Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
3D cell culture approaches
Extracellular components of the...
Cellular components of the...
Conclusions and future...
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(11):5074-5089. doi:10.7150/thno.42441 This issue Cite
Review
3D approaches to model the tumor microenvironment of pancreatic cancer
Elena Tomás-Bort1, Markus Kieler2, Shreya Sharma1, Juliana B Candido1, Daniela Loessner1,3,4 ![]()
1. Centre for Tumor Microenvironment, Barts Cancer Institute, Queen Mary University of London, EC1M 6BQ London, United Kingdom;
2. Institute of Vascular Biology, Medical University of Vienna, 1090 Vienna, Austria;
3. Department of Chemical Engineering and Department of Materials Science and Engineering, Faculty of Engineering, Monash University, Melbourne, VIC 3800, Australia;
4. Department of Anatomy and Developmental Biology, Faculty of Medicine, Monash University, Melbourne, VIC 3800, Australia.
Received 2019-11-25; Accepted 2020-2-19; Published 2020-4-6
Abstract

In tumor engineering, 3D approaches are used to model components of the tumor microenvironment and to test new treatments. Pancreatic cancers are a cancer of substantial unmet need and survival rates are lower compared to any other cancer. Bioengineering techniques are increasingly applied to understand the unique biology of pancreatic tumors and to design patient-specific models. Here we summarize how extracellular and cellular elements of the pancreatic tumor microenvironment and their interactions have been studied in 3D cell cultures. We review selected clinical trials, assess the benefits of therapies interfering with the tumor microenvironment and address their limitations and future perspectives.
Introduction
Pancreatic cancer is one of the deadliest cancer and less than 9% of patients will survive for 5 years [1]. Despite decades of research, this statistic has remained unchanged and emphasizes its substantial unmet need. Pancreatic cancer is predicted to be the second cause of cancer-related death by 2030 [2]. One reason for the high mortality of this disease is its asymptomatic early stages. Hence, the majority of patients are presented at the time of diagnoses at advanced stages, when the tumor has progressed and treatment options are very limited [3]. In comparison to other malignant diseases, such as melanoma [4], lung cancer [5] and breast cancer [6], that have witnessed the implementation of targeted therapies or immunotherapies, systemic chemotherapy remains the standard treatment of metastatic pancreatic cancer. Advances have been made in our understanding of the genetic drivers of this disease. Yet, a therapeutic breakthrough in pancreatic cancer treatment is still missing [7].
Only 6.7% of oncology drugs in clinical development entering phase I achieve approval by the US Food and Drug Association [8]. One reason for the lack of success of progressing from phase I is the inappropriate biodistribution and off-target toxicities observed with oncology drugs in patients. At the preclinical stage, 2D cell culture, animal and xenograft approaches are the most popular systems [9]. However, these approaches fail to reflect the human tumor microenvironment (TME) and its molecular components accurately and can lead to non-translatable results [10, 11].
In recent years, the possibility of mimicking the TME in vitro by using tumor cells combined with a matrix or scaffold has helped researchers to narrow the uncertainty of the effectiveness of tested compounds [12]. These TME in vitro models have been shown superior to 2D cell culture approaches, allowing 3D culture of multiple cell populations, cell-matrix interactions, treatment responses and the tumor heterogeneity as seen in patients diagnosed with pancreatic cancer [13-15].
The most common type of pancreatic cancer is pancreatic ductal adenocarcinoma (PDAC) [16]. PDAC tissues have a high stroma content, which accounts for up to 90% of the total tumor volume [17-20]. Extracellular matrix (ECM) components, including collagen and hyaluronic acid (HA), and cellular components, including cancer-associated fibroblasts (CAFs) and immune cells, promote pancreatic cancer cell proliferation, immunosuppression and metastasis through a range of molecular factors [19, 21, 22] (Figure 1). These TME components form a fibrotic area that can block chemotherapeutics from reaching the tumor [23]. The number of therapeutics targeting the pancreatic TME is steadily increasing. Therefore, there is a demand for preclinical models that can accurately mimic the TME as seen in patients [24-29] (Table 1).
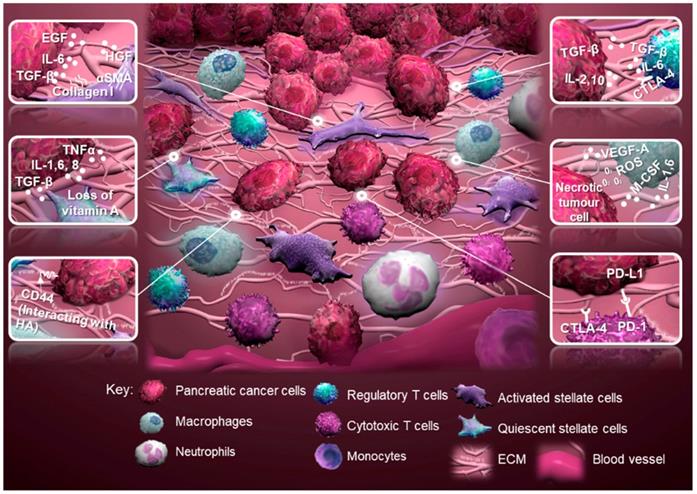
Schematic of the tumor microenvironment of pancreatic cancer and tumor-stromal interactions. Regulatory T cells produce an anti-inflammatory milieu through expression of cytotoxic T-lymphocyte antigen 4 (CTLA-4), while promoting tumor progression through transforming growth factor-beta (TGF-β). Activated stellate cells, characterized by alpha-smooth muscle actin (α-SMA) expression, contribute to tumor progression through multiple factors, for example collagen I, epidermal growth factor (EGF), hepatocyte growth factor (HGF) and expression of CD44 to interact with hyaluronic acid (HA). Quiescent stellate cells lose their capacity to store vitamin A caused by secretion of TGF-β, interleukin 1 (IL-1), IL-6, IL-8 and tumor necrosis factor (TNFα) by cancer and immune cells. Macrophages promote tumor progression through vascular endothelial growth factor-A (VEGF-A), IL-1, IL-6 and macrophage colony-stimulating factor (M-CSF), while increasing the mutational load of the tumor through the expression of reactive oxygen species (ROS). Cytotoxic T cells can be deprived from their tumoricidal activity by expression of CTLA-4 or programmed cell death protein 1 (PD-1; adopted from [19, 21, 22, 130]). Abbreviation: ECM, extracellular matrix.
Selection of 3D approaches that incorporate elements of the tumor microenvironment of pancreatic cancer.
| 3D model | Element of the TME | Cell types | Advantage | Disadvantage | Reference |
|---|---|---|---|---|---|
| Hydrogel (collagen oligomer) | Collagen fibrillar structure | BxPC-3, PANC-1, MIAPaCa-2 | Fibrillar structure used to simulate interstitial matrix | Physical properties, rather soft matrix | [13] |
| Hydrogel, 3D co-culture (collagen/HA, microfluidic culture) | Stroma: PSCs | PANC-1, neonatal human dermal fibroblasts, PSCs | Fusion of three channels into one to form tri-layer patterning of cells and matrix components | Limited to matrix components with compatible crosslinking methods, culture time limited | [14] |
| Spheroids (collagen coating, microfluidic culture) | Continuous perfusion | BxPC-3, PANC-1, MIAPaCa-2 | 24 cell culture regions per device | Dependent on cell spreading and adhesion to collagen | [15] |
| Organoids, 3D co-culture (Matrigel) | Stroma: PSCs, CAFs | Murine and patient- derived pancreatic cancer cells, PSCs, CAFs | Patient-derived multicellular 3D cultures, basement membrane mixture | Physical properties, rather soft matrix | [20] |
| Spheroids, 3D co-culture (modified hanging drop method) | Stroma: PSCs | AsPC-1, BxPC-3, Capan-1, PANC-1, MIAPaCa-2, PSCs | Reproducibility, uniformity | 2:1 ratio of PDAC cells and PSCs, not representative of stroma content | [24] |
| Spheroids, 3D co-culture (gelatin porous microbeads, spinner flask culture) | Stroma: CAFs | PT45, NFs, CAFs | Microbeads provide a scaffold during microtissue formation and matrix production | Spheroid size is dependent on size of microbeads, cells need to be tagged and sorted prior to 3D co-culture for subsequent analysis | [25] |
| Organoids (Matrigel) | Stroma: CAFs, PBMCs | PANC-1, T cells, resected primary and metastatic tumor tissues, ascites, rapid autopsy specimen, murine xenografts, CAFs, PBMCs | Patient-derived multicellular 3D cultures, basement membrane mixture | Physical properties, rather soft matrix | [26] |
| Spheroids, hydrogel, 3D co-culture (type-I collagen, microfluidic culture) | Stroma: PSCs | AsPC-1, PANC-1, MIAPaCa-2, PSCs | Multichannel device with inter-channel cell migration and separation of different cell populations for subsequent analysis | Indirect 3D co-cultures, no direct cell-cell contacts, low cell numbers | [27] |
| Hydrogel, 3D co-culture (type-I collagen, microfluidic culture) | Blood vessel: HUVECs | BxPC-3, PANC-1, murine pancreatic cancer cells, HUVECs | Two-channel device with inter-channel cell invasion and separation of different cell populations for subsequent analysis, analysis of capillary-like networks | Physical properties, rather stiff matrix | [28] |
| Hydrogel (gelatin/HA) | Matrix stiffness | Colo-357 | On-demand matrix stiffening and softening | Effects on remodeling induced by stromal cells, such as CAFs, not included | [47] |
| Hydrogel (polyacrylamide) | Matrix stiffness | AsPC-1, BxPC-3, Suit2-007 | Control and reproducibility of mechanical properties with wide stiffness range (1-25 kPa) | Synthetic material, no biological cues provided | [60] |
| Spheroids, hydrogel, 3D co-culture (type-I collagen, microchannel device) | Stroma: PSCs | PANC-1, patient-derived PSCs | Visualization of collagen fibers and alignment | 1:1 ratio of PDAC cells and PSCs, not representative of stroma content | [82] |
| Hydrogel, 3D co-culture (type-I collagen/Matrigel) | Stroma: PSCs Blood vessel: HUVECs | AsPC-1, Capan-1, Colo-357, primary PSCs, HUVECs | Organotypic multicellular 3D cultures, basement membrane mixture | Physical properties, rather soft matrix | [83] |
| Spheroids, 3D co-culture (poly-HEMA-coated multi-well dishes) | Stroma: CAFs, PBMCs | BxPC-3, HPAC, MIAPaCa-2, Pa-Tu 8902, fetal lung fibroblasts, CAFs, PBMCs | Reproducibility, uniformity | No consideration of physical properties and matrix components | [87] |
Abbreviations: CAFs, cancer-associated fibroblasts; HA, hyaluronic acid; poly-HEMA, poly-2-hydroxyethyl methacrylate; HUVECs, human umbilical vein endothelial cells; NFs, normal fibroblasts; PBMCs, peripheral blood mononuclear cells; PDAC, pancreatic ductal adenocarcinoma; PSCs, pancreatic stellate cells.
