Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(15):6638-6660. doi:10.7150/thno.44793 This issue Cite
Research Paper
Genetic and pharmacological activation of Hedgehog signaling inhibits osteoclastogenesis and attenuates titanium particle-induced osteolysis partly through suppressing the JNK/c-Fos-NFATc1 cascade
Liwei Zhang1,2#, Yanjun Yang1,2#, Zirui Liao2, Qingbai Liu1, Xinhuan Lei3, Meng Li1,2, Saijilafu1,2, Zunyi Zhang4, Dun Hong3, Min Zhu3, Bin Li1,2, Huilin Yang1,2, Jianquan Chen1,2 ![]()
1. Department of Orthopaedics, the First Affiliated Hospital of Soochow University, Suzhou, Jiangsu 215006, China
2. Orthopedic Institute, Medical College, Soochow University, Suzhou, Jiangsu 215007, China
3. Orthopedic Department, Taizhou Hospital Affiliated to Wenzhou Medical University, Linhai, Zhejiang 317000, China
4. Institute of Life Sciences, College of Life and Environmental Science, Key Laboratory of Mammalian Organogenesis and Regeneration, Hangzhou Normal University, Hangzhou, Zhejiang 310036, China
#These authors contributed equally to this study.
Received 2020-2-11; Accepted 2020-5-7; Published 2020-5-17
Abstract

Rationale: Wear particle-induced periprosthetic osteolysis (PPO) is a common long-term complication of total joint arthroplasty, and represents the major cause of aseptic loosening and subsequent implant failure. Previous studies have identified the central role of osteoclast-mediated bone resorption in the pathogenesis of PPO. Thus, therapeutic approaches of inhibiting osteoclast formation and activity are considered to be of great potential to prevent and treat this osteolytic disease. Hedgehog (Hh) signaling has been shown to play an important role in promoting osteoblast differentiation and bone formation. While Hh signaling is also implicated in regulating osteoclastogenesis, whether it can directly inhibit osteoclast differentiation and bone resorption remains controversial. Moreover, its potential therapeutic effects on PPO have never been assessed. In this study, we explored the cell-autonomous role of Hh signaling in regulating osteoclastogenesis and its therapeutic potential in preventing wear particle-induced osteolysis.
Methods: Hh signaling was activated in macrophages by genetically ablating Sufu in these cells using LysM-Cre or by treating them with purmorphamine (PM), a pharmacological activator of Smoothened (Smo). In vitro cell-autonomous effects of Hh pathway activation on RANKL-induced osteoclast differentiation and activity were evaluated by TRAP staining, phalloidin staining, qPCR analyses, and bone resorption assays. In vivo evaluation of its therapeutic efficacy against PPO was performed in a murine calvarial model of titanium particle-induced osteolysis by μCT and histological analyses. Mechanistic details were explored in RANKL-treated macrophages through Western blot analyses.
Results: We found that Sufu deletion or PM treatment potently activated Hh signaling in macrophages, and strongly inhibited RANKL-induced TRAP+ osteoclast production, F-actin ring formation, osteoclast-specific gene expression, and osteoclast activity in vitro. Furthermore, we found that Sufu deletion or PM administration significantly attenuated titanium particle-induced osteoclast formation and bone loss in vivo. Our mechanistic study revealed that activation of Hh signaling suppressed RANKL-induced activation of JNK pathway and downregulated protein levels of two key osteoclastic transcriptional factors, c-Fos and its downstream target NFATc1.
Conclusions: Both genetic and pharmacological activation of Hh signaling can cell-autonomously inhibit RANKL-induced osteoclast differentiation and activity in vitro and protect against titanium particle-induced osteolysis in vivo. Mechanistically, Hh signaling hinders osteoclastogenesis partly through suppressing the JNK/c-Fos-NFATc1 cascade. Thus, Hh signaling may serve as a promising therapeutic target for the prevention and treatment of PPO and other osteolytic diseases.
Keywords: peri-prosthetic osteolysis, titanium particle, Hh signaling, osteoclatogenesis
Introduction
Total joint arthroplasty (TJA) has been performed widely to treat arthralgia, joint instability and deformity caused by severe trauma, osteoarthritis, rheumatoid arthritis and osteoporotic fracture. It was estimated that more than one million cases of TJA were performed every year in the U.S.A., and this number was expected to reach two million by the year 2030 [1, 2]. Although TJA is considered to be one of the most successful operations, aseptic loosening resulting from periprosthetic osteolysis (PPO) is still a common and serious complication, which often leads to implant failure and revision surgery [3-5]. While the precise mechanism underlying PPO remains elusive, increasing evidence has pointed to wear particles released from implant surfaces as a major causative factor [6]. Wear particles stimulate several different types of cells such as macrophages and T lymphocytes to secrete a variety of proinflammatory chemokines and cytokines [7, 8]. These molecules in turn recruit osteoclast precursors, and at the same time act on other target cells, mainly osteoblasts and marrow stromal cells, to promote their expression of receptor activator of nuclear factor-κB ligand (RANKL), a critical regulator of osteoclastogenesis. Upon binding to its receptor RANK on the surface of osteoclasts and their precursors, RANKL triggers activation of multiple intracellular signaling pathways to promote osteoclast differentiation and activity whereas suppressing its apoptosis. As a result, osteoclasts are excessively formed and activated at the peri-implant sites, eventually leading to extensive bone destruction [7, 8]. Given the central role of osteoclast-mediated bone resorption in the pathogenesis of PPO, the therapeutic approach inhibiting osteoclast formation and activity is considered to be of great potential to prevent and treat this osteolytic disease. However, clinical studies testing the efficacy of several anti-resorption agents, including bisphosphonate and denosumab, only yielded limited success [9-11]. Thus, an alternative agent or approach that can effectively prevent and treat PPO-triggered aseptic loosening is still urgently needed.
In recent years, the therapeutic approaches simultaneously inhibiting bone destruction and promoting bone regeneration have received increasing attention for prevention and treatment of osteolytic diseases [12, 13]. Hh pathway, an evolutionarily conserved signaling pathway, plays critical roles in skeletal development and homeostasis, and is regarded as a promising anabolic pathway for treating osteoporosis and promoting bone regeneration [14, 15]. This pathway is triggered by binding Hh ligands to their receptor Patched1 (Ptch1), thus relieving its suppression on Smoothened (Smo), a seven-pass transmembrane protein. Active Smo accumulates in the primary cilia, where it reverses the inhibitory effect of the Sufu-containing suppressive complex on formation of the activator form of Gli family transcriptional factors (GliA). Consequently, GliA is produced and subsequently enters the nucleus to activate transcription of Hh target genes, including Gli1 and Ptch1, two Hh pathway components that are commonly used as readouts of Hh signaling activity [14, 16]. Another way to activate Hh signaling is to genetically remove either Ptch1 or Sufu [17, 18]. In addition, several pharmacological agents, including purmorphamine, can also directly act on Smo protein to activate signaling independently of Hh molecules [19, 20].
While Hh signaling has been firmly established as an important positive regulator of osteoblast differentiation and bone formation [14, 15, 21], its role in inhibiting osteoclastogenesis and bone resorption still needs to be determined, which has greatly limited its potential application as a therapeutic target with the dual effects of promoting bone formation and suppressing bone resorption in treating osteolytic diseases. In fact, genetic activation of Hh signaling by either Ptch1 haploinsufficiency or conditional deletion of Ptch1 in mature osteoblasts was shown to indirectly stimulate osteoclastogenesis through upregulating RANKL expression in osteoblasts both in vitro and in vivo [22-24]. Similarly, some types of tumor cells were reported to secrete Hh ligands to activate Hh signaling in osteoblasts to stimulate their production of RANKL, which aberrantly increased the number of osteoclasts to cause metastatic osteolysis [25, 26]. The above in vitro and in vivo studies have clearly demonstrated that Hh signaling can promote osteoclastogenesis via a non-cell-autonomous mechanism under physiological and certain pathological conditions. In contrast, the cell-autonomous role of Hh signaling in osteoclastogenesis remains unclear. While a few in vitro studies did explore the direct effect of Hh pathway activation on osteoclast differentiation, these studies reported inconsistent results [27-32], none of which was validated in vivo. Interestingly, a recent genetic study revealed that conditional ablation of Ihh in limb mesenchymal cells, which potentially disrupted Hh signaling in both osteoblastic and osteoclastic lineage cells, led to increased osteoclast formation [17]. Although the exact mechanism underlying this inhibitory effect of Ihh signaling on osteoclastogenesis remains to be elucidated, this study raised the possibility that Hh signaling can cell-autonomously inhibit RANKL-induced osteoclast differentiation despite its stimulatory effect on RANKL expression in osteoblasts. However, direct evidence to support such a suppressive role of Hh activation in osteoclastogenesis is still lacking. Given this uncertainty, it remains to be determined whether activation of Hh signaling can be utilized to prevent osteolytic diseases, such as wear particle-induced osteolysis.
In this study, we explored the cell-autonomous role of Hh signaling in regulating osteoclastogenesis and its therapeutic potential in preventing wear particle-induced osteolysis using both genetic and pharmacological approaches. Our results demonstrated that activation of Hh signaling either by conditionally deleting Sufu (a key negative regulator of Hh signaling) in osteoclast precursors or by treatment with purmorphamine (a pharmacological activator of Smo protein) inhibited RANKL-stimulated osteoclast differentiation in vitro and attenuated Ti particle-induced osteoclastogenesis and bone loss in vivo. Mechanistically, we found that Hh signaling directly inhibited osteoclast differentiation through suppressing the JNK/c-Fos-NFATc1 cascade. Thus, our study revealed a previously unrecognized role of Hh signaling in inhibiting osteoclastogenesis and provided the first preclinical evidence to support activating Hh pathway as a potential therapeutic approach to preventing and treating PPO and aseptic loosening.
Materials and Methods
Antibodies and other reagents
Rabbit primary antibodies against p-p65 (#3033, 1:1000), AKT (#9272, 1:1000), p-AKT (#9271, 1:1000), JNK (#9252, 1:1000), p-JNK (#4668, 1:1000), p38 (#8690, 1:1000), ERK (#4695, 1:1000), p-ERK (#4370, 1:1000), and GAPDH (#5174, 1:2000) were obtained from Cell Signaling Technology (Cambridge, MA, USA). Rabbit primary antibodies for p-p38 (#ab4822, 1:1000), NFATc1 (#ab25916, 1:1000) and c-fos (#ab190289, 1:1000) were purchased from Abcam (San Francisco, CA, USA). Rabbit primary antibodies against total p65 (#10745-1-AP,1:1000), Sufu (#26759-1-AP,1:1000), and PCNA (#10205-2-AP,1:1000) were purchased from proteintech (Wuhan, Hubei, China). HRP-conjugated anti-rabbit secondary antibody (#7074, 1:2000) was provided by Cell Signaling Technology (Cambridge, MA, USA). Recombinant mouse macrophage-colony stimulating factor (M-CSF) and receptor activator for nuclear factor-κB ligand (RANKL) were purchased from R&D Systems (Minneapolis, MN, USA), and used at 30 ng/ml and 50 ng/ml, respectively. Purmorphamine (PM) was purchased from Selleck Chemicals (Houston, TX, USA) and dissolved in DMSO with a concentration of 40 mM. FITC-phalloidin and DAPI were purchased from MesGen Biotech (Shanghai, China), and used at 1:100 and 1:2000 dilutions, respectively. The α-minimum essential medium (α-MEM), fetal bovine serum (FBS), penicillin/streptomycin (P/S), and 2.5% trypsin were obtained from Gibco-BRL (Sydney, Australia). RIPA buffer was provided by Beyotime Biotechnology (Shanghai, China). cOmplete™ protease and PhosSTOP™ phosphatase inhibitors were purchased from Roche (Indianapolis, IN, USA). Cell Counting Kit-8 (CCK-8) was supplied by Dojindo laboratories (Mashiki-machi, Kumamoto, Japan).
Mouse strains
Mice carrying Sufu conditional allele (Sufuflox) were generated by flanking Exon 7 with two repeated LoxP sites as described previously [18]. LysM-Cre, a knockin mouse line expressing Cre recombinase under the control of the endogenous M lysozyme locus, was previously reported [33] and obtained from the Jackson Laboratories (Bar Harbor, ME, USA). To conditionally delete Sufu in monocyte/macrophage lineage cells, LysM-Cre+/+; Sufuflox/+ mice were intercrossed to obtain progenies carrying two copies of both LysM-Cre and Sufuflox alleles (LysM-Cre+/+; Sufuflox/flox, hereafter referred to as SufuCKO). Their gender-matched littermates with LysM-Cre+/+ genotype were used as controls. For experiments involving only wildtype mice, 8 to 10-week-old male C57BL/6 mice were used. All mice were raised in the specific pathogen-free (SPF) animal facility at Laboratory Animal Center of Soochow University. All experimental protocols involving the use of animals were approved by the Ethics Committee of the First Affiliated Hospital of Soochow University (#201810A044).
