Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Cancer Stemness, Cellular Stress...
Markers Identifying Cancer...
Role of stress-induced...
Metabolic remodeling during...
Crosstalk between metabolic...
Conclusion
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(14):6261-6277. doi:10.7150/thno.42523 This issue Cite
Review
Epigenetics and metabolism at the crossroads of stress-induced plasticity, stemness and therapeutic resistance in cancer
Dinoop Ravindran Menon1*, Heinz Hammerlindl2*, Joachim Torrano2, Helmut Schaider2 ![]() , Mayumi Fujita1,3,4
, Mayumi Fujita1,3,4 ![]()
1. Department of Dermatology, University of Colorado School of Medicine, Aurora, CO, USA
2. The University of Queensland Diamantina Institute, The University of Queensland, Brisbane, OLD, Australia
3. Eastern Colorado VA Health Care System, Aurora CO, USA
4. Department of Immunology and Microbiology, University of Colorado School of Medicine, Aurora, CO, USA
*These authors contributed equally to the study
Received 2019-12-27; Accepted 2020-2-13; Published 2020-5-15
Abstract

Despite the recent advances in the treatment of cancers, acquired drug resistance remains a major challenge in cancer management. While earlier studies suggest Darwinian factors driving acquired drug resistance, recent studies point to a more dynamic process involving phenotypic plasticity and tumor heterogeneity in the evolution of acquired drug resistance. Chronic stress after drug treatment induces intrinsic cellular reprogramming and cancer stemness through a slow-cycling persister state, which subsequently drives cancer progression. Both epigenetic and metabolic mechanisms play an important role in this dynamic process. In this review, we discuss how epigenetic and metabolic reprogramming leads to stress-induced phenotypic plasticity and acquired drug resistance, and how the two reprogramming mechanisms crosstalk with each other.
Keywords: Epigenetics, metabolism, stress-induced plasticity, stemness
Introduction
Acquired drug resistance is one of the major causes of mortality in cancer. This phenomenon is wide spread for distinct kind of treatment strategies which include standard chemotherapy, targeted therapy and immune therapy. Multiple mechanisms have been implicated in the development of drug resistance, which include reactivation of targeted pathways, activation of parallel pathways, drug efflux, and immune evasive mechanisms [1] . While earlier studies have suggested a Darwinian model of evolution with a rigid non-reversible phenotype caused by genetic alterations and natural selection, others have shown a reversible resistance phenotypic state and the involvement of non-Darwinian factors [2-6]. The latter model points to a more dynamic process that involves tumor microenvironment, cellular heterogeneity and phenotypic plasticity, all of which play an important role in the evolution of acquired drug resistance [7, 8]. These non-Darwinian factors may be distinct, but are often interconnected with phenotypic plasticity. This process of phenotypic plasticity has many similarities to genetic accommodation or “organic selection”, a term coined by Baldwin [9], except that some of these adaptations are heritable in cancer cells, which could be explained by the process of epigenetic imprinting. Phenotypic plasticity provides cancer cells an initial survival advantage that allows them to accumulate genetic changes or imprint epigenetic memory, leading to a resistant phenotype. The initial underlying mechanisms aiding this process could be our own pre-existing cellular programs that allow our body to cope with a stressful or unfavorable microenvironment, which is adapted by cancer cells to acclimate to natural or drug-induced challenges [10].
This process of cellular plasticity is not only the privilege of cancer cells but also shared by non-cancerous cells. While differentiation and heterogeneity of human stem cells were initially thought to be unidirectional with embryonic stem cells differentiating into adult stem cells and differentiated tissues, growing evidence suggests that each of these phenotypes is semi-stable and interchangeable [11]. Accordingly, differentiated cells from normal tissue can acquire distinct phenotypes, as shown by trans-differentiation experiments where distinct cell types are inter-convertible without reprogramming into an adult stem cell or embryonic stem cell phenotype [12]. Similarly, cancer cells could be in diverse semi-stable epigenetic states with more intrinsic plasticity, allowing them to switch between phenotypes, depending on fitness landscape or mutation-selection balance [13-17].
Whether it is a cancer or non-cancerous cell, the primary factor that defines a metastable (stable states of a dynamic system) or differentiated state is the pattern of gene expression. Hence, epigenetic memory plays an important role in stabilizing each state or transitioning from one state to another. Metabolic factors also trigger and support the transition. Both processes of epigenetic memory and metabolic changes are mutually regulated in nature rather than unidirectionally controlled [18]. These two mechanisms are also important in developing and maintaining cancer subpopulation with stem-like characters, such as high tumor growth potential, drug resistance and tumor heterogeneity [19, 20]. In this review, we will discuss how epigenetic and metabolic factors drive phenotypic plasticity in cancer, and highlight their importance in cancer stemness, disease progression and drug resistance.
Cancer Stemness, Cellular Stress and Senescence
Stem cells are classically defined by two basic principles; the ability to self-propagate and the potency to differentiate into multiple cell types [21]. Similarly, cancer stem cells (CSCs) are historically defined by two fundamental stem cell properties; self-propagation and restoration of the original tumor with phenotypic heterogeneity [22, 23]. Another feature of CSCs is their resilience to unfavorable microenvironments and drugs, which allows them to survive in disadvantageous conditions and re-establish the tumor [24]. The initial CSC model of a unidirectional hierarchical system was established from the studies of hematological cancers where rare cell populations propagated after serial transplantations [25]. Accordingly, a variety of markers were identified in each cancer type that met the defining criteria for CSCs [23]. However, recent studies have provided evidence for a dynamic, non-hierarchical cancer stemness model in multiple cancer types including glioblastoma, breast cancer, pancreatic cancer and melanoma by accommodating the concept of phenotypic plasticity [26-31]. Therefore, subpopulations with rapid tumor-initiating potency can rise from stochastic switching of cancer cell populations [31]. While it is clear that subpopulations of cells have growth advantage or drug resistance capability when compared to the bulk of tumor cells, it is likely that these populations are in flux through constantly switching phenotypes.
These studies also raise another interesting question about cancer stemness. Can a particular phenotype qualify as stemness or can the process of plasticity itself better explain the stemness? While some of these phenotypes have a growth or survival advantage in certain conditions, this advantage could be rather contextual, entailing cells to switch between these states to grow and thrive. Often, cells in a slow-cycling or semi-quiescent phenotype are resistant to a wide variety of treatments and described as cancer persister cells [32]. They are also reported to have an advantage in tumor growth and tumor-initiating potential [33]. This observation is rather counterintuitive as it is difficult to directly explain how a slow-cycling population can lead to higher tumorigenicity. A possible explanation would be that these cells enter a state that is more tolerant to a wide variety of stressful conditions, which allow their initial successful engraftment and subsequent swift switch into a proliferative state once the environment becomes favorable. On the other hand, cells already in a proliferative state may need to switch into a slow-cycling state to allow their engraftment and then switch back to a proliferative state for tumor growth. This explanation points to a general mechanism whereby a slow-cycling state, described as a pseudo-senescent or quiescent phenotype, can be associated with cancer stemness.
Cellular senescence is broadly classified into three types: replicative senescence, embryonic senescence and stress-induced premature senescence (SIPS) [34]. Replicative senescence is characterized by shortening of telomeres due to cellular replication, a closely regulated process to ensure tissue homeostasis [35]. Embryonic senescence, on the other hand, plays a central role in early embryonic development. While embryonic senescence was thought to be terminal, recent studies suggest it is temporal in nature and cells exit out of senescence at later stages [36]. SIPS, as the name suggests, is a stress-induced premature senescence phenotype that does not affect telomere function [37]. SIPS is induced by cellular stress from extrinsic and intrinsic factors such as oncogenic stress, chemotherapeutic agents, ionizing radiation and nutrient starvation [38-40]. Hence, SIPS have characteristics that resemble the senescent-like phenotype observed in cancer cells that arise due to oncogenic stress or treatment with therapeutic agents.
The underlying process relating these seemingly paradoxical phenotypes of senescence and cancer stemness had remained elusive. However, a recent study illustrated that the process of therapy-induced senescence promoted cancer stemness by reprogramming cells and enhancing canonical WNT signaling [41]. This phenotypic shift from senescence into a stem-like state could be a universal phenomenon not exclusive to cancer cells, since even 'Yamanaka factors' [42] induces an initial transition of primary cells into senescence that is required for efficient reprogramming [43, 44]. Because cancer cells are heterogeneous in their efficacy to switch between slow-cycling persisters and proliferative states, the cells that are highly dynamic and switch effortlessly between these states could have growth and survival advantages over others.
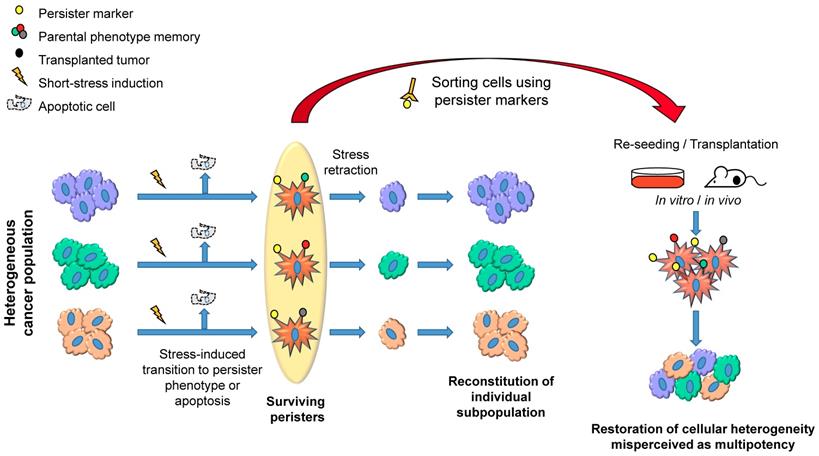
In line with these observations, we have shown that acute stress-induced phenotypic switching of distinct melanoma subpopulations into a slow-cycling state [45]. This switching led to the upregulation of multiple melanoma stem cell markers including nerve growth factor receptor (NGFR) [46], ATP-binding cassette sub-family B member 5 (ABCB5) [47] and aldehyde dehydrogenase (ALDH) activity [48]. However, this process did not involve direct reprogramming, and the slow-cycling cells reverted back to their respective phenotype once the stress was removed, suggesting that the cancer persister cells serve as a reservoir for many distinct subpopulations, thereby maintaining tumor heterogeneity (Figure 1, left). We have also shown that this switching into persister cells provided stem-like characteristics such as high tumorigenic potential and drug resistance [45]. Therefore, the markers of a slow-cycling state fulfill the definitions of the hierarchical CSC model except lineage hierarchy (Figure 1, right). This concept is supported by many studies, which show that unfavorable conditions, such as hypoxia, drug treatment and nutrient starvation, induce phenotypic plasticity that leads to the induction of a stem-like state in cancer cells [45, 49-54]. The transition into a slow-cycling state is also closely associated with an epithelial-mesenchymal transition (EMT) that leads to drug resistance and metastasis [55-57]. Drug holiday studies have shown that the slow-cycling populations swiftly switch back into a proliferative state in the absence of drug and display phenotypes corresponding to cancer stemness [2, 3, 45, 55, 58, 59]. Accordingly, CSCs are often reported to be in a slow-cycling state, providing them with drug resistance and tumor growth advantage [60].
A schematic representation describing how distinct subpopulations of cancer cells transiently shift into a persister state (star-shaped red cells) under stress and maintain tumor heterogeneity. The short-term stress exposure causes a shift of multiple subpopulations into persister phenotype both in vitro and in vivo. The efficiency of transition could vary depending on the stress-inducing conditions and sensitivity of subpopulations to the stress. When the factor inducing stress is removed, the surviving persisters would reverse back to their original phenotypes and proliferate to reconstitute their subpopulations (left). When persister populations are sorted using persister markers and reseeded in vitro or implanted in vivo, each of the populations reverse back to the original phenotype, resulting in the restoration of phenotypic heterogeneity, which could be misperceived as pluripotency or multipotency of cancer cells, because it looks as if the persister cells differentiate into multiple phenotypes (right).