3D cell culture approaches
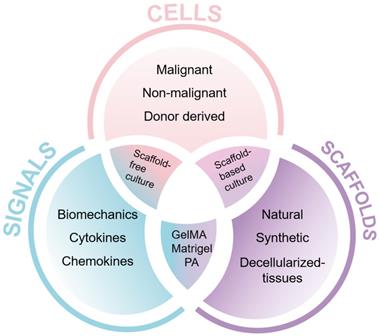
The use of 3D cell cultures is an innovative approach that narrows the gap between traditional 2D cell cultures and animal models. It allows the control and manipulation of individual components of the TME in order to decipher their contribution to disease progression and treatment responses [30]. Genetically engineered mouse models for PDAC, for example the LSL-KrasG12D/+, LSL-p53R172H/+, Pdx1-Cre (KPC) model, are resource-intensive and time-consuming [31]. However, 3D cell cultures permit a validation step prior to animal testing, and when used in combination with animal studies reduce the number of animals needed [32]. Immune cells play a role in tumor progression and response to treatment [33]. 3D approaches allow the inclusion of human immune cells compared to patient-derived xenografts that are established in immunodeficient animals. With the possibility of humanized mouse models, xenograft approaches help us to assess the interaction of human immune cells with human tumors and circumvent difficulties which arise from interspecies differences [34, 35]. When building a physiological mimic of a patient's tumor in the laboratory, the individual cell types [36], biomechanical and molecular signals and the type of scaffold need to be carefully considered (Figure 2).
Key components of 3D cell culture approaches. The three major components to build a 3D cell culture model are cells, signals and scaffolds. Abbreviations: GelMA, gelatin methacryloyl [46]; PA, peptide amphiphile [131].
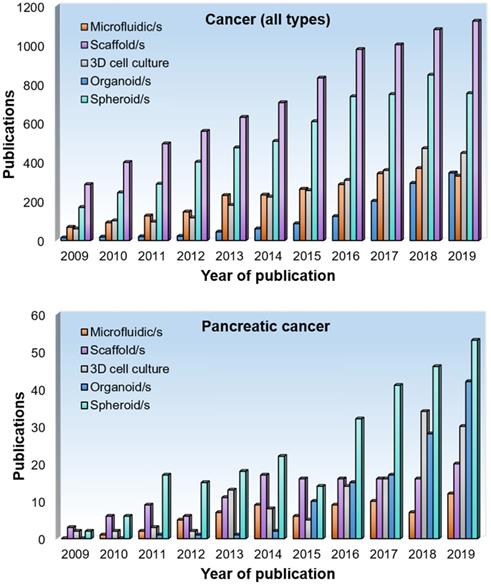
We conducted a literature research on different types of 3D cell culture methods used in cancer research (Figure 3). We found that scaffolds represent the most popular option in terms of the number of papers published per year according to PubMed containing the search terms 'scaffold(s)' and 'cancer', 'tumo(u)r' or 'neoplasm' in the title or abstract, accounting for 37% of our search results. For pancreatic cancer research, we found that spheroids are the most popular method, accounting for 42% of our search results. This is perhaps due to the relatively simple set-up of spheroid cultures and the low numbers of scaffold designs that are specific for this cancer compared to other cancer types. Other popular methods in pancreatic cancer research are organoids (33%), scaffolds (16%) and microfluidic systems (9%). Microfluidic devices arose in the early 2000's and are steadily gaining popularity in cancer research, accounting for 11% of our search results.
Number of publications per year on 3D cell culture approaches in pancreatic cancer research. A literature search was performed using PubMed using the following terms 'microfluidic/s', 'scaffold/s', '3D cell culture', 'organoid/s' and 'spheroid/s' in combination with 'cancer', 'tumor/tumor' or 'neoplasm' in the title or abstract. For the bottom graph, the word 'pancreatic' and 'pancreas' was included.
Spheroids and organoids
Since the development of the hanging drop cell culture technique, spheroids have been used to study morphogenesis and the architecture and composition of malignant tissues [37]. This method includes a coverslip or multi-well plastic lid to suspend cells in a drop of medium. The absence of direct contact with the surface forces cells to aggregate. This aggregation creates a cell cluster with limited oxygen diffusion in the center, leading to a hypoxic or apoptotic area that resembles the chemoresistant core of a tumor [38]. Other traditional methods to grow spheroids from PDAC cells are non-adhesive plastic plates and spinner flask cultures [25]. Bioengineering strategies, such as the use of methylcellulose [38], gelatin/ fibronectin multilayers [39], gelatin/polyvinyl alcohol scaffolds [40] or fibrous gelatin/polyglyconate scaffolds [41], delivered more uniform and reproducible methods.
These spheroid formation methods only partially provide PDAC cells with the characteristic dense stroma, tissue-like or cancer stem cell-like features found in patient tissues. To incorporate stromal components into tumor spheroids, non-malignant cells have been added to PDAC spheroids. Using a modified hanging drop method incorporating methylcellulose, the co-culture of PDAC cells with pancreatic stellate cells (PSCs) led to the production of a desmoplastic reaction, tumor-like cell morphology and tissue architecture [24]. This model had high reproducibility and uniformity, however, spheroids were grown over a time frame of 7-10 days and had a low PSC seeding ratio, which needs to be considered when assessing treatment efficacy.
The surrounding matrix stiffness affects tumor spheroid formation and malignant behavior [42]. Culture methods that allow spheroid growth with a physiological stiffness are crucial in modeling the TME properties of patient tissues. At early tumor stages, PDAC cells adhere on the basement membrane, which is mostly composed of type-IV collagen, fibronectin and laminin [37]. Matrigel is a laminin-rich protein mixture derived from Engelbreth-Holm-Swarm mouse sarcoma and is the gold standard matrix for spheroid and organoid cultures. It has been used to mimic normal as well as diseased pancreatic tissue [43, 44]. Because of its variable biological composition and animal-derived origin, Matrigel has limitations to truly recapitulate the physical and biochemical properties of the TME [45].
Alternatives include hydrogels formed from natural materials, such as collagen, fibrin or alginate, with improved tuneability and control over cell-matrix interactions. Semi-synthetic hydrogels that combine elements of synthetic and natural polymers, such as gelatin methacryloyl (GelMA) [46] or modified gelatin and HA [47], provide a higher control of the mechanical properties of the hydrogel while maintaining the biological cues to mimic cell-matrix interactions (Figure 2).
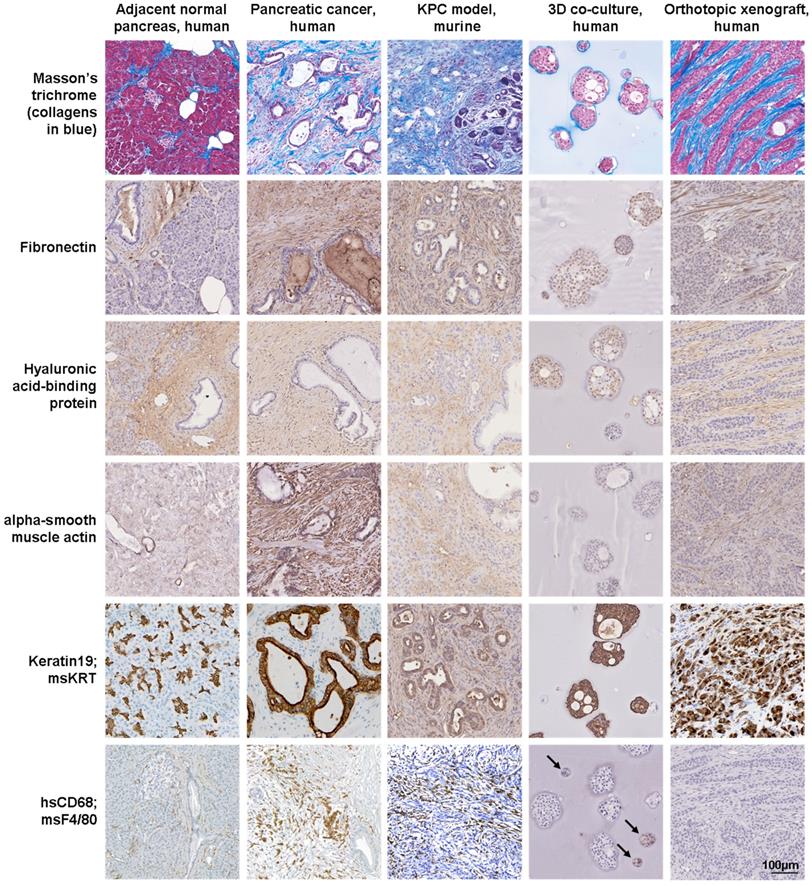
Although still rather ambiguous and disputed, the term organoid mostly refers to a cell cluster capable of self-renewal and self-organization [37]. Organoids are established from normal or malignant tissue fragments, cell lines, embryonic or induced pluripotent stem cells, grown in 3D culture using an animal-derived ECM substitute, typically Matrigel or collagen gels, to produce an organ-like structure [37]. Organoid cultures have been used to study human development and disease, as well as preclinical screening platforms for drug discovery and drug testing and as models of the TME [26, 43, 45]. Spheroids are either self-assembling or are forced to grow as cell clusters or aggregates from a single cell suspension in the absence or presence of exogenous ECM components and recapitulate the 3D structure and organization of tissues or organs. The 3D co-culture of tumor spheroids with non-malignant cells, such as fibroblasts, endothelial or immune cells, has been used as preclinical platform for drug discovery and drug testing and as model of the TME [10, 24, 27, 38]. Over the past decade, both 3D approaches have been increasingly used for pancreatic research (Figure 3). Our group uses tumor spheroids grown embedded within different hydrogel matrices, including collagen and semi-synthetic gels, for the 3D co-culture of PDAC cells with myeloid cells over 14-28 days and as an orthotopic xenograft approach to study the extracellular and cellular components of the pancreatic TME (Figure 4).
Protein expression in pancreatic and xenograft tissues and 3D cell cultures. Masson's trichome and hyaluronic acid-binding protein staining and immunoexpression of fibronectin, alpha-smooth muscle actin (fibroblasts), keratin19 (epithelial cells) and CD68 (macrophages) in normal pancreas and pancreatic cancer tissues, KPC tissues, 3D co-culture of pancreatic cancer cells with myeloid cells and orthotopic xenograft tissues. Human-specific (hs) antibodies were used for human-derived tissue, 3D co-culture (arrows) and xenograft tissue samples, while a mouse-specific (ms) pan-keratin (KRT) antibody and F4/80 were used for KPC tissue samples.