Preparation and culture of mouse bone marrow-derived macrophages (BMMs)
To prepare mouse primary BMMs, total bone marrow cells were isolated from the femur and tibia of 8 to 10-week-old mice as previously described [34, 35]. Isolated bone marrow cells were then plated in 10 cm cell culture dishes, and cultured in BMM maintenance medium (α-MEM containing 10% FBS, 1% P/S, and 30 ng/ml recombinant mouse M-CSF) for 24 h. Following complete aspiration of old media, cultures were briefly rinsed with DPBS to remove non-adherent cells. The remaining attached BMMs were further expanded in BMM maintenance medium for additional 2-3 days until they reached nearly 100% confluence. Confluent BMMs were subsequently trypsinized and re-seeded in cell culture plates at a density of 2×104 cells/cm2 unless otherwise indicated. All cell cultures were maintained in a 37 °C humidified incubator with 5% CO2 (v/v), and medium was changed every other day.
In vitro osteoclast differentiation assays
For in vitro osteoclast differentiation, BMMs were seeded in a 24-well plate (for TRAP or F-actin ring staining) or 6-well plate (for protein or RNA analysis) at the density of 2×104 cells/cm2, and incubated in BMM maintenance medium for 18 h. Osteoclastic differentiation of BMMs was then induced with differentiation medium (BMM maintenance medium supplemented with 50 ng/ml recombinant mouse RANKL) for 5-6 days with the medium changed every 2 days. In experiments involving PM treatment, vehicle or different concentrations of PM (0.5, 1, or 2 μM) was included in the differentiation medium. At the end of osteoclastic induction, status of osteoclast differentiation was evaluated by TRAP staining, F-actin ring immunofluorescence, qPCR, or Western blot analyses as indicated.
To assess the effect of PM on protein expression of key osteoclastic transcriptional factors during the course of osteoclastogenesis, BMMs were seeded and cultured as described above. Afterwards, BMMs were stimulated with 50 ng/ml RANKL in the presence of vehicle or 2 μM PM for 0,1 or 3 days. At each time point, cells were harvested for protein extraction.
To determine the effect of PM on RANKL-mediated activation of intracellular signaling pathways, a higher number (4×105 cells/well) of BMMs were planted in 6-well plates, and cultured in BMM maintenance medium for 18 h. BMMs were then pretreated with vehicle (DMSO) or 2 μM PM for 4 h, followed by stimulation with 50 ng/ml RANKL for 0, 5, 15, 30 or 60 min. At each time point, cells were harvested for isolation of total RNA, total protein or cytoplasmic/nuclear protein.
Cell viability assay
The effect of PM on viability of BMMs was determined using Cell Counting Kit-8 (CCK-8) (Dojindo Laboratories, Japan) according to the manufacturer's instruction. Briefly, BMMs were plated into 96-well plates at a density of 8×103 cells/well in triplicate and cultured in BMM maintenance medium for 18 h. Afterwards, the cells were treated with vehicle (DMSO) or different concentrations of PM (0.0625, 0.125, 0.25, 0.5, 1, 2, 4, 8, 16 and 32 μM) for 48, 72 and 96 h. At each of these time points, 10 μl CCK-8 solution was added to each well, and the plates were then incubated for 1 h at 37 °C. The optical density at 450 nm (OD450) was measured by an ELX800 absorbance microplate reader (Bio-Tek, Winooski, USA). The OD450 values were plotted against the corresponding PM concentrations using GraphPad Prism.
TRAP staining
TRAP staining of cultured cells was performed as we previously described [36]. Briefly, cells were washed once with phosphate-buffered saline (PBS), and then fixed with 4% paraformaldehyde (PFA) for 10 min. After being rinsed twice with PBS, cells were incubated with tartrate buffer at 37 °C for 30 min. Subsequently, cells were stained with TRAP staining solution (tartrate buffer containing naphthol AS-BI phosphate and pararosaniline chloride) at 37 °C for 1 h in darkness. Images of stained cells were then acquired using Axiovert 40 CFL microscope (Carl Zeiss Microscopy, Thornwood, NY). TRAP-positive cells with more than three nuclei were considered as mature osteoclasts. The number and relative size of mature osteoclasts in each well were quantified by Image J software (v.1.51, National Institutes of Health, USA). A minimum of 3 wells were analyzed for each group.
Quantitative PCR (qPCR)
Total RNA was isolated from whole cells with Total RNA Extractor (Sangon Biotech, Shanghai, China) in accordance with the instructions provided in the manual. 500 ng of total RNA was reverse-transcribed into cDNA using HiScript II Q RT SuperMix (Vazyme, Nanjing, China). PCR reaction was set up in triplicates in a 20 µl volume comprised of 10 µl SYBR Green qPCR Mix (Novoprotein, Shanghai, China), 7.5 µl ddH2O, 2 µl cDNA, and 0.5 µl of primer pairs. Real time PCR was performed on the CFX Connect™ Real-Time PCR Detection System (Bio-Rad Laboratories, CA, USA), with the following cycling conditions: 40 cycles of denaturation at 95 °C for 10 s and amplification at 60 °C for 30 s, followed by melting curve analysis from 65 °C to 95 °C at 0.5 °C/step. Expression of each target gene was normalized by 18S ribosomal RNA. The relative changes in mRNA level were analyzed by 2-ΔΔCT method as previously reported [37]. The nucleotide sequences for the primers used in this study are provided in Table S1.
Western Blot analyses
Total protein was extracted from cells using 1×RIPA buffer supplemented with protease and phosphatase inhibitors as we previously described [35]. Cytoplasmic and nuclear protein extracts were isolated using a Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime Biotechnology, Shanghai, China) according to the manufacturer's instructions. Concentrations of protein samples were measured using a BCA Protein Assay Kit (Beyotime Biotechnology, Shanghai, China) as per instructions. Thirty-microgram (30 μg) protein lysates were separated on 10% SDS-PAGE gels by electrophoresis, and subsequently transferred onto 0.2 μm NC membranes (GE Healthcare, Freiburg, Germany) by electroblotting. After being blocked with 5% nonfat milk in 1×TBST for 1 h, the blots were incubated with primary antibodies at 4 °C overnight. The next day, the membranes were incubated with HRP-conjugated secondary antibodies at RT for 1 h. Afterwards, the immunoreactivity was revealed using the chemiluminescence reagent (Millipore, Billerica, MA) and then imaged with ChemiDoc™ Touch Imaging System (Bio-Rad Laboratories, CA, USA). Quantification of the band intensity was performed using Image J software.
Staining of F-actin rings
F-actin ring formation was visualized by phalloidin staining as previously described with minor modifications [38]. In brief, BMMs were washed with PBS prior to fixation with 4% paraformaldehyde for 10 min at RT. The fixed cells were then permeabilized with 0.1% Triton X-100 for 5 min, followed by incubation with 3% BSA in PBS for 20 min. Subsequently, cells were stained with FITC-conjugated phalloidin (1:100) at RT in darkness for 20 min. After washing, cells were stained with DAPI (1:2000) for 5 min, rinsed with PBS, and then mounted with anti-fade mounting medium (Beyotime Biotechnology, Shanghai, China). Fluorescent staining was observed and photographed using the Axiovert 40 CFL fluorescent microscope installed with Zeiss ZEN software (Carl Zeiss Microscopy, Thornwood, NY). The number and relative size of F-actin rings in each group were measured using Image J software (National Institutes of Health, MD, USA). A minimum of 3 wells per group were analyzed.
Bone resorption assay
The effect of PM on bone-resorbing activity of osteoclasts was evaluated using bovine bone slices as previously reported [39]. In brief, bone marrow cells were seeded in 6-well plates at a density of 2×104 cells/cm2 and stimulated with 30 ng/ml M-CSF and 50 ng/ml RANKL for 3 days. After trypsinization, differentiating osteoclasts were re-seeded onto bovine bone slices placed in individual wells of a 96-well plate at a density of 8×103 cells/well, and further cultured in osteoclast differentiation medium containing 0, 0.5, 1 or 2 μM PM until they became fully differentiated. Subsequently, bone slices were treated with 5% sodium hypochlorite for 5 min to eliminate any attached cell, and then washed twice with ddH2O. After that, resorption pits on bone slices were visualized and imaged by a FEI Quanta 250 scanning electron microscope (Hillsboro, OR, USA). The number and relative resorption area were analyzed using the Image J software (National Institutes of Health, MD, USA).
Characterizations of Ti particles
Pure titanium (Ti) particles (>93% in purity and < 20 μm in diameter) were supplied by Alfa Aesar (#000681, Heysham, UK). Characterization of their morphology and size has been previously reported [40, 41]. The sizes of these particles were in the range of 0.9-5.7 μm and their average size was 3.3 μm [40, 41]. To remove endotoxins, the particles were baked at 180 °C for 6 h, followed by immersing in 75% ethanol for 48 h as previously described [42]. After being washed four times with sterile ultra-pure water, Ti particles were re-suspended in sterile PBS at a concentration of 1 g/ml for further use. Morphology of Ti particles before and after being baked for 6 h at 180 °C was observed and imaged by a scanning electron microscope (FEI Quanta 250, Hillsboro, OR, USA). The effect of the baking treatment on the phase composition of Ti particles was analyzed by X-ray diffraction (XRD) using a SmartLab 9kW X-ray diffractometer (Rigaku, Tokyo, Japan) with CuKa radiation (40Kv, 150mA) in a 2θ range of 5-90° at a step size of 0.02° and a scanning speed of 6°/min.
Ti particle-induced murine calvarial osteolysis model
Two independent sets of experiments were performed to determine the potential effect of activation of Hh signaling on Ti particle-induced osteolysis. In the first experiment, 10-week-old male LysM-Cre+/+ (Control) mice or LysM-Cre+/+; Sufuflox/flox (SufuCKO) mice were assigned to the following groups (n=5 mice per group): control mice with sham operation (Sham-Ctrl group), SufuCKO mice with sham operation (Sham-SufuCKO group), control mice with Ti implantation (Ti-Ctrl group), and SufuCKO mice with Ti implantation (Ti-SufuCKO group). In the second experiment, twenty-eight C57BL/6 mice (male, 10-week-old) were randomly separated into 4 groups (n= 7 mice per group): sham operation+vehicle treatment group (Sham group), Ti implantation+vehicle treatment group (vehicle group), Ti implantation+ 2.5 mg/kg PM treatment group (low-PM group), and Ti implantation+ 10 mg/kg PM treatment group (high-PM group). In both sets of experiments, Ti particle-induced calvarial osteolysis model was established in all Ti-implantation groups. Briefly, mice were anesthetized by intraperitoneal injection of ketamine and xylazine following the standard protocol. Following anesthesia, skin on the calvariae was shaved, disinfected and then incised with a sharp scalpel along the middle line. Subsequently, 30 mg of Ti particles (30 μl) were evenly spread on the surfaces of the bilateral parietal bones, followed by closure of the surgical skin incision. In all sham-operated groups, mice were subjected to the same above surgical procedures except that equal volume of pure PBS (30 μl) was used to replace Ti particle suspension. For pharmacological activation of Hh signaling in mice, PM was diluted in a vehicle solution consisting of 2% DMSO, 30% PEG 300 and 5% Tween 80, and subcutaneously injected to mice in low- and high-PM groups in the center of the calvariae at a dosage of 2.5 mg/kg and 10 mg/kg, respectively. The PM dosage of 10 mg/kg used in this study was converted from a dose of 5 mg/kg previously used in rats based on relative body surface area of rats and mice [43, 44]. As control, the same amount of vehicle solution was given to mice in sham and vehicle groups. Both vehicle and PM were administered once daily for consecutive 14 days. Two weeks after the surgery, all mice were sacrificed, and their calvariae were isolated and fixed with 4% PFA for 2 days. After fixation, calvarial samples were washed three times with PBS and then stored in 75% ethanol at 4 °C until further analysis.
Micro-computed Tomography (μCT) analyses
For μCT analysis, implanted-Ti particles were carefully removed from each calvaria to avoid metal artifacts. After that, the specimens were scanned with a SkyScan 1176 scanner (Bruker, Aartselaar, Belgium) at an isometric resolution of 9 μm with X-ray energy source set at 50 kV and 500 μA. Raw data obtained from μCT scanning were reconstructed with NRecon software and then reoriented with Dataviewer software (Bruker, Aartselaar, Belgium) to generate two-dimensional coronal images of μCT slices. For quantitative analysis, 100 μCT slices in the center of each calvaria were used, and region of interest (ROI) was selected as previously described [13]. CTAn software (SkyScan, Aartselaar, Belgium) was utilized to generate three-dimensional (3D) images and calculate the percentage of bone volume out of total tissue volume (BV/TV). 3D μCT images were visualized using Mimics v10.01 software (Materialise, Leuven, Belgium). The number of pores and percentage of porosity were counted by Image J software.
