Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
History of long peptide-based...
Advantages of long peptide-based...
Current studies of long...
Future perspectives
Conclusion
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(13):6011-6023. doi:10.7150/thno.38742 This issue Cite
Review
Personalized neoantigen vaccination with synthetic long peptides: recent advances and future perspectives
Xiaotong Chen, Ju Yang, Lifeng Wang ![]() , Baorui Liu
, Baorui Liu ![]()
The Comprehensive Cancer Centre of Drum Tower Hospital, Medical School of Nanjing University & Clinical Cancer Institute of Nanjing University, Nanjing, China
Received 2019-7-25; Accepted 2020-4-22; Published 2020-5-15
Abstract

Therapeutic cancer vaccines are one of the most promising strategies of immunotherapy. Traditional vaccines consisting of tumor-associated antigens have met with limited success. Recently, neoantigens derived from nonsynonymous mutations in tumor cells have emerged as alternatives that can improve tumor-specificity and reduce on-target off-tumor toxicity. Synthetic peptides are a common platform for neoantigen vaccines. It has been suggested that extending short peptides into long peptides can overcome immune tolerance and induce both CD4+ and CD8+ T cell responses. This review will introduce the history of long peptide-based neoantigen vaccines, discuss their advantages, summarize current preclinical and clinical developments, and propose future perspectives.
Keywords: Neoantigen, cancer vaccine, long peptide, solid tumor, immunotherapy
Introduction
Immunotherapy represents a significant breakthrough in the field of cancer treatment, which aims to harness the immune system to recognize tumor antigens and destroy tumors while leaving normal tissues undamaged [1]. Therapeutic cancer vaccines are one of the most promising strategies of immunotherapy [2]. In contrast to prophylactic vaccines, therapeutic vaccines are intended to induce robust cell-mediated immunity rather than antibody protection [3]. This can be achieved through increasing tumor antigen presentation of the major histocompatibility complex (MHC) expressed on antigen-presenting cells (APCs), allowing a larger number of T lymphocytes to identify and eradicate tumor cells [4]. Despite numerous efforts to develop cancer vaccines, their translation into efficacious clinical therapies has been challenging, with less than 7% objective clinical responses and an overall rate of clinical benefit around 20% [5]. To achieve the full potential of cancer vaccines, personalized neoantigen vaccines have been introduced [6].
Personalized neoantigen vaccines utilize neoantigens derived from nonsynonymous mutations of tumor cells, which are an important class of tumor antigens mediating anti-tumor immunity in addition to tumor-associated antigens (TAAs) [1]. While TAAs are present on tumor cells as well as normal cells, the expression of neoantigens is restricted to tumor tissues [7]. As such, improved tumor-specificity and lower levels of on-target off-tumor toxicity can be expected for neoantigen vaccines compared to TAA vaccines [8]. In addition, vaccines targeting self-antigens have been shown to elicit T cells with low avidity due to thymic selection and central tolerance [9]. However, neoantigens are new to the immune system. High-avidity T cells targeting neoantigens are more likely to exist [10]. From this perspective, neoantigen vaccines represent an attractive approach for therapeutic cancer vaccines. Recent clinical trials have demonstrated that T cell responses can be augmented or induced de novo by vaccination with predicted neoantigens in melanoma and glioblastoma patients, highlighting their potential as anti-cancer therapeutics [11-15].
Dendritic cell (DC) vaccines, DNA vaccines, RNA vaccines and synthetic peptide vaccines constitute the four platforms of personalized neoantigen vaccines [2]. Due to their relatively simple manufacturing process and stable storage [16], synthetic peptide vaccines represent a cost-effective way to generate anti-tumor responses, thereby remaining the therapy of choice for most studies (Table 1). In recent decades, the optimal design of peptide-based vaccines, particularly the size of the vaccinated peptides, has been intensively studied [3]. Short peptides typically refer to peptides of 8-10 amino acids in length, which represent the exact minimal CD8+ T cell epitopes. They can be extended by natural flanking amino acids to form long peptides, which are generally 15-31 amino acids in length. After vaccination, short peptides directly bind to MHC class I (MHC-I) molecules expressed by all nucleated cells, most of which are not specialized for antigen presentation, thus causing suboptimal T-cell priming or tolerance [17]. However, long peptides must be taken up and processed by professional APCs for presentation and T cell activation, thus alleviating potential immune tolerance and enhancing vaccine potency [17, 18]. In addition, long peptides involve both CD4+ and CD8+ T cell responses and have prolonged antigen presentation compared to short peptides [10, 19, 20]. Here, we review the history of long peptide-based neoantigen vaccines, elucidate the possible advantages of long peptide vaccines and their mechanisms, summarize their current preclinical and clinical developments, and propose future perspectives.
Published clinical trials of personalized neoantigen vaccines
| Year | Cancer type | Phase | Formulation | Additional intervention | Vaccine platform | Patient number | Response |
|---|---|---|---|---|---|---|---|
| 2015 [11] | Melanoma | I | / | / | DC vaccine | 3 | 1 CR 2 SD |
| 2017 [12] | Melanoma | I | / | / | RNA vaccine | 13 | 8 recurrence free 12-23m; 5 relapse:2 CR with pembrolizumab, 1 PR, 1 mixed response, 1 SD |
| 2017 [13] | Melanoma | I | Poly-ICLC | / | Long peptide vaccine | 6 | 4 recurrence free 20-32m; 2 relapse: CR with pembrolizumab |
| 2019 [14] | Glioblastoma | I/Ib | Poly-ICLC | / | Long peptide vaccine | 8 | 8 PD, died; PFS 7.6m, OS 16.8m; |
| 2019 [15] | Glioblastoma | I | Poly-ICLC GM-CSF | Chemotherapy | Long peptide and short peptide vaccine | 15 | 8 PD, died; PFS 14.2m, OS 29.0m; |
CR: complete response; DC: dendritic cell; GM-CSF: granulocyte-macrophage colony-stimulating factor; OS: overall survival; PD: progressive disease; PFS: progression-free survival; Poly-ICLC: polyinosinic-polycytidylic acid stabilized with polylysine and carboxymethylcellulose; PR: partial response; SD: stable disease.
History of long peptide-based neoantigen vaccines
CD8+ T lymphocytes have long been regarded as the predominant effector cells in tumor-rejection activities [21]. Since the binding grooves of MHC-I molecules are closed at both ends, CD8+ T cell epitopes binding to MHC-I molecules are typically restricted to 8-10 amino acids in length. In MHC class II (MHC-II) molecules, the binding grooves are open, which allows the peptides to extend out of the binding grooves, resulting in length diversity (13-25 amino acids) and binding promiscuity of MHC-II ligands. It is much more complicated to predict MHC-II epitopes [22]. As a result, researchers initially focused on short peptide vaccines targeting CD8+ T cells [23-25]. The first two experiments demonstrating the protective effects of peptide vaccines emerged in 1991 [24, 25]. Immunizing mice with free synthetic peptides could not only generate cytotoxic T lymphocyte (CTL) responses but also induce protection against lethal virus infections. However, it was soon appreciated that not all peptide vaccines could induce strong T cell responses [26-28]. In some cases, tumor growth was accelerated after vaccination [29]. The outcomes of clinical trials were also disappointing, highlighting the need for alternative strategies to improve therapeutic efficiency (Figure 1) [5, 30].
Historical overview of long peptide-based neoantigen vaccines. Since the first demonstration that free synthetic peptides could induce protective CTL responses in 1991, considerable efforts have been put into developing peptide-based cancer vaccines, most of which focused on short peptides (8-10 mer) exactly representing the tumor-specific CTL epitopes. However, the clinical translation has met with limited success, and in some cases, peptide vaccination could even accelerate tumor growth. Further exploration revealed that short peptides can lead to immune tolerance, and long peptides (15-31 mer) may act as a more effective platform for therapeutic cancer vaccines. Recent advances in high-throughput sequencing technologies have facilitated the development of personalized vaccines targeting neoantigens derived from nonsynonymous mutations in tumor cells, where long peptides are extensively used. In 2017, the first clinical trial of long peptide-based neoantigen vaccines reported encouraging outcomes in melanoma patients. Subsequent clinical trials have indicated the feasibility in immunologically cold tumors with a relatively low TMB. Emerging data has suggested that neoantigen vaccination with long peptides is a promising strategy to induce potent anti-tumor immunity. CLT: cytotoxic T lymphocyte; SLPs: synthetic long peptides; TMB: tumor mutation burden.