Scaffold-based 3D cell culture
Scaffolds are materials with a 3D architecture of fibers and pores that act as the natural ECM structure where tumor and stromal cells reside, proliferate and migrate. Scaffold-based 3D cell cultures allow cells to grow in a 3D microenvironment without the need for cells to aggregate or form spheroids [30]. With the use of new technologies, for example 3D bioprinting, different types of cells, materials and/or biological factors can be positioned with the scaffolding material in order to generate a tissue-specific TME [48]. Although 3D bioprinting is still in its early stages, it has vast potential for drug discovery due to its spatiotemporal control and automation of 3D cell culture [49, 50]. While 3D bioprinted scaffold-based models for pancreatic cancer have not yet been reported, 3D bioprinting has been used for example to produce models for cervical tumor [51], glioblastoma [52], breast cancer [53] and cancer metastasis [54]. One of the challenges of 3D bioprinting is the development of bioinks. Bioinks are materials that are biocompatible and rapidly crosslinkable at ambient or body temperature without being cytotoxic [55].
Different types of scaffold materials have been used for 3D pancreatic cancer models, for example biocompatible materials, such as soft agar with a layer of Matrigel [56], polymeric scaffolds including a polyvinyl alcohol/gelatin mixture [40] and gelatin [25]. The latter was used for the 3D co-culture of PDAC cells with CAFs, resulting in increased tumor cell proliferation and production of ECM components, such as collagen and glycosaminoglycans. Nonetheless, a major drawback of these scaffold-based cultures is the lack of vascularization, which is crucial for cancer metastasis.
Microfluidic systems
Microfluidic 'organ-on-a-chip' approaches are used to maintain cells in 3D with the addition of perfused microchannels. This serves as a substitute for the vascular perfusion of the body required for the continuous supply of oxygen and nutrients and waste removal in an automated manner [57]. However, the collection of cells at the end of experiments may be challenging and the fixed timescale design does not allow for scale-up cultures.
There are several microfluidic devices for PDAC using polydimethylsiloxane (PDMS) soft lithography [14, 27, 28]. Hydrogels containing type-I collagen and HA were mixed with PDAC cells, fibroblasts or PSCs and loaded as a droplet in the inlets of the device. These 3D co-cultures were maintained for 3 days to allow cell-mediated ECM remodeling and to determine the cells' response to paclitaxel-based chemotherapy [14]. In another study, a collagen-coated cyclic olefin polymer was used to culture PDAC cells, which showed an increased chemoresistance to cisplatin compared to spheroid cultures [15]. To incorporate the physiological hemodynamics experienced by endothelial cells in the TME, a rotatory shearing motion was added. Endothelial cells were separated by a porous polycarbonate membrane from the 3D co-culture of PDAC cells and patient-derived PSCs. Transcriptome analysis revealed that the microfluidic 3D co-cultures were similar to the patient transcriptome [58]. Using a different microfluidic system, pancreatic tumor spheroids were co-cultured with PSCs in a type-I collagen matrix. Each cell type was placed in a different channel separated by a porous PDMS membrane, allowing tumor cell migration towards the channel seeded with PSCs through the porous membrane [27].
It is well-known that cells in the TME sense the surrounding stiffness, which leads to the induction of an epithelial-to-mesenchymal transition (EMT) [59, 60]. Therefore, it is possible that microfluidic cell culture systems based on PDMS and without the inclusion of cytocompatible materials or matrices may alter the behavior of PDAC cells, given that PDAC tissue has a stiffness of 4-8 kPa [60, 61]. To study cell-matrix interactions, the properties and components of the ECM have to be systematically integrated into 3D approaches. We have provided an overview of the advantages and disadvantages of 3D approaches that incorporate key components of the pancreatic TME (Table 1). The different extracellular and cellular components of the pancreatic TME will be discussed in the following sections.
Extracellular components of the pancreatic TME
The ECM of pancreatic tumors is composed of collagens, glycoproteins, proteoglycans, ECM regulators, ECM-affiliated proteins and secreted factors [62], which are released by stromal and tumor cells (Figure 4). The ECM not only contributes to the cellular architecture but also to homeostasis. In PDAC, ECM components impact tumor cell behavior, progression, metastasis, resistance to chemotherapy and are associated with poor clinical outcomes [63, 64].
Collagen and promotion of a mesenchymal phenotype
Collagens are the main components of the PDAC-specific ECM [62]. Type-I collagen is associated with enhanced signaling through the activation of focal adhesion kinase and Smad-interacting protein 1 and a loss of E-cadherin in PDAC cells, which are key factors during EMT [65, 66]. High protein levels of type-I collagen were linked to shorter patient survival (6.4 months) compared to low levels (14.6 months), with expression found in primary PDAC tissues and metastatic lesions [67].
Due to the abundance of collagens in the ECM of PDAC and its implications in tumor biology, 3D cell cultures using collagen gels are widely used to study cell-matrix interactions (Table 1). In one study, type-I collagen was mixed with Matrigel by embedding PDAC cells into hydrogels with different collagen/Matrigel ratios to mimic the interstitial matrix and basement membrane. PDAC cells cultured in Matrigel exhibited an epithelial phenotype, while type-I collagen induced EMT and an invasive phenotype, which was due to an increase in matrix stiffness and fibril density [13]. However, the highest stiffness tested was 1 kPa, which is not close to the reported PDAC tissue stiffness of 4-8 kPa [60, 61]. One solution is to use collagen-like composite materials, for example GelMA, that permit a greater stiffness range [46].
In another study, the 3D culture of PDAC cells in a collagen matrix led to the upregulation of membrane type-I matrix metalloprotease (MT1- MMP), which was associated with resistance to the chemotherapeutic gemcitabine. Importantly, these effects were only seen in 3D not in 2D cell cultures, reinforcing the importance of 3D approaches for drug screening [23].
Cell adhesion proteins and apoptosis resistance
Laminin is abundant in the basement membrane and mediates cell adhesion, while fibronectin is mainly in the interstitial matrix and secreted by PSCs to promote cell adhesion [19]. In a simple study, plastic plates were coated with laminin or fibronectin. Both ECM proteins orchestrated resistance to apoptosis which occurred after cell detachment through cytochrome c-mediated caspase activation. However, the experiments did not include the fibrous collagen matrix of PDAC, in which laminin and fibronectin reside [68]. One advantage of 3D models that integrate ECM proteins is that they facilitate long-term cell cultures. Fibronectin has been shown to maintain survival of T cells isolated from peripheral blood from patients with advanced cancer by stimulating them with anti-CD3 [69]. Laminin and fibronectin also limit early necrosis and apoptosis of PDAC cells [68]. This may explain the correlation between high levels of fibronectin and a larger tumor size seen in patients [70].
Hyaluronic acid as a tumor shield
HA is a non-sulphated glycosaminoglycan secreted by cancer cells and CAFs. PDAC is characterized by an accumulation of HA [71]. Low molecular weight HA has been associated with aggressive and metastatic phenotypes through interaction with CD44, altered cell spreading as well as impaired vascularization and drug delivery [47, 72]. High protein levels of HA were linked to shorter patient survival (9.3 months) compared to low levels (24.3 months), with expression found in primary PDAC tissues and metastatic lesions [67].
To improve drug diffusion, pegylated recombinant human PH20 hyaluronidase (PEGPH20) is being used to deplete HA enzymatically and is currently in clinical trials (Table 2). Studies in animal models showed an increase in survival when using a combination of PEGPH20 and gemcitabine compared to gemcitabine alone due to a normalization of the fluid pressure, reduced vascular collapse and increased permeability. This demonstrates the importance of HA in drug delivery as it may act as a shield that impedes chemotherapy delivery to tumor cells [72].
Selection of ongoing clinical trials that target the tumor microenvironment of pancreatic cancer.