Markers Identifying Cancer Persisters
The slow-cycling, drug-resistant persister population has been detected in multiple cancer types and often shares previously described markers of cancer stem cells. However, it is unclear whether these markers are related with the persister phenotype. Below, we will discuss some of the markers that have been shown to identify cancer persisters. Members of the jumonji AT-rich interactive domain-1 (JARID1) family, particularly lysine-specific demethylase 5A (KDM5A) and lysine-specific demethylase 5B (KDM5B) [2, 3, 45, 58, 59, 61], demethylate histone 3 lysine 4 trimethylation (H3K4me3). KDM5A and KDM5B expression correlates with the expression of stem cell markers in various cancer types [2, 45, 62]. Many of these studies explain the rise of a slow-cycling population as an outcome of random stochastic switching within the population [2, 3]. However, we have shown that chronic stress and drug treatments induce the upregulation of KDM5A and KDM5B in multiple cancer types [45, 63]. KDM5 gene is shown to modulate oxidative stress response in a Drosophila model, therefore its upregulation would illustrate an evolutionarily conserved mechanism for coping with stress [64]. Indeed, KDM5A plays an important role in hypoxia-induced chromatin reprogramming [65] while hypoxia-inducible factor HIF1α is a direct transcriptional regulator of KDM5B [66]. In line with the correlation of KDM5 with a slow-cycling state, H3K4me3 demethylation by KDM5A and KDM5B is associated with cellular senescence [67], indicating a conserved mechanism for KDM5 to cope with stressful environments. However, KDM5B was also shown to prevent terminal differentiation of embryonic bodies [68]. Therefore, KDM5 could be a double-edged sword, acting initially as tumor suppressor and senescence inducer but later driving cancer progression.
Another key marker is NGFR, which was shown to be associated with stemness in melanoma [46, 69], breast cancer [70], colon cancer [71] and squamous cell carcinoma [72, 73]. NGFR expression is transient [45, 74], similar to that of KDM5A and KDM5B, and represents a slow-cycling phenotype in multiple cancer types including melanoma [45], breast cancer [75], lung cancer [63] and squamous cell carcinoma [73]. The induction of NGFR by interferon gamma (IFN-γ) in cancer cells [76] suggests a generic stress-induced phenotype. We have shown that NGFR expression is indeed induced by multiple stress factors such as drug exposure, hypoxia and glucose starvation in multiple cancer types [45, 63] and correlates with KDM5A/B expression. In many cancers, NGFR expression correlates with ALDH activity [45, 70, 77], which is upregulated in cancer persisters [78, 79]. However, since ALDH activity is regulated by a family of ALDH genes [80], its applicability to consistently identify persister population needs further testing. NGFR inhibits p53 activity [81] and protects cells from reactive oxygen species [82], which would account for the drug resistant mechanism of NGFR. NGFR expression is associated with resistance to MAPK inhibitors (MAPKi) in melanoma [45, 83] and chemotherapy in multiple cancer types [81, 84]. NGFR expression is also associated with downregulation of melanoma antigens, suppression of cytotoxic T cell responses [76] and resistance to adoptive T cell transfer therapy in melanoma patients [85] and mouse models [86]. Taken together, these studies indicate that a stress-induced shift to a slow-cycling state of cells expressing KDM5 and NGFR serves as a crucial step for cancer progression and therapy resistance. We will further discuss how the stress-induced expression of these factors contribute to disease progression through epigenetic reprogramming in the section below.
Role of stress-induced epigenetic reprogramming in cellular plasticity and drug resistance
Epigenetics modifies genes via several mechanisms including DNA methylation and histone modifications. Epigenetic modulation plays a central role in phenotypic plasticity, and determines cell fate in many processes such as senescence induction and maintenance, important steps for slow-cycling cancer cells. The stress-induced, slow-cycling subpopulation of cancer cells carries a resemblance to a SIPS phenotype of non-cancerous cells. SIPS is regulated through temporal histone modifications rather than DNA methylation processes [40, 87], whereas replicative senescence is controlled by heritable DNA methylation changes such as global hypomethylation [88] of DNA and focal DNA hypermethylation [89]. The modifications in SIPS include a loss of transcriptional activator mark, H3K4me3 [67, 90], and a gain of transcriptional repressor mark, histone 3 lysine 9 trimethylation (H3K9me3) [91, 92], both of which result in repressive chromatin formation and gene silencing. H3K9me3 plays an important role in the formation of senescence-associated heterochromatin foci in replicative senescence, embryonic senescence and SIPS [92-94], suggesting their generic role in multiple senescent phenotypes. Accordingly, a shift to a slow-cycling stem-like state in cancer cells is accompanied by a loss of [2, 45, 58, 63]. The H3K4me3 demethylation is carried out by KDM5A and KDM5B [2, 45, 58], whereas the modification of H3K9me3 is achieved by two histone methyltransferases, SET domain bifurcated histone lysine methyltransferase 1 (SETDB1) and SET domain bifurcated histone lysine methyltransferase 2 (SETDB2) [63, 90]. SETDB1 is involved in cellular senescence through formation of heterochromatin structures, which then leads to alternative lengthening of telomeres and cancer cell immortality [95]. In line with this, SETDB1 has been shown to contribute to melanoma progression and therapy resistance [96]. On the other hand, the role and function of SETDB2 in the process of senescence and slow-cycling cancer cells remain largely unknown. In addition, despite the above-mentioned important roles for histone modifications in a slow-cycling stem-like state in cancer cells, no correlation exists between DNA methylation patterns and gene expression in these slow-cycling cancer cells nor are the DNA methylation patterns associated with this shift [63]. This is not surprising, as changes in DNA methylation patterns are considered to be more stable and heritable than histone modifications, which would explain the transient and reversible phenotype of a slow-cycling state. Subsequently, the H3K4me3 and H3K9me3 epigenetic patterns are reversible when the stress exerted on the cells is released. Furthermore, while epigenetic phenotypes of low H3K4me3 and high H3K9me3 are commonly observed in various cancer types, the gene expression profiles altered by the shared epigenetic patterns are cancer type-specific rather than universal [63]. This is expected, as different cancer types need to upregulate or downregulate a unique set of genes to switch into a slow-cycling cancer persister state.
When cancer cells are treated with drugs continuously, the slow-cycling phase leads to an acquired drug-resistant state that is phenotypically stable compared to the transient, slow-cycling state [4, 45, 83]. This observation suggests that the slow-cycling state is important for two distinct functions: The first being its role in ensuring survival of the cell population after acute stress and the second being its capacity to develop a stable drug resistant phenotype. This could happen either through the gain of genetic changes or epigenetic cellular reprogramming in the slow-cycling state. Recent studies have demonstrated the involvement of the latter process in the development of acquired drug resistance that is phenotypically stable. For example, when melanoma cells were treated with MAPKi for a long period, they underwent global epigenetic reprogramming and upregulated the expression of drug resistant genes such as APC down-regulated 1 (APCDD1), epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor beta (PDGFRB) and neuregulin 1 (NRG1). This process was gradual and dependent on the continuous exposure to the drug, resulting in a shift from a transient transcriptional state to a stable resistant state [83]. Accordingly, escape of cancer cells from drug-induced senescence was associated with a gain of specific H3K4me3 peaks that lead to CSC gene activation [41], which could aid global epigenetic reprogramming. The involvement of transcriptional reprogramming in the escape of cancer cells from a slow-cycling state to a stable resistant state was also reported in other melanoma studies [97, 98], one of which shows an association of this reprogramming with histone 3 lysine 27 (H3K27) acetylation and a mesenchymal phenotype [97]. H3K27 acetylation is a transcriptional activator and negatively correlates with DNA-methylated CpG islands that correspond to gene inactivation [99]. These studies suggest that even though transient subpopulations of preexisting cells have an advantage against inhibitors during the initial selection process, they further undergo a time-dependent epigenetic reprogramming to acquire a stable resistant state. Interestingly, while the stable resistant phenotype showed substantial changes in the global access to chromatin, the epigenetic changes in transient resistant cells were only marginal in comparison to untreated cells, suggesting its gradual shift over the course of treatment [83]. These observations indicate that multiple interconnected epigenetic mechanisms could be involved in global epigenetic remodeling. They also suggest that DNA methylation could have a decisive role in determining stable resistant phenotypes, even though its involvement is limited in a transient resistance mechanism. Non-genetic progressive transformations leading to drug resistance were also reported in lung cancer patients who underwent EGFR inhibitor therapy [100, 101]. The underlying mechanism that leads to epigenetic reprogramming is not known. Furthermore, while the early transient phenotype is well documented in certain cancer types, whether this early phenotype directly contributes to acquired stable resistance is still a topic under investigation [102].
When cancer cells are treated with drugs continuously until they acquire drug resistance, stopping the drugs thereafter can lead to sudden hyperactivation of pathways and fitness deficit in the resistant populations [4, 5, 103, 104]. However, this growth disadvantage of resistant cancer cells could be overcome by epigenetic or transcriptional remodeling, resulting in the development of a stable drug-resistant phenotype and tumor progression. For example, dual specificity phosphatase 5 (DUSP5) is required for recalibrating MAPK activation in cancer cells that have accrued B-Raf proto-oncogene, serine/threonine kinase (BRAF) mutations [105]. Similarly, SHOC2 scaffold protein is needed for NRAS Q61K-induced resistance to BRAF inhibitors [106]. These observations suggest that epigenetic reprogramming is utilized by resistant cancer cells to negate the fitness deficit induced by genetic changes, which, cells gradually evolve to defeat through factors that help them to recalibrate the activation of signaling pathways. Therefore, in order to control drug-resistant cancer cells that have acquired resistant genetic changes, the strategy to administer drugs intermittently would be effective, because 'drug holidays' would result in the loss of fitness in the resistant cells and subsequent outgrowth of drug-sensitive parental cells [4]. These mechanisms attribute to the success of 'drug holidays' in MAPKi resistance in melanoma and EGFRi resistance in lung cancer patients [4, 5, 103, 104, 107]. Furthermore, 'drug holidays' could prevent transiently resistant cancer cells from acquiring a stable resistant phenotype, as stable epigenetic/transcriptional reprogramming is usually achieved through continuous drug exposure [83, 108].
In addition to the resistance to signaling inhibitors, stress-induced phenotypic plasticity may be involved in resistance to PD-1 checkpoint inhibition. Melanoma tumors that acquire BRAF/MEK inhibitor resistance are shown to display a mesenchymal phenotype and an immunologically-cold, M2 macrophage signature with decreased infiltration of cytotoxic CD8 T cells compared to the pre-treatment tumor [109, 110]. Accordingly, BRAF/MEK inhibitor resistance signatures correspond with an innate resistance to anti-PD-1 therapy signature, and share many factors that induce immune escape, including AXL receptor tyrosine kinase (AXL) [111], vascular endothelial growth factor (VEGF) [112], IL8 [113] and C-C motif chemokine ligand 2 (CCL2) [114, 115]. These observations could explain why patients who progress under BRAF/MEK inhibitors respond poorly to immune checkpoint inhibition [116-118]. In line with these reports, the expression of KDM5B in cancer cells, corresponding to the slow-cycling phenotype, leads to the suppression of anti-tumor immune responses by inducing EMT, suppressing interferon response signaling pathways and downregulating cancer testis antigen expression, all of which are important for immune surveillance [119, 120].
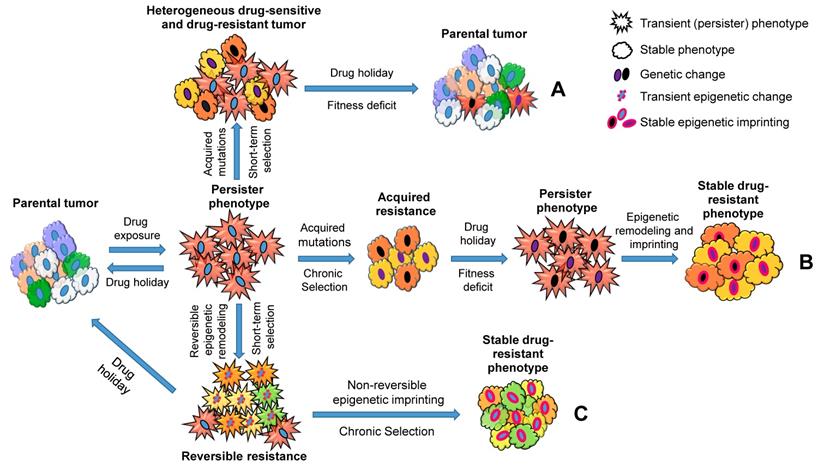
Taken together, these studies suggest a central role of stress-induced phenotypic plasticity that contributes to acquired drug resistance (Figure 2). This could be achieved through slow-cycling persister cells either gaining mutations (Figure 2; A and B) or undergoing epigenetic reprogramming (Figure 2; C). When cancer cells receive a short-term drug treatment, 'drug holidays' induce persister cells to differentiate back to the parental tumor phenotype and outgrow the cancer cells that acquired drug-resistant genetic changes, because the latter cells lose fitness and will acquire a persister phenotype in the absence of the drug (Figure 2; A). However, when cancer cells receive a long-term treatment, the fitness deficit induced by 'drug holidays' would be overcome by epigenetic modelling, and cancer cells regain cellular homeostasis (Figure 2; B). In addition, long-term exposure of drugs would induce phenotypic plasticity and stable drug resistance in cancer cells without genetic changes (Figure 2; C). Overall, a ubiquitous stress-induced epigenetic plasticity plays a critical role in the evolution of cancer drug resistance and disease progression. The mechanisms driving this important shift still remains to be elucidated.