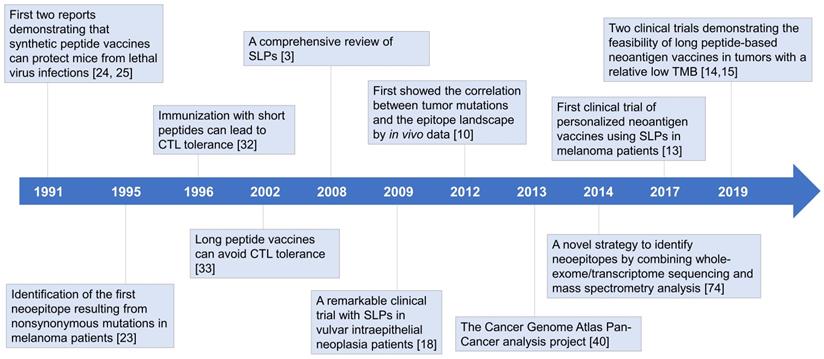
In the early 21st century, a team from Leiden University Medical Center made a conceptual breakthrough that the size of peptides matters [3]. They pointed out that some successful studies did not necessarily use the exact short peptides of 8-10 amino acids in length but may be longer [24, 31]. Subsequent experiments elucidated that short peptide vaccines could induce CTL tolerance (discussed below) [17, 19, 29, 32, 33], explaining the inconsistency between studies. Great success was achieved in 2009 in vulvar intraepithelial neoplasia patients, further highlighting the potential of synthetic long peptides (SLPs) as a vaccine platform (Figure 1) [18]. In this clinical trial, 15 of 19 patients showed clinical responses, with complete responses in 9 of them. These complete responses were maintained at 24 months of follow-up [18].
Likewise, “neoantigen” is not a new concept. Since the identification of the src oncogene in the 1970s [34], scientists gradually realized that cancer is a genetic disease, and that malignant transformation was caused by mutations in proto-oncogenes or tumor suppressor genes, resulting in their abnormal expression. The protein products of these mutated genes became candidate cancer neoantigens [35]. It was first shown in 1995 that neoantigens purified from murine lung carcinoma could be converted into peptide vaccines that were therapeutically effective, enhancing the lifespan of mice by protecting them from metastasis [23, 36]. Similar conclusions were achieved in melanoma patients [37]. However, traditional neoantigen vaccines typically consisted of a single mutant peptide corresponding to only one hotspot mutation (e.g. KRAS codon 12 mutations) [38]. Despite their improved tumor-specificity and lower on-target off-tumor toxicity, the clinical translation of such vaccines remains in its infancy [39].
Rapid developments in high-throughput sequencing and bioinformatics have facilitated the comprehensive mapping of all mutations in a tumor, termed the “mutanome” [40]. In 2012, Matsushita et al. validated the feasibility of combining next-generation sequencing (NGS) and predictive algorithms to identify MHC class I-restricted neoepitopes in a mouse sarcoma model [41]. Soon the mode of manufacturing personalized neoantigen vaccines was established, which incorporated exome and RNA sequencing of tumor and normal tissues to identify somatic mutations, followed by computational prediction and prioritization of neoepitopes (Figure 1) [10]. Generally, neoepitopes are predicted according to their HLA binding affinity [42]. Other factors accounting for immunogenicity are also considered in neoantigen prediction and prioritization, including proteasome cleavage preference [43], gene expression and peptide abundance [44], and structural and physicochemical features of peptide-MHC complexes [45]. Neoantigen vaccines derived from this strategy differ from the traditional version in that they consist of several top ranked epitopes rather than a single mutant peptide to deal with tumor heterogeneity and avoid immune escape [13-15]. Moreover, they are truly personalized due to the diversity of each individual's mutanome. The multi-epitope neoantigen vaccines are customized for each individual patient. Recent studies have shown attractive prospects, where SLPs are extensively used [10, 13, 14]. SLPs vary from 15 to 31 amino acids in length, composed of a predicted MHC-I epitope elongated at both ends with natural residues [10, 13-15]. This design enables all potential MHC-I or MHC-II epitopes of 8-15 amino acids in length carrying the mutation to be processed from the precursor peptide [10]. Peptides binding to the same MHC molecules are separated into different pools, administered in a non-rotating fashion to one of up to four extremities, avoiding potential antigen competition in the draining lymph nodes (dLNs) [13, 14, 46].
Advantages of long peptide-based neoantigen vaccines
Overcoming potential CTL tolerance
The substantial differences between long and short peptide vaccines originate from their presentation to APCs. The most distinctive feature of minimal CTL epitope vaccines is their direct binding to MHC molecules. Theoretically, minimal CTL epitopes are expected to be loaded directly onto MHC-I molecules expressed by local submucosal DCs at the injection site [47]. These DCs migrate to the dLNs where they present antigens to naive T cells, stimulating them to differentiate into antigen-specific CD8+ effector T lymphocytes [17]. Chemokines and other signaling factors further recruit these CTLs to tumor sites [48]. Thus, tumor-specific CTL responses are generated and anti-tumor responses are initiated.
However, the ability of a short peptide to be presented in dLNs depends on its ability to remain bound to MHC molecules. Short peptides displaying low MHC-binding affinity often show difficulties in eliciting a robust CTL response (Figure 2). Furthermore, peptides can diffuse from the vaccine site and spread systemically, and MHC-I molecules are expressed on the surface of all nucleated cells [49]. Consequently, short peptides can be directly loaded onto the MHC-I molecules of various types of professional APCs and non-professional APCs, most of which lack a full range of costimulatory molecules required for optimal CD8+ T cell activation [19]. As a result, short peptides often activate CTLs transiently or even induce CTL tolerance [17, 19, 33, 50]. For example, short peptide vaccination was shown to result in antigen presentation by circulating lymphocytes (including B cells and T cells) in not only dLNs but also non-draining lymph nodes (ndLNs) in the absence of a strong pro-inflammatory context (Figure 2A) [19, 51]. Even when activated by CpG oligodeoxynucleotides (CpG-ODN), they still failed to generate therapeutically efficient anti-tumor immunity [19].
Possible mechanisms for the superior performance of long peptide-based neoantigen vaccines vs short peptides. (A) Short peptide neoantigen vaccines (green) bind to MHC class I molecules expressed on local submucosal DCs once injected. These DCs migrate to the dLNs to present and activate naive T cells. However, short peptides with low MHC-binding affinity may fail to elicit a robust CTL response. In addition, short peptides can be presented systemically in not only dLNs but also ndLNs by all nucleated cells, most of which are not specialized for antigen presentation. Lack of costimulatory molecules on those non-professional APCs and improper stimulating environments (ndLNs) can both result in impaired T cell function. (B) Long peptides (red) must be endocytosed and processed for their transport to the cell surface in a DC-focused pattern. They are presented predominantly in dLNs. In addition, long peptide neoantigen vaccines may cover CD4+ T cell epitopes, involving CD4+ Th responses which play an important role in neoantigen anti-tumor immunity. Subsequently, they exhibit superior performance over short peptide neoantigen vaccines. APCs: antigen-presenting cells; CTL: cytotoxic T lymphocyte; DC: dendritic cell; dLNs: draining lymph nodes; MHC: major histocompatibility complex; ndLNs: non-draining lymph nodes.
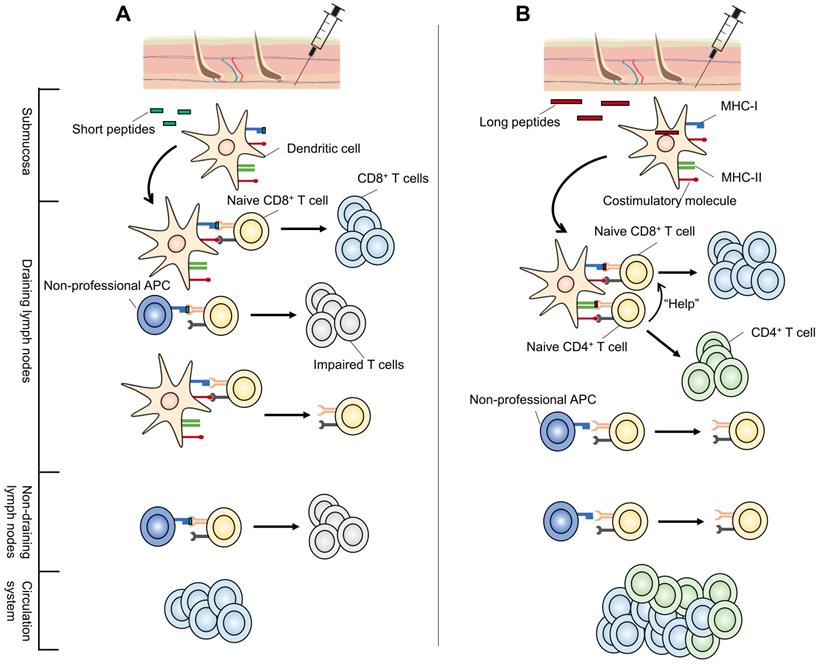
Elongating short peptides with natural flanking amino acids into SLPs alters this procedure (Figure 2B). SLPs elicit stronger effector CTL responses with greater tumoricidal potential in a DC-focused pattern [51-53], which is regarded as the main cross-presentation APC in vivo [54]. SLPs must be endocytosed, processed by DCs, and transported to cell surface rather than directly binding to MHC molecules [51]. Following internalization, a proportion of SLPs are degraded through the endosomal pathway and loaded onto MHC-II molecules, permitting their recognition by CD4+ T helper cells (Th cells) [47]. Another part of endocytosed SLPs enter either the cytosol or vacuolar pathway and are cross-presented by MHC-I molecules, activating CD8+ CTLs [54, 55]. This processing-dependency of SLPs to generate antigen-specific CD8+ T cell responses circumvents the possible CTL tolerance mechanisms [51]. Besides, data showed that, in contrast to systemically presented short peptides, long peptides are presented predominantly in dLNs [19]. These findings make it reasonable to believe that a stronger and more effective response can be induced with long peptide vaccines. In fact, the efficacy of this superior tumor-specific immunity has been demonstrated in a preclinical model of human papillomavirus (HPV) 16-induced cervical cancer. The eradication of large, established HPV16-expressing tumors was accomplished using a 35-mer long peptide admixed with the DC-activating adjuvant CpG-ODN, but not with a 9-mer short peptide containing the same CTL epitope [33].