| Trial ID | Target(s) | Therapeutics | Phase | Mode of action | References |
|---|---|---|---|---|---|
| NCT02715804 | ECM | PEGPH20 (HA degradation) + gemcitabine/ nab-paclitaxel | III | A major component of the ECM is HA, which raises the IFP within tumors and reduces drug delivery to malignant cells. PEGPH20 is a compound that degrades HA and normalizes IFP to enhance the delivery of cytotoxic agents. | [85, 104] |
| NCT02436668, completed | Immune cells | Ibrutinib (BTK inhibitor) + Gemcitabine /nab-paclitaxel | III | Bregs, mast cells and macrophages contribute to desmoplasia and an immunosuppressive TME. These three cell populations can be effectively targeted by BTK inhibitors like ibrutinib. | [105] |
| NCT02923921 | Immune cells | AM0010 (activates T cells) + FOLFOX | III | AM0010 is a pegylated form of recombinant human IL-10. Preclinical studies showed that pegylated IL-10 has immunostimulatory effects that induce the activation, proliferation and survival of CD8+ T cells in the TME of PDAC. | [106] |
| NCT03126435 | Tumor endothelial cells | EndoTAG-1 (liposome-embedded paclitaxel) + gemcitabine | III | Tumor endothelial cells lack the glycocalyx of the normal endothelium and therefore become negatively charged. This allows selective attachment and internalization of EndoTAG-1, which contains a positively charged lipid-based complex and leads to enhanced delivery of chemotherapeutic drugs. | [107, 108] |
| NCT03214250 | Immune cells | APX005M (agonistic CD40 mAb) + gemcitabine/ nab-paclitaxel ± nivolumab (anti-PD-1 mAb) | II | CD40 is a costimulatory receptor and mainly found on antigen-presenting cells, in particular B lymphocytes, DCs and macrophages. Binding of CD40 ligands activates these cells, which have a crucial role in activating CTLs. In preclinical models, treatment with APX005M, an agonistic CD40 antibody, is associated with an influx of CTLs into tumors and subsequent tumor regression. A previous phase I study has shown immune activation and that the therapy was well tolerated. | [109, 110] |
| NCT02983578 | Immune cells, PDAC cells, PSCs | AZD9150 (antisense STAT3) + durvalumab (anti-PD-L1 mAb) | II | The role of the transcription factor STAT3 is complex and it has diverse functions in different cell populations of the TME including PDAC cells and PSCs. Inhibition of STAT3 in preclinical models leads to reduced tumor growth and desmoplasia. There is conflicting evidence regarding the role of STAT3 inhibition in immune cells, particularly in the myeloid compartment. | [111, 112] |
| NCT02301130, completed | Immune cells | Mogamulizumab (anti-CCR4 mAb) + durvalumab (anti-PD-L1 mAb) or tremelimumab (anti-CTLA-4 mAb) | II | Tregs have a detrimental effect on anti-tumor immunity. These cells are attracted to the tumor by binding of ligands to CCR4. It has been shown that tremelimumab, an anti-CTLA-4 mAb, can eliminate Tregs in the TME, thus enhancing the effect of the CCR4-inhibitory antibody. | [113] |
| NCT03336216 | Immune cells | Cabiralizumab (anti-CSF1R mAb) + nivolumab (anti-PD-1 mAb) or either investigator's choice chemotherapy | II | Inhibition of CSF1R signaling decreases the population of anti-inflammatory TAMs and furthermore functionally reprograms remaining macrophages to enhance antigen presentation and induce anti-tumor T cell responses in an animal model of PDAC. Investigations of this response revealed that CSF1R blockade also upregulates T cell checkpoint molecules, including PD-L1 and CTLA-4, thereby restraining beneficial therapeutic effects, which suggests a combination with checkpoint blockade. | [88, 114] |
| NCT02907099 | Immune cells | BL-8040 (peptidic CXCR4 antagonist) + pembrolizumab (anti-PD-1 mAb) | II | Activated PSCs secrete CXCL12, a ligand for CXCR4. This attracts CD8+ T cells towards the juxta-tumoral stromal compartment and prevents their access to PDAC cells. In another study, FAP-positive stromal cells were identified as a source of CXCL12. Both studies reported that inhibition of the CXCR4-CXCL12 axis increases the number of intra-tumoral CTLs and improves anti-tumor responses. | [95, 115, 116] |
| NCT02758587 | Immune cells, PDAC cells | Defactinib (FAK inhibitor) + pembrolizumab (anti-PD-1 mAb) | II | Signaling through the protein kinase FAK has been identified as a key pathway in PDAC cells regulating the fibrotic and immunosuppressive TME in PDAC. FAK inhibitors delayed tumor progression that was dependent on the presence of immune cells. A synergistic effect with anti-PD-1/PD-L1 therapy was observed in preclinical models. | [117, 118] |
| NCT03006302 | Immune cells | Epacadostat (IDO inhibitor) + pembrolizumab (anti-PD-1 mAb) + CRS-207 ± GVAX and cyclophosphamide | II | IDO catalyzes the reaction from L-tryptophan to N-formylkynurenine and its overexpression in the TME leads to depletion of this amino acid. As L-tryptophan is essential for metabolic programming of T cells towards Th1 effector cells and natural killer cells functioning, IDO overexpression inhibits anti-tumor immune responses. In this trial, an IDO inhibitor is combined with an anti-PD-1 mAb, anti-cancer vaccines (CRS-207, GVAX) and a potent Treg depleting drug (cyclophosphamide). | [119] |
| NCT02210559 | PSCs | FG-3019 (anti-CTGF mAb) +gemcitabine/ nab-paclitaxel | II | The pleiotropic matricellular signaling protein CTGF plays an important role in the development of desmoplasia by modulating integrin α5β1-dependent adhesion, cell migration, and type-I collagen synthesis. CTGF is overexpressed in PDAC cells and PSCs. Results from preclinical models suggest that the observed anti-neoplastic effect goes beyond enhanced drug delivery. The US Food and Drug Association has granted a fast track designation to pamrevlumab (FG-3019) for the treatment of patients with locally advanced, unresectable pancreatic cancer. | [120-122] |
| NCT03184870 | Immune cells | BMS-813160 (CCR2/CCR5 antagonist) + nivolumab (anti-PD-1 mAb) | I/II | The G-protein coupled receptors CCR2 and CCR5 are expressed on the cell surface of monocytes and macrophages to stimulate their migration and infiltration into tumors. A preclinical study showed that dual targeting of CCR2+ TAMs and CXCR2+ TANs improves anti-tumor immunity and chemotherapeutic response in PDAC compared to either strategy alone. | [123, 124] |
| NCT02807844 | Immune cells | Lacnotuzumab (anti-M-CSF-1 mAb) + spartalizumab (anti-PD-1 mAb) | I/II | TAMs mediate resistance to PD-1 inhibitors via upregulation of several anti-inflammatory mechanisms. These cells can be reduced by inhibiting the M-CSF-1 pathway with lacnotuzumab, a humanized anti-M-CSF-1 mAb, and spartalizumab, a humanized anti-PD-1 mAb, which may have synergistic anti-tumor activity. | [114, 125] |
| NCT03168139 | Immune cells | Olaptesed pegol (CXCL12 inhibitor) ± pembrolizumab (anti-PD-1 mAb) | I/II | Olaptesed pegol blocks a key chemokine in the TME, CXCL12, which is involved in the homeostasis of blood and immune cells. In PDAC, CXCL12 acts as a communication point between tumor cells and the TME. In particular, it confers resistance to checkpoint inhibitors through T cell exclusion in preclinical models. | [95, 116] |
| NCT03307148 | PSCs | ATRA + gemcitabine/ nab-paclitaxel | I | ATRA reduces the ability of PSCs to generate high traction forces, adapt to extracellular mechanical cues and force-mediated ECM remodeling which blocks PDAC cell invasion in 3D organotypic models. | [126, 127] |
| NCT02947165 | Immune cells, PSCs | NIS793 (anti-TGF-β mAb) + PDR001 (anti-PD-1 mAb) | I | The robust desmoplastic reaction that accompanies PDAC progression is caused by TGF-β release from activated macrophages that stimulate PSCs to synthesize collagen type-I and fibronectin. Furthermore, TGF-β attenuates tumor response to PD-L1 blockade by contributing to exclusion of T cells. Synergistic effects of blocking these two pathways have shown promising preclinical results. | [128, 129] |
Abbreviations: AM0010, pegylated human IL-10; ATRA, all-trans retinoic acid; Bregs, B regulatory cells; BTK, Bruton's tyrosine kinase; CCR2, C-C chemokine receptor 2; CCR4, C-C chemokine receptor 4; CCR5, C-C chemokine receptor 5; CD40, cluster of differentiation 40; CSF1R, colony stimulating factor 1 receptor; CTGF, connective tissue growth factor; CTLA-4, cytotoxic T-lymphocyte antigen 4; CTLs, cytotoxic T-lymphocytes; CXCL12, C-X-C chemokine ligand 12; CXCR2, C-X-C chemokine receptor 2; CXCR4, C-X-C chemokine receptor 4; DCs, dendritic cells; ECM, extracellular matrix; FAK, focal adhesion kinase; FAP, fibroblast activation protein; HA, hyaluronic acid; IDO, indoleamine 2,3-dioxygenase; IL-10, interleukin 10; IFP, interstitial fluid pressure; mAb, monoclonal antibody; nab-paclitaxel, nanoparticle albumin-bound paclitaxel; M-CSF1, macrophage colony-stimulating factor 1; PD-1, programmed cell death protein 1; PD-L1, programmed cell death-ligand 1; PDAC, pancreatic ductal adenocarcinoma; PEG, polyethylene glycol; PEGPH20, pegylated recombinant human PH20 hyaluronidase; PSCs, pancreatic stellate cells; STAT3, signal transducer and activator of transcription 3; TAMs, tumor-associated macrophages; TAN, tumor-associated neutrophils; TGF-β, transforming growth factor-beta; Th1, T helper 1; TME, tumor microenvironment; Tregs, T-regulatory cells.
HA promotes mobility and drug resistance of tumor cells through the expression of hyaluronan-mediated motility (RHAMM) and interaction with CD44. The latter is a known stem cell marker and promotes metastasis through the loss of E-cadherin and accumulation of β-catenin. It also induces the expression of the transcription factor NANOG and stem cell regulators, which leads to the activation of the multidrug resistance protein 1 and chemoresistance in CD44-positive cells [73]. Tumor cells expressing high levels of CD44 have been associated with gemcitabine resistance [74]. RHAMM is overexpressed in poorly differentiated PDAC with high metastatic potential [75], indicating a role in epithelial transformation and a migratory phenotype.
A 3D system based on gelatin/HA hybrid hydrogels was developed that can be stiffened on demand using tyrosinase (Table 1). In this study, hydrogels of different stiffness, 1 kPa and 3 kPa, were prepared, which were then used to mimic the stiffness of normal and diseased pancreas tissues, respectively. Soft HA-containing hydrogels inhibited PDAC cell proliferation. In contrast, stiff HA-containing hydrogels promoted cell spreading and migration, which was attributed to EMT-induced changes [47]. These results imply a synergic relationship between HA and stiffness in promoting a malignant cell behavior.
Tumor-promoting effects of proteoglycans
Proteoglycans bind to different ECM components and influence protein activation and inhibition. One of the most commonly overexpressed proteoglycans in PDAC is Sparc/osteonectin, Cwcv and Kazal‐like domains proteoglycan (SPOCK1), which was characterized using co-cultured organoids. PDAC cells and fibroblasts were placed on top of mixed type-I collagen/Matrigel matrices using a 1:2 cell ratio. In response to transforming growth factor-beta (TGF-β), SPOCK1 had tumor-promoting effects by enhancing PDAC cell proliferation and modulation of collagen composition, which was not observed in 2D cell cultures [76].
Lumican is another proteoglycan found in PDAC and stromal tissues [77]. In a retrospective study including PDAC patients, those with lumican-positive tumor cells survived longer than those with lumican- negative cells, whereas patients with lumican-positive stromal tissue had a lower survival than those with lumican-negative stroma. In contrast, when tumor cell monolayers and patient-derived xenografts were exposed to lumican they entered a quiescent state [77]. This illustrates the complexity of PDAC-specific stromal proteins as lumican has both a tumor- suppressing and tumor-promoting role dependent on its location within the TME [78].
Cellular components of the pancreatic TME
Previous research has identified key cellular components that are essential to the tumor biology of PDAC (Figure 4). The matrisome analysis of PDAC tumor tissues revealed that ECM proteins secreted by stromal cells are either positively or negatively correlated with patient survival [62]. By using 3D approaches, the tumor-suppressing functions of stromal cells and therapeutic targets that interfere with their tumor-supporting properties can be explored by manipulating ECM proteins and cells.
Cancer-associated fibroblasts
CAFs have a key role in PDAC development, progression and chemoresistance. They secrete multiple ECM components, forming a dense fibrous matrix characteristic of PDAC. CAFs are a mixed population of cells originating from resident fibroblasts, bone marrow-derived cells and PSCs. PSCs are the most studied fibroblast subtype in pancreatic cancer and have been used in 3D co-cultures with tumor cells [79].
Named after their star-like shape, PSCs are the key factors of the desmoplastic reaction in PDAC. In the normal pancreas, quiescent PSCs are found in the periacinar region and store vitamin A. Quiescent PSCs form only 4-7% of the total cell population [19]. In PDAC, loss of vitamin A, triggered by cytokines secreted by tumor cells, chronically activates PSCs into their myofibroblast-like phenotype and modifies their star-like shape into a spindle shape. Activated PSCs consequently start expressing alpha-smooth muscle actin (α-SMA), type-I collagen, TGF-β and other proteins involved in cell proliferation, migration, ECM remodeling, EMT and inflammation [79].