Histological and histomorphometric analyses
Following μCT scanning, calvariae were decalcified in 14% EDTA for 14 days. The EDTA solution was changed daily. After decalcification, calvarial samples were dehydrated in increasing concentrations of ethanol, cleared in xylene, and embedded in paraffin prior to being sectioned coronally at 6 μm with a Leica microtome (Leica Biosystems, Buffalo Grove, IL, USA). For assessing the degree of bone erosion and the number of osteoclasts, coronal sections at the middle level were subjected to standard H&E and TRAP staining, respectively. Images of stained sections were captured using the Axiovert 40 CFL microscope with Zeiss ZEN software (Carl Zeiss Microscopy, Thornwood, NY). The regions containing the discontinuous and non-osseous tissues were considered as the osteolytic areas, and selected for histomorphometric analyses. The proportion of eroded surface as well as the number of TRAP-positive multinucleated osteoclasts in the selected regions of each group were quantified by Image J software.
Statistical analyses
Data were presented as average ± standard deviation (S.D.) from at least three independent samples. SPSS 19.0 software (SPSS Inc, Chicago, IL, USA) was used for statistical analyses. Statistical differences between two groups were determined by two-tailed Student's t-test. A p value less than 0.05 was considered to be statistically significant.
Results
LysM-Cre-mediated ablation of Sufu activated Hh signaling in macrophages
Sufu is a key intracellular suppressor of Hh signaling, and its inactivation was known to result in constitutively activated Hh signaling [45, 46]. To explore the potential role of Sufu in regulating osteoclast differentiation, we first performed qPCR to examine the mRNA expression of Sufu gene during RANKL-induced osteoclastogenesis. The results showed that Sufu was abundantly expressed in BMMs and its mRNA level was modestly but statistically significantly increased with the progression of osteoclast differentiation (Figure 1A), implying that Hh signaling needed to be repressed by Sufu for osteoclast differentiation to proceed. To genetically assess the direct role of Hh activation in osteoclastogenesis, we next crossed mice carrying a conditional allele of Sufu (Sufuflox) to LysM-Cre mice to generate LysM-Cre+/+; Sufuflox/flox (hereafter SufuCKO) mice (Figure 1B-D) in which Sufu will be specifically deleted in monocyte/macrophage lineage cells, including osteoclasts and their precursors (Figure 1E). qPCR analysis of RNA directly isolated from macrophage-abundant spleens showed significantly reduced expression of Sufu gene in SufuCKO mice compared with their littermate controls (Figure 1F). To further verify the efficiency of LysM-Cre-mediated deletion of Sufu gene in macrophages, we isolated bone marrow macrophages (BMMs) from SufuCKO and Ctrl (LysM-Cre+/+) mice, and cultivated them in complete culture medium containing 30 ng/ml M-CSF for 2 days prior to RNA isolation. qPCR experiment showed that the mRNA level of Sufu in BMM isolated from SufuCKO mice was reduced by 82% when compared with that of control mice (Figure 1G). In contrast, the mRNA expression of Gli1 and Ptch1, two well-established target genes of Hh signaling pathway, was elevated to 840.3 ± 106.9% and 175.3 ± 13.9%, respectively (Figure 1H, I). Taken together, our data demonstrated that Sufu gene was efficiently deleted in the macrophages of SufuCKO mice, and that Sufu ablation in these cells resulted in activation of Hh signaling pathway.
LysM-Cre-mediated ablation of Sufu activated Hh signaling in macrophages. (A) qPCR analyses of relative mRNA levels of Nfatc1, c-Fos, Oscar, and Sufu in BMMs at 0, 1, 3, and 5 days after RANKL stimulation. Expression of each target gene was normalized by 18S ribosomal RNA. The relative changes in mRNA level were analyzed by 2-ΔΔCT method. All values were calculated from three independent biological replicates and presented as mean ± SD. *P<0.05, **P<0.01, for comparison between two indicated groups. (B) Schematic diagram of Sufu conditional allele (Sufuflox) which was created by flanking Exon 7 of the mouse Sufu gene with two parallel LoxP sites by gene targeting. (C) Schematic diagram of LysM-Cre knockin allele, in which Cre recombinase was under the control of the endogenous M lysozyme locus. (D) Schematic illustration of the mating strategy to obtain SufuCKO (LysM-Cre+/+; Sufuflox/flox) mice and their littermate control mice (LysM-Cre+/+). (E) Schematic illustration of the strategy to disrupt Sufu gene in osteoclast precursors. (F) qPCR analyses of Sufu mRNA expression in macrophage-abundant spleens in SufuCKO mice relative to their littermate controls (Ctrl). (G-I) qPCR analyses of mRNA expression of Sufu (G), Gli1 (H), and Ptch1 (I) in BMMs from SufuCKO mice relative to their littermate controls (Ctrl). n=3 per group. All bar graphs were presented as mean ± SD, **P<0.01, compared with control group.
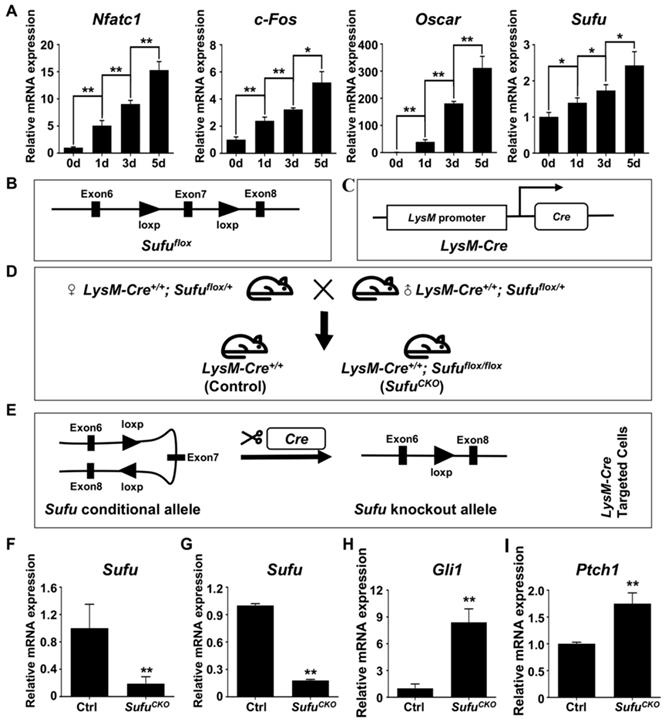
Genetic activation of Hh signaling by Sufu ablation inhibited RANKL-induced osteoclastogenesis and F-actin ring formation
After confirming activation of Hh signaling following Sufu ablation, we next investigated the effects of Sufu ablation on osteoclast formation. To this end, BMMs were isolated from SufuCKO and control mice, and subjected to osteoclastic induction in the presence of 50 ng/ml RANKL. As expected, abundant TRAP-positive multinucleated cells, indicative of mature osteoclasts, appeared in the control cultures after 6 days of RANKL treatment (Figure 2A). In contrast, formation of TRAP-positive osteoclasts was severely inhibited in the RANKL-treated SufuCKO cultures (Figure 2A). Quantitative analyses showed that an average of 259 ± 15.4/well TRAP-positive osteoclasts was formed in the control cultures, whereas this number dropped to 72.3 ± 7.1/well in the SufuCKO cultures (Figure 2B). Similarly, the size of TRAP-positive osteoclasts was significantly decreased in the control cultures compared with that in the SufuCKO cultures (Figure 2C). These results clearly indicated that activation of Hh signaling by Sufu ablation directly suppressed osteoclastogenesis in vitro.
Genetic activation of Hh signaling by Sufu ablation inhibited RANKL-induced osteoclastogenesis and F-actin ring formation in vitro. (A) BMMs isolated from control and SufuCKO mice were stimulated with 30 ng/ml M-CSF and 50 ng/ml RANKL for 6 days and then subjected to TRAP staining. Images shown were representative results from three independent experiments. Scale bar, 400 μm. (B) Quantification of number of TRAP+ multinucleated osteoclasts/well. (C) Quantification of relative size of TRAP+ multinucleated osteoclasts. Values were normalized to average size of TRAP+ osteoclasts in control group. (D) Representative images of F-actin rings. After stimulation with M-CSF and RANKL for 6 days, control and SufuCKO BMM cultures were fixed with 4% PFA and then sequentially stained with FITC-phalloidin and DAPI. Green, F-actin; Blue, nuclei. Scale bar, 200 μm. (E-G) Quantitative analyses of F-actin rings showing number of F-actin rings per well (E), relative size of F-actin rings (F), and number of nuclei per osteoclast (G). All values were calculated from three independent replicates, and presented as mean ± SD. ** indicated P<0.01, relative to control group.
F-actin ring is a characteristic feature of mature osteoclasts, and required for their bone-resorbing activity [7, 38]. To further validate that Sufu ablation impaired osteoclastogenesis, we analyzed F-actin ring formation in RANKL-treated control and SufuCKO BMM cultures. Consistent with results from TRAP staining, phalloidin staining revealed that SufuCKO BMMs formed significantly fewer number of F-actin rings than control BMMs after RANKL treatment (Figure 2D, E). Moreover, the average size of F-actin rings in multinucleated osteoclasts also significantly decreased by 93.3% in the SufuCKO group when compared with that of the control group, and number of nuclei per osteoclast dropped from 45.3 ± 5.1 to 6.7 ± 1.1 (Figure 2F, G). Collectively, our data demonstrated that Sufu ablation inhibited RANKL-induced F-actin ring formation in BMMs, and therefore provided additional evidence to support the role of Hh activation in suppressing osteoclastogenesis.
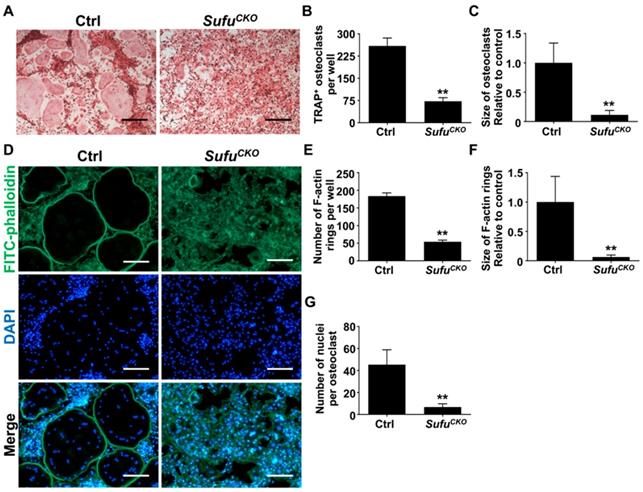
Genetic activation of Hh signaling by Sufu ablation suppressed expression of osteoclast-specific genes in vitro
The expression of a group of osteoclast-associated genes, including Nfatc1, c-Fos, Acp5, Ctsk, Oscar, Dcstamp, Atp6v0a3 and Atp6v0d2, was upregulated during osteoclast differentiation [39, 47, 48]. To further verify the inhibitory effect of Sufu ablation on osteoclast differentiation, we utilized quantitative RT-PCR to assess mRNA expression of these osteoclast differentiation markers in control and SufuCKO BMMs that were induced toward osteoclasts with 6 days of RANKL stimulation. As shown in Figure 3A&B, mRNA levels of Nfatc1 and c-Fos, two key transcriptional regulators of osteoclast differentiation, significantly decreased to 29.3 ± 1.2% and 22.7 ± 1.2%, respectively, in the RANKL-stimulated SufuCKO BMMs relative to control cells. Similarly, Sufu ablation dramatically suppressed expression of Acp5, Oscar, Ctsk, Dcstamp, Atp6v0a3 and Atp6v0d2 (Figure 3C-H), osteoclast marker genes that are known to be involved in various stages of osteoclast differentiation or their bone-resorbing activity. Collectively, these results demonstrated that activation of Hh signaling by Sufu ablation suppressed expression of osteoclast-specific genes, and thus osteoclast differentiation in vitro.
Genetic activation of Hh signaling by Sufu ablation suppressed expression of osteoclast-specific genes in vitro. qPCR analysis of relative mRNA levels of osteoclast-specific genes, including Nfatc1 (A), c-Fos (B), Acp5 (C), Oscar (D), Ctsk (E), Dcstamp (F), Atp6v0a3 (G), and Atp6v0d2 (H), in BMMs isolated from control and SufuCKO mice at 6 days after RANKL stimulation. Expression of each target gene was normalized by 18S ribosomal RNA. The relative changes in mRNA level were analyzed by 2-ΔΔCT method. All values were calculated from three independent biological replicates and presented as mean ± SD. **P<0.01, compared with control group.