Involving CD4+ Th cell responses
CD8+ cytotoxic T cells (Tc cells) have been intensely studied in anti-tumor immunity [21]. However, over recent decades, emerging evidence has shown that a deficiency of CD4+ Th cells impairs CTL responses, indicating their indispensable role [56-58]. CD4+ Th cells can license DCs through CD40-CD40L interaction, generating more efficient antigen presentation to CTLs [59]. Despite their traditional role in the immune response through cytokine secretion [60], CD4+ T cells exhibit cytotoxic features and can directly eliminate tumors in the absence of an MHC-I restricted CD8+ T cell response [57, 58, 61] . Surprisingly, when it comes to personalized neoantigen vaccines, it has been suggested that the majority of the immunogenic mutanome is recognized by CD4+ T cells in tumor-bearing C57BL/6 mice [62], further emphasizing the necessity of involving CD4+ T cell responses in anti-tumor immunity. Meanwhile, prominent CD4+ T cell responses were also shown in recently published neoantigen vaccine clinical trials, despite the use of MHC-I binding prediction algorithms [12-14].
It seems that short peptides can activate CD8+ T cells more easily than SLPs since short peptides can be directly presented on MHC-I molecules after vaccination, skipping the endocytosis and intracellular processing steps [51, 63]. However, this superior antigen presentation efficacy of short peptides to that of SLPs can only be observed early after vaccination, and this phenomenon is reversed with time due to the long-lasting cross-presentation of SLPs (discussed below) [63]. Furthermore, SLPs allow the generation of various combinations of the Th or Tc epitopes containing the mutated amino acid[10]. While vaccination with mixed Tc and Th epitopes can achieve the same goal [64, 65], chemically linked Th and Tc epitopes further increased the magnitude of the CTL response, suggesting that it is more efficient to provoke anti-tumor immunity by presenting the two epitopes on the same APC rather than on different APCs [59, 66, 67]. But this method still risks failure as alterations in the amino acid terminus to link the two epitopes together may lead to inappropriate cleavage by the proteasome, directly affecting epitope presentation [68]. In addition, algorithms to predict MHC-II restricted neoepitopes are still in their infancy [69]. Therefore, there is a tendency to vaccinate with SLPs to provide a potential class II epitope(s) so as to involve both CD4+ and CD8+ T cell responses [59, 70].
Prolonging antigen cross-presentation
Short peptides directly bind to MHC-I molecules expressed on local DCs once injected, forming MHC class I-peptide complexes to prime antigen-specific CD8+ T cells [47]. However, MHC class I-peptide complexes have a high turnover at the cell surface of mature DCs. Most MHC class I-peptide complexes can hardly be detected on the cell surface of mature DCs after 24 h, while MHC class II-peptide complexes are stable for several days [20]. This is because that the function of MHC-I molecules is to continuously present ligands derived from cytosolic proteins, newly synthesized mis-folded proteins and/or viral proteins for the timely elimination of abnormal tissue cells and pathogens [71]. As a result, the duration of antigen presentation of short peptide vaccines is limited, which is considered to partly account for their suboptimal efficiency [19].
However, antigen cross-presentation by MHC-I molecules of SLPs remained detectable for at least 3 days, correlating with an increased magnitude of anti-tumor responses [19, 20]. Further investigations showed that antigens which need internalization and intracellular processing to release MHC-I ligands (such as SLPs) can be conserved in intracellular storage depots of DCs for several days. This ensures a continuous supply of antigens and contributes to their sustained cross-presentation by DCs, despite the high turnover of MHC class I-peptide complexes at the cell surface [20]. Storage organelles were characterized as lysosome-like compartments [20]. Additional experiments are required for more detailed descriptions regarding these antigen depots. Taken together, SLPs which can be persistently cross-presented to activate CTLs may be superior to short peptides that are rapidly lost from MHC-I molecules in vaccine formulations.
Current studies of long peptide-based neoantigen vaccines
Preclinical studies
Bijker et al. performed a comparison of different peptide vaccination strategies with the highly immunogenic model antigen OVA, and showed a superior performance of long peptides over short peptides [17]. The minimal Tc epitope OVA257-264 (OVA8), the Th epitope OVA323-339 (OVA17), the extended Tc epitope OVA241-270 (OVA30) and the extended Th epitope OVA317-347 (OVA31) were prepared. Injecting OVA8 alone in incomplete Freund's adjuvant (IFA) induced a transient CD8+ response but failed to undergo a secondary expansion 30 days later, while OVA17 alone was sufficient to induce both short-term (10 days) and long-term (30 days) responses. The addition of the Th epitope OVA17 to OVA8 retained CD8+ T cell functionality, indicating an important role of CD4+ T cells in generating memory T cells. Long-lasting CD8+ T cell immunity was also observed when the extended Tc epitope OVA30 was injected alone. Depletion of CD4+ T cells did not influence the cytotoxic capacity of OVA30-induced CTLs. The number of OVA-specific CD8+ CTLs after expansion was higher when combined with OVA31 or an agonistic CD40 antibody. The greater capability of long peptides to induce anti-tumor immunity was related but not limited to CD4+ T cell responses. Similar results were confirmed in other preclinical models [19, 33, 72].
Based on these observations, Castle et al. made the first attempt to manufacture personalized neoantigen vaccines employing the long peptide platform in a high-throughput way [10]. DNA and RNA of matched tumor and normal tissues were extracted from B16F10 mice. Whole-exome sequencing (WES) was performed and their expression were further validated via RNA sequencing (RNA-Seq). In total, 563 mutations in expressed genes were identified, 50 of which were selected for synthesis into peptides of 27 amino acids long with the mutated or wild-type amino acid on position 14. By vaccinating C57BL/6 mice subcutaneously with polyinosinic-polycytidylic acid stabilized with polylysine and carboxymethylcellulose (poly-ICLC) as adjuvants, 16 out of 50 mutations were immunogenic and 60% in this group evoked strong immune responses directed preferentially against the mutated sequence rather than the wild-type sequence. These neoantigen vaccines also confirmed in vivo tumor control in protective and therapeutic settings. Furthermore, compared to vaccines comprised of the minimal 8-mer epitopes, long peptide-based personalized neoantigen vaccines showed improved protection during a tumor rechallenge analysis [73].
To simplify the laborious screening of immunogenic mutant epitopes, Yadav et al. established a new strategy to identify neoepitopes [74]. After WES and RNA-seq, mass spectrometry (MS) analysis was used to recognize the truly presented peptides by MHC-I molecules in MC-38 and TRAMP-C1 mice tumor cell lines, followed by a structural prediction algorithm to predict MHC-I peptide immunogenicity. Compared to the 170 and 6 neoepitopes respectively suggested in 2 tumor cell lines by direct algorithm prediction after RNA-seq, 5 and 0 neoepitopes were predicted using this strategy. Of these, 3 were confirmed as immunogenic through T cell analysis. The authors also employed the long peptide platform and successfully validated the feasibility and effectiveness of the neoantigen vaccines developed by this method [74].
Other than the highly personalized neoantigens identified through the two strategies described above, some neoantigens are encoded by recurrent driver mutations and hence are shared between patients [75]. Through literature reviews and database analysis, one can also identify such shared neoantigens to develop corresponding vaccines. Schumacher et al. selected the mutation IDH1(R132H) that is expressed in more than 70% diffuse grade II and III glioma patients as a target for neoantigen vaccines [61]. They generated peptide libraries encompassing this mutation and demonstrated that, the long peptide p123-142 (R132H) vaccine exhibited an anti-tumor immunity that was equivalent to another peptide vaccine targeting a well-studied tumor-associated antigen: NY-ESO-1. This study also represented the strong CD4+ immune responses which can reject tumors independent of CD8+ T cell responses [61, 76, 77]. The three approaches mentioned above represent the major screening methods to develop neoantigen vaccines, all of which have been validated through long peptide vaccines, demonstrating their recognition, immunogenicity, tumor-specificity and in vivo protective effects.
Clinical studies
Currently, five clinical trials of personalized neoantigen vaccines have been published (Table 1), three of which employed long peptides [13-15]. The other two were DC [11] and RNA vaccines [12]. Sahin et al. engineered two synthetic RNAs, each encoding five linker-connected 27-mer peptides with mutations at position 14 [12]. These RNA vaccines were eventually translated into long peptides, and to some extent, were consistent with the idea of long peptide vaccines, wherein the translated peptides are loaded on to the MHC intracellularly, and then exported to the cell surface for presentation to T cells [78].