In a microfluidic co-culture system PSCs were embedded in a type-I collagen matrix to study the distance at which tumor cells trigger their activation. PSC activation occurred over a distance of 1 mm away from tumor cells via secreted factors [27]. Considering the heterogeneity of activated PSCs, the behavior of PSCs in proximity to tumor cells was investigated using co-cultured organoids in a 1:6 ratio of tumor cells to PSCs. The contact-dependent activation of PSCs by PDAC cells resulted in increased α-SMA levels, while paracrine activation led to increased interleukin 6 (IL-6) levels and other pro-inflammatory cytokines [20]. Although PSCs contribute to the tumor biology of PDAC, targeting PSCs and reduction of the stromal compartment led to increased invasive tumor cell behavior in animal models of PDAC [80, 81]. This is evidence of their tumor-modulating function. The use of 3D approaches helps us to understand why therapeutics fail and how to target specific subtypes of PSCs.
Multiple studies reported an increase in the metastatic phenotypes of PDAC cells when co-cultured with PSCs. Using a PDMS microchannel device, PDAC cells were co-cultured with PSCs as multicellular spheroids in a type-I collagen matrix (Table 1). 3D co-cultures promoted the alignment of collagen fibers and enhanced migration of both cell types. While PSCs exhibited F-actin stress fibers, which aligned with the collagen fibers and through activity of Rho-associated kinase, PDAC cells trailed PSCs along the collagen fibers [82]. These findings suggest that PSCs assist tumor cells in their navigation outside the bulk tumor. PSCs also promote angiogenesis by assisting tumor cells to reach the blood vessels and metastasize to distant sites [83].
Endothelial cells
PDAC is known for its poor vascularization which can cause a hypoxic microenvironment. Immature blood vessels are formed and require constant stimulation by the vascular endothelial growth factor (VEGF), which is secreted by PSCs [83]. In contrast, blood vessel maturation was linked to better overall survival and cytotoxic immune cell infiltration in PDAC patients [84].
In the panstromal compartment (non-adjacent to tumor), which surrounds the juxta-tumoral stroma (<100 µm from tumor), a higher density of blood vessels has been observed [83]. This increase in vascularization may be due to the lower endostatin concentration derived from the low number of PDAC cells and the high number of PSCs and macrophages that secrete VEGF to maintain endothelial cell survival. The expression of VEGF can also be triggered by a response to hypoxic conditions [85]. In addition, the dense fibrous stroma and abundant HA content lead to a high interstitial pressure which causes compression of capillaries in PDAC tissues [85]. In the juxta-tumoral stroma, the expression of endostatin, an angiogenic inhibitor derived from type-VIII collagen degradation, causes a hypovascular microenvironment [83]. This is crucial when designing capillary-like networks in 3D cell cultures, as PDAC cells will inhibit vascular growth in a theoretical radius of 100 µm. Of note, PDAC cells and CAFs are known to secrete HA, which can also inhibit vessel formation [72].
The phenomenon of vascular inhibition has been studied using a microfluidic device, where two channels, one containing PDAC cells and the other one containing human umbilical vein endothelial cells (HUVECs), represent pancreatic ducts and blood vessels (Table 1). In proximity to PDAC cells, apoptotic endothelial cells appeared, which was attributed to the activin-ALK7 pathway and endothelial ablation [28].
PDAC cells, PSCs and HUVECs were co-cultured in a mixed type-I collagen/Matrigel matrix. When HUVECs were co-cultured with PDAC cells only, HUVECs survival and sprouting was decreased after 48-72 hours. In contrast, HUVECs co-cultured with activated PSCs formed luminal structures. These structures were suppressed when all-trans retinoic acid (ATRA)-induced quiescent PSCs were used instead of activated PSCs. These results demonstrate a pro- and anti-angiogenic role modulated by the activation of PSCs [83]. ATRA is currently in clinical trials for PDAC and may be an effective stromal modulator used in combination with chemotherapy or immunotherapy (Table 2).
Immune cells
Immunotherapy is achieving remarkable results in some solid tumors, including melanoma [4], lung cancer [5] and breast cancer [6]. Because of the immunosuppressive microenvironment of PDAC and the immunological heterogeneity between patients, it is challenging to study and to treat patients with immunotherapies [86]. Our understanding of the immune landscape of PDAC is constrained by the lack of appropriate experimental systems that can mimic the tumor immune microenvironment. 3D approaches hold great potential in providing tools to research the PDAC-specific immune system, even in a patient- specific manner.
Macrophages
In solid tumors, tumor-associated macrophages (TAMs) constitute the main population of immune cells. In the presence of apoptotic cells, macrophages secrete chemokines and cytokines assisting in the immune response. Monocytes differentiate into two simplified macrophage phenotypes: M1-like, associated with a pro-inflammatory and tumor-suppressing activity, or M2-like, linked to an anti-inflammatory and tumor-promoting activity [87].
3D cell culture models combining monocytes, fibroblasts and cancer cells have been valuable to understand monocyte differentiation into TAMs (Table 1). Tumor spheroids were co-cultured with fibroblasts and monocytes, leading to monocyte- derived macrophages with a M2-like phenotype. This was accompanied by the secretion of different cytokines, chemokines and growth factors by PDAC cells and fibroblasts. Cytokines like IL-6, IL-8 and IL-10 are known for their immunosuppressive role and involvement in M2 differentiation. The addition of macrophages did not influence the proliferation or survival of PDAC cells in the 3D co-cultures [87].
The concept of targeting TAMs arises from their role in angiogenesis, VEGF expression, ECM stiffening and suppression of T cells in PDAC. The influence of colony stimulating factor 1 receptor- positive (CSF1R+) macrophages in the pancreatic TME was studied in the KPC model. Inhibition of CSF1R showed a reduction in tumorigenesis and MYC gene programs. In addition, T cell genes were upregulated, promoting an adaptive immune response [88]. This study demonstrated the profound effect of macrophages in the tumor immune microenvironment and their potential as a target in a clinical setting (Table 2).
Neutrophils
Neutrophils are some of the earliest immune cells recruited to the inflammatory TME and their accumulation is linked to poor prognosis [89]. When tumor-associated neutrophils (TANs) phagocytize, they release reactive oxygen species and cytotoxic factors that impact the surrounding cells in the TME [89]. Similarly to macrophages, neutrophils have a tumor-inhibitory N1-like phenotype and a pro-tumorigenic N2-like phenotype [90].
Spheroid cultures with HUVECs embedded in a type-I collagen matrix were used to study the pro-angiogenic effect of neutrophils. MMP9 added to spheroid cultures stimulated capillary-like networks. Neutrophils secreted MMP9 but not HUVECs or PDAC cells, the latter secreting VEGF instead. Antibodies against VEGF did not reduce the capillary-like networks of MMP9-exposed spheroid cultures, which demonstrates the need to study neutrophil-derived angiogenesis independent of VEGF [91]. Neutrophils form neutrophil extracellular traps and expel their DNA, intracellular proteins and histones into the extracellular space [92]. Neutrophil extracellular traps can also sequester circulating tumor cells and facilitate distant metastasis [93]. These traps are important for inflammation and PDAC growth [92].
Tumor-infiltrating lymphocytes
A focus of immunotherapeutics has been tumor-infiltrating lymphocytes (TILs) in order to boost their cytotoxic effect through checkpoint blockades or with adoptive cell transfer. However, one major difficulty in PDAC is its immunosuppressive microenvironment that hampers the function of cytotoxic CD8+ T cells [94].
Activated PSCs have been associated with sequestration of CD8+ T cells in the panstromal compartment (non-adjacent to tumor), thus preventing T cell migration to the juxta-tumoral stroma (<100 µm from tumor) and isolating them from PDAC cells [95]. This behavior was further investigated using 3D cell cultures. Organoids derived from patient-derived primary and metastatic tumor tissues and matched CAFs were grown in Matrigel. These organoid cultures were then placed in medium with suspended lymphocytes and analyzed for lymphocytic infiltration and migration towards the organoids. This tool may be used to validate immunotherapeutics aiming to improve lymphocyte infiltration into PDAC tissues [26].
The ability to study immunosuppressive targets in PDAC in a patient-specific manner is crucial for the improvement of immunotherapeutic responses. At present, several preclinical models are being used to screen combination strategies with chimeric antigen receptor (CAR) T cells and stroma-targeting therapies. However, these studies mostly use 2D cell cultures and animal models [96]. Bioengineered 3D approaches can advance these studies by enabling real-time analysis of CAR T cell efficacy in a medium to high throughput manner, with tunable physical and biochemical properties and the inclusion of patient-derived immune cells [97].
Conclusions and future perspective
In PDAC, the stroma accounts for most of the tumor volume, which is critical for tumor cell survival, proliferation and metastasis. Here we have discussed several 3D approaches that successfully incorporated key elements of the PDAC-specific TME into 3D cell cultures, including ECM constituents and non-malignant cell types. It is an exciting time for pancreatic cancer research, with the increase of stroma-targeting therapies and patient-specific 3D models that recreate a patient's unique TME. In spite of this, PDAC continues to be a cancer of substantial unmet need, and this is where 3D cell culture models can aid drug discovery and biological therapies.
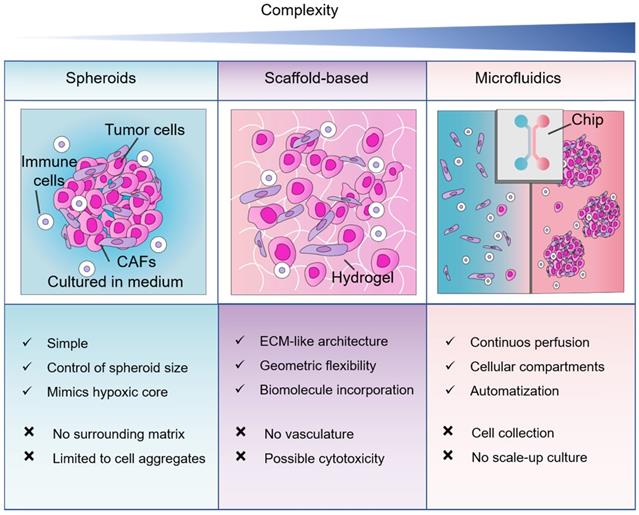
Our PubMed search results demonstrated that spheroids are the most popular 3D method in pancreatic cancer research. Depending on the terminology used, organoid cultures are as popular. In terms of mimicking tumor-stromal interactions, the literature reports a higher expression of ECM molecules in PDAC cells and CAFs in scaffold-based models in comparison to spheroids. Therefore, spheroid models may not entirely represent the ECM features of the pancreatic TME. However, the optimal method will always depend on the research question and the advantages and disadvantages of the 3D cell culture method considered (Figure 5).
Comparison between 3D cell culture approaches for co-culture of non-malignant and pancreatic cancer cells. Three methods of 3D cell cultures, spheroids, scaffold-based and microfluidics in order of complexity. Abbreviations: CAFs, cancer-associated fibroblasts; ECM, extracellular matrix.