Genetic activation of Hh signaling by LysM-Cre-mediated Sufu deletion suppressed osteoclastogenesis and prevented Ti particle-induced bone loss in vivo
Since genetic ablation of Sufu inhibited osteoclast formation in vitro, we next explored whether it could exert protective effects on wear particle-induced periprosthetic osteolysis, which is known to be primarily caused by excessive osteoclast formation and activity. To this end, we employed a murine calvarial model of Ti particle-induced osteolysis. Specifically, we first baked Ti particles for 6 h at 180 °C to remove endotoxins, and then confirmed that the baking treatment did not overtly alter the morphology and phase composition of Ti particles using SEM and XRD analyses, respectively (Figure S1). We implanted Ti particles on the calvariae of SufuCKO and their littermate control mice, and analyzed calvarial bones two weeks after implantation. As revealed by 2D and 3D μCT imaging, Ti particles induced profound bone loss on the calvariae of the control mice, which was largely prevented in the SufuCKO mice (Figure 4A). Quantitative μCT analyses further revealed that bone volume/total tissue volume (BV/TV) was markedly reduced, whereas both number of pores and the percentage of porosity were notably increased, in Ti particle-treated control mice when compared with those in sham-operated control mice (Figure 4B-D). Of note, these Ti particle-induced changes were significantly diminished in the SufuCKO mice (Figure 4B-D). Taken together, these results suggested that Sufu ablation exerted protective effects on Ti particle-induced osteolysis.
Genetic activation of Hh signaling by LysM-Cre-mediated deletion of Sufu gene prevented Ti particle-induced bone loss and suppressed osteoclastogenesis in vivo. (A) Representative 2D (bottom panels) and 3D (top panels) μCT images of calvariae from sham-operated control mice (Sham-Ctrl), SufuCKO mice with sham operation (Sham-SufuCKO), Ti particle-implanted control mice (Ti-Ctrl), and Ti particle-implanted SufuCKO mice (Ti-SufuCKO). (B-D) Quantitative μCT analyses of bone architecture parameters including bone volume/total tissue volume (BV/TV) (B), number of pores (C) and percentage of porosity (D). (E) Representative images of H&E staining of calvarial sections from Sham-Ctrl, Sham-SufuCKO, Ti-Ctrl, and Ti-SufuCKO mice. Scale bar, 100 μm. (F) Quantification of eroded surface proportion on H&E-stained calvarial sections. (G) Representative images of TRAP staining of calvarial sections from Sham-Ctrl, Sham-SufuCKO, Ti-Ctrl, and Ti-SufuCKO mice. Scale bar, 100 μm. (H) Quantitative analyses of number of TRAP-positive multinucleated cells. All bar graphs were presented as mean ± SD. n=5 mice per group, **P<0.01, determined by statistical analyses between two indicated groups. Sham-Ctrl, sham-operated control mice; Sham-SufuCKO, SufuCKO mice with sham operation; Ti-Ctrl, Ti particle-implanted control mice; Ti-SufuCKO, Ti particle-implanted SufuCKO mice.
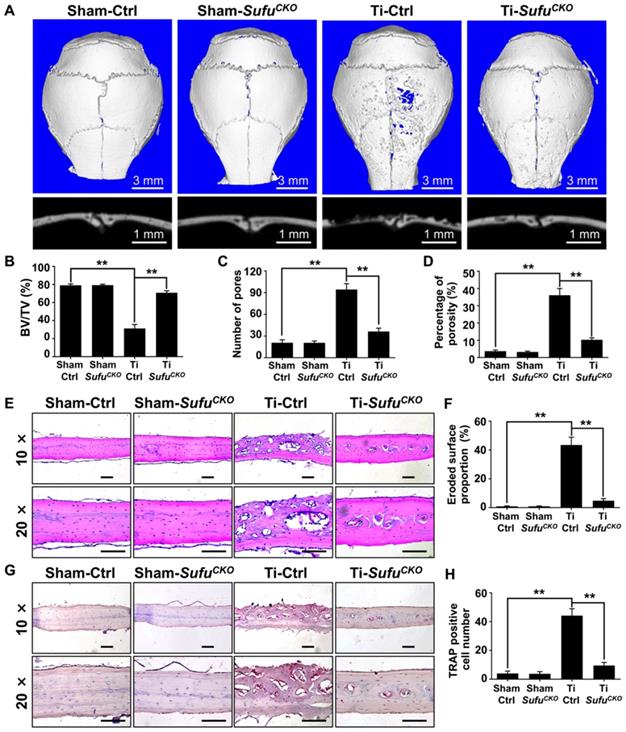
To confirm the above μCT results, we next performed histological analyses on calvarial sections. H&E staining showed that extensive bone destruction occurred in Ti particle-treated control mice, but was rarely detected in sham-operated mice (Figure 4E). More importantly, such osteolytic changes were obviously reduced upon Sufu deletion (Figure 4E). Consistent with these morphological observations, histomorphometric analysis revealed that the percentage of eroded surface caused by Ti particles was significantly decreased in SufuCKO mice, when compared with that of control mice (Figure 4F). Similarly, TRAP staining showed that Ti particles profoundly increased the number of osteoclasts, as evidenced by a large number of TRAP-positive osteoclasts lining eroded bone surfaces in Ti particle-stimulated mice (Figure 4G). However, significantly fewer osteoclasts were formed in SufuCKO mice than in control mice in response to Ti particles (Figure 4G, H). Taken together, our data indicated that conditional deletion of Sufu in macrophage lineage cells can alleviate Ti particle-induced bone loss by suppressing osteoclastogenesis in vivo.
Pharmacological activation of Hh signaling suppressed RANKL-induced osteoclastogenesis and osteoclast-specific gene expression without causing cytotoxicity
The above in vitro and in vivo genetic studies have established a protective role of Hh activation in Ti particle-induced osteolysis. These results inspired us to further test therapeutic efficacy of pharmacological activation of Hh signaling against osteolytic bone loss. To this end, we utilized purmorphamine (PM), a pharmacological activator of Smoothened (Smo) that is known to potently stimulate Hh signaling in a variety of cell types [19, 49-51]. The molecular structure, formula, and CAS number of PM were shown in Figure 5A. To determine the potential cytotoxicity of PM, we performed CCK-8 assays to assess the viability of BMMs after being treated with different concentrations of PM for 48, 72, or 96 h. The results showed that PM at concentrations up to 2 μM did not exert any discernible effect on the viability of BMMs at all time points examined (Figure 5B-D). To confirm the stimulatory effect of PM on Hh signaling, we treated BMMs with PM at non-toxic concentrations (0, 0.5, 1 or 2 μM) for 2 days, and then examined their mRNA expression of Gli1, a direct Hh target gene commonly used as a readout of signaling activity. Indeed, qPCR analyses showed that PM treatments dose-dependently induced the transcription of Gli1 in BMMs, therefore confirming activation of Hh signaling by PM in these cells (Figure 5E).
Pharmacological activation of Hh signaling by PM suppressed RANKL-induced osteoclast formation without causing cytotoxicity in vitro. (A) The molecular structure, formula and CAS number of PM. (B-D) BMMs were cultured with M-CSF in the presence of indicated concentrations of PM for 48 (B), 72 (C), or 96 h (D), then cell viability was measured by Cell Counting Kit-8 (CCK-8) assays. (E) qPCR analysis of relative Gli1 mRNA expression in BMMs treated with 0, 0,5, 1, or 2 μM PM for 48 h. (F) BMMs were stimulated with 30 ng/ml M-CSF and 50 ng/ml RANKL for 6 days and then subjected to TRAP staining. Images shown were representative results from three independent replicates. Scale bar, 400 μm. (G) Quantification of number of TRAP+ multinucleated osteoclasts per well. (H) Quantification of relative size of TRAP+ multinucleated osteoclasts. All values were calculated from three independent biological replicates and presented as mean ± SD. *P<0.05, **P<0.01, relative to vehicle-treated group.
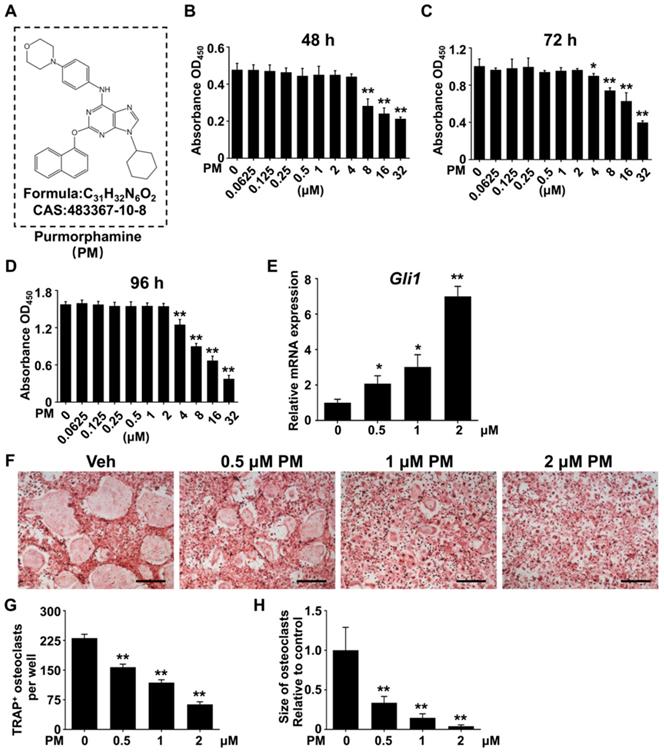
We next investigated the effect of Hh activation by PM on osteoclastogenesis. Osteoclastic differentiation of BMMs were induced with 50 ng/ml RANKL in the presence of vehicle (DMSO) or increasing concentrations of PM (0.5, 1 or 2 μM PM). After 6 days of induction, many large TRAP-positive multinucleated mature osteoclasts formed in the vehicle group (Figure 5F), whereas PM-treated groups were found to exhibit dose-dependent decreases in both number and size of these cells (Figure 5F). Quantitative measurement of TRAP+ osteoclasts further confirmed the above observations. While an average of 230.7 ± 5.6 osteoclasts were induced by RANKL stimulation in control BMM cultures, this number decreased to 157.3 ± 4.4, 118 ± 4.1, and 63.3 ± 3.7 in cells treated with 0.5, 1 and 2 μM PM, respectively (Figure 5G). Similarly, the average size of osteoclasts was also markedly diminished in PM-treated groups to 33.7 ± 2.1% at 0.5 μM, 14.6 ± 1.3% at 1 μM, and 4.1 ± 0.4% at 2 μM, relative to that of the vehicle-treated group (Figure 5H).
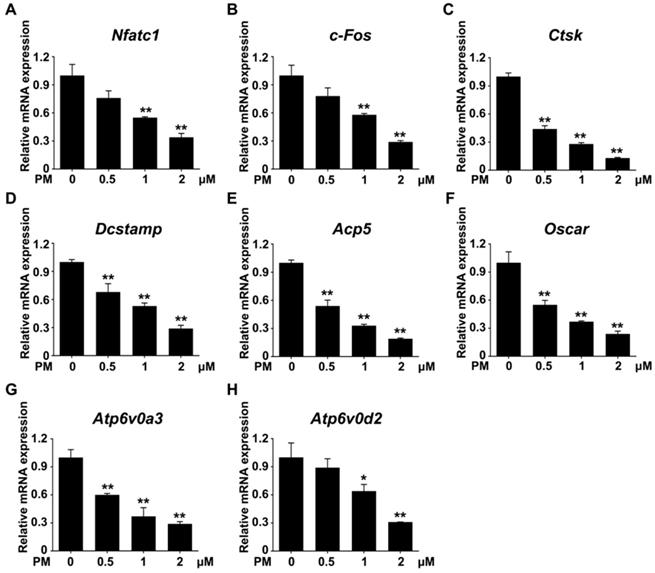
Furthermore, the transcription levels of genes associated with osteoclast differentiation, including Nfatc1, c-Fos, Acp5, Oscar, Ctsk, Dcstamp, Atp6v0a3, and Atp6v0d2, were all markedly downregulated by PM treatment in a dose-dependent fashion (Figure 6). Thus, pharmacological activation of Hh signaling by PM can dose-dependently suppress RANKL-induced osteoclastogenesis and osteoclast-specific gene expression without causing cytotoxicity.
Pharmacological activation of Hh signaling by PM restrained osteoclast-specific genes expressions under RANKL stimulation in vitro. qPCR analysis of relative mRNA levels of osteoclast-specific genes, including Nfatc1 (A), c-Fos (B), Ctsk (C), Dcstamp (D), Acp5 (E), Oscar (F), Atp6v0a3 (G), and Atp6v0d2 (H), in BMMs stimulated with RANKL for 6 days in the presence of 0, 0.5, 1, or 2 μM PM. Expression of each target gene was normalized by 18S ribosomal RNA. The relative changes in mRNA level were analyzed by 2-ΔΔCT method. All values were calculated from three independent biological replicates and presented as mean ± SD. **P<0.01, compared with vehicle-treated group.