Ott et al. prepared up to 20 long peptides for each melanoma patient that were 15-30 amino acids in length and divided into 4 pools [13]. Of the six vaccinated patients, four remained recurrence-free at 25 months post-vaccination, while two with progresssive disease were subsequently treated with anti-PD-1 therapy and experienced complete tumor regression. These results were astonishing and provided a strong rationale for further exploration to combine immune checkpoint inhibitors with neoantigen vaccines [13]. Keskin et al. from the same team of Dana-Farber Cancer Institute adopted a similar vaccination scheme in methylguanine methyltransferase (MGMT)-unmethylated glioblastoma patients [14]. Their results showed that long peptide-based neoantigen vaccines were a feasible therapeutic strategy for immunologically cold tumors with a relatively low tumor mutation burden (TMB) [14]. However, all patients died of progressive disease with a median progression-free survival of 7.6 months and overall survival of 16.8 months. Hilf et al. investigated glioblastoma therapeutics with a different design [15]. They used short peptides targeting unmutated TAAs derived from a premanufactured library, followed by the 19-mer neoepitope vaccination. The median progression-free survival was 14.2 months and the overall survival was 29.0 months [15]. This study investigated the clinical outcome of short TAA-targeting epitopes incorporating long neoantigen-targeting epitopes with encouraging results. However, all these trials revealed that considerable challenges still remain and further exploration is required to achieve the optimal design of neoantigen vaccines for ideal therapeutic effects.
Future perspectives
Despite the remarkable anti-tumor potential of long peptide-based neoantigen vaccines shown in both preclinical and clinical settings, the total results remain far from satisfying. Generally, there are four critical issues during vaccine design: (1) antigen selection; (2) adjuvant utilization; (3) vaccine delivery methods; (4) immune suppression reversion. We herein propose some improving approaches according to these four aspects.
Improving antigen prediction
The common workflow to create a personalized neoantigen vaccine includes exome and transcriptome sequencing of matched tumor-normal tissues, followed by in silico prediction and prioritization of neoepitopes [79]. Great progress has been made in the methodologies employed by neoantigen predictors, shifting from scoring function-based tools to machine learning-based tools [80], but there is still significant room for improvement.
Firstly, neoantigens can be generated from various sources beyond single nucleotide variants (SNVs), including frameshift mutations [81], gene fusions [82], intron retentions [83], non-coding expressed regions [84] and post-translational modifications [85]. However, most predictors only identify neoantigens from SNVs, leaving many highly immunogenic neoepitopes undiscovered [69]. Secondly, MHC-II alleles are inadequately supported in current prediction tools due to the variable length and binding promiscuity of MHC-II ligands and lack of binding data for model training [86]. However, it has been recently appreciated that neoantigen-specific responses are mediated predominantly by CD4+ T cells, highlighting the essential role of accurate MHC-II predictions [12-14]. In addition, a majority of predictors predict candidate neoepitopes according to their binding affinity of MHC molecules [69], without the consideration of other factors contributing to the immunogenicity such as proteasomal cleavage and peptide transportation [43], stability and T-cell receptor (TCR) recognition of the peptide-MHC complexes [87], and structural and physicochemical features [45], etc.
In recent years, while significant technological improvements have extended neoantigen identification to indels (tools like Strelka [88], EBCall [89]) and gene fusions (tools like JAFFA [90], INTEGRATE [91]), other newly-emerged sources remain to be involved. Specialized bioinformatics tools have also been developed to predict MHC-II antigen presentation. Two recently published algorithms (MARIA [86] and MixMHC2pred [92]) were reported to outperform existing methods, including NetMHCIIpan and SMM Align, which are commonly used in MHC-II restricted neoepitope prediction at present [69]. Moreover, MHC-I and MHC-II algorithms can be combined for more accurate predictions [44]. pVACtools is a comprehensive and extensible toolkit that can identify neoantigens from SNVs, indels and gene fusions. It integrates eight MHC-I and four MHC-II algorithms, supporting stability and cleavage predictions. pVACtools can be used for the design of long peptide-based vaccines, assessing candidate SLPs by evaluating their manufacturability (NCT03122106) [44]. Other specialized algorithms include NetChop for peptide processing prediction [43], DeepHLApan for TCR recognition prediction [87], and TRUST for TCR repertoire profiling [93], all of which have been well summarized in other reviews [94, 95]. With these fast updating bioinformatics tools, personalized neoantigen vaccines will be more accessible to patients, especially those with low TMB.
Engaging novel adjuvants
Since their first description by Ramon in 1924, diverse classes of adjuvants have been developed. Examples in current stages of development are listed in Table 3 (data from clinicaltrials.gov). Recent studies have reported that traditional adjuvants such as IFA and aluminum salts may induce T cell retention, exhaustion and deletion based on observations using short peptide vaccines [96]. Although this may not be the case for long peptides [3], more powerful adjuvants have been explored.
Common cancer vaccine adjuvants and their development stages
| Classification | Examples under investigation | Stage of development | |
|---|---|---|---|
| Emulsions | Incomplete Freund's Adjuvant | Montanide ISA51 | Phase III |
| Montanide ISA720 | Phase I | ||
| Mineral salts | Aluminum salts | Aluminum hydroxide (Alhydrogel™) | FDA approved |
| Aluminum phosphate (Adjut-phos™) | FDA approved | ||
| Cytokines | IL-2 | Aldesleukin | FDA approved |
| GM-CSF | Sargramostim | FDA approved | |
| IFNs | Intron A | FDA approved | |
| Sylatron | FDA approved | ||
| Saponin-based adjuvants | QS-21 | Phase III | |
| ISCOMATRIX | Phase II | ||
| TLR agonists | TLR2 agonist | Pam3CSK4 | Preclinical |
| TLR3 agonist | Poly-ICLC | Phase II | |
| TLR4 agonist | MPLA | Phase II | |
| TLR7/8 agonist | Imiquimod | FDA approved | |
| Resiquimod | Phase II | ||
| TLR9 agonist | CpG-ODN | Phase II | |
| DC-targeted monoclonal antibodies | Agonist anti-CD40 antibody | APX005M | Phase II |
| CFZ533 | Phase II | ||
| CP-870893 | Phase I | ||
| ADC-1013 | Phase I | ||
| Selicrelumab | Phase I | ||
| Chi Lob 7/4 | Phase I | ||
| STING agonists | MIW815 | Phase I | |
CpG-ODN: CpG oligodeoxynucleotides; DC: dendritic cell; GM-CSF: granulocyte-macrophage colony-stimulating factor; IFN: interferon; IL-2: interleukin-2; MPLA: monophosphoryl lipid A; Poly-ICLC: polyinosinic-polycytidylic acid stabilized with polylysine and carboxymethylcellulose; STING: stimulator of interferon genes; TLR: Toll-like receptor.
Toll-like receptor (TLR) agonists have been extensively investigated (Table 3). Poly-ICLC acts as a TLR3 agonist and has shown promising prospects. In 3 out of 5 published neoantigen clinical trials used poly-ICLC, strong T cell responses were demonstrated [13-15]. Among 36 ongoing clinical trials of peptide neoantigen vaccines, 20 selected poly-ICLC as adjuvants (Table 2). Agonist antibodies targeting CD40 expressed on DCs represent another attractive approach to improve the activation of DCs and induce superior immune responses. APX005M is among the six CD40 agonists currently under development (Table 3). It is in a phase I study of melanoma patients, combined with neoantigen peptide vaccines (NEO-PV-01) and immune checkpoint inhibitors (NCT03597282). Stimulator of interferon genes (STING) is an endoplasmic reticulum adaptor first described in 2008 [97]. Subsequent elucidation of downstream signaling pathways highlighted its potential as a target for cancer immunotherapy that can activate innate immunity [98]. Clinical studies incorporating STING agonists and peptide vaccines are still lacking , but their potent ability to function as an adjuvant with a whole-cell tumor cell vaccine have been demonstrated in mice [99].