The main advantage of bioengineered 3D approaches is their ability to provide a high degree of control and flexibility [30]. The stiffness of the culture matrix can be precisely tuned by using semi-synthetic and synthetic hydrogels and varying their polymer concentration and crosslinking parameters [45]. The stiffness of the PDAC-specific ECM is known to vary with time and space as well as with disease progression, which influences malignant and non-malignant cell behaviors. Nonetheless, a bioengineered 3D pancreatic cancer model with a physiologically relevant stiffness for both the extracellular and cellular components has yet to be developed. One possible reason for the lack of such a 3D approach may be that this area of research is dominated by tumor biologists and not by tissue engineers.
There are still many challenges to overcome in 3D approaches and to establish experimental models for translational research. For example, how to effectively mimic T cell behavior in vitro for 3D PDAC-specific immunotherapy assays or how to mimic the heterogenic vascular network of PDAC with epithelial cells to improve drug delivery. Maybe we can learn from the advances that have been made in 3D bioprinting human tissues. The emergence of 3D bioprinting is based on recent advances in material sciences and polymer chemistry. 3D bioprinting allows the generation of an artificial pancreas for the treatment of type 1 diabetes. Pancreatic islets are manufactured with supporting vascular and immune cells, biomimetic materials and bioactive factors for transplantation [98]. Maybe we can also learn from the advances that have been made in regenerative medicine. Biological scaffolds can be derived from the ECM from decellularized and delipidized human pancreas tissues. After enzymatic digestions, the protein mixture forms a hydrogel allowing for 3D cell cultures [99]. Biomimetic tissue engineering is a powerful approach to generate 3D cancer models. However, only a few scientists use these technologies. We found that most 3D cultures of human PDAC cells utilize soft reconstituted matrices that originate from murine tumors or tissues and contain undefined amounts of ECM proteins and growth factors. Other pitfalls of organoid cultures in Matrigel include a high batch-to-batch variation [100], overstimulation of cellular activity [101] and lack of tagging individual cell types for separate analysis after 3D culture [45], which pose risks for long-term cell expansion and controlled drug testing.
Another challenge is the design and analysis of cellular responses in 3D models and how we use them to understand the stromal response of a tumor and to measure the efficacy of stroma-targeting therapies. For example, model systems used to measure the interstitial fluid pressure and vascular collapse in pancreatic cancer have to be carefully considered depending on the research question asked and subsequent analysis. The physiology of fluid homeostasis in genetically engineered mouse models, xenograft and in vitro approaches, as well as pancreatic tumor tissues presents with different vascular features. Besides interstitial fluid pressure and hyaluronan, solid stress also contributes to the impaired perfusion in pancreatic cancer [12, 102]. In terms of the complex epithelial-stromal interactions, maybe we can use 3D approaches to better understand the molecular mechanisms that are linked to the failure of some stroma-targeting therapies. For example, hedgehog inhibitors had dual activity in clinical trials with tumor-promoting and tumor-suppressing effects. Although hedgehog inhibitors had promising results at the preclinical stage, clinical studies failed to show a benefit and resulted in increased tumor growth and aggressiveness [81, 103].
Future bioengineered 3D approaches with control over patient-specific and biomechanical characteristics of the pancreatic TME have enormous potential to display features of desmoplasia and fibrosis that are important drivers of disease progression and immune escape mechanisms. Hybrid material approaches integrating fibrous scaffolds may recreate the matrix composition and architecture of primary tumor tissues and metastatic lesions. Modifications to these hybrid material approaches will make this new technology platform applicable to other stroma-rich cancers.
Abbreviations
α-SMA: alpha smooth muscle actin; ATRA: all-trans retinoic acid; CAFs: cancer-associated fibroblasts; CAR: chimeric antigen receptor; CSF1R: colony stimulating factor 1 receptor; ECM: extracellular matrix; EGF: epidermal growth factor; EMT: epithelial-to-mesenchymal transition; GelMA: gelatin methacryloyl; HA: hyaluronic acid; HUVECs: human umbilical vein endothelial cells; IL-6: interleukin 6; KPC: LSL-KrasG12D/+: LSL-p53R172H/+: Pdx1-Cre; MT1-MMP: membrane type-I matrix metalloprotease; PDAC: pancreatic ductal adenocarcinoma; PDMS: polydimethylsiloxane; PEGPH20: pegylated recombinant human PH20 hyaluronidase; RHAMM: receptor for hyaluronan-mediated motility; PSCs: pancreatic stellate cells; SPOCK1: Sparc/osteonectin: Cwcv and Kazal‐like domains proteoglycan; TAMs: tumor-associated macrophages; TANs: tumor-associated neutrophils; TGF-β: transforming growth factor-beta; TILs: tumor-infiltrating lymphocytes; TME: tumor microenvironment; VEGF: vascular endothelial growth factor.
Acknowledgements
The authors acknowledge the Barts Pancreas Tissue Bank for providing the patient-derived samples (REC 18/SC/0630) and the staff of the pathology and microscopy core facilities and animal housing facility (license number PBE3719B3).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7-34
2. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913-21
3. Wolfgang CL, Herman JM, Laheru DA, Klein AP, Erdek MA, Fishman EK. et al. Recent progress in pancreatic cancer. CA Cancer J Clin. 2013;63:318-48
4. Eggermont AMM, Blank CU, Mandala M, Long GV, Atkinson V, Dalle S. et al. Adjuvant Pembrolizumab versus Placebo in Resected Stage III Melanoma. N Engl J Med. 2018;378:1789-801
5. Horn L, Mansfield AS, Szczesna A, Havel L, Krzakowski M, Hochmair MJ. et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N Engl J Med. 2018;379:2220-9
6. Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H. et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med. 2018;379:2108-21
7. Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC. et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47-52
8. Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J. Clinical development success rates for investigational drugs. Nat Biotechnol. 2014;32:40-51
9. Weeber F, Ooft SN, Dijkstra KK, Voest EE. Tumor Organoids as a Pre-clinical Cancer Model for Drug Discovery. Cell Chem Biol. 2017;24:1092-100
10. Barros AS, Costa EC, Nunes AS, de Melo-Diogo D, Correia IJ. Comparative study of the therapeutic effect of Doxorubicin and Resveratrol combination on 2D and 3D (spheroids) cell culture models. Int J Pharm. 2018;551:76-83
11. Ben-David U, Ha G, Tseng YY, Greenwald NF, Oh C, Shih J. et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat Genet. 2017;49:1567-75
12. Mitchell MJ, Jain RK, Langer R. Engineering and physical sciences in oncology: challenges and opportunities. Nat Rev Cancer. 2017;17:659-75
13. Puls TJ, Tan X, Whittington CF, Voytik-Harbin SL. 3D collagen fibrillar microstructure guides pancreatic cancer cell phenotype and serves as a critical design parameter for phenotypic models of EMT. PLoS One. 2017;12:e0188870
14. Drifka CR, Eliceiri KW, Weber SM, Kao WJ. A bioengineered heterotypic stroma-cancer microenvironment model to study pancreatic ductal adenocarcinoma. Lab Chip. 2013;13:3965-75
15. Beer M, Kuppalu N, Stefanini M, Becker H, Schulz I, Manoli S. et al. A novel microfluidic 3D platform for culturing pancreatic ductal adenocarcinoma cells: comparison with in vitro cultures and in vivo xenografts. Sci Rep. 2017;7:1325
16. Kleeff J, Korc M, Apte M, La Vecchia C, Johnson CD, Biankin AV. et al. Pancreatic cancer. Nat Rev Dis Primers. 2016;2:16022
17. Neesse A, Algul H, Tuveson DA, Gress TM. Stromal biology and therapy in pancreatic cancer: a changing paradigm. Gut. 2015;64:1476-84
18. Djurec M, Grana O, Lee A, Troule K, Espinet E, Cabras L. et al. Saa3 is a key mediator of the protumorigenic properties of cancer-associated fibroblasts in pancreatic tumors. PNAS. 2018;115:E1147-E56
19. Veenstra VL, Garcia-Garijo A, van Laarhoven HW, Bijlsma MF. Extracellular Influences: Molecular Subclasses and the Microenvironment in Pancreatic Cancer. Cancers. 2018;10:E34
20. Ohlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M. et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. 2017;214:579-96
21. Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK. et al. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front Pharmacol. 2017;8:561
22. Whatcott C, Han H, Posner RG, Von Hoff DD. Tumor-stromal interactions in pancreatic cancer. Crit Rev Oncog. 2013;18:135-51
23. Dangi-Garimella S, Krantz SB, Barron MR, Shields MA, Heiferman MJ, Grippo PJ. et al. Three-dimensional collagen I promotes gemcitabine resistance in pancreatic cancer through MT1-MMP-mediated expression of HMGA2. Cancer Res. 2011;71:1019-28
24. Ware MJ, Keshishian V, Law JJ, Ho JC, Favela CA, Rees P. et al. Generation of an in vitro 3D PDAC stroma rich spheroid model. Biomaterials. 2016;108:129-42
25. Brancato V, Comunanza V, Imparato G, Cora D, Urciuolo F, Noghero A. et al. Bioengineered tumoral microtissues recapitulate desmoplastic reaction of pancreatic cancer. Acta Biomater. 2017;49:152-66
26. Tsai S, McOlash L, Palen K, Johnson B, Duris C, Yang Q. et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer. 2018;18:335
27. Lee JH, Kim SK, Khawar IA, Jeong SY, Chung S, Kuh HJ. Microfluidic co-culture of pancreatic tumor spheroids with stellate cells as a novel 3D model for investigation of stroma-mediated cell motility and drug resistance. J Exp Clin Cancer Res. 2018;37:4