Pharmacological activation of Hh signaling by PM inhibited RANKL-induced F-actin ring formation and osteoclastic bone resorption
To further confirm the effect of PM on osteoclastogenesis, we assessed RANKL-induced osteoclast F-actin ring formation. RANKL-treated BMMs were stained with phalloidin and DAPI to visualize F-actin ring and nuclei, respectively. The results showed that RANKL effectively induced formation of large F-actin ring in multinucleated cells, which was notably suppressed by PM supplementation (Figure 7A). Compared with control group, PM-treated groups exhibited significant reductions in both number and size of F-actin rings in a dose-dependent manner (Figure 7B-D). Of note, in cells treated with 2 μM PM, formation of F-actin rings was nearly completely disrupted (Figure 7A). Collectively, these findings suggested that pharmacological activation of Hh signaling by PM impaired osteoclast F-actin ring formation in vitro.
Pharmacological activation of Hh signaling by PM inhibited RANKL-induced F-actin ring formation and osteoclastic bone resorption in vitro. (A) Representative images of phalloidin staining of BMM clutures treated with RANKL for 6 days in the presence of 0, 0.5, 1, or 2 μM PM. F-actin rings. Green, F-actin; Blue, nuclei. Scale bar, 200 μm. (B-D) Quantitative analyses of F-actin rings showing number of F-actin rings per well (B), relative size of F-actin rings (C), and number of nuclei per osteoclast (D) in BMM clutures treated with RANKL for 6 days in the presence of 0, 0.5, 1, or 2 μM PM. (E) Representative scanning electron microscope (SEM) images of resorption pits on bovine slices seeded with differentiating osteoclasts in the presence of 0, 0.5, 1, or 2 μM PM. (F-G) Quantification of number (F) and average area (G) of bone resorption pits on SEM images. All values were calculated from three independent replicates, and presented as mean ± SD. ** indicated P<0.01, compared with vehicle-treated group.
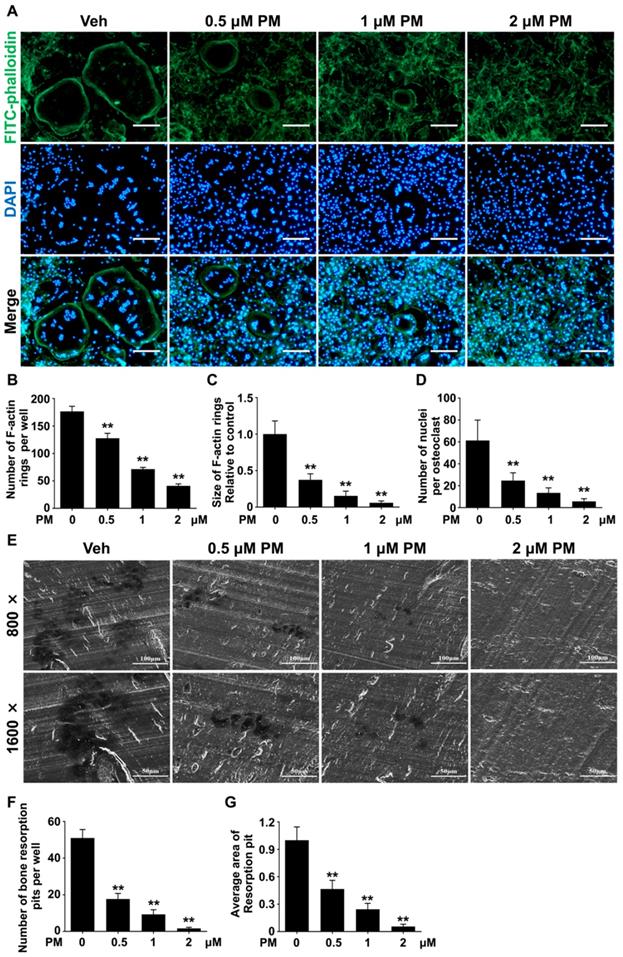
Since PM severely impaired osteoclastogenesis and F-actin ring formation, we predicted that osteoclastic bone resorption would also be inhibited by PM. To test this hypothesis, we performed an osteoclastic bone resorption assay. In this assay, differentiating BMMs were cultured on bovine bone slices, and then further induced to differentiate into mature osteoclast with RANKL stimulation. The resorption pits created by these osteoclasts were then analyzed by scanning electron microscopy (SEM). As shown in Figure 7E, several large resorption pits clearly formed in the vehicle group whereas fewer and smaller resorption pits were observed in PM groups. In line with impaired osteoclast formation, the number of resorption pits decreased significantly upon PM treatment, from 51 ± 2.7 in vehicle-treated group to 17.7 ± 1.8, 9.3 ± 1.5, and 1.7 ± 0.3 in groups treated with 0.5 μM, 1 μM, and 2 μM PM, respectively.
Furthermore, groups treated with 0.5, 1, and 2 μM PM exhibited 53.3 ± 3.9%, 75.6 ± 2.7%, and 94.4 ± 1.0% decreases in the average area of per bone resorption pit, respectively, when compared to that of vehicle-treated cells (Figure 7F, G). Taken together, these results demonstrated that activation of Hh signaling by PM treatment not only impaired osteoclastogeneis, but also inhibits osteoclast bone resorption activity in vitro.
Pharmacological activation of Hh signaling by PM attenuated Ti particle-induced osteolysis in vivo
Having established that pharmacological activation of Hh signaling by PM suppressed RANKL-induced osteoclastogenesis in vitro, we then investigated whether PM could protect against particle-induced bone loss in vivo. Mice were implanted with Ti particles on the calvariae before being subjected to daily injections of vehicle, low or high dosages of PM (2.5 mg/kg and 10 mg/kg, respectively) for 2 weeks. As shown in Figure 8A, Ti particles induced massive bone loss in vehicle-treated calvariae, which was clearly suppressed by treatment of PM in a dose-dependent manner. Consistently, quantification of μCT images further revealed that BV/TV of the vehicle group was significantly decreased compared with that of the sham group (32.3 ± 1.9% vs. 79.1 ± 0.9%), whereas such reduction in bone mass is considerably reversed in low- and high-PM groups (59 ± 1.9% and 73.3 ± 1.2%, respectively) (Figure 8B). Similarly, PM treatments dose-dependently decreased the number of pores as well as the percentage of porosity in Ti particle-treated calvariae (Figure 8C, D).
Pharmacological activation of Hh signaling by PM attenuated Ti particle-induced osteolysis in vivo. (A) Representative 2D (bottom panels) and 3D (top panels) μCT images of calvariae from sham-operated mice (Sham) and Ti particle-implanted mice subjected to administration with vehicle (Veh), 2.5 mg/kg PM (Low-PM), or 10 mg/kg PM (High-PM). (B-D) Quantitative μCT analyses of bone architecture parameters including bone volume/total tissue volume (BV/TV) (B), number of pores (C) and percentage of porosity (D). (E) Representative images of H&E staining of calvarial sections from sham-operated mice (Sham) and Ti particle-implanted mice subjected to administration with vehicle (Veh), 2.5 mg/kg PM (Low-PM), or 10 mg/kg PM (High-PM). Scale bar, 100 μm. (F) Quantification of eroded surface proportion on H&E-stained calvarial sections. (G) Representative images of TRAP staining of calvarial sections from sham-operated mice (Sham) and Ti particle-implanted mice subjected to administration with vehicle (Veh), 2.5 mg/kg PM (Low-PM), or 10 mg/kg PM (High-PM). Scale bar, 100 μm. (H) Quantitative analyses of number of TRAP-positive multinucleated cells. All bar graphs were presented as mean ± SD. n=7 mice per group, *P<0.05, **P<0.01, determined by statistical analyses between two indicated groups.
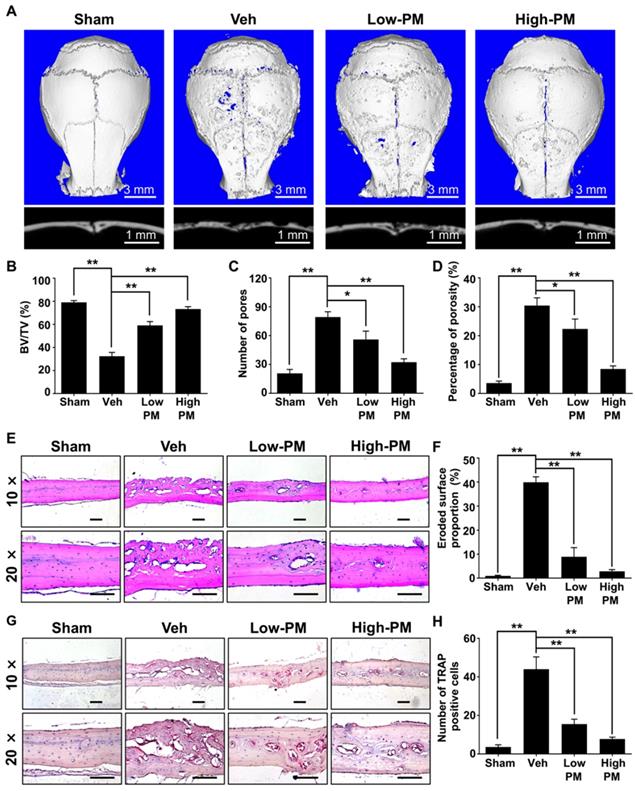
To further verify the therapeutic effect of PM on Ti particle-induced osteolysis in vivo, we performed histological and histomorphometric analysis on calvarial sections. H&E staining revealed that Ti particles caused severe bone erosion in the calvariae of vehicle group, which was significantly attenuated in low-PM group, and almost completely blocked in high-PM group (Figure 8E). Statistical analyses further revealed that PM markedly reduced the percentage of eroded bone surface out of total bone surface, an indicator reflecting degrees of bone destruction, from 38.9 ± 1.1% in the vehicle group to 8.9 ± 1.9% and 2.9 ± 0.4% in low- and high-PM groups, respectively (Figure 8F). Furthermore, TRAP staining showed that TRAP-positive multinucleated cells accumulated in the osteolytic regions predominantly along the eroded bone surfaces in Ti particle-implanted mice (Figure 8G). Importantly, the average numbers of osteoclasts in low- and high-PM groups reduced to 15.5 ± 1.3 and 7.8 ± 0.5, respectively, which were significantly less than 44 ± 3.2 in the vehicle group (Figure 8H).
Taken together, our results demonstrated that pharmacological activation of Hh signaling by PM protected against Ti particle-induced osteolysis in a dose-dependent manner.
Hh signaling negatively regulates the JNK/c-Fos-NFATc1 cascade during osteoclast differentiation
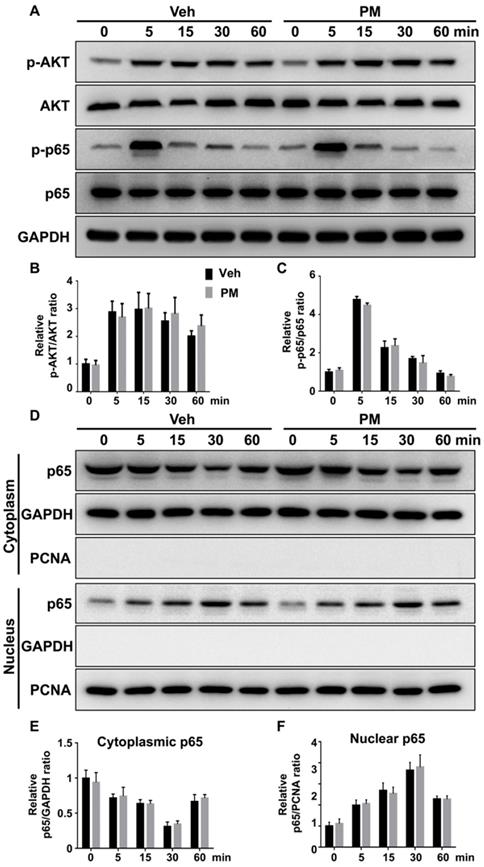
Multiple signaling pathways, including NF-κB, PI3K-Akt, and MAPKs, are activated by RANKL and involved in various aspects of osteoclastogenesis [48, 52-55]. To explore the mechanism underlying the inhibitory effect of Hh signaling on osteoclast formation, we investigated the effects of PM on RANKL-induced activation of these pathways. BMMs were pretreated with vehicle or 2 µM PM for 4 h, then stimulated with 50 ng/ml RANKL for 0, 5, 15, 30 and 60 min before being subjected to qPCR and Western blot analyses. qPCR analysis revealed an increase in mRNA expression of Gli1 in PM-treated BMMs compared to control cells, therefore verifying activation of Hh signaling in these cells (Figure S2). Western blot analysis further showed that RANKL potently increased levels of phosphorylated NF-κB p65 and AKT, which was not significantly affected by PM treatment at all time points examined (Figure 9A-C). In addition to phosphorylation of NF-κB p65, its nuclear translocation is a critical event for activation of NF-κB signaling [56]. To determine whether PM affected the RANKL-induced nuclear translocation of p65 in BMMs, we further examined protein levels of p65 in the cytoplasmic and nuclear extracts from BMMs treated as described above. As shown in Figure 9D-F, RANKL-induced nuclear translocation of p65 was not significantly altered in PM-treated BMMs compared to vehicle-treated cells. Thus, the inhibitory effect of Hh signaling on osteoclast differentiation was unlikely mediated by NF-κB and AKT pathways.
