-Q. et al. Intraperitoneal Administration of Monoclonal Antibody Against Pathologic Aβ 42 Aggregates Alleviated Cognitive Deficits and Synaptic Lesions in APP/PS1 Mice. J Alzheimer's Dis. 2020;73:657-670
95. Cuadrado-Tejedor M, Hervias I, Ricobaraza A, Puerta E, Pérez-Roldán JM, García-Barroso C. et al. Sildenafil restores cognitive function without affecting β-amyloid burden in a mouse model of Alzheimer's disease. Br J Pharmacol. 2011;164(8):2029-2041
96. Gerenu G, Martisova E, Ferrero H, Carracedo M, Rantamäki T, Ramirez MJ. et al. Modulation of BDNF cleavage by plasminogen-activator inhibitor-1 contributes to Alzheimer's neuropathology and cognitive deficits. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2017;1863(4):991-1001
97. Merkouris S, Barde Y-A, Binley KE, Allen ND, Stepanov AV, Wu NC. et al. Fully human agonist antibodies to TrkB using autocrine cell-based selection from a combinatorial antibody library. Proc Natl Acad Sci U S A. 2018;115(30):E7023
98. Caccamo A, Branca C, Piras IS, Ferreira E, Huentelman MJ, Liang WS. et al. Necroptosis activation in Alzheimer's disease. Nat Neurosci. 2017;20:1236
99. Milnerwood AJ, Raymond LA. Early synaptic pathophysiology in neurodegeneration: insights from Huntington's disease. Trends Neurosci. 2010;33(11):513-523
100. Picconi B, Piccoli G, Calabresi P. Synaptic dysfunction in Parkinson's disease. Adv Exp Med Biol. 2012;970:553-572
101. Ji Y, Pang PT, Feng L, Lu B. Cyclic AMP controls BDNF-induced TrkB phosphorylation and dendritic spine formation in mature hippocampal neurons. Nat Neurosci. 2005;8(2):164-172
102. Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA. et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13(2):159-170
103. Han F, Guan X, Guo W, Lu B. Therapeutic potential of a TrkB agonistic antibody for ischemic brain injury. Neurobiol Dis. 2019;127:570-581
104. Targowska-Duda KM, Ozawa A, Bertels Z, Cippitelli A, Marcus JL, Mielke-Maday HK. et al. NOP receptor agonist attenuates nitroglycerin-induced migraine-like symptoms in mice. Neuropharmacology. 2020;170:108029
105. Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1(2):848-858
Author contact
![]() Corresponding authors: Dr. Bai Lu. School of Pharmaceutical Sciences, Room B303, Tsinghua University, Beijing 100084, China, Tel: +86 62785101, E-mail: bai_luedu.cn; Dr. Wei Guo. School of Pharmaceutical Sciences, Room B302, Tsinghua University, Beijing, China, 100084 Tel: +86 62787837, E-mail: wguoedu.cn.
Corresponding authors: Dr. Bai Lu. School of Pharmaceutical Sciences, Room B303, Tsinghua University, Beijing 100084, China, Tel: +86 62785101, E-mail: bai_luedu.cn; Dr. Wei Guo. School of Pharmaceutical Sciences, Room B302, Tsinghua University, Beijing, China, 100084 Tel: +86 62787837, E-mail: wguoedu.cn.