28. Nguyen DT, Lee E, Alimperti S, Norgard RJ, Wong A, Lee JJ. et al. A biomimetic pancreatic cancer on-chip reveals endothelial ablation via ALK7 signaling. Sci Adv. 2019;5:eaav6789
29. Hundeyin M, Kurz E, Mishra A, Rossi JAK, Liudahl SM, Leis KR. et al. Innate alphabeta T Cells Mediate Antitumor Immunity by Orchestrating Immunogenic Macrophage Programming. Cancer Discov. 2019;9:1288-305
30. Loessner D, Rockstroh A, Shokoohmand A, Holzapfel BM, Wagner F, Baldwin J. et al. A 3D tumor microenvironment regulates cell proliferation, peritoneal growth and expression patterns. Biomaterials. 2019;190-191:63-75
31. Coleman SJ, Watt J, Arumugam P, Solaini L, Carapuca E, Ghallab M. et al. Pancreatic cancer organotypics: High throughput, preclinical models for pharmacological agent evaluation. World J Gastroenterol. 2014;20:8471-81
32. Ishiguro S, Kawabata A, Zulbaran-Rojas A, Monson K, Uppalapati D, Ohta N. et al. Co-treatment with a C1B5 peptide of protein kinase Cgamma and a low dose of gemcitabine strongly attenuated pancreatic cancer growth in mice through T cell activation. Biochem Biophys Res Commun. 2018;495:962-8
33. Qiu W, Su GH. Challenges and advances in mouse modeling for human pancreatic tumorigenesis and metastasis. Cancer Metastasis Rev. 2013;32:83-107
34. Wagner F, Holzapfel BM, McGovern JA, Shafiee A, Baldwin JG, Martine LC. et al. Humanization of bone and bone marrow in an orthotopic site reveals new potential therapeutic targets in osteosarcoma. Biomaterials. 2018;171:230-46
35. Herndler-Brandstetter D, Shan L, Yao Y, Stecher C, Plajer V, Lietzenmayer M. et al. Humanized mouse model supports development, function, and tissue residency of human natural killer cells. PNAS. 2017;114:E9626-E34
36. Gradiz R, Silva HC, Carvalho L, Botelho MF, Mota-Pinto A. MIA PaCa-2 and PANC-1 - pancreas ductal adenocarcinoma cell lines with neuroendocrine differentiation and somatostatin receptors. Sci Rep. 2016;6:21648
37. Simian M, Bissell MJ. Organoids: A historical perspective of thinking in three dimensions. J Cell Biol. 2017;216:31-40
38. Longati P, Jia X, Eimer J, Wagman A, Witt MR, Rehnmark S. et al. 3D pancreatic carcinoma spheroids induce a matrix-rich, chemoresistant phenotype offering a better model for drug testing. BMC Cancer. 2013;13:95
39. Hosoya H, Kadowaki K, Matsusaki M, Cabral H, Nishihara H, Ijichi H. et al. Engineering fibrotic tissue in pancreatic cancer: a novel three-dimensional model to investigate nanoparticle delivery. Biochem Biophys Res Commun. 2012;419:32-7
40. Ricci C, Mota C, Moscato S, D'Alessandro D, Ugel S, Sartoris S. et al. Interfacing polymeric scaffolds with primary pancreatic ductal adenocarcinoma cells to develop 3D cancer models. Biomatter. 2014;4:e955386
41. He Q, Wang X, Zhang X, Han H, Han B, Xu J. et al. A tissue-engineered subcutaneous pancreatic cancer model for antitumor drug evaluation. Int J Nanomedicine. 2013;8:1167-76
42. Laklai H, Miroshnikova YA, Pickup MW, Collisson EA, Kim GE, Barrett AS. et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nature Med. 2016;22:497-505
43. Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V. et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160:324-38
44. Tiriac H, Belleau P, Engle DD, Plenker D, Deschenes A, Somerville TDD. et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov. 2018;8:1112-29
45. Gjorevski N, Sachs N, Manfrin A, Giger S, Bragina ME, Ordonez-Moran P. et al. Designer matrices for intestinal stem cell and organoid culture. Nature. 2016;539:560-4
46. Loessner D, Meinert C, Kaemmerer E, Martine LC, Yue K, Levett PA. et al. Functionalization, preparation and use of cell-laden gelatin methacryloyl-based hydrogels as modular tissue culture platforms. Nat Protoc. 2016;11:727-46
47. Liu HY, Korc M, Lin CC. Biomimetic and enzyme-responsive dynamic hydrogels for studying cell-matrix interactions in pancreatic ductal adenocarcinoma. Biomaterials. 2018;160:24-36
48. Albritton JL, Miller JS. 3D bioprinting: improving in vitro models of metastasis with heterogeneous tumor microenvironments. Dis Model Mech. 2017;10:3-14
49. Knowlton S, Onal S, Yu CH, Zhao JJ, Tasoglu S. Bioprinting for cancer research. Trends Biotechnol. 2015;33:504-13
50. Pati F, Gantelius J, Svahn HA. 3D Bioprinting of Tissue/Organ Models. Angew Chem Int Ed Engl. 2016;55:4650-65
51. Zhao Y, Yao R, Ouyang L, Ding H, Zhang T, Zhang K. et al. Three-dimensional printing of Hela cells for cervical tumor model in vitro. Biofabrication. 2014;6:035001
52. Tabriz AG, Hermida MA, Leslie NR, Shu W. Three-dimensional bioprinting of complex cell laden alginate hydrogel structures. Biofabrication. 2015;7:045012
53. Grolman JM, Zhang D, Smith AM, Moore JS, Kilian KA. Rapid 3D Extrusion of Synthetic Tumor Microenvironments. Adv Mater. 2015;27:5512-7
54. Zhou X, Zhu W, Nowicki M, Miao S, Cui H, Holmes B. et al. 3D Bioprinting a Cell-Laden Bone Matrix for Breast Cancer Metastasis Study. ACS Appl Mater Interfaces. 2016;8:30017-26
55. Jungst T, Smolan W, Schacht K, Scheibel T, Groll J. Strategies and Molecular Design Criteria for 3D Printable Hydrogels. Chem Rev. 2016;116:1496-539
56. Sempere LF, Gunn JR, Korc M. A novel 3-dimensional culture system uncovers growth stimulatory actions by TGFbeta in pancreatic cancer cells. Cancer Biol Ther. 2011;12:198-207
57. Hassell BA, Goyal G, Lee E, Sontheimer-Phelps A, Levy O, Chen CS. et al. Human Organ Chip Models Recapitulate Orthotopic Lung Cancer Growth, Therapeutic Responses, and Tumor Dormancy In Vitro. Cell Rep. 2017;21:508-16
58. Gioeli D, Snow CJ, Simmers MB, Hoang SA, Figler RA, Allende JA. et al. Development of a multicellular pancreatic tumor microenvironment system using patient-derived tumor cells. Lab Chip. 2019;19:1193-204
59. Alonso-Nocelo M, Raimondo TM, Vining KH, Lopez-Lopez R, de la Fuente M, Mooney DJ. Matrix stiffness and tumor-associated macrophages modulate epithelial to mesenchymal transition of human adenocarcinoma cells. Biofabrication. 2018;10:035004
60. Rice AJ, Cortes E, Lachowski D, Cheung BCH, Karim SA, Morton JP. et al. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis. 2017;6:e352
61. Rubiano A, Delitto D, Han S, Gerber M, Galitz C, Trevino J. et al. Viscoelastic properties of human pancreatic tumors and in vitro constructs to mimic mechanical properties. Acta Biomater. 2018;67:331-40
62. Tian C, Clauser KR, Ohlund D, Rickelt S, Huang Y, Gupta M. et al. Proteomic analyses of ECM during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. PNAS. 2019;116:19609-18
63. Zhang H, Pan YZ, Cheung M, Cao M, Yu C, Chen L. et al. LAMB3 mediates apoptotic, proliferative, invasive, and metastatic behaviors in pancreatic cancer by regulating the PI3K/Akt signaling pathway. Cell Death Dis. 2019;10:230
64. Amrutkar M, Aasrum M, Verbeke CS, Gladhaug IP. Secretion of fibronectin by human pancreatic stellate cells promotes chemoresistance to gemcitabine in pancreatic cancer cells. BMC Cancer. 2019;19:596
65. Koenig A, Mueller C, Hasel C, Adler G, Menke A. Collagen type I induces disruption of E-cadherin-mediated cell-cell contacts and promotes proliferation of pancreatic carcinoma cells. Cancer Res. 2006;66:4662-71
66. Imamichi Y, Konig A, Gress T, Menke A. Collagen type I-induced Smad-interacting protein 1 expression downregulates E-cadherin in pancreatic cancer. Oncogene. 2007;26:2381-5
67. Whatcott CJ, Diep CH, Jiang P, Watanabe A, LoBello J, Sima C. et al. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancer. Clin Cancer Res. 2015;21:3561-8
68. Vaquero EC, Edderkaoui M, Nam KJ, Gukovsky I, Pandol SJ, Gukovskaya AS. Extracellular matrix proteins protect pancreatic cancer cells from death via mitochondrial and nonmitochondrial pathways. Gastroenterology. 2003;125:1188-202
69. Ishikawa T, Kokura S, Enoki T, Sakamoto N, Okayama T, Ideno M. et al. Phase I clinical trial of fibronectin CH296-stimulated T cell therapy in patients with advanced cancer. PLoS One. 2014;9:e83786
70. Hu D, Ansari D, Zhou Q, Sasor A, Said Hilmersson K, Andersson R. Stromal fibronectin expression in patients with resected pancreatic ductal adenocarcinoma. World J Surg Oncol. 2019;17:29
71. Gebauer F, Kemper M, Sauter G, Prehm P, Schumacher U. Is hyaluronan deposition in the stroma of pancreatic ductal adenocarcinoma of prognostic significance? PLoS One. 2017;12:e0178703
72. Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK. et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62:112-20
73. Hong SP, Wen J, Bang S, Park S, Song SY. CD44-positive cells are responsible for gemcitabine resistance in pancreatic cancer cells. Int J Cancer. 2009;125:2323-31
74. Zhao S, Chen C, Chang K, Karnad A, Jagirdar J, Kumar AP. et al. CD44 Expression Level and Isoform Contributes to Pancreatic Cancer Cell Plasticity, Invasiveness, and Response to Therapy. Clin Cancer Res. 2016;22:5592-604
75. Cheng XB, Sato N, Kohi S, Koga A, Hirata K. Receptor for Hyaluronic Acid-Mediated Motility is Associated with Poor Survival in Pancreatic Ductal Adenocarcinoma. J Cancer. 2015;6:1093-8
76. Veenstra VL, Damhofer H, Waasdorp C, Steins A, Kocher HM, Medema JP. et al. Stromal SPOCK1 supports invasive pancreatic cancer growth. Mol Oncol. 2017;11:1050-64
77. Li X, Kang Y, Roife D, Lee Y, Pratt M, Perez MR. et al. Prolonged exposure to extracellular lumican restrains pancreatic adenocarcinoma growth. Oncogene. 2017;36:5432-8
78. Ishiwata T, Cho K, Kawahara K, Yamamoto T, Fujiwara Y, Uchida E. et al. Role of lumican in cancer cells and adjacent stromal tissues in human pancreatic cancer. Oncol Rep. 2007;18:537-43
79. Nielsen MF, Mortensen MB, Detlefsen S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J Gastroenterol. 2016;22:2678-700
80. Lee JJ, Perera RM, Wang H, Wu DC, Liu XS, Han S. et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. PNAS. 2014;111:E3091-100
81. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA. et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735-47
82. Drifka CR, Loeffler AG, Esquibel CR, Weber SM, Eliceiri KW, Kao WJ. Human pancreatic stellate cells modulate 3D collagen alignment to promote the migration of pancreatic ductal adenocarcinoma cells. Biomed Microdevices. 2016;18:105
83. Di Maggio F, Arumugam P, Delvecchio FR, Batista S, Lechertier T, Hodivala-Dilke K. et al. Pancreatic stellate cells regulate blood vessel density in the stroma of pancreatic ductal adenocarcinoma. Pancreatology. 2016;16:995-1004
84. Katsuta E, Qi Q, Peng X, Hochwald SN, Yan L, Takabe K. Pancreatic adenocarcinomas with mature blood vessels have better overall survival. Sci Rep. 2019;9:1310
85. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418-29
86. Johansson H, Andersson R, Bauden M, Hammes S, Holdenrieder S, Ansari D. Immune checkpoint therapy for pancreatic cancer. World J Gastroenterol. 2016;22:9457-76
87. Kuen J, Darowski D, Kluge T, Majety M. Pancreatic cancer cell/fibroblast co-culture induces M2 like macrophages that influence therapeutic response in a 3D model. PLoS One. 2017;12:e0182039
88. Candido JB, Morton JP, Bailey P, Campbell AD, Karim SA, Jamieson T. et al. CSF1R(+) Macrophages Sustain Pancreatic Tumor Growth through T Cell Suppression and Maintenance of Key Gene Programs that Define the Squamous Subtype. Cell Rep. 2018;23:1448-60
89. Wang Y, Fang T, Huang L, Wang H, Zhang L, Wang Z. et al. Neutrophils infiltrating pancreatic ductal adenocarcinoma indicate higher malignancy and worse prognosis. Biochem Biophys Res Commun. 2018;501:313-9
90. Shaul ME, Levy L, Sun J, Mishalian I, Singhal S, Kapoor V. et al. Tumor-associated neutrophils display a distinct N1 profile following TGFbeta modulation: A transcriptomics analysis of pro- vs. antitumor TANs. Oncoimmunology. 2016;5:e1232221
91. Bausch D, Pausch T, Krauss T, Hopt UT, Fernandez-del-Castillo C, Warshaw AL. et al. Neutrophil granulocyte derived MMP-9 is a VEGF independent functional component of the angiogenic switch in pancreatic ductal adenocarcinoma. Angiogenesis. 2011;14:235-43
92. Boone BA, Orlichenko L, Schapiro NE, Loughran P, Gianfrate GC, Ellis JT. et al. The receptor for advanced glycation end products (RAGE) enhances autophagy and neutrophil extracellular traps in pancreatic cancer. Cancer Gene Ther. 2015;22:326-34
93. Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B. et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest. 2013 67484
94. Thind K, Padrnos LJ, Ramanathan RK, Borad MJ. Immunotherapy in pancreatic cancer treatment: a new frontier. Therap Adv Gastroenterol. 2017;10:168-94
95. Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC. et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology. 2013;145:1121-32
96. DeSelm CJ, Tano ZE, Varghese AM, Adusumilli PS. CAR T-cell therapy for pancreatic cancer. J Surg Oncol. 2017;116:63-74
97. Hickey JW, Dong Y, Chung JW, Salathe SF, Pruitt HC, Li X. et al. Engineering an Artificial T-Cell Stimulating Matrix for Immunotherapy. Adv Mater. 2019;31:e1807359
98. Kim J, Kang K, Drogemuller CJ, Wallace GG, Coates PT. Bioprinting an Artificial Pancreas for Type 1 Diabetes. Curr Diab Rep. 2019;19:53
99. Sackett SD, Tremmel DM, Ma F, Feeney AK, Maguire RM, Brown ME. et al. Extracellular matrix scaffold and hydrogel derived from decellularized and delipidized human pancreas. Sci Rep. 2018;8:10452
100. Hughes CS, Postovit LM, Lajoie GA. Matrigel: a complex protein mixture required for optimal growth of cell culture. Proteomics. 2010;10:1886-90
101. Vukicevic S, Kleinman HK, Luyten FP, Roberts AB, Roche NS, Reddi AH. Identification of multiple active growth factors in basement membrane Matrigel suggests caution in interpretation of cellular activity related to extracellular matrix components. Exp Cell Res. 1992;202:1-8
102. DuFort CC, DelGiorno KE, Hingorani SR. Mounting Pressure in the Microenvironment: Fluids, Solids, and Cells in Pancreatic Ductal Adenocarcinoma. Gastroenterology. 2016;150:1545-57 e2
103. Raleigh DR, Reiter JF. Misactivation of Hedgehog signaling causes inherited and sporadic cancers. J Clin Invest. 2019;129:465-75
104. Hingorani SR, Zheng L, Bullock AJ, Seery TE, Harris WP, Sigal DS. et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients With Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J Clin Oncol. 2018;36:359-66
105. Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B. et al. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-talk Drives Pancreas Cancer. Cancer Discov. 2016;6:270-85
106. Batchu RB, Gruzdyn OV, Mahmud EM, Chukr F, Dachepalli R, Manmari SK. et al. Inhibition of Interleukin-10 in the tumor microenvironment can restore mesothelin chimeric antigen receptor T cell activity in pancreatic cancer in vitro. Surgery. 2018;163:627-32
107. Fasol U, Frost A, Buchert M, Arends J, Fiedler U, Scharr D. et al. Vascular and pharmacokinetic effects of EndoTAG-1 in patients with advanced cancer and liver metastasis. Ann Oncol. 2012;23:1030-6
108. Eichhorn ME, Ischenko I, Luedemann S, Strieth S, Papyan A, Werner A. et al. Vascular targeting by EndoTAG-1 enhances therapeutic efficacy of conventional chemotherapy in lung and pancreatic cancer. Int J Cancer. 2010;126:1235-45
109. Beatty GL, Winograd R, Evans RA, Long KB, Luque SL, Lee JW. et al. Exclusion of T Cells From Pancreatic Carcinomas in Mice Is Regulated by Ly6C(low) F4/80(+) Extratumoral Macrophages. Gastroenterology. 2015;149:201-10
110. Winograd R, Byrne KT, Evans RA, Odorizzi PM, Meyer AR, Bajor DL. et al. Induction of T-cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunol Res. 2015;3:399-411
111. Kumar V, Cheng P, Condamine T, Mony S, Languino LR, McCaffrey JC. et al. CD45 Phosphatase Inhibits STAT3 Transcription Factor Activity in Myeloid Cells and Promotes Tumor-Associated Macrophage Differentiation. Immunity. 2016;44:303-15
112. Nagathihalli NS, Castellanos JA, Shi C, Beesetty Y, Reyzer ML, Caprioli R. et al. Signal Transducer and Activator of Transcription 3, Mediated Remodeling of the Tumor Microenvironment Results in Enhanced Tumor Drug Delivery in a Mouse Model of Pancreatic Cancer. Gastroenterology. 2015;149:1932-43 e9
113. Rapp M, Grassmann S, Chaloupka M, Layritz P, Kruger S, Ormanns S. et al. C-C chemokine receptor type-4 transduction of T cells enhances interaction with dendritic cells, tumor infiltration and therapeutic efficacy of adoptive T cell transfer. Oncoimmunology. 2016;5:e1105428
114. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J. et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74:5057-69
115. Morimoto M, Matsuo Y, Koide S, Tsuboi K, Shamoto T, Sato T. et al. Enhancement of the CXCL12/CXCR4 axis due to acquisition of gemcitabine resistance in pancreatic cancer: effect of CXCR4 antagonists. BMC Cancer. 2016;16:305
116. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS. et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. PNAS. 2013;110:20212-7
117. Symeonides SN, Anderton SM, Serrels A. FAK-inhibition opens the door to checkpoint immunotherapy in Pancreatic Cancer. J Immunother Cancer. 2017;5:17
118. Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA. et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nature Med. 2016;22:851-60
119. Peng YP, Zhang JJ, Liang WB, Tu M, Lu ZP, Wei JS. et al. Elevation of MMP-9 and IDO induced by pancreatic cancer cells mediates natural killer cell dysfunction. BMC Cancer. 2014;14:738
120. Aikawa T, Gunn J, Spong SM, Klaus SJ, Korc M. Connective tissue growth factor-specific antibody attenuates tumor growth, metastasis, and angiogenesis in an orthotopic mouse model of pancreatic cancer. Mol Cancer Ther. 2006;5:1108-16
121. Neesse A, Frese KK, Bapiro TE, Nakagawa T, Sternlicht MD, Seeley TW. et al. CTGF antagonism with mAb FG-3019 enhances chemotherapy response without increasing drug delivery in murine ductal pancreas cancer. PNAS. 2013;110:12325-30
122. Picozzi VJ, Rocha FG, Helton S, Pishvaian MJ, Jackson PG, Mody K. et al. Randomized, open-label trial of gemcitabine/nab-paclitaxel (G/NP) ± pamrevlumab (P) as neoadjuvant chemotherapy in locally advanced, unresectable pancreatic cancer (LAPC). J Clin Oncol. 2017;35:365 -
123. Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D. et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res. 2013;19:3404-15
124. Nywening TM, Belt BA, Cullinan DR, Panni RZ, Han BJ, Sanford DE. et al. Targeting both tumor-associated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut. 2018;67:1112-23
125. Waghray M, Yalamanchili M, Dziubinski M, Zeinali M, Erkkinen M, Yang H. et al. GM-CSF Mediates Mesenchymal-Epithelial Cross-talk in Pancreatic Cancer. Cancer Discov. 2016;6:886-99
126. Chronopoulos A, Robinson B, Sarper M, Cortes E, Auernheimer V, Lachowski D. et al. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodeling and inhibit cancer cell invasion. Nat Commun. 2016;7:12630
127. Carapuca EF, Gemenetzidis E, Feig C, Bapiro TE, Williams MD, Wilson AS. et al. Anti-stromal treatment together with chemotherapy targets multiple signaling pathways in pancreatic adenocarcinoma. J Pathol. 2016;239:286-96
128. Principe DR, DeCant B, Mascarinas E, Wayne EA, Diaz AM, Akagi N. et al. TGFbeta Signaling in the Pancreatic Tumor Microenvironment Promotes Fibrosis and Immune Evasion to Facilitate Tumorigenesis. Cancer Res. 2016;76:2525-39
129. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y. et al. TGFbeta attenuates tumor response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544-8
130. Canli O, Nicolas AM, Gupta J, Finkelmeier F, Goncharova O, Pesic M. et al. Myeloid Cell-Derived Reactive Oxygen Species Induce Epithelial Mutagenesis. Cancer Cell. 2017;32:869-83 e5
131. Inostroza-Brito KE, Collin E, Siton-Mendelson O, Smith KH, Monge-Marcet A, Ferreira DS. et al. Co-assembly, spatiotemporal control and morphogenesis of a hybrid protein-peptide system. Nat Chem. 2015;7:897-904
Author contact
![]() Corresponding author: Dr. Daniela Loessner. d.loessnerac.uk
Corresponding author: Dr. Daniela Loessner. d.loessnerac.uk