Activation of Hh signaling did not affect RANKL-induced activation of AKT and NF-κB pathways. (A) Western blot analyses of protein levels of phosphorylated AKT (p-AKT), total AKT, phosphorylated NF-κB p65 (p-p65), total NF-κB p65 (p65), and GAPDH in BMMs treated with 50 ng/ml RANKL for indicate times in the presence of vehicle (Veh) or 2 µM PM (PM). Representative images from three independent biological replicates were shown. (B-C) Quantitative analyses of relative ratios of p-AKT/AKT (B) and p-p65/p65 (C). n=3 per group. (D) Western blot analyses of protein levels of NF-κB p65 (p65) in the cytoplasmic and nuclear extracts from BMMs treated with 50 ng/ml RANKL in the presence of vehicle (Veh) or 2 µM PM (PM) for indicate times. Representative images from three independent biological replicates were shown. (E-F) Quantitative analyses of relative ratios of cytoplasmic p65/GAPDH (E), and nuclear p65/PCNA (F). n=3 per group. All bar graphs were presented as mean ± SD.
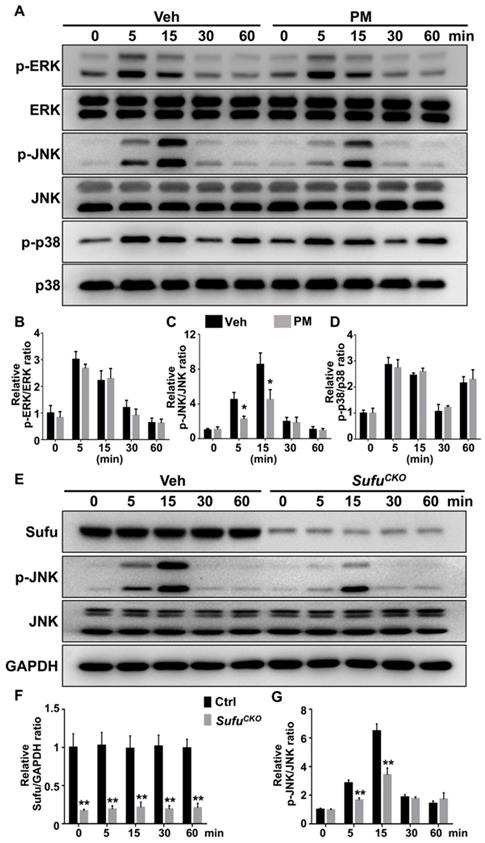
For analyzing the MAPK pathway, we examined phosphorylation status of its family members including ERK, JNK and p38. After RANKL stimulation, levels of the phosphorylated ERK, JNK, and p38 were rapidly induced, and peaked at 5, 15, and 5 min, respectively (Figure 10A-D). Interestingly, PM notably decreased phosphorylation of JNK at 5 and 15 min after RANKL treatment (Figure 10A-D), whereas it did not significantly alter RANKL-induced phosphorylation of p38 kinase and ERK (Figure 10A-D). Similarly, Sufu deletion significantly impaired RANKL-induced phosphorylation of JNK (Figure 10E-G). Thus, Hh signaling appeared to exert a specific inhibitory effect on RANKL-induced activation of JNK signaling.
Activation of Hh signaling inhibited RANKL-induced activation of JNK pathway. (A) Western blot analyses of protein levels of phosphorylated ERK (p-ERK), total ERK, phosphorylated JNK (p-JNK), total JNK, phosphorylated p38 (p-p38), and total p38 in BMMs treated with 50 ng/ml RANKL in the presence of vehicle (Veh) or 2 µM PM (PM) for indicate times. Representative images from three independent biological replicates were shown. (B-D) Quantitative analyses of relative ratios of p-ERK/ERK (B), p-JNK/JNK(C) and p-p38/p38 (D). n=3 per group. All bar graphs were presented as mean ± SD. *P<0.05, compared with vehicle-treated (Veh) group. (E) Western blot analyses of protein levels of Sufu, phosphorylated JNK (p-JNK), total JNK, and GAPDH in BMMs from control and SufuCKO (CKO) mice after treatment with 50 ng/ml RANKL for indicate times. Representative images from three independent biological replicates were shown. (F-G) Quantitative analyses of relative ratios of Sufu/GAPDH (F), and p-JNK/JNK(G). **P<0.01, compared with control groups.
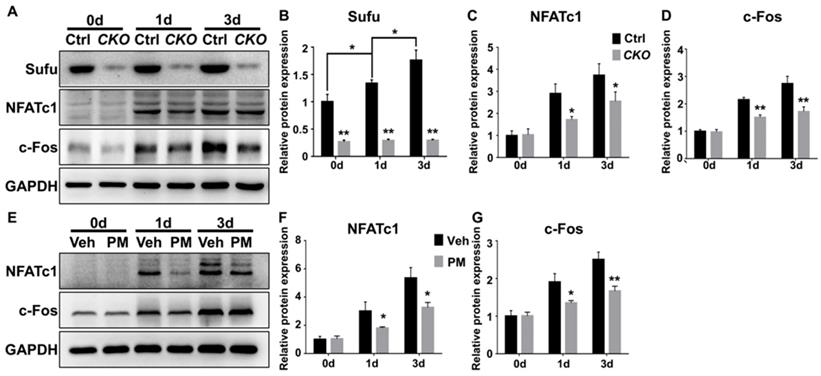
NFATc1 is a master transcriptional regulator of osteoclast differentiation that functions downstream of JNK signaling and c-Fos protein, another transcriptional regulator essential for osteoclast differentiation and function [52, 55, 57]. JNK pathway phosphorylates and thus activates activity of c-Jun, which forms the activator protein 1 (AP-1) complex with c-Fos to induce and amplify Nfatc1 expression [52, 55, 57, 58]. Of note, the above qPCR analyses have shown that genetic or pharmacological activation of Hh signaling significantly down-regulated mRNA levels of both Nfatc1 and c-Fos in RANKL-treated BMMs. To further confirm these results, we performed Western blot analyses to analyze the effect of Hh activation on RANKL-induced expression of NFATc1 and c-Fos proteins. As shown in Figure 11, levels of NFATc1 and c-Fos proteins were highly elevated in BMMs following 1 and 3 days of RANKL stimulation. However, such inductions were significantly reduced in BMMs upon Sufu deletion or PM treatment. Taken together, our results strongly suggested that Hh signaling cell-autonomously hindered osteoclastogenesis through suppressing JNK- and c-Fos-mediated regulation of NFATc1.
Activation of Hh signaling suppressed NFATc1 and c-Fos expression during osteoclastogenesis. (A) Western blot analyses of protein levels of Sufu, NFATc1 and c-Fos in BMMs from control and SufuCKO (CKO) mice at 0, 1, and 3 days after RANKL stimulation. Images shown were representative results from three independent biological replicates. (B-D) Quantification of relative protein levels of Sufu (B), NFATc1 (C) and c-Fos (D). All values were normalized to protein level of GAPDH, and shown as mean ± S.D. n=3 per group. *P<0.05, **P<0.01, compared with control or indicated group. (E) Western blot analyses of protein levels of NFATc1 and c-Fos in vehicle- (Veh) or PM-treated (PM) BMMs at 0, 1, and 3 days after RANKL stimulation. Images shown were representative results from three independent biological replicates. (F-G) Quantification of relative protein levels of NFATc1(F) and c-Fos (G). GAPDH was used as a loading control. All bar graphs were presented as mean ± SD. n=3 per group. *P<0.05, **P<0.01, compared with vehicle-treated (Veh) group.
Discussion
PPO is a common long-term complication of TJA operations, and represents the major cause of aseptic loosening and subsequent implant failure, which often leads to revision surgery and thus causes tremendous economic as well as psychological burdens on patients and their families. Since PPO is predominantly caused by hyperactive bone resorption activated by wear particles liberated from implant surfaces, current clinical treatments are mainly focused on inhibiting osteoclast formation and/or activity. However, several anti-resorption drugs, such as bisphosphonates and denosumab, exhibited limited therapeutic efficacy against this disease [9-11], not to mention that long-term use of these drugs could potentially cause severe side effects, such as osteonecrosis of the jaw and atypical femur fracture [59, 60]. Moreover, these anti-resorption drugs cannot promote bone regeneration to repair pre-existing osteolytic lesions. Therefore, it is urgent to search for novel alternative agents or approaches that can better prevent and treat PPO. Here, we demonstrated that genetic or pharmacological activation of Hh signaling potently inhibited RANKL-induced osteoclastogenesis in vitro and strongly protected against Ti particle-induced bone loss in a murine calvarial model in vivo. Combined with the established bone anabolic role of Hh pathway, our findings suggested that activation of Hh signaling could be a promising therapeutic approach with both bone anabolic and anti-resorptive properties for battling PPO and other osteolytic diseases.
A few previous studies have explored the cell-autonomous effect of Hh pathway activation on osteoclast differentiation in vitro, but reported inconsistent results. Shimo et al. reported that Shh treatment notably enhanced RANKL-induced osteoclast differentiation of RAW264.7 cells, BMMs, and CD11b+ bone marrow cells [29]. Similarly, Li et al. showed that pharmacological activation of Hh signaling promoted RANKL-induced formation of osteoclasts from RAW264.7 cells in the condition of estrogen deficiency [27]. However, Heller et al. demonstrated that genetic activation of Hh signaling by removing one copy of Ptch1 in BMMs did not affect the number of osteoclasts formed upon RANKL stimulation [28]. In contrast, Tibullo et al. showed that Shh signaling mediated the inhibitory effect of Ixazomib (a third-generation proteasome inhibitor) on RANKL-induced osteoclastogenesis in human monocytes [30]. Thus, these studies did not reach a consensus about the cell-autonomous role of Hh signaling in regulating osteoclastogenesis in vitro. Moreover, none of these studies evaluated the direct effects of enhanced Hh signaling on osteoclast formation in vivo under either a physiological or a pathological condition. Recently, a genetic study showed that specific ablation of Ihh in limb mesenchymal cells resulted in increased osteoclast formation, suggesting that Ihh signaling could play a physiological role in inhibiting osteoclastogenesis in vivo [17]. However, ablation of Ihh in limb mesenchymal cells could potentially inactivate Ihh signaling in both osteoblastic and osteoclastic linage cells. Thus, it remains unclear whether Ihh signaling functions within osteoclast precursors to negatively regulate osteoclastogenesis. To clarify the cell-autonomous role of Hh signaling in osteoclastogenesis, we activated Hh signaling in primary BMMs by utilizing LysM-Cre to specifically ablate Sufu, a key negative regulator of Hh signaling in these cells or by directly treating them with PM, a pharmacological agonist of Smo. We then performed comprehensive analyses to evaluate direct effects of Hh activation on RANKL-induced osteoclastogenesis. We found that activation of Hh signaling by either approach suppressed osteoclast formation, F-actin ring formation, and osteoclast-specific gene expression. In addition, we observed the reduced number and area of bone resorption pits upon PM treatment in bone resorption assays. Taken together, our data have clearly demonstrated that enhancing Hh signaling cannot only directly restrain RANKL-induced formation of osteoclasts, but also impair their bone-resorbing capacity in vitro. Our results are seemingly at odds with the earlier findings described above. Although the reason for these conflicting results is still unclear, the following differences between our study and others may account for these discrepancies. First, we activated Hh signaling at the level of Smo or its downstream component Sufu, while studies by Shimo et al. and Heller et al. modulated this pathway by inhibiting Ptch1 activity or reducing its expression, respectively. It is possible that manipulation of Hh-Ptch1 signaling can exert a stimulatory impact on osteoclastogenesis via a Smo/Sufu-independent mechanism, while Smo/Sufu-mediated canonical Hh signaling acts to suppress bone resorption. Second, we used primary BMMs in the entire study, whereas others performed some experiments with RAW264.7 cells and CD11b+ bone marrow cells. Different types of osteoclast precursors may respond differently to RANKL induction upon Hh stimulation. Third, we used 50 ng/ml RANKL to induce osteoclast differentiation in the normal condition, while Li et al. performed osteoclastogenesis with 100 ng/ml RANKL in the condition of estrogen depletion. Hh signaling may play opposite roles in osteoclastogenesis under different conditions. While the discrepancies between our study and others surely need to be resolved, our studies have clearly demonstrated that Sufu deletion or PM treatment not only potently inhibited osteoclast differentiation in vitro, but also strongly suppressed Ti particle-induced osteoclastogenesis in vivo. Thus, enhanced Hh signaling can play a cell-autonomous role in inhibiting osteoclastogenesis.