Ongoing clinical trials of peptide-based neoantigen vaccines (data from ClinicalTrials.gov)
| ClinicalTrials.gov Identifier | Cancer type | Phase | Recruitment status | Formulation | Additional intervention |
|---|---|---|---|---|---|
| NCT03662815 | Advanced Malignant Solid Tumor | I | Recruiting | GM-CSF | / |
| NCT03645148 | Pancreatic Cancer | I | Recruiting | GM-CSF | / |
| NCT03558945 | Pancreatic Tumor | I | Recruiting | Poly-ICLC | / |
| NCT03715985 | Melanoma/NSCLC /Kidney Cancer | I | Recruiting | CAF09b | Anti-PD-1/anti-PD-L1 |
| NCT01970358 | Melanoma | I | Active, not recruiting | Poly-ICLC | / |
| NCT03422094 | Glioblastoma | I | Recruiting | Poly-ICLC | Nivolumab/ipilimumab |
| NCT03068832 | Pediatric Brain Tumor | I | Not yet recruiting | Poly-ICLC | / |
| NCT03361852 | Follicular Lymphoma | I | Not yet recruiting | Poly-ICLC | Rituximab |
| NCT02287428 | Glioblastoma | I | Active, not recruiting | / | Radiation/pembrolizumab/temozolomide |
| NCT02950766 | Kidney Cancer | I | Not yet recruiting | Poly-ICLC | Ipilimumab |
| NCT03606967 | TNBC | II | Not yet recruiting | Poly-ICLC | Durvalumab/nab-paclitaxel |
| NCT03219450 | chronic lymphocytic leukemia | I | Not yet recruiting | Poly-ICLC | Cyclophosphamide |
| NCT03359239 | Urothelial/Bladder Cancer | I | Recruiting | Poly-ICLC | Atezolizumab |
| NCT03559413 | Acute lymphoblastic leukemia | I/II | Recruiting | GM-CSF/Imiquimod | |
| NCT03380871 | NSCLC | I | Recruiting | Poly-ICLC | Pembrolizumab/carboplatin/pemetrexed |
| NCT03597282 | Melanoma | I | Recruiting | Poly-ICLC | Ipilimumab/nivolumab/APX005M |
| NCT02897765 | Urinary Bladder Cancer/NSCLC/Melanoma | I | Active, not recruiting | Poly-ICLC | Nivolumab |
| NCT02992977 | Advanced Cancer | I | Active, not recruiting | QS-21 Stimulon® | / |
| NCT03673020 | Solid Tumor, Adult | I | Recruiting | QS-21 Stimulon® | / |
| NCT03633110 | Melanoma/NSCLC/HNSCC /Urothelial Carcinoma/Renal Cell Carcinoma | I/II | Recruiting | Poly-ICLC | Nivolumab |
| NCT03631043 | Smoldering Plasma Cell Myeloma | I | Recruiting | / | / |
| NCT02600949 | Pancreatic /Colorectal Cancer | I | Active, not recruiting | / | Pembrolizumab |
| NCT02721043 | Solid Tumors | I | Recruiting | Poly-ICLC | Lenalidomide |
| NCT02933073 | Ovarian Cancer | I | Recruiting | / | / |
| NCT03929029 | Melanoma | I | Not yet recruiting | Montanide | Ipilimumab/ Nivolumab |
| NCT04087252 | Cancer | I | Recruiting | / | / |
| NCT03956056 | Pancreatic Cancer | I | Not yet recruiting | Poly-ICLC | / |
| NCT04117087 | Pancreatic /Colorectal Cancer | I | Not yet recruiting | Poly-ICLC | Ipilimumab/ Nivolumab |
| NCT04072900 | Melanoma | I | Not yet recruiting | rhGM-CSF | Toripalimab/ Imiquimod |
| NCT03953235 | NSCLC/ Pancreatic /Colorectal Cancer | I/II | Recruiting | / | Ipilimumab/ Nivolumab |
| NCT03639714 | NSCLC/ Colorectal Cancer /Gastroesophageal Adenocarcinoma/Urothelial Carcinoma | I/II | Recruiting | / | Ipilimumab/ Nivolumab |
| NCT04024878 | Ovarian Cancer | I | Not yet recruiting | Poly-ICLC | Nivolumab |
| NCT03568058 | Advanced Cancer | I | Recruiting | / | Pembrolizumab |
| NCT03121677 | Follicular Lymphoma | I | Recruiting | Poly-ICLC | Rituximab |
| NCT04266730 | NSCLC/HNSCC | I | Not yet recruiting | Poly-ICLC | / |
| NCT04248569 | Fibrolamellar Hepatocellular Carcinoma | I | Not yet recruiting | Poly-ICLC | Ipilimumab/ Nivolumab |
GM-CSF: granulocyte-macrophage colony-stimulating factor; HNSCC: head and neck squamous cell carcinoma; NSCLC: non-small cell lung cancer; Poly-ICLC: polyinosinic-polycytidylic acid stabilized with polylysine and carboxymethylcellulose; TNBC: triple-negative breast carcinoma.
Employing nanodelivery systems
Nanoparticles (NPs) for drug delivery have long been an attractive therapeutic strategy [100]. By employing the nanovaccine delivery system, we can: (1) protect peptides from rapid degradation to prolong their presentation time; (2) increase the accumulation of peptides in lymphatic tissue and improve the co-delivery of antigen peptides and adjuvants to dLNs; (3) deliver antigens and adjuvants simultaneously to DCs and promote their internalization [101]. Intracellular delivery is particularly important for long peptide-based vaccines as they must undergo endocytosis [3]. Moreover, some pattern recognition receptors (PRRs) are expressed inside the cell, such as TLR3, TLR7, TLR8, TLR9 and STING [102]. Delivering vaccines in the form of NPs can therefore improve immune activation and achieve optimal results.
Kuai et al. designed a synthetic high-density lipoprotein (sHDL) nanodisc, of which the surface was decorated with neoantigen long peptides and the TLR9 agonist CpG motif. This nanodisc generated 47-fold greater frequencies of neoantigen-specific CTLs than soluble vaccines with CpG as an adjuvant. Moreover, established MC-38 and B16F10 tumors were eliminated when combined with anti-PD-1 and anti-CTLA-4 therapies [103]. Recently, Li et al. reported a simple adsorption strategy using polyethyleneimine (PEI) in a mesoporous silica micro-rod (MSR) vaccine approach to enhance antigen response for neoantigen vaccines, with granulocyte-macrophage colony-stimulating factor (GM-CSF) and CpG-ODN as adjuvants. A single injection of this vaccine using a synthetic long peptide derived from the HPV E7 oncoprotein completely eradicated large established TC-1 tumors in ~80% of mice and generated immunological memory [104]. Furthermore, special biomaterials or novel designs are now under rapid development, which allow nanovaccines to respond to certain environmental triggers such as pH, redox, light or ultrasound. Improved DC-targeting, cytosolic delivery and therapeutic efficiency have been demonstrated [105]. Wang et al. designed a carrier-free nanovaccine with a high antigen density. Intermolecular disulfide cross-linking between antigens formed a nanoscale network. CpG bearing a thiol group was further incorporated into this network as a “danger signal” to activate DCs. Upon taken up by DCs, intracellular enriched glutathione (GSH) mediated the cleavage of the disulfide bonds, resulting in the release of antigens and CpG. This nanovaccine significantly promoted antigen-specific T cell activation with enhanced dLN retention, showing a higher survival rate of C57BL/6 mice and successful induction of tumor prevention [106].
Combining with other therapies
While great efforts have been made in perfecting the design of neoantigen vaccines, immune escape remains a problem that restricts clinical efficacy [107]. Tumors may evolve through a set of complex resistance mechanisms under the strong selection pressure of neoantigen-targeting immunotherapies [108], leading to the need for a combination of different therapeutic strategies.
One challenge is the loss or decreased expression of the recognized neoantigen in tumor cells [109]. This may be addressed through delivering vaccines consisting of multiple neoepitopes to induce polyclonal immune responses, as performed in many studies of neoantigen vaccines [13-15]. DNA-damaging chemoradiotherapies can act as powerful mutagens to introduce new somatic mutations and convert the tumor into an in situ vaccine, adding to the efficiency of neoantigen vaccines [110, 111]. Furthermore, vaccines targeting TAAs may also serve as a complement especially in patients with low TMB, as shown by Hilf et al [15].
The downregulation of components of antigen presentation machinery such as MHC-I molecules and the transporter associated with antigen processing (TAP) is the most frequently observed immune evasion mechanism that results in impaired antigen presentation [112].
Therapeutic kinase inhibitors targeting MEK and EGFR may have synergistic effects with neoantigen vaccines since they can upregulate MHC-I and TAP expression and enhance antigen presentation [113]. Epigenetic modulators such as DNA methyltransferase inhibitors can be considered for combination as well according to the epigenetic repression mechanisms of MHC expression [107]. Moreover, tumor cells lacking antigen presentation can be additionally eliminated in an MHC-independent fashion either by adoptive transfer of chimeric antigen receptor T (CAR-T) cells [114] or through the induction of antibody-mediated activation of natural killer cells [115].
Immune-inhibitory tumor microenvironment (TME) is another important factor hampering the performance of neoantigen vaccines [116]. Although lymphocytes can be efficiently activated by long peptides in the peripheral blood, reduced adhesion molecules due to abnormal angiogenesis and increased extracellular matrix density in tumor tissues prevent effective T cell migration and infiltration. Local immunosuppressive cells and molecules also compromise neoantigen recognition and T cell activation [117]. Combination strategies incorporating anti-angiogenesis therapies can normalize tumor vessels and reprogram suppressive TME, promoting T cell infiltration [118]. Immunomodulatory antibodies, including immune checkpoint inhibitors and costimulatory molecule agonists, hold great promise to reverse immune suppression and are under rapid development (Table 4). Complete remission has been achieved in progressed melanoma patients by combining PD-1 inhibitors (described above) [12, 13]. In addition, some chemotherapeutic agents (e.g., cyclophosphamide) that deplete immunosuppressive cells are actively being investigated as complementary therapies (NCT03219450, NCT03380871, NCT03606967) [119].