A schematic representation describing how cancer persister cells contribute to therapy resistance through genetic and epigenetic changes. Parental cell populations under drug treatment acquire a dormant persister phenotype (star-shaped red cells). The transition is transient in nature and dependent on continuous drug exposure. The persister cells can acquire mutations that produce resistant subpopulations (A and B: Mutant subpopulations are shown using the changes in nuclear and cytoplasmic colors, representing genetic and phenotypic changes, respectively). Under a short-term drug selection followed by a drug holiday, the mutant drug-resistant subpopulations shift their phenotypes into dormant persisters due to the lack of fitness in the absence of drug, and non-mutant cells in turn switch back to parental subpopulations (A). On the other hand, under a chronic drug selection, the mutant subpopulations outgrow the non-mutant populations; however, under a drug holiday, the lack of fitness in the mutant populations leads to a persister phenotype that could be overcome by epigenetic reprogramming (Epigenetically reprogrammed subpopulations are shown using the change in nuclear membrane colors to pink). The final phenotype observed in this process is stable and does not respond to drug treatments or drug holiday (B). In contrast to the genetic changes observed in A and B, the dormant persister cells could also undergo epigenetic remodeling to acquire a semi-proliferative transient resistant state (multi-colored star-shaped population with pink dotted nuclear membrane). When these cells are treated with drugs continuously, they can further undergo epigenetic imprinting to transition into a resistant phenotype (C, right. Epigenetically reprogrammed subpopulations are shown using the change in their nuclear membrane color to pink). Similar to the resistant cells caused by genetic changes (B), the final phenotype observed in the process (C) is stable and does not respond to drug treatments or drug holiday. However, a drug holiday before establishing this stable phenotypes could prevent semi-proliferative transiently resistant cells from acquiring stable resistance (C, left).
Metabolic remodeling during phenotypic plasticity and therapy resistance
Metabolic adaption is a hallmark of cancer development [121] and significantly contributes to stem cell reprogramming and epigenetic regulation [122-124]. The importance of metabolism for cellular plasticity is evident by the vastly different metabolic states of embryonic stem cells (ESCs) compared to most differentiated cells. ESCs rely on high rates of glycolysis and have poorly developed, highly fragmented mitochondria that mature during differentiation, coinciding with a switch from glycolysis to oxidative phosphorylation [125]. The critical role of metabolism in ESCs is also evident in stemness transcription factor-induced cellular reprogramming [126]. In fact, the generation of induced pluripotent stem cell (iPSC) is accompanied by mitochondrial remodeling, resulting in immature spherical and cristae-poor structures and a glycolysis-dependent metabolic phenotype [126] that is, at least in part, regulated by de novo fatty acid synthesis to facilitate mitochondria fission [127].
Despite the morphologic evidence that suggests underdeveloped mitochondria in stem cells, human pluripotent stem cells (hPSC) mitochondria are capable of respiration at maximal capacity, with the mitochondria uncoupling protein UCP2 preventing glucose-derived pyruvate oxidation [128]. Moreover, glutamine oxidation is crucial for the maintenance of TCA cycle intermediates and ultimately hPSC survival [129]. Overall, hPSC show a substantial plasticity in their metabolic program whereas their energy requirements mainly depend on glycolysis, with mitochondria metabolism playing crucial roles for survival and cell fate decisions [124, 130]. Similarly, cells that transition to a senescent phenotype undergo extensive metabolic remodeling that can lead to increased glycolysis and/or oxidative phosphorylation [131].
One of the key events in oncogene-induced senescence is a specific shift of pyruvate utilization toward the TCA cycle [132]. BRAFV600E-induced senescence in human fibroblasts activates pyruvate dehydrogenase (PDH) through suppression of pyruvate dehydrogenase kinase 1 (PDK1) and induction of pyruvate dehydrogenase phosphatase 2 (PDP2), resulting in cell cycle arrest and senescence [132]. Overexpression of PDK1 allows p53-depleted BRAFV600E-expressing melanocytes to develop tumors, whereas PDK1 knockdown in melanoma reduces tumor initiation, progression and maintenance [132], supporting a tumor-promoting role of PDK1. The requirement of a PDK1-mediated switch, in and out of a senescence-like state, to promote tumor initiation and maintenance is reminiscent of the KDM5Bhigh slow-cycling melanoma subpopulation that drives tumor initiation, propagation and drug resistance [3, 58]. KDM5Bhigh cells are also dependent on mitochondria metabolism with increased dependence on glucose and fatty acids to fuel their oxidative phosphorylation [3, 133]. Interestingly, PDK1 knockdown sensitizes BRAF-mutant melanoma to BRAF inhibitor treatment [132], suggesting the role of senescence PDK1-mediated drug resistance. It would be interesting to test how PDK1 affects markers of cancer persisters, such as KDM5B and NGFR, and whether the knockdown cells are still capable of stochastic phenotypic switching. What we do know is that the emergence of drug tolerant melanoma cells is accompanied by a BRAF inhibitor-induced suppression of glycolysis [134] as well as dependence on mitochondrial biogenesis and oxidative phosphorylation [3, 135]. A similar reliance is found in stem-like drug resistant subpopulations of chronic myeloid leukemia [136], acute myeloid leukemia [137], breast cancer [138] and glioblastoma [139], suggesting that a metabolic switch toward a mitochondria-dependent phenotype is common for therapy-resistant stem-like cancer cells. A consequence of increased oxidative phosphorylation is, among others, tissue hypoxia due to increased oxygen consumption, which is associated with T-cell exhaustion and therefore, decreased anti-PD-1 response in melanoma mouse models [140]. This effect can be overcome by oxidative phosphorylation inhibitors such as metformin [141] that target metabolic adaptions to boost immunogenicity. A similar shift toward oxidative phosphorylation is observed in melanoma brain metastases, and linked to immunosuppression and brain metastasis incidence [142]. It is worth noting that glycolysis-related gene expression patterns and tumor glycolytic activity have been negatively correlated with T-cell infiltration, leading to adoptive T-cell therapy failure in melanoma and lung cancer [143]. This is in part mediated by tumor-derived lactate, a byproduct of anaerobic glycolysis in the tumor microenvironment, that results in the suppression of natural killer cell cytolytic function [144] and polarization of tumor-promoting macrophages [145], suggesting that a balance between oxidative and glycolytic metabolism is important to evoke an effective anti-tumor immune response.
An intriguing feature of slow-cycling CSCs in glioblastoma and ovarian cancer is a specific increase in unsaturated lipid metabolites [139, 146] that was later found to be a therapeutic vulnerability [146]. This resembles the recently identified increase of unsaturated fatty acids in the persister cells of various cancer types after treatment with anti-cancer drugs [55, 147], which leads to a reliance on enzymes involved in the detoxification of lipid peroxides, mainly glutathione peroxidase 4 (GPX4) to prevent ferroptosis [55, 56, 147, 148]. This vulnerability to iron-dependent oxidative stress is also found in response to immune therapy [98]. Mechanistically, ferroptosis requires high levels of polyunsaturated fatty acids (PUFA) containing phospholipids in the cell membrane, which are prone to oxygenation [149], suggesting that the observed increase in unsaturated fatty acids and the resulting GPX4 dependence is caused by increased unsaturation of cancer persister cell membranes. In clear cell carcinoma, a highly aggressive form of kidney cancer, HIF-2α was identified as a driver of PUFA generation, a factor that has been described to stabilize β-catenin that mediates lipid desaturation [148, 150-153]. Interestingly, WNT-beta-catenin signaling and a permissive H3K4me3 epigenetic landscape at WNT and stemness-related genes are key mechanisms involved in senescence-associated cancer cell reprogramming [41]. Accordingly, fatty acid unsaturation has been described to be increased in replicative senescence [154] and oncogene-induced senescence [155], which is also reflected by increased lipid peroxidation in therapy-induced senescent cells [156], suggesting that a senescence-like metabolic remodeling process is involved in cancer persistence.
One functional consequence of the ferroptosis-sensitive state is immunoevasion, possibly through the release of immunosuppressive eicosanoids such as prostaglandin E2 [157]. Furthermore, the presence of oxidatively-truncated lipids has been shown to interfere with major histocompatibility complex (MHC)-mediated antigen cross-presentation by dendritic cells in cancer [158], and alterations of the membrane lipid composition, specifically increased PUFA content, have been shown to negatively affect antigen presentation and modulate the inflammatory eicosanoid metabolites [159], suggesting decreased immunogenicity of cancer persister cells. Besides changes in lipid metabolism, alterations in amino acid metabolism, specifically tryptophan, have significant immunosuppressive consequences. Tryptophan is an essential amino acid that shows increased uptake in tumor tissue [160] . Recently, the MYC-dependent upregulation of the tryptophan transporters SLC7A5 and SLC1A5 and the tryptophan catabolizing enzyme arylformamidase (AFMID), involved in the conversion of tryptophan into kynurenine, has been identified as a mechanism underlying increased tryptophan uptake in colon cancer cells [161]. Within tumor cells, indoleamine-2,3-dioxygenase (IDO) and tryptophan-2,3-dioxygenase (TDO)-mediated tryptophan catabolism produces the active metabolite kynurenine, an agonist of the aryl hydrocarbon receptor (AhR) that exerts immunosuppressive functions [162]. The kynurenine-AhR metabolic circuit has been implicated in the dormancy of stem-like tumor repopulating cells (TRC) in response to IFN-γ stimulation [163]. These TRCs actively import tryptophan and produce kynurenine, which is released into the tumor microenvironment where it stimulates PD-1 expression in adjacent CD8+ T-cells in a kynurenine-AhR-dependent manner to decrease the immune response [164] and has therefore attracted vast interest in the development of combination therapies including anti-PD-1 therapy [165]. This is in line with the previously mentioned transcriptomic signatures of residual MAPKi-treated melanoma that resembles an innate anti-PD-1 resistance (IPRES) signature [109, 110] and the clinical observations that anti-PD-1 therapy is less effective immediately following BRAF inhibition [116]. Overall, these studies suggest that stress-induced phenotypic switching to a drug resistant state mediates cross-resistance to PD-1 immune checkpoint inhibitors, which may be mediated, at least in part, by metabolic remodeling.
Crosstalk between metabolic remodeling and epigenetic regulation
As previously mentioned, a hallmark of cancer persisters is remodeling of their epigenetic landscape [45, 63, 79], a process that is inherently linked to metabolism [166]. As described above, the persister phenotype contributing to cancer stemness may require multiple metabolic shifts, one allowing the senescence phenotype and the other allowing the escape from senescence. Hence, dynamic regulation of metabolism, which is linked to epigenetics, could be a key factor driving this plasticity. The shift toward oxidative phosphorylation-dependent metabolism in cancer persister cells can have a strong influence on epigenetics, as multiple TCA cycle intermediates directly affect epigenetic reactions. Among the most important metabolites involved in epigenetic regulation is alpha ketoglutarate (α-KG), which is a co-factor of α-KG-dependent dioxygenases of the Jumonji C (JmjC)-family of histone demethylases and ten-eleven translocation (TET)-family of DNA demethylases, both important to maintain stem cell self-renewal [167] and influence early differentiation of human pluripotent stem cells [168]. These reactions convert α-KG to succinate, whereas succinate and the structurally similar fumarate, both TCA cycle intermediates, act as competitive inhibitors [169]. For example, accumulation of fumarate has been shown to drive EMT through inhibition of TET enzyme-mediated DNA demethylation and subsequent decreased expression of miR-200 [170]. Accordingly, KDM5 family H3K4 demethylases belonging to JmjC family are known to be dependent on α-KG for their function [171]. Considering the central role of KDM5A and KDM5B for the development of cancer persisters [2, 3], it is very likely that the observed shift toward oxidative phosphorylation is, at least in part, necessary to supporting epigenetic remodeling. Besides glucose-derived carbon, glutamine is a key amino acid for cellular energetics that is often used to feed TCA cycle intermediates in cancer cells [172], a phenomenon reported during BRAF inhibitor resistance [173]. In contrast, localized glutamine deficiency in solid tumors results in histone hypermethylation of H3K4me3, H3K9me3, histone 3 lysine 27 trimethylation (H3K27me3) and histone 3 lysine 36 trimethylation (H3K36me3) [174]. This shift in epigenetic state leads to cancer cell dedifferentiation and BRAF inhibitor resistance because of decreased α-KG availability and consequently, inhibition of JmjC-family histone demethylases. These cells, subjected to α-KG deficiency, show increased expression of cancer persister marker, NGFR. This observation suggests that a delicate balance in α-KG levels is necessary to maintain the histone epigenetic patterns of cells. A shift in this balance seems to trigger a similar response either through histone hyper- or hypo- methylation, which is quite intriguing. For example, this effect could be due to the upregulation of transcriptional repressor marks, H3K9me3, H3K27me3 and H3K36me3, which could override the effect of H3K4me3 upregulation. Further understanding of the role of KDM5 enzymes in the context of histone hypermethylation is needed.
The one carbon donor, S-adenosylmethionine (SAM), regulates histone and DNA methylation, and H3K4me3 is specifically sensitive to fluctuations in SAM concentrations [175]. SAM is the main product of the one carbon cycle and its synthesis and regeneration requires amino acid metabolism [176]. Changes in methionine availability, the amino acid that is directly converted to SAM, have been shown to influence H3K4me3 peak width and subsequently, gene expression [177]. Depletion of methionine is a vulnerability of tumor-initiating lung CSCs as they are characterized by high methionine cycle activity, which, if not maintained, presumably leads to alterations of the epigenetic landscape, blocking tumor initiation and survival of tumor-initiating cells (TICs) [178]. This suggests that the availability of SAM may positively regulate the capacity of cancer persisters to exit the slow-cycling state and gain expression of stem cell factors, which could be dependent on H3K4me3 [41]. Similarly, CSCs specifically require SAM biosynthesis to maintain viability and H3K4me3 marking [179], exemplifying the fundamental connection of metabolism and epigenetic plasticity. Beside methionine, serine has been reported to contribute to the maintenance of the one carbon cycle, supporting de novo nucleotide synthesis by fueling the folate cycle [180], also known as serine, glycine, one-carbon pathway [181]. This phenomenon has been shown to occur specifically in TICs [182], and has been described during the development of neuroendocrine prostate cancer, the most lethal subtype of castration-resistant prostate cancer. De novo serine synthesis that might rely on retrograde flux through glycolysis [183], together with glucose-derived ribose, are major contributors that fuel de novo ATP synthesis to drive SAM generation in inflammatory macrophages [184], highlighting the importance of glycolysis to maintain one carbon pools. Beyond that, one carbon metabolism and SAM levels are strongly influenced by mitochondria dysfunctions, which increase serine biosynthesis and affect polyamine and methionine metabolism as a direct result of changes in TCA flux, resulting in DNA hypermethylation and transcriptional changes [185, 186]. In general, metabolic flux through the TCA cycle, the pentose phosphate pathway and the serine, glycine, once carbon pathway (SGOCP) are interconnected and it appears that phosphoglycerate dehydrogenase (PHGDH), the enzyme that commits carbon units to de novo serine biosynthesis, coordinates this central carbon metabolism [187]. An intriguing example for the interplay between these pathways is the identification of the serine-responsive SAM-containing metabolic enzyme complex in yeast [188]. This complex consists of the yeast analogs of pyruvate kinase M2, serine metabolic enzymes, SAM synthetases, and an acetyl-CoA synthetase, that interacts with the H3K4 methyltransferase complex SET1 to regulate H3K4me3, amongst other histone modifications [188].
Similar to histone and DNA methylation, histone acetylation and deacetylation are dependent on the availability of metabolic co-factors. Glucose-derived acetyl-CoA is required as a substrate for protein acetylation and is generated in an ATP-citrate lyase (ACL)-dependent manner [189]. AKT activation, which is found in response to treatment with anti-cancer drugs [45, 63], facilitates ACL-dependent acetyl-CoA production in low glucose conditions, possibly aiding increased H3K27 acetylation of cis-regulatory elements found in slow-cycling drug tolerant glioblastoma stem cells [190]. However, in addition to glucose, acetyl-CoA derived from fatty acids [191] or acetate recycling [192] has been shown to fuel histone acetylation. Detailed analysis of metabolic mechanisms that fuel acetylation reactions during the development of drug resistance is warranted. The reverse reaction, histone deacetylation, is also in partly dependent on the availability of the metabolite nicotinamide adenine dinucleotide (NAD+). Skeletal muscle stem cells undergoing a transition from a quiescent to a proliferative state reprogram their metabolism from oxidative phosphorylation dependent to glycolysis dependent, which results in decreased NAD+ availability and subsequently, increased histone acetylation [193]. The importance for NAD+ metabolism for therapy resistance is highlighted by the key roles of NAD+ metabolism for SAM-dependent methylation reactions and glioblastoma stem cell maintenance [194] as well as the dependence of self-renewal and radiation resistance of glioblastoma stem-like cells on nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting step in NAD+ synthesis [195]. Whether or not NAD+ metabolism is important for cancer persistence is currently unclear but the high degree of lipid desaturation discussed previously would be an intriguing avenue that has been shown to contribute to NAD+ recycling [196].
Another interesting caveat is the local synthesis of metabolites to support enzymatic reactions, which has been demonstrated for the nuclear synthesis of fumarate [197] and acetyl-CoA [198] as well as the previously mentioned SAM-containing metabolic enzyme complex [188]. A more comprehensive nuclear translocation of TCA cycle enzymes occurs during zygotic genome activation (ZGA) in early embryogenesis, a shift depending on protein O-GlcNAc transferase (OGT) ultimately promoting epigenetic remodeling [199]. Interestingly, OGT is known to play an important role in multiple stress responses including oxidative, ER, and genotoxic stresses [200]. Chromatin-associated fumarase is phosphorylated by AMPK at Ser75 to maintain di-methylation at the 36th lysine residue of the histone H3 (H3K36me2) and a gene expression profile that facilitates cell cycle arrest [201]. This is counteracted by O-GlcNAcylation of the same serine residue to avoid cell growth arrest, even in glucose-deprived conditions [201], and is overall reminiscent of the key role of the hexosamine biosynthesis pathway, OGT and protein O-GlcNAcylation to overcome KRAS proto-oncogene, GTPase (KRAS)-induced [202] or radiation-induced senescence [203]. Furthermore, O-GlcNAcylation has been shown to be important to maintain acute myeloid leukemia (AML) in an undifferentiated state [204] and has a key role in maintaining pluripotency [205], suggesting that the metabolic reprogramming that fuels the hexosamine biosynthesis pathway might be important to reactivate the genome and initiate a stem-like cell state following senescence-like reprogramming. It remains to be investigated if a nuclear shift of metabolic enzymes is involved in the epigenetic regulation of CSCs or cancer persister cells, which might represent an intriguing aspect of metabolic and epigenetic crosstalk in cancer plasticity that is yet to be explored.
Similar to the profound consequences of metabolic alteration that shape the epigenetic landscape, many metabolic pathways are regulated by epigenetic mechanisms. A quite intriguing and less explored function of KDM5 proteins is activation of genes against its well-documented gene-repressive function. This function of KDM5 in gene activation has been reported in humans and drosophila pointing to an evolutionarily conserved mechanism [206-208]. Interestingly, KDM5 directly upregulates genes required for mitochondrial function in drosophila through its PHD reader motif which is distinct from its classical JmjC domain-mediated functions [207]. However, this particular function of KDM5B in cancer persisters still remains ambiguous, even though KDM5B expression is shown to correlate with dependency on oxidative phosphorylation. The interdependency of epigenetic and metabolism regulation is exemplified by the direct phosphorylation of the DNA methyltransferase 1 (DNMT1), the histone acetyltransferase 1 (HAT1), and the HAT co-activator RB binding protein 7 (RBBP7) by the metabolic master regulator AMPK [209]. Phosphorylation of these proteins facilitates mitochondria biogenesis by increasing the expression of peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α), nuclear respiratory factor 1 and 2 (NRF1 and NRF2), and the mitochondrial transcription factor A (TFAM), and controls the mitochondrial membrane potential through expression of uncoupling proteins 2 and 3 (UCP2 and UCP3) [209]. Furthermore, studies in C. elegans showed that the H3K27 demethylases JMJD-1.2 and JMJD-3.1 regulate the mitochondrial unfolded protein response to maintain mitochondrial proteostasis, which also modulates longevity in mouse models [210].
Glycolytic pathways are also subject to extensive epigenetic regulation. The EMT regulatory complex Snail-G9a-Dnmt1 was found to suppress expression of fructose-1,6-biphosphatase in basal-like breast cancer through increased histone 3 lysine 9 dimethylation (H3K9me2) and DNA methylation [211]. Loss of fructose-1,6-biphosphatase, rate-limiting enzyme in gluconeogenesis, drives a shift towards increased glucose uptake and glycolysis, while inhibiting oxygen consumption and mediating the development of CSC-like characteristics [211]. Furthermore, glucose uptake has been shown to be regulated by CpG island hypermethylation-linked inactivation of DERL3, a member of the protein degradation pathway, which leads to the accumulation of GLUT1 and contribution to glycolytic phenotypes in cancer cells [212]. The glycolytic phenotype of cancer cell appears to be strongly mediated by epigenetic regulation as sirtuin (SIRT) 6, a H3K9 deacetylase that also controls cellular senescence [213], is a tumor suppressor that regulates aerobic glycolysis in cancer cells [214].
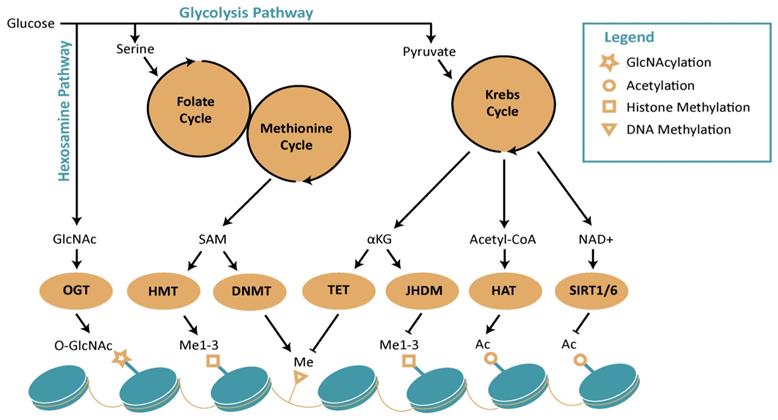
In conclusion, the literature suggests intertwound mechanisms that connect metabolic changes to epigenetic reprogramming, which could play a central role in stress-induced phenotypic plasticity (Figure 3).
Schematic diagram representing interplay between metabolism and epigenetics. The figure represents how distinct metabolic pathways, which includes hexosamine pathway, Krebs cycle, Folate and methionine cycle, contribute to epigenetic remodeling. Abbreviations: DNMT: DNA methyl transferase; GlcNAc: N-acetylglucosamine; HAT: histone acetyltransferases; HMT: histone methyltransferases; NAD+: nicotinamide adenine dinucleotide; OGT: O-GlcNAc transferase; SAM: S-adenosylmethionine; SIRT: sirtuins; TET: ten-eleven translocation methylcytosine dioxygenase; α-KG: alpha ketoglutarate
Conclusion
Growing evidence indicates a central role for a stress-induced phenotypic plasticity in cancer progression and drug resistance. The drug-induced phenotypic plasticity leads to an initial shift of cells into a slow-cycling persister phenotype, which acts as a reservoir for the accumulation of genetic or epigenetic changes that drive the process of acquired drug resistance. This process could be critical for drug resistance to not only chemotherapy but also targeted and immune checkpoint inhibitors. Phenotypic and epigenetic plasticity is closely linked to metabolic remodeling, suggesting a close interplay between epigenetic and metabolic pathways. Furthermore, epigenetic changes are progressive rather than stochastic, suggesting a tightly regulated time-bound mechanism, which determines transient or permanent epigenetic marks contributing to the respective phenotype.
Multiple strategies have been proposed and tested to target this process of drug-induced phenotypic plasticity, which have shown promise to varying degrees. Drug holidays and intermittent treatment strategy have shown clinical efficacy [215-218]. Most studies were conducted in patients who relapsed after continuous treatment of the drug. Although the patients regained partial sensitivity to the same drug after discontinuation of the drug, the precise regimen, its safety and efficacy still need further testing. Clinical trials (NCT03352947, NCT02196181) are ongoing to address these questions in melanoma patients. In addition, histone deacetylase inhibition is reported to deplete quiescent stem-like cells in multiple cancer types [219, 220], and a strategy to combine drug holidays with vorinostat, a histone deacetylase inhibitor, has shown better effects than the combination of BRAF-MEK inhibitors in a melanoma model [221]. A clinical trial is ongoing in melanoma patients who showed signs of progression on BRAF-MEK inhibitor therapy (NCT02836548). A similar strategy of combining drug holidays with chemotherapeutic agent dacarbazine has also shown efficacy in melanoma pre-clinical models [104]. The success of these strategies could be due to the time-dependent vulnerability induced by drug holidays during which the cells undergo phenotypic switching. Strategies targeting the metabolic reprogramming through inducing ferroptosis have also shown promise and await further testing in clinical settings [55, 98]. Although many strategies that target phenotypic plasticity, epigenetics and metabolism are promising, the underlying mechanisms that determine a transient or permanent phenotype remain elusive and need further investigation. Single-cell omics analysis could be employed to dissect the heterogeneity and the transition state of stress-induced phenotypic plasticity, and further decipher transcriptional, epigenetic and metabolic crosstalks. A deeper understanding of the mechanisms that contribute to this plasticity triggered by epigenetic and metabolic changes will open a plethora of therapeutic opportunities.
Abbreviations
ABCB5: ATP-binding cassette subfamily B member 5
ACL: ATP-citrate lyase
AFMID: arylformamidase
AhR: aryl hydrocarbon receptor
ALDH: aldehyde dehydrogenase
AML: acute myeloid leukemia
AMPK: AMP-activated protein kinase
APCDD1: APC down-regulated 1
AXL: AXL receptor tyrosine kinase
BRAF: B-Raf proto-oncogene, serine/threonine kinase
CCL2: C-C motif chemokine ligand 2
CSCs: cancer stem cells
DUSP5: dual specificity phosphatase 5
DNMT1: DNA methyl transferase 1
EGFR: epidermal growth factor receptor
EMT: epithelial-mesenchymal transition
ESC: embryonic stem cells
GPX4: Glutathione Peroxidase 4
GlcNAc: N-acetylglucosamine
HAT1: histone acetyltransferases 1
HMT: histone methyltransferases
hPSC: human pluripotent stem cells
H3K4me3: histone 3 lysine 4 trimethylation
H3K9me3: histone 3 lysine 9 trimethylation
H3K9me2: histone 3 lysine 9 dimethylation
H3K27: histone 3 lysine 27
H3K27me3: histone 3 lysine 27 trimethylation
H3K36me3: histone 3 lysine 36 trimethylation
H3K36me2: histone 3 lysine 36 dimethylation
IDO: indoleamine-2,3-dioxygenase
IFN-γ: interferon-γ
IPRES: innate anti-PD-1 resistance signature
iPSC: induced pluripotent stem cell
JARID1: Jumonji AT-rich interactive domain 1
JmjC: Jumonji C
KDM5A: lysine-specific demethylase 5A
KDM5B: lysine-specific demethylase 5B
KRAS: KRAS Proto-Oncogene, GTPase
MAPK: mitogen-activated protein kinase
MHC: major histocompatibility complex
NAD+: nicotinamide adenine dinucleotide
NAMPT: nicotinamide phosphoribosyltransferase
NGFR: nerve growth factor receptor
NRAS: neuroblastoma RAS Viral (V-Ras) Oncogene Homolog
NRF1: nuclear respiratory factor 1
NRF2: nuclear respiratory factor 2
NRG1: neuregulin 1
NSG: NOD scid gamma mouse
OGT: O-GlcNAc transferase
OXPHOS: oxidative phosphorylation
PD1: programmed cell death protein 1
PDGFRB: platelet-derived growth factor receptor beta
PDH: pyruvate dehydrogenase
PDK1: pyruvate dehydrogenase kinase 1
PDP2: pyruvate dehydrogenase phosphatase 2
PGC-1α: peroxisome proliferator-activated receptor gamma coactivator-1α
PHGDH: phosphoglycerate dehydrogenase
PUFA: polyunsaturated fatty acids
RBBP7: RB binding protein 7
SAHF: senescent associated heterochromatin foci
SAM: S-adenosylmethionine
SCID: severe combined immunodeficiency
SETDB1: SET domain bifurcated histone lysine methyltransferase 1
SETDB2: SET domain bifurcated histone lysine methyltransferase 2
SGOCP: serine, glycine, one-carbon pathway
SIPS: stress-induced premature senescence
SIRT: sirtuin
TDO: tryptophan-2,3-dioxygenase
TET: ten-eleven translocation methylcytosine dioxygenase
TFAM: mitochondrial transcription factor A
TIC: tumor initiating lung cancer stem cells
TRC: tumor repopulating cells
VEGF: vascular endothelial growth factor
ZGA: zygotic genome activation
α-KG: alpha ketoglutarate
Acknowledgements
We thank Ms. Joanne Domenico and Dr. Zhai Zilli (Dermatology, UCD) for editing the manuscript. This work was supported by an NIH/NCI R01CA197919 (to M. Fujita), Veterans Affairs Merit Review Award 5I01BX001228 (to M. Fujita), Cancer League of Colorado (to M. Fujita), Tadamitsu Cancer Research Fund (to M. Fujita), Cancer Council Queensland (to H.Schaider), the Epiderm Foundation, and the Princess Alexandra Hospital Research Foundation (PARSS2016_NearMiss to H.Schaider). H. Hammerlindl is funded by the International Postgraduate Research Scholarship (IPRS) and UQ Centennial Scholarship (UQCent).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sarmento-Ribeiro AB, Scorilas A, Goncalves AC, Efferth T, Trougakos IP. The emergence of drug resistance to targeted cancer therapies: Clinical evidence. Drug Resist Updat. 2019;47:100646
2. Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S. et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69-80
3. Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D. et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell. 2013;23:811-25
4. Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP. et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494:251-5
5. Becker A, Crombag L, Heideman DA, Thunnissen FB, van Wijk AW, Postmus PE. et al. Retreatment with erlotinib: Regain of TKI sensitivity following a drug holiday for patients with NSCLC who initially responded to EGFR-TKI treatment. Eur J Cancer. 2011;47:2603-6
6. Mittal K, Derosa L, Albiges L, Wood L, Elson P, Gilligan T. et al. Drug Holiday in Metastatic Renal-Cell Carcinoma Patients Treated With Vascular Endothelial Growth Factor Receptor Inhibitors. Clin Genitourin Cancer. 2018;16:e663-e7
7. Sheng S, Margarida Bernardo M, Dzinic SH, Chen K, Heath EI, Sakr WA. Tackling tumor heterogeneity and phenotypic plasticity in cancer precision medicine: our experience and a literature review. Cancer Metastasis Rev. 2018;37:655-63
8. Son B, Lee S, Youn H, Kim E, Kim W, Youn B. The role of tumor microenvironment in therapeutic resistance. Oncotarget. 2017;8:3933-45
9. Crispo E. The Baldwin effect and genetic assimilation: revisiting two mechanisms of evolutionary change mediated by phenotypic plasticity. Evolution. 2007;61:2469-79
10. Poljsak B, Milisav I. Clinical implications of cellular stress responses. Bosn J Basic Med Sci. 2012;12:122-6
11. Sanchez Alvarado A, Yamanaka S. Rethinking differentiation: stem cells, regeneration, and plasticity. Cell. 2014;157:110-9
12. Cieslar-Pobuda A, Knoflach V, Ringh MV, Stark J, Likus W, Siemianowicz K. et al. Transdifferentiation and reprogramming: Overview of the processes, their similarities and differences. Biochim Biophys Acta Mol Cell Res. 2017;1864:1359-69
13. Ashcroft P, Michor F, Galla T. Stochastic tunneling and metastable states during the somatic evolution of cancer. Genetics. 2015;199:1213-28
14. Jin R, Chen X, Han D, Luo X, Li H. Clusterin modulates transdifferentiation of non-small-cell lung cancer. BMC Cancer. 2017;17:661
15. Sinha S, Nelson PS. The Path of Most Resistance: Transdifferentiation Underlies Exceptional Nonresponses to Androgen Receptor Pathway Inhibition in Prostate Cancer. Cancer Discov. 2017;7:673-4
16. Huang Z, Wu T, Liu AY, Ouyang G. Differentiation and transdifferentiation potentials of cancer stem cells. Oncotarget. 2015;6:39550-63
17. Ishay-Ronen D, Diepenbruck M, Kalathur RKR, Sugiyama N, Tiede S, Ivanek R. et al. Gain Fat-Lose Metastasis: Converting Invasive Breast Cancer Cells into Adipocytes Inhibits Cancer Metastasis. Cancer Cell. 2019;35:17-32 e6
18. Etchegaray JP, Mostoslavsky R. Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol Cell. 2016;62:695-711
19. Wainwright EN, Scaffidi P. Epigenetics and Cancer Stem Cells: Unleashing, Hijacking, and Restricting Cellular Plasticity. Trends Cancer. 2017;3:372-86
20. Snyder V, Reed-Newman TC, Arnold L, Thomas SM, Anant S. Cancer Stem Cell Metabolism and Potential Therapeutic Targets. Front Oncol. 2018;8:203
21. Zakrzewski W, Dobrzynski M, Szymonowicz M, Rybak Z. Stem cells: past, present, and future. Stem Cell Res Ther. 2019;10:68
22. Valent P, Bonnet D, De Maria R, Lapidot T, Copland M, Melo JV. et al. Cancer stem cell definitions and terminology: the devil is in the details. Nat Rev Cancer. 2012;12:767-75
23. Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer. 2012;12:133-43
24. Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275-84
25. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J. et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645-8
26. Auffinger B, Tobias AL, Han Y, Lee G, Guo D, Dey M. et al. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014;21:1119-31
27. Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci U S A. 2011;108:1397-402
28. Vlashi E, Pajonk F. Cancer stem cells, cancer cell plasticity and radiation therapy. Semin Cancer Biol. 2015;31:28-35
29. Giannoni E, Bianchini F, Masieri L, Serni S, Torre E, Calorini L. et al. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 2010;70:6945-56
30. Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F. et al. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell. 2013;154:61-74
31. Ravindran Menon D, Luo Y, Arcaroli JJ, Liu S, KrishnanKutty LN, Osborne DG. et al. CDK1 Interacts with Sox2 and Promotes Tumor Initiation in Human Melanoma. Cancer Res. 2018;78:6561-74
32. Recasens A, Munoz L. Targeting Cancer Cell Dormancy. Trends Pharmacol Sci. 2019;40:128-41
33. Chen W, Dong J, Haiech J, Kilhoffer MC, Zeniou M. Cancer Stem Cell Quiescence and Plasticity as Major Challenges in Cancer Therapy. Stem Cells Int. 2016;2016:1740936
34. de Magalhaes JP, Passos JF. Stress, cell senescence and organismal ageing. Mech Ageing Dev. 2018;170:2-9
35. Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB. et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349-52
36. Li Y, Zhao H, Huang X, Tang J, Zhang S, Li Y. et al. Embryonic senescent cells re-enter cell cycle and contribute to tissues after birth. Cell Res. 2018;28:775-8
37. Takahashi A, Ohtani N, Yamakoshi K, Iida S, Tahara H, Nakayama K. et al. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat Cell Biol. 2006;8:1291-7
38. Koch CM, Reck K, Shao K, Lin Q, Joussen S, Ziegler P. et al. Pluripotent stem cells escape from senescence-associated DNA methylation changes. Genome Res. 2013;23:248-59
39. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593-602
40. Bielak-Zmijewska A, Wnuk M, Przybylska D, Grabowska W, Lewinska A, Alster O. et al. A comparison of replicative senescence and doxorubicin-induced premature senescence of vascular smooth muscle cells isolated from human aorta. Biogerontology. 2014;15:47-64
41. Milanovic M, Fan DNY, Belenki D, Dabritz JHM, Zhao Z, Yu Y. et al. Senescence-associated reprogramming promotes cancer stemness. Nature. 2018;553:96-100
42. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663-76
43. Chiche A, Le Roux I, von Joest M, Sakai H, Aguin SB, Cazin C. et al. Injury-Induced Senescence Enables In Vivo Reprogramming in Skeletal Muscle. Cell Stem Cell. 2017;20:407-14 e4
44. Mosteiro L, Pantoja C, Alcazar N, Marion RM, Chondronasiou D, Rovira M. et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science. 2016 354
45. Ravindran Menon D, Das S, Krepler C, Vultur A, Rinner B, Schauer S. et al. A stress-induced early innate response causes multidrug tolerance in melanoma. Oncogene. 2015;34:4448-59
46. Boiko AD, Razorenova OV, van de Rijn M, Swetter SM, Johnson DL, Ly DP. et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature. 2010;466:133-7
47. Schatton T, Murphy GF, Frank NY, Yamaura K, Waaga-Gasser AM, Gasser M. et al. Identification of cells initiating human melanomas. Nature. 2008;451:345-9
48. Luo Y, Dallaglio K, Chen Y, Robinson WA, Robinson SE, McCarter MD. et al. ALDH1A isozymes are markers of human melanoma stem cells and potential therapeutic targets. Stem Cells. 2012;30:2100-13
49. Kang N, Choi SY, Kim BN, Yeo CD, Park CK, Kim YK. et al. Hypoxia-induced cancer stemness acquisition is associated with CXCR4 activation by its aberrant promoter demethylation. BMC Cancer. 2019;19:148
50. Li Z, Rich JN. Hypoxia and hypoxia inducible factors in cancer stem cell maintenance. Curr Top Microbiol Immunol. 2010;345:21-30
51. Xiang L, Semenza GL. Hypoxia-inducible factors promote breast cancer stem cell specification and maintenance in response to hypoxia or cytotoxic chemotherapy. Adv Cancer Res. 2019;141:175-212
52. Hou PC, Li YH, Lin SC, Lin SC, Lee JC, Lin BW. et al. Hypoxia-Induced Downregulation of DUSP-2 Phosphatase Drives Colon Cancer Stemness. Cancer Res. 2017;77:4305-16
53. Peng Q, Qin J, Zhang Y, Cheng X, Wang X, Lu W. et al. Autophagy maintains the stemness of ovarian cancer stem cells by FOXA2. J Exp Clin Cancer Res. 2017;36:171
54. Wang L, Shang Z, Zhou Y, Hu X, Chen Y, Fan Y. et al. Autophagy mediates glucose starvation-induced glioblastoma cell quiescence and chemoresistance through coordinating cell metabolism, cell cycle, and survival. Cell Death Dis. 2018;9:213
55. Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A. et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551:247-50
56. Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B. et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547:453-7
57. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY. et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704-15
58. Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A. et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141:583-94
59. Lu Y, Liu Y, Oeck S, Glazer PM. Hypoxia Promotes Resistance to EGFR Inhibition in NSCLC Cells via the Histone Demethylases, LSD1 and PLU-1. Mol Cancer Res. 2018;16:1458-69
60. Moore N, Lyle S. Quiescent, slow-cycling stem cell populations in cancer: a review of the evidence and discussion of significance. J Oncol. 2011. 2011
61. Puig I, Tenbaum SP, Chicote I, Arques O, Martinez-Quintanilla J, Cuesta-Borras E. et al. TET2 controls chemoresistant slow-cycling cancer cell survival and tumor recurrence. J Clin Invest. 2018;128:3887-905
62. Kuo KT, Huang WC, Bamodu OA, Lee WH, Wang CH, Hsiao M. et al. Histone demethylase JARID1B/KDM5B promotes aggressiveness of non-small cell lung cancer and serves as a good prognostic predictor. Clin Epigenetics. 2018;10:107
63. Al Emran A, Marzese DM, Menon DR, Stark MS, Torrano J, Hammerlindl H. et al. Distinct histone modifications denote early stress-induced drug tolerance in cancer. Oncotarget. 2018;9:8206-22
64. Liu X, Greer C, Secombe J. KDM5 interacts with Foxo to modulate cellular levels of oxidative stress. PLoS Genet. 2014;10:e1004676
65. Batie M, Frost J, Frost M, Wilson JW, Schofield P, Rocha S. Hypoxia induces rapid changes to histone methylation and reprograms chromatin. Science. 2019;363:1222-6
66. Krieg AJ, Rankin EB, Chan D, Razorenova O, Fernandez S, Giaccia AJ. Regulation of the histone demethylase JMJD1A by hypoxia-inducible factor 1 alpha enhances hypoxic gene expression and tumor growth. Mol Cell Biol. 2010;30:344-53
67. Chicas A, Kapoor A, Wang X, Aksoy O, Evertts AG, Zhang MQ. et al. H3K4 demethylation by Jarid1a and Jarid1b contributes to retinoblastoma-mediated gene silencing during cellular senescence. Proc Natl Acad Sci U S A. 2012;109:8971-6
68. Dey BK, Stalker L, Schnerch A, Bhatia M, Taylor-Papidimitriou J, Wynder C. The histone demethylase KDM5b/JARID1b plays a role in cell fate decisions by blocking terminal differentiation. Mol Cell Biol. 2008;28:5312-27
69. Redmer T, Welte Y, Behrens D, Fichtner I, Przybilla D, Wruck W. et al. The nerve growth factor receptor CD271 is crucial to maintain tumorigenicity and stem-like properties of melanoma cells. PLoS One. 2014;9:e92596
70. Tomellini E, Touil Y, Lagadec C, Julien S, Ostyn P, Ziental-Gelus N. et al. Nerve growth factor and proNGF simultaneously promote symmetric self-renewal, quiescence, and epithelial to mesenchymal transition to enlarge the breast cancer stem cell compartment. Stem Cells. 2015;33:342-53
71. Kudo-Saito C, Yura M, Yamamoto R, Kawakami Y. Induction of immunoregulatory CD271+ cells by metastatic tumor cells that express human endogenous retrovirus H. Cancer Res. 2014;74:1361-70
72. Murillo-Sauca O, Chung MK, Shin JH, Karamboulas C, Kwok S, Jung YH. et al. CD271 is a functional and targetable marker of tumor-initiating cells in head and neck squamous cell carcinoma. Oncotarget. 2014;5:6854-66
73. Kojima H, Okumura T, Yamaguchi T, Miwa T, Shimada Y, Nagata T. Enhanced cancer stem cell properties of a mitotically quiescent subpopulation of p75NTR-positive cells in esophageal squamous cell carcinoma. Int J Oncol. 2017;51:49-62
74. Restivo G, Diener J, Cheng PF, Kiowski G, Bonalli M, Biedermann T. et al. low neurotrophin receptor CD271 regulates phenotype switching in melanoma. Nat Commun. 2017;8:1988
75. Verbeke S, Meignan S, Lagadec C, Germain E, Hondermarck H, Adriaenssens E. et al. Overexpression of p75(NTR) increases survival of breast cancer cells through p21(waf1). Cell Signal. 2010;22:1864-73
76. Furuta J, Inozume T, Harada K, Shimada S. CD271 on melanoma cell is an IFN-gamma-inducible immunosuppressive factor that mediates downregulation of melanoma antigens. J Invest Dermatol. 2014;134:1369-77
77. Li S, Yue D, Chen X, Wang L, Li J, Ping Y. et al. Epigenetic regulation of CD271, a potential cancer stem cell marker associated with chemoresistance and metastatic capacity. Oncol Rep. 2015;33:425-32
78. Raha D, Wilson TR, Peng J, Peterson D, Yue P, Evangelista M. et al. The cancer stem cell marker aldehyde dehydrogenase is required to maintain a drug-tolerant tumor cell subpopulation. Cancer Res. 2014;74:3579-90
79. Guler GD, Tindell CA, Pitti R, Wilson C, Nichols K, KaiWai Cheung T. et al. Repression of Stress-Induced LINE-1 Expression Protects Cancer Cell Subpopulations from Lethal Drug Exposure. Cancer Cell. 2017;32:221-37 e13
80. Vassalli G. Aldehyde Dehydrogenases: Not Just Markers, but Functional Regulators of Stem Cells. Stem Cells Int. 2019;2019:3904645
81. Zhou X, Hao Q, Liao P, Luo S, Zhang M, Hu G. et al. Nerve growth factor receptor negates the tumor suppressor p53 as a feedback regulator. Elife. 2016 5
82. Mi Z, Rogers DA, Mirnics ZK, Schor NF. p75NTR-dependent modulation of cellular handling of reactive oxygen species. J Neurochem. 2009;110:295-306
83. Shaffer SM, Dunagin MC, Torborg SR, Torre EA, Emert B, Krepler C. et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature. 2017;546:431-5
84. Chakravarthy R, Mnich K, Gorman AM. Nerve growth factor (NGF)-mediated regulation of p75(NTR) expression contributes to chemotherapeutic resistance in triple negative breast cancer cells. Biochem Biophys Res Commun. 2016;478:1541-7
85. Mehta A, Kim YJ, Robert L, Tsoi J, Comin-Anduix B, Berent-Maoz B. et al. Immunotherapy Resistance by Inflammation-Induced Dedifferentiation. Cancer Discov. 2018;8:935-43
86. Landsberg J, Kohlmeyer J, Renn M, Bald T, Rogava M, Cron M. et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature. 2012;490:412-6
87. Sakaki M, Ebihara Y, Okamura K, Nakabayashi K, Igarashi A, Matsumoto K. et al. Potential roles of DNA methylation in the initiation and establishment of replicative senescence revealed by array-based methylome and transcriptome analyses. PLoS One. 2017;12:e0171431
88. Cruickshanks HA, McBryan T, Nelson DM, Vanderkraats ND, Shah PP, van Tuyn J. et al. Senescent cells harbour features of the cancer epigenome. Nat Cell Biol. 2013;15:1495-506
89. Smallwood A, Esteve PO, Pradhan S, Carey M. Functional cooperation between HP1 and DNMT1 mediates gene silencing. Genes Dev. 2007;21:1169-78
90. Torrano J, Al Emran A, Hammerlindl H, Schaider H. Emerging roles of H3K9me3, SETDB1 and SETDB2 in therapy-induced cellular reprogramming. Clin Epigenetics. 2019;11:43
91. Raghuram GV, Mishra PK. Stress induced premature senescence: a new culprit in ovarian tumorigenesis? Indian J Med Res. 2014;140(Suppl):S120-9
92. Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA. et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703-16
93. Chandra T, Kirschner K, Thuret JY, Pope BD, Ryba T, Newman S. et al. Independence of repressive histone marks and chromatin compaction during senescent heterochromatic layer formation. Mol Cell. 2012;47:203-14
94. Aird KM, Zhang R. Detection of senescence-associated heterochromatin foci (SAHF). Methods Mol Biol. 2013;965:185-96
95. Gauchier M, Kan S, Barral A, Sauzet S, Agirre E, Bonnell E. et al. SETDB1-dependent heterochromatin stimulates alternative lengthening of telomeres. Sci Adv. 2019;5:eaav3673
96. Orouji E, Federico A, Larribere L, Novak D, Lipka DB, Assenov Y. et al. Histone methyltransferase SETDB1 contributes to melanoma tumorigenesis and serves as a new potential therapeutic target. Int J Cancer. 2019
97. Song C, Piva M, Sun L, Hong A, Moriceau G, Kong X. et al. Recurrent Tumor Cell-Intrinsic and -Extrinsic Alterations during MAPKi-Induced Melanoma Regression and Early Adaptation. Cancer Discov. 2017;7:1248-65
98. Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J. et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell. 2018;33:890-904 e5
99. Verfaillie A, Imrichova H, Atak ZK, Dewaele M, Rambow F, Hulselmans G. et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat Commun. 2015;6:6683
100. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P. et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26
101. Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T. et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44:852-60
102. Hammerlindl H, Schaider H. Tumor cell-intrinsic phenotypic plasticity facilitates adaptive cellular reprogramming driving acquired drug resistance. J Cell Commun Signal. 2018;12:133-41
103. Sale MJ, Balmanno K, Saxena J, Ozono E, Wojdyla K, McIntyre RE. et al. MEK1/2 inhibitor withdrawal reverses acquired resistance driven by BRAFV600E amplification whereas KRASG13D amplification promotes EMT-chemoresistance. Nature Communications. 2019;10:2030
104. Kong X, Kuilman T, Shahrabi A, Boshuizen J, Kemper K, Song JY. et al. Cancer drug addiction is relayed by an ERK2-dependent phenotype switch. Nature. 2017;550:270-4
105. Kidger AM, Rushworth LK, Stellzig J, Davidson J, Bryant CJ, Bayley C. et al. Dual-specificity phosphatase 5 controls the localized inhibition, propagation, and transforming potential of ERK signaling. Proc Natl Acad Sci U S A. 2017;114:E317-E26
106. Kaplan FM, Kugel CH 3rd, Dadpey N, Shao Y, Abel EV, Aplin AE. SHOC2 and CRAF mediate ERK1/2 reactivation in mutant NRAS-mediated resistance to RAF inhibitor. J Biol Chem. 2012;287:41797-807
107. Oxnard GR, Janjigian YY, Arcila ME, Sima CS, Kass SL, Riely GJ. et al. Maintained sensitivity to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer recurring after adjuvant erlotinib or gefitinib. Clin Cancer Res. 2011;17:6322-8
108. Soucheray M, Capelletti M, Pulido I, Kuang Y, Paweletz CP, Becker JH. et al. Intratumoral Heterogeneity in EGFR-Mutant NSCLC Results in Divergent Resistance Mechanisms in Response to EGFR Tyrosine Kinase Inhibition. Cancer Res. 2015;75:4372-83
109. Hugo W, Shi H, Sun L, Piva M, Song C, Kong X. et al. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell. 2015;162:1271-85
110. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S. et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165:35-44
111. Guo Z, Li Y, Zhang D, Ma J. Axl inhibition induces the antitumor immune response which can be further potentiated by PD-1 blockade in the mouse cancer models. Oncotarget. 2017;8:89761-74
112. Ziogas AC, Gavalas NG, Tsiatas M, Tsitsilonis O, Politi E, Terpos E. et al. VEGF directly suppresses activation of T cells from ovarian cancer patients and healthy individuals via VEGF receptor Type 2. Int J Cancer. 2012;130:857-64
113. Alfaro C, Teijeira A, Onate C, Perez G, Sanmamed MF, Andueza MP. et al. Tumor-Produced Interleukin-8 Attracts Human Myeloid-Derived Suppressor Cells and Elicits Extrusion of Neutrophil Extracellular Traps (NETs). Clin Cancer Res. 2016;22:3924-36
114. Kudo-Saito C, Shirako H, Ohike M, Tsukamoto N, Kawakami Y. CCL2 is critical for immunosuppression to promote cancer metastasis. Clin Exp Metastasis. 2013;30:393-405
115. Wang Y, Zhang X, Yang L, Xue J, Hu G. Blockade of CCL2 enhances immunotherapeutic effect of anti-PD1 in lung cancer. J Bone Oncol. 2018;11:27-32
116. Amini-Adle M, Khanafer N, Le-Bouar M, Duru G, Dalle S, Thomas L. Ineffective anti PD-1 therapy after BRAF inhibitor failure in advanced melanoma. BMC Cancer. 2018;18:705
117. Ackerman A, Klein O, McDermott DF, Wang W, Ibrahim N, Lawrence DP. et al. Outcomes of patients with metastatic melanoma treated with immunotherapy prior to or after BRAF inhibitors. Cancer. 2014;120:1695-701
118. Simeone E, Grimaldi AM, Festino L, Giannarelli D, Vanella V, Palla M. et al. Correlation between previous treatment with BRAF inhibitors and clinical response to pembrolizumab in patients with advanced melanoma. Oncoimmunology. 2017;6:e1283462
119. Wu L, Cao J, Cai WL, Lang SM, Horton JR, Jansen DJ. et al. KDM5 histone demethylases repress immune response via suppression of STING. PLoS Biol. 2018;16:e2006134
120. Rao M, Chinnasamy N, Hong JA, Zhang Y, Zhang M, Xi S. et al. Inhibition of histone lysine methylation enhances cancer-testis antigen expression in lung cancer cells: implications for adoptive immunotherapy of cancer. Cancer Res. 2011;71:4192-204
121. Vander Heiden MG, DeBerardinis RJ. Understanding the Intersections between Metabolism and Cancer Biology. Cell. 2017;168:657-69
122. Ryall JG, Cliff T, Dalton S, Sartorelli V. Metabolic Reprogramming of Stem Cell Epigenetics. Cell Stem Cell. 2015;17:651-62
123. Kinnaird A, Zhao S, Wellen KE, Michelakis ED. Metabolic control of epigenetics in cancer. Nat Rev Cancer. 2016;16:694-707
124. Intlekofer AM, Finley LWJNM. Metabolic signatures of cancer cells and stem cells. 2019; 1: 177.
125. Shyh-Chang N, Daley GQ. Metabolic switches linked to pluripotency and embryonic stem cell differentiation. Cell Metab. 2015;21:349-50
126. Folmes CD, Nelson TJ, Martinez-Fernandez A, Arrell DK, Lindor JZ, Dzeja PP. et al. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 2011;14:264-71
127. Wang L, Zhang T, Wang L, Cai Y, Zhong X, He X. et al. Fatty acid synthesis is critical for stem cell pluripotency via promoting mitochondrial fission. EMBO J. 2017;36:1330-47
128. Zhang J, Khvorostov I, Hong JS, Oktay Y, Vergnes L, Nuebel E. et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011;30:4860-73
129. Tohyama S, Fujita J, Hishiki T, Matsuura T, Hattori F, Ohno R. et al. Glutamine Oxidation Is Indispensable for Survival of Human Pluripotent Stem Cells. Cell Metab. 2016;23:663-74
130. Khacho M, Clark A, Svoboda DS, Azzi J, MacLaurin JG, Meghaizel C. et al. Mitochondrial Dynamics Impacts Stem Cell Identity and Fate Decisions by Regulating a Nuclear Transcriptional Program. Cell Stem Cell. 2016;19:232-47
131. Wiley CD, Campisi J. From Ancient Pathways to Aging Cells-Connecting Metabolism and Cellular Senescence. Cell Metab. 2016;23:1013-21
132. Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G. et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature. 2013;498:109-12
133. Vogel FCE, Bordag N, Zugner E, Trajkovic-Arsic M, Chauvistre H, Shannan B. et al. Targeting the H3K4 Demethylase KDM5B Reprograms the Metabolome and Phenotype of Melanoma Cells. J Invest Dermatol. 2019
134. Parmenter TJ, Kleinschmidt M, Kinross KM, Bond ST, Li J, Kaadige MR. et al. Response of BRAF-mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cancer Discov. 2014;4:423-33
135. Zhang G, Frederick DT, Wu L, Wei Z, Krepler C, Srinivasan S. et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J Clin Invest. 2016;126:1834-56
136. Kuntz EM, Baquero P, Michie AM, Dunn K, Tardito S, Holyoake TL. et al. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat Med. 2017;23:1234-40
137. Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R. et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017;7:716-35
138. Lee KM, Giltnane JM, Balko JM, Schwarz LJ, Guerrero-Zotano AL, Hutchinson KE. et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 2017;26:633-47 e7
139. Hoang-Minh LB, Siebzehnrubl FA, Yang C, Suzuki-Hatano S, Dajac K, Loche T. et al. Infiltrative and drug-resistant slow-cycling cells support metabolic heterogeneity in glioblastoma. EMBO J. 2018 37
140. Najjar YG, Menk AV, Sander C, Rao U, Karunamurthy A, Bhatia R. et al. Tumor cell oxidative metabolism as a barrier to PD-1 blockade immunotherapy in melanoma. JCI Insight. 2019 4
141. Scharping NE, Menk AV, Whetstone RD, Zeng X, Delgoffe GM. Efficacy of PD-1 Blockade Is Potentiated by Metformin-Induced Reduction of Tumor Hypoxia. Cancer Immunol Res. 2017;5:9-16
142. Fischer GM, Jalali A, Kircher DA, Lee WC, McQuade JL, Haydu LE. et al. Molecular Profiling Reveals Unique Immune and Metabolic Features of Melanoma Brain Metastases. Cancer Discov. 2019;9:628-45
143. Cascone T, McKenzie JA, Mbofung RM, Punt S, Wang Z, Xu C. et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab. 2018;27:977-87 e4
144. Husain Z, Huang Y, Seth P, Sukhatme VP. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J Immunol. 2013;191:1486-95
145. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V. et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559-63
146. Li J, Condello S, Thomes-Pepin J, Ma X, Xia Y, Hurley TD. et al. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell. 2017;20:303-14 e5
147. Liu H, Schreiber SL, Stockwell BR. Targeting Dependency on the GPX4 Lipid Peroxide Repair Pathway for Cancer Therapy. Biochemistry. 2018;57:2059-60
148. Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W. et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun. 2019;10:1617
149. Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell. 2019
150. Yao Y, Sun S, Wang J, Fei F, Dong Z, Ke AW. et al. Canonical Wnt Signaling Remodels Lipid Metabolism in Zebrafish Hepatocytes following Ras Oncogenic Insult. Cancer Res. 2018;78:5548-60
151. Kim H, Rodriguez-Navas C, Kollipara RK, Kapur P, Pedrosa I, Brugarolas J. et al. Unsaturated Fatty Acids Stimulate Tumor Growth through Stabilization of beta-Catenin. Cell Rep. 2015;13:495-503
152. Zhang Q, Lou Y, Zhang J, Fu Q, Wei T, Sun X. et al. Hypoxia-inducible factor-2α promotes tumor progression and has crosstalk with Wnt/β-catenin signaling in pancreatic cancer. 2017; 16: 119.
153. Sun X, Pang L, Shi M, Huang J, Wang YJJotm. HIF2α induces cardiomyogenesis via Wnt/β-catenin signaling in mouse embryonic stem cells. 2015; 13: 88.
154. Lizardo DY, Lin Y-L, Gokcumen O, Atilla-Gokcumen GEJMB. Regulation of lipids is central to replicative senescence. 2017; 13: 498-509.
155. Sagini K, Urbanelli L, Costanzi E, Mitro N, Caruso D, Emiliani C. et al. Oncogenic H-Ras Expression Induces Fatty Acid Profile Changes in Human Fibroblasts and Extracellular Vesicles. Int J Mol Sci. 2018 19
156. Flor AC, Wolfgeher D, Wu D, Kron SJ. A signature of enhanced lipid metabolism, lipid peroxidation and aldehyde stress in therapy-induced senescence. Cell Death Discov. 2017;3:17075
157. Friedmann Angeli JP, Krysko DV, Conrad M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer. 2019
158. Veglia F, Tyurin VA, Mohammadyani D, Blasi M, Duperret EK, Donthireddy L. et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat Commun. 2017;8:2122
159. Shaikh SR, Edidin M. Polyunsaturated fatty acids and membrane organization: elucidating mechanisms to balance immunotherapy and susceptibility to infection. Chem Phys Lipids. 2008;153:24-33
160. Badawy AA. Targeting tryptophan availability to tumors: the answer to immune escape? Immunol Cell Biol. 2018;96:1026-34
161. Venkateswaran N, Lafita-Navarro MC, Hao YH, Kilgore JA, Perez-Castro L, Braverman J. et al. MYC promotes tryptophan uptake and metabolism by the kynurenine pathway in colon cancer. Genes Dev. 2019;33:1236-51
162. Gutierrez-Vazquez C, Quintana FJ. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity. 2018;48:19-33
163. Liu Y, Liang X, Yin X, Lv J, Tang K, Ma J. et al. Blockade of IDO-kynurenine-AhR metabolic circuitry abrogates IFN-gamma-induced immunologic dormancy of tumor-repopulating cells. Nat Commun. 2017;8:15207
164. Liu Y, Liang X, Dong W, Fang Y, Lv J, Zhang T. et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8(+) T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell. 2018;33:480-94 e7
165. Lemos H, Huang L, Prendergast GC, Mellor AL. Immune control by amino acid catabolism during tumorigenesis and therapy. Nat Rev Cancer. 2019;19:162-75
166. Lu C, Thompson CB. Metabolic regulation of epigenetics. Cell Metab. 2012;16:9-17
167. Carey BW, Finley LW, Cross JR, Allis CD, Thompson CB. Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature. 2015;518:413-6
168. TeSlaa T, Chaikovsky AC, Lipchina I, Escobar SL, Hochedlinger K, Huang J. et al. alpha-Ketoglutarate Accelerates the Initial Differentiation of Primed Human Pluripotent Stem Cells. Cell Metab. 2016;24:485-93
169. Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H. et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26:1326-38
170. Sciacovelli M, Gonçalves E, Johnson TI, Zecchini VR, da Costa ASH, Gaude E. et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. 2016; 537: 544.
171. Horton JR, Engstrom A, Zoeller EL, Liu X, Shanks JR, Zhang X. et al. Characterization of a Linked Jumonji Domain of the KDM5/JARID1 Family of Histone H3 Lysine 4 Demethylases. J Biol Chem. 2016;291:2631-46
172. Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16:749
173. Baenke F, Chaneton B, Smith M, Van Den Broek N, Hogan K, Tang H. et al. Resistance to BRAF inhibitors induces glutamine dependency in melanoma cells. Mol Oncol. 2016;10:73-84
174. Pan M, Reid MA, Lowman XH, Kulkarni RP, Tran TQ, Liu X. et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat Cell Biol. 2016;18:1090-101
175. Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D. et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metab. 2015;22:861-73
176. Yang M, Vousden KH. Serine and one-carbon metabolism in cancer. Nat Rev Cancer. 2016;16:650-62
177. Dai Z, Mentch SJ, Gao X, Nichenametla SN, Locasale JW. Methionine metabolism influences genomic architecture and gene expression through H3K4me3 peak width. Nat Commun. 2018;9:1955
178. Wang Z, Yip LY, Lee JHJ, Wu Z, Chew HY, Chong PKW. et al. Methionine is a metabolic dependency of tumor-initiating cells. Nat Med. 2019;25:825-37
179. Strekalova E, Malin D, Weisenhorn EMM, Russell JD, Hoelper D, Jain A. et al. S-adenosylmethionine biosynthesis is a targetable metabolic vulnerability of cancer stem cells. Breast Cancer Res Treat. 2019;175:39-50
180. Maddocks OD, Labuschagne CF, Adams PD, Vousden KH. Serine Metabolism Supports the Methionine Cycle and DNA/RNA Methylation through De Novo ATP Synthesis in Cancer Cells. Mol Cell. 2016;61:210-21
181. Reina-Campos M, Diaz-Meco MT, Moscat J. The complexity of the serine glycine one-carbon pathway in cancer. J Cell Biol. 2020 219
182. Zhang WC, Shyh-Chang N, Yang H, Rai A, Umashankar S, Ma S. et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell. 2012;148:259-72
183. Li Y, Luo S, Ma R, Liu J, Xu P, Zhang H. et al. Upregulation of cytosolic phosphoenolpyruvate carboxykinase is a critical metabolic event in melanoma cells that repopulate tumors. Cancer Res. 2015;75:1191-6
184. Yu W, Wang Z, Zhang K, Chi Z, Xu T, Jiang D. et al. One-Carbon Metabolism Supports S-Adenosylmethionine and Histone Methylation to Drive Inflammatory Macrophages. Mol Cell. 2019;75:1147-60 e5
185. Lozoya OA, Martinez-Reyes I, Wang T, Grenet D, Bushel P, Li J. et al. Mitochondrial nicotinamide adenine dinucleotide reduced (NADH) oxidation links the tricarboxylic acid (TCA) cycle with methionine metabolism and nuclear DNA methylation. PLoS Biol. 2018;16:e2005707
186. Bao XR, Ong SE, Goldberger O, Peng J, Sharma R, Thompson DA. et al. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. Elife. 2016 5
187. Reid MA, Allen AE, Liu S, Liberti MV, Liu P, Liu X. et al. Serine synthesis through PHGDH coordinates nucleotide levels by maintaining central carbon metabolism. Nat Commun. 2018;9:5442
188. Li S, Swanson SK, Gogol M, Florens L, Washburn MP, Workman JL. et al. Serine and SAM Responsive Complex SESAME Regulates Histone Modification Crosstalk by Sensing Cellular Metabolism. Mol Cell. 2015;60:408-21
189. Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076-80
190. Liau BB, Sievers C, Donohue LK, Gillespie SM, Flavahan WA, Miller TE. et al. Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell. 2017;20:233-46 e7
191. McDonnell E, Crown SB, Fox DB, Kitir B, Ilkayeva OR, Olsen CA. et al. Lipids Reprogram Metabolism to Become a Major Carbon Source for Histone Acetylation. Cell Rep. 2016;17:1463-72
192. Bulusu V, Tumanov S, Michalopoulou E, van den Broek NJ, MacKay G, Nixon C. et al. Acetate Recapturing by Nuclear Acetyl-CoA Synthetase 2 Prevents Loss of Histone Acetylation during Oxygen and Serum Limitation. Cell Rep. 2017;18:647-58
193. Ryall JG, Dell'Orso S, Derfoul A, Juan A, Zare H, Feng X. et al. The NAD(+)-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell. 2015;16:171-83
194. Jung J, Kim LJ, Wang X, Wu Q, Sanvoranart T, Hubert CG. et al. Nicotinamide metabolism regulates glioblastoma stem cell maintenance. JCI Insight. 2017 2
195. Gujar AD, Le S, Mao DD, Dadey DY, Turski A, Sasaki Y. et al. An NAD+-dependent transcriptional program governs self-renewal and radiation resistance in glioblastoma. Proc Natl Acad Sci U S A. 2016;113:E8247-E56
196. Kim W, Deik A, Gonzalez C, Gonzalez ME, Fu F, Ferrari M. et al. Polyunsaturated fatty acid desaturation is a mechanism for glycolytic NAD+ recycling. 2019; 29: 856-70. e7.
197. Jiang Y, Qian X, Shen J, Wang Y, Li X, Liu R. et al. Local generation of fumarate promotes DNA repair through inhibition of histone H3 demethylation. Nat Cell Biol. 2015;17:1158-68
198. Sutendra G, Kinnaird A, Dromparis P, Paulin R, Stenson TH, Haromy A. et al. A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell. 2014;158:84-97
199. Nagaraj R, Sharpley MS, Chi F, Braas D, Zhou Y, Kim R. et al. Nuclear Localization of Mitochondrial TCA Cycle Enzymes as a Critical Step in Mammalian Zygotic Genome Activation. Cell. 2017;168:210-23 e11
200. Groves JA, Lee A, Yildirir G, Zachara NE. Dynamic O-GlcNAcylation and its roles in the cellular stress response and homeostasis. Cell Stress Chaperones. 2013;18:535-58
201. Wang T, Yu Q, Li J, Hu B, Zhao Q, Ma C. et al. O-GlcNAcylation of fumarase maintains tumour growth under glucose deficiency. Nat Cell Biol. 2017;19:833-43
202. Taparra K, Wang H, Malek R, Lafargue A, Barbhuiya MA, Wang X. et al. O-GlcNAcylation is required for mutant KRAS-induced lung tumorigenesis. J Clin Invest. 2018;128:4924-37
203. Efimova EV, Appelbe OK, Ricco N, Lee SS, Liu Y, Wolfgeher DJ. et al. O-GlcNAcylation Enhances Double-Strand Break Repair, Promotes Cancer Cell Proliferation, and Prevents Therapy-Induced Senescence in Irradiated Tumors. Mol Cancer Res. 2019;17:1338-50
204. Asthana A, Ramakrishnan P, Vicioso Y, Zhang K, Parameswaran R. Hexosamine Biosynthetic Pathway Inhibition Leads to AML Cell Differentiation and Cell Death. Mol Cancer Ther. 2018;17:2226-37
205. Jang H, Kim TW, Yoon S, Choi SY, Kang TW, Kim SY. et al. O-GlcNAc regulates pluripotency and reprogramming by directly acting on core components of the pluripotency network. Cell Stem Cell. 2012;11:62-74
206. Brier AB, Loft A, Madsen JGS, Rosengren T, Nielsen R, Schmidt SF. et al. The KDM5 family is required for activation of pro-proliferative cell cycle genes during adipocyte differentiation. Nucleic Acids Res. 2017;45:1743-59
207. Liu X, Secombe J. The Histone Demethylase KDM5 Activates Gene Expression by Recognizing Chromatin Context through Its PHD Reader Motif. Cell Rep. 2015;13:2219-31
208. Xu J, Kidder BL. KDM5B decommissions the H3K4 methylation landscape of self-renewal genes during trophoblast stem cell differentiation. Biol Open. 2018 7
209. Marin TL, Gongol B, Zhang F, Martin M, Johnson DA, Xiao H. et al. AMPK promotes mitochondrial biogenesis and function by phosphorylating the epigenetic factors DNMT1, RBBP7, and HAT1. Sci Signal. 2017 10
210. Merkwirth C, Jovaisaite V, Durieux J, Matilainen O, Jordan SD, Quiros PM. et al. Two Conserved Histone Demethylases Regulate Mitochondrial Stress-Induced Longevity. Cell. 2016;165:1209-23
211. Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S. et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell. 2013;23:316-31
212. Lopez-Serra P, Marcilla M, Villanueva A, Ramos-Fernandez A, Palau A, Leal L. et al. A DERL3-associated defect in the degradation of SLC2A1 mediates the Warburg effect. Nat Commun. 2014;5:3608
213. Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M. et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008;452:492-6
214. Sebastian C, Zwaans BM, Silberman DM, Gymrek M, Goren A, Zhong L. et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell. 2012;151:1185-99
215. Dooley AJ, Gupta A, Bhattacharyya M, Middleton MR. Intermittent dosing with vemurafenib in BRAF V600E-mutant melanoma: review of a case series. Ther Adv Med Oncol. 2014;6:262-6
216. Russo D, Malagola M, Skert C, Cancelli V, Turri D, Pregno P. et al. Managing chronic myeloid leukaemia in the elderly with intermittent imatinib treatment. Blood Cancer J. 2015;5:e347
217. Schreuer M, Jansen Y, Planken S, Chevolet I, Seremet T, Kruse V. et al. Combination of dabrafenib plus trametinib for BRAF and MEK inhibitor pretreated patients with advanced BRAF(V600)-mutant melanoma: an open-label, single arm, dual-centre, phase 2 clinical trial. Lancet Oncol. 2017;18:464-72
218. Djebbari F, Stoner N, Lavender VT. A systematic review of non-standard dosing of oral anticancer therapies. BMC Cancer. 2018;18:1154
219. Jin Y, Yao Y, Chen L, Zhu X, Jin B, Shen Y. et al. Depletion of gamma-catenin by Histone Deacetylase Inhibition Confers Elimination of CML Stem Cells in Combination with Imatinib. Theranostics. 2016;6:1947-62
220. Wang D, Li W, Zhao R, Chen L, Liu N, Tian Y. et al. Stabilized Peptide HDAC Inhibitors Derived from HDAC1 Substrate H3K56 for the Treatment of Cancer Stem-Like Cells In Vivo. Cancer Res. 2019;79:1769-83
221. Wang L, Leite de Oliveira R, Huijberts S, Bosdriesz E, Pencheva N, Brunen D. et al. An Acquired Vulnerability of Drug-Resistant Melanoma with Therapeutic Potential. Cell. 2018;173:1413-25 e14
Author contact
![]() Corresponding authors: Mayumi Fujita, University of Colorado School of Medicine, 12801 E.17th Ave, MS 8127, RC-1S, Rm L18-4124, Aurora, CO 80045. Phone: 303-724-4045; Fax: 303-724-4048; E-mail: mayumi.fujitaedu. Helmut Schaider, The University of Queensland Diamantina Institute, The University of Queensland, Translational Research Institute, 37 Kent Street, Woolloongabba QLD 4102, Australia. T 61 7 3443 7395; F 61 7 3443 7799; E-mail: h.schaideredu.au
Corresponding authors: Mayumi Fujita, University of Colorado School of Medicine, 12801 E.17th Ave, MS 8127, RC-1S, Rm L18-4124, Aurora, CO 80045. Phone: 303-724-4045; Fax: 303-724-4048; E-mail: mayumi.fujitaedu. Helmut Schaider, The University of Queensland Diamantina Institute, The University of Queensland, Translational Research Institute, 37 Kent Street, Woolloongabba QLD 4102, Australia. T 61 7 3443 7395; F 61 7 3443 7799; E-mail: h.schaideredu.au