The role of Hh signaling in regulating osteoclastogenesis is likely complicated. While our study provided solid evidence to support a cell-autonomous role of Hh signaling in suppressing osteoclast differentiation and activity, previous studies have demonstrated that Hh signaling can indirectly promote osteoclast differentiation by activating the PTHrP-PKA-CREB cascade in mature osteoblasts to upregulate their expression of RANKL, the potent inducer of osteoclastogenesis [22-24]. Similarly, Hh ligands secreted by some tumor cells can stimulate osteoblast expression of RANKL to indirectly promote osteoclastogenesis and subsequent bone destruction [25, 26]. Collectively, these studies indicated that Hh signaling can negatively and positively regulate osteoclastogenesis via cell-autonomous and non-cell autonomous mechanisms, respectively. Clearly, the ultimate effect of systemic Hh pathway activation on osteoclastogenesis is determined by the balance between these two distinct mechanisms, and probably varies depending on different physiological or pathological conditions. Therefore, it should be individually tested for each osteolytic disease whether Hh activation can be used to inhibit bone destruction. Using a murine calvarial model of Ti-particle-induced osteolysis, we showed that PM administration, which presumably activated Hh signaling in both osteoblasts and osteoclasts, led to suppression of Ti particle-induced osteoclast formation. Thus, the direct inhibitory effect of Hh signaling on osteoclastogenesis appeared to be dominant over its indirect stimulatory effect in this particular osteolytic condition.
Our results showed that inhibition of osteoclastogenesis by conditional ablation of Sufu in osteoclast precursors was sufficient to effectively alleviate Ti particle-induced bone loss, confirming the predominant role of osteoclasts in this osteolytic disease. In addition to enhanced bone resorption, recent studies indicated that impaired bone formation is also a contributing factor of this disease. These studies showed that wear particles cannot only directly impair osteogenesis and bone-forming capacity of osteoblasts [61, 62], but also indirectly suppress osteoblast differentiation and function through stimulating production of proinflammatory factors by macrophages [62, 63]. Based on these facts, simultaneously suppressing bone resorption and promoting bone formation is likely a better strategy to prevent and treat PPO. In addition to its anti-resorption activity, our data suggested that activating Hh signaling in osteoclast precursors probably also exerted its therapeutic effect on Ti particle-induced bone loss through indirectly enhancing bone formation. Indeed, TRAP staining revealed that activation of Hh pathway in osteoclast precursors by either Sufu deletion or PM treatment markedly suppressed RANKL-induced formation of TRAP+ multinucleated osteoclasts, resulting in accumulation of TRAP+ mononuclear preosteoclasts. Preosteoclasts were known to secrete platelet-derived growth factor-BB (PDGF-BB) to stimulate formation of type-H vessels, which in turn promoted osteogenesis and bone formation [64, 65]. Thus, accumulated preosteoclasts in Sufu-ablated or PM-treated mice could indirectly enhance bone formation through promoting type-H vessel formation.
Previous studies have shown that Hh signaling played a crucial role in osteoblastogenesis, and that forced activation of this pathway by both genetic or pharmacological approaches can potently stimulate osteogenic differentiation and bone formation in vitro and in vivo [14, 15, 20, 37, 49, 66-69]. In line with these earlier findings, we also found that ablation of Sufu in BMSCs or treatment of these cells with PM caused accelerated osteoblast differentiation (data not shown). Thus, the protective effects of PM on Ti particle-induced bone loss observed in this study are probably in part attributed to its direct action on osteoblast differentiation and bone formation. In summary, our study and others collectively indicated that activation of Hh signaling cannot only suppress bone resorption, but may also promote bone formation through both indirect and direct mechanisms, therefore representing a promising therapeutic approach to prevent and treat PPO.
We should acknowledge that our study did have several limitations. Firstly, we used Ti particle-induced osteolysis as a model of PPO, despite the fact that the majority of wear particles present around metal-on-polyethylene implants are polyethylene particles [41, 70]. However, titanium particles do represent one of the pathological factors eliciting osteolysis. Moreover, the pathogenesis of Ti particle- and polyethylene particle-induced osteolysis is similar, and both types of particles cause bone destruction predominantly through activating osteoclast formation and function [41, 70]. In addition, Ti particle-induced osteolysis is the most commonly-used model for evaluating the effects of pharmaceuticals on wear particle-induced PPO, thus allowing comparison of our findings with others. Based on the above facts, we utilized this animal model in our study despite its potential limitation. It is no doubt that further studies are warranted to confirm our findings using osteolytic animal model that closely mimics the clinical features of PPO. Secondly, our study did not address whether targeting Hh pathway can treat pre-existing bone destruction since Sufu ablation and PM administration in this study were performed before osteolytic bone destruction has occurred. However, our findings did demonstrate that targeted activation of Hh signaling can inhibit osteoclastogenesis both in vitro and in vivo. Moreover, previous studies have established an anabolic role of Hh signaling in bone formation. Based on these findings, we predicted that activation of Hh signaling could not only prevent but also treat wear particle-induced PPO. Clearly, additional experiments should be performed to further verify this prediction.
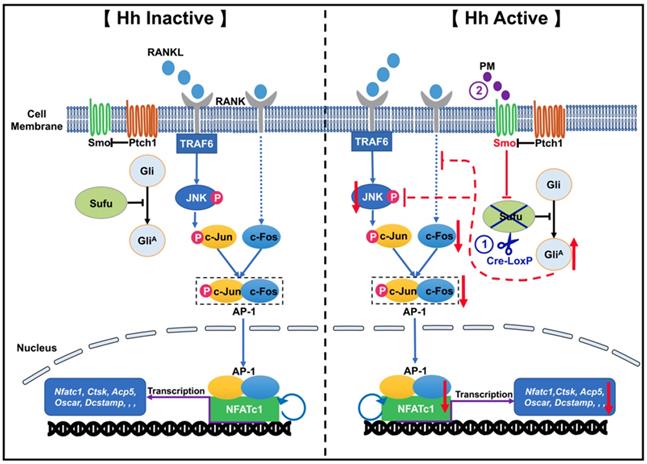
In conclusion, by utilizing both genetic and pharmacological approaches to activate Hh signaling in osteoclast precursors, we proved that Hh signaling strongly inhibited RANKL-mediated osteoclast differentiation in vitro through suppression of the JNK/c-Fos/NFATc1 signaling cascade (Figure 12). Furthermore, we showed that activation of Hh signaling significantly reduced osteoclast formation and bone destruction in a murine calvarial model of Ti particle-induced osteolysis. To our knowledge, our study is the first to demonstrate that Hh pathway activation could be used as a promising therapeutic approach to prevent and treat PPO and other osteolytic diseases.
A proposed model for the mechanism underlying the inhibitory effect of Hh signaling on osteoclastogenesis. When Hh signaling is inactive (Left panel), binding of RANKL to RANK activates JNK signaling, which in turn phosphorylates and thus stimulates the activity of c-Jun. In addition, RANKL induces expression of c-Fos through an unknown mechanism. c-Jun forms the activator protein 1 (AP-1) complex with c-Fos to induce and amplify expression of Nfatc1, which further upregulates the transcription of osteoclast-specific genes such as Ctsk, Acp5, and Oscar and Dcstamp. Our results demonstrated for the first time that activation of Hh signaling by Sufu deletion or PM treatment impairs osteoclastogenesis via suppression of NFATc1 expression by inhibiting RANKL-indued actvation of JNK signaling and expression of c-Fos (Right panel).
Abbreviations
Acp5: acid phosphatase 5; AKT: protein kinase B; Atp6v0a3: ATPase H+ transporting V0 subunit A3; Atp6v0d2: ATPase H+ transporting V0 subunit D2; BMMs: bone marrow-derived macrophages; BSA: bovine serum albumin; BV/TV: bone volume/total tissue volume; CCK-8: cell counting kit-8; c-Fos: cellular protooncogene Fos; Ctsk: cathepsin K; DAPI: 4′,6-diamidino-2-phenylindole; Dcstamp: dendritic cell-specific transmembrane protein; ddH2O: double distilled water; DMSO: dimethyl sulfoxide; DPBS: Dulbecco's phosphate-buffered saline; EDTA: ethylenediaminetetraacetic acid; ERK: extracellular signal-regulated kinase; F-actin: fibrous actin; FBS: fetal bovine serum; FITC: fluorescein isothiocyanate; Gapdh: glyceraldehyde-3-phosphate dehydrogenase; GliA: Gli activator; H: hour; H&E: hematoxylin and eosin; Hh: Hedgehog; JNK: c-Jun N-terminal kinase; MAPKs: mitogen-activated protein kinases; M-CSF: macrophage-colony stimulating factor; Min: minute; NFATc1: nuclear factor of activated T cells c1; NF-κB: nuclear factor kappa-B; Oscar: osteoclast-associated immunoglobulin-like receptor; P/S: penicillin/streptomycin; PFA: paraformaldehyde; PM: purmorphamine; PPO: periprosthetic osteolysis; Ptch1: patched1; qPCR: quantitative real-time polymerase chain reaction; RANK: receptor activator of nuclear factor-κB; RANKL: receptor activator of nuclear factor-κB ligand; RIPA buffer: radioimmunoprecipitation assay buffer; RT: room temperature; S: second; Smo: smoothened; Sufu: suppressor of fused; Ti: titanium; TJA: total joint arthroplasty; TRAP: tartrate-resistant acid phosphatase; α-MEM: α-modified Eagle's medium; μCT: micro-computed tomography.
Acknowledgements
This study was funded by the National Natural Science Foundation of China (81974344, 81772294), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), the Experimental Animal Science Project of Zhejiang Province (2018C37123), the Natural Science Foundation of Zhejiang Province (LY16H070008), the Medical and Health Science and Technology Program Project of Zhejiang Province (2015KYB435), and the Basic and Public Research Project of Zhejiang Province (LGF20H250004).