Examples of current immunomodulatory antibodies targeting T cells
| Receptor | Ligand | Antibody | Stage of development |
|---|---|---|---|
| Costimulation molecules | |||
| 4-1BB | 4-1BBL | Urelumab | Phase II |
| Utomilumab | Phase I | ||
| ADG106 | Phase I | ||
| OX40 | OX40L | MEDI6469 | Phase II |
| PF-04518600 | Phase II | ||
| GSK3174998 | Phase I | ||
| BMS 986178 | Phase I | ||
| MOXR0916 | Phase I | ||
| INBRX-106 | Phase I | ||
| BGB-A445 | Phase I | ||
| CD27 | CD70 | Varlilumab | Phase II |
| GITR | GITRL | TRX518 | Phase II |
| BMS-986156 | Phase II | ||
| INCAGN01876 | Phase II | ||
| GWN323 | Phase I | ||
| MEDI1873 | Phase I | ||
| OMP-336B11 | Phase I | ||
| MK-4166 | Phase I | ||
| ICOS | ICOSL | GSK3359609 | Phase II |
| Vopratelimab | Phase I/II | ||
| KY1044 | Phase I/II | ||
| TNFRSF25 | TL1A | Preclinical | |
| Inhibitory molecules | |||
| PD1 | PD-L1/PD-L2 | Pembrolizumab | Approved |
| Nivolumab | Approved | ||
| Cemiplimab | Approved | ||
| Sintilimab | Approved | ||
| JS001 | Approved | ||
| Camrelizumab | Phase III | ||
| BCD-100 | Phase III | ||
| Tislelizumab | Phase III | ||
| Spartalizumab | Phase III | ||
| Dostarlimab | Phase III | ||
| REGN2810 | Phase III | ||
| CTLA4 | CD80/CD86 | Ipilimumab | FDA approved |
| Tremelimumab | Phase III | ||
| LAG3 | MHC-II | Relatlimab | Phase II |
| LAG525 | Phase II | ||
| REGN3767 | Phase I | ||
| TSR-033 | Phase I | ||
| Sym022 | Phase I | ||
| TIM3 | Phosphatidylserine | TSR-022 | Phase II |
| BGB-A425 | Phase I/II | ||
| MBG453 | Phase I/II | ||
| LY3321367 | Phase I | ||
| Sym023 | Phase I | ||
Conclusion
Personalized neoantigen vaccines show improved tumor specificity and immunogenicity compared to conventional TAA vaccines. Long peptides are widely employed in neoantigen vaccines as a substitute for short peptide-based vaccines to overcome potential immunological tolerance, elicit not only CD8+ T cell responses but also CD4+ lymphocyte responses and prolong the antigen cross-presentation. Although preclinical experiments and clinical trials of long peptide-based neoantigen vaccines have indicated promising results, additional efforts are warranted to meet the expectations of therapeutic cancer vaccines. Improvements can be made through optimizing antigen prediction, engaging novel adjuvants, employing advanced nanodelivery systems and combining with immunomodulatory antibodies and/or traditional therapies. In summary, a new era of long peptide-based neoantigen vaccines has come and the results of ongoing clinical trials are eagerly anticipated.
Abbreviations
APCs: antigen-presenting cells; CAR-T cells: chimeric antigen receptor T cells; CpG-ODN: CpG oligodeoxynucleotides; CR: complete response; CTL: cytotoxic T lymphocyte; DC: dendritic cell; dLNs: draining lymph nodes; GM-CSF: granulocyte-macrophage colony-stimulating factor; GSH: glutathione; HNSCC: head and neck squamous cell carcinoma; HPV: human papillomavirus; IFA: incomplete Freund's adjuvant; IFN: interferon; IL-2: interleukin-2; MGMT: methylguanine methyltransferase; MHC: major histocompatibility complex; MHC-I molecules: MHC class I molecules; MHC-II molecules: MHC class II molecules; MPLA: monophosphoryl lipid A; MS: mass spectrometry; MSR: mesoporous silica micro-rod; ndLNs: non-draining lymph nodes; NGS: next-generation sequencing; NPs: nanoparticles; NSCLC: non-small cell lung cancer; OS: overall survival; PD: progressive disease; PEI: polyethyleneimine; PFS: progression-free survival; poly-ICLC: polyinosinic-polycytidylic acid stabilized with polylysine and carboxymethylcellulose; PR: partial response; PRR: pattern recognition receptor; RNA-Seq: RNA sequencing; SD: stable disease; sHDL: synthetic high-density lipoprotein; SLPs: synthetic long peptides; SNVs: single nucleotide variants; STING: stimulator of interferon genes; TAAs: tumor-associated antigens; TAP: transporter associated with antigen processing; Tc cells: cytotoxic T cells; TCR: T-cell receptor; Th cells: T helper cells; TLR: Toll-like receptor; TMB: tumor mutation burden; TME: tumor microenvironment; TNBC: triple-negative breast carcinoma; WES: whole-exome sequencing.
Acknowledgements
Baorui Liu was funded by grants from the National Natural Science Foundation of China (Grant No. 81930080). Lifeng Wang was supported by the Medical Research Project of Jiangsu Commission of Health (H2018111). The funding sources had no role in the study design, data collection, data analysis, data interpretation, or writing of the report.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sahin U, Tureci O. Personalized vaccines for cancer immunotherapy. Science. 2018;359:1355-60
2. Li L, Goedegebuure SP, Gillanders WE. Preclinical and clinical development of neoantigen vaccines. Ann Oncol. 2017;28:xii11-xii7
3. Melief CJ, van der Burg SH. Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat Rev Cancer. 2008;8:351-60
4. Tran T, Blanc C, Granier C, Saldmann A, Tanchot C, Tartour E. Therapeutic cancer vaccine: building the future from lessons of the past. Semin Immunopathol. 2019;41:69-85
5. Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909-15
6. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69-74
7. Wirth TC, Kuhnel F. Neoantigen targeting-dawn of a new era in cancer immunotherapy? Front Immunol. 2017;8:1848
8. Lu YC, Parker LL, Lu T, Zheng Z, Toomey MA, White DE. et al. Treatment of patients with metastatic cancer using a major histocompatibility complex class II-restricted T-cell receptor targeting the cancer germline antigen MAGE-A3. J Clin Oncol. 2017;35:3322-9
9. Bos R, van Duikeren S, van Hall T, Kaaijk P, Taubert R, Kyewski B. et al. Expression of a natural tumor antigen by thymic epithelial cells impairs the tumor-protective CD4+ T-cell repertoire. Cancer Res. 2005;65:6443-9
10. Castle JC, Kreiter S, Diekmann J, Lower M, van de Roemer N, de Graaf J. et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012;72:1081-91
11. Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA. et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348:803-8
12. Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M. et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547:222-6
13. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ. et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217-21
14. Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S. et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 2019;565:234-9
15. Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanovic S, Gouttefangeas C. et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature. 2019;565:240-5
16. Skwarczynski M, Toth I. Peptide-based synthetic vaccines. Chem Sci. 2016;7:842-54
17. Bijker MS, van den Eeden SJ, Franken KL, Melief CJ, Offringa R, van der Burg SH. CD8+ CTL priming by exact peptide epitopes in incomplete Freund's adjuvant induces a vanishing CTL response, whereas long peptides induce sustained CTL reactivity. J Immunol. 2007;179:5033-40
18. Kenter GG, Welters MJ, Valentijn AR, Lowik MJ, Berends-van der Meer DM, Vloon AP. et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med. 2009;361:1838-47
19. Bijker MS, van den Eeden SJ, Franken KL, Melief CJ, van der Burg SH, Offringa R. Superior induction of anti-tumor CTL immunity by extended peptide vaccines involves prolonged, DC-focused antigen presentation. Eur J Immunol. 2008;38:1033-42
20. van Montfoort N, Camps MG, Khan S, Filippov DV, Weterings JJ, Griffith JM. et al. Antigen storage compartments in mature dendritic cells facilitate prolonged cytotoxic T lymphocyte cross-priming capacity. Proc Natl Acad Sci U S A. 2009;106:6730-5
21. Appay V, Douek DC, Price DA. CD8+ T cell efficacy in vaccination and disease. Nat Med. 2008;14:623-8
22. Wieczorek M, Abualrous ET, Sticht J, Alvaro-Benito M, Stolzenberg S, Noe F. et al. Major histocompatibility complex (MHC) class I and MHC class II proteins: conformational plasticity in antigen presentation. Front Immunol. 2017;8:292
23. Mandelboim O, Vadai E, Fridkin M, Katz-Hillel A, Feldman M, Berke G. et al. Regression of established murine carcinoma metastases following vaccination with tumour-associated antigen peptides. Nat Med. 1995;1:1179-83
24. Schulz M, Zinkernagel RM, Hengartner H. Peptide-induced antiviral protection by cytotoxic T cells. Proc Natl Acad Sci U S A. 1991;88:991-3
25. Kast WM, Roux L, Curren J, Blom HJ, Voordouw AC, Meloen RH. et al. Protection against lethal Sendai virus infection by in vivo priming of virus-specific cytotoxic T lymphocytes with a free synthetic peptide. Proc Natl Acad Sci U S A. 1991;88:2283-7
26. Knutson KL, Schiffman K, Cheever MA, Disis ML. Immunization of cancer patients with a HER-2/neu, HLA-A2 peptide, p369-377, results in short-lived peptide-specific immunity. Clin Cancer Res. 2002;8:1014-8
27. Overwijk WW, Tsung A, Irvine KR, Parkhurst MR, Goletz TJ, Tsung K. et al. gp100/pmel 17 is a murine tumor rejection antigen: induction of "self"-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J Exp Med. 1998;188:277-86
28. Widmann C, Romero P, Maryanski JL, Corradin G, Valmori D. T helper epitopes enhance the cytotoxic response of mice immunized with MHC class I-restricted malaria peptides. J Immunol Methods. 1992;155:95-9
29. Toes RE, Blom RJ, Offringa R, Kast WM, Melief CJ. Enhanced tumor outgrowth after peptide vaccination. Functional deletion of tumor-specific CTL induced by peptide vaccination can lead to the inability to reject tumors. J Immunol. 1996;156:3911-8
30. Mocellin S, Mandruzzato S, Bronte V, Lise M, Nitti D. Part I: Vaccines for solid tumours. Lancet Oncol. 2004;5:681-9
31. Aichele P, Hengartner H, Zinkernagel RM, Schulz M. Antiviral cytotoxic T cell response induced by in vivo priming with a free synthetic peptide. J Exp Med. 1990;171:1815-20
32. Toes RE, Offringa R, Blom RJ, Melief CJ, Kast WM. Peptide vaccination can lead to enhanced tumor growth through specific T-cell tolerance induction. Proc Natl Acad Sci U S A. 1996;93:7855-60
33. Zwaveling S, Ferreira Mota SC, Nouta J, Johnson M, Lipford GB, Offringa R. et al. Established human papillomavirus type 16-expressing tumors are effectively eradicated following vaccination with long peptides. J Immunol. 2002;169:350-8
34. Stehelin D, Varmus HE, Bishop JM, Vogt PK. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature. 1976;260:170-3
35. Urban JL, Schreiber H. Tumor antigens. Annu Rev Immunol. 1992;10:617-44
36. Mandelboim O, Berke G, Fridkin M, Feldman M, Eisenstein M, Eisenbach L. CTL induction by a tumour-associated antigen octapeptide derived from a murine lung carcinoma. Nature. 1994;369:67-71
37. Wolfel T, Hauer M, Schneider J, Serrano M, Wolfel C, Klehmann-Hieb E. et al. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science. 1995;269:1281-4
38. Gjertsen MK, Buanes T, Rosseland AR, Bakka A, Gladhaug I, Soreide O. et al. Intradermal ras peptide vaccination with granulocyte-macrophage colony-stimulating factor as adjuvant: Clinical and immunological responses in patients with pancreatic adenocarcinoma. Int J Cancer. 2001;92:441-50
39. Bezu L, Kepp O, Cerrato G, Pol J, Fucikova J, Spisek R. et al. Trial watch: Peptide-based vaccines in anticancer therapy. Oncoimmunology. 2018;7:e1511506
40. Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K. et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45:1113-20
41. Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ. et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482:400-4
42. Vita R, Overton JA, Greenbaum JA, Ponomarenko J, Clark JD, Cantrell JR. et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2015;43:D405-12
43. Nielsen M, Lundegaard C, Lund O, Kesmir C. The role of the proteasome in generating cytotoxic T-cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics. 2005;57:33-41
44. Hundal J, Kiwala S, McMichael J, Miller CA, Xia H, Wollam AT. et al. pVACtools: A computational toolkit to identify and visualize cancer neoantigens. Cancer Immunol Res. 2020;8:409-20
45. Riley TP, Keller GLJ, Smith AR, Davancaze LM, Arbuiso AG, Devlin JR. et al. Structure based prediction of neoantigen immunogenicity. Front Immunol. 2019;10:2047
46. Kenter GG, Welters MJ, Valentijn AR, Lowik MJ, Berends-van der Meer DM, Vloon AP. et al. Phase I immunotherapeutic trial with long peptides spanning the E6 and E7 sequences of high-risk human papillomavirus 16 in end-stage cervical cancer patients shows low toxicity and robust immunogenicity. Clin Cancer Res. 2008;14:169-77
47. Rock KL, Reits E, Neefjes J. Present yourself! By MHC class I and MHC class II molecules. Trends Immunol. 2016;37:724-37
48. van der Woude LL, Gorris MAJ, Halilovic A, Figdor CG, de Vries IJM. Migrating into the tumor: a roadmap for T cells. Trends Cancer. 2017;3:797-808
49. Weijzen S, Meredith SC, Velders MP, Elmishad AG, Schreiber H, Kast WM. Pharmacokinetic differences between a T cell-tolerizing and a T cell-activating peptide. J Immunol. 2001;166:7151-7
50. Aichele P, Brduscha-Riem K, Zinkernagel RM, Hengartner H, Pircher H. T cell priming versus T cell tolerance induced by synthetic peptides. J Exp Med. 1995;182:261-6
51. Hambach L, Aghai Z, Pool J, Kroger N, Goulmy E. Peptide length extension skews the minor HA-1 antigen presentation toward activated dendritic cells but reduces its presentation efficiency. J Immunol. 2010;185:4582-9
52. Kitamura H, Sedlik C, Jacquet A, Zaragoza B, Dusseaux M, Premel V. et al. Long peptide vaccination can lead to lethality through CD4+ T cell-mediated cytokine storm. J Immunol. 2010;185:892-901
53. Rosalia RA, Quakkelaar ED, Redeker A, Khan S, Camps M, Drijfhout JW. et al. Dendritic cells process synthetic long peptides better than whole protein, improving antigen presentation and T-cell activation. Eur J Immunol. 2013;43:2554-65
54. Ma W, Stroobant V, Heirman C, Sun Z, Thielemans K, Mulder A. et al. The vacuolar pathway of long peptide cross-presentation can be TAP dependent. J Immunol. 2019;202:451-9
55. Menager J, Ebstein F, Oger R, Hulin P, Nedellec S, Duverger E. et al. Cross-presentation of synthetic long peptides by human dendritic cells: a process dependent on ERAD component p97/VCP but Not sec61 and/or Derlin-1. PLoS One. 2014;9:e89897
56. Borst J, Ahrends T, Babala N, Melief CJM, Kastenmuller W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat Rev Immunol. 2018;18:635-47
57. Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R. et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med. 2008;358:2698-703
58. Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X. et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. 2010;207:637-50
59. Perez SA, von Hofe E, Kallinteris NL, Gritzapis AD, Peoples GE, Papamichail M. et al. A new era in anticancer peptide vaccines. Cancer. 2010;116:2071-80
60. Bos R, Sherman LA. CD4+ T-cell help in the tumor milieu is required for recruitment and cytolytic function of CD8+ T lymphocytes. Cancer Res. 2010;70:8368-77
61. Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J. et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature. 2014;512:324-7
62. Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J. et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520:692-6
63. Faure F, Mantegazza A, Sadaka C, Sedlik C, Jotereau F, Amigorena S. Long-lasting cross-presentation of tumor antigen in human DC. Eur J Immunol. 2009;39:380-90
64. Li F, Chen C, Ju T, Gao J, Yan J, Wang P. et al. Rapid tumor regression in an Asian lung cancer patient following personalized neo-epitope peptide vaccination. Oncoimmunology. 2016;5:e1238539
65. Tobias J, Jasinska J, Baier K, Kundi M, Ede N, Zielinski C. et al. Enhanced and long term immunogenicity of a Her-2/neu multi-epitope vaccine conjugated to the carrier CRM197 in conjunction with the adjuvant Montanide. BMC Cancer. 2017;17:118
66. Riccione KA, He LZ, Fecci PE, Norberg PK, Suryadevara CM, Swartz A. et al. CD27 stimulation unveils the efficacy of linked class I/II peptide vaccines in poorly immunogenic tumors by orchestrating a coordinated CD4/CD8 T cell response. Oncoimmunology. 2018;7:e1502904
67. Shirai M, Pendleton CD, Ahlers J, Takeshita T, Newman M, Berzofsky JA. Helper-cytotoxic T lymphocyte (CTL) determinant linkage required for priming of anti-HIV CD8+ CTL in vivo with peptide vaccine constructs. J Immunol. 1994;152:549-56
68. Rabu C, Rangan L, Florenceau L, Fortun A, Charpentier M, Dupre E. et al. Cancer vaccines: designing artificial synthetic long peptides to improve presentation of class I and class II T cell epitopes by dendritic cells. Oncoimmunology. 2019;8:e1560919
69. Richters MM, Xia H, Campbell KM, Gillanders WE, Griffith OL, Griffith M. Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome Med. 2019;11:56
70. van der Burg SH, Bijker MS, Welters MJ, Offringa R, Melief CJ. Improved peptide vaccine strategies, creating synthetic artificial infections to maximize immune efficacy. Adv Drug Deliv Rev. 2006;58:916-30
71. Yewdell JW. Plumbing the sources of endogenous MHC class I peptide ligands. Curr Opin Immunol. 2007;19:79-86
72. Wick DA, Martin SD, Nelson BH, Webb JR. Profound CD8+ T cell immunity elicited by sequential daily immunization with exogenous antigen plus the TLR3 agonist poly(I:C). Vaccine. 2011;29:984-93
73. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T. et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577-81
74. Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S. et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515:572-6
75. Chheda ZS, Kohanbash G, Okada K, Jahan N, Sidney J, Pecoraro M. et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J Exp Med. 2018;215:141-57
76. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME. et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344:641-5
77. Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ. et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med. 2015;21:81-5
78. Hellmann MD, Snyder A. Making it personal: neoantigen vaccines in metastatic melanoma. Immunity. 2017;47:221-3
79. Chu Y, Liu Q, Wei J, Liu B. Personalized cancer neoantigen vaccines come of age. Theranostics. 2018;8:4238-46
80. Mei S, Li F, Leier A, Marquez-Lago TT, Giam K, Croft NP. et al. A comprehensive review and performance evaluation of bioinformatics tools for HLA class I peptide-binding prediction. Brief Bioinform. 2019
81. Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL. et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol. 2017;18:1009-21
82. Yang W, Lee KW, Srivastava RM, Kuo F, Krishna C, Chowell D. et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat Med. 2019;25:767-75
83. Smart AC, Margolis CA, Pimentel H, He MX, Miao D, Adeegbe D. et al. Intron retention is a source of neoepitopes in cancer. Nat Biotechnol. 2018;36:1056-8
84. Laumont CM, Vincent K, Hesnard L, Audemard E, Bonneil E, Laverdure JP. et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci Transl Med. 2018 10
85. Malaker SA, Penny SA, Steadman LG, Myers PT, Loke JC, Raghavan M. et al. Identification of glycopeptides as posttranslationally modified neoantigens in leukemia. Cancer Immunol Res. 2017;5:376-84
86. Chen B, Khodadoust MS, Olsson N, Wagar LE, Fast E, Liu CL. et al. Predicting HLA class II antigen presentation through integrated deep learning. Nat Biotechnol. 2019;37:1332-43
87. Wu J, Wang W, Zhang J, Zhou B, Zhao W, Su Z. et al. DeepHLApan: A deep learning approach for neoantigen prediction considering both HLA-peptide binding and immunogenicity. Front Immunol. 2019;10:2559
88. Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811-7
89. Shiraishi Y, Sato Y, Chiba K, Okuno Y, Nagata Y, Yoshida K. et al. An empirical Bayesian framework for somatic mutation detection from cancer genome sequencing data. Nucleic Acids Res. 2013;41:e89
90. Davidson NM, Majewski IJ, Oshlack A. JAFFA: High sensitivity transcriptome-focused fusion gene detection. Genome Med. 2015;7:43
91. Zhang J, White NM, Schmidt HK, Fulton RS, Tomlinson C, Warren WC. et al. INTEGRATE: gene fusion discovery using whole genome and transcriptome data. Genome Res. 2016;26:108-18
92. Racle J, Michaux J, Rockinger GA, Arnaud M, Bobisse S, Chong C. et al. Robust prediction of HLA class II epitopes by deep motif deconvolution of immunopeptidomes. Nat Biotechnol. 2019;37:1283-6
93. Li B, Li T, Wang B, Dou R, Zhang J, Liu JS. et al. Ultrasensitive detection of TCR hypervariable-region sequences in solid-tissue RNA-seq data. Nat Genet. 2017;49:482-3
94. Chen R, Fulton KM, Twine SM, Li J. Identification of MHC peptides using mass spectrometry for neoantigen discovery and cancer vaccine development. Mass Spectrom Rev. 2019
95. Boegel S, Castle JC, Kodysh J, O'Donnell T, Rubinsteyn A. Bioinformatic methods for cancer neoantigen prediction. Prog Mol Biol Transl Sci. 2019;164:25-60
96. Hailemichael Y, Dai Z, Jaffarzad N, Ye Y, Medina MA, Huang XF. et al. Persistent antigen at vaccination sites induces tumor-specific CD8(+) T cell sequestration, dysfunction and deletion. Nat Med. 2013;19:465-72
97. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674-8
98. Shae D, Becker KW, Christov P, Yun DS, Lytton-Jean AKR, Sevimli S. et al. Endosomolytic polymersomes increase the activity of cyclic dinucleotide STING agonists to enhance cancer immunotherapy. Nat Nanotechnol. 2019;14:269-78
99. Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E. et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med. 2015;7:283ra52
100. Lim S, Park J, Shim MK, Um W, Yoon HY, Ryu JH. et al. Recent advances and challenges of repurposing nanoparticle-based drug delivery systems to enhance cancer immunotherapy. Theranostics. 2019;9:7906-23
101. Mi Y, Hagan CTt, Vincent BG, Wang AZ. Emerging nano-/microapproaches for cancer immunotherapy. Adv Sci (Weinh). 2019;6:1801847
102. Khong H, Overwijk WW. Adjuvants for peptide-based cancer vaccines. J Immunother Cancer. 2016;4:56
103. Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A, Moon JJ. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat Mater. 2017;16:489-96
104. Li AW, Sobral MC, Badrinath S, Choi Y, Graveline A, Stafford AG. et al. A facile approach to enhance antigen response for personalized cancer vaccination. Nat Mater. 2018;17:528-34
105. Zhao Y, Guo Y, Tang L. Engineering cancer vaccines using stimuli-responsive biomaterials. Nano Res. 2018;11:5355-71
106. Wang K, Wen S, He L, Li A, Li Y, Dong H. et al. "Minimalist" nanovaccine constituted from near whole antigen for cancer immunotherapy. ACS nano. 2018;12:6398-409
107. Rosenthal R, Cadieux EL, Salgado R, Bakir MA, Moore DA, Hiley CT. et al. Neoantigen-directed immune escape in lung cancer evolution. Nature. 2019;567:479-85
108. Iorgulescu JB, Braun D, Oliveira G, Keskin DB, Wu CJ. Acquired mechanisms of immune escape in cancer following immunotherapy. Genome Med. 2018;10:87
109. Nejo T, Matsushita H, Karasaki T, Nomura M, Saito K, Tanaka S. et al. Reduced neoantigen expression revealed by longitudinal multiomics as a possible immune evasion mechanism in glioma. Cancer Immunol Res. 2019;7:1148-61
110. Lhuillier C, Rudqvist N-P, Elemento O, Formenti SC, Demaria S. Radiation therapy and anti-tumor immunity: exposing immunogenic mutations to the immune system. Genome Med. 2019;11:40
111. Ozpiskin OM, Zhang L, Li JJ. Immune targets in the tumor microenvironment treated by radiotherapy. Theranostics. 2019;9:1215-31
112. Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res. 2015;21:687-92
113. Brea EJ, Oh CY, Manchado E, Budhu S, Gejman RS, Mo G. et al. Kinase regulation of human MHC class I molecule expression on cancer cells. Cancer Immunol Res. 2016;4:936-47
114. Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. 'Off-the-shelf' allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov. 2020;19:185-99
115. Georgiev H, Ravens I, Papadogianni G, Bernhardt G. Coming of age: CD96 emerges as modulator of immune responses. Front Immunol. 2018;9:1072
116. Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S. et al. Cold tumors: A therapeutic challenge for immunotherapy. Front Immunol. 2019;10:168
117. Debets R, Donnadieu E, Chouaib S, Coukos G. TCR-engineered T cells to treat tumors: Seeing but not touching? Semin Immunol. 2016;28:10-21
118. Yi M, Jiao D, Qin S, Chu Q, Wu K, Li A. Synergistic effect of immune checkpoint blockade and anti-angiogenesis in cancer treatment. Mol Cancer. 2019;18:60
119. Buccione C, Fragale A, Polverino F, Ziccheddu G, Arico E, Belardelli F. et al. Role of interferon regulatory factor 1 in governing Treg depletion, Th1 polarization, inflammasome activation and antitumor efficacy of cyclophosphamide. Int J Cancer. 2018;142:976-87
Author contact
![]() Corresponding authors: Baorui Liu, MD, Ph.D, The Comprehensive Cancer Centre of Drum Tower Hospital, Medical School of Nanjing University, Clinical Cancer Institute of Nanjing University, 321 Zhongshan Road, Nanjing 210008, China. Tel: +86-25-83107081; Fax: +86-25-83317016; E-mail: baoruiliuedu.cn; Lifeng Wang, MD, Ph. D, The Comprehensive Cancer Centre of Drum Tower Hospital, Medical School of Nanjing University, Clinical Cancer Institute of Nanjing University, 321 Zhongshan Road, Nanjing 210008, China. Tel: +86-25-83304616-61346; E-mail: lifengwangedu.cn
Corresponding authors: Baorui Liu, MD, Ph.D, The Comprehensive Cancer Centre of Drum Tower Hospital, Medical School of Nanjing University, Clinical Cancer Institute of Nanjing University, 321 Zhongshan Road, Nanjing 210008, China. Tel: +86-25-83107081; Fax: +86-25-83317016; E-mail: baoruiliuedu.cn; Lifeng Wang, MD, Ph. D, The Comprehensive Cancer Centre of Drum Tower Hospital, Medical School of Nanjing University, Clinical Cancer Institute of Nanjing University, 321 Zhongshan Road, Nanjing 210008, China. Tel: +86-25-83304616-61346; E-mail: lifengwangedu.cn