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hall MJ, DeFrances CJ, Williams SN, Golosinskiy A, Schwartzman A. National hospital discharge survey: 2007 summary. Natl Health Stat Report. 2010;29:1-24
2. Sloan M, Premkumar A, Sheth NP. Projected volume of primary total joint arthroplasty in the U.S, 2014 to 2030. J Bone Joint Surg Am. 2018;100:1455-60
3. Harris WH. Wear and periprosthetic osteolysis: the problem. Clin Orthop Relat Res. 2001;393:66-70
4. Abu-Amer Y, Darwech I, Clohisy JC. Aseptic loosening of total joint replacements: mechanisms underlying osteolysis and potential therapies. Arthritis Res Ther. 2007;9(Suppl 1):S6
5. Sadoghi P, Liebensteiner M, Agreiter M, Leithner A, Bohler N, Labek G. Revision surgery after total joint arthroplasty: a complication-based analysis using worldwide arthroplasty registers. J Arthroplasty. 2013;28:1329-32
6. Purdue PE, Koulouvaris P, Nestor BJ, Sculco TP. The central role of wear debris in periprosthetic osteolysis. HSS J. 2006;2:102-13
7. Wu C, Wang W, Tian B, Liu X, Qu X, Zhai Z. et al. Myricetin prevents titanium particle-induced osteolysis in vivo and inhibits rankl-induced osteoclastogenesis in vitro. Biochem Pharmacol. 2015;93:59-71
8. Purdue PE, Koulouvaris P, Potter HG, Nestor BJ, Sculco TP. The cellular and molecular biology of periprosthetic osteolysis. Clin Orthop Relat Res. 2007;454:251-61
9. Smith RL, Schwarz EM. Are biologic treatments a potential approach to wear- and corrosion-related problems? Clin Orthop Relat Res. 2014;472:3740-6
10. Shi M, Chen L, Xin Z, Wang Y, Wang W, Yan S. Bisphosphonates for the preservation of periprosthetic bone mineral density after total joint arthroplasty: a meta-analysis of 25 randomized controlled trials. Osteoporos Int. 2018;29:1525-37
11. Ledin H, Good L, Aspenberg P. Denosumab reduces early migration in total knee replacement. Acta Orthop. 2017;88:255-8
12. Ping Z, Hu X, Wang L, Shi J, Tao Y, Wu X. et al. Melatonin attenuates titanium particle-induced osteolysis via activation of wnt/beta-catenin signaling pathway. Acta Biomater. 2017;51:513-25
13. Xiong L, Liu Y, Zhu F, Lin J, Wen D, Wang Z. et al. Acetyl-11-keto-beta-boswellic acid attenuates titanium particle-induced osteogenic inhibition via activation of the gsk-3beta/beta-catenin signaling pathway. Theranostics. 2019;9:7140-55
14. Alman BA. The role of hedgehog signalling in skeletal health and disease. Nat Rev Rheumatol. 2015;11:552-60
15. Yang J, Andre P, Ye L, Yang YZ. The hedgehog signalling pathway in bone formation. Int J Oral Sci. 2015;7:73-9
16. Bangs F, Anderson KV. Primary cilia and mammalian hedgehog signaling. Cold Spring Harb Perspect Biol. 2017;9:a028175
17. Amano K, Densmore MJ, Lanske B. Conditional deletion of indian hedgehog in limb mesenchyme results in complete loss of growth plate formation but allows mature osteoblast differentiation. J Bone Miner Res. 2015;30:2262-72
18. Li J, Wang Q, Cui Y, Yang X, Li Y, Zhang X. et al. Suppressor of fused is required for determining digit number and identity via gli3/fgfs/gremlin. PLoS One. 2015;10:e0128006
19. Sinha S, Chen JK. Purmorphamine activates the hedgehog pathway by targeting smoothened. Nat Chem Biol. 2006;2:29-30
20. Dwyer JR, Sever N, Carlson M, Nelson SF, Beachy PA, Parhami F. Oxysterols are novel activators of the hedgehog signaling pathway in pluripotent mesenchymal cells. J Biol Chem. 2007;282:8959-68
21. Lv WT, Du DH, Gao RJ, Yu CW, Jia Y, Jia ZF. et al. Regulation of hedgehog signaling offers a novel perspective for bone homeostasis disorder treatment. Int J Mol Sci. 2019;20:3981
22. Mak KK, Bi Y, Wan C, Chuang PT, Clemens T, Young M. et al. Hedgehog signaling in mature osteoblasts regulates bone formation and resorption by controlling pthrp and rankl expression. Dev Cell. 2008;14:674-88
23. Ohba S, Kawaguchi H, Kugimiya F, Ogasawara T, Kawamura N, Saito T. et al. Patched1 haploinsufficiency increases adult bone mass and modulates gli3 repressor activity. Dev Cell. 2008;14:689-99
24. Mundy GR, Yang X. Hedgehog coordination of postnatal osteoclast and osteoblast activities. Dev Cell. 2008;14:637-8
25. Wu JB, Yin L, Shi C, Li Q, Duan P, Huang JM. et al. Maoa-dependent activation of shh-il6-rankl signaling network promotes prostate cancer metastasis by engaging tumor-stromal cell interactions. Cancer Cell. 2017;31:368-82
26. Das S, Tucker JA, Khullar S, Samant RS, Shevde LA. Hedgehog signaling in tumor cells facilitates osteoblast-enhanced osteolytic metastases. PLoS One. 2012;7:e34374
27. Li X, Jie Q, Zhang H, Zhao Y, Lin Y, Du J. et al. Disturbed mek/erk signaling increases osteoclast activity via the hedgehog-gli pathway in postmenopausal osteoporosis. Prog Biophys Mol Biol. 2016;122:101-11
28. Heller E, Hurchla MA, Xiang J, Su X, Chen S, Schneider J. et al. Hedgehog signaling inhibition blocks growth of resistant tumors through effects on tumor microenvironment. Cancer Res. 2012;72:897-907
29. Shimo T, Matsumoto K, Takabatake K, Aoyama E, Takebe Y, Ibaragi S. et al. The role of sonic hedgehog signaling in osteoclastogenesis and jaw bone destruction. PLoS One. 2016;11:e0151731
30. Tibullo D, Longo A, Vicario N, Romano A, Barbato A, Di Rosa M. et al. Ixazomib improves bone remodeling and counteracts sonic hedgehog signaling inhibition mediated by myeloma cells. Cancers (Basel). 2020;12:323
31. Das S, Samant RS, Shevde LA. Hedgehog signaling induced by breast cancer cells promotes osteoclastogenesis and osteolysis. J Biol Chem. 2011;286:9612-22
32. Matsumoto K, Shimo T, Kurio N, Okui T, Obata K, Masui M. et al. Expression and role of sonic hedgehog in the process of fracture healing with aging. In Vivo. 2016;30:99-105
33. Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using lysmcre mice. Transgenic Res. 1999;8:265-77
34. Chen J, Holguin N, Shi Y, Silva MJ, Long F. Mtorc2 signaling promotes skeletal growth and bone formation in mice. J Bone Miner Res. 2015;30:369-78
35. Gu X, Fu X, Lu J, Saijilafu, Li B, Luo ZP. et al. Pharmacological inhibition of s6k1 impairs self-renewal and osteogenic differentiation of bone marrow stromal cells. J Cell Biochem. 2018;119:1041-9
36. Chen J, Long F. Beta-catenin promotes bone formation and suppresses bone resorption in postnatal growing mice. J Bone Miner Res. 2013;28:1160-9
37. Li K, Xiu C, Zhou Q, Ni L, Du J, Gong T. et al. A dual role of cholesterol in osteogenic differentiation of bone marrow stromal cells. J Cell Physiol. 2019;234:2058-66
38. Sun X, Zhang J, Wang Z, Liu B, Zhu S, Zhu L. et al. Licorice isoliquiritigenin-encapsulated mesoporous silica nanoparticles for osteoclast inhibition and bone loss prevention. Theranostics. 2019;9:5183-99
39. Chen K, Qiu P, Yuan Y, Zheng L, He J, Wang C. et al. Pseurotin a inhibits osteoclastogenesis and prevents ovariectomized-induced bone loss by suppressing reactive oxygen species. Theranostics. 2019;9:1634-50
40. Geng D, Xu Y, Yang H, Wang J, Zhu X, Zhu G. et al. Protection against titanium particle induced osteolysis by cannabinoid receptor 2 selective antagonist. Biomaterials. 2010;31:1996-2000
41. Shao H, Shen J, Wang M, Cui J, Wang Y, Zhu S. et al. Icariin protects against titanium particle-induced osteolysis and inflammatory response in a mouse calvarial model. Biomaterials. 2015;60:92-9
42. Rakshit DS, Ly K, Sengupta TK, Nestor BJ, Sculco TP, Ivashkiv LB. et al. Wear debris inhibition of anti-osteoclastogenic signaling by interleukin-6 and interferon-gamma. Mechanistic insights and implications for periprosthetic osteolysis. J Bone Joint Surg Am. 2006;88:788-99
43. Hu Q, Li T, Wang L, Xie Y, Liu S, Bai X. et al. Neuroprotective effects of a smoothened receptor agonist against early brain injury after experimental subarachnoid hemorrhage in rats. Front Cell Neurosci. 2016;10:306
44. Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016;7:27-31
45. Li J, Cui Y, Xu J, Wang Q, Yang X, Li Y. et al. Suppressor of fused restraint of hedgehog activity level is critical for osteogenic proliferation and differentiation during calvarial bone development. J Biol Chem. 2017;292:15814-25
46. Svard J, Heby-Henricson K, Persson-Lek M, Rozell B, Lauth M, Bergstrom A. et al. Genetic elimination of suppressor of fused reveals an essential repressor function in the mammalian hedgehog signaling pathway. Dev Cell. 2006;10:187-97
47. Asagiri M, Takayanagi H. The molecular understanding of osteoclast differentiation. Bone. 2007;40:251-64
48. Yuan Q, Gao F, Yao Y, Cai P, Zhang X, Yuan J. et al. Gold clusters prevent inflammation-induced bone erosion through inhibiting the activation of nf-kappab pathway. Theranostics. 2019;9:1825-36
49. Wu X, Walker J, Zhang J, Ding S, Schultz PG. Purmorphamine induces osteogenesis by activation of the hedgehog signaling pathway. Chem Biol. 2004;11:1229-38
50. Gu D, Wang S, Zhang S, Zhang P, Zhou G. Directed transdifferentiation of muller glial cells to photoreceptors using the sonic hedgehog signaling pathway agonist purmorphamine. Mol Med Rep. 2017;16:7993-8002
51. Namchaiw P, Wen H, Mayrhofer F, Chechneva O, Biswas S, Deng W. Temporal and partial inhibition of gli1 in neural stem cells (nscs) results in the early maturation of nsc derived oligodendrocytes in vitro. Stem Cell Res Ther. 2019;10:272
52. Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337-42
53. Liu Y, Wang C, Wang G, Sun Y, Deng Z, Chen L. et al. Loureirin b suppresses rankl-induced osteoclastogenesis and ovariectomized osteoporosis via attenuating nfatc1 and ros activities. Theranostics. 2019;9:4648-62
54. Kikuta J, Ishii M. Osteoclast migration, differentiation and function: novel therapeutic targets for rheumatic diseases. Rheumatology (Oxford). 2013;52:226-34
55. Boyce BF. Advances in the regulation of osteoclasts and osteoclast functions. J Dent Res. 2013;92:860-7
56. Novack DV. Role of nf-kappab in the skeleton. Cell Res. 2011;21:169-82
57. Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H. et al. Induction and activation of the transcription factor nfatc1 (nfat2) integrate rankl signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889-901
58. David JP, Sabapathy K, Hoffmann O, Idarraga MH, Wagner EF. Jnk1 modulates osteoclastogenesis through both c-jun phosphorylation-dependent and -independent mechanisms. J Cell Sci. 2002;115:4317-25
59. Khan AA, Morrison A, Hanley DA, Felsenberg D, McCauley LK, O'Ryan F. et al. Diagnosis and management of osteonecrosis of the jaw: a systematic review and international consensus. J Bone Miner Res. 2015;30:3-23
60. Lo JC, Grimsrud CD, Ott SM, Chandra M, Hui RL, Ettinger B. Atypical femur fracture incidence in women increases with duration of bisphosphonate exposure. Osteoporos Int. 2019;30:2515-20
61. Gu Y, Wang Z, Shi J, Wang L, Hou Z, Guo X. et al. Titanium particle-induced osteogenic inhibition and bone destruction are mediated by the gsk-3beta/beta-catenin signal pathway. Cell Death Dis. 2017;8:e2878
62. O'Neill SC, Queally JM, Devitt BM, Doran PP, O'Byrne JM. The role of osteoblasts in peri-prosthetic osteolysis. Bone Joint J. 2013 95-B: 1022-6
63. Lee SS, Sharma AR, Choi BS, Jung JS, Chang JD, Park S. et al. The effect of tnfalpha secreted from macrophages activated by titanium particles on osteogenic activity regulated by wnt/bmp signaling in osteoprogenitor cells. Biomaterials. 2012;33:4251-63
64. Huang J, Yin H, Rao SS, Xie PL, Cao X, Rao T. et al. Harmine enhances type h vessel formation and prevents bone loss in ovariectomized mice. Theranostics. 2018;8:2435-46
65. Xie H, Cui Z, Wang L, Xia Z, Hu Y, Xian L. et al. Pdgf-bb secreted by preosteoclasts induces angiogenesis during coupling with osteogenesis. Nat Med. 2014;20:1270-8
66. Regard JB, Malhotra D, Gvozdenovic-Jeremic J, Josey M, Chen M, Weinstein LS. et al. Activation of hedgehog signaling by loss of gnas causes heterotopic ossification. Nat Med. 2013;19:1505-12
67. Hojo H, Ohba S, Yano F, Saito T, Ikeda T, Nakajima K. et al. Gli1 protein participates in hedgehog-mediated specification of osteoblast lineage during endochondral ossification. J Biol Chem. 2012;287:17860-9
68. Spinella-Jaegle S, Rawadi G, Kawai S, Gallea S, Faucheu C, Mollat P. et al. Sonic hedgehog increases the commitment of pluripotent mesenchymal cells into the osteoblastic lineage and abolishes adipocytic differentiation. J Cell Sci. 2001;114:2085-94
69. Shi Y, Chen J, Karner CM, Long F. Hedgehog signaling activates a positive feedback mechanism involving insulin-like growth factors to induce osteoblast differentiation. Proc Natl Acad Sci U S A. 2015;112:4678-83
70. Hu X, Ping Z, Gan M, Tao Y, Wang L, Shi J. et al. Theaflavin-3,3'-digallate represses osteoclastogenesis and prevents wear debris-induced osteolysis via suppression of erk pathway. Acta Biomater. 2017;48:479-88
Author contact
![]() Corresponding author: Jianquan Chen, Ph.D. Address: Orthopedic Institute, Soochow University, 708 Renmin Rd, Suzhou, Jiangsu 215007, People's Republic of China. E-mail: chenjianquanedu.cn; Tel: +86-0512-67780781
Corresponding author: Jianquan Chen, Ph.D. Address: Orthopedic Institute, Soochow University, 708 Renmin Rd, Suzhou, Jiangsu 215007, People's Republic of China. E-mail: chenjianquanedu.cn; Tel: +86-0512